 Thai Minh Duy Le
Thai Minh Duy Le A-Rum Yoon
A-Rum Yoon Thavasyappan Thambi
Thavasyappan Thambi Chae-Ok Yun
Chae-Ok Yun- 1Department of Bioengineering, College of Engineering, Hanayang University, Seoul, South Korea
- 2Institute of Nano Science and Technology (INST), Hanayang University, Seoul, South Korea
- 3Hanyang Institute of Bioscience and Biotechnology (HY-IBB), Hanyang University, Seoul, South Korea
- 4GeneMedicine CO., Ltd., Seoul, South Korea
Immunotherapy holds enormous promise to create a new outlook of cancer therapy by eliminating tumors via activation of the immune system. In immunotherapy, polymeric systems play a significant role in improving antitumor efficacy and safety profile. Polymeric systems possess many favorable properties, including magnificent biocompatibility and biodegradability, structural and component diversity, easy and controllable fabrication, and high loading capacity for immune-related substances. These properties allow polymeric systems to perform multiple functions in immunotherapy, such as immune stimulants, modifying and activating T cells, delivery system for immune cargos, or as an artificial antigen-presenting cell. Among diverse immunotherapies, immune checkpoint inhibitors, chimeric antigen receptor (CAR) T cell, and oncolytic virus recently have been dramatically investigated for their remarkable success in clinical trials. In this report, we review the monotherapy status of immune checkpoint inhibitors, CAR-T cell, and oncolytic virus, and their current combination strategies with diverse polymeric systems.
1 Introduction
According to the World Health Organization (WHO), cancer is among the top leading causes of nearly 10.0 million deaths worldwide in 2020 (1, 2). Even though traditional treatment methods, such as invasive surgery, chemotherapy, targeted therapy, and radiation, have prevailed and made tremendous progress in the clinical setting, these regimens still face some inherent limitations in terms of therapeutic efficacy and safety (3–6). To address these issues, cancer immunotherapy, also known as immuno-oncology, has stepped into the spotlight and many kinds of immune therapeutics are investigated in the research and development stage (7, 8). Some of them have even been commercialized (9, 10). Cancer immunotherapy is a type of biological therapy that utilizes the body’s immune system to generate the attacking response of the tumor cells and thus produce an anti-tumor effect (11, 12). It can train the immune system to recognize and strike specific cancer cells and boost immune cells to help them eliminate cancer. It is noteworthy that cancer immunotherapy targets not only the primary tumor but also the secondary tumor metastasis by stimulating systemic immune response (13, 14). Further, it can inhibit tumor recurrence through cancer-specific memory immune response which will be reactive when encountering tumor associate antigens (TAA) (15, 16).
There have been diverse cancer immune therapeutics, such as cancer vaccines, antibody therapy, cytokines, immune checkpoint inhibitors (ICI), adoptive cell transfer (ACT), and oncolytic viruses (OV) (17, 18). Among those immune therapeutics, remarkable success in the commercialization of ICI and chimeric antigen receptor (CAR)-T cells have driven cancer immunotherapy into the limelight (19–22). ICI, which blocks the binding of checkpoint proteins with their partner proteins and allows T cells to kill cancer cells, has been revolutionarily developed, since Yervoy, the first ICI and CTLA-4 inhibitor, was approved by the U.S. FDA in 2011. CAR-T is a genetically engineered T cell to express artificial T cell receptors and specifically target tumor cells. In addition to ICI and CAR-T, the utilizations of OV have rapidly expanded in the past few years since the U.S. FDA approved the first OV, Imlygic, in 2015 (23). OV has been recognized as a novel therapeutic platform due to its unique feature, selectively replicating in and eradicating cancer cells through a domino-like cascading infection of tumor cells, which other conventional therapeutics cannot mimic (24). Moreover, it is also highlighted with its capacity to target cancer immunity in multiple steps, leading to potent clinical benefits (25, 26).
Even though these immunotherapies possess dramatic breakthroughs in the last decade, manifold obstacles regarding therapeutic efficacy and safety remain to be overcome. To further elaborate, previous research has shown that less than 50% of the solid tumor types have positive responses to ICI therapy (27). Moreover, regarding safety issues, many researchers have demonstrated the association of ICI therapy with several side effects such as colitis, fatigue, pneumonitis, endocrinopathies, and dermatitis (28, 29). Meanwhile, for the CAR-T cell therapy, safety is also still an utmost concerning issue according to neurotoxicity and severe side effects, which include death, cytokine release syndrome, hyperuricemia, hyperkalemia, acute anaphylaxis, and B-cell aplasia (30, 31). These obstacles are also found in systemically administered OV, which demonstrates limited therapeutic efficacy due to its hepatotoxicity and immunogenicity that respectively trigger nonspecific liver toxicity and inflammatory responses, making it difficult to cure inaccessible tumors (32, 33). Further, the complex tumor microenvironment (TME) is also a difficult hurdle that limits the application of current immunotherapies (34, 35). In addition, the above immunotherapies required a large dose of therapeutic drugs regarding their instability and short half-life, which exhibits toxicity and severe side effects in a certain number of patients.
To lower the negative effects and enhance the targeting of immunotherapies, advanced delivery systems have been exploited. In recent years, diverse delivery systems have been developed for immunotherapeutic drugs, both for local, systemic delivery and sustained release in vivo (36–38). Among these materials, diverse polymeric systems have been exploited both as excellent carriers for therapeutic immunogenic agents and favorable adjuvants. These polymeric systems obtain various advantages including superior biocompatibility and biodegradability, effective activity for immune stimulation, tunable size, designable structure, and large loading capacity for immune-inducing factors (39–43). Moreover, advanced polymeric systems with different functions can be utilized to carry immune pharmaceuticals to targeting organs by different routes of administration such as subcutaneously, intranasally, and intravenously (44–47). To elaborate, these pharmaceutical drugs are delivered through polymeric systems in various forms such as polymer–drug conjugates or drug-loaded micelles (48, 49). These efficient delivery systems are therapeutically promising due to several reasons. Most polymer backbones consist of diverse functional groups, enabling the easy conjugation of pharmaceutical drugs to polymer systems through specific ligands. Moreover, the fabricable nano size system can allow the polymeric system to stay in the blood circulation and induce passive tumor targeting. Due to these advantages, the polymeric systems are considered a promising strategy to efficiently apply for diverse immunotherapies (Figure 1).
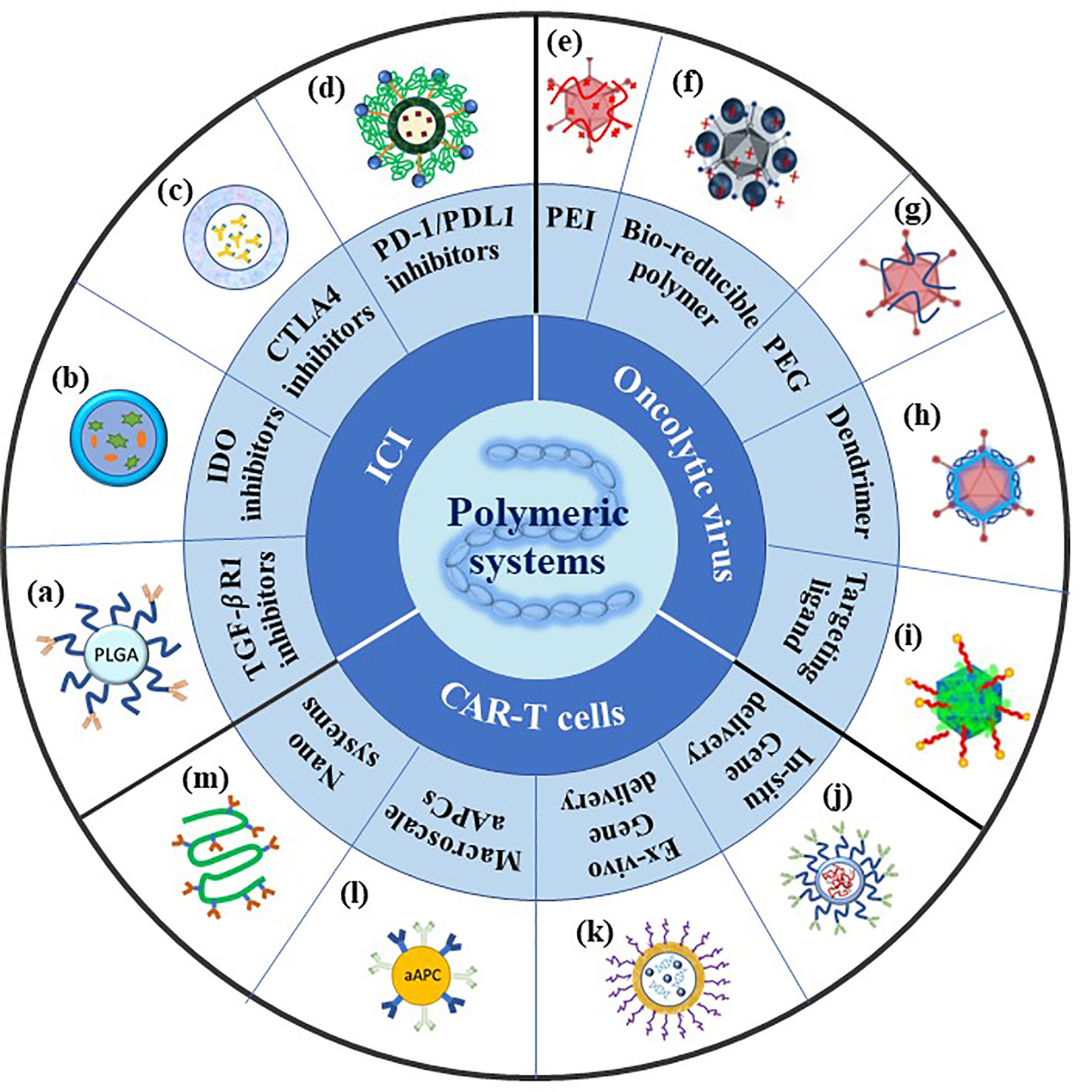
Figure 1 Summary of polymeric systems for cancer immunotherapy. ICI: (A) Scheme of antibody conjugated PEG-PLGA polymeric nanoparticles recreated referencing from (50); (B) scheme of NLG919(IODI)/IR780 coloaded micelles recreated referencing from (51); (C) scheme of polymeric micelles containing ICI antibodies recreated referencing from (52); (D) scheme of PEG sheddable, anti-PD-1 antibody (aPD-1)-conjugated, and PTX-loaded micelle recreated referencing from (53); OV: (E) Scheme of cationic PEI-Ad complex created referencing from (54); (F) scheme of PPSA-Ad complex reused referencing from our group (55); (G) scheme of PEG conjugated-Ad created referencing from (56); (H) scheme of amphiphilic dendrimer binding Ad recreated referencing from (57); (I) scheme of Ad/chitosan-PEG-FA nanocomplex reused referencing from our group (58); CAR-T cells: (J) Scheme of targeted mRNA-carrying polymeric nanoparticle recreated referencing from (59); (K) scheme of targeted pDNA-carrying supramolecular self-assemble nanoparticles recreated referencing from (60); (L) scheme of artificial antigen presenting cell recreated referencing from (61); (M) scheme of semi-stiff synthetic dendritic cells recreated referencing from (62).
In this review, we focus on representing immunotherapeutic strategies including immune checkpoint inhibitors, CAR-T cells, and oncolytic viruses, and their combined application with polymeric systems. First, we would like to introduce the recent development of immune checkpoint inhibitor strategies and then discuss relevant polymeric systems applied with each ICI type. Second, we describe adoptive cell transfer immunotherapy and its representative which is a CAR-T cell. We analyze the current manufacturing methods of the CAR-T cell and its limitations. We then demonstrate how application of polymeric systems can reduce these limitations and innovate CAR-T cell to a new clinical efficacy. Finally, we will present the ongoing possibility of the oncolytic viruses in immunotherapy. We intensify on adenoviruses which are the most extensively studied virus type. We begin by describing basic oncolytic adenovirus biology and analyze their advantages as well as their limitations. We then introduce some typical modifications with polymeric systems that can be used to promote better anti-tumor inhibition.
2 Different Immunotherapies and Polymeric Systems Applying for These Strategies
2.1 Immunological Checkpoint Inhibitors
2.1.1 Monotherapy of ICI
Immunological checkpoint inhibitors currently have been dramatically investigated and the most reported inhibitors are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death receptors 1/programmed death receptor-ligand 1 blockade (PD-1/PD-L1). The body’s immune system uses the immune checkpoints to control the corporeal immune balance and maintain self-tolerance (63). In normal conditions, the activated T cells express PD-1 to recognize abnormal or cancerous cells and then eliminate them to protect the body from their development (64, 65). However, the tumor cells might up-regulate the expression of PD-L1 or PD-L2 that bind to PD-1 to evade recognition and attack of immune cells (66, 67). Therefore, anticancer immunotherapy can be achieved by using blocking inhibitors of PD-1 or its ligand. Another immune checkpoint is CTLA-4, which diminishes the T-cell activity and assists the maintenance of self-tolerance (68). The anti-CTLA-4 antibody has been used to block the CTLA-4 to induce T cell activation, which inhibits tumor growth.
Nonetheless, many challenges remain with these checkpoint blockade strategies that limit their application for complete cancer treatment. As mentioned above, the administration of immunological checkpoint inhibitors can cause severe harm to different normal organs (69, 70). Moreover, only a small portion of patients show positive effectiveness of checkpoint inhibitor treatments, this phenomenon is still under study (71, 72). In addition, the extracellular matrix and microenvironment of different tumors suppress the immune recognition and activation (73). With the new development of polymers and biomaterials science, these challenges can be overcome by utilizing various polymers to obtain targeting tumor delivery (Table 1).
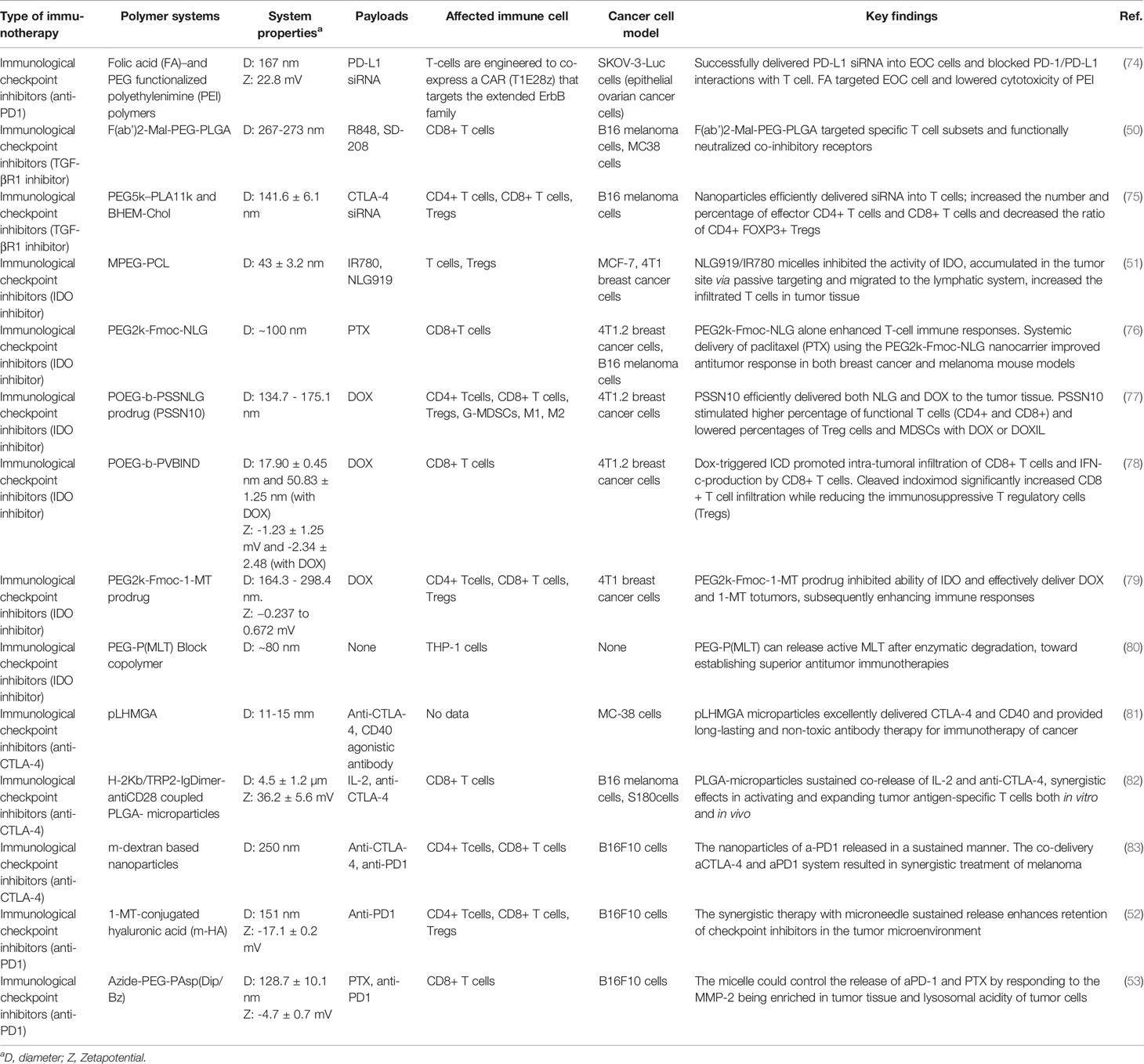
Table 1 Summary of recent research on different polymeric systems for immunological checkpoint inhibitors.
2.1.2 Combination ICI Therapy With Polymer Systems
One of the typical strategies in ICI is applying siRNA gene transfection to express the protein knockout of the PD-1/PD-L1 immunosuppressive pathway. This strategy has performed promising results for cancer treatment (84). Specifically, even though adoptive T cell immunotherapy performs promising results in epithelial ovarian cancer (EOC) treatment, the EOC cell-expressed PD-L1 can interact with PD-1 from T cells and induce undesired immunosuppression resulting in low therapeutic effect (85). To overcome this problem, Teo et al. have designed a folic acid (FA) functionalized PEI polymer complexed with PD-L1 siRNA. The research showed the polyplex, which consisted of FA/polymer/siRNA, has successfully blocked the PD-1 and PD-L1 pathways and repelled the immunosuppression of T cells, leading to promote the recognition of T cells toward EOC cells (74). It is important that the FA not only lower the cytotoxicity of the PEI but also effectively enhance specificity of uptake into EOC tumor cells.
