 Yuhong Pan
Yuhong Pan Wenjun Cai
Wenjun Cai Anchun Cheng
Anchun Cheng Mingshu Wang
Mingshu Wang Zhongqiong Yin
Zhongqiong Yin Renyong Jia
Renyong Jia- 1Research Center of Avian Disease, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
- 2Institute of Preventive Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
- 3Key Laboratory of Animal Disease and Human Health of Sichuan Province, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
The innate immune system is the host’s first line of defense against the invasion of pathogens including flavivirus. The programmed cell death controlled by genes plays an irreplaceable role in resisting pathogen invasion and preventing pathogen infection. However, the inflammatory cell death, which can trigger the overflow of a large number of pro-inflammatory cytokines and cell contents, will initiate a severe inflammatory response. In this review, we summarized the current understanding of the innate immune response, inflammatory cell death pathway and cytokine secretion regulation during Dengue virus, West Nile virus, Zika virus, Japanese encephalitis virus and other flavivirus infections. We also discussed the impact of these flavivirus and viral proteins on these biological processes. This not only provides a scientific basis for elucidating the pathogenesis of flavivirus, but also lays the foundation for the development of effective antiviral therapies.
1 Innate Immunity and Flaviviruses
1.1 Innate Immunity and Inflammasomes
As the first hurdle to protect the host from microbial invasion, the innate immune system can not only establish a rapid, broadly response to control infection, but also plays a key role in the establishment of an adaptive immune response, which can lead to pathogen-specific and durable immune memory (1). In order to quickly detect and resist a variety of pathogens, host cells have evolved many pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and nucleotide-binding oligomerization domain (NOD)-like receptor family proteins (NLRs). When dealing with specific danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), members of the NLRs family are able to assemble large multiprotein complexes called inflammasomes (2). The NLRP3 (NLR family pyrin domain-containing 3) inflammasome is the most widely studied inflammasome that has been identified. NLRP3 is a cytoplasmic protein with three domains: the carboxy-terminal leucine-rich repeat sequence, the central nucleotide binding and oligomerization domain (NACHT) with ATPase activity, and the amino-termina pyrin domain (PYD) (3). Since the basal level of NLRP3 expression is usually not enough to activate the NLRP3 inflammasome. Therefore, a two-step process is required for priming and activation (4, 5). The priming step is induced by TLRs and cytokine receptors, such as the tumor necrosis factor receptor (TNFR) or IL-1 receptor (IL-1R), which recognize PAMPs or DAMPs and upregulate the transcription of NLRP3 and IL-1β. During the activation step, PAMPs and DAMPs promote NLRP3 inflammasome assembly. After assembly, the inflammasome induces the formation of membrane pores and the release of pro-inflammatory cytokines, which ultimately leads to a form of inflammatory cell death called pyroptosis (6, 7).
Innate immune response and inflammasome activation are recognized key obstacles in the process of virus invasion. On the other hand, the initiation of the innate immune response needs to be strictly regulated, since excessive activation could cause harmful tissue damage and systemic inflammation (8). Thence, the balance regulation between host’s innate immune responses and virus invasion is considered as a potential method for the treatment of viral infection. The balance should be well regulated to maintain antiviral function and avoid excessive inflammation.
1.2 Flaviviruses
The Flavivirus genus is composed of more than 70 positive-stranded RNA viruses transmitted by arthropods, especially mosquitoes and ticks. Flavivirus include pathogens of global concern such as Dengue virus (DENV), West Nile virus (WNV), Zika virus (ZIKV), Japanese encephalitis virus (JEV), Yellow Fever virus (YFV), Tick-borne encephalitis virus (TBEV) and Langat virus (LGTV). These viruses are arboviruses that can cause serious human infections, they pose a threat to global health and may cause serious outbreaks. These are demonstrated by the global distribution of DENV (9), the spread of ZIKV in South America (10), the outbreak of YFV in Brazil and Africa (11, 12), and the WNV outbreak in North America (13). The symptoms of flavivirus infection range from mild fever to joint pain to life-threatening hemorrhagic and encephalitis (14). Although vaccines against several of the viruses including DENV, JEV and YFV have been licensed, the outbreak is still happening. There is currently no clinical antiviral treatment for flavivirus infection, highlighting the challenge in implementing an effective vaccination program (15).
The approximately 11 kb flavivirus genome has only one open reading frame (ORF) flanked by a 5’-untranslated region (UTR) and a 3’-UTR, encoding 3 structural proteins [capsid (C); precursor of M (prM) and envelope (E)] and 7 non-structural (NS) proteins (NS1, NS2A/2B, NS3, NS4A/4B and NS5) (Figure 1A). Structural proteins mainly constitute virions, while NS proteins are involved in viral genome replication and viral particle assembly, and are involved in initiating host innate immunity (16). The nucleocapsid is composed of C protein wrapped RNA genome (17), nucleocapsid is surrounded by a lipid bilayer, M and E proteins are inserted into the lipid bilayer. In immature virus particles, M protein exists in the form of precursor protein (prM), the immature particle contains 60 trimeric spikes of prM-E heterodimers (Figure 1B). PrM is cleaved when the mature virus particle is formed (18), the mature flavivirus particle is composed of 90 E homodimers and 90 M homodimers on the surface, and the E protein is responsible for receptor binding, attachment, membrane fusion and viral entry.
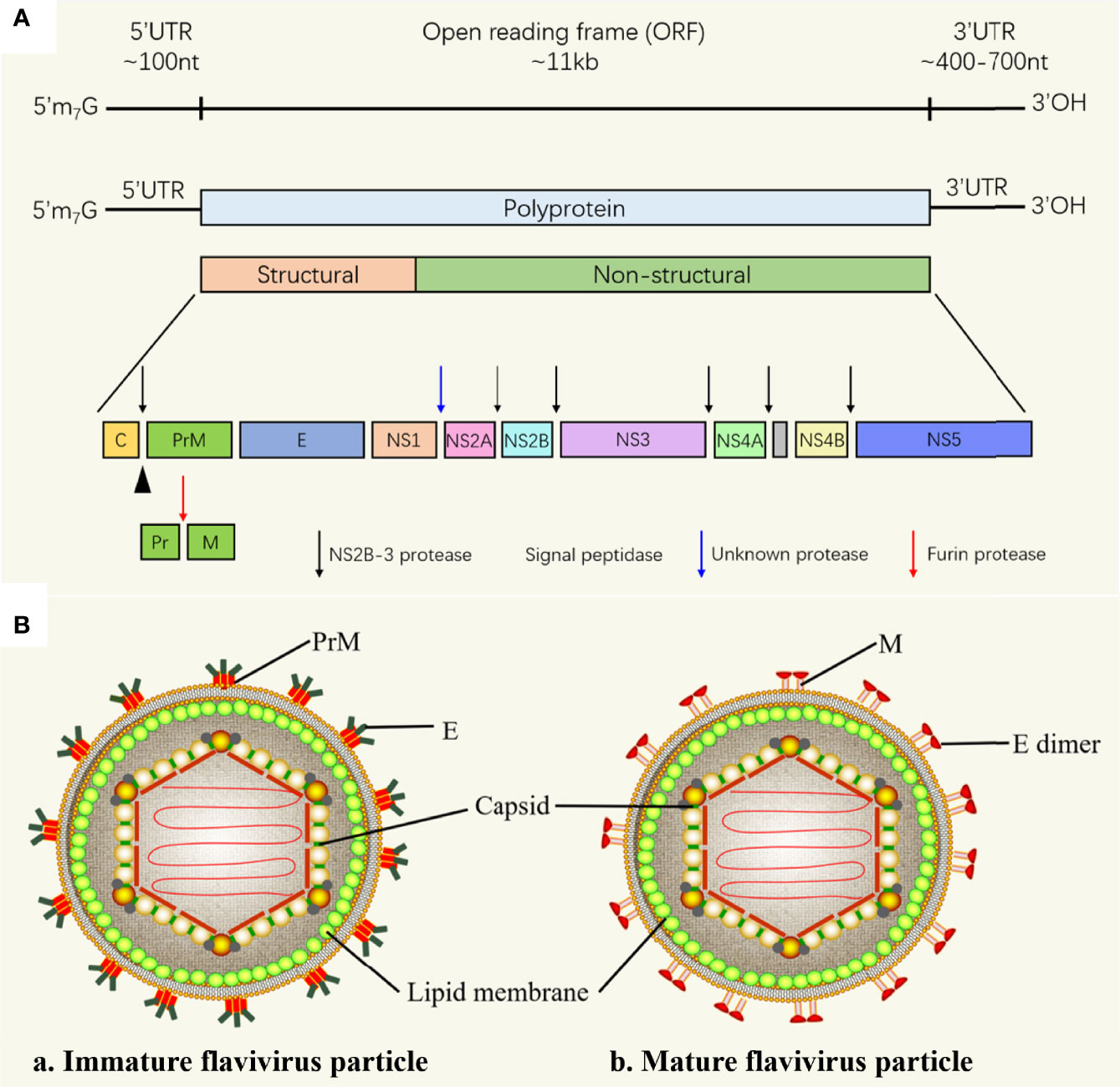
Figure 1 Flavivirus RNA genome and flavivirus particle. (A) Schematic representation of flavivirus genome. The polyprotein encoded by the genome is cleaved by the host and virus proteases to form 3 structural proteins: capsid (C), membrane (M) and envelope (E), and 7 nonstructural proteins (NS-1, 2A, 2B, 3, 4A, 4B and 5) (16). (B) Schematic diagram of the flavivirus particle model. (a) Immature flavivirus particles. The surface of immature virus particles is covered with 60 trimeric spikes, each of which is composed of a prM-E heterodimer (17). (b) Mature flavivirus particles. prM of is cleaved into M to form mature particles, there are 90 E homodimers and 90 M homodimers on the surface of each particle (18).
In the process of infecting the host, flavivirus first attaches to the cell surface and enter host cell through endocytosis mediated by cell surface receptors (Figure 2). The acidic environment of endosome triggers the membrane fusion of the virus with the cell. Through membrane fusion, nucleocapsid is released to cytoplasm, capsid protein and the viral RNA are dissociated, the viral RNA genome begins to replicate, the viral proteins are expressed, and the viral particles begin to assemble. Initially, immature, non-infectious virus particles are formed in the endoplasmic reticulum, which cannot yet induce fusion with the cell membrane (19). Subsequently, immature virus particles are transported to Golgi apparatus, prM is cleaved to form mature M protein, E protein is rearranged, and mature infectious virus particles begin to form. Mature virus particles are released from host cell through cellular exocytosis.
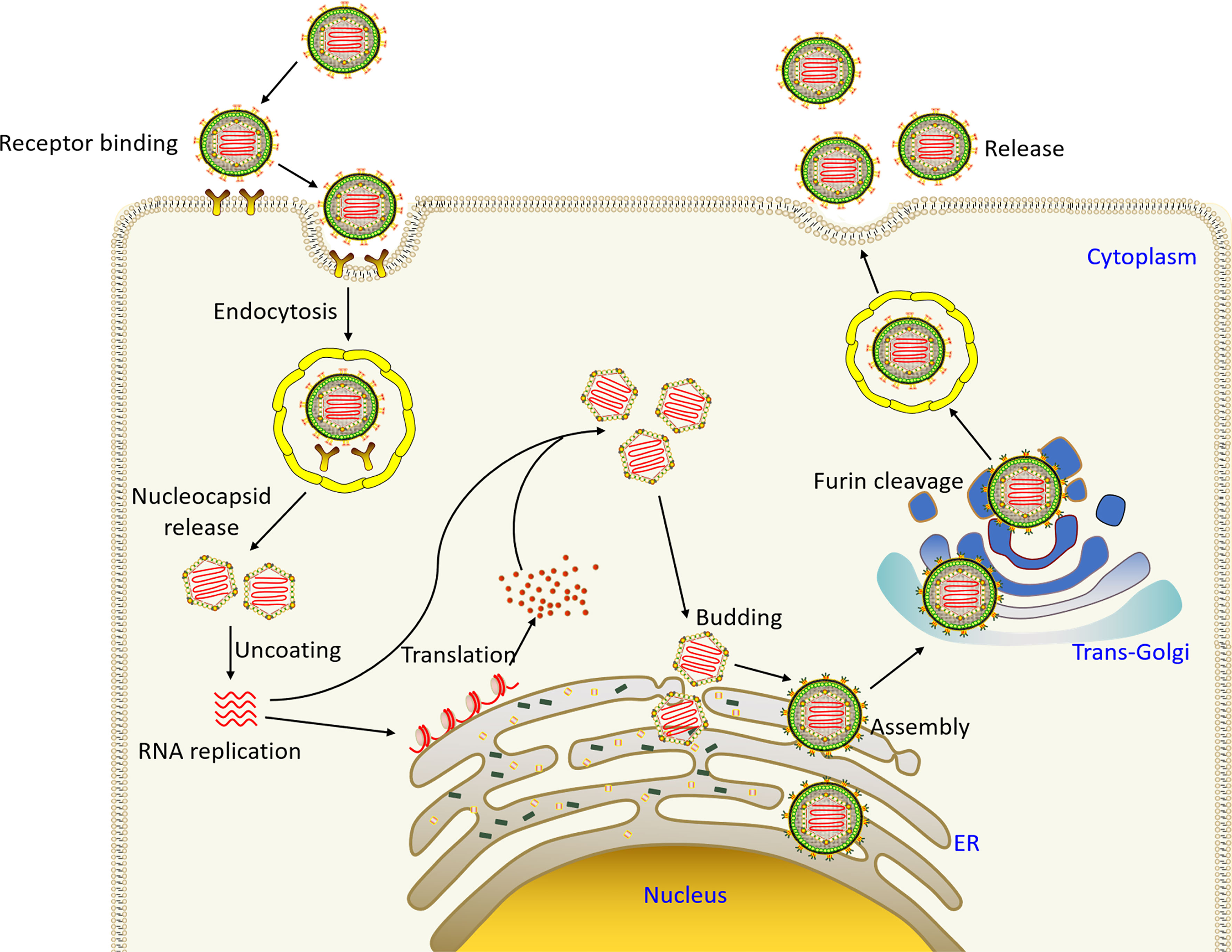
Figure 2 The flavivirus life cycle. Virus particles bind to receptors on the surface of the host cell membrane and enter the cell through endocytosis. The acid environment of endosomal vesicle induces the conformational changes of virus particles, and the fusion of virus and vesicle membrane leads to the release of virus particles. Subsequently, the viral genome is released into cytoplasm, and the positive-sense RNA is directly translated into a polyprotein, which is cleaved and processed by the virus and host proteases. Virus assembly occurs on the surface of the ER, and then non-infectious, immature virus particles carrying prM and E are transported to trans-Golgi network (TGN). The host protease furin then cleaves prM to M, producing mature infectious particles. Eventually mature virus particles are released by exocytosis (19).
