 Ravichandra Tagirasa
Ravichandra Tagirasa Euna Yoo
Euna Yoo- Chemical Biology Laboratory, National Cancer Institute, Frederick, MD, United States
During tumor development, invasion and metastasis, the intimate interaction between tumor and stroma shapes the tumor microenvironment and dictates the fate of tumor cells. Stromal cells can also influence anti-tumor immunity and response to immunotherapy. Understanding the molecular mechanisms that govern this complex and dynamic interplay, thus is important for cancer diagnosis and therapy. Proteolytic enzymes that are expressed and secreted by both cancer and stromal cells play important roles in modulating tumor-stromal interaction. Among, several serine proteases such as fibroblast activation protein, urokinase-type plasminogen activator, kallikrein-related peptidases, and granzymes have attracted great attention owing to their elevated expression and dysregulated activity in the tumor microenvironment. This review highlights the role of serine proteases that are mainly derived from stromal cells in tumor progression and associated theranostic applications.
Introduction
The tumor microenvironment (TME) is a highly complex system that is comprised of a heterogenous population of cancer cells and associated stromal cells, and the extracellular matrix (ECM). The ECM not only provides structural support in the extracellular space but also regulates multiple cellular signaling in tumor tissues (1, 2). The tumor stroma, as a critical component of the TME, actively contributes to cancer proliferation, angiogenesis, invasion and metastasis, immune evasion, and resistance to cancer therapy (3, 4). The function of stromal cells and their interaction with cancer cells within the TME are modulated by expression and secretion of various signaling molecules such as growth factors (5–7), chemokines (8–10), cytokines (11–14), and proteolytic enzymes (15–17). Although once thought to be limited to the degradation of ECM, the role of proteases in tumors is now better understood to be significantly more complicated and critical. In addition to cancer cells, stromal cells including fibroblasts, endothelial cells and infiltrating immune cells all contribute proteases in developing tumors such as matrix metalloproteinases (MMPs), cysteine cathepsins, and serine proteases. Proteases are involved in proteolytic networks, remodeling of ECM, regulation of growth factor and cytokine signaling, and modulation of inflammatory responses and immunosuppressive effects (Figure 1). Depending on the cellular context, activity and interaction of these proteases can have either tumor-promoting or -suppressing effects. Among, serine proteases, which make up approximately one-third of human proteases, are important class of proteases in carcinomas. Dysregulated expression and activity of several serine proteases that are derived from stromal cells have been associated with tumor development and metastasis. In this review, we aim to summarize our current understanding of functional roles that key serine proteases play in the transformed stroma and discuss their theranostic potential.
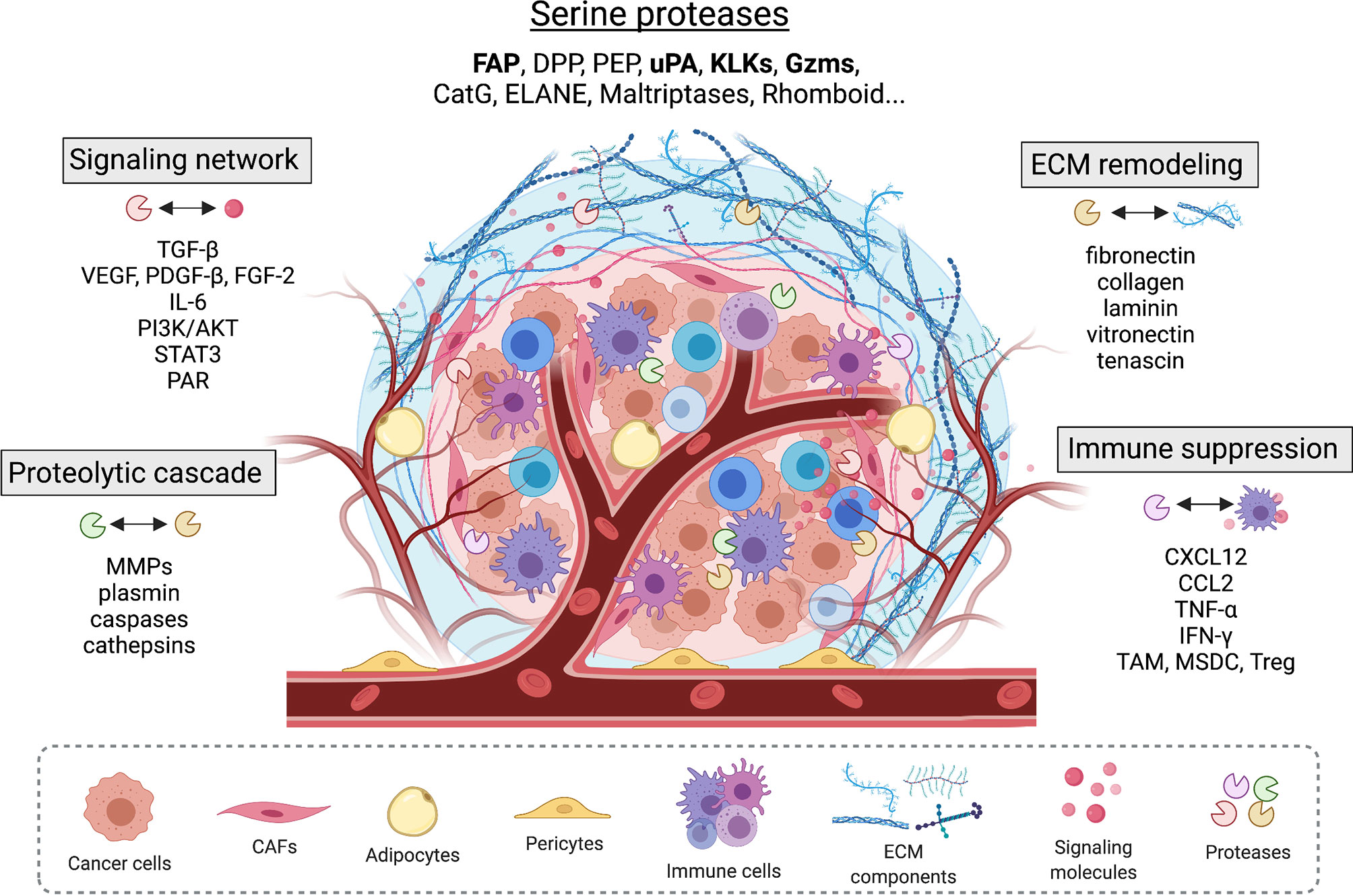
Figure 1 Serine proteases found in the tumor microenvironment modulate tumor-stromal interplay by interacting with other proteases, extracellular matrix proteins, and important signaling pathways that involve growth factors, cell surface receptors, cytokines and chemokines.
Fibroblast Activation Protein α (FAPα, FAP or Seprase)
Cancer-associated fibroblasts (CAFs), which are perpetually activated and distinct from normal fibroblasts in their morphological and functional features, are extremely abundant in the tumor microenvironment including breast, prostate, and pancreatic carcinomas with potent tumorigenic effects (18). CAFs are highly heterogeneous, proliferative, and are resistant to apoptotic cell death. Among several surface bound markers identified in CAFs, fibroblast activation protein α (FAP) is shown to be selectively upregulated on reactive stromal fibroblasts of more than 90% of human epithelial carcinomas (19). It has been demonstrated that altered tumor microenvironment or inflammation induces FAP expression through stimulation of cytokines such as TGF-β1 and TNF-α (20, 21), chemical substances (22), or physical stimulants (23). FAP is a type II transmembrane glycoprotein that displays both dipeptidyl peptidase and endopeptidase activities, removing two amino acids from the N-terminus of the substrate as well as hydrolyzing peptide bonds of nonterminal amino acids after a proline residue. The postprolyl peptidase activity of FAP requires homodimerization of the protein and is carried out by the catalytic triad consisting of Ser, Asp, and His residues (24). Physiological substrates of FAP include gelatin, denatured type I collagen, α-antitrypsin, α2-antiplasmin, fibroblast growth factor 21, fibrillin-2, extracellular matrix protein 1, C-X-C motif chemokine 5, tumor necrosis factor related protein 6, lysyl oxidase homolog 1, and several neuropeptides (25–27). Protease-independent activity of FAP has been associated with activation of MMP2/9, phosphoinositide 3-kinases (PI3Ks), and STAT signaling pathways (28, 29).
Given its high expression on CAFs and ability to degrade major ECM proteins, the role of FAP in remodeling and patterning of ECM, which in turn affects cellular response and tumorigenesis, has been investigated. In both syngeneic transplant and endogenous mouse tumor models, genetic deletion of FAP or pharmacologic inhibition of its enzymatic activity led to excessive accumulation and disorganization of collagen and decrease in myofibroblast content and blood vessel density in tumors, thus inhibiting tumor growth (30). Utilizing an in vivo-like 3-dimensional matrix system, Lee et al. demonstrated that FAP overexpression on fibroblasts modifies architecture and composition of ECM through inducing tumor stromal-like parallel organization of fibronectin and collagen I fibers and modulating protein levels of tenascin C, collagen I, fibronectin and α-smooth muscle actin (31). Enhanced velocity and directionality of pancreatic cancer cells invading through FAP+ matrices were observed, which was effectively reversed by inhibition of FAP enzymatic activity. In vitro, a study showed that compared to control transfectants that do not express FAP and form slow growing tumors, cells expressing not only wild type FAP but also catalytic mutant of FAP degrade fibronectin matrices more extensively, accumulate higher levels of MMP-9, invade type I collagen gels to a significantly higher degree, and have altered pattern of tyrosine phosphorylated proteins, suggesting the functions of FAP independent of its enzymatic activity (32). FAP is found to form a protease complex with dipeptidyl peptidase IV (DPP4), which is another type II transmembrane protein with serine protease activity, at the endothelial cells of capillary-like micro-vessels within invasive breast ductal carcinoma. In vitro experiments showed that gelatin-binding domain of DPP4 brings this DPP4-FAP complex together with gelatin substrates at the migratory endothelial cells facilitating the local degradation of the extracellular matrix and subsequent cell migration and invasion (33). Analysis of gastric cancer (GC) patient tissue samples showed that FAP expression is positively correlated with micro-vessel density indicating the role of FAP expression in angiogenesis and metastasis (34). Many studies have demonstrated that FAP expression regulates signaling pathways that control cell cycle, proliferation, migration, and invasion (29, 35–37).