On the other hand, transforming growth factor-β (TGF-β) mediated tumor microenvironment also plays an important role in immune suppression besides the PD-1/PD-L1 pathway (86, 87). Unfortunately, the TGF-β signaling is necessary for many cellular processes so utilizing TGF-βR1 inhibitors can cause severe side effects such as hemorrhagic, degenerative, and inflammatory lesions in heart valves (88). To overcome this problem, diverse polymeric nonviral vectors have been generated to encapsulate TGF-βR1 inhibitors and target them to the tumor site. In 2017, Schmid et al. introduced a CD8+ T cell-specific nanoparticle system based on PLGA-PEG polymer conjugated with anti-CD8a F(ab’)2 fragments to encapsulate TGF-βR1 inhibitor SD-208 (50). The nanoparticles system successfully reduced the cytotoxicity of TGF-βR1 inhibitor SD-208 to normal cells and recover the immune function of T cells. Moreover, the PD-1-PLGA-PEG nanoparticle co-delivery of Toll-like receptor (TLR7/8) agonist (R848) showed recruited a significantly high number of T-lymphocytes at the tumors. In addition, a nanoparticle system containing poly(ethylene glycol)-block-poly(D,L-lactide) (PEG5k–PLA11k) and the cationic lipidN,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryoxycarbonyl-aminoethyl) ammonium bromide (BHEM-Chol) has been used to deliver immunosuppressive factor siRNA to a tumor (75). The siRNA encapsulated in the copolymer-based nanoparticles not only was protected from enzymatic degradation but also enhanced the cell internalization compared with negatively charged bare siRNAs. The system exhibited a favorable modulation implement in tumor invasive CTL. The loaded CTL-associated molecule-4-siRNA nanoparticles (NPsiCTLA-4) effectively stimulate T cells’ activation and hinder tumor growth in melanoma mice.
To overcome the limitations of checkpoint blockers, the polymeric micelles have also been a promising system. According to their special structure which consists of polymeric amphiphiles, micelles can carry and deliver manifold hydrophobic drugs to the target tumor. Recently, Peng et al. introduced a polymeric micelle system for tumor immunity post photothermal therapy (PTT) based on amphipathic polymer MPEG-PCL to co-deliver photosensitizer IR780 and NLG919 (an indoleamine 2,3-dioxygenase (IDO) inhibitor) (51). This nano system has shown sufficient accumulation at the tumor site and shifts to lymph nodes to promote the activation of T lymphocytes. In another research, an immunostimulatory dual-functional nanocarrier based on a prodrug conjugate of PEG with NLG919 was studied by Chen et al. The system was also equipped with a Fmoc group, a drug-interactive motif for enhancing drug loading capacity and formulation stability. The PEG2k-Fmoc-NLG alone showed greatly stimulated T-cell immune responses and excellent tumor inhibition in vivo. It is noteworthy to mention that the systemic administration of paclitaxel (PTX) loaded PEG2k-Fmoc-NLG nano system exhibited significant tumor inhibition in both melanoma and breast cancer mouse models (76). A redox-responsive immunostimulatory polymeric prodrug carrier which can controllably co-deliver chemotherapeutic DOX and immune checkpoint inhibitor NLG was introduced by Sun et al. (77). The system, which is called POEG-b-PSSNLG prodrug (PSSN10), was a synthesized poly(oligo(ethylene glycol) methacrylate)-poly(N,N′-(tbutyoxycarbonyl)cystamine) copolymer conjugated NLG919 prodrug which can self-assemble into nano-sized micelles. The PSSN10 carrier can improve cell immune responses in the lymphocyte-Panc02 co-culture experiments and exhibited significant anti-tumor activity in vivo. Moreover, the DOX/PSSN10 micelles displayed higher efficacy in the tumor growth inhibition and longer survival rate of 4T1.2 tumor-bearing mouse model compared with free DOX or a clinical formulation of liposomal DOX (DOXIL). Another IDO inhibitor, indoximod, was also co-delivered with DOX in a synthesized copolymer POEG-b-PVBIND micelles by Wan et al. in 2019 (78). The indoximod conjugating copolymer micelles effectively promoted the anti-tumor immunity resulting in an extreme tumor inhibition effect in a preclinical breast cancer model. In a different concept, Lan et al. developed a dual functional indoximod-based carrier PEG2K-Fmoc-1-MT also for breast cancer chemo and immunochemotherapy (79). The polymeric micelle itself successfully inhibited IDO effect with decreased kynurenine (KYN) production leading to the proliferation of CD4+ and CD8+ T cells. The DOX/PEG2kFmoc-1-MT micelles can generate an immunogenic cell death process, subsequently secreting many cytokines [such as interferon (IFN)-γ, IL-2, and tumor necrosis factor-alpha (TNF-α)] inducing later T cell-mediated immunity. The 4T1 murine breast cancer model tumor inhibition profile of DOX/PEG2kFmoc-1-MT micelles was dramatically high with long survival time compared with other groups. Another amphiphilic PEGpoly-1-Methyl-l-Tryptophan (MLT) block of copolymer self-assembled polymeric micelles was investigated by Huang et al. which showed effectively reduced levels of KYN in activated macrophages (80).
As mentioned above, CTL-4 can suppress the activation of T cells and promote self-tolerance. The anti-CTLA-4 antibody can be used for blocking the CTLA-4 and stimulating the activity of T cells toward the tumor. CTLA-4 antibodies have been loaded in poly(lactic-co-hydroxymethyl-glycolic acid) (PLHMGA) by Sima et al. for cancer immunotherapy. The nanoparticle system has been proven to block inhibitory receptors on T cells and obtained promising therapeutic efficacy than the IFA formulation in colon carcinoma tumor model (MC-38) (81). Lei Zhang et al. reported a study on PLGA microparticles to co-deliver IL-2 and CTLA-4 antibodies. The surface of the PLGA microparticles was conjugated with a H-2Kb/TRP2-Ig dimer and anti-CD28. The polymeric macroparticles successfully co-released IL-2 and anti-CTLA-4 inducing dual effects in activating and promoting tumor antigen-specific T cells. The systems exhibited enhancement in anti-tumor efficacy in a mouse melanoma model (82).
Similar to CTL-4, diverse PD-1/PDL-1 inhibitors have been used for combinatorial therapy in recent clinical trials (89). Different kinds of polymers have been exploited with PD-1/PD-L1 blockade for cancer immunotherapy presently. Especially, utilizing the synergistic effect of diverse types of delivery systems can exploit the maximized potential of immune therapeutics. For example, Chao et al. reported a hyaluronic acid (a biocompatible natural polymer) microneedle integrated with pH-sensitive dextran nanoparticles (NPs) that can encapsulate and release PD-1 antibodies in a controlled manner to melanoma tissue. The report showed that this self-degradable microneedle encapsulated PD-1 antibody system generated a higher robust immune response compared to free anti-PD-1 antibody at the same dose in a B16F10 melanoma model (83). Another study conducted by Ye et al. has also produced immunotherapeutic nanoparticles from hyaluronic acid but modified with 1-methyl-DL-tryptophan (1MT) to deliver anti-PD-1 antibody. The particles combined with microneedle successfully sustain release and increased the accumulation of anti-PD-1 antibodies in the TME. The system indicated the improvement in tumor growth inhibition and lowered the immunosuppression in a B16F10 melanoma model (52). In addition, the specific tumor targeting nanoparticles can possibly enhance the performance of antitumor immunity. For example, a pH and matrix metalloproteinase dual-sensitive micellar nanocarrier which can spatiotemporally control the release of anti-PD-1 and PTX in solid tumors has been developed by Su et al. (53). The report interestingly indicated that the PTX-induced immunogenic cell death (ICD) can activate the antitumor immunity along with the blocking of the PD-1/PD-L1 axis from anti-PD-1. Together, they hinder the immune escape of tumor cells due to PTX-induced PD-L1 up-regulation. Of note, the pH-sensitive polyethylene glycol (PEG) shell could be sheddable at the tumor acidity site resulting in release of anti-PD-1 and PTX. In general, polymer-based material can be utilized as a superlative and effective delivery system to sustain biocompatible antibodies and other immunological checkpoint inhibitors in cancer immunotherapy (Figure 2).
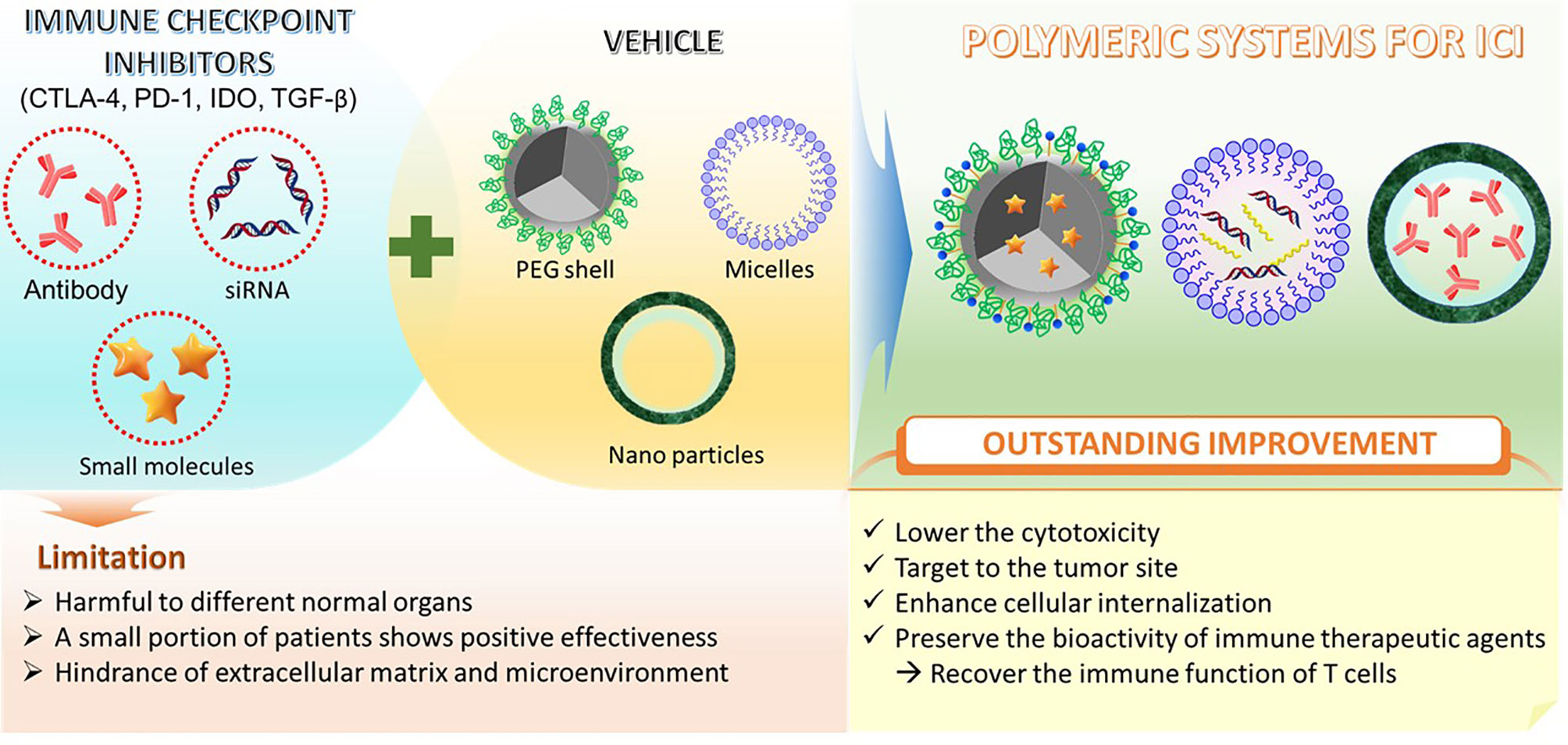
Figure 2 Schematic of different polymeric systems designed for ICI therapy.
2.2 Chimeric Antigen Receptor-T Cells
2.2.1 Monotherapy of CAR-T Cells
The term “adoptive cell transfer” (ACT) involves a group of cell-based anticancer immunotherapies and is very attractive for its smart and patient-tailored strategies (90, 91). ACT contains different steps to induce immune-mediated clearance of cancer. First, the circulating or tumor-infiltrating lymphocytes are collected from the patient. Then the cells are elected, activated ex vivo, genetically modified to express a cancer-targeting receptor, and multiplied to a therapeutic quantity. Finally, the cells are reinjected into the treated patient to recognize and eliminate cancer cells.
CAR-T cell is the most representative of ACT recently and has obtained optimistic clinical success. This therapy utilizes CAR to engineer autologous T cells for achieving immune activation without major histocompatibility complex (MHC)-restriction. The CAR is mainly composed of an extracellular antibody-derived antigen binding domain for cancer targeting and a one or more linked intracellular signaling domain. The extracellular binding domains are commonly constructed from single-chain variable fragments (scFvs) derived from tumor antigen-reactive antibodies (92). The intracellular signaling domain comprises CD3ζ chain domain and co-stimulatory domains, such as CD28 and/or 4-1BB to provide costimulatory signals for promoting CAR-T cell expansion, persistence, and function (93).
However, the manufacturing of CAR-T cell is time-consuming, expensive, and technically complex compared to other small therapeutic drugs. There are multiple aspects that need to be carefully controlled to archive a secure, therapeutically safe and effective CAR-T cell therapy, such as balanced CD4/CD8 ratio, the viability of differentiated CD3+CAR+ cells, and in vitro cytotoxicity and cytokine release against cells expressing the target antigen, which makes it costly and limits its widespread use (94). For instance, the list cost of Kymriah is US$475,000 and of Yescarta is US$373,000 which is higher than the current common cancer therapies’ cost (95). The manufacturing time to archive enough therapeutic cell numbers is from 3 to 4 weeks at least depending on different methods (96–98). It is noteworthy that the consistency of cell products plays a significant role in the tumor clearance effect of the patient. An optimum manufacturing of engineered T-cell process is required for reducing the cost and escalating clinical translation of CAR-T cell therapy.
Notably, utilizing engineered polymers can improve and streamline CAR-T cell manufacturing process. Diverse kinds of polymers have been synthesized and modified to apply in many medical engineering processes including cell culture (99), tissue engineering (100), separation (101), and drug and gene delivery (102). Similarly, many polymeric systems have been used to optimize streamlined CAR-T cell manufacturing process, mainly focusing on activation and genetic modification of the CAR-T cell.
2.2.2 Polymeric Systems Utilized in CAR-T Cell Activation
The ex vivo CAR T-cell activation consists of three central signaling steps, which are T cell receptor (TCR) stimulation, CD28 co-stimulation, and cytokine signaling. Usually the T cell receptor (TCR) stimulation and CD28 co-stimulation utilize independent anti-CD3 and anti-CD28 monoclonal antibodies (mAbs) adhered to solid materials for receptor clustering. On the other hand, the cytokine signaling commonly uses soluble cytokines dissolved in the culture medium. Therefore, an activation platform should match the above requirements and these following criteria. The platform first needs to be “friendly” and can promote T cells to multiply to around 1 to 5 × 108 CAR+ T cells for one patient (103), and maintain the stably therapeutic state in vivo. It is favorable that the platform can keep the balance number of CD4+ and CD8+ T cells in expansion (104). One more crucial point is the activation materials should be convenient to process and easily separate from T cells.
In this part, we discuss different polymeric T cells activation systems and their properties for adequately activating and expanding T cells.
2.2.2.1 Nanoscale Activation Polymers and Particles
A commercial reagent to activate and expand human T cells via CD3 and CD28 was developed by Miltenyi Biotech which called it TransAct. According to the Miltenyi Biotech specification, TransAct has a nano core-shell structure where the core is iron oxide crystal and the shell is biodegradable polysaccharide matrix conjugated to humanized CD3 and CD28 agonists (105, 106). The size of TransAct is around 100 nm and can be filtered sterilized, the excess reagent can be cleared by centrifugation with the following conventional supernatant replacement or simply by a medium wash. Many studies have investigated and compared the effectiveness in expansion, differentiation, CAR transduction, and functions of activated T cells by TransAct and the results suggest that TransAct can be used for clinical-scale T-cell activation (107, 108).
In 2013, Mandal et al. synthesized a semiflexible synthetic dendritic cell for T cell activation (62). The semi-stiff poly(isocyano peptide) has been coupled with BCN-functionalized streptavidin (Sav) and then conjugated with biotinylated αCD3 antibodies to produce αCD3– “synthetic dendritic cells” (sDCs). The flexibility of this system allows the effector molecules on it which can effectively bind to the receptors of the targeted T cell. With the size around 150−200 nm, the αCD3–sDCs showed higher efficacy in expanding T cells compared to spherical αCD3–PLGA particles (1.8 μm diameter) or free αCD3 antibodies with the same antibody amount. The group then continuously developed this semiflexible synthetic mimic dendritic cells system with co-carrying anti-CD3 antibodies (αCD3) for triggering the T cell receptor and anti-CD28 antibodies (αCD28) as a costimulatory signal (109). The bifunctional αCD3/αCD28-sDC significantly stimulated T cell activation at a considerably lower antibody concentration than free soluble antibodies. Interestingly, the highest level of polyclonal T cell activation was only achieved when the sDCs carry both αCD3 and αCD28 antibodies on the same polymer. Utilizing different polymers αCD3-sDC and αCD28-sDC did not improve any T cell activation compared with αCD3-sDC alone. These results suggested that polymer flexibility and multiple signals equipped on one polymer are crucial for mimicking the endogenous receptor clustering needed for optimal T-cell activation.
