 Hongjuan You1Suping Qin1Fulong Zhang2Wei Hu3Xiaocui Li1Dongsheng Liu3Fanyun Kong1*Xiucheng Pan4Kuiyang Zheng1,5Renxian Tang1,5*
Hongjuan You1Suping Qin1Fulong Zhang2Wei Hu3Xiaocui Li1Dongsheng Liu3Fanyun Kong1*Xiucheng Pan4Kuiyang Zheng1,5Renxian Tang1,5*- 1Jiangsu Key Laboratory of Immunity and Metabolism, Department of Pathogenic Biology and Immunology, Xuzhou Medical University, Xuzhou, China
- 2Imaging Department, The Second Affiliated Hospital of Shandong First Medical University, Taian, China
- 3Nanjing Drum Tower Hospital Group Suqian Hospital, The Affiliate Suqian Hospital of Xuzhou Medical University, Suqian, China
- 4Department of Infectious Diseases, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, China
- 5National Demonstration Center for Experimental Basic Medical Sciences Education, Xuzhou Medical University, Xuzhou, China
As a small DNA virus, hepatitis B virus (HBV) plays a pivotal role in the development of various liver diseases, including hepatitis, cirrhosis, and liver cancer. Among the molecules encoded by this virus, the HBV X protein (HBX) is a viral transactivator that plays a vital role in HBV replication and virus-associated diseases. Accumulating evidence so far indicates that pattern recognition receptors (PRRs) are at the front-line of the host defense responses to restrict the virus by inducing the expression of interferons and various inflammatory factors. However, depending on HBX, the virus can control PRR signaling by modulating the expression and activity of essential molecules involved in the toll-like receptor (TLR), retinoic acid inducible gene I (RIG-I)-like receptor (RLR), and NOD-like receptor (NLR) signaling pathways, to not only facilitate HBV replication, but also promote the development of viral diseases. In this review, we provide an overview of the mechanisms that are linked to the regulation of PRR signaling mediated by HBX to inhibit innate immunity, regulation of viral propagation, virus-induced inflammation, and hepatocarcinogenesis. Given the importance of PRRs in the control of HBV replication, we propose that a comprehensive understanding of the modulation of cellular factors involved in PRR signaling induced by the viral protein may open new avenues for the treatment of HBV infection.
Introduction
Although the hepatitis B virus (HBV), a small DNA virus, has been known for more than 50 years, chronic infection caused by this virus remains a global cause of hepatitis, cirrhosis, and liver cancer, especially hepatocellular carcinoma (HCC) (1–3). It has been widely demonstrated that after recognition by pattern recognition receptors (PRRs), the virus can be restrained by the innate immune system (4–7). To date, four types of PRRs, including toll-like receptors (TLRs), cytosolic DNA sensors, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and NOD-like receptors (NLRs), have been discovered (8–10). Generally, once sensitized by the virus, these PRRs can initiate the activation of multiple intracellular signaling pathways to produce interferons (IFNs), which further stimulate the sensitization of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling to participate in the inherent antiviral immune response. Additionally, these PRRs can stimulate the production of inflammatory cytokines induced by the nuclear factor κB (NF-κB) pathway to resist the virus (11, 12). However, accumulating evidence suggests that to maintain persistent infection, the virus has evolved a variety of strategies to overcome the host antiviral responses mediated by PRR components, such as TLRs, cytosolic DNA sensors, RLRs, and NLRs, as well as their downstream pathways, including the JAK-STAT and NF-κB pathways, to facilitate viral replication and liver pathogenesis (13–16). Moreover, the exact mechanisms by which this virus evades the immune response mediated by PRRs to facilitate its persistent infection and development of different diseases remain elusive.
HBV genome contains four overlapping open reading frames (ORFs), namely S, P, C, and X. S ORF contributes to the production of large, middle, and small envelope proteins that are composed of HBsAg, preS1, or preS2 antigens; P and C ORFs encode the HBV polymerase protein, core protein, and HBeAg. Additionally, X ORF is responsible for the expression of HBX, a highly conserved nonstructural protein with 154-amino acids (17–19). Based on the studies on cell and mouse models, HBX is considered to be important for initiating and maintaining HBV replication (20, 21). Mechanistically, the viral protein in the cell cytoplasm can stimulate signal transduction pathways to facilitate HBV replication. In the nucleus, HBX can bind to the HBV covalently closed circular DNA (cccDNA) and activate the transcription of viral promoters with the help of different transcription factors and epigenetic regulatory molecules (22–24). At different stages of HBV infection, the viral protein performs various biological functions (22), participate in the growth, migration, autophagy, apoptosis, and epigenetic regulation of virus-infected hepatocytes, and play prominent roles in the development of different liver diseases, especially HCC (25–28). More importantly, there is growing evidence that HBX can regulate the expression and activity of numerous molecules in different PRR subfamilies, including TLRs, RLRs, and NLRs (29–32). In the present review, we summarize the current research on the modulation of PRR signaling induced by HBX to regulate the innate immune responses that aid in HBV replication and various biological functions that facilitate the development of HCC.
Effect of HBX on TLR-mediated Signaling Pathways
TLRs are a large group of conserved type I transmembrane molecules. So far, 10 humans TLRs have been discovered that identify specific pathogen-associated molecular patterns (PAMPs) (33, 34). Among the identified TLRs, TLR1-2 and TLR4-6 are expressed on the cell surface, whereas TLR3 and TLR7-9 are situated on the endosomal surface. Upon activation, the TLRs (except TLR3) can activate the adaptor protein, myeloid differentiation primary response 88 (MyD88), to enhance the sensitization of the tumor necrosis factor (TNF) receptor-associated factor (TRAF) 6, which further promotes phosphorylation of the transforming growth factor-activated kinase-1 (TAK1). Next, TAK1 activates NF-κB signaling and then allows NF-κB to translocate into the nucleus to initiate the gene expression of inflammatory factors. Additionally, TAK1 activation also prompts the sensitization of mitogen-activated protein kinases (MAPK), which can cause the recruitment of the transcription factor activator protein 1 (AP-1) to upregulate the induction of inflammatory cytokines. TLR3 is unique and uses MyD88-independent signaling pathways to initiate the immune response. After TLR3 activation is triggered by double-stranded RNA (dsRNA), it can sensitize toll-interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF) to activate the protein complex composed of TRAF3 and TANK-binding kinase 1 (TBK1). These molecules activate IFN regulatory factor (IRF)-3/7 and NF-κB. TLR3 also recruits TRAF6 to phosphorylate TAK1. TAK1 sensitizes AP-1 via the activation of the MAPK pathway and activate NF-κB via the IKK-IκB complex to modulate the inflammatory reaction (33–38).
Accumulating data shows that initiating the host TLR response is a new therapeutic strategy for HBV infection (36, 37). Several studies have demonstrated that the stimulation of TLR2-5, TLR7, and TLR9 with their specific ligands can inhibit HBV replication in the cell and animal models (38). Furthermore, the potential of TLR agonists, including GS-9620, RO7020531, JNJ-64794964 (TLR7 agonists), GS-9688 (TLR8 agonist), and AIC649 (TLR9 agonist), has been investigated in clinical trials at different stages (38, 39). HBV is considered as a “stealth virus,” and the suppression of TLR signaling molecules has been reported in different kinds of cells in patients with HBV infection (40, 41). For example, decreased expression levels of TLR2 and TLR9 have been reported in the peripheral monocytes of patients with chronic hepatitis B (CHB) (42, 43). Suppressed TLR signaling molecules, such as TLR3 (44), TLR7, TLR9 (45), TRAF3, IRAK4, and IRF7 (46), were observed in the peripheral blood mononuclear cells (PBMCs) of patients with chronic HBV infection. Additionally, downregulation of TLR7 and TLR9 was also observed in the plasmacytoid dendritic cells (pDCs) in HBV-infected patients (47–49).
Furthermore, several studies have shown that multiple virus-encoded proteins, including HBsAg, HBeAg, and HBV polymerase, contribute to HBV persistence by inhibiting TLR-mediated antiviral responses (38). For instance, HBsAg can inhibit TLR3-mediated immune response in murine Kupffer cells and sinusoidal endothelial cells (50). HBeAg has been shown to restrict TLR2 expression and interact with the TIR proteins, TRAM and Mal, to suppress TLR-mediated immune responses in hepatocytes (51, 52). Meanwhile, HBV polymerase suppresses TLR3-mediated induction of IFN-β in hepatocytes by interfering with IRF3 activation (53) and inhibits MyD88 expression by blocking the nuclear translocation of STAT1 (54). Additionally, HBsAg and HBeAg can control the major vault protein (MVP)-induced IFN production by suppressing the interaction between MVP and MyD88 in liver cells (55).
In addition to viral replication, molecules involved in TLR signaling also contribute to the pathological changes in the liver caused by the virus. For example, the virus can activate B cells via the TLR2 signaling pathway, which may be associated with the activation of antiviral responses mediated by B cells in patients with CHB (56). Among the proteins encoded by the virus, HBeAg sensitizes macrophages via the TLR2 signaling pathway to exacerbate hepatic fibrosis (57). Depending on the TLR2 signaling pathway, the HBV core protein promotes the production of the inflammatory cytokines, IL-6 and TNF-α, from M2 macrophages (58). After HBeAg stimulation, upregulation of TLR4 was also observed in the monocytes of patients with CHB. Moreover, overexpression of TLR4 on monocytes mediated by HBeAg may regulate the activity of regulatory T cells, which is related to the immunotolerance of the virus infection (59). Additionally, HBsAg can enhance the invasion of HBV-associated HCC cells by upregulating TLR2 (60). HBsAg also inhibits the production of IFN-α by pDCs by decreasing TLR9 expression in pDCs, and this effect may be related to the reduced capacity antiviral immune response of pDCs in patients with CHB (61).
HBX has a critical role in suppressing host innate immune response by disrupting TLR signaling to regulate viral replication. Moreover, the molecules regulated by HBX in TLR signaling contribute to virus-mediated inflammation and hepatocarcinogenesis. Among the identified TLRs, HBX was found to upregulate TLR4 in immortalized proximal tubule epithelial cells (Figure 1), which may be associated with the dysregulated expression of cytokines, including IL-6, TNF-α, IFN-γ, and IL-4, mediated by the viral protein (62). TLR4 is upregulated in HBV-related hepatoma cells, and promotes their growth, while inhibiting the apoptosis of these cells, by activating the extracellular signal-regulated kinase (ERK)-1/2 signaling pathway (29). HBX can interact with TLR4 in HBV-related hepatoma cells, and the physical interaction of HBX with TLR4 may contribute to the activation of ERK1/2 in hepatoma cells. To date, the effect of HBX-mediated TLR4 on HBV replication has not been well investigated. However, in response to TLR4 stimulation, HBX facilitates the migration of liver cancer cells by enhancing the interaction of vacuolar protein sorting 34 (VPS34) with the TRAF6-Beclin 1 (BECN1) complex (63), thereby increasing BECN1 ubiquitination and autophagy, a physiological process that contributes to HBV replication (64).