Growing evidence has also suggested that FAP+CAFs can promote the immunosuppressive tumor microenvironment. When FAP+ cells were depleted in lung or pancreatic cancers, it caused immediate growth arrest of immunogenic tumor through TNF-α- and IFN-γ-mediated mechanism (38). The study by Feig et al. showed that production of chemokine (C-X-C motif) ligand 12 (CXCL12) by FAP+CAFs mediates the immune suppressive activity and accounts for the failure of T cell checkpoint inhibitors in pancreatic ductal adenocarcinoma (PDA) (39). Combining with administration of α-CTLA-4 or α-PD-L1, depletion of FAP+ cells or inhibiting CXCR4 (CXCL12 receptor) diminished PDA growth. FAP also induces inflammatory CAFs by STAT3 activation leading to increased expression of CCL2, which promotes the tumor recruitment of myeloid-derived suppressor cells (MDSCs) and immunosuppression (40, 41). In murine models of pancreatic adenocarcinoma, inhibition of FAP proteolytic activity resulted in decreased macrophage recruitment, and genetic knockout of FAP enhanced T cell infiltration and cytotoxicity (42). However, in another study in non-small cell lung cancer (NSCLC), high density of FAP+CAFs was found to be associated with improved prognosis in patients with high expression of CD8 and CD3 T lymphocytes (43, 44). This result is contrary to previous finding in a small study (n= 59) that higher levels of FAP expressing stromal cells are associated with worse overall survival and increased peripheral neutrophil and lymphocyte count ratio (NLR) in NSCLC patients (45). In invasive ductal carcinoma of the breast, FAP+CAFs have been associated with longer survival (46), while in pancreatic adenocarcinoma and rectum they are found to be associated with worse clinical outcome (47–49). Thus, the cellular context and mechanism of FAP on the antitumor immune responses in the TME would require further investigation.
Urokinase Plasminogen Activator (Urokinase-Type Plasminogen Activator, Urokinase, uPA)
Urokinase plasminogen activator (uPA) is a serine protease that is extracellularly localized and involved in the plasminogen activator system. It was first found in the urine (50), but later identified also in plasma, seminal fluid, and the extracellular matrix (51). It is synthesized and secreted as an inactive zymogen known as pro-uPA and different proteases such as cathepsin B and L, trypsin, kallikrein, and mast cell tryptase convert pro-uPA into an active uPA (52, 53). Binding of uPA to uPA receptor (uPAR), which is a glycosyl-phosphatidylinositol (GPI)-anchored cell membrane receptor highly expressed in most types of solid tumors such as breast, prostate, brain, and head and neck cancers, localizes the active uPA to the cell surface and converts its major substrate plasminogen into plasmin. Once activated, non-specific protease plasmin is involved in degradation of collagen IV, laminin, fibronectin, vitronectin, fibrin, and several blood clotting factors either directly or through activation of other proteases such as MMPs (54). The inhibitory proteins, plasminogen activator inhibitor-1 (PAI-1) and PAI-2, regulate the activity of uPA (55).
The role of uPA in tumor invasion and metastasis has been widely investigated. Depending on cancer type, uPA and uPAR are expressed both by cancer and stromal cells. uPA is mainly expressed by tumor-associated macrophages (TAMs) and CAFs, and on tumor endothelial cells (TECs) to lesser extent. Through interaction between uPAR and integrins as well as ECM components such as vitronectin, uPA system regulates cell adhesion and migration. uPA supports tumor cell proliferation by proteolytically activating various growth factors that include epidermal growth factor (EGF), fibroblast growth factor-2 (FGF-2) and hepatocyte growth factor/scatter factor (HGF-SF) (56). TGF-β, which is a predominant and multifunctional cytokine found in the TME, regulates the expression of uPA in several types of transformed cells (57–59). uPA-activated plasmin, in turn, activates the secreted TGF-β precursor by a proteolytic cleavage within the N-terminal region of latency-associated peptide (60–62). This loop contributes to tumor growth, cancer cell migration, epithelial to mesenchymal transition (EMT), and metastasis. uPA also mediates the effects of vascular endothelial growth factor (VEGF), a key factor in angiogenesis (63). A study showed that downregulation of uPA/uPAR inhibits angiogenesis in glioblastoma and endothelial cells by regulating tissue inhibitors of metalloproteinase-1 (TIMP-1) secretion, subsequently enhancing secretion of sVEGFR1, a known scavenger of VEGF (64). In vitro, uPA derived from CAFs was found to promote esophageal squamous cell carcinoma (ESCC) cell proliferation, migration, and invasion by activating PI3K/AKT and ERK signaling pathways (65). In multiple myeloma, increased expression and activity of both uPA and uPAR on CAF cells with higher proliferative rate and invasion potential were observed, suggesting the potential role of uPA/uPAR system in promoting metastasis of malignant plasma cells (66). uPA/uPAR system has been implicated in suppression of apoptotic cell death. RNAi-mediated downregulation of uPA and uPAR led to dephosphorylation of focal adhesion kinase (FAK), p38 MAPK, janus kinase (JNK) and ERK1/2 which in turn activates caspase-8, cytochrome c release, PARP cleavage, and subsequent apoptosis of human glioma cells (67). The uPA/uPAR system also plays a critical role in macrophage infiltration (68). TAMs, one of most abundant types of tumor-infiltrating immune cells found in the TME, exhibit important functions in tumor growth, metastasis, angiogenesis, and immune regulation. For example, by producing cytokines, chemokines, growth factors, and triggering the inhibitory immune checkpoint proteins release in T cells, TAMs promote immunosuppression (69). In addition, expression of uPAR and PAI-1 in TAMs has been correlated with vessel remodeling and node status and tumor grade, indicating that TAMs have an important role in the expression and regulation of uPA system for establishing the vascular network in tumors (70).
Kallikrein Related Peptidases (KLKs)
Kallikrein or kallikrein related peptidases are a family of secreted serine proteases that play important roles in ECM remodeling, angiogenesis, skin homeostasis, innate immunity, male reproduction, tooth enamel formation, and neural development. In humans, there are 15 secreted KLKs (KLK1-15). KLK1-2, KLK4-6, KLK8, and KLK10-15 have trypsin-like and KLK3, KLK7, and KLK9 have chymotrypsin-like activities. Aberrant expression of KLKs has been associated with a variety of malignancies, thus the potential of KLKs as cancer markers has been suggested for several members of this protease family. In particular, because of the restricted expression in prostate, KLK3, also known as a prostate-specific antigen (PSA), has been widely employed as a clinical biomarker for prostate cancer. Laser cell microdissection analysis and immunochemistry in human breast cancer surrounding stromal cells showed significant upregulation of KLK4 but downregulation of all other KLKs (71). Immunohistochemistry analysis of tissue sections of ovarian and melanoma patients found the overexpression of KLK6 in tumor associated stromal cells and keratinocytes (72, 73). In pancreatic ductal adenocarcinoma, immunostaining analysis of epithelial tumor cells and the surrounding stroma and immune cells showed that high KLK6 protein levels in the tumor and immune cells are significantly associated with shorter survival compared to low protein levels (74). mRNA analysis of colorectal cancer (CRC) tissue samples from 136 patients showed upregulated KLK10 expression (75). Significantly increased expression of KLK10 has been also found in ovarian cancer tissues (76).
During the tumor progression, KLKs from cancer and stromal cells are released into the TME, where they can exert their proteolytic activity, mainly activating signaling networks and modulating the expression of genes and proteins important to tumor growth and invasion. For example, KLKs activate EGFR and protease-activated receptor (PAR), resulting in the stimulation of ERK1/2 signaling and enhanced subsequent cell proliferation (77–79). Analyzing the secretome of endothelial cells, a study showed that KLK12 can catalyze the release of PDGF-β from ECM components and cell surface. The released PDGF-β then mediates secretion of VEGF and angiogenesis. In addition, KLK12 has been shown to cleave the ECM proteins fibronectin and tenascin (80). KLK12-mediated remodeling of fibronectin matrix led to an increase in endothelial cell migration which was inhibited by a polyclonal antibody directed against the KLK12 cleavage site (81). An in vitro study showed that KLK14 recognizes and hydrolyzes pro-MMPs to active MMPs, especially the membrane-type MMPs (82). Combined expression of KLK4-7 in ovarian cancer has been associated with increased level of TGF-β1, neural cell adhesion molecule L1 (L1CAM), and other tumor-associated factors such as keratin 19 and moesin, indicating the impact of KLK proteases on the secreted proteomes shaping tumor microenvironment (83, 84). KLK4, highly expressed in prostate cancer, promotes CAF differentiation. Through PAR1 activation, KLK4 regulates FGF-1, tasgelin, and lysyl oxidase, and several soluble factors in the prostate stromal cell secretome (85). KLK4 can also stimulate uPA/uPAR system and activate MMPs, leading to the ECM degradation. Overexpression of KLK7 in melanoma cells was shown to induce a decrease in cell proliferation and colony formation but an increase in cell motility and invasion possibly through modulating cell adhesion molecules such as E-cadherin and MCAM/CD146 (86). Kallikreins have been also associated with infiltration of immune cells (87). Although many more studies are needed to better define the pathophysiological functions of each KLK in cancer, emerging evidence suggest that KLKs have significant effect on the tumor-microenvironment interaction and targeting specific KLK may provide therapeutic benefits in cancer therapy.
