 Maren von Köckritz-Blickwede
Maren von Köckritz-Blickwede Volker Winstel
Volker Winstel- 1Department of Biochemistry, University of Veterinary Medicine Hannover, Hannover, Germany
- 2Research Center for Emerging Infections and Zoonoses (RIZ), University of Veterinary Medicine Hannover, Hannover, Germany
- 3Research Group Pathogenesis of Bacterial Infections, TWINCORE, Centre for Experimental and Clinical Infection Research, a joint venture between the Hannover Medical School and the Helmholtz Centre for Infection Research, Hannover, Germany
- 4Institute of Medical Microbiology and Hospital Epidemiology, Hannover Medical School, Hannover, Germany
NETosis is a multi-facetted cellular process that promotes the formation of neutrophil extracellular traps (NETs). NETs as web-like structures consist of DNA fibers armed with granular proteins, histones, and microbicidal peptides, thereby exhibiting pathogen-immobilizing and antimicrobial attributes that maximize innate immune defenses against invading microbes. However, clinically relevant pathogens often tolerate entrapment and even take advantage of the remnants of NETs to cause persistent infections in mammalian hosts. Here, we briefly summarize how Staphylococcus aureus, a high-priority pathogen and causative agent of fatal diseases in humans as well as animals, catalyzes and concurrently exploits NETs during pathogenesis and recurrent infections. Specifically, we focus on toxigenic and immunomodulatory effector molecules produced by staphylococci that prime NET formation, and further highlight the molecular and underlying principles of suicidal NETosis compared to vital NET-formation by viable neutrophils in response to these stimuli. We also discuss the inflammatory potential of NET-controlled microenvironments, as excessive expulsion of NETs from activated neutrophils provokes local tissue injury and may therefore amplify staphylococcal disease severity in hospitalized or chronically ill patients. Combined with an overview of adaptation and counteracting strategies evolved by S. aureus to impede NET-mediated killing, these insights may stimulate biomedical research activities to uncover novel aspects of NET biology at the host-microbe interface.
Introduction
Polymorphonuclear leukocytes (PMNs or neutrophils) are highly abundant immune cells found in human or animal blood (1). As part of the innate immune response, neutrophils represent crucial effector cells that substantially contribute to immune surveillance and the clearance of microbial infections (1, 2). Of note, neutrophils are recruited in large numbers to infectious foci within minutes following pathogen entry as these cells rapidly sense microbial signatures as well as inflammatory signals released by injured or damaged host tissues (1–3). Specifically, neutrophils express an array of cellular receptors including pattern recognition receptors (PRRs), G protein-coupled receptors (GPCRs), and Fc receptors that mediate recognition of danger signals, thereby governing chemotactic routing and extravasation of PMNs from blood vessel into infected tissues (1, 4). Concurrently, subcellular signaling cascades downstream of these receptors initiate pathogen-eradicating processes that encompass phagocytosis, degranulation, and the biogenesis of reactive oxygen species (ROS) (4). Moreover, neutrophils may kill pathogens by the formation of neutrophil extracellular DNA traps (NETs), an extracellular matrix composed of nuclear and mitochondrial DNA loaded with cell-specific proteases, antimicrobial peptides, and granular proteins (5, 6). This mechanism is of particular importance for controlling infectious diseases as NETs not only display antimicrobial properties, but also exhibit pathogen-capturing features that help to limit the dissemination of microbes in the mammalian host (5, 7). Nevertheless, clinically relevant pathogens have evolved refined counteracting strategies that mediate tolerance or even evasion from extracellular trap-mediated killing (6, 8). A prominent example is Staphylococcus aureus, a deadly and multidrug-resistant Gram-positive bacterium that colonizes approximately 30% of the human population (9, 10). This microbe is a very frequent cause of superficial skin and soft tissue infections as well as life-threatening diseases including sepsis, septic arthritis, endocarditis, or pneumonia (10, 11). Notably, S. aureus infection triggers a conspicuously strong infiltration of neutrophils into infectious foci which is typically coupled with excessive NET-formation and the development of large abscesses (7, 12, 13). Surprisingly though, the antimicrobial repertoire of NETs often fails to promote clearance of persistent S. aureus infections in humans or animal hosts (14–17). On the contrary, S. aureus readily outsmarts neutrophil responses and even exploits NET-formation to kill bystander immune cells, highlighting the cunning lifestyle of this microbe (18, 19).
Herein, we briefly summarize the molecular prerequisites of suicidal NETosis, a distinct form of programmed cell death, and vital NET-formation by viable neutrophils in response to various staphylococcal effector molecules. Particularly, we describe how S. aureus simultaneously provokes and takes advantage of NET formation during acute and relapsing infections. Further, we review adaptation strategies of staphylococci that confer tolerance to NET-mediated entrapment and killing. Finally, we discuss the pathophysiological potential of NET-controlled infectious foci in the context of severe and chronic S. aureus diseases, with the overall aim to stimulate scientific investigations that may lead to the conception of new therapeutic approaches to fight antibiotic-resistant S. aureus and other NET-evading pathogens.
Molecular Mechanisms of Suicidal NETosis and Vital NET Formation
The formation of NETs can be initiated by a variety of stimuli including microorganisms, antibodies, immune complexes, microcrystals, and certain chemicals (6, 20, 21). Various pro-inflammatory cytokines have also been reported to interfere with the release of NETs from PMNs (20, 21). For example, interleukin 1-β (IL-1β), IL-6, IL-8, as well as tumor necrosis factor alpha (TNF-α) elicit NETs and thus may represent crucial immunoregulatory effector molecules of the host that presumably modulate NETosis during specific inflammatory or infectious diseases (5, 20, 22–27).
To date, two predominant pathways have been described in detail that lead to the expulsion of NETs from neutrophils: suicidal (lytic) NETosis, which causes neutrophil cell death, and vital NET formation that results in the release of NET-loaded vesicles from viable PMNs (Figure 1) (21, 28). Suicidal NETosis, a process that provokes NET formation within 3-4 hours post-stimulation (23), largely depends on calcium signaling along with the protein kinase C (PKC)- or Raf-MEK-ERK pathway-dependent activation of the membrane-bound NADPH oxidase, which represents a multisubunit protein complex that synthesizes ROS for the subsequent activation of PAD4 (protein-arginine deiminase type 4) (Figure 1) (6, 21, 23, 29, 30). PAD4 is a Ca2+-binding protein and key driver of NETosis as it causes citrullination of core histone (6, 31). Particularly, activated PAD4 migrates to the nucleus where it catalyzes the conversion of positively charged arginine residues of histones into citrullines, thereby inducing the process of chromatin decondensation (6, 31). Concurrently, ROS break down the azurosome, a serine protease- as well as myeloperoxidase (MPO)-encompassing protein complex found in neutrophil granules (32). In this manner, azurophilic proteases (e.g. azurocidin, cathepsin G, and neutrophil elastase (NE)) as well as MPO are released into the cytosol, where NE disrupts the actin cytoskeleton and simultaneously traffics together with MPO to the nucleus, further contributing to histone degradation and chromatin decondensation (6, 32, 33). Interestingly, recent work demonstrated that mitochondrial ROS (mtROS) can also contribute to the phenotype of suicidal NETosis, particularly in response to S. aureus (34, 35). Subsequent steps involve disruption of the nuclear cell envelope and electrostatic interaction of core-derived decondensed chromatin with cytosolic and granular proteins in the cytoplasm (Figure 1) (23). In this scenario, the human cathelicidin LL-37 and its mouse analogue mCRAMP (mouse cathelicidin related antimicrobial peptide) have been shown to contribute to the perforation of nuclear membranes in NET-forming neutrophils (36). Moreover, emerging literature suggests that NE-processed gasdermin D (GSDM-D), a crucial executor protein of the pyroptotic cell death pathway (37–39), may also alter the membrane integrity of PMNs at this stage (30, 40, 41). Specifically, cleaved GSDM-D displays potent pore-forming and membrane-damaging capacities and is therefore believed to puncture the core as well as the plasma membranes of netting neutrophils, ultimately culminating in the expulsion of antimicrobial NETs from dying PMNs into the extracellular space (Figure 1) (30, 37–41). Of note, processed GSDM-D may further contribute to a non-canonical form of suicidal NETosis, which occurs exquisitely in response to intracellular Gram-negative bacteria (42). In summary, suicidal NETosis of neutrophils differs from other cell death mechanisms based on the specific phenomenon that chromatin decondensation and disintegration of the nuclear membrane occurs concomitant with cytoplasmic granule dissolution, allowing the NET components to mix in the cytoplasm prior to their extracellular release (23).
