 Peter Gullickson
Peter Gullickson Yunwen W. Xu
Yunwen W. Xu Laura J. Niedernhofer
Laura J. Niedernhofer Elizabeth L. Thompson
Elizabeth L. Thompson Matthew J. Yousefzadeh
Matthew J. Yousefzadeh- Department of Biochemistry, Institute on the Biology of Aging and Metabolism, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, United States
An effective humoral immune response necessitates the generation of diverse and high-affinity antibodies to neutralize pathogens and their products. To generate this assorted immune repertoire, DNA damage is introduced at specific regions of the genome. Purposeful genotoxic insults are needed for the successful completion of multiple immunological diversity processes: V(D)J recombination, class-switch recombination, and somatic hypermutation. These three processes, in concert, yield a broad but highly specific immune response. This review highlights the importance of DNA repair mechanisms involved in each of these processes and the catastrophic diseases that arise from DNA repair deficiencies impacting immune system function. These DNA repair disorders underline not only the importance of maintaining genomic integrity for preventing disease but also for robust adaptive immunity.
Introduction
A functional immune system is defined by a diverse repertoire of cells, surface receptors, and antibodies needed to effectively respond to pathogenic challenges (1). Endogenous DNA damage is a potent driver of disease and aging (2), can trigger innate immune responses, and drive loss of cells via apoptosis, necrosis, and senescence (3–5). However, deliberate DNA damage is necessary for vertebrates to respond to the limitless variability of pathogen-related antigens (6, 7). Programmed DNA double-strand breaks (DSB) that occur in B and T cell receptor genes are necessary for lymphocyte development and maturation (6, 8, 9). These programmed DNA breaks occur at specific sites and serve as critical intermediates for rearrangements required for V(D)J recombination (Figure 1) (9). Through this process, the nearly 1012 B and T cells in an individual express millions of unique combinations of antibody and T-cell receptor genes (10). Immune repertoires of any two individuals may overlap by only a fraction of a percent even though these repertoires are formed by Variable, Diversity, and Joining gene segments that are shared by all humans (11, 12). The diversity between two individuals at the immunoglobulin loci is greater than their germline diversity.
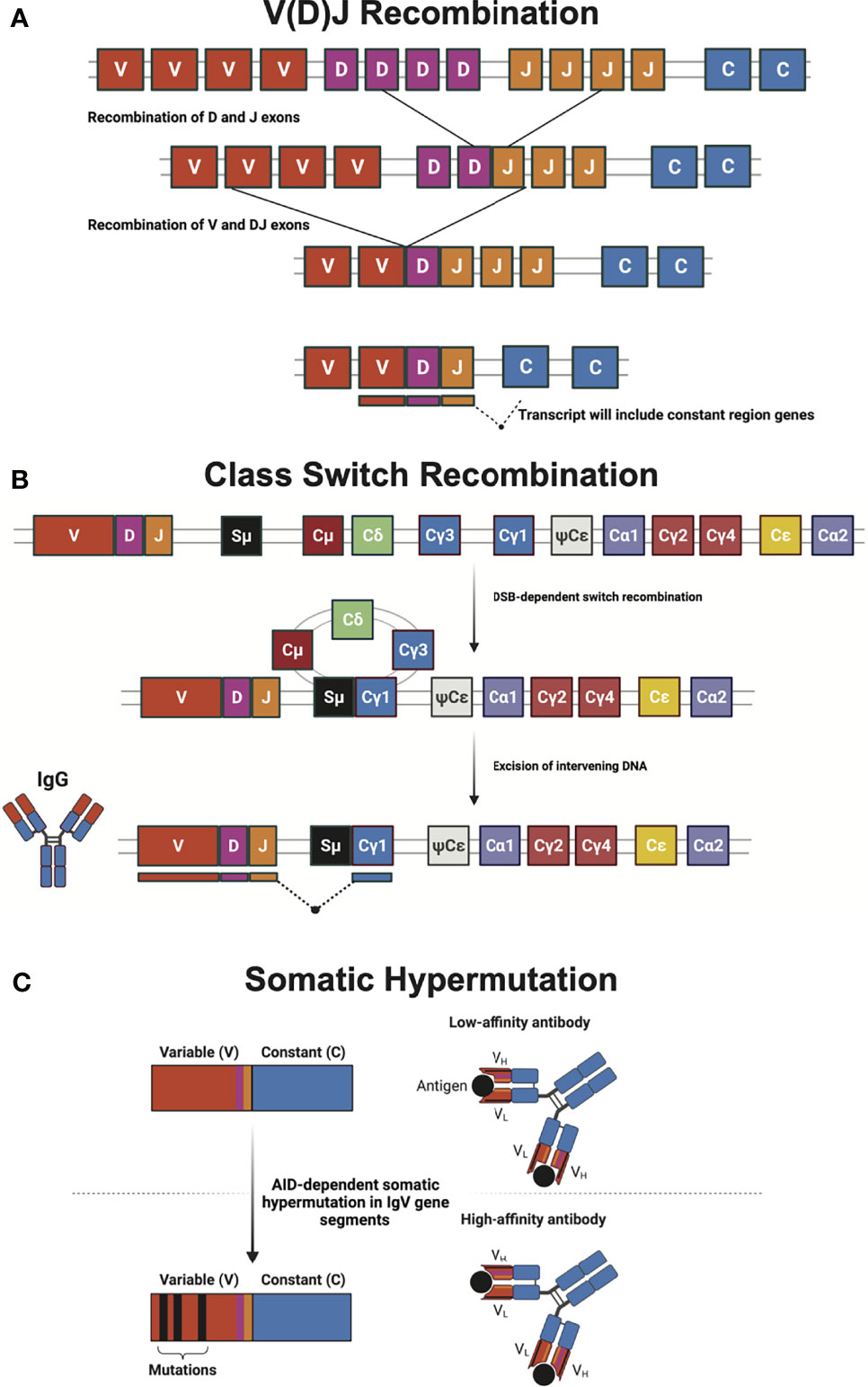
Figure 1 Mechanisms of generating diversity in adaptive immunity. (A) V(D)J recombination relies upon RAG-mediated recombination for the rearrangement of immunoglobulin and T cell receptor variable (V), diversity (D), and joining (J) gene segments during lymphocyte development. Many enzymes involved in non-homologous end joining (NHEJ) and other DNA repair mechanisms are required to correct the programmed DNA double-strand breaks (DSB) that initiate gene segment rearrangement. (B) Class-switch recombination (CSR) of the immunoglobulin heavy chain locus swaps antibody isotype via recombination of different constant (C) regions. CSR requires activation-induced cytidine deaminase (AID) to initiate a DNA DSB break at the switch (S) region, which is subsequently repaired by classical and alternative NHEJ. The schematic shows a CSR event that leads to the production of IgG antibody isotype. (C) Somatic hypermutation (SHM) utilizes AID-dependent programmed mutations in the variable region of antibody gene segments to create a large number of antibodies with goal of creating greater affinity for antigen. Antibody heavy (VH) and light (VL) chains, as well as antigen (black circle) are illustrated. Figure created with BioRender.com.
