 Saulo Brito Silva
Saulo Brito Silva Carlos Wagner S. Wanderley
Carlos Wagner S. Wanderley Leandro Machado Colli
Leandro Machado Colli- 1Department of Medical Imaging, Hematology, and Oncology, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, Brazil
- 2Department of Pharmacology, Ribeirao Preto Medical School, University of Sao Paulo, Ribeirao Preto, Brazil
Cancer cells harbor genomic instability due to accumulated DNA damage, one of the cancer hallmarks. At least five major DNA Damage Repair (DDR) pathways are recognized to repair DNA damages during different stages of the cell cycle, comprehending base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end joining (NHEJ). The unprecedented benefits achieved with immunological checkpoint inhibitors (ICIs) in tumors with mismatch repair deficiency (dMMR) have prompted efforts to extend this efficacy to tumors with HR deficiency (HRD), which are greatly sensitive to chemotherapy or PARP inhibitors, and also considered highly immunogenic. However, an in-depth understanding of HRD’s molecular underpinnings has pointed to essential singularities that might impact ICIs sensitivity. Here we address the main molecular aspects of HRD that underlie a differential profile of efficacy and resistance to the treatment with ICIs compared to other DDR deficiencies.
Introduction
The central DNA Damage Repair (DDR) pathways comprise base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end joining (NHEJ), which are collectively responsible for repairing DNA damages during different stages of the cell cycle (1). In tumor cells, defects on DRR pathways, by one hand, works as a source of genetic diversity and mutations that are beneficial for tumor evolution. On the other hand, it exposes the tumor cell to fragilities not observed in normal cells. In this context, the functional status of the DDR system has long been recognized as a biomarker for a broad range of treatments (2).
Different therapies could take advantage of DDR pathways’ defects to induce additional tumor genetic structural damage, as with radiotherapy, cytotoxic chemotherapies, or targeted DNA repair mechanisms such as PARP inhibitors, to enhance tumors cells’ lethality (3). In addition, recently, mismatch repair-deficient (dMMR) tumors have consistently been shown to harbor greater immunogenicity and be highly effective to immune checkpoint inhibitors (ICIs) (4, 5). Consequently, dMMR granted accelerated approval by the FDA to ICIs agnostic use to treat advanced solid tumors (6).
From this point onwards, understanding whether this effect also extends to other DDR pathways started to be deeply investigated. Although any type of DDR dysfunction can lead to the accumulation of tumoral mutations, there is a wide variety in burden and type of mutations, depending on the DNA level each repair mechanism actuates (7). However, the impact of those different pattern of mutations on immunogenicity and, consequently, on the response to immunotherapy is still a matter of debate.
The benefits achieved with ICIs in tumors with dMMR have prompted efforts to extend this efficacy to tumors with HRD, which are highly sensitive to chemotherapy or PARP inhibitors and expected to be highly immunogenic. Nonetheless, molecular underpinnings of HR defects have pointed to singularities that might impact antitumor immune response and ICIs effectiveness. This review will summarize the main molecular aspects of HRD that underlie a differential profile of efficacy and resistance to the treatment with ICIs compared to other DDR deficiencies.
DDR in Current Clinical Practice
Mismatch Repair
The most significant evidence linking DNA repair deficiency with ICIs activity stems from tumors with a deficiency in mismatch repair (MMR) (dMMR). Roughly 18% of endometrial cancers, 11% of ovarian cancers, and 4% of metastatic colorectal cancer present with mutations or epigenetic silencing in genes comprising the MMR system (8). In a phase II clinical trial evaluating pembrolizumab in a set of treatment-refractory dMMR tumors, the response rates were as high as 40% to 70% (9). The studies Checkmate 142 (10) and Keynote 164 (11), which evaluated nivolumab and pembrolizumab, respectively, led to ICIs’ first approval, in dMMR tumors, for colorectal cancer previously submitted to chemotherapy. In addition, the Keynote 177 study (12) currently supports pembrolizumab use in the first-line setting of colorectal cancers. Finally, the Keynote 158 study (6) led to pembrolizumab approval for previously treated dMMR tumors irrespective of histology. Such an efficacy led to MMR status evaluation in current clinical practice for a broad set of other tumor histologies wherein this DDR deficiency can also be noticed, such as stomach, biliary tract, pancreas, prostate, and small intestine cancer (13).
Homologous Recombination
Homologous recombination (HR) is the most likely DDR mechanism found when considering a non-selected histology-based population (14). It is a crucial pathway to repair double-strand DNA breaks due to its error-free repairing system that relies on an intact sister chromatid instead of the non-homologous end joining (NHEJ) process (7). The incidence of pathogenic HRD varies according to histology, staging, and previous treatment burden (15). Notwithstanding, HRD is currently most recognized in tumors for which PARP inhibitors are currently approved based on a biomarker-guided BRCA or HR loss of function: ovarian cancer (40-50% with HRD), prostate cancer (20-25%), breast cancer (18%), and pancreatic cancer (12%) (16–20). Recently, many other malignancies were also shown to have a high incidence of HRD, such as endometrial (34%), biliary tract (28%), bladder (23%), hepatocellular (20%), and gastroesophageal cancer (20%) (14).