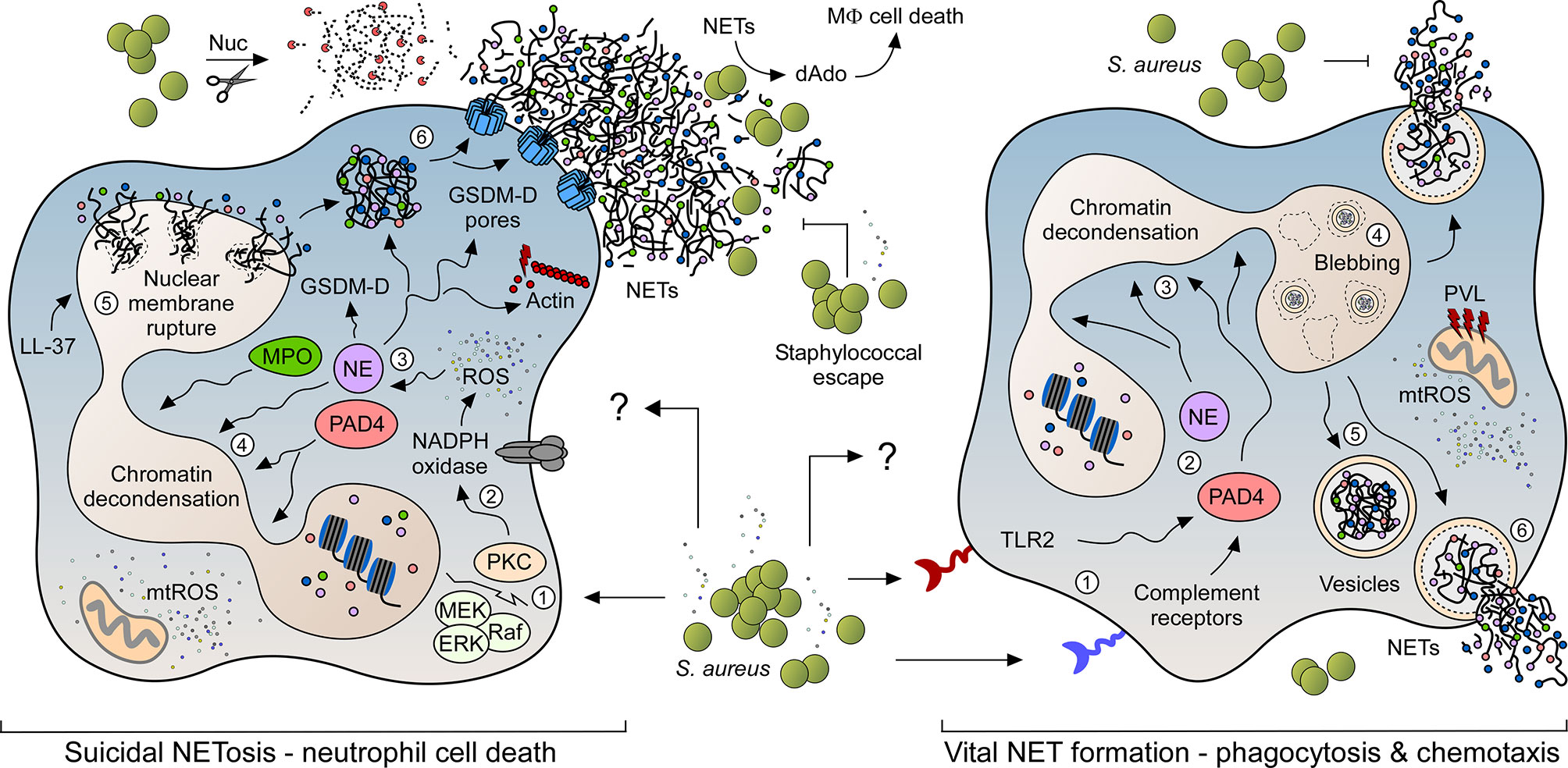
Figure 1 NET formation pathways in response to S. aureus. NET formation in response to live staphylococci and their exoproducts may occur via two predominant signaling pathways. While suicidal NETosis leads to neutrophil cell death (left panel), rapidly occurring vital NET formation retains the ability of PMNs to migrate and phagocytose bacterial invaders (right panel). Numbers 1-6 in each panel indicate the order of events during NET formation. S. aureus readily escapes from NET-mediated entrapment and killing by secreting multiple virulence determinants (e.g. thermonuclease (Nuc)), thereby boosting staphylococcal persistence and dissemination of disease. Characteristic features and key host signaling molecules including toll-like receptor 2 (TLR2), myeloperoxidase (MPO), neutrophil elastase (NE), protein-arginine deiminase type 4 (PAD4), cathelicidin LL-37, reactive or mitochondrial reactive oxygen species (ROS; mtROS), protein kinase C (PKC), Raf–MEK–ERK cascade (Raf; MEK; ERK), membrane-bound NADPH oxidase, processed gasdermin D (GSDM-D) and associated pores are highlighted.
NET formation can also be induced via a secondary mechanism which retains PMN integrity and viability by an active release of DNA-containing vesicles (Figure 1). This process (i.e. vital NET formation; sometimes referred to as vesicular NETosis) is mainly NADPH oxidase-independent and rapidly occurs within less than hour upon stimulation (43, 44). Activation of vital NET formation involves complement-mediated pathogen opsonization or sensing via toll-like receptors (TLRs) (44). Particularly, Clark and colleagues have initially shown that TLR-4-activated platelets trigger vital NET formation in order to capture bacteria in septic blood (45), while subsequent in vitro and in vivo work by the same laboratory demonstrated vital NET release and involvement of TLR-2 and complement receptor 3 upon neutrophil stimulation with Gram-positive bacteria (7, 43). Similar to suicidal NETosis, vital NET formation has also been shown to be partially dependent on calcium influx and activated PAD4 and NE, both of which migrate to the nucleus to initiate unpacking of histones and chromatin decondensation (Figure 1) (28, 46). Subsequently, nuclear envelope blebbing leads to the formation of DNA-containing vesicles that eventually fuse with the plasma membrane to expel their antimicrobial content into the extracellular space (Figure 1) (44). Notably, this process does not damage the plasma membrane so that neutrophils initially maintain their ability to migrate and phagocytose microbial invaders (7, 43). At later stages, however, the nuclear membrane may rupture thereby causing an accumulation of chromatin fibers in the cytosol (43). Thus, vital NET formation may indirectly be coupled with suicidal NETosis and lytic NET formation respectively. In this regard, we finally note that a specific form of vital NET formation involves a rapid release of mitochondrial DNA (mtDNA) from viable PMNs (47). Opposed to the canonical pathway of vital NET formation, this form of vital NET release requires the activity of the NADPH oxidase along with granulocyte-macrophage colony-stimulating factor (GM-CSF)-mediated priming of neutrophils, followed by subsequent stimulation by complement factor 5a (C5a) or lipopolysaccharide (LPS) (47). Yet, mechanistic details and the exact role of this form of vital NET release during infectious diseases or other pathophysiological conditions remain largely unknown.
Staphylococcal Catalysts of NET Formation
Neutrophils release NETs in response to multiple infectious agents such as Gram-negative and Gram-positive bacteria (20, 43). Intriguingly, S. aureus is considered one of the most potent inducers of NETosis, irrespective of whether PMNs sense live or metabolically inactive (dead) staphylococci (43). Nevertheless, the magnitude of NET induction is significantly increased when PMNs are exposed to viable S. aureus cells or staphylococcal culture supernatants, suggesting that secreted exoproteins together with intact structural components of the bacterial cell envelope substantially affect S. aureus-triggered NET formation (Figure 2) (43). In fact, various studies revealed that stimulation with live bacteria or S. aureus pore-forming toxins contribute to this phenomenon (Table 1) (23, 34, 43, 51, 56). For example, the staphylococcal bi-component toxin PVL (Panton-Valentine leukocidin) has been shown to prime vital NET formation in freshly isolated human neutrophils (43, 56). In this NADPH-independent process, endocytosed PVL particularly targets mitochondria thereby triggering the formation of mtROS from these organelles (Figure 1) (56). Further, PVL-mediated release of NETs involves MPO, Ca2+-signaling and PAD4 activation, as well as citrullination of histone H3 (56). However, PVL-induced NET formation seems to be highly dose-dependent as elevated levels of PVL may promote necroptotic or apoptotic cell death rather than classical NETosis (61, 62). Dose-dependency may also play a crucial role during NET formation induced by LukAB (also known as LukGH), another staphylococcal pore-forming toxin that lineage-dependently targets human CD11b or the hydrogen voltage-gated channel 1 (HVCN1) (51, 63, 64). Like PVL, LukAB consists of two subunits and promotes NET release from PMNs exclusively at sublytic concentrations in vitro (Table 1) (51). LukAB-induced NETs, however, did not display enhanced bactericidal activity towards staphylococci suggesting that S. aureus may systematically induce or even exploit NET formation during persistent infections (51). In agreement with this idea, recent studies by Bhattacharya et al. uncovered that extracellular traps augment chronic staphylococcal infections (15). Specifically, the combined activity of LukAB and PVL provokes NET formation within biofilms, a rather detrimental effect that affects the clinical outcome of chronic burn wounds in pigs (15). In this regard, we note that many staphylococcal toxins and virulence determinants are controlled by the global accessory regulatory system Agr (65). Agr is a quorum sensing system that consists of several structural components including AgrD, the precursor molecule of the autoinducing peptide AIP which facilitates staphylococcal communication and target gene regulation via activation of AgrC-AgrA two component system (65). Surprisingly, truncated and formylated peptide variants of AgrD solely exhibit immunomodulatory and NETosis-promoting capacities, a phenomenon which raises the question of whether staphylococci directly process components of the Agr system to enhance NET formation in infectious foci (Table 1) (50). Agr is also controlling an array of amphipathic, α-helical peptides designated phenol-soluble modulins (PSMs) (65, 66). These small peptides have strong lytic properties and usually trigger necroptotic or pyroptotic cell death in target cells (66, 67). At micromolar concentrations, purified PSM-α peptides were also found to initiate a very rapidly occurring and NADPH oxidase-independent form of NETosis in purified PMNs, a process that is reminiscent of vital NET formation (57). Moreover, PSM-α peptides govern budding of lipoprotein-containing extracellular membrane vesicles from staphylococcal cytoplasmic membranes (68, 69). Since S. aureus-derived lipoproteins constitute potent inducers of TLR-2 (70–72), and TLR-2 signaling along with complement-mediated opsonization correlates with vital NETosis in human neutrophils (7), lipoprotein-comprising plasma membrane preparations of the methicillin-resistant S. aureus (MRSA) strain USA300 have recently been identified as another potent driver of vital and PAD4-depenent NET formation (Table 1) (52).