Within this article, we discussed innate immune recognition, the activation of inflammatory cell death pathways, and the release of cytokines during flavivirus infection to promote resistance to viral infections; and described how flavivirus evade host innate immune response to promote viral infection.
2 Innate Immune Recognition of Flavivirus
The innate immune system is a strong barrier to prevent flavivirus infection. In a typical flavivirus infection process, viral RNA could be identified by a variety of PRRs, such as TLRs (20), RLRs (21, 22), cyclic GMP-AMP synthase (cGAS) and NLRs (23), which ultimately produce pro-inflammatory cytokines and induce antiviral status (Figure 3 and Table 1). Typically, pro-inflammatory cytokines can trigger the infiltration of immune cells and eliminate infectious viral factors, which is beneficial to the host to a large extent. However, excessive pro-inflammatory cytokines can cause harmful tissue damage and systemic inflammation (53).
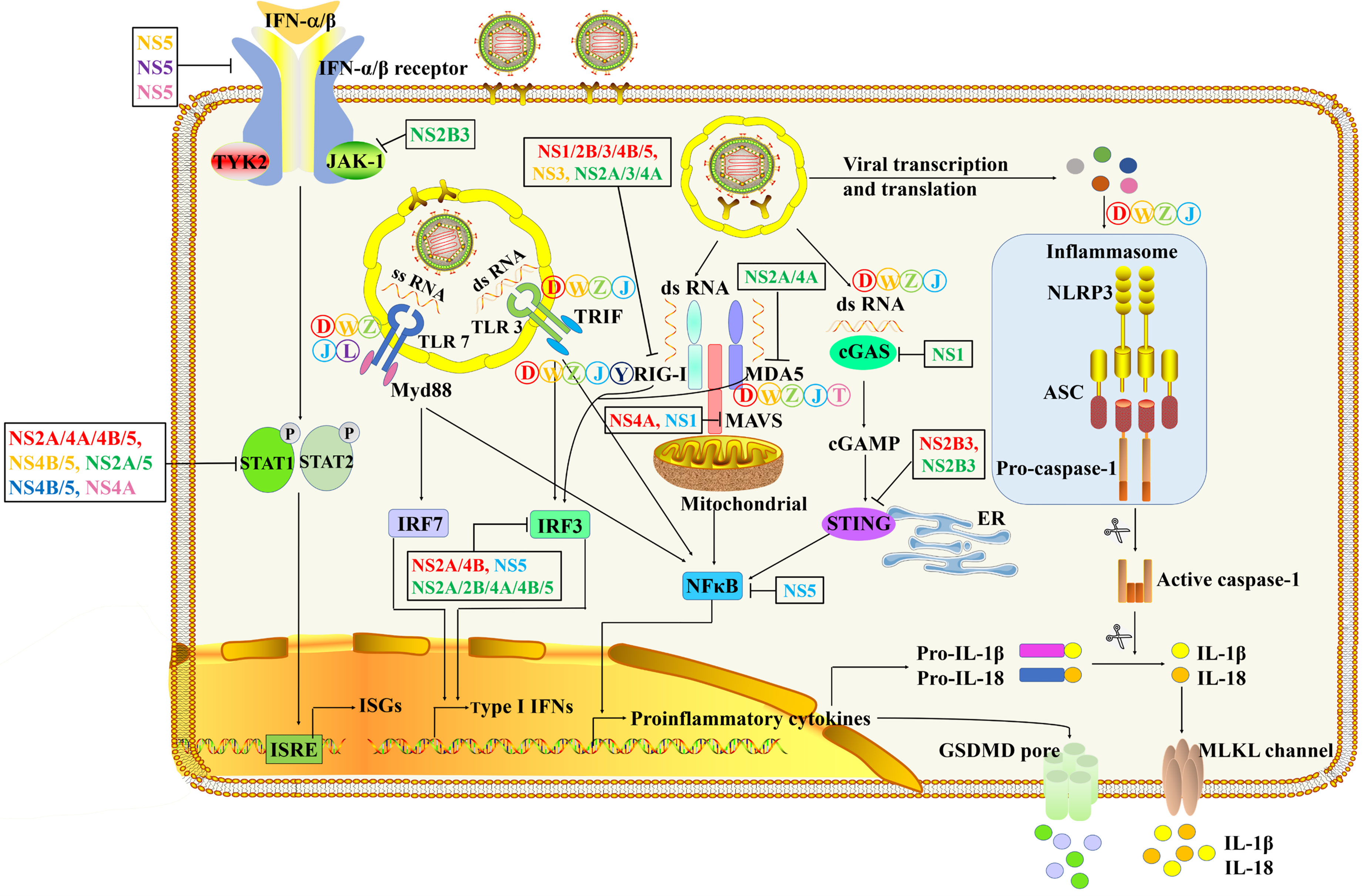
Figure 3 The innate immune pathways in the process of flavivirus infection and immune evasion. After flavivirus infection, TLR3 and TLR7 recognize double-stranded RNA (dsRNA) and single-stranded RNA (ssRNA), respectively. It then triggers a signal cascade downstream of the TLRs to induce the activation of NF-κB to produce pro-inflammatory cytokines and phosphorylation of IRF/7 to drive the production of type I IFN (24–32). The dsRNA RNA in the cytoplasm is recognized by RIG-I and MDA5 (33–36). The combination of RIG-I and MDA5 with MAVS leads to the activation of NF-κB and the phosphorylation of IRF3 (37–41). Severe flavivirus can also stimulate the formation of inflammasomes, leading to caspase-1 activation and release of the cytokines IL-1β and IL-18 (42–45).
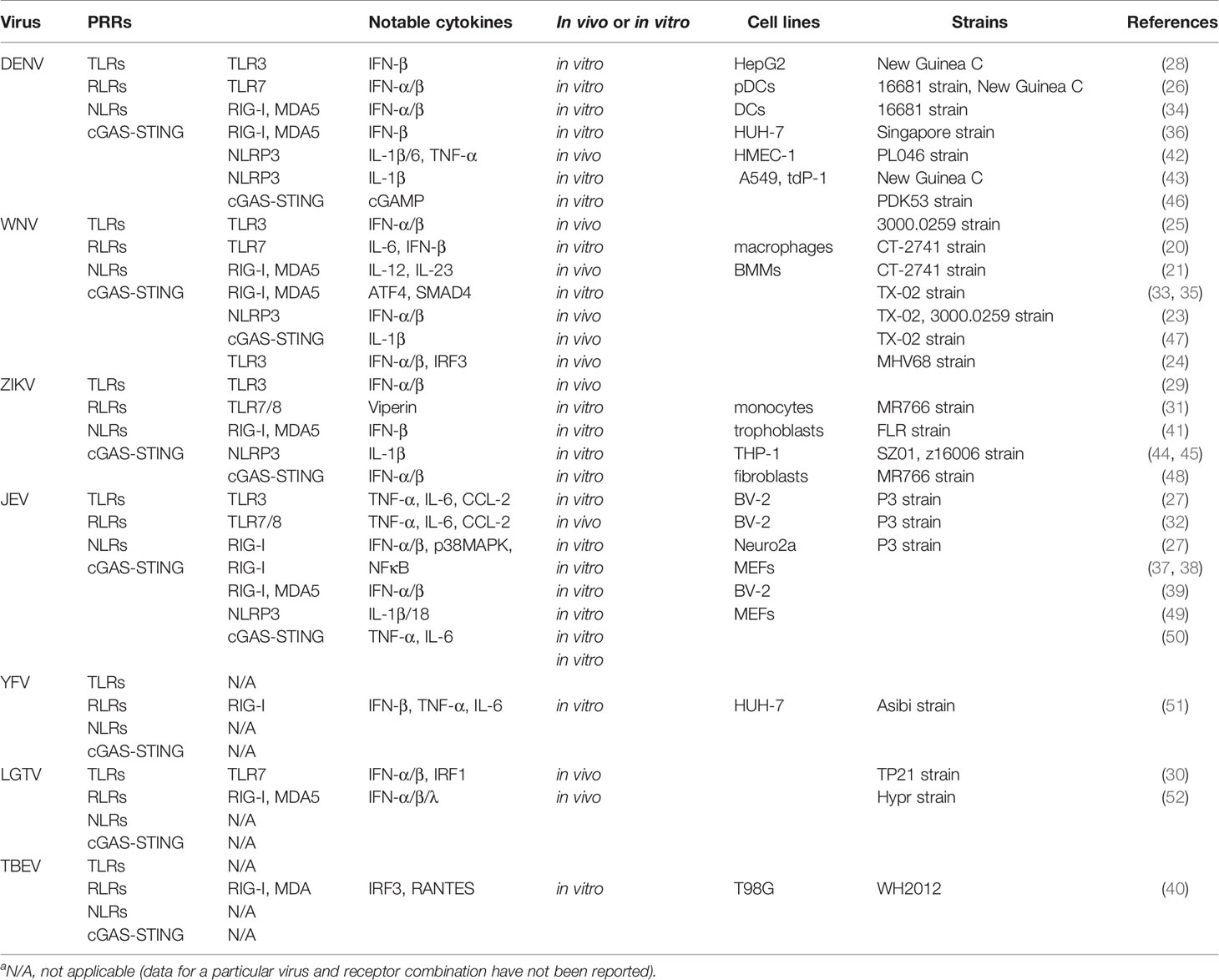
Table 1 Engaged innate immune sensors and notable cytokines released in response to specific flavivirusa.
2.1 TLRs- and RLRs-Mediated Flavivirus Recognition
Signal transduction mediated by TLRs and RLRs results in the secretion of type I interferon (IFN-I), which subsequently stimulates the expression of IFN-stimulating genes (ISGs) to establish an antiviral state (24–26). The TLRs and RLRs signaling cascade also induce the production of pro-inflammatory cytokines, such as interleukin 1β (IL-1β) and interleukin 18 (IL-18) (20).
TLR3 is a membrane-bound PRR located in the endosome, which could recognize dsRNA from DENV, WNV ZIKV and JEV (24, 25, 27–29). TLR3 activates TRIF in turn, and TRIF activates TRAF3/TBK-1/IKKϵ kinase complex through RIP-1. The complex then phosphorylates interferon regulatory factor 3/7 (IRF-3/7) causing their activation. The activated IRF3/7 then induce the transcription of IFN-I genes (24, 25, 28). RIP-1 can also activate IKKα/β kinase and then activate nuclear factor-κB (NFκB) to induce the expression of pro-inflammatory cytokines. TLR7, as another membrane-bound PRR, has also been shown to be involved in regulating the expression of IFN-I and pro-inflammatory cytokines in response to DENV, WNV, ZIKV, JEV and LGTV infection (20, 26, 30–32). After TLR7 recognizes ssRNA, it dimerizes and recruits the myeloid differentiation primary response 88 (MYD88) adaptor protein (20, 26). MYD88 then activates the transcription factors IRF7 and NF-κB, which stimulate the production of IFN-I and pro-inflammatory cytokines for host defense, respectively (30). Indeed, compared with wild-type mice, TLR7-/- and Myd88-/- mice have more severe disease after WNV infection, with reduced survival and increased virus transmission (20).