Granzymes (Gzms)
Granzymes are cell death-inducing serine proteases primarily known for their role in eliminating infected and transformed cells through cytotoxic T cells and natural killer cells. Among five granzymes identified in humans (A, B, H, K, and M), GzmA and GzmB have been most widely studied. The expression of granzymes is regulated by many factors including receptor engagement and stimulation with cytokines. Granzymes are expressed as an inactive pro-enzyme that requires cleavage of N-terminal dipeptide inside secretory granules by cathepsin C (88). As granzyme is optimally active at neutral pH, granzymes stored in acidic granules are quiescent and become enzymatically active following release from the granules into the cytoplasm. In addition, the presence of protein inhibitors such as serpins regulates the proteolytic activity of granzymes (89).
Infiltrating immune cells within the TME significantly contribute to tumor suppression (immune surveillance) or tumor promotion (inflammation and angiogenesis) either directly or through the interplay with other stromal components. In regard to anti-tumor immunity, the intracellular role of granzymes is well characterized and appreciated. Upon activation, the pore-forming protein perforin helps deliver GzmB into the cytosol of target cells, where it induces apoptosis by caspase-dependent and -independent mechanisms. Activation of gasdermin B (GSDMB) by GzmA (90) or GSDME by GzmB (and indirectly by caspase-3) initiates pyroptotic cell death in tumors (91). Mounting evidence suggests that granzymes are also active players in immune regulatory cells and tumor cells. Regulatory T cells (Tregs), MDSCs, dendritic cells (DCs), mast cells, and Bregs are found to express granzymes. If not controlled, granzymes can cause self-inflicted damage of expressing cells. It has been suggested that GzmB is involved in Treg-mediated suppression and elimination of activated CTL/NK cells and antigen-presenting cells, indicating Treg cells utilize GzmB to suppress immune responses and tumor clearance, thus depending on the relative abundance of these cells in the tumor, GzmB can have either detrimental or protective function in antitumor immunity (92, 93).
In addition, extracellular (perforin-independent) functions of granzymes are emerging. Although it remains unclear what stimuli and signaling pathways regulate granzyme release, studies have long shown that patients with infectious diseases and certain proinflammatory conditions have elevated levels of extracellular GzmA/B. GzmB is also found to be constitutively released from CTL/NK cells in vivo (94). Once released, they can mediate the cleavage of extracellular matrix, cell surface receptors, cytokines, and act as proinflammatory proteases. The ECM substrates of GzmB include fibronectin, vitronectin, aggrecan, laminin, and decorin. D’Eliseo et al. showed that GzmB expressed in bladder cancer cell lines and urothelial carcinoma tissues is active in catalyzing vitronectin cleavage, and inhibition of GzmB activity suppresses bladder cancer cell invasion (95). A recent study also demonstrated that active GzmB released from migrating CTLs contribute to extravasation and homing of CTLs via basement membrane cleavage and remodeling. GzmB-null CTLs exhibited impaired homing and decreased transmigration through the vessel wall in mouse models of viral infection and inflammation. In vitro migration assays using Matrigel or Madin-Darby canine kidney (MDCK) cell basement membrane showed that active GzmB released from migrating CTLs enabled chemokine-driven movement and cleavage of basement membrane components (96). GzmM expressed in carcinomas has been implicated in promoting tumor growth, metastasis, and EMT dependent on STAT3 signaling (97). GzmM expression is positively related to IL-6 and VEGF release from cancer cells. Recently, extracellular GzmA was found to be engaged in gut inflammation in colorectal cancer by inducing NF-kB-dependent IL-6 production in macrophages leading to STAT3 activation (98). In mouse models, GzmA knockout or inhibition reduced inflammation and CRC development, suggesting that development of effective GzmA inhibitors could offer therapeutic benefits treating gut inflammation and CRC.
Targeting Serine Proteases in Tumor Stroma
With respect to the clinical applications in cancer diagnosis and therapy, a number of tools that either detect the protein expression or harness and leverage the specific protease activity have been developed (Table 1).

Table 1 Theranostic targeting of serine proteases.
Given their high and selective expression on tumor-associated stromal cells, inhibition of serine proteases by active-site targeting small molecules has been extensively investigated in cancer treatment (30, 42, 102, 103, 115–121, 126–130). Antibody-based approaches that can inhibit the activity of serine proteases aim to achieve better selectivity. For example, a humanized version of monoclonal antibody F19, sibrotuzumab has been developed to target the cell surface bound FAP on tumor stromal fibroblasts and explored for its anti-tumor response (99). Protease biology in cancer offers opportunities for diagnostic profiling as well. Radiolabeled antibodies targeting FAP (106), uPA (122, 123), and KLK3 (135, 136) have been evaluated in single photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging of primary and metastatic tumors. In vivo imaging tools exploit the cleavage activity of serine proteases. Incorporating the optimal peptidic sequence, substrate-based, activatable probes are designed either as quenched probes or as FRET probes that produce a fluorescent signal only after cleavage as a measure for protease activity (112, 113, 124, 142, 144–148). On the other hand, activity-based probes contain an electrophilic warhead that reacts with catalytic serine of the protease and a reporter group that generates a signal upon covalent interaction with the protease (137, 141, 149–152). In that case not only peptides but also non-peptidic small molecules can serve as a recognition motif targeting the protease active site (153–156).
Taking advantage of upregulation of specific serine protease activity in tumors, protease-activatable prodrug approach has been widely used in cancer therapy for selective delivery of drugs while minimizing toxicity to normal tissues. Chemotherapeutic compounds such as doxorubicin are conjugated to cleavable linkers sensitive to active serine proteases (104, 105, 131–134). For example, the FAP-cleavable GP linker and KLK3-cleavable SSKYQSL linker have been successfully used in prodrug approach. Antibody-drug conjugates that incorporate tumor-targeting antibodies into protease-activatable prodrug format further enhance tumor-specific activation. A new technology termed as Probody masks antibody binding using linkers cleaved by extracellular proteases. EGFR Probody PB1 only binds EGFR following cleavage by matriptase, uPA, or legumain (125).
While cancer immunotherapies have shown significant clinical outcomes, only a small subset of patients respond to the treatment, calling for reliable biomarkers and therapeutic strategies to maximize the benefits of the immunotherapy. Targeting the immunomodulatory, tumor stroma-associated serine proteases may provide a potential therapeutic option that complements and/or synergizes with the currently available immuno-oncology therapeutics. In cancer patients that high FAP expression in CAFs restricts T cell distribution and promotes immune checkpoint blockades (ICBs) resistance (41, 157, 158), treatment with FAP inhibitor could neutralize the immunosuppressive function of CAFs and reverse anti-PD1 drug resistance. Conditional depletion of FAP+CAFs could result in enhanced T cell infiltration and better response to anti-CTLA4 and anti-PD-L1 treatment. Injection of chimeric antigen receptor (CAR) T-cells constructed with anti-FAP mAb effectively depleted FAPhi stromal cells and inhibited tumor growth. More importantly, FAP-specific CAR T cells augmented the antitumor responses of endogenous CD8+ T cells (159). The FAP-specific CAR T cell therapy lately advanced into a phase I clinical trial for treatment of patients with malignant pleural mesothelioma (NCT01722149).
Tumor-specific protease-activation can also be exploited to limit systemic exposure of immunotherapeutic agents and minimize unwanted immune toxicity and side effects. Recently, a dual variable domain (DVD) immunoglobulin of anti-CTLA4 antibody, that an outer domain (tumor targeting) is connected by serine protease cleavable linkers to an inner domain (CTLA4 targeting), has been developed (160). During systemic circulation, the CTLA4-binding site is shielded by the outer domain. Upon reaching the tumor, the outer domain is cleaved by membrane type-serine protease 1 (MT-SP1) present in the tumor microenvironment, leading to enhanced localization of anti-CTLA4 without causing significant treatment-associated toxicity. To deliver functionally active cytokines, that are found to be critical for establishing and maintaining the immune response to tumors, preferentially at the tumor site, the Frelinger group developed an activatable IL-2 fusion protein consisting of IL-2 joined to a specific IL-2 binder that blocks its function connected by a KLK3 cleavage sequence (161). This PACman (for protease activated cytokine) approach has been also applied to the generation of IL-12 fusion protein (162). Tumor-specific protease-activation strategy should be applicable to other TME proteases and immunomodulators allowing site-specific activation in immunotherapy.
A granule-associated serine protease GzmB has attracted significant attention as an early predictive biomarker for monitoring of immunotherapy responses. In particular, in vivo imaging approaches with activatable probes utilizing GzmB-cleavable IEPD or IEFD linker allow to directly report functional readouts of immune cell (e.g. CTLs and NK cells) infiltration, activation, and cytotoxicity in tumors after immunotherapy treatments (138–140, 143–146). Advanced protease-responsive technologies would enable more comprehensive exploration of proteolytic networks in cancer and provide the next generation of clinical modalities for cancer chemotherapy and immunotherapy.