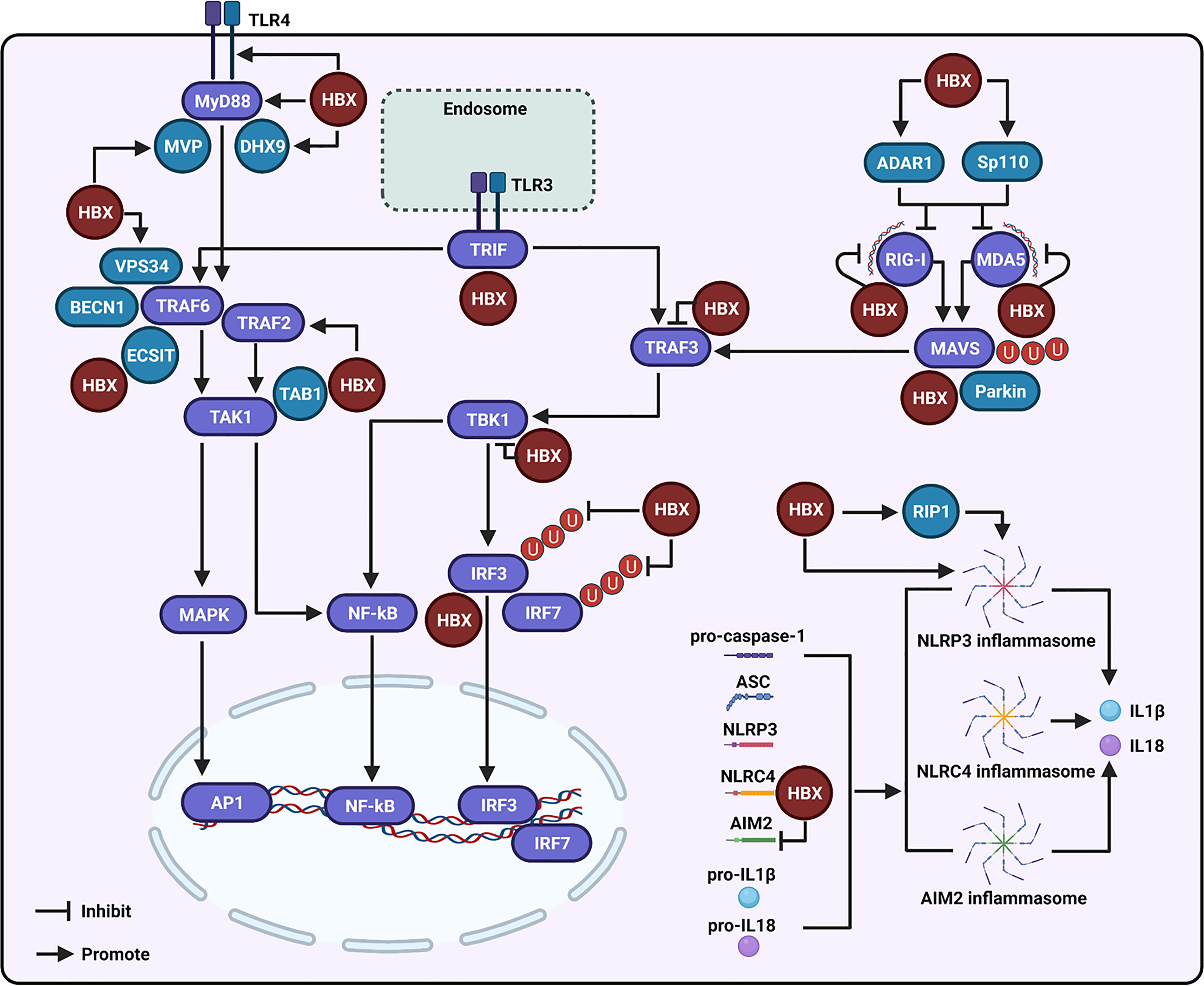
Figure 1 Regulation of the hepatitis B virus X protein (HBX) on the toll-like receptor (TLR), retinoic acid inducible gene I (RIG-I)-like receptor (RLR), and NOD-like receptor (NLR) signaling-associated molecules. During the modulation of TLR signaling mediated by HBX, the viral protein can promote the expression of TLR4 and myeloid differentiation primary response 88 (MyD88), and enhance the levels of the major vault protein (MVP) and DExH-box RNA helicase 9 (DHX9), which may further interact with MyD88 to regulate the innate immune response. HBX can activate the tumor necrosis factor (TNF) receptor-associated factor (TRAF)-2/transforming growth factor-activated kinase-1 (TAK1) signaling pathway. HBX is also able to interact with TAB1, and the interaction may affect the function of TAK1. The viral protein is capable of interacting with the evolutionarily conserved signaling intermediate in Toll pathway (ECSIT), which is a partner of TRAF6. The viral protein also contributes to the interaction of vacuolar protein sorting 34 (VPS34) with Beclin 1 (BECN1) and TRAF6. HBX interacts with toll-interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β (TRIF), a downstream molecule of TLR3. HBX can bind to RIG-I, melanoma differentiation-associated 5 (MDA5), and mitochondrial antiviral signaling (MAVS) to inhibit RLR signaling. Adenosine deaminase acting on RNA 1 (ADAR1) and speckled at 110 kDa (Sp110) may participate in the inhibition of RIG-I and MDA5 mediated by HBX. Parkin is involved in the degradation of MAVS induced by HBX. Besides these, HBX also interacts with TRAF3 and TBK1 to inhibit their activities. HBX can bind to IRF3 and inhibit the ubiquitination of IRF3 and IRF7 at the lysine 63 sites to suppress their activation. During the modulation of NLR signaling, HBX can activate the NLR family pyrin domain-containing 3 (NLRP-3) inflammasome. RIP1 may be involved in the activation of NLRP3 inflammasome mediated by HBX. Moreover, HBX can interact with the NLR with CARD domain-containing 4 (NLRC4) and through this interaction, HBX may influence the NLRC4 inflammasome. The viral protein can also inhibit the expression of absent-in-melanoma-2 (AIM2) and may further affect the AIM2 inflammasome.
MyD88 can accelerate the degradation of HBV pre-genomic RNA to restrain its replication (65, 66). HBX can promote the expression of MyD88 at the transcriptional level in both liver and hepatoma cells. To date, the effect of MyD88 on HBX-mediated HBV replication has not been assessed. However, the role and associated molecular mechanisms of viral proteins in the regulation of MyD88 and its related proteins in TLR signaling to facilitate HCC development have been investigated. In particular, HBX activates the downstream signaling molecules of MyD88, including IRAK1, ERK/p38, and NF-κB, to induce the production of IL-6, a major inflammatory cytokine that facilitates the development of HCC (67). However, Wu et al. suggested that HBX was able to inhibit MyD88 promoter activity in hepatocytes during IFN-α stimulation (54). The reasons for the inconsistent results regarding the effect of HBX on the expression of MyD88 with or without exogenous stimulation should be further assessed in future studies.
MVP interacts with MyD88 to stimulate IFN production (3, 55). Liu et al. observed high levels of MVP in the liver tissues of HBV-infected patients. Researchers have also elucidated that HBX can stimulate MVP promoter activity to enhance its expression in hepatocytes. Functionally, Yu et al. found that MVP is involved in the proliferation, migration, and invasion of cells mediated by HBX, by sequestering IRF2 and enhancing the HDM2-dependent loss of P53 (68). In addition to MVP, DExH-box RNA helicase 9 (DHX9) directly binds to MyD88 to facilitate IFN production (69). A recent study showed that HBX can enhance the expression of DHX9 by inhibiting its degradation, which is regulated by MDM2. Moreover, the interaction between DHX9 and Nup98 contributes to HBX-mediated HBV replication (70). In addition, HBX is able to interact with DHX9 to downregulate the circular RNA circSFMBT2 and then release miR-665 to suppress TIMP3 expression and enhance HCC metastasis (71). However, the importance of DHX9 in modulating the HBX-mediated innate immune response has not been well investigated.
In TLR signaling, stimulation of MyD88 can induce the activation of TAK1 to initiate the inflammatory reaction and IFN production (33–35). Zhou et al. showed that HBX is capable of inducing the activation of TAK1 through TRAF2 to stimulate NF-κB and cause the upregulation of IP-10, and TRAF2 may participate in the regulation of TAK1 mediated by MyD88. Furthermore, HBX-mediated increase in IP-10 is associated with the migration of leukocytes, which may cause pathological immune injury of the liver during HBV infection (72). Activation of TAK1 is dependent on its interaction with its binding partners, TAK1-binding proteins (TAB1, TAB2, and TAB3). Based on immunoprecipitation and mass spectrometric analyses, HBX interacts with TAB1 (73). However, based on this interaction, whether HBX could regulate the function of TAK1 and then modulate the innate immune response has not been assessed so far.
Activation of TRAF6 requires oligomerization, and HBX significantly enhances TRAF6 activation by promoting its oligomerization. Furthermore, TRAF6 oligomerization facilitates its interaction with histone deacetylase 3 (HDAC3) and then promotes gene expression and protein stability of c-Myc to promote hepatocarcinogenesis (74). The evolutionarily conserved signaling intermediate in Toll pathway (ECSIT) is a partner of TRAF6 and activates both NF-κB and AP-1. Chen et al. revealed that HBX could interact with ECSIT to increase NF-κB activation, leading to the induction of IL-10, an inflammation-related cytokine (75). In addition, TRIF is a vital adaptor protein that initiates TLR3-mediated innate immune signaling. Hong et al. showed that HBX can enhance TRIF protein degradation to evade the innate immune response and facilitate HBV replication (76). Taken together, these studies suggest that HBX can regulate the expression of multiple molecules in TLR signaling to inhibit the immune response and facilitate viral replication. Moreover, the viral protein is also capable of utilizing molecules in the TLR signaling pathway to regulate inflammation and enhance the development of HCC.
Role of HBX in RLR-mediated Signaling Pathways
The DExD/H box RNA helicases, RIG-I and melanoma differentiation-associated gene 5 (MDA5), participate in the activation of RLR signaling (77, 78). These two molecules recognize dsRNAs from various viruses in the cytoplasm. Once RIG-I and MDA5 are sensitized by dsRNA, they can activate mitochondrial antiviral signaling (MAVS), which occurs on the mitochondrial membrane. MAVS further initiates TRAF3 activation. TRAF3 sensitizes TBK1 to activate IRF3/7 and leads to the production of IFN (8, 79).
Current evidence shows that RIG-I can recognize HBV pregenomic RNA and suppress HBV cccDNA, thereby activating the innate immune response to inhibit its replication (80–82). Similar to TLR, drugs activating RLR signaling also underwent a phase II trial to investigate the therapeutic effects of PLR agonists on HBV inhibition in clinical settings (13). It was found that the virus could escape immune reactions by regulating RLR signaling (13). For example, no significant increase in RIG-I and MDA5 levels was found in the liver tissues of patients with CHB (83). To benefit from persistent HBV infection, the virus uses N6-methyladenosine modification to block the recognition of viral RNA mediated by RIG-I signaling (84). The virus also restricts immune signaling mediated by RIG-I by inducing miR146a (85). Besides these, based on its polymerase protein, HBV can inhibit RIG-I-mediated IFN induction by inhibiting the interaction between TBK1 and DDX3 (53).
HBX plays a central role in the regulation of viral replication by dysregulating RLR signaling. For example, to facilitate HBV replication, HBX can suppress RIG-I, MDA5, and MAVS-mediated activation of IFN-β promoters (31, 86) (Figure 1). Furthermore, residues Asn118 and Glu119 of HBX were found to be critical for the viral protein-mediated suppression of RIG-I-MAVS signaling (87). Mechanistically, HBX binds to RIG-I and MDA5 (31, 86). Similarly, HBX also interacts with MAVS and disrupts the interactions between MAVS and other proteins within the MAVS-associated complex, including RIG-I, MDA5, and TBK1, to suppress IFN-β promoter sensitization (31). Furthermore, the interaction of HBX with MAVS promoted protein degradation to block IFN-β production (88). In addition, HBX-induced Parkin is capable of binding to the accumulating unanchored linear polyubiquitin chains on MAVS via the linear ubiquitin assembly complex (LUBAC) to disrupt MAVS signalosome and abate IRF3 sensitization (89).