In contrast to the high clinical efficacy of ICIs in MMR deficient tumors, the clinical benefits are not consistent with an HRD. In phase II KEYNOTE 100 study, response rates with pembrolizumab in patients with advanced ovarian cancer were less than 10% among those harboring an HRD, with no statistical difference found when comparing BRCA-mutated versus wild-type counterparts (21). Despite other HRD genes being currently tested in ovarian cancer through NGS platforms, no prospective clinical data have evaluated their differential effectiveness, such that all available clinical data stem from BRCA-mutated tumors. Moreover, in phase III Keynote 119, patients with previously treated triple-negative breast cancers - approximately 50% of whom have HRD – derived no benefit from pembrolizumab compared to chemotherapy concerning response rate or survival (22). Although this study was not designed to evaluate patients with breast cancer having HRD specifically, both those ovarian and breast early clinical data shed light on a significant difference in clinical efficacy compared to what is seen early on with ICIs for dMMR tumors. In addition, those evidence has ultimately contributed to shifting strives for various ICIs combinations that are now undergoing prospectively to overcome such immune restoration mitigation – through anti-PD-1/PD-L1 –, which is taking place in the presence of HRD and remain underrecognized.
DDR and Immunogenicity
Deficient DDR processes that predispose to genetic alterations at the DNA sequence level, such as in dMMR, have the highest potential to elicit antigenicity due to the vast number of mutation-associated neoantigens (23). Since it has been shown that only a tiny fraction of predicted neo-epitopes are presented through MHC-I to enable T-cell responses (24, 25), it seems likely that tumors with a higher number of tumor mutation burden (TMB) are more likely to present with neoantigens that effectively stimulate the immune system (26).
Extensive mutational assessments have demonstrated enrichment in single- and multi-nucleotide variants (SNVs and MNVs) in tumors with dMMR, resulting in a high TMB, generally higher than 17 mutations/Mb (27). In the rare inherent genetic condition of bi-allelic germline dMMR, tumors can display >250 mutations/Mb (28). Due to dMMR tumors’ high immunogenicity, ICIs are substantially effective in various settings, thus warranting approval on an agnostic indication basis. Regarding other hypermutated tumors, yet non-dMMR, a TCGA analysis has shown that somatic mutations in polymerase epsilon (POLE) or delta POLD1 also comprise a DDR deficient group with high TMB (29). Like in dMMR, impairment in the proofreading capability of POLE and POLD1 leads to genetic alterations at the DNA sequence level. Pathogenic somatic mutations in the proofreading exonuclease domain of POLE confer similar phenotypes regardless of the tumor tissue type, resulting in a large mutation rate, especially TCT>TAT and TCG>TTG transversions and, more rarely, concomitant microsatellite instability (30). Although somatic mutations in POLE have been identified in 2–8% of colorectal cancer and 7–15% of endometrial carcinoma (31), there are little data available reporting ICIs efficacy in these DDR populations due to their low incidence and the absence of systematic screening in daily practice (32, 33). Interestingly, extensive mutational profiling of 21.074 patients from 23 cancer types and subtypes suggested that POLE/POLD1 mutation was not independently associated with survival to ICIs treatment after adjusting for TMB. The study concludes that mutations in the proofreading domain of POLE/POLD1 are more likely to result in DNA repair defects featuring extremely high TMB, which contribute to more significant benefits from ICI treatment (34).
Tumors with HRD also have a higher mutational load and predicted neo-epitopes than those without DDR deficiencies (35). Intriguingly, when considering patients with high TMB tumors that are not MMR, POLE, or POLD1 deficient, there is no difference in survival compared to patients with low (<10 mut/Mb) TMB tumors also submitted to ICIs therapy (36). Although such an analysis did not specifically evaluate HRD, it emphasizes that TMB alone should not be considered a biomarker of sensibility to ICIs. Furthermore, the accumulation of genetic errors at the DNA strands’ breaks level leads to a different set of a mutational landscape than DNA sequence alterations that characterize dMMR tumors (37), thus supporting that a high TMB in the presence of HRD may not correlate with the same efficacy seen in MMR or POLE/POLD1 deficiencies.
Pan-tumor studies have shown that patients with genetic alterations classified as HR deficient frequently present with a high number of small deletions (indels) with flanking microhomology at the breakpoint, in addition to copy number variations (CNVs) (38). Notably, a pan-cancer TCGA analysis demonstrated that the levels of CNVs inversely correlated with a cytotoxic immune signature and clinical benefit from ICIs therapy (39). Moreover, when comparing tumors having a similar oncogenetic driver background but differing with respect to a BRCA1 or BRCA2 mutation, there is a significant difference in the levels of CNVs between each of these different HR deficient subtypes, in addition to a distinct set of immunoregulatory genes and ICIs efficacy (40). Conversely to BRCA2 tumors, those with BRCA1 deficiency presented with an immunoregulatory infiltrate and a limited response to ICIs. Moreover, another in-depth TCGA analysis also pointed to the coexistence of anti-tumoral immune transcripts downregulation, such as IFN-γ related genes, with the upregulation of immunosuppressive markers related to myeloid tolerogenic cells activity in BRCA1 mutated breast cancers (41). Altogether, these data suggest that the tumoral HR-related genetic modifications could differentially regulate immune responses.