Conclusion
Understanding molecular mechanisms that orchestrate the complex and dynamic interplay between tumor cells and stromal microenvironment is important to provide insight into cancer biology. During malignant progression, several serine proteases appear to be key players at the tumor-stroma interface. These serine proteases function in multidirectional way by interacting not only with other proteases but also with important signaling pathways that involve activation or inactivation of cytokines, chemokines, growth factors, and kinases. The functions of serine proteases described in this review are not intended to be exhaustive but rather representative examples of recent discoveries. It is necessary to continue investigating the multifaceted roles proteases play within the tumor microenvironment in addition to their effects on the degradation of extracellular matrix to find more relevant diagnostic markers and therapeutic targets. Especially, in the context of anti-tumor immunity either naturally occurring or induced by immunotherapy, elucidating how these serine proteases on stromal cells are involved in modulating immune responses will help advance pharmaceutical strategies. Given the functional redundancy of proteases and cellular heterogeneity within the TME, advanced technologies that allow to dissect and manipulate specific serine protease with a spatiotemporal control will be highly required.
Author Contributions
RT and EY made substantial contributions to conception and design of the review, contributed to the manuscript writing and revision, read and approved the submitted version.
Funding
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, the Center for Cancer Research (ZIA BC011962).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Aumailley M, Gayraud B. Structure and Biological Activity of the Extracellular Matrix. J Mol Med (Berl) (1998) 76(3-4):253–65. doi: 10.1007/s001090050215
2. Kim SH, Turnbull J, Guimond S. Extracellular Matrix and Cell Signalling: The Dynamic Cooperation of Integrin, Proteoglycan and Growth Factor Receptor. J Endocrinol (2011) 209(2):139–51. doi: 10.1530/JOE-10-0377
3. Paget S. The Distribution of Secondary Growths in Cancer of the Breast. 1889. Cancer Metastasis Rev (1989) 8(2):98–101. doi: 10.1016/S0140-6736(00)49915-0
4. de Groot AE, Roy S, Brown JS, Pienta KJ, Amend SR. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Mol Cancer Res (2017) 15(4):361–70. doi: 10.1158/1541-7786.MCR-16-0436
5. Yang F, Strand DW, Rowley DR. Fibroblast Growth Factor-2 Mediates Transforming Growth Factor-Beta Action in Prostate Cancer Reactive Stroma. Oncogene (2008) 27(4):450–9. doi: 10.1038/sj.onc.1210663
6. Wong YC, Wang YZ. Growth Factors and Epithelial-Stromal Interactions in Prostate Cancer Development. Int Rev Cytol (2000) 199:65–116. doi: 10.1016/s0074-7696(00)99002-8
7. Ellis MJ, Singer C, Hornby A, Rasmussen A, Cullen KJ. Insulin-Like Growth Factor Mediated Stromal-Epithelial Interactions in Human Breast Cancer. Breast Cancer Res Treat (1994) 31(2-3):249–61. doi: 10.1007/BF00666158
8. Naito Y, Yamamoto Y, Sakamoto N, Shimomura I, Kogure A, Kumazaki M, et al. Cancer Extracellular Vesicles Contribute to Stromal Heterogeneity by Inducing Chemokines in Cancer-Associated Fibroblasts. Oncogene (2019) 38(28):5566–79. doi: 10.1038/s41388-019-0832-4
9. Hussain S, Peng B, Cherian M, Song JW, Ahirwar DK, Ganju RK. The Roles of Stroma-Derived Chemokine in Different Stages of Cancer Metastases. Front Immunol (2020) 11:598532. doi: 10.3389/fimmu.2020.598532
10. Singh AJ, Gray JW. Chemokine Signaling in Cancer-Stroma Communications. J Cell Commun Signal (2021) 15(3):361–81. doi: 10.1007/s12079-021-00621-7
11. Leuning DG, Beijer NRM, du Fosse NA, Vermeulen S, Lievers E, van Kooten C, et al. The Cytokine Secretion Profile of Mesenchymal Stromal Cells is Determined by Surface Structure of the Microenvironment. Sci Rep (2018) 8(1):7716. doi: 10.1038/s41598-018-25700-5
12. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-Associated Stromal Cells as Key Contributors to the Tumor Microenvironment. Breast Cancer Res (2016) 18(1):84. doi: 10.1186/s13058-016-0740-2
13. Malone MK, Smrekar K, Park S, Blakely B, Walter A, Nasta N, et al. Cytokines Secreted by Stromal Cells in TNBC Microenvironment as Potential Targets for Cancer Therapy. Cancer Biol Ther (2020) 21(6):560–69. doi: 10.1080/15384047.2020.1739484
14. Dosch AR, Singh S, Dai XZ, Mehra S, Silva ID, Bianchi A, et al. Targeting Tumor-Stromal IL6/STAT3 Signaling Through IL1 Receptor Inhibition in Pancreatic Cancer. Mol Cancer Ther (2021) 20(11):2280–90. doi: 10.1158/1535-7163.Mct-21-0083
15. Sevenich L, Joyce JA. Pericellular Proteolysis in Cancer. Genes Dev (2014) 28(21):2331–47. doi: 10.1101/gad.250647.114
16. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of Tumour- and Stroma-Supplied Proteolytic Networks Reveals a Brain-Metastasis-Promoting Role for Cathepsin S. Nat Cell Biol (2014) 16(9):876–88. doi: 10.1038/ncb3011
17. Breznik B, Motaln H, Turnsek TL. Proteases and Cytokines as Mediators of Interactions Between Cancer and Stromal Cells in Tumours. Biol Chem (2017) 398(7):709–19. doi: 10.1515/hsz-2016-0283
18. Scanlan MJ, Raj BK, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, et al. Molecular Cloning of Fibroblast Activation Protein Alpha, a Member of the Serine Protease Family Selectively Expressed in Stromal Fibroblasts of Epithelial Cancers. Proc Natl Acad Sci USA (1994) 91(12):5657–61. doi: 10.1073/pnas.91.12.5657
19. Garinchesa P, Old LJ, Rettig WJ. Cell-Surface Glycoprotein of Reactive Stromal Fibroblasts as a Potential Antibody Target in Human Epithelial Cancers. Proc Natl Acad Sci USA (1990) 87(18):7235–39. doi: 10.1073/pnas.87.18.7235
20. Rovedatti L, Di Sabatino A, Knowles CH, Sengupta N, Biancheri P, Corazza GR, et al. Fibroblast Activation Protein Expression in Crohn's Disease Strictures. Inflamm Bowel Dis (2011) 17(5):1251–3. doi: 10.1002/ibd.21446
21. Brokopp CE, Schoenauer R, Richards P, Bauer S, Lohmann C, Emmert MY, et al. Fibroblast Activation Protein is Induced by Inflammation and Degrades Type I Collagen in Thin-Cap Fibroatheromata. Eur Heart J (2011) 32(21):2713–22. doi: 10.1093/eurheartj/ehq519
22. Rettig WJ, Garin-Chesa P, Healey JH, Su SL, Ozer HL, Schwab M, et al. Regulation and Heteromeric Structure of the Fibroblast Activation Protein in Normal and Transformed Cells of Mesenchymal and Neuroectodermal Origin. Cancer Res (1993) 53(14):3327–35.
23. Mentlein R, Hattermann K, Hemion C, Jungbluth AA, Held-Feindt J. Expression and Role of the Cell Surface Protease Seprase/Fibroblast Activation Protein-Alpha (FAP-Alpha) in Astroglial Tumors. Biol Chem (2011) 392(3):199–207. doi: 10.1515/BC.2010.119
24. Edosada CY, Quan C, Tran T, Pham V, Wiesmann C, Fairbrother W. Peptide Substrate Profiling Defines Fibroblast Activation Protein as an Endopeptidase of Strict Gly(2)-Pro(1)-Cleaving Specificity. FEBS Lett (2006) 580(6):1581–86. doi: 10.1016/j.febslet.2006.01.087
25. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast Activation Protein, a Dual Specificity Serine Protease Expressed in Reactive Human Tumor Stromal Fibroblasts. J Biol Chem (1999) 274(51):36505–12. doi: 10.1074/jbc.274.51.36505
26. Dunshee DR, Bainbridge TW, Kljavin NM, Zavala-Solorio J, Schroeder AC, Chan R, et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J Biol Chem (2016) 291(11):5986–96. doi: 10.1074/jbc.M115.710582
27. Keane FM, Nadvi NA, Yao TW, Gorrell MD. Neuropeptide Y, B-Type Natriuretic Peptide, Substance P and Peptide YY are Novel Substrates of Fibroblast Activation Protein-Alpha. FEBS J (2011) 278(8):1316–32. doi: 10.1111/j.1742-4658.2011.08051.x
28. Lv BB, Xie F, Zhao PX, Ma XM, Jiang WG, Yu J, et al. Promotion of Cellular Growth and Motility is Independent of Enzymatic Activity of Fibroblast Activation Protein-Alpha. Cancer Genom Proteom (2016) 13(3):201–08.