Studies shown that adenosine deaminases acting on RNA 1 (ADAR1), RNA-editing enzymes that convert adenosine to inosine in duplex RNA regions, are vital cellular factors controlling the innate immune response mediated by endogenous RNAs. Relying on the transcription factor YY1, HBX was observed to accelerate ADAR1 expression in a dose-dependent manner to block the transcriptional levels of MDA5 and RIG-I, and inhibit the recognition of HBV RNA mediated by these two molecules in hepatocytes (90). The transcription factor, speckled at 110 kDa (Sp110), can control the levels of RIG-I and MDA5. HBX interacts with Sp110 and may modulate the production of RIG-I and MDA5 to control the innate immune response (91). In addition to RIG-I, MDA5, and MAVS, HBX interacts with TRAF3, TRIF, TBK1, and IRF3 to inhibit IFN induction (86). Furthermore, HBX acts as a deubiquitinating enzyme to suppress IRF3 and IRF7 ubiquitination with lysine 63-linked chains and attenuates their activities (86). The expression levels of TBK1 are increased in the liver cancer tissue samples. In particular, HBX upregulates the expression of TBK1 to enhance NF-κB activation (92). However, the biological role of HBX-mediated TBK1 has not yet been investigated.
Function of HBX in NLR-mediated Signaling Pathways
The NLR family contains a variety of cytoplasmic sensors, such as the NLR family pyrin domain-containing (NLRP)-1, NLR with CARD domain-containing 4 (NLRC4), and NLRP3 (93). Among all the NLR molecules discovered, the NLRP3-inflammasome has been extensively studied in recent years. It can be activated by mitochondrial dysfunction, ionic influx, reactive oxygen species (ROS) production, and stimuli from pathogens or damaged cells (94). In general, sensitization of the NRLP3 inflammasome involves two steps. In the first step, dependent on NF-κB signaling, the production of NLRP3, pro-IL1β, and pro-IL18 is induced by other PRRs. In the second step, NLRP3 recruits the apoptosis-associated speck-like protein (ASC), which results in the formation of ASC prion-like oligomer that further binds to pro-caspase-1 and lead to its activation. Sequentially, activated pro-caspase-1 cleaves pro-IL1β and pro-IL18 into IL1β and IL18, respectively, to initiate the innate immune response and inflammatory reaction (8, 94).
NLRP3 plays a vital role in the inflammation caused by the virus. For example, it has been found that the levels of NLRP3 are low in normal liver tissues; however, activation of the NLRP3 inflammasome is implicated in hepatic injury. The NLRP3 inflammasome mediates liver failure by sensitizing procaspase-1 and pro-IL-1 β in HBV-related acute-on-chronic liver failure (ACLF) (95, 96). To date, the molecular mechanisms associated with virus-mediated sensitization of the NLRP3 inflammasome have not been well examined. However, a recent study by Ding et al. showed that the HBV core protein is capable of enhancing the lipopolysaccharide-induced activation of the NLRP3 inflammasome and promoting IL-1β production to cause liver inflammation (97). However, dependent on HBeAg, the virus is discovered to suppress LPS-induced activation of NLRP3 inflammasome and IL-1β production via inhibiting the sensitization of NF-κB pathway and the production of reactive oxygen species. The inhibition of NLRP3 inflammasome mediated by HBeAg may be responsible for HBV-mediated restrain of innate immune response (98).
Dependent on the activation of the NLRP3 inflammasome, HBX is found to promote hepatocyte pyroptosis by triggering the production of ASC, IL-1β, IL-18, and HMGB1 in hydrogen peroxide-stimulated hepatocytes (32) (Figure 1). Additionally, NLRP3 inflammasome sensitization stimulated by the viral protein may be related to liver inflammation caused by viral infection. Current evidence indicates that RIP1 contributes to the regulation of IL-1β maturation by activating the NLRP3 inflammasome. Xie et al. found that the overexpression of HBX increased the expression of RIP1 in hepatocytes. Mechanistically, AFB1 participates in the HBX-induced upregulation of RIP1 (99, 100). Although RIP1 contributes to the activation of AP-1 and NF-κB to induce inflammation in HBX-positive cells, its effect on NLRP3 inflammasome sensitization mediated by HBX has not yet been assessed; therefore, further investigation is required to assess the effect of RIP1 on the regulation of inflammasome activation. In addition to NLRP3, NLRC4 was also found to interact with HBX based on substrate-trapping proteomics analysis (101). However, whether HBX affects NLRC4-associated inflammasomes has not yet been well assessed.
Similar to NLRP3, the absent-in-melanoma-2 (AIM2) inflammasome can recognize cytoplasmic DNA, resulting in the production of IL-1β and IL-18 and the induction of target cell pyroptosis (102). Although the mRNA expression levels of AIM2 are upregulated in PBMCs of patients with acute and chronic hepatitis B (103), Chen et al. found that in HBV-related HCC cells, HBX not only suppressed the expression of AIM2 at the gene level by promoting the stability of the enhancer of zeste homolog 2 (EZH2), but also interacted with AIM2, resulting in AIM2 degradation via the ubiquitin-proteasome pathway. Functionally, knockdown of AIM2 enhances HBX-mediated migration and metastasis of hepatoma cells (104). Nevertheless, based on the current studies, whether HBX can influence the inflammasomes via AIM2 remains unclear. Moreover, the effect of HBX on the inhibition of AIM2-associated inflammasomes needs to be explored further in future studies.
Role of HBX in the JAK-STAT Pathway
JAK-STAT signaling is a vital downstream pathway of IFN receptors that stimulates IFN-stimulated gene (ISG) production (8, 105). The JAK family comprises of JAK1-3 and TYK2. The STAT family has seven members, including STAT1-4, STAT5a, STAT5b, and STAT6 (106). JAK-STAT signaling is known to play a critical role in IFN-mediated inhibition of HBV replication (3). To facilitate the persistence of HBV infection, the virus can promote MMP-9 and CTHRC1 expression to repress JAK-STAT signaling (107, 108). HBeAg can also inhibit JAK-STAT signaling to enhance HBV replication (109).
The JAK-STAT pathway is a highly conserved signaling pathway that can affect various biological processes including immune response, apoptosis, inflammation, tissue repair, and adipogenesis (110). Currently, the link between HBX and sensitization of JAK-STAT signaling has been well established (Table 1). Current evidence shows that the regulation of JAK-STAT signaling activation mediated by HBX plays a vital role in not only regulating cellular proliferation, apoptosis, epithelial-mesenchymal transition (EMT), and migration, but also in modulating the innate immune response and viral replication. HBX regulates JAK1, JAK2, and TYK2. For example, HBX interacts with JAK1 to facilitate its activation (111). Furthermore, HBX-mediated JAK1 activation is associated with sensitization of the Ras-Raf1 signaling axis (112). In addition to JAK1, HBX also contributes to the activation of JAK2 to induce apoptosis of renal tubular epithelial cells (113). However, Cho et al. showed that HBX can inhibit the activation of TYK2 to decrease the expression of IFN-α receptor 1 (IFNAR1) to inhibit extracellular IFN-mediated signal transduction (114).
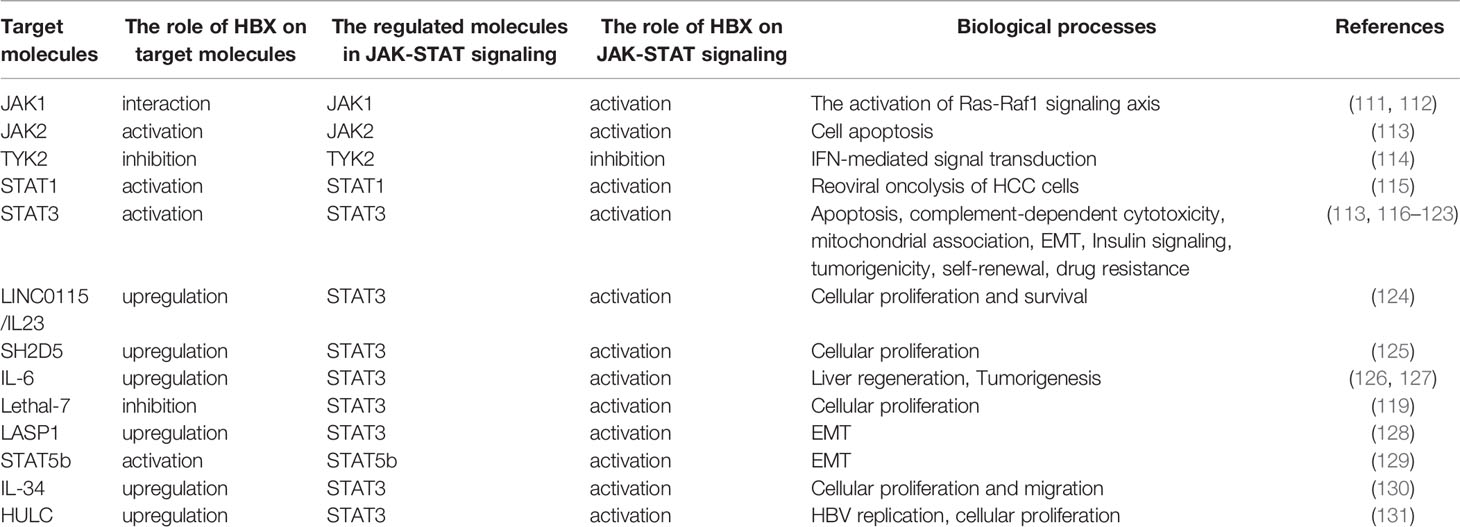
Table 1 The detailed information on the regulation of JAK-STAT signaling mediated by HBX.
HBX also plays a vital role in STAT activation. Especially, among the STAT molecules, the effect of HBX on the sensitization of STAT3 has been wildly investigated. For example, viral proteins inhibit reoviral oncolysis of hepatoma cells by activating STAT1 (115). In human renal proximal tubular epithelial cells, HBX modulates apoptosis by activating the STAT3 signaling pathway (113). Additionally, HBX decreases nephrin expression and induces podocyte apoptosis by activating STAT3 (116). However, a recent study revealed that depending on STAT3, HBX protects hepatoma cells and hepatocytes from complement-dependent cytotoxicity by increasing the membrane-bound complement regulatory protein CD46 (117). In particular, HBX-mediated STAT3 activation may be associated with oxidative stress in HBV-associated hepatoma cells (118). In addition, HBX can increase the transcription of LncRNA LINC01152 to enhance IL-23 expression and then initiate the activation of STAT3 to promote the proliferation and survival of hepatoma cells (124). In HCC cells, HBX was also found to trigger SH2D5 expression, and based on the HBX-mediated interaction of SH2 domain-containing 5 (SH2D5) with transketolase (TKT), STAT3 can be activated to promote HCC cell proliferation (125). IL-6 plays a vital role in the activation of STAT3. Current research indicates that HBX can promote IL-6 expression in hepatoma cells (126). In response to IL-6, HBX recovers the dephosphorylation of STAT3 mediated by PP2Cα, a protein that interacts with HBX to facilitate hepatocarcinogenesis (127). Additionally, downregulation of miRNA Lethal-7 mediated by HBX also activates STAT3 to regulate cellular proliferation (119).