The molecular mechanisms supporting why CNVs or other specific genetic features associated with HRD mitigate immune responses remain unclear. Speculative hypothesis resides on large-scale mutational alterations leading to protein imbalance that impair tumor signals needed for cytotoxic immune cell infiltration or to deregulation of tumor signaling pathways that ultimately regulate immune cell recruitment (39). For a proper tumor antigen presentation, extensive integrity within the large HLA complex and the whole antigen processing machinery should be met (42). That complexity highlights the various vulnerable points that might lead to a dysfunctional tumor antigen presentation. The presence of CNVs can be associated with impaired antigen presentation owing to proteotoxic stress. Accordingly, the increased flux of unstable wild-type proteins may saturate critical chaperones and the proteasome complex while generating more self-peptides that ultimately place neoantigens at a further competitive disadvantage for loading onto limiting MHC protein (43).
Somatic copy number variation may also hinder tumor antigenic recognition through the downregulation of MHC I molecules. Extensive TCGA analyses demonstrate that loss of heterozygosis (LOH) in any MHC I genetic complex loci frequently accompanies tumors harboring chromosomal instability owing to alterations in cell cycle checkpoint genes such as TP53, in addition to HR deficient genes. Furthermore, tumor models with genomic instability frequently evolve with DNA hypermethylation silencing of genes belonging to the antigen presentation through MHC class I pathways (44). It is also noteworthy that a non-linear correlation between HLA-I LOH, TMB, and neoantigen burden has been suggested, such that HLA-I LOH is a frequent immune evasion mechanism in tumors overall, except for those with an either low or high (>30 mut/Mb) TMB, the latter of which are commonly represented by MMRd tumors (45).
In order to leverage neoantigen load and, thus, tumor recognition by immune cells, ongoing prospective studies are now evaluating PARPi added to ICIs in various HR deficient scenarios. Although it is attempting to speculate that further inducing inflammation in a somewhat immune-excluded tumor might restore anti-tumoral immune responses, some concerns may still be set. As aforementioned, neoantigen presentation’s multifaceted and complex processes may hamper tumor recognition despite efforts to enhance immunogenicity by fostering tumor mutations, particularly in settings where at least a non-low tumor mutation burden and neoantigen load predominates. In such conditions, immune-tolerance likely occurs due to multiple coexisting mechanisms such as dysfunctional neoantigen presentation and CD8+ cells exhaustion mediated by cell-cell interactions and other non-ligand-receptor interactions that lead to immune resistance. As such, none of these mechanisms would be reversed by the primary intention of using iPARP to enhance tumor neoantigen load. Furthermore, PARP inhibitors in the presence of HR defects could foster the emergence of subclonal mutations that contribute to establishing intratumor heterogeneity under the pressure of the immunoediting process (46, 47). Indeed, intratumor heterogeneity has also been associated with ICIs resistance (48).
DDR and PD-L1 Expression
Cancer cells with dysfunction at the DNA strand break repairing apparatus increase the rate of DNA repair basal activity to establish genome stability, particularly in the presence of constant cell proliferation. When molecular cascades featuring the homologous repair system are operating, checkpoint kinase 1 (Chk1) activation can also trigger the STAT1 – STAT3 – IRF1 signaling pathway, inducing PD-L1 expression in tumor cells (49). This model of tumor intrinsic PD-L1 expression, which is dependent on oncogenetic tumor features, has been defined as constitutive to distinguish the so-called acquired expression, in which tumor cells express PD-L1 in response to IFN-γ expression mediated by antitumor lymphocyte activity (50). The DNA repair signaling pathway ATR/Chk1/STAT3 can also upregulate CD47 and, through the engagement of SIRPα, suppress the capacity of antigen-presenting cells (APCs) to phagocytose and cross-present (51).
Concerning the constitutive tumor PD-L1 expression, ICIs may poorly correlate with response and survival, paradoxically predicting less benefit with the anti-PD-1/PD-L1 blockade in some tumors. Although PD-L1 expression is strongly correlated with clinical benefit in non-small cell lung cancer, tumors with EGFR activated-mutations, which can upregulate PD-L1 expression (52), do not derive benefit from ICIs’ treatment (53). Likewise, in the context of PD-L1 expression mediated by HRD, BRCA1 mutated breast cancer has been demonstrated to have a higher PD-L1 tumor score than BRCA2 mutated, even though clinical efficacy is inferior (41, 54). Not only do these data support that the PD-L1 expression does not represent a perfect biomarker for ICIs response across all tumor settings, but also suggest that a non-canonical tumor PD-L1 expression (i.e., constitutive) might even associate with mechanisms of immune resistance.