Melanoma differentiation-associated gene 5 (MDA5) is a cytoplasmic PRR belonging to RLRs family, it typically binds to double-stranded long RNA. Studies have found that it plays a vital role in responding to various flavivirus infections (33, 34). MDA5 is composed of two caspase recruitment domain (CARD) domains at the N-terminus and a DExD/H-box helicase domain at the C-terminus. When the C terminal domain binds to the viral ligand, MDA5 undergoes a conformational change, which allows the CARD domain to bind to mitochondrial antiviral signaling protein (MAVS), and ultimately induces the expression of interferon and pro-inflammatory cytokines (21, 35, 36). RIG-I, another member of the RLRs, can sense 5′-triphosphate-double-stranded RNA (22, 54). So far, RIG-I is participated in identifying almost every member of the flavivirus genus (51, 52). Similar to MDA5, after RIG-I recognizes cytoplasmic dsRNA, it interacts with MAVS located on mitochondria (55). This complex can activate both IRF-3/7 and NF-κB (37, 38), eventually increase the release of IFN-I and pro-inflammatory cytokines, and establish an antiviral state in the cell (39, 40). Accordingly, IFN-β induced by ZIKV decreased in RIG-I–/– and MDA5–/– cells, and completely abolished in MAVS–/– cells, indicating RIG-I and MDA5 play an indispensable role in this process (41).
Interestingly, it’s recently reported that PARPs (poly-adenosine 5′-diphosphate (ADP)-ribose polymerases) family member PARP9 is a non-canonical sensor for RNA virus in dendritic cells (56). Additionally, DHX15 is identified as RNA sensor for RNA viruses and is required to control RNA virus-induced inflammation by activating NLRP6-mediated inflammasome (57, 58). Studies have identified PARP1 and PARP12 as strong inhibitors of ZIKV replication (59, 60). They inhibit virus replication by reducing intracellular ATP and NAD+ concentrations or mediating the degradation of NS1 and NS3 by the proteasome pathway, respectively. Whether the RNA sensors PARP9 and DHX15 have similar functions to control flavivirus infections requires more in-depth research.
2.2 cGAS-Mediated Flavivirus Recognition
Besides the RNA sensors, the DNA sensor cGAS has also been proven to detect and limit flavivirus (46, 47). cGAS is activated after binding to DNA in the cytoplasm, and cyclizes AMP and GMP in the cytoplasm to produce 2’, 3’-cGAMP. As the second signal, cGAMP continues to activate STING, which in turn promotes the expression of interferon and pro-inflammatory cytokines to exert antiviral effects. Studies have found that the disturbance of mitochondrial membrane induced by DENV leads to the leakage of mitochondrial DNA into the cytoplasm, which finally triggers the activation of the cGAS-STING signaling pathway and promotes downstream IFN gene expression (46). In addition, DTMUV infection of a variety of cell lines lacking STING found enhanced replication of DENV (61). ZIKV replication in STING–/– human fibroblasts is enhanced, ZIKV also promotes infection by actively antagonizing STING in the cGAS pathway (48). Besides, JEV can also activate the cGAS-STING axis after infecting the mouse embryonic fibroblasts (MEFs) (50).
2.3 Type I IFN Response During Flavivirus Infection
Mammalian cells mainly sense flavivirus infections through PRRs (including TLRs, RLRs and cGAS), and then their downstream signaling pathways are activated, ultimately inducing the production of IFN-I. Then, the released IFN-I binds to the IFN-I receptors (IFNAR1/IFNAR2) to activate the JAK/STAT signaling cascade to initiate antiviral status.
First, IFN-I plays an important role in resisting DENV infection and generating an immune response (62). Other studies found that IFN-α/β plays a leading role in resisting WNV by limiting cell and tissue tropism infection (63), and TRIM6 helps establish IFN-I-induced antiviral response against WNV (64). In addition, IFN-I is essential to resist ZIKV, since IFN-I-mediated strong antiviral effects on ZIKV replication (>100-fold reduction) (65), while IFNAR-deficient mice are highly susceptible to ZIKV (66). Meanwhile, autophagy induced by ZIKV is conducive to activating host immunity through IFN-I signaling (67). The IFN-I response has also been shown to be a major obstacle to the viscerotropism and pathogenicity of JEV (68), such as IFN-I can limit hemorrhage-like disease after infection with JEV (69). Moreover, IFN-I is also essential to protect against LGTV and TBEV in mice at two different stages. The first stage inhibits virus replication and prevents its spread to the central nervous system (CNS) at the periphery. In the second stage, local IFN responses of the CNS can prevent the development of inflammation and encephalitis caused by the virus (52, 70, 71).
2.4 Flavivirus Infection Stimulates NLRP3 Inflammasome Activation
The innate immune response has significant effects on antiviral immunity, inflammatory signal transduction and cytokine production. Within pro-inflammatory cytokines, IL-1β and IL-18 are crucial factors that trigger inflammatory response. The inflammasome processes inactive pro-caspase-1 into active caspase-1, which cleaves pro-IL-1β/18 into mature IL-1β/18, leading to inflammation (72). The inflammasome sensors can identify PAMPs and DAMPs produced after pathogen infection (72). The elucidation of the NLRs family pyrin domain-containing 3 (NLRP3) is the most thorough among these sensors, and it is related to a variety of diseases, such as autoinflammatory diseases, obesity and colitis (73–79).
DENV can activate NLRP3-specific inflammasome in human patients and mice; specifically including human peripheral blood mononuclear cells (PBMCs), keratinocytes and platelets, as well as mouse bone marrow derived macrophages (BMDMs), endothelial and dendritic cells (80). After DENV infection, caspase-1 is activated by inflammasomes, which cleaves IL-1β and IL-18 to induce inflammatory response (42). In addition, DENV NS2A and NS2B proteins activate the NLRP3 inflammasome, which then increases the oligomerization of apoptosis-associated speck-like protein containing a CARD (ASC), and promotes caspase-1 activation and IL-1β secretion (43). Ramos et al. (23) found that NLRP3 signaling pathway is the most important way to trigger IL-1β production after WNV infection. ZIKV can stimulate human PBMCs, macrophages and mice BMDCs to secret IL-1β through NLRP3 inflammasome (44). Moreover, the combination of ZIKV NS5 protein and NLRP3 promotes the assemble of inflammasome complex to promote the secretion of IL-1β (45). However, overexpression of ZIKV NS3 protein can reduce the activation of caspase-1 and even degrade NLRP3, which ultimately inhibits IL-1β secretion (81). The efflux of K+ ion and the release of reactive oxygen species (ROS) mediated by JEV infection can also induce NLRP3 inflammasome activation (49).
In the host antiviral responses, the activation of NLRP3 inflammasome plays a crucial role (76, 77). However, excessive activation of NLRP3 inflammasome can also cause severe pathological damage. For example, the interaction of DENV M protein and NLRP3 causes over-activation of NLRP3 inflammasomes and excessive release of pro-inflammatory cytokine IL-1β, which ultimately leads to increased endothelial permeability and vascular leakage (82). Additionally, DENV E protein domain III (EIII) induces neutrophil death in vitro and in vivo, which also depends on NLRP3 and caspase-1 (83, 84). During ZIKV infection, acute kidney injury can also be induced by activating the NLRP3 inflammasome (85). The above findings confirm that proper activation of NLRP3 inflammasome is beneficial to the host, but abnormal activation may cause unfavorable results.
3 PANoptosis and Proinflammatory Cytokines During Flavivirus Infections
3.1 PANoptosis
Cell death plays a vital role in resisting pathogen invasion. On the other hand, inflammatory cell death can lead to the release of pro-inflammatory cytokines, cell contents, PAMPs and DAMPs, which will induce a severe inflammatory response (86, 87). The immune system has evolved a variety of mechanisms to limit microbial infections and regulate inflammation. The innate immune system recognizes microbial molecules that are conserved in many pathogens, and responds quickly by producing inflammatory mediators and activating programmed cell death pathways, including pyroptosis, apoptosis, and necroptosis. The activation of pattern recognition receptors, combined with inflammatory cytokine-induced signal transduction through receptors containing death domains, initiates a highly interrelated cell death process called PANoptosis (pyroptosis, apoptosis, necroptosis) (Figure 4).
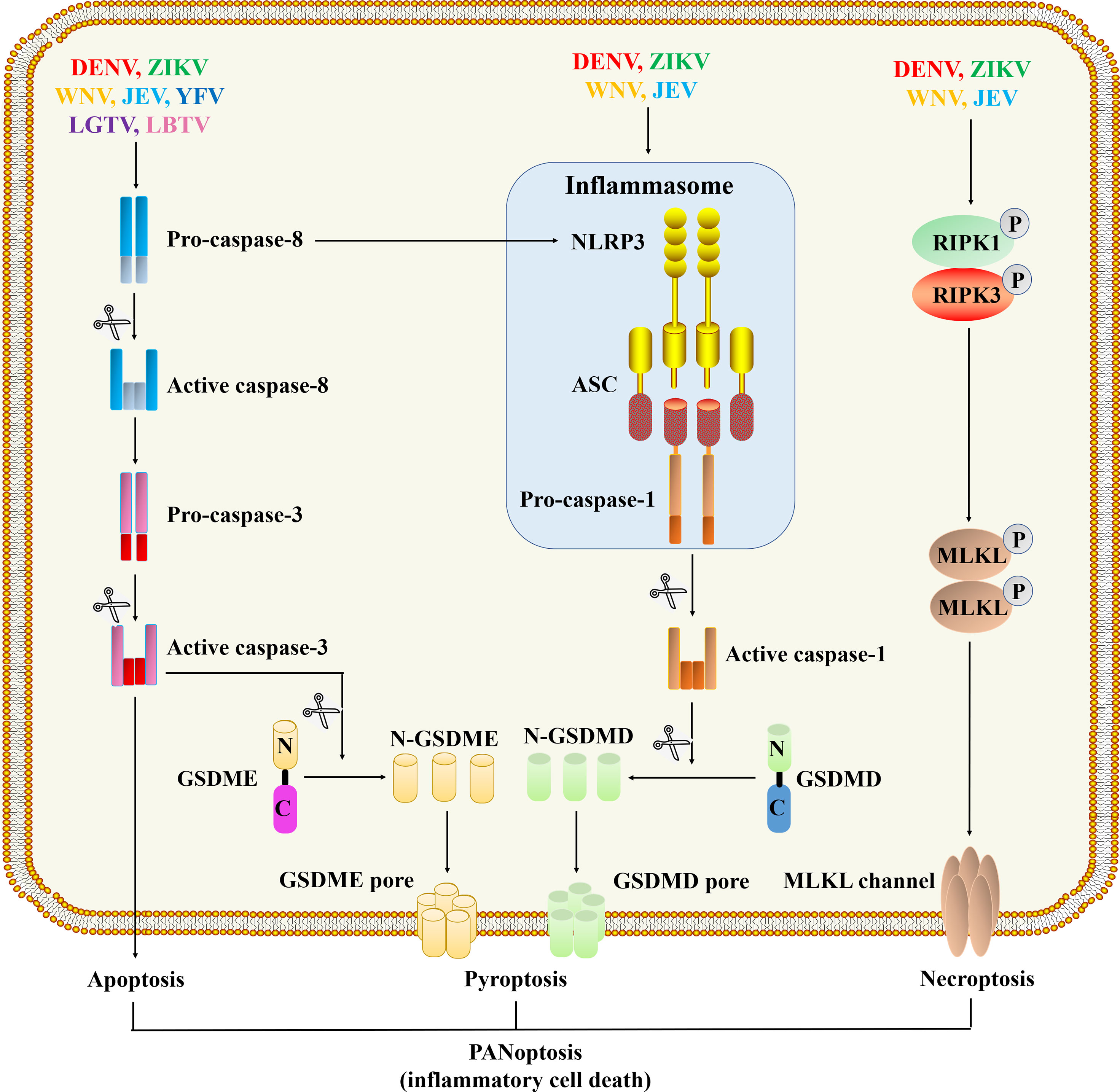
Figure 4 Programmed cell death pathways during flavivirus infection. The viral proteins of DENV (88–90), WNV (91), ZIKA (92, 93) and JEV (94) can act as cytoplasmic PAMPs to stimulate the assembly of inflammasomes, thereby activating caspase-1. Activated caspase-1 cleaves GSDMD, and then the N-terminal fragment of GSDMD oligomerizes in the membrane to form membrane pores and initiate pyroptosis. Flavivirus infections also initiate a signal cascade mediated by caspase-8, activated caspase-8 cleaves and activates caspase-3 to initiate apoptosis (95–98). WNV (68), ZIKA (99, 100) and JEV (101) infections can trigger necroptosis, which depends on the formation of RIPK1 and RIPK3 complexes and the activation of downstream MLKL proteins to form channels in the membrane.