Antibodies (immunoglobulins) directly neutralize pathogens and their gene products (13). In addition, antibodies recruit cellular effectors of immunity to eliminate pathogens and tumor cells. During development, variable regions of the immunoglobulin (Ig) locus undergo V(D)J recombination of both the heavy (IGH) and light chains (IGL) to generate 1011 to 1014 novel combinations of genetic material (13–15). Upon stimulation, further diversifications of Ig genes can be induced by Class Switch Recombination (CSR) and Somatic Hypermutation (SHM) (Figure 1). Antibody effector function is governed by its antibody class or isotype. In response to antigen stimulation and costimulatory signaling, programmed DNA damage in the constant region of the IGH locus of mature B cells initiates CSR, causing cells to undergo antibody class switching (13, 16). This allows antigen-activated B cells, which are initially IgM+ or IgD+, to change heavy chain constant domains and express one of the other isotypes encoded downstream in the locus, thus altering antibody function and tissue distribution (6).
Germinal center B cells undergo affinity maturation in lymphoid tissue germinal centers to generate high-affinity antibodies that enable a more effective humoral immune response (17, 18). This process relies upon SHM to generate single point mutations in the IGH and IGL loci (19, 20). While CSR acts on the constant region of the IGH and IGL loci, SHM is directed at the variable region. CSR and SHM act in concert to create high-affinity immune responses to each pathogen encountered. VDJ, CSR, and SHM are absolutely dependent on intentional but tightly regulated induction of DNA damage at discrete areas of the genome (20). Multiple components of the DNA repair machinery: sensors, binding proteins, kinases, helicases, recombinases, nucleases, polymerases, and ligases are required for the resolution of the programmed DNA damage that occurs in each of these processes (19, 21). This review highlights the pathophysiological consequences caused by mutations in genes encoding these DNA repair enzymes required for immune diversification.
V(D)J Recombination
V(D)J recombination is the process that assembles the variable domain of immunoglobulin and T-Cell Receptor (TCR) genes via DNA rearrangements (22). V(D)J recombination increases the sequence heterogeneity of a defined gene fragment during the early stages of lymphocyte development. It shapes the immune system repertoire by forming T-cell receptors and immunoglobulins in immature B cells. V(D)J recombination involves multiple DNA repair proteins, including DNA-PKcs, Ku70, Ku80, XRCC4, DNA Ligase IV, and the Cernunnos-XLF protein, all required for non-homologous end-joining of DSBs. Initiation of V(D)J recombination requires lymphoid-specific DNA recombinases RAG1 and RAG2, which recognize recombination signal sequences that flank all V, D, and J gene units and as a complex introduce site-specific DSBs (23–26). MRE11, RAD50, and EXO1 repair proteins are then needed to join the broken DNA ends and resolve the DSB. Mutations in DNA repair factors that participate in V(D)J recombination can severely impact immune function. Mutations in the above genes encoding the above DNA modifying proteins cause varied effects on T and B cell immune cell repertoires. Immunological diseases that arise from DNA repair defects impacting V(D)J recombination are discussed below.
Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) is a rare genetic disorder characterized by impaired development of the immune system and absence of T and B lymphocytes. Mutations in human DNA repair genes RAG1, RAG2, DCLRE1C, PRKDC, NHEJ1, and LIG4 cause SCID (27). These genes all encode proteins that incise (RAG1-RAG2 complex), excise (DCLRE1C Artemis protein) or participate in NHEJ DSB repair (PRKDC/DNA-PKcs, NHEJ1/XLF4, and LIG4). Loss of function mutations in RAG1/2, PRKDC, KU70, or KU80 preclude T and B cell development, leading to SCID (26, 28) (Table 1).
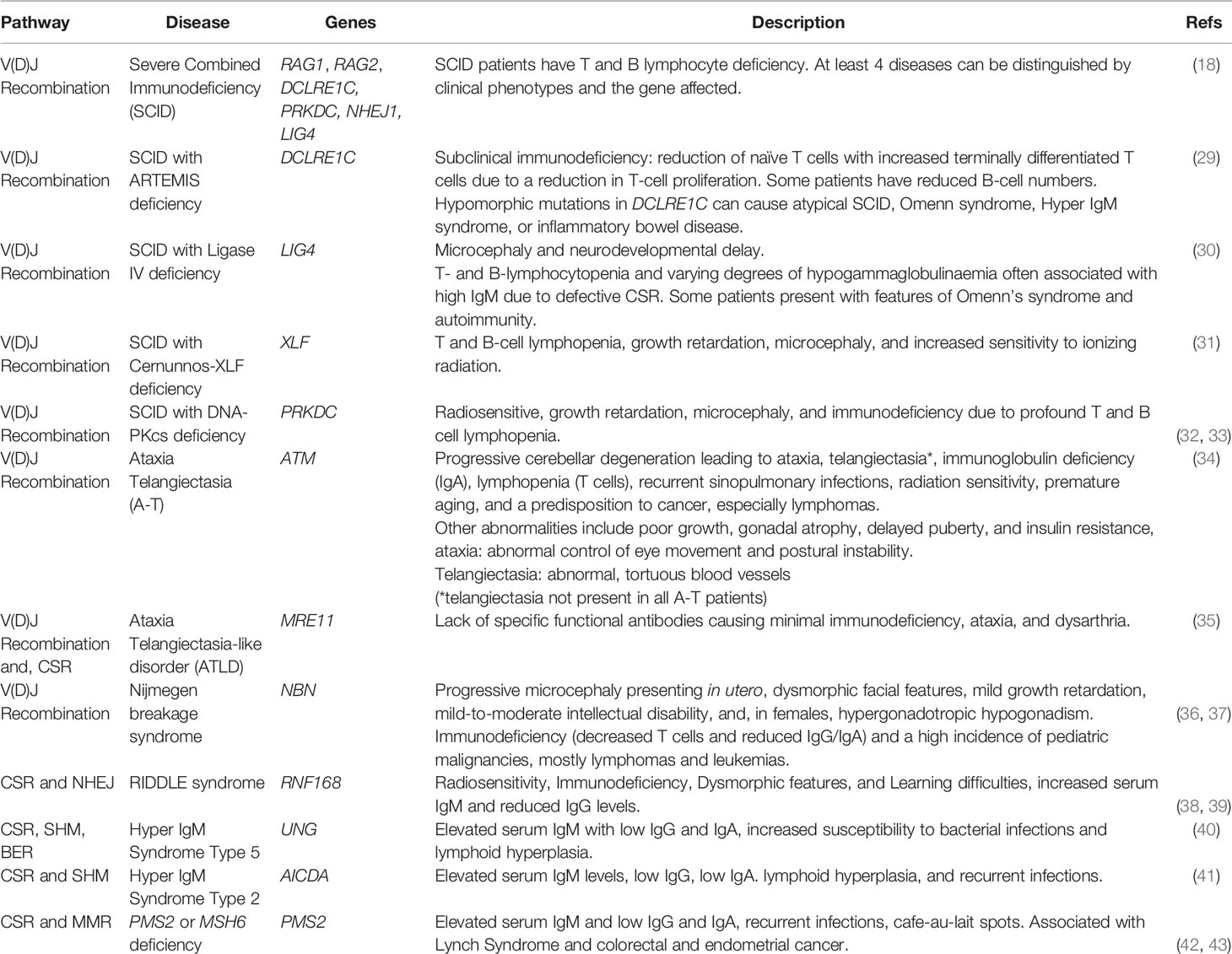
Table 1 DNA repair deficiency-induced immunological disorders.