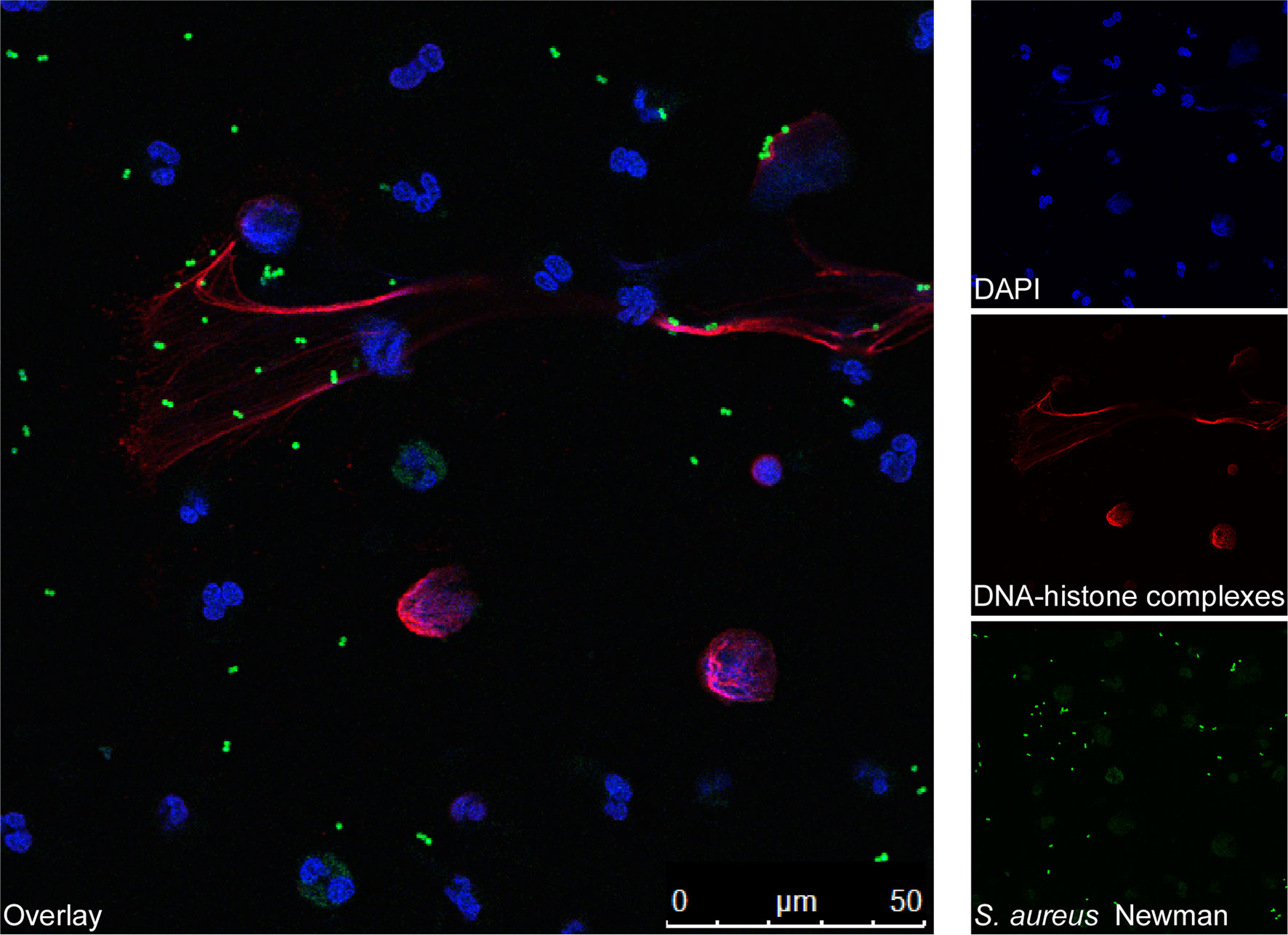
Figure 2 Fluorescence microscopy images of human blood-derived neutrophils forming NETs during incubation with S. aureus. Human blood-derived neutrophils were stimulated with FITC-labelled S. aureus Newman for 90 min (green). Next, the formation of NETs was visualized using confocal fluorescence microscopy with antibodies against DNA-histone-complexes (red) as previously described (48). Nuclei were stained with DAPI (blue). The main image shows staphylococci (green) entrapped by NETs (red).
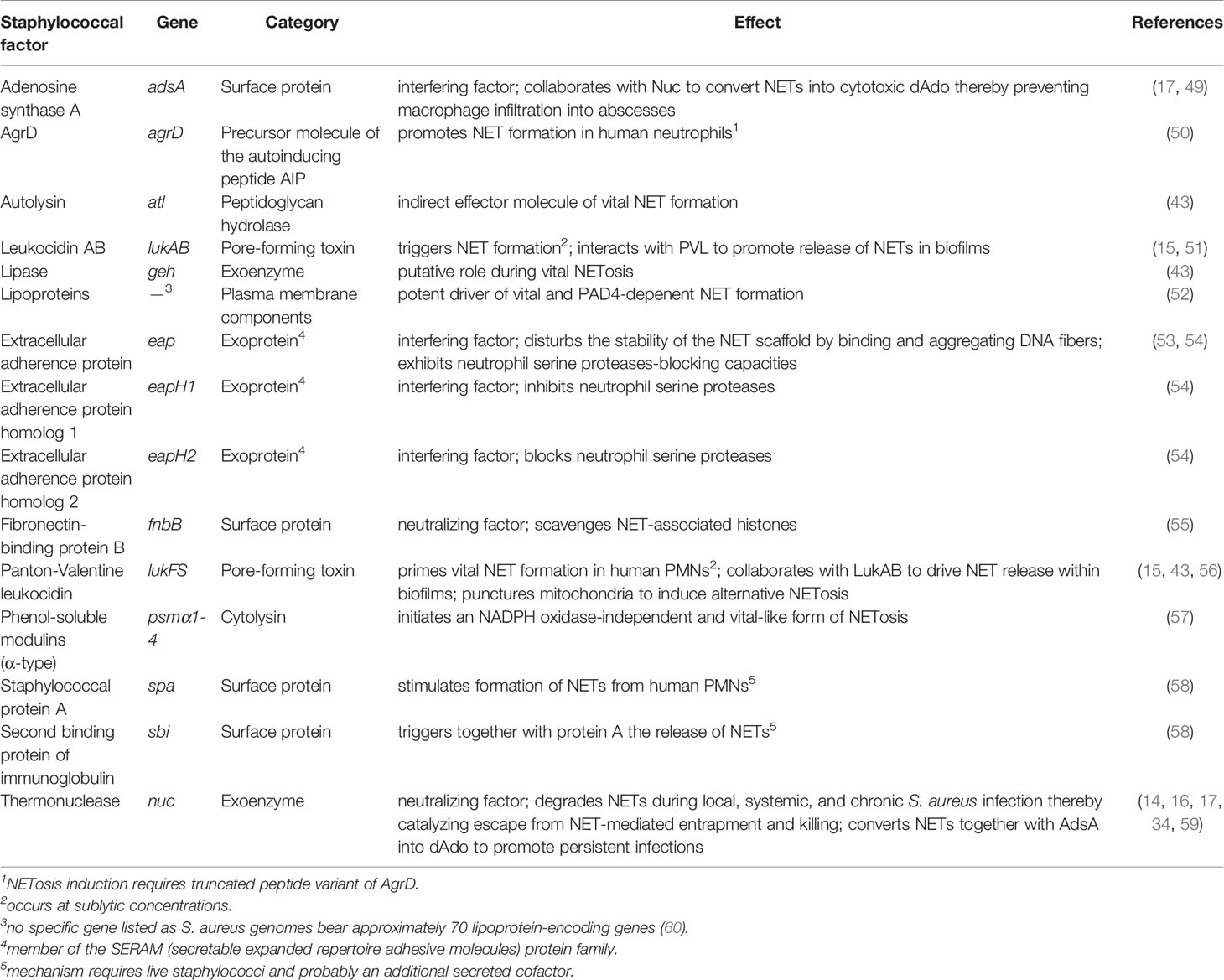
Table 1 Selected staphylococcal factors that induce or interfere with NET formation.
Apart from toxin- or lipoprotein-mediated induction of NET formation, certain peptidoglycan-associated molecules synthesized by staphylococci contribute to the release of extracellular traps from PMNs (Table 1). For example, staphylococcal protein A (SpA) and Sbi (second binding protein of immunoglobulin), both cell surface-displayed proteins that exhibit strong immunoglobulin-binding properties (73), affect the generation of NETs as a S. aureus Newman Δspa Δsbi double-mutant lost the ability to promote release of NETs from human neutrophils in tissue culture-based assays (58). In this context, Hoppenbrouwers et al. discovered that the amount of SpA in staphylococcal cell walls correlates with the capacity of S. aureus to provoke NETosis (58). Isolates that display elevated amounts of SpA in the cell wall (SpAhigh) activated NET formation more efficiently as compared to strains that exhibit a SpAlow phenotype (58). However, purified SpA alone failed to stimulate NETosis in purified PMNs indicating that other, presumably secreted factors of live staphylococci along with a unique mechanism are required to promote SpA-associated NET formation (58). This may also hold true for the major autolysin Atl, which has been identified as another possible effector molecule of vital NET formation (43). Albeit release of NETs from human PMNs occurred in response to high concentrations of recombinant autolysin, Atl may rather contribute indirectly to vital NETosis (43). Specifically, Wang et al. demonstrated that the peptidoglycan-hydrolyzing activity of Atl and Sle1, an N-acetylmuramyl-L-alanine amidase (74), impacts trafficking of S. aureus-derived membrane vesicles across murein layers (68), and thus may contribute to lipoprotein-induced stimulation of TLR-2 and vital NET formation. Finally, we note that a staphylococcal lipase has also been implicated in vital NETosis, although mechanistic details remain elusive (43). Together, these comprehensive studies demonstrate that S. aureus together with its exoproducts predominantly elicits vital NET formation, albeit viable staphylococci may also skew neutrophils toward suicidal NETosis in a manner requiring NADPH oxidase-derived ROS or even mtROS, especially when co-cultivated with PMNs for longer time periods or during infection of the heart (Figure 1) (23, 34, 35).
