 Alain E. Andrea1†
Alain E. Andrea1† Andrada Chiron2,3†Sarah Mallah4Stéphanie Bessoles2Guillaume Sarrabayrouse2
Andrada Chiron2,3†Sarah Mallah4Stéphanie Bessoles2Guillaume Sarrabayrouse2 Salima Hacein-Bey-Abina2,3*
Salima Hacein-Bey-Abina2,3*- 1Laboratoire de Biochimie et Thérapies Moléculaires, Faculté de Pharmacie, Université Saint Joseph de Beyrouth, Beirut, Lebanon
- 2Université de Paris, CNRS, INSERM, UTCBS, Unité des technologies Chimiques et Biologiques pour la Santé, Paris, France
- 3Clinical Immunology Laboratory, Groupe Hospitalier Universitaire Paris-Sud, Hôpital Kremlin-Bicêtre, Assistance Publique-Hôpitaux de Paris, Le-Kremlin-Bicêtre, France
- 4Faculty of Arts and Sciences, Lebanese American University, Beirut, Lebanon
During this last decade, adoptive transfer of T lymphocytes genetically modified to express chimeric antigen receptors (CARs) emerged as a valuable therapeutic strategy in hematological cancers. However, this immunotherapy has demonstrated limited efficacy in solid tumors. The main obstacle encountered by CAR-T cells in solid malignancies is the immunosuppressive tumor microenvironment (TME). The TME impedes tumor trafficking and penetration of T lymphocytes and installs an immunosuppressive milieu by producing suppressive soluble factors and by overexpressing negative immune checkpoints. In order to overcome these hurdles, new CAR-T cells engineering strategies were designed, to potentiate tumor recognition and infiltration and anti-cancer activity in the hostile TME. In this review, we provide an overview of the major mechanisms used by tumor cells to evade immune defenses and we critically expose the most optimistic engineering strategies to make CAR-T cell therapy a solid option for solid tumors.
1 Introduction
Chimeric antigen receptor (CAR)-T cells are genetically engineered T lymphocytes with an extracellular antibody-like domain (consisting of a single chain variable fragment or scFv), a transmembrane domain and an intracellular signaling domain. Four main generations of CAR-T cells have been designed to date. The main driver of genetically engineered enhancements across all these generations is the improvement of the anticancer potential of this innovative immunotherapy. First-generation CAR-T cells are engineered with a single activating intracellular domain, CD3ζ, (known as signal 1), without any additional costimulatory domains. As these CAR-T cells cannot produce enough interleukin (IL)-2 –vital for proliferation and growth- exogenous administration of IL-2 (IL-2 immunotherapy) is necessary to enhance CAR-T cells persistence in vivo and, thus, anticancer activity. Second- and third-generation CAR-T cells are genetically engineered with one or more intracellular costimulatory domains (known as signal 2), which increases CAR-T cell efficacy and persistence (1). In the case of fourth-generation CAR-T cells, also known as T cell redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs), an additional cassette coding for a transgenic protein (such as a cytokine) is expressed. This protein is released by the genetically modified lymphocytes and modulates their anti-cancer response (2).
Adoptive transfer of CAR-T cells has shown immense success in treating B cell malignancies. In the contrary, the response rates of CAR-T cell immunotherapy among solid cancer patients are less favorable. Major obstacles in solid tumor immunotherapy with CAR-T cells are, first, difficulties in tumor targeting and second an insufficient trafficking and fitness of genetically modified T cells, especially in the hostile tumor microenvironment (TME). Because of the lack of tumor-specific antigens (TSA) or the heterogeneous expression of tumor associated antigens (TAA) with overlapping expression between healthy tissues and tumor cells, one of the roadblocks to effective CAR-T immunotherapy is specific tumor targeting. Hurdles in solid tumor targeting make it a challenge to develop safe immunotherapies devoid of on-target/off-tumor toxicities. Moreover, TAAs can be lost in case of tumor antigen escape (as the case of proliferating tumor subclones), with CAR-T cell immunotherapy becoming ineffective. Other drawbacks, some inherent to CAR-T cells, are represented by limited tumor trafficking and tumor infiltration, as well as an insufficient expansion and persistence of genetically modified T cells in the homeostatic cytokine-deprived TME. All these challenges have been addressed by various preclinical models recently and efforts to improve engineering are still ongoing. In this review, we expose the major obstacles that CAR-T cells face in solid tumors, especially the decrease of T lymphocytes infiltration to the tumor site, the immunosuppressive milieu and the inhibition of CAR-T cell activity by the negative immune checkpoints, and we propose, by reviewing the literature, an extensive list of solutions to each of the mentioned obstacles.
2 Challenges and Engineering Strategies to Overcome CAR-T Cells’ Limitations in Solid Tumors
2.1 Enhancing CAR-T Cells Tumor Trafficking and Penetration
Solid tumors are organ-like, disorganized structures composed of proliferating tumor cells surrounded by supporting stromal cells and by nourishing blood vessels of the tumor neovasculature and associated to a cellular immune infiltrate composed of both innate and adaptive immune cells. Tumor growth can be controlled by both the innate and adaptive components of the immune system. Therefore, the infiltrating cell populations in solid tumors are comprised of both innate immune cells: neutrophils, macrophages, dendritic cells (DCs), mast cells, natural killer cells (NK cells), and myeloid derived suppressor cells (MDSCs) and of adaptive immune cells: T and B lymphocytes, and regulatory T cells (Tregs). All these immune cells are associated with non-tumor-stromal cells composing the TME: endothelial cells, fibroblasts, pericytes, and mesenchymal cells. All these cells, as well as their secreted factors and molecules compose the TME, an immunosuppressive, hostile milieu for tumor-infiltrating T cells (TILs) and a physical barrier for T cell migration and tumor infiltration.
Among all aforementioned cells, the key player of the anti-tumor response are TILs, their capacity to infiltrate the tumor bed being related to tumor outgrowth and extension (3–5). It is well acknowledged that TILs are a trademark of ongoing tumor immunosurveillance as they have shown both therapeutic and prognostic significance in animals and in humans. Indeed, higher density of TILs in patients’ TME correlates with improved clinical outcomes (6), whereas fail to respond to immunotherapy is associated with a low post-treatment infiltration of T cells (7).
Therefore, the key for successful therapeutic strategies is the switch from a poorly infiltrated “cold” TME or from an “immune-excluded” TME [i.e limited presence of T cells at the periphery of tumor nests without intra-tumoral infiltration (8)] to a “hot” TME, with a rich, active, immune cell infiltrate in the tumor core, especially including functional TILs (9).
Despite initial expectations in solid tumor treatment with CAR-T cell therapies, one major roadblock in treating solid tumors turned out to be the limited access of cellular therapies to the tumor bed, as T cells must face additional barriers before inducing their antitumor activity (10). Indeed, great response to systemically infused CAR-T cells in hematological cancers is due, at least in part, to the easy access of CAR-T cells to malignant cells residing in hematologic organs readily accessible to the blood flow (bone marrow, lymph nodes, spleen) (10).
2.1.1 T cell Trafficking and Homing to Tumor Sites
T cell trafficking to both lymphoid organs and peripheral tissues is tightly regulated by chemotactic cues and controlled by chemokine/chemokine receptors axis and adhesion molecules interactions. T cell migration from the bloodstream and homing into peripheral tissues is a regulated, three-step process starting with 1) an initial transitory attachment and selectin-mediated rolling on the endothelium, followed by 2) chemokine-receptor mediated activation of integrins and finally by 3) integrin-dependent transmigration and extravasation (11, 12). Homing and retention of naïve T cells to lymph nodes is regulated by the expression of CD62L and of the CC chemokine receptor 7 (CCR7, which binds lymph-nodes chemokines CCL19 and CCL21), accompanied by the activation of LFA-1 (13). After T cell priming by antigen presentation, central memory T cells (TCM) lose the expression of both CCR7 and CD62L to acquire an effector memory (CD45RO+) phenotype (TEM), thereby losing their ability to access lymph nodes through the high endothelial venules (HEV).
Therefore, TEMs recirculate in the bloodstream to migrate to peripheral tissues and their migration back to the lymphoid organs is inhibited. Instead, activated T cells gain expression of a cohort of homing molecules that enable them to migrate to diseased/inflamed tissues (14). The T cell effector population presenting with homing capacity to tumor sites expresses homing molecules including ligands for E-selectin (CD62E) and P-selectin (CD62P) expressed on activated endothelial cells as well as chemokine receptors for inflammatory chemokines, such as CXCR3 which binds inflammatory chemokines CXCL9 and CXCL10 (14) and CCR5 which binds respectively to CCL3/CCL3L1/CCL4/CCL5/CCL8/CCL11/CCL13/CCL16 ligands produced by tumor tissues (15, 16). Moreover, the activation of chemokine receptors enables adhesion to the endothelium by inducing the expression of two integrins: β2-integrin leukocyte function-associated antigen-1 (LFA-1), and very late antigen-4 (VLA-4, also known as α4β1), which bind respectively to ICAM-1 and VCAM-1 receptors expressed on the endothelium (17). Upon activation, integrins express binding sites that interact with cell adhesion molecules on the blood vessel walls, leading to T cell firm adhesion and transmigration into the tumor site (18) (Figure 1).
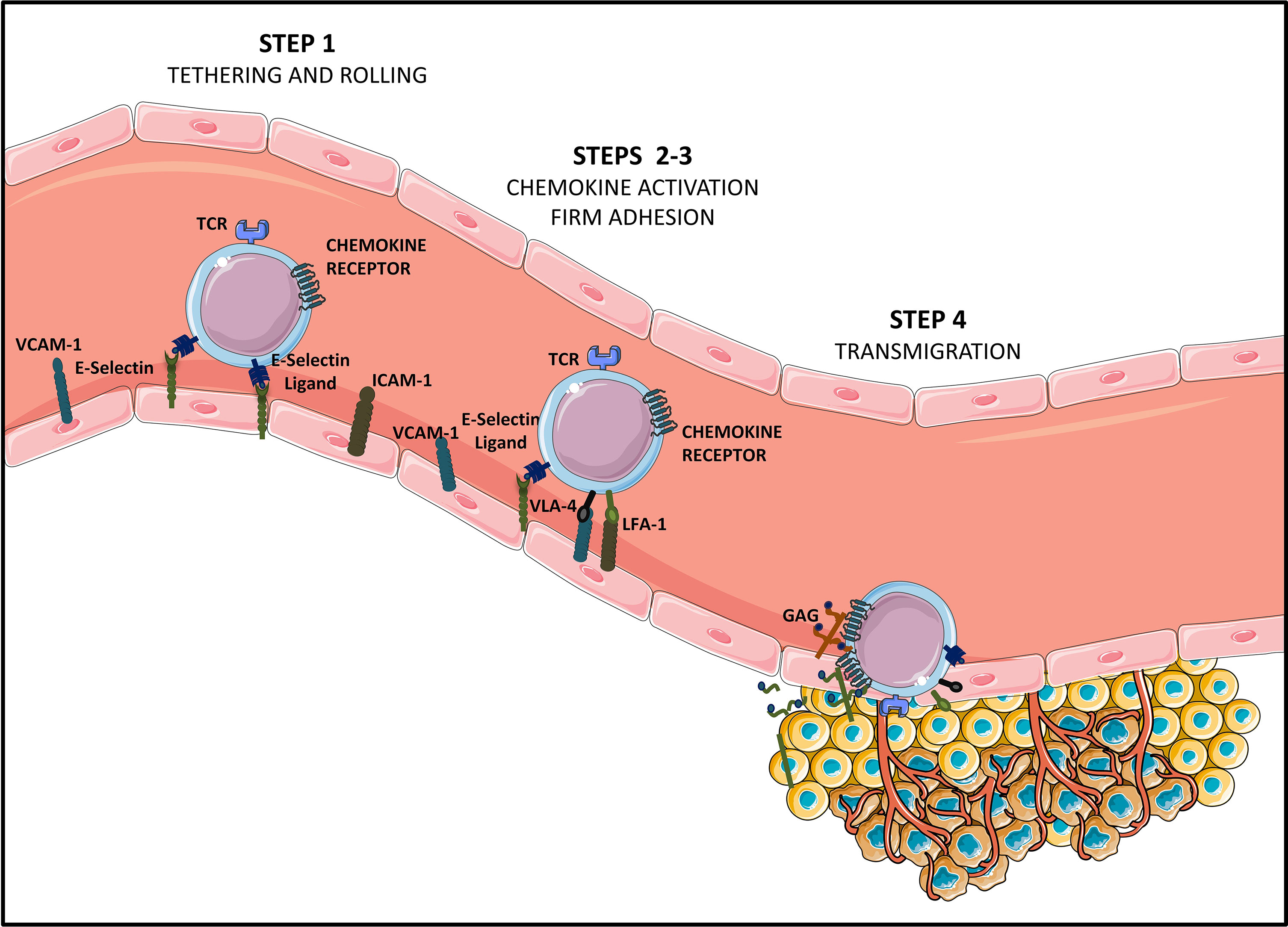
Figure 1 Steps of T cell homing to tumor tissues [Adapted from Sackstein et al. (11)]. Tumor infiltrating CD8+ effector T lymphocytes (Teffs) presenting a specific tumor antigen circulate in the blood stream. They express homing molecules allowing for their oriented migration towards the tumor (like CXCR3 and CCR5-chemokine receptors), as well as ligands allowing binding to endothelial cells (E-selectin ligands and VLA-4 and LFA-1 integrins at suboptimal levels). Circulating Teffs tether and roll on the endothelium (STEP 1) via engagement of E-Selectin ligands with endothelial E-Selectin, which slows down Teffs, and allows firm adhesion to the endothelium (STEP 2). In this second step of Teffs entry into tumoral tissues, chemokines produced by cancer cells or by stromal cells from the TME (CXCL9, CXCL10, CCL5…) bind chemokine receptors. This binding of chemokine receptors to their ligands elicits activation of VLA-4 and LFA-1, allowing for VLA-4/VCAM-1 and LFA-1/ICAM-1 firm adhesion (STEP 3). Firmly adherent Teffs undergo transendothelial migration (STEP 4), to infiltrate the TME and establish cell-to-cell contact with tumor cells, via TCR-based recognition of cancer antigens presented on HLA molecules.
Peripheral tissues are the homing site for specialized memory T cell subsets identified and characterized extensively in the context of infectious diseases, called tissue resident memory T cells or TRM, and whose presence in solid cancer is associated with better outcomes. TRMs are localized in non-lymphoid peripheral tissues, do not recirculate and have a unique surface phenotype characterized by the lack of expression of receptors/transcription factors enabling egress from the tissues and lymph node homing (CCR7, CD62L, S1pr1 and Klf2). TRMs express the activation marker CD69, and the integrins CD103 (αEβ7) and CD49a (α1β1), which bind to E-cadherin and type IV collagen on epithelial and endothelial cells, respectively. They also upregulate the LFA1 integrin (αL(CD11a) β2), which binds to the ICAM-1 adhesion molecule on endothelial cells (19). Moreover, CD8+ TILs with a TRM phenotype expressing the adenosine producing ectonucleotidase CD39 and the CD103 integrin are a unique, specific tumor-reactive population found exclusively in the TME, both in primary and metastatic tumors, and whose frequencies are associated with overall survival (OS) in some cancer patients (20). Furthermore, it has recently been shown that a high density of CD8+CD103+CD49a+CD69+ TRM TILs correlates with an improved response to anti-programmed death 1 (PD-1) immune checkpoint blockade (19). Immune checkpoint inhibitors (i.e. ICI) represent a new Nobel-Prize worth immunotherapy with immense success in some incurable cancers, which target s inhibitory costimulatory molecules on the surface of T cells (like PD-1 or CTLA-4).
Barriers limiting access of CAR-T cells to the tumor bed are both physical (represented by surrounding blood vessels and the tumor stroma) and functional (represented by immunosuppressive molecules and soluble factors in the TME). Briefly, infused CAR-T cells need, in order to exert their cytotoxic effect, to: 1) traffic through the blood stream and migrate to the tumor tissue, in a chemokine directed manner, 2) cross the limiting blood vessels during the transmigration step 3) infiltrate the tumor and migrate to the vicinity of tumor cells by degrading TME components and 4) generate stable cell to cell contacts with tumor cells. Finally, success of adoptive cell therapy (ACT) is warranted by an increased persistence of infused CAR-T cells, which is dictated by their capacity to proliferate and survive in the hostile (acidic, hypoxic and nutrient and cytokine derived) tumor environment. Moreover, tumors have developed “escape mechanisms” in order to divert the immune-patrol process (21).
Therefore, CAR-T cell trafficking to and infiltration of the tumor is the first roadblock that needs to be overcome. Defective CAR-T cell infiltration is caused by: (i) chemokine/chemokine receptors mismatch or downregulated tumor-derived chemokines (22), (ii) an aberrant vasculature with downregulated or deficient adhesion molecules (23) and (iii) a remodeled tumor stroma, mainly composed of extracellular matrix (ECM) and cancer associated fibroblasts (CAFs) (24, 25).
2.1.2 Overcoming the Mismatch or the Dysregulation of Chemokine Receptor/Ligand Axes
Recent studies have shown that endothelial cells lining the tumor vasculature are able to prevent the trafficking, the adhesion and to eventually hijack anti-tumor activity of T cells (26). Some tumors block T cell homing by reducing the expression of adhesion molecules such as ICAM-1, VCAM-1, and CD34 on the tumor endothelium (14). For instance, the overexpression of endothelin B receptors (ETBR) on the tumor vasculature in ovarian cancer represses T cell trafficking by preventing ICAM-1 clustering on endothelial cells, which has a central role in T cell arrest and migration (27). Furthermore, as CXCR3 and CCR5 are often used by activated T cell to infiltrate tumors that should express their respective ligands (28), an insufficient expression of CXCR3 and CCR5 ligands by some tumors leads to a decrease in T cell recruitment (29, 30).
Since efficient trafficking is the first critical step for CAR-T cells to mediate their anti-tumor activity, several strategies targeting chemokine‐chemokine receptor signaling are currently being explored in solid tumors. Some of them have already been tested in preclinical and clinical studies. To this end, CAR-T cells were genetically modified to co-express either chemokine receptors, among which we can cite: CCR2b, CXCR1/CXCR2, CCR4, CX3CL1, CSF-1R and CCR8 or to produce various chemokines: CCL19, CCL21 or CXCL11 (Figure 2). In more recent studies, co-expression of tissue homing molecules, as CD103 or CD39 was used to direct CAR-T cells to the tumor sites more efficiently
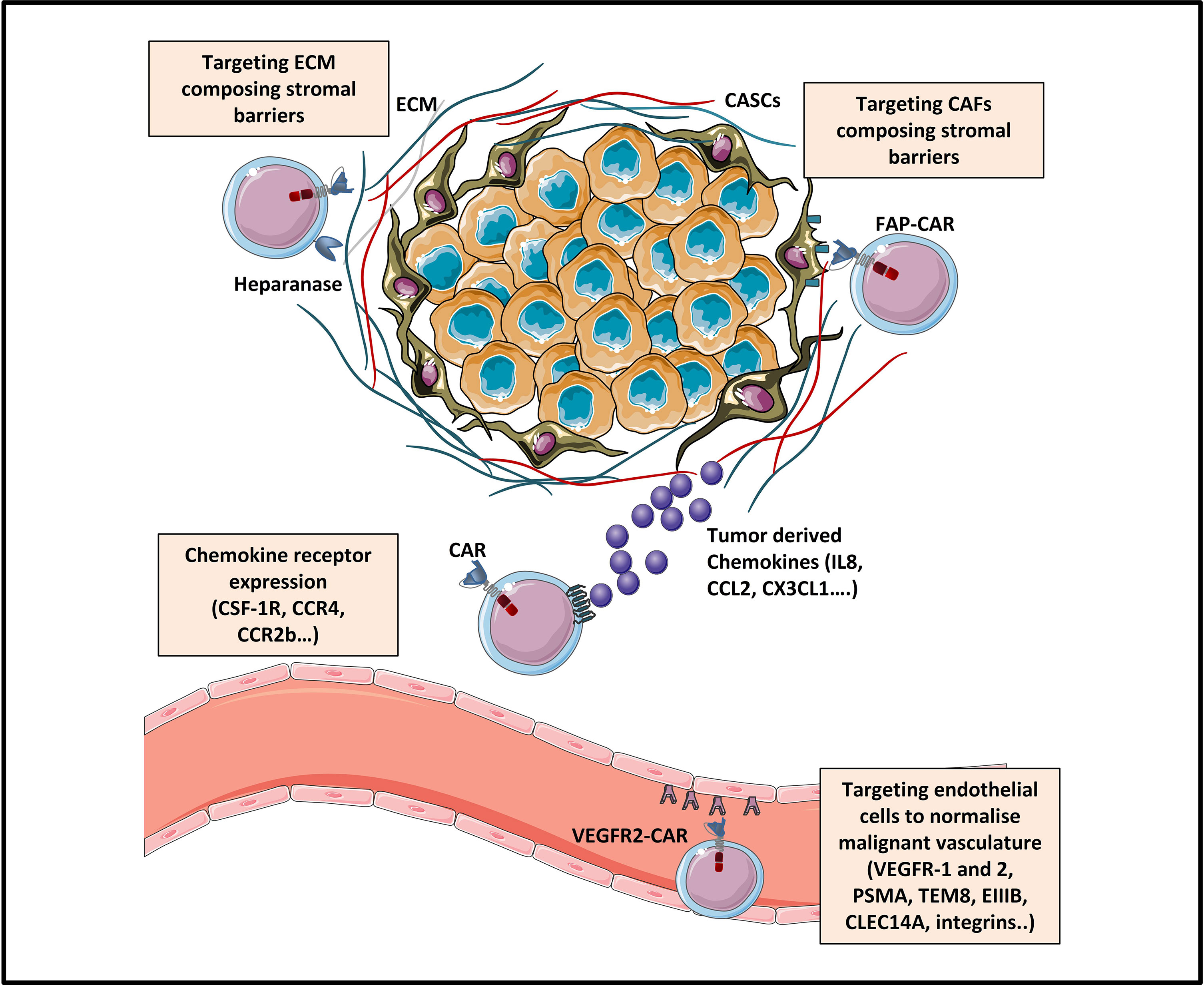
Figure 2 Strategies enhancing tumor trafficking and penetration [Adapted from Rafiq et al. (31)]. The trafficking of CAR-T cells towards tumor sites can be enhanced by engineering CAR-T cells expressing chemokine receptors (as for example CSF-1R, CCR4 or CCR2b) specific for tumor-derived chemokine ligands (IL8, CCL2, CXCL1…). Tumor penetration of CAR-T cells can be enhanced by various strategies: (1) normalizing the malignant vasculature by targeting tumor blood vessels via CAR targeting of endothelial/tumoral antigens (like VEGFR, EIIIB, TEM8, integrins.), and (2) targeting physical barriers in the tumor microenvironment (TME) like the extracellular matrix (ECM) or the cancer associated fibroblasts (CAFs).
The chemokine ligand CCL2 or monocyte chemoattractant protein 1 (MCP-1) mediates the trafficking of immune cells into the TME in many types of malignancies, such as melanoma, colorectal, breast, prostate and pancreatic cancer (32). Therefore, co-expression of the CCL2 chemokine receptor, CCR2b, in CAR-T cells improves their anti-tumor activity, by enhancing their ability to traffic to the tumor bed. Craddock et al. demonstrated an improved homing (>10-fold) of GD2-specific CAR-T cells co-expressing CCR2b to CCL2-secreting neuroblastoma, as compared to CCR2-negative CAR-T cells (33). Likewise, co-expression of CCR2b was also associated with an increased migration (12.5-fold) of mesothelin (MSLN)-targeted CAR-T cells toward malignant pleural mesothelioma, in a study conducted by Moon et al. (34).
Furthermore, IL-8/CXCL8 was shown to be a pro-inflammatory chemokine that plays an important role in a variety of human cancers, including melanoma (35), prostate (36), colon (37), breast (38) and ovarian (39) cancers, by mediating tumorigenesis and angiogenesis. Some researchers took advantage of tumor-produced IL-8 in order to guide the IL-8 receptor (CXCR1 or CXCR2)-expressing CAR-T cells to infiltrate solid tumors (glioblastoma, hepatocellular carcinoma (HCC), ovarian and pancreatic cancer), and stimulate an antitumor immune response. Results showed a significantly enhanced tumor trafficking and persistence of genetically modified T cells, which triggered tumor regression, durable immunologic memory and better toxicity profile in mice (40–42). A clinical trial (NCT01740557) was initiated to evaluate the efficacy of T cells transduced with CXCR2 and with nerve growth factor receptor (NGFR), associated with Recombinant Human IL-2 (Aldesleukin) infusion in melanoma (Table 10). Exogenous supplementation of the Il2 vital support cytokine is widely used in the clinical setting (See Targeting Fibroblast Activation Protein (FAP) and Tregs), and is resumed in the table dedicated to CAR-T cells clinical trials (Table 10).
Moreover, it has been noted that two CCR4 ligands -CCL17 and CCL22-, are overexpressed in lymphoid malignancies such as Hodgkin’s lymphoma (HL) (43), and in many other types of human cancers (44) including ovarian (45), breast (46), esophageal (47) and gastric (48) cancers. The aberrant overexpression of those ligands at the tumor site plays a central role in recruiting CCR4+ Th2 and regulatory T cells (Tregs) to such malignancies, resulting in an immunosuppressive TME (43). Since CCR4- effector T cells are barely present at the tumor site, the forced co-expression of surface CCR4 in CAR-T cells appears to be a promising therapeutic strategy in the treatment of certain types of lymphomas. Taking advantage of a mouse model of HL, Di Stasi et al. demonstrated that CAR-T cells engineered to co-express the chemokine receptor CCR4 together with the effector antigen receptor CD30 (CAR-CD30 T cells), had improved migration towards the tumor and enhanced anti-lymphoma activity as compared to CD30 CAR-T cells lacking CCR4 expression (49). A clinical trial (NCT03602157) was initiated to ascertain the effectiveness of CAR-T cells co-expressing CD30 and CCR4 in relapsed/refractory CD30+ HL and cutaneous T cell lymphoma (CTCL) (Table 10).
More recently, mesothelin specific CAR-T cells (MSLN-CAR) transduced to express either CCR2b or CCR4 chemokine receptors of Mcp-1 were engineered by Wang et al. and tested in vitro and in vivo in a non-small-cell lung carcinoma (NSCLC) model. MSLN-CCR2b-CAR-T cells displayed superior anti-tumor function due to enhanced migration and infiltration into tumor tissues as well as no obvious toxicity (no organ damage). The MSLN-CCR4-CAR-T cells showed enhanced migration and potent cytotoxic function and cytokine production in vitro but were not further tested in vivo (50).
As previously mentioned, CXCR3 is highly expressed on effector T cells and plays a key role in their trafficking (51). Therefore, tumors expressing chemokines such as interferon-γ (IFN-γ)-inducible CXCR3 ligands would attract effector lymphocytes. CXCR3 binds three ligands: CXCL9 (monokine induced by IFN-γ), CXCL10 (interferon-induced protein-10) and CXCL11 (interferon-inducible T cell alpha chemoattractant) (52). Moon et al. used CAR-T cells as vehicles to deliver CXCL11 to the cancer site in order to increase its expression within the tumor and therefore recruit effector TILs. Unfortunately, this approach was not able to improve T cell tumor infiltration, despite of the local increase in CXCL11 (53). Given the success of oncolytic vaccinia viruses (VVs) expressing CXCL11 in increasing the numbers of effector T lymphocytes in specific murine tumors (54, 55), the same team combined the use of a VV engineered to produce CXCL11 with MSLN CAR-T cells administration. Results showed increased efficacy in CAR-T cells trafficking and tumor progression control of this combined strategy, as compared to VV.CXCL11 alone (53).