29. Wang H, Wu Q, Liu Z, Luo X, Fan Y, Liu Y, et al. Downregulation of FAP Suppresses Cell Proliferation and Metastasis Through PTEN/PI3K/AKT and Ras-ERK Signaling in Oral Squamous Cell Carcinoma. Cell Death Dis (2014) 5(4):e1155. doi: 10.1038/cddis.2014.122
30. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting Fibroblast Activation Protein Inhibits Tumor Stromagenesis and Growth in Mice. J Clin Invest (2009) 119(12):3613–25. doi: 10.1172/JCI38988
31. Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-Overexpressing Fibroblasts Produce an Extracellular Matrix That Enhances Invasive Velocity and Directionality of Pancreatic Cancer Cells. BMC Cancer (2011) 11:245. doi: 10.1186/1471-2407-11-245
32. Huang Y, Simms Ae, Mazur A, Wang S, León NR, Jones B, et al. Fibroblast Activation Protein-α Promotes Tumor Growth and Invasion of Breast Cancer Cells Through non-Enzymatic Functions. Clin Exp Metastasis (2011) 28(6):567–79. doi: 10.1007/s10585-011-9392-x
33. Ghersi G, Zhao Q, Salamone M, Yeh Y, Zucker S, Chen W-T. The Protease Complex Consisting of Dipeptidyl Peptidase IV and Seprase Plays a Role in the Migration and Invasion of Human Endothelial Cells in Collagenous Matrices. Cancer Res (2006) 66(9):4652–61. doi: 10.1158/0008-5472.CAN-05-1245
34. Gao LM, Wang F, Zheng Y, Fu ZZ, Zheng L, Chen LL. Roles of Fibroblast Activation Protein and Hepatocyte Growth Factor Expressions in Angiogenesis and Metastasis of Gastric Cancer. Pathol Oncol Res (2019) 25(1):369–76. doi: 10.1007/s12253-017-0359-3
35. Jia J, Martin TA, Ye L, Jiang WG. FAP-α (Fibroblast Activation Protein-α) is Involved in the Control of Human Breast Cancer Cell Line Growth and Motility via the FAK Pathway. BMC Cell Biol (2014) 15(1):16. doi: 10.1186/1471-2121-15-16
36. Kawase T, Yasui Y, Nishina S, Hara Y, Yanatori I, Tomiyama Y, et al. Fibroblast Activation Protein-Alpha-Expressing Fibroblasts Promote the Progression of Pancreatic Ductal Adenocarcinoma. BMC Gastroenterol (2015) 15:109. doi: 10.1186/s12876-015-0340-0
37. Jia J, Martin TA, Ye L, Meng L, Xia N, Jiang WG, et al. Fibroblast Activation Protein-Alpha Promotes the Growth and Migration of Lung Cancer Cells via the PI3K and Sonic Hedgehog Pathways. Int J Mol Med (2018) 41(1):275–83. doi: 10.3892/ijmm.2017.3224
38. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein-Alpha. Science (2010) 330(6005):827–30. doi: 10.1126/science.1195300
39. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 From FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes With Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc Natl Acad Sci USA (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
40. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res (2016) 76(14):4124–35. doi: 10.1158/0008-5472.CAN-15-2973
41. Chen L, Qiu X, Wang X, He J. FAP Positive Fibroblasts Induce Immune Checkpoint Blockade Resistance in Colorectal Cancer via Promoting Immunosuppression. Biochem Biophys Res Commun (2017) 487(1):8–14. doi: 10.1016/j.bbrc.2017.03.039
42. Gunderson AJ, Yamazaki T, McCarty K, Phillips M, Alice A, Bambina S, et al. Blockade of Fibroblast Activation Protein in Combination With Radiation Treatment in Murine Models of Pancreatic Adenocarcinoma. PloS One (2019) 14(2):e0211117. doi: 10.1371/journal.pone.0211117
43. Kilvaer TK, Rakaee M, Hellevik T, Ostman A, Strell C, Bremnes RM, et al. Tissue Analyses Reveal a Potential Immune-Adjuvant Function of FAP-1 Positive Fibroblasts in Non-Small Cell Lung Cancer. PloS One (2018) 13(2):e0192157. doi: 10.1371/journal.pone.0192157
44. Kilvaer TK, Khanehkenari MR, Hellevik T, Al-Saad S, Paulsen EE, Bremnes RM, et al. Cancer Associated Fibroblasts in Stage I-IIIA NSCLC: Prognostic Impact and Their Correlations With Tumor Molecular Markers. PloS One (2015) 10(8):e0134965. doi: 10.1371/journal.pone.0134965
45. Liao Y, Ni Y, He R, Liu W, Du J. Clinical Implications of Fibroblast Activation Protein-Alpha in non-Small Cell Lung Cancer After Curative Resection: A New Predictor for Prognosis. J Cancer Res Clin Oncol (2013) 139(9):1523–8. doi: 10.1007/s00432-013-1471-8
46. Ariga N, Sato E, Ohuchi N, Nagura H, Ohtani H. Stromal Expression of Fibroblast Activation Protein/Seprase, a Cell Membrane Serine Proteinase and Gelatinase, is Associated With Longer Survival in Patients With Invasive Ductal Carcinoma of Breast. Int J Cancer (2001) 95(1):67–72. doi: 10.1002/1097-0215(20010120)95:1<67::Aid-Ijc1012>3.3.Co;2-L
47. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, Xu Z, et al. Fibroblast Activation Protein and its Relationship to Clinical Outcome in Pancreatic Adenocarcinoma. Pancreas (2008) 37(2):154–8. doi: 10.1097/MPA.0b013e31816618ce
48. Wikberg ML, Edin S, Lundberg IV, Van Guelpen B, Dahlin AM, Rutegard J, et al. High Intratumoral Expression of Fibroblast Activation Protein (FAP) in Colon Cancer Is Associated With Poorer Patient Prognosis. Tumor Biol (2013) 34(2):1013–20. doi: 10.1007/s13277-012-0638-2
49. Paulsson J, Micke P. Prognostic Relevance of Cancer-Associated Fibroblasts in Human Cancer. Semin Cancer Biol (2014) 25:61–8. doi: 10.1016/j.semcancer.2014.02.006
50. Macfarlane RG, Pilling J. Fibrinolytic Activity of Normal Urine. Nature (1947) 159(4049):779. doi: 10.1038/159779a0
51. Dano K, Andreasen PA, Grondahl-Hansen J, Kristensen P, Nielsen LS, Skriver L. Plasminogen Activators, Tissue Degradation, and Cancer. Adv Cancer Res (1985) 44:139–266. doi: 10.1016/s0065-230x(08)60028-7
52. Stack MS, Johnson DA. Human Mast Cell Tryptase Activates Single-Chain Urinary-Type Plasminogen Activator (Pro-Urokinase). J Biol Chem (1994) 269(13):9416–9. doi: 10.1016/S0021-9258(17)36896-5
53. Goretzki L, Schmitt M, Mann K, Calvete J, Chucholowski N, Kramer M, et al. Effective Activation of the Proenzyme Form of the Urokinase-Type Plasminogen Activator (pro-uPA) by the Cysteine Protease Cathepsin L. FEBS Lett (1992) 297(1-2):112–8. doi: 10.1016/0014-5793(92)80339-i
54. Irigoyen JP, Munoz-Canoves P, Montero L, Koziczak M, Nagamine Y. The Plasminogen Activator System: Biology and Regulation. Cell Mol Life Sci (1999) 56(1-2):104–32. doi: 10.1007/pl00000615
55. Andreasen PA, Georg B, Lund LR, Riccio A, Stacey SN. Plasminogen Activator Inhibitors: Hormonally Regulated Serpins. Mol Cell Endocrinol (1990) 68(1):1–19. doi: 10.1016/0303-7207(90)90164-4
56. Duffy MJ. The Urokinase Plasminogen Activator System: Role in Malignancy. Curr Pharm Des (2004) 10(1):39–49. doi: 10.2174/1381612043453559
57. Santibanez JF, Iglesias M, Frontelo P, Martinez J, Quintanilla M. Involvement of the Ras/MAPK Signaling Pathway in the Modulation of Urokinase Production and Cellular Invasiveness by Transforming Growth Factor-Beta(1) in Transformed Keratinocytes. Biochem Biophys Res Commun (2000) 273(2):521–7. doi: 10.1006/bbrc.2000.2946
58. Villar V, Kocic J, Santibanez JF. Spred2 Inhibits TGF-Beta1-Induced Urokinase Type Plasminogen Activator Expression, Cell Motility and Epithelial Mesenchymal Transition. Int J Cancer (2010) 127(1):77–85. doi: 10.1002/ijc.25045
59. Santibanez JF. JNK Mediates TGF-Beta1-Induced Epithelial Mesenchymal Transdifferentiation of Mouse Transformed Keratinocytes. FEBS Lett (2006) 580(22):5385–91. doi: 10.1016/j.febslet.2006.09.003
60. Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of Activation of Latent Recombinant Transforming Growth Factor Beta 1 by Plasmin. J Cell Biol (1990) 110(4):1361–7. doi: 10.1083/jcb.110.4.1361
61. Odekon LE, Blasi F, Rifkin DB. Requirement for Receptor-Bound Urokinase in Plasmin-Dependent Cellular Conversion of Latent TGF-Beta to TGF-Beta. J Cell Physiol (1994) 158(3):398–407. doi: 10.1002/jcp.1041580303
