
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 May 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.835711
This article is part of the Research Topic Insights in Parasite Immunology: 2021 View all 9 articles
 Juliana C. Costa-Madeira1
Juliana C. Costa-Madeira1 Gabrielly B. Trindade1
Gabrielly B. Trindade1 Paulo H. P. Almeida1
Paulo H. P. Almeida1 João S. Silva1,2
João S. Silva1,2 Vanessa Carregaro1*
Vanessa Carregaro1*A key point of immunity against protozoan Leishmania parasites is the development of an optimal T cell response, which includes a low apoptotic rate, high proliferative activity and polyfunctionality. During acute infection, antigen-specific T cells recognize the pathogen resulting in pathogen control but not elimination, promoting the development and the maintenance of a population of circulating effector cells that mount rapid response quickly after re-exposure to the parasite. However, in the case of visceral disease, the functionality of specific T cells is lost during chronic infection, resulting in inferior effector functions, poor response to specific restimulation, and suboptimal homeostatic proliferation, a term referred to as T cell exhaustion. Multiple factors, including parasite load, infection duration and host immunity, affect T lymphocyte exhaustion. These factors contribute to antigen persistence by promoting inhibitory receptor expression and sustained production of soluble mediators, influencing suppressive cell function and the release of endogenous molecules into chronically inflamed tissue. Together, these signals encourage several changes, reprogramming cells into a quiescent state, which reflects disease progression to more severe forms, and development of acquired resistance to conventional drugs to treat the disease. These points are discussed in this review.
Leishmaniasis is a set of anthropozoonotic diseases caused by several species of trypanosomatids of the Leishmania genus, comprising species responsible for different pathologies (1). Clinically, leishmaniasis can manifest in tegumentary (localized, disseminated or diffuse), mucocutaneous and visceral forms. The taxonomy of Leishmania correlates a particular species with a particular clinical manifestation in humans. The parasite species of the Leishmania mexicana and Leishmania braziliensis complex (in the New World) and Leishmania major and Leishmania tropica (in the Old World) are related to cutaneous and mucocutaneous forms, while the parasites of the Leishmania donovani complex, the species Leishmania donovani and Leishmania infantum, are associated with the visceral form of the disease. Visceral leishmaniasis (VL), or kala azar, is the most severe form of disease. The disease can be caused by L. donovani in India and sub-Saharan Africa and L. infantum in southern Europe, North Africa and Brazil (2, 3). The disease is characterized by spreading of parasites to the viscera (mainly spleen, liver and bone marrow). The clinical forms are diverse, and affected individuals may present symptoms ranging from spontaneous cure and oligo and asymptomatic forms to severe manifestations, which may lead to death (4). The symptomatological spectrum of the disease is characterized by dissemination of parasites to the organs (5). Symptoms such as prolonged fever, asthenia, anorexia, weight loss, hepatosplenomegaly, pancytopenia, hypergammaglobulinemia and severe anemia are present in individuals with the disease. In the most severe form of VL, alopecia and edema of the lower limbs can be observed, as well as hemorrhagic manifestations, such as epistaxis, ecchymosis and petechiae, as a result of liver alterations and thrombocytopenia resulting from pathological liver changes (6). Another important aspect is the development of neutropenia, which promotes host susceptibility to bacterial infections (7). Together, these signs and symptoms may intensify the severity of disease and lead to patient death if not properly treated. Current treatment options are far from ideal, with diverse outcomes based on multiple factors, including geographic location, immune status, disease stage, and patient comorbidities. Current first-line treatments for VL, such as amphotericin B (liposomal or deoxycholate formulations), miltefosine, paromomycin, and antimonials, are far from ideal for use in resource-poor settings due to issues such as teratogenicity, clinical resistance and/or relapse, the long-term nature of the treatment regimens and parenteral administration (4, 8). These findings suggest that exploring the therapeutic potential of immunomodulatory drugs for the treatment of VL, mainly in areas of anthroponotic transmission (transmitted by only humans), is warranted (9).
It has been well established in a murine model that host resistance is related to CD4+T cells producing IFN-γ (T helper 1 (Th1) subset). Dendritic cells (DCs) capture parasites at the site of infection and migrate to draining lymphoid organs, where they stimulate the differentiation of naïve CD4+T cells to differentiate into the Th1 subset in a manner dependent on IL-12 production (10). Along with CD4+T lymphocytes, natural killer (NK) cells and CD8+T cells are also important sources of IFN-γ (11). Such cytokines act on macrophages, activating inducible nitric oxide synthase (iNOS) with concomitant production of nitric oxide (NO), leading to the death of phagocytosed parasites (12). TNF is produced by infected macrophages and acts with IFN-γ, increasing iNOS activation and thus leading to NO-mediated parasite death (13–15). Furthermore, Th17 response controls infection by acting in synergism with IFN-γ, increasing NO production by macrophages, and promoting neutrophil influx into the target organs of the disease (16, 17). Conversely, the Th2-type response is related to susceptibility and disease progression (18–20). A microenvironment with a predominance of IL-4, IL-13 and IL-10 induces the differentiation of naïve CD4+ T cells into the Th2 subtype, promoting parasite survival (21). Leishmania parasites can use monocytes to induce the infection establishment into the host, as mechanisms of subversion of the immune response and promoting parasites survival. In this sense, it has been demonstrated that L. donovani induces the expansion of hematopoietic stem cells (HSCs) and differentiation of GMP (granulocyte-monocyte progenitors) cells into Ly6Chi (an inflammatory monocytes) with a more permissive profile to infection, characterized by the expression of regulatory markers such as Sca1, Galactin-3, MHC-II and IL-10 (22). In addition to Kupffer cells and macrophages from the marginal zone of the spleen, L. donovani also infects these Ly6Chi recruited to the spleen and liver during infection and compromises the ability of these cells to control parasites, which contributes to susceptibility to disease (23, 24).
As mentioned above, hepatosplenomegaly is a hallmark of VL. This symptom results from an imbalance between the immune response that controls parasite replication and those that allow the pathogen to persist. In experimental models, the infection resolves within 3 months. This resolution is related to the development of well-formed granulomas by Th1 cells (25). Neutrophils and macrophages surround infected cells (macrophages and Kupfer cells) and are activated by IFN-γ, leading to NO release from phagocytes and promoting parasite restriction (26, 27).
Both Th1 and Th17 immune responses are driven by innate molecules, receptors such as TLRs (TLR2, TLR7, TLR8, and TLR9) and NLRs (NOD2 and NLRP3), and cytokines (IL-1β, IL-6, IL-12, IL-23 and IL-18) expressed by DCs, which influence parasite restriction (28–30). Other innate receptors, such as TLR4, are exploited by parasites to both subvert the Th1 immune response and successfully establish themselves in the vertebrate host (31). Moreover, the signaling triggered through NOD2-RIP2 induces a Th1 response but inhibits genes related to the Th17 subtype in a murine model and in leukocytes recovered from VL patients (32). An interesting issue is that, in general, VL-causing species are less inflammatory than cutaneous leishmaniasis-causing species (33). Thus, in the early stages of the disease, the liver is less affected and suffers damage with chronicity. This promotes disease progression without promoting parasite elimination.
During the acute phase of Leishmania infection, specific T cell populations undergo a dramatic numerical increase and differentiate to cells with appropriate effector functions for declining parasite number. This process is usually followed by a substantial loss of effector cells but maintains an elevated number of memory T cells, which can be efficiently deployed when an individual is reinfected by the parasite (Figure 1A). However, several patients become immunosuppressed and may succumb to secondary infections. This inability to control visceral infection has been attributed to a defect in generating a cellular immune response, which enables parasite survival (34, 35). Persistent exposure to antigens and/or chronic exuberant inflammation alters several T cell functions, mainly those of memory cells, and is one of the causes of immunopathology development that occurs during the active form of the disease. This process is known as cell exhaustion, and it precludes an effector immune response for long periods. The cell exhaustion process is followed by an increase in parasite load. In such scenarios, the impairment of T cell effector functions is supported by the increase and coexpression of several inhibitory receptors, cytokines and endogenous molecules that promote several alterations, reprogramming cells to a quiescent state (Figure 1B).
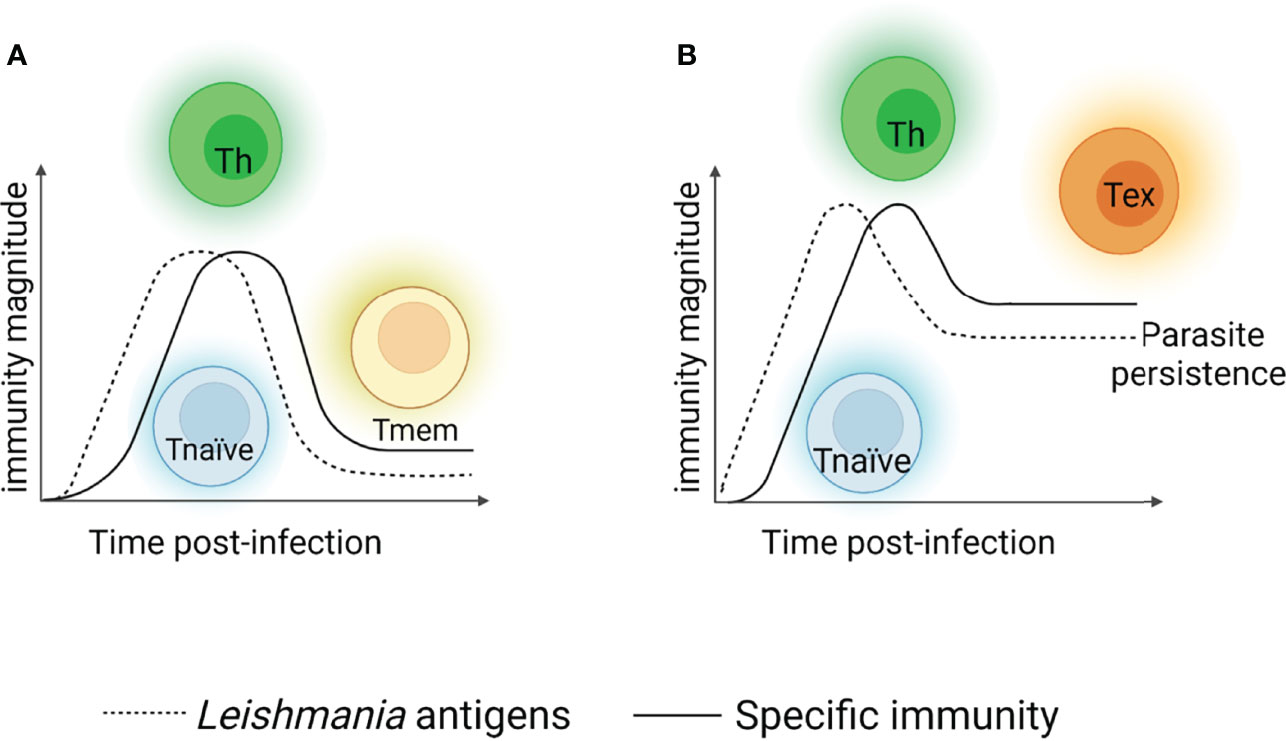
Figure 1 T cell response after viscerotropic leishmania parasites recognition. When L. donovani or L. infantum antigens are recognized, specific naïve CD4+ T cells (blue cells) are activated and differentiate into distinct subtypes of T helper lymphocytes (Th1, Th17 – green cells), restricting the parasites replication. After parasite declining, the most of Th cells die and the remaining cells differentiate into memory T cells (yellow cells). In the case of secondary exposure to the microorganism, memory T cells may be reactivated and promoting parasite control (A). Situations where the specific immune response generated in the host cannot control the parasite, a chronic infection process is established. The parasite’s persistence induces Thelper cells to enter a non-functional state, a term referred as exhaustion (represented by orange cells), rather than developing a classical memory cells. Exhausted T cells present a compromised effector functions and maintain the persistence of the parasites in the host for long periods, which reflects disease progression to more severe forms, but it was not found it in asymptomatic individual or patients with mild form of disease (B). Created with BioRender.com (Agreement number: IK23QKCBAC).