3.2 PANoptosis During Flavivirus Infection
3.2.1 Pyroptosis
Pyroptosis is a way of inflammatory cell death mediated by inflammasome and gasdermin (7). After receiving the pyroptosis signal, the inflammasome assembly causes the activation of inflammatory caspases (caspase-1/4/5/11), and the N-terminal fragment of gasdermin D (GSDMD) produced by the activated caspase is transported to the plasma membrane to form pores, resulting in the production of pro-inflammatory cytokines (2, 6, 7). Among them, the release of IL-1β and IL-18 caused by GSDMD cleavage is closely related to the activation of caspase-1 (2). Recent findings indicate that when infecting monocytes or macrophages, DENV activates the inflammasome and caspase-1, followed by the release of IL-1β and cellular contents, which ultimately induces pyroptosis (88–90). Another study showed that during DENV infection of macrophages, caspase-4 is located upstream of caspase-1 to regulate pyroptosis (102). In addition, both DENV EIII and NS1 proteins can induce pyroptosis through the inflammasome NLRP3, it further causes the typical manifestations of DHF (dengue hemorrhagic fever) such as vascular damage, liver dysfunction, thrombocytopenia, and hemorrhage (84, 103, 104). The pyroptosis induced by ZIKV infection directly affects the development of neural progenitor cells, which is closely related to the development of microcephaly (92, 93). ZIKV’s protease NS2B3 can directly cleave GSDMD in a caspase-independent manner to trigger cell pyroptosis, indicating a new mechanism for ZIKV to directly induce cell death and inflammation (105). Transcriptome analysis of JEV-infected peritoneal macrophages found that almost all PCD pathways, including pyroptosis, were activated after JEV infection (94) (Table 1).
3.2.2 Necroptosis
The activation of necroptosis depends on the phosphorylation of MLKL (mixed-lineage kinase domain-like pseudokinase) regulated by RIPK3 (receptor-interacting serine/threonine protein kinase 3), which causes conformational changes and activation of MLKL. The activated MLKL translocate and form channels in the plasma membrane (106). Increased expression of RIPK1, RIPK3 and MLKL proteins was detected in ZIKV-infected astrocytes, indicating that ZIKV induced necroptosis; after inhibiting necroptosis, virus replication increased significantly, indicating that necroptosis has a resistance to virus replication (99). In addition, retinopathy caused by ZIKV is associated with inflammation mediated by necroptosis (100). Bian et al. (101) demonstrated that necroptosis is related to neuronal loss during JEV infection, providing evidence for necroptosis to participate in the pathogenesis of JEV infection. Transcriptomics analysis of JEV-infected macrophages revealed that the necroptotic pathway was activated, which was confirmed by the immunofluorescent staining with specific markers (94). Similarly, transcriptomics has also found evidence of differential expression of markers of pyroptosis and necroptosis during WNV and CHIKV neuroinvasive diseases, but more research is needed to explain the role of inflammatory cell death in viral neuroinvasive diseases (91). Alternatively, necroptosis can also be activated by the sensor Z-DNA-binding protein 1 (ZBP1), which is an ISGs containing the RHIM domain to recruit and activate RIPK3-induced MLKL phosphorylation, leading to cell death. It has been reported that ZBP1-mediated cell death is involved in various viral infections including WNV (107) and ZIKV (108).
3.2.3 Apoptosis
Pyroptosis and necroptosis-mediated lytic forms of cell death are driven by GSDMD pores or MLKL channels, respectively. They release inflammatory factors and other cytokines to alert nearby cells of danger and recruit innate and adaptive immune cells (86, 87). Apoptosis was originally thought to be a way of cell death that does not cause inflammation. It breaks down cells by forming apoptotic bodies that wrap the cellular contents, which is finally cleared by phagocytes (109). But new research shows that apoptosis is not always immune-silent because of the signal crossing between it and the lytic cell death pathways (109). Apoptosis is driven by the initiator caspase-8, -9 and -10 cleavage executor caspase-3 and -7. Apoptosis is induced by the “initiator” caspase-8, -9 and -10 cleavage downstream “executor” caspase-3 and -7. According to reports, caspase-3 and caspase-8 can cleave and activate GSDME (gasdermin E) or GSDMD, respectively, leading to inflammatory cell death (109, 110).
DENV can induce apoptosis in a variety of cells, such as mouse neuroblastoma cells [Neuro 2a (111)), liver cancer cells (HepG2 (112), Hep3B (113)], endothelial cells (114) and PBMCs (115), and human monocyte-derived dendritic cells (Mo-DC) (116). Among them, DENV2 induces vascular endothelial cell apoptosis through FasL/Fas and XIAP-related factor 1 (XAF1)-dependent pathways, which is related to vascular endothelial dysfunction in the pathogenesis of DHF (117). And sphingosine kinase 2 (SPHK2) plays a pro-apoptotic effect in DENV-infected liver cells, which is associated with liver dysfunction (118). Within 2 hours of being infected with DENV or ZIKV, apoptosis of Aedes aegypti midgut epithelial cells was rapidly induced to prevent virus proliferation (119), similar results were observed in the midgut cells of WNV-infected C. p. pipiens (120). In addition, DENV C protein induces apoptosis by localizing to the nucleus and interacting with Fas death domain associated protein xx (DAXX) (121). The DENV-M ectodomain can activate the mitochondrial-dependent apoptotic pathway after being transported from the Golgi apparatus to the plasma membrane, the M ectodomain of JEV, WNV and YFV also has pro-apoptotic properties (122, 123). In addition, DENV-EIII inhibits megakaryopoiesis by activating the apoptosis of its progenitors, which is associated with thrombocytopenia that is frequently observed in patients with dengue fever (124). However, DENV-NS1 can interact with the key autophagy gene Beclin-1 to inhibit the degradation of Beclin-1, and ultimately promote autophagy and prevent cell apoptosis (125). The proteases NS2B3 and NS3 can trigger apoptosis via the caspase-8 or NF-κB pathway (95, 126). Another study found that DENV-NS2B3 caused endothelial cell apoptosis by activating NF-κB pathway, indicating that NS2B3 is involved in the pathogenesis of DHF (127). Similarly, NS3 of JEV and WNV can also induce apoptosis by activating caspase-3 or caspase-8, leading to extensive damage to the nervous system (95, 96).
WNV induces cell death of various cells with the participation of extrinsic and the intrinsic apoptotic pathways (128, 129). Cellular microRNA Hs_154 was significantly up-regulated after WNV infection, and subsequently caused apoptosis by targeting anti-apoptotic protein (130). WNV C protein interacts with importin-α and triggers phosphorylation of protein kinase C to induce apoptosis (131). On the contrary, WNV-C can activate PI3K/AKT signaling pathway to inhibit the activation of caspase-3 and 8 (97), and other flavivirus capsid proteins can also protect cells from apoptosis by activating Akt (132). WNV-NS2A is also involved in apoptosis and pathogenesis, because after NS2A mutation (converting alanine 30 to proline (A30P)), the quantity of TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) positive cells is greatly decreased (133).
ZIKV infection triggers apoptosis of human neural progenitor cells (134), as evidenced by the activation of caspase 3, 7, 8 and 9, leading to cortical thinning and microcephaly (98). Inhibition of tumor suppressor protein p53 prevents ZIKV-mediated apoptosis of neural progenitor cells, confirming p53 is involved in ZIKV-induced apoptosis (135). The ZIKV-C induces ribosomal stress and apoptosis, and the C protein of DENV has the same function (136). ZIKV-M oligopeptide ZAMP can induce apoptosis by activating caspase-3/7 (137). Interestingly, the subgenomic flaviviral RNA (sfRNA) of ZIKV promotes the spread of ZIKV by inhibiting cell apoptosis in mosquito tissues (138).
JEV can induce apoptosis through multiple signal pathways. It can activate IRE1/JNK pathway of endoplasmic reticulum stress (ERS) to induce apoptosis (139), it can also inhibit the STAT3-Foxo-Bcl-6/p21 pathway to trigger apoptosis (140). What’s more, JEV-NS4B induces apoptosis through the PERK-ATF4-CHOP pathway in response to ER stress (141). Lee et al. showed that the PI3K/Akt pathway triggered by JEV and DENV-2 has an anti-apoptotic effect to protect infected cells from early apoptosis (142). In addition, LGTV-E also induces apoptosis (143), and LGTV-NS3 is a multifunctional protein that binds to caspase-8 and induces apoptosis (144). Activation of the apoptotic pathway can also be observed in TBEV-infected DCs (145). Collectively, these findings indicate that flavivirus can regulate cell apoptosis (Table 1).
3.3 Excessive Pro-Inflammatory Cytokines and Diseases
Excessive activation of the inflammatory cell death can cause severe inflammation and tissue damage (86). For instance, in patients with dengue fever, the severity of the disease is connected with high levels of IL-1RA and CXCL10 in the plasma (146). The detection of severe cases of dengue fever found that the expression of pro-inflammatory cytokines (IL-1, TNFα), anti-inflammatory cytokines IL-10 and chemokines (IL-8, CXCL10) were significantly up-regulated, and this cytokine storm was associated with plasma leakage and hemorrhage (147). The recognition of DENV NS1 by TLR4 can also lead to the pro-inflammatory cytokines production, which contributes to vascular damage (148). In addition, some pro-inflammatory cytokines and IFN-stimulated chemokines are closely related to the severity of ZIKV. Studies have found that ZIKV patients with moderate symptoms and viremia have higher levels of IL-8, IL-1RA, CXCL10 and CCL2 compared with patients with mild symptoms or no viremia (149). This means that immunopathology is an important part of flavivirus pathogenesis, and we need further research to fully clarify the pathways and functional consequences of these pro-inflammatory cytokines released during flavivirus infection.
4 Flavivirus Immune Evasion
The innate immune response uses PRRs to identify pathogens and triggers inflammatory response and programmed cell death to prevent virus invasion and promote its clearance. On the other hand, flavivirus have evolved the ability to limit the innate immune responses to promote viral replication. Among them, the most in-depth research on the immune evasion mechanism of flavivirus is the regulation of type I interferon signals (Figure 3).
DENV NS1 protein interacts with ApoA1, a key component of HDL (high-density lipoprotein), to change the sensitivity of the membrane to viral infections, and ultimately evade the immune response (150). ZIKV-NS1 can inhibit IFN-α response mediated by CD303 (151), Xia et al. (152) found that ZIKV-NS1 inhibits RIG-I-mediated activation of IFN-β promoter, the same author proved that ZIKV NS2A/B, NS4A/B and NS5 could inhibit IFN-β by reducing the phosphorylation of IRF3 at Ser-396. New research shows that cGAS is cleaved by caspase-1 downstream of the NLRP3 inflammasome activated by ZIKV-NS1 (153). Furthermore, ZIKV NS1 and NS4B interact with TBK1 to prevent TBK1 oligomerization, phosphorylation and its mediated activation of IFN-I (154). The NS1 of JEV could inhibit the production of IFN-I by targeting MAVS (155).
In addition, DENV NS2A, NS4A and NS4B can suppress the transcription of ISRE promoter and ISGs by blocking the phosphorylation of STAT1/STAT2 and hinder their nuclear localization to inhibit the IFN response (156), and the NS2A of ZIKV and the NS4B of WNV and YFV have similar functions (157–159). NS2A and NS4B from multiple DENV serotypes can also inhibit IFN production by targeting IRF3 and TBK1, but only NS4A from serotype-1 can inhibit TBK1 (160). ZIKV NS2A and NS4A proteins antagonize the production of IFN-β mediated by MDA5/RIG-I (161). The conserved phosphomimetic motif in NS3 of DENV, WNV and ZIKV competes with RIG-I to bind 14-3-3ϵ, and finally prevents RIG-I from translocating to mitochondria (162). The expression of DENV NS2B can suppress the cGAS/STING-dependent IFN-β promoter activity and down-regulate the level of cGAS protein (163), DENV NS2B3 protease inhibits the production of type I IFN by cleaving STING (61, 164). Similar to DENV NS2B3, ZIKV NS2B3 also cleaves STING to inhibit IFN production (48). Furthermore, ZIKV NS2B3 degrades JAK1 in a proteasome-dependent manner, which ultimately leads to the down-regulation of IFN-mediated ISGs expression (165). According to reports, DENV NS4A binds to MAVS and blocks its interaction with RIG-I and downstream innate immune signals (166). In addition, TBEV antagonizes IRF-1 and IFN-I signaling to suppress dendritic cells function (167), and TBEV-NS4A can inhibit the phosphorylation of STAT1 and STAT2 to block type I and II IFN signaling (168). DENV NS4B can also trigger mitochondria elongation, causing altered MAMs (mitochondria-associated membranes) and reducing IFN production-possibly by preventing the recruitment of activated RIG-I to MAMs (169).
The NS5 of DENV and other flavivirus (WNV, ZIKV, YFV, JEV, TBEV, LGTV) can inhibit IFN signaling by targeting different steps and participants of the IFN-I signaling pathway (170–173). The NS5 protein of DENV and ZIKV binds to STAT2 and degrades STAT2 via the proteasome pathway, thereby inhibiting IFN-α signal transduction (170, 174–177), while YFV NS5 interacts with STAT2 and inhibits downstream ISRE activation (178). The DENV NS5 2′-O-methylation of 5′ also protects the virus from detection by RIG-I (179). Further experiments showed that ZIKV NS5 localizes to the nucleus and inhibits IRF3-mediated IFN-I transcriptional activation, and independent of its effect on STAT2 degradation (180). Additionally, the interaction between ZIKV NS5 and IKKϵ leads to a decrease in IKKϵ protein level and phosphorylation, thus blocking the activation of IRF3 (181). Meanwhile, WNV-NS5 has been proven to effectively prevent STAT1 phosphorylation and translocation to the nucleus (158, 182); JEV NS5 protein blocks IFN-I signaling and antiviral response by inhibiting the activation of transcription factors IRF3 and NF-κB (183). Moreover, the NS5 protein of TBEV, LGTV and WNV can interfere with the maturation of the IFNAR1 receptor and thus affect the IFN-I signaling pathway (184–186). In addition, DENV’s sfRNA can interact with TRIM25 to prevent its activation and interaction with RIG-I (187). ZIKV sfRNA can inhibit IFN-β promoter activation mediated by RIG-I or MDA5 (188), while JEV sfRNA reduces IRF3 phosphorylation and nuclear translocation, as well as downstream IFN-β expression (189).