On another front, data showed that the unique member of the CX3-chemokine subfamily, termed fractalkine or CX3CL1, can be exploited to help overcome the poor homing of CAR-T cells to tumor sites. The CX3CL1-CX3CR1 axis is involved in chemotaxis and adhesion of leukocytes and in the recruitment of immune cell subpopulation such as NK cells, Th1 lymphocytes and macrophages (56). CX3CL1 is expressed in breast (57), pancreatic (58), gastric (59) and colon (60, 61) cancers. Siddiqui et al. demonstrated that CAR-T cells engineered to express CX3CR1 have increased infiltration towards CX3CL1-producing tumors in mice as well as decreased tumor growth (62).
In a proof-of-concept in vitro model, Lo et al. induced forced expression of the macrophage colony-stimulating factor 1 receptor (CSF-1R) to render CAR-T cells sensitive to CSF1, a monocyte recruiting chemokine enriched in various tumor tissues. Forced expression of CSF-1R exploits the T cell signaling machinery to enhance CAR-T cells Il-2 driven proliferation and costimulate production of IFN-γ, without reducing cytotoxicity and without inducing transdifferentiation to the monocytic/macrophagic lineage. CSF-1 forced expression is a cytokine engineering strategy which could improve both CAR-T cell effector function (i.e. persistence/proliferation and cytokine production) and CAR-T chemotaxis to the tumor site (63).
More recently, Cadilha et al. employed a combined CAR-T cells engineering strategy enabling enhanced recruitment by CCR8 expression together with shielding from immunosuppression by the expression of a dominant-negative TGF-β receptor 2 (TGF-β DNR). The team exploited the CCR8-CCL1 recruitment axis, by which various tumors with poor prognosis attract Tregs, to empower effector CAR-T cells with enhanced chemotaxis. The team validated this strategy in a murine model of pancreatic cancer and in human xenograft tumor models. Furthermore, this strategy exploits activated T cell derived CCL1 to potentialize a positive feedback loop in CCR8+ cells recruitment to the tumor site (64).
Two other teams designed fourth generation CAR-T cells producing/co-expressing both IL-7 and CCL19 or CCL21 (65, 66). These combinatorial strategies associating co-expression of chemokine receptors/ligands with production of homeostatic cytokines could enhance both migration of CAR-T cells to the tumor site and proliferation/persistence of CAR-T cells in the hostile TME. Adachi et al. engineered CAR-T cells specific to fluorescein isothiocyanate (FITC) co-expressing IL-7 and CCL19 (7 × 19 CAR-T cells), two factors produced by T-zone fibroblastic reticular cells and essential for the maintenance of T cell zones in lymphoid organs. Treated mice achieved complete remission of pre-established tumors and 7 × 19 CAR-T cells showed superior anti-tumor activity than conventional CAR-T cells, as well as an improved ability of both migration and proliferation in the TME. Response to 7 × 19 CAR-T cells was dependent on the recipient’s immune system (i.e activation and recruitment of dendritic cells and of tumor-reactive recipient T cells). Moreover, recipient conventional T cells also generated tumor –antigen-specific memory, probably due to epitope spreading. The authors raised security concerns about this engineering strategy, as gain of function (GOF) mutations of the IL-7 receptor (CD127) are frequent in pediatric T cell acute lymphoblastic leukemia (T-ALL) and as CCR7 could play a role in tumor metastasis. This engineering strategy could, therefore, benefit from the integration of a suicide gene system in order to prevent an eventual leukemic change of 7 × 19 CAR-T cells before clinical application (65). The same team validated the use of anti-mesothelin IL-7/CCL19-producing human CAR-T cells in a preclinical model of orthotopic pre-established malignant mesothelioma, as well as in patient derived xenograft (PTX) models of mesothelin-positive pancreatic cancer. As in the previous study, IL-7/CCL19-producing human CAR-T cells exerted a significant inhibition of tumor growth and prolonged survival of treated mice. Tumors showed increased infiltration with T recipient no-CAR-T cells as well as downregulation of exhaustion markers PD-1 and T cell immunoreceptor with Ig and ITIM domains (TIGIT) on T cells (67). Similar results were obtained with 7 × 19 CAR-T cells in vivo in the context of hepatocellular carcinoma (HCC) and pancreatic carcinoma (68).
There are two clinical trials on CAR-T cells co-expressing IL-7 and CCL19. Results from a first six-case cohort preliminary phase I clinical study (NCT03198546) in advanced HCC/PC/ovarian carcinoma (OC) patients with glypican-3 (GPC3) or MSLN expression have been published recently and show encouraging results: two complete responses (CR), two partial responses (PR) and 2 steady diseases (SD). There were no grade 2–4 adverse events or major complications (68). Another ongoing clinical trial (NCT03932565) evaluates intratumoral injection of Nectin4/FAP-targeted fourth-generation CAR-T cells (expressing IL-7 and CCL19, or IL12) for the treatment of Nectin4-positive advanced malignant solid tumors (NSCLC, breast, ovarian, bladder or pancreatic cancer). This represents an engineering strategy designed to enhance migration (CCL19), proliferation/maintenance (IL-7/IL-12) of CAR-T cells and to simultaneously target the stromal CAFs (anti-FAP). Three other clinical trials are evaluating the efficacy of this type of immunotherapy in the context of B cell lymphoma/multiple myeloma: NCT04833504 evaluating CD19-CAR-T expressing IL-7 and CCL19 in the context of relapsed/refractory B cell lymphoma, NCT04381741 evaluating CD19 CAR-T expressing IL-7 and CCL19 combined with PD-1 mAb for relapsed or refractory diffuse large B cell Lymphoma (DLBCL) and NCT03778346 evaluating fourth generation CAR-T cells simultaneously expressing IL-7 and CCL19 and directed against single or compound targets (Integrin β7, BCMA, CS1, CD38 and/or CD138) in the context of refractory/recurrent multiple myeloma (R/R MM). Results for the first two patients treated with BCMA-7 × 19 CAR-T cells (NCT03778346) in the context of R/R MM show encouraging results: an objective response within 1 month after BCMA-7 × 19 CAR-T cell infusion with one patient reaching CR and one a very good partial response (VGPR) and responses lasted more than 12-months (Table 10). There was no clinically significant toxicity. It is worth noticing that this CAR-T cell therapy was associated with a high proportion of stem cell memory (TSCM) among produced CAR-T cells, possibly due to IL-7 production (69). Indeed, several clinical studies have shown that the modifications to induce differentiation toward a TCM/TSCM profile improve CAR-T cell responses in subjects (70–72).
Another similar approach was to engineer Claudin18.2 (CLDN18.2)-specific CAR-T cells to co-express IL-7 together with the chemokine receptor CCR7 ligand CCL21 (7 × 21 CAR-T cells). CLDN18.2-specific second-generation CAR-T cells coexpressing IL-7 and CCL21 were tested in vitro and in vivo in three tumor models (breast, pancreatic and hepatocellular carcinoma) and revealed superior therapeutic effects to either conventional CAR-T cells or 7 × 19 CAR-T cells, without preconditioned lymphodepletion. As for 7 × 19 CAR-T cells, 7 × 21 CAR-T cells showed significantly improved survival and tumor infiltration. Treated mice showed increased infiltration of DCs as well as an inhibition of the tumor angiogenesis (presumed effect of CCL21) (66). No clinical trials evaluating the efficacy of CCL21 expressing CAR-T cells have been designed to date. However, various clinical trials use CCL21 gene modified dendritic cells (DCs-adenovirus CCL21) as anticancer vaccination strategies in lung cancer or melanoma (NCT00601094, NCT01433172, NCT01574222, NCT03546361 and NCT00798629).
Genetically engineered expression of TRM-type markers CD103 or CD39 on CAR-T cells has recently been evaluated as a strategy to overcome insufficient trafficking and infiltration of solid tumors (HCC) or hematologic cancers (human Raji lymphoma). In a HCC model, hepatitis B virus (HBV) surface protein-specific CAR-T cells (HBVsCAR-T cells) were genetically manipulated to express CD39 and showed increased cytotoxicity in an in vitro model of HCC organoids and T lymphocytes coculture and in a PDX mouse model. To prevent an exhausted phenotype of CD39+ CAR-T cells, the team used a combinatorial strategy of CD39 expression on CAR-T cells, together with knockdown of inhibitory immune-checkpoints (triple knockdown of PD-1, T cell immunoglobulin domain and mucin domain-3 (TIM-3), and lymphocyte-activation gene 3 (LAG-3) with shRNAs). CD39+ CAR-T cells showed enhanced cytokine production and antitumor effect. According to the authors, CD39 can serve as a biomarker to identify both personalized tumor-reactive CD8+ T cells as well as active CAR-T cells. Besides phenotypic identification, CD39 expression is also necessary for the cytotoxic effect of CD8 CARs and positively regulates antitumor activity (73). The TRM marker CD103 is a tissue homing molecule important for effector T cell trafficking as well as a promising prognosis biomarker for assessment of tumor-reactive TILS in various types of cancer, such as lung cancer, ovarian cancer and cervical cancers. CD103 is an integrin protein (αE) that binds integrin β7 to form the heterodimeric integrin complex αEβ7. Sun et al. used an E-Cadherin positive human lymphoma preclinical model (human Raji leukemia/lymphoma cells injected in NSG mice) to test therapeutic effects of CD103 expression on CD19-specific human CAR T cells. The gene encoding for the αE integrin was incorporated in the CD19-specific CAR structure to generate CD103-CD19-BBz-CAR T cells. These CAR-T cells showed more immature phenotypes (expressing high levels of CD62L and CD45RA), as compared to conventional CD19-BBz-CAR T cells, an increased production of IL-2 and greater expansion in culture, as well as improved anti-tumor efficacy (increased persistence, infiltration and eradication of lymphoma distant metastasis) upon adoptive transfer in immunodeficient mice (74).
The aforementioned preclinical studies on chemokine receptors expressing CAR-T cells are summarized in the table below (Table 1).
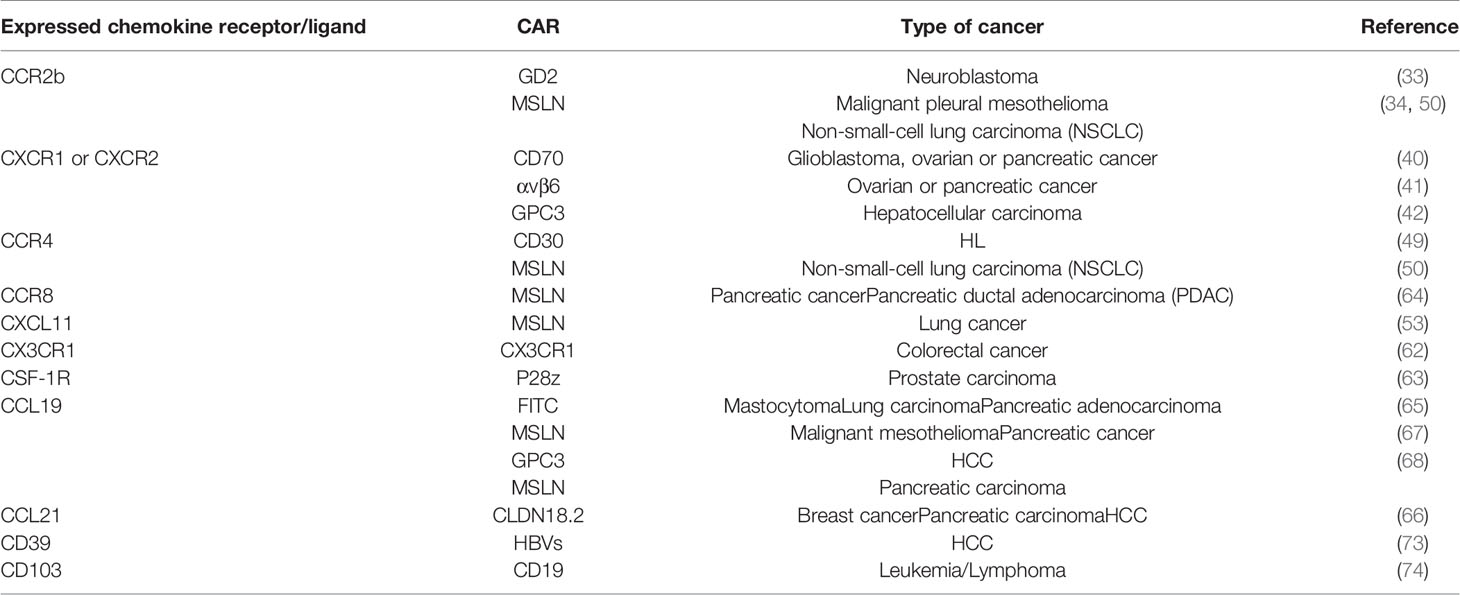
Table 1 Summary of preclinical studies on chemokine receptors/ligands or homing molecules expressing CAR-T cells.
2.1.3 Handling Neovasculature Aberrancies
Tumor angiogenesis is a hallmark of cancer growth and progression (75). The generation of a tumor-associated neovasculature enables the growing tumor mass to obtain nutriments and oxygen. Moreover, the tumor uses these new vessels as a principal route to enter the circulation and to metastasize and proliferate to distant areas (76). Tumor neovasculature is a disorganized labyrinth of vessels at risk of vascular collapse. It lacks a hierarchical vessel division, which gives rise to abnormal blood flow and permeability, diffusion-limited nutrient delivery, oxygen deprivation, and an increased interstitial fluid pressure in the tumor (77). Tumor-induced angiogenesis is induced by the imbalanced production of proangiogenic factors by the tumor cells, including vascular endothelial growth factor-A (VEGF), platelet-derived endothelial growth factor (PDGF), transforming growth factor (TGF)-α, angiopoietin (Ang), basic fibroblast growth factor (bFGF), fibroblast growth factor (FGF), and placental growth factor (PGF) (78, 79). These soluble factors bind to and activate diverse tyrosine kinase (TK) receptors, such as VEGFR1, VEGFR2, PDGFRA, and endothelial growth factor receptor (EGFR), promoting angiogenesis, among other biological events (80).
As stated previously, in order to reach the tumor site, T cells encounter a physical barrier, represented by this abnormal vasculature, which operates though as a first obstacle for lymphocyte recruitment into the tumor. Therefore, vascular targeting, using anti-angiogenic molecules, has been proposed as a novel strategy to block tumor growth. This approach aims at correcting the structural and functional abnormalities of the tumor vasculature, in order to improve T cell infiltration and immunotherapy efficacy (81). The first Food and Drug Administration (FDA) approval of an anti-angiogenic monoclonal antibody (mAb) (Bevacizumab) dates back to more than a decade ago (82). In more recent studies, substantial efforts were deployed to develop CAR-T cells with a chimeric receptor comprising a scFv antibody against specific angiogenic growth factors/receptors or adhesion molecules abnormally expressed on the tumor vasculature (83). To this end, Kershaw et al. were the first to suggest an indirect strategy to target stromal tumors by the usage of CAR-T cells targeting the vascular stroma instead of the cancer cell itself (84).
In order to inhibit tumor angiogenesis, Chinnasamy et al. genetically modified murine and human T cells to express a CAR targeted against VEGFR-2 (85). VEGFR-2 is overexpressed in tumor vasculature and is known to be critical for both physiological and pathological/tumor angiogenesis, as well as for VEGF-mediated tumor progression (86). VEGFR-2 is overexpressed in many types of solid tumors, including breast cancer, cervical cancer, NSCLC, hepatocellular carcinoma, and renal carcinoma (87). Chinnasamy et al. demonstrated that the antitumor effect of VEGFR-2 targeting CAR- T cells was not mediated through their direct cytotoxicity on the tumor cells but rather through their ability to eliminate VEGFR-2-expressing cells in the tumor vasculature. A single dose of VEGFR-2 CAR-T cells was effective in increasing tumor infiltration, and inhibiting the growth of 5 vascularized syngeneic tumors of various histological origins (85). The same group showed, in another study, that the coadministration of anti-VEGFR-2 CAR-T cells along with tumor-specific TCR transduced T cells (premelanosome (Pmel) TCR, tyrosinase-related-protein-1 (TRP-1) TCR, and tyrosinase-related-protein-2 (TRP2) TCR traduced T cells) resulted in a synergic anti-tumor effect and an extended tumor-free survival (TFS) of mice with metastatic melanoma tumors. These results emphasize the advantageous effects of dual targeting adoptive therapy including an anti-angiogenic strategy (88). Recently, Englisch et al. suggested that VEGFR-2 expressed on tumor vasculature could be a potential CAR target in Ewing sarcoma (EwS) (89), especially that this type of cancer is characterized by a limited TSA expression on cancer cells (90). Contact with their target triggered a powerful antigen‐specific degranulation response, increased proliferation and cytokine secretion of VEGFR-2 CAR-T cells. Data showed that VEGFR-2 CAR-T cells with short‐length or medium‐length hinge domains effectively destroyed VEGFR-2-expressing tumor‐associated endothelial cells (89). Similarly, in a study from Taheri et al. nanobody‐based anti-VEGFR2 CAR showed effective activation, degranulation and lysis of VEGFR2+ cell lines in an in vitro model (91). Unfortunately, adoptive transfer of VEGFR-2 CAR-T cells in a clinical setting was devoid of great success in a phase 1 clinical trial NCT01218867 on patients with metastatic cancer. The trial was terminated due to lack of objective responses: out of 24 infused patients, only one reached a PR and another one had a stable disease (SD) after CAR-T cell injection. There were no CR (Table 10).
While VEGFR-2 plays a critical role both in physiological and pathological angiogenesis, VEGFR-1, another member of the VEGFR family, is strictly involved in pathological angiogenesis (92). Even though both are abnormally expressed at high levels on tumor vasculature, their signaling characteristics are different (93). However, VEGFR-1 is not restricted to endothelial cells as expression has also been proven on monocyte/macrophages, and on various types of tumor cells (92). VEGFR-1 has been shown to be a key regulator of macrophage’ function and of cancer metastasis, among others, which makes it an interesting target in the development of novel approaches for cancer ACT (94). Wang et al. demonstrated that VEGFR-1 CAR-T cells can be a promising solution to break the resistance to traditional anti-angiogenic therapies, with higher efficacy than strategies blocking separately cancer growth or angiogenesis. This study also showed that co-administration of IL-5 producing CAR-T cells enhanced the anti-metastasis activity mediated by VEGFR-1 CAR-T cells (95).
Prostate-specific membrane antigen (PSMA) is another transmembrane protein highly expressed on the tumor-associated endothelium of a great variety of solid tumors - including bladder, oral, hepatocellular, gastric, colorectal, breast, ovarian, renal, and pancreatic ductal carcinoma as well as NSCLC and melanoma - (96, 97). Although not expressed by the normal endothelium, like it is the case of VEGFR, PSMA is still expressed at low levels in normal tissues as the brain, liver, kidney, intestine, colon and the prostate (98). Moreover, PSMA has a crucial role in tumor neovascularization. Santoro et al. directed a proof-of-concept study showing that PSMA CAR-T cells can recognize primary tumor PSMA-expressing endothelial cells and disrupt the tumor vasculature both in vitro and in vivo. Contrary to traditional anti-angiogenic agents, anti-PSMA CAR-T cells showed long-term in vivo persistence. However, in order to improve the safety profile of PSMA CAR-T cells, toxicity control mechanisms like the use of split-signaling CAR-T cells should be needed (97). PSMA has especially been targeted in prostate cancer patients, with various ongoing clinical trials (NCT01140373 (99, 100), NCT01929239, NCT00664196; and NCT03089203) (see Tregs and Table 10) (101).
Tumor endothelial marker 8 (TEM8), also known as anthrax receptor 1 (ANTRX1), is another cell membrane glycoprotein consistently overexpressed in the tumor vasculature and in many types of cancer, including breast (102), gastric (103),, skin (104), colon (105), and lung (106) cancers. Blocking or knocking out TEM8 inhibited pathological angiogenesis in several preclinical cancer models (104, 107). Moreover, anti-TEM8 CAR-T cells can serve as a potential targeted therapy for triple-negative breast cancers (TNBC). Results from Byrd et al. showed that TEM8-targeted CAR-T cells were able to concomitantly destroy TNBC tumor cells, breast cancer stem-like cells (BCSC) as well as tumor endothelial cells, and to cause regression of lung metastatic TNBC cell line-derived xenograft tumors (108). Unfortunately, a study published by Petrovic et al. raised concerns over possible on-target/off-tumor toxicities of TEM8-specific CAR-T cells (109).
It has been shown that fibronectin (FN) splice variants EIIIA and EIIIB are overexpressed in the vasculature of many types of tumors, including breast, lung and prostate cancers and high-grade glioma, whereas absent in normal tissues (110–112). These properties make EIIIA and EIIIB ideal targets for CAR-T cell therapy. Genetically engineered CAR-T cells targeting EIIIB were able to inhibit the growth of solid cancers in immunocompetent mice by compromising the blood supply of the tumor (113). Based on three tumor models, Wagner et al. reported similar results using immunodeficient mice treated with anti-EIIIB CAR-T cells (114).
Recently, C-type lectin domain family 14 member A (CLEC14A) has been identified as part of a molecular gene signature for tumor angiogenesis based on a meta-analysis on breast cancer, head and neck squamous cell carcinoma (HNSCC), and clear-cell renal cell carcinoma (ccRCC) (115). This protein is mainly overexpressed in the three aforementioned cancers (116, 117). CLEC14A could be a promising target for antiangiogenic therapy. A single injection of CLEC14A-specific CAR-T cells was sufficient for a significant suppression of tumor growth in 3 distinct tumor models. Use of anti-CLEC14A CAR-T cells could be combined with CAR-T cells targeting another tumor endothelial marker, in order to increase tumor vessel targeting capacities (118).
The integrin αvβ3 emerges as another potential target for cancer immunotherapy. Integrin αvβ3 is expressed on different types of cancer, including glioblastoma (119), melanoma (120), pancreatic (121), breast (122) and prostate cancers (123). Even though expressed on activated endothelial cells and newly formed vessels, it is not detectable in resting endothelial cells and normal tissues, making it a valid target for the treatment of many solid tumors (124). Wallstabe et al. generated αvβ3 targeted CAR-T cells and investigated antitumor effects of such approach in preclinical models in vitro and in vivo. They concluded that this strategy was able to inhibit tumor growth, but without achieving tumor eradication. Presence of haematomas in the tumor tissues proved that engineered T cells damaged tumor vessels, due to αvβ3-expression on tumor endothelium. Results also showed that adoptive therapy with αvβ3 CAR-T cells was more effective than immunotherapy with anti-αvβ3 mAbs (125).
Another integrin, the integrin αvβ6, whose expression on endothelial cells is restricted to development and remodeling processes (like wound healing, chronic inflammation and cancer), is upregulated in various cancers (126) and associated with more invasive tumor phenotypes, characterized by high tumor invasion and shorten survival in colon and cervix cancers or in NSCLC (127). This integrin emerged as an interesting target for immunotherapy with CAR-T cells in cholangiocarcinoma (CCA), a lethal bile duct cancer with poor responses to classic therapy. Targeting of CCA with anti- αvβ6 fourth generation CAR-T cells showed anti-tumor function against αvβ6 expressing CCA tumor spheroids, in vitro (128). In a previous study from Whilding et al., anti-αvβ6 CAR-T cells showed in vivo efficacy in other solid tumors expressing intermediate to high levels of this integrin (ovarian, breast, and pancreatic tumor xenografts in SCID beige mice). For selective expansion, CAR-T cells were engineered to co-express the IL-4-responsive fusion gene (4αβ, obtained by fusing the human IL-4 receptor α ectodomain to the shared human IL-2/IL-15 receptor β transmembrane and endodomain regions). Moreover, despite expression of this integrin in non-tumor endothelium, toxicities related to anti-αvβ6 CAR-T infusion were mild and reversible and only associated to systemic infusion of supra-therapeutic doses (129). There is no ongoing clinical trial with anti-αvβ6 CAR-T cells in cancer patients, but anti-αvβ6 cancer targeting, either by monoclonal antibodies or by peptides has already been tested in in vitro or preclinical animal models of breast (130) and pancreatic cancers (131, 132).
The aforementioned preclinical studies on proangiogenic factors/receptors-targeting CAR-T cells are summarized in the table below (Table 2).

Table 2 Summary of preclinical studies on proangiogenic factors/receptors-targeting CAR-T cells.
2.1.4 Targeting the Tumor Stroma
Besides strategies aiming at targeting tumor blood vessels, engineering modifications targeting stromal cells may also be promising strategies for CAR-based immunotherapy. Targeting non-cancer cell components of the tumor stroma could help to enhance the anti-cancer effect of this immunotherapy for many reasons. First, stromal cells are less prone to immune-escape from the CAR-T cells attack as they show higher genetic stability than tumor cells, and are less likely to lose antigen expression via immunoediting (133). Second, since tumor stroma can be found in almost all human adenocarcinomas, CAR-T cells targeting the extracellular matrix (ECM) and/or the nonmalignant cancer-associated stromal cells (CASCs) could generate “broad-spectrum” CAR-T cells (134). Finally, tumor stroma plays a major role in tumor survival, growth, invasion, and angiogenesis, by producing growth factors, and chemotactic factors that attract immunosuppressive cells, and by expressing inhibitory surface checkpoint proteins (135). However, as extracellular matrix components are vital components of connective tissues, this targeting strategy needs identification and usage of specific tumor-ECM targets, in order to avoid on-target/off-tumor toxicities. To this regard, some studies focused on targeting ECM components by using CAR-T cells expressing ECM degrading enzymes while others chose as an attractive stromal candidate the fibroblast activation protein (FAP) expressed in CASCs (Figure 2).
a. ECM components modifying enzymes
Another strategy aiming at facilitating cellular penetration into solid tumors is genetic manipulation of CAR-T cells to secrete ECM-modifying enzymes. Indeed, the ECM is a complex structural component of the TME and the main physical barrier that hinders T cell-cancer cell contacts. The ECM is synthesized by malignant cells and cancer associated fibroblasts (CAFs) and can constitute up to 60/% of the tumor mass (136). Different ECM molecules, such as fibrillar collagen, hyaluronan (HA), proteoglycans (chondroitin sulfate, dermatan sulfate, heparan sulfate, and keratan sulfate), elastin, fibronectin and laminins are highly expressed in many solid cancers (136). Therefore, in order to access the tumor sites and mediate their anti-tumor functions, T cells must be able to degrade the main components of the ECM. Lymphocytes secrete specific enzymes to disrupt the ECM, including: (i) heparinase (HPSE), an endoglucuronidase that cleaves heparan sulfate side chains of heparan sulfate proteoglycans (137), (ii) hyaluronidase, an endoglycosidase that cleaves glycosidic bonds of hyaluronic acid (138), and (iii) matrix metalloproteinases (MMPs), endopeptidase proteases that cleave the majority of ECM and non-ECM components (Figure 2). Caruana et al. noted that in vitro-engineered and cultured T cells lose heparinase expression following TP53 binding to the HPSE gene promoter, which may restrict CAR-T cell infiltration in stroma-rich solid tumors. To this regard, the authors engineered CAR-T cells to express heparinase and demonstrated that it’s expression led to improved cell migration in neuroblastoma xenograft models (139). Xiong et al. studied, in vitro and in vivo, the ability of GPC3 (Glypican 3 protein)-targeted CAR-T cells co-expressing IL-7 and the PH20 hyaluronidase to infiltrate hepatocellular carcinoma (HCC) xenograft models. Their results showed that the co-expression of the two aforementioned genes improved CAR-T cells trafficking, which may significantly enhance their efficacy in solid tumors (140).