Artemis deficiency, caused by null mutations in DCLRE1C, also causes SCID. Artemis is an exonuclease essential for the repair of DSBs via non-homologous end-joining (NHEJ) and plays a critical role in V(D)J recombination. Artemis mutations create a broad spectrum of phenotypes that range from SCID to antibody deficiency (29, 44, 45). NK cell number and function are unaffected in Artemis-deficient SCID patients. However, these patients commonly have radiation sensitivity consistent with a DSB repair defect (44). The impact of a mutation on NHEJ repair and capacity can vary between individuals with mutations in DCLRE1C and do not correlate well with clinical severity (46).
DNA-PKcs is a key component of DNA Protein Kinase complex (DNA-PK), which plays a critical role in NHEJ. Artemis is a substrate for DNA-PKcs kinase activity and phosphorylation is required for its nuclease activity that cleans up broken DNA ends. Artemis binds DNA-PKcs and the Artemis-DNA-PK complex cleaves 5’ and 3’ overhangs of hairpins generated by the RAG complex. Mutations in PRKDC can also impair Artemis activation or its ability to bind DNA ends during DSB repair. DNA-PK also has a role in recruiting other NHEJ proteins like XRCC4 and LIG4 to DSBs. As the NHEJ pathway is critical in V(D)J recombination, hypomorphic and null mutations in PRKDC lead to dysfunction in the development of T and B cells. PRKDC mutations were only discovered relatively recently in a SCID patient that exhibited symptoms similar to patients with RAG or DCLRE1C mutations (32). The patient was practically devoid of B and T cells while NK cell numbers were normal. The patient did not display signs of microcephaly or intellectual disability observed in other DNA repair disorders impacting the immune system (32).
DNA ligase IV syndrome, which has features of SCID, is caused by a LIG4 deficiency. This rare autosomal recessive disorder is characterized by microcephaly, abnormal facial features, sensitivity to ionizing radiation, and SCID (30). Only 30 patients with Ligase IV syndrome have been described, and while they all are sensitive to ionizing radiation (47), they exhibit a broad spectrum of clinical features. Patients typically exhibit low T and B cell numbers and low serum Ig levels, resulting in immunodeficiency (30).
Ataxia Telangiectasia
Ataxia Telangiectasia (A-T) is a genetic neurodegenerative disorder that is characterized by progressively cerebellar atrophy with impaired coordination of voluntary movements (ataxia), the development of reddish lesions of the skin and mucous membranes due to dilation of blood vessels (telangiectasia), and immune dysfunction (cellular and humoral immunodeficiency resulting in increased susceptibility to infections, cancer and malignancies, in particular lymphoid malignancies) (34). A-T is caused by mutations in the AT-mutated (ATM) gene, the gene product of which is a key component of the DNA damage response. Mutations in ATM cause aberrant V(D)J recombination and apoptosis during lymphocyte development, resulting in patients having immunoglobulin deficiencies and lymphopenia (48–50). A-T patients with inactivating mutations in ATM sporadically have T cell prolymphocytic leukemia (T-PLL), B cell chronic lymphocytic leukemia (B-CLL), and mantle cell lymphoma (MCL) (51). CSR deficiency is also characteristic of A-T, resulting in high serum IgM levels, with low IgA and IgG levels (35).
Nijmegen Breakage Syndrome
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive syndrome of chromosomal instability mainly characterized by microcephaly at birth, SCID, and a predisposition to malignancies. It is caused by mutations in NBN, which encodes NBS1 (36, 52). NBS1 forms a multimeric complex with MRE11 and RAD50 nuclease (MRN complex) via its C-terminus. The function of NBS1 is to recruit and retain the complex at sites of DNA damage by directly binding to histone H2AX, a histone phosphorylated by PI3-kinase family members such as ATM, in response to DNA damage. The MRN complex facilitates the rejoining of DBSs predominantly by homologous recombination repair rather than NHEJ (52, 53). NBS patients have variability in immunodeficiency, as the number of CD8+ T cells could be normal, elevated, or considerably reduced, with decreased CD4+ T cell counts. However, universally there is an increase in unresolved recombination-mediated breaks in IGH and a compensatory proliferation of mature B cells as absolute B cell numbers are decreased, consistent with a V(D)J recombination defect (36, 37).
Class Switch Recombination
The ability of the immune system to fight and eliminate a wide array of pathogens is made possible by the production of a variety of antibody isotypes, each with unique effector functions. Naïve B-cells produce only membrane-bound antibodies IgM and IgD. Following infection, naïve B cells are activated and can be induced to undergo CSR (13, 18, 54). CSR occurs in the DNA encoding the constant region of IGH (16). Here, deletional recombination occurs between DSBs intentionally introduced at switch (S) regions between IGH constant region genes (18) (Figure 1).