Although the exhaustion term was first described during viral infections, we recognize that parasites evoke a robust immune response in the host that often fails to eliminate the pathogen (33). In this sense, the participation of exhausted T cells in the immunopathogenesis of leishmaniasis has been investigated. The presence of T cells with exhaustion phenotypes has been described in infections caused by species that cause visceral disease and those that cause the cutaneous manifestations. Interestingly, the exhausted T cells seem to affect the outcome of the disease differently depending on the species of Leishmania involved. During L. mexicana infection, dendritic cells produce high amounts of TNF, contributing to the T cells exhaustion, compromising the proliferation and functionality of these cells, which favors the progression of the disease (36). Patients infected by L. braziliensis show increased expression of exhaustion markers on CD4+ T cells and CD8+ T cells in the skin and in the bloodstream as well. However, the extent of the lesion is not related to the expression of inhibitory molecules such as PD-1, suggesting that exhausted T cells do not interfere in the pathogenesis of the disease (37). In L. major infection, characterized by lesions that heal spontaneously in most affected individuals, the infection is controlled within 3-8 weeks. However, no sterile cure is observed and the persistence of remaining parasites, controlled in part by CD4+CD25+ regulatory T cells, appears to be crucial for the maintenance of short-lived CD4+ effector T cells, protecting the individual from reinfection. This protection is lost when the parasites are controlled, suggesting that antigenic persistence is necessary for long-lasting immunity, although it can induce exhaustion phenotype in antigen-specific cells (38–40). Conversely, the persistence of the parasites in visceral leishmaniasis promotes a dysfunctional response of CD8+T lymphocytes, which encourages parasite survival and replication. Even though CD8+T cells are increased in the blood and lesions of chronically infected patients, individuals with severe VL show impaired cellular proliferation and cytokine production (i.e., IL-2 and IFN-γ by these cells. In a VL experimental model induced by L. donovani, CD8+T lymphocytes do not undergo significant proliferation or activation, maintaining antigen persistence (41, 42). The presence of the parasite for a long period promotes the exhaustion of CD8+T lymphocytes, generating cells with limited capacity to produce IFN-γ, which leads to cell death (43). CD8+T cells from transgenic L. donovani expressing ovoalbumin cells exhibited a biphasic wave of activation, with the first wave being involved in a limited expansion and the second wave resulting in the death and exhaustion of CD8+T lymphocytes. Peripheral blood-derived mononuclear cells (PBMCs) recovered from L. infantum-infected patients with the severe disease neither proliferate nor produce IFN-γ after in vitro soluble Leishmania antigen (SLA) stimulation. PBMCs isolated from healed VL patients respond to specific antigens and produce IFN-γ, suggesting that reversal of the profile of exhausted cells can lead to a favorable clinical outcome (44–46). It is important to consider that the field of CD4+T lymphocyte exhaustion is less explored, as from an evolutionary point of view, CD8+T lymphocytes are more vulnerable than CD4+T cells to tolerogenic stimuli, including prolonged persistence of specific antigen stimulation during chronic infection. This scenario protects the infected cells from cytotoxic responses and significantly reduces the inflammatory stimuli that could cause immunopathology in the host (47), and these mechanisms are promising topics to explore to increase the understanding of the physiopathogenesis of VL. Interestingly, different from cutaneous disease, where C57BL/6 or BALB/c strain may be employ as resistance and susceptible model of infection, respectively, the murine VL model is a clear example of organ-restricted immunity, where the parasites are naturally declined in long-term, without the development of clinical manifestations in mice strain-independent (26, 48). Although the most susceptible mouse strain, including BALB/c, developed a specific immunity, they may control parasites growth at longer term of infection. Thus, it may be plausible to consider the process of exhausted T cells could occur in mice strain independently in murine model of disease.
We do not discard the possibility that the failure to detect cellular-mediated immunity (CMI) in some sample of patients with the classical VL disease could be also an interference of the assay systems employed versus a lack of a CMI response. Although it has been demonstrated that the development of a Th1 response is necessary for the control of the parasite, recently it was described that patients with active VL present high levels of IFN-γ in the bloodstream, bone marrow, in addition to increased expression of such cytokine in organs such as the spleen and liver, suggesting that there is no defect in cell-mediated immunity in these patients (41, 49, 50). Furthermore, studies using whole blood assays show that, unlike from PBMC assays, CD4+ T cells from patients with active VL produce IFN-γ when re-stimulated in vitro with parasite antigens, in example, the absence of a response cell-mediated immune response observed in patients with active VL appears to be more due to the detection method employed than an actual failure of cellular immunity in VL generated (51, 52). In this sense, in humans, the disease progression appears to be unrelated to the production of IFN-γ and other mechanisms may be involved in the immunopathogenesis of the VL disease, such as cytokines, inhibitory molecules, endogenous mediators and suppressive cells. All these issues will be covered in this Review.
Given their ability to produce anti-inflammatory cytokines, to express high levels of inhibitory receptors, Tregs block the activation and function of innate and adaptive immune cells, promoting immune response control and maintenance of self-tolerance (53, 54). Despite the beneficial roles of Tregs in the host, including preventing the development of immunopathologies, their suppressive function can be exploited by some types of microorganisms to promote escape of the host immune response (55). This scenario is commonly seen during infections caused by Leishmania parasites (56).
The presence of both IL-10 and TGF-β cytokines in the infected tissue promotes the suppression of the protective immune response (i.e., Th1 cells), causing persistence of the parasite and chronicity of the disease, strongly suggesting the involvement of Tregs in the disease establishment (57–59). CD4+T cells often coproduce IL-10 and IFN-γ, designated type 1 regulatory T cells (Tr1s), and promote the onset of infection by suppressing Th1 cell-mediated immunity (50, 59, 60). The role of IL-10 and TGF-β in immunosuppression and disease progression is well documented in both experimental and human VL (9, 61). Regarding L. donovani infection, studies report that the increase in parasite number is related to the presence of Tregs and heightened levels of systemic IL-10 and TGF-β in patients with active disease. Corroborating the role of Tregs in disease progression, a reduced hepatic parasite load was observed in animals depleted of Tregs using both anti-CD25 and anti-FR4 neutralizing antibodies (62, 63). It was identified that such cells produce only TGF-β in a sustained manner, while the production of IL-10 was attributed to CD4+CD25- T cells and DCs. TGF-β produced by Tregs was also observed in an experimental model of VL caused by L. infantum.
Despite not directly influencing parasite replication, Tregs play other roles, mainly in tissue protection and controlling leukocyte activation in both the initial and chronic phases of infection. These roles are independent of IL-10. In contrast, infection caused by L. infantum and TGF-β production by Tregs are related to the growth and persistence of the parasite, in addition to acting in the control of immunopathologies developed during infection (50, 59, 60, 62, 64). In experimental models of VL with L. infantum or L. donovani, treatment with CXCL10 promotes the protection of infected mice by stimulating the Th1 response, which decreases the population of Tregs and CD4+CD25- T cells producing TGF-β and IL-10, leading to a reduction in the number of parasites in the spleen of treated animals and a consequent reduction in organ size (65, 66). Thus, Tregs contribute to T cell exhaustion by suppressing the effector functions of T lymphocytes and contributing to the persistence of the parasite.
Inhibitory receptors are negative regulators that control autoreactivity and prevent the development of immunopathology. Although some of these receptors are transiently expressed on functional effector T cells during activation, the high and sustained expression of inhibitory receptors is a hallmark of exhausted T cells. In experimental VL, it has been shown that chronic infection promotes the upregulation of several inhibitory receptor genes and some of their ligands. Programmed cell death protein 1 (PD1)-mediated inhibitory signaling in response to PDL1 and PDL2 provides a classic example by which this pathway manages T lymphocyte exhaustion. PD-1 is an inhibitory receptor and a member of the B-7 costimulatory receptor family expressed by all activated T cells, although it can also be expressed by other cell types (67). PD-1 regulates lymphocyte activation by binding to PD-L1 (B7-H1) and PD-L2 (B7-H2) ligands on lymphocytes and is essential for the maintenance of tolerance and homeostasis and the prevention (67, 68). Although the PD-1/PD-L1 or PD-L2 pathway plays an role in regulating the immune response magnitude, it may limit protective immunity against persistent antigens, a response observed in both cancer and chronic infection (69). It has been shown that experimental VL induced by L. donovani results in increased PD-1 expression by CD8+T cells with a phenotype of cell exhaustion, characterized by low expression of IL-12, IFN-γ and TNF, and blockade of PD-L1 partially recovers cell function (70). Spleen cell cultures from hamsters infected with L. donovani do not respond to parasite antigens. This unresponsiveness of CD4+T lymphocytes correlates with an increased production of regulatory cytokines, such as IL-10, TGF-β, IL-27, IFN-I, and inhibitory receptors, such as PD1 (71). In vivo blockade of PD1 using specific antibodies decreases arginase-1 expression in macrophages, resulting in a reduction in the parasite load of the organ (72). In addition to restoring the function of both CD4+ T and CD8+ T lymphocytes and decreasing parasite numbers, blocking the PD1/PDL1 pathway with anti-PDL1 antibodies reverses inhibition of caused by L. donovani, a mechanism used by the parasite to subvert the host’s autophagic machinery to encourage survival and induce the establishment of infection (73). In canine VL caused by L. infantum, PD1 blockade restores the effector functions of both CD4+ T and CD8+ T lymphocytes and the production of reactive oxygen species (ROS) by monocytes recovered from dogs with active VL, thus controlling the parasites replication (72). The interaction between PD1 and PDL1 increases the expression of FOXP3 and enhances the immunosuppressive activity of Tregs (74, 75). Furthermore, PDL1 converts naïve CD4+T cells into Tregs by downregulating AKT, mTOR and ERK2 and simultaneously upregulating PTEN (74). Thus, blocking the PD-1/PD-L1 axis reverses cellular exhaustion, exerts effects on peripheral tolerance by interfering with Treg induction and function, and may be a potential strategy for the treatment of patients with VL, mainly those unresponsive to conventional treatments.
There is currently a debate regarding the effect of immunotherapy that blocks the PD1/PDL1 or PDL2 axis in clinical oncology. Although highly effective for different types of cancer, rapid tumor progression was observed in approximately 10% of patients with advanced gastric cancer using an anti-PD1 monoclonal antibody (76–78). These patients had FOXP3+ Tregs with a high proliferation rate and increased suppressive function. Likewise, PD1-deficient murine Tregs are more proliferative and suppressive than wild-type Tregs isolated from the tumor (79). Evidence on the PD1/PDL1 interaction during Treg induction and function in VL is still scarce, but it is a topic that warrants investigation considering that blocking this pathway may generate a subversive response, maintain the persistence of the parasite, or increase the effector response and promote immunopathologies.
Cytotoxic T lymphocyte antigen 4 (CTLA-4) is another inhibitory receptor related to T lymphocyte expansion. It is a transmembrane glycoprotein homologous to the CD28 costimulatory receptor and essential for the regulation of the immune system. Both CD28 and CTLA-4 receptors share the same ligands, B7-1 (CD80) and B7-2 (CD86), expressed by antigen-presenting cells (APCs) during antigen presentation to T cells (80, 81). Competition between receptors for ligands reduces the number of specific T cells, which constitutes an important strategy for controlling the magnitude of responses in peripheral tissues to prevent the tissue damage (80, 82). The increased expression of such molecules has been demonstrated during acute infections and chronic infections, in which the antigen persists. Although this regulatory mechanism minimizes tissue damage, it can also compromise the elimination of pathogens, favoring their persistence in the host (83). During chronic VL infections, CTLA-4 is one of the markers of T cell exhaustion (84), and its role in susceptibility to infection in murine models, humans and dogs has been reported. CTLA-4 blockade during L. donovani infection increases parasite resistance in BALB/c mice, characterized by increased IFN-γ- and IL-4-producing cells and the rapid development of hepatic granulomas, which contain the spread of the parasite (85). Mice infected with L. donovani anti-CTLA-4 antibodies showed induction of increased leishmanicidal activity, IFN-γ production and increased hepatic granulomas compared to the untreated group (86, 87). Likewise, CD4+T cells recovered from the spleen of L. infantum-infected BALB/c mice show a poor proliferative rate in response to anti-CD3 stimuli and do not respond to Leishmania Lcr1 antigen, but in vitro, the blockade of CTLA-4 partially recovers the response (88). One suppressive mechanism mediated by CTLA-4 is the induction of TGF-β production by T cells, which inhibits the production of IFN-γ (89, 90), it has been shown that the stimulation of CD4+T cells derived from the spleen of animals infected with L. infantum results in prominent production of TGF-β. Such a response is not seen when CTLA-4 is blocked, suggesting in vitro growth of L. infantum depends on the expression of both CTLA-4 and TGF-β, suggesting that the induction of the CTLA-4/TGF-β pathway is important for the replication of the parasite (91). Regarding human disease, the timing of lymphocyte exhaustion is controversial. While Clarencio et al. (92) demonstrated a lower frequency of CTLA-4+ T cells in patients with VL, Gautam et al. (43) observed greater expression of CTLA-4 and PD-1 mRNA, important markers of T cell exhaustion, in CD8+ T cells. Similar results were observed by Viana et al. (93) and Clarêncio et al. (92). However, CTLA-4 blockade did not alter IFN-γ levels or parasite survival in culture, suggesting that CTLA-4 is not the only molecule responsible for T cell dysfunction during human VL (43). Analysis of the transcriptional profile in CD8+T cells recovered from peripheral blood from patients also revealed increased expression of CTLA-4 (94). Polymorphisms in the gene encoding CTLA-4 (CTLA-4+49-A/G) may be a risk factor for VL development, it was also demonstrated that VL patients who had the polymorphism presented higher anti-Leishmania antibody titers (95). Together, these data indicate that CTLA-4 plays an important role in the T cell exhaustion process observed in VL.