5 Concluding Remarks
We discussed the perception and recognition of various members of flavivirus by the innate immune system, and the inflammatory cell death pathways initiated by the host in response to flavivirus infections. Appropriate cell death and inflammatory cytokines release are beneficial for host to resist virus invasion. On the contrary, excessive cell death and inflammation can cause harmful cytokine storms and tissue damage. Therefore, the innate immune response and cell death induced by flavivirus need to be strictly regulated to avoid excessive inflammatory response while maintaining antiviral function.
The reduction of innate antiviral defense ability and the excessive production of inflammatory cytokines may be some of the driving characteristics of flavivirus-mediated diseases. Therefore, we should design treatment strategies based on the mechanisms by which different flavivirus regulate innate immune responses, and evaluate the clinical efficacy of targeted innate immune pathways. This article will help us deeply understand the recognition and response of the innate immune system after flavivirus infection, as well as the regulation of different cell death pathways, and lay the foundation for further development of antiviral strategies.
Author Contributions
YP and WC contributed ideas for the review and wrote the manuscript and produced the figures. AC, MW, ZY, and RJ edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (32172833), and the Program Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (SCCXTD-2021-18), and by China Agriculture Research System of MOF and MARA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We apologize to the authors of articles reporting relevant research that were not cited in this manuscript due to limited space.
Abbreviations
PRRs, pattern recognition receptors; TLRs, Toll-like receptors; RLRs, retinoic acid-inducible gene I (RIG-I)-like receptors; NLRs, nucleotide-binding oligomerization domain (NOD)-like receptor family proteins; PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular patterns; DENV, Dengue virus; WNV, West Nile virus; ZIKV, Zika virus; JEV, Japanese encephalitis virus; YFV, Yellow Fever virus; TBEV, Tick-borne encephalitis virus; LGTV, Langat virus; cGAS, cyclic GMP-AMP synthase; IFN, interferon; ISG, IFN-stimulating genes; IL-1β/18, interleukin 1β/18; IRF-3/7, interferon regulatory factor 3/7; NFκB, nuclear factor-κB; MYD88, myeloid differentiation primary response 88; MDA5, melanoma differentiation-associated gene 5; CARD, caspase recruitment domain; MAVS, mitochondrial antiviral signaling protein; CNS, central nervous system; NLRP3, NLR family pyrin domain-containing 3; PCD, programmed cell death; GSDMD/GSDME, gasdermin D/E; MLKL, mixed-lineage kinase domain-like pseudokinase; RIPK, receptor-interacting serine/threonine protein kinase.
References
1. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
2. Man SM, Karki R, Kanneganti T-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol Rev (2017) 277(1):61–75. doi: 10.1111/imr.12534
3. Malik A, Kanneganti T-D. Inflammasome Activation and Assembly at a Glance. J Cell Sci (2017) 130(23):3955–63. doi: 10.1242/jcs.207365
4. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting Edge: NF-kappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol (Baltimore Md 1950) (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
5. Christgen S, Kanneganti T-D. Inflammasomes and the Fine Line Between Defense and Disease. Curr Opin Immunol (2020) 62:39–44. doi: 10.1016/j.coi.2019.11.007
6. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
7. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
8. Nathan C. Points of Control in Inflammation. Nature (2002) 420(6917):846–52. doi: 10.1038/nature01320
9. Messina JP, Brady OJ, Scott TW, Zou C, Pigott DM, Duda KA, et al. Global Spread of Dengue Virus Types: Mapping the 70 Year History. Trends Microbiol (2014) 22(3):138–46. doi: 10.1016/j.tim.2013.12.011
10. Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, et al. Zika Virus: History, Emergence, Biology, and Prospects for Control. Antiviral Res (2016) 130:69–80. doi: 10.1016/j.antiviral.2016.03.010
11. Kraemer MUG, Faria NR, Reiner RC Jr, Golding N, Nikolay B, Stasse S, et al. Spread of Yellow Fever Virus Outbreak in Angola and the Democratic Republic of the Congo 2015-16: A Modelling Study. Lancet Infect Dis (2017) 17(3):330–8. doi: 10.1016/S1473-3099(16)30513-8
12. Hamer DH, Angelo K, Caumes E, van Genderen PJJ, Florescu SA, Popescu CP, et al. Fatal Yellow Fever in Travelers to Brazil, 2018. MMWR Morb Mortal Wkly Rep (2018) 67(11):340–1. doi: 10.15585/mmwr.mm6711e1
13. Hayes EB, Gubler DJ. West Nile Virus: Epidemiology and Clinical Features of an Emerging Epidemic in the United States. Annu Rev Med (2006) 57:181–94. doi: 10.1146/annurev.med.57.121304.131418
14. Gould EA, Solomon T. Pathogenic Flaviviruses. Lancet (Lond Engl) (2008) 371(9611):500–9. doi: 10.1016/S0140-6736(08)60238-X
15. Collins MH, Metz SW. Progress and Works in Progress: Update on Flavivirus Vaccine Development. Clin Ther (2017) 39(8):1519–36. doi: 10.1016/j.clinthera.2017.07.001
16. Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, et al. Strategies for Development of Dengue Virus Inhibitors. Antiviral Res (2010) 85(3):450–62. doi: 10.1016/j.antiviral.2009.12.011
17. Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, et al. The 3.8 Å Resolution Cryo-EM Structure of Zika Virus. Science (New York NY) (2016) 352(6284):467–70. doi: 10.1126/science.aaf5316
18. Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, et al. Cryo-EM Structure of the Mature Dengue Virus at 3.5-Å Resolution. Nat Struct Mol Biol (2013) 20(1):105–10. doi: 10.1038/nsmb.2463
19. Stiasny K, Fritz R, Pangerl K, Heinz FX. Molecular Mechanisms of Flavivirus Membrane Fusion. Amino Acids (2011) 41(5):1159–63. doi: 10.1007/s00726-009-0370-4
20. Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, et al. Toll-Like Receptor 7 Mitigates Lethal West Nile Encephalitis via Interleukin 23-Dependent Immune Cell Infiltration and Homing. Immunity (2009) 30(2):242–53. doi: 10.1016/j.immuni.2008.11.012
21. Stone AEL, Green R, Wilkins C, Hemann EA, Gale M Jr. RIG-I-Like Receptors Direct Inflammatory Macrophage Polarization Against West Nile Virus Infection. Nat Commun (2019) 10(1):3649. doi: 10.1038/s41467-019-11250-5
22. Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, et al. 5'-Triphosphate RNA Is the Ligand for RIG-I. Science (New York NY) (2006) 314(5801):994–7. doi: 10.1126/science.1132505
23. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β Signaling Promotes CNS-Intrinsic Immune Control of West Nile Virus Infection. PloS Pathog (2012) 8(11):e1003039. doi: 10.1371/journal.ppat.1003039
24. Daffis S, Samuel MA, Suthar MS, Gale M Jr, Diamond MS. Toll-Like Receptor 3 has a Protective Role Against West Nile Virus Infection. J Virol (2008) 82(21):10349–58. doi: 10.1128/JVI.00935-08
25. Kong KF, Delroux K, Wang X, Qian F, Arjona A, Malawista SE, et al. Dysregulation of TLR3 Impairs the Innate Immune Response to West Nile Virus in the Elderly. J Virol (2008) 82(15):7613–23. doi: 10.1128/JVI.00618-08
26. Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus Activation of Plasmacytoid Dendritic Cells Delineates Key Elements of TLR7 Signaling Beyond Endosomal Recognition. J Immunol (Baltimore Md 1950) (2006) 177(10):7114–21. doi: 10.4049/jimmunol.177.10.7114
27. Jiang R, Ye J, Zhu B, Song Y, Chen H, Cao S. Roles of TLR3 and RIG-I in Mediating the Inflammatory Response in Mouse Microglia Following Japanese Encephalitis Virus Infection. J Immunol Res (2014) 2014:787023. doi: 10.1155/2014/787023
28. Liang Z, Wu S, Li Y, He L, Wu M, Jiang L, et al. Activation of Toll-Like Receptor 3 Impairs the Dengue Virus Serotype 2 Replication Through Induction of IFN-β in Cultured Hepatoma Cells. PloS One (2011) 6(8):e23346. doi: 10.1371/journal.pone.0023346
29. da Silva MHM, Moises RNC, Alves BEB, Pereira HWB, de Paiva AAP, Morais IC, et al. Innate Immune Response in Patients With Acute Zika Virus Infection. Med Microbiol Immunol (2019) 208(6):703–14. doi: 10.1007/s00430-019-00588-8
30. Baker DG, Woods TA, Butchi NB, Morgan TM, Taylor RT, Sunyakumthorn P, et al. Toll-Like Receptor 7 Suppresses Virus Replication in Neurons But Does Not Affect Viral Pathogenesis in a Mouse Model of Langat Virus Infection. J Gen Virol (2013) 94(Pt 2):336–47. doi: 10.1099/vir.0.043984-0
31. Vanwalscappel B, Tada T, Landau NR. Toll-Like Receptor Agonist R848 Blocks Zika Virus Replication by Inducing the Antiviral Protein Viperin. Virology (2018) 522:199–208. doi: 10.1016/j.virol.2018.07.014
32. Awais M, Wang K, Lin X, Qian W, Zhang N, Wang C, et al. TLR7 Deficiency Leads to TLR8 Compensative Regulation of Immune Response Against JEV in Mice. Front Immunol (2017) 8:160. doi: 10.3389/fimmu.2017.00160
33. Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M Jr. The Essential, Nonredundant Roles of RIG-I and MDA5 in Detecting and Controlling West Nile Virus Infection. J Virol (2013) 87(21):11416–25. doi: 10.1128/JVI.01488-13
34. Sprokholt JK, Kaptein TM, van Hamme JL, Overmars RJ, Gringhuis SI, Geijtenbeek TBH. RIG-I-Like Receptor Activation by Dengue Virus Drives Follicular T Helper Cell Formation and Antibody Production. PloS Pathog (2017) 13(11):e1006738. doi: 10.1371/journal.ppat.1006738
35. Lazear HM, Pinto AK, Ramos HJ, Vick SC, Shrestha B, Suthar MS, et al. Pattern Recognition Receptor MDA5 Modulates CD8+ T Cell-Dependent Clearance of West Nile Virus From the Central Nervous System. J Virol (2013) 87(21):11401–15. doi: 10.1128/JVI.01403-13
36. Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX. RIG-I, MDA5 and TLR3 Synergistically Play an Important Role in Restriction of Dengue Virus Infection. PloS Negl Trop Dis (2011) 5(1):e926. doi: 10.1371/journal.pntd.0000926
37. Nazmi A, Dutta K, Basu A. RIG-I Mediates Innate Immune Response in Mouse Neurons Following Japanese Encephalitis Virus Infection. PloS One (2011) 6(6):e21761. doi: 10.1371/journal.pone.0021761
38. Nazmi A, Mukhopadhyay R, Dutta K, Basu A. STING Mediates Neuronal Innate Immune Response Following Japanese Encephalitis Virus Infection. Sci Rep (2012) 2:347. doi: 10.1038/srep00347
39. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature (2006) 441(7089):101–5. doi: 10.1038/nature04734
40. Zheng Z, Yang J, Jiang X, Liu Y, Zhang X, Li M, et al. Tick-Borne Encephalitis Virus Nonstructural Protein NS5 Induces RANTES Expression Dependent on the RNA-Dependent RNA Polymerase Activity. J Immunol (Baltimore Md 1950) (2018) 201(1):53–68. doi: 10.4049/jimmunol.1701507
41. Ma J, Ketkar H, Geng T, Lo E, Wang L, Xi J, et al. Zika Virus Non-Structural Protein 4a Blocks the RLR-MAVS Signaling. Front Microbiol (2018) 9:1350. doi: 10.3389/fmicb.2018.01350