Staphylococcal Evasion From NET-Mediated Entrapment and Killing
Pioneering work by Brinkmann et al. uncovered that NETs have pathogen-capturing and antimicrobial properties (5). Multiple microbes including bacteria, viruses, and fungi can be entangled, disarmed, or neutralized by these extracellular structures, readily preventing pathogen spread and dissemination of disease (5, 27, 44). Of note, not only various NET-associated factors such as calprotectin, histones, cathepsin G, or the DNA itself have been discussed to mediate antimicrobial activity of NETs (5, 75–77), but also the boosting of NET-formation has been demonstrated to be protective in some disease conditions, e.g. during systemic S. aureus infections in mitochondrial calcium uptake 1 (MICU1)-deficient (MICU1-/-) mice or during lung infections upon treatment with statins that block cholesterol biosynthesis (35, 78). However, NETs often fail to eradicate replicating S. aureus during persistent infections as this pathogen releases a plethora of virulence factors into the extracellular milieu that antagonize NET-mediated entrapment and killing (Table 1) (79). For example, S. aureus secretes a robust thermonuclease (Nuc) which rapidly dismantles NETs thereby affecting local, systemic, as well as chronic infections (Figure 1) (14, 16, 17, 34, 59). In that regard, we note that Nuc-mediated degradation of NETs may further restrict the communication of PMNs and macrophages (34). This striking observation seems to be of particular importance during slowly-occurring suicidal NETosis since an accelerated and mtROS-controlled form of lytic NETosis, as observed in S100A9-deficient neutrophils, abrogated Nuc-derived effects and simultaneously enhanced macrophage-mediated killing of S. aureus and other extracellular bacterial pathogens (34). Nonetheless, macrophages often reside at the periphery of infectious foci in wild-type animals, a pathophysiological phenomenon that also involves the activity of Nuc (17). Particularly, Nuc-mediated degradation of NETs together with the activity of AdsA (adenosine synthase A), a cell wall anchored 5’-3’-nucleotidase, leads to the biosynthesis of deoxyadenosine (dAdo) (Figure 1) (17). dAdo is a cytotoxic deoxyribonucleoside which exquisitely kills macrophages during abscess formation by targeting the purine salvage pathway and apoptotic signaling cascade (17, 49, 80). In this manner, S. aureus and related staphylococci not only prevent NET-mediated killing within abscesses, but rather exploit excretion of NETs to suppress phagocyte entry into deeper cavities of infectious foci, ultimately enhancing pathogen survival and establishment of persistent infections (17, 49, 81). Overall, staphylococcal Nuc represents a key determinant utilized by staphylococci to prevent NET-associated enmeshment and killing.
S. aureus evolved additional virulence determinants to block NET-mediated entrapment (Table 1). Most of these Nuc-independent mechanisms target the structural backbone of NETs along with NET-associated proteins such as histones, NE, or cathepsin G (53–55). Staphylococcal extracellular adherence protein (Eap), for example, binds and aggregates NET fibers in vitro and therefore affects the formation and stability of neutrophil-derived DNA traps (53). Together with two orphan Eap homologes (EapH1 and EapH2), Eap has also neutrophil serine proteases-blocking capacities, as proteins of the Eap family efficiently bind to the catalytic domains of NE, cathepsin G, or proteinase 3 (54). Accordingly, S. aureus variants lacking the Eap protein-encoding genes are attenuated in a mouse model of blood stream infection (54). More recent in vitro work showed that staphylococcal fibronectin-binding protein B (FnBPB) contributes to the neutralization of NETs (55). In this model, FnBPB binds with very high affinity to histones thereby suppressing their bactericidal activity (55). Scavenging histones further enhances survival of live S. aureus exposed to NETs since FnBPB-deficient mutant cells were more prone to NET-mediated killing as compared to the enmeshed parental S. aureus strain (55). Finally, we note that S. aureus alters the net charge of the bacterial cell surface by lysinylating membrane phosphatidylglycerol and alanylating teichoic acids (TAs) (82, 83). Interestingly, D-alanylation of TAs by the DltABCD machinery together with the formation of a capsule mediates tolerance to the microbicidal attributes of NETs in Streptococcus pneumoniae (84), raising the possibility that alanylated TAs and encapsulation may similarly protect S. aureus from NET-mediated killing. In this context, it should also be considered that D-alanylation of TAs confers resistance to several cationic antimicrobial peptides including the NET-associated cathelicidin LL-37 (82, 85). However, Jann et al. uncovered that S. aureus wild type bacteria and their dltA variants were killed by NETs in a similar fashion, indicating that neutrophils use the cathelicidins mainly for the phagolysosomal but not NET-associated antimicrobial defense (86). In agreement with this data, previous in vitro work found that particularly LL-37 lost its antimicrobial properties when bound to NETs, and rather represents a stabilization factor that antagonizes Nuc-mediated degradation of extracellular traps (48). This may also explain why S. aureus secretes further nucleases including the membrane-bound Nuc2 enzyme as well as EssD (or EsaD), a substrate of the staphylococcal type VII secretion apparatus (87–89). Direct evidence that these enzymes significantly contribute to the escape from NET-mediated entrapment has, however, not been provided so far. Collectively, S. aureus secretes multiple virulence factors to incapacitate the microbicidal features of NETs, presumably to promote immune evasion and dissemination in the mammalian host.
Immunopathological Consequences of Aberrant NET Formation During Staphylococcal Infections
Recent advances suggest that excessive or dysregulated NETosis may cause severe pathologies during various human diseases (6, 90). During acute or chronic infections, for example, aberrant NET formation has been linked to inflammatory processes, tissue and organ injury, and negative disease outcomes along with increased mortality rates (6, 91). This is exemplified in the context of pulmonary diseases and cystic fibrosis (CF), a genetic disorder caused by a mutation in the CF transmembrane conductance regulator-encoding gene CFTR (91, 92). CF patients develop a highly viscous mucus which favors pathogen colonization and infection, especially by NETosis-triggering microbes such as Pseudomonas aeruginosa or S. aureus (92, 93). Not surprisingly though that sputum samples derived of P. aeruginosa- or S. aureus-infected CF patients contain elevated amounts of NETs together with NET-bound peptides (16, 94). Yet, NET-rich microenvironments of CF lungs that are often associated with hypoxia (95), a condition that retains the ability of PMNs to form NETs in response to staphylococci (96), typically fail to clear microbial invaders and rather initiate detrimental events that affect lung pathology and chronic airway inflammation (16, 91). Particularly, several NET-associated proteins exhibit cytotoxic and tissue destructive capacities (97–100). For example, sputum specimens from CF patients were found to contain large amounts of MPO along with MPO-derived oxidizing and nitrating species (94, 100). These factors may contribute to respiratory dysfunction and poor disease prognosis in S. aureus-infected CF patients, as MPO and MPO-derived oxidants confer tracheobronchial or alveolar epithelial cell damage (99, 100). Epithelial injury and NET-associated cytotoxicity toward CF lung tissue may further occur in response to NET-assembled histones or NE that have also been identified in sputum samples of affected individuals (94, 101). Especially extracellular histones display toxigenic properties toward epithelial and endothelial cells due to their membrane-binding and damaging attributes (97, 99). Similarly, uncontrolled release of NE and NE-DNA complexes during MRSA-induced pneumonia is considered as a substantial mediator of acute lung injury and may therefore exacerbate disease outcomes of staphylococcal pulmonary infections (Table 2) (98, 113). In light of this, increased concentrations of histones, NE, and calprotectin, a NET-bound alarmin (75), could potentiate the release of cytokines and chemokines into lung fluids (102, 116, 117), presumably explaining hyper-inflammatory responses and the non-ending recruitment of neutrophils along with excessive NETosis in S. aureus-infected CF lungs. Thus, dysregulated NETosis potentially mediates adverse and harmful effects during CF and S. aureus-caused infections of the respiratory tract (Table 2).
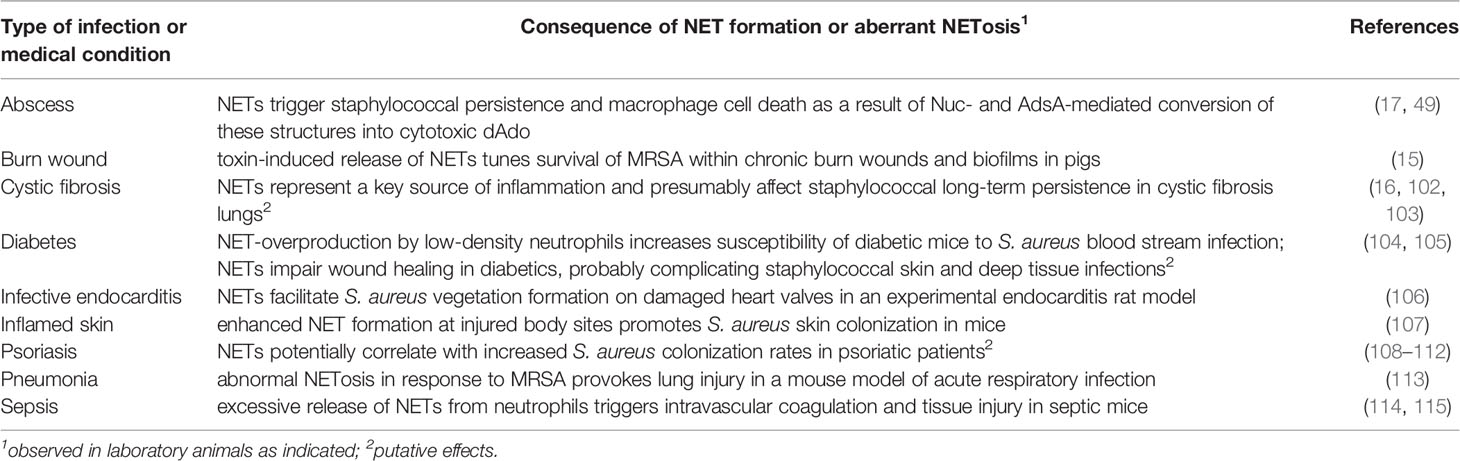
Table 2 Detrimental effects of NETs during infection with S. aureus.