62. Santibanez JF, Krstic J. Transforming Growth Factor-Beta and Urokinase Type Plasminogen Interplay in Cancer. Curr Protein Pept Sci (2018) 19(12):1155–63. doi: 10.2174/1389203718666171030103801
63. Stepanova V, Jayaraman PS, Zaitsev SV, Lebedeva T, Bdeir K, Kershaw R, et al. Urokinase-Type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-Rich Homeodomain Protein (PRH) Transcription Factor Activity and De-Repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J Biol Chem (2016) 291(29):15029–45. doi: 10.1074/jbc.M115.678490
64. Raghu H, Nalla AK, Gondi CS, Gujrati M, Dinh DH, Rao JS. uPA and uPAR shRNA Inhibit Angiogenesis via Enhanced Secretion of SVEGFR1 Independent of GM-CSF But Dependent on TIMP-1 in Endothelial and Glioblastoma Cells. Mol Oncol (2012) 6(1):33–47. doi: 10.1016/j.molonc.2011.11.008
65. Tian B, Chen X, Zhang H, Li X, Wang J, Han W, et al. Urokinase Plasminogen Activator Secreted by Cancer-Associated Fibroblasts Induces Tumor Progression via PI3K/AKT and ERK Signaling in Esophageal Squamous Cell Carcinoma. Oncotarget (2017) 8(26):42300–13. doi: 10.18632/oncotarget.15857
66. Ciavarella S, Laurenzana A, De Summa S, Pilato B, Chilla A, Lacalamita R, et al. U-PAR Expression in Cancer Associated Fibroblast: New Acquisitions in Multiple Myeloma Progression. BMC Cancer (2017) 17(1):215. doi: 10.1186/s12885-017-3183-y
67. Besch R, Berking C, Kammerbauer C, Degitz K. Inhibition of Urokinase-Type Plasminogen Activator Receptor Induces Apoptosis in Melanoma Cells by Activation of P53. Cell Death Differ (2007) 14(4):818–29. doi: 10.1038/sj.cdd.4402065
68. Zhang J, Sud S, Mizutani K, Gyetko MR, Pienta KJ. Activation of Urokinase Plasminogen Activator and its Receptor Axis Is Essential for Macrophage Infiltration in a Prostate Cancer Mouse Model. Neoplasia (2011) 13(1):23–30. doi: 10.1593/neo.10728
69. Lin Y, Xu J, Lan H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J Hematol Oncol (2019) 12(1):76. doi: 10.1186/s13045-019-0760-3
70. Hildenbrand R, Dilger I, Horlin A, Stutte HJ. Urokinase and Macrophages in Tumour Angiogenesis. Br J Cancer (1995) 72(4):818–23. doi: 10.1038/bjc.1995.419
71. Mange A, Desmetz C, Berthes ML, Maudelonde T, Solassol J. Specific Increase of Human Kallikrein 4 mRNA and Protein Levels in Breast Cancer Stromal Cells. Biochem Biophys Res Commun (2008) 375(1):107–12. doi: 10.1016/j.bbrc.2008.07.138
72. Seiz L, Dorn J, Kotzsch M, Walch A, Grebenchtchikov NI, Gkazepis A, et al. Stromal Cell-Associated Expression of Kallikrein-Related Peptidase 6 (KLK6) Indicates Poor Prognosis of Ovarian Cancer Patients. Biol Chem (2012) 393(5):391–401. doi: 10.1515/hsz-2011-0264
73. Krenzer S, Peterziel H, Mauch C, Blaber SI, Blaber M, Angel P, et al. Expression and Function of the Kallikrein-Related Peptidase 6 in the Human Melanoma Microenvironment. J Invest Dermatol (2011) 131(11):2281–8. doi: 10.1038/jid.2011.190
74. Candido JB, Maiques O, Boxberg M, Kast V, Peerani E, Tomas-Bort E, et al. Kallikrein-Related Peptidase 6 Is Associated With the Tumour Microenvironment of Pancreatic Ductal Adenocarcinoma. Cancers (Basel) (2021) 13(16):3969. doi: 10.3390/cancers13163969
75. Alexopoulou DK, Papadopoulos IN, Scorilas A. Clinical Significance of Kallikrein-Related Peptidase (KLK10) mRNA Expression in Colorectal Cancer. Clin Biochem (2013) 46(15):1453–61. doi: 10.1016/j.clinbiochem.2013.03.002
76. Dong Y, Loessner D, Irving-Rodgers H, Obermair A, Nicklin JL, Clements JA. Metastasis of Ovarian Cancer is Mediated by Kallikrein Related Peptidases. Clin Exp Metastasis (2014) 31(1):135–47. doi: 10.1007/s10585-013-9615-4
77. Gao L, Chao L, Chao J. A Novel Signaling Pathway of Tissue Kallikrein in Promoting Keratinocyte Migration: Activation of Proteinase-Activated Receptor 1 and Epidermal Growth Factor Receptor. Exp Cell Res (2010) 316(3):376–89. doi: 10.1016/j.yexcr.2009.10.022
78. Mize GJ, Wang WB, Takayama TK. Prostate-Specific Kallikreins-2 and-4 Enhance the Proliferation of DU-145 Prostate Cancer Cells Through Protease-Activated Receptors-1 and-2. Mol Cancer Res (2008) 6(6):1043–51. doi: 10.1158/1541-7786.Mcr-08-0096
79. Gratio V, Loriot C, Virca GD, Oikonomopoulou K, Walker F, Diamandis EP, et al. Kallikrein-Related Peptidase 14 Acts on Proteinase-Activated Receptor 2 to Induce Signaling Pathway in Colon Cancer Cells. Am J Pathol (2011) 179(5):2625–36. doi: 10.1016/j.ajpath.2011.07.016
80. Kryza T, Achard C, Parent C, Marchand-Adam S, Guillon-Munos A, Iochmann S, et al. Angiogenesis Stimulated by Human Kallikrein-Related Peptidase 12 Acting via a Platelet-Derived Growth Factor B-Dependent Paracrine Pathway. FASEB J (2014) 28(2):740–51. doi: 10.1096/fj.13-237503
81. Kryza T, Parent C, Pardessus J, Petit A, Burlaud-Gaillard J, Reverdiau P, et al. Human Kallikrein-Related Peptidase 12 Stimulates Endothelial Cell Migration by Remodeling the Fibronectin Matrix. Sci Rep (2018) 8(1):6331. doi: 10.1038/s41598-018-24576-9
82. Falkowski K, Bielecka E, Thogersen IB, Bochenska O, Plaza K, Kalinska M, et al. Kallikrein-Related Peptidase 14 Activates Zymogens of Membrane Type Matrix Metalloproteinases (MT-MMPs)-A CleavEx Based Analysis. Int J Mol Sci (2020) 21(12):4383. doi: 10.3390/ijms21124383
83. Shahinian H, Loessner D, Biniossek ML, Kizhakkedathu JN, Clements JA, Magdolen V, et al. Secretome and Degradome Profiling Shows That Kallikrein-Related Peptidases 4, 5, 6, and 7 Induce Tgfβ-1 Signaling in Ovarian Cancer Cells. Mol Oncol (2014) 8(1):68–82. doi: 10.1016/j.molonc.2013.09.003
84. Wang P, Magdolen V, Seidl C, Dorn J, Drecoll E, Kotzsch M, et al. Kallikrein-Related Peptidases 4, 5, 6 and 7 Regulate Tumour-Associated Factors in Serous Ovarian Cancer. Br J Cancer (2018) 119(7):1–9. doi: 10.1038/s41416-018-0260-1
85. Kryza T, Silva LM, Bock N, Fuhrman-Luck RA, Stephens CR, Gao J, et al. Kallikrein-Related Peptidase 4 Induces Cancer-Associated Fibroblast Features in Prostate-Derived Stromal Cells. Mol Oncol (2017) 11(10):1307–29. doi: 10.1002/1878-0261.12075
86. Delaunay T, Deschamps L, Haddada M, Walker F, Soosaipillai A, Soualmia F, et al. Aberrant Expression of Kallikrein-Related Peptidase 7 Is Correlated With Human Melanoma Aggressiveness by Stimulating Cell Migration and Invasion. Mol Oncol (2017) 11(10):1330–47. doi: 10.1002/1878-0261.12103
87. Sotiropoulou G, Pampalakis G. Kallikrein-Related Peptidases: Bridges Between Immune Functions and Extracellular Matrix Degradation. Biol Chem (2010) 391(4):321–31. doi: 10.1515/Bc.2010.036
88. McGuire MJ, Lipsky PE, Thiele DL. Generation of Active Myeloid and Lymphoid Granule Serine Proteases Requires Processing by the Granule Thiol Protease Dipeptidyl Peptidase I. J Biol Chem (1993) 268(4):2458–67. doi: 10.1016/S0021-9258(18)53798-4
89. Wowk ME, Trapani JA. Cytotoxic Activity of the Lymphocyte Toxin Granzyme B. Microbes Infect (2004) 6(8):752–8. doi: 10.1016/j.micinf.2004.03.008
90. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A From Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
91. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
92. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
93. Sun B, Liu M, Cui M, Li T. Granzyme B-Expressing Treg Cells are Enriched in Colorectal Cancer and Present the Potential to Eliminate Autologous T Conventional Cells. Immunol Lett (2020) 217:7–14. doi: 10.1016/j.imlet.2019.10.007
94. Prakash MD, Bird CH, Bird PI. Active and Zymogen Forms of Granzyme B are Constitutively Released From Cytotoxic Lymphocytes in the Absence of Target Cell Engagement. Immunol Cell Biol (2009) 87(3):249–54. doi: 10.1038/icb.2008.98