EMT has been implicated in HCC development. It has been demonstrated that HBX contributes to the activation of STAT3 to control the EMT of hepatoma cells (120). Depending on STAT3, HBX promotes the expression of HMGB1 to enhance EMT in liver cancer cells (121). Additionally, our results have shown that HBX can activate STAT3 through LASP1 to facilitate vimentin expression and enhance EMT (128). Similar to STAT3, STAT5b participates in the induction of EMT mediated by HBX (129).
Suppressor of cytokine signaling (SOCS) mediates insulin resistance in the liver. Kim et al. found that HBX could induce SOCS3 expression via STAT3 to impair hepatic insulin signaling (122). Up to now, more and more evidence has demonstrated that C-terminally truncated HBX contributes to the development of HCC (2). In particular, Ching et al. found that relying on STAT3, C-terminal truncated HBX can regulate tumorigenicity, self-renewal, and drug resistance (123). Additionally, our previous study showed that HBX was capable of promoting the levels of IL-34 to activate STAT3 and further facilitate the growth and migration of liver cancer cells (130).
Interestingly, although JAK-STAT signaling participates in the inhibition of HBV replication, the current study showed that STAT3 also contributes to HBX-mediated HBV replication. Long noncoding RNA (lncRNA) is highly upregulated in liver cancer (HULC) and has been identified to be significantly upregulated in HCC. Liu et al. found that HULC could elevate the expression of HBX, which in turn sensitizes STAT3 to stimulate the miR-539 promoter. miR-539 decreased the expression of APOBEC3B and then enhanced HBV replication. Furthermore, HULC mediated by HBX can also enhance the proliferation of hepatoma cells in in vitro and in vivo models (131).
Influence of HBX on the NF-κB Pathway
As mentioned above, NF-κB contributes to the innate immune responses induced by different PRRs. In the cytoplasm, IκB-α and IκB-β bind to NF-κB p50/p65 and form an inactivated protein complex. Depending on the upstream signal transduction from the IKK complex, which is composed of IKKα, IKKβ, and IKKγ, IκB proteins are degraded to free NF-κB p65/p50. Next, NF-κB p65/p50 is transported into the nucleus, resulting in the transcription of different inflammatory cytokines (132, 133).
The published reports have suggested that the activation of NF-κB facilitates the inhibition of HBV replication (134–136). To accelerate replication, the virus promotes fibronectin expression to restrain the activation of NF-κB and sensitize HBV enhancers (137). Wang et al. showed that HBeAg can interrupt the ubiquitination of NEMO to suppress NF-κB activity and enhance HBV replication (138). HBeAg also suppresses NF-κB signaling mediated by IL-18 and IL-1β in natural killer (NK) cells and hepatocytes, and the inhibition of NF-κB signaling may contribute to the maintenance of persistent HBV infection (139, 140). However, current evidence from other groups has shown that NF-κB signaling also facilitates HBX-mediated HBV replication. For instance, HBX can promote HBV replication by inhibiting the miR-192-3p-XIAP axis to activate the NF-κB pathway (141). Xu et al. found that HBX can activate NF-κB to promote the expression of IFIT3 and then enhance viral replication (142). HBX increases gp96 expression via NF-κB signaling to facilitate HBV replication (143). HBX upregulates miR-146a-5p via NF-κB to increase autophagy and enhance HBV replication (144) (Table 2). To date, the reasons for the contradictory function of NF-κB signaling in viral replication in different studies are unknown and need to be explored in the future.

Table 2 The detailed information related to the modulation of NF-κB signaling mediated by HBX.
In addition to viral replication, many studies have suggested that sensitization of NF-κB induced by HBX plays a vital role in cellular growth, apoptosis, EMT, and migration of hepatoma cells (156, 157). Mechanistically, HBX can interact with NF-κB signaling molecules to initiate the signal transduction activation (Table 2). For example, HBX can inhibit IkBα to induce sustained activation of this signaling pathway (145). In addition, HBX can bind to NF-κB (p65) to facilitate its activation and nuclear localization and induce the expression of metastasis-associated protein 1 (MTA1), which is involved in inflammation and tumorigenesis (146). Furthermore, the activation of NF-κB signaling induced by HBX is mediated by the interactions between HBX and IKKγ (86, 147). In addition, HBX can enhance the expression of IKKα and modulate the activity of IKKβ to activate NF-κB signaling to facilitate inflammatory cytokine production and cell proliferation (148, 149).
In addition, accumulating evidence has also verified that multiple molecules participate in NF-κB activation initiated by HBX (Table 2). HBX enhances NF-κB activation by interacting with ECSIT (75). HBX can directly interact with VHL-binding protein (VBP1) and synergistically promote NF-κB activation to facilitate cellular proliferation (150). Viral proteins can enhance the activation of NF-κB through interactions with the valosin-containing protein (VCP) (151). In addition, HBX binds to amplification in breast cancer 1 (AIB1) protein and stabilizes the protein to enhance the sensitization of NF-κB signaling (152). Additionally, the reports from Lim et al. showed that dependent on the chaperoning activity, ribosomal protein S3α (RPS3α) can stabilize HBX and enhance the activation of NF-κB signaling (158).
In recent years, increasing evidence has shown that HBX can influence various signaling pathways to initiate hepatocarcinogenesis. Interestingly, HBX can activate NF-κB through a variety of distinct signaling pathways involving PI3-K (153, 154), and ERK (155), contributing to the regulation of motility and inflammatory responses in HCC cells. Autophagy is a vital metabolic process that is dependent on the degradation of damaged proteins and organelles in lysosomes and can maintain homeostasis of the intracellular environment. Luo et al. suggested that HBX can induce autophagy to stimulate the activation of NF-κB in hepatocytes (148).
Conclusions
Although the innate immune system plays a central role in targeting HBV, the virus triggers only a small innate response and has evolved a variety of escape strategies to block the antiviral response in host cells. As a viral nonstructural protein, HBX is considered a vital therapeutic target for HBV infection because the viral protein not only contributes to the replication of the virus and participates in the development of liver cancer induced by HBV, but also protects HBV-infected cells from immune-mediated clearance (22, 159). In the present review, we summarized the recent progress regarding the role and associated mechanisms of HBX in PPR signaling during HBV infection. Our reviewed studies indicate that HBX can regulate the expression and function of various vital molecules in TLR, RLR, and NLR signaling, as well as their downstream pathways, including the JAK-STAT and NF-κB pathways. Interestingly, the effect of molecules in these signaling pathways mediated by HBX is different during HBV infection. First, many molecules involved in TLR signaling can be regulated by HBX to inhibit the immune response, aid in viral replication, and regulate a variety of biological processes to modulate inflammation and facilitate the development of HCC. Second, regulation of RLR signaling mediated by HBX can inhibit the immune response to benefit viral replication. Third, HBX may promote the activation of NLR signaling to cause inflammation during HBV infection. Fourth, the regulation of the JAK-STAT and NF-κB pathways stimulated by viral proteins controls viral replication and modulates different biological processes to accelerate the development of HCC. In particular, current data show that targeting HBX could suppress viral replication by enhancing the immune response mediated by PRR signaling in HBV-expressing liver cells (160–162). Additionally, inhibition of innate immune-related molecules regulated by HBX also blocks HCC development (29). Therefore, a deeper understanding of the regulation of PRR signaling by HBX may facilitate the treatment of HBV infections and related illnesses.
Cyclic GMP-AMP synthase (cGAS) and DNA-dependent activator of IFN regulatory factors (DAI) participate in DNA-dependent immune responses (163, 164). In particular, after cytoplasmic DNA is activated by cGAS or DAI, these molecules can induce the activation of the stimulator of interferon genes (STING) to further induce the expression of IFN and inflammatory cytokines (8, 165). It has been found that the cGAS-STING pathway facilitates the inhibition of HBV replication and attenuates hepatocyte injury and fibrosis induced by the virus (166–168). However, the expression levels of cGAS and its effector genes have been shown to decline in hepatocytes infected with HBV (169). Additionally, STING expression levels in peripheral monocytes were dramatically decreased in patients with CHB (170). To maintain persistent infection, the virus can evade the antiviral activity of the cGAS-STING pathway by various strategies (166, 171). For example, HBV polymerase protein can decrease the ubiquitination of STING and inhibit the production of IFN by interacting with STING (172). HBsAg can inhibit STING expression and suppress the immune response of NK cells (173). HBX has also been found to restrain the dsDNA-induced immune response (30) and suppress STING-mediated activation of IFN-β (31). However, the cellular factors that contribute to the suppression of STING-associated signaling mediated by HBX have not yet been well elucidated. Given the importance of STING-mediated immune response in the restriction of HBV (171), further studies are needed to assess the effect of regulation of HBX on the immune response mediated by STING in the future.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
The study was supported by Xuzhou Technology Bureau Foundation (KC21065), the Natural Science Foundation of Jiangsu Province (BK20211347), the Natural Science Foundation of the Jiangsu Higher Education Institutions (21KJA310004), Suqain Sci&Tech Program (K202015), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The figure used in this review was created by BioRender (https://biorender.com/).