The process of introducing DSBs begins with activation-induced cytidine deaminase (AID), which demethylates cytosines to uracil at immunoglobulin switch regions (55, 56). Next, uracil-DNA glycosylase (UNG), a component of the base excision repair (BER) pathway, excises the uracils, leaving abasic sites that are further processed to create DNA single strand breaks (SSB) (57, 58). If SSBs occur in both strands of the DNA in close proximity, then a DSB results. DNA mismatch repair (MMR) can also create DSBs following AID-induced demethylation (16). MMR recognizes U:G mismatches and resects single-stranded DNA created by mismatch-induced DNA unwinding. If there is a SSB on the opposite strand in the resected region, then a DSB is introduced. The DSBs at the switch regions are recognized, recombined, and then repaired using primarily NHEJ, similar to VDJ recombination (59). In CSR, alternative end-joining (A-EJ) also plays a role in repairing DSBs (60). In contrast to the classical NHEJ (c-NHEJ), A-EJ is a relatively slower and more error-prone process that relies upon annealing at microhomologies. A-EJ is also considered as a prominent source of genome instability (59). A-EJ is substantially less efficient than NHEJ but enables CSR in c-NHEJ-deficient cells (60). Many factors including, stage of the cell cycle, also influence which repair pathway is utilized (61). Some DNA repair factors have distinct contributions in A-EJ versus c-NHEJ. For example, 5-Hydroxymethylcytosine binding, ES cell-specific-protein (HMCES) is dispensable for c-NHEJ but the significant CSR defect observed in HMCES-deficient primary B cells is due to its downstream role in A-EJ (62). Elevated end-resection, non-productive interchromosomal translocations and inversions were observed during sequence analysis of CSR junctions of kinase-dead DNA-PKcs but not DNA-PKcs-deficient B cells (63). ERCC1-XPF, whose role in CSR is not fully understood, removes non-homologous 3’ overhangs that result from annealing at microhomologies during A-EJ (64).
While most CSR-related diseases (discussed below) result from non-functional CSR proteins, the initiation of AID-induced damage outside of the IGH locus can lead to translocations and B cell lymphomas (65–68). Beyond AID’s role in CSR, it also participates in a phenomenon called locus suicide recombination (LSR) which abolishes B cell function (69, 70). In LSR, AID initiates recombination between the most upstream IGH switch region (Sμ) and a “switch-like” region near the 3’ regulatory region resulting in the deletion of the IGH constant region, rendering the B cell non-viable. Although its regulation is not well understood, the balance between CSR and LSR may play a critical role in B cell fate. These studies illustrate the deleterious aspects of AID-mediated recombination that yield non-productive antibodies and B cell death (69, 71).
DNA Repair Syndromes Affecting CSR
DNA repair is critical for antibody diversification through CSR, which is evident in the numerous CSR-related diseases caused by mutations in DNA repair proteins (Table 1). When CSR is not functioning properly, individuals exhibit immunodeficiency due to an impaired ability of B-cells to switch to IgA, IgG, and/or IgE production. The characteristic phenotype of CSR-related diseases is elevated serum IgM levels with low IgA, IgG, and/or IgE levels (6, 13). There is substantial variation in clinical phenotypes both within a disease and between diseases with impaired CSR. For example, a study of patients with MSH6 deficiency found that one patient had elevated IgM levels and reduced IgG, four had elevated IgM and normal IgG, two had normal IgM and reduced IgG, and one had normal IgM and normal IgG (72).
Although AID is not technically a DNA repair protein, its intentional introductions of DNA damage are crucial for the initiation of CSR. Mutations in AICDA, the gene that encodes AID, cause hyper-IgM syndrome (HIGM) type 2 (41). Patients with HIGM type 2 typically present with elevated serum IgM levels with low IgA and IgG levels (73, 74). Following the replacement of cytosine DNA bases in switch regions with uracil by AID, BER and MMR proteins play critical roles in producing DSBs. Mutations in UNG, coding for the BER protein UNG, result in the HIGM Type 5 (Table 1). Like HIGM type 2, this syndrome is characterized by high serum IgM levels, low IgG levels, and low IgA levels (40, 42). Additionally, patients with PMS2- and MSH6-driven MMR deficiency exhibit defective CSR, which is also the case in MLH1 and MSH5-deficient mice (Table 1) (43, 72, 75, 76).
Defects in DSB recognition and signaling proteins can cause CSR-related immunodeficiency. The MRN complex (MRE11-RAD50-NBS1) recognizes DSBs and activates ATM, the key transducer of signaling in response to DSBs. Ataxia Telangiectasia (A-T), Ataxia Telangiectasia-Like Disorder (A-TLD), and NBS, caused by mutations in ATM, MRE11, and NBS1, respectively, all lead to CSR defects (34, 35). NBS patients have a defect in CSR as well as VDJ recombination. A-T and A-TLD share many clinical phenotypes such as ataxia, dysarthria, and abnormal eye movements. However, A-T and NBS result in more similar immunodeficiency phenotypes than A-T and A-TLD. Patients with A-T and NBS often exhibit elevated serum IgM levels, low IgA levels, and low IgG levels (Table 1) (77–79). In contrast, A-TLD patients exhibit very mild immunodeficiency, with reductions in some specific antibody isotypes observed (35, 80). RNF168 is another protein involved in signaling and repair protein recruitment following recognition of DSBs (38). RIDDLE syndrome is caused by RNF168 mutations and is characterized by defective CSR resulting in low serum IgG levels (39). Mutations affecting critical NHEJ proteins often cause CSR-related immunodeficiency. Low or absent serum IgA and IgG levels are common in Cernunnos-XLF- and DNA-PKcs-deficient patients (81, 82). In addition, DNA Ligase IV deficiency often results in low serum IgG levels (30) (Table 1).