42. Lien TS, Sun DS, Chang CM, Wu CY, Dai MS, Chan H, et al. Dengue Virus and Antiplatelet Autoantibodies Synergistically Induce Haemorrhage Through Nlrp3-Inflammasome and Fcγriii. Thromb Haemostasis (2015) 113(5):1060–70. doi: 10.1160/TH14-07-0637
43. Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, Leon-Juarez M, Chavez-Munguia B, Lopez T, et al. Dengue Virus Serotype 2 and Its Non-Structural Proteins 2A and 2B Activate NLRP3 Inflammasome. Front Immunol (2020) 11:352. doi: 10.3389/fimmu.2020.00352
44. He Z, Chen J, Zhu X, An S, Dong X, Yu J, et al. NLRP3 Inflammasome Activation Mediates Zika Virus-Associated Inflammation. J Infect Dis (2018) 217(12):1942–51. doi: 10.1093/infdis/jiy129
45. Wang W, Li G, Wu D, Luo Z, Pan P, Tian M, et al. Zika Virus Infection Induces Host Inflammatory Responses by Facilitating NLRP3 Inflammasome Assembly and Interleukin-1β Secretion. Nat Commun (2018) 9(1):106. doi: 10.1038/s41467-017-02645-3
46. Sun B, Sundström KB, Chew JJ, Bist P, Gan ES, Tan HC, et al. Dengue Virus Activates cGAS Through the Release of Mitochondrial DNA. Sci Rep (2017) 7(1):3594. doi: 10.1038/s41598-017-03932-1
47. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Pan-Viral Specificity of IFN-Induced Genes Reveals New Roles for cGAS in Innate Immunity. Nature (2014) 505(7485):691–5. doi: 10.1038/nature12862
48. Ding Q, Gaska JM, Douam F, Wei L, Kim D, Balev M, et al. Species-Specific Disruption of STING-Dependent Antiviral Cellular Defenses by the Zika Virus NS2B3 Protease. Proc Natl Acad Sci USA (2018) 115(27):E6310–8. doi: 10.1073/pnas.1803406115
49. Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 Inflammasome: Key Mediator of Neuroinflammation in Murine Japanese Encephalitis. PloS One (2012) 7(2):e32270. doi: 10.1371/journal.pone.0032270
50. Sharma KB, Chhabra S, Aggarwal S, Tripathi A, Banerjee A, Yadav AK, et al. Proteomic Landscape of Japanese Encephalitis Virus-Infected Fibroblasts. J Gen Virol (2021) 102(9):. doi: 10.1099/jgv.0.001657
51. Beauclair G, Streicher F, Chazal M, Bruni D, Lesage S, Gracias S, et al. Retinoic Acid Inducible Gene I and Protein Kinase R, But Not Stress Granules, Mediate the Proinflammatory Response to Yellow Fever Virus. J Virol (2020) 94(22):e00403–20. doi: 10.1128/JVI.00403-20
52. Kurhade C, Zegenhagen L, Weber E, Nair S, Michaelsen-Preusse K, Spanier J, et al. Type I Interferon Response in Olfactory Bulb, the Site of Tick-Borne Flavivirus Accumulation, Is Primarily Regulated by IPS-1. J Neuroinflamm (2016) 13:22. doi: 10.1186/s12974-016-0487-9
53. Malavige GN, Jeewandara C, Ogg GS. Dysfunctional Innate Immune Responses and Severe Dengue. Front Cell Infect Microbiol (2020) 10:590004. doi: 10.3389/fcimb.2020.590004
54. Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, et al. The C-Terminal Regulatory Domain Is the RNA 5'-Triphosphate Sensor of RIG-I. Mol Cell (2008) 29(2):169–79. doi: 10.1016/j.molcel.2007.10.032
55. Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, et al. IPS-1 Is Essential for the Control of West Nile Virus Infection and Immunity. PloS Pathog (2010) 6(2):e1000757. doi: 10.1371/journal.ppat.1000757
56. Xing J, Zhang A, Du Y, Fang M, Minze LJ, Liu YJ, et al. Identification of Poly(ADP-Ribose) Polymerase 9 (PARP9) as a Noncanonical Sensor for RNA Virus in Dendritic Cells. Nat Commun (2021) 12(1):2681. doi: 10.1038/s41467-021-23003-4
57. Lu H, Lu N, Weng L, Yuan B, Liu YJ, Zhang Z. DHX15 Senses Double-Stranded RNA in Myeloid Dendritic Cells. J Immunol (Baltimore Md 1950) (2014) 193(3):1364–72. doi: 10.4049/jimmunol.1303322
58. Xing J, Zhou X, Fang M, Zhang E, Minze LJ, Zhang Z. DHX15 Is Required to Control RNA Virus-Induced Intestinal Inflammation. Cell Rep (2021) 35(12):109205. doi: 10.1016/j.celrep.2021.109205
59. Xu G, Li S, Liu X, Gao P, Chen X, Wang H, et al. PARP-1 Mediated Cell Death Is Directly Activated by ZIKV Infection. Virology (2019) 537:254–62. doi: 10.1016/j.virol.2019.08.024
60. Li L, Zhao H, Liu P, Li C, Quanquin N, Ji X, et al. Suppresses Zika Virus Infection Through PARP-Dependent Degradation of NS1 and NS3 Viral Proteins. Sci Signal (2018) 11(535):eaas9332. doi: 10.1126/scisignal.aas9332
61. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PloS Pathog (2012) 8(10):e1002934. doi: 10.1371/journal.ppat.1002934
62. Züst R, Toh YX, Valdés I, Cerny D, Heinrich J, Hermida L, et al. Type I Interferon Signals in Macrophages and Dendritic Cells Control Dengue Virus Infection: Implications for a New Mouse Model to Test Dengue Vaccines. J Virol (2014) 88(13):7276–85. doi: 10.1128/JVI.03827-13
63. Daffis S, Suthar MS, Gale M Jr, Diamond MS. Measure and Countermeasure: Type I IFN (IFN-Alpha/Beta) Antiviral Response Against West Nile Virus. J Innate Immun (2009) 1(5):435–45. doi: 10.1159/000226248
64. van Tol S, Atkins C, Bharaj P, Johnson KN, Hage A, Freiberg AN. VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response During West Nile Virus Infection. J Virol (2020) 94(2):e01454–1. doi: 10.1128/JVI.01454-19
65. Gobillot TA, Humes D, Sharma A, Kikawa C, Overbaugh J. The Robust Restriction of Zika Virus by Type-I Interferon in A549 Cells Varies by Viral Lineage and Is Not Determined by IFITM3. Viruses (2020) 12(5):503. doi: 10.3390/v12050503
66. Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, et al. Type I Interferons Instigate Fetal Demise After Zika Virus Infection. Sci Immunol (2018) 3(19):eaao1680. doi: 10.1126/sciimmunol.aao1680
67. Huang Y, Wang Y, Meng S, Chen Z, Kong H, Pan T, et al. Autophagy Contributes to Host Immunity and Protection Against Zika Virus Infection via Type I IFN Signaling. Mediators Inflamm (2020) 2020p:9527147. doi: 10.1155/2020/9527147
68. Li XF, Li XD, Deng CL, Dong HL, Zhang QY, Ye Q, et al. Visualization of a Neurotropic Flavivirus Infection in Mouse Reveals Unique Viscerotropism Controlled by Host Type I Interferon Signaling. Theranostics (2017) 7(4):912–25. doi: 10.7150/thno.16615
69. Patil AM, Choi JY, Park SO, Uyangaa E, Kim B, Kim K, et al. Type I IFN Signaling Limits Hemorrhage-Like Disease After Infection With Japanese Encephalitis Virus Through Modulating a Prerequisite Infection of CD11bLy-6C Monocytes. J Neuroinflamm (2021) 18(1):136. doi: 10.1186/s12974-021-02180-5
70. Weber E, Finsterbusch K, Lindquist R, Nair S, Lienenklaus S, Gekara NO, et al. Type I Interferon Protects Mice From Fatal Neurotropic Infection With Langat Virus by Systemic and Local Antiviral Responses. J Virol (2014) 88(21):12202–12. doi: 10.1128/JVI.01215-14
71. Lindqvist R, Mundt F, Gilthorpe JD, Wölfel S, Gekara NO, Kröger A, et al. Fast Type I Interferon Response Protects Astrocytes From Flavivirus Infection and Virus-Induced Cytopathic Effects. J Neuroinflamm (2016) 13(1):277. doi: 10.1186/s12974-016-0748-7
72. Kanneganti T-D. Central Roles of NLRs and Inflammasomes in Viral Infection. Nat Rev Immunol (2010) 10(10):688–98. doi: 10.1038/nri2851
73. Gurung P, Kanneganti T-D. Autoinflammatory Skin Disorders: The Inflammasomme in Focus. Trends Mol Med (2016) 22(7):545–64. doi: 10.1016/j.molmed.2016.05.003
74. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 Inflammasome Protects Against Loss of Epithelial Integrity and Mortality During Experimental Colitis. Immunity (2010) 32(3):379–91. doi: 10.1016/j.immuni.2010.03.003
75. Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome Is a Central Player in the Induction of Obesity and Insulin Resistance. Proc Natl Acad Sci USA (2011) 108(37):15324–9. doi: 10.1073/pnas.1100255108
76. Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and Small Antiviral Compounds Activate Caspase-1 Through Cryopyrin/Nalp3. Nature (2006) 440(7081):233–6. doi: 10.1038/nature04517
77. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical Role for Cryopyrin/Nalp3 in Activation of Caspase-1 in Response to Viral Infection and Double-Stranded RNA. J Biol Chem (2006) 281(48):36560–8. doi: 10.1074/jbc.M607594200
78. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, et al. Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection Against Aspergillus Infection. Cell Host Microbe (2015) 17(3):357–68. doi: 10.1016/j.chom.2015.01.006
79. Sharma BR, Kanneganti T-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat Immunol (2021) 22(5):550–9. doi: 10.1038/s41590-021-00886-5
80. Shrivastava G, Valenzuela Leon PC, Calvo E. Inflammasome Fuels Dengue Severity. Front Cell Infect Microbiol (2020) 10:489. doi: 10.3389/fcimb.2020.00489
81. Gim E, Shim DW, Hwang I, Shin OS, Yu JW. Zika Virus Impairs Host NLRP3-Mediated Inflammasome Activation in an NS3-Dependent Manner. Immune Netw (2019) 19(6):e40. doi: 10.4110/in.2019.19.e40
82. Pan P, Zhang Q, Liu W, Wang W, Lao Z, Zhang W, et al. Dengue Virus M Protein Promotes NLRP3 Inflammasome Activation To Induce Vascular Leakage in Mice. J Virol (2019) 93(21):e00996–19. doi: 10.1128/JVI.00996-19
83. Lien TS, Sun DS, Hung SC, Wu WS, Chang HH. Dengue Virus Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent NETosis-Mediated Inflammation in Mice. Front Immunol (2021) 12:618577. doi: 10.3389/fimmu.2021.618577
84. Lien TS, Chan H, Sun DS, Wu JC, Lin YY, Lin GL, et al. Exposure of Platelets to Dengue Virus and Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Platelet Cell Death and Thrombocytopenia in Mice. Front Immunol (2021) 12:616394. doi: 10.3389/fimmu.2021.616394
85. Liu T, Tang L, Tang H, Pu J, Gong S, Fang D, et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front Immunol (2019) 10:1925. doi: 10.3389/fimmu.2019.01925
86. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
87. Pasparakis M, Vandenabeele P. Necroptosis and Its Role in Inflammation. Nature (2015) 517(7534):311–20. doi: 10.1038/nature14191
88. Tan TY, Chu JJH. Dengue Virus-Infected Human Monocytes Trigger Late Activation of Caspase-1, Which Mediates Pro-Inflammatory IL-1β Secretion and Pyroptosis. J Gen Virol (2013) 94(Pt 10):2215–20. doi: 10.1099/vir.0.055277-0
89. Castillo JA, Urcuqui-Inchima S. Mechanisms of Monocyte Cell Death Triggered by Dengue Virus Infection. Apoptosis an Int J Programmed Cell Death (2018) 23(11-12):576–86. doi: 10.1007/s10495-018-1488-1
90. Wu MF, Chen ST, Yang AH, Lin WW, Lin YL, Chen NJ, et al. CLEC5A Is Critical for Dengue Virus-Induced Inflammasome Activation in Human Macrophages. Blood (2013) 121(1):95–106. doi: 10.1182/blood-2012-05-430090
91. Lim SM, van den Ham HJ, Oduber M, Martina E, Zaaraoui-Boutahar F, Roose JM, et al. Transcriptomic Analyses Reveal Differential Gene Expression of Immune and Cell Death Pathways in the Brains of Mice Infected With West Nile Virus and Chikungunya Virus. Front Microbiol (2017) 8:1556. doi: 10.3389/fmicb.2017.01556
92. He Z, An S, Chen J, Zhang S, Tan C, Yu J, et al. Neural Progenitor Cell Pyroptosis Contributes to Zika Virus-Induced Brain Atrophy and Represents a Therapeutic Target. Proc Natl Acad Sci USA (2020) 117(38):23869–78. doi: 10.1073/pnas.2007773117