The HR-driven constitutive PD-L1 expression, which occurs in a non-canonical fashion, irrespective of effector T cells activity, might mitigate ICIs efficacy by hypothetical mechanisms. Firstly, and simplistically, a sufficient lymphocyte infiltration to be restored by ICIs is essential for an effective immune response to take place. Indeed, tumor immune infiltrates (TIL) are a known biomarker predicting clinical efficacy to ICIs in various tumors (55, 56). In this regard, the simple fact of witnessing PD-L1 expression does not guarantee that this results from the positive pressure (i.e., INF-γ driven) of the presence of an immune infiltrate. Secondly, even in the presence of an adequately primed effector immune infiltrate, the constitutive tumor PD-L1 expression fomented by HRD could provide an overwhelming pool of ligands to the PD-1-expressing immune cells that might occupy the tumor microenvironment. Therefore, this could help to polarize immune responses towards a suppressive spectrum, as exemplified by the PD-L1 persistent inducement of FOXP3 expression (FOXP3 high) in PD-1+ T-cells (57), which are characteristically associated with a decreased capacity to reinvigorate into anti-tumoral responses despite ICIs’ activity (58, 59). Lastly, the constitutive expression of PD-L1 may also provide evolutionary metabolic advantages to cancer cells by fostering tumor glycolysis and, in turn, impacting immune cells’ metabolic fate (60). As such, the PD-L1 expression in cancer cells can directly regulate tumor metabolism through Akt/mTOR signaling, independently of the PD-1 engagement, therefore upregulating tumor glycolysis that leads to microenvironment glucose deprivation and lactic acid concentration (Figure 1).
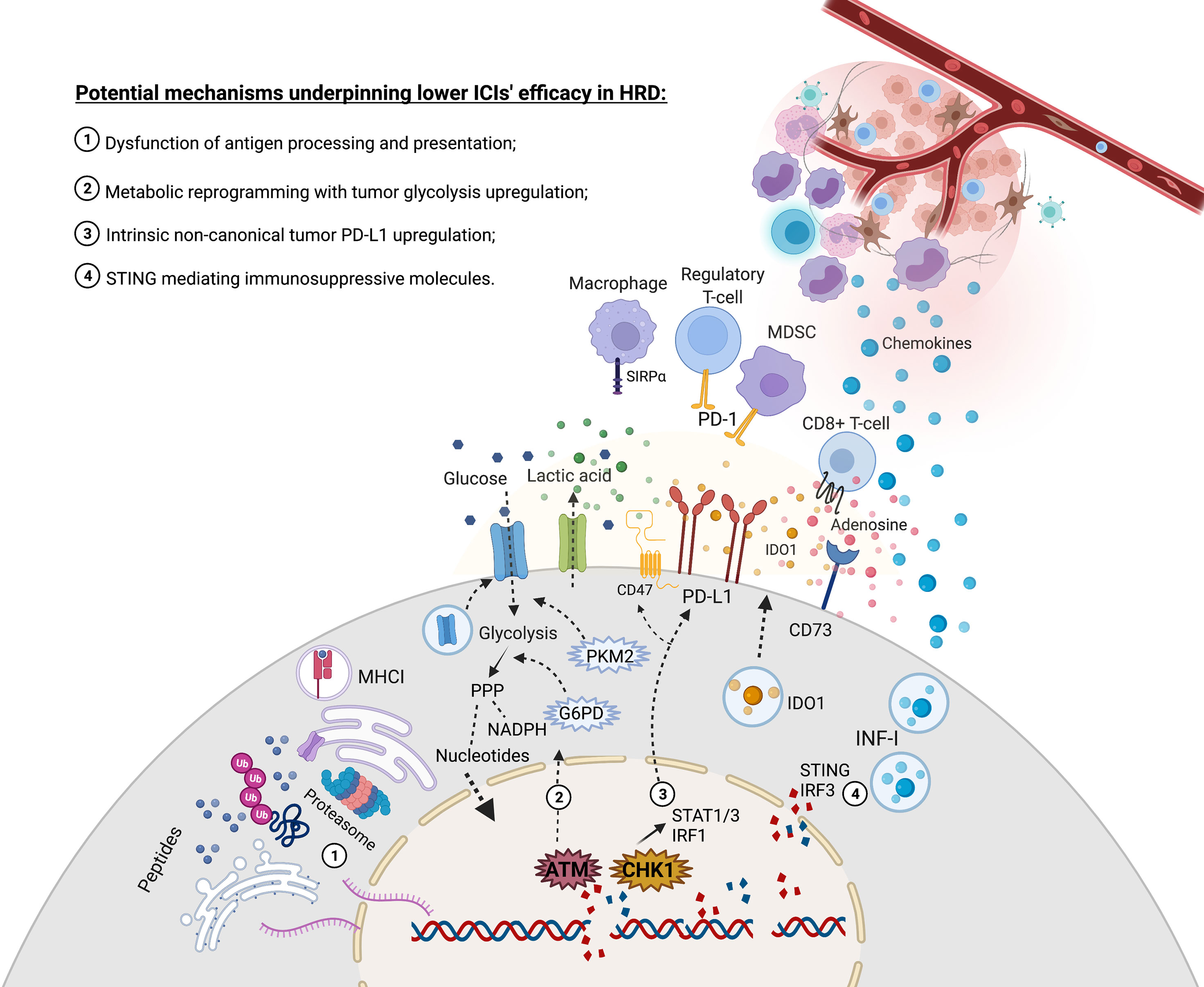
Figure 1 Increased rate of DNA double strands breaks due to deficiency in HR might evolve with molecular events that lead to challenges in restoring immune responses through immune checkpoint inhibitors. (1) DNA double-strand breaks association with CNV and large structural genetic alterations contribute to an increased flux of unstable mRNA and, ultimately, proteins that may saturate critical chaperones and the proteasome complex, thus leading to a dysfunctional tumor antigen presentation. (2) ATM plays a central role in recognizing DNA strand breaks but can also upregulate glycolysis and PPP to replenish nucleotides and NADPH supply for the upcoming anabolic reactions to restore DNA damages. This metabolic regulation might deprive glucose in the tumor microenvironment and export acid lactic, impacting immune responses. (3) CHK1 is crucial to repair strands breaks but may also activate the STAT1-STAT3-IRF1 signaling pathway that contributes to upregulating PD-L1 expression. (4) Cytosolic DNA censoring can lead to STING-IRF3 production of IFN-I, which might recruit monocytes that will be further exposed to a range of tolerogenic stimuli in the tumor microenvironment. Moreover, the STING signaling pathway might induce IDO1, and the expression of IFN-I might upregulate CD73, thus contributing to producing inhibitory molecules in the tumor microenvironment. Created with BioRender.com.