2.2.2.2 Microscale Artificial Antigen-Presenting Cells
On the other hand, different from nanoscale platforms, the microscale activation platforms imitate the original scale of endogenous antigen presenting cells and their immunological synapses with T cells. One of the first synthetic aAPCs design was based on latex (polystyrene) beads in the 1990s (110, 111). They have been used for exploring elemental features of T cell biology (112–114), and also as translational platforms for adoptive immunotherapy (115–118). These latex platforms are generated by chemically functionalized polystyrene surfaces with soluble proteins, or by binding avidin coated particles with biotin-labeled T cell activating proteins. Furthermore, MHC presented on the solid microparticle platforms by glutaraldehyde can induce stronger activation signal for T cells than the MHC presented on a cell membrane (119). Interestingly, hybrid lipid-latex particles, which utilize both the potential advantages of a solid particle and a flexible membrane, can be incorporated by coating polystyrene microsphere with plasma membrane vesicles (120–122). The studies suggested that this type of platform might effectively improve the efficacy of tumor immunotherapy with antigen. However, the main application of these polystyrene-based microparticle systems was for expansion of T cells in vitro because of its biodegradation and biocompatibility problems. The intravenous administration of larger 3-5 μm solid particles can accommodate in small capillary beds and cause capillary infarction (123, 124). Because of these issues, more advanced microparticle systems which are removable or biodegradable after the culturing period need to be developed.
In recent years, many biodegradable polymer-based microspheres have been utilized as vehicles for drug delivery (125, 126). The variety of these systems is varied in sizes in the range of hundreds of nanometers to 10 μm and can be generated from various polymers, such as poly(glycolic acid) (PGA), poly(lactic acid) (PLA), or their copolymer, poly(lactic-co-glycolic acid). After administration to the body, the biodegradable polymer particles are degraded to nontoxic substances and release the encapsulated drugs in several hours or weeks, depending on their design. Based on their favorable biocompatibility and biodegradability, the biodegradable polymer particles are an attractive candidate for aAPCs platforms that can deliver in vivo (127, 128). Notably, they can be formulated to release cytokine signals as they degrade and integrate these signals into designed aAPC (129, 130)
An ellipsoidal PLGA microparticle system was generated by Sunshine et al. as an aAPCs with 4.3 μm average diameter. Different aspect ratios have been investigated in T cells expansion efficacy (61). Compared with a spherical shape, ellipsoidal aAPCs dramatically improved T cell proliferation both in vitro and in vivo, especially at higher aspect ratios. The same tendency was observed with nanoscale aAPCs (131). On the aspect of surface topography, Fadel et al. developed an aAPC platform based on carbon nanotube−polymer composite (CNP) with the average size around 20−40 μm (132). The surface of the CNP possessed many defects that induced high surface area for attaching the stimuli for T cells. IL-2, a cytokine for T-cell cluster initiation and persistence after antigen priming, was encapsulated in biotinylated PLGA nanoparticles together with magnetite. These CNP aAPCs facilitated T cell expansion, differentiation, and the number of obtained T cells was proportionated to a level that would require at least 1000-fold less soluble IL-2 under conventional culture conditions. In addition, the magnetite in the PLGA nanoparticles allowed magnetic removal of CNP aAPCs, which contained the utmost physically and chemically stable carbon nanotubes.
2.2.3 Polymeric Systems Utilized in CAR-T Cell Genetic Modification
Compared to viral transduction and nucleofection with expensive cost and safety issues, polymer-based nonviral gene delivery systems have recently become a potential candidate for CAR T-cell genetic modification. Cationic polymers which can electrostatically interact with negatively charged DNA and RNA to form polyplexes are the most favorable polymers for this purpose. Moreover, the positive charge of polyplex facilitates cellular uptake by the ionic interaction with negatively charged proteoglycans on the cell surface (133). Especially, if the cationic polymers contain amine groups, the amine groups will be protonated in the pH range of 5.0 to 6.8 and induce the proton sponge effect resulting in endosomal escape (134). Numerous polymers have been exploited such as poly(β-amino ester) (PBAE) (135), poly(2-dimethyl)aminoethyl methacrylate) (pDMAEMA) (136), polyamidoamine (PAMAM) (137), and branched polyethylenimine (bPEI) (138).
In this part, we report different polymer architectures for nonviral gene delivery to T cells, arranging them by ex vivo and in situ application. We also review main key barriers that require further improvement to achieve more efficient transfection with these systems.
2.2.3.1 Ex Vivo Gene Delivery With Polymeric System
In 2018, Pun’s group investigated the transfection efficiencies of different synthesized polymers and their concomitant toxicity to T cells (139). They compared the transfection efficiency and cytotoxicity of branched polyethylenimine (bPEI), VIPER (virus-inspired polymer for endosomal release), linear pDMAEMA290, linear-branched (comb), and cyclic-branched (sunflower) polymers with varying pHEMA core sizes and pDMAEMA branch lengths in the Jurkat human T cell line. VIPER is an -block copolymer that consists of a hydrophilic cationic block for nucleic acid loading and colloidal stability, and a pH sensitive membrane lytic block for endosomal release in acidic pH (e.g., pH < 6.4) which was developed by their group (140, 141). Between the distinct polymer structures of pHEMA and pDMAEMA, sunflower and comb polymers obtained the most effective greatest transfection of Jurkat cells (25−50%) with low toxicity compared to bPEI, VIPER, or linear pDMAEMA290. The change in brand length of comb and sunflower polymer did not affect much in performance, however, reducing the core size of the comb polymer significantly lowers the transfection and cell viability. The best formula for the comb polymer was with core size of degree of polymerization 25 and branch length of degree of polymerization 16, this helped transfection efficiencies in primary T cells achieve an average 20% with mRNA and 10% with plasmid DNA (pDNA) and with cell viability persistently above 75%.
Many studies suggested that a decrease in branching of polymer can have a negative effect on transfection and viability. Christopher and Anja et al. developed multi-arm star-shaped pDMAEMA polycations for DNA transection and the result showed that the 5-arm star-shape exhibited lower cytotoxic than 3-arm counterparts with the similar transfection (142). Their research group continuously generated higher-branched star polycations, 20-arm pDMAEMA conjugated on a silsesquioxane initiator core and 120-arm self-assembled nanoparticles comprised of amphiphilic polybutadiene-block-pDMAEMA block copolymers showed 10 to 15% transfection efficiency of pDNA in primary T cells with more than 80% viability (143, 144). These data suggested that high polycation branching can be suitable for enhancing T-cell transfection. Notably, currently Yu et al. synthesized supramolecular self-assembled nanoparticles from adamantane-grafted PEG, adamantane-grafted PAMAM dendrimers, and cyclodextrin-grafted low-molecular-weight branched PEI which achieved more efficient pDNA transfection in Jurkat T cells than high-molecular-weight branched PEI (60).
Importantly, equipping cationic polymers with T-cell targeting ligands to facilitate cell surface binding and internalization could significantly enhance transfection. For example, Moffett et al. fabricated biodegradable poly(β-amino ester) (PBAE)-mRNA polyplex with poly(glutamic acid) (PGA)-modified antibodies (anti-CD3 or CD8) to form particular cell targeting mRNA nanocarriers (59). The nanocarriers successfully internalized into primary T cells within 2 h and effectively transfected more than 80% T cells with mRNA. Especially the lyophilized nanocarriers also showed the same result without any serve interference on T cell viability and expansion.
2.2.3.2 In Situ Gene Delivery With Polymeric System
It is worth mentioning that for covering the expensive and laborious ex vivo CAR-T cell generation, in situ polymer-based gene delivery strategies have been developed. In 2017, Matthias T. Stephan’s research group utilized the polyglutamic acid (PGA)-conjugated antibody decorated poly(β-amino ester) (PBAE)-DNA polyplexes to target the T cell and genetically modify host T cells with leukemia-specific CAR genes in vivo (145). The nanocarrier has the core-shell structure, where the core was plasmid DNA complexed with PBAE polymers grafted peptides containing microtubule-associated sequences (MTAS) and nuclear localization signals (NLS) to facilitate nuclear plasmid import of their genetic cargo via the microtubule transport machinery under resting T-cell conditions. They used two plasmids, one with the leukemia-specific 194-1BBz CAR and one for coding the hyperactive iPB7 transposase for stable CAR integration. The shell of the nanocarrier was poly(glutamic acid) (PGA)-modified anti-CD3e f(ab′)2 for T cell targeting. The nanocarriers were systemically delivered into mice and the result indicated 34% (± 5.1%) of the circulating T lymphocytes bound CD3-targeted nanoparticles after 4 h, whereas the signals from off-target cells were 5.9 ± 2.8% after 4 h. To investigate the in-situ reprogramming circulating T cells ability of the nanocarrier system, five sequential doses of 3 × 1011 nanoparticles were intravenously injected into mice bearing B-cell acute lymphoblastic leukemia. Interestingly, only the injected nanoparticles co-encapsulated 194-1BBz and iPB7 transgenes groups showed rapidly and efficiently programmed peripheral T cells to recognize leukemia cells. After that, these T cells robustly replicated and differentiated to effector phenotypes while keeping a high-level expression of the CAR transgene over 24 d, then achieved a CD44high CD62L+ memory phenotype. Remarkably, the tumor-inhibition profile of in situ nanocarrier-programmed CAR-T cells was commensurate with high-dose adoptively transferred CAR-T cells.
In summary, the engineering and manufacturing of CAR-T remain a challenge for the widespread adoption of this technology. To overcome these challenges, we have introduced two main strategies using polymer systems to engineer T cells into CAR-T cells. As T cells need to be expanded for the engineering to CAR-T cells, novel biodegradable polymeric systems that act as synthetic dendritic cells can be utilized. Further, the use of branched cationic polymers and advanced polymeric nanocarriers can improve the genetic transfection of CAR genes into T cells (Figure 3).
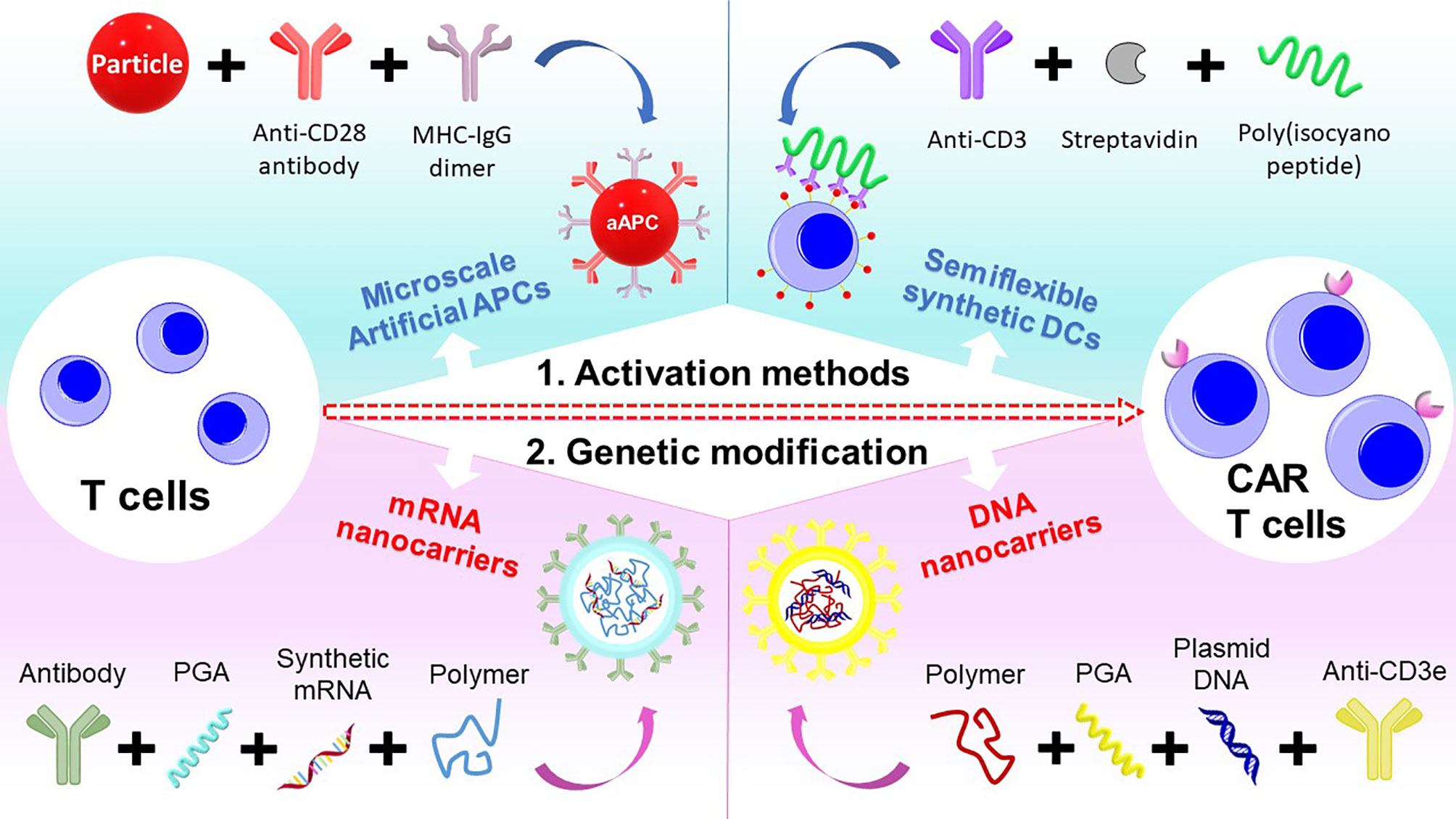
Figure 3 Schematic of different polymeric systems designed for CAR-T cell therapy.
2.3 Oncolytic Virus
2.3.1 Monotherapy of Oncolytic Virus
Oncolytic virotherapy is the most auspicious access for tumor immunotherapy. The vital advantage of oncolytic virotherapy is based on the ability of replication-competent viruses that can proliferate selectively at tumor cells (146). Among many different viruses, adenoviruses can be represented as an example for illustrating oncolytic viruses as a whole. In 2005, the State Food and Drug Administration of China approved Oncorine, a replicative, oncolytic recombinant ad5 (rAd5-H101) for treating refractory nasopharyngeal cancer. This was considered the first approved oncolytic virotherapy for clinical use in the world (147, 148). Many types of cancer cells lose the p53 gene which causes drug resistance and lower survival rates in cancer patients (149). The p53 gene inactivation cell can halt the activation of apoptotic pathway. The Oncorine is a human serotype 5 adenovirus which is deleted by the E1B 55K gene. The elimination of the E1B 55K gene prohibits viral proliferation in normal cells, tolerating only multiples in the p53-lacking host cells. Therefore, the rAd5-H101 selectively proliferates in tumor cells and causes cancer cell lysis. The newly generated viruses release and infect surrounding cancer cells which leads to a chain-reaction of ultimate cancer cell destruction (150). The Oncolytic Adenovirus (oAd) recently become one of the most interesting generic immunotherapies for cancer in numerous phases of clinical trials (151, 152). The adenovirus (Ad) possesses several advantages that are beneficial for generic immunotherapy, such as the transduction ability of dividing and non-dividing cells with high efficacy, high loading capacity, easy modification of Ad genome, high production of viral progenies, and subsequent spreading of progenies to adjacent cancer cells (153–156).
It is noteworthy that the virus propagation inducing cell lysis is an extremely immunogenic process (157). This aspect is crucially relevant considering that the cancer cells usually disguise themselves from the host immune systems. The cell lysis releases multiple immunogenic molecules, such as abundant tumor-associated antigens for presenting to dendritic cells. The released virus genomes induce immunological danger signals through pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) receptors. These simultaneous actions stimulate the adaptive immune system, including helper CD4+ T cells and cytotoxic CD8+ T cells, toward the tumor resulting in disabling the tumor immunosuppression (158). Moreover, the T cell immunity toward the replicated adenovirus can enhance the overall antitumor process (159). The adenovirus infection can also indirectly activate the nature killing cells response for further immunogenic tumor inhibition (160). Indeed, many reports have investigated and confirmed the ability to inhibit the growth of different tumors of locally administered oAds, both in preclinical and clinical cases (161–166). The Oncorine is representative of oAds that have been used for clinical cancer treatment. However, the oAds still face certain challenges that narrow the therapeutic effectiveness for clinical trials.
One of the major hurdles for oAds’ efficacy is the pre-existing humoral immunity of the host body. Nowadays the human adenovirus serotype 5 (Ad5) is the most popularly used for adenoviral virotherapy and many reports showed that high percentages of the general population possess anti-Ad5 neutralizing antibodies (Nabs) which can easily terminate the bioactivity of Ad5. On the other hand, another utmost limitation of utilizing oAds for cancer therapy is the internalization of oAds dramatically depends on appropriate receptors, such as the Coxsackievirus and adenovirus receptor (CAR) on the surface of targeted cells. The CAR is a protein that belongs to a type I membrane receptor for subgroup C adenoviruses. CAR protein is expressed in many human tissues, including brain, heart, and some endothelial and epithelial cells (167–169). The efficiency of adenoviral transgene expression and CAR expression have been corresponded in numerous studies, suggesting that adenoviral binding and entry into target cells play an important role in achieving successful adenoviral gene expression (170, 171). Unfortunately, several cancer cell lines and clinical cancer tumors have been frequently observed in the loss of CAR expression, preventing attempts to achieve adequate oncolytic adenovirus virotherapy for cancer patients (172–175). It is impossible for oAds to obtain satisfactory remedial efficacy without overwhelming this CAR-dependent internalization. To overcome this, most currently ongoing clinical trials of oAds have been genetically modified to equip the beneficial fiber region. This adjusted fiber region can improve cellular internalization of the adenovirus and is independent of CAR expression level in heterogenic clinical tumor or tumor-specific internalization (176–178). However, processing genetic engineering for optimization of fiber-modified virus is risky and contains various disadvantages such as excessive cost, time, and labor-consuming. Inappropriate genetic editing can cause viral replicability loss, viral inactivity, and inadequate gene sequence expression (163).