Similarly, Zhao et al. reported the construction of MSLN (mesothelin)-targeted CAR-T cells with the overexpression of a secreted form of the human hyaluronidase (sPH20-IgG2) and found that this enzyme can promote the antitumor activity of these CAR-T cells in vitro and in vivo in gastric cancer cell xenografts, by promoting their infiltration (141). Use of a pegylated form of the human recombinant hyaluronidase (PEGPH20) has already been tested in the clinical setting in two randomized trials, as an adjuvant to chemotherapy in metastatic pancreatic adenocarcinoma (142, 143). Results of one of the trials (NCT01959139) could claim certain caution as adjuvant PEGPH20 therapy resulted in a diminished OS as well as an increased toxicity (gastrointestinal and thromboembolic events) (142). The other clinical trial (NCT01839487) did not confirm the reduction in survival (143) (Table 10). Even so, both studies confirmed an increased thromboembolic risk of the PEGPH20 therapy and imposed the adjunction of an anticoagulant prophylaxis with low molecular weight heparin in the study from Hingorani et al. (143). In a more recent phase III trial adjunction of PEGPH20 to chemotherapy had no benefits in terms of OS or progression free survival (PFS) in the case of metastatic pancreatic carcinoma (144).
Not least, another strategy to enhance CAR-T cells migration through the collagen barriers of the ECM could be CAR-T cell production of another ECM-modifying enzyme, the MMP8 metalloproteinases (also known as collagenase-2), as suggested by Mardomi and Abediankenari (145). However, transgenic production of MMPs has not been applied yet to CAR-T cells engineering. This type of engineering strategy, can also seam tempting for genetically engineered Macrophages (CAR-Macrophages) (146).
The aforementioned preclinical studies on CAR-T cells expressing ECM degrading enzymes are summarized in the table below (Table 3).
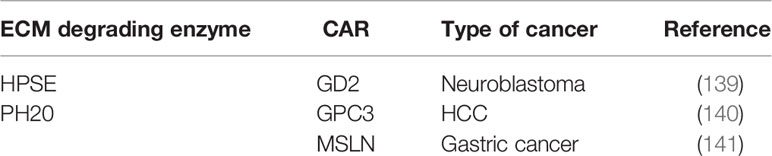
Table 3 Summary of preclinical studies on CAR-T expressing ECM degrading enzymes.
b. Targeting Fibroblast Activation Protein (FAP)
Growing evidence proves that many cell types within the TME play a key role in oncogenesis. Among them, CAFs, a major component of the tumor stroma, represent a reactive tumor-associated fibroblast population that secretes various active factors promoting tumor development, progression, metastasis, and therapeutic resistance (147) (Figure 3). CAFs express various molecules that can be targeted by immunotherapies. Among them, FAP has recently emerged as the most promising target (149). FAP is a cell surface serine protease that is highly expressed on the CASCs of various human cancer types (150), such as lung (151), prostate (152), pancreatic (153), colorectal (154), and ovarian cancer (155). In contrast, the expression of this proteolytic enzyme on normal quiescent adult stromal cells and benign tumors is reported to be low to undetectable. Moreover, several studies have shown that tumors expressing FAP are associated with poor prognosis (150), enhanced tumorigenesis (150) and an increased neo-angiogenesis (156). Therefore, different strategies have been used to target FAP using antibodies (157, 158), vaccines (159, 160), immunoconjugates (161, 162), peptide-drug complexes (163–166), FAP gene knock-down by siRNA delivery (167), and CAR-T cells (168).
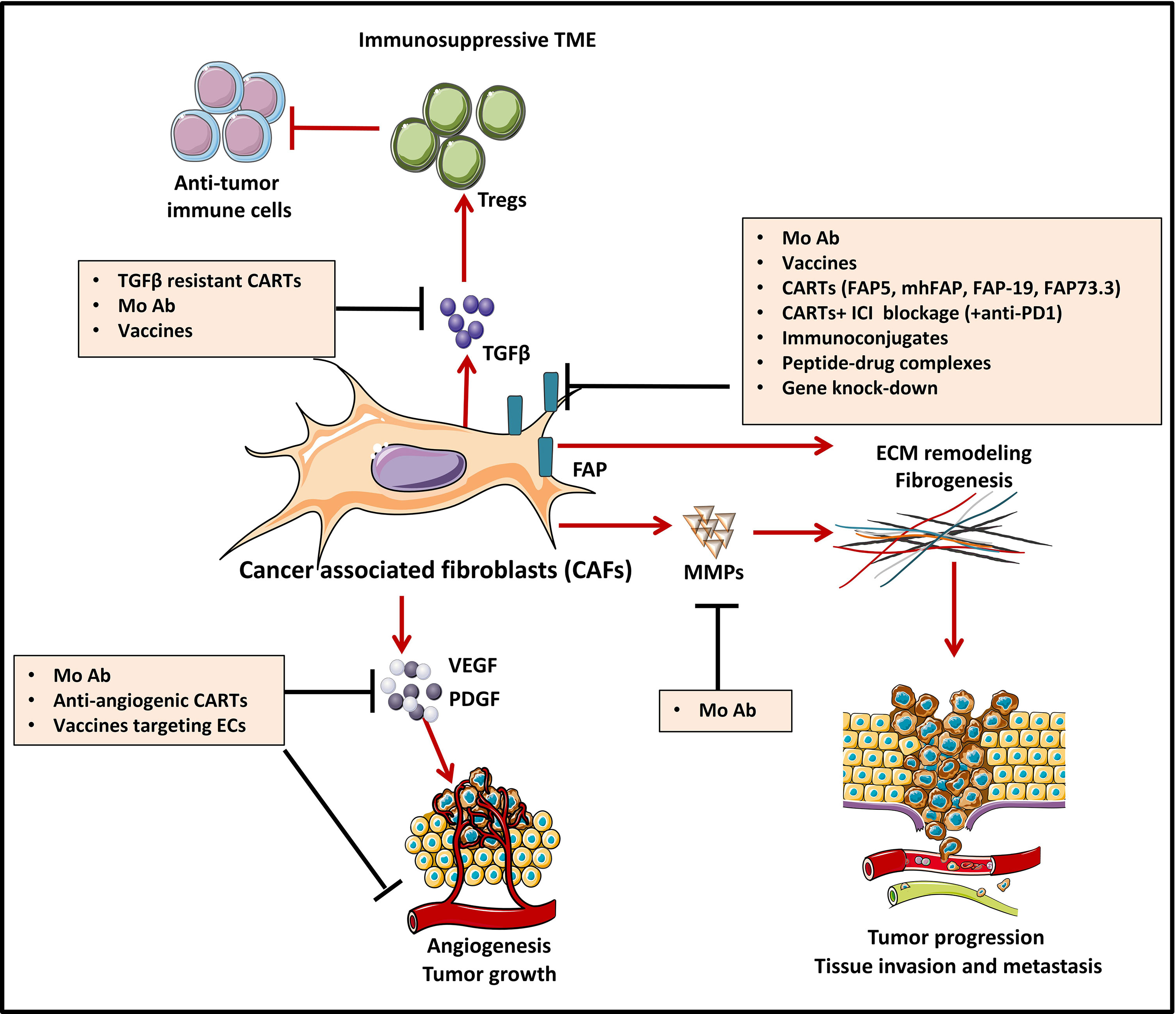
Figure 3 Strategies to counteract protumorigenic effects of CAFs [Adapted from Kakarla et al. (148)]. Cancer associated fibroblasts (CAF)-directed anti-cancer therapies are one of the weapons of tumor targeting which is directed against the stromal compartment. Strategies depicted in this figure aim at inhibiting cancer associated fibroblasts (CAFs) functions and are based on targeting crucial signals and effectors of CAFs such as cytokines (TGFβ) and growth factor pathways (VEGF, PDGF…). For instance, CAF-derived extracellular matrix proteins (MMPs) and associated signaling can be targeted with monoclonal antibodies (MAb), to induce stromal depletion and increase immune T cell infiltration. Blocking some targets like TGFβ, can act both upstream and downstream, by blocking CAF formation and attenuating downstream signaling in CAFs that are already established. FAP targeting aims at blocking CAFs ability to exert tumor promoting effects in the TME. Targeting FAP can be done by using either MAb/antibody-drug conjugates, immunoconjugates or peptide-drug complexes, FAP-specific CAR-T cells or strategies of gene-knock out. Some other strategies, not depicted in this figure aim at CAFs direct depletion or CAFs normalization towards an inactive phenotype.
A large number of preclinical studies using FAP-targeted CAR mouse T cells have been reported to date (Table 4). Tran et al. genetically modified T cells to express a scFv from the FAP-specific monoclonal antibody (MAb) FAP5, reactive both to human and mouse FAP. They report effective cytotoxic effect of FAP-reactive CAR T cells in vitro. However, adoptive transfer of FAP5-CAR-T cells into mice bearing a variety of subcutaneous tumors mediated limited antitumor effects and induced significant cachexia and lethal bone toxicities in two mouse strains, due to low-level expression of FAP in multipotent bone marrow stromal cells (BMSCs) (169). Moreover, low level expression of FAP has been documented in other healthy tissues like: adipose tissue, skin, muscle and pancreas (150). Other on-target/off tumor toxicities after FAP+ stromal cell depletion with CAR-T cells, reported by Roberts et al., were bone marrow hypoplasia, anemia, pancreatic toxicity and loss of muscle mass (175). In an established lung cancer model, Kakarla et al. generated a CAR specific for both murine and human FAP (mhFAP) using the scFv from MO36 (previously generated by phage display from an immunized FAP/knock-out mouse) (176). They noted that mhFAP CAR-T cells were able to significantly reduce FAP+ stromal cells and tumor growth, with no toxicity or negative effects on wound healing. This study shed the light on the advantage of co-targeting CAFs and cancer cells since the authors demonstrated that combining mhFAP CAR-T cells with EphA2 CAR-T cells increased overall antitumor activity (170). Schuberth et al. developed FAP CAR-T cells using F19 CAR that only recognizes the human version of FAP. They removed the binding site of lck from the CD28 intracellular signaling domain in order to impede IL-2 secretion upon FAP CAR-T cell engagement with its target, and thus reduce Tregs persistence. The authors found that the redirected T cells successfully lysed FAP+ mesothelioma cells in an antigen-specific manner in vitro and in vivo. However, the authors could not evaluated the on-target/off-tumor toxicity of their CAR-T cells since F19-FAP antibody targets only the human version of FAP, with no cross-reactivity with the mouse version (171).
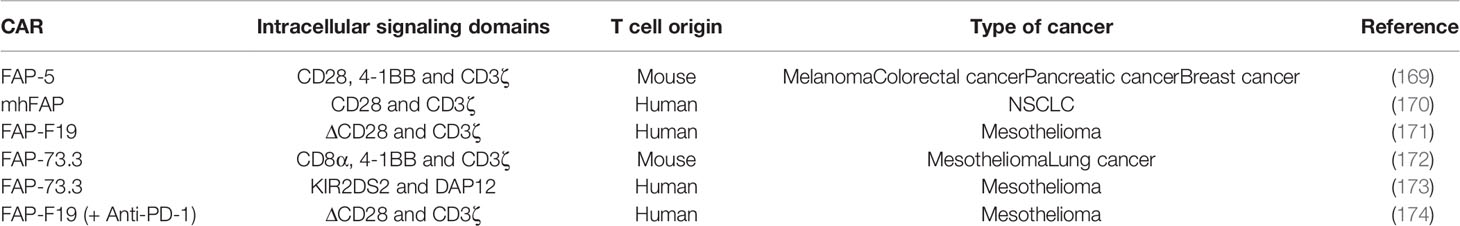
Table 4 Summary of preclinical studies on FAP-targeted CAR-T cells.
Wang et al. developed FAP-73.3 CAR mouse T cells against mouse FAP and demonstrated that depletion of FAP+ cells reduced tumor growth in an immune-dependent manner, as the antitumor effect was only seen in fully immunocompetent mice. Moreover, no clinical toxicities have been observed in mice following the administration of FAP-73.3 CAR mouse T cells in vivo. In order to enhance the antitumor activity, the authors successfully increased the efficacy of their FAP CAR-T cells either by reinfusing a second dose one week later or by combining the redirected T cells with an HPV-E7 vaccine (Ad.E7) (172). The same group designed an alternative chimeric immunoreceptor by fusing the FAP CAR to the transmembrane and cytoplasmic domains of KIR2DS2, a stimulatory killer immunoglobulin-like receptor (KIR), instead of the conventional cytoplasmic domain of CD28 used previously. The aim of this study was to evaluate whether KIR-based CAR-T cells expressing FAP-KIR2DS2 and DAP12 (an immunoreceptor tyrosine-based activation motif (ITAM)-bearing transmembrane adaptor associated with NK-activating receptors) can exhibit a more powerful antitumor response as compared to CD3ζ-based CAR-T cells. Therefore, they generated murine FAP-KIRS2/DAP12-modified T cells using the same scFv from the FAP-73.3 hybridoma. Results showed an enhanced antitumor effect with a complete inhibition of tumor growth, as compared to the significant but minimal slowing of tumor growth with CD3ζ-based CAR-T cells. However, despite the lack of toxicity of the CD3ζ-based FAP-specific CAR-T cells, FAP-KIRS2/DAP12 CAR-T cells showed similar toxicity to the one reported by Roberts et al. in the aforementioned study, suggesting that higher efficacy of FAP targeting is also associated with higher risk of on-target/off-tumor toxicity (173). This issue prompted Gulati et al. to investigate which intracellular signaling domains should be combined with FAP CAR for malignant pleural mesothelioma treatment. When comparing CAR-T cells expressing the CD28/CD3ζ, ΔCD28/CD3ζ and 4-1BB/CD3ζ CAR, the authors noted that 4-1BB/CD3ζ CAR-T cells persisted the most (until day 44) in the peripheral blood of humanized mice, and that the deletion of lck in ΔCD28/CD3ζ CAR enhanced antigen-specific proliferation. Despite higher persistence of 4-1BB/CD3ζ CAR-T cells, statistically significant tumor control in vivo was only obtained when combining FAP-ΔCD28/CD3ζ CAR-T cells with the immune checkpoint PD-1 inhibitor antibodies (174).
To date, two clinical trials using FAP CAR-T cells have already been conducted. The first one is a phase I clinical trial (NCT01722149) using CD3ζ/CD28-based FAP-specific CAR-T cells in three patients with malignant pleural mesothelioma (Table 10). A single dose of 1x 106 CAR-T cells was administered through a pleural catheter. This therapy was well tolerated without any significant toxicity. In addition, one of the three patients received an anti-PD-1 checkpoint inhibitor antibody 8 months after FAP CAR-T cell administration; no clinical toxicity has been reported and 2 out of 3 patients were still alive after a follow-up of 18 months (177, 178).
The second one, cited earlier, is a phase I clinical trial (NCT03932565) using fourth-generation CAR-T cells coproducing IL-7 and CCL19/IL-2 in patients with Nectin4-positive advanced malignant solid tumors such as NSCLC, breast, bladder, pancreatic and ovarian cancer. An approach of intravenous infusion combined with intratumoral injection of Nectin4/FAP-targeted CAR-T cells will be undertaken. The clinical trial is ongoing and still recruiting (Table 10).
2.2 Counteracting the Immunosuppressive TME
The solid tumor microenvironment is composed, as stated previously, by stromal cells (including CAFs), surrounded by the tumor vasculature and by an immune infiltrate of immunosuppressive cells, among which myeloid cells (myeloid-derived suppressor cells or MDSCs), tumor-associated macrophages (TAMs) and Tregs (Figure 4). As previously shown (see Targeting the Tumor Stroma), stromal cells strongly impact the TME as well as the interactions between the immune system and the tumor. Various cell types of hematopoietic origin contribute to the generation of an immunosuppressive TME. This immunosuppressive TME is maintained both by contact mechanisms as cancer cells and stromal cells express a broad range of inhibitory immune-checkpoint ligands (for PD-1, TIGIT, LAG-3 and TIM-3) and by suppressive soluble factors produced by immune cells or by CAFs (cytokines like TGF-β or IL-10). Moreover, other soluble factors with known effects on angiogenesis and produced by this pro-tumorigenic cells, like VEGFA and prostaglandin E2 (PGE2), also induce immunosuppression by inhibiting cytotoxic T lymphocytes (CTLs) and NK cells and by inducing accumulation and proliferation of Tregs (179–181). Therefore, targeting immunosuppressive cells in the TME could improve the efficacy of immunotherapies by increasing tumor recognition by the immune system. In this section, we will discuss the mechanisms by which pro-tumorigenic immune cells from the TME hijack T cell function as well as the different molecular strategies deployed to enhance the efficacy of genetically modified T cells to surmount these roadblocks.
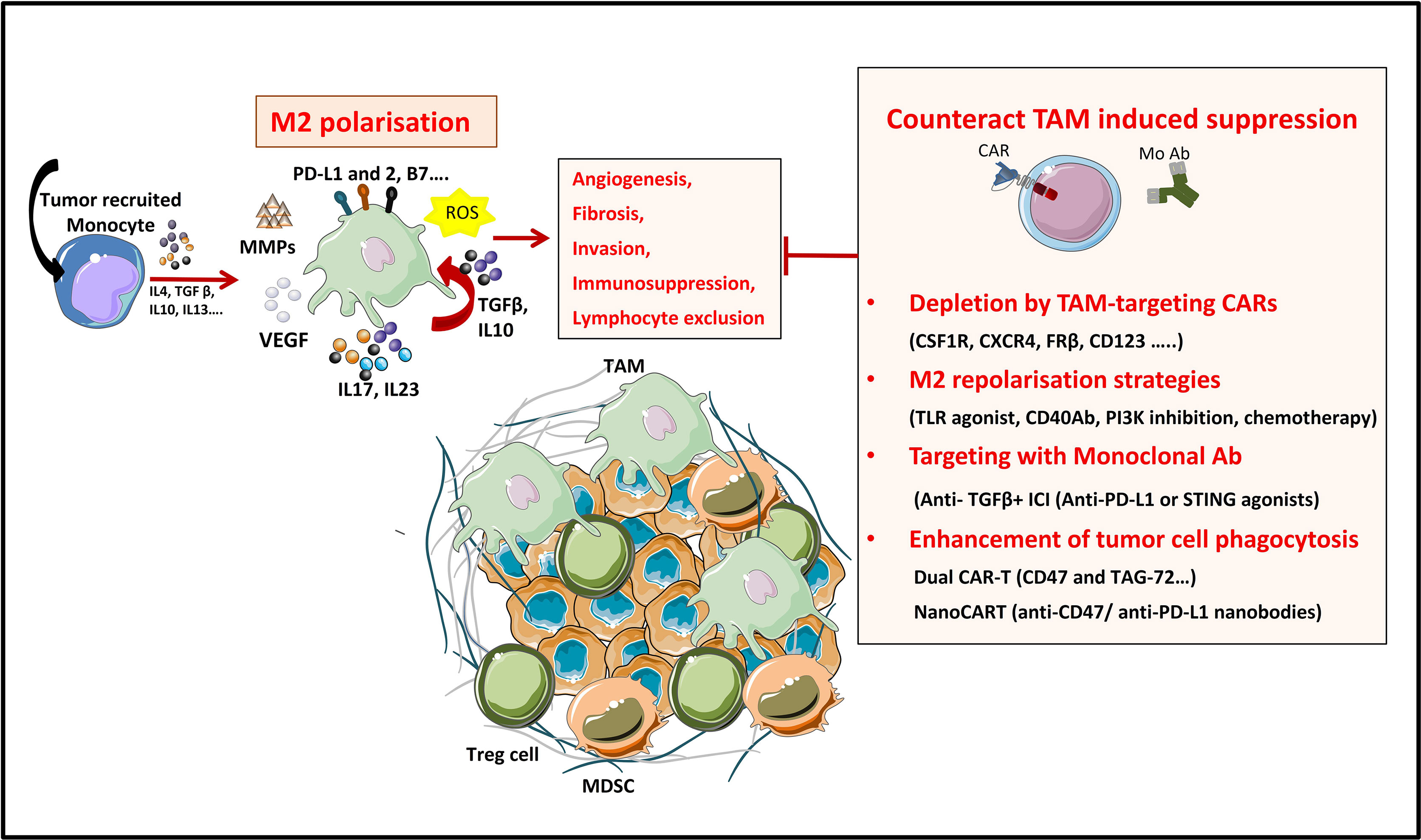
Figure 4 Strategies to overcome TAM’s induced suppression in the TME. TAMs are a tumor promoting immune populations derived under a specific cytokine milieu either from blood circulating monocytes or from tumor resident macrophages. TAMs exert their tumor promoting and immunosuppressive role by means of cell-to cell contact (inhibitory check point ligands), by secreting soluble factors (like cytokines IL10, IL17, L23), by producing ECM-modifying enzymes (MMPs) or by producing reactive species of oxygen (ROS). All these factors promote tumor progression. TAMs directed therapies in the TME aim either at (1) specifically depleting the TAM population, at (2) reprogramming M2 towards proinflammatory M1 phenotypes, at (3) targeting TAM-secreted factors or 4) at enhancing TAM’s phagocytic functions in the TME.
2.2.1 TAMs
Macrophages, one of the main effector cells of the immune system, play a key role in both innate and adaptive immune responses. They constitute the first line of defense against foreign pathogens and help trigger an adaptive antigen-specific response. Macrophages are potent immune effector cells with extensive plasticity and heterogeneity. Some types of macrophages play a crucial role in maintaining tissue homeostasis, by promoting wound healing, whereas others promote inflammation (182). Moreover, impaired macrophage function may lead to the development of many pathologies such as cancer (183). Macrophages are polarized into two contrasting groups: classically activated macrophages or M1 macrophages (pro-inflammatory and usually anti-tumor) and alternatively activated macrophages or M2 macrophages (anti-inflammatory and pro-tumor). This polarization is induced by exposure to soluble factors or pathogen derived molecules in the tissues. M1 macrophage polarization is driven by GMCSF, IFN-γ, TNF-α, lipopolysaccharide (LPS), or other pathogen-associated molecular patterns (PAMPs). M1 macrophages are proinflammatory and play an important role in anti-tumor immunity by: (i) orienting cellular immunity towards à TH1 type response by secreting TNFα, IL-1β, and IL-12, (ii) recruiting Th1 lymphocytes to sites of inflammation through secretion of CXCL9 and CXCL10 chemokines and (iii) presenting processed antigens and expressing costimulatory molecules which enhance T cell responses (184). M2 polarization on the other hand, occurs in the presence of cytokines like MCSF, IL-4, IL-10, IL-13, or TGF-β. Despite their role in tissue homeostasis (stimulating Th2 responses to eliminate parasites, immune regulation, wound healing and tissue repair), M2 macrophages can also promote tumor progression.
Tumors secrete and produce a variety of soluble and mechanical factors to recruit both circulating monocytes and tissue resident macrophages to the TME and convert them to TAMs. TAMs are a specialized population of M2-like macrophages, located in the TME, that share some phenotypic characteristics with M1 and M2 macrophages but have a particular transcriptional profile which is distinct from both types. TAMs enhance tumor progression and metastasis by promoting genetic instability and by enhancing angiogenesis, fibrosis, invasion, immunosuppression and lymphocyte exclusion (185, 186).
On the one hand, TAMs produce inflammatory cytokines like IL-17 and IL-23, which increase genetic instability and on the other hand they can impede tumor immunosurveillance, and thus T cell-mediated antitumor immunity, by secreting immunosuppressive cytokines like TGF-β and IL-10, by expressing immune checkpoint ligands such as PD-L1, PD-L2, B7-H4, or VISTA (4, 187) or by producing reactive oxygen species (ROS). Furthermore, immunosuppressive cytokines produced by TAMs have a role in Treg recruitment. Nonetheless, other factors produced by TAMs are VEGF and MMP enzymes, which promote tumor angiogenesis and metastasis by inducing TME remodeling, increased blood vessel formation, and tumor cell migration (184). All these characteristics make TAMs targeting a promising strategy for cancer treatment (188).
Up to date, various therapeutic strategies targeting TAMs have already been tested in preclinical studies and clinical trials (Figure 4 and Table 10) (189). Macrophage-focused immunotherapeutic strategies aimed either to deplete or to repolarize TAMs. Therefore, the first approach was to reduce or deplete TAMs by eliminating existent TAMs or by inhibiting further TAM recruitment, by targeting: (i) colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R) signaling pathway (190), (ii) chemokines/chemokines receptors axis such as CCL2/CCR2, CCL5/CCR5 (191, 192), (iii) IL-8/CXCR2 (193) or (iv) CXCL12/CXCR4 axis (194).
Second approach is to repolarize TAMs toward an M1-like phenotype, by inhibiting the PI3Kγ signaling pathway (195), by triggering inflammatory activating toll-like receptor (TLR: TLR3, TLR4, TLR7/8 and TLR9) signaling pathway with TLR agonists (196), or by using agonistic CD40 antibodies (197). Third approach of TAM reprogramming is to promote antigen presentation and phagocytosis of TAMs by blocking anti-phagocytic surface proteins called “don’t eat me” signals, like SIRPα or Siglec-10, with antibodies blocking CD47 or CD24 expressed on cancer cells (198, 199) (Figure 4).
Nonetheless, another molecule of interest for targeting TAMs is TGF-β, an anti-inflammatory cytokine typically expressed by macrophages during injury resolution. Macrophages are both a source and a target for TGF-β, causing a positive feedback loop for TAMs and maintaining the immunosuppressive TME by promoting the secretion of additional TGF-β. TAM targeting by TGF-β blockade has already been employed, either in association with STING agonists or with anti-PDL1 blockade and showed tumor regression in preclinical models (200–202). For example, STING agonists DMXAA and cGAMP promote CAR-T cell persistence in the TME of immunocompetent mice in a breast cancer preclinical model. Association of STING agonists with CAR-T cell immunotherapy reprograms macrophagic and myeloid immunosuppressive populations in the TME. This is proven by an increased expression of CXCL9 and CXCL10 by myeloid cells within the TME, with increased recruitment of CXCR3+ TH1 to the tumor as well as by the enhanced expression of genes associated with M1-like macrophages and a marked loss of genes associated with M2-like macrophages and MDSC-like cells (201).
Additional potential molecular targets are discussed by Li et al. in a recent review (189). An increased research aimed at identifying TAM-associated or even TAM-specific targets and some have been used to redirect CAR-T cells against TAMs. Lynn et al. identified, in 2015, folate receptor beta (FRβ), a glycophosphatidylinositol-anchored receptor, as a potential target, as it is highly expressed in monocyte-derived TAMs from primary ovarian cancer. They, thereby, developed mouse FRβ-specific CAR-T cells to target the immunosuppressive M2-like subset of TAMs, while sparing M1-like subpopulations. The preliminary data showed that adoptive transfer of FRβ CAR-T cells into ID8 tumor-bearing mice depleted FRβ+ TAMs and delayed tumor development (203). Similarly, in the study from Rodriguez-Garcia et al., infusion of FRβ-specific CAR-T cells resulted in depletion of FRβ+ TAMs and controlled tumor progression in ovarian cancer, melanoma and colon adenocarcinoma (204). Ruella et al. found that CD123, the α chain of the receptor for IL-3, is expressed within Hodgkin lymphoma (HL) tumor masses both on cancer cells and on the M2-like TAMs. They demonstrated that CD123-specific CAR-T cells target both malignant cells and the surrounding immunosuppressive TME, and lead to the eradication of HL tumor xenografts. Moreover, anti-CD123 CAR-T immunotherapy induced long-term remission and the generation of an antitumor memory response. However, the use of immunodeficient mouse models in this studies does not enable for an accurate evaluation of the role of all endogenous immune system components (205).