DDR and Metabolic Reprogramming
Tumor cells with DDR defects have a high requirement to restore DNA damage through compensatory pathways. Ataxia-telangiectasia mutated (ATM) and DNA-dependent kinases (DNA-PK) are crucial proteins to recognize DNA damage and initiate repair signaling cascades. Besides their function in DNA strand-break repair, these proteins can remodel cancer metabolism through upregulation of glucose transporter (GLUT) channels and pyruvate kinase M2 (PKM2) enzyme, thus fostering tumor glycolysis (61). Hyperactivation of glycolysis is one of the hallmarks of cancers and has been implicated in immune evasion owing to nutrient competition and toxic metabolites accumulation, such as lactic acid (60). Furthermore, ATM activity can also induce glucose-6- phosphate dehydrogenase (G6PD) expression, which is fundamental to enable the pentose phosphate pathway (PPP) (62). The oxidative PPP generates ribose-5-phosphate, a precursor for nucleotide synthesis, and reduces the potential in the form of NADPH, which is needed for nucleotide biosynthesis and lipogenesis (Figure 1). Previous studies also demonstrated that BRCA1 mutation and PARP1 activity also influence tumor metabolism. The BRCA1 lack in breast cancer was associated with increased glycolytic metabolism than BRCA1-WT (63, 64). Furthermore, it was demonstrated that PARP1 works as a transcriptional coactivator for PKM2 driving the expression of glycolytic genes (GLUT and LDH) in tumor cells (65). However, the role of metabolic changes induced by BRCA1 and PARP1 on primary resistance to ICIs remains unknown.
DDR and STING
A dysfunctional HR status predisposes cancer cells to DNA strands fragmentation in the presence of additional DNA damaging factors, such as radiotherapy, chemotherapy, or PARP inhibitors. Furthermore, DNA instability can occur spontaneously owing to the high tumor cell turn-over coupled with cell cycle checkpoints suppression and enhanced metabolic stress due to tumor metabolism deregulation and microenvironmental hypoxia. This background predisposes to frequent cytosolic DNA exposure in cancer cells. The cytosolic DNA activates the stimulator of IFN genes pathway (STING pathway) and IRF3 activity, thus inducing the transcription of IFN type I and chemoattractive cytokines (CXCL10 and CCL5), which mediates monocytes and neutrophil recruitment in an antigen-independent manner (66). Although type I IFN is a known contributor to T cell priming by inducing MHC I antigen cross-presentation in APCs, there have been growing insights linking STING-IFN molecular pathways to mechanisms mitigating effective immune responses (67, 68). Accordingly, an enhanced baseline STING-IRF3 activity can promote the sustained recruitment of monocytes in response to CXCL10 and CCL5 chemokines, inducing a chronic myelocytic inflammatory infiltrate that could further contribute to establishing an immune tolerogenic state (69). Those constant levels of DNA damage featuring HR deficient tumors can also activate an alternative STING pathway through ATM-TRAF6 driven transcription of TGF-β that promotes protumor M2-like macrophage and Treg cell differentiation, respectively (70). Lastly, the STING signaling pathway can also contribute to establishing a tolerogenic tumor microenvironment by inducing immune-suppressive soluble factors. An increase in IDO expression was shown to occur in STING mediated fashion when in the presence of mild tumor antigenicity (71). Moreover, the augmented IFN-α expression has been shown to upregulate the ectonucleotidase CD73 and leverage adenosine production in a tumor microenvironment wherein DDR might be fostering ATP production (72) (Figure 1).