To overcome these severe limitations of both local and systemic administration of oAd, many advanced polymer-based delivery techniques have been developed in recent years. The polymer-based delivery techniques can enhance the bioavailability while at the same time provide the necessary (Figure 4) protection of oAd. The delivery and tumoral targeting profile of encapsulated oAd can be adapted to the specific medical goals by choosing a proper encapsulation polymer (179, 180). By covering the outer surface of oAd, the polymer-based delivery systems can avoid adverse problems caused by the viral capsid. Furthermore, the applied materials can sufficiently equip the oAd with beneficial properties and override the natively disadvantageous attributes of viral vectors, which efficiently contribute to improving the tumor-specific accumulation of oAd (58, 181–183) (Table 2).
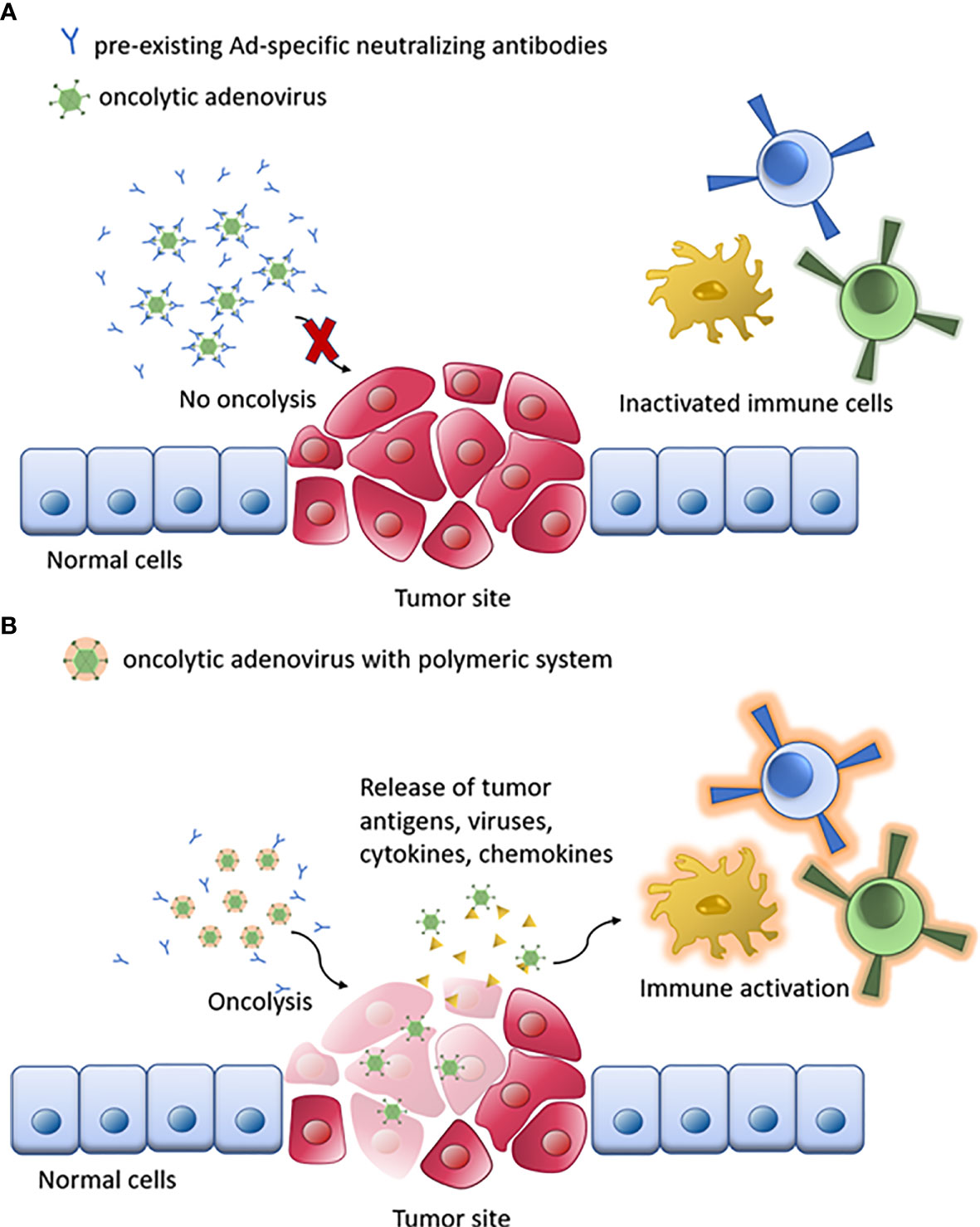
Figure 4 Schematic of oAd immunotherapy with polymeric system. (A) The naked oAd is disabled by pre-existing Ad-specific neutralizing antibodies. (B) Oncolysis by polymer/oAd system stimulates the immune system response against tumor cells, enhancing the therapeutic response.
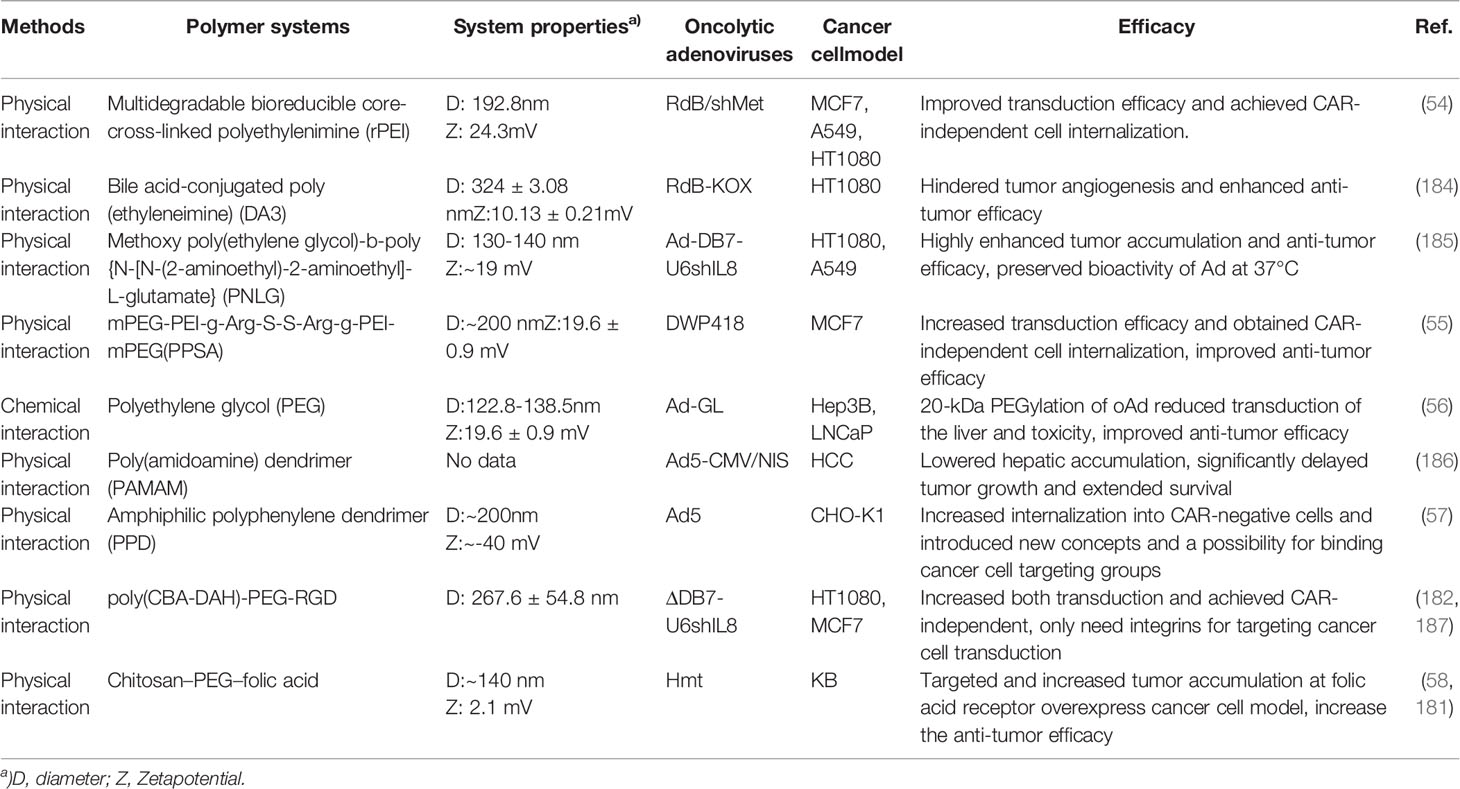
Table 2 Summary of recent research on different polymeric systems for oAd.
2.3.2 Combination Oncolytic Adenovirus Therapy With Polymer Systems
2.3.2.1 Overcoming CAR-Dependent oAd Uptake by Cationic Polymers
The Ad surface interestingly possesses negative charge (188, 189). Therefore, the anionic surface of Ad can electrostatically interact with the cationic polymer to form an Ad/polymer complex. Because of the cationic charge of the polymer, the complex surface is positive, therefore, the complex can enhance the cellular uptake and transgene expression of Ad. Various kinds of polymers have been used for complexing with Ad, most of them based on the positively charged amine groups in the backbone of the polymers. It is noteworthy that the secondary and tertiary amines additionally have high buffering capacity, which compellingly promotes the escape from endosomes into the cytosol of the virus due to the proton sponge effect (190, 191). The polymer structure can be beneficially designed and easily controlled to obtain the advanced bio-function for improving oAd tumor-inhibition efficacy. The method to generate oAd/polymer complex is also straightforward and effortless compared to genetic editing or chemical modification of the virus structure. The oAd/polymer complex can be formed in an aqueous buffer without any additional steps or chemicals. Notably, the original bioactivity of the oAd does not change and is preserved in the complex. On the other hand, the remaining limitations of this method are that the cationic oAd/polymer complex not only can specifically internalize to the cancer cells but also uptake to the body’s healthy cells, subsequently increasing cytotoxicity toward the body. Moreover, the electrostatic interaction of the complex can easily be dissociated in the bloodstream through intravenous injection via associating the cationic polymer with the negatively charged serum protein. The association with some specific serum protein can also trigger the interaction with macrophages and monocytes (192, 193).
2.3.2.1.1 Engineered Poly(Ethyleneimine) for oAd
In the history of developing transfection reagents, beside the polylysine, the poly(ethyleneimine) (PEI) was the second polymeric transfection agent discovered (194–196). The repeating unit of PEI consists of the amine group and two carbon aliphatic spacers. Depending on whether the PEI is linear, branched, or dendrimer form, the structure of PEI can contain primary, secondary, or tertiary amino groups. The PEI (25 kDa) is considered as a standard model for transfection reagent because of its high transgene expression (197, 198). Though the PEI performs high cytotoxicity by two mechanisms, the cationic charge of PEI can possibly disrupt the cell membrane and lead to necrotic cell death or disrupt the mitochondrial membrane after cytosol internalization leading to apoptosis. Many attempts have been made to reduce the PEI cytotoxicity, including modified with non-toxic polymers such as polyethylene glycol (PEG) or other biopolymers, or cholesterol (199–203). Among these attempts, various bioreducible PEIs have been studied for applying cancer therapy (204–206). The bioreducible PEI contains the disulfide moiety in the copolymer blocks, which will degrade via a reductive environment in the cytoplasm and release the therapeutic materials. The degraded fragments can be clearly excreted by the host body’s excretory system producing low systemic cytotoxicity. In 2015, a new low molecular weight PEI multi cross-linked to bioreducible disulfide cystamine core (rPEI) had been generated by Choi et al. and complexed with Ad (54). The Ad/rPEI complex showed remarkably higher transduction efficiency compared to naked Ad in both CAR-positive and -negative cancer cells, which suggests that the complex can independently transduce to CAR expression cancer cells. Moreover, the GFP intensity of GFP-expressing Ad in the 16kDa rPEI complex was manifold higher than Ad/25 kDa PEI in all A549 (7.7-fold), HT1080 (2.9-fold), and MCF7 cells (2.0-fold), which exhibited the remarkable transduction efficiency of the Ad/rPEI. The oncolytic Ad expressing short hairpin RNA against c-Met mRNA complexed with rPEI, demonstrated more efficient cancer cell killing effect, suppression of Met and VEGF level, and viral production than naked Ad. In recent times, Lee et al. developed a bile acid-conjugated 1.8 kDa PEI (DA3). The VEGF inhibitory gene (KOX) expressing-oncolytic Ad was complexed with DA3 (KOX/DA3) and showed a higher transduction efficiency than naked oAd in both CAR-positive and -negative cancer cells (184). Interestingly, the internalization mechanism of the Ad/DA3 complex and naked Ad were investigated and the results indicated that the mechanism of cellular uptake of the Ad/DA3 complex differed from that of naked Ad. The Ad/DA3 complex appeared to be transduced via clathrin-, caveolae-, and macropinocytosis-mediated endocytosis, whereas the naked Ad appeared to internalize cells mainly by clathrin-mediated endocytosis. The KOX/DA3 exhibited an improved antitumor efficacy compared with naked KOX. The data suggest the DA3 can facilitate the amplification and active replication of KOX.
2.3.2.1.2 Biodegradable/Reducible Polymers Coated oAd
In 2013, a biodegradable methoxy poly(ethylene glycol)-b-poly{N-[N-(2-aminoethyl)-2-aminoethyl]- L-glutamate (PNLG) polymer was generated and used to coat oncolytic Ad (Ad-ΔB7-U6shIL8) (oAd/PNLG) by our group (185). The GFP-expressing Ad complexed PNLG showed improvement in transgene expression in both positive and negative CAR-expressing cells than naked Ad and Ad/bPEI in vitro. In addition, the oAd/PNLG showed better cancer cell killing efficacy in vitro than naked Ad, Ad/PEI. The biodistribution result demonstrated higher tumor accumulation when systemically administered oAd/PNLG compared with naked Ad and Ad/PEI. The oAd/PNLG showed 1229-fold higher tumor-to-liver ratio than the naked oAd. It is noteworthy that the oAd/PNLG also showed significantly lower innate and adaptive immune responses than the naked Ad. Another cationic polymer was introduced by our group in 2014 especially containing arginine moieties that enable promotion of cellular internalization in both low and high CAR-expressing cells (55). The polymer, mPEG-PEI-g-Arg-S-S-Arg-gPEI-mPEG (PPSA), contains multiple arginine functional moieties for increasing transgene expression and introduced a bioreducible disulfide bond to lower cytotoxicity. The oncolytic Ad (DWP418) was complexed with PPSA (DWP418/PPSA) and intratumorally injected into CAR negative MCF7 xenograft mice. The result revealed that the DWP418/PPSA provided more effective anti-tumor responses compared with naked DWP418. Moreover, the results also indicated the DWP418/PPSA-treated mice produced less innate immune response and oAd-specialized neutralizing antibodies than the only DWP418-treated group but produced more viral replication and viral cancer cell lysis in tumor tissues. The optimistic results demonstrate the advantages of utilizing the bioreducible and biodegradable polymer masked oAd in cancer treatment.
2.3.2.2 Tumor Targeting by Oncolytic Ad/Polymer
On the other hand, the complexes of oAd with cationic polymer still face diverse obstacles for clinical translation, such as the cationic oAd/polymer complex not only non-specifically internalizing the cancer cells but also uptaking the normal tissues, subsequently causing cytotoxicity. The electrostatic interaction of the complex can also easily be dissociated in the bloodstream through intravenous injection via associating the cationic polymer with the negatively charged serum protein (207–209).
2.3.2.2.1 PEGylation of Oncolytic Ad
Polyethylene glycol (PEG) is a biocompatible, synthetic, hydrophilic polyether, composed of CH2CH2O repeat units. Abundant therapeutic drugs are frustrated by short half-lives in vivo. The PEG is considered the first material that successfully improves both physiochemical, pharmacodynamic, and pharmacokinetic properties of biological drugs such as proteins, peptides, enzymes, synthetic drugs, etc. and has been approved by the FDA in 1990 (210–212). By interacting with PEG, the therapeutic agents can increase water solubility, and reduce renal clearance and immunogenicity. In 1999, the first research on conjugating Ad with PEG was recorded (212). The PEGylation did not show any damaging effect on the bioactivity of Ad. Also it was shown that it can enhance serum stability, blood circulation, and tumor-specific accumulation. The PEGylation has been proved to improve remarkably the Ad to avoid antibody neutralization and also unfavorable immune response in vitro and in vivo (213, 214).
In 2009, Doronin et al. investigated the effect of PEGylation on oncolytic Ad by using 5-kDa and 20-kDa PEG (56). Interestingly, the therapeutic efficacy of PEGylated oncolytic Ad improves with increasing molecular weight of the PEG. The 20kDa PEG conjugated oAd dramatically reduced liver accumulation and hepatotoxicity by systemic administration. Compared with naked and 5-kDa PEGylated oAd, the 20-kDa PEGylated oAd showed reduction hepatocyte transduction by 19- or 90-fold, respectively, in hepatocarcinoma xenograft tumor models. Moreover, the 20-kDa PEGylated oAd administered mice possessed average survival rate double to only the naked oAd group. The effectiveness of higher molecular weight PEGylated oAd can be explained by PEGylated oAd that has a higher molecular weight and larger hydrodynamic radius than the naked oAd and cleared from the body at a much slower rate by kidney or Kupffer cells in the liver, subsequently increasing the half-life and passive tumor targeting through EPR effect. These positive advantages promise that high molecular weight PEGylation can benefit the therapeutic and survival efficacy of oncolytic adenovirus.