Dysfunctional NETosis can also complicate staphylococcal infections in the context of other chronic diseases (Table 2). In diabetic mice, staphylococcal α-toxin drives the transforming growth factor β (TGF-β)-signaling-dependent expansion of low-density neutrophils (LDN) (104). LDN in turn excrete large amounts of NETs, an adverse feature that has been linked to increased mortality rates in mice challenged with the community-acquired MRSA strain USA300 (104). These effects may clarify why diabetic patients are more vulnerable to S. aureus bacteremia as compared to non-diabetic individuals (118). In this regard, we further note that NETs alter the wound healing process in patients with diabetes (105), probably explaining why this population often suffers from complicated S. aureus-mediated skin or foot ulcer infections (119). Moreover, recent work by Bitschar et al. revealed that NETs interact with keratinocytes at injured or inflamed skin sites thereby promoting S. aureus vegetation formation on body sites that are typically not colonized by this microbe (107). This appears highly relevant as S. aureus is frequently isolated from the skin of patients with atopic dermatitis or psoriasis (108–110, 120), both chronic inflammatory skin diseases that affect large segments of the human population (121, 122). At least skin lesion sites of psoriatic patients are characterized by increased amounts of NETs (111, 112), which could correlate with increased S. aureus colonization rates. Nonetheless, aberrant NETosis can also affect staphylococcal disease pathogenesis in otherwise healthy individuals and immunocompetent laboratory animals as, for example, enhanced neutrophil influx and NET formation boost biofilm and implant-associated infections in wild-type mice (15, 123). Further, it is worth noting that a massive release of NETs in foci of infection causes tissue damage and organ injury during staphylococcal systemic infection (Table 2) (114). Innovative in vivo imaging technologies uncovered that NETs along with NET-bound peptides accumulated in the liver vasculature of septic mice (114). Here, NET-associated NE and histones were found to co-localize with necrotic tissue sites suggesting that NET components exhibit organ-damaging attributes during severe staphylococcal diseases (114). In line with these observations, McDonald and colleagues discovered that a sepsis-provoked release of NETs into the vasculature triggers networking of platelets and extracellular traps, ultimately leading to intravascular coagulation and injury of hepatic tissues (115). Finally, the platelet-NET axis impairs S. aureus-induced infective endocarditis (106). Specifically, NETs have been found to amplify S. aureus vegetation formation on injured heart valves in an experimental rat model of infective endocarditis (106), a fatal side-effect of NETs that is also exploited by other endocarditis-promoting pathogens such as Streptococcus mutans (124). Collectively, these compelling studies demonstrate that excessive formation of NETs along with elevated levels of NET-associated peptides in response to acute or chronic staphylococcal infections can be detrimental to the mammalian host, particularly in the context of systemic or pulmonary diseases.
Concluding Remarks
NETs as part of the innate immune response are generally believed to correlate with clinical outcomes of many infectious diseases. As long as the magnitude of NET formation is coordinated and tightly balanced, these extracellular structures exhibit beneficial properties and contribute to the entrapment, disarming, and killing of microorganisms (5, 6). This may also hold true for local infections caused by S. aureus as NETs are not only formed within abscesses but also diminish the risk of pathogen entry into circulating body fluids and the development of invasive diseases (7). Paradoxically, NETs may exacerbate staphylococcal infections and disease progression, specifically when excessively synthesized during acute or chronic infections (16, 104, 106, 114, 115). This raises the question of whether S. aureus selectively induces or even gains advantage of NET formation under certain pathophysiological conditions. In line with this model, earlier work demonstrated that induction of NETosis promotes intra-abscess survival of S. aureus, colonization of injured skin sites, and biofilm formation (15, 17, 107). As aberrant NETosis is also linked to severe staphylococcal infections and the establishment of pulmonary infections in chronically ill patients (16, 114), it is further tempting to speculate that S. aureus may take advantage of the organ-damaging capacities of NETs or NET-associated components to traverse endothelial or epithelial barriers for subsequent penetration of deep tissues. If so, concurrent stimulation and exploitation of excessive NETosis may represent a refined immune-evasive maneuver evolved by S. aureus to create new proliferative niches in the mammalian host, a fact that may clarify why staphylococci excrete a plethora of NET-inducing effector molecules into the extracellular space. In this context, it should also be taken into account that NETosis-catalyzing molecules released by S. aureus represent predominant immune evasion molecules, most of which the pathogen secretes in any way to manipulate or kill host cells (19, 125, 126). This is exemplified by staphylococcal LukAB which primarily lyses various immune cells but concurrently has the capacity to trigger NET formation in human neutrophils (51, 127). Expulsion of microbicidal NETs from viable or dying neutrophils may therefore simply reflect an inadvertent side-effect within foci of infection that S. aureus readily tolerates or even exploits due to the biogenesis of numerous virulence and entrapment-protective factors such as Nuc. Nevertheless, owing to the expression of these evasion molecules, it appears irrelevant at first glance whether the host induces vital or suicidal NETosis during S. aureus-mediated infections. When compared to suicidal NETosis, however, a rapid release of vital NETs might be more advantageous for mammalian hosts in terms of combating staphylococci as this route of NET generation not only mediates prompt trapping of the microbe at very early infection stages, but also retains the ability of PMNs to crawl and phagocytose the bacterial invader (7, 43). Further, vital NET formation is known to maintain the membrane integrity of netting PMNs (7, 43) and thus presumably evolved to limit a release of inflammation-promoting and otherwise-sequestered intracellular molecules, ultimately minimizing the risk of developing a hyper-inflammatory milieu that eventually potentiates tissue injury and staphylococcal disease severity. However, it remains elusive whether S. aureus-infected hosts are capable of selectively activating a specific type of NET formation during infection. Assuming a particular form of NETosis is indeed more effective against S. aureus but concomitantly less detrimental to the host, the discovery of appropriate host factors controlling specific NET-forming events along with the optimization of individual therapeutics or antibiotics, some of which are known to interfere with NETosis or NET-mediated killing of MRSA (128–131), may help to better manage staphylococcal infectious diseases in the future.
Overall, the formation of NETs shapes staphylococcal disease pathogenesis and clinical manifestations in many aspects. Whilst NETs display antimicrobial properties and to some extend reduce pathogen spread, these web-like matrices may also unfold adverse characteristics and constitute a bio-scaffold utilized by staphylococci to establish persistent infections in humans or animal hosts. Thus, deciphering all molecular facets and mechanistic details by which clinical S. aureus isolates stimulate or manipulate various forms of NET formation, along with the discovery of contributing host signaling cascades and NET-stabilizing factors, may help to conceive innovative and selective therapeutic approaches to improve staphylococcal infection outcomes, especially in hospitalized or critically ill patients.
Ethics Statement
Blood samples were drawn from healthy donors in agreement with the local ethical board. The study was approved by the medical ethics committee of the Hannover Medical School (Hannover, Germany) under the permission number 3295-2016.
Author Contributions
VW performed the literature review as well as data collection and prepared the manuscript draft. MK-B provided revisions, additional literature review, comments, and essential materials. Both authors substantially contributed to the article and approved the submitted version.
Funding
We are grateful for support from the German Research Foundation (project grant WI4582/2-1 to VW, project number 449712894; project grant KO 3552/4-1 to MK-B) and from Else Kröner-Fresenius-Stiftung (award 2021_EKEA.16 to VW). Furthermore, this work was partially supported by the R2N project under grant (74ZN1574) provided to MK-B, which is funded by the Federal State of Lower Saxony.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all members of our laboratories for their constant support and insights and for sharing their enthusiasm for the study of host pathogen interactions. We apologize to authors whose work was either not referenced or not discussed.