Summary and Future Perspectives
The data summarized in this review suggest that HRD tumors have a differential profile of efficacy and resistance to ICIs’ treatment compared to other dMMR. Each DDR deficient pathway could lead to the emergence of a singular tumor mutational background, but the correlation between such a range of mutational patterns and the response to ICIs remains unclear. Furthermore, various mechanisms potentially impacting immune responses could emerge from the increased DDR pathways activity, which leads to tumor metabolic rearrangements and microenvironmental recruitment of immune-suppressive factors. The TMB status may not be a pan-cancer predictive biomarker for immunotherapy response, and the incorporation of tumor DDR pathways might be necessary for future genomic biomarker refinements. As such, it would be interesting to carry out studies on tumors harboring different defects in DNA repair pathways.
Author Contributions
SBS and LMC conceived the work. SBS, CW, SW and LMC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grant 2020/10960-5, São Paulo Research Foundation (FAPESP).
Conflict of Interest
LMC reports grants from Novartis and BMS.
The remaining authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency Across The Cancer Genome Atlas. Cell Rep (2018) 23(1):239–54.e6. doi: 10.1016/j.celrep.2018.03.076
2. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating Genetic Approaches Into the Discovery of Anticancer Drugs. Science (1997) 278(5340):1064–8. doi: 10.1126/science.278.5340.1064
3. Madhusudan S, Middleton MR. The Emerging Role of DNA Repair Proteins as Predictive, Prognostic and Therapeutic Targets in Cancer. Cancer Treat Rev (2005) 31(8):603–17. doi: 10.1016/j.ctrv.2005.09.006
4. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
5. Ma J, Setton J, Lee NY, Riaz N, Powell SN. The Therapeutic Significance of Mutational Signatures From DNA Repair Deficiency in Cancer. Nat Commun (2018) 9(1):1–12. doi: 10.1038/s41467-018-05228-y
6. Marabelle A, Le DT, Ascierto PA, di Giacomo AM, de Jesus-Acosta A, Delord J-P, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
7. Alhmoud JF, Woolley JF, al Moustafa A-E, Malki MI. DNA Damage/Repair Management in Cancers. Cancers (2020) 12(4):1–22. doi: 10.3390/cancers12041050
8. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and Characterization of Microsatellite Instability Across 18 Cancer Types. Nat Med (2016) 22(11):1342–50. doi: 10.1038/nm.4191
9. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
10. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9
11. Le DT, Kim TW, van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability–High/Mismatch Repair–Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol (2020) 38(1):11–9. doi: 10.1200/JCO.19.02107
12. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N Engl J Med (2020) 383(23):2207–18. doi: 10.1056/NEJMoa2017699
13. Williams AS, Huang W-Y. The Analysis of Microsatellite Instability in Extracolonic Gastrointestinal Malignancy. Pathology (2013) 45(6):540–52. doi: 10.1097/PAT.0b013e3283653307
14. Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen W-J, et al. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis Oncol (2018) 2018:1–13. doi: 10.1200/PO.17.00286
15. Ye F, He M, Huang L, Lang G, Hu X, Shao Z, et al. Insights Into the Impacts of BRCA Mutations on Clinicopathology and Management of Early-Onset Triple-Negative Breast Cancer. Front Oncol (2021) 10:574813. doi: 10.3389/fonc.2020.574813
16. González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2019) 381(25):2391–402. doi: 10.1056/NEJMoa1910962
17. Moore K, Colombo N, Scambia G, Kim B-G, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients With Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med (2018) 379(26):2495–505. doi: 10.1056/NEJMoa1810858
18. Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients With a Germline BRCA Mutation. N Engl J Med (2017) 377(6):523–33. doi: 10.1056/NEJMoa1706450
19. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med (2020) 382(22):2091–102. doi: 10.1056/NEJMoa1911440
20. Golan T, Hammel P, Reni M, van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA -Mutated Metastatic Pancreatic Cancer. N Engl J Med (2019) 381(4):317–27. doi: 10.1056/NEJMoa1903387
21. Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor Activity and Safety of Pembrolizumab in Patients With Advanced Recurrent Ovarian Cancer: Results From the Phase II KEYNOTE-100 Study. Ann Oncol (2019) 30(7):1080–7. doi: 10.1093/annonc/mdz135
22. Winer EP, Lipatov O, Im SA, Goncalves A, Muñoz-Couselo E, Lee KS, et al. Pembrolizumab Versus Investigator-Choice Chemotherapy for Metastatic Triple-Negative Breast Cancer (KEYNOTE-119): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2021) 22(4):499–511. doi: 10.1016/S1470-2045(20)30754-3