2.3.2.2.2 Dendrimer-Coated Ad
Dendrimers are highly ordered, symmetric, branched polymeric molecules. Many reports have shown that dendrimers potentially transfer genes into cells without damaging or deactivating the DNA (215–217). Poly(amidoamine), or PAMAM, is the most popular dendrimer, which used ethylene diamine or ammonium as a core molecule. In 2013, amine-terminated generation 5 PAMAM dendrimer was used to coat sodium iodide symporter (NIS) expressing oncolytic Ad (Ad5-CMV/NIS) (186). This research conducted by Gruanwald et al. showed the complex increased the transduction efficacy in CAR-negative cells and preserved the activity of Ad against neutralizing Abs. Moreover, the 123I scintigraphy of mice from biodistribution results demonstrated systemically administered PAMAM-complexed Ad5-CMV/NIS significantly diminished transgene expression and induced lower liver toxicity than naked Ad5-CMV/NIS. Further, the in vivo antitumor study indicated the PAMAM-complexed Ad5-CMV/NIS possessed higher tumor inhibition efficacy and survival rate than the naked Ad. Through the research, the PAMAM complex displayed improvement in tumor targeting and reducing liver accumulation of oAd, therefore suggesting potential application of oncolytic virotherapy by systemic administration. Interestingly, in 2020 Wagner et al. reported an amphiphilic polyphenylene dendrimer (dendron) to complex with adenovirus which contains a propargyl-modified triethylene glycol linker at the core (57). This linker of the dendron provides for the complex system high aqueous solubility and the possibility to introduce chemical modifications on the viral surface without directly covalently modifying the virus particles. The research showed that the dendrons link to the surface of the adenovirus through their polar and nonpolar surface groups. The report indicated the masking dendrons can promote the internalization of the Ad/dendron complexes into CAR-negative CHO-K1 cells. Even though the research did not proceed any further in vivo experiments, it did introduce a new concept and a possibility for binding cancer cell targeting groups, subsequently expanding the therapeutic potential of oAd.
2.3.2.2.3 Oncolytic Ad Complexed CD-PEG-cRGD
Even though utilizing cationic polymer masking oAd can improve the therapeutic efficacy, there are still several limitations for the clinical setting. Some nano complexes have low diffusion and the positive charge of the complex can induce normal cell internalization or interact with RES, leading to low tumoral accumulation (207).
Our group has developed an Arg-Glye-Asp (RGD) peptide domain conjugated poly(cystaminebisacrylamine-diaminohexane) [poly(CBA-DAH)] (CD) for modifying oAd to target the tumor side (187). The new biodegradable polymer CD possesses the disulfide bonds which allow the polymer to be smoothly cleaved into harmless fragments when reaching the reductive environment of the cytoplasm. The cyclic RGD, on the other hand, can specifically target avb3 and avb5 integrins which were overexpressed in abundant types of tumor cells (218, 219). The oAd/cRGD-conjugated CD complex increased both transduction and cancer cell killing effect with high specificity in a dose-dependent manner in vitro. The competition assay, which used anti-CAR and anti-integrin antibodies, demonstrated that the oAd/cRGD-conjugated CD complex achieved CAR-independence and only needed integrins for targeting cancer cell transduction, contracted to naked Ad which needs both of CAR and integrins to infect. Furthermore, the oAd/cRGD-conjugated CD complex also showed dramatically induced apoptosis and necrosis besides reduced VEGF and IL-8 secreting in cancer cells compared to naked oAd. The in vivo antitumor efficacy of the oncolytic Ad/CD-PEG-cRGD complex was further reported in a lung orthotopic tumor model in 2014 (182). The oAd was successfully covered by the bioreducible CD-PEG-cRGD polymer help to avoid anti-viral immune responses resulting in decreased hepatotoxicity. More importantly, the length of PEG moiety showed a large impact on the therapeutic effect of the system. The CD-PEG2000-cRGD, which has 2000 Da PEG length, coated with oAd obtained a better tumor growth inhibition than the CD-PEG500-cRGD coated oncolytic Ad, illustrating that PEG with longer length could improve the pharmacokinetic and tumoral accumulation of the complex. These reports have indicated the cRGD and CD-PEG-cRGD potentially enhanced the transduction and tumoral accumulation of the oAd according to the interaction between tumor homing peptides and integrins.
2.3.2.2.4 Folate Receptor Overexpressed Cancer Therapy With Oncolytic Ad/Chitosan PEG-FA
In recent years, folic acid (FA) has been wildly investigated as an active targeting moiety for cancer therapy according to a large number of cancer cells overexpress folate receptors on their surface (220–222). To utilize the tumoral targeting property of FA, our group has developed an oAd/chitosan-PEG-FA complex by the advanced electrospinning technique (58, 181). After the electrospinning process, the chitosan-PEG-FA coating on the Ad was confirmed by the size and the ζ-potential of the complex as well as the biological activity of the Ad was preserved. Furthermore, the oAd/chitosan–PEG–FA obtained the blood retention time 48.9-fold higher than the naked oAd and the liver uptake was also 378-fold reduced. This suggested the PEG moiety can lower the non-specific liver uptake dramatically. It is noteworthy that the oAd/chitosan–PEG–FA significantly increased the tumor-to-liver ratio by 1.08 × 105-fold compared to naked oAd. This strongly exhibited the excellence in tumor selectivity of oAd/chitosan–PEG–FA complex in vivo. The antitumor efficacy result confirmed the oncolytic Ad/chitosan–PEG possessed more novel therapeutic effect than the oncolytic Ad/chitosan–PEG.
2.3.2.3 Combined With Immune Cell Therapy by Polymeric Hydrogel
Another interesting utilization of a polymeric system is hydrogel. The hydrogel is the polymer matrix which contains a high amount of water or biological fluid. Therefore, the hydrogel can be utilized to deliver several types of therapeutic agents, including cells or viruses. One researcher of our group in 2017 applied gelatin-hydroxyphenyl propionic acid (GHPA)-based hydrogel to co-deliver oAd and dendritic cells (DCs) for combined immunotherapy (223). The DCs can present TAA to cytotoxic T cells to induce tumor-specific immunity while the oAd can co-express interleukin (IL)-12 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to elicit synergistic tumor growth inhibition. The hydrogel system successfully protected the biological activity and released both oAd and DCs in a controlled manner, leading to a long retention time of both therapeutics in the tumor site. Moreover, the expression level of IL-12, GM-CSF, and interferon-γ (IFN-γ) in the tumor treated with oAd- and DC-loaded gel (oAd + DC/gel) was dramatically higher than oAd or DC only, or the dual injection without gel (oAd + DC). As a result, the number of both activated endogenous and exogenous DCs, the number of DCs migrated to lymph nodes, and the tumor infiltration of CD4+ and CD8+ T cells are remarkably high in the (oAd + DC/gel) samples. Further, the tumor inhibition profile of the (oAd + DC/gel) group indicated the best antitumor performant which demonstrated the novelty of this method. By utilizing gelatin-based hydrogel, the research showed the potential of co-delivery oAd and DCs to the tumor tissue not only can preserve but also induce synergistic immune response with a single dose for a relatively long time.
In summary, systemic delivery of oAd has shown limited therapeutic efficacy due to hepatotoxicity, immunogenicity, and CAR-dependent transduction of Ad. Therefore, surface modification of oAd with polymeric systems provides a novel delivery strategy which can reduce immunogenicity, nonspecific liver sequestration and hepatotoxicity, and enhance transduction efficacy. Furthermore, it prolongs blood retention time and enhances overall intratumoral accumulation of Ad, ultimately leading to potent therapeutic efficacy.
3 Conclusion and Perspectives
Cancer immunotherapies using ICI, CAR-T, and OV are very new and promising treatment strategies to eradicate tumors and inhibit tumor metastasis by activating the immune system. These strategies can be considered as a game-changer for modern cancer treatment in the next coming time. Despite abundant advancements and excellent clinical outcomes, many challenges are remaining and need to be overcome, relating to low antitumor efficiency, costly processes, and side effects (224, 225). In this review, we have summarized divergent polymeric systems for improving the overall therapeutic efficacy of mentioned cancer immunotherapies. Through utilization of appropriate polymeric systems, the above-mentioned disadvantages of immunotherapies can be resolved and can further enhance therapeutic efficacy, biocompatibility, and high specificity. Polymeric systems provide a novel delivery method with numerous benefits such as low toxicity, excellent biodegradability, and flexible surface and size modification for the conjugation of immune ligands and the loading of immunotherapeutic agents. It is notable that polymeric systems can protect and preserve the bioactivity of bioactive agents, insulating them from the unfavorable immune reaction or stimulate the favorable one in the body condition.
Regardless of these polymeric systems-mediated delivery of immunotherapy methods, there are still hurdles that remain before the application to patients in the clinic, such as low treatment efficacy, resistance to cancer immunotherapies, patient safety issues, and expensive treatment costs. Therefore, further research must be conducted to improve current delivery strategies. Delivery systems must yield a more effective and reliable approach for the delivery of immunotherapy agents. New methods to proliferate and engineer immune cell therapies ex vivo should also be developed with lower-cost manufacturing methods. For these purposes, it is expected that in the future, polymeric systems will be more extensively and ingeniously fabricated for cancer immunotherapies, hence enhancing their efficacy, and lowering immune-related side effects. Moreover, for the development of the clinical translation of nanomedicine, extra investigation on whole-body biocompatibility and the effects of various polymer systems on different organs is necessary. Furthermore, the next generation of polymer nanotechnology-based immunotherapy should supremely possess multiple functions, including targeting capability, smart responsiveness, and convenient applicability. Especially the personalized immune treatment with the assistant of polymer nanoparticles will be a critical and promising research trend in the future.
Author Contributions
Conceptualization, TL, A-RY, and C-OY. Methodology, TL. Formal analysis, TL, A-RY and TT. data curation, TL and TT. Writing—original draft preparation, TL and A-RY. Writing—review and editing, TT and C-OY. Supervision, C-OY. Funding acquisition, C-OY. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
C-OY is CEO of GeneMedicine, Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This research was supported by grants from the National Research Foundation of Korea (2016M3A9B5942352 and 2021R1A2C301016611, C-OY).
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Pineros M, Znaor A, et al. Cancer Statistics for the Year 2020: An Overview. Int J Cancer (2021) 149(4):778–89. doi: 10.1002/ijc.33588
3. Wang SM, Ping M, Zhang GH, Guo YR, Li YF, Jia JM. Clinical Observation on Prognosis Survival Rate and Side Effects of Sequential Radiotherapy and Chemotherapy in Patients With Stage Ii-Iii Gastric Cancer. Acta Med Mediterr (2021) 37(2):903–7. doi: 10.19193/0393-6384_2021_2_137
4. Olver I, Carey M, Boyes A, Hall A, Noble N, Bryant J, et al. The Timeliness of Patients Reporting the Side Effects of Chemotherapy. Support Care Cancer (2018) 26(10):3579–86. doi: 10.1007/s00520-018-4225-y
5. Burstein HJ. Side Effects of Chemotherapy. Case 1. Radiation Recall Dermatitis From Gemcitabine. J Clin Oncol (2000) 18(3):693-4. doi: 10.1200/JCO.2000.18.3.693
6. Bergelt C, Lehmann C, Beierlein V, Koch U. Quality of Life and Treatment Side Effects Before and After Radiation Therapy. Psychooncology (2008) 17:S288–9. doi: 10.1007/s11136-008-9349-y
7. Wang L, Ma Q, Yao R, Liu J. Current Status and Development of Anti-PD-1/PD-L1 Immunotherapy for Lung Cancer. Int Immunopharmacol (2020) 79:106088. doi: 10.1016/j.intimp.2019.106088
8. Kiaie SH, Sanaei MJ, Heshmati M, Asadzadeh Z, Azimi I, Hadidi S, et al. Immune Checkpoints in Targeted-Immunotherapy of Pancreatic Cancer: New Hope for Clinical Development. Acta Pharm Sin B (2021) 11(5):1083–97. doi: 10.1016/j.apsb.2020.12.011
9. Velcheti V, Hu X, Piperdi B, Burke T. Real-World Outcomes of First-Line Pembrolizumab Plus Pemetrexed-Carboplatin for Metastatic Nonsquamous NSCLC at US Oncology Practices. Sci Rep (2021) 11(1):9222. doi: 10.1038/s41598-021-88453-8
10. Twomey JD, Zhang B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J (2021) 23(2):39. doi: 10.1208/s12248-021-00574-0
11. Eskander RN, Powell MA. Immunotherapy as a Treatment Strategy in Advanced Stage and Recurrent Endometrial Cancer: Review of Current Phase III Immunotherapy Clinical Trials. Ther Adv Med Oncol (2021) 13:17588359211001199. doi: 10.1177/17588359211001199
12. Cheng G, Dong H, Yang C, Liu Y, Wu Y, Zhu L, et al. A Review on the Advances and Challenges of Immunotherapy for Head and Neck Cancer. Cancer Cell Int (2021) 21(1):406. doi: 10.1186/s12935-021-02024-5
13. Shaikh SS, Kumthekar PU, Mohindra NA. A Review of Eligibility for Patients With Central Nervous System (CNS) Metastases From non-Small Cell Lung Cancer (NSCLC) in Immunotherapy Clinical Trials. J Neurooncol (2019) 144(1):235–7. doi: 10.1007/s11060-019-03219-7
14. Chen WX, Li GX, Hu ZN, Zhu P, Zhang BX, Ding ZY. Significant Response to Anti-PD-1 Based Immunotherapy Plus Lenvatinib for Recurrent Intrahepatic Cholangiocarcinoma With Bone Metastasis: A Case Report and Literature Review. Medicine (Baltimore) (2019) 98(45):e17832. doi: 10.1097/MD.0000000000017832
15. Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus-Specific Memory T Cells Populate Tumors and can be Repurposed for Tumor Immunotherapy. Nat Commun (2019) 10(1):567. doi: 10.1038/s41467-019-08534-1
16. Byrne A, Savas P, Sant S, Li R, Virassamy B, Luen SJ, et al. Tissue-Resident Memory T Cells in Breast Cancer Control and Immunotherapy Responses, Nature Reviews. Clin Oncol (2020) 17(6):341–8. doi: 10.1038/s41571-020-0333-y
17. Yousefi H, Yuan JD, Keshavarz-Fathi M, Murphy JF, Rezaei N. Immunotherapy of Cancers Comes of Age. Expert Rev Clin Immunol (2017) 13(10):1001–15. doi: 10.1080/1744666X.2017.1366315
18. Finck A, Gill SI, June CH. Cancer Immunotherapy Comes of Age and Looks for Maturity. Nat Commun (2020) 11(1):3325. doi: 10.1038/s41467-020-17140-5
19. Wang JL, Yang T, Xu J. Therapeutic Development of Immune Checkpoint Inhibitors. In: Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy New York: Springer Nature/Advances in Experimental Medicine and Biology. vol. 1248. (2020). p. 619–49.
20. Jaspers JE, Brentjens RJ. Development of CAR T Cells Designed to Improve Antitumor Efficacy and Safety. Pharmacol Ther (2017) 178:83–91. doi: 10.1016/j.pharmthera.2017.03.012
21. Singh AK, McGuirk JP. CAR T Cells: Continuation in a Revolution of Immunotherapy. Lancet Oncol (2020) 21(3):e168–78. doi: 10.1016/S1470-2045(19)30823-X
22. Mishra AK, Kemler I, Dingli D. Preclinical Development of CD126 CAR-T Cells With Broad Antitumor Activity. Blood Cancer J (2021) 11(1):3. doi: 10.1038/s41408-020-00405-z
23. Pol J, Kroemer G, Galluzzi L. First Oncolytic Virus Approved for Melanoma Immunotherapy. Oncoimmunology (2015) 5(1):e1115641. doi: 10.1080/2162402X.2015.1115641
24. Carpenter AB, Carpenter AM, Aiken R, Hanft S. Oncolytic Virus in Gliomas: A Review of Human Clinical Investigations. Ann Oncol (2021) 32(8):968–82. doi: 10.1016/j.annonc.2021.03.197
25. Evgin L, Huff AL, Wongthida P, Thompson J, Kottke T, Tonne J, et al. Oncolytic Virus-Derived Type I Interferon Restricts CAR T Cell Therapy. Nat Commun (2020) 11(1):3187. doi: 10.1038/s41467-020-17011-z
26. Msaouel P, Opyrchal M, Dispenzieri A, Peng KW, Federspiel MJ, Russell SJ, et al. Clinical Trials With Oncolytic Measles Virus: Current Status and Future Prospects. Curr Cancer Drug Targets (2018) 18(2):177–87. doi: 10.2174/1568009617666170222125035
27. Centanni M, Moes DJAR, Troconiz IF, Ciccolini J, van Hasselt JGC. Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clin Pharmacokinet (2019) 58(7):835–57. doi: 10.1007/s40262-019-00748-2
28. Kourie HR, Klastersky J. Immune Checkpoint Inhibitors Side Effects and Management. Immunotherapy-Uk (2016) 8(7):799–807. doi: 10.2217/imt-2016-0029
29. Hussaini S, Chehade R, Boldt RG, Raphael J, Blanchette P, Maleki Vareki S, et al. Association Between Immune-Related Side Effects and Efficacy and Benefit of Immune Checkpoint Inhibitors - A Systematic Review and Meta-Analysis. Cancer Treat Rev (2021) 92:102134. doi: 10.1016/j.ctrv.2020.102134
30. Smith LT, Venella K. Cytokine Release Syndrome Inpatient Care for Side Effects of CAR T-Cell Therapy. Clin J Oncol Nurs (2017) 21(2):29–34. doi: 10.1188/17.CJON.S2.29-34
31. Ying Z, Huang XF, Xian X, Liu Y, Kang X, Song Y, et al A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat Med (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
32. Su C, Peng L, Sham J, Wang X, Zhang Q, Chua D, et al. Immune Gene-Viral Therapy With Triplex Efficacy Mediated by Oncolytic Adenovirus Carrying an Interferon-Gamma Gene Yields Efficient Antitumor Activity in Immunodeficient and Immunocompetent Mice. Mol Ther (2006) 13(5):918–27. doi: 10.1016/j.ymthe.2005.12.011
33. van Beusechem VW, van den Doel PB, Grill J, Pinedo HM, Gerritsen WR. Conditionally Replicative Adenovirus Expressing P53 Exhibits Enhanced Oncolytic Potency. Cancer Res (2002) 62(21):6165–71.