Somatic Hypermutation
SHM is another example of intentional DNA damage being induced to enable antibody diversification in germinal center B cells. SHM introduces point mutations in the Ig locus primarily in the antibody variable (V) region that codes for the antigen-binding site of immunoglobulin heavy and light chains (Figure 1). This allows for the production and selection of B cells with high-affinity antibodies (17, 83, 84). The mutation frequency in SHM is a million-fold higher than the basal genome mutation rate. How B cells restrict SHM to the V region while maintaining genome-wide integrity is not well understood. AID initiates antibody affinity maturation through SHM, analogous to initiating CSR. Centroblast B cells in the germinal centers of lymphoid organs express large amounts of AID to initiate SHM (85). Numerous point mutations occur at both the site of uracil incorporation and proximal nucleotides through three predominant mechanisms: replication, BER, and MMR. Uracil incorporated by AID can persist into the S phase during which DNA replication can result in C to T (or G to A) transition mutations (86). However, replication accounts for less than half of all the mutations incorporated during SHM (83). Error-prone non-canonical BER and MMR can combine to diversify mutations introduced during SHM (87, 88). Similar to CSR, SHM-associated uracils are excised by UNG creating an abasic site during BER. Abasic sites are then bypassed by an error-prone translesion synthesis (TLS) DNA polymerase, like Rev1, which can introduce C:G transversion (88, 89). Alternatively, a non-canonical MMR pathway can recognize and repair AID-induced U:G mispairs. This pathway utilizes the error-prone TLS DNA polymerase η, which primarily creates mutations at A:T base pairs (88, 90, 91). Inactivating mutations in AID can result in HIGM type 2 and UNG mutations can result in HIGM type 5 (41, 57, 92). In both conditions, the patients have defects in CSR and SHM and are susceptible to infections (Table 1).
Conclusion
While genotoxic injury is looked upon as unfavorable, it is quite beneficial for certain processes like meiotic recombination and immunological diversity. Deliberate induction and repair of DNA damage serve as a catalyst to expand our immune repertoire. V(D)J and class-switch recombination yield unique antibody combinations and establish effector function (6). Both pathways incorporate many components of the DNA damage response, recombinases, and enzymes from NHEJ repair pathway in addition to other components of the DNA repair machinery, including helicases, nucleases, polymerases, and ligases. Lastly, intentional de novo mutations in the variable region of immunoglobulin genes by SHM create high-affinity antibodies. While DNA repair-deficient murine models have been used to explore disease mechanisms and driver events in tumorigenesis, samples from DNA repair disorder patients have provided great insight into the functional consequences of impaired DNA damage on diversification and development of the adaptive immune system. Future exploration to investigate immune perturbations in other monogenic diseases of DNA repair may provide insight into other DNA repair mechanisms that contribute to immune responses.
Author Contributions
PG, YWX, and MJY prepared the figure and table. PG, YWX, MJY, ELT, and LJN wrote the manuscript. The order of co-first authorship was chosen alphabetically. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by funds from the NIH (U01 ES029603, R01 AG063543, P01 AG062413, and U19 AG056278) to LJN. ELT is supported by funds from the NIH T32 AG029796. MJY is supported by the Irene Diamond Fund/American Federation for Aging Research Postdoctoral Transition Award.
Conflict of Interest
LJN is a co-founder of NRTK Biosciences, a start-up biotechnology company developing senolytic drugs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and Its Role in Health and Disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
2. Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JHJ, Robbins PD. Nuclear Genomic Instability and Aging. Annu Rev Biochem (2018) 87:295–322. doi: 10.1146/annurev-biochem-062917-012239
3. Xu Y. DNA Damage: A Trigger of Innate Immunity But a Requirement for Adaptive Immune Homeostasis. Nat Rev Immunol (2006) 6(4):261–70. doi: 10.1038/nri1804
4. Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, et al. Cytoplasmic DNA: Sources, Sensing, and Role in Aging and Disease. Cell (2021) 184(22):5506–26. doi: 10.1016/j.cell.2021.09.034
5. Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA Damage-How and Why We Age? Elife (2021) 10. doi: 10.7554/eLife.62852
6. Bednarski JJ, Sleckman BP. At the Intersection of DNA Damage and Immune Responses. Nat Rev Immunol (2019) 19(4):231–42. doi: 10.1038/s41577-019-0135-6
7. Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet (2016) 7:147. doi: 10.3389/fgene.2016.00147
8. Rooney S, Chaudhuri J, Alt FW. The Role of the non-Homologous End-Joining Pathway in Lymphocyte Development. Immunol Rev (2004) 200:115–31. doi: 10.1111/j.0105-2896.2004.00165.x
9. Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG Proteins and V(D)J Recombination: Complexes, Ends, and Transposition. Annu Rev Immunol (2000) 18:495–527. doi: 10.1146/annurev.immunol.18.1.495
10. Jiang N, He J, Weinstein JA, Penland L, Sasaki S, He XS, et al. Lineage Structure of the Human Antibody Repertoire in Response to Influenza Vaccination. Sci Transl Med (2013) 5(171):171ra19. doi: 10.1126/scitranslmed.3004794
11. Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, et al. Overlap and Effective Size of the Human CD8+ T Cell Receptor Repertoire. Sci Transl Med (2010) 2(47):47ra64. doi: 10.1126/scitranslmed.3001442