23. Schumacher TN, Schreiber RD. Neoantigens in Cancer Immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
24. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
25. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-Throughput Epitope Discovery Reveals Frequent Recognition of Neo-Antigens by CD4+ T Cells in Human Melanoma. Nat Med (2015) 21(1):81–5. doi: 10.1038/nm.3773
26. Colli LM, Machiela MJ, Myers TA, Jessop L, Yu K, Chanock SJ. Burden of Nonsynonymous Mutations Among TCGA Cancers and Candidate Immune Checkpoint Inhibitor Responses. Cancer Res (2016) 76(13):3767–72. doi: 10.1158/0008-5472.CAN-16-0170
27. Salem ME, Puccini A, Grothey A, Xiu J, Goldberg R, Kim ES, et al. Comparative Molecular Analysis Between Microsatellite Instability-High (MSI-H) Tumors With High Tumor Mutational Burden (TMB-H) Versus MSI-H Tumors With TMB-Intermediate/Low. Ann Oncol (2018) 29:1. doi: 10.1093/annonc/mdy303.005
28. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol (2016) 34(19):2206–11. doi: 10.1200/JCO.2016.66.6552
29. Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A Panoply of Errors: Polymerase Proofreading Domain Mutations in Cancer. Nat Rev Cancer (2016) 16(2):71–81. doi: 10.1038/nrc.2015.12
30. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin A, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
31. Kim T-M, Laird PW, Park PJ. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell (2013) 155(4):858–68. doi: 10.1016/j.cell.2013.10.015
32. Rousseau BJ-C, Bieche I, Pasmant E, Simmet V, Hamzaoui N, Masliah-Planchon J, et al. 526o High Activity of Nivolumab in Patients With Pathogenic Exonucleasic Domain POLE (edPOLE) Mutated Mismatch Repair Proficient (MMRp) Advanced Tumours. Ann Oncol (2020) 31:S463. doi: 10.1016/j.annonc.2020.08.640
33. Schenck K, Masetti M, Pfarr N, Lorenzen S. PD-1 Blockade Elicits Ongoing Remission in Two Cases of Refractory Microsatellite-Stable Cancer Harboring a POLE Mutation. Oncol Res Treat (2021) 1–5. doi: 10.1159/000521332
34. He J, Ouyang W, Zhao W, Shao L, Li B, Liu B, et al. Distinctive Genomic Characteristics in POLE/POLD1-Mutant Cancers can Potentially Predict Beneficial Clinical Outcomes in Patients Who Receive Immune Checkpoint Inhibitor. Ann Trans Med (2021) 9(2):129–9. doi: 10.21037/atm-20-7553
35. Guo N, Guo T, Ge M, Yan L, Guo H. 119p A Pan-Cancer Study on Difference of Homologous Recombination Deficiency and Tumour Mutational Burden Between Chinese and Western Patients. Ann Oncol (2020) 31:1. doi: 10.1016/j.annonc.2020.08.240
36. Rousseau B, Foote MB, Maron SB, Diplas BH, Lu S, Argilés G, et al. The Spectrum of Benefit From Checkpoint Blockade in Hypermutated Tumors. N Engl J Med (2021) 384(12):1168–70. doi: 10.1056/NEJMc2031965
37. van Wilpe S, Tolmeijer SH, Koornstra RHT, de Vries IJM, Gerritsen WR, Ligtenberg M, et al. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers (2021) 13(9):1–19. doi: 10.3390/cancers13092249
38. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature (2016) 534(7605):47–54. doi: 10.1038/nature17676
39. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor Aneuploidy Correlates With Markers of Immune Evasion and With Reduced Response to Immunotherapy. Science (2017) 355(6322):1–14. doi: 10.1126/science.aaf8399
40. Samstein RM, Krishna C, Ma X, Pei X, Lee K-W, Makarov V, et al. Mutations in BRCA1 and BRCA2 Differentially Affect the Tumor Microenvironment and Response to Checkpoint Blockade Immunotherapy. Nat Cancer (2020) 1(12):1188–203. doi: 10.1038/s43018-020-00139-8
41. Wen WX, Leong C-O. Association of BRCA1- and BRCA2-Deficiency With Mutation Burden, Expression of PD-L1/PD-1, Immune Infiltrates, and T Cell-Inflamed Signature in Breast Cancer. PloS One (2019) 14(4):1–16. doi: 10.1371/journal.pone.0215381
42. Khong HT, Restifo NP. Natural Selection of Tumor Variants in the Generation of “Tumor Escape” Phenotypes. Nat Immunol (2002) 3(11):999–1005. doi: 10.1038/ni1102-999
43. Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional Consequences of Aneuploidy. Proc Natl Acad Sci (2012) 109(31):1–6. doi: 10.1073/pnas.1209227109
44. Tripathi R, Modur V, Senovilla L, Kroemer G, Komurov K. Suppression of Tumor Antigen Presentation During Aneuploid Tumor Evolution Contributes to Immune Evasion. OncoImmunology (2019) 8(11):1657374. doi: 10.1080/2162402X.2019.1657374
45. Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov (2021) 11(2):282–92. doi: 10.1158/2159-8290.CD-20-0672
46. Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating Subclonal Evolution of Resistant Cancer Phenotypes. Nat Commun (2017) 8(1):1–15. doi: 10.1038/s41467-017-01174-3
47. Jacob SL, Kiedrowski LA, Chae YK. The Dynamic Landscape of BRCA1 Reversion Mutations From Indel to SNV in a Patient With Ovarian Cancer Treated With PARP-Inhibitors and Immunotherapy. Heliyon (2020) 6(5):1–5. doi: 10.1016/j.heliyon.2020.e03841
48. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
49. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat Commun (2017) 8(1):1–11. doi: 10.1038/s41467-017-01883-9
50. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
51. Hsieh RC-E, Krishnan S, Wu R-C, Boda A, Liu A, Winkler M, et al. 592 ATR-Mediated CD47 and PD-L1 Upregulation Restricts Radiotherapy-Induced Immune Priming and Abscopal Responses in Colorectal Cancer. J ImmunoTherapy Cancer (2021) 9(Suppl 2):A622–2. doi: 10.1136/jitc-2021-SITC2021.592
52. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients With EGFR Mutation. J Thorac Oncol (2015) 10(6):910–23. doi: 10.1097/JTO.0000000000000500
53. Gainor JF, Shaw AT, Sequist L, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR Mutations and ALK Rearrangements Are Associated With Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res (2016) 22(18):4585–93. doi: 10.1158/1078-0432.CCR-15-3101
54. Zhou Z, Li M. Evaluation of BRCA1 and BRCA2 as Indicators of Response to Immune Checkpoint Inhibitors. JAMA Network Open (2021) 4(5):e217728. doi: 10.1001/jamanetworkopen.2021.7728
55. Loi S, Drubay D, Adams S, Pruneri G, Francis PA, Lacroix-Triki M, et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J Clin Oncol (2019) 37:559–69. doi: 10.1200/JCO.18
56. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International Validation of the Consensus Immunoscore for the Classification of Colon Cancer: A Prognostic and Accuracy Study. Lancet (2018) 391(10135):2128–39. doi: 10.1016/S0140-6736(18)30789-X
57. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
58. Wherry EJ, Kurachi M. Molecular and Cellular Insights Into T Cell Exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
59. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted Deletion of PD-1 in Myeloid Cells Induces Antitumor Immunity. Sci Immunol (2020) 5(43):1–14. doi: 10.1126/sciimmunol.aay1863
60. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
61. Yang D, Peng M, Hou Y, Qin Y, Wan X, Zhu P, et al. Oxidized ATM Promotes Breast Cancer Stem Cell Enrichment Through Energy Metabolism Reprogram-Mediated Acetyl-CoA Accumulation. Cell Death Dis (2020) 11(7):1–17. doi: 10.1038/s41419-020-2714-7
62. Cosentino C, Grieco D, Costanzo V. ATM Activates the Pentose Phosphate Pathway Promoting Anti-Oxidant Defence and DNA Repair. EMBO J (2011) 30(3):546–55. doi: 10.1038/emboj.2010.330
63. Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, et al. BRCA1 Induces Major Energetic Metabolism Reprogramming in Breast Cancer Cells. PloS One (2014) 9(7):e102438. doi: 10.1371/journal.pone.0102438
64. Chiyoda T, Hart PC, Eckert MA, McGregor SM, Lastra RR, Hamamoto R, et al. Loss of BRCA1 in the Cells of Origin of Ovarian Cancer Induces Glycolysis: A Window of Opportunity for Ovarian Cancer Chemoprevention. Cancer Prev Res (2017) 10(4):255–66. doi: 10.1158/1940-6207.CAPR-16-0281
65. Li N, Feng L, Liu H, Wang J, Kasembeli M, Tran MK, et al. PARP Inhibition Suppresses Growth of EGFR-Mutant Cancers by Targeting Nuclear Pkm2. Cell Rep (2016) 15(4):843–56. doi: 10.1016/j.celrep.2016.03.070
66. Pantelidou C, Sonzogni O, de Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8 + T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov (2019) 9(6):723–37. doi: 10.1158/2159-8290.CD-18-1218
67. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-Driven Carcinogenesis Is Mediated Through STING. Nat Commun (2014) 5(1):1–9. doi: 10.1038/ncomms6166
68. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-Tumor Immunity to Resistance. Front Oncol (2018) 8:322. doi: 10.3389/fonc.2018.00322
69. Nowarski R, Gagliani N, Huber S, Flavell RA. Innate Immune Cells in Inflammation and Cancer. Cancer Immunol Res (2013) 1(2):77–84. doi: 10.1158/2326-6066.CIR-13-0081
70. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κb Signaling After Nuclear DNA Damage. Mol Cell (2018) 71(5):745–60. doi: 10.1016/j.molcel.2018.07.034
71. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res (2016) 76(8):2076–81. doi: 10.1158/0008-5472.CAN-15-1456
Keywords: immune checkpoint inhibitors, homologous recombination, DNA damage repair, mismatch repair, oncology
Citation: Silva SB, Wanderley CWS and Colli LM (2022) Immune Checkpoint Inhibitors in Tumors Harboring Homologous Recombination Deficiency: Challenges in Attaining Efficacy. Front. Immunol. 13:826577. doi: 10.3389/fimmu.2022.826577
Received: 01 December 2021; Accepted: 14 January 2022;
Published: 08 February 2022.
Edited by:
Erik A. L. Biessen, Maastricht University, NetherlandsReviewed by:
Sara Cingarlini, Integrated University Hospital Verona, ItalyCopyright © 2022 Silva, Wanderley and Colli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leandro Machado Colli, bGVhbmRyb2NAZm1ycC51c3AuYnI=