34. Han S, Huang K, Gu Z, Wu J. Tumor Immune Microenvironment Modulation-Based Drug Delivery Strategies for Cancer Immunotherapy. Nanoscale (2020) 12(2):413–36. doi: 10.1039/C9NR08086D
35. Economopoulou P, Kotsantis I, Psyrri A. Tumor Microenvironment and Immunotherapy Response in Head and Neck Cancer. Cancers (2020) 12(11):3377. doi: 10.3390/cancers12113377
36. Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Biopolymer Implants Enhance the Efficacy of Adoptive T-Cell Therapy. Nat Biotechnol (2015) 33(1):97–U277. doi: 10.1038/nbt.3104
37. Liang J-L, Luo G-F, Chen W-H, Zhang X-Z. Recent Advances in Engineered Materials for Immunotherapy-Involved Combination Cancer Therapy. Adv Mater (2021) 33(31):2007630. doi: 10.1002/adma.202007630
38. Shields CW IV, Wang LL-W, Evans MA, Mitragotri S. Materials for Immunotherapy. Adv Mater (2020) 32(13):1901633. doi: 10.1002/adma.201901633
39. Lv M, Li S, Zhao HJ, Wang KW, Chen QQ, Guo Z, et al. Redox-Responsive Hyperbranched Poly(Amido Amine) and Polymer Dots as a Vaccine Delivery System for Cancer Immunotherapy. J Mater Chem B (2017) 5(48):9532–45. doi: 10.1039/C7TB02334K
40. Yoshizaki Y, Yuba E, Sakaguchi N, Koiwai K, Harada A, Kono K. Effect of Molecular Adjuvant Inclusion in pH-Sensitive Polymer-Modified Liposomes on Their Performance as Antigen Delivery Carriers for Cancer Immunotherapy. J Control Release (2015) 213:E143–3. doi: 10.1016/j.jconrel.2015.05.242
41. Yuba E, Kono Y, Harada A, Yokoyama S, Arai M, Kubo K, et al. The Application of pH-Sensitive Polymer-Lipids to Antigen Delivery for Cancer Immunotherapy. Biomaterials (2013) 34(22):5711–21. doi: 10.1016/j.biomaterials.2013.04.007
42. Liu L, Wang Y, Guo X, Zhao J, Zhou S. A Biomimetic Polymer Magnetic Nanocarrier Polarizing Tumor-Associated Macrophages for Potentiating Immunotherapy. Small (2020) 16(38):e2003543. doi: 10.1002/smll.202003543
43. Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer Nanoparticles for Immunotherapy From Encapsulated Tumor-Associated Antigens and Whole Tumor Cells. Mol Pharm (2007) 4(1):47–57. doi: 10.1021/mp060107e
44. Fu JR, Cai J, Ling GX, Li AN, Zhao JW, Guo XJ, et al. Cationic Polymers for Enhancing CpG Oligodeoxynucleotides-Mediated Cancer Immunotherapy. Eur Polym J (2019) 113:115–32. doi: 10.1016/j.eurpolymj.2018.12.044
45. Rodes KR, Isser A, Hickey JW, Ben-Akiva E, Meyer RA, Kosmides AK, et al. Biodegradable Cationic Polymer Blends for Fabrication of Enhanced Artificial Antigen Presenting Cells to Treat Melanoma. ACS Appl Mater Interfaces (2021) 13(7):7913-23. doi: 10.1021/acsami.0c19955
46. Wu YB, Wei W, Zhou M, Wang YQ, Wu J, Ma GH, et al. Thermal-Sensitive Hydrogel as Adjuvant-Free Vaccine Delivery System for H5N1 Intranasal Immunization. Biomaterials (2012) 33(7):2351–60. doi: 10.1016/j.biomaterials.2011.11.068
47. Amani H, Shahbazi MA, D'Amico C, Fontana F, Abbaszadeh S, Santos HA. Microneedles for Painless Transdermal Immunotherapeutic Applications. J Control Release (2021) 330:185–217. doi: 10.1016/j.jconrel.2020.12.019
48. Zhou L, Hou B, Wang D, Sun F, Song R, Shao Q, et al. Engineering Polymeric Prodrug Nanoplatform for Vaccination Immunotherapy of Cancer. Nano Lett (2020) 20(6):4393–402. doi: 10.1021/acs.nanolett.0c01140
49. Hwang D, Ramsey JD, Kabanov AV. Polymeric Micelles for the Delivery of Poorly Soluble Drugs: From Nanoformulation to Clinical Approval. Adv Drug Deliv Rev (2020) 156:80–118. doi: 10.1016/j.addr.2020.09.009
50. Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, et al. T Cell-Targeting Nanoparticles Focus Delivery of Immunotherapy to Improve Antitumor Immunity. Nat Commun (2017) 8(1):1747. doi: 10.1038/s41467-017-01830-8
51. Peng J, Xiao Y, Li W, Yang Q, Tan L, Jia Y, et al. Photosensitizer Micelles Together With IDO Inhibitor Enhance Cancer Photothermal Therapy and Immunotherapy. Adv Sci (2018) 5(5):(2198–3844 (Print)). doi: 10.1002/advs.201700891
52. Ye YQ, Wang JQ, Hu QY, Hochu GM, Xin HL, Wang C, et al. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses Through Delivery of Checkpoint Inhibitors. ACS Nano (2016) 10(9):8956–63. doi: 10.1021/acsnano.6b04989
53. Su Z, Xiao Z, Wang Y, Huang J, An Y, Wang X, et al. Codelivery of Anti-PD-1 Antibody and Paclitaxel With Matrix Metalloproteinase and pH Dual-Sensitive Micelles for Enhanced Tumor Chemoimmunotherapy. Small (2020) 16(7):1906832. doi: 10.1002/smll.201906832
54. Choi JW, Nam JP, Nam K, Lee YS, Yun CO, Kim SW. Oncolytic Adenovirus Coated With Multidegradable Bioreducible Core-Cross-Linked Polyethylenimine for Cancer Gene Therapy. Biomacromolecules (2015) 16(7):2132–43. doi: 10.1021/acs.biomac.5b00538
55. Jung SJ, Kasala D, Choi JW, Lee SH, Hwang JK, Kim SW, et al. Safety Profiles and Antitumor Efficacy of Oncolytic Adenovirus Coated With Bioreducible Polymer in the Treatment of a CAR Negative Tumor Model. Biomacromolecules (2015) 16(1):87–96. doi: 10.1021/bm501116x
56. Doronin K, Shashkova EV, May SM, Hofherr SE, Barry MA. Chemical Modification With High Molecular Weight Polyethylene Glycol Reduces Transduction of Hepatocytes and Increases Efficacy of Intravenously Delivered Oncolytic Adenovirus. Hum Gene Ther (2009) 20(9):975–88. doi: 10.1089/hum.2009.028
57. Wagner J, Li LJ, Simon J, Krutzke L, Landfester K, Mailander V, et al. Amphiphilic Polyphenylene Dendron Conjugates for Surface Remodeling of Adenovirus 5. Angew Chem Int Edit (2020) 59(14):5712–20. doi: 10.1002/anie.201913708
58. Park Y, Kang E, Kwon OJ, Hwang T, Park H, Lee JM, et al. Ionically Crosslinked Ad/chitosan Nanocomplexes Processed by Electrospinning for Targeted Cancer Gene Therapy. J Control Release (2010) 148(1):75–82. doi: 10.1016/j.jconrel.2010.06.027
59. Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, et al. Hit-And-Run Programming of Therapeutic Cytoreagents Using mRNA Nanocarriers. Nat Commun (2017) 8(1):389. doi: 10.1038/s41467-017-00505-8
60. Yu QR, Zhang MX, Chen YT, Chen XL, Shi SY, Sun K, et al. Self-Assembled Nanoparticles Prepared From Low-Molecular-Weight PEI and Low-Generation PAMAM for EGFRvIII-Chimeric Antigen Receptor Gene Loading and T-Cell Transient Modification. Int J Nanomedicine (2020) 15:483–95. doi: 10.2147/IJN.S229858
61. Sunshine JC, Perica K, Schneck JP, Green JJ. Particle Shape Dependence of CD8+T Cell Activation by Artificial Antigen Presenting Cells. Biomaterials (2014) 35(1):269–77. doi: 10.1016/j.biomaterials.2013.09.050
62. Mandal S, Eksteen-Akeroyd ZH, Jacobs MJ, Hammink R, Koepf M, Lambeck AJA, et al. Therapeutic Nanoworms: Towards Novel Synthetic Dendritic Cells for Immunotherapy. Chem Sci (2013) 4(11):4168–74. doi: 10.1039/c3sc51399h
63. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
64. Munn DH, Bronte V. Immune Suppressive Mechanisms in the Tumor Microenvironment. Curr Opin Immunol (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
65. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The Immune Checkpoint PD-1 in Natural Killer Cells: Expression, Function and Targeting in Tumour Immunotherapy. Cancers (2020) 12(11):3285. doi: 10.3390/cancers12113285
66. Setordzi P, Chang X, Liu Z, Wu Y, Zuo D. The Recent Advances of PD-1 and PD-L1 Checkpoint Signaling Inhibition for Breast Cancer Immunotherapy. Eur J Pharmacol (2021) 895:173867. doi: 10.1016/j.ejphar.2021.173867
67. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol (2017) 8(561). doi: 10.3389/fphar.2017.00561
68. Ma XT, Zhang YJ, Wang S, Wei HM, Yu J. Immune Checkpoint Inhibitor (ICI) Combination Therapy Compared to Monotherapy in Advanced Solid Cancer: A Systematic Review. J Cancer (2021) 12(5):1318–33. doi: 10.7150/jca.49174
69. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors A Review. JAMA Oncol (2016) 2(10):1346–53. doi: 10.1001/jamaoncol.2016.1051
70. Heleno CT, Mustafa A, Gotera NA, Tesar A. Myasthenia Gravis as an Immune-Mediated Side Effect of Checkpoint Inhibitors. Cureus (2021) 13(7):e16316. doi: 10.7759/cureus.16316
71. Restifo NP, Smyth MJ, Snyder A. Acquired Resistance to Immunotherapy and Future Challenges. Nat Rev Cancer (2016) 16(2):121–6. doi: 10.1038/nrc.2016.2
72. Bashash D, Zandi Z, Kashani B, Pourbagheri-Sigaroodi A, Salari S, Ghaffari SH. Resistance to Immunotherapy in Human Malignancies: Mechanisms, Research Progresses, Challenges, and Opportunities. J Cell Physiol (2021) 237:346–72. doi: 10.1002/jcp.30575
73. Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
74. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJT, et al. Ovarian Cancer Immunotherapy Using PD-L1 siRNA Targeted Delivery From Folic Acid-Functionalized Polyethylenimine: Strategies to Enhance T Cell Killing. Adv Healthc Mater (2015) 4(8):1180–9. doi: 10.1002/adhm.201500089
75. Li SY, Liu Y, Xu CF, Shen S, Sun R, Du XJ, et al. Restoring Anti-Tumor Functions of T Cells via Nanoparticle-Mediated Immune Checkpoint Modulation. J Control Release (2016) 231:17–28. doi: 10.1016/j.jconrel.2016.01.044
76. Chen Y, Xia R, Huang Y, Zhao W, Li J, Zhang X, et al. An Immunostimulatory Dual-Functional Nanocarrier That Improves Cancer Immunochemotherapy. Nat Commun (2016) 7(1):13443. doi: 10.1038/ncomms13443
77. Sun JJ, Chen YC, Huang YX, Zhao WC, Liu YH, Venkataramanan R, et al. Programmable Co-Delivery of the Immune Checkpoint Inhibitor NLG919 and Chemotherapeutic Doxorubicin via a Redox-Responsive Immunostimulatory Polymeric Prodrug Carrier. Acta Pharmacol Sin (2017) 38(6):823–34. doi: 10.1038/aps.2017.44
78. Wan ZY, Sun JJ, Xu JN, Moharil P, Chen J, Xu JC, et al. Dual Functional Immunostimulatory Polymeric Prodrug Carrier With Pendent Indoximod for Enhanced Cancer Immunochemotherapy. Acta Biomater (2019) 90:300–13. doi: 10.1016/j.actbio.2019.03.048
79. Lan Y, Liang QW, Sun Y, Cao AC, Liu L, Yu SY, et al. Codelivered Chemotherapeutic Doxorubicin via a Dual-Functional Immunostimulatory Polymeric Prodrug for Breast Cancer Immunochemotherapy. ACS Appl Mater Interfaces (2020) 12(28):31904–21. doi: 10.1021/acsami.0c06120
80. Huang GL, Tao A, Miyazaki T, Khan T, Hong T, Nakagawa Y, et al. PEG-Poly(1-Methyl-L-Tryptophan)-Based Polymeric Micelles as Enzymatically Activated Inhibitors of Indoleamine 2,3-Dioxygenase. Nanomaterials (2019) 9(5):719. doi: 10.3390/nano9050719
81. Rahimian S, Fransen MF, Kleinovink JW, Amidi M, Ossendorp F, Hennink WE. Polymeric Microparticles for Sustained and Local Delivery of Anticd40 and antiCTLA-4 in Immunotherapy of Cancer. Biomaterials (2015) 61:33–40. doi: 10.1016/j.biomaterials.2015.04.043
82. Zhang L, Wang LM, Shahzad KA, Xu T, Wan X, Pei WY, et al. Paracrine Release of IL-2 and Anti-CTLA-4 Enhances the Ability of Artificial Polymer Antigen-Presenting Cells to Expand Antigen-Specific T Cells and Inhibit Tumor Growth in a Mouse Model. Cancer Immunol Immun (2017) 66(9):1229–41. doi: 10.1007/s00262-017-2016-9
83. Wang C, Ye YQ, Hochu GM, Sadeghifar H, Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett (2016) 16(4):2334–40. doi: 10.1021/acs.nanolett.5b05030
84. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-Cell Engineering Using non-Viral Sleeping Beauty Transposition From Minicircle Vectors. Leukemia (2017) 31(1):186–94. doi: 10.1038/leu.2016.180
85. Maine CJ, Aziz NH, Chatterjee J, Hayford C, Brewig N, Whilding L, et al. Programmed Death Ligand-1 Over-Expression Correlates With Malignancy and Contributes to Immune Regulation in Ovarian Cancer. Cancer Immunol Immunother (2014) 63(3):215–24. doi: 10.1007/s00262-013-1503-x
86. Papageorgis P, Stylianopoulos T. Role of TGF Beta in Regulation of the Tumor Microenvironment and Drug Delivery. Int J Oncol (2015) 46(3):933–43. doi: 10.3892/ijo.2015.2816
87. Pickup M, Novitskiy S, Moses HL. The Roles of TGF Beta in the Tumour Microenvironment. Nat Rev Cancer (2013) 13(11):788–99. doi: 10.1038/nrc3603
88. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical Development of Galunisertib (LY2157299 Monohydrate), a Small Molecule Inhibitor of Transforming Growth Factor-Beta Signaling Pathway. Drug Des Dev Ther (2015) 9:4479–99. doi: 10.2147/DDDT.S86621
89. Mahoney KM, Rennert PD, Freeman GJ. Combination Cancer Immunotherapy and New Immunomodulatory Targets. Nat Rev Drug Discov (2015) 14(8):561–84. doi: 10.1038/nrd4591
90. Restifo NP, Dudley ME, Rosenberg SA. Adoptive Immunotherapy for Cancer: Harnessing the T Cell Response. Nat Rev Immunol (2012) 12(4):269–81. doi: 10.1038/nri3191
91. Obajdin J, Davies DM, Maher J. Engineering of Chimeric Natural Killer Cell Receptors to Develop Precision Adoptive Immunotherapies for Cancer. Clin Exp Immunol (2020) 202(1):11–27. doi: 10.1111/cei.13478
92. Rafiq S, Jackson HJ, Yeku O, Purdon TJ, van Leeuwen DG, Curran KJ, et al. Enhancing CAR T Cell Anti-Tumor Efficacy Through Secreted Single Chain Variable Fragment (scFv) Immune Checkpoint Blockade. Blood (2017) 130(Supplement 1):842–2. doi: 10.1182/blood.V130.Suppl_1.842.842
93. Weinkove R, George P, Dasyam N, McLellan AD. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin Transl Immunol (2019) 8(5):e1049–9. doi: 10.1002/cti2.1049
94. Lipsitz YY, Timmins NE, Zandstra PW. Quality Cell Therapy Manufacturing by Design. Nat Biotechnol (2016) 34(4):393–400. doi: 10.1038/nbt.3525
95. Yip A, Webster RM. The Market for Chimeric Antigen Receptor T Cell Therapies. Nat Rev Drug Discov (2018) 17(3):161–2. doi: 10.1038/nrd.2017.266
96. Tyagarajan S, Spencer T, Smith J. Optimizing CAR-T Cell Manufacturing Processes During Pivotal Clinical Trials. Mol Ther Methods Clin Dev (2020) 16:136–44. doi: 10.1016/j.omtm.2019.11.018
97. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
98. Perica K, Curran KJ, Brentjens RJ, Giralt SA. Building a CAR Garage: Preparing for the Delivery of Commercial CAR T Cell Products at Memorial Sloan Kettering Cancer Center. Biol Blood Marrow Transplant (2018) 24(6):1135–41. doi: 10.1016/j.bbmt.2018.02.018
99. Sponchioni M, Manfredini N, Zanoni A, Scibona E, Morbidelli M, Moscatelli D. Readily Adsorbable Thermoresponsive Polymers for the Preparation of Smart Cell-Culturing Surfaces on Site. ACS Biomater Sci Eng (2020) 6(9):5337–45. doi: 10.1021/acsbiomaterials.0c01029
100. Janouskova O. Synthetic Polymer Scaffolds for Soft Tissue Engineering. Physiol Res (2018) 67(Suppl 2):S335–48. doi: 10.33549/physiolres.933983
101. Zhu ZX, Wu D, Li S, Han Y, Xiang N, Wang CL, et al. A Polymer-Film Inertial Microfluidic Sorter Fabricated by Jigsaw Puzzle Method for Precise Size-Based Cell Separation. Anal Chim Acta (2021) 1143:306–14. doi: 10.1016/j.aca.2020.11.001
102. Xia W, Tao Z, Zhu B, Zhang W, Liu C, Chen S, et al. Targeted Delivery of Drugs and Genes Using Polymer Nanocarriers for Cancer Therapy. Int J Mol Sci (2021) 22(17):9118. doi: 10.3390/ijms22179118
103. Hay KA, Turtle CJ. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned From Targeting of CD19 in B-Cell Malignancies. Drugs (2017) 77(3):237–45. doi: 10.1007/s40265-017-0690-8
104. Olden BR, Perez CR, Wilson AL, Cardle II, Lin Y-S, Kaehr B, et al. Cell-Templated Silica Microparticles With Supported Lipid Bilayers as Artificial Antigen-Presenting Cells for T Cell Activation. Adv Healthc Mater (2019) 8(2):1801188. doi: 10.1002/adhm.201801188
105. Mockel-Tenbrinck N, Barth C, Mauer D, Meyer M, Klumb C, Schenk T, et al. Safety Assessment of the MACS GMP T Cell TransAct, a Robust and Potent Polyclonal T Cell Activation and Expansion Tool for Clinical Scale Manufacturing of Gene-Modified T Cells. Mol Ther (2017) 25(5):227–7.