95. D'Eliseo D, Pisu P, Romano C, Tubaro A, De Nunzio C, Morrone S, et al. Granzyme B is Expressed in Urothelial Carcinoma and Promotes Cancer Cell Invasion. Int J Cancer (2010) 127(6):1283–94. doi: 10.1002/ijc.25135
96. Prakash MD, Munoz MA, Jain R, Tong PL, Koskinen A, Regner M, et al. Granzyme B Promotes Cytotoxic Lymphocyte Transmigration via Basement Membrane Remodeling. Immunity (2014) 41(6):960–72. doi: 10.1016/j.immuni.2014.11.012
97. Wang H, Sun Q, Wu Y, Wang L, Zhou C, Ma W, et al. Granzyme M Expressed by Tumor Cells Promotes Chemoresistance and EMT In Vitro and Metastasis In Vivo Associated With STAT3 Activation. Oncotarget (2015) 6(8):5818–31. doi: 10.18632/oncotarget.3461
98. Santiago L, Castro M, Sanz-Pamplona R, Garzon M, Ramirez-Labrada A, Tapia E, et al. Extracellular Granzyme A Promotes Colorectal Cancer Development by Enhancing Gut Inflammation. Cell Rep (2020) 32(1):107847. doi: 10.1016/j.celrep.2020.107847
99. Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients With Metastatic Colorectal Cancer. Onkologie (2003) 26(1):44–8. doi: 10.1159/000069863
100. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clin Cancer Res (2008) 14(14):4584–92. doi: 10.1158/1078-0432.CCR-07-5211
101. Wuest T, Moosmayer D, Pfizenmaier K. Construction of a Bispecific Single Chain Antibody for Recruitment of Cytotoxic T Cells to the Tumour Stroma Associated Antigen Fibroblast Activation Protein. J Biotechnol (2001) 92(2):159–68. doi: 10.1016/s0168-1656(01)00355-8
102. Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ, et al. Phase II Trial of Single Agent Val-Boropro (Talabostat) Inhibiting Fibroblast Activation Protein in Patients With Metastatic Colorectal Cancer. Cancer Biol Ther (2007) 6(11):1691–99. doi: 10.4161/cbt.6.11.4874
103. Jackson KW, Christiansen VJ, Yadav VR, Silasi-Mansat R, Lupu F, Awasthi V, et al. Suppression of Tumor Growth in Mice by Rationally Designed Pseudopeptide Inhibitors of Fibroblast Activation Protein and Prolyl Oligopeptidase. Neoplasia (2015) 17(1):43–54. doi: 10.1016/j.neo.2014.11.002
104. Huang S, Fang R, Xu J, Qiu S, Zhang H, Du J, et al. Evaluation of the Tumor Targeting of a Fapα-Based Doxorubicin Prodrug. J Drug Target (2011) 19(7):487–96. doi: 10.3109/1061186X.2010.511225
105. Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting Carcinoma-Associated Fibroblasts Within the Tumor Stroma With a Fibroblast Activation Protein-Activated Prodrug. Jnci-J Natl Cancer I (2012) 104(17):1320–34. doi: 10.1093/jnci/djs336
106. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody Targeting in Metastatic Colon Cancer: A Phase I Study of Monoclonal Antibody F19 Against a Cell-Surface Protein of Reactive Tumor Stromal Fibroblasts. J Clin Oncol (1994) 12(6):1193–203. doi: 10.1200/JCO.1994.12.6.1193
107. Lindner T, Altmann A, Kramer S, Kleist C, Loktev A, Kratochwil C, et al. Design and Development of 99mtc-Labeled FAPI Rracers for SPECT Imaging and 188Re Therapy. J Nucl Med (2020) 61(10):1507–13. doi: 10.2967/jnumed.119.239731
108. Kratochwil C, Flechsig P, Lindner T, Abderrahim L, Altmann A, Mier W, et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J Nucl Med (2019) 60(6):801–05. doi: 10.2967/jnumed.119.227967
109. Giesel FL, Adeberg S, Syed M, Lindner T, Jiménez-Franco LD, Mavriopoulou E, et al. FAPI-74 PET/CT Using Either 18F-AlF or Cold-Kit 68Ga Labeling: Biodistribution, Radiation Dosimetry, and Tumor Delineation in Lung Cancer Patients. J Nucl Med (2021) 62(2):201. doi: 10.2967/jnumed.120.245084
110. Qin C, Shao F, Gai Y, Liu Q, Ruan W, Liu F, et al. 68Ga-DOTA-FAPI-04 PET/MR in the Evaluation of Gastric Carcinomas: Comparison With 18F-FDG PET/CT. J Nucl Med (2021) 63(1):81–8. doi: 10.2967/jnumed.120.258467
111. Baum RP, Schuchardt C, Singh A, Chantadisai M, Robiller FC, Zhang J, et al. Feasibility, Biodistribution and Preliminary Dosimetry in Peptide-Targeted Radionuclide Therapy (PTRT) of Diverse Adenocarcinomas Using 177Lu-FAP-2286: First-In-Human Results. J Nucl Med (2021) 62(11):jnumed.120.259192. doi: 10.2967/jnumed.120.259192
112. Li J, Chen K, Liu H, Cheng K, Yang M, Zhang J, et al. Activatable Near-Infrared Fluorescent Probe for In Vivo Imaging of Fibroblast Activation Protein-Alpha. Bioconjug Chem (2012) 23(8):1704–11. doi: 10.1021/bc300278r
113. Xing J, Gong Q, Zou R, Li Z, Xia Y, Yu Z, et al. A Novel Fibroblast Activation Protein-Targeted Near-Infrared Fluorescent Off-on Probe for Cancer Cell Detection, In Vitro and In Vivo Imaging. J Mater Chem B (2018) 6(10):1449–51. doi: 10.1039/c7tb03303f
114. Mazar AP, Ahn RW, O'Halloran TV. Development of Novel Therapeutics Targeting the Urokinase Plasminogen Activator Receptor (uPAR) and Their Translation Toward the Clinic. Curr Pharm Des (2011) 17(19):1970–78. doi: 10.2174/138161211796718152
115. Jankun J, Skrzypczak-Jankun E. Molecular Basis of Specific Inhibition of Urokinase Plasminogen Activator by Amiloride. Cancer Biochem Biophys (1999) 17(1-2):109–23.
116. Buckley BJ, Aboelela A, Minaei E, Jiang LGX, Xu ZH, Ali U, et al. 6-Substituted Hexamethylene Amiloride (HMA) Derivatives as Potent and Selective Inhibitors of the Human Urokinase Plasminogen Activator for Use in Cancer. J Med Chem (2018) 61(18):8299–320. doi: 10.1021/acs.jmedchem.8b00838
117. Towle MJ, Lee A, Maduakor EC, Schwartz CE, Bridges AJ, Littlefield BA. Inhibition of Urokinase by 4-Substituted Benzo[B]Thiophene-2-Carboxamidines: An Important New Class of Selective Synthetic Urokinase Inhibitor. Cancer Res (1993) 53(11):2553–9.
118. Rabbani SA, Harakidas P, Davidson DJ, Henkin J, Mazar AP. Prevention of Prostate-Cancer Metastasis In Vivo by a Novel Synthetic Inhibitor of Urokinase-Type Plasminogen Activator (uPA). Int J Cancer (1995) 63(6):840–5. doi: 10.1002/ijc.2910630615
119. Goldstein LJ. Experience in Phase I Trials and an Upcoming Phase II Study With uPA Inhibitors in Metastatic Breast Cancer. Breast Care (Basel) (2008) 3(s2):25–8. doi: 10.1159/000151733
120. Meyer JE, Brocks C, Graefe H, Mala C, Thans N, Burgle M, et al. The Oral Serine Protease Inhibitor WX-671 - First Experience in Patients With Advanced Head and Neck Carcinoma. Breast Care (Basel) (2008) 3(s2):20–4. doi: 10.1159/000151736
121. Zhu M, Gokhale VM, Szabo L, Munoz RM, Baek H, Bashyam S, et al. Identification of a Novel Inhibitor of Urokinase-Type Plasminogen Activator. Mol Cancer Ther (2007) 6(4):1348–56. doi: 10.1158/1535-7163.MCT-06-0520
122. LeBeau AM, Sevillano N, Markham K, Winter MB, Murphy ST, Hostetter DR, et al. Imaging Active Urokinase Plasminogen Activator in Prostate Cancer. Cancer Res (2015) 75(7):1225–35. doi: 10.1158/0008-5472.CAN-14-2185
123. Yang D, Severin GW, Dougherty CA, Lombardi R, Chen D, Van Dort ME, et al. Antibody-Based PET of uPA/uPAR Signaling With Broad Applicability for Cancer Imaging. Oncotarget (2016) 7(45):73912–24. doi: 10.18632/oncotarget.12528
124. Li Q, Li S, He S, Chen W, Cheng P, Zhang Y, et al. An Activatable Polymeric Reporter for Near-Infrared Fluorescent and Photoacoustic Imaging of Invasive Cancer. Angew Chem Int Ed Engl (2020) 59(18):7018–23. doi: 10.1002/anie.202000035
125. Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Sci Transl Med (2013) 5(207):207ra144. doi: 10.1126/scitranslmed.3006682
126. Wolf WC, Evans DM, Chao L, Chao J. A Synthetic Tissue Kallikrein Inhibitor Suppresses Cancer Cell Invasiveness. Am J Pathol (2001) 159(5):1797–805. doi: 10.1016/S0002-9440(10)63026-X
127. Deperthes D, Kündig C. 7 Kallikrein-Related Peptidases as Pharmaceutical Targets. In: Magdolen V, Sommerhoff CP, Fritz H, Schmitt M, editors. Volume 1 Characterization, Regulation, and Interactions Within the Protease Web: Characterization, Regulation, and Interactions Within the Protease Web. Berlin, Boston: De Gruyter (2012). p. 161–86.