References
1. Mayadas TN, Cullere X, Lowell CA. The Multifaceted Functions of Neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
2. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: New Insights and Open Questions. Sci Immunol (2018) 3(30):eaat4579. doi: 10.1126/sciimmunol.aat4579
3. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil Migration in Infection and Wound Repair: Going Forward in Reverse. Nat Rev Immunol (2016) 16(6):378–91. doi: 10.1038/nri.2016.49
4. Futosi K, Fodor S, Mocsai A. Neutrophil Cell Surface Receptors and Their Intracellular Signal Transduction Pathways. Int Immunopharmacol (2013) 17(3):638–50. doi: 10.1016/j.intimp.2013.06.034
5. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
6. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
7. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking. Vivo Nat Med (2012) 18(9):1386–93. doi: 10.1038/nm.2847
8. von Kockritz-Blickwede M, Blodkamp S, Nizet V. Interaction of Bacterial Exotoxins With Neutrophil Extracellular Traps: Impact for the Infected Host. Front Microbiol (2016) 7:402. doi: 10.3389/fmicb.2016.00402
9. Kluytmans J, van Belkum A, Verbrugh H. Nasal Carriage of Staphylococcus Aureus: Epidemiology, Underlying Mechanisms, and Associated Risks. Clin Microbiol Rev (1997) 10(3):505–20. doi: 10.1128/CMR.10.3.505
10. Lowy FD. Staphylococcus Aureus Infections. New Engl J Med (1998) 339(8):520–32. doi: 10.1056/NEJM199808203390806
11. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus Aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin Microbiol Rev (2015) 28(3):603–61. doi: 10.1128/CMR.00134-14
12. Kobayashi SD, Malachowa N, DeLeo FR. Pathogenesis of Staphylococcus Aureus Abscesses. Am J Pathol (2015) 185(6):1518–27. doi: 10.1016/j.ajpath.2014.11.030
13. Cheng AG, DeDent AC, Schneewind O, Missiakas D. A Play in Four Acts: Staphylococcus Aureus Abscess Formation. Trends Microbiol (2011) 19(5):225–32. doi: 10.1016/j.tim.2011.01.007
14. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Kockritz-Blickwede M. Nuclease Expression by Staphylococcus Aureus Facilitates Escape From Neutrophil Extracellular Traps. J Innate Immun (2010) 2(6):576–86. doi: 10.1159/000319909
15. Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, et al. Staphylococcus Aureus Biofilms Release Leukocidins to Elicit Extracellular Trap Formation and Evade Neutrophil-Mediated Killing. Proc Natl Acad Sci USA (2018) 115(28):7416–21. doi: 10.1073/pnas.1721949115
16. Herzog S, Dach F, de Buhr N, Niemann S, Schlagowski J, Chaves-Moreno D, et al. High Nuclease Activity of Long Persisting Staphylococcus Aureus Isolates Within the Airways of Cystic Fibrosis Patients Protects Against NET-Mediated Killing. Front Immunol (2019) 10:2552. doi: 10.3389/fimmu.2019.02552
17. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus Aureus Degrades Neutrophil Extracellular Traps to Promote Immune Cell Death. Science (2013) 342(6160):863–6. doi: 10.1126/science.1242255
18. Spaan AN, Surewaard BG, Nijland R, van Strijp JA. Neutrophils Versus Staphylococcus Aureus: A Biological Tug of War. Annu Rev Microbiol (2013) 67:629–50. doi: 10.1146/annurev-micro-092412-155746
19. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal Manipulation of Host Immune Responses. Nat Rev Microbiol (2015) 13(9):529–43. doi: 10.1038/nrmicro3521
20. Hoppenbrouwers T, Autar ASA, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, et al. In Vitro Induction of NETosis: Comprehensive Live Imaging Comparison and Systematic Review. PloS One (2017) 12(5):e0176472. doi: 10.1371/journal.pone.0176472
21. Vorobjeva NV, Chernyak BV. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochem Biokhimiia (2020) 85(10):1178–90. doi: 10.1134/S0006297920100065
22. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PloS One (2012) 7(10):e48111. doi: 10.1371/journal.pone.0048111
23. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
24. Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of Neutrophil Extracellular DNA Lattices by Placental Microparticles and IL-8 and Their Presence in Preeclampsia. Hum Immunol (2005) 66(11):1146–54. doi: 10.1016/j.humimm.2005.11.003
25. Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, Satyamoorthy K. High Glucose Modulates IL-6 Mediated Immune Homeostasis Through Impeding Neutrophil Extracellular Trap Formation. FEBS Lett (2013) 587(14):2241–6. doi: 10.1016/j.febslet.2013.05.053
26. Barbu EA, Mendelsohn L, Samsel L, Thein SL. Pro-Inflammatory Cytokines Associate With NETosis During Sickle Cell Vaso-Occlusive Crises. Cytokine (2020) 127:154933. doi: 10.1016/j.cyto.2019.154933
27. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol (2017) 8:81. doi: 10.3389/fimmu.2017.00081
28. Ronchetti L, Boubaker NS, Barba M, Vici P, Gurtner A, Piaggio G. Neutrophil Extracellular Traps in Cancer: Not Only Catching Microbes. J Exp Clin Cancer Res: CR (2021) 40(1):231. doi: 10.1186/s13046-021-02036-z
29. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK Pathway Is Required for Neutrophil Extracellular Trap Formation. Nat Chem Biol (2011) 7(2):75–7. doi: 10.1038/nchembio.496
30. Castanheira FVS, Kubes P. Neutrophils and NETs in Modulating Acute and Chronic Inflammation. Blood (2019) 133(20):2178–85. doi: 10.1182/blood-2018-11-844530
31. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J Exp Med (2010) 207(9):1853–62. doi: 10.1084/jem.20100239
32. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A. Papayannopoulos V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics During NETosis. Cell Rep (2014) 8(3):883–96. doi: 10.1016/j.celrep.2014.06.044
33. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
34. Monteith AJ, Miller JM, Maxwell CN, Chazin WJ, Skaar EP. Neutrophil Extracellular Traps Enhance Macrophage Killing of Bacterial Pathogens. Sci Adv (2021) 7(37):eabj2101. doi: 10.1126/sciadv.abj2101
35. Monteith AJ, Miller JM, Beavers WN, Maloney KN, Seifert EL, Hajnoczky G, et al. Mitochondrial Calcium Uniporter Affects Neutrophil Bactericidal Activity During Staphylococcus Aureus Infection. Infect Immun (2021) IAI0055121. doi: 10.1128/IAI.00551-21
36. Neumann A, Berends ET, Nerlich A, Molhoek EM, Gallo RL, Meerloo T, et al. The Antimicrobial Peptide LL-37 Facilitates the Formation of Neutrophil Extracellular Traps. Biochem J (2014) 464(1):3–11. doi: 10.1042/BJ20140778
37. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
38. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
39. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
40. Chen KW, Demarco B, Broz P. Beyond Inflammasomes: Emerging Function of Gasdermins During Apoptosis and NETosis. EMBO J (2020) 39(2):e103397. doi: 10.15252/embj.2019103397
41. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
42. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
43. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J Immunol (2010) 185(12):7413–25. doi: 10.4049/jimmunol.1000675
44. Yipp BG, Kubes P. NETosis: How Vital is it? Blood (2013) 122(16):2784–94. doi: 10.1182/blood-2013-04-457671
45. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
46. Burgener SS, Schroder K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harbor Perspect Biol (2020) 12(7):a037028. doi: 10.1101/cshperspect.a037028
47. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differentiation (2009) 16(11):1438–44. doi: 10.1038/cdd.2009.96
48. Neumann A, Vollger L, Berends ET, Molhoek EM, Stapels DA, Midon M, et al. Novel Role of the Antimicrobial Peptide LL-37 in the Protection of Neutrophil Extracellular Traps Against Degradation by Bacterial Nucleases. J Innate Immun (2014) 6(6):860–8. doi: 10.1159/000363699
49. Winstel V, Schneewind O, Missiakas D. Staphylococcus Aureus Exploits the Host Apoptotic Pathway To Persist During Infection. mBio (2019) 10(6):e02270-19. doi: 10.1128/mBio.02270-19
50. Gonzalez DJ, Corriden R, Akong-Moore K, Olson J, Dorrestein PC. Nizet V. N-Terminal ArgD Peptides From the Classical Staphylococcus Aureus Agr System Have Cytotoxic and Proinflammatory Activities. Chem Biol (2014) 21(11):1457–62. doi: 10.1016/j.chembiol.2014.09.015
51. Malachowa N, Kobayashi SD, Freedman B, Dorward DW, DeLeo FR. Staphylococcus Aureus Leukotoxin GH Promotes Formation of Neutrophil Extracellular Traps. J Immunol (2013) 191(12):6022–9. doi: 10.4049/jimmunol.1301821
52. Hook JS, Patel PA, O’Malley A, Xie L, Kavanaugh JS, Horswill AR, et al. Lipoproteins From Staphylococcus Aureus Drive Neutrophil Extracellular Trap Formation in a TLR2/1- and PAD-Dependent Manner. J Immunol (2021) 207(3):966–73. doi: 10.4049/jimmunol.2100283