106. Casati A, Varghaei-Nahvi A, Feldman SA, Assenmacher M, Rosenberg SA, Dudley ME, et al. Clinical-Scale Selection and Viral Transduction of Human Na < Ve and Central Memory CD8(+) T Cells for Adoptive Cell Therapy of Cancer Patients. Cancer Immunol Immun (2013) 62(10):1563–73. doi: 10.1007/s00262-013-1459-x
107. Wang X, Stefanski J, Borquez-Ojeda O, Qu J, Hack A, He Q, et al. Comparison of CTS (TM) Dynabeads (R) CD3/CD28, Miltenyi TransAct CD3/28 and ExpAct Beads for Large-Scale CAR T Cell Manufacturing. Hum Gene Ther (2015) 26(10):A31–1.
108. Wang XY, Qu JR, Stefanski J, Borquez-Ojeda O, Hack A, He Q, et al. Evaluation of Miltenyi ExpAct and TransAct CD3/28 Beads for CAR-T Cell Manufacturing. Mol Ther (2016) 24:S182–2. doi: 10.1016/S1525-0016(16)33268-3
109. Mandal S, Hammink R, Tel J, Eksteen-Akeroyd ZH, Rowan AE, Blank K, et al. Polymer-Based Synthetic Dendritic Cells for Tailoring Robust and Multifunctional T Cell Responses. ACS Chem Biol (2015) 10(2):485–92. doi: 10.1021/cb500455g
110. Mescher MF. Surface-Contact Requirements for Activation of Cytotoxic Lymphocytes-T. J Immunol (1992) 149(7):2402–5.
111. Curtsinger J, Deeths MJ, Pease P, Mescher MF. Artificial Cell Surface Constructs for Studying Receptor-Ligand Contributions to Lymphocyte Activation. J Immunol Methods (1997) 209(1):47–57. doi: 10.1016/S0022-1759(97)00146-4
112. Curtsinger JM, Lins DC, Mescher MF. CD8(+) Memory T Cells (CD44(high), Ly-6C(+)) are More Sensitive Than Naive Cells (CD44(low), Ly-6C(-)) to TCR/CD8 Signaling in Response to Antigen. J Immunol (1998) 160(7):3236–43.
113. Deeths MJ, Mescher MF. B7-1-Dependent Co-Stimulation Results in Qualitatively and Quantitatively Different Responses by CD4(+) and CD8(+) T Cells. Eur J Immunol (1997) 27(3):598–608. doi: 10.1002/eji.1830270305
114. Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, et al. Inflammatory Cytokines Provide a Third Signal for Activation of Naive CD4(+) and CD8(+) T Cells. J Immunol (1999) 162(6):3256–62.
115. Jiang XB, Lu XL, Liu R, Zhang FC, Zhaol HY. HLA Tetramer-Based Artificial Antigen-Presenting Cells Efficiently Stimulate CTLs Specific for Malignant Glioma. Clin Cancer Res (2007) 13(24):7329–34. doi: 10.1158/1078-0432.CCR-07-1025
116. Xiaobing J, Xiaoling L, Ruen L, Fangcheng Z, Hongyang Z. Induction of Cytotoxic T-Lymphocytes Specific for Malignant Glioma by HLA Dimer-Based Artificial Antigen-Presenting Cells. Cancer Biother Radiopharm (2007) 22(6):826–35. doi: 10.1089/cbr.2007.0406
117. Lu XL, Jiang XB, Liu R, Zhao HY, Liang ZH. Adoptive Transfer of Ptrp2-Specific CTLs Expanding by Bead-Based Artificial Antigen-Presenting Cells Mediates Anti-Melanoma Response. Cancer Lett (2008) 271(1):129–39. doi: 10.1016/j.canlet.2008.05.049
118. Shen CL, Cheng K, Miao SW, Wang W, He Y, Meng FY, et al. Latex Bead-Based Artificial Antigen-Presenting Cells Induce Tumor-Specific CTL Responses in the Native T-Cell Repertoires and Inhibit Tumor Growth. Immunol Lett (2013) 150(1-2):1–11. doi: 10.1016/j.imlet.2013.01.003
119. Luxembourg AT, Brunmark A, Kong Y, Jackson MR, Peterson PA, Sprent J, et al. Requirements for Stimulating Naive CD8(+) T Cells via Signal 1 Alone. J Immunol (1998) 161(10):5226–35.
120. Mescher MF, Rogers JD. Immunotherapy of Established Murine Tumors With Large Multivalent Immunogen and Cyclophosphamide. J Immunother (1996) 19(2):102–12. doi: 10.1097/00002371-199603000-00003
121. Goldberg J, Shrikant P, Mescher MF. In Vivo Augmentation of Tumor-Specific CTL Responses by Class I/peptide Antigen Complexes on Microspheres (Large Multivalent Immunogen). J Immunol (2003) 170(1):228–35. doi: 10.4049/jimmunol.170.1.228
122. Mitchell MS, Kan-Mitchell J, Morrow PR, Darrah D, Jones VE, Mescher MF. Phase I Trial of Large Multivalent Immunogen Derived From Melanoma Lysates in Patients With Disseminated Melanoma. Clin Cancer Res (2004) 10(1):76–83. doi: 10.1158/1078-0432.CCR-0689-3
123. Slack JD, Kanke M, Simmons GH, Deluca PP. Acute Hemodynamic-Effects and Blood Pool Kinetics of Polystyrene Microspheres Following Intravenous Administration. J Pharm Sci-Us (1981) 70(6):660–4. doi: 10.1002/jps.2600700621
124. Kutscher HL, Chao P, Deshmukh M, Singh Y, Hu P, Joseph LB, et al. Threshold Size for Optimal Passive Pulmonary Targeting and Retention of Rigid Microparticles in Rats. J Control Release (2010) 143(1):31–7. doi: 10.1016/j.jconrel.2009.12.019
125. Gomzyak VI, Sedush NG, Puchkov AA, Polyakov DK, Chvalun SN. Linear and Branched Lactide Polymers for Targeted Drug Delivery Systems. Polym Sci Ser B+ (2021) 63(3):257–71. doi: 10.1134/S1560090421030064
126. Ravivarapu HB, Burton K, DeLuca PP. Polymer and Microsphere Blending to Alter the Release of a Peptide From PLGA Microspheres. Eur J Pharm Biopharm (2000) 50(2):263–70. doi: 10.1016/S0939-6411(00)00099-0
127. Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, et al. In Vivo Evaluation of the Biodistribution and Safety of PLGA Nanoparticles as Drug Delivery Systems. Nanomed-Nanotechnol (2010) 6(5):662–71. doi: 10.1016/j.nano.2010.02.002
128. Trinh TA, Duy Le TM, Ho HGV, To TCT, Nguyen VVL, Huynh DP, et al. A Novel Injectable pH-Temperature Sensitive Hydrogel Containing Chitosan-Insulin Electrosprayed Nanosphere Composite for an Insulin Delivery System in Type I Diabetes Treatment. Biomater Sci (2020) 8(14):3830–43. doi: 10.1039/D0BM00634C
129. Steenblock ER, Wrzesinski SH, Flavell RA, Fahmy TM. Antigen Presentation on Artificial Acellular Substrates: Modular Systems for Flexible, Adaptable Immunotherapy. Expert Opin Biol Ther (2009) 9(4):451–64. doi: 10.1517/14712590902849216
130. Shalaby WSW, Yeh H, Woo E, Corbett JT, Gray H, June CH, et al. Absorbable Microparticulate Cation Exchanger for Immunotherapeutic Delivery. J BioMed Mater Res B (2004) 69B(2):173–82. doi: 10.1002/jbm.b.20040
131. Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, et al. Biodegradable Nanoellipsoidal Artificial Antigen Presenting Cells for Antigen Specific T-Cell Activation. Small (2015) 11(13):1519–25. doi: 10.1002/smll.201402369
132. Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, et al. A Carbon Nanotube-Polymer Composite for T-Cell Therapy. Nat Nanotechnol (2014) 9(8):639–47. doi: 10.1038/nnano.2014.154
133. Mannisto M, Reinisalo M, Ruponen M, Honkakoski P, Tammi M, Urtti A. Polyplex-Mediated Gene Transfer and Cell Cycle: Effect of Carrier on Cellular Uptake and Intracellular Kinetics, and Significance of Glycosaminoglycans. J Gene Med (2007) 9(6):479–87. doi: 10.1002/jgm.1035
134. Behr JP. The Proton Sponge: A Trick to Enter Cells the Viruses did Not Exploit. Chimia (1997) 51(1-2):34–6.
135. Fornaguera C, Castells-Sala C, Lázaro MA, Cascante A, Borrós S. Development of an Optimized Freeze-Drying Protocol for OM-PBAE Nucleic Acid Polyplexes. Int J Pharm (2019) 569:118612. doi: 10.1016/j.ijpharm.2019.118612
136. Lin S, Du F, Wang Y, Ji S, Liang D, Yu L, et al. An Acid-Labile Block Copolymer of PDMAEMA and PEG as Potential Carrier for Intelligent Gene Delivery Systems. Biomacromolecules (2008) 9(1):109–15. doi: 10.1021/bm7008747
137. Wang D, Wang J, Song J, Shen Q, Wang R, Lu W, et al. Guanidyl and Imidazolyl Integration Group-Modified PAMAM for Gastric Adenocarcinoma Gene Therapy. J Gene Med (2020) 22(10):e3240. doi: 10.1002/jgm.3240
138. Wightman L, Kircheis R, Rossler V, Carotta S, Ruzicka R, Kursa M, et al. Different Behavior of Branched and Linear Polyethylenimine for Gene Delivery In Vitro and In Vivo. J Gene Med (2001) 3(4):362–72. doi: 10.1002/jgm.187
139. Olden BR, Cheng YL, Yu JL, Pun SH. Cationic Polymers for non-Viral Gene Delivery to Human T Cells. J Control Release (2018) 282:140–7. doi: 10.1016/j.jconrel.2018.02.043
140. Cheng YL, Yumul RC, Pun SH. Virus-Inspired Polymer for Efficient In Vitro and In Vivo Gene Delivery. Angew Chem Int Edit (2016) 55(39):12013–7. doi: 10.1002/anie.201605958
141. Peeler DJ, Thai SN, Cheng YL, Horner PJ, Sellers DL, Pun SH. pH-Sensitive Polymer Micelles Provide Selective and Potentiated Lytic Capacity to Venom Peptides for Effective Intracellular Delivery. Biomaterials (2019) 192:235–44. doi: 10.1016/j.biomaterials.2018.11.004
142. Synatschke CV, Schallon A, Jerome V, Freitag R, Muller AHE. Influence of Polymer Architecture and Molecular Weight of Poly(2-(Dimethylamino)Ethyl Methacrylate) Polycations on Transfection Efficiency and Cell Viability in Gene Delivery. Biomacromolecules (2011) 12(12):4247–55. doi: 10.1021/bm201111d
143. Schallon A, Synatschke CV, Jerome V, Muller AHE, Freitag R. Nanoparticulate Nonviral Agent for the Effective Delivery of pDNA and siRNA to Differentiated Cells and Primary Human T Lymphocytes. Biomacromolecules (2012) 13(11):3463–74. doi: 10.1021/bm3012055
144. Raup A, Stahlschmidt U, Jérôme V, Synatschke CV, Müller AHE, Freitag R. Influence of Polyplex Formation on the Performance of Star-Shaped Polycationic Transfection Agents for Mammalian Cells. Polymers (Basel) (2016) 8(6):224. doi: 10.3390/polym8060224
145. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In Situ Programming of Leukaemia-Specific T Cells Using Synthetic DNA Nanocarriers. Nat Nanotechnol (2017) 12(8):813–20. doi: 10.1038/nnano.2017.57
146. Sostoa Jd, Dutoit V, Migliorini D. Oncolytic Viruses as a Platform for the Treatment of Malignant Brain Tumors. Int J Mol Sci (2020) 21(20):7449. doi: 10.3390/ijms21207449
147. Liang M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr Cancer Drug Targets (2018) 18(2):171–6.
148. Zhang QN, Li Y, Zhao Q, Tian M, Chen LL, Miao LY, et al. Recombinant Human Adenovirus Type 5 (Oncorine) Reverses Resistance to Immune Checkpoint Inhibitor in a Patient With Recurrent non-Small Cell Lung Cancer: A Case Report. Thorac Cancer (2021) 12(10):1617–9. doi: 10.1111/1759-7714.13947
149. Zhang WW, Li LJ, Li DG, Liu JL, Li XQ, Li W, et al. The First Approved Gene Therapy Product for Cancer Ad-P53 (Gendicine): 12 Years in the Clinic. Hum Gene Ther (2018) 29(2):160–79. doi: 10.1089/hum.2017.218
150. Fang JB, Zhu GZ, Zhu YL, Li YQ, Shang C, Bai B, et al. Antitumor Effects of Apoptin Expressed by the Dual Cancer-Specific Oncolytic Adenovirus - a Review. Eur Rev Med Pharmacol Sci (2020) 24(21):11334–43. doi: 10.26355/eurrev_202011_23624
151. Lee CS, Bishop ES, Zhang RY, Yu XY, Farina EM, Yan SJ, et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis (2017) 4(2):43–63. doi: 10.1016/j.gendis.2017.04.001
152. Farrera-Sal M, Moya-Borrego L, Bazan-Peregrino M, Alemany R. Evolving Status of Clinical Immunotherapy With Oncolytic Adenovirus. Clin Cancer Res (2021) 27(11):2979–88. doi: 10.1158/1078-0432.CCR-20-1565
153. Volpers C, Kochanek S. Adenoviral Vectors for Gene Transfer and Therapy. J Gene Med (2004) 6:S164–71. doi: 10.1002/jgm.496
154. Bramson JL, Graham FL, Gauldie J. The Use of Adenoviral Vectors for Gene-Therapy and Gene-Transfer in-Vivo. Curr Opin Biotech (1995) 6(5):590–5. doi: 10.1016/0958-1669(95)80097-2
155. Saito M, Murata T, Watanabe K, Kawakami K, Suzuki M, Koji T, et al. Adenoviral Vector-Mediated Gene Transfer of IL-13R Alpha 2 Chain Followed by IL-13 Cytotoxin Treatment Offers Potent Targeted Therapy for Cytotoxin-Resistant Cancers. Int J Cancer (2005) 116(1):1–8. doi: 10.1002/ijc.20995
156. Joung I, Kim HS, Hong JS, Kwon H, Kwon YK. Effective Gene Transfer Into Regenerating Sciatic Nerves by Adenoviral Vectors: Potentials for Gene Therapy of Peripheral Nerve Injury. Mol Cells (2000) 10(5):540–5. doi: 10.1007/s10059-000-0540-4
157. Zhao Y, Liu Z, Li L, Wu J, Zhang H, Zhang H, et al. Oncolytic Adenovirus: Prospects for Cancer Immunotherapy. Front Microbiol (2021) 12:707290. doi: 10.3389/fmicb.2021.707290
158. Ranki T, Joensuu T, Jäger E, Karbach J, Wahle C, Kairemo K, et al. Local Treatment of a Pleural Mesothelioma Tumor With ONCOS-102 Induces a Systemic Antitumor CD8+ T-Cell Response, Prominent Infiltration of CD8+ Lymphocytes and Th1 Type Polarization. Oncoimmunology (2014) 3(10):e958937. doi: 10.4161/21624011.2014.958937
159. Seder RA, Darrah PA, Roederer M. T-Cell Quality in Memory and Protection: Implications for Vaccine Design. Nat Rev Immunol (2008) 8(4):247–58. doi: 10.1038/nri2274
160. Leung EYL, Ennis DP, Kennedy PR, Hansell C, Dowson S, Farquharson M, et al. NK Cells Augment Oncolytic Adenovirus Cytotoxicity in Ovarian Cancer. Mol Ther Oncolytics (2020) 16:289–301. doi: 10.1016/j.omto.2020.02.001
161. Huang JH, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee M, et al. Therapeutic and Tumor-Specific Immunity Induced by Combination of Dendritic Cells and Oncolytic Adenovirus Expressing IL-12 and 4-1BBL. Mol Ther (2010) 18(2):264–74. doi: 10.1038/mt.2009.205
162. Yang YF, Xu WD, Neill T, Hu ZB, Wang CH, Xiao XH, et al. Systemic Delivery of an Oncolytic Adenovirus Expressing Decorin for the Treatment of Breast Cancer Bone Metastases. Hum Gene Ther (2015) 26(12):813–25. doi: 10.1089/hum.2015.098
163. Yoon AR, Hong J, Yun CO. A Vesicular Stomatitis Virus Glycoprotein Epitope-Incorporated Oncolytic Adenovirus Overcomes CAR-Dependency and Shows Markedly Enhanced Cancer Cell Killing and Suppression of Tumor Growth. Oncotarget (2015) 6(33):34875–91. doi: 10.18632/oncotarget.5332
164. Le TMD, Jung BK, Li Y, Duong HTT, Nguyen TL, Hong JW, et al. Physically Crosslinked Injectable Hydrogels for Long-Term Delivery of Oncolytic Adenoviruses for Cancer Treatment. Biomater Sci (2019) 7(10):4195–207. doi: 10.1039/C9BM00992B
165. Pesonen S, Kangasniemi L, Hemminki A. Oncolytic Adenoviruses for the Treatment of Human Cancer: Focus on Translational and Clinical Data. Mol Pharm (2011) 8(1):12–28. doi: 10.1021/mp100219n
166. Tejada S, Díez-Valle R, Domínguez PD, Patiño-García A, González-Huarriz M, Fueyo J, et al. DNX-2401, an Oncolytic Virus, for the Treatment of Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Case Report. Front Oncol (2018) 8:61–1. doi: 10.3389/fonc.2018.00061
167. Cohen CJ, Shieh JTC, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The Coxsackievirus and Adenovirus Receptor is a Transmembrane Component of the Tight Junction. Proc Natl Acad Sci U S A (2001) 98(26):15191–6. doi: 10.1073/pnas.261452898
168. Freimuth P, Philipson L, Carson SD. The Coxsackievirus and Adenovirus Receptor, Group B Coxsackieviruses. Current Topics Microbiol Immunol (2008) 323:67–87. doi: 10.1007/978-3-540-75546-3_4
169. Ito M, Kodama M, Masuko M, Yamaura M, Fuse K, Uesugi Y, et al. Expression of Coxsackievirus and Adenovirus Receptor in Hearts of Rats With Experimental Autoimmune Myocarditis. Circ Res (2000) 86(3):275–80. doi: 10.1161/01.RES.86.3.275
170. Buttgereit P, Weineck S, Ropke G, Marten A, Brand K, Heinicke T, et al. Efficient Gene Transfer Into Lymphoma Cells Using Adenoviral Vectors Combined With Lipofection. Cancer Gene Ther (2000) 7(8):1145–55. doi: 10.1038/sj.cgt.7700209
171. Li YM, Pong RC, Bergelson JM, Hall MC, Sagalowsky AI, Tseng CP, et al. Loss of Adenoviral Receptor Expression in Human Bladder Cancer Cells: A Potential Impact on the Efficacy of Gene Therapy. Cancer Res (1999) 59(2):325–30. doi: 10.1097/00005392-199904010-00449
172. Hidaka C, Milano E, Leopold PL, Bergelson JM, Hackett NR, Finberg RW, et al. CAR-Dependent and CAR-Independent Pathways of Adenovirus Vector-Mediated Gene Transfer and Expression in Human Fibroblasts. J Clin Invest (1999) 103(4):579–87. doi: 10.1172/JCI5309
173. Kim JS, Lee SH, Cho YS, Choi JJ, Kim YH, Lee JH. Enhancement of the Adenoviral Sensitivity of Human Ovarian Cancer Cells by Transient Expression of Coxsackievirus and Adenovirus Receptor (CAR). Gynecol Oncol (2002) 85(2):260–5. doi: 10.1006/gyno.2002.6607
174. Anders M, Vieth M, Rocken C, Ebert M, Pross M, Gretschel S, et al. Loss of the Coxsackie and Adenovirus Receptor Contributes to Gastric Cancer Progression. Br J Cancer (2009) 100(2):352–9. doi: 10.1038/sj.bjc.6604876
175. Ma YY, Wang XJ, Han Y, Li G, Wang HJ, Wang SB, et al. Loss of Coxsackie and Adenovirus Receptor Expression in Human Colorectal Cancer: A Potential Impact on the Efficacy of Adenovirus-Mediated Gene Therapy in Chinese Han Population. Mol Med Rep (2016) 14(3):2541–7. doi: 10.3892/mmr.2016.5536
176. Eager RM, Nemunaitis J. Clinical Development Directions in Oncolytic Viral Therapy. Cancer Gene Ther (2011) 18(5):305–17. doi: 10.1038/cgt.2011.7
177. Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, et al. A Phase I Trial of Intravenous CG7870, a Replication-Selective, Prostate-Specific Antigen-Targeted Oncolytic Adenovirus, for the Treatment of Hormone-Refractory, Metastatic Prostate Cancer. Mol Ther (2006) 14(1):107–17. doi: 10.1016/j.ymthe.2006.02.011
178. Yu DC. The Addition of Adenovirus Type 5 Region E3 Enables Calydon Virus 787 to Eliminate Distant Prostate Tumor Xenografts (Vol 60, Pg 4200, 1999). Cancer Res (2000) 60(4):1150–0.