New studies have underlined the importance of multiple antigen targeting as a means to both enhance the effectiveness of CAR-T cell therapy and to reduce off-target reactivity (206). To this end, Shu et al. generated CAR-T cells with two tandem CARs targeting CD47 and TAG-72 (Tumor-Associated Glycoprotein 72) (207). CD47 is a cell surface antigen highly expressed in ovarian tumors that functions equally as a macrophage “don’t eat me” signal enabling malignant cells to escape cell phagocytosis and thus detection by the immune system, by interacting with macrophage’ surface signal-regulatory protein-α (SIRPα) (208). TAG-72 is a pancarcinoma antigen and a tumor marker highly expressed in ovarian cancer (209). Blocking both CD47 and TAG-72 with CAR-T cells was associated with increased levels of macrophage-inflammatory protein (MIP)-1α and MIP-1β chemotactic factors in breast cancers, indicating functionality of the CD47 receptor in this model. The dual targeting strategy demonstrated enhanced ability of CAR-T cells to destroy tumor cells expressing low antigen levels, in favor of an increased binding avidity of the tandem CARs to the tumor cell. Another study, conducted by Xie et al., indicated that NanoCAR-T cells engineered to secrete anti-CD47 nanobodies (variable domain of heavy chain-only antibodies or VHH) were able to inhibit tumor growth, while avoiding toxicity encountered with systemic anti-CD47 therapy. This strategy of TAM reprogramming showed superior antitumor activity compared with standard CAR-T cells (210). The team also engineered anti-PD-L1 or anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) nanobodies secreting NanoCAR-T cells, which showed increased persistence. Moreover, this strategy to modify the intra-tumoral immune landscape by nanobody/VHH secretion can offer antitumor agents for multiple targets, has the advantage of being applied to immunocompetent animals and could limit systemic toxicity by means of local delivery at the tumor site (210).
Another approach to increase CAR-T cell infiltration and counteract the immunosuppressive TME is to induce tumor remodeling with adjuvant therapies (like chemotherapy or immune-checkpoint blockade). Srivastava et al. demonstrate that adding oxaliplatin to the lymphodepletion regimen given before ROR1 CAR-T cell infusion activates lung tumor macrophages to produce T cell-recruiting chemokines (reprogramming of TAMs to M1-macrophage). This results in improved CAR-T cell infiltration, tumor remodeling, and response to anti-PD-L1 checkpoint blockade, providing a strategy to improve CAR-T cell efficacy in the clinic. Moreover, a positive control loop has been noted in this model: CAR-T cells remodel the tumor microenvironment to amplify recruitment of endogenous T cells (211).
The aforementioned preclinical studies on CAR-T cells engineered to overcome the immunosuppressive effect of TAMs are summarized in the table below (Table 5).

Table 5 Summary of preclinical studies on CAR-T cells engineered to overcome the immunosuppressive effect of TAMs.
2.2.2 Tregs
For years, regulatory T cells have been known to participate in the immunosuppressive environment of tumors. Due to their suppressive functions, Tregs are able to inhibit the effector functions of tumor-specific cells and reduce the effectiveness of active immunotherapy strategies based on the adoptive transfer of cytotoxic effectors. Therefore, several approaches have been developed to reduce the negative impact of Tregs in CAR-T cell therapies (Figure 5), and evaluated further on in various in vitro and pre-clinical studies (Table 6). Strategies to overcome immunosuppressive impact of the Treg population can be resumed as follows: (i) depletion strategies aiming to reduce cellular density of Tregs in the tumor, (ii) expression of interleukin receptors, hybrid interleukin receptors or switch receptors (iii) optimization of costimulatory domains of CAR-T cell, (iv) transgenic production of various cytokines by TRUCKs and, not least, (v) shielding of CARs from the suppressive effect of TGF-β by gene editing (Figure 5).
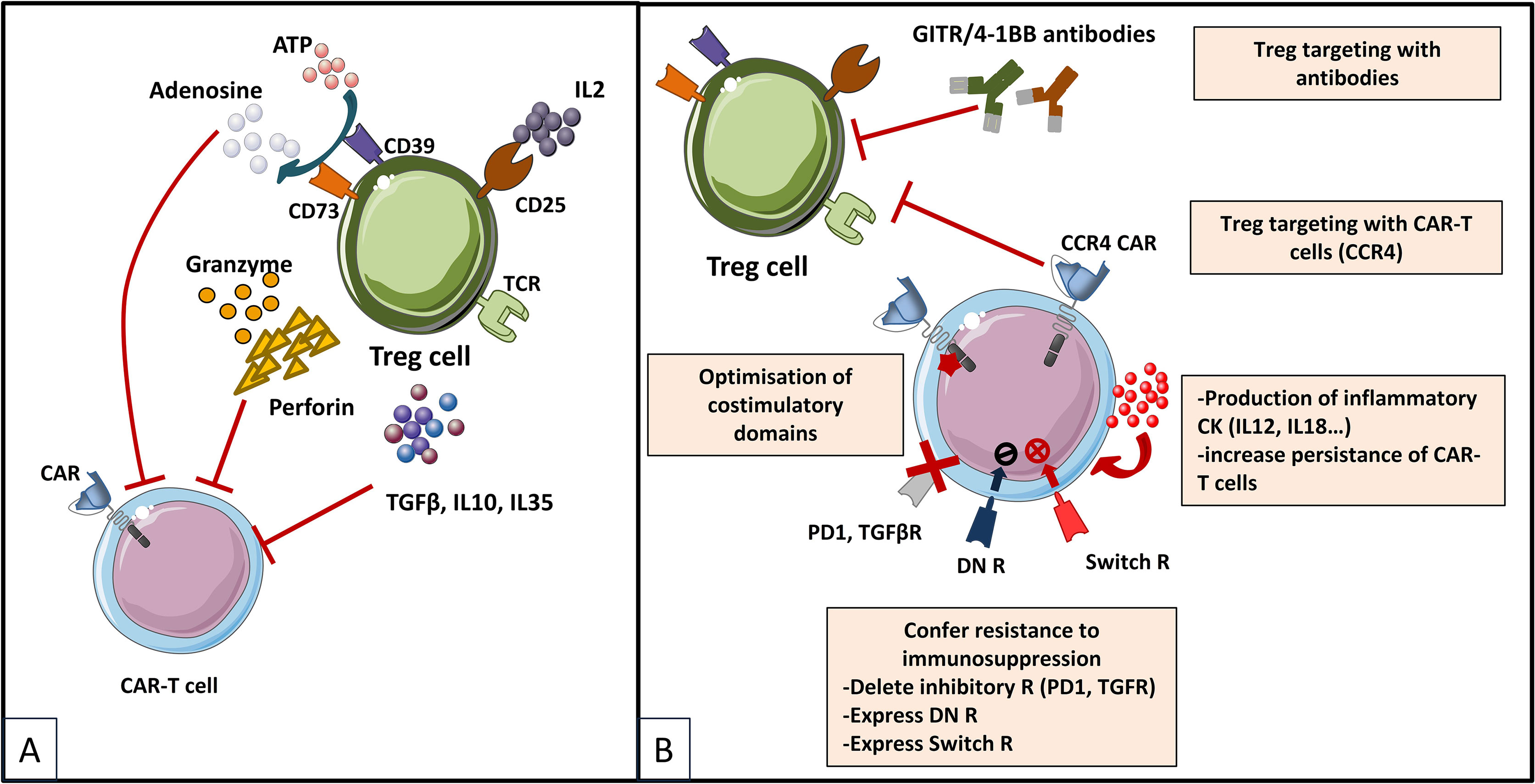
Figure 5 Immunosuppressive mechanisms exerted by Tregs in the TME (A) and engineering strategies to surmount Treg-induced immunosuppression (B) [Adapted from Togashi et al. (212), and Rodriguez-Garcia et al. (213)]. (A) depicts mechanisms for regulatory T (Treg) cells immunosuppressive effects on CAR-T cells based on their physiologic roles. Tregs are immunosuppressive cells highly dependent on IL-2. They bind to and deplete IL-2 from their surroundings, thus reducing availability to effector T (Teff) cells by constitutively expressing the high affinity IL-2 receptor (IL2R) subunit-α (CD25). Treg cells also produce immunosuppressive cytokines (IL-10, IL-35 and TGFβ), which can downregulate the activity of both Teffs and antigen presenting cells (APCs) and they exert direct cytotoxic effects by secreting granzymes and perforin. Moreover, Treg cells release large amounts of ATP, which is converted to adenosine (by CD39 and CD73) that can provide immunosuppressive signals to Teff cells and APCs. Other indirect mechanisms not depicted in the figure by which Tregs exert immunosuppressive effects are mediated by APC, as for instance Tregs expression of cytotoxic T lymphocyte antigen 4 (CTLA-4), which binds to CD80/CD86 on APCs, thereby transmitting suppressive signals to these cells and reducing their capacity to activate Teff cells. (B) shows therapeutic strategies to overcome the immunosuppressive TME sustained by Tregs. Some strategies are based on elimination of Tregs by CAR-T cells or combinations of CAR-T cells with monoclonal antibodies (mAbs) or drugs. CAR-T cells have been designed to target antigens expressed by Tregs for direct depletion. Other strategies are based on immunomodulation of the TME in order to increase CAR-T cells performance: 1) expression of proinflammatory cytokines by CAR-T cells and 2) optimization of costimulatory signaling domains in order to reduce IL-2 secretion and impair Treg expansion and tumor infiltration. Last type of strategies are meant to confer an intrinsic resistance to immunosuppression to CAR-T cells, either by endowing them with 1) dominant-negative receptors (DN R) meant to disrupt signaling, or 2) a chimeric switch receptor (CSR or Switch R) to convert negative signaling into a positive one, or by abrogating the expression of inhibitory receptors (like PD1 of TGFβ receptors) using genome-editing tools (knock out).
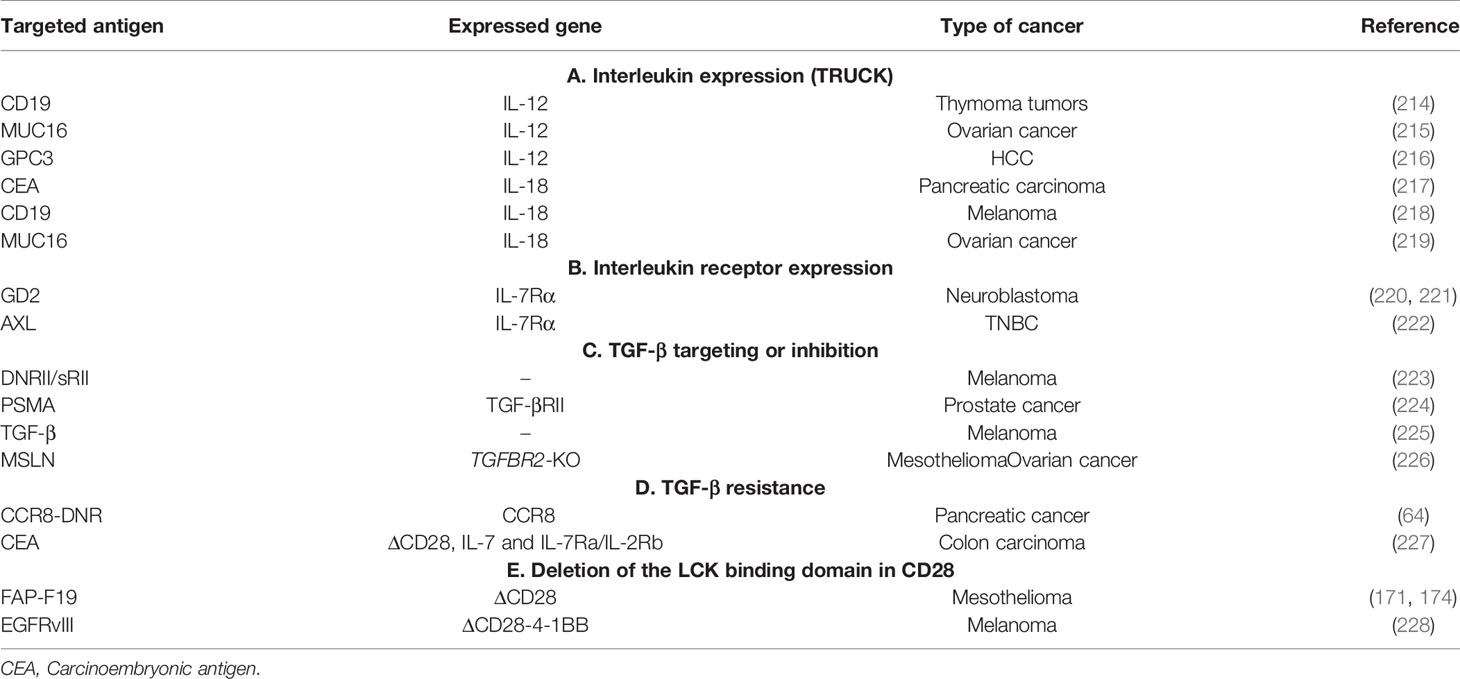
Table 6 Summary of preclinical studies on CAR-T cells to overcome the immunosuppressive effect of Tregs.
For instance, the modification of CAR-T cells to produce IL-12 resulted in improved anti-tumor immune response by different mechanisms and in particular by decreasing CAR-T cells sensitivity to inhibition by regulatory T cells (214) but also by reduction of Tregs densities in the TME (215, 216). In a similar way, it was shown that CAR-T cells producing IL-18 promote antitumor immune responses (218, 219) by modifying the tumor environment notably by increasing the density of M1 macrophages and NK cells and by decreasing Treg infiltration, CD103+ suppressive DCs and M2 macrophages frequency (217, 219). It was also demonstrated that these two cytokines improve the antitumor response by increasing in vivo the survival and the proliferation of CAR-T cells that produce them (215, 216, 218). Promoting the proliferation of CAR-T cells in vivo is an important issue and initial strategies were based on injection of IL-2, a stimulator of T cells proliferation. Unfortunately, this adjuvant treatment has the major inconvenient of inducing the proliferation of Tregs in cancer patients. In order to overcome this side effect, some approaches have sought to increase CAR-T cells dependency on proliferative cytokines different from IL-2, such as IL-7. In various murine solid cancer models, the use of CAR-T cells expressing a constitutively active IL-7 receptor (IL7R) promotes in vitro activation, proliferation and cytotoxicity of CAR-T cells and increases survival of animals by eliciting a protective immune response (220–222). Moreover, unlike the case of IL-2 utilization, IL-7 conjoint injection upon CAR-T infusion does not result in increased proliferation and immunosuppressive function of Tregs, which have low expression of the IL-7R (220). On the other hand, endogenous production of IL-2 by activated T cells has a similar effect as exogenous IL-2 administration, and participates in the generation of Tregs in the TME. Therefore, in order to abrogate production of IL-2 by CAR-T cells, optimization of costimulatory domains of CARs, like the genetic modifications of the intra-cytoplasmic part of the CD28 molecule were designed (171, 174, 228). Preliminary results showed that the elimination of CD28-mediated IL-2 induction impairs CAR engraftment in vivo. However, when impairment of the IL-2 autocrine signaling is compensated for by another costimulatory molecule such as 4-1BB, the CARs accumulate in the bloodstream, suppress tumor growth and resist Tregs-induced immunosuppression (228). As IL-2 release and autocrine IL-2 receptor signaling seemed crucial in counteracting TGF-β repression, but CAR-T cell-released IL-2 negatively impacts the anti-tumor activity through sustaining survival and function of Treg cells, another elegant strategy to improve resistance to TGF-β is the engineering of a hybrid IL7/IL2 receptor to provide IL2 signaling upon IL7 binding. Therefore, Golumba-Nagy et al. designed TRUCKs releasing IL-7 and co-expressing hybrid IL-7Rα/IL-2Rβ receptor, which showed improved survival over a prolonged period and improved activity against TGF-β+ tumors (227).
Indeed, Tregs represent one of the major sources of TGF-β, an immunosuppressive cytokine which impacts the efficiency of immune effectors in the TME. Therefore, a different strategy to resist to TGF-β-induced immunosuppression is to inhibit or delete its receptor on the surface of CAR-T cells. Accordingly, the absence of a functional TGF-β receptor in CAR-T cells promotes proliferation as well as cytokine secretion, resistance to exhaustion and long-term in vivo persistence. Engineering methods for TGF-β receptor deletion/inhibition are: (i) the expression of a dominant negative (DN) receptor, and (ii) genetic disruption by gene-editing techniques like the CRISPR/Cas9 technology. To this regard, CAR-T cells expressing a dominant negative (DN) TGF-β receptor gene are more efficient at inducing protective responses (224, 225) and elimination of endogenous TGF-β receptor II (TGFBR2) in CAR-T cells using CRISPR/Cas9 technology reduces the induction of Treg cells and prevents CAR T cell depletion (226). These strategies aiming to use CAR-T cells modified to resist the immunosuppression induced by the TME derived TGF-β are currently undergoing clinical trials in hematological cancers and solid tumors such as prostate cancer (NCT03089203, NCT04227275.). The first clinical trial (NCT03089203) showed encouraging preliminary results (101) (Table 10).
In contrast to these strategies, which aim to reduce the negative impact of TGF-β on the anti-tumor response of CAR-T cells by inhibiting the expression of its receptor, other approaches are currently evaluating the therapeutic benefit of CAR-T cells modified to express a chimeric TGF-β receptor (switch receptor) whose activation by the cytokine would promote their functions (229). The aforementioned preclinical studies on CAR-T cells engineered to overcome the immunosuppressive effect of Tregs are summarized in the table below (Table 6).
2.2.3 MDSC
MDSCs are a heterogeneous group of immature myeloid cells at various stages of differentiation and which differ from differentiated mature myeloid cells, such as neutrophils, macrophages, and dendritic cells (Dcs). As their name implies, MDSCs are a major group of immunosuppressive cells abundant in different types of cancers (230–233). Recent reports have suggested that MDSCs exert their immunosuppressive activity both on the innate and the adaptive immune system, both by cell-to-cell contact and by the secretion of soluble factors. MDSCs can also facilitate cancer progression by regulating cell mobility or even angiogenesis (234). MDSCs induce immunosuppression of T cell immune responses by various mechanisms: (i) degradation of amino acids essential for activation and proliferation (such as arginine, cysteine, or tryptophan) by production of arginase 1 (Arg1) and indoleamine 2,3-dioxygenase 1 (IDO1) enzymes (235), (ii) Treg induction via IL-10 and TGF- β secretion (236), (iii) suppression of T cell proliferation by MDSC-derived nitric oxide (NO) inhibition of the Jak/STAT5 pathway (237), (iv) impairment of T cell migration into tumor sites by the cleavage of the ectodomain of L-selectin by a disintegrin and metalloproteinase 17 (ADAM17) expressed by MDSCs, and reduction of E-selectin expression on endothelial cells caused by MDSC-derived NO (238), and (v) release of MDSC-derived reactive oxygen species (ROS) implicated in MDSC-mediated T cell suppression (239). In addition, accumulating research showed that MDSCs have potent mechanisms to promote cancer growth (via downregulation of IFN-γ and expression of MMP9) and metastasis (via TNFα, TGFβ, CXCL2 and S100A8/9) by establishing an immunotolerant environment (240). A meta-analysis, published by Zhang et al., concluded that the presence of MDSCs was correlated with poor prognosis in patients with solid cancer (241). Recent reports have also indicated that MDSCs may play a role in resistance to immunotherapy and CAR-T cell therapy (242).
Several strategies have been deployed to increase CAR-T cells resistance to the immunosuppressive effects of MDSCs (Table 7). Among these various strategies, some are based on combinatorial therapies. Briefly, strategies aiming at counteracting MDSC are based on: (1) preventing differentiation and recruitment of MDSCs to the tumor bed, (2) depleting tumor infiltrating MDSCs or (3) mitigating MDSCs immunosuppressive effects (Figure 6). For instance, blocking of MDSC differentiation could be obtained by using a multitargeted TK inhibitor (TKI), Sunitinib, which inhibits STAT3 signaling and induces apoptosis of murine MDSCs. Li et al. demonstrated that coadministration of carbonic anhydrase IX (CAIX)-CAR-T cells with Sunitinib significantly enhanced therapeutic efficacy against a mouse model of human metastatic renal cancer and resulted in prolonged survival of mice, as well as reduction in the number of MDSCs at the cancer site (246). Sunitinib has already shown positive results in a clinical trial (NCT03277924), in combination with anti-PD1 blocking in advanced sarcoma (251) (Table 10).

Table 7 Summary of preclinical studies on CAR-T cells strategies counteracting the immunosuppressive effect of MDSCs.
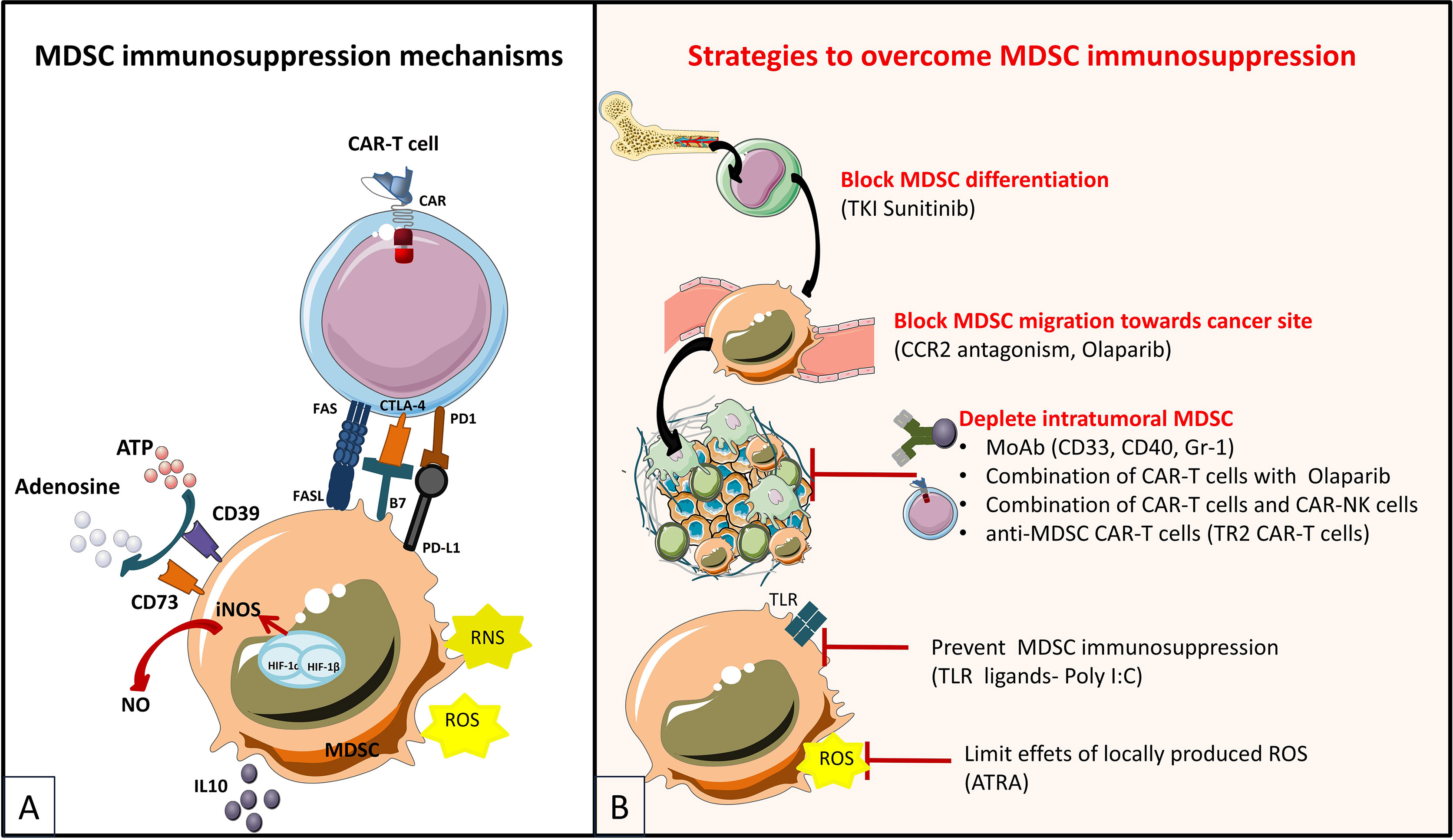
Figure 6 Immunosuppressive mechanisms exerted by MDSCs in the tumor microenvironment (A) and engineering strategies to surmount MDSC-induced immunosuppression (B) [Adapted from Krishnamoorthy et al. (250)]. (A) MDSC exert immunosuppressive effects in the TME (tumor microenvironment) by secretion of IL10 (which activates other immunosuppressive cells such as Tregs). Moreover, MSDCs can induce upregulation of checkpoint molecules (CTLA4, PD1) on T-cells, further inducing T-cell anergy, or can upregulate Fas which induces T-cell apoptosis by contact Fas/Fas-L mechanism. As an effect of hypoxia in the TME, MDSC can contribute to adenosine production by upregulation of CD73 and CD39. MDSCs also produce reactive oxygen (ROS) and nitrogen (RNS) species that can decrease T-cell proliferation and alter antigen recognition capabilities. (B) Strategies for targeting MDSCs in cancer are for example the prevention of MDSC differentiation from hematopoietic stem cells by the usage of Sunitinib, a tyrosine kinase inhibitor (TKI) that inhibits crucial factors for MDSC differentiation (VEGF and STAT3 activity). Second type of strategy is to prevent MDCS migrating to the tumor by targeting chemokine/chemokine receptor axes (CCR2/CCL2). Third, MDSCs depletion from the tumor can be achieved by using immunotherapy (depleting antibodies targeting CD33/gemtuzumab ozogamicin (GO), CD40 or Gr1) or chemotherapy. And least, mitigating the immunosuppressive effects of MDSCs at the tumor site can be realized by reducing the local effects of ROS (with ATRA or all-trans-retinoic acid) or by using TLR stimulation with specific ligands (TLR3 ligand polyinosinic-polycytidylic acid Poly I:C)
Blocking recruitment of MDSC to the tumor bed has been obtained for example by impeding the chemotaxis axis SDF1α/CXCR4 by Sun et al. They proved that olaparib, a Poly(ADP-ribose) polymerase inhibitor (PARPi), could enhance the anti-tumor immune response of EGFRvIII-specific CAR-T cells as well as the recruitment of CD8+ T cells in mouse breast cancer models. Moreover, mice treated with a combination of olaparib and EGFRvIII-specific CAR-T cells showed decreased expression of SDF1α (CXCL12), one of the main MDSC chemoattractant, as well as decreased MDSC recruitment. In parallel, a decrease of the CXCL12 receptor, CXCR4, expression has been noted in MDSCs treated with olaparib, proving that Olaparib might reduce MDSC recruitment by interfering with the SDF1α/CXCR4 axis (245). Other strategies to mitigate MDSCs recruitment to the tumor bed could by the blocking of MDSCs chemokine receptors, as it is the case of CCR2. The CCR2 blocking technique has already been employed but, it can, unfortunately also impede TILs recruitment to the tumor bed (252, 253). Even so, combination of CCR2 blockade with anti-PD1 therapy enhanced anti-tumor responses in many preclinical cancer models (melanoma, breast cancer) (254). Moreover, CCR2 antagonism decreased MDSC counts in pancreatic cancer patients (NCT02345408) (255) (Table 10).