53. Eisenbeis J, Saffarzadeh M, Peisker H, Jung P, Thewes N, Preissner KT, et al. The Staphylococcus Aureus Extracellular Adherence Protein Eap Is a DNA Binding Protein Capable of Blocking Neutrophil Extracellular Trap Formation. Front Cell Infect Microbiol (2018) 8:235. doi: 10.3389/fcimb.2018.00235
54. Stapels DA, Ramyar KX, Bischoff M, von Kockritz-Blickwede M, Milder FJ, Ruyken M, et al. Staphylococcus Aureus Secretes a Unique Class of Neutrophil Serine Protease Inhibitors. Proc Natl Acad Sci USA (2014) 111(36):13187–92. doi: 10.1073/pnas.1407616111
55. Pietrocola G, Nobile G, Alfeo MJ, Foster TJ, Geoghegan JA, De Filippis V, et al. Fibronectin-Binding Protein B (FnBPB) From Staphylococcus Aureus Protects Against the Antimicrobial Activity of Histones. J Biol Chem (2019) 294(10):3588–602. doi: 10.1074/jbc.RA118.005707
56. Mazzoleni V, Zimmermann K, Smirnova A, Tarassov I, Prevost G. Staphylococcus Aureus Panton-Valentine Leukocidin Triggers an Alternative NETosis Process Targeting Mitochondria. FASEB J: Off Publ Fed Am Societies Exp Biol (2021) 35(2):e21167. doi: 10.1096/fj.201902981R
57. Bjornsdottir H, Dahlstrand Rudin A, Klose FP, Elmwall J, Welin A, Stylianou M, et al. Phenol-Soluble Modulin Alpha Peptide Toxins From Aggressive Staphylococcus Aureus Induce Rapid Formation of Neutrophil Extracellular Traps Through a Reactive Oxygen Species-Independent Pathway. Front Immunol (2017) 8:257. doi: 10.3389/fimmu.2017.00257
58. Hoppenbrouwers T, Sultan AR, Abraham TE, Lemmens-den Toom NA, Hansenova Manaskova S, van Cappellen WA, et al. Staphylococcal Protein A Is a Key Factor in Neutrophil Extracellular Traps Formation. Front Immunol (2018) 9:165. doi: 10.3389/fimmu.2018.00165
59. Yu J, Jiang F, Zhang F, Hamushan M, Du J, Mao Y, et al. Thermonucleases Contribute to Staphylococcus Aureus Biofilm Formation in Implant-Associated Infections-A Redundant and Complementary Story. Front Microbiol (2021) 12:687888. doi: 10.3389/fmicb.2021.687888
60. Stoll H, Dengjel J, Nerz C, Gotz F. Staphylococcus Aureus Deficient in Lipidation of Prelipoproteins is Attenuated in Growth and Immune Activation. Infect Immun (2005) 73(4):2411–23. doi: 10.1128/IAI.73.4.2411-2423.2005
61. Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus Aureus Panton-Valentine Leukocidin Directly Targets Mitochondria and Induces Bax-Independent Apoptosis of Human Neutrophils. J Clin Invest (2005) 115(11):3117–27. doi: 10.1172/JCI22684
62. Loffler B, Hussain M, Grundmeier M, Bruck M, Holzinger D, Varga G, et al. Staphylococcus Aureus Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils. PloS Pathog (2010) 6(1):e1000715. doi: 10.1371/journal.ppat.1000715
63. Boguslawski KM, McKeown AN, Day CJ, Lacey KA, Tam K, Vozhilla N, et al. Exploiting Species Specificity to Understand the Tropism of a Human-Specific Toxin. Sci Adv (2020) 6(11):eaax7515. doi: 10.1126/sciadv.aax7515
64. Perelman SS, James DBA, Boguslawski KM, Nelson CW, Ilmain JK, Zwack EE, et al. Genetic Variation of Staphylococcal LukAB Toxin Determines Receptor Tropism. Nat Microbiol (2021) 6(6):731–45. doi: 10.1038/s41564-021-00890-3
65. Le KY, Otto M. Quorum-Sensing Regulation in Staphylococci-an Overview. Front Microbiol (2015) 6:1174. doi: 10.3389/fmicb.2015.01174
66. Peschel A, Otto M. Phenol-Soluble Modulins and Staphylococcal Infection. Nat Rev Microbiol (2013) 11(10):667–73. doi: 10.1038/nrmicro3110
67. Missiakas D, Winstel V. Selective Host Cell Death by Staphylococcus Aureus: A Strategy for Bacterial Persistence. Front Immunol (2021) 11:621733. doi: 10.3389/fimmu.2020.621733
68. Wang X, Thompson CD, Weidenmaier C, Lee JC. Release of Staphylococcus Aureus Extracellular Vesicles and Their Application as a Vaccine Platform. Nat Commun (2018) 9(1):1379. doi: 10.1038/s41467-018-03847-z
69. Schlatterer K, Beck C, Hanzelmann D, Lebtig M, Fehrenbacher B, Schaller M, et al. The Mechanism Behind Bacterial Lipoprotein Release: Phenol-Soluble Modulins Mediate Toll-Like Receptor 2 Activation via Extracellular Vesicle Release From Staphylococcus Aureus. mBio (2018) 9(6):e01851-18. doi: 10.1128/mBio.01851-18
70. Hanzelmann D, Joo HS, Franz-Wachtel M, Hertlein T, Stevanovic S, Macek B, et al. Toll-Like Receptor 2 Activation Depends on Lipopeptide Shedding by Bacterial Surfactants. Nat Commun (2016) 7:12304. doi: 10.1038/ncomms12304
71. Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, et al. Not Lipoteichoic Acid But Lipoproteins Appear to be the Dominant Immunobiologically Active Compounds in Staphylococcus Aureus. J Immunol (2006) 177(5):3162–9. doi: 10.4049/jimmunol.177.5.3162
72. Mohammad M, Na M, Hu Z, Nguyen MT, Kopparapu PK, Jarneborn A, et al. Staphylococcus Aureus Lipoproteins Promote Abscess Formation in Mice, Shielding Bacteria From Immune Killing. Commun Biol (2021) 4(1):432. doi: 10.1038/s42003-021-01947-z
73. Foster TJ. Surface Proteins of Staphylococcus Aureus. Microbiol Spectr (2019) 7(4). doi: 10.1128/microbiolspec.GPP3-0046-2018
74. Kajimura J, Fujiwara T, Yamada S, Suzawa Y, Nishida T, Oyamada Y, et al. Identification and Molecular Characterization of an N-Acetylmuramyl-L-Alanine Amidase Sle1 Involved in Cell Separation of Staphylococcus Aureus. Mol Microbiol (2005) 58(4):1087–101. doi: 10.1111/j.1365-2958.2005.04881.x
75. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil Extracellular Traps Contain Calprotectin, A Cytosolic Protein Complex Involved in Host Defense Against Candida Albicans. PloS Pathog (2009) 5(10):e1000639. doi: 10.1371/journal.ppat.1000639
76. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil Extracellular Traps Capture and Kill Candida Albicans Yeast and Hyphal Forms. Cell Microbiol (2006) 8(4):668–76. doi: 10.1111/j.1462-5822.2005.00659.x
77. Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U. In Vitro Resistance Mechanisms of Neisseria Meningitidis Against Neutrophil Extracellular Traps. Mol Microbiol (2013) 89(3):433–49. doi: 10.1111/mmi.12288
78. Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe (2010) 8(5):445–54. doi: 10.1016/j.chom.2010.10.005
79. Speziale P, Pietrocola G. Staphylococcus Aureus Induces Neutrophil Extracellular Traps (NETs) and Neutralizes Their Bactericidal Potential. Comput Struct Biotechnol J (2021) 19:3451–7. doi: 10.1016/j.csbj.2021.06.012
80. Winstel V, Missiakas D, Schneewind O. Staphylococcus Aureus Targets the Purine Salvage Pathway to Kill Phagocytes. Proc Natl Acad Sci USA (2018) 115(26):6846–51. doi: 10.1073/pnas.1805622115
81. Bunsow D, Tantawy E, Ostermeier T, Bahre H, Garbe A, Larsen J, et al. Methicillin-Resistant Staphylococcus Pseudintermedius Synthesizes Deoxyadenosine to Cause Persistent Infection. Virulence (2021) 12(1):989–1002. doi: 10.1080/21505594.2021.1903691
82. Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the Dlt Operon in Staphylococcus Aureus Confers Sensitivity to Defensins, Protegrins, and Other Antimicrobial Peptides. J Biol Chem (1999) 274(13):8405–10. doi: 10.1074/jbc.274.13.8405
83. Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, et al. Staphylococcus Aureus Resistance to Human Defensins and Evasion of Neutrophil Killing via the Novel Virulence Factor MprF Is Based on Modification of Membrane Lipids With L-Lysine. J Exp Med (2001) 193(9):1067–76. doi: 10.1084/jem.193.9.1067
84. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-Alanylated Lipoteichoic Acids Protect Streptococcus Pneumoniae Against Neutrophil Extracellular Traps. Cell Microbiol (2007) 9(5):1162–71. doi: 10.1111/j.1462-5822.2006.00857.x
85. Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, et al. D-Alanylation of Lipoteichoic Acids Confers Resistance to Cationic Peptides in Group B Streptococcus by Increasing the Cell Wall Density. PloS Pathog (2012) 8(9):e1002891. doi: 10.1371/journal.ppat.1002891
86. Jann NJ, Schmaler M, Kristian SA, Radek KA, Gallo RL, Nizet V, et al. Neutrophil Antimicrobial Defense Against Staphylococcus Aureus Is Mediated by Phagolysosomal But Not Extracellular Trap-Associated Cathelicidin. J Leukocyte Biol (2009) 86(5):1159–69. doi: 10.1189/jlb.0209053
87. Kiedrowski MR, Crosby HA, Hernandez FJ, Malone CL, McNamara JO 2nd, Horswill AR. Staphylococcus Aureus Nuc2 is a Functional, Surface-Attached Extracellular Nuclease. PloS One (2014) 9(4):e95574. doi: 10.1371/journal.pone.0095574
88. Cao Z, Casabona MG, Kneuper H, Chalmers JD, Palmer T. The Type VII Secretion System of Staphylococcus Aureus Secretes a Nuclease Toxin That Targets Competitor Bacteria. Nat Microbiol (2016) 2:16183. doi: 10.1038/nmicrobiol.2016.183
89. Ohr RJ, Anderson M, Shi M, Schneewind O, Missiakas D. EssD, A Nuclease Effector of the Staphylococcus Aureus ESS Pathway. J Bacteriol (2017) 199(1):e00528-16. doi: 10.1128/JB.00528-16
90. Cahilog Z, Zhao H, Wu L, Alam A, Eguchi S, Weng H, et al. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation (2020) 43(6):2021–32. doi: 10.1007/s10753-020-01294-x
