 Darko Stojkov1,2
Darko Stojkov1,2 Lea Gigon1
Lea Gigon1 Shuang Peng1
Shuang Peng1 Robert Lukowski2
Robert Lukowski2 Peter Ruth2
Peter Ruth2 Alexander Karaulov3
Alexander Karaulov3 Albert Rizvanov4
Albert Rizvanov4 Nickolai A. Barlev5,6
Nickolai A. Barlev5,6 Shida Yousefi1
Shida Yousefi1 Hans-Uwe Simon1,3,4,7*
Hans-Uwe Simon1,3,4,7*- 1Institute of Pharmacology, University of Bern, Bern, Switzerland
- 2Department of Pharmacology, Toxicology and Clinical Pharmacy, Institute of Pharmacy, University of Tübingen, Tübingen, Germany
- 3Department of Clinical Immunology and Allergology, Sechenov University, Moscow, Russia
- 4Institute of Fundamental Medicine and Biology, Kazan Federal University, Kazan, Russia
- 5Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russia
- 6Regulation of Cell Signaling Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Russia
- 7Institute of Biochemistry, Brandenburg Medical School, Neuruppin, Germany
Neutrophils are the most numerous cells in the leukocyte population and essential for innate immunity. To limit their effector functions, neutrophils are able to modulate glycolysis and other cellular metabolic pathways. These metabolic pathways are essential not only for energy usage, but also for specialized effector actions, such as the production of reactive oxygen species (ROS), chemotaxis, phagocytosis, degranulation, and the formation of neutrophil extracellular traps (NETs). It has been demonstrated that activated viable neutrophils can produce NETs, which consists of a DNA scaffold able to bind granule proteins and microorganisms. The formation of NETs requires the availability of increased amounts of adenosine triphosphate (ATP) as it is an active cellular and therefore energy-dependent process. In this article, we discuss the glycolytic and other metabolic routes in association with neutrophil functions focusing on their role for building up NETs in the extracellular space. A better understanding of the requirements of metabolic pathways for neutrophil functions may lead to the discovery of molecular targets suitable to develop novel anti-infectious and/or anti-inflammatory drugs.
Introduction
Neutrophils are the most abundant white blood cells in the human peripheral blood with an estimated daily turnover of 1010 to 1011 cells (1, 2). They exhibit a characteristic nucleus with 3 to 5 segmented lobes which are connected to a thin strip of nuclear material (3). Neutrophils are generated in the bone marrow in a regulated process of granulopoiesis. They are terminally differentiated cells with a short half-life time under physiological conditions (4–6). Therefore, they need to be constantly replenished from bone marrow precursor cells (3). The key role of neutrophils is to respond and diminish extracellular pathogens and to participate in the initiation of the adaptive immune response (2, 5). In addition, neutrophils perform diverse cellular functions, including oxidative burst, phagocytosis, degranulation, production of different cytokines and chemokines, and neutrophil extracellular trap (NET) formation (2, 7). Many neutrophil functions depend on energy and cytoskeleton reorganization (8, 9).
Neutrophils are equipped with a specialized enzyme system, so-called nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (often referred to as NOX2), which is able to produce reactive oxygen species (ROS) in higher quantity as compared to ROS generated by mitochondria. The NADPH oxidase reduces molecular oxygen to superoxide, that serves as a precursor for formation of hydrogen peroxide contributing to bacterial killing in order to dampen the inflammatory response (10). During their anti-microbial activity, neutrophils release granule proteins (3). Three main types of granules can be distinguished in neutrophils. During the promyelocyte stage the azurophilic granules are formed. These primary granules are large and dense, and contain myeloperoxidase (MPO) (11). Lactoferrin is found in specific or secondary granules, which develop during the myelocyte-metamyelocyte phase (12). At the band-stage of neutrophil development, the tertiary granules are formed that contain gelatinase (13). Neutrophils release many cytokines, including interleukin (IL)-6, IL-1, and tumor necrosis factor alpha (TNF-α), which aid in the inflammatory process by attracting additional white blood cells to the inflammation site (14, 15).
When activated, neutrophils migrate through chemotaxis towards the inflammatory sites (16) to combat the invading microorganisms. The precise mechanisms of anti-microbial inflammatory responses of neutrophils are still under intense investigations. Neutrophils are capable of killing microorganisms both intracellularly by phagocytosis and extracellularly by forming NETs containing released DNA and granule proteins (17–19). We and others have reported that the released web-like structure is composed of mitochondrial DNA (mtDNA) and granule proteins (8, 9, 19–26). NETs ensure the elimination of pathogens by preventing their spread and increase the local concentration of antimicrobial and toxic factors (27). Eosinophils, basophils, mast cells, and monocytes/macrophages can also form extracellular DNA traps (28–35).
Since the discovery of NETs, several microbial and noninfectious stimuli have been identified to activate neutrophils to induce NET formation (24, 36–39).
The production of ROS seems to be essential for NET formation (19, 40–42). Increased intracellular ROS activates actin and tubulin glutathionylation, tightly regulated by glutaredoxin 1 (Grx1), an enzyme required for deglutathionylation of actin and microtubules. Hence, an active cytoskeleton movement is vital for the formation of NETs (8). Moreover, the re-organization of the microtubule arrangements and consequently NET formation have been shown to depend on glycolytic ATP (9). To execute effector functions, neutrophils rely on simple metabolic pathways, such as glycolysis (43). Despite the fact that glycolysis remains the major metabolic pathway in the cytosol of neutrophils, more recent studies have identified additional metabolic routes in these cells (3). For instance, neutrophils utilize the pentose-phosphate pathway (PPP) to produce NADPH and ribose. In certain conditions, mainly due to limited availability of glucose, neutrophils are also able to adapt to fatty acid metabolism and glutaminolysis. On the contrary, when glucose is abundant, they can store small amounts of glycogen (44). In this review, we discuss the metabolic pathways used by neutrophils and explore the metabolic changes during NET formation.
Neutrophil Functions and Associated Metabolic Pathways
In the last decade, there has been a great interest to investigate the impact of metabolic activity on regulatory pathways in immune cells, including neutrophils (45–48). Compared to other immune cells such as macrophages and lymphocytes, neutrophils have modest numbers of mitochondria and they obtain their energy mainly from glycolysis (49, 50). Although initial analyses using electron microscopy suggested a few functional mitochondria in mature neutrophils only (51, 52), recently developed mitochondrial probes and new methods to quantify both oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) resulted in novel insights regarding the functional role of mitochondria in neutrophils (3, 9). It is now well established that neutrophils express components of the oxidative phosphorylation (OXPHOS) complex (50) and exhibit an active mitochondrial network (53). They exhibit functional mitochondria (54) and changes in mitochondrial potential (ΔΨm) were detected in activated mature neutrophils (50, 53). It has also been reported that the optic atrophy 1 (OPA1) protein, known to be responsible for controlling mitochondrial fusion and maintaining the mitochondrial cristae junctions tight to allow efficient oxidative respiration (55–57), is also required for ATP production through glycolysis in neutrophils (9). Moreover, functional mitochondria and glycolytic ATP production were necessary to form an intact microtubule network and functional NETs, both contributing to the anti-microbial defense mediated by neutrophils in an in vivo model of Pseudomonas aeruginosa (P. aeruginosa) lung infection (9).
Metabolic activities in neutrophils are not yet completely understood, and several metabolic pathways, including glycolysis, via tricarboxylic acid (TCA) cycle (also known as Krebs cycle), pentose phosphate pathway (PPP) via OXPHOS, glycogenolysis, glutaminolysis, and fatty acid β-oxidation (FAO) are considered to be utilized by neutrophils to fulfill the energetic, biosynthetic, and functional requirements of neutrophils (3, 43, 58–62) (Figure 1).