93. de Sousa JR, Azevedo RDSDS, Martins Filho AJ, de Araujo MTF, Cruz EDRM, Vasconcelos BCB, et al. In Situ Inflammasome Activation Results in Severe Damage to the Central Nervous System in Fatal Zika Virus Microcephaly Cases. Cytokine (2018) 111:255–64. doi: 10.1016/j.cyto.2018.08.008
94. Wang Z-Y, Zhen ZD, Fan DY, Wang PG, An J. Transcriptomic Analysis Suggests the M1 Polarization and Launch of Diverse Programmed Cell Death Pathways in Japanese Encephalitis Virus-Infected Macrophages. Viruses (2020) 12(3):356. doi: 10.3390/v12030356
95. Ramanathan MP, Chambers JA, Pankhong P, Chattergoon M, Attatippaholkun W, Dang K, et al. Host Cell Killing by the West Nile Virus NS2B-NS3 Proteolytic Complex: NS3 Alone Is Sufficient to Recruit Caspase-8-Based Apoptotic Pathway. Virology (2006) 345(1):56–72. doi: 10.1016/j.virol.2005.08.043
96. Yang T-C, Shiu SL, Chuang PH, Lin YJ, Wan L, Lan YC, et al. Japanese Encephalitis Virus NS2B-NS3 Protease Induces Caspase 3 Activation and Mitochondria-Mediated Apoptosis in Human Medulloblastoma Cells. Virus Res (2009) 143(1):77–85. doi: 10.1016/j.virusres.2009.03.007
97. Urbanowski MD, Hobman TC. The West Nile Virus Capsid Protein Blocks Apoptosis Through a Phosphatidylinositol 3-Kinase-Dependent Mechanism. J Virol (2013) 87(2):872–81. doi: 10.1128/JVI.02030-12
98. Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, et al. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell (2016) 19(1):120–6. doi: 10.1016/j.stem.2016.04.017
99. Wen C, Yu Y, Gao C, Qi X, Cardona CJ, Xing Z. RIPK3-Dependent Necroptosis Is Induced and Restricts Viral Replication in Human Astrocytes Infected With Zika Virus. Front Cell Infect Microbiol (2021) 11:637710. doi: 10.3389/fcimb.2021.637710
100. Li Y, Shi S, Xia F, Shan C, Ha Y, Zou J, et al. Zika Virus Induces Neuronal and Vascular Degeneration in Developing Mouse Retina. Acta Neuropathologica Commun (2021) 9(1):97. doi: 10.1186/s40478-021-01195-6
101. Bian P, Zheng X, Wei L, Ye C, Fan H, Cai Y, et al. MLKL Mediated Necroptosis Accelerates JEV-Induced Neuroinflammation in Mice. Front Microbiol (2017) 8:303. doi: 10.3389/fmicb.2017.00303
102. Cheung KT, Sze DM, Chan KH, Leung PH. Involvement of Caspase-4 in IL-1 Beta Production and Pyroptosis in Human Macrophages During Dengue Virus Infection. Immunobiology (2018) 223(4-5):356–64. doi: 10.1016/j.imbio.2017.10.044
103. Lien T-S, Sun DS, Wu CY, Chang HH. Exposure to Dengue Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Endothelial Dysfunction and Hemorrhage in Mice. Front Immunol (2021) 12:617251. doi: 10.3389/fimmu.2021.617251
104. Suwanmanee S, Luplertlop N. Immunopathogenesis of Dengue Virus-Induced Redundant Cell Death: Apoptosis and Pyroptosis. Viral Immunol (2017) 30(1):13–9. doi: 10.1089/vim.2016.0092
105. Yamaoka Y, Matsunaga S, Jeremiah SS, Nishi M, Miyakawa K, Morita T, et al. Zika Virus Protease Induces Caspase-Independent Pyroptotic Cell Death by Directly Cleaving Gasdermin D. Biochem Biophys Res Commun (2021) 534:666–71. doi: 10.1016/j.bbrc.2020.11.023
106. Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep (2014) 7(4):971–81. doi: 10.1016/j.celrep.2014.04.026
107. Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, et al. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell (2017) 169(2):301–13. doi: 10.1016/j.cell.2017.03.011
108. Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity (2019) 50(1):64–76. doi: 10.1016/j.immuni.2018.11.017
109. Place DE, Lee S, Kanneganti T-D. PANoptosis in Microbial Infection. Curr Opin Microbiol (2021) 59:42–9. doi: 10.1016/j.mib.2020.07.012
110. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation During Apoptosis and Inflammasome Activation. Nat Commun (2019) 10(1):1689. doi: 10.1038/s41467-019-09397-2
111. Desprès P, Flamand M, Ceccaldi PE, Deubel V. Human Isolates of Dengue Type 1 Virus Induce Apoptosis in Mouse Neuroblastoma Cells. J Virol (1996) 70(6):4090–6. doi: 10.1128/jvi.70.6.4090-4096.1996
112. Thepparit C, Khakpoor A, Khongwichit S, Wikan N, Fongsaran C, Chingsuwanrote P, et al. Dengue 2 Infection of HepG2 Liver Cells Results in Endoplasmic Reticulum Stress and Induction of Multiple Pathways of Cell Death. BMC Res Notes (2013) 6:372. doi: 10.1186/1756-0500-6-372
113. Thongtan T, Panyim S, Smith DR. Apoptosis in Dengue Virus Infected Liver Cell Lines HepG2 and Hep3B. J Med Virol (2004) 72(3):436–44. doi: 10.1002/jmv.20004
114. Huang J, Li Y, Qi Y, Zhang Y, Zhang L, Wang Z, et al. Coordinated Regulation of Autophagy and Apoptosis Determines Endothelial Cell Fate During Dengue Virus Type 2 Infection. Mol Cell Biochem (2014) 397(1-2):157–65. doi: 10.1007/s11010-014-2183-3
115. Cherupanakkal C, Samadanam DM, Muthuraman KR, Ramesh S, Venkatesan A, Balakrishna Pillai AK, et al. Lipid Peroxidation, DNA Damage, and Apoptosis in Dengue Fever. IUBMB Life (2018) 70(11):1133–43. doi: 10.1002/iub.1925
116. Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, et al. Cellular Oxidative Stress Response Controls the Antiviral and Apoptotic Programs in Dengue Virus-Infected Dendritic Cells. PloS Pathog (2014) 10(12):e1004566. doi: 10.1371/journal.ppat.1004566
117. Long X, Li Y, Qi Y, Xu J, Wang Z, Zhang X, et al. XAF1 Contributes to Dengue Virus-Induced Apoptosis in Vascular Endothelial Cells. FASEB J Off Publ Fed Am Societies Exp Biol (2013) 27(3):1062–73. doi: 10.1096/fj.12-213967
118. Morchang A, Lee RCH, Yenchitsomanus PT, Sreekanth GP, Noisakran S, Chu JJH, et al. RNAi Screen Reveals a Role of SPHK2 in Dengue Virus-Mediated Apoptosis in Hepatic Cell Lines. PloS One (2017) 12(11):e0188121. doi: 10.1371/journal.pone.0188121
119. Ayers JB, Coatsworth HG, Kang S, Dinglasan RR, Zhou L. Clustered Rapid Induction of Apoptosis Limits ZIKV and DENV-2 Proliferation in the Midguts of Aedes Aegypti. Commun Biol (2021) 4(1):69. doi: 10.1038/s42003-020-01614-9
120. Vaidyanathan R, Scott TW. Apoptosis in Mosquito Midgut Epithelia Associated With West Nile Virus Infection. Apoptosis an Int J Programmed Cell Death (2006) 11(9):1643–51. doi: 10.1007/s10495-006-8783-y
121. Netsawang J, Panaampon J, Khunchai S, Kooptiwut S, Nagila A, Puttikhunt C, et al. Dengue Virus Disrupts Daxx and NF-κb Interaction to Induce CD137-Mediated Apoptosis. Biochem Biophys Res Commun (2014) 450(4):1485–91. doi: 10.1016/j.bbrc.2014.07.016
122. Catteau A, Kalinina O, Wagner MC, Deubel V, Courageot MP, Desprès P. Dengue Virus M Protein Contains a Proapoptotic Sequence Referred to as ApoptoM. J Gen Virol (2003) 84(Pt 10):2781–93. doi: 10.1099/vir.0.19163-0
123. Brabant M, Baux L, Casimir R, Briand JP, Chaloin O, Porceddu M, et al. A Flavivirus Protein M-Derived Peptide Directly Permeabilizes Mitochondrial Membranes, Triggers Cell Death and Reduces Human Tumor Growth in Nude Mice. Apoptosis an Int J programmed Cell Death (2009) 14(10):1190–203. doi: 10.1007/s10495-009-0394-y
124. Lin G-L, Chang HH, Lien TS, Chen PK, Chan H, Su MT, et al. Suppressive Effect of Dengue Virus Envelope Protein Domain III on Megakaryopoiesis. Virulence (2017) 8(8):1719–31. doi: 10.1080/21505594.2017.1343769
125. Lu Z-Y, Cheng MH, Yu CY, Lin YS, Yeh TM, Chen CL, et al. Dengue Nonstructural Protein 1 Maintains Autophagy Through Retarding Caspase-Mediated Cleavage of Beclin-1. Int J Mol Sci (2020) 21(24):9702. doi: 10.3390/ijms21249702
126. Shafee N, AbuBakar S. Dengue Virus Type 2 NS3 Protease and NS2B-NS3 Protease Precursor Induce Apoptosis. J Gen Virol (2003) 84(Pt 8):2191–5. doi: 10.1099/vir.0.19022-0
127. Lin J-C, Lin SC, Chen WY, Yen YT, Lai CW, Tao MH, et al. Dengue Viral Protease Interaction With NF-κb Inhibitor α/β Results in Endothelial Cell Apoptosis and Hemorrhage Development. J Immunol (Baltimore Md 1950) (2014) 193(3):1258–67. doi: 10.4049/jimmunol.1302675
128. Kleinschmidt MC, Michaelis M, Ogbomo H, Doerr HW, Cinatl J Jr. Inhibition of Apoptosis Prevents West Nile Virus Induced Cell Death. BMC Microbiol (2007) 7:49. doi: 10.1186/1471-2180-7-49
129. Parquet MC, Kumatori A, Hasebe F, Morita K, Igarashi A. West Nile Virus-Induced Bax-Dependent Apoptosis. FEBS Lett (2001) 500(1-2):17–24. doi: 10.1016/S0014-5793(01)02573-X
130. Smith JL, Grey FE, Uhrlaub JL, Nikolich-Zugich J, Hirsch AJ. Induction of the Cellular microRNA, Hs_154, by West Nile Virus Contributes to Virus-Mediated Apoptosis Through Repression of Antiapoptotic Factors. J Virol (2012) 86(9):5278–87. doi: 10.1128/JVI.06883-11
131. Bhuvanakantham R, Cheong YK, Ng M-L. West Nile Virus Capsid Protein Interaction With Importin and HDM2 Protein Is Regulated by Protein Kinase C-Mediated Phosphorylation. Microbes Infect (2010) 12(8-9):615–25. doi: 10.1016/j.micinf.2010.04.005
132. Airo AM, Urbanowski MD, Lopez-Orozco J, You JH, Skene-Arnold TD, Holmes C, et al. Expression of Flavivirus Capsids Enhance the Cellular Environment for Viral Replication by Activating Akt-Signalling Pathways. Virology (2018) 516:147–57. doi: 10.1016/j.virol.2018.01.009
133. Melian EB, Edmonds JH, Nagasaki TK, Hinzman E, Floden N, Khromykh AA. West Nile Virus NS2A Protein Facilitates Virus-Induced Apoptosis Independently of Interferon Response. J Gen Virol (2013) 94(Pt 2):308–13. doi: 10.1099/vir.0.047076-0
134. Carod-Artal FJ. Neurological Complications of Zika Virus Infection. Expert Rev Anti-Infect Ther (2018) 16(5):399–410. doi: 10.1080/14787210.2018.1466702
135. Ghouzzi VE, Bianchi FT, Molineris I, Mounce BC, Berto GE, Rak M, et al. ZIKA Virus Elicits P53 Activation and Genotoxic Stress in Human Neural Progenitors Similar to Mutations Involved in Severe Forms of Genetic Microcephaly and P53. Cell Death Dis (2017) 8(1):e2567. doi: 10.1038/cddis.2016.446
136. Slomnicki LP, Chung DH, Parker A, Hermann T, Boyd NL, Hetman M. Ribosomal Stress and Tp53-Mediated Neuronal Apoptosis in Response to Capsid Protein of the Zika Virus. Sci Rep (2017) 7(1):16652. doi: 10.1038/s41598-017-16952-8
137. Vanwalscappel B, Haddad JG, Almokdad R, Decotter J, Gadea G, Desprès P, et al. Zika M Oligopeptide ZAMP Confers Cell Death-Promoting Capability to a Soluble Tumor-Associated Antigen Through Caspase-3/7 Activation. Int J Mol Sci (2020) 21(24):9578. doi: 10.3390/ijms21249578
138. Slonchak A, Hugo LE, Freney ME, Hall-Mendelin S, Amarilla AA, Torres FJ, et al. Zika Virus Noncoding RNA Suppresses Apoptosis and Is Required for Virus Transmission by Mosquitoes. Nat Commun (2020) 11(1):2205. doi: 10.1038/s41467-020-16086-y
139. Huang M, Xu A, Wu X, Zhang Y, Guo Y, Guo F, et al. Japanese Encephalitis Virus Induces Apoptosis by the IRE1/JNK Pathway of ER Stress Response in BHK-21 Cells. Arch Virol (2016) 161(3):699–703. doi: 10.1007/s00705-015-2715-5
140. Guo F, Yu X, Xu A, Xu J, Wang Q, Guo Y, et al. Japanese Encephalitis Virus Induces Apoptosis by Inhibiting Foxo Signaling Pathway. Vet Microbiol (2018) 220:73–82. doi: 10.1016/j.vetmic.2018.05.008
141. Wang Q, Xin X, Wang T, Wan J, Ou Y, Yang Z, et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J Virol (2019) 93(17):e00887–19. doi: 10.1128/JVI.00887-19