References
1. Liu B, Wen X, Huang C, Wei Y. Unraveling the Complexity of Hepatitis B Virus: From Molecular Understanding to Therapeutic Strategy in 50 Years. Int J Biochem Cell Biol (2013) 45:1987–96. doi: 10.1016/j.biocel.2013.06.017
2. Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, Fatima K, et al. Hepatitis B Virus, HBx Mutants and Their Role in Hepatocellular Carcinoma. World J Gastroenterol (2014) 20:10238–48. doi: 10.3748/wjg.v20.i30.10238
3. Tan G, Song H, Xu F, Cheng G. When Hepatitis B Virus Meets Interferons. Front Microbiol (2018) 9:1611. doi: 10.3389/fmicb.2018.01611
4. Ye J, Chen J. Interferon and Hepatitis B: Current and Future Perspectives. Front Immunol (2021) 12:733364. doi: 10.3389/fimmu.2021.733364
5. Gehring AJ, Protzer U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology (2019) 156:325–37. doi: 10.1053/j.gastro.2018.10.032
6. Rehermann B, Thimme R. Insights From Antiviral Therapy Into Immune Responses to Hepatitis B and C Virus Infection. Gastroenterology (2019) 156:369–83. doi: 10.1053/j.gastro.2018.08.061
7. Hennessy C, McKernan DP. Anti-Viral Pattern Recognition Receptors as Therapeutic Targets. Cells (2021) 10:2258. doi: 10.3390/cells10092258
8. Kong F, You H, Zheng K, Tang R, Zheng C. The Crosstalk Between Pattern-Recognition Receptor Signaling and Calcium Signaling. Int J Biol Macromol (2021) 192:745–56. doi: 10.1016/j.ijbiomac.2021.10.014
9. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
10. Bowie AG, Unterholzner L. Viral Evasion and Subversion of Pattern-Recognition Receptor Signalling. Nat Rev Immunol (2008) 8:911–22. doi: 10.1038/nri2436
11. Nosratabadi R, Alavian SM, Zare-Bidaki M, Shahrokhi VM, Arababadi MK. Innate Immunity Related Pathogen Recognition Receptors and Chronic Hepatitis B Infection. Mol Immunol (2017) 90:64–73. doi: 10.1016/j.molimm.2017.07.002
12. Golsaz-Shirazi F, Shokri F. Cross Talk Between Hepatitis B Virus and Innate Immunity of Hepatocytes. Rev Med Virol (2021) 32:e2256. doi: 10.1002/rmv.2256
13. Chiale C, Marchese AM, Robek MD. Innate Immunity and HBV Persistence. Curr Opin Virol (2021) 49:13–20. doi: 10.1016/j.coviro.2021.04.003
14. Kuipery A, Gehring AJ, Isogawa M. Mechanisms of HBV Immune Evasion. Antiviral Res (2020) 179:104816. doi: 10.1016/j.antiviral.2020.104816
15. Thomas E, Baumert TF. Hepatitis B Virus-Hepatocyte Interactions and Innate Immune Responses: Experimental Models and Molecular Mechanisms. Semin Liver Dis (2019) 39:301–14. doi: 10.1055/s-0039-1685518
16. Tsai KN, Kuo CF, Ou JJ. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol (2018) 26:33–42. doi: 10.1016/j.tim.2017.07.006
17. Zhao F, Xie X, Tan X, Yu H, Tian M, Lv H, et al. The Functions of Hepatitis B Virus Encoding Proteins: Viral Persistence and Liver Pathogenesis. Front Immunol (2021) 12:691766. doi: 10.3389/fimmu.2021.691766
18. Wei L, Ploss A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses (2021) 13:1463. doi: 10.3390/v13081463
19. Xu R, Hu P, Li Y, Tian A, Li J, Zhu C. Advances in HBV Infection and Replication Systems In Vitro. Virol J (2021) 18:105. doi: 10.1186/s12985-021-01580-6
20. Slagle BL, Bouchard MJ. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb Perspect Med (2016) 6:a021402. doi: 10.1101/cshperspect.a021402
21. Feitelson MA, Bonamassa B, Arzumanyan A. The Roles of Hepatitis B Virus-Encoded X Protein in Virus Replication and the Pathogenesis of Chronic Liver Disease. Expert Opin Ther Targets (2014) 18:293–306. doi: 10.1517/14728222.2014.867947
22. Slagle BL, Bouchard MJ. Role of HBx in Hepatitis B Virus Persistence and Its Therapeutic Implications. Curr Opin Virol (2018) 30:32–8. doi: 10.1016/j.coviro.2018.01.007
23. Kong F, Zhang F, Liu X, Qin S, Yang X, Kong D, et al. Calcium Signaling in Hepatitis B Virus Infection and Its Potential as a Therapeutic Target. Cell Commun Signal (2021) 19:82. doi: 10.1186/s12964-021-00762-7
24. Kong F, Li Q, Zhang F, Li X, You H, Pan X, et al. Sirtuins as Potential Therapeutic Targets for Hepatitis B Virus Infection. Front Med (Lausanne) (2021) 8:751516. doi: 10.3389/fmed.2021.751516
25. Kong F, You H, Tang R, Zheng K. The Regulation of Proteins Associated With the Cytoskeleton by Hepatitis B Virus X Protein During Hepatocarcinogenesis. Oncol Lett (2017) 13:2514–20. doi: 10.3892/ol.2017.5757
26. Chaturvedi VK, Singh A, Dubey SK, Hetta HF, John J, Singh MP. Molecular Mechanistic Insight of Hepatitis B Virus Mediated Hepatocellular Carcinoma. Microb Pathog (2019) 128:184–94. doi: 10.1016/j.micpath.2019.01.004
27. Geng M, Xin X, Bi LQ, Zhou LT, Liu XH. Molecular Mechanism of Hepatitis B Virus X Protein Function in Hepatocarcinogenesis. World J Gastroenterol (2015) 21:10732–8. doi: 10.3748/wjg.v21.i38.10732
28. Jiang Y, Han Q, Zhao H, Zhang J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J Hepatocell Carcinoma (2021) 8:435–50. doi: 10.2147/JHC.S307962
29. Wang Y, Cai J, Zeng X, Chen Y, Yan W, Ouyang Y, et al. Downregulation of Toll-Like Receptor 4 Induces Suppressive Effects on Hepatitis B Virus-Related Hepatocellular Carcinoma via ERK1/2 Signaling. BMC Cancer (2015) 15:821. doi: 10.1186/s12885-015-1866-9
30. Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B Virus Regulatory HBx Protein Binds to Adaptor Protein IPS-1 and Inhibits the Activation of Beta Interferon. J Virol (2011) 85:987–95. doi: 10.1128/JVI.01825-10
31. Wang X, Li Y, Mao A, Li C, Tien P. Hepatitis B Virus X Protein Suppresses Virus-Triggered IRF3 Activation and IFN-Beta Induction by Disrupting the VISA-Associated Complex. Cell Mol Immunol (2010) 7:341–8. doi: 10.1038/cmi.2010.36
32. Xie WH, Ding J, Xie XX, Yang XH, Wu XF, Chen ZX, et al. Hepatitis B Virus X Protein Promotes Liver Cell Pyroptosis Under Oxidative Stress Through NLRP3 Inflammasome Activation. Inflamm Res (2020) 69:683–96. doi: 10.1007/s00011-020-01351-z
33. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
34. Zhou R, Liu L, Wang Y. Viral Proteins Recognized by Different TLRs. J Med Virol (2021) 93:6116–23. doi: 10.1002/jmv.27265
35. Nie L, Cai SY, Shao JZ, Chen J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front Immunol (2018) 9:1523. doi: 10.3389/fimmu.2018.01523
36. Pei RJ, Chen XW, Lu MJ. Control of Hepatitis B Virus Replication by Interferons and Toll-Like Receptor Signaling Pathways. World J Gastroenterol (2014) 20:11618–29. doi: 10.3748/wjg.v20.i33.11618
37. Zhang E, Lu M. Toll-Like Receptor (TLR)-Mediated Innate Immune Responses in the Control of Hepatitis B Virus (HBV) Infection. Med Microbiol Immunol (2015) 204:11–20. doi: 10.1007/s00430-014-0370-1
38. Kayesh MEH, Kohara M, Tsukiyama-Kohara K. Toll-Like Receptor Response to Hepatitis B Virus Infection and Potential of TLR Agonists as Immunomodulators for Treating Chronic Hepatitis B: An Overview. Int J Mol Sci (2021) 22:10462. doi: 10.3390/ijms221910462
39. Meng Z, Chen Y, Lu M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front Immunol (2019) 10:3127. doi: 10.3389/fimmu.2019.03127
40. Ma Z, Cao Q, Xiong Y, Zhang E, Lu M. Interaction Between Hepatitis B Virus and Toll-Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis B. Vaccines (Basel) (2018) 6:6. doi: 10.3390/vaccines6010006
41. Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, et al. Hepatitis B Virus Suppresses Toll-Like Receptor-Mediated Innate Immune Responses in Murine Parenchymal and Nonparenchymal Liver Cells. Hepatology (2009) 49:1132–40. doi: 10.1002/hep.22751
42. Riordan SM, Skinner N, Kurtovic J, Locarnini S, Visvanathan K. Reduced Expression of Toll-Like Receptor 2 on Peripheral Monocytes in Patients With Chronic Hepatitis B. Clin Vaccine Immunol (2006) 13:972–4. doi: 10.1128/CVI.00396-05
43. Huang YW, Hsu CK, Lin SC, Wei SC, Hu JT, Chang HY, et al. Reduced Toll-Like Receptor 9 Expression on Peripheral CD14+ Monocytes of Chronic Hepatitis B Patients and Its Restoration by Effective Therapy. Antivir Ther (2014) 19:637–43. doi: 10.3851/IMP2762
44. Huang YW, Lin SC, Wei SC, Hu JT, Chang HY, Huang SH, et al. Reduced Toll-Like Receptor 3 Expression in Chronic Hepatitis B Patients and Its Restoration by Interferon Therapy. Antivir Ther (2013) 18:877–84. doi: 10.3851/IMP2630
45. Xu N, Yao HP, Sun Z, Chen Z. Toll-Like Receptor 7 and 9 Expression in Peripheral Blood Mononuclear Cells From Patients With Chronic Hepatitis B and Related Hepatocellular Carcinoma. Acta Pharmacol Sin (2008) 29:239–44. doi: 10.1111/j.1745-7254.2008.00711.x
46. Momeni M, Zainodini N, Bidaki R, Hassanshahi G, Daneshvar H, Khaleghinia M, et al. Decreased Expression of Toll Like Receptor Signaling Molecules in Chronic HBV Infected Patients. Hum Immunol (2014) 75:15–9. doi: 10.1016/j.humimm.2013.09.015
47. Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P, et al. Hepatitis B Virus Impairs TLR9 Expression and Function in Plasmacytoid Dendritic Cells. PloS One (2011) 6:e26315. doi: 10.1371/journal.pone.0026315
48. Xu N, Yao HP, Lv GC, Chen Z. Downregulation of TLR7/9 Leads to Deficient Production of IFN-Alpha From Plasmacytoid Dendritic Cells in Chronic Hepatitis B. Inflamm Res (2012) 61:997–1004. doi: 10.1007/s00011-012-0493-z
49. Xie Q, Shen HC, Jia NN, Wang H, Lin LY, An BY, et al. Patients With Chronic Hepatitis B Infection Display Deficiency of Plasmacytoid Dendritic Cells With Reduced Expression of TLR9. Microbes Infect (2009) 11:515–23. doi: 10.1016/j.micinf.2009.02.008
50. Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G, et al. Toll-Like Receptor-Mediated Immune Responses Are Attenuated in the Presence of High Levels of Hepatitis B Virus Surface Antigen. J Viral Hepat (2014) 21:860–72. doi: 10.1111/jvh.12216
51. Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. The Hepatitis B E Antigen (HBeAg) Targets and Suppresses Activation of the Toll-Like Receptor Signaling Pathway. J Hepatol (2011) 55:762–9. doi: 10.1016/j.jhep.2010.12.042
52. Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, et al. Regulation of Toll-Like Receptor-2 Expression in Chronic Hepatitis B by the Precore Protein. Hepatology (2007) 45:102–10. doi: 10.1002/hep.21482
53. Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. Hepatitis B Virus Polymerase Inhibits RIG-I- and Toll-Like Receptor 3-Mediated Beta Interferon Induction in Human Hepatocytes Through Interference With Interferon Regulatory Factor 3 Activation and Dampening of the Interaction Between TBK1/IKKepsilon and DDX3. J Gen Virol (2010) 91:2080–90. doi: 10.1099/vir.0.020552-0
54. Wu M, Xu Y, Lin S, Zhang X, Xiang L, Yuan Z. Hepatitis B Virus Polymerase Inhibits the Interferon-Inducible MyD88 Promoter by Blocking Nuclear Translocation of Stat1. J Gen Virol (2007) 88:3260–9. doi: 10.1099/vir.0.82959-0
55. Liu S, Peng N, Xie J, Hao Q, Zhang M, Zhang Y, et al. Human Hepatitis B Virus Surface and E Antigens Inhibit Major Vault Protein Signaling in Interferon Induction Pathways. J Hepatol (2015) 62:1015–23. doi: 10.1016/j.jhep.2014.11.035
56. Li Q, Wang J, Islam H, Kirschning C, Lu H, Hoffmann D, et al. Hepatitis B Virus Particles Activate B Cells Through the TLR2-MyD88-mTOR Axis. Cell Death Dis (2021) 12:34. doi: 10.1038/s41419-020-03284-1
57. Xie X, Lv H, Liu C, Su X, Yu Z, Song S, et al. HBeAg Mediates Inflammatory Functions of Macrophages by TLR2 Contributing to Hepatic Fibrosis. BMC Med (2021) 19:247. doi: 10.1186/s12916-021-02085-3
58. Yi H, Zhang Y, Yang X, Li M, Hu H, Xiong J, et al. Hepatitis B Core Antigen Impairs the Polarization While Promoting the Production of Inflammatory Cytokines of M2 Macrophages via the TLR2 Pathway. Front Immunol (2020) 11:535. doi: 10.3389/fimmu.2020.00535
59. Zhang Y, Lian JQ, Huang CX, Wang JP, Wei X, Nan XP, et al. Overexpression of Toll-Like Receptor 2/4 on Monocytes Modulates the Activities of CD4(+)CD25(+) Regulatory T Cells in Chronic Hepatitis B Virus Infection. Virology (2010) 397:34–42. doi: 10.1016/j.virol.2009.11.007
60. Cheng S, Zhang B, Du JY, Jin YH, Lang HY, Zeng LH. Hepatitis B Surface Antigen Promotes the Invasion of Hepatitis B Virus-Related Hepatocellular Carcinoma Cells by Upregulation of Toll-Like Receptor 2. Viral Immunol (2017) 30:232–9. doi: 10.1089/vim.2016.0162
61. Shi B, Ren G, Hu Y, Wang S, Zhang Z, Yuan Z. HBsAg Inhibits IFN-Alpha Production in Plasmacytoid Dendritic Cells Through TNF-Alpha and IL-10 Induction in Monocytes. PloS One (2012) 7:e44900. doi: 10.1371/journal.pone.0044900
62. Wang X, Zhou Y, Zhu N, Yuan WJ. Effects of Hepatitis B Virus X Gene on Apoptosis and Expression of Immune Molecules of Human Proximal Tubular Epithelial Cells. Arch Virol (2013) 158:2479–85. doi: 10.1007/s00705-013-1759-7
63. Son J, Kim MJ, Lee JS, Kim JY, Chun E, Lee KY. Hepatitis B Virus X Protein Promotes Liver Cancer Progression Through Autophagy Induction in Response to TLR4 Stimulation. Immune Netw (2021) 21:e37. doi: 10.4110/in.2021.21.e37
64. Zhang L. Autophagy in Hepatitis B or C Virus Infection: An Incubator and a Potential Therapeutic Target. Life Sci (2020) 242:117206. doi: 10.1016/j.lfs.2019.117206
65. Li J, Lin S, Chen Q, Peng L, Zhai J, Liu Y, et al. Inhibition of Hepatitis B Virus Replication by MyD88 Involves Accelerated Degradation of Pregenomic RNA and Nuclear Retention of Pre-s/S RNAs. J Virol (2010) 84:6387–99. doi: 10.1128/JVI.00236-10
66. Xiong W, Wang X, Liu X, Xiang L, Zheng L, Yuan Z. Interferon-Inducible MyD88 Protein Inhibits Hepatitis B Virus Replication. Virology (2004) 319:306–14. doi: 10.1016/j.virol.2003.11.005
67. Xiang WQ, Feng WF, Ke W, Sun Z, Chen Z, Liu W. Hepatitis B Virus X Protein Stimulates IL-6 Expression in Hepatocytes via a MyD88-Dependent Pathway. J Hepatol (2011) 54:26–33. doi: 10.1016/j.jhep.2010.08.006
68. Yu H, Li M, He R, Fang P, Wang Q, Yi Y, et al. Major Vault Protein Promotes Hepatocellular Carcinoma Through Targeting Interferon Regulatory Factor 2 and Decreasing P53 Activity. Hepatology (2020) 72:518–34. doi: 10.1002/hep.31045
69. Cavlar T, Ablasser A, Hornung V. Induction of Type I IFNs by Intracellular DNA-Sensing Pathways. Immunol Cell Biol (2012) 90:474–82. doi: 10.1038/icb.2012.11
70. Shen B, Chen Y, Hu J, Qiao M, Ren J, Chen J, et al. Hepatitis B Virus X Protein Modulates Upregulation of DHX9 to Promote Viral DNA Replication. Cell Microbiol (2020) 22:e13148. doi: 10.1111/cmi.13148
71. Liu H, Yan Y, Lin J, He C, Yu Z, Liu Q, et al. Circular RNA Circsfmbt2 Downregulation by HBx Promotes Hepatocellular Carcinoma Metastasis Via the miR-665/TIMP3 Axis. SSRN. doi: 10.2139/ssrn.3882480
72. Zhou Y, Wang S, Ma JW, Lei Z, Zhu HF, Lei P, et al. Hepatitis B Virus Protein X-Induced Expression of the CXC Chemokine IP-10 Is Mediated Through Activation of NF-KappaB and Increases Migration of Leukocytes. J Biol Chem (2010) 285:12159–68. doi: 10.1074/jbc.M109.067629
73. Hu B, Yu L, Zhu N, Xie J. Cellular UAP56 Interacts With the HBx Protein of the Hepatitis B Virus and Is Involved in Viral RNA Nuclear Export in Hepatocytes. Exp Cell Res (2020) 390:111929. doi: 10.1016/j.yexcr.2020.111929
74. Wu H, Yang TY, Li Y, Ye WL, Liu F, He XS, et al. Tumor Necrosis Factor Receptor-Associated Factor 6 Promotes Hepatocarcinogenesis by Interacting With Histone Deacetylase 3 to Enhance C-Myc Gene Expression and Protein Stability. Hepatology (2020) 71:148–63. doi: 10.1002/hep.30801
75. Chen WN, Liu LL, Jiao BY, Lin WS, Lin XJ, Lin X. Hepatitis B Virus X Protein Increases the IL-1beta-Induced NF-KappaB Activation via Interaction With Evolutionarily Conserved Signaling Intermediate in Toll Pathways (ECSIT). Virus Res (2015) 195:236–45. doi: 10.1016/j.virusres.2014.10.025
76. Hong Y, Zhou L, Xie H, Zheng S. Innate Immune Evasion by Hepatitis B Virus-Mediated Downregulation of TRIF. Biochem Biophys Res Commun (2015) 463:719–25. doi: 10.1016/j.bbrc.2015.05.130
77. Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The Molecular Mechanism of RIG-I Activation and Signaling. Immunol Rev (2021) 304:154–68. doi: 10.1111/imr.13022
78. Rehwinkel J, Gack MU. RIG-I-Like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
79. Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG-I-Like Receptor-Mediated Signaling: Interaction Between Host and Viral Factors. Cell Mol Immunol (2021) 18:539–55. doi: 10.1038/s41423-020-00602-7
80. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity (2015) 42:123–32. doi: 10.1016/j.immuni.2014.12.016
81. Lee S, Goyal A, Perelson AS, Ishida Y, Saito T, Gale M Jr. Suppression of Hepatitis B Virus Through Therapeutic Activation of RIG-I and IRF3 Signaling in Hepatocytes. iScience (2021) 24:101969. doi: 10.1016/j.isci.2020.101969
82. Chen X, Qian Y, Yan F, Tu J, Yang X, Xing Y, et al. 5'-triphosphate-siRNA Activates RIG-I-Dependent Type I Interferon Production and Enhances Inhibition of Hepatitis B Virus Replication in HepG2.2.15 Cells. Eur J Pharmacol (2013) 721:86–95. doi: 10.1016/j.ejphar.2013.09.050
83. Yoneda M, Hyun J, Jakubski S, Saito S, Nakajima A, Schiff ER, et al. Hepatitis B Virus and DNA Stimulation Trigger a Rapid Innate Immune Response Through NF-Kappab. J Immunol (2016) 197:630–43. doi: 10.4049/jimmunol.1502677
84. Kim GW, Imam H, Khan M, Siddiqui AN. (6)-Methyladenosine Modification of Hepatitis B and C Viral RNAs Attenuates Host Innate Immunity via RIG-I Signaling. J Biol Chem (2020) 295:13123–33. doi: 10.1074/jbc.RA120.014260
85. Hou Z, Zhang J, Han Q, Su C, Qu J, Xu D, et al. Hepatitis B Virus Inhibits Intrinsic RIG-I and RIG-G Immune Signaling via Inducing Mir146a. Sci Rep (2016) 6:26150. doi: 10.1038/srep26150
86. Jiang J, Tang H. Mechanism of Inhibiting Type I Interferon Induction by Hepatitis B Virus X Protein. Protein Cell (2010) 1:1106–17. doi: 10.1007/s13238-010-0141-8
87. Wang F, Shen F, Wang Y, Li Z, Chen J, Yuan Z. Residues Asn118 and Glu119 of Hepatitis B Virus X Protein Are Critical for HBx-Mediated Inhibition of RIG-I-MAVS Signaling. Virology (2020) 539:92–103. doi: 10.1016/j.virol.2019.10.009
88. Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, et al. The Hepatitis B Virus X Protein Disrupts Innate Immunity by Downregulating Mitochondrial Antiviral Signaling Protein. J Immunol (2010) 185:1158–68. doi: 10.4049/jimmunol.0903874
89. Khan M, Syed GH, Kim SJ, Siddiqui A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PloS Pathog (2016) 12:e1005693. doi: 10.1371/journal.ppat.1005693
90. Wang L, Sun Y, Song X, Wang Z, Zhang Y, Zhao Y, et al. Hepatitis B Virus Evades Immune Recognition via RNA Adenosine Deaminase ADAR1-Mediated Viral RNA Editing in Hepatocytes. Cell Mol Immunol (2021) 18:1871–82. doi: 10.1038/s41423-021-00729-1
91. Sengupta I, Das D, Singh SP, Chakravarty R, Das C. Host Transcription Factor Speckled 110 kDa (Sp110), a Nuclear Body Protein, Is Hijacked by Hepatitis B Virus Protein X for Viral Persistence. J Biol Chem (2017) 292:20379–93. doi: 10.1074/jbc.M117.796839