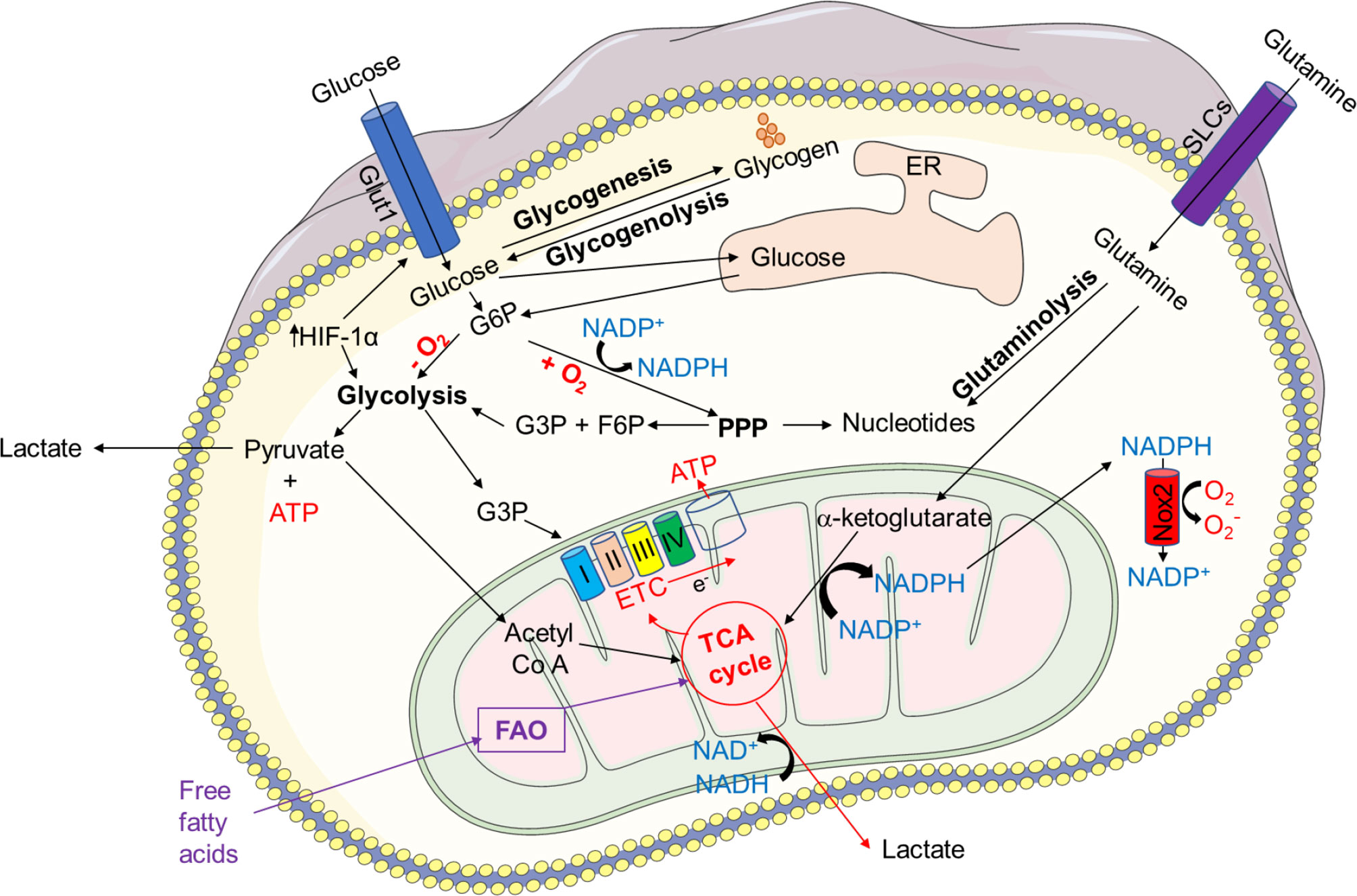
Figure 1 Overview of key metabolic pathways in neutrophils. In neutrophil cytosol, glycolysis is a primary metabolic route that transforms glucose to pyruvate via a number of enzymes and reactions. Pyruvate is converted to lactate and released out of the cells during anaerobic glycolysis in absence of oxygen. Pyruvate contributes to the TCA cycle after conversion to acetyl-coenzyme A (acetyl Co A) in the presence of oxygen, which results in reducing energy intermediates NADH and dihydroflavine-adenine dinucleotide (FADH2) to generate ATP via the electron transport chain (ETC). Neutrophils also use the pentose-phosphate pathway (PPP) to generate NADPH and riboses, which are then used to build nucleotides, by employing glucose-6-phosphate, a glycolytic pathway intermediate, as an entry point during the oxidative and non-oxidative phases to manufacture NADPH and riboses. NADPH modulates redox signaling and is required for NADPH oxidase-dependent ROS production in neutrophils. Glycogen reserves become concentrated in neutrophils when glucose levels rise, providing glucose-based glycolytic intermediate supply on demand. Through fatty acid synthesis (FAS), the TCA cycle intermediate citrate can be converted to free fatty acids, which can then be transported from the extracellular environment through the FAO pathway to produce acetyl-CoA, which fuels the TCA cycle and produces significantly more energy in the form of ATP. Glutamine helps the TCA cycle by producing α-ketoglutarate through glutaminolysis. Neutrophils also use the glycerol-3-phosphate shuttle to create NAD+ from NADH, which aids in mitochondrial membrane potential. HIF-1α augments the activity and expression of GLUT1 and GLUT3, resulting in increased glucose uptake, elevated hexokinase 2 (HK2) and phosphofructokinase B3 (PFKFB3) enzymatic activities, and enhanced ATP production.
Even though glycolysis is a dominant metabolic pathway for neutrophils, under glucose depletion, neutrophils mostly rely on the breakdown of stored glycogen (glycogenolysis) (63, 64), and certain functions of neutrophils, such as phagocytosis, require stored glycogen (65). The PPP or hexose monophosphate (HMP) shunt, which uses the glycolytic intermediate glucose-6-phosphate (G6P) to create ribose-5-phosphate (R5P) and NADPH in the cytosol, is required for the formation of ROS by the NADPH oxidase in neutrophils (58). Furthermore, in case of limited glucose availability, neutrophils activate a compensatory metabolism of fatty acids (66). Mitochondrial FAO transforms fatty acids to acyl-CoAs, which then enters the TCA cycle as acetyl-CoA, and energy in form of ATP is generated through the electron transport chain (3, 47, 67). Glutamine is another metabolic substrate used by neutrophils, especially when glucose levels are low (3, 68–70). Glutamate dehydrogenase produces α-ketoglutarate, which feeds the TCA cycle (70).
Neutrophils are frequently found in microenvironments with low oxygen levels. To survive under hypoxia condition, neutrophils retain hypoxia-inducible factor-1α (HIF-1α) and factor inhibiting HIF (FIH) hydroxylase oxygen-sensing pathway to diminish neutrophil apoptosis (71, 72). It has been described that hypoxia inhibits apoptosis by inducing a number of glycolytic enzymes, including GAPDH, allowing for the ongoing production of ATP, which is critical for neutrophil survival and function (73). Under hypoxic conditions, HIF-1α knockout mouse neutrophils are more prone to apoptosis (71). Moreover, HIF-1α and mammalian target of rapamycin (mTOR) have been also identified as important regulators of glycolysis in neutrophils (74, 75). HIF-1α upregulates the enzymes hexokinase 2 (HK2) and phosphofructokinase B3 (PFKFB3) to enhance the activity and expression of glucose transporter (GLUT) 1 and GLUT3 on neutrophils, resulting in increased glucose absorption, glucose metabolism, and ATP generation (74, 76, 77). HIF-1α has also been implicated in NET formation (78, 79). Increased NET formation has been associated with HIF-1α stabilization. Thus, reduced NET formation and NET-mediated extracellular bacteria killing have been described in case of pharmacologic or genetic knockdown of HIF-1α (80). Additionally, HIF-2α, which has overlapping activities with HIF-1α, is also expressed by neutrophils, and its expression has been up-regulated under inflammatory circumstances (81).
Taken together, metabolic flexibility under a variety of stress conditions aids in meeting energy demands by efficiently diverting intermediate metabolites created by the operating metabolic pathways (82). In Figure 1, we describe major metabolic pathways, which supply the required energy for diverse neutrophil functions. In addition, we summarize the recent publications reporting defect in NET formation under pathological conditions where the metabolic activities of neutrophils are compromised (Table 1).
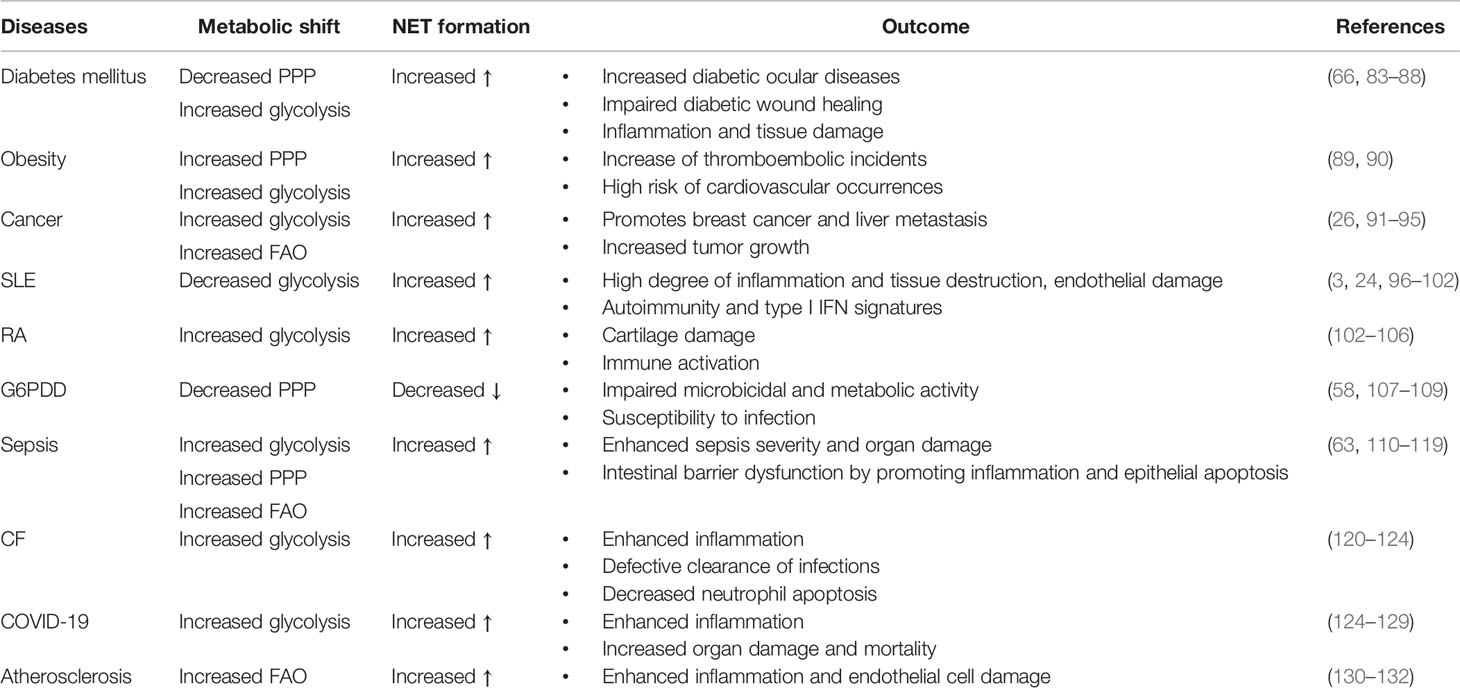
Table 1 Metabolic changes in NET-associated diseases.
Glycolysis
Glycolysis is a key metabolic pathway that converts glucose to pyruvate to generate small quantities of ATP and NADPH (3, 49, 50, 133, 134). Glycolysis is the foremost metabolic pathway in most immune cells, including neutrophils, which depend on glycolysis as a source of ATP (43). This fundamental metabolic pathway does not require oxygen and represents a sequence of ten reactions catalyzed by enzymes (43, 65). During glycolysis, neutrophils uptake extracellular glucose through glucose transporters (GLUTs), and express various GLUTs, such as GLUT1, GLUT3, and GLUT4 (135). Upon activation, increased surface expression of GLUTs is observed followed by an increase in the uptake of glucose. Once inside the cell, glucose is immediately converted into glucose-6-phosphate by the hexokinase enzymes (136). Each G6P molecule is converted into two molecules of pyruvate and two molecules of ATP as well as NADH. In the presence of oxygen (aerobic conditions), pyruvate can be used to make acetyl-Co A, which can then be oxidized via the TCA cycle to produce energy (ATP through ETC) (3, 47, 67, 134). Many studies have demonstrated the essential role of glucose in neutrophils, whereby the depletion of glucose completely terminated most of neutrophil effector functions (43, 49, 58, 63) (Figure 1)
Pentose-Phosphate Pathway (PPP)
An alternative glucose-dependent metabolic pathway in neutrophils is the PPP, also known as the hexose monophosphate shunt, which has been seen in both resting and activated neutrophils as well as during NET formation (58). PPP is involved in the NADPH oxidase (NOX)-dependent ROS production and consequently contribute to NET formation (58). Superoxide production via the PPP is an essential catalysator in the process of bacteria killing by neutrophils (137). Defects in the PPP pathway or inhibition of the PPP key enzyme, glucose-6-phosphate dehydrogenase (G6PD), under high glucose concentrations led to reduced ROS production (138).
The PPP encloses an oxidative and a non-oxidative phase (Figure 2). During the oxidative phase, G6PD, 6-phosphogluconolactonase, and 6-phosphogluconate dehydrogenase (PGD) convert G6P into CO2, ribulose-5-phosphate, and NADPH, to maintain redox equilibrium during cell stress. Several enzymes are involved in the non-oxidative phase, including ribose-5-phosphate isomerase, ribulose-5-phosphate 3-epimerase, a transketolase (TKT), and a transaldolase (TAL), which convert ribulose-5-phosphate to nucleic acids, sugar phosphate precursors, or glycolytic precursors like fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphat (G3P). PPP and glycolysis share a pool of G3P and F6P, which results in lactate or pyruvate production, respectively.
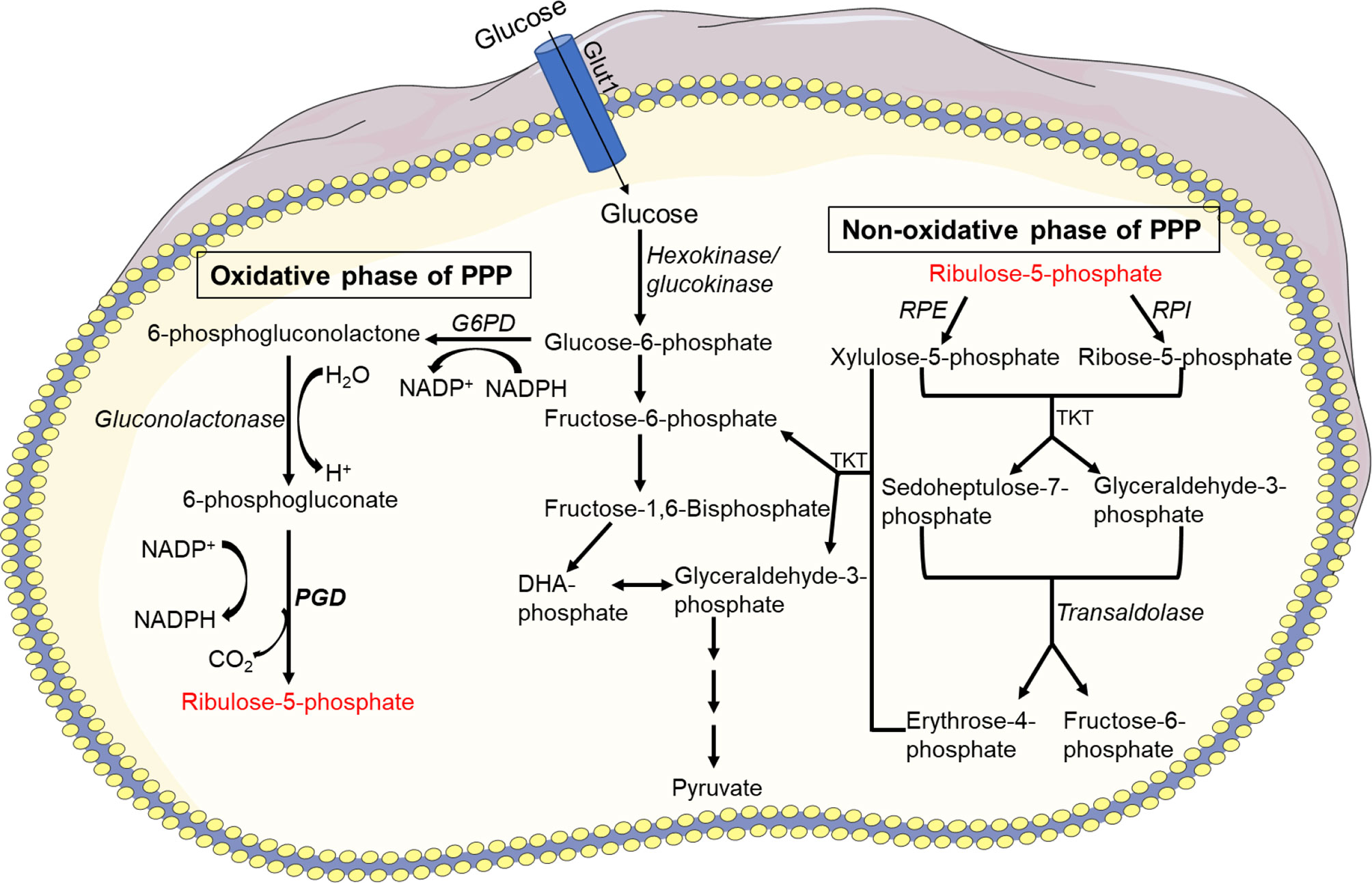
Figure 2 Pentose phosphate pathway (PPP). There are two phases in the PPP: oxidative and non-oxidative. G6P-dehydrogenase (G6PD), 6-phosphogluconolactonase, and 6-phosphogluconate dehydrogenase (PGD) convert G6P into CO2, ribulose-5-phosphate, and NADPH during the oxidative phase to maintain redox balance under stressful conditions. In the non-oxidative phase, enzymes such as ribose-5-phosphate isomerase, ribulose-5-phosphate 3-epimerase, a transketolase, and a transaldolase convert ribulose-5-phosphate to nucleic acids, sugar phosphate precursors, or glycolytic precursors such as fructose-6-phosphate (F6P) and glyceraldehyde-3-phosphate (G3P). PPP and glycolysis share a pool of G3P and F6P, resulting in lactate or pyruvate production.
Glycogen Metabolism
Glycogen, a multibranched polysaccharide stored in the neutrophil cytosol, is the central storage of glucose, and glycogenolysis, a breakdown of glycogen to glucose-1-phosphate, is a signal of limited availability of glucose and ATP (63, 139). Neutrophils appear to be the only myeloid cell type capable of gluconeogenesis and glycogenesis, which reveals their capacity to create ATP under low-glucose and low-oxygen conditions (140). Glucose-1-phosphate is converted to glucose-6-phosphate, which is often hydrolyzed to glucose using glucose-6-phosphatases (G6Pase) via gluconeogenesis (141). In the absence of glucose, neutrophils rely on glycogenolysis to break down stored glycogen as a source of glucose (64). Neutrophils isolated from inflammatory sites accumulate more glycogen than neutrophils from the peripheral circulation (64, 139). As a result, during inflammation, glycolysis appears to be a prominent and the critical metabolic route for neutrophils. Glycogen content raises as neutrophils mature (142). Once sufficient glucose is available, and normal intracellular levels of glucose are re-established, synthesis of glycogen is resumed (49, 143).
The interplay of glucose between endoplasmic reticulum (ER) and cytoplasm regulates the neutrophil glycogen level (Figure 3). Glucose cycling in the ER plays an essential role in neutrophil apoptosis and other neutrophil functions (141). Moreover, it has been reported that deficiency of the ER glucose-6-phosphatase-β (G6Pase-β also known as G6PC3) underlies the G6PC3–deficient congenital neutropenia syndrome. Neutrophils from these patients exhibited increased ER stress and apoptosis. G6PC3 converts G6P into glucose and phosphate, which enters the ER via the G6P transporter (G6PT). G6PC3 deficiency in these patients leads to reduced cytoplasmic concentrations of glucose, G6P, lactate, and ATP, leading to increased apoptosis and typical neutropenia (144). Increased enzymatic activity of glycogen phosphorylase leads to further accumulation of intracellular G6P which can be utilized in different metabolic pathways in neutrophils (145). Neutrophils are susceptible to fluctuations of intracellular and extracellular glucose levels or impaired glucose absorption and respond accordingly (146). Deficiency of glucose-6-phosphate translocase (G6PT/SLC37A4) leads to glycogen storage disease type Iβ (GSD-1β) (147). GSD-1β−deficient neutrophils accumulate glycolysis-inhibiting analog of glucose in the cytoplasm that affect protein glycosylation (148), ATP generation, NADPH synthesis. Subsequently, neutrophils of glycogen storage disease patients exhibit impaired chemotaxis, oxidative burst and bactericidal activity (147, 149, 150), and increased apoptosis (151). Interestingly, a recent study discovered that treating GSD-1β patients with a sodium glucose cotransporter 2 (SGLT2) inhibitor, an anti-diabetic drug inhibits renal reabsorption of glucose while facilitating excretion of the inhibitory glucose analog, resolving the neutropenia and neutrophil dysfunctions (152). These studies indicate the importance of glucose in energy metabolism and NADPH oxidase-dependent neutrophil functional responses, hinting the essential need to better understand the glucose flux in neutrophils (Figure 3).
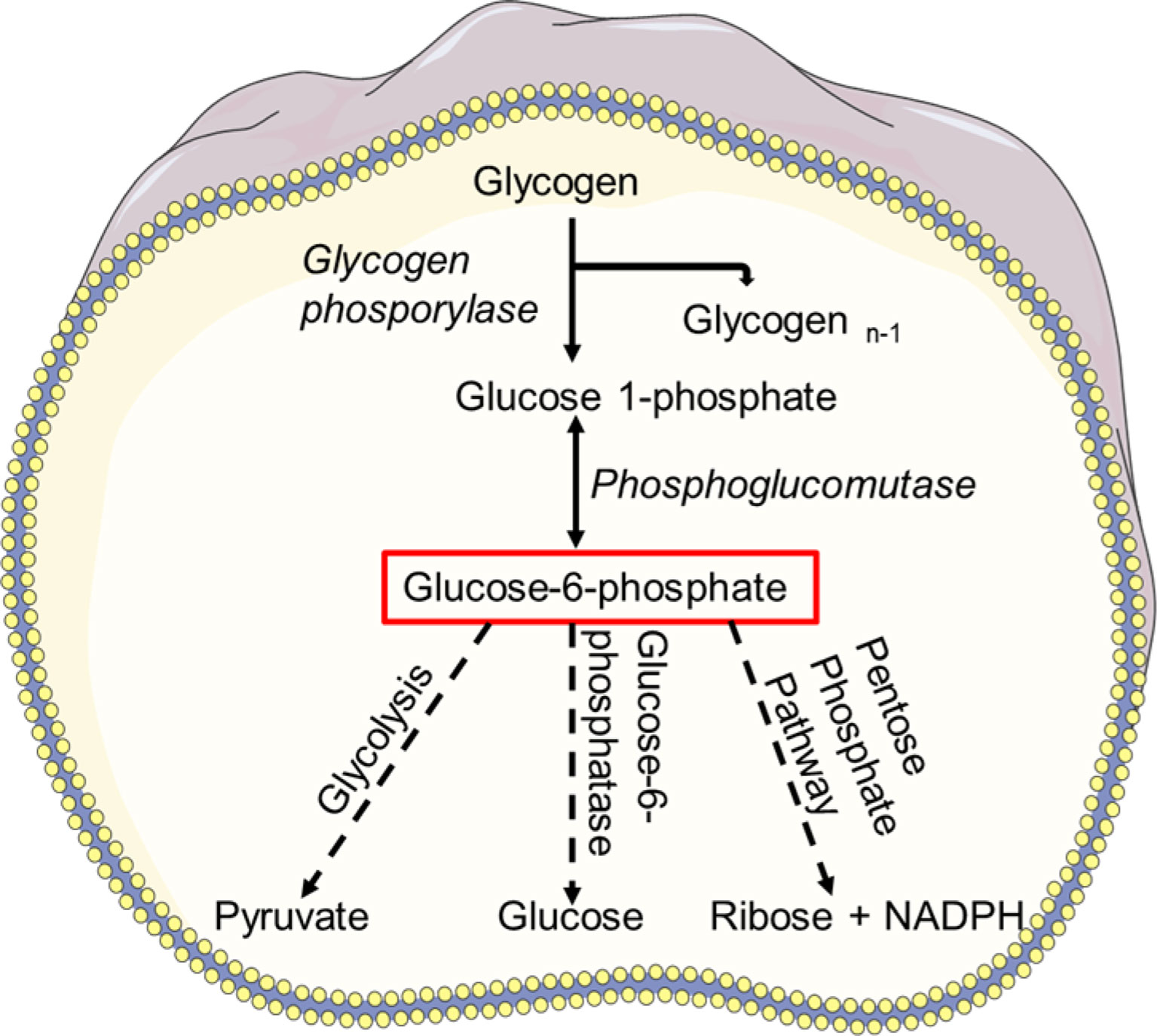
Figure 3 Glycogen metabolism. Breakdown of glycogen results in glucose-1 phosphate which is then converted to glucose-6-phosphate (G6P) by phosphoglucomutase. G6P can enter the glycolysis pathway and used as a source of energy. Using glucose-6-phosphatase (G6Pase), G6P can be hydrolyzed to glucose via gluconeogenesis and subsequently released into the blood. Glucose-6-phosphate can also be taken by PPP and converted to NADPH or ribose in a variety of tissues.
Glutamine Metabolism
Glutamine is one of the most abundant amino acids in the body and can be used as a substrate in the biosynthesis of proteins, antioxidants, NADPH and other metabolic pathways involved in cellular integrity and cell functions (153). In a series of biochemical reactions, glutamine metabolism results in glutamate, aspartate, lactate and ammonia production (134). Glutamine is required for the synthesis of nucleotide precursors, such as RNA and DNA, in physiological conditions. When glucose availability is restricted under pathophysiological settings, many cells, including neutrophils, can shift to glutaminolysis metabolism to meet their energy requirements (70, 154–156). Once it enters the cell, glutamine needs to be oxidized in order to be transformed into glutamate (Figure 4). Glutamate then enters the mitochondria and becomes oxidized and metabolized to α-ketoglutarate, which allows NAD+ to be oxygenated and converted to NADH. The TCA cycle may accept α-ketoglutarate and create malate, which is subsequently transformed to pyruvate by the malate dehydrogenase, which oxygenates NADP+.
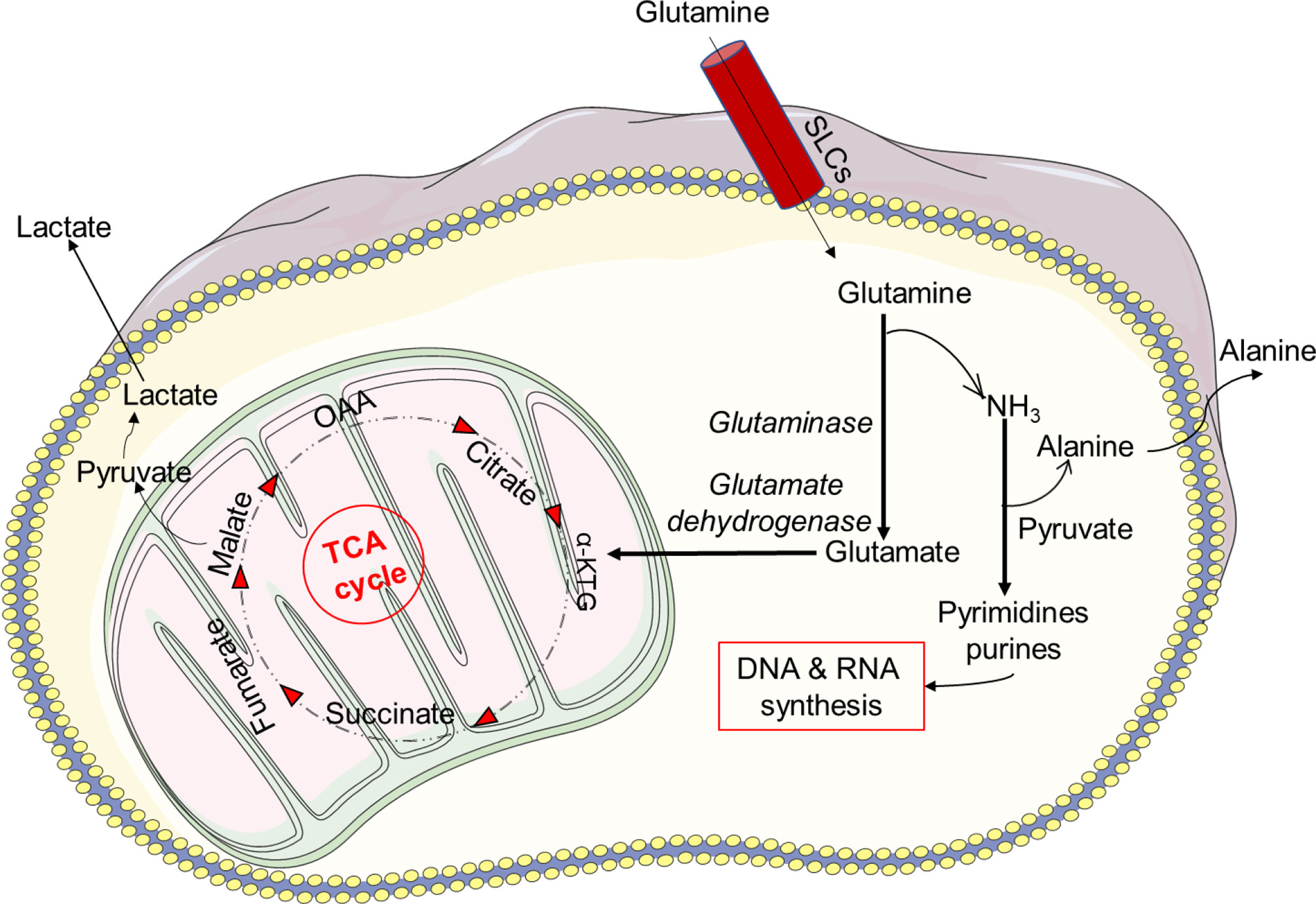
Figure 4 Glutamine metabolism. Glutamine metabolism results in glutamate, aspartate, lactate and ammonia production. Under physiological conditions glutamine is required for the synthesis of nucleotide precursors, such as RNA and DNA. The sodium-coupled neutral amino acid transporter (SNAT) family of proteins is one of several carrier type transporters (SLCs) responsible for bringing glutamine into the cell. Once inside the cell, glutamine is oxidized and transformed into glutamate. Furthermore, glutamate enters the mitochondria and becomes oxidized and metabolized to α-ketoglutarate, which allows NAD+ to be oxygenated and converted to NADH. The TCA cycle may accept α-ketoglutarate and create malate, which is subsequently transformed to pyruvate by the malate dehydrogenase, which oxygenates NADP+.
Pyruvate is subsequently converted into lactate in the cytosol or used by the mitochondria for OXPHOS. NAD+ can be restored through the generation of lactate in conditions of inflammation and low oxygen. How a cytosolic NAD+ enters mitochondria is still not known, especially because mammalian mitochondria do not generate NAD+ and are thought to be impermeable to pyridine nucleotides (134, 157). However, it has recently been shown that cytosolic NAD+ or NADH may be delivered directly into mammalian mitochondria (134, 157). Neutrophils may also consume glutamine at a faster rate than glucose (68). In addition, glutaminolysis, like PPP, plays a key role in the formation of NADPH and the expression of the NOX complex (158). In vitro studies have indicated that glutamine enhances bacterial killing by neutrophils from postoperative patients. The addition of 2 mM extracellular glutamine to neutrophils attenuated the adrenaline-induced suppression of superoxide generation, resulting in enhanced NADPH producing activity via glutamine metabolism (159, 160).
Fatty Acids
Fatty acids are energy substrates for tissues and cells, including leukocytes, and involved in the maintenance of glucose and fuel homeostasis (161). The metabolism of fatty acids has an important role in the functional activity of neutrophils (162). The fatty acid chain length can vary from 3 to 30 carbon atoms, and, depending on the number of carbon atoms, the fatty acids are characterized as short-, medium, and long-chain fatty acids. Short-chain amino acids contain less than six carbon atoms. Those with six to ten carbon atoms are medium-chain fatty acids and long-chain fatty acids exhibit over twelve carbon atoms. The fatty acids can be monounsaturated with one carbon - carbon bond or polyunsaturated with two or more double bonds. In addition, the fatty acids with no double bonds in the molecule are known as saturated fatty acids (144, 163). The metabolism of fatty acids is increased when cells are exposed to limited glucose conditions. Utilization and oxidation of fatty acids occur in neutrophils and are important for their activation (164). Moreover, during neutrophil differentiation and maturation, fatty acids enter into the FAO pathway to produce ATP through OXPHOS (165). It has also been shown that fatty acids impaired glucose uptake and glucose metabolism (166).
Fatty acids are oxidized to produce acetyl-Co A, which is then converted to citrate via citrate synthase. The activation of pyruvate dehydrogenase kinase (PDK), which inactivates pyruvate dehydrogenase (PDH), is promoted by high acetyl-Co A/Co A and NADH/NAD ratios. ATP and citrate block phosphofructokinase (PFK), resulting in the buildup of G6P and inhibition of hexokinase, and therefore glycolysis is hindered. The mitochondrial matrix also produces acetyl-Co A, which can be oxidized in the TCA cycle (69) (Figure 5). Furthermore, carnitine, a quaternary ammonium compound, plays an important task in energy production due to its role in FAO by transporting long-chain fatty acids into mitochondria to be oxidized for energy production. Carnitine also participates in removing products of metabolism from cells. Additionally, acylcarnitines located in the outer mitochondrial membrane, are generated by carnitine palmitoyltransferase-1 (CPT-1) that catalyzes an ester bond of carnitine with long-chain fatty acids. Carnitine-acylcarnitine translocase (CACT) translocates acylcarnitines across the inner mitochondrial membrane (167). Acylcarnitines renew acyl-Co A inside the mitochondrion via the carnitine palmitoyltransferase-2 (CPT-2), which is located in the inner mitochondrial membrane (168). Carnitine returns to the cytoplasm for another cycle, whereas acyl-Co A enters the β-oxidation pathway, creating acetyl-Co A (under aerobic circumstances and low ATP levels) (167). The utilization of fatty acids by neutrophils and consequently the outcome of the interaction depends on different aspects, including the presence and absence of inflammation and the concentration of the fatty acids. Under in vivo conditions, fatty acids also interact with other cells, such as macrophages and endothelial cells (162).
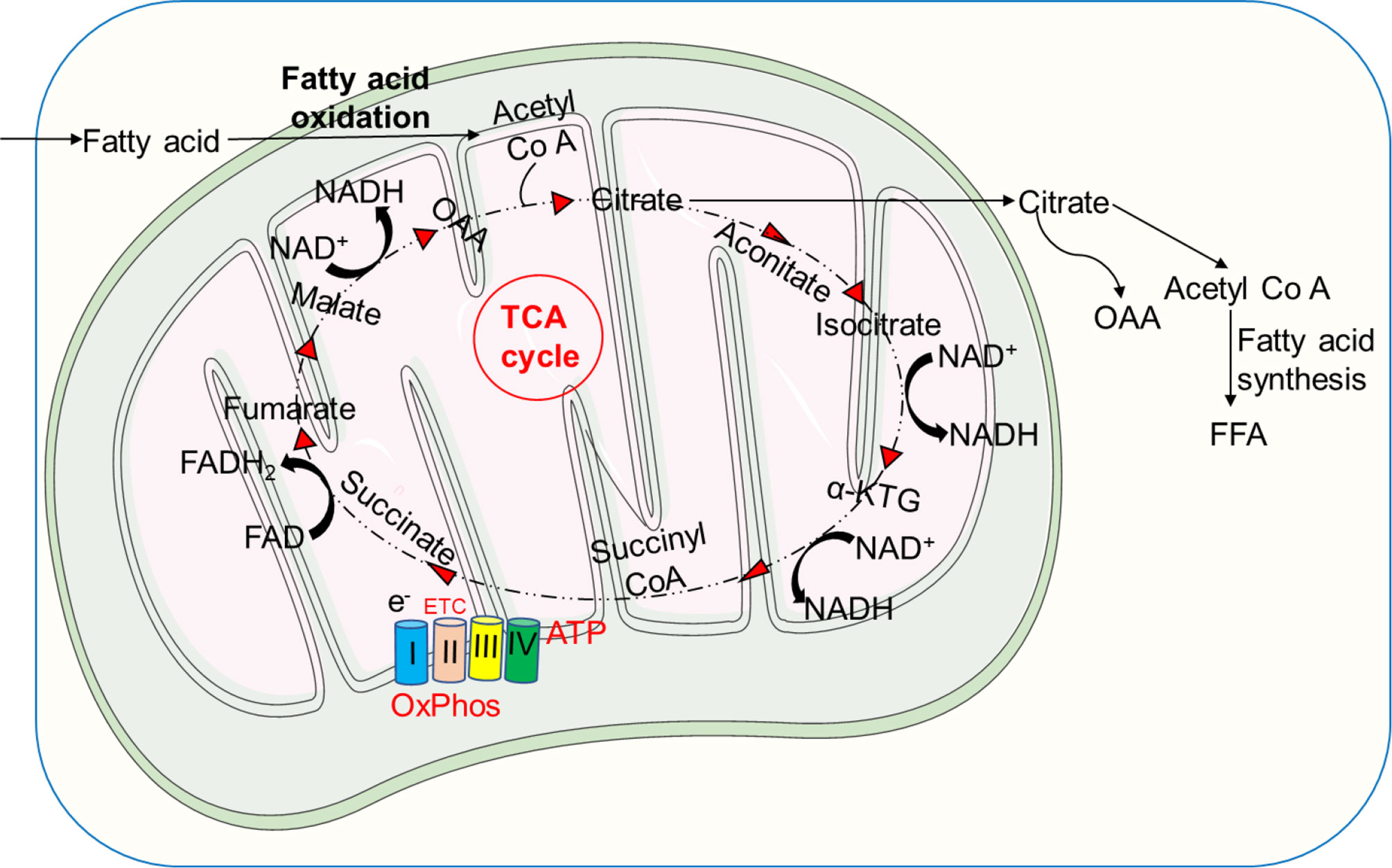
Figure 5 Metabolism of fatty acids. Fatty acids are oxidized to form acetyl-Co A, which is then converted to citrate by the activity of citrate synthase. Acetyl-Co A can be also produced by the mitochondrial matrix and further oxidized in the TCA cycle. Through FAS, the TCA cycle intermediate citrate can be converted to free fatty acids (FFA), which can then be transported from the extracellular environment via the FAO pathway to produce acetyl-Co A, fueling the TCA cycle to produce significantly more energy in the form of ATP.
Metabolite Changes During Net Formation
To form NETs, neutrophils release dsDNA as a scaffold decorated with toxic granule proteins (8, 9, 17, 19, 169). Currently, several groups have studied the metabolic requirements of NET formation (9, 43, 58). NET formation requires glycolytic ATP production in order to rearrange cytoskeleton for catapult-like release of cytoplasmic granules together with mitochondrial DNA (8, 9, 17, 62, 169). It has been reported that glycolytic ATP production aids the microtubule network assembly to form NETs (9, 169). Using genetic and pharmacologic approaches, it has been demonstrated that lack of Opa1 reduces the mitochondrial ETC I activity in mouse neutrophils. As a result, the ATP production via glycolysis was reduced due to lowered level of NAD+ availability. Moreover, we demonstrated that mice lacking Opa1 in neutrophil populations (Opa1NΔ) exhibited a less antibacterial defense capability, providing direct evidence for the role of mitochondria in NET formation (9).
NET formation has been suggested to depend on glucose, but not on glutamine (43). It was reported that glucose uptake increased upon phorbol myristate acetate (PMA) to induce NETs (43). Moreover, NET formation depends on increased intracellular ROS production (19, 40, 170, 171). Patients exhibiting chronic granulomatous disease (CGD) suffer from an increased susceptibility to bacterial and fungal infections. The genetic disorder is characterized by defects of the NADPH oxidase which results in impaired ROS production (19, 172). The lack of sufficient ROS production leads subsequently to defects in cytoskeleton reorganization, degranulation, DNA release and ultimately in reduced formation of NETs (8). However, under certain circumstances in absence of NADPH-oxidase activity mitochondria could provide sufficient mitochondrial ROS (mitoROS) as a compensatory mechanism to generate NETs (24). However, the increase in ROS production occurs much faster and seems not to be energy-dependent (43). Additionally, 2-deoxy-d-glucose (2-DG), an inhibitor of glycolysis, can block NET formation (9, 43) by inhibiting glycolysis and PPP.
The NADPH used by NOX originates from the PPP, which demonstrates the importance of a tightly regulated glucose metabolism in neutrophil functions. Hyperglycemia has been demonstrated to enhance the production of NETs (88, 134), which might explain the higher incidence of spontaneous NETs seen in type 2 diabetes patients (88). For example, in high glucose concentration, neutrophils were spontaneously activated, but had less ability to respond to bacterial antigen stimulation such as lipopolysaccharide (LPS) (134). On the other hand, hyperglycemia was also shown to reduce key functions of neutrophils, such as phagocytosis, ROS production and bacterial killing (134, 173), perhaps owing to unstable NETs which in addition contain decreased amounts of anti-microbial peptides compared to NETs released in the presence of physiological glucose concentrations (174, 175).
Withdrawal of glucose from medium leads to complete eradication of NETs (43, 58, 64). When glucose is replenished, the formation of NETs is rescued (58). Upon neutrophil activation, GLUT1 expression is increased on the cell surface of neutrophils in order to uptake glucose for glycolysis (43). Moreover, increases in the extracellular acidification rate and lactate dehydrogenase (LDH) activity were also reported (176), supporting the notion that glycolysis is important in the process of NET formation. Similarly, 6-aminonicotinamide [6-AN, a glucose-6-phosphate dehydrogenase (G6PDH) inhibitor] can disrupt PPP metabolism, which leads to NET formation (58). Subsequently, a metabolic flip toward PPP occurs during NET formation (3, 58), accompanied by increased G6PD activity, which diverts the glycolytic intermediate G6P. In PMA and amyloid fibrils triggered neutrophils, the G6PD inhibitor 6-aminonicotinamide (6-AN) blocked ROS generation and dsDNA release, suggesting that G6P is a fuel for NOX2 activation (58). However, the inhibition of ROS and NETs was incomplete, indicating that there might be additional sources of energy such as an active glycolytic pathway (3, 43, 176).
Furthermore, TKT, a thiamine pyrophosphate (vitamin B1)-dependent enzyme that connects the pentose phosphate pathway and the glycolytic pathway, was shown to be important for the NADPH oxidase activity and consequently for the NET formation. Pharmacological suppression of TKT or a genetic deficit in transaldolase 1, non-oxidative PPP enzymes that aid in the conversion of oxPPP reaction products back into glucose 6-phosphate, reduce oxidative burst and NET formation (177).
Although neutrophils are regularly found under hypoxic inflammatory conditions in tissues with enriched lactate, the role of lactate in neutrophil activity has received so far little attention only. Given the current paradigm shift in the role of lactate in metabolism, it has recently been reported that lactate plays a unique role in NET formation. Moreover, human neutrophils exogenously treated with lactate exhibit NET formation, albeit NET formation was dramatically reduced when LDH activity was inhibited (3, 9, 43, 176). In addition, the lactic acid produced by bacteria could also induce NET formation by bovine neutrophils (178), and inhibition of its transporter, the monocarboxylate or LDH enzyme, blocks the lactate-induced NET formation (64, 178). LDH has the property to convert both pyruvate and lactate, suggesting that neutrophils exploiting lactate during NET formation and glycolysis bypass the PPP to act as a major metabolic pathway during NET formation. Therefore, the modulation of lactate level under inflammatory conditions, such as diabetes and sepsis, could control neutrophil activation and NET formation. Additionally, oxidized low-density lipoprotein (oxLDL) upon activation of the TLR pathway cause NET formation in human PMNs (131) (Figure 6).
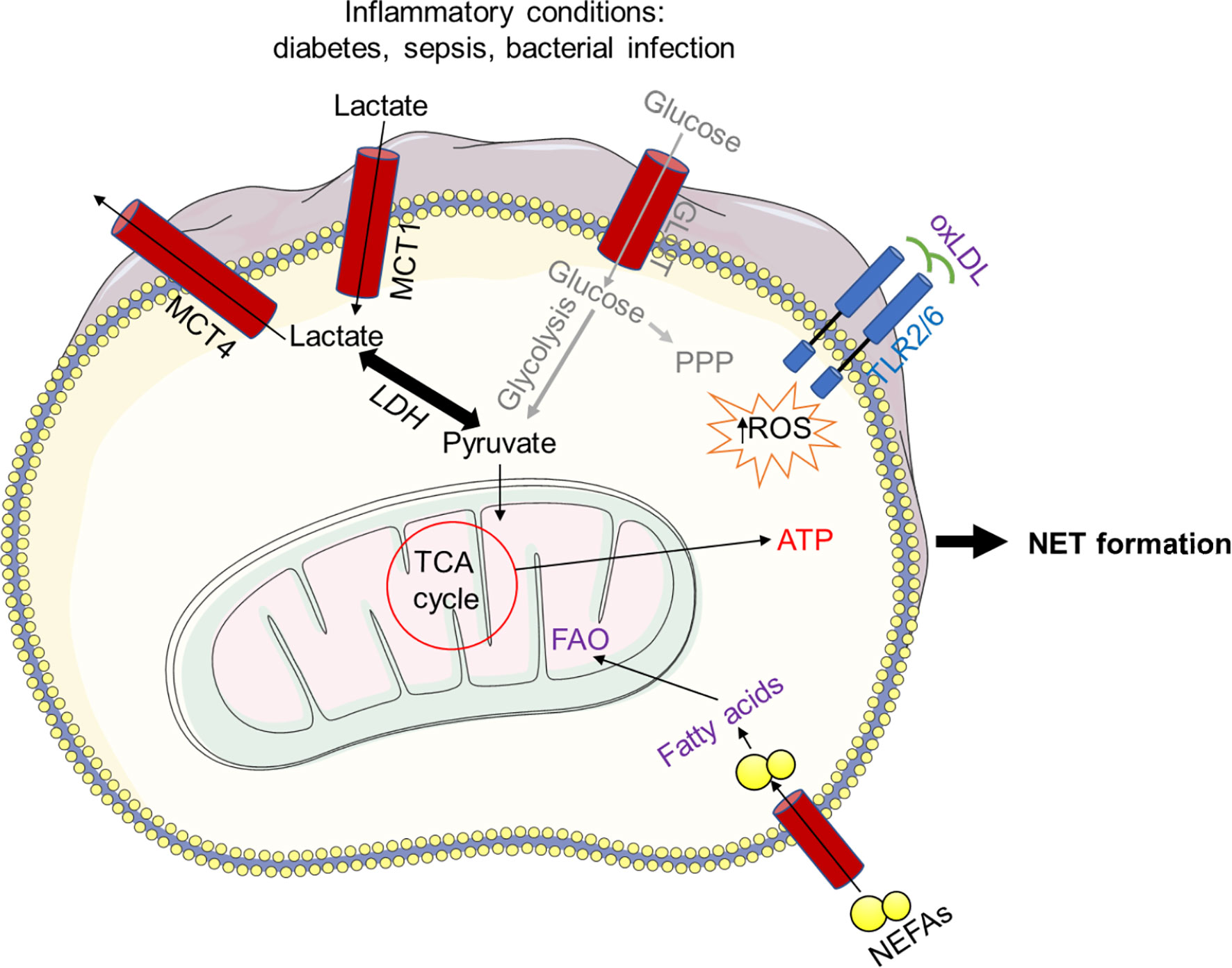
Figure 6 Bacterial lactic acid generated during infection can induce NET formation. Bacteria-produced lactic acid can cause NET formation in bovine neutrophils, and inhibiting the lactate transporter, the mono-carboxylate or lactate dehydrogenase (LDH) enzyme, prevents this. Because LDH can convert both pyruvate and lactate, it is possible that neutrophils use lactate during NET formation and glycolysis to circumvent the PPP. Furthermore, NET formation in human neutrophils is caused by oxidized low-density lipoprotein (oxLDL) in a ROS-dependent manner upon TLR activation. In addition, non-esterified fatty acids (NEFAs), such as oleic acid (OA) and linoleic acid (LA), initiate FAO, allowing increased ATP production and consequently NET formation.
Evidence for an important role of the mitochondrial metabolism has recently been obtained when platelet-activating factor (PAF) triggered mitochondrial hyperpolarization and extracellular ATP release via pannexin-1, resulting in NET formation. Interestingly, P2X1 purinergic receptor blockage hindered mitochondrial hyperpolarization and NET formation. Hence, ATP and ATP channel pannexin 1 (Panx1) contribute to NET formation and may represent therapeutic targets (179). It has been further concluded that PAF-induced NET release is dependent on glycolysis, mitochondrial ATP synthesis and purinergic signaling (180). Recent studies have revealed the importance of glutamine in NET formation. For example, lack of glutamine or inhibition of complex V to disrupt the mitochondrial ATPase function partially inhibited NET formation, indicating a glutamine-driven mitochondria-dependent metabolism is also utilized for neutrophils to form NETs (3, 64). Mitochondrial translocation toward the cell surface upon activation (8, 9) and its hyperpolarization to release mitochondrial ROS (180), as well as oxidized mitochondrial DNA (24) point to a direct role of mitochondria in NET formation (169, 181, 182).
NETs could potentially be formed independent of the NOX-NADPH oxidase, depending on the availability of sufficient amounts of mitochondrial ROS (24, 33, 134). Some pathogens increase mitochondrial ROS production (183) that may result in reduced dependency on glucose metabolism.
Oxygen level and the pH seem to have an important role in NOX-independent NET formation as well, as it has been demonstrated that, under acidic conditions, glycolysis and ROS production are reduced and therefore less NETs are detected, indicating that conditions at the site of inflammation can also influence the extent of NET formation (184, 185). The role of the metabolism of fatty acids on NET formation is less studied, although it has been reported that non-esterified fatty acids (NEFAs) such as oleic acid (OA) and linoleic acid (LA), that are often associated with infectious diseases, are associated with a higher incidence of infections and NEFAs induced the formation of NETs through extracellular release of ATP via Pannexin 1 and subsequent activation of purinergic receptors. Specific blocking of P2X1, a non-selective cation channel, eliminated NET formation upon NEFAs exposure (186) (Figure 6). Moreover, other fatty acids, such as Furanoid F-Acid F6, can also activate neutrophils to induce ROS via both NADPH oxidase (NOX) and mitochondrial activity, resulting in rapid NET formation. Moreover, Furanoid F-Acid F6 also required protein kinase B (AKT) to induce NET formation (187). Common long-chain fatty acids, such as palmitic acid (PA), palmitoleic acid (PO), stearic acid (SA), and oleic acid caused NET formation via NOX, ERK (extracellular signal-regulated kinase) and JNK (c-Jun N-terminal kinase)-dependent pathways. Others reports suggested that chlorinated lipids, 2-chlorofatty aldehyde and 2-chlorofatty acid (2-ClFA), which is the byproduct of MPO release in the phagosome during neutrophil activation, can convert hydrogen peroxide and chloride to hypochlorous acid (HOCl), leading to NET formation (188). This study also demonstrated that human neutrophils exposed to physiological levels of 2-ClFAs induced functional NETs, albeit in the absence of degranulation, and reduced Escherichia coli colony forming units. NETs formed by 2-ClFA were calcium and protein arginine deiminase 4 (PAD4)-dependent (188). NET formation can also be induced by oxidized lipoprotein in correlation with ROS production (131), which is supported by the tumor infiltrating c-kit+ neutrophils that utilize mitochondrial FAO (91). Important role of diverse metabolic pathways on NET formation have been intensely studied and summarized in Table 1.
Metabolic Pathways in Net-Related Diseases
Diabetes
Diabetes is a systemic metabolic disorder that is characterized by increased glucose levels (3). Several studies have described impaired neutrophil functions in diabetic patients and animal models, including neutrophil adhesion, chemotaxis, phagocytosis, ROS production, microbicidal activity, mitochondrial potential and ATP levels (66, 189, 190). Hyperglycemia results in decreased G6PD and glutaminase enzymatic activities (66). Consequently, putative compensatory FAO utilization is observed in neutrophils during hyperglycemia due to defective glucose and glutamine metabolism (66). High glucose levels found in diabetic patients are reported to increase NET formation in vitro and in vivo in association with increased NADPH oxidase activity (84, 86, 88), demonstrating the role of NETs in the pathogenesis of diabetes and its complications like diabetic retinopathy (DR) (83, 87). The development of high glucose-induced NETs was found to be affiliated with the NADPH oxidase machinery. Furthermore, long-term hyperglycemia allows peripheral neutrophils to cross the blood–retinal barrier (BRB) form NETs in the vitreous body and retina. Monitoring NET circulating products in diabetic patients’ serum might be used to assess the severity of DR in order to minimize disease progression and offer prompt therapy (88). Additionally, improved neutrophils phagocytosis and ROS production is reported after treatment of insulin, a common blood glucose lowering therapy (66). Furthermore, GLUT4 is translocated to the cell surface upon activation with PMA in an insulin-sensitive manner and may play a role in the redistribution of glucose during hypoglycemia. In contrast, GLUT1 and GLUT3 are expressed on the surface independent of neutrophil activation and insulin levels (135) (Table 1).
Obesity
Obesity is associated with a risk of higher mortality due to low-grade chronic inflammation (90). Moreover, enhanced neutrophil activity, increased superoxide radicals and NET formation are observed in association with obesity (89, 191). Weight loss of obese patients results in decrease of NET formation (192). While normal neutrophils utilize glycolysis and PPP under both physiological and inflammatory conditions, neutrophils from obese mice exhibit diminished NET release, perhaps owing to “exhaustion” (90). Moreover, the ability to form NETs under in vivo condition was reduced in obese mice. Consequently, reduced NET formation and suppressed overactivation of neutrophils due to obesity may promote infections and increase the severity of sepsis (90). Furthermore, GLUT1 surface expression was reduced in LPS-activated neutrophils of high-fat diet mice, while GLUT1 expression was unaffected in resting neutrophils of these mice (90) (Table 1).
Cystic Fibrosis
Cystic Fibrosis (CF) is a genetic syndrome characterized by mutations in the CF transmembrane conductance regulator gene (193). High levels of free glucose and amino acids are found in CF airway lumen of CF patients (122). CF neutrophils exhibit suppressed apoptosis compared to normal neutrophils (193). Furthermore, an increased capacity of NET formation is observed in CF neutrophils (123), a phenomenon, which has been linked with impairment of the obstructive lung function (120). CF airway neutrophils, but not blood neutrophils, from CF patients exhibit increased expression of GLUT1, neutral amino acids transporter 2 (ASCT2) and inorganic phosphate transporter 1 & 2 (PiT1, PiT2) compared to normal neutrophils, suggesting that metabolic adaption processes occur in neutrophils upon their recruitment from blood into CF airway lumen (122). ROS production is decreased in airway CF neutrophils in contrast to blood neutrophils (121). Furthermore, GLUT1 and PiT2 levels in blood neutrophils are diminished upon oral treatment with corticosteroids (122) (Table 1).
Cancer
Tumor-associated neutrophils (TANs) can exhibit both pro- and antitumor properties (3). In mouse lung adenocarcinoma, TANs display increased GLUT1 expression and glucose metabolism. These findings have been associated with a decelerated neutrophil turnover in tumors, increased tumor growth, and diminished efficacy of radiotherapy (94). Immature low-density neutrophils (iLDNs) rely on mitochondria-dependent ATP production through the catabolism of glutamate and proline. It has been shown that they can exhibit a pro-metastatic function under nutrient-deprived conditions (92). Additionally, neutrophils are able to adapt an oxidative phenotype through c-KIT signaling in response to tumor. Subsequent ROS production by NADPH oxidase allows ROS-mediated suppression of T cells even in glucose-limited environment and favors tumor growth (91). Furthermore, neutrophils are attracted by tumor growth to infiltrate and form NETs that modulate the metabolic profile of the tumors leading to its increased survival (93). NETs formed from TANs and their associated proteins elastase and matrixmetalloproteinase-9 also facilitate the reestablishment of the proliferative capacity of dormant lung cancer cells through extracellular matrix remodeling (95). Moreover, NETs are reported to trap circulating tumor cells leading to increased metastatic formation (194). Neutrophil infiltration in human thyroid cancer (TC) has been recently reported to correlate with tumor growth. Moreover, TC-derived conditioned media (CM) attracted neutrophils, increased their survival, caused morphological alterations in neutrophils, and triggered and altered neutrophil kinetic characteristics. Interestingly, anaplastic (ATC) CM-primed neutrophils produced NETs containing mitochondrial DNA in a mitochondrial ROS-dependent and cell death-independent manner (26, 195) (Table 1).
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by a high degree of immune inflammation and self-destruction, as well as defective clearance of NETs (97, 101). Neutrophils of SLE patients exhibit dysregulated aggregation, phagocytosis, oxidative burst and increased NET formation (3). Deficient GLUT3 and GLUT6 expression in SLE neutrophils results in increased intracellular basal lactate production and decreased ATP production. The decreased expression of GLUT3 and GLUT6 results in a decreased ability to uptake glucose, resulting in decreased glycolytic activity (100). Furthermore, intracellular glutathione (