12. Arnaout R, Lee W, Cahill P, Honan T, Sparrow T, Weiand M, et al. High-Resolution Description of Antibody Heavy-Chain Repertoires in Humans. PLoS One (2011) 6(8):e22365. doi: 10.1371/journal.pone.0022365
13. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin Class-Switch DNA Recombination: Induction, Targeting and Beyond. Nat Rev Immunol (2012) 12(7):517–31. doi: 10.1038/nri3216
14. Stavnezer J, Guikema JE, Schrader CE. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
15. Dauba A, Khamlichi AA. Long-Range Control of Class Switch Recombination by Transcriptional Regulatory Elements. Front Immunol (2021) 12:738216. doi: 10.3389/fimmu.2021.738216
16. Stavnezer J, Schrader CE. IgH Chain Class Switch Recombination: Mechanism and Regulation. J Immunol (2014) 193(11):5370–8. doi: 10.4049/jimmunol.1401849
17. Pilzecker B, Jacobs H. Mutating for Good: DNA Damage Responses During Somatic Hypermutation. Front Immunol (2019) 10:438. doi: 10.3389/fimmu.2019.00438
18. Chi X, Li Y, Qiu X. V(D)J Recombination, Somatic Hypermutation and Class Switch Recombination of Immunoglobulins: Mechanism and Regulation. Immunology (2020) 160(3):233–47. doi: 10.1111/imm.13176
19. Martomo SA, Gearhart PJ. Somatic Hypermutation: Subverted DNA Repair. Curr Opin Immunol (2006) 18(3):243–8. doi: 10.1016/j.coi.2006.03.007
20. Methot SP, Di Noia JM. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
21. Goodman MF. Better Living With Hyper-Mutation. Environ Mol Mutagen (2016) 57(6):421–34. doi: 10.1002/em.22023
22. Mansilla-Soto J, Cortes P. VDJ Recombination: Artemis and its In Vivo Role in Hairpin Opening. J Exp Med (2003) 197(5):543–7. doi: 10.1084/jem.20022210
23. Soulas-Sprauel P, Rivera-Munoz P, Malivert L, Le Guyader G, Abramowski V, Revy P, et al. V(D)J and Immunoglobulin Class Switch Recombinations: A Paradigm to Study the Regulation of DNA End-Joining. Oncogene (2007) 26(56):7780–91. doi: 10.1038/sj.onc.1210875
24. Rivera-Munoz P, Malivert L, Derdouch S, Azerrad C, Abramowski V, Revy P, et al. DNA Repair and the Immune System: From V(D)J Recombination to Aging Lymphocytes. Eur J Immunol (2007) 37(Suppl 1):S71–82. doi: 10.1002/eji.200737396
25. Schatz DG, Oettinger MA, Baltimore D. The V(D)J Recombination Activating Gene, RAG-1. Cell (1989) 59(6):1035–48. doi: 10.1016/0092-8674(89)90760-5
26. Schatz DG, Swanson PC. V(D)J Recombination: Mechanisms of Initiation. Annu Rev Genet (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
27. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38(1):96–128. doi: 10.1007/s10875-017-0464-9
28. Schroeder HW Jr., Cavacini L. Structure and Function of Immunoglobulins. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S41–52. doi: 10.1016/j.jaci.2009.09.046
29. Volk T, Pannicke U, Reisli I, Bulashevska A, Ritter J, Bjorkman A, et al. DCLRE1C (ARTEMIS) Mutations Causing Phenotypes Ranging From Atypical Severe Combined Immunodeficiency to Mere Antibody Deficiency. Hum Mol Genet (2015) 24(25):7361–72. doi: 10.1093/hmg/ddv437
30. Altmann T, Gennery AR. DNA Ligase IV Syndrome; a Review. Orphanet J Rare Dis (2016) 11(1):137. doi: 10.1186/s13023-016-0520-1
31. Recio MJ, Dominguez-Pinilla N, Perrig MS, Rodriguez Vigil-Iturrate C, Salmon-Rodriguez N, Martinez Faci C, et al. Extreme Phenotypes With Identical Mutations: Two Patients With Same Non-Sense NHEJ1 Homozygous Mutation. Front Immunol (2018) 9:2959. doi: 10.3389/fimmu.2018.02959
32. van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs Mutation in a Radiosensitive T-B- SCID Patient Inhibits Artemis Activation and Nonhomologous End-Joining. J Clin Invest (2009) 119(1):91–8. doi: 10.1172/JCI37141
33. van der Burg M, van Dongen JJ, van Gent DC. DNA-PKcs Deficiency in Human: Long Predicted, Finally Found. Curr Opin Allergy Clin Immunol (2009) 9(6):503–9. doi: 10.1097/ACI.0b013e3283327e41
34. Weitering TJ, Takada S, Weemaes CMR, van Schouwenburg PA, van der Burg M. ATM: Translating the DNA Damage Response to Adaptive Immunity. Trends Immunol (2021) 42(4):350–65. doi: 10.1016/j.it.2021.02.001
35. Taylor AM, Groom A, Byrd PJ. Ataxia-Telangiectasia-Like Disorder (ATLD)-Its Clinical Presentation and Molecular Basis. DNA Repair (Amst) (2004) 3(8-9):1219–25. doi: 10.1016/j.dnarep.2004.04.009
36. Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. Nijmegen Breakage Syndrome (NBS). Orphanet J Rare Dis (2012) 7:13. doi: 10.1186/1750-1172-7-13
37. van der Burg M, Pac M, Berkowska MA, Goryluk-Kozakiewicz B, Wakulinska A, Dembowska-Baginska B, et al. Loss of Juxtaposition of RAG-Induced Immunoglobulin DNA Ends is Implicated in the Precursor B-Cell Differentiation Defect in NBS Patients. Blood (2010) 115(23):4770–7. doi: 10.1182/blood-2009-10-250514
38. Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell (2009) 136(3):420–34. doi: 10.1016/j.cell.2008.12.042
39. Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, et al. RIDDLE Immunodeficiency Syndrome is Linked to Defects in 53BP1-Mediated DNA Damage Signaling. Proc Natl Acad Sci USA (2007) 104(43):16910–5. doi: 10.1073/pnas.0708408104
40. Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human Uracil-DNA Glycosylase Deficiency Associated With Profoundly Impaired Immunoglobulin Class-Switch Recombination. Nat Immunol (2003) 4(10):1023–8. doi: 10.1038/ni974
41. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell (2000) 102(5):565–75. doi: 10.1016/S0092-8674(00)00079-9
42. de la Morena MT. Clinical Phenotypes of Hyper-IgM Syndromes. J Allergy Clin Immunol Pract (2016) 4(6):1023–36. doi: 10.1016/j.jaip.2016.09.013