92. Kim HR, Lee SH, Jung G. The Hepatitis B Viral X Protein Activates NF-KappaB Signaling Pathway Through the Up-Regulation of TBK1. FEBS Lett (2010) 584:525–30. doi: 10.1016/j.febslet.2009.11.091
93. Pandey A, Shen C, Feng S, Man SM. Cell Biology of Inflammasome Activation. Trends Cell Biol (2021) 31:924–39. doi: 10.1016/j.tcb.2021.06.010
94. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
95. Jia Y, Ma L, Wang Y, Wang W, Shen C, Wang X, et al. NLRP3 Inflammasome and Related Cytokines Reflect the Immune Status of Patients With HBV-ACLF. Mol Immunol (2020) 120:179–86. doi: 10.1016/j.molimm.2020.01.011
96. Li Z, Jiang J. The NLRP3 Inflammasome Mediates Liver Failure by Activating Procaspase-1 and Pro-IL-1 Beta and Regulating Downstream CD40-CD40L Signaling. J Int Med Res (2021) 49:3000605211036845. doi: 10.1177/03000605211036845
97. Ding X, Lei Q, Li T, Li L, Qin B. Hepatitis B Core Antigen can Regulate NLRP3 Inflammasome Pathway in HepG2 Cells. J Med Virol (2019) 91:1528–36. doi: 10.1002/jmv.25490
98. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1beta Production via Suppressing the NF-kappaB Pathway and ROS Production. J Hepatol (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
99. Xie L, Huang Y. Antagonism of RIP1 Using Necrostatin-1 (Nec-1) Ameliorated Damage and Inflammation of HBV X Protein (HBx) in Human Normal Hepatocytes. Artif Cells Nanomed Biotechnol (2019) 47:1194–9. doi: 10.1080/21691401.2019.1575231
100. Chen YY, Lin Y, Han PY, Jiang S, Che L, He CY, et al. HBx Combined With AFB1 Triggers Hepatic Steatosis via COX-2-Mediated Necrosome Formation and Mitochondrial Dynamics Disorder. J Cell Mol Med (2019) 23:5920–33. doi: 10.1111/jcmm.14388
101. Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, et al. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep (2016) 16:2846–54. doi: 10.1016/j.celrep.2016.08.026
102. Sharma BR, Karki R, Kanneganti TD. Role of AIM2 Inflammasome in Inflammatory Diseases, Cancer and Infection. Eur J Immunol (2019) 49:1998–2011. doi: 10.1002/eji.201848070
103. Chen H, He G, Chen Y, Zhang X, Wu S. Differential Activation of NLRP3, AIM2, and IFI16 Inflammasomes in Humans With Acute and Chronic Hepatitis B. Viral Immunol (2018) 31:639–45. doi: 10.1089/vim.2018.0058
104. Chen SL, Liu LL, Lu SX, Luo RZ, Wang CH, Wang H, et al. HBx-Mediated Decrease of AIM2 Contributes to Hepatocellular Carcinoma Metastasis. Mol Oncol (2017) 11:1225–40. doi: 10.1002/1878-0261.12090
105. Shi LZ, Bonner JA. Bridging Radiotherapy to Immunotherapy: The IFN-JAK-STAT Axis. Int J Mol Sci (2021) 22:12295. doi: 10.3390/ijms222212295
106. Yin Q, Wang L, Yu H, Chen D, Zhu W, Sun C. Pharmacological Effects of Polyphenol Phytochemicals on the JAK-STAT Signaling Pathway. Front Pharmacol (2021) 12:716672. doi: 10.3389/fphar.2021.716672
107. Chen J, Xu W, Chen Y, Xie X, Zhang Y, Ma C, et al. Matrix Metalloproteinase 9 Facilitates Hepatitis B Virus Replication Through Binding With Type I Interferon (IFN) Receptor 1 To Repress IFN/JAK/STAT Signaling. J Virol (2017) 91:e01824–16. doi: 10.1128/JVI.01824-16
108. Bai L, Zhang W, Tan L, Yang H, Ge M, Zhu C, et al. Hepatitis B Virus Hijacks CTHRC1 to Evade Host Immunity and Maintain Replication. J Mol Cell Biol (2015) 7:543–56. doi: 10.1093/jmcb/mjv048
109. Yu Y, Wan P, Cao Y, Zhang W, Chen J, Tan L, et al. Hepatitis B Virus E Antigen Activates the Suppressor of Cytokine Signaling 2 to Repress Interferon Action. Sci Rep (2017) 7:1729. doi: 10.1038/s41598-017-01773-6
110. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Signal Transduct Target Ther (2021) 6:402. doi: 10.1038/s41392-021-00791-1
111. Lee YH, Yun Y. HBx Protein of Hepatitis B Virus Activates Jak1-STAT Signaling. J Biol Chem (1998) 273:25510–5. doi: 10.1074/jbc.273.39.25510
112. Kim H, Lee YH, Won J, Yun Y. Through Induction of Juxtaposition and Tyrosine Kinase Activity of Jak1, X-Gene Product of Hepatitis B Virus Stimulates Ras and the Transcriptional Activation Through AP-1, NF-Kappab, and SRE Enhancers. Biochem Biophys Res Commun (2001) 286:886–94. doi: 10.1006/bbrc.2001.5496
113. He P, Zhang D, Li H, Yang X, Li D, Zhai Y, et al. Hepatitis B Virus X Protein Modulates Apoptosis in Human Renal Proximal Tubular Epithelial Cells by Activating the JAK2/STAT3 Signaling Pathway. Int J Mol Med (2013) 31:1017–29. doi: 10.3892/ijmm.2013.1295
114. Cho IR, Oh M, Koh SS, Malilas W, Srisuttee R, Jhun BH, et al. Hepatitis B Virus X Protein Inhibits Extracellular IFN-Alpha-Mediated Signal Transduction by Downregulation of Type I IFN Receptor. Int J Mol Med (2012) 29:581–6. doi: 10.3892/ijmm.2012.879
115. Park EH, Koh SS, Srisuttee R, Cho IR, Min HJ, Jhun BH, et al. Expression of HBX, an Oncoprotein of Hepatitis B Virus, Blocks Reoviral Oncolysis of Hepatocellular Carcinoma Cells. Cancer Gene Ther (2009) 16:453–61. doi: 10.1038/cgt.2008.95
116. Lei XY, Chen XX, Sun YH, Gao MD, Hu XX, Suo YH. Hepatitis B Virus X Protein Decreases Nephrin Expression and Induces Podocyte Apoptosis via Activating STAT3. Exp Ther Med (2019) 17:4223–9. doi: 10.3892/etm.2019.7453
117. Zhang S, Shan C, Cui W, You X, Du Y, Kong G, et al. Hepatitis B Virus X Protein Protects Hepatoma and Hepatic Cells From Complement-Dependent Cytotoxicity by Up-Regulation of CD46. FEBS Lett (2013) 587:645–51. doi: 10.1016/j.febslet.2013.01.019
118. Waris G, Huh KW, Siddiqui A. Mitochondrially Associated Hepatitis B Virus X Protein Constitutively Activates Transcription Factors STAT-3 and NF-Kappa B via Oxidative Stress. Mol Cell Biol (2001) 21:7721–30. doi: 10.1128/MCB.21.22.7721-7730.2001
119. Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, et al. Lethal-7 Is Down-Regulated by the Hepatitis B Virus X Protein and Targets Signal Transducer and Activator of Transcription 3. J Hepatol (2010) 53:57–66. doi: 10.1016/j.jhep.2009.12.043
120. Teng J, Wang X, Xu Z, Tang N. HBx-Dependent Activation of Twist Mediates STAT3 Control of Epithelium-Mesenchymal Transition of Liver Cells. J Cell Biochem (2013) 114:1097–104. doi: 10.1002/jcb.24450
121. Zhang Y, Ren H, Li J, Xue R, Liu H, Zhu Z, et al. Elevated HMGB1 Expression Induced by Hepatitis B Virus X Protein Promotes Epithelial-Mesenchymal Transition and Angiogenesis Through STAT3/miR-34a/NF-kappaB in Primary Liver Cancer. Am J Cancer Res (2021) 11:479–94.
122. Kim K, Kim KH, Cheong J. Hepatitis B Virus X Protein Impairs Hepatic Insulin Signaling Through Degradation of IRS1 and Induction of SOCS3. PloS One (2010) 5:e8649. doi: 10.1371/journal.pone.0008649
123. Ching RHH, Sze KMF, Lau EYT, Chiu YT, Lee JMF, Ng IOL, et al. C-Terminal Truncated Hepatitis B Virus X Protein Regulates Tumorigenicity, Self-Renewal and Drug Resistance via STAT3/Nanog Signaling Pathway. Oncotarget (2017) 8:23507–16. doi: 10.18632/oncotarget.15183
124. Chen T, Pei J, Wang J, Luo R, Liu L, Wang L, et al. HBx-Related Long Non-Coding RNA 01152 Promotes Cell Proliferation and Survival by IL-23 in Hepatocellular Carcinoma. BioMed Pharmacother (2019) 115:108877. doi: 10.1016/j.biopha.2019.108877
125. Zheng Y, Ming P, Zhu C, Si Y, Xu S, Chen A, et al. Hepatitis B Virus X Protein-Induced SH2 Domain-Containing 5 (SH2D5) Expression Promotes Hepatoma Cell Growth via an SH2D5-Transketolase Interaction. J Biol Chem (2019) 294:4815–27. doi: 10.1074/jbc.RA118.005739
126. Quetier I, Brezillon N, Duriez M, Massinet H, Giang E, Ahodantin J, et al. Hepatitis B Virus HBx Protein Impairs Liver Regeneration Through Enhanced Expression of IL-6 in Transgenic Mice. J Hepatol (2013) 59:285–91. doi: 10.1016/j.jhep.2013.03.021
127. Kim JS, Rho B, Lee TH, Lee JM, Kim SJ, Park JH. The Interaction of Hepatitis B Virus X Protein and Protein Phosphatase Type 2 Calpha and Its Effect on IL-6. Biochem Biophys Res Commun (2006) 351:253–8. doi: 10.1016/j.bbrc.2006.10.028
128. You H, Yuan D, Bi Y, Zhang N, Li Q, Tu T, et al. Hepatitis B Virus X Protein Promotes Vimentin Expression via LIM and SH3 Domain Protein 1 to Facilitate Epithelial-Mesenchymal Transition and Hepatocarcinogenesis. Cell Commun Signal (2021) 19:33. doi: 10.1186/s12964-021-00714-1
129. Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng IO, et al. Signal Transducers and Activators of Transcription 5b Activation Enhances Hepatocellular Carcinoma Aggressiveness Through Induction of Epithelial-Mesenchymal Transition. Cancer Res (2006) 66:9948–56. doi: 10.1158/0008-5472.CAN-06-1092
130. Kong F, Zhou K, Zhu T, Lian Q, Tao Y, Li N, et al. Interleukin-34 Mediated by Hepatitis B Virus X Protein via CCAAT/enhancer-Binding Protein Alpha Contributes to the Proliferation and Migration of Hepatoma Cells. Cell Prolif (2019) 52:e12703. doi: 10.1111/cpr.12703
131. Liu Y, Feng J, Sun M, Yang G, Yuan H, Wang Y, et al. Long Non-Coding RNA HULC Activates HBV by Modulating HBx/STAT3/miR-539/APOBEC3B Signaling in HBV-Related Hepatocellular Carcinoma. Cancer Lett (2019) 454:158–70. doi: 10.1016/j.canlet.2019.04.008
132. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
133. Hayden MS, Ghosh S. Shared Principles in NF-kappaB Signaling. Cell (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
134. Liu L, Zhu J, Yang J, Li X, Yuan J, Wu J, et al. GP73 Facilitates Hepatitis B Virus Replication by Repressing the NF-kappaB Signaling Pathway. J Med Virol (2020) 92:3327–35. doi: 10.1002/jmv.25718