Other strategies already deployed to deplete intra-tumoral MDSC were antagonism or inhibition of specific receptors, like Gr1, CD40 (with mAbs) or CD33 (256, 257). CD33 was identified as a common surface marker of MDSCs and Fultang et al. provided evidence that Gemtuzumab ozogamicin (GO), an antibody-drug conjugate consisting of a humanized mAb targeting CD33 linked to an intracellular toxin named calichamicin, can eliminate MDSCs, leading to CAR-T cell reactivation against multiple cancers. In this study, coadministration of GO restored GD2, MSLN and EGFRvIII CAR-T cell proliferation, leading to increased tumor cell death (244). Moreover, depletion or expansion reduction of MDSC in combination with CAR-T cell therapy has also been employed by using GM-CSF or PD-L1 neutralization as it has been recently shown the immunosuppressive capacities of MDSCs are modulated by GM-CSF through the PD1-PD-L1 axis (258). In a different approach, Parihar et al. demonstrated the effectiveness of specific CAR-NK cells in suppressing MDSCs (248). It has been reported that MDSCs overexpress NKG2D ligands, which are able to activate the NKG2D cytotoxicity receptor on NK cells (259). The immunosuppressive TME restricts, however, NK cell activation. To overcome this obstacle and enhance MDSC depletion in vivo, they generated a CAR that fuses the NKG2D receptor to CD3ζ, the NKG2D.ζ CAR-NK cells. Coadministration of these NKG2D.ζ CAR-NK cells increased the anti-tumor activity of GD2 CAR-T cells in a xenograft model of neuroblastoma (248).
In a 2021 ASCO annual meeting’s abstract, Nalawade et al. demonstrated that MUC1-specific CAR-T cells engineered with a novel chimeric co-stimulatory receptor, TR2.4-1BB, comprising a ScFv derived from a TNF-related apoptosis-inducing ligand receptor 2 (TR2) mAb with a 4-1BB endodomain., induced MDSC apoptosis. As MDSCs express TR2, this strategy could warrant selective MDSC depletion. MUC1.TR2.4-1BB CAR-T cells showed increased cytotoxic activity against breast cancer tumors and inhibited tumor growth more effectively than either MUC1 CAR-T cells or TR2.4-1BB T cells. Similar results have been observed with HER2.TR2.4-1BB CAR-T cells in a HER2+ breast cancer model (249).
Furthermore, Di et al. proved that administration of the toll-like receptor 3 (TLR3) ligand polyinosinic-polycytidylic acid (poly I:C) can increase EGFRvIII-specific CAR-T cell efficacy in immune competent mice bearing colon and breast cancers by enhancing specific lysis of cancer cells and cytokine release upon antigen stimulation. Poly I:C also impeded the suppressive effect of MDSCs on T cell proliferation (247)
And finally, Long et al. identified all-trans-retinoic acid (ATRA) as an effective agent in decreasing the suppressive effect of MDSCs. Co-treatment with ATRA and GD2 CAR-T cells led to an increased antitumor activity compared to ATRA or CAR-T cells treatment alone. These positive effects of ATRA treatment can be explained by an augmented expression of glutathione synthase in MDSCs resulting in higher glutathione synthesis and neutralization of ROS (which contribute to T cell depletion and impede MDSC differentiation) (243).
2.2.4 Enhancing Persistence/Fitness of Genetically Modified T Cells by Interleukin Production
In order to increase persistence and/or maintenance in the immunosuppressive TME, CAR-T cells can be genetically engineered to produce vital cytokines. This engineering strategy is meant to complete the third signal of the immunological synapse (i.e. cytokine stimulation), which is lacking or insufficient in the TME. Various cytokines have already been transgenically expressed in CAR-T cells, the most common being: IL-7, IL-12, IL-15, IL-18, IL-21 and IL-23 (Table 8). Some of them hace been addressed in the Treg section (see section 2.2.2 on Tregs). For an extensive review on gene-edited interleukin CAR-T cells published recently, see Zhang et al. (268).

Table 8 Summary of preclinical studies on CAR-T cells engineered to produce vital cytokines or to express transgenic cytokine receptors.
Therefore, another strategy to overcome the immunosuppressive TME, is the use of fourth generation CAR-T cells genetically redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs), modified to secrete immune stimulatory cytokines (2). Yeku et al. reported the efficacy of IL-12 secreting TRUCKs directed against mucin-16 (MUC16), known as 4H1128ζ-IL-12 T cells, in an aggressive disseminated mouse ovarian cancer model. Indeed, these CAR-T cells induced the eradication of TAMs via Fas/FasL pathway, secreted more inflammatory cytokines (such as IFN-γ) and exhibited increased cytotoxicity in vitro and in vivo. Moreover, cytokine stimulation of 4H1128ζ-IL-12 T cells was associated with increased resistance of CAR-Ts in the TME. The armored CAR-T cells also showed a decreased expression of eomesodermin (Eomes), forkhead box P3 (FOXP3), CTLA-4, LAG-3, TIM-3 and programmed death-ligand 1 (PD-L1), all of which play a major role in the establishment of an immunosuppressive environment (260). Similarly, Chmielewski et al. found that treatment with IL-18 secreting TRUCKs directed against carcinoembryonic antigen (CEA), enhanced CAR-T cell function and survival of mice with advanced pancreatic and lung cancers, and induced an acute Th1 inflammatory response. Data showed an increase in the number of NK cells and M1-like macrophages, and a decrease in the number of M2-like macrophages, Tregs, and inhibitory DCs, allowing for an enhanced antitumor activity (217). The interleukin IL-15 emerged as an immunomodulatory cytokine with anti-tumor effects thanks to its roles in inducing expansion and activation of NK, natural killer T (NKT) cells, and long-lasting memory CD8+ T cells (CTLs). Indeed, IL-15 promotes memory CTL survival and effector function (cytotoxic activity and IFN γ release) and could prevent Tregs from influencing the effector functions of CD4 and CD8 T cells. Moreover, IL-15 can inhibit IL-2 activation induced cell death of effector lymphocytes (269). To this regard, many groups found that preconditioning with IL-7 and IL-15 resulted in better in vitro expansion of CAR-T cells as well as superior antitumor effects in vivo and even increased efficacy upon immune-checkpoint blockade (increased CAR T cell responses to anti-PD-1 adjuvant therapy) (262, 270–272).
Therefore, transgenic expression of IL-15 seemed like an appealing strategy to enhance CAR-T cell effector function, by enhancing proliferation and persistence of CAR-T cells in the TME and has been used in preclinical models of acute myeloid leukemia (AML), melanoma, glioblastoma or neuroblastoma (261–265, 273). In preclinical models of AML, CLL1-directed CAR-T cells with transgenic expression of IL-15 showed increased expansion, survival and antileukemic potency. Unfortunately, co-expression of IL-15 was associated with lethal cytokine release syndrome (CRS), a fatal adverse effect that could be prevented with anti-TNF-antibodies pretreatment and depletion of IL-15 secreting CARs by the inducible caspase-9 (iCas-9) suicide switch (261).
Another fourth generation CAR targeting VEGFR2 on the tumor vasculature and co-expressing murine IL-15 has been evaluated as a tool to overcome TME immunosuppression in immunocompetent, syngeneic melanoma-bearing mice. As in previous studies, expanded CAR-T cells transduced to co-express IL-15 cultivated with both IL-7 and IL-15 showed enhanced expansion as well as a TCM cell phenotype predominantly. In vivo, CAR-T cells up-regulated the antiapoptotic marker Bcl-2 and down-regulated the inhibitory receptor PD-1. These CARs showed enhanced effector functions, engraftment and tumor control, in part through reprograming of the TME in favor of protective endogenous immunity, including NK cell activation and reduced presence of M2 macrophages (262). IL-15 co-expression was also used in a model of IL-13Rα2-positive glioma (IL-13Rα2-CAR.IL-15 T cells) and results showed better in vivo persistence and greater antiglioma activity. Unfortunately, despite enhanced recognition of glioma cells, greater proliferative capacity and increased production of cytokines, the improved T cell persistence was associated with recurrence of gliomas with down-regulated IL-13Rα2 expression. Therefore, at least in GBM treatment, this engineering strategy should be coupled with multiple antigen targeting techniques (263). Expression of IL-15 as a reinforcement proliferative signal was also used in a neuroblastoma model. As in previous studies, GD2 specific CARs armored with IL-15 (GD2.CAR.15-Ts) showed reduced expression of the PD-1 receptor as well as superior antitumor activity. IL-15 forced expression resulted in enrichment in stem cell-like cells (TSCM-like). CAR-Ts were engineered to contain the inducible caspase 9 (iCas9) safety switch (264).
Another method of interleukin gene-editing of CAR-T cells is the co-expression of two simultaneous cytokines. Batra et al. engineered fourth generation CAR-T directed against GPC3+ composed of a 4-1BB costimulatory motif and co-expressing both IL-21 and IL-15 and found superior expansion and antitumor activity against HCC in a preclinical model. IL-21 and IL-15 armored GPC3+ CAR-T cells showed higher proliferation at least in part by maintaining the expression of T cell factor-1 (TCF-1), a transcription factor critical for T cell development and survival. Moreover, manufacturing outcome showed a higher percentage of TSCM and TCM populations. For effective management of toxicity risk in the clinical setting, the authors also proved that the iCas-9 “suicide switch” can effectively eliminate these CAR-T cells. Two clinical trials were open to explore anti-tumoral benefit: NCT02932956 and NCT02905188 (265) (Table 10).
Nonetheless, co-expression of cytokines by genetically engineered CAR-T cells as a mean of boosting anti-tumor activity has the inconvenience of constitutive cytokine signaling in T cells and activation of bystander cells which may cause toxicity. To prevent hyper-activation and excessive cytokine production of CAR-T cells, another team engineered CAR-Ts containing signaling TCR-responsive nanoparticles containing human IL-15 superagonist complex. These protein nanogels embedded in CARs were associated with enhanced selective expansion of CAR-T cells within tumors and improved therapeutic efficacy (273).
In order to circumvent interleukin forced expression, Xincong et al. engineered CAR-T cells expressing the p40 subunit of the IL-23 receptor (p40-Td cells). The p40 subunit of the IL-23R is the only subunit which is not up-regulated upon TCR stimulation. Over-expression of the p40 subunit induced selective proliferation of CAR-T cells via an IL-23 autocrine loop. Moreover, p40-Td cells showed improved antitumor capacity both in vitro and in vivo as well as attenuated side effects in comparison to CAR T cells expressing IL-18 or IL-15 (266).
The interleukin IL-23 has also been targeted with a different strategy, in the context of prostate cancer, by designing CAR-Ts targeting both PSMA and IL-23 (IL-23 mAb). The inflammatory cytokine IL-23 plays an active role in tumorigenesis, by upregulating certain MMPs (MMP9), by increasing angiogenesis and infiltration of M2 macrophages and neutrophils and by reducing CD8 T cell infiltration in the TME. Moreover, it has been proven that IL-23 secreted by MDSCs drives castration-resistant prostate cancer by activating the androgen receptor pathway (274). Therefore, Wang et al. designed three types of CAR-T cells in order to simultaneously target PSMA expressing cells and capture local soluble IL-23 produced by tumor cells or by MDSCs: either (i) dual or duo CAR-Ts expressing 2 CARs at the surface (IL-23mAb-T2A-PSMA), or (ii) a tandem CAR IL-23mAb/PSMA or (iii) PSMA-CARs secreting anti-Il-23 Ab. IL-23 and PSMA targeted duo-CAR-Ts (IL-23mAb-T2A-PSMA) were more efficient in prostate cancer eradication than PSMA CARs only and induced stronger T cell activation, and increased cytokine production when compared to single-molecule tandem CAR IL-23mAb/PSMA (267).
In a different approach, Shum et al. engineered CAR-T cells constitutively expressing the IL-7 receptor (C7R CAR-T cells), in order to deliver potent stimulation and increase CAR-T persistence and antitumor activity (221). An ongoing clinical trial evaluates efficacy of C7R-GD2.CAR T cells in the treatment of brain tumors (NCT04099797) (Table 10). Except armored CAR-T cells secreting IL7 and/or IL2 cited above (see Overcoming the Mismatch or the Dysregulation of Chemokine Receptor/Ligand Axes), armored CAR-T cells secreting other survival cytokines have also gone to the clinic (like IL-15 and/or IL-21: NCT04715191, NCT05155189, NCT02932956 and NCT02905188, NCT03721068, NCT04377932, and NCT05103631) (Table 10).
2.2.5 Overcoming Inhibition by Negative Immune Checkpoints
The tumor-related immune response is regulated by various stimulating and inhibitory signals. Immune checkpoints (ICs) insure the maintenance of immune homeostasis, and thus self-tolerance, by regulating the time course and the intensity of the immune reaction. However, receptor-based signal cascades emerging from ICs play a negative regulatory role in T cells, by inducing immune tolerance and therefore tumor escape from immunosurveillance (275). The first main ICs identified as essential receptors for T cell and CAR-T cell inhibition and apoptosis are CTLA-4 and PD-1 (276). Other immunoreceptors extensively studied in cancer are LAG-3, TIGIT, T-cell immunoglobulin and mucin containing protein-3 (TIM3) and B and T lymphocyte attenuator (BTLA). Since, many different monoclonal Abs (mAbs) and bispecific antibodies (BsAbs) that prevent ligand- inhibitory IC receptor engagement have been used to block immune checkpoints. IC receptors use mono-tyrosine signaling motifs, such as ITIM and immunoreceptor tyrosine-based switch motifs (ITSM), to exert their inhibitory activity (276). IC inhibition either in monotherapy or as supplementary therapy turned out to be a very efficient weapon to fight cancer (277). As PD1/PD-L1 inhibition is the most studied axis, this chapter mainly focuses on PD-1/PD-L1 inhibition in CAR-T cell therapy (Table 9).

Table 9 Summary of preclinical studies on CAR-T cells to overcome the inhibitory effect of ICs.
PD-1 is a member of the B7/CD28 family which exerts its role in modulating T cell activity by interacting with two ligands- PD-L1 and PD-L2. PD-1/PD-L1 binding impedes the synthesis of IFN-γ and IL-2, which decreases T cell proliferation (290). Furthermore, the overexpression of PD-L1 is correlated with poor prognosis in many cancers (291–294). In order to block the interaction between PD-1 and PD-L1, various mAbs, such as nivolumab, pembrolizumab, avelumab, lambrolizumab, and atezolizumab, have been developed (295). After encouraging results from different clinical trials, the FDA granted accelerated approval to nivolumab for the treatment of advanced melanoma in 2014 (296), and advanced squamous NSCLC in 2015 (297). Nivolumab has also shown positive results in many other cancers, such as R/R HL (298) and HCC (299). Furthermore, other anti-PD-1 mAb, pembrolizumab (humanized IgG4 kappa anti-PD-1 mAb), has been approved by the FDA for the treatment of many types of cancer, including unresectable or metastatic melanoma in 2014 (300), advanced NSCLC in 2015 (301), recurrent or metastatic HNSCC in 2016 (302), and locally advanced or metastatic urothelial carcinoma in 2017 (303).
Given acknowledged CAR-T cell dysfunction following engagement of IC receptors and especially spectacular results obtained with anti-PD1 and anti-PD-L1 ICI mAbs, different strategies were deployed in order to enhance CAR-T cell efficacy (Figure 7).
Figure 7 CAR-T cell engineering strategies to overcome inhibition form negative immune checkpoint regulation – Example of PD1/PD-L1 axis targeting in CAR-T cells [Adapted from Rafiq et al. (31)]. In order to prevent CAR-T cell exhaustion and immunosuppression in the TME, different strategies can be used, like combination of CAR-T cells with immune checkpoint inhibitors (ICIs like anti-PD1 or PD-L1 antibodies). PD1-medited inhibition can also be surmounted by designing CAR-T cells that secrete either PD-1-blocking or PD-L1 blocking scFv. Other means of shielding CAR-T cells from the inhibitory effect of the PD1/PD-L1 interaction is to design genetically modified CAR-T cells that express a dominant negative PD-1 receptor (PD-1 DNR) which interferes with PD1 downstream signaling or a PD-1 chimeric switch receptor (CSR), which converts an inhibitory signaling into an activating one. Last type of strategy is based on PD1 expression deletion either by genetic knock-out or by means of shRNA (short hairpin RNA) inhibition.
John et al. were the first to demonstrate that inhibiting an important immunosuppressive pathway such as PD-1 can significantly increase adoptive immunotherapy efficacy using genetically modified T cells (278). To this regard, PD-1 expression was shown to be significantly enhanced on CAR-T cells cocultured with PD-L1+ HER-2+ tumor cells, whereas PD-1 inhibition enhanced CAR-T cell proliferation and activity in vitro and in vivo. Moreover, the coadministration of anti-PD-1 mAbs together with HER-2 specific CAR-T cells enhanced HER-2+ tumor regression and mice survival in a transgenic animal model without any sign of autoimmunity. Interestingly, CAR-T cell and anti-PD-1 mAbs combined therapy significantly depleted MDSCs but not Tregs at the cancer site, as compared to untreated control mice. Similar results have been observed by Gargett et al. with GD2-specific CAR-T cells combined to pembrolizumab against neuroblastoma and melanoma cell lines (279), and Gulati et al. with FAP-specific CAR-T cells combined to anti-PD-1 mAbs against a malignant pleural mesothelioma cell line (174). Many ongoing clinical trials are evaluating the efficacy of the administration of CAR-T cells with anti-PD-1/PD-L1 blocking antibodies in patients with solid tumors (NCT04995003, NCT02414269, NCT01822652, NCT04003649, NCT03980288, and NCT03726515).
In order to avoid repeated anti-PD-1/PD-L1 mAbs administration and the toxicity associated with it, new approaches have been proposed to inhibit the PD-1/PD-L1 axis, by engineering CAR-T cells expressing PD-1-blocking scFv (280, 281) or PD-L1-blocking scFv (282), expressing PD-1 DNR (283, 284), or chimeric switch receptors (CSR, like PD1/CD28 CSR) (285) (Figure 7). Various ongoing clinical trials are evaluating anti-PD-1 antibodies secreting (NCT03030001, NCT03615313, NCT04489862, and NCT02873390), anti-PD-L1 scFv secreting (NCT04556669) or PD-1 nanobodies secreting CAR-T cells (NCT04489862, NCT04503980 and NCT05089266) (Table 10). Genetic engineering strategies aiming at counteracting immunosuppressive signaling by the design of PD1/CD28 CSR are also evaluated in the clinical setting (clinical trials NCT03932955, NCT04850560, NCT02937844). Other clinical trials evaluate efficiency of CAR-T cells designed to resist immunosuppression by various other mechanisms, employing mutant PD-1 proteins, PD-1 dominant negative receptors (DNR), cytoplasmic activated PD-1 or PD-1 downregulation (NCT03540303, NCT04768608, NCT04577326, NCT04163302, NCT04162119, and NCT04836507) (Table 10).
Table 10 Summary of clinical trials on CAR-T cells designed to improve tumor homing and penetration, CAR-T cell persistence and resistance to immunossupresssion in solid tumors.
A different strategy to provide an enhanced version of genetically modified T cells with increased antitumor activity against solid tumors was to engineer PD-1 knockout (KO) CAR-T cells. The breakthrough of gene editing techniques has, thus, allowed to genetically disrupt PD-1 function in CAR-T-cells (320). As a strategy that increases lymphocyte fitness in the immunosuppressive TME, PD-1 KO engineering in CAR-T cells proved enhanced antitumor activity in preclinical models, both in vitro and in vivo (283, 321). PD-1 deletion was first applied to autologous T cells and its efficacy was variable, depending on gene editing techniques. To this regard, Beane et al. used the zinc finger nuclease-mediated (ZFN) gene editing technology to KO PD1 expression in TILs drawn from melanoma patients, with an average reduction of 76% in PD-1 surface-expression. In addition, PD-1 KO TILs showed enhanced in vitro activity and a significantly superior polyfunctional cytokine profile (IFNγ, TNFα, and GM-CSF) as compared to unmodified TILs in 66.67% (2 of 3) patients tested (322). Menger et al. knocked out the PD-1 gene in melanoma-reactive CTLs and in fibrosarcoma-reactive polyclonal T cells, using the TALEN technology and noticed that modified T cells had better persistence at the cancer site and were able to control the tumor progression more efficiently than non-modified T cells (323). Other groups have also succeeded in inactivating the PD-1 gene in TILs by using the CRISPR-Cas9 technology (321, 324, 325).
Considering immune-related adverse events related to anti-PD-1 mAbs administration and the success of PD-1 gene inactivation in primary human T cells, this strategy was extended to CAR-T cell therapy (326). Hu B. et al., designed CAR-T cells directed against CD133 and KO for the PD-1 gene by using the CRISPR/Cas9 technology, with an average of 91.5% of inactivated gene sites. This disruption enhanced both in vitro cytotoxicity against a glioma cell line and in vivo antitumor activity in an orthotopic glioma mouse model. No significant toxicity was observed, confirming the safety profile of PD-1 KO CD133-specific CAR-T cells. Moreover, PD-1 KO did not impede cytokine production and CAR-T functionality as PD-1-deficient lymphocytes secreted similar amounts of cytokines (IFN-γ, IL-2, TNF-α, and GM-CSF) as conventional CAR-T cells (286). The team used this technology to disrupt the PD-1 gene in MSLN-specific CAR-T cells also. Despite a significant effect on CAR-T cell proliferation, this strategy greatly increased CAR-T cell cytokine synthesis and cytotoxicity towards PD-L1+ tumor cells in vitro. The antitumor activity of PD-1 KO MSLN-specific CAR-T cells was also increased in a TNBC animal model. Moreover, PD-1 KO could improve the CAR-T cells antitumor effect more efficiently than the combination of CAR-T cells with PD-1 blocking (287). Similar results have been published by Guo et al. with PD-1 KO GPC3-specific CAR-T cells in a HCC preclinical study (288), and Choi et al. with PD-1 KO EGFRvIII-specific CAR-T cells in a glioblastoma preclinical study (289). Many ongoing clinical trials are evaluating the efficacy of PD-1 deficient T cells/CAR-T cells in patients with solid tumors or hematological cancers (NCT03545815, NCT03525782, NCT03298828, NCT03525652, NCT02793856, NCT03081715, NCT03399448, NCT02867332, NCT03044743, NCT03030001, NCT02863913, NCT03747965, NCT03706326, ChiCTR1800020306, ChiCTR1800018713, ChiCTR1900022620, ChiCTR-OIC-1701131, ChiCTR-OIN-17012136, ChiCTR1800016023, ChiCTR1900025088, NCT03208556 and NCT04213469) (Table 10 for clinical tials on solid tumors).
Last but not least, new reports have suggested the feasibly of targeting other inhibitory receptors, such as CTLA-4 (327, 328), LAG-3 (329, 330) or TIM-3 (330, 331). However, additional studies are required to determine whether these novel strategies are as effective as CAR-T cells engineered to overcome PD1 inhibition. Results from ongoing clinical trials with CAR-T engineered to block simultaneously PD-1 and CTLA-4 +/- TIGIT (by antibody or ScFv secretion) will establish the eventual benefit of combinatorial ICI blockade strategies (NCT03179007, NCT03182816, NCT03182803, NCT04842812, NCT03198052).
3 Conclusion and Future Perspectives
CAR-T cell based cell therapy is a moving field, which showed impressive results in hematopoietic cancer management and brought hope to incurable patients. Unfortunately, success in managing solid cancers was less outstanding. Assiduous research has been done to overcome unexpected roadblocks which impede CAR-T cells trafficking, infiltration, persistence or function in the unwelcoming tumor environment. Indeed, research focused on identifying target antigens and avoiding on-target-off tumor toxicity (206), improving CAR-T cell trafficking and entry into the tumor site, promoting better signaling, less exhaustion, and memory phenotypes in solid tumors. Preclinical models propose various engineering strategies, some of which have already advanced from bench to bedside, with encouraging preliminary results.
As reviewed herein, trafficking and infiltration have been addressed by genetically manipulating chemotaxis and tissue homing. Moreover, tumor stroma targeting emerged as a promising strategy, based either on depletion of stromal cells/immunosuppressive cells or at reprogramming strategies directed at regulating TME plasticity. To this regard, a new generation of CAR-T cells has been designed to directly target stroma components like fibroblasts and immunosuppressive cells (Tregs, TAMs or MDSCs). However, a remaining challenge for the development of both effective and safe CAR-T cell therapies is the insufficient clinical relevance of preclinical mouse models. Indeed, these models sometimes failed to predict clinical level toxicities or, on the other hand, inefficient tumor targeting when translated to the clinic. Further research is still needed to overcome this hurdle and develop advanced preclinical models able to address tumor heterogeneity and TME complexity in order to accomplish a perfect balance between efficacy and safety of CAR-T cell therapies in solid tumors.
Furthermore, exciting new opportunities emerged thanks to gene editing/gene ablation techniques based on the revolutionary, highly specific and efficient CRISPR/Cas9 tools (332, 333), which have been used not only to generate immune-checkpoint knock-outs (PD-1 KO) but also to design “universal” CARs, edited for TCR and/or HLA molecules expression (206, 332, 333), which could pave the road towards cost-effective allogeneic CAR-T cells for an “off-the-shelf” ACT with a broader spectrum (334, 335). This technique can even be used for multiplexed genome editing (336). To this regard, feasibility of targeting multiple genes in T cells by multiplex CRISPR-Cas9 has recently been proven in a small interventional study in patients with advanced, refractory cancer (NCT03399448) (312). Further improvements of this technology are awaited as recent advances seem to insure increased precision and minimized side effects both in case of gene deletion and gene insertion (336). As allogeneic (allo)-CAR-T cells could offer readily available ACT sources that could expand the usage of CAR-cells based immunotherapy, other recent strategies for allo-CAR-T cells generation emerged, like the NKG2D (an NK-based activating receptor) expression. NKG2D expression in allogeneic CAR-T cells offers a non-TCR edited cellular therapy with broad solid tumor targeting, and two clinical trials are ongoing in metastatic colon cancer (NCT04991948 and NCT03692429, Celyad Oncology) with encouraging preliminary results in the second one (2/15 PR and 9/15 SD). Another allo-CAR-T cells product, the CD70-targeting ALLO-316 cells (Allogene Therapeutics) is under evaluation in a clinical trial on renal cell carcinoma (together with anti-CD52 mAb, NCT04696731).
On the other hand, a part from innovations in CAR design addressed in this review, advances in transduction techniques, cell culture and amplification conditions (like IL-7/IL-15 media) as well as identification of the most suitable stage of T cell differentiation (TCM/TSCM) to use for adoptive transfer represent additional steps towards effective CAR-T cell therapy in solid tumors (337, 338). To this regard, the need for large-scale CAR-T manufacturing persists and could limit cancer patients’ accessibility to CAR-T cell-based ACT. Therefore, the already engaged transition from academic to industrial manufacturing could ensure increased availability and reproducibility as well as shorter delays thanks to Good Manufacturing Practice (GMP)-compliant automated, closed systems (339–341). Contrary to large scale, commercial in vitro manufacturing, Smith and colleagues recently described an in-vivo manufacturing technique of CAR-T cells, by programming circulating, bloodstream T cells with DNA-carrying polymer nanoparticles, which efficiently introduced leukemia-targeting CAR genes into T-cell nuclei (342, 343).Accumulating knowledge on efficacy, toxicity and resistance drawn from clinical trials as well as fundamental research data on TILs interaction with the TME will allow for the identification of novel molecular targets in CAR-T cells design (344). To this regard, and pointing out once more the role of the hypoxia response in cancer, the VHL-HIF axis and particularly HIF’s activity, has recently been identified as a tool to potentiate tissue residency of CD8+ CTLs, as well as a potential molecular candidate to modulate CAR-T cell therapy efficacy (345). Genetic targeting of precise molecular or metabolic pathways critical for TILs survival in the TME emerge therefore as novel strategies to overcome insufficient amplification and persistence of CAR-T cells in solid tumors (31, 213, 346, 347).