In addition to PD1 and CTLA-4, increased mRNA expression of T cell immunoglobulin and mucin-domain containing-3 (TIM-3) and lymphocyte activation gene-3 (LAG3) is also observed in peripheral blood samples from patients with active VL compared to asymptomatic individuals and endemic controls, which suggests a relationship between the expression of exhaustion markers and disease severity (96). Such receptors are expressed on the lymphocyte membrane after their activation, and when interacting with costimulatory receptors, they interrupt the TCR-dependent cell signaling pathway (97–99), thus maintaining the suppression of the effector response against the parasite and parasite persistence. Studies also demonstrate that LAG-3 and TIM3 are differentially expressed in both natural and induced regulatory T cells (iTregs) and are necessary for their suppressive function (98, 100, 101). In this sense, evaluation of these inhibitory markers on the cell surface is important, as their presence represents the exact moment that T cell exhaustion appears and interferes with the development of protective immunity against the parasite; therefore, they could be biomarkers of disease progression.
The chronic inflammation developed during VL is associated with characteristics that commonly result in a hypoxic microenvironment, such as compromised blood microcirculation and energy demand (102). In this context, there is a reduction in the tissue oxygen supply, leading to the activation of the transcription factor hypoxia inducible factor 1-α (HIF-1α) (103, 104). The role of HIF-1α has been demonstrated during L. donovani infection. Parasite infection promotes the increased expression and activation of HIF-1α (105, 106), and macrophages in which HIF-1α is silenced using siRNA control intracellular parasite replication, suggesting that protozoa can use this pathway to survive and proliferate inside the cell (106). The transcription factor IRF-5 compromises the expansion of CD8+T cells during L. donovani infection, and this effect is dependent on HIF1-α in DCs (107). The absence of HIF-1α in DCs results in increased CD8+T cell proliferation, enhanced CD4+T cell recruitment to the spleen and a pronounced Th1 response, ideal for parasite control, suggesting that HIF-1α may be involved in T cell exhaustion (107, 108). Corroborating these findings, Hammami et al. (109) demonstrated that myeloid cells from L. donovani-infected spleens present a phenotype of myeloid-derived suppressor cells (MDSCs) mediated by HIF-1α. Furthermore, HIF-1α appears to be important in the polarization of macrophages into the M2 phenotype, with M2 macrophages being less efficient in controlling the parasite. Conversely, the data suggest that HIF-1α is involved in the persistence of the parasite in the host. Mesquita et al. (110) showed that the absence of HIF-1α increases the susceptibility to infection by L. donovani, demonstrating a protective role of HIF-1α during VL. The levels of HIF-1α were higher in both infected C57BL/6 and 129/Sv mice, strains that are naturally resistant to the parasite, than in BALB/c animals, which are susceptible to the disease. Likewise, the absence of HIF1-α results in metabolic dysregulation and increased lipogenesis, which seem to favor parasite growth (110). Although there are contradictory data in the literature, most evidence indicates that HIF1-α favors the persistence of the parasite in the host, acting as a molecule that suppresses protective immunity against pathogens. HIF1-α could also contribute to the persistence of the parasite by acting on the differentiation and functions of Tregs. The role of HIF1-α in regulatory T cells has already been explored by others (111). HIF1-α-/- Tregs lack suppressor function and produce IFN-γ in an excessive manner. The FOXP3 gene promoter in HIF1-α-/- Tregs fails to protect the animal from colitis caused by effector T cells, further demonstrating the role of HIF1-α in the suppressive functions of Tregs (109, 112). To date, the role of HIF1-α in Treg differentiation and functions during VL is not known.
Another endogenous molecule released under conditions of cellular hypoxia is adenosine. Derived from ATP degradation by the action of the ectonucleotidases CD39 and CD73, adenosine is a critical immunosuppressive metabolite released during chronic inflammation and involved in T lymphocyte exhaustion (113–116). In cutaneous leishmaniasis (CL), ADO and AMP act via the A2AR adenosine receptor to induce tolerogenic dendritic cells (tDCs) through the sequential production of prostaglandin E2 (PGE2) and IL-10. As a consequence, both mediators inhibit the proliferative ability of CD4+ T lymphocytes and IFN-γ production to hinder to the induction of a regulatory profile in such leucocytes, promoting the suppression of the effector immune response against parasites (117). Furthermore, VL patients present elevated serum levels of adenosine, linking ectonucleotidase activity to disease progression (118). Under inflammatory conditions, the A2B receptor is also expressed on monocytes recovered from patients with VL (119), suggesting that during disease, Leishmania parasites may use the adenosinergic signaling pathway to avoid the host’s immune response and promote to their own silent growth, thus ensuring their survival inside the cell. In L. infantum infection, the parasite benefits from the A2AR signaling pathway and promotes the development of an immunosuppressive response mediated by Tregs and IL-10; this inhibits specific Th1 responses, thus allowing the escape of parasites and establishment of infection (58). Increased expression of CD39 and CD73 is observed in effectors and memory T cells with pronounced IFN-γ production and serves to downregulate lymphocyte activation, preventing the host from developing immunopathologies. Soluble factors such as TGF-β and IL-10 can increase the frequency of ectonucleotidases expressed in T cells (117, 120). However, this phenomenon remains to be investigated during VL. The increased and maintained expression of endogenous molecules related to tissue hypoxia, such as HIF-1α and/or the CD39/CD73 ectonucleotidases, may contribute to patient unresponsiveness to conventional drugs applied in the treatment of VL. Such gold-standard drugs not only kill the parasite but also alter host immunity. Antimonial drugs stimulate the production of ROS and NO, while miltefosine and AmBisome induce the secretion of IFN-у, TNF, IL-12, IL-6 and IL1β from leukocytes and reduced anti-inflammatory cytokines (121–124). Thus, both HIF-1α and increased adenosine in the chronic inflammatory microenvironment maintain the constant production of anti-inflammatory cytokines such as IL-10 and TGF-β, promoting the induction of T lymphocyte apoptosis and generation of Tregs; these effects inhibit host immunity and favors the persistence of the parasite, factors that contribute to ineffectiveness of antiparasitic drugs.
TGF-β is an anti-inflammatory cytokine produced by antigen-specific T cells and by phagocytic mononuclear cells (125). It presents several immunosuppressive effects during infectious diseases, including inhibition of T lymphocyte proliferation, proinflammatory cytokine release and macrophage activation (126). TGF-β inhibits both TNF and IFN-γ function and controls iNOS expression and the development of naïve CD4+ T lymphocytes into Th1, Th2, and Th17 cells. TGF-β acts as a facilitator of parasitic growth by modulating both innate and adaptive responses (127) and increasing arginase expression in macrophages (128, 129). In a murine model, TGF-β secreted by lymphocytes in response to Leishmania antigens shifts the arginine pool from iNOS to arginase as a source of polyamines, which support parasite growth (130). TGF-β is able to induce apoptosis in lymphocytes isolated from lymph nodes of hamsters infected with L. donovani (131). During L. infantum infection, IL-12-deficient C57BL/6 mice show an increased parasite load due to the sustained production of TGF-β in those mice (132).
Another cytokine that plays an important suppressive role in VL is IL-10, and its role in the progression of visceral disease is already well established in both murine and human models. High levels of IL-10 are associated with a higher parasite load and are related to the development of the most severe form of disease (61, 133, 134). Both BALB/c and C57BL/6 mice lacking IL-10 due to either gene deletion or blockade using specific antibodies are resistant to Leishmania and do not develop the clinical manifestations of the disease (135–137). In line with these findings, it has been observed that splenic cells aspirated from patients with active VL present higher levels of TNF and IFN-γ under conditions of in vitro IL-10 neutralization. Furthermore, such cells present a greater ability to kill parasites (138). Such effects occur because IL-10 is one of the main factors responsible for attenuating the proliferation and activation of T cells, compromising the microbicidal function of macrophages during infection. Impaired immunity is mainly characterized by reduced iNOS expression and NO release (57, 139). Furthermore, the production of IL-10 is associated with T cell differentiation into Th2 cells. Several leucocytes can be sources of IL-10, such as conventional Tregs, Tr1s, CD8+ T cells, B cells, NK cells, DCs, macrophages, and neutrophils (60, 61). Among these cells, conventional Tregs, characterized as CD4+CD25+FOXP3+ T cells, and conventional effector T cells, characterized as CD4+CD25-FOXP3- T cells, are the main sources of IL-10 and are involved in susceptibility to disease. It has been shown that the expansion of IL-10-producing CD4+CD25+FOXP3+ T cells in the plasma of patients with active VL is related to a higher parasite load (64), and there is significant enrichment of this population in bone marrow aspirates from patients with a high parasite load (63). Conditional knockout mice, in which only conventional Tregs do not produce IL-10, present better control of the parasite, although they also display greater disorganization of the splenic microarchitecture (60). Therefore, IL-10 plays a critical role in limiting the antiparasitic immune response by inhibiting the function of important cells involved in parasite restriction.
Several immunoregulatory molecules and pathways, most notably those associated with IL-10 production, are activated following infection by L. infantum and L. donovani and suppress CD4+ T cell functions. One such mechanism includes the induction of IL-10 by BLIMP-1 expressed on Tr1s during clinical and experimental VL. B lymphocyte-induced maturation protein 1 (Blimp-1) is a transcription factor that plays crucial roles in regulating B and T lymphocyte function (140, 141). In different models of inflammatory disease (i.e., asthma, colitis, and infection), mice with specific Blimp-1 gene deletion in T lymphocytes show pronounced production of cytokines, which contributes to worsening inflammation (142–145). IL-10 produced by Tr1s through the BLIMP-1 pathway suppresses specific immunity to the parasite but plays a critical role in protecting the tissue against inflammatory insults induced during the chronic process caused by the parasite (146).
Type I IFNs, including IFN-α and IFN-β, bind to the IFNAR receptor and display regulatory functions preventing pathogen control. IFNα/β remains elevated during chronic infectious processes and induces the expression of IL-10, indoleamine 2,3-dioxygenase 1 (IDO-1), PDL1 and other negative regulators of T cell responses, such as TIM3, in CD4+ T lymphocytes, in addition to their role in promoting apoptosis of T cells via Fas/FasL (147, 148). In experimental infection caused by L. infantum, type I IFNs (IFN-α and IFN-β) produced during parasite recognition via sequential signaling mediated by TLR4/TRIF/IRF-1 suppress the development of Th1 responses via mechanisms depending on IL-10, which contributes to antigen persistence. Patients with classical symptoms of VL present reduced expression of genes associated with TLR4 and IFN-I compared to asymptomatic individuals and endemic controls, suggesting that failure of the regulatory mechanisms of the immune response favors the exacerbation of inflammation and, consequently, leads to the most severe form of disease (31). Likewise, during L. donovani infection, IFN-I contributes to parasite persistence in the target organs of visceral disease by suppressing the development of the Th1 response and promoting Tr1 expansion. Interestingly, temporary blockade of the type I IFN-mediated signaling pathway ameliorated the therapeutic efficacy of antiparasitic drugs by increasing tolerance of the parasite (149). In both mentioned studies, the improvement of parasite replication control in the absence of the type-I IFN signaling pathway was dependent on a pronounced Th1 response generated in the infected tissues. However, this control was accompanied by an enormous cost to the host, since the aberrant inflammation generated promoted liver damage while the infection progressed (31, 149).
IFN-γ also displays an immunosuppressive effect. Persistent presence of the parasite maintains high levels of IFN-γ, contributing to cell exhaustion by inducing IDO. This enzyme catalyzes the breakdown of tryptophan into kynurenine, inducing apoptosis of T lymphocytes by activating the caspase-8 pathway and releasing mitochondrial cytochrome c (150). Conventional Tregs expressing IDO, PDL1 and CTLA4 are present in the peripheral blood of cancer patients and are strongly related to the severe forms of the disease (151, 152). Interestingly, IDO enzymatic activity in human VL is associated with immunosuppression and can be used as a biomarker of human disease caused by L. infantum (153). In the model of CL caused by L. major, IDO is a key factor attenuating the inflammatory response and increasing parasite replication. Administering IDO inhibitors to mice with well-established infection reduces the parasite load associated with inflammation (154), suggesting that IDO inhibitors may offer a new therapeutic tool for patients with chronic leishmaniasis. IDO inhibitors are already being used in clinical oncology as adjuncts to broaden the therapeutic window and limit the autoimmune side effects that immunobiological therapy can cause in cancer patients (155, 156).