91. Porto BN, Stein RT. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol (2016) 7:311. doi: 10.3389/fimmu.2016.00311
92. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic Fibrosis. Lancet (2021) 397(10290):2195–211. doi: 10.1016/S0140-6736(20)32542-3
93. Cohen TS, Prince A. Cystic Fibrosis: A Mucosal Immunodeficiency Syndrome. Nat Med (2012) 18(4):509–19. doi: 10.1038/nm.2715
94. Papayannopoulos V, Staab D, Zychlinsky A. Neutrophil Elastase Enhances Sputum Solubilization in Cystic Fibrosis Patients Receiving DNase Therapy. PloS One (2011) 6(12):e28526. doi: 10.1371/journal.pone.0028526
95. Montgomery ST, Mall MA, Kicic A, Stick SM, Arest CF. Hypoxia and Sterile Inflammation in Cystic Fibrosis Airways: Mechanisms and Potential Therapies. Eur Respir J (2017) 49(1):1600903. doi: 10.1183/13993003.00903-2016
96. Branitzki-Heinemann K, Mollerherm H, Vollger L, Husein DM, de Buhr N, Blodkamp S, et al. Formation of Neutrophil Extracellular Traps Under Low Oxygen Level. Front Immunol (2016) 7:518. doi: 10.3389/fimmu.2016.00518
97. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating Histones Are Mediators of Trauma-Associated Lung Injury. Am J Respir Crit Care Med (2013) 187(2):160–9. doi: 10.1164/rccm.201206-1037OC
98. Kawabata K, Hagio T, Matsuoka S. The Role of Neutrophil Elastase in Acute Lung Injury. Eur J Pharmacol (2002) 451(1):1–10. doi: 10.1016/s0014-2999(02)02182-9
99. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PloS One (2012) 7(2):e32366. doi: 10.1371/journal.pone.0032366
100. Van Der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP, Marelich GP, Cross CE. Myeloperoxidase and Protein Oxidation in Cystic Fibrosis. Am J Physiol Lung Cell Mol Physiol (2000) 279(3):L537–46. doi: 10.1152/ajplung.2000.279.3.L537
101. Dwyer M, Shan Q, D’Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic Fibrosis Sputum DNA has NETosis Characteristics and Neutrophil Extracellular Trap Release is Regulated by Macrophage Migration-Inhibitory Factor. J Innate Immun (2014) 6(6):765–79. doi: 10.1159/000363242
102. Law SM, Gray RD. Neutrophil Extracellular Traps and the Dysfunctional Innate Immune Response of Cystic Fibrosis Lung Disease: A Review. J Inflamm (2017) 14:29. doi: 10.1186/s12950-017-0176-1
103. Martinez-Aleman SR, Campos-Garcia L, Palma-Nicolas JP, Hernandez-Bello R, Gonzalez GM, Sanchez-Gonzalez A. Understanding the Entanglement: Neutrophil Extracellular Traps (NETs) in Cystic Fibrosis. Front Cell Infect Microbiol (2017) 7:104. doi: 10.3389/fcimb.2017.00104
104. Cohen TS, Takahashi V, Bonnell J, Tovchigrechko A, Chaerkady R, Yu W, et al. Staphylococcus Aureus Drives Expansion of Low-Density Neutrophils in Diabetic Mice. J Clin Invest (2019) 129(5):2133–44. doi: 10.1172/JCI126938
105. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat Med (2015) 21(7):815–9. doi: 10.1038/nm.3887
106. Hsu CC, Hsu RB, Ohniwa RL, Chen JW, Yuan CT, Chia JS, et al. Neutrophil Extracellular Traps Enhance Staphylococcus Aureus Vegetation Formation Through Interaction With Platelets in Infective Endocarditis. Thromb Haemostasis (2019) 119(5):786–96. doi: 10.1055/s-0039-1678665
107. Bitschar K, Staudenmaier L, Klink L, Focken J, Sauer B, Fehrenbacher B, et al. Staphylococcus Aureus Skin Colonization Is Enhanced by the Interaction of Neutrophil Extracellular Traps With Keratinocytes. J Invest Dermatol (2020) 140(5):1054–65 e4. doi: 10.1016/j.jid.2019.10.017
108. Ng CY, Huang YH, Chu CF, Wu TC, Liu SH. Risks for Staphylococcus Aureus Colonization in Patients With Psoriasis: A Systematic Review and Meta-Analysis. Br J Dermatol (2017) 177(4):967–77. doi: 10.1111/bjd.15366
109. Chang HW, Yan D, Singh R, Liu J, Lu X, Ucmak D, et al. Alteration of the Cutaneous Microbiome in Psoriasis and Potential Role in Th17 Polarization. Microbiome (2018) 6(1):154. doi: 10.1186/s40168-018-0533-1
110. Marples RR, Heaton CL, Kligman AM. Staphylococcus Aureus in Psoriasis. Arch Dermatol (1973) 107(4):568–70. doi: 10.1001/archderm.107.4.568
111. Hu SC, Yu HS, Yen FL, Lin CL, Chen GS, Lan CC. Neutrophil Extracellular Trap Formation is Increased in Psoriasis and Induces Human Beta-Defensin-2 Production in Epidermal Keratinocytes. Sci Rep (2016) 6:31119. doi: 10.1038/srep31119
112. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast Cells and Neutrophils Release IL-17 Through Extracellular Trap Formation in Psoriasis. J Immunol (2011) 187(1):490–500. doi: 10.4049/jimmunol.1100123
113. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive Role of Neutrophil Extracellular Traps in Pathogen-Induced Lung Injury. JCI Insight (2018) 3(3):e98178. doi: 10.1172/jci.insight.98178
114. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular Mechanisms of NET Formation and Degradation Revealed by Intravital Imaging in the Liver Vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
115. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and Neutrophil Extracellular Traps Collaborate to Promote Intravascular Coagulation During Sepsis in Mice. Blood (2017) 129(10):1357–67. doi: 10.1182/blood-2016-09-741298
116. Doring G. The Role of Neutrophil Elastase in Chronic Inflammation. Am J Respir Crit Care Med (1994) 150(6 Pt 2):S114-7. doi: 10.1164/ajrccm/150.6_Pt_2.S114
117. Hoeksema M, van Eijk M, Haagsman HP, Hartshorn KL. Histones as Mediators of Host Defense, Inflammation and Thrombosis. Future Microbiol (2016) 11(3):441–53. doi: 10.2217/fmb.15.151
118. Hansen MU, Gotland N, Mejer N, Petersen A, Larsen AR, Benfield T, et al. Diabetes Increases the Risk of Disease and Death Due to Staphylococcus Aureus Bacteremia. A Matched Case-Control and Cohort Study. Infect Dis (2017) 49(9):689–97. doi: 10.1080/23744235.2017.1331463
119. Dunyach-Remy C, Ngba Essebe C, Sotto A, Lavigne JP. Staphylococcus Aureus Toxins and Diabetic Foot Ulcers: Role in Pathogenesis and Interest in Diagnosis. Toxins (2016) 8(7):209. doi: 10.3390/toxins8070209
120. Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus Aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol (2018) 26(6):484–97. doi: 10.1016/j.tim.2017.11.008
121. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet (2021) 397(10281):1301–15. doi: 10.1016/S0140-6736(20)32549-6
122. Weidinger S, Novak N. Atopic Dermatitis. Lancet (2016) 387(10023):1109–22. doi: 10.1016/S0140-6736(15)00149-X
123. Gutierrez Jauregui R, Fleige H, Bubke A, Rohde M, Weiss S, Forster R. IL-1beta Promotes Staphylococcus Aureus Biofilms on Implants. Vivo Front Immunol (2019) 10:1082. doi: 10.3389/fimmu.2019.01082
124. Jung CJ, Yeh CY, Hsu RB, Lee CM, Shun CT, Chia JS. Endocarditis Pathogen Promotes Vegetation Formation by Inducing Intravascular Neutrophil Extracellular Traps Through Activated Platelets. Circulation (2015) 131(6):571–81. doi: 10.1161/CIRCULATIONAHA.114.011432
125. Foster TJ. Immune Evasion by Staphylococci. Nat Rev Microbiol (2005) 3(12):948–58. doi: 10.1038/nrmicro1289
126. Tam K, Torres VJ. Staphylococcus Aureus Secreted Toxins and Extracellular Enzymes. Microbiol Spectr (2019) 7(2):10. doi: 10.1128/microbiolspec.GPP3-0039-2018
127. Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, et al. Characterization of a New Cytotoxin That Contributes to Staphylococcus Aureus Pathogenesis. Mol Microbiol (2011) 79(3):814–25. doi: 10.1111/j.1365-2958.2010.07490.x
128. Mutua V. Gershwin LJ. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin Rev Allergy Immunol (2021) 61(2):194–211. doi: 10.1007/s12016-020-08804-7
129. Manda-Handzlik A, Bystrzycka W, Sieczkowska S, Demkow U, Ciepiela O. Antibiotics Modulate the Ability of Neutrophils to Release Neutrophil Extracellular Traps. Adv Exp Med Biol (2017) 944:47–52. doi: 10.1007/5584_2016_59
130. Bystrzycka W, Manda-Handzlik A, Sieczkowska S, Moskalik A, Demkow U, Ciepiela O. Azithromycin and Chloramphenicol Diminish Neutrophil Extracellular Traps (NETs) Release. Int J Mol Sci (2017) 18(12):2666. doi: 10.3390/ijms18122666
Keywords: Staphylococcus aureus, immune evasion, neutrophil extracelluar traps, pathogenesis, NETosis
Citation: von Köckritz-Blickwede M and Winstel V (2022) Molecular Prerequisites for Neutrophil Extracellular Trap Formation and Evasion Mechanisms of Staphylococcus aureus. Front. Immunol. 13:836278. doi: 10.3389/fimmu.2022.836278
Received: 15 December 2021; Accepted: 19 January 2022;
Published: 14 February 2022.
Edited by:
Bart Tummers, St. Jude Children’s Research Hospital, United StatesReviewed by:
Nathan K. Archer, Johns Hopkins Medicine, United StatesYudong Liu, Chinese Academy of Medical Sciences, China
Copyright © 2022 von Köckritz-Blickwede and Winstel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Volker Winstel, d2luc3RlbC52b2xrZXJAbWgtaGFubm92ZXIuZGU=