135. Lin J, Gu C, Shen Z, Liu Y, Wang W, Tao S, et al. Hepatocyte Nuclear Factor 1alpha Downregulates HBV Gene Expression and Replication by Activating the NF-kappaB Signaling Pathway. PloS One (2017) 12:e0174017. doi: 10.1371/journal.pone.0174017
136. Lin S, Wu M, Xu Y, Xiong W, Yi Z, Zhang X, et al. Inhibition of Hepatitis B Virus Replication by MyD88 Is Mediated by Nuclear factor-kappaB Activation. Biochim Biophys Acta (2007) 1772:1150–7. doi: 10.1016/j.bbadis.2007.08.001
137. Ren S, Wang J, Chen TL, Li HY, Wan YS, Peng NF, et al. Hepatitis B Virus Stimulated Fibronectin Facilitates Viral Maintenance and Replication Through Two Distinct Mechanisms. PloS One (2016) 11:e0152721. doi: 10.1371/journal.pone.0152721
138. Wang Y, Cui L, Yang G, Zhan J, Guo L, Chen Y, et al. Hepatitis B E Antigen Inhibits NF-kappaB Activity by Interrupting K63-Linked Ubiquitination of NEMO. J Virol (2019) 93:e00667–18. doi: 10.1128/JVI.00667-18
139. Jegaskanda S, Ahn SH, Skinner N, Thompson AJ, Ngyuen T, Holmes J, et al. Downregulation of Interleukin-18-Mediated Cell Signaling and Interferon Gamma Expression by the Hepatitis B Virus E Antigen. J Virol (2014) 88:10412–20. doi: 10.1128/JVI.00111-14
140. Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, et al. The Hepatitis B E Antigen Suppresses IL-1beta-Mediated NF-kappaB Activation in Hepatocytes. J Viral Hepat (2011) 18:e499–507. doi: 10.1111/j.1365-2893.2011.01484.x
141. Wang J, Chen J, Liu Y, Zeng X, Wei M, Wu S, et al. Hepatitis B Virus Induces Autophagy to Promote Its Replication by the Axis of miR-192-3p-XIAP Through NF Kappa B Signaling. Hepatology (2019) 69:974–92. doi: 10.1002/hep.30248
142. Xu F, Song H, An B, Xiao Q, Cheng G, Tan G. NF-kappaB-Dependent IFIT3 Induction by HBx Promotes Hepatitis B Virus Replication. Front Microbiol (2019) 10:2382. doi: 10.3389/fmicb.2019.02382
143. Fan H, Yan X, Zhang Y, Zhang X, Gao Y, Xu Y, et al. Increased Expression of Gp96 by HBx-Induced NF-kappaB Activation Feedback Enhances Hepatitis B Virus Production. PloS One (2013) 8:e65588. doi: 10.1371/journal.pone.0065588
144. Fu L, Fu X, Mo J, Li X, Li R, Peng S. miR-146a-5p Enhances Hepatitis B Virus Replication Through Autophagy to Promote Aggravation of Chronic Hepatitis B. IUBMB Life (2019) 71:1336–46. doi: 10.1002/iub.2044
145. Su F, Schneider RJ. Hepatitis B Virus HBx Protein Activates Transcription Factor NF-kappaB by Acting on Multiple Cytoplasmic Inhibitors of Rel-Related Proteins. J Virol (1996) 70:4558–66. doi: 10.1128/JVI.70.7.4558-4566.1996
146. Bui-Nguyen TM, Pakala SB, Sirigiri RD, Xia W, Hung MC, Sarin SK, et al. NF-kappaB Signaling Mediates the Induction of MTA1 by Hepatitis B Virus Transactivator Protein HBx. Oncogene (2010) 29:1179–89. doi: 10.1038/onc.2009.404
147. Lim KH, Choi HS, Park YK, Park ES, Shin GC, Kim DH, et al. HBx-Induced NF-kappaB Signaling in Liver Cells Is Potentially Mediated by the Ternary Complex of HBx With P22-FLIP and NEMO. PloS One (2013) 8:e57331. doi: 10.1371/journal.pone.0057331
148. Luo MX, Wong SH, Chan MT, Yu L, Yu SS, Wu F, et al. Autophagy Mediates HBx-Induced Nuclear Factor-kappaB Activation and Release of IL-6, IL-8, and CXCL2 in Hepatocytes. J Cell Physiol (2015) 230:2382–9. doi: 10.1002/jcp.24967
149. Yen CJ, Lin YJ, Yen CS, Tsai HW, Tsai TF, Chang KY, et al. Hepatitis B Virus X Protein Upregulates mTOR Signaling Through IKKbeta to Increase Cell Proliferation and VEGF Production in Hepatocellular Carcinoma. PloS One (2012) 7:e41931. doi: 10.1371/journal.pone.0041931
150. Kim SY, Kim JC, Kim JK, Kim HJ, Lee HM, Choi MS, et al. Hepatitis B Virus X Protein Enhances NFkappaB Activity Through Cooperating With VBP1. BMB Rep (2008) 41:158–63. doi: 10.5483/bmbrep.2008.41.2.158
151. Jiao BY, Lin WS, She FF, Chen WN, Lin X. Hepatitis B Virus X Protein Enhances Activation of Nuclear Factor kappaB Through Interaction With Valosin-Containing Protein. Arch Virol (2011) 156:2015–21. doi: 10.1007/s00705-011-1099-4
152. Hong A, Han DD, Wright CJ, Burch T, Piper J, Osiowy C, et al. The Interaction Between Hepatitis B Virus X Protein and AIB1 Oncogene Is Required for the Activation of NFkappaB Signal Transduction. Biochem Biophys Res Commun (2012) 423:6–12. doi: 10.1016/j.bbrc.2012.05.021
153. Liang DY, Sha S, Yi Q, Shi J, Chen Y, Hou Y, et al. Hepatitis B X Protein Upregulates Decoy Receptor 3 Expression via the PI3K/NF-kappaB Pathway. Cell Signal (2019) 62:109346. doi: 10.1016/j.cellsig.2019.109346
154. Huang WC, Chen WS, Chen YJ, Wang LY, Hsu SC, Chen CC, et al. Hepatitis B Virus X Protein Induces IKKalpha Nuclear Translocation via Akt-Dependent Phosphorylation to Promote the Motility of Hepatocarcinoma Cells. J Cell Physiol (2012) 227:1446–54. doi: 10.1002/jcp.22860
155. Xia L, Tian D, Huang W, Zhu H, Wang J, Zhang Y, et al. Upregulation of IL-23 Expression in Patients With Chronic Hepatitis B Is Mediated by the HBx/ERK/NF-kappaB Pathway. J Immunol (2012) 188:753–64. doi: 10.4049/jimmunol.1101652
156. Shokri S, Mahmoudvand S, Taherkhani R, Farshadpour F, Jalalian FA. Complexity on Modulation of NF-kappaB Pathways by Hepatitis B and C: A Double-Edged Sword in Hepatocarcinogenesis. J Cell Physiol (2019) 234:14734–42. doi: 10.1002/jcp.28249
157. Kong F, You H, Zhao J, Liu W, Hu L, Luo W, et al. The Enhanced Expression of Death Receptor 5 (DR5) Mediated by HBV X Protein Through NF-kappaB Pathway Is Associated With Cell Apoptosis Induced by (TNF-Alpha Related Apoptosis Inducing Ligand) TRAIL in Hepatoma Cells. Virol J (2015) 12:192. doi: 10.1186/s12985-015-0416-z
158. Lim KH, Kim KH, Choi SI, Park ES, Park SH, Ryu K, et al. RPS3a Over-Expressed in HBV-Associated Hepatocellular Carcinoma Enhances the HBx-Induced NF-kappaB Signaling via Its Novel Chaperoning Function. PloS One (2011) 6:e22258. doi: 10.1371/journal.pone.0022258
159. Medhat A, Arzumanyan A, Feitelson MA. Hepatitis B X Antigen (HBx) Is an Important Therapeutic Target in the Pathogenesis of Hepatocellular Carcinoma. Oncotarget (2021) 12(24):2421–33. doi: 10.18632/oncotarget.28077
160. Han Q, Zhang C, Zhang J, Tian Z. Reversal of Hepatitis B Virus-Induced Immune Tolerance by an Immunostimulatory 3p-HBx-siRNAs in a Retinoic Acid Inducible Gene I-Dependent Manner. Hepatology (2011) 54:1179–89. doi: 10.1002/hep.24505
161. Han Q, Zhang C, Zhang J, Tian Z. Involvement of Activation of PKR in HBx-siRNA-Mediated Innate Immune Effects on HBV Inhibition. PloS One (2011) 6:e27931. doi: 10.1371/journal.pone.0027931
162. Han Q, Hou Z, Yin C, Zhang C, Zhang J. 5'-Triphosphate siRNA Targeting HBx Elicits a Potent Anti-HBV Immune Response in pAAV-HBV Transfected Mice. Antiviral Res (2019) 161:36–45. doi: 10.1016/j.antiviral.2018.11.006
163. Bao T, Liu J, Leng J, Cai L. The cGAS-STING Pathway: More Than Fighting Against Viruses and Cancer. Cell Biosci (2021) 11:209. doi: 10.1186/s13578-021-00724-z
164. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
165. Vashi N, Bakhoum SF. The Evolution of STING Signaling and Its Involvement in Cancer. Trends Biochem Sci (2021) 46:446–60. doi: 10.1016/j.tibs.2020.12.010
166. Chen B, Rao X, Wang X, Luo Z, Wang J, Sheng S, et al. cGAS-STING Signaling Pathway and Liver Disease: From Basic Research to Clinical Practice. Front Pharmacol (2021) 12:719644. doi: 10.3389/fphar.2021.719644
167. Li Y, He M, Wang Z, Duan Z, Guo Z, Gong R, et al. STING Signaling Activation Inhibits HBV Replication and Attenuates the Severity of Liver Injury and HBV-Induced Fibrosis. Cell Mol Immunol (2021) 19:92–107. doi: 10.1038/s41423-021-00801-w
168. Chen H, He G, Chen Y, Zhang X. Hepatitis B Virus Might Be Sensed by STING-Dependent DNA Sensors and Attenuates the Response of STING-Dependent DNA Sensing Pathway in Humans With Acute and Chronic Hepatitis B Virus Infection. Viral Immunol (2020) 33:642–51. doi: 10.1089/vim.2020.0096
169. Verrier ER, Yim SA, Heydmann L, El Saghire H, Bach C, Turon-Lagot V, et al. Hepatitis B Virus Evasion From Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthase Sensing in Human Hepatocytes. Hepatology (2018) 68:1695–709. doi: 10.1002/hep.30054
170. Karimi-Googheri M, Daneshvar H, Khaleghinia M, Bidaki R, Kazemi Arababadi M. Decreased Expressions of STING But Not IRF3 Molecules in Chronic HBV Infected Patients. Arch Iran Med (2015) 18:351–4.
171. Chen R, Du J, Zhu H, Ling Q. The Role of cGAS-STING Signalling in Liver Diseases. JHEP Rep (2021) 3:100324. doi: 10.1016/j.jhepr.2021.100324
172. Liu Y, Li J, Chen J, Li Y, Wang W, Du X, et al. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING to Block Innate Cytosolic DNA-Sensing Pathways. J Virol (2015) 89:2287–300. doi: 10.1128/JVI.02760-14
Keywords: hepatitis B virus, retinoic acid inducible gene I (RIG-I)-like receptors, HBx, pattern recognition receptors, toll-like receptors, nod-like receptors
Citation: You H, Qin S, Zhang F, Hu W, Li X, Liu D, Kong F, Pan X, Zheng K and Tang R (2022) Regulation of Pattern-Recognition Receptor Signaling by HBX During Hepatitis B Virus Infection. Front. Immunol. 13:829923. doi: 10.3389/fimmu.2022.829923
Received: 06 December 2021; Accepted: 24 January 2022;
Published: 17 February 2022.
Edited by:
Cai Zhang, Shandong University, ChinaReviewed by:
Xiaohong Liang, Shandong University, ChinaKyun-Hwan Kim, Sungkyunkwan University, South Korea
Copyright © 2022 You, Qin, Zhang, Hu, Li, Liu, Kong, Pan, Zheng and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renxian Tang, tangrenxian-t@163.com; Fanyun Kong, kong.fanyun@163.com