142. Lee C-J, Liao C-L, Lin Y-L. Flavivirus Activates Phosphatidylinositol 3-Kinase Signaling to Block Caspase-Dependent Apoptotic Cell Death at the Early Stage of Virus Infection. J Virol (2005) 79(13):8388–99. doi: 10.1128/JVI.79.13.8388-8399.2005
143. Prikhod'ko GG, Prikhod'ko EA, Cohen JI, Pletnev AG. Infection With Langat Flavivirus or Expression of the Envelope Protein Induces Apoptotic Cell Death. Virology (2001) 286(2):328–35. doi: 10.1006/viro.2001.0980
144. Prikhod'ko GG, Prikhod'ko EA, Pletnev AG, Cohen JI. Langat Flavivirus Protease NS3 Binds Caspase-8 and Induces Apoptosis. J Virol (2002) 76(11):5701–10. doi: 10.1128/JVI.76.11.5701-5710.2002
145. Lieskovská J, Páleníková J, Langhansová H, Chmelař J, Kopecký J. Saliva of Ixodes Ricinus Enhances TBE Virus Replication in Dendritic Cells by Modulation of Pro-Survival Akt Pathway. Virology (2018) 514:98–105. doi: 10.1016/j.virol.2017.11.008
146. Her Z, Kam YW, Gan VC, Lee B, Thein TL, Tan JJ, et al. Severity of Plasma Leakage Is Associated With High Levels of Interferon γ-Inducible Protein 10, Hepatocyte Growth Factor, Matrix Metalloproteinase 2 (MMP-2), and MMP-9 During Dengue Virus Infection. J Infect Dis (2017) 215(1):42–51. doi: 10.1093/infdis/jiw494
147. Pandey N, Jain A, Garg RK, Kumar R, Agrawal OP, Lakshmana Rao PV. Serum Levels of IL-8, Ifnγ, IL-10, and TGF β and Their Gene Expression Levels in Severe and Non-Severe Cases of Dengue Virus Infection. Arch Virol (2015) 160(6):1463–75. doi: 10.1007/s00705-015-2410-6
148. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue Virus NS1 Protein Activates Cells via Toll-Like Receptor 4 and Disrupts Endothelial Cell Monolayer Integrity. Sci Trans Med (2015) 7(304):304ra142. doi: 10.1126/scitranslmed.aaa3863
149. Lum F-M, Lye DCB, Tan JJL, Lee B, Chia PY, Chua TK, et al. Longitudinal Study of Cellular and Systemic Cytokine Signatures to Define the Dynamics of a Balanced Immune Environment During Disease Manifestation in Zika Virus-Infected Patients. J Infect Dis (2018) 218(5):814–24. doi: 10.1093/infdis/jiy225
150. Coelho DR, Carneiro PH, Mendes-Monteiro L, Conde JN, Andrade I, Cao T, et al. ApoA1 Neutralizes Proinflammatory Effects of Dengue Virus NS1 Protein and Modulates Viral Immune Evasion. J Virol (2021) 95(13):e0197420. doi: 10.1128/JVI.01974-20
151. Bos S, Poirier-Beaudouin B, Seffer V, Manich M, Mardi C, Desprès P, et al. Zika Virus Inhibits IFN-α Response by Human Plasmacytoid Dendritic Cells and Induces NS1-Dependent Triggering of CD303 (BDCA-2) Signaling. Front Immunol (2020) 11:582061. doi: 10.3389/fimmu.2020.582061
152. Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA, et al. An Evolutionary NS1 Mutation Enhances Zika Virus Evasion of Host Interferon Induction. Nat Commun (2018) 9(1):414. doi: 10.1038/s41467-017-02816-2
153. Zheng Y, Liu Q, Wu Y, Ma L, Zhang Z, Liu T, et al. Zika Virus Elicits Inflammation to Evade Antiviral Response by Cleaving cGAS via NS1-Caspase-1 Axis. EMBO J (2018) 37(18):e99347. doi: 10.15252/embj.201899347
154. Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, et al. Erratum: Zika Virus Evades Interferon-Mediated Antiviral Response Through the Co-Operation of Multiple Nonstructural Proteins. Cell Discov (2017) 3:17014. doi: 10.1038/celldisc.2017.14
155. Zhou D, Li Q, Jia F, Zhang L, Wan S, Li Y, et al. The Japanese Encephalitis Virus NS1' Protein Inhibits Type I IFN Production by Targeting MAVS. J Immunol (Baltimore Md 1950) (2020) 204(5):1287–98. doi: 10.4049/jimmunol.1900946
156. Castillo Ramirez JA, Urcuqui-Inchima S. Dengue Virus Control of Type I IFN Responses: A History of Manipulation and Control. J Interf Cytokine Res Off J Int Soc Interf Cytokine Res (2015) 35(6):421–30. doi: 10.1089/jir.2014.0129
157. Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, et al. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J Virol (2005) 79(13):8004–13. doi: 10.1128/JVI.79.13.8004-8013.2005
158. Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J Virol (2005) 79(3):1934–42. doi: 10.1128/JVI.79.3.1934-1942.2005
159. Fanunza E, Carletti F, Quartu M, Grandi N, Ermellino L, Milia J, et al. Zika Virus NS2A Inhibits Interferon Signaling by Degradation of STAT1 and STAT2. Virulence (2021) 12(1):1580–96. doi: 10.1080/21505594.2021.1935613
160. Dalrymple NA, Cimica V, Mackow ER. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio (2015) 6(3):e00553–15. doi: 10.1128/mBio.00553-15
161. Ngueyen TTN, Kim SJ, Lee JY, Myoung J. Zika Virus Proteins NS2A and NS4A Are Major Antagonists That Reduce IFN-β Promoter Activity Induced by the MDA5/RIG-I Signaling Pathway. J Microbiol Biotechnol (2019) 29(10):1665–74. doi: 10.4014/jmb.1909.09017
162. Riedl W, Acharya D, Lee JH, Liu G, Serman T, Chiang C, et al. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe (2019) 26(4):493–503. doi: 10.1016/j.chom.2019.09.012
163. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue Virus NS2B Protein Targets cGAS for Degradation and Prevents Mitochondrial DNA Sensing During Infection. Nat Microbiol (2017) 2:17037. doi: 10.1038/nmicrobiol.2017.37
164. Yu C-Y, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, et al. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PloS Pathog (2012) 8(6):e1002780. doi: 10.1371/journal.ppat.1002780
165. Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, et al. Zika Virus Evades Interferon-Mediated Antiviral Response Through the Co-Operation of Multiple Nonstructural Proteins. Cell Discov (2017) 3:17006. doi: 10.1038/celldisc.2017.6
166. He Z, Zhu X, Wen W, Yuan J, Hu Y, Chen J, et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J Virol (2016) 90(16):7219–30. doi: 10.1128/JVI.00221-16
167. Robertson SJ, Lubick KJ, Freedman BA, Carmody AB, Best SM. Tick-Borne Flaviviruses Antagonize Both IRF-1 and Type I IFN Signaling to Inhibit Dendritic Cell Function. J Immunol (Baltimore Md 1950) (2014) 192(6):2744–55. doi: 10.4049/jimmunol.1302110
168. Yang Q, You J, Zhou Y, Wang Y, Pei R, Chen X, et al. Tick-Borne Encephalitis Virus NS4A Ubiquitination Antagonizes Type I Interferon-Stimulated STAT1/2 Signalling Pathway. Emerging Microbes Infect (2020) 9(1):714–26. doi: 10.1080/22221751.2020.1745094
169. Chatel-Chaix L, Cortese M, Romero-Brey I, Bender S, Neufeldt CJ, Fischl W, et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe (2016) 20(3):342–56. doi: 10.1016/j.chom.2016.07.008
170. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe (2016) 19(6):882–90. doi: 10.1016/j.chom.2016.05.009
171. Thurmond S, Wang B, Song J, Hai R. Suppression of Type I Interferon Signaling by NS5. Viruses (2018) 10(12):712. doi: 10.3390/v10120712
172. Coldbeck-Shackley RC, Eyre NS, Beard MR. The Molecular Interactions of ZIKV and DENV With the Type-I IFN Response. Vaccines (2020) 8(3):530. doi: 10.3390/vaccines8030530
173. Best SM. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J Virol (2017) 91(3):e01970–16. doi: 10.1128/JVI.01970-16
174. Bowen JR, Quicke KM, Maddur MS, O'Neal JT, McDonald CE, Fedorova NB, et al. Zika Virus Antagonizes Type I Interferon Responses During Infection of Human Dendritic Cells. PloS Pathog (2017) 13(2):e1006164. doi: 10.1371/journal.ppat.1006164
175. Ashour J, Laurent-Rolle M, Shi PY, García-Sastre A. NS5 of Dengue Virus Mediates STAT2 Binding and Degradation. J Virol (2009) 83(11):5408–18. doi: 10.1128/JVI.02188-08
176. Parisien J-P, Lenoir JJ, Alvarado G, Horvath Cm. The Human STAT2 Coiled-Coil Domain Contains a Degron for Zika Virus Interferon Evasion. J Virol (2021) 96(1):JVI0130121. doi: 10.1101/2021.10.01.462781
177. Shu J, Ma X, Zhang Y, Zou J, Yuan Z, Yi Z. NS5-Independent Ablation of STAT2 by Zika Virus to Antagonize Interferon Signalling. Emerg Microbes Infect (2021) 10(1):1609–25. doi: 10.1080/22221751.2021.1964384
178. Laurent-Rolle M, Morrison J, Rajsbaum R, Macleod JML, Pisanelli G, Pham A, et al. The Interferon Signaling Antagonist Function of Yellow Fever Virus NS5 Protein Is Activated by Type I Interferon. Cell Host Microbe (2014) 16(3):314–27. doi: 10.1016/j.chom.2014.07.015
179. Chang DC, Hoang LT, Mohamed Naim AN, Dong H, Schreiber MJ, Hibberd ML, et al. Evasion of Early Innate Immune Response by 2'-O-Methylation of Dengue Genomic RNA. Virology (2016) 499:259–66. doi: 10.1016/j.virol.2016.09.022
180. Zhao Z, Tao M, Han W, Fan Z, Imran M, Cao S, et al. Nuclear Localization of Zika Virus NS5 Contributes to Suppression of Type I Interferon Production and Response. J Gen Virol (2021) 102(3):001376. doi: 10.1099/jgv.0.001376
181. Lundberg R, Melén K, Westenius V, Jiang M, Österlund P, Khan H, et al. Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses (2019) 11(11):1024. doi: 10.3390/v11111024
182. Laurent-Rolle M, Boer EF, Lubick KJ, Wolfinbarger JB, Carmody AB, Rockx B, et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J Virol (2010) 84(7):3503–15. doi: 10.1128/JVI.01161-09
183. Ye J, Chen Z, Li Y, Zhao Z, He W, Zohaib A, et al. Japanese Encephalitis Virus NS5 Inhibits Type I Interferon (IFN) Production by Blocking the Nuclear Translocation of IFN Regulatory Factor 3 and NF-κb. J Virol (2017) 91(8):e00039–17. doi: 10.1128/JVI.00039-17
184. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol Manipulation by West Nile Virus Perturbs the Cellular Immune Response. Cell Host Microbe (2007) 2(4):229–39. doi: 10.1016/j.chom.2007.09.003
185. Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, et al. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J Virol (2005) 79(20):12828–39. doi: 10.1128/JVI.79.20.12828-12839.2005
186. Lubick KJ, Robertson SJ, McNally KL, Freedman BA, Rasmussen AL, Taylor RT, et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe (2015) 18(1):61–74. doi: 10.1016/j.chom.2015.06.007
187. Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, et al. Dengue Subgenomic RNA Binds TRIM25 to Inhibit Interferon Expression for Epidemiological Fitness. Science (New York NY) (2015) 350(6257):217–21. doi: 10.1126/science.aab3369
188. Donald CL, Brennan B, Cumberworth SL, Rezelj VV, Clark JJ, Cordeiro MT, et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus From Recife, Brazil. PloS Negl Trop Dis (2016) 10(10):e0005048. doi: 10.1371/journal.pntd.0005048
Keywords: innate immune, inflammasome, inflammatory cell death, cytokines, flaviviruses
Citation: Pan Y, Cai W, Cheng A, Wang M, Yin Z and Jia R (2022) Flaviviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Front. Immunol. 13:829433. doi: 10.3389/fimmu.2022.829433
Received: 05 December 2021; Accepted: 10 January 2022;
Published: 28 January 2022.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Xin Li, China Agricultural University, ChinaBhesh Raj Sharma, St. Jude Children’s Research Hospital, United States
Copyright © 2022 Pan, Cai, Cheng, Wang, Yin and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renyong Jia, amlhcnlAc2ljYXUuZWR1LmNu; Anchun Cheng, Y2hlbmdhbmNodW5AdmlwLjE2My5jb20=
†These authors have contributed equally to this work