128. Dong Y, Stephens C, Walpole C, Swedberg JE, Boyle GM, Parsons PG, et al. Paclitaxel Resistance and Multicellular Spheroid Formation are Induced by Kallikrein-Related Peptidase 4 in Serous Ovarian Cancer Cells in an Ascites Mimicking Microenvironment. PloS One (2013) 8(2):e57056. doi: 10.1371/journal.pone.0057056
129. Sananes A, Cohen I, Shahar A, Hockla A, De Vita E, Miller AK, et al. A Potent, Proteolysis-Resistant Inhibitor of Kallikrein-Related Peptidase 6 (KLK6) for Cancer Therapy, Developed by Combinatorial Engineering. J Biol Chem (2018) 293(33):12663–80. doi: 10.1074/jbc.RA117.000871
130. De Vita E, Schuler P, Lovell S, Lohbeck J, Kullmann S, Rabinovich E, et al. Depsipeptides Featuring a Neutral P1 are Potent Inhibitors of Kallikrein-Related Peptidase 6 With on-Target Cellular Activity. J Med Chem (2018) 61(19):8859–74. doi: 10.1021/acs.jmedchem.8b01106
131. Janssen S, Rosen DM, Ricklis RM, Dionne CA, Lilja H, Christensen SB, et al. Pharmacokinetics, Biodistribution, and Antitumor Efficacy of a Human Glandular Kallikrein 2 (Hk2)-Activated Thapsigargin Prodrug. Prostate (2006) 66(4):358–68. doi: 10.1002/pros.20348
132. DeFeo-Jones D, Garsky VM, Wong BK, Feng DM, Bolyar T, Haskell K, et al. A Peptide-Doxorubicin 'Prodrug' Activated by Prostate-Specific Antigen Selectively Kills Prostate Tumor Cells Positive for Prostate-Specific Antigen In Vivo. Nat Med (2000) 6(11):1248–52. doi: 10.1038/81351
133. DiPaola RS, Rinehart J, Nemunaitis J, Ebbinghaus S, Rubin E, Capanna T, et al. Characterization of a Novel Prostate-Specific Antigen-Activated Peptide-Doxorubicin Conjugate in Patients With Prostate Cancer. J Clin Oncol (2002) 20(7):1874–9. doi: 10.1200/JCO.2002.07.001
134. Tai W, Shukla RS, Qin B, Li B, Cheng K. Development of a Peptide-Drug Conjugate for Prostate Cancer Therapy. Mol Pharm (2011) 8(3):901–12. doi: 10.1021/mp200007b
135. Ulmert D, Evans MJ, Holland JP, Rice SL, Wongvipat J, Pettersson K, et al. Imaging Androgen Receptor Signaling With a Radiotracer Targeting Free Prostate-Specific Antigen. Cancer Discovery (2012) 2(4):320–7. doi: 10.1158/2159-8290.CD-11-0316
136. Timmermand OV, Ulmert D, Evans-Axelsson S, Pettersson K, Bjartell A, Lilja H, et al. Preclinical Imaging of Kallikrein-Related Peptidase 2 (Hk2) in Prostate Cancer With a In-111-Radiolabelled Monoclonal Antibody, 11B6. Ejnmmi Res (2014) 4(1):51. doi: 10.1186/s13550-014-0051-5
137. Lovell S, Zhang L, Kryza T, Neodo A, Bock N, De Vita E, et al. A Suite of Activity-Based Probes to Dissect the KLK Activome in Drug-Resistant Prostate Cancer. J Am Chem Soc (2021) 143(23):8911–24. doi: 10.1021/jacs.1c03950
138. Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K, et al. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res (2017) 77(9):2318–27. doi: 10.1158/0008-5472.Can-16-3346
139. Goggi JL, Hartimath SV, Xuan TY, Khanapur S, Jieu B, Chin HX, et al. Granzyme B PET Imaging of Combined Chemotherapy and Immune Checkpoint Inhibitor Therapy in Colon Cancer. Mol Imaging Biol (2021) 23(5):714–23. doi: 10.1007/s11307-021-01596-y
140. Zhao N, Bardine C, Lourenco AL, Wang YH, Huang Y, Cleary SJ, et al. In Vivo Measurement of Granzyme Proteolysis From Activated Immune Cells With PET. ACS Cent Sci (2021) 7(10):1638–49. doi: 10.1021/acscentsci.1c00529
141. Kolt S, Janiszewski T, Kaiserman D, Modrzycka S, Snipas SJ, Salvesen G, et al. Detection of Active Granzyme A in NK92 Cells With Fluorescent Activity-Based Probe. J Med Chem (2020) 63(6):3359–69. doi: 10.1021/acs.jmedchem.9b02042
142. Janiszewski T, Kolt S, Kaiserman D, Snipas SJ, Li S, Kulbacka J, et al. Noninvasive Optical Detection of Granzyme B From Natural Killer Cells With Enzyme-Activated Fluorogenic Probes. J Biol Chem (2020) 295(28):9567–82. doi: 10.1074/jbc.RA120.013204
143. Scott JI, Gutkin S, Green O, Thompson EJ, Kitamura T, Shabat D, et al. A Functional Chemiluminescent Probe for In Vivo Imaging of Natural Killer Cell Activity Against Tumours. Angew Chem Int Ed Engl (2021) 60(11):5699–703. doi: 10.1002/anie.202011429
144. Nguyen A, Ramesh A, Kumar S, Nandi D, Brouillard A, Wells A, et al. Granzyme B Nanoreporter for Early Monitoring of Tumor Response to Immunotherapy. Sci Adv (2020) 6(40):eabc2777. doi: 10.1126/sciadv.abc2777
145. He S, Li J, Lyu Y, Huang J, Pu K. Near-Infrared Fluorescent Macromolecular Reporters for Real-Time Imaging and Urinalysis of Cancer Immunotherapy. J Am Chem Soc (2020) 142(15):7075–82. doi: 10.1021/jacs.0c00659
146. Zhang Y, He S, Chen W, Liu Y, Zhang X, Miao Q, et al. Activatable Polymeric Nanoprobe for Near-Infrared Fluorescence and Photoacoustic Imaging of T Lymphocytes. Angew Chem Int Ed Engl (2021) 60(11):5921–27. doi: 10.1002/anie.202015116
147. Weissleder R, Tung CH, Mahmood U, Bogdanov A Jr. In Vivo Imaging of Tumors With Protease-Activated Near-Infrared Fluorescent Probes. Nat Biotechnol (1999) 17(4):375–8. doi: 10.1038/7933
148. Hu HY, Gehrig S, Reither G, Subramanian D, Mall MA, Plettenburg O, et al. FRET-Based and Other Fluorescent Proteinase Probes. Biotechnol J (2014) 9(2):266–81. doi: 10.1002/biot.201300201
149. Kahler JP, Vanhoutte R, Verhelst SHL. Activity-Based Protein Profiling of Serine Proteases in Immune Cells. Arch Immunol Ther Exp (Warsz) (2020) 68(4):23. doi: 10.1007/s00005-020-00586-2
150. Sanman LE, Bogyo M. Activity-Based Profiling of Proteases. Annu Rev Biochem (2014) 83:249–73. doi: 10.1146/annurev-biochem-060713-035352
151. Pan Z, Jeffery DA, Chehade K, Beltman J, Clark JM, Grothaus P, et al. Development of Activity-Based Probes for Trypsin-Family Serine Proteases. Bioorg Med Chem Lett (2006) 16(11):2882–5. doi: 10.1016/j.bmcl.2006.03.012
152. Kasperkiewicz P, Altman Y, D'Angelo M, Salvesen GS, Drag M. Toolbox of Fluorescent Probes for Parallel Imaging Reveals Uneven Location of Serine Proteases in Neutrophils. J Am Chem Soc (2017) 139(29):10115–25. doi: 10.1021/jacs.7b04394
153. Serim S, Haedke U, Verhelst SH. Activity-Based Probes for the Study of Proteases: Recent Advances and Developments. ChemMedChem (2012) 7(7):1146–59. doi: 10.1002/cmdc.201200057
154. Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K, et al. A Nonpeptidic Cathepsin S Activity-Based Probe for Noninvasive Optical Imaging of Tumor-Associated Macrophages. Chem Biol (2012) 19(5):619–28. doi: 10.1016/j.chembiol.2012.03.012
155. Brak K, Doyle PS, McKerrow JH, Ellman JA. Identification of a New Class of Nonpeptidic Inhibitors of Cruzain. J Am Chem Soc (2008) 130(20):6404–10. doi: 10.1021/ja710254m
156. Kooij R, Liu S, Sapmaz A, Xin BT, Janssen GMC, van Veelen PA, et al. Small-Molecule Activity-Based Probe for Monitoring Ubiquitin C-Terminal Hydrolase L1 (UCHL1) Activity in Live Cells and Zebrafish Embryos. J Am Chem Soc (2020) 142(39):16825–41. doi: 10.1021/jacs.0c07726
157. Fearon DT. The Carcinoma-Associated Fibroblast Expressing Fibroblast Activation Protein and Escape From Immune Surveillance. Cancer Immunol Res (2014) 2(3):187–93. doi: 10.1158/2326-6066.CIR-14-0002
158. Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
159. Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting Fibroblast Activation Protein in Tumor Stroma With Chimeric Antigen Receptor T Cells can Inhibit Tumor Growth and Augment Host Immunity Without Severe Toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.Cir-13-0027
160. Pai CS, Simons DM, Lu X, Evans M, Wei J, Wang YH, et al. Tumor-Conditional Anti-CTLA4 Uncouples Antitumor Efficacy From Immunotherapy-Related Toxicity. J Clin Invest (2019) 129(1):349–63. doi: 10.1172/JCI123391
161. Puskas J, Skrombolas D, Sedlacek A, Lord E, Sullivan M, Frelinger J. Development of an Attenuated Interleukin-2 Fusion Protein That Can Be Activated by Tumour-Expressed Proteases. Immunology (2011) 133(2):206–20. doi: 10.1111/j.1365-2567.2011.03428.x
Keywords: tumor-stromal interaction, serine protease, fibroblast activation protein, urokinase plasminogen activator, kallikrein, granzyme, extracellular matrix remodeling, signaling pathways
Citation: Tagirasa R and Yoo E (2022) Role of Serine Proteases at the Tumor-Stroma Interface. Front. Immunol. 13:832418. doi: 10.3389/fimmu.2022.832418
Received: 09 December 2021; Accepted: 24 January 2022;
Published: 11 February 2022.
Edited by:
Jaewoo Hong, Catholic University of Daegu, South KoreaReviewed by:
Juan Francisco Santibanez, University of Belgrade, SerbiaJun-O Jin, Yeungnam University, South Korea
Copyright © 2022 Tagirasa and Yoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Euna Yoo, ZXVuYS55b29AbmloLmdvdg==