This review focuses less on engineering strategies aiming at enhancing tumor recognition and preventing antigen escape. Such combinatorial targeting strategies employ bispecific/dual CARs or Tandem CARs, trivalent or pool CARs and have already been reviewed by us (206) and others (31, 213, 346, 348). Bispecific CARs have gained an important place in hematologic cancers management, with numerous ongoing clinical trials (NCT04662099, NCT0327115, NCT03919526, NCT03879382, NCT03881761, NCT03706547, NCT04303520, NCT04412174, NCT03825731, NCT04499573, NCT05098613, NCT04034446, NCT04007029, and NCT04215016). However, usage of bispecific CARs in solid tumors is still at its beginning (NCT03672305, NCT04483778, NCT03618381 and NCT04684459) (Table 10). Nonetheless, multiple antigen targeting by employing universal immune receptors CAR also gains increasing interest (349) and a universal CAR is being tested in a clinical trial on prostate cancer patients (UniCAR02, NCT04633148). Moreover, toxicity management strategies and especially prevention of on-target off tumor effects were not thoroughly described here-in but were reviewed previously (206). Switchable CARs for instance emerge as valuable safety strategies, like is the case of the iCas9 safety suicide switch employed in some ongoing clinical trials (NCT04715191, NCT03721068). Moreover, orthogonal switchable CARs or dual-switch CAR-T cells capable of both regulated costimulation/inducible activation to drive CAR-T cell expansion and activity and regulated iCasp9 safety switch for CAR elimination have recently been described (350). Co-activated switchable CAR-T cells also advanced to clinical testing (ongoing clinical trial NCT02744287 of BPX-601 CAR-T cells expressing a PSCA specific CAR and a rimiducid-inducible MyD88/CD40 co-activation switch, Bellicum Pharmaceuticals), with encouraging preliminary results (307) (Table 10).
Besides optimization of costimulatory domains discussed here-in, modulation of scFv avidity could be another strategy to increase antigen recognition and CAR-T cell engagement. Surprisingly, lower avidity CAR-T cells (the 8F8-BBz CAR-T cells) could show greater therapeutic potential, by increased resistance to exhaustion and apoptosis in an HCC context (351).
Considering all the aforementioned hurdles in CAR-T cells homing, as well as the diversity and plasticity of cells composing the tumor microenvironment, the best engineering option could be based on a combination of strategies that enhance at the same time trafficking, penetration, persistence and/or CAR-T cell function. Some combinations have already been tested, like it is the case of armored CAR-T cells/TRUCKs engineered to co-express chemokine receptors and secrete vital cytokines. As monotherapeutic approaches are rarely effective, strategies targeting multiple antigens, combinations of different genetic engineering strategies or combinations based on CAR-T cells and innovative immunotherapies (like ICIs) could represent a turning point in a still ongoing revolution in solid cancer management. Nonetheless, CAR-T cells could also be combined with other therapeutic modalities, such as standard chemotherapy and/or radiation therapy, tyrosine kinase inhibitors, epigenetic modulators, other small molecule drugs or vaccines.
All in all, CAR-T cell immunotherapy stands out as a promising, evolving weapon in the fight against solid cancer.
Beside CAR-T cell based ACT, novel genetic engineering techniques, such as gene-editing and cellular reprogramming allowed for the emergence of new ACT strategies employing various innate killer cells (IKC) (like NK cells, NKT cells, and γδ T-cells (CAR-IKC) (352–358), macrophages (CAR-M) (146), and even B lymphocytes (CAR-B cells). A combination of “classic” CAR-T cells and CAR- IKC/CAR-Macrophages as bridging therapy could potentially increase efficiency in solid tumors by increasing the cross-talk between various immune cells or by TME remodeling effects (248, 359, 360). Unfortunately, CAR-NK also have some limitations, as for example high-dosage conditioned efficacy and decreased persistence. On the other hand, co-administration of cord-blood derived-NK cells (CB-NKs) proved to be a potent immunoregulatory strategy, promoting early activation and migration, enhanced fitness and increased anti-tumor efficacy of CAR-T cells (360). Surprisingly, chimeric receptor engineered Tregs (CAR-Tregs), which emerged as potential immune-tolerance inducers in autoimmunity or transplantation (361), also showed potent anti-tumor effect (362). Moreover, CAR-B cells, which could represent safe and controllable vehicles for local delivery of monoclonal antibodies emerged in preclinical studies as potential candidates for infectious diseases and protein deficiencies and might therefore be interesting candidates for cancer therapy as well.
Author Contributions
AA and AC contributed equally. All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Parts of the figures were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
References
1. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T Cells. biomark Res (2017) 5:22. doi: 10.1186/s40364-017-0102-y
2. Chmielewski M, Abken H. TRUCKS, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv IN Cell AND Gene Ther (2020) 3:e84. doi: 10.1002/acg2.84
3. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
4. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
5. Chow MT, Luster AD. Chemokines in Cancer. Cancer Immunol Res (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
6. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The Immune Contexture in Cancer Prognosis and Treatment. Nat Rev Clin Oncol (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
7. Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, et al. Adoptive Transfer of Autologous, HER2-Specific, Cytotoxic T Lymphocytes for the Treatment of HER2-Overexpressing Breast Cancer. Cancer Immunol Immunother (2008) 57:271–80. doi: 10.1007/s00262-007-0355-7
8. Pai SI, Cesano A, Marincola FM. The Paradox of Cancer Immune Exclusion: Immune Oncology Next Frontier. Cancer Treat Res (2020) 180:173–95. doi: 10.1007/978-3-030-38862-1_6
9. Haanen JBAG. Converting Cold Into Hot Tumors by Combining Immunotherapies. Cell (2017) 170:1055–6. doi: 10.1016/j.cell.2017.08.031
10. Donnadieu E, Dupré L, Pinho LG, Cotta-de-Almeida V. Surmounting the Obstacles That Impede Effective CAR T Cell Trafficking to Solid Tumors. J Leukocyte Biol (2020) 108:1067–79. doi: 10.1002/JLB.1MR0520-746R
11. Sackstein R, Schatton T, Barthel SR. T-Lymphocyte Homing: An Underappreciated Yet Critical Hurdle for Successful Cancer Immunotherapy. Lab Invest (2017) 97:669–97. doi: 10.1038/labinvest.2017.25
12. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
13. Girard J-P, Moussion C, Förster R. HEVs, Lymphatics and Homeostatic Immune Cell Trafficking in Lymph Nodes. Nat Rev Immunol (2012) 12:762–73. doi: 10.1038/nri3298
14. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T Cells Into Tumors. Cancer Res (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
15. Humblin E, Kamphorst AO. CXCR3-CXCL9: It’s All in the Tumor. Immunity (2019) 50:1347–9. doi: 10.1016/j.immuni.2019.05.013
16. Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res (2019) 79:4801–7. doi: 10.1158/0008-5472.CAN-19-1167
17. Masopust D, Schenkel JM. The Integration of T Cell Migration, Differentiation and Function. Nat Rev Immunol (2013) 13:309–20. doi: 10.1038/nri3442
18. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
19. Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched With Tc17. Cell Rep Med (2020) 1:100127. doi: 10.1016/j.xcrm.2020.100127
20. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-Expression of CD39 and CD103 Identifies Tumor-Reactive CD8 T Cells in Human Solid Tumors. Nat Commun (2018) 9:2724. doi: 10.1038/s41467-018-05072-0
21. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
22. Nagarsheth N, Wicha MS, Zou W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat Rev Immunol (2017) 17:559–72. doi: 10.1038/nri.2017.49
23. Lanitis E, Irving M, Coukos G. Targeting the Tumor Vasculature to Enhance T Cell Activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
24. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
25. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
26. Schaaf MB, Garg AD, Agostinis P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis (2018) 9:115. doi: 10.1038/s41419-017-0061-0
27. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B Receptor Mediates the Endothelial Barrier to T Cell Homing to Tumors and Disables Immune Therapy. Nat Med (2008) 14:28–36. doi: 10.1038/nm1699
28. Guirnalda P, Wood L, Goenka R, Crespo J, Paterson Y. Interferon γ-Induced Intratumoral Expression of CXCL9 Alters the Local Distribution of T Cells Following Immunotherapy With Listeria Monocytogenes. Oncoimmunology (2013) 2(8):e25752. doi: 10.4161/onci.25752
29. Mulligan AM, Raitman I, Feeley L, Pinnaduwage D, Nguyen LT, O’Malley FP, et al. Tumoral Lymphocytic Infiltration and Expression of the Chemokine CXCL10 in Breast Cancers From the Ontario Familial Breast Cancer Registry. Clin Cancer Res (2013) 19:336–46. doi: 10.1158/1078-0432.CCR-11-3314
30. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527:249–53. doi: 10.1038/nature15520
31. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
32. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
33. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
34. Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin - Specific Chimeric Antibody Receptor. Clin Cancer Res (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
35. Tobin RP, Jordan KR, Kapoor P, Spongberg E, Davis D, Vorwald VM, et al. IL-6 and IL-8 Are Linked With Myeloid-Derived Suppressor Cell Accumulation and Correlate With Poor Clinical Outcomes in Melanoma Patients. Front Oncol (2019) 9:1223. doi: 10.3389/fonc.2019.01223
36. Maynard JP, Ertunc O, Kulac I, Valle J.A.B.-D., Marzo AMD, Sfanos KS. IL8 Expression Is Associated With Prostate Cancer Aggressiveness and Androgen Receptor Loss in Primary and Metastatic Prostate Cancer. Mol Cancer Res (2020) 18:153–65. doi: 10.1158/1541-7786.MCR-19-0595
37. Najdaghi S, Razi S, Rezaei N. An Overview of the Role of Interleukin-8 in Colorectal Cancer. Cytokine (2020) 135:155205. doi: 10.1016/j.cyto.2020.155205
38. Todorović-Raković N, Milovanović J. Interleukin-8 in Breast Cancer Progression. J Interferon Cytokine Res (2013) 33:563–70. doi: 10.1089/jir.2013.0023
39. Wang Y, Xu RC, Zhang XL, Niu XL, Qu Y, Li LZ, et al. Interleukin-8 Secretion by Ovarian Cancer Cells Increases Anchorage-Independent Growth, Proliferation, Angiogenic Potential, Adhesion and Invasion. Cytokine (2012) 59:145–55. doi: 10.1016/j.cyto.2012.04.013
40. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
41. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-Cells Targeting the Integrin Avβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy Against Several Solid Malignancies. Cancers (Basel) (2019) 11(5):674. doi: 10.3390/cancers11050674
42. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-Modified CAR-T Cells Have Enhanced Trafficking Ability That Improves Treatment of Hepatocellular Carcinoma. Eur J Immunol (2020) 50:712–24. doi: 10.1002/eji.201948457
43. Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific Recruitment of CC Chemokine Receptor 4-Positive Regulatory T Cells in Hodgkin Lymphoma Fosters Immune Privilege. Cancer Res (2006) 66:5716–22. doi: 10.1158/0008-5472.CAN-06-0261
44. Kohli K, Pillarisetty VG, Kim TS. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther (2021) 29:10–21. doi: 10.1038/s41417-021-00303-x
45. Fialová A, Partlová S, Sojka L, Hromádková H, Brtnický T, Fučíková J, et al. Dynamics of T-Cell Infiltration During the Course of Ovarian Cancer: The Gradual Shift From a Th17 Effector Cell Response to a Predominant Infiltration by Regulatory T-Cells. Int J Cancer (2013) 132:1070–9. doi: 10.1002/ijc.27759
46. Jafarzadeh A, Fooladseresht H, Minaee K, Bazrafshani MR, Khosravimashizi A, Nemati M, et al. Higher Circulating Levels of Chemokine CCL22 in Patients With Breast Cancer: Evaluation of the Influences of Tumor Stage and Chemokine Gene Polymorphism. Tumour Biol (2015) 36:1163–71. doi: 10.1007/s13277-014-2739-6
47. Maruyama T, Kono K, Izawa S, Mizukami Y, Kawaguchi Y, Mimura K, et al. CCL17 and CCL22 Chemokines Within Tumor Microenvironment Are Related to Infiltration of Regulatory T Cells in Esophageal Squamous Cell Carcinoma. Dis Esophagus (2010) 23:422–9. doi: 10.1111/j.1442-2050.2009.01029.x
48. Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 Chemokines Within Tumor Microenvironment Are Related to Accumulation of Foxp3+ Regulatory T Cells in Gastric Cancer. Int J Cancer (2008) 122:2286–93. doi: 10.1002/ijc.23392
49. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. Lymphocytes Coexpressing CCR4 and a Chimeric Antigen Receptor Targeting CD30 Have Improved Homing and Antitumor Activity in a Hodgkin Tumor Model. Blood (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
50. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine Receptor CCR2b Enhanced Anti-Tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-Small-Cell Lung Carcinoma Model. Front Immunol (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
51. Groom JR, Luster AD. CXCR3 in T Cell Function. Exp Cell Res (2011) 317:620–31. doi: 10.1016/j.yexcr.2010.12.017
52. Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the Mechanisms That May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front Immunol (2018) 8:1970. doi: 10.3389/fimmu.2017.01970
53. Moon EK, Wang L-CS, Bekdache K, Lynn RC, Lo A, Thorne SH, et al. Intra-Tumoral Delivery of CXCL11 via a Vaccinia Virus, But Not by Modified T Cells, Enhances the Efficacy of Adoptive T Cell Therapy and Vaccines. Oncoimmunology (2018) 7(3):e1395997. doi: 10.1080/2162402X.2017.1395997
54. Francis L, Guo ZS, Liu Z, Ravindranathan R, Urban JA, Sathaiah M, et al. Modulation of Chemokines in the Tumor Microenvironment Enhances Oncolytic Virotherapy for Colorectal Cancer. Oncotarget (2016) 7:22174–85. doi: 10.18632/oncotarget.7907
55. Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed Oncolytic Poxvirus Elicits Potent Antitumor Immunity and Shows Enhanced Therapeutic Efficacy. Oncoimmunology (2016) 5:e1091554. doi: 10.1080/2162402X.2015.1091554
56. D’Haese JG, Demir IE, Friess H, Ceyhan GO. Fractalkine/CX3CR1: Why a Single Chemokine-Receptor Duo Bears a Major and Unique Therapeutic Potential. Expert Opin Ther Targets (2010) 14:207–19. doi: 10.1517/14728220903540265
57. Tsang JYS, Ni Y-B, Chan S-K, Shao M-M, Kwok Y-K, Chan K-W, et al. CX3CL1 Expression Is Associated With Poor Outcome in Breast Cancer Patients. Breast Cancer Res Treat (2013) 140:495–504. doi: 10.1007/s10549-013-2653-4
58. Xu X, Wang Y, Chen J, Ma H, Shao Z, Chen H, et al. High Expression of CX3CL1/CX3CR1 Axis Predicts a Poor Prognosis of Pancreatic Ductal Adenocarcinoma. J Gastrointest Surg (2012) 16:1493–8. doi: 10.1007/s11605-012-1921-7
59. Hyakudomi M, Matsubara T, Hyakudomi R, Yamamoto T, Kinugasa S, Yamanoi A, et al. Increased Expression of Fractalkine Is Correlated With a Better Prognosis and an Increased Number of Both CD8+ T Cells and Natural Killer Cells in Gastric Adenocarcinoma. Ann Surg Oncol (2008) 15:1775–82. doi: 10.1245/s10434-008-9876-3
60. Erreni M, Siddiqui I, Marelli G, Grizzi F, Bianchi P, Morone D, et al. The Fractalkine-Receptor Axis Improves Human Colorectal Cancer Prognosis by Limiting Tumor Metastatic Dissemination. J Immunol (2016) 196:902–14. doi: 10.4049/jimmunol.1501335
61. Brueckmann M, Borggrefe M. Therapeutic Potential of Fractalkine: A Novel Approach to Metastatic Colon Cancer. Gut (2007) 56:314–6. doi: 10.1136/gut.2006.103317
62. Siddiqui I, Erreni M, van Brakel M, Debets R, Allavena P. Enhanced Recruitment of Genetically Modified CX3CR1-Positive Human T Cells Into Fractalkine/CX3CL1 Expressing Tumors: Importance of the Chemokine Gradient. J Immunother Cancer (2016) 4:21. doi: 10.1186/s40425-016-0125-1
63. Lo ASY, Taylor JR, Farzaneh F, Kemeny DM, Dibb NJ, Maher J. Harnessing the Tumour-Derived Cytokine, CSF-1, to Co-Stimulate T-Cell Growth and Activation. Mol Immunol (2008) 45:1276–87. doi: 10.1016/j.molimm.2007.09.010
64. Cadilha BL, Benmebarek M-R, Dorman K, Oner A, Lorenzini T, Obeck H, et al. Combined Tumor-Directed Recruitment and Protection From Immune Suppression Enable CAR T Cell Efficacy in Solid Tumors. Sci Adv (2021) 7:eabi5781. doi: 10.1126/sciadv.abi5781
65. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36:346–51. doi: 10.1038/nbt.4086
66. Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, et al. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors Without Requiring Preconditioned Lymphodepletion. Clin Cancer Res (2020) 26:5494–505. doi: 10.1158/1078-0432.CCR-20-0777
67. Goto S, Sakoda Y, Adachi K, Sekido Y, Yano S, Eto M, et al. Enhanced Anti-Tumor Efficacy of IL-7/CCL19-Producing Human CAR-T Cells in Orthotopic and Patient-Derived Xenograft Tumor Models. Cancer Immunol Immunother (2021) 70:2503–15. doi: 10.1007/s00262-021-02853-3
68. Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-Secreting CAR-T Cell Therapy for Tumors With Positive Glypican-3 or Mesothelin. J Hematol Oncol (2021) 14:118. doi: 10.1186/s13045-021-01128-9
69. Duan D, Wang K, Wei C, Feng D, Liu Y, He Q, et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front Immunol (2021) 12:609421. doi: 10.3389/fimmu.2021.609421
70. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
71. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
72. Meyran D, Zhu J, Butler J, Macdonald S, Tantalo D, Thio N, et al. 126 Early-Phenotype Lewis Y CAR-T Cells Persist Better in Vivo and Induce Solid Tumor Regression in Combination With Anti-Pd1. J Immunother Cancer (2020) 8(Suppl 3):A76–8. doi: 10.1136/jitc-2020-SITC2020.0126
73. Zou F, Tan J, Liu T, Liu B, Tang Y, Zhang H, et al. The CD39+ HBV Surface Protein-Targeted CAR-T and Personalized Tumor-Reactive CD8+ T Cells Exhibit Potent Anti-HCC Activity. Mol Ther (2021) 29:1794–807. doi: 10.1016/j.ymthe.2021.01.021
74. Sun H, He S, Meng L, Wang Y, Zhang H, Liu Y, et al. Engineering of CD19-Specific Chimeric Antigen Receptor T Cells With the Integrin CD103 Results in Augmented Therapeutic Efficacy Against Human Lymphoma in a Preclinical Model. Blood (2018) 132:2050. doi: 10.1182/blood-2018-99-117354
76. Saman H, Raza SS, Uddin S, Rasul K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers (Basel) (2020) 12(5):1172. doi: 10.3390/cancers12051172
77. Welti J, Loges S, Dimmeler S, Carmeliet P. Recent Molecular Discoveries in Angiogenesis and Antiangiogenic Therapies in Cancer. J Clin Invest (2013) 123:3190–200. doi: 10.1172/JCI70212
78. Dvorak HF. Vascular Permeability Factor/Vascular Endothelial Growth Factor: A Critical Cytokine in Tumor Angiogenesis and a Potential Target for Diagnosis and Therapy. J Clin Oncol (2002) 20:4368–80. doi: 10.1200/JCO.2002.10.088
79. Huang Z, Bao S-D. Roles of Main Pro- and Anti-Angiogenic Factors in Tumor Angiogenesis. World J Gastroenterol (2004) 10:463–70. doi: 10.3748/wjg.v10.i4.463
80. Weis SM, Cheresh DA. Tumor Angiogenesis: Molecular Pathways and Therapeutic Targets. Nat Med (2011) 17:1359–70. doi: 10.1038/nm.2537
81. Song Y, Fu Y, Xie Q, Zhu B, Wang J, Zhang B. Anti-Angiogenic Agents in Combination With Immune Checkpoint Inhibitors: A Promising Strategy for Cancer Treatment. Front Immunol (2020) 11:1956. doi: 10.3389/fimmu.2020.01956
83. Annan DA-M, Kikuchi H, Maishi N, Hida Y, Hida K. Tumor Endothelial Cell—A Biological Tool for Translational Cancer Research. Int J Mol Sci (2020) 21:3238. doi: 10.3390/ijms21093238
84. Kershaw MH, Westwood JA, Zhu Z, Witte L, Libutti SK, Hwu P. Generation of Gene-Modified T Cells Reactive Against the Angiogenic Kinase Insert Domain-Containing Receptor (KDR) Found on Tumor Vasculature. Hum Gene Ther (2000) 11:2445–52. doi: 10.1089/10430340050207939
85. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene Therapy Using Genetically Modified Lymphocytes Targeting VEGFR-2 Inhibits the Growth of Vascularized Syngenic Tumors in Mice. J Clin Invest (2010) 120:3953–68. doi: 10.1172/JCI43490
86. Zhao Y, Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist (2015) 20:660–73. doi: 10.1634/theoncologist.2014-0465
87. Modi SJ, Kulkarni VM. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med Drug Discov (2019) 2:100009. doi: 10.1016/j.medidd.2019.100009
88. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous Targeting of Tumor Antigens and the Tumor Vasculature Using T Lymphocyte Transfer Synergize to Induce Regression of Established Tumors in Mice. Cancer Res (2013) 73:3371–80. doi: 10.1158/0008-5472.CAN-12-3913
89. Englisch A, Altvater B, Kailayangiri S, Hartmann W, Rossig C. VEGFR2 as a Target for CAR T Cell Therapy of Ewing Sarcoma. Pediatr Blood Cancer (2020) 67:e28313. doi: 10.1002/pbc.28313
90. Morales E, Olson M, Iglesias F, Dahiya S, Luetkens T, Atanackovic D. Role of Immunotherapy in Ewing Sarcoma. J Immunother Cancer (2020) 8(2):e000653. doi: 10.1136/jitc-2020-000653
91. Taheri FH, Hassani M, Sharifzadeh Z, Behdani M, Arashkia A, Abolhassani MT. Cell Engineered With a Novel Nanobody-Based Chimeric Antigen Receptor Against VEGFR2 as a Candidate for Tumor Immunotherapy. IUBMB Life (2019) 71:1259–67. doi: 10.1002/iub.2019
92. Ceci C, Atzori MG, Lacal PM, Graziani G. Role of VEGFs/VEGFR-1 Signaling and Its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int J Mol Sci (2020) 21(4):1388. doi: 10.3390/ijms21041388
93. Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct Roles of VEGFR-1 and VEGFR-2 in the Aberrant Hematopoiesis Associated With Elevated Levels of VEGF. Blood (2007) 110:624–31. doi: 10.1182/blood-2007-01-065714
94. Shibuya M. Vascular Endothelial Growth Factor Receptor-1 (VEGFR-1/Flt-1): A Dual Regulator for Angiogenesis. Angiogenesis (2006) 9:225–30. doi: 10.1007/s10456-006-9055-8
95. Wang W, Ma Y, Li J, Shi H-S, Wang L-Q, Guo F-C, et al. Specificity Redirection by CAR With Human VEGFR-1 Affinity Endows T Lymphocytes With Tumor-Killing Ability and Anti-Angiogenic Potency. Gene Ther (2013) 20:970–8. doi: 10.1038/gt.2013.19
96. Ss C, Ds O, Dj B, Ve R, Wd H, Pb G. Prostate-Specific Membrane Antigen Is Produced in Tumor-Associated Neovasculature. Clin Cancer Res (1999) 5(10):2674–81.
97. Santoro SP, Kim S, Motz GT, Alatzoglou D, Li C, Irving M, et al. Cells Bearing a Chimeric Antigen Receptor Against Prostate-Specific Membrane Antigen Mediate Vascular Disruption and Result in Tumor Regression. Cancer Immunol Res (2015) 3:68–84. doi: 10.1158/2326-6066.CIR-14-0192
98. Kinoshita Y, Kuratsukuri K, Landas S, Imaida K, Rovito PM, Wang CY, et al. Expression of Prostate-Specific Membrane Antigen in Normal and Malignant Human Tissues. World J Surg (2006) 30:628–36. doi: 10.1007/s00268-005-0544-5
99. Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, et al. Chimeric Antigen Receptor (CAR+) Modified T Cells Targeting Prostate-Specific Membrane Antigen (PSMA) in Patients (Pts) With Castrate Metastatic Prostate Cancer (CMPC). JCO (2013) 31:72–2. doi: 10.1200/jco.2013.31.6_suppl.72
100. Slovin SF, Wang X, Borquez-Ojeda O, Stefanski J, Olszewska M, Taylor C, et al. 81. Targeting Castration Resistant Prostate Cancer (CRPC) With Autologous PSMA-Directed Chimeric Antigen Receptor T Cells. Mol Ther (2012) 20:S33. doi: 10.1016/S1525-0016(16)35885-3
101. Narayan V, Barber-Rotenberg J, Fraietta J, Hwang W-T, Lacey SF, Plesa G, et al. A Phase I Clinical Trial of PSMA-Directed/Tgfβ-Insensitive CAR-T Cells in Metastatic Castration-Resistant Prostate Cancer. JCO (2021) 39:125–5. doi: 10.1200/JCO.2021.39.6_suppl.125
102. Gutwein LG, Al-Quran SZ, Fernando S, Fletcher BS, Copeland EM, Grobmyer SR. Tumor Endothelial Marker 8 Expression in Triple-Negative Breast Cancer. Anticancer Res (2011) 31:3417–22.
103. Sotoudeh M, Shakeri R, Dawsey SM, Sharififard B, Ahmadbeigi N, Naderi M. ANTXR1 (TEM8) Overexpression in Gastric Adenocarcinoma Makes the Protein a Potential Target of Immunotherapy. Cancer Immunol Immunother (2019) 68:1597–603. doi: 10.1007/s00262-019-02392-y
104. Cullen M, Seaman S, Chaudhary A, Yang MY, Hilton MB, Logsdon D, et al. Host-Derived Tumor Endothelial Marker 8 (TEM8) Promotes the Growth of Melanoma. Cancer Res (2009) 69:6021–6. doi: 10.1158/0008-5472.CAN-09-1086
105. Rmali KA, Watkins G, Harrison G, Parr C, Puntis MCA, Jiang WG. Tumour Endothelial Marker 8 (TEM-8) in Human Colon Cancer and Its Association With Tumour Progression. Eur J Surg Oncol (2004) 30:948–53. doi: 10.1016/j.ejso.2004.07.023
106. Gong Q, Liu C, Wang C, Zhuang L, Zhang L, Wang X. Effect of Silencing TEM8 Gene on Proliferation, Apoptosis, Migration and Invasion of XWLC−05 Lung Cancer Cells. Mol Med Rep (2018) 17:911–7. doi: 10.3892/mmr.2017.7959
107. Chaudhary A, Hilton MB, Seaman S, Haines DC, Stevenson S, Lemotte PK, et al. TEM8/ANTXR1 Blockade Inhibits Pathological Angiogenesis and Potentiates Tumoricidal Responses Against Multiple Cancer Types. Cancer Cell (2012) 21:212–26. doi: 10.1016/j.ccr.2012.01.004
108. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res (2018) 78:489–500. doi: 10.1158/0008-5472.CAN-16-1911
109. Petrovic K, Robinson J, Whitworth K, Jinks E, Shaaban A, Lee SP. TEM8/ANTXR1-Specific CAR T Cells Mediate Toxicity in Vivo. PLoS One (2019) 14(10):e0224015. doi: 10.1371/journal.pone.0224015
110. Astrof S, Crowley D, George EL, Fukuda T, Sekiguchi K, Hanahan D, et al. Direct Test of Potential Roles of EIIIA and EIIIB Alternatively Spliced Segments of Fibronectin in Physiological and Tumor Angiogenesis. Mol Cell Biol (2004) 24:8662–70. doi: 10.1128/MCB.24.19.8662-8670.2004
111. Kaspar M, Zardi L, Neri D. Fibronectin as Target for Tumor Therapy. Int J Cancer (2006) 118:1331–9. doi: 10.1002/ijc.21677
112. Midulla M, Verma R, Pignatelli M, Ritter MA, Courtenay-Luck NS, George AJT. Source of Oncofetal ED-B-Containing Fibronectin: Implications of Production by Both Tumor and Endothelial Cells. Cancer Res (2000) 60:164–9.
113. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-Based CAR T Cells That Target the Tumor Microenvironment Inhibit the Growth of Solid Tumors in Immunocompetent Mice. Proc Natl Acad Sci U S A (2019) 116:7624–31. doi: 10.1073/pnas.1817147116
114. Wagner J, Wickman E, Shaw TI, Anido AA, Langfitt D, Zhang J, et al. Antitumor Effects of CAR T Cells Redirected to the EDB Splice Variant of Fibronectin. Cancer Immunol Res (2021) 9:279–90. doi: 10.1158/2326-6066.CIR-20-0280
115. Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, et al. A Core Human Primary Tumor Angiogenesis Signature Identifies the Endothelial Orphan Receptor ELTD1 as a Key Regulator of Angiogenesis. Cancer Cell (2013) 24:229–41. doi: 10.1016/j.ccr.2013.06.004
116. Mura M, Swain RK, Zhuang X, Vorschmitt H, Reynolds G, Durant S, et al. Identification and Angiogenic Role of the Novel Tumor Endothelial Marker CLEC14A. Oncogene (2012) 31:293–305. doi: 10.1038/onc.2011.233
117. Noy PJ, Lodhia P, Khan K, Zhuang X, Ward DG, Verissimo AR, et al. Blocking CLEC14A-MMRN2 Binding Inhibits Sprouting Angiogenesis and Tumour Growth. Oncogene (2015) 34:5821–31. doi: 10.1038/onc.2015.34
118. Zhuang X, Maione F, Robinson J, Bentley M, Kaul B, Whitworth K, et al. CAR T Cells Targeting Tumor Endothelial Marker CLEC14A Inhibit Tumor Growth. JCI Insight (2020) 5(19):e138808. doi: 10.1172/jci.insight.138808
119. Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, et al. Expression of Integrin Avβ3 in Gliomas Correlates With Tumor Grade and Is Not Restricted to Tumor Vasculature. Brain Pathol (2008) 18:378–86. doi: 10.1111/j.1750-3639.2008.00137.x
120. Petitclerc E, Strömblad S, von Schalscha TL, Mitjans F, Piulats J, Montgomery AMP, et al. Integrin Avβ3 Promotes M21 Melanoma Growth in Human Skin by Regulating Tumor Cell Survival. Cancer Res (1999) 59:2724–30.
121. Hosotani R, Kawaguchi M, Masui T, Koshiba T, Ida J, Fujimoto K, et al. Expression of Integrin AlphaVbeta3 in Pancreatic Carcinoma: Relation to MMP-2 Activation and Lymph Node Metastasis. Pancreas (2002) 25:e30–35. doi: 10.1097/00006676-200208000-00021
122. Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, et al. Integrin Activation Controls Metastasis in Human Breast Cancer. Proc Natl Acad Sci U S A (2001) 98:1853–8. doi: 10.1073/pnas.98.4.1853
123. McCabe N, De S, Vasanji A, Brainard J, Byzova T. Prostate Cancer Specific Integrin Avβ3 Modulates Bone Metastatic Growth and Tissue Remodeling. Oncogene (2007) 26:6238–43. doi: 10.1038/sj.onc.1210429
124. Liu Z, Wang F, Chen X. Integrin Avβ3-Targeted Cancer Therapy. Drug Dev Res (2008) 69:329–39. doi: 10.1002/ddr.20265
125. Wallstabe L, Mades A, Frenz S, Einsele H, Rader C, Hudecek M. CAR T Cells Targeting Avβ3 Integrin Are Effective Against Advanced Cancer in Preclinical Models. Adv Cell Gene Ther (2018) 1(2):e11. doi: 10.1002/acg2.11
126. Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RMG, Kao YV, et al. Targeting of Aberrant Avβ6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol Ther (2017) 25:259–73. doi: 10.1016/j.ymthe.2016.10.012
127. Saha A, Ellison D, Thomas GJ, Vallath S, Mather SJ, Hart IR, et al. High-Resolution in Vivo Imaging of Breast Cancer by Targeting the Pro-Invasive Integrin Alphavbeta6. J Pathol (2010) 222:52–63. doi: 10.1002/path.2745
128. Phanthaphol N, Somboonpatarakun C, Suwanchiwasiri K, Chieochansin T, Sujjitjoon J, Wongkham S, et al. Chimeric Antigen Receptor T Cells Targeting Integrin Avβ6 Expressed on Cholangiocarcinoma Cells. Front Oncol (2021) 11:657868. doi: 10.3389/fonc.2021.657868
129. Whilding LM, Vallath S, Maher J. The Integrin Avβ6: A Novel Target for CAR T-Cell Immunotherapy? Biochem Soc Trans (2016) 44:349–55. doi: 10.1042/BST20150249
130. Moore KM, Thomas GJ, Duffy SW, Warwick J, Gabe R, Chou P, et al. Therapeutic Targeting of Integrin Avβ6 in Breast Cancer. JNCI: J Natl Cancer Inst (2014) 106(8):dju169. doi: 10.1093/jnci/dju169
131. Moore KM, Desai A, de Delgado BL, Trabulo SMD, Reader C, Brown NF, et al. Integrin Avβ6-Specific Therapy for Pancreatic Cancer Developed From Foot-And-Mouth-Disease Virus. Theranostics (2020) 10:2930–42. doi: 10.7150/thno.38702
132. Reader CS, Vallath S, Steele CW, Haider S, Brentnall A, Desai A, et al. The Integrin Avβ6 Drives Pancreatic Cancer Through Diverse Mechanisms and Represents an Effective Target for Therapy. J Pathol (2019) 249:332–42. doi: 10.1002/path.5320
133. Belli C, Trapani D, Viale G, D’Amico P, Duso BA, Vigna PD, et al. Targeting the Microenvironment in Solid Tumors. Cancer Treat Rev (2018) 65:22–32. doi: 10.1016/j.ctrv.2018.02.004
134. Walter SG, Scheidt S, Nißler R, Gaisendrees C, Zarghooni K, Schildberg FA. In-Depth Characterization of Stromal Cells Within the Tumor Microenvironment Yields Novel Therapeutic Targets. Cancers (Basel) (2021) 13:1466. doi: 10.3390/cancers13061466
135. Ni Y, Zhou X, Yang J, Shi H, Li H, Zhao X, et al. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front Cell Dev Biol (2021) 9:637675. doi: 10.3389/fcell.2021.637675
136. Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
137. Coombe DR, Gandhi NS. Heparanase: A Challenging Cancer Drug Target. Front Oncol (2019) 9:1316. doi: 10.3389/fonc.2019.01316
138. Jung H. Hyaluronidase: An Overview of Its Properties, Applications, and Side Effects. Arch Plast Surg (2020) 47:297–300. doi: 10.5999/aps.2020.00752
139. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat Med (2015) 21:524–9. doi: 10.1038/nm.3833
140. Xiong X, Xi J, Liu Q, Wang C, Jiang Z, Yue S-Y, et al. Co-Expression of IL-7 and PH20 Promote Anti-GPC3 CAR-T Tumour Suppressor Activity in Vivo and in Vitro. Liver Int (2021) 41:1033–43. doi: 10.1111/liv.14771
141. Zhao R, Cui Y, Zheng Y, Li S, Lv J, Wu Q, et al. Human Hyaluronidase PH20 Potentiates the Antitumor Activities of Mesothelin-Specific CAR-T Cells Against Gastric Cancer. Front Immunol (2021) 12:660488. doi: 10.3389/fimmu.2021.660488
142. Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J Clin Oncol (2019) 37:1062–9. doi: 10.1200/JCO.18.01295
143. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol (2018) 36:359–66. doi: 10.1200/JCO.2017.74.9564
144. Van Cutsem E, Tempero MA, Sigal D, Oh D-Y, Fazio N, Macarulla T, et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol (2020) 38:3185–94. doi: 10.1200/JCO.20.00590
145. Mardomi A, Abediankenari S. Matrix Metalloproteinase 8: Could It Benefit the CAR-T Cell Therapy of Solid Tumors?- A- Commentary on Therapeutic Potential. Cancer Microenviron (2018) 11:93–6. doi: 10.1007/s12307-018-0208-2
146. Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-Macrophage: A New Immunotherapy Candidate Against Solid Tumors. Biomed Pharmacother (2021) 139:111605. doi: 10.1016/j.biopha.2021.111605
147. Ping Q, Yan R, Cheng X, Wang W, Zhong Y, Hou Z, et al. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene Ther (2021) 28:984–99. doi: 10.1038/s41417-021-00318-4
148. Kakarla S, Song X-T, Gottschalk S. Cancer-Associated Fibroblasts as Targets for Immunotherapy. Immunotherapy (2012) 4:1129–38. doi: 10.2217/imt.12.112
149. Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting Fibroblast Activation Protein in Cancer - Prospects and Caveats. Front Biosci (Landmark Ed) (2018) 23:1933–68. doi: 10.2741/4682
150. Puré E, Blomberg R. Pro-Tumorigenic Roles of Fibroblast Activation Protein in Cancer: Back to the Basics. Oncogene (2018) 37:4343–57. doi: 10.1038/s41388-018-0275-3
151. Shi J, Hou Z, Yan J, Qiu W, Liang L, Meng M, et al. The Prognostic Significance of Fibroblast Activation Protein-α in Human Lung Adenocarcinoma. Ann Transl Med (2020) 8:224. doi: 10.21037/atm.2020.01.82
152. Kesch C, Yirga L, Dendl K, Handke A, Darr C, Krafft U, et al. High Fibroblast-Activation-Protein Expression in Castration-Resistant Prostate Cancer Supports the Use of FAPI-Molecular Theranostics. Eur J Nucl Med Mol Imaging (2021) 49(1):385–9. doi: 10.1007/s00259-021-05423-y
153. Helms E, Onate MK, Sherman MH. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discovery (2020) 10:648–56. doi: 10.1158/2159-8290.CD-19-1353
154. Coto-Llerena M, Ercan C, Kancherla V, Taha-Mehlitz S, Eppenberger-Castori S, Soysal SD, et al. High Expression of FAP in Colorectal Cancer Is Associated With Angiogenesis and Immunoregulation Processes. Front Oncol (2020) 10:979. doi: 10.3389/fonc.2020.00979
155. Li M, Cheng X, Rong R, Gao Y, Tang X, Chen Y. High Expression of Fibroblast Activation Protein (FAP) Predicts Poor Outcome in High-Grade Serous Ovarian Cancer. BMC Cancer (2020) 20:1032. doi: 10.1186/s12885-020-07541-6
156. Wang F-T, Sun W, Zhang J-T, Fan Y-Z. Cancer−associated Fibroblast Regulation of Tumor Neo−angiogenesis as a Therapeutic Target in Cancer (Review). Oncol Lett (2019) 17:3055–65. doi: 10.3892/ol.2019.9973
157. Hofheinz R-D, al-Batran S-E, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients With Metastatic Colorectal Cancer. Onkologie (2003) 26:44–8. doi: 10.1159/000069863
158. Scott AM, Wiseman G, Welt S, Adjei A, Lee F-T, Hopkins W, et al. A Phase I Dose-Escalation Study of Sibrotuzumab in Patients With Advanced or Metastatic Fibroblast Activation Protein-Positive Cancer. Clin Cancer Res (2003) 9:1639–47.
159. Lee J, Fassnacht M, Nair S, Boczkowski D, Gilboa E. Tumor Immunotherapy Targeting Fibroblast Activation Protein, a Product Expressed in Tumor-Associated Fibroblasts. Cancer Res (2005) 65:11156–63. doi: 10.1158/0008-5472.CAN-05-2805
160. Loeffler M, Krüger JA, Niethammer AG, Reisfeld RA. Targeting Tumor-Associated Fibroblasts Improves Cancer Chemotherapy by Increasing Intratumoral Drug Uptake. J Clin Invest (2006) 116:1955–62. doi: 10.1172/JCI26532
161. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clin Cancer Res (2008) 14:4584–92. doi: 10.1158/1078-0432.CCR-07-5211
162. Fischer E, Chaitanya K, Wüest T, Wadle A, Scott AM, van den Broek M, et al. Radioimmunotherapy of Fibroblast Activation Protein Positive Tumors by Rapidly Internalizing Antibodies. Clin Cancer Res (2012) 18:6208–18. doi: 10.1158/1078-0432.CCR-12-0644
163. Brennen WN, Isaacs JT, Denmeade SR. Rationale Behind Targeting Fibroblast Activation Protein–Expressing Carcinoma-Associated Fibroblasts as a Novel Chemotherapeutic Strategy. Mol Cancer Ther (2012) 11:257–66. doi: 10.1158/1535-7163.MCT-11-0340
164. Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting Carcinoma-Associated Fibroblasts Within the Tumor Stroma With a Fibroblast Activation Protein-Activated Prodrug. J Natl Cancer Inst (2012) 104:1320–34. doi: 10.1093/jnci/djs336
165. Chen M, Lei X, Shi C, Huang M, Li X, Wu B, et al. Pericyte-Targeting Prodrug Overcomes Tumor Resistance to Vascular Disrupting Agents. J Clin Invest (2017) 127:3689–701. doi: 10.1172/JCI94258
166. Wang J, Li Q, Li X, Yuan W, Huang S, Cai S, et al. Novel Fapα-Based Z-Gly-Pro Epirubicin Prodrug for Improving Tumor-Targeting Chemotherapy. Eur J Pharmacol (2017) 815:166–72. doi: 10.1016/j.ejphar.2017.09.016
167. Teichgräber V, Monasterio C, Chaitanya K, Boger R, Gordon K, Dieterle T, et al. Specific Inhibition of Fibroblast Activation Protein (FAP)-Alpha Prevents Tumor Progression in Vitro. Adv Med Sci (2015) 60:264–72. doi: 10.1016/j.advms.2015.04.006
168. Bughda R, Dimou P, D’Souza RR, Klampatsa A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. Immunotargets Ther (2021) 10:313–23. doi: 10.2147/ITT.S291767
169. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee C-CR, Restifo NP, et al. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. J Exp Med (2013) 210:1125–35. doi: 10.1084/jem.20130110
170. Kakarla S, Chow KK, Mata M, Shaffer DR, Song X-T, Wu M-F, et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol Ther (2013) 21:1611–20. doi: 10.1038/mt.2013.110
171. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of Malignant Pleural Mesothelioma by Fibroblast Activation Protein-Specific Re-Directed T Cells. J Transl Med (2013) 11:187. doi: 10.1186/1479-5876-11-187
172. Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting Fibroblast Activation Protein in Tumor Stroma With Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity Without Severe Toxicity. Cancer Immunol Res (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
173. Wang E, Wang L-C, Tsai C-Y, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-Cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res (2015) 3:815. doi: 10.1158/2326-6066.CIR-15-0054
174. Gulati P, Rühl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, et al. Aberrant Lck Signal via CD28 Costimulation Augments Antigen-Specific Functionality and Tumor Control by Redirected T Cells With PD-1 Blockade in Humanized Mice. Clin Cancer Res (2018) 24:3981–93. doi: 10.1158/1078-0432.CCR-17-1788
175. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of Stromal Cells Expressing Fibroblast Activation Protein-α From Skeletal Muscle and Bone Marrow Results in Cachexia and Anemia. J Exp Med (2013) 210:1137–51. doi: 10.1084/jem.20122344
176. Brocks B, Garin-Chesa P, Behrle E, Park JE, Rettig WJ, Pfizenmaier K, et al. Species-Crossreactive ScFv Against the Tumor Stroma Marker “Fibroblast Activation Protein” Selected by Phage Display From an Immunized FAP-/- Knock-Out Mouse. Mol Med (2001) 7:461–9. doi: 10.1007/BF03401851
177. Curioni A, Britschgi C, Hiltbrunner S, Bankel L, Gulati P, Weder W, et al. 1226p - A Phase I Clinical Trial of Malignant Pleural Mesothelioma Treated With Locally Delivered Autologous Anti-FAP-Targeted CAR T-Cells. Ann Oncol (2019) 30:v501. doi: 10.1093/annonc/mdz253.052
178. Hiltbrunner S, Britschgi C, Schuberth P, Bankel L, Nguyen-Kim TDL, Gulati P, et al. Local Delivery of CAR T Cells Targeting Fibroblast Activation Protein Is Safe in Patients With Pleural Mesothelioma: First Report of FAPME, a Phase I Clinical Trial. Ann Oncol (2021) 32:120–1. doi: 10.1016/j.annonc.2020.10.474
179. Zalfa C, Paust S. Natural Killer Cell Interactions With Myeloid Derived Suppressor Cells in the Tumor Microenvironment and Implications for Cancer Immunotherapy. Front Immunol (2021) 12:633205. doi: 10.3389/fimmu.2021.633205
180. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
181. Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front Immunol (2021) 12:616837. doi: 10.3389/fimmu.2021.616837
182. Zhang C, Yang M, Ericsson AC. Function of Macrophages in Disease: Current Understanding on Molecular Mechanisms. Front Immunol (2021) 12:620510. doi: 10.3389/fimmu.2021.620510
183. Lin Y, Xu J, Lan H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J Hematol Oncol (2019) 12:76. doi: 10.1186/s13045-019-0760-3
184. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res (2021) 81:1201–8. doi: 10.1158/0008-5472.CAN-20-2990
185. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front Oncol (2020) 9:1512. doi: 10.3389/fonc.2019.01512
186. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
187. Ge Z, Ding S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front Oncol (2020) 10:590941. doi: 10.3389/fonc.2020.590941
188. Tan Y, Wang M, Zhang Y, Ge S, Zhong F, Xia G, et al. Tumor-Associated Macrophages: A Potential Target for Cancer Therapy. Front Oncol (2021) 11:693517. doi: 10.3389/fonc.2021.693517
189. Li C, Xu X, Wei S, Jiang P, Xue L, Wang J. Senior Correspondence Tumor-Associated Macrophages: Potential Therapeutic Strategies and Future Prospects in Cancer. J Immunother Cancer (2021) 9:e001341. doi: 10.1136/jitc-2020-001341
190. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-In-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients With Advanced Solid Tumors. Clin Cancer Res (2017) 23:5703–10. doi: 10.1158/1078-0432.CCR-16-3261
191. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 Promotes Breast Cancer Recurrence Through Macrophage Recruitment in Residual Tumors. Elife (2019) 8:e43653. doi: 10.7554/eLife.43653
192. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 Axis Recruits Tumor Associated Macrophages to Induce Immune Evasion Through PD-1 Signaling in Esophageal Carcinogenesis. Mol Cancer (2020) 19:41. doi: 10.1186/s12943-020-01165-x
193. Alfaro C, Teijeira A, Oñate C, Pérez G, Sanmamed MF, Andueza MP, et al. Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs). Clin Cancer Res (2016) 22:3924–36. doi: 10.1158/1078-0432.CCR-15-2463
194. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-Dependent Immunosuppressive Ly6Clow Monocytes Improves Antiangiogenic Therapy in Colorectal Cancer. Proc Natl Acad Sci U S A (2017) 114:10455–60. doi: 10.1073/pnas.1710754114
195. Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of Macrophage Plasticity in the Tumor Environment: Manipulating Activation State to Improve Outcomes. Front Immunol (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
196. Zeng Q, Jewell CM. Directing Toll-Like Receptor Signaling in Macrophages to Enhance Tumor Immunotherapy. Curr Opin Biotechnol (2019) 60:138–45. doi: 10.1016/j.copbio.2019.01.010
197. Richards DM, Sefrin JP, Gieffers C, Hill O, Merz C. Concepts for Agonistic Targeting of CD40 in Immuno-Oncology. Hum Vaccin Immunother (2019) 16:377–87. doi: 10.1080/21645515.2019.1653744
198. Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H, et al. Advances in Anti-Tumor Treatments Targeting the CD47/Sirpα Axis. Front Immunol (2020) 11:18. doi: 10.3389/fimmu.2020.00018
199. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 Signalling Through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
200. Guerin MV, Regnier F, Feuillet V, Vimeux L, Weiss JM, Bismuth G, et al. Tgfβ Blocks Ifnα/β Release and Tumor Rejection in Spontaneous Mammary Tumors. Nat Commun (2019) 10:4131. doi: 10.1038/s41467-019-11998-w
201. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING Agonist Promotes CAR T Cell Trafficking and Persistence in Breast Cancer. J Exp Med (2021) 218:e20200844. doi: 10.1084/jem.20200844
202. Dodagatta-Marri E, Meyer DS, Reeves MQ, Paniagua R, To MD, Binnewies M, et al. α-PD-1 Therapy Elevates Treg/Th Balance and Increases Tumor Cell PSmad3 That Are Both Targeted by α-Tgfβ Antibody to Promote Durable Rejection and Immunity in Squamous Cell Carcinomas. J Immunother Cancer (2019) 7:62. doi: 10.1186/s40425-018-0493-9
203. Lynn RC, Matsuyama T, Powell DJ. Targeting Frβ+ Tumor Associated Macrophages With Car T Cells in Ovarian Cancer. J Immunother Cancer (2015) 3:32. doi: 10.1186/2051-1426-3-S2-P32
204. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T Cell-Mediated Depletion of Immunosuppressive Tumor-Associated Macrophages Promotes Endogenous Antitumor Immunity and Augments Adoptive Immunotherapy. Nat Commun (2021) 12:877. doi: 10.1038/s41467-021-20893-2
205. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov (2017) 7:1154–67. doi: 10.1158/2159-8290.CD-16-0850
206. Andrea AE, Chiron A, Bessoles S, Hacein-Bey-Abina S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. Int J Mol Sci (2020) 21:8620. doi: 10.3390/ijms21228620
207. Shu R, Evtimov VJ, Hammett MV, Nguyen N-YN, Zhuang J, Hudson PJ, et al. Engineered CAR-T Cells Targeting TAG-72 and CD47 in Ovarian Cancer. Mol Ther Oncolytics (2021) 20:325–41. doi: 10.1016/j.omto.2021.01.002
208. Wang C-L, Lin M-J, Hsu C-Y, Lin H-Y, Tsai H-P, Long C-Y, et al. CD47 Promotes Cell Growth and Motility in Epithelial Ovarian Cancer. BioMed Pharmacother (2019) 119:109105. doi: 10.1016/j.biopha.2019.109105
209. Minnix M, Li L, Yazaki P, Chea J, Poku E, Colcher D, et al. Improved Targeting of an Anti-TAG-72 Antibody Drug Conjugate for the Treatment of Ovarian Cancer. Cancer Med (2020) 9:4756–67. doi: 10.1002/cam4.3078
210. Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved Antitumor Efficacy of Chimeric Antigen Receptor T Cells That Secrete Single-Domain Antibody Fragments. Cancer Immunol Res (2020) 8:518–29. doi: 10.1158/2326-6066.CIR-19-0734
211. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined With Checkpoint Blockade. Cancer Cell (2021) 39:193–208.e10. doi: 10.1016/j.ccell.2020.11.005
212. Togashi Y, Shitara K, Nishikawa H. Regulatory T Cells in Cancer Immunosuppression — Implications for Anticancer Therapy. Nat Rev Clin Oncol (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
213. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, Guedan S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front Immunol (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
214. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
215. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors in Vivo. Oncoimmunology (2015) 4:e994446. doi: 10.4161/2162402X.2014.994446
216. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol (2019) 203:198–207. doi: 10.4049/jimmunol.1800033
217. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
218. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep (2017) 20:3025–33. doi: 10.1016/j.celrep.2017.09.002
219. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23:2130–41. doi: 10.1016/j.celrep.2018.04.051
220. Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner M, Savoldo B, et al. Interleukin-7 Mediates Selective Expansion of Tumor-Redirected Cytotoxic T Lymphocytes Without Enhancement of Regulatory T-Cell Inhibition. Clin Cancer Res (2014) 20:131–9. doi: 10.1158/1078-0432.CCR-13-1016
221. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered IL-7 Receptor Promotes Durable Tumor Elimination by Tumor Redirected T-Cells. Cancer Discov (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
222. Zhao Z, Li Y, Liu W, Li X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res Int (2020) 2020:4795171. doi: 10.1155/2020/4795171
223. Zhang L, Yu Z, Muranski P, Palmer D, Restifo N, Rosenberg S, et al. Inhibition of TGF-β Signaling in Genetically Engineered Tumor Antigen-Reactive T Cells Significantly Enhances Tumor Treatment Efficacy. Gene Ther (2013) 20:575–80. doi: 10.1038/gt.2012.75
224. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
225. Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-β–Responsive CAR-T Cells Promote Anti-Tumor Immune Function. Bioeng Transl Med (2018) 3:75–86. doi: 10.1002/btm2.10097
226. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β Inhibition via CRISPR Promotes the Long-Term Efficacy of CAR T Cells Against Solid Tumors. JCI Insight (2020) 5:e133977. doi: 10.1172/jci.insight.133977
227. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-ζ CAR T Cells Resist TGF-β Repression Through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol Ther (2018) 26:2218–30. doi: 10.1016/j.ymthe.2018.07.005
228. Suryadevara CM, Desai R, Farber SH, Choi BD, Swartz AM, Shen SH, et al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance But Requires 4-1bb Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin Cancer Res (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
229. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-Cell Responses to Soluble Factors With Chimeric Antigen Receptors. Nat Chem Biol (2018) 14:317–24. doi: 10.1038/nchembio.2565
230. Farshidpour M, Ahmed M, Junna S, Merchant JL. Myeloid-Derived Suppressor Cells in Gastrointestinal Cancers: A Systemic Review. World J Gastrointest Oncol (2021) 13:1–11. doi: 10.4251/wjgo.v13.i1.1
231. Wang Y, Ding Y, Deng Y, Zheng Y, Wang S. Role of Myeloid-Derived Suppressor Cells in the Promotion and Immunotherapy of Colitis-Associated Cancer. J Immunother Cancer (2020) 8:e000609. doi: 10.1136/jitc-2020-000609
232. Shibata M, Nakajima T, Mimura K, Shimura T, Kono K, Takenoshita S. MDSC (Myeloid-Derived Suppressor Cells) Is an Important Immunosuppressing Factor and Functionally Related With VEGF and IL-17 in Patients With Gastrointestinal Cancer. Ann Oncol (2019) 30:xi4. doi: 10.1093/annonc/mdz447.011
233. Yang Z, Guo J, Weng L, Tang W, Jin S, Ma W. Myeloid-Derived Suppressor Cells—New and Exciting Players in Lung Cancer. J Hematol Oncol (2020) 13:10. doi: 10.1186/s13045-020-0843-1
234. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) During Tumour Progression. Br J Cancer (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
235. Le Bourgeois T, Strauss L, Aksoylar H-I, Daneshmandi S, Seth P, Patsoukis N, et al. Targeting T Cell Metabolism for Improvement of Cancer Immunotherapy. Front Oncol (2018) 8:237. doi: 10.3389/fonc.2018.00237
236. Haist M, Stege H, Grabbe S, Bros M. The Functional Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells Within the Immunosuppressive Tumor Microenvironment. Cancers (2021) 13:210. doi: 10.3390/cancers13020210
237. Tumino N, Di Pace AL, Besi F, Quatrini L, Vacca P, Moretta L. Interaction Between MDSC and NK Cells in Solid and Hematological Malignancies: Impact on HSCT. Front Immunol (2021) 12:638841. doi: 10.3389/fimmu.2021.638841
238. Yang Y, Li C, Liu T, Dai X, Bazhin AV. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front Immunol (2020) 11:1371. doi: 10.3389/fimmu.2020.01371
239. Ohl K, Tenbrock K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
240. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells (2020) 9:561. doi: 10.3390/cells9030561
241. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The Role of Myeloid-Derived Suppressor Cells in Patients With Solid Tumors: A Meta-Analysis. PLoS One (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