Other cytokines have been implicated in inducing the expression of inhibitory receptors on leukocytes. In this context, the role of IL-27 in increasing TIM3 expression in CD4+ T cells has been illustrated (148). In vivo, the maintained expression of IL-27 promotes the enhancement of inhibitory receptors on T cells, such as PDL1, LAG3, TIGIT and TIM3 (97, 157, 158). IL-27 is composed of the EBI-3 and p28 subunits, belongs to the IL-6/12 cytokine family and was originally described as a cofactor for Th1 lymphocyte differentiation, together with IL-12 (159, 160). The mechanism by which IL-27 induces inhibitory receptors remains to be determined, but it is proposed that stimulation of TCRs by persistent antigens increases chromatin accessibility, allowing IL-27-induced STAT1 to bind directly upstream of the PD-L1, LAG-3, CTLA-4, TIGIT and TIM3 gene promoters (161). Furthermore, IL-27-mediated signaling through IL-27Rα (an IL-27 receptor) is responsible for the upregulation of TIGIT and PD1 in memory T cells during infectious processes, such as toxoplasmosis and malaria, and cancer (97, 158). In a sepsis model, IL-27Rα expression was associated with TIGIT but not with PD1 expression in memory CD4+ T cells and the loss of IFN-γ production (162). During VL caused by L. infantum, the absence of IL-27 promotes prominent Th1 inflammation that causes immunopathology (163). Quirino et al. (163) demonstrated that IL-27 is able to induce IL-10-producing Tr1s and plays an important regulatory role in mediating susceptibility to infection. IL-27 inhibits the production of IL-17A in infected tissue and controls neutrophil influx to target organs, preventing the development of liver injury but favoring parasite growth (163). Similarly, IL-27R-deficient mice infected with L. donovani develop immunopathology due to an increased Th1 response in VL organs (164). Patients with active VL show an increase in systemic IL-27 (163, 165), and IL-27 in the presence of IL-21 promotes the expansion of specific IL-10-producing T cells (45, 166). However, it remains unclear whether IL-27 suppresses the specific immune response of T cells in VL by promoting the expression of multiple inhibitory receptors on CD4+ T and CD8+ T lymphocytes.
IL-27, composed of p28 and EBI3, shares common subunits with IL-35 (p35 and EBI3) and IL-39 (p19 and EBI3) (167, 168). IL-35 has been reported to have immunosuppressive activity in autoimmune diseases, cancer and infectious diseases (169–172). In VL caused by L. donovani, the chronic inflammatory response is accompanied by enhanced Tregs, TGF-β and IL-35, factors that suppress all T cell effector functions. TGF-β and EBI-3 act synergistically to inhibit the Th1 response and maintain parasite persistence. Double neutralization using specific antibodies against EBI3 and TGF-β limits progression of the infection by promoting remarkable effector immunity against the parasite (172). In the chronic phase of VL caused by L. donovani, Tregs suppress the proliferation and functions of Th17 cells via mechanisms that depend on TGF-β and IL-35, preventing disease progression but maintaining parasite persistence. Restoration of the IL-17 response during in vivo neutralization of TGF-β and IL-35 is accompanied by hepatic and splenic resistance consistent with the low parasite load in such organs (173). IL-35 production by Tregs has been shown to play a central role in T cell exhaustion, as indicated by its ability to induce the expression of LAG3, TIM3, and PD1 on the cell surface of tumor-infiltrating CD4+ T and CD8+ T cells (174), and it is a topic that warrants exploration in experimental and human VL.
VL is a serious public health problem, causing high morbidity and mortality in several countries. Among infected individuals, 85% remain asymptomatic, while the other 15% present clinical manifestations, ranging from oligosymptomatic (mild) forms to more severe symptomatic forms that can lead to death if not properly treated. In the absence of effective vaccine strategies, therapy with pentavalent antimonial and amphotericin B is routinely used for VL treatment. However, these drugs present adverse reactions and side effects and require several rounds of treatment, promoting the acquisition of resistance with long-term use (8, 175, 176). An important question that arises is whether the refractoriness of critically ill patients is a consequence of combined and redundant actions of factors that promote the exhaustion of T lymphocytes. The absence of the proliferative response of T lymphocytes to parasite antigens and the inability to produce IFN-γ indicate that cell exhaustion is occurring, and a more accurate assessment should be performed when these features manifest (177). As such, combining immunotherapy, specifically drugs targeting inhibitory molecules and regulatory cytokines, with drugs routinely used earlier in the process when T cell exhaustion appears could reduce the chances of VL patients acquiring resistance to conventional treatments. Such strategies have been gaining attention from the scientific and medical communities (124, 178–180). The optimization of treatments combining typical drugs with immunotherapy might help to delay the emergence of resistance and increase the therapeutic lifespan of the respective drugs. Immunotherapy focused on inhibitory molecules of the immune system, also known as modulators of immune checkpoints, has revolutionized the oncology field, and responses far beyond the remarkable clinical effectiveness in some patients have been achieved. The prevalence of immunotherapy has generated a dramatic change in how the efficacy and toxicity of antineoplastic treatment are evaluated, with a more holistic view of cancer patients. An example of a drug used successfully in clinical oncology is ipilimumab, the first immunological checkpoint-blocking antibody (blocking CTLA4) to be authorized for clinical use. The approval of ipilimumab was quickly followed by the development of monoclonal antibodies targeting PD1 (pembrolizumab and nivolumab) and PDL1 (atezolizumab and durvalumab). Currently, anti-PD1/PDL1 antibodies have become one of the most widely prescribed anticancer therapies (181). The first studies of immunotherapies as treatments for experimental VL came from Murray et al. (57), who reported that anti-IL-10 receptor monoclonal antibodies (mAbs) promote parasite death via iNOS-dependent mechanisms and increase IFN-γ expression. The results obtained from studies of experimental disease models and in human samples suggest that immunotherapy involving different antagonists of the inhibitory molecules involved in the course of disease progression is a promising treatment option for VL. As explored throughout this review, blocking the inhibitory targets of the immune system rescues the host from the subversion of the immune response imposed by the parasite to successfully establish the infection in the organism, leading to healing (Figure 2). Thus, combining inhibitory molecules that block the immune system with conventional drugs used for the treatment of VL could lead to promising results and increase the identification of therapeutic targets that restore the host’s initial defense mechanisms, preventing disease progression. We recognize that the clinical potential in this field is broad and that enormous research efforts and rapid refinement of the understanding of the role of immune system inhibitory molecules in experimental and human diseases are needed.
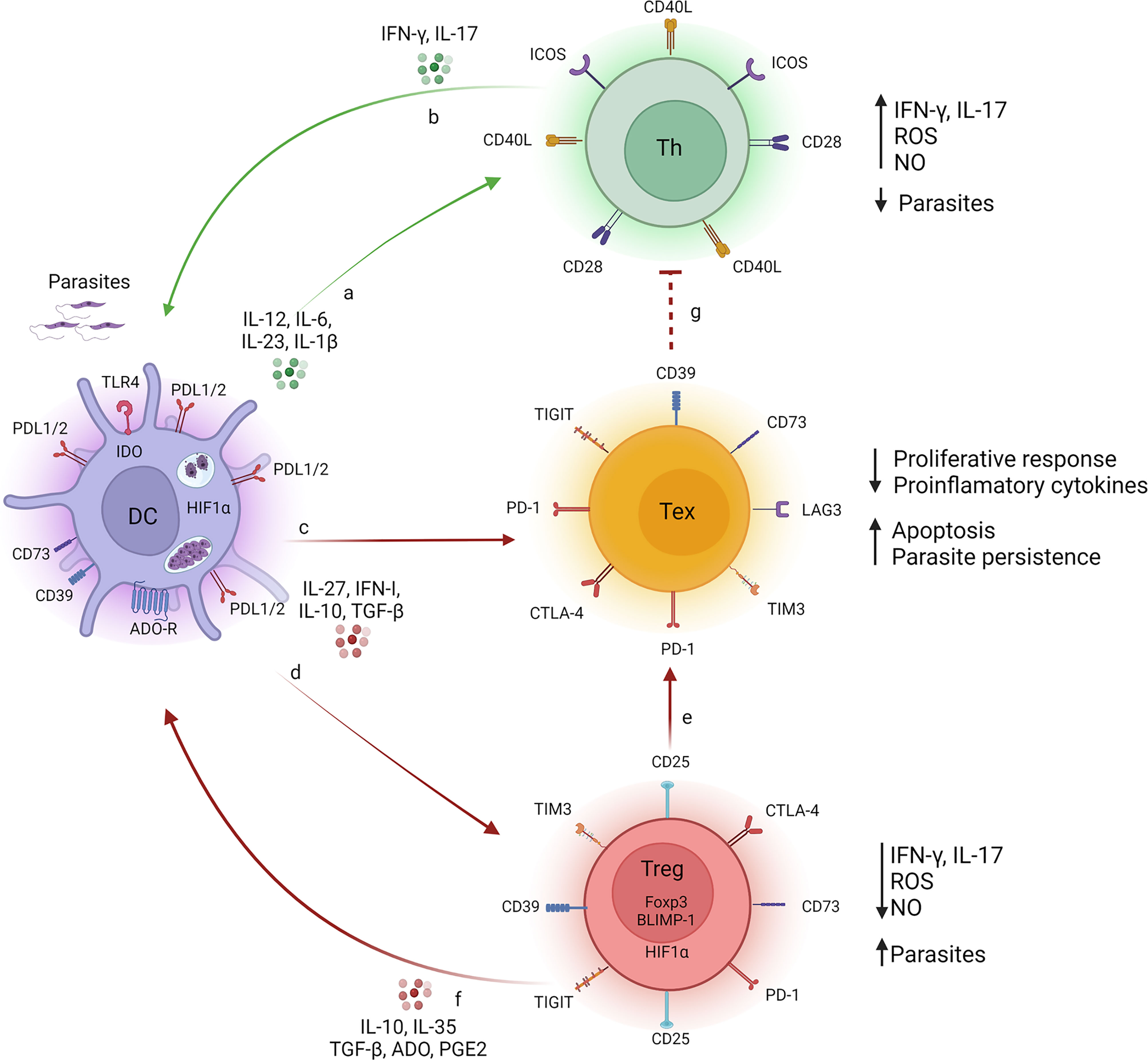
Figure 2 T cell fate during Visceral Leishmaniasis. During infection with L. donovani or L. infantum, monocyte-derived cell (i.e. dendritic cell or macrophage) (purple cells) produce pro-inflammatory cytokines (IL-1β, IL-6, IL-12 and IL-23), which act on naive T lymphocytes, targeting the cells to Th1 and Th17 subtypes (green cells), resulting in the production of IFN-γ/IL-17 (A). Secreted IFN-γ and IL-17 promote the phagocytes microbicidal function of monocyte-derived cell trough the release of ROS and NO, reducing the parasite load (B). The persistent presence of parasites induces a sustained and maintained production of regulatory molecules, such as TGF-β, IL-10, IL-27, PGE2, which promote exhaustion of T lymphocytes (yellow cells), a process characterized by increased expression of inhibitory molecules, such as CTLA-4, PD-1, TIM3, TIGIT, CD39, CD73, LAG-3, on T cell surface. As a consequence, the proliferative response and production of pro-inflammatory cytokines are suboptimal, mechanisms by which the parasite uses to survive in the host-vertebrate (C). Such regulatory molecules influence the inhibitory receptors on the surface of Treg cells (red cells) impacting on their both differentiation and suppressive function (D) and upon DCs functions (F). Such factors repress the protective immune response mediated by IFN-γ/IL-17 and ROS/NO (G). Tregs also respond to regulatory molecules, and contribute to the exhaustion process of T lymphocytes by sustaining the release of regulatory mediators (TGF-β, IL-10, IL-35, PGE2 and adenosine) in the microenvironment and maintaining high levels of inhibitory molecules on the surface of exhausted T lymphocytes (E). Together, these signs favor the persistence of the parasite. Legends: green arrow (induction of effector response), red arrow (induction of suppressor response), red dotted arrow (inhibition of effector response). Created with BioRender.com (Agreement number: LC23QKDV88).
Conceptualization: VC; Writing - Original Draft: JC-M, GT, PA, and VC; Writing - Review and Editing: JS and VC; Funding Acquisition: JS and VC; Resources: JS and VC; and Supervision: VC. All authors contributed to the article and approved the submitted version.
We are thankful to FAPESP, CNPq for their financial support. The research leading to these results has received funding from the São Paulo Research Foundation (FAPESP) under grant agreements N 2019/12991-8 and N.° 2013/08216-2 (Center for Research in Inflammatory Disease) and Universal Project (CNPq) under agreement N° 302419/2018-7 from the University of São Paulo.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are thankful to the São Paulo Research Foundation (FAPESP), CNPq and CAPES for their financial support.
1. dos Santos Marques LH, Rocha DAIC, Reis IA, Cunha DAGM, Oliveira E, Pfeilsticker TR, et al. Leishmania Infantum: Illness, Transmission Profile and Risk Factors for Asymptomatic Infection in an Endemic Metropolis in Brazil. Parasitol (2017) 144(4):546–56. doi: 10.1017/S0031182016002134
2. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, et al. Visceral Leishmaniasis: What Are the Needs for Diagnosis, Treatment and Control? Nat Rev Microbiol (2007) 5(11):873–82. doi: 10.1038/nrmicro1748
4. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet (2018) 392(10151):951–70. doi: 10.1016/S0140-6736(18)31204-2
5. Mauricio IL, Stothard JR, Miles MA. The Strange Case of Leishmania Chagasi. Parasitol Today (2000) 16(5):188–9. doi: 10.1016/S0169-4758(00)01637-9
6. Croft AM, Taylor NA, Rodenhurst KE. Sandflies and Leishmaniasis. Lancet (2006) 367(9505):112. doi: 10.1016/S0140-6736(06)67960-9
7. Costa DL, Rocha RL, Carvalho RM, Lima-Neto AS, Harhay MO, Costa CH, et al. Serum Cytokines Associated With Severity and Complications of Kala-Azar. Pathog Glob Health (2013) 107(2):78–87. doi: 10.1179/2047773213Y.0000000078
8. van Griensven J, Balasegaram M, Meheus F, Alvar J, Lynen L, Boelaert M. Combination Therapy for Visceral Leishmaniasis. Lancet Infect Dis (2010) 10(3):184–94. doi: 10.1016/S1473-3099(10)70011-6
9. Dayakar A, Chandrasekaran S, Kuchipudi SV, Kalangi SK. Cytokines: Key Determinants of Resistance or Disease Progression in Visceral Leishmaniasis: Opportunities for Novel Diagnostics and Immunotherapy. Front Immunol (2019) 10:670. doi: 10.3389/fimmu.2019.00670
10. Alexander J, Bryson K. T Helper (H)1/Th2 and Leishmania: Paradox Rather Than Paradigm. Immunol Lett (2005) 99(1):17–23. doi: 10.1016/j.imlet.2005.01.009
11. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The Role of Interleukin (IL)-10 in the Persistence of Leishmania Major in the Skin After Healing and the Therapeutic Potential of Anti-IL-10 Receptor Antibody for Sterile Cure. J Exp Med (2001) 194(10):1497–506. doi: 10.1084/jem.194.10.1497
12. Bogdan C. Nitric Oxide and the Immune Response. Nat Immunol (2001) 2(10):907–16. doi: 10.1038/ni1001-907
13. Green SJ, Crawford RM, Hockmeyer JT, Meltzer MS, Nacy CA. Leishmania Major Amastigotes Initiate the L-Arginine-Dependent Killing Mechanism in IFN-Gamma-Stimulated Macrophages by Induction of Tumor Necrosis Factor-Alpha. J Immunol (1990) 145(12):4290–7.
14. Bogdan C, Moll H, Solbach W, Rollinghoff M. Tumor Necrosis Factor-Alpha in Combination With Interferon-Gamma, But Not With Interleukin 4 Activates Murine Macrophages for Elimination of Leishmania Major Amastigotes. Eur J Immunol (1990) 20(5):1131–5. doi: 10.1002/eji.1830200528
15. Scott P, Novais FO. Cutaneous Leishmaniasis: Immune Responses in Protection and Pathogenesis. Nat Rev Immunol (2016) 16(9):581–92. doi: 10.1038/nri.2016.72
16. Sacramento LA, Cunha FQ, de Almeida RP, da Silva JS, Carregaro V. Protective Role of 5-Lipoxigenase During Leishmania Infantum Infection is Associated With Th17 Subset. BioMed Res Int (2014) 2014:264270. doi: 10.1155/2014/264270
17. Nascimento MS, Carregaro V, Lima-Junior DS, Costa DL, Ryffel B, Duthie MS, et al. Interleukin 17A Acts Synergistically With Interferon Gamma to Promote Protection Against Leishmania Infantum Infection. J Infect Dis (2015) 211(6):1015–26. doi: 10.1093/infdis/jiu531
18. Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of Cutaneous Leishmaniasis. T Cell Lines That Transfer Protective Immunity or Exacerbation Belong to Different T Helper Subsets and Respond to Distinct Parasite Antigens. J Exp Med (1988) 168(5):1675–84. doi: 10.1084/jem.168.5.1675
19. Nigg AP, Zahn S, Ruckerl D, Holscher C, Yoshimoto T, Ehrchen JM, et al. Dendritic Cell-Derived IL-12p40 Homodimer Contributes to Susceptibility in Cutaneous Leishmaniasis in BALB/c Mice. J Immunol (2007) 178(11):7251–8. doi: 10.4049/jimmunol.178.11.7251
20. Sacks D, Noben-Trauth N. The Immunology of Susceptibility and Resistance to Leishmania Major in Mice. Nat Rev Immunol (2002) 2(11):845–58. doi: 10.1038/nri933
21. Thakur CP, Mitra DK, Narayan S. Skewing of Cytokine Profiles Towards T Helper Cell Type 2 Response in Visceral Leishmaniasis Patients Unresponsive to Sodium Antimony Gluconate. Trans R Soc Trop Med Hyg (2003) 97(4):409–12. doi: 10.1016/S0035-9203(03)90071-X
22. Abidin BM, Hammami A, Stäger S, Heinonen KM. Infection-Adapted Emergency Hematopoiesis Promotes Visceral Leishmaniasis. PloS Pathog (2017) 13(8):e1006422. doi: 10.1371/journal.ppat.1006422
23. Terrazas C, Varikuti S, Oghumu S, Steinkamp HM, Ardic N, Kimble J, et al. Ly6C(hi) Inflammatory Monocytes Promote Susceptibility to Leishmania Donovani Infection. Sci Rep (2017) 7(1):14693. doi: 10.1038/s41598-017-14935-3
24. Matte C, Arango Duque G, Descoteaux A. Leishmania Donovani Metacyclic Promastigotes Impair Phagosome Properties in Inflammatory Monocytes. Infect Immun (2021) 89(7):e0000921. doi: 10.1128/IAI.00009-21
25. McElrath MJ, Murray HW, Cohn ZA. The Dynamics of Granuloma Formation in Experimental Visceral Leishmaniasis. J Exp Med (1988) 167(6):1927–37. doi: 10.1084/jem.167.6.1927
26. Engwerda CR, Ato M, Kaye PM. Macrophages, Pathology and Parasite Persistence in Experimental Visceral Leishmaniasis. Trends Parasitol (2004) 20(11):524–30. doi: 10.1016/j.pt.2004.08.009
27. Wilson ME, Jeronimo SM, Pearson RD. Immunopathogenesis of Infection With the Visceralizing Leishmania Species. Microb Pathog (2005) 38(4):147–60. doi: 10.1016/j.micpath.2004.11.002
28. Gurung P, Kanneganti TD. Innate Immunity Against Leishmania Infections. Cell Microbiol (2015) 17(9):1286–94. doi: 10.1111/cmi.12484
29. de Carvalho RVH, Zamboni DS. Inflammasome Activation in Response to Intracellular Protozoan Parasites. Trends Parasitol (2020) 36(5):459–72. doi: 10.1016/j.pt.2020.02.006
30. Sacramento LA, da Costa JL, de Lima MH, Sampaio PA, Almeida RP, Cunha FQ, et al. Toll-Like Receptor 2 Is Required for Inflammatory Process Development During Leishmania Infantum Infection. Front Microbiol (2017) 8:262. doi: 10.3389/fmicb.2017.00262
31. Sacramento LA, Benevides L, Maruyama SR, Tavares L, Fukutani KF, Francozo M, et al. TLR4 Abrogates the Th1 Immune Response Through IRF1 and IFN-Beta to Prevent Immunopathology During L. Infantum Infection. PloS Pathog (2020) 16(3):e1008435. doi: 10.1371/journal.ppat.1008435
32. Nascimento MS, Ferreira MD, Quirino GF, Maruyama SR, Krishnaswamy JK, Liu D, et al. NOD2-RIP2-Mediated Signaling Helps Shape Adaptive Immunity in Visceral Leishmaniasis. J Infect Dis (2016) 214(11):1647–57. doi: 10.1093/infdis/jiw446
33. Gigley JP, Bhadra R, Moretto MM, Khan IA. T Cell Exhaustion in Protozoan Disease. Trends Parasitol (2012) 28(9):377–84. doi: 10.1016/j.pt.2012.07.001
34. Sacks DL, Lal SL, Shrivastava SN, Blackwell J, Neva FA. An Analysis of T Cell Responsiveness in Indian Kala-Azar. J Immunol (1987) 138(3):908–13.
35. Faleiro RJ, Kumar R, Hafner LM, Engwerda CR. Immune Regulation During Chronic Visceral Leishmaniasis. PloS Negl Trop Dis (2014) 8(7):e2914. doi: 10.1371/journal.pntd.0002914
36. González-Tafoya E, Diupotex M, Zamora-Chimal J, Salaiza-Suazo N, Ruiz-Remigio A, Becker I. TNF Contributes to T-Cell Exhaustion in Chronic L. Mexicana Infections of Mice Through PD-L1 Up-Regulation. Cell Immunol (2020) 358:104196. doi: 10.1016/j.cellimm.2020.104196
37. Garcia de Moura R, Covre LP, Fantecelle CH, Gajardo VAT, Cunha CB, Stringari LL, et al. PD-1 Blockade Modulates Functional Activities of Exhausted-Like T Cell in Patients With Cutaneous Leishmaniasis. Front Immunol (2021) 12:632667. doi: 10.3389/fimmu.2021.632667
38. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ Regulatory T Cells Control Leishmania Major Persistence and Immunity. Nat (2002) 420(6915):502–7. doi: 10.1038/nature01152
39. Peters NC, Pagán AJ, Lawyer PG, Hand TW, Henrique Roma E, Stamper LW, et al. Chronic Parasitic Infection Maintains High Frequencies of Short-Lived Ly6C+CD4+ Effector T Cells That Are Required for Protection Against Re-Infection. PloS Pathog (2014) 10(12):e1004538. doi: 10.1371/journal.ppat.1004538
40. Zaph C, Uzonna J, Beverley SM, Scott P. Central Memory T Cells Mediate Long-Term Immunity to Leishmania Major in the Absence of Persistent Parasites. Nat Med (2004) 10(10):1104–10. doi: 10.1038/nm1108
41. Karp CL, el-Safi SH, Wynn TA, Satti MM, Kordofani AM, Hashim FA, et al. In Vivo Cytokine Profiles in Patients With Kala-Azar. Marked Elevation of Both Interleukin-10 and Interferon-Gamma. J Clin Invest (1993) 91(4):1644–8. doi: 10.1172/JCI116372
42. Hernandez-Ruiz J, Salaiza-Suazo N, Carrada G, Escoto S, Ruiz-Remigio A, Rosenstein Y, et al. CD8 Cells of Patients With Diffuse Cutaneous Leishmaniasis Display Functional Exhaustion: The Latter Is Reversed, In Vitro, by TLR2 Agonists. PloS Negl Trop Dis (2010) 4(11):e871. doi: 10.1371/journal.pntd.0000871
43. Gautam S, Kumar R, Singh N, Singh AK, Rai M, Sacks D, et al. CD8 T Cell Exhaustion in Human Visceral Leishmaniasis. J Infect Dis (2014) 209(2):290–9. doi: 10.1093/infdis/jit401
44. Singh OP, Gidwani K, Kumar R, Nylen S, Jones SL, Boelaert M, et al. Reassessment of Immune Correlates in Human Visceral Leishmaniasis as Defined by Cytokine Release in Whole Blood. Clin Vaccine Immunol (2012) 19(6):961–6. doi: 10.1128/CVI.00143-12
45. Ansari NA, Kumar R, Gautam S, Nylen S, Singh OP, Sundar S, et al. IL-27 and IL-21 are Associated With T Cell IL-10 Responses in Human Visceral Leishmaniasis. J Immunol (2011) 186(7):3977–85. doi: 10.4049/jimmunol.1003588
46. Gidwani K, Picado A, Rijal S, Singh SP, Roy L, Volfova V, et al. Serological Markers of Sand Fly Exposure to Evaluate Insecticidal Nets Against Visceral Leishmaniasis in India and Nepal: A Cluster-Randomized Trial. PloS Negl Trop Dis (2011) 5(9):e1296. doi: 10.1371/journal.pntd.0001296
47. Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, et al. Intestinal Tolerance Is Converted to Autoimmune Enteritis Upon PD-1 Ligand Blockade. J Immunol (2009) 182(4):2102–12. doi: 10.4049/jimmunol.0802769
48. Giunchetti RC, Silveira P, Resende LA, Leite JC, Melo-Júnior OAO, Rodrigues-Alves ML, et al. Canine Visceral Leishmaniasis Biomarkers and Their Employment in Vaccines Role of Cytokines in the Pathogenesis of Visceral Leishmaniasis Visceral Leishmaniasis: An Overview of Vaccine Adjuvants and Their Applications Vaccines for Visceral Leishmaniasis: A Review Canine Leishmaniasis Control in the Context of One Health. Vet Parasitol (2019) 271:87–97. Netherlands Germany: © 2019 Elsevier B.V © 2019. Published by Elsevier Ltd. © 2015 Elsevier B.V. doi: 10.1016/j.vetpar.2019.05.006
49. Teles LF, Viana AG, Cardoso MS, Pinheiro GRG, Bento GA, Lula JF, et al. Evaluation of Medullary Cytokine Expression and Clinical and Laboratory Aspects in Severe Human Visceral Leishmaniasis. Parasite Immunol (2021) 43(12):e12880. doi: 10.1111/pim.12880
50. Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D. Splenic Accumulation of IL-10 mRNA in T Cells Distinct From CD4+CD25+ (Foxp3) Regulatory T Cells in Human Visceral Leishmaniasis. J Exp Med (2007) 204(4):805–17. doi: 10.1084/jem.20061141
51. Gidwani K, Jones S, Kumar R, Boelaert M, Sundar S. Interferon-Gamma Release Assay (Modified QuantiFERON) as a Potential Marker of Infection for Leishmania Donovani, a Proof of Concept Study. PloS Negl Trop Dis (2011) 5(4):e1042. doi: 10.1371/journal.pntd.0001042
52. Kumar R, Singh N, Gautam S, Singh OP, Gidwani K, Rai M, et al. Leishmania Specific CD4 T Cells Release Ifnγ That Limits Parasite Replication in Patients With Visceral Leishmaniasis. PloS Negl Trop Dis (2014) 8(10):e3198. doi: 10.1371/journal.pntd.0003198
53. Corthay A. How do Regulatory T Cells Work? Scand J Immunol (2009) 70(4):326–36. doi: 10.1111/j.1365-3083.2009.02308.x
54. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
55. Boer MC, Joosten SA, Ottenhoff TH. Regulatory T-Cells at the Interface Between Human Host and Pathogens in Infectious Diseases and Vaccination. Front Immunol (2015) 6:217. doi: 10.3389/fimmu.2015.00217
56. Ghosh S, Roy K, Rajalingam R, Martin S, Pal C. Cytokines in the Generation and Function of Regulatory T Cell Subsets in Leishmaniasis. Cytokine (2021) 147:155266. doi: 10.1016/j.cyto.2020.155266
57. Murray HW, Moreira AL, Lu CM, DeVecchio JL, Matsuhashi M, Ma X, et al. Determinants of Response to Interleukin-10 Receptor Blockade Immunotherapy in Experimental Visceral Leishmaniasis. J Infect Dis (2003) 188(3):458–64. doi: 10.1086/376510
58. Lima MHF, Sacramento LA, Quirino GFS, Ferreira MD, Benevides L, Santana AKM, et al. Leishmania Infantum Parasites Subvert the Host Inflammatory Response Through the Adenosine A2A Receptor to Promote the Establishment of Infection. Front Immunol (2017) 8:815. doi: 10.3389/fimmu.2017.00815
59. Rodrigues OR, Marques C, Soares-Clemente M, Ferronha MH, Santos-Gomes GM. Identification of Regulatory T Cells During Experimental Leishmania Infantum Infection. Immunobiol (2009) 214(2):101–11. doi: 10.1016/j.imbio.2008.07.001
60. Bunn PT, Montes de Oca M, de Labastida Rivera F, Kumar R, Ng SS, Edwards CL, et al. Distinct Roles for CD4+ Foxp3+ Regulatory T Cells and IL-10-Mediated Immunoregulatory Mechanisms During Experimental Visceral Leishmaniasis Caused by Leishmania Donovani. J Immunol (2018) 201(11):3362–72. doi: 10.4049/jimmunol.1701582
61. Nylen S, Sacks D. Interleukin-10 and the Pathogenesis of Human Visceral Leishmaniasis. Trends Immunol (2007) 28(9):378–84. doi: 10.1016/j.it.2007.07.004
62. Tiwananthagorn S, Iwabuchi K, Ato M, Sakurai T, Kato H, Katakura K. Involvement of CD4+ Foxp3+ Regulatory T Cells in Persistence of Leishmania Donovani in the Liver of Alymphoplastic Aly/Aly Mice. PloS Negl Trop Dis (2012) 6(8):e1798. doi: 10.1371/journal.pntd.0001798
63. Kumar P, Misra P, Thakur CP, Saurabh A, Rishi N, Mitra DK. T Cell Suppression in the Bone Marrow of Visceral Leishmaniasis Patients: Impact of Parasite Load. Clin Exp Immunol (2018) 191(3):318–27. doi: 10.1111/cei.13074
64. Bhattacharya P, Ghosh S, Ejazi SA, Rahaman M, Pandey K, Ravi Das VN, et al. Induction of IL-10 and Tgfβ From CD4+CD25+FoxP3+ T Cells Correlates With Parasite Load in Indian Kala-Azar Patients Infected With Leishmania Donovani. PloS Negl Trop Dis (2016) 10(2):e0004422. doi: 10.1371/journal.pntd.0004422
65. Gupta G, Majumdar S, Adhikari A, Bhattacharya P, Mukherjee AK, Majumdar SB. Treatment With IP-10 Induces Host-Protective Immune Response by Regulating the T Regulatory Cell Functioning in Leishmania Donovani-Infected Mice. Med Microbiol Immunol (2011) 200(4):241–53. doi: 10.1007/s00430-011-0197-y
66. Eufrásio de Figueiredo WM, Heredia FF, Santos AS, da Rocha Braga R, Marciano Fonseca FR, Lúcia de Castro Rodrigues N, et al. CXCL10 Treatment Promotes Reduction of IL-10+ Regulatory T (Foxp3+ and Tr1) Cells in the Spleen of BALB/c Mice Infected by Leishmania Infantum. Exp Parasitol (2019) 207:107789. doi: 10.1016/j.exppara.2019.107789
67. Sharpe AH, Pauken KE. The Diverse Functions of the PD1 Inhibitory Pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
68. Han Y, Liu D, Li L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am J Cancer Res (2020) 10(3):727–42.
69. Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 Pathway. Sci Adv (2020) 6(38):eabd2712. doi: 10.1126/sciadv.abd2712
70. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stager S. B7-H1 Blockade Increases Survival of Dysfunctional CD8+ T Cells and Confers Protection Against Leishmania Donovani Infections. PloS Pathog (2009) 5(5):e1000431. doi: 10.1371/journal.ppat.1000431
71. Medina-Colorado AA, Osorio EY, Saldarriaga OA, Travi BL, Kong F, Spratt H, et al. Splenic CD4+ T Cells in Progressive Visceral Leishmaniasis Show a Mixed Effector-Regulatory Phenotype and Impair Macrophage Effector Function Through Inhibitory Receptor Expression. PloS One (2017) 12(1):e0169496. doi: 10.1371/journal.pone.0169496
72. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed Death 1-Mediated T Cell Exhaustion During Visceral Leishmaniasis Impairs Phagocyte Function. J Immunol (2013) 191(11):5542–50. doi: 10.4049/jimmunol.1301810
73. Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, et al. PDL-1 Blockade Prevents T Cell Exhaustion, Inhibits Autophagy, and Promotes Clearance of Leishmania Donovani. Infect Immun (2018) 86(6):e00019–18. doi: 10.1128/IAI.00019-18
74. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
75. Cai J, Wang D, Zhang G, Guo X. The Role of PD-1/PD-L1 Axis in Treg Development and Function: Implications for Cancer Immunotherapy. Onco Targets Ther (2019) 12:8437–45. doi: 10.2147/OTT.S221340
76. Champiat S, Besse B, Marabelle A. Hyperprogression During Immunotherapy: Do We Really Want to Know? Ann Oncol (2019) 30(7):1028–31. doi: 10.1093/annonc/mdz184
77. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, et al. Hyperprogressive Disease: Recognizing a Novel Pattern to Improve Patient Management. Nat Rev Clin Oncol (2018) 15(12):748–62. doi: 10.1038/s41571-018-0111-2
78. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive Disease is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res (2017) 23(8):1920–8. doi: 10.1158/1078-0432.CCR-16-1741
79. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1+ Regulatory T Cells Amplified by PD-1 Blockade Promote Hyperprogression of Cancer. Proc Natl Acad Sci USA (2019) 116(20):9999–10008. doi: 10.1073/pnas.1822001116
80. Sullivan RJ, Weber JS. Immune-Related Toxicities of Checkpoint Inhibitors: Mechanisms and Mitigation Strategies. Nat Rev Drug Discovery (2021). doi: 10.1038/s41573-021-00259-5
81. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting Co-Stimulatory Molecules in Autoimmune Disease. Nat Rev Drug Discovery (2020) 19(12):860–83. doi: 10.1038/s41573-020-0081-9
82. Wykes MN, Lewin SR. Immune Checkpoint Blockade in Infectious Diseases. Nat Rev Immunol (2018) 18(2):91–104. doi: 10.1038/nri.2017.112
83. Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immun (2016) 44(5):1052–68. doi: 10.1016/j.immuni.2016.04.022
84. Andrews LP, Yano H, Vignali DAA. Inhibitory Receptors and Ligands Beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or Backups. Nat Immunol (2019) 20(11):1425–34. doi: 10.1038/s41590-019-0512-0
85. Murphy ML, Cotterell SE, Gorak PM, Engwerda CR, Kaye PM. Blockade of CTLA-4 Enhances Host Resistance to the Intracellular Pathogen, Leishmania Donovani. J Immunol (1998) 161(8):4153–60.
86. Murray HW, Lu CM, Brooks EB, Fichtl RE, DeVecchio JL, Heinzel FP. Modulation of T-Cell Costimulation as Immunotherapy or Immunochemotherapy in Experimental Visceral Leishmaniasis. Infect Immun (2003) 71(11):6453–62. doi: 10.1128/IAI.71.11.6453-6462.2003
87. Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy With OX40L-Fc or Anti-CTLA-4 Enhances Local Tissue Responses and Killing of Leishmania Donovani. Eur J Immunol (2004) 34(5):1433–40. doi: 10.1002/eji.200324021
88. Gomes NA, Barreto-de-Souza V, Wilson ME, DosReis GA. Unresponsive CD4+ T Lymphocytes From Leishmania Chagasi-Infected Mice Increase Cytokine Production and Mediate Parasite Killing After Blockade of B7-1/CTLA-4 Molecular Pathway. J Infect Dis (1998) 178(6):1847–51. doi: 10.1086/314520
89. Chen W, Jin W, Wahl SM. Engagement of Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4) Induces Transforming Growth Factor Beta (TGF-Beta) Production by Murine CD4+ T Cells. J Exp Med (1998) 188(10):1849–57. doi: 10.1084/jem.188.10.1849
90. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional Immune Checkpoint-Targeted Antibody-Ligand Traps That Simultaneously Disable Tgfβ Enhance the Efficacy of Cancer Immunotherapy. Nat Commun (2018) 9(1):741. doi: 10.1038/s41467-017-02696-6
91. Gomes NA, Gattass CR, Barreto-de-Souza V, Wilson ME, DosReis GA. TGF-Beta Mediates CTLA-4 Suppression of Cellular Immunity in Murine Kalaazar. J Immunol (2000) 164(4):2001–8. doi: 10.4049/jimmunol.164.4.2001
92. Clarêncio J, de Oliveira CI, Favali C, Medina O, Caldas A, Costa CH, et al. Could the Lower Frequency of CD8+CD18+CD45RO+ Lymphocytes be Biomarkers of Human VL? Int Immunol (2009) 21(2):137–44. doi: 10.1093/intimm/dxn131
93. Viana AG, Magalhães LMD, Giunchetti RC, Dutra WO, Gollob KJ. Leishmania Infantum Induces Expression of the Negative Regulatory Checkpoint, CTLA-4, by Human Naïve CD8(+) T Cells. Parasite Immunol (2019) 41(9):e12659. doi: 10.1111/pim.12659
94. Singh B, Bhushan Chauhan S, Kumar R, Singh SS, Ng S, Amante F, et al. A Molecular Signature for CD8+ T Cells From Visceral Leishmaniasis Patients. Parasite Immunol (2019) 41(11):e12669. doi: 10.1111/pim.12669
95. Hajilooi M, Lotfi P, Seif F, Bazmani A, Momeni M, Ravary A, et al. The Cytotoxic T Lymphocyte Antigen-4 +49A/G Single Nucleotide Polymorphism Association With Visceral Leishmaniasis. Jundishapur J Microbiol (2014) 7(10):e12143. doi: 10.1016/j.vaccine.2016.09.016
96. Schaut RG, Grinnage-Pulley TL, Esch KJ, Toepp AJ, Duthie MS, Howard RF, et al. Recovery of Antigen-Specific T Cell Responses From Dogs Infected With Leishmania (L.) Infantum by Use of Vaccine Associated TLR-Agonist Adjuvant. Vaccine (2016) 34(44):5225–34. doi: 10.1016/j.vaccine.2016.09.016
97. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and Transcriptional Regulation of the Co-Inhibitory Gene Module in T Cells. Nat (2018) 558(7710):454–9. doi: 10.1038/s41586-018-0206-z
98. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors With Specialized Functions in Immune Regulation. Immun (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
99. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernandez-Rubio L, et al. Understanding LAG-3 Signaling. Int J Mol Sci (2021) 22(10):5282. doi: 10.3390/ijms22105282
100. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in Regulatory T Cells. Immun (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
101. Banerjee H, Nieves-Rosado H, Kulkarni A, Murter B, McGrath KV, Chandran UR, et al. Expression of Tim-3 Drives Phenotypic and Functional Changes in Treg Cells in Secondary Lymphoid Organs and the Tumor Microenvironment. Cell Rep (2021) 36(11):109699. doi: 10.1016/j.celrep.2021.109699
102. Charpentier T, Hammami A, Stäger S. Hypoxia Inducible Factor 1α: A Critical Factor for the Immune Response to Pathogens and Leishmania. Cell Immunol (2016) 309:42–9. doi: 10.1016/j.cellimm.2016.06.002
103. Gunton JE. Hypoxia-Inducible Factors and Diabetes. J Clin Invest (2020) 130(10):5063–73. doi: 10.1172/JCI137556
104. Balamurugan K. HIF-1 at the Crossroads of Hypoxia, Inflammation, and Cancer. Int J Cancer (2016) 138(5):1058–66. doi: 10.1002/ijc.29519
105. Kumar V, Kumar A, Das S, Abhishek K, Verma S, Mandal A, et al. Leishmania Donovani Activates Hypoxia Inducible Factor-1α and miR-210 for Survival in Macrophages by Downregulation of NF-κb Mediated Pro-Inflammatory Immune Response. Front Microbiol (2018) 9:385. doi: 10.3389/fmicb.2018.00385
106. Singh AK, Mukhopadhyay C, Biswas S, Singh VK, Mukhopadhyay CK. Intracellular Pathogen Leishmania Donovani Activates Hypoxia Inducible Factor-1 by Dual Mechanism for Survival Advantage Within Macrophage. PloS One (2012) 7(6):e38489. doi: 10.1371/journal.pone.0038489
107. Hammami A, Charpentier T, Smans M, Stäger S. IRF-5-Mediated Inflammation Limits CD8+ T Cell Expansion by Inducing HIF-1α and Impairing Dendritic Cell Functions During Leishmania Infection. PloS Pathog (2015) 11(6):e1004938. doi: 10.1371/journal.ppat.1004938
108. Hammami A, Abidin BM, Heinonen KM, Stäger S. HIF-1α Hampers Dendritic Cell Function and Th1 Generation During Chronic Visceral Leishmaniasis. Sci Rep (2018) 8(1):3500. doi: 10.1038/s41598-018-21891-z
109. Hammami A, Abidin BM, Charpentier T, Fabié A, Duguay AP, Heinonen KM, et al. HIF-1α is a Key Regulator in Potentiating Suppressor Activity and Limiting the Microbicidal Capacity of MDSC-Like Cells During Visceral Leishmaniasis. PloS Pathog (2017) 13(9):e1006616. doi: 10.1371/journal.ppat.1006616
110. Mesquita I, Ferreira C, Moreira D, Kluck GEG, Barbosa AM, Torrado E, et al. The Absence of HIF-1α Increases Susceptibility to Leishmania Donovani Infection via Activation of BNIP3/mTOR/SREBP-1c Axis. Cell Rep (2020) 30(12):4052–64.e7. doi: 10.1016/j.celrep.2020.02.098
111. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai AC, et al. HIF-2alpha Is Indispensable for Regulatory T Cell Function. Nat Commun (2020) 11(1):5005. doi: 10.1038/s41467-020-18731-y
112. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-Inducible Factor-1 Alpha-Dependent Induction of FoxP3 Drives Regulatory T-Cell Abundance and Function During Inflammatory Hypoxia of the Mucosa. Proc Natl Acad Sci U S A (2012) 109(41):E2784–E93. doi: 10.1073/pnas.1202366109
113. Peres-Sampaio CE, Palumbo ST, Meyer-Fernandes JR. An Ecto-ATPase Activity Present in Leishmania Tropica Stimulated by Dextran Sulfate. Z Naturforsch C J Biosci (2001) 56(9-10):820–5. doi: 10.1515/znc-2001-9-1023
114. de Souza MC, de Assis EA, Gomes RS, Marques da Silva Ede A, Melo MN, Fietto JL, et al. The Influence of Ecto-Nucleotidases on Leishmania Amazonensis Infection and Immune Response in C57B/6 Mice. Acta Trop (2010) 115(3):262–9. doi: 10.1016/j.actatropica.2010.04.007
115. Gomes RS, de Carvalho LC, de Souza Vasconcellos R, Fietto JL, Afonso LC. E-NTPDase (Ecto-Nucleoside Triphosphate Diphosphohydrolase) of Leishmania Amazonensis Inhibits Macrophage Activation. Microbes Infect (2015) 17(4):295–303. doi: 10.1016/j.micinf.2014.12.009
116. Takenaka MC, Robson S, Quintana FJ. Regulation of the T Cell Response by CD39. Trends Immunol (2016) 37(7):427–39. doi: 10.1016/j.it.2016.04.009
117. Carregaro V, Ribeiro JM, Valenzuela JG, Souza-Junior DL, Costa DL, Oliveira CJ, et al. Nucleosides Present on Phlebotomine Saliva Induce Immunossuppression and Promote the Infection Establishment. PloS Negl Trop Dis (2015) 9(4):e0003600. doi: 10.1371/journal.pntd.0003600
118. Rai AK, Thakur CP, Velpandian T, Sharma SK, Ghosh B, Mitra DK. High Concentration of Adenosine in Human Visceral Leishmaniasis Despite Increased ADA and Decreased CD73. Parasite Immunol (2011) 33(11):632–6. doi: 10.1111/j.1365-3024.2011.01315.x
119. Vijayamahantesh, Amit A, Kumar S, Dikhit MR, Jha PK, Singh AK, et al. Up Regulation of A2B Adenosine Receptor on Monocytes are Crucially Required for Immune Pathogenicity in Indian Patients Exposed to Leishmania Donovani. Cytokine (2016) 79:38–44. doi: 10.1016/j.cyto.2015.12.016
120. Shevchenko I, Mathes A, Groth C, Karakhanova S, Muller V, Utikal J, et al. Enhanced Expression of CD39 and CD73 on T Cells in the Regulation of Anti-Tumor Immune Responses. Oncoimmunol (2020) 9(1):1744946. doi: 10.1080/2162402X.2020.1744946
121. Mukhopadhyay D, Das NK, Roy S, Kundu S, Barbhuiya JN, Chatterjee M. Miltefosine Effectively Modulates the Cytokine Milieu in Indian Post Kala-Azar Dermal Leishmaniasis. J Infect Dis (2011) 204(9):1427–36. doi: 10.1093/infdis/jir551
122. Wadhone P, Maiti M, Agarwal R, Kamat V, Martin S, Saha B. Miltefosine Promotes IFN-Gamma-Dominated Anti-Leishmanial Immune Response. J Immunol (2009) 182(11):7146–54. doi: 10.4049/jimmunol.0803859
123. Tokuda Y, Tsuji M, Yamazaki M, Kimura S, Abe S, Yamaguchi H. Augmentation of Murine Tumor Necrosis Factor Production by Amphotericin B In Vitro and In Vivo. Antimicrob Agents Chemother (1993) 37(10):2228–30. doi: 10.1128/AAC.37.10.2228
124. Kumar R, Chauhan SB, Ng SS, Sundar S, Engwerda CR. Immune Checkpoint Targets for Host-Directed Therapy to Prevent and Treat Leishmaniasis. Front Immunol (2017) 8:1492. doi: 10.3389/fimmu.2017.01492
125. Tripathi P, Singh V, Naik S. Immune Response to Leishmania: Paradox Rather Than Paradigm. FEMS Immunol Med Microbiol (2007) 51(2):229–42. doi: 10.1111/j.1574-695X.2007.00311.x
126. Hirsch CS, Ellner JJ, Blinkhorn R, Toossi Z. In Vitro Restoration of T Cell Responses in Tuberculosis and Augmentation of Monocyte Effector Function Against Mycobacterium Tuberculosis by Natural Inhibitors of Transforming Growth Factor Beta. Proc Natl Acad Sci U S A (1997) 94(8):3926–31. doi: 10.1073/pnas.94.8.3926
127. Gantt KR, Schultz-Cherry S, Rodriguez N, Jeronimo SM, Nascimento ET, Goldman TL, et al. Activation of TGF-Beta by Leishmania Chagasi: Importance for Parasite Survival in Macrophages. J Immunol (2003) 170(5):2613–20. doi: 10.4049/jimmunol.170.5.2613
128. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase Induction by Suppressors of Nitric Oxide Synthesis (IL-4, IL-10 and PGE2) in Murine Bone-Marrow-Derived Macrophages. Biochem Biophys Res Commun (1995) 206(2):667–73. doi: 10.1006/bbrc.1995.1094
129. Omer FM, Kurtzhals JA, Riley EM. Maintaining the Immunological Balance in Parasitic Infections: A Role for TGF-Beta? Parasitol Today (2000) 16(1):18–23. doi: 10.1016/S0169-4758(99)01562-8
130. Iniesta V, Gomez-Nieto LC, Corraliza I. The Inhibition of Arginase by N(omega)-Hydroxy-L-Arginine Controls the Growth of Leishmania Inside Macrophages. J Exp Med (2001) 193(6):777–84. doi: 10.1084/jem.193.6.777
131. Banerjee R, Kumar S, Sen A, Mookerjee A, Mukherjee P, Roy S, et al. TGF-Beta-Regulated Tyrosine Phosphatases Induce Lymphocyte Apoptosis in Leishmania Donovani-Infected Hamsters. Immunol Cell Biol (2011) 89(3):466–74. doi: 10.1038/icb.2010.108
132. Wilson ME, Recker TJ, Rodriguez NE, Young BM, Burnell KK, Streit JA, et al. The TGF-Beta Response to Leishmania Chagasi in the Absence of IL-12. Eur J Immunol (2002) 32(12):3556–65. doi: 10.1002/1521-4141(200212)32:12<3556::AID-IMMU3556>3.0.CO;2-Q
133. Ghalib HW, Piuvezam MR, Skeiky YA, Siddig M, Hashim FA, el-Hassan AM, et al. Interleukin 10 Production Correlates With Pathology in Human Leishmania Donovani Infections. J Clin Invest (1993) 92(1):324–9. doi: 10.1172/JCI116570
134. Moulik S, Karmakar J, Joshi S, Dube A, Mandal C, Chatterjee M. Status of IL-4 and IL-10 Driven Markers in Experimental Models of Visceral Leishmaniasis. Parasite Immunol (2021) 43(1):e12783. doi: 10.1111/pim.12783
135. Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 Mediates Susceptibility to Leishmania Donovani Infection. Eur J Immunol (2001) 31(10):2848–56. doi: 10.1002/1521-4141(2001010)31:10<2848::AID-IMMU2848>3.0.CO;2-T
136. Murray HW, Lu CM, Mauze S, Freeman S, Moreira AL, Kaplan G, et al. Interleukin-10 (IL-10) in Experimental Visceral Leishmaniasis and IL-10 Receptor Blockade as Immunotherapy. Infect Immun (2002) 70(11):6284–93. doi: 10.1128/IAI.70.11.6284-6293.2002
137. Bhattacharjee S, Gupta G, Bhattacharya P, Adhikari A, Majumdar SB, Majumdar S. Anti-IL-10 mAb Protection Against Experimental Visceral Leishmaniasis via Induction of Th1 Cytokines and Nitric Oxide. Indian J Exp Biol (2009) 47(6):489–97.
138. Gautam S, Kumar R, Maurya R, Nylen S, Ansari N, Rai M, et al. IL-10 Neutralization Promotes Parasite Clearance in Splenic Aspirate Cells From Patients With Visceral Leishmaniasis. J Infect Dis (2011) 204(7):1134–7. doi: 10.1093/infdis/jir461
139. Singh OP, Sundar S. Immunotherapy and Targeted Therapies in Treatment of Visceral Leishmaniasis: Current Status and Future Prospects. Front Immunol (2014) 5:296. doi: 10.3389/fimmu.2014.00296
140. Nutt SL, Fairfax KA, Kallies A. BLIMP1 Guides the Fate of Effector B and T Cells. Nat Rev Immunol (2007) 7(12):923–7. doi: 10.1038/nri2204
141. Nutt SL, Kallies A, Belz GT. Blimp-1 Connects the Intrinsic and Extrinsic Regulation of T Cell Homeostasis. J Clin Immunol (2008) 28(2):97–106. doi: 10.1007/s10875-007-9151-6
142. Benevides L, Costa RS, Tavares LA, Russo M, Martins GA, da Silva LLP, et al. B Lymphocyte-Induced Maturation Protein 1 Controls TH9 Cell Development, IL-9 Production, and Allergic Inflammation. J Allergy Clin Immunol (2019) 143(3):1119–30.e3. doi: 10.1016/j.jaci.2018.06.046
143. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-Driven CD4+ Tissue-Resident Memory T Cells Control Chronic Intestinal Inflammation. Nat Immunol (2019) 20(3):288–300. doi: 10.1038/s41590-018-0298-5
144. Lin MH, Yeh LT, Chen SJ, Chiou HY, Chu CC, Yen LB, et al. T Cell-Specific BLIMP-1 Deficiency Exacerbates Experimental Autoimmune Encephalomyelitis in Nonobese Diabetic Mice by Increasing Th1 and Th17 Cells. Clin Immunol (2014) 151(2):101–13. doi: 10.1016/j.clim.2014.02.006
145. Salehi S, Bankoti R, Benevides L, Willen J, Couse M, Silva JS, et al. B Lymphocyte-Induced Maturation Protein-1 Contributes to Intestinal Mucosa Homeostasis by Limiting the Number of IL-17-Producing CD4+ T Cells. J Immunol (2012) 189(12):5682–93. doi: 10.4049/jimmunol.1201966
146. Montes de Oca M, Kumar R, de Labastida Rivera F, Amante FH, Sheel M, Faleiro RJ, et al. Blimp-1-Dependent IL-10 Production by Tr1 Cells Regulates TNF-Mediated Tissue Pathology. PloS Pathog (2016) 12(1):e1005398. doi: 10.1038/s41467-020-18731-y
147. Papatriantafyllou M. Regulatory T Cells: Distilling Regulatory T Cell Inducers. Nat Rev Immunol (2013) 13(8):546. doi: 10.1038/nri3506
148. Boivin N, Baillargeon J, Doss PM, Roy AP, Rangachari M. Interferon-Beta Suppresses Murine Th1 Cell Function in the Absence of Antigen-Presenting Cells. PloS One (2015) 10(4):e0124802. doi: 1371/journal.pone.0124802
149. Kumar R, Bunn PT, Singh SS, Ng SS, Montes de Oca M, de Labastida Rivera F, et al. Type I Interferons Suppress Anti-Parasitic Immunity and Can be Targeted to Improve Treatment of Visceral Leishmaniasis. Cell Rep (2020) 30(8):2512–25.e9. doi: 10.1016/j.celrep.2020.01.099
150. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ (2002) 9(10):1069–77. doi: 10.1038/sj.cdd.4401073
151. Chevolet I, Speeckaert R, Schreuer M, Neyns B, Krysko O, Bachert C, et al. Characterization of the In Vivo Immune Network of IDO, Tryptophan Metabolism, PD-L1, and CTLA-4 in Circulating Immune Cells in Melanoma. Oncoimmunol (2015) 4(3):e982382. doi: 10.4161/2162402X.2014.982382
152. Gocher AM, Workman CJ, Vignali DAA. Interferon-Gamma: Teammate or Opponent in the Tumour Microenvironment? Nat Rev Immunol (2021). doi: 10.1038/s41577-021-00566-3
153. Gangneux JP, Poinsignon Y, Donaghy L, Amiot L, Tarte K, Mary C, et al. Indoleamine 2,3-Dioxygenase Activity as a Potential Biomarker of Immune Suppression During Visceral Leishmaniasis. Innate Immun (2013) 19(6):564–8. doi: 10.1177/1753425912473170
154. Makala LH, Baban B, Lemos H, El-Awady AR, Chandler PR, Hou DY, et al. Leishmania Major Attenuates Host Immunity by Stimulating Local Indoleamine 2,3-Dioxygenase Expression. J Infect Dis (2011) 203(5):715–25. doi: 10.1093/infdis/jiq095
155. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res (2017) 77(24):6795–811. doi: 10.1158/0008-5472.CAN-17-2285
156. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-Dioxygenase and its Therapeutic Inhibition in Cancer. Int Rev Cell Mol Biol (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
157. Sauer KA, Maxeiner JH, Karwot R, Scholtes P, Lehr HA, Birkenbach M, et al. Immunosurveillance of Lung Melanoma Metastasis in EBI-3-Deficient Mice Mediated by CD8+ T Cells. J Immunol (2008) 181(9):6148–57. doi: 10.4049/jimmunol.181.9.6148
158. DeLong JH, O'Hara Hall A, Rausch M, Moodley D, Perry J, Park J, et al. IL-27 and TCR Stimulation Promote T Cell Expression of Multiple Inhibitory Receptors. ImmunoHorizons (2019) 3(1):13–25. doi: 10.4049/immunohorizons.1800083
159. Stumhofer JS, Hunter CA. Advances in Understanding the Anti-Inflammatory Properties of IL-27. Immunol Lett (2008) 117(2):123–30. doi: 10.1016/j.imlet.2008.01.011
160. Yoshida H, Miyazaki Y. Interleukin 27 Signaling Pathways in Regulation of Immune and Autoimmune Responses. Int J Biochem Cell Biol (2008) 40(11):2379–83. doi: 10.1016/j.biocel.2008.05.020
161. Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 Priming of T Cells Controls IL-17 Production in Trans via Induction of the Ligand PD-L1. Immun (2012) 36(6):1017–30. doi: 10.1016/j.immuni.2012.03.024
162. Morrow KN, Liang Z, Xue M, Chihade DB, Sun Y, Chen CW, et al. The IL-27 Receptor Regulates TIGIT on Memory CD4(+) T Cells During Sepsis. iScience (2021) 24(2):102093.
163. Quirino GFS, Nascimento MSL, Davoli-Ferreira M, Sacramento LA, Lima MHF, Almeida RP, et al. Interleukin-27 (IL-27) Mediates Susceptibility to Visceral Leishmaniasis by Suppressing the IL-17-Neutrophil Response. Infect Immun (2016) 84(8):2289–98. doi: 10.1128/IAI.00283-16
164. Rosas LE, Satoskar AA, Roth KM, Keiser TL, Barbi J, Hunter C, et al. Interleukin-27r (WSX-1/T-Cell Cytokine Receptor) Gene-Deficient Mice Display Enhanced Resistance to Leishmania Donovani Infection But Develop Severe Liver Immunopathology. Am J Pathol (2006) 168(1):158–69. doi: 10.2353/ajpath.2006.050013
165. dos Santos PL, de Oliveira FA, Santos ML, Cunha LC, Lino MT, de Oliveira MF, et al. The Severity of Visceral Leishmaniasis Correlates With Elevated Levels of Serum IL-6, IL-27 and Scd14. PloS Negl Trop Dis (2016) 10(1):e0004375. doi: 10.1371/journal.pntd.0004375
166. Jafarzadeh A, Nemati M, Chauhan P, Patidar A, Sarkar A, Sharifi I, et al. Interleukin-27 Functional Duality Balances Leishmania Infectivity and Pathogenesis. Front Immunol (2020) 11:1573. doi: 10.3389/fimmu.2020.01573
167. Jones GW, Hill DG, Cardus A, Jones SA. IL-27: A Double Agent in the IL-6 Family. Clin Exp Immunol (2018) 193(1):37–46. doi: 10.1111/cei.13116
168. Vignali DA, Kuchroo VK. IL-12 Family Cytokines: Immunological Playmakers. Nat Immunol (2012) 13(8):722–8. doi: 10.1038/ni.2366
169. Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, et al. Interleukin-35 Limits Anti-Tumor Immunity. Immun (2016) 44(2):316–29. doi: 10.1016/j.immuni.2016.01.013
170. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The Inhibitory Cytokine IL-35 Contributes to Regulatory T-Cell Function. Nat (2007) 450(7169):566–9. doi: 10.1038/nature06306
171. Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-Producing B Cells Are Critical Regulators of Immunity During Autoimmune and Infectious Diseases. Nat (2014) 507(7492):366–70. doi: 10.1038/nature12979
172. Asad M, Sabur A, Shadab M, Das S, Kamran M, Didwania N, et al. EB1-3 Chain of IL-35 Along With TGF-Beta Synergistically Regulate Anti-Leishmanial Immunity. Front Immunol (2019) 10:616. doi: 10.3389/fimmu.2019.00616
173. Asad M, Sabur A, Kamran M, Shadab M, Das S, Ali N. Effector Functions of Th17 Cells Are Regulated by IL-35 and TGF-Beta in Visceral Leishmaniasis. FASEB J (2021) 35(9):e21755. doi: 10.1096/fj.202002356RR
174. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive Plasticity of IL-10+ and IL-35+ Treg Cells Cooperatively Promotes Tumor T Cell Exhaustion. Nat Immunol (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
175. Dorlo TPC, Ostyn BA, Uranw S, Dujardin JC, Boelaert M. Treatment of Visceral Leishmaniasis: Pitfalls and Stewardship. Lancet Infect Dis (2016) 16(7):777–8. doi: 10.1016/S1473-3099(16)30091-3
176. Bhattacharya SK, Dash AP. Treatment of Visceral Leishmaniasis: Options and Choice. Lancet Infect Dis (2016) 16(2):142–3. doi: 10.1016/S1473-3099(15)00528-9
177. Adem E, Tajebe F, Getahun M, Kiflie A, Diro E, Hailu A, et al. Successful Treatment of Human Visceral Leishmaniasis Restores Antigen-Specific IFN-Gamma, But Not IL-10 Production. PloS Negl Trop Dis (2016) 10(3):e0004468. doi: 10.1371/journal.pntd.0004468
178. de Freitas ESR, von Stebut E. Unraveling the Role of Immune Checkpoints in Leishmaniasis. Front Immunol (2021) 12:620144. doi: 10.3389/fimmu.2021.620144
179. Rodrigues V, Cordeiro-da-Silva A, Laforge M, Ouaissi A, Akharid K, Silvestre R, et al. Impairment of T Cell Function in Parasitic Infections. PloS Negl Trop Dis (2014) 8(2):e2567. doi: 10.1371/journal.pntd.0002567
180. Ali N, Bhattacharya P. Translating Immune Cell Cross-Talk Into a Treatment Opportunity for Visceral Leishmaniasis. Immunother (2013) 5(10):1025–7. doi: 10.2217/imt.13.104
Keywords: visceral leishmanaisis, T cell exhaustion, Th subsets, inflammation, inhibitory receptor
Citation: Costa-Madeira JC, Trindade GB, Almeida PHP, Silva JS and Carregaro V (2022) T Lymphocyte Exhaustion During Human and Experimental Visceral Leishmaniasis. Front. Immunol. 13:835711. doi: 10.3389/fimmu.2022.835711
Received: 14 December 2021; Accepted: 07 April 2022;
Published: 02 May 2022.
Edited by:
Christoph Hölscher, Research Center Borstel (LG), GermanyCopyright © 2022 Costa-Madeira, Trindade, Almeida, Silva and Carregaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanessa Carregaro, dmNhcnJlZ2Fyb0B1c3AuYnI=; dmNhcnJlZ2Fyb0BmbXJwLnVzcC5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.