43. Peron S, Metin A, Gardes P, Alyanakian MA, Sheridan E, Kratz CP, et al. Human PMS2 Deficiency is Associated With Impaired Immunoglobulin Class Switch Recombination. J Exp Med (2008) 205(11):2465–72. doi: 10.1084/jem.20080789
44. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary Immunodeficiency Diseases: An Update on the Classification From the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol (2014) 5:162. doi: 10.3389/fimmu.2014.00162
45. Punwani D, Kawahara M, Yu J, Sanford U, Roy S, Patel K, et al. Lentivirus Mediated Correction of Artemis-Deficient Severe Combined Immunodeficiency. Hum Gene Ther (2017) 28(1):112–24. doi: 10.1089/hum.2016.064
46. Lee PP, Woodbine L, Gilmour KC, Bibi S, Cale CM, Amrolia PJ, et al. The Many Faces of Artemis-Deficient Combined Immunodeficiency - Two Patients With DCLRE1C Mutations and a Systematic Literature Review of Genotype-Phenotype Correlation. Clin Immunol (2013) 149(3):464–74. doi: 10.1016/j.clim.2013.08.006
47. Iyengar JJ, Quinonez SC, Razumilava N, Soyster B, Smith YR, Vander Lugt MT, et al. DNA Ligase Iv Syndrome: A Rare Cause of Growth Failure & Hypogonadism. AACE Clin Case Rep (2019) 5(2):e154–e8. doi: 10.4158/ACCR-2018-0291
48. Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, et al. Biallelic Mutations in the ATM Gene in T-Prolymphocytic Leukemia. Nat Med (1997) 3(10):1155–9. doi: 10.1038/nm1097-1155
49. Stoppa-Lyonnet D, Soulier J, Lauge A, Dastot H, Garand R, Sigaux F, et al. Inactivation of the ATM Gene in T-Cell Prolymphocytic Leukemias. Blood (1998) 91(10):3920–6. doi: 10.1182/blood.V91.10.3920.3920_3920_3926
50. Yamaguchi M, Yamamoto K, Miki T, Mizutani S. And Miura O. T-Cell Prolymphocytic Leukemia With Der(11)T(1;11)(Q21;Q23) and ATM Deficiency. Cancer Genet Cytogenet (2003) 146(1):22–6. doi: 10.1016/s0165-4608(03)00104-3
51. Boultwood J. Ataxia Telangiectasia Gene Mutations in Leukaemia and Lymphoma. J Clin Pathol (2001) 54(7):512–6. doi: 10.1136/jcp.54.7.512
52. Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K. Nijmegen Breakage Syndrome and Functions of the Responsible Protein, NBS1. Genome Dyn (2006) 1:191–205. doi: 10.1159/000092508
53. Brugmans L, Verkaik NS, Kunen M, van Drunen E, Williams BR, Petrini JH, et al. NBS1 Cooperates With Homologous Recombination to Counteract Chromosome Breakage During Replication. DNA Repair (Amst) (2009) 8(12):1363–70. doi: 10.1016/j.dnarep.2009.09.002
54. Yu K, Lieber MR. Current Insights Into the Mechanism of Mammalian Immunoglobulin Class Switch Recombination. Crit Rev Biochem Mol Biol (2019) 54(4):333–51. doi: 10.1080/10409238.2019.1659227
55. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell (2000) 102(5):553–63. doi: 10.1016/S0092-8674(00)00078-7
56. Feng Y, Seija N, Di Noia JM, Martin A. AID in Antibody Diversification: There and Back Again. Trends Immunol (2020) 41(7):586–600. doi: 10.1016/j.it.2020.04.009
57. Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, et al. B Cells From Hyper-IgM Patients Carrying UNG Mutations Lack Ability to Remove Uracil From ssDNA and Have Elevated Genomic Uracil. J Exp Med (2005) 201(12):2011–21. doi: 10.1084/jem.20050042
58. Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, et al. APE1- and APE2-Dependent DNA Breaks in Immunoglobulin Class Switch Recombination. J Exp Med (2007) 204(12):3017–26. doi: 10.1084/jem.20071289
59. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat Rev Mol Cell Biol (2017) 18(8):495–506. doi: 10.1038/nrm.2017.48
60. Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH Class Switching and Translocations Use a Robust non-Classical End-Joining Pathway. Nature (2007) 449(7161):478–82. doi: 10.1038/nature06020
61. Yu W, Lescale C, Babin L, Bedora-Faure M, Lenden-Hasse H, Baron L, et al. Repair of G1 Induced DNA Double-Strand Breaks in S-G2/M by Alternative NHEJ. Nat Commun (2020) 11(1):5239. doi: 10.1038/s41467-020-19060-w
62. Shukla V, Halabelian L, Balagere S, Samaniego-Castruita D, Feldman DE, Arrowsmith CH, et al. HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair During Class Switch Recombination in B Cells. Mol Cell (2020) 77(5):1154. doi: 10.1038/s41467-020-19060-w
63. Crowe JL, Shao Z, Wang XS, Wei PC, Jiang W, Lee BJ, et al. Kinase-Dependent Structural Role of DNA-PKcs During Immunoglobulin Class Switch Recombination. Proc Natl Acad Sci USA (2018) 115(34):8615–20. doi: 10.1016/j.molcel.2020.02.008
64. Bai W, Zhu G, Xu J, Chen P, Meng F, Xue H, et al. The 3’-Flap Endonuclease XPF-ERCC1 Promotes Alternative End Joining and Chromosomal Translocation During B Cell Class Switching. Cell Rep (2021) 36(13):109756. doi: 10.1016/j.celrep.2021.109756
65. Ramiro A, Reina San-Martin B, McBride K, Jankovic M, Barreto V, Nussenzweig A, et al. The Role of Activation-Induced Deaminase in Antibody Diversification and Chromosome Translocations. Adv Immunol (2007) 94:75–107. doi: 10.1016/S0065-2776(06)94003-6
66. Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is Required for the Chromosomal Breaks in C-Myc That Lead to C-Myc/IgH Translocations. Cell (2008) 135(6):1028–38. doi: 10.1016/j.cell.2008.09.062
67. Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-Capture Sequencing Reveals the Extent and Nature of Chromosomal Rearrangements in B Lymphocytes. Cell (2011) 147(1):95–106. doi: 10.1016/j.cell.2011.07.048
68. Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, Kataegis and Translocations in B Cells: Understanding AID Promiscuous Activity. Nat Rev Immunol (2016) 16(3):164–76. doi: 10.1038/nri.2016.2
69. Dalloul I, Boyer F, Dalloul Z, Pignarre A, Caron G, Fest T, et al. Locus Suicide Recombination Actively Occurs on the Functionally Rearranged IgH Allele in B-Cells From Inflamed Human Lymphoid Tissues. PLoS Genet (2019) 15(6):e1007721. doi: 10.1371/journal.pgen.1007721