242. Cervantes EV, Boucher JC, Lee SB, Spitler K, Reid K, Davila ML. MDSC Suppression of CAR T Cells Can Be Reduced By Targeted Signaling Disruption. Blood (2019) 134:4438. doi: 10.1182/blood-2019-122752
243. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs With All-Trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol Res (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
244. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. MDSC Targeting With Gemtuzumab Ozogamicin Restores T Cell Immunity and Immunotherapy Against Cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
245. Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-Tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol Ther (2021) 29:60–74. doi: 10.1016/j.ymthe.2020.09.034
246. Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. CAIX-Specific CAR-T Cells and Sunitinib Show Synergistic Effects Against Metastatic Renal Cancer Models. J Immunother (2020) 43:16–28. doi: 10.1097/CJI.0000000000000301
247. Di S, Zhou M, Pan Z, Sun R, Chen M, Jiang H, et al. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front Oncol (2019) 9:241. doi: 10.3389/fonc.2019.00241
248. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity Against Solid Tumors. Cancer Immunol Res (2019) 7:363–75. doi: 10.1158/2326-6066.CIR-18-0572
249. Nalawade SA, Shafer P, Bajgain P, McKenna K, Ali A, Kelly L, et al. Overcoming the Breast Tumor Microenvironment by Targeting MDSCs Through CAR-T Cell Therapy. JCO (2021) 39:1032–2. doi: 10.1200/JCO.2021.39.15_suppl.1032
250. Krishnamoorthy M, Gerhardt L, Maleki Vareki S. Immunosuppressive Effects of Myeloid-Derived Suppressor Cells in Cancer and Immunotherapy. Cells (2021) 10:1170. doi: 10.3390/cells10051170
251. Martin-Broto J, Hindi N, Grignani G, Martinez-Trufero J, Redondo A, Valverde C, et al. Nivolumab and Sunitinib Combination in Advanced Soft Tissue Sarcomas: A Multicenter, Single-Arm, Phase Ib/II Trial. J Immunother Cancer (2020) 8:e001561. doi: 10.1136/jitc-2020-001561
252. Homey B, Müller A, Zlotnik A. Chemokines: Agents for the Immunotherapy of Cancer? Nat Rev Immunol (2002) 2:175–84. doi: 10.1038/nri748
253. Lança T, Costa MF, Gonçalves-Sousa N, Rei M, Grosso AR, Penido C, et al. Protective Role of the Inflammatory CCR2/CCL2 Chemokine Pathway Through Recruitment of Type 1 Cytotoxic Γδ T Lymphocytes to Tumor Beds. J Immunol (2013) 190:6673–80. doi: 10.4049/jimmunol.1300434
254. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, et al. Inhibition of the CCL2 Receptor, CCR2, Enhances Tumor Response to Immune Checkpoint Therapy. Commun Biol (2020) 3:1–12. doi: 10.1038/s42003-020-01441-y
255. Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, et al. Overall Survival in a Trial of Orally Administered CCR2 Inhibitor CCX872 in Locally Advanced/Metastatic Pancreatic Cancer: Correlation With Blood Monocyte Counts. JCO (2018) 36:92–2. doi: 10.1200/JCO.2018.36.5_suppl.92
256. Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy Between T-Cell Immunity and Inhibition of Paracrine Stimulation Causes Tumor Rejection. Proc Natl Acad Sci U S A (1995) 92:6254–8. doi: 10.1073/pnas.92.14.6254
257. Weiss JM, Subleski JJ, Back T, Chen X, Watkins SK, Yagita H, et al. Regulatory T Cells and Myeloid-Derived Suppressor Cells in the Tumor Microenvironment Undergo Fas-Dependent Cell Death During IL-2/Acd40 Therapy. J Immunol (2014) 192:5821–9. doi: 10.4049/jimmunol.1400404
258. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver Myeloid-Derived Suppressor Cells Expand in Response to Liver Metastases in Mice and Inhibit the Anti-Tumor Efficacy of Anti-CEA CAR-T. Cancer Immunol Immunother (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
259. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear Myeloid-Derived “Suppressor” Cells Express RAE-1 and Activate Natural Killer Cells. Blood (2008) 112:4080–9. doi: 10.1182/blood-2008-03-143776
260. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci Rep (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
261. Ataca Atilla P, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating Tnfα Activity Allows Transgenic IL15-Expressing CLL-1 CAR T Cells to Safely Eliminate Acute Myeloid Leukemia. J Immunother Cancer (2020) 8:e001229. doi: 10.1136/jitc-2020-001229
262. Lanitis E, Rota G, Kosti P, Ronet C, Spill A, Seijo B, et al. Optimized Gene Engineering of Murine CAR-T Cells Reveals the Beneficial Effects of IL-15 Coexpression. J Exp Med (2021) 218:e20192203. doi: 10.1084/jem.20192203
263. Krenciute G, Prinzing BL, Yi Z, Wu M-F, Liu H, Dotti G, et al. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells But Results in Antigen Loss Variants. Cancer Immunol Res (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
264. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected With an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin Cancer Res (2019) 25:2915–24. doi: 10.1158/1078-0432.CCR-18-1811
265. Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity Against Hepatocellular Carcinoma. Cancer Immunol Res (2020) 8:309–20. doi: 10.1158/2326-6066.CIR-19-0293
266. Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 Engineering Improves CAR T Cell Function in Solid Tumors. Nat Biotechnol (2020) 38:448–59. doi: 10.1038/s41587-019-0398-2
267. Wang D, Shao Y, Zhang X, Lu G, Liu B. IL-23 and PSMA-Targeted Duo-CAR T Cells in Prostate Cancer Eradication in a Preclinical Model. J Transl Med (2020) 18:23. doi: 10.1186/s12967-019-02206-w
268. Zhang Z, Miao L, Ren Z, Tang F, Li Y. Gene-Edited Interleukin CAR-T Cells Therapy in the Treatment of Malignancies: Present and Future. Front Immunol (2021) 12:718686. doi: 10.3389/fimmu.2021.718686
269. Perera P-Y, Lichy JH, Waldmann TA, Perera LP. The Role of Interleukin-15 in Inflammation and Immune Responses to Infection: Implications for Its Therapeutic Use. Microbes Infect (2012) 14:247–61. doi: 10.1016/j.micinf.2011.10.006
270. Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric Antigen Receptor T (CAR-T) Cells Expanded With IL-7/IL-15 Mediate Superior Antitumor Effects. Protein Cell (2019) 10:764–9. doi: 10.1007/s13238-019-0643-y
271. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely Related T-Memory Stem Cells Correlate With in Vivo Expansion of CAR.CD19-T Cells and Are Preserved by IL-7 and IL-15. Blood (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
272. Giuffrida L, Sek K, Henderson MA, House IG, Lai J, Chen AXY, et al. IL-15 Preconditioning Augments CAR T Cell Responses to Checkpoint Blockade for Improved Treatment of Solid Tumors. Mol Ther (2020) 28:2379–93. doi: 10.1016/j.ymthe.2020.07.018
273. Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie Y-Q, et al. Enhancing T Cell Therapy Through TCR-Signaling-Responsive Nanoparticle Drug Delivery. Nat Biotechnol (2018) 36:707–16. doi: 10.1038/nbt.4181
274. Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, et al. IL-23 Secreted by Myeloid Cells Drives Castration-Resistant Prostate Cancer. Nature (2018) 559:363–9. doi: 10.1038/s41586-018-0266-0
275. Russell BL, Sooklal SA, Malindisa ST, Daka LJ, Ntwasa M. The Tumor Microenvironment Factors That Promote Resistance to Immune Checkpoint Blockade Therapy. Front Oncol (2021) 11:641428. doi: 10.3389/fonc.2021.641428
276. He X, Xu C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res (2020) 30:660–9. doi: 10.1038/s41422-020-0343-4
277. Golay J, Andrea AE. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies (Basel) (2020) 9:E17. doi: 10.3390/antib9020017
278. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin Cancer Res (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
279. Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-Specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter But Can Be Protected From Activation-Induced Cell Death by PD-1 Blockade. Mol Ther (2016) 24:1135–49. doi: 10.1038/mt.2016.63
280. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
281. Zhou J-T, Liu J-H, Song T-T, Ma B, Amidula N, Bai C. EGLIF-CAR-T Cells Secreting PD-1 Blocking Antibodies Significantly Mediate the Elimination of Gastric Cancer. Cancer Manage Res (2020) 12:8893. doi: 10.2147/CMAR.S260915
282. Suarez ER, Chang D-K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
283. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/JCI83092
284. Kiesgen S, Linot C, Quach HT, Saini J, Bellis R, Banerjee S, et al. Abstract LB-378: Regional Delivery of Clinical-Grade Mesothelin-Targeted CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade: Translation to a Phase I Trial. Cancer Res (2020) 80:LB–378. doi: 10.1158/1538-7445.AM2020-LB-378
285. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
286. Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection With Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum Gene Ther (2019) 30:446–58. doi: 10.1089/hum.2017.234
287. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol Immunother (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
288. Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 Enhanced the Anti-Tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front Pharmacol (2018) 9:1118. doi: 10.3389/fphar.2018.01118
289. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J Immunother Cancer (2019) 7:304. doi: 10.1186/s40425-019-0806-7
290. Butte MJ, Keir ME, Phamduy TB, Freeman GJ, Sharpe AH. PD-L1 Interacts Specifically With B7-1 to Inhibit T Cell Proliferation. Immunity (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
291. Vrankar M, Zwitter M, Kern I, Stanic K. PD-L1 Expression Can Be Regarded as Prognostic Factor for Survival of Non-Small Cell Lung Cancer Patients After Chemoradiotherapy. Neoplasma (2018) 65:140–6. doi: 10.4149/neo_2018_170206N77
292. Hu Y, Chen W, Yan Z, Ma J, Zhu F, Huo J. Prognostic Value of PD-L1 Expression in Patients With Pancreatic Cancer. Med (Baltimore) (2019) 98:e14006. doi: 10.1097/MD.0000000000014006
293. Litvin IE, Paganella MP, Wendland EM, Roehe AV. Prognosis of PD-L1 in Human Breast Cancer: Protocol for a Systematic Review and Meta-Analysis. Syst Rev (2020) 9:66. doi: 10.1186/s13643-020-01306-9
294. Tashima Y, Kuwata T, Yoneda K, Hirai A, Mori M, Kanayama M, et al. Prognostic Impact of PD-L1 Expression in Correlation With Neutrophil-To-Lymphocyte Ratio in Squamous Cell Carcinoma of the Lung. Sci Rep (2020) 10:1243. doi: 10.1038/s41598-019-57321-x
295. Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front Pharmacol (2021) 12:731798. doi: 10.3389/fphar.2021.731798
296. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med. (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
297. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
298. Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J Clin Oncol (2018) 36:1428–39. doi: 10.1200/JCO.2017.76.0793
299. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in Patients With Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
300. Raedler LA. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am Health Drug Benefits (2015) 8:96–100.
301. Sul J, Blumenthal GM, Jiang X, He K, Keegan P, Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. Oncologist (2016) 21:643–50. doi: 10.1634/theoncologist.2015-0498
302. Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S, et al. FDA Approval Summary: Pembrolizumab for the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma With Disease Progression on or After Platinum-Containing Chemotherapy. Oncologist (2017) 22:873–8. doi: 10.1634/theoncologist.2016-0496
303. Suzman DL, Agrawal S, Ning Y-M, Maher VE, Fernandes LL, Karuri S, et al. FDA Approval Summary: Atezolizumab or Pembrolizumab for the Treatment of Patients With Advanced Urothelial Carcinoma Ineligible for Cisplatin-Containing Chemotherapy. Oncologist (2019) 24:563–9. doi: 10.1634/theoncologist.2018-0084
304. Disis ML, Patel MR, Pant S, Hamilton EP, Lockhart AC, Kelly K, et al. Avelumab (MSB0010718C; Anti-PD-L1) in Patients With Recurrent/Refractory Ovarian Cancer From the JAVELIN Solid Tumor Phase Ib Trial: Safety and Clinical Activity. JCO (2016) 34:5533–3. doi: 10.1200/JCO.2016.34.15_suppl.5533
305. Scher HI, Fizazi K, Saad F, Chi KN, Taplin M-E, Sternberg CN, et al. Impact of On-Study Corticosteroid Use on Efficacy and Safety in the Phase III AFFIRM Study of Enzalutamide (ENZA), an Androgen Receptor Inhibitor. JCO (2013) 31:6–6. doi: 10.1200/jco.2013.31.6_suppl.6
306. Motzer RJ, Eisen T, Hutson TE, Szczylik C, Krygowski M, Strahs AL, et al. Overall Survival Results From a Phase III Study of Tivozanib Hydrochloride Versus Sorafenib in Patients With Renal Cell Carcinoma. JCO (2013) 31:350–0. doi: 10.1200/jco.2013.31.6_suppl.350
307. Becerra CR, Manji GA, Kim DW, Gardner O, Malankar A, Shaw J, et al. Ligand-Inducible, Prostate Stem Cell Antigen (PSCA)-Directed GoCAR-T Cells in Advanced Solid Tumors: Preliminary Results With Cyclophosphamide (Cy) ± Fludarabine (Flu) Lymphodepletion (Ld). JCO (2019) 37:2536–6. doi: 10.1200/JCO.2019.37.15_suppl.2536
308. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I Study of CAR-T Cells With PD-1 and TCR Disruption in Mesothelin-Positive Solid Tumors. Cell Mol Immunol (2021) 18:2188–98. doi: 10.1038/s41423-021-00749-x
309. Lin Y, Chen S, Zhong S, An H, Yin H, McGowan E. 35o - Phase I Clinical Trial of PD-1 Knockout Anti-MUC1 CAR-T Cells in the Treatment of Patients With Non-Small Cell Lung Cancer. Ann Oncol (2019) 30:xi12. doi: 10.1093/annonc/mdz448
310. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Publisher Correction: Safety and Feasibility of CRISPR-Edited T Cells in Patients With Refractory Non-Small-Cell Lung Cancer. Nat Med (2020) 26:1149–9. doi: 10.1038/s41591-020-0973-6
311. Jing Z, Zhang N, Ding L, Wang X, Hua Y, Jiang M, et al. Safety and Activity of Programmed Cell Death-1 Gene Knockout Engineered T Cells in Patients With Previously Treated Advanced Esophageal Squamous Cell Carcinoma: An Open-Label, Single-Arm Phase I Study. JCO (2018) 36:3054–4. doi: 10.1200/JCO.2018.36.15_suppl.3054
312. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367:eaba7365. doi: 10.1126/science.aba7365
313. Saleh K, Daste A, Martin N, Pons-Tostivint E, Auperin A, Herrera-Gómez RG, et al. Response to Salvage Chemotherapy After Progression on Immune Checkpoint Inhibitors in Patients With Squamous Cell Carcinoma of the Head and Neck. JCO (2018) 36:6015–5. doi: 10.1200/JCO.2018.36.15_suppl.6015
314. Wang Z, Chen M, Zhang Y, Liu Y, Yang Q, Nie J, et al. Phase I Study of CRISPR-Engineered CAR-T Cells With PD-1 Inactivation in Treating Mesothelin-Positive Solid Tumors. JCO (2020) 38:3038–8. doi: 10.1200/JCO.2020.38.15_suppl.3038
315. Chen S, Lin Y, Cui Y, Zhou C, An H, Huang J, et al. McGowan, E. P-258 Clinical Study of PD-1 Disrupted Anti-MUC1 CAR-T Cells in Patients With Advanced Oesophageal Cancer. Ann Oncol (2021) 32:S186. doi: 10.1016/j.annonc.2021.05.312
316. Fang J, Ding N, Guo X, Sun Y, Zhang Z, Xie B, et al. Apd-1-MesoCAR-T Cells Partially Inhibit the Growth of Advanced/Refractory Ovarian Cancer in a Patient Along With Daily Apatinib. J Immunother Cancer (2021) 9:e001162. doi: 10.1136/jitc-2020-001162
317. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I Clinical Trial of EGFR-Specific CAR-T Cells Generated by the PiggyBac Transposon System in Advanced Relapsed/Refractory Non-Small Cell Lung Cancer Patients. J Cancer Res Clin Oncol (2021) 147:3725–34. doi: 10.1007/s00432-021-03613-7
318. Adams S, Dieras V, Barrios CH, Winer EP, Schneeweiss A, Iwata H, et al. Patient-Reported Outcomes (PROs) From the Phase III IMpassion130 Trial of Atezolizumab (Atezo) Plus Nabpaclitaxel (NP) in Metastatic Triple-Negative Breast Cancer (MTNBC). JCO (2019) 37:1067–7. doi: 10.1200/JCO.2019.37.15_suppl.1067
319. El-Khoueiry AB, Kim RD, Harris WP, Sung MW, Waldschmidt D, Cabrera R, et al. Updated Results of a Phase 1b Study of Regorafenib (REG) 80 Mg/Day or 120 Mg/Day Plus Pembrolizumab (PEMBRO) for First-Line Treatment of Advanced Hepatocellular Carcinoma (HCC). JCO (2021) 39:4078–8. doi: 10.1200/JCO.2021.39.15_suppl.4078
320. McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 Disrupted CAR-T Cells in the Treatment of Solid Tumors: Promises and Challenges. Biomed Pharmacother (2020) 121:109625. doi: 10.1016/j.biopha.2019.109625
321. Zhao Z, Shi L, Zhang W, Han J, Zhang S, Fu Z, et al. CRISPR Knock Out of Programmed Cell Death Protein 1 Enhances Anti-Tumor Activity of Cytotoxic T Lymphocytes. Oncotarget (2017) 9:5208–15. doi: 10.18632/oncotarget.23730
322. Beane JD, Lee G, Zheng Z, Mendel M, Abate-Daga D, Bharathan M, et al. Clinical Scale Zinc Finger Nuclease-Mediated Gene Editing of PD-1 in Tumor Infiltrating Lymphocytes for the Treatment of Metastatic Melanoma. Mol Ther (2015) 23:1380–90. doi: 10.1038/mt.2015.71
323. Menger L, Sledzinska A, Bergerhoff K, Vargas FA, Smith J, Poirot L, et al. TALEN-Mediated Inactivation of PD-1 in Tumor-Reactive Lymphocytes Promotes Intratumoral T-Cell Persistence and Rejection of Established Tumors. Cancer Res (2016) 76:2087–93. doi: 10.1158/0008-5472.CAN-15-3352
324. Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 Mediated Efficient PD-1 Disruption on Human Primary T Cells From Cancer Patients. Sci Rep (2016) 6:20070. doi: 10.1038/srep20070
325. Seki A, Rutz S. Optimized RNP Transfection for Highly Efficient CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. J Exp Med (2018) 215:985–97. doi: 10.1084/jem.20171626
326. Liu X, Shi Y, Zhang D, Zhou Q, Liu J, Chen M, et al. Risk Factors for Immune-Related Adverse Events: What Have We Learned and What Lies Ahead? biomark Res (2021) 9:79. doi: 10.1186/s40364-021-00314-8
327. Lin S, Cheng L, Ye W, Li S, Zheng D, Qin L, et al. Chimeric CTLA4-CD28-CD3z T Cells Potentiate Antitumor Activity Against CD80/CD86–Positive B Cell Malignancies. Front Immunol (2021) 12:642528. doi: 10.3389/fimmu.2021.642528
328. Shi LZ, Goswami S, Fu T, Guan B, Chen J, Xiong L, et al. Blockade of CTLA-4 and PD-1 Enhances Adoptive T-Cell Therapy Efficacy in an ICOS-Mediated Manner. Cancer Immunol Res (2019) 7:1803–12. doi: 10.1158/2326-6066.CIR-18-0873
329. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 Mediated LAG-3 Disruption in CAR-T Cells. Front Med (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
330. Zou F, Lu L, Liu J, Xia B, Zhang W, Hu Q, et al. Engineered Triple Inhibitory Receptor Resistance Improves Anti-Tumor CAR-T Cell Performance. via CD56 Nat Commun (2019) 10:4109. doi: 10.1038/s41467-019-11893-4
331. He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, et al. Bispecific and Split CAR T Cells Targeting CD13 and TIM3 Eradicate Acute Myeloid Leukemia. Blood (2020) 135:713–23. doi: 10.1182/blood.2019002779
332. Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic T Cells. Blood (2018) 131:311–22. doi: 10.1182/blood-2017-05-787598
333. Ruella M, Kenderian SS. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going Off the Shelf. BioDrugs (2017) 31:473–81. doi: 10.1007/s40259-017-0247-0
334. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-The-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat Rev Drug Discov (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
335. Martínez Bedoya D, Dutoit V, Migliorini D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front Immunol (2021) 12:640082. doi: 10.3389/fimmu.2021.640082
336. Mo F, Heslop HE, Mamonkin M. CRISPR-Edited Immune Effectors: The End of the Beginning. Mol Ther (2020) 28:995–6. doi: 10.1016/j.ymthe.2020.03.009
337. Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, et al. Enhancing Chimeric Antigen Receptor T Cell Anti-Tumor Function Through Advanced Media Design. Mol Ther - Methods Clin Dev (2020) 18:595–606. doi: 10.1016/j.omtm.2020.07.008
338. Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-Generation Manufacturing Protocols Enriching TSCM CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front Immunol (2020) 11:1217. doi: 10.3389/fimmu.2020.01217
339. Abou-el-Enein M, Elsallab M, Feldman SA, Fesnak AD, Heslop HE, Marks P, et al. Scalable Manufacturing of CAR T Cells for Cancer Immunotherapy. Blood Cancer Discov (2021) 2(5):408–22. doi: 10.1158/2643-3230.BCD-21-0084
340. Levine BL, Miskin J, Wonnacott K, Keir C. Global Manufacturing of CAR T Cell Therapy. Mol Ther - Methods Clin Dev (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
341. Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T Cell Manufacturing Processes During Pivotal Clinical Trials. Mol Ther - Methods Clin Dev (2020) 16:136–44. doi: 10.1016/j.omtm.2019.11.018
342. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In Situ Programming of Leukaemia-Specific T Cells Using Synthetic DNA Nanocarriers. Nat Nanotech (2017) 12:813–20. doi: 10.1038/nnano.2017.57
343. Olweus J. Manufacture of CAR-T Cells in the Body. Nat Biotechnol (2017) 35:520–1. doi: 10.1038/nbt.3898
344. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
345. Liikanen I, Lauhan C, Quon S, Omilusik K, Phan AT, Bartrolí LB, et al. Hypoxia-Inducible Factor Activity Promotes Antitumor Effector Function and Tissue Residency by CD8+ T Cells. J Clin Invest (2021) 131:e143729. doi: 10.1172/JCI143729
346. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28:2320–39. doi: 10.1016/j.ymthe.2020.09.015
347. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
348. Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol (2018) 9:2593. doi: 10.3389/fimmu.2018.02593
349. Minutolo NG, Hollander EE, Powell DJ. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front Oncol (2019) 9:176. doi: 10.3389/fonc.2019.00176
350. Duong MT, Collinson-Pautz MR, Morschl E, Lu A, Szymanski SP, Zhang M, et al. Two-Dimensional Regulation of CAR-T Cell Therapy With Orthogonal Switches. Mol Ther Oncolytics (2019) 12:124–37. doi: 10.1016/j.omto.2018.12.009
351. Caraballo Galva LD, Jiang X, Hussein MS, Zhang H, Mao R, Brody P, et al. Novel Low-Avidity Glypican-3 Specific CARTs Resist Exhaustion and Mediate Durable Antitumor Effects Against HCC. Hepatology (2021) 00:1–15. doi: 10.1002/hep.32279
352. Zhang C, Hu Y, Xiao W, Tian Z. Chimeric Antigen Receptor- and Natural Killer Cell Receptor-Engineered Innate Killer Cells in Cancer Immunotherapy. Cell Mol Immunol (2021) 18:2083–100. doi: 10.1038/s41423-021-00732-6
353. Elahi R, Heidary AH, Hadiloo K, Esmaeilzadeh A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev Rep (2021) 17:2081–106. doi: 10.1007/s12015-021-10246-3
354. Chaudhry K, Dowlati E, Bollard CM. Chimeric Antigen Receptor-Engineered Natural Killer Cells: A Promising Cancer Immunotherapy. Expert Rev Clin Immunol (2021) 17:643–59. doi: 10.1080/1744666X.2021.1911648
355. Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric Antigen Receptor-Engineered Natural Killer Cells for Cancer Immunotherapy. J Hematol Oncol (2020) 13:168. doi: 10.1186/s13045-020-00998-9
356. Lin C-Y, Gobius I, Souza-Fonseca-Guimaraes F. Natural Killer Cell Engineering - a New Hope for Cancer Immunotherapy. Semin Hematol (2020) 57:194–200. doi: 10.1053/j.seminhematol.2020.10.002
357. Lin C, Zhang J. Chimeric Antigen Receptor Engineered Innate Immune Cells in Cancer Immunotherapy. Sci China Life Sci (2019) 62:633–9. doi: 10.1007/s11427-018-9451-0
358. Makkouk A, Yang XC, Barca T, Lucas A, Turkoz M, Wong JTS, et al. Off-The-Shelf Vδ1 Gamma Delta T Cells Engineered With Glypican-3 (GPC-3)-Specific Chimeric Antigen Receptor (CAR) and Soluble IL-15 Display Robust Antitumor Efficacy Against Hepatocellular Carcinoma. J Immunother Cancer (2021) 9:e003441. doi: 10.1136/jitc-2021-003441
359. Qin VM, D’Souza C, Neeson PJ, Zhu JJ. Chimeric Antigen Receptor Beyond CAR-T Cells. Cancers (2021) 13:404. doi: 10.3390/cancers13030404
360. Bachiller M, Perez-Amill L, Battram AM, Carné SC, Najjar A, Verhoeyen E, et al. NK Cells Enhance CAR-T Cell Antitumor Efficacy by Enhancing Immune/Tumor Cells Cluster Formation and Improving CAR-T Cell Fitness. J Immunother Cancer (2021) 9:e002866. doi: 10.1136/jitc-2021-002866
361. Zhang Q, Lu W, Liang C-L, Chen Y, Liu H, Qiu F, et al. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
Keywords: CAR-T cell immunotherapy, Tumor microenvironment, Solid tumor, Tumor Homing, Chemokines, Angiogenesis, Tumor stroma, Immune checkpoints
Citation: Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G and Hacein-Bey-Abina S (2022) Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front. Immunol. 13:830292. doi: 10.3389/fimmu.2022.830292
Received: 06 December 2021; Accepted: 18 January 2022;
Published: 08 February 2022.
Edited by:
Ken Young, Duke University, United StatesReviewed by:
Jamie Berta Spangler, Johns Hopkins University, United StatesVita Golubovskaya, University of Oklahoma Health Sciences Center, United States
Copyright © 2022 Andrea, Chiron, Mallah, Bessoles, Sarrabayrouse and Hacein-Bey-Abina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salima Hacein-Bey-Abina, c2FsaW1hLmhhY2Vpbi1iZXktYWJpbmFAdS1wYXJpcy5mcg==; c2FsaW1hLmhhY2Vpbi1iZXlAYXBocC5mcg==
†These authors have contributed equally to this work