179. Singleton DC, Li D, Bai SY, Syddall SP, Smaill JB, Shen Y, et al. The Nitroreductase Prodrug SN 28343 Enhances the Potency of Systemically Administered Armed Oncolytic Adenovirus ONYX-411(NTR). Cancer Gene Ther (2007) 14(12):953–67. doi: 10.1038/sj.cgt.7701088
180. Choi JW, Lee YS, Yun CO, Kim SW. Polymeric Oncolytic Adenovirus for Cancer Gene Therapy. J Control Release (2015) 219:181–91. doi: 10.1016/j.jconrel.2015.10.009
181. Kwon OJ, Kang E, Choi JW, Kim SW, Yun CO. Therapeutic Targeting of Chitosan-PEG-Folate-Complexed Oncolytic Adenovirus for Active and Systemic Cancer Gene Therapy. J Control Release (2013) 169(3):257–65. doi: 10.1016/j.jconrel.2013.03.030
182. Kim J, Nam HY, Choi JW, Yun CO, Kim SW. Efficient Lung Orthotopic Tumor-Growth Suppression of Oncolytic Adenovirus Complexed With RGD-Targeted Bioreducible Polymer. Gene Ther (2014) 21(5):476–83. doi: 10.1038/gt.2014.18
183. Kasala D, Hong J, Yun CO. Overcoming the Barriers to Optimization of Adenovirus Delivery Using Biomaterials: Current Status and Future Perspective. J Control Release (2021) 332:285–300. doi: 10.1016/j.jconrel.2021.02.018
184. Lee CH, Kasala D, Na Y, Lee MS, Kim SW, Jeong JH, et al. Enhanced Therapeutic Efficacy of an Adenovirus-PEI-Bile-Acid Complex in Tumors With Low Coxsackie and Adenovirus Receptor Expression. Biomaterials (2014) 35(21):5505–16. doi: 10.1016/j.biomaterials.2014.03.060
185. Kim J, Li Y, Kim SW, Lee DS, Yun CO. Therapeutic Efficacy of a Systemically Delivered Oncolytic Adenovirus - Biodegradable Polymer Complex. Biomaterials (2013) 34(19):4622–31. doi: 10.1016/j.biomaterials.2013.03.004
186. Grunwald GK, Vetter A, Klutz K, Willhauck MJ, Schwenk N, Senekowitsch-Schmidtke R, et al. Systemic Image-Guided Liver Cancer Radiovirotherapy Using Dendrimer-Coated Adenovirus Encoding the Sodium Iodide Symporter as Theranostic Gene. J Nucl Med (2013) 54(8):1450–7. doi: 10.2967/jnumed.112.115493
187. Kim J, Nam HY, Kim TI, Kim PH, Ryu J, Yun CO, et al. Active Targeting of RGD-Conjugated Bioreducible Polymer for Delivery of Oncolytic Adenovirus Expressing shRNA Against IL-8 mRNA. Biomaterials (2011) 32(22):5158–66. doi: 10.1016/j.biomaterials.2011.03.084
188. Bajpayee NS, McGrath WJ, Mangel WF. Interaction of the Adenovirus Proteinase With Protein Cofactors With High Negative Charge Densities. Biochemistry-Us (2005) 44(24):8721–9. doi: 10.1021/bi0502240
189. Yoon AR, Hong J, Kim SW, Yun CO. Redirecting Adenovirus Tropism by Genetic, Chemical, and Mechanical Modification of the Adenovirus Surface for Cancer Gene Therapy. Expert Opin Drug Deliv (2016) 13(6):843–58. doi: 10.1517/17425247.2016.1158707
190. Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal Escape Pathways for Delivery of Biologicals. J Control Release (2011) 151(3):220–8. doi: 10.1016/j.jconrel.2010.11.004
191. Lee DU, Park JY, Kwon S, Park JY, Kim YH, Khang D, et al. Apoptotic Lysosomal Proton Sponge Effect in Tumor Tissue by Cationic Gold Nanorods. Nanoscale (2019) 11(42):19980–93. doi: 10.1039/C9NR04323C
192. Moghimi SM, Hunter AC, Murray JC. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol Rev (2001) 53(2):283–318.
193. Furumoto K, Nagayama S, Ogawara K, Takakura Y, Hashida M, Higaki K, et al. Hepatic Uptake of Negatively Charged Particles in Rats: Possible Involvement of Serum Proteins in Recognition by Scavenger Receptor. J Control Release (2004) 97(1):133–41. doi: 10.1016/j.jconrel.2004.03.004
194. Patnaik S, Aggarwal A, Nimesh S, Goel A, Ganguli M, Saini N, et al. PEI-Alginate Nanocomposites as Efficient In Vitro Gene Transfection Agents. J Control Release (2006) 114(3):398–409. doi: 10.1016/j.jconrel.2006.06.025
195. Bischof J, Vietor I, Cotten M, Huber LA. Transient Transfection of Mammary Epithelial Cells With a PEI/DNA/adenovirus System. Biol Chem (1999) 380(2):269–73. doi: 10.1515/BC.1999.036
196. Pack DW, Hoffman AS, Pun S, Stayton PS. Design and Development of Polymers for Gene Delivery. Nat Rev Drug Discov (2005) 4(7):581–93. doi: 10.1038/nrd1775
197. Baker A, Saltik M, Lehrmann H, Killisch I, Mautner V, Lamm G, et al. Polyethylenimine (PEI) is a Simple, Inexpensive and Effective Reagent for Condensing and Linking Plasmid DNA to Adenovirus for Gene Delivery. Gene Ther (1997) 4(8):773–82. doi: 10.1038/sj.gt.3300471
198. Sakae M, Ito T, Yoshihara C, Iida-Tanaka N, Yanagie H, Eriguchi M, et al. Highly Efficient In Vivo Gene Transfection by Plasmid/PEI Complexes Coated by Anionic PEG Derivatives Bearing Carboxyl Groups and RGD Peptide. BioMed Pharmacother (2008) 62(7):448–53. doi: 10.1016/j.biopha.2007.12.009
199. Bettinger T, Remy JS, Erbacher P. Size Reduction of Galactosylated PEI/DNA Complexes Improves Lectin-Mediated Gene Transfer Into Hepatocytes. Bioconjug Chem (1999) 10(4):558–61. doi: 10.1021/bc990006h
200. Cheng H, Zhu JL, Sun YX, Cheng SX, Zhang XZ, Zhuo RX. Novel Thermoresponsive Nonviral Gene Vector: P(NIPAAm-Co-NDAPM)-B-PEI With Adjustable Gene Transfection Efficiency. Bioconjug Chem (2008) 19(7):1368–74. doi: 10.1021/bc700478s
201. Brownlie A, Uchegbu IF, Schatzlein AG. PEI-based Vesicle-Polymer Hybrid Gene Delivery System With Improved Biocompatibility. Br J Cancer (2002) 86:S104–5. doi: 10.1016/j.ijpharm.2003.12.029
202. Brownlie A, Uchegbu IF, Schatzlein AG. PEI-Based Vesicle-Polymer Hybrid Gene Delivery System With Improved Biocompatibility. Int J Pharm (2004) 274(1-2):41–52. doi: 10.1016/j.ijpharm.2003.12.029
203. Furgeson DY, Cohen RN, Mahato RI, Kim SW. Novel Water Insoluble Lipoparticulates for Gene Delivery. Pharm Res (2002) 19(4):382–90. doi: 10.1023/A:1015166806366
204. Gaspar VM, Baril P, Costa EC, de Melo-Diogo D, Foucher F, Queiroz JA, et al. Bioreducible Poly(2-Ethyl-2-Oxazoline)-PLA-PEI-SS Triblock Copolymer Micelles for Co-Delivery of DNA Minicircles and Doxorubicin. J Control Release (2015) 213:175–91. doi: 10.1016/j.jconrel.2015.07.011
205. Xiao JS, Duan XP, Yin Q, Miao ZH, Yu HJ, Chen CY, et al. The Inhibition of Metastasis and Growth of Breast Cancer by Blocking the NF-Kappa B Signaling Pathway Using Bioreducible PEI-Based/P65 shRNA Complex Nanoparticles. Biomaterials (2013) 34(21):5381–90. doi: 10.1016/j.biomaterials.2013.03.084
206. Zhang L, Chen Z, Li Y. Dual-Degradable Disulfide-Containing PEI-Pluronic/DNA Polyplexes: Transfection Efficiency and Balancing Protection and DNA Release. Int J Nanomedicine (2013) 8:3689–701. doi: 10.2147/IJN.S49595
207. Jain RK. Barriers to Drug-Delivery in Solid Tumors. Sci Am (1994) 271(1):58–65. doi: 10.1038/scientificamerican0794-58
208. Nakano K, Egashira K, Masuda S, Funakoshi K, Zhao G, Kimura S, et al. Formulation of Nanoparticle-Eluting Stents by a Cationic Electrodeposition Coating Technology Efficient Nano-Drug Delivery via Bioabsorbable Polymeric Nanoparticle-Eluting Stents in Porcine Coronary Arteries. JACC Cardiovasc Interv (2009) 2(4):277–83. doi: 10.1016/j.jcin.2008.08.023
209. Rajput SM, Gangele K, Kumar S, Aswal VK, Mata JP, Malek NI, et al. Nano-Vehicles for Drug Delivery Using Low-Cost Cationic Surfactants: A Drug Induced Structural Transitions. ChemistrySelect (2018) 3(32):9454–63. doi: 10.1002/slct.201801111
210. Zhang XW, Wang H, Ma ZG, Wu BJ. Effects of Pharmaceutical PEGylation on Drug Metabolism and its Clinical Concerns. Expert Opin Drug Metab Toxicol (2014) 10(12):1691–702. doi: 10.1517/17425255.2014.967679
211. Swierczewska M, Lee KC, Lee S. What is the Future of PEGylated Therapies? Expert Opin Emerg Drugs (2015) 20(4):531–6. doi: 10.1517/14728214.2015.1113254
212. O'Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, et al. PEGylation of Adenovirus With Retention of Infectivity and Protection From Neutralizing Antibody In Vitro and In Vivo. Hum Gene Ther (1999) 10(8):1349–58. doi: 10.1089/10430349950018021
213. Croyle MA, Chirmule N, Zhang Y, Wilson JM. "Stealth" Adenoviruses Blunt Cell-Mediated and Humoral Immune Responses Against the Virus and Allow for Significant Gene Expression Upon Readministration in the Lung. J Virol (2001) 75(10):4792–801. doi: 10.1128/JVI.75.10.4792-4801.2001
214. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene Glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chem Int Edit (2010) 49(36):6288–308. doi: 10.1002/anie.200902672
215. Liu LA, Han LC, Wu QH, Sun Y, Li KH, Liu Y, et al. Multifunctional DNA Dendrimer Nanostructures for Biomedical Applications. J Mater Chem B (2021) 9(25):4991–5007. doi: 10.1039/D1TB00689D
216. Kurtoglu YE, Navath RS, Wang B, Kannan S, Romero R, Kannan RM. Poly(amidoamine) Dendrimer-Drug Conjugates With Disulfide Linkages for Intracellular Drug Delivery. Biomaterials (2009) 30(11):2112-21. doi: 10.1016/j.biomaterials.2008.12.054
217. Heitz M, Kwok A, Eggimann GA, Hollfelder F, Darbre T, Reymond JL. Peptide Dendrimer-Lipid Conjugates as DNA and siRNA Transfection Reagents: Role of Charge Distribution Across Generations. Chimia (2017) 71(4):220–5. doi: 10.2533/chimia.2017.220
218. Kim WJ, Yockman JW, Lee M, Jeong JH, Kim YH, Kim SW. Soluble Flt-1 Gene Delivery Using PEI-G-PEG-RGD Conjugate for Anti-Angiogenesis. J Control Release (2005) 106(1-2):224–34. doi: 10.1016/j.jconrel.2005.04.016
219. Pike DB, Ghandehari H. HPMA Copolymer-Cyclic RGD Conjugates for Tumor Targeting. Adv Drug Deliv Rev (2010) 62(2):167–83. doi: 10.1016/j.addr.2009.11.027
220. Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor Cell Targeting of Liposome-Entrapped Drugs With Phospholipid-Anchored Folic Acid-PEG Conjugates. Adv Drug Deliv Rev (2004) 56(8):1177–92. doi: 10.1016/j.addr.2004.01.011
221. Zhang GL, Gao JL, Qian JC, Zhang LL, Zheng K, Zhong K, et al. Hydroxylated Mesoporous Nanosilica Coated by Polyethylenimine Coupled With Gadolinium and Folic Acid: A Tumor-Targeted T-1 Magnetic Resonance Contrast Agent and Drug Delivery System. ACS Appl Mater Interfaces (2015) 7(26):14192–200. doi: 10.1021/acsami.5b04294
222. Han MM, Ji XM, Li JF, Ge ZM, Luo B, Zhou K, et al. Lipoprotein-Inspired Nanocarrier Composed of Folic Acid-Modified Protein and Lipids: Preparation and Evaluation of Tumor-Targeting Effect. Int J Nanomedicine (2020) 15:3433–45. doi: 10.2147/IJN.S241448
223. Oh E, Oh JE, Hong J, Chung Y, Lee Y, Park KD, et al. Optimized Biodegradable Polymeric Reservoir-Mediated Local and Sustained Co-Delivery of Dendritic Cells and Oncolytic Adenovirus Co-Expressing IL-12 and GM-CSF for Cancer Immunotherapy. J Control Release (2017) 259:115–27. doi: 10.1016/j.jconrel.2017.03.028
224. Cao PX, Bae Y. Polymer Nanoparticulate Drug Delivery and Combination Cancer Therapy. Future Oncol (2012) 8(11):1471–80. doi: 10.2217/fon.12.139
Keywords: immune checkpoint inhibitor, immunotherapy, polymeric system, oncolytic adenovirus, CAR-T cell
Citation: Le TMD, Yoon A-R, Thambi T and Yun C-O (2022) Polymeric Systems for Cancer Immunotherapy: A Review. Front. Immunol. 13:826876. doi: 10.3389/fimmu.2022.826876
Received: 10 December 2021; Accepted: 24 January 2022;
Published: 22 February 2022.
Edited by:
Zong Sheng Guo, Roswell Park Comprehensive Cancer Center, United StatesReviewed by:
Qing Zhang, Xuzhou Medical University, ChinaChu Lin, Peking University People’s Hospital, China
Copyright © 2022 Le, Yoon, Thambi and Yun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chae-Ok Yun, Y2hhZW9rQGhhbnlhbmcuYWMua3I=