70. Peron S, Laffleur B, Denis-Lagache N, Cook-Moreau J, Tinguely A, Delpy L, et al. AID-Driven Deletion Causes Immunoglobulin Heavy Chain Locus Suicide Recombination in B Cells. Science (2012) 336(6083):931–4. doi: 10.1126/science.1218692
71. Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, et al. The Microanatomic Segregation of Selection by Apoptosis in the Germinal Center. Science (2017) 358(6360). doi: 10.1126/science.aao2602
72. Gardes P, Forveille M, Alyanakian MA, Aucouturier P, Ilencikova D, Leroux D, et al. Human MSH6 Deficiency is Associated With Impaired Antibody Maturation. J Immunol (2012) 188(4):2023–9. doi: 10.4049/jimmunol.1102984
73. Imai K, Zhu Y, Revy P, Morio T, Mizutani S, Fischer A, et al. Analysis of Class Switch Recombination and Somatic Hypermutation in Patients Affected With Autosomal Dominant Hyper-IgM Syndrome Type 2. Clin Immunol (2005) 115(3):277–85. doi: 10.1016/j.clim.2005.02.003
74. Trotta L, Hautala T, Hamalainen S, Syrjanen J, Viskari H, Almusa H, et al. Enrichment of Rare Variants in Population Isolates: Single AICDA Mutation Responsible for Hyper-IgM Syndrome Type 2 in Finland. Eur J Hum Genet (2016) 24(10):1473–8. doi: 10.1038/ejhg.2016.37
75. Wu X, Tsai CY, Patam MB, Zan H, Chen JP, Lipkin SM, et al. A Role for the MutL Mismatch Repair Mlh3 Protein in Immunoglobulin Class Switch DNA Recombination and Somatic Hypermutation. J Immunol (2006) 176(9):5426–37. doi: 10.4049/jimmunol.176.9.5426
76. Sekine H, Ferreira RC, Pan-Hammarstrom Q, Graham RR, Ziemba B, de Vries SS, et al. Role for Msh5 in the Regulation of Ig Class Switch Recombination. Proc Natl Acad Sci USA (2007) 104(17):7193–8. doi: 10.1073/pnas.0700815104
77. Demuth I, Digweed M. The Clinical Manifestation of a Defective Response to DNA Double-Strand Breaks as Exemplified by Nijmegen Breakage Syndrome. Oncogene (2007) 26(56):7792–8. doi: 10.1038/sj.onc.1210876
78. Noordzij JG, Wulffraat NM, Haraldsson A, Meyts I, van’t Veer LJ, Hogervorst FB, et al. Ataxia-Telangiectasia Patients Presenting With Hyper-IgM Syndrome. Arch Dis Child (2009) 94(6):448–9. doi: 10.1136/adc.2008.149351
79. de Miranda NF, Bjorkman A, Pan-Hammarstrom Q. DNA Repair: The Link Between Primary Immunodeficiency and Cancer. Ann N Y Acad Sci (2011) 1246:50–63. doi: 10.1111/j.1749-6632.2011.06322.x
80. Lahdesmaki A, Taylor AM, Chrzanowska KH, Pan-Hammarstrom Q. Delineation of the Role of the Mre11 Complex in Class Switch Recombination. J Biol Chem (2004) 279(16):16479–87. doi: 10.1074/jbc.M312796200
81. Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, et al. Lymphocyte-Specific Compensation for XLF/cernunnos End-Joining Functions in V(D)J Recombination. Mol Cell (2008) 31(5):631–40. doi: 10.1016/j.molcel.2008.07.017
82. Bjorkman A, Du L, Felgentreff K, Rosner C, Pankaj Kamdar R, Kokaraki G, et al. DNA-PKcs Is Involved in Ig Class Switch Recombination in Human B Cells. J Immunol (2015) 195(12):5608–15. doi: 10.4049/jimmunol.1501633
83. Peled J, Kuang F, Iglesias-Ussel M, Roa S, Kalis S, Goodman M, et al. The Biochemistry of Somatic Hypermutation. Annu Rev Immunol (2008) 26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236
84. Heltzel J, Gearhart P. What Targets Somatic Hypermutation to the Immunoglobulin Loci? Viral Immunol (2020) 33(4):277–81. doi: 10.1089/vim.2019.0149
85. Muramatsu M, Sankaranand V, Anant S, Sugai M, Kinoshita K, Davidson N, et al. Specific Expression of Activation-Induced Cytidine Deaminase (AID), a Novel Member of the RNA-Editing Deaminase Family in Germinal Center B Cells. J Biol Chem (1999) 274(26):18470–6. doi: 10.1074/jbc.274.26.18470
86. Xue K, Rada C, Neuberger MS. The In Vivo Pattern of AID Targeting to Immunoglobulin Switch Regions Deduced From Mutation Spectra in Msh2-/- Ung-/- Mice. J Exp Med (2006) 203(9):2085–94. doi: 10.1084/jem.20061067
87. Safavi S, Larouche A, Zahn A, Patenaude AM, Domanska D, Dionne K, et al. The Uracil-DNA Glycosylase UNG Protects the Fitness of Normal and Cancer B Cells Expressing AID. NAR Cancer (2020) 2(3):zcaa019. doi: 10.1093/narcan/zcaa019
88. Thientosapol ES, Sharbeen G, Lau KKE, Bosnjak D, Durack T, Stevanovski I, et al. Proximity to AGCT Sequences Dictates MMR-Independent Versus MMR-Dependent Mechanisms for AID-Induced Mutation via UNG2. Nucleic Acids Res (2017) 45(6):3146–57. doi: 10.1093/nar/gkw1300
89. Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-Biased Defect in C/G Transversions in Hypermutating Immunoglobulin Genes in Rev1-Deficient Mice. J Exp Med (2006) 203(2):319–23. doi: 10.1084/jem.20052227
90. Neuberger MS, Rada C. Somatic Hypermutation: Activation-Induced Deaminase for C/G Followed by Polymerase Eta for a/T. J Exp Med (2007) 204(1):7–10. doi: 10.1084/jem.20062409
91. Zanotti KJ, Gearhart PJ. Antibody Diversification Caused by Disrupted Mismatch Repair and Promiscuous DNA Polymerases. DNA Repair (Amst) (2016) 38:110–6. doi: 10.1016/j.dnarep.2015.11.011
Keywords: immunological diversity, immunodeficiency, antibodies, DNA damage, DNA repair
Citation: Gullickson P, Xu YW, Niedernhofer LJ, Thompson EL and Yousefzadeh MJ (2022) The Role of DNA Repair in Immunological Diversity: From Molecular Mechanisms to Clinical Ramifications. Front. Immunol. 13:834889. doi: 10.3389/fimmu.2022.834889
Received: 13 December 2021; Accepted: 02 March 2022;
Published: 01 April 2022.
Edited by:
Erik A. L. Biessen, Maastricht University, NetherlandsReviewed by:
Michel Cogne, University of Rennes 1, FranceMatthew Scharff, Albert Einstein College of Medicine, United States
Copyright © 2022 Gullickson, Xu, Niedernhofer, Thompson and Yousefzadeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Yousefzadeh, bXlvdXNlZnpAdW1uLmVkdQ==
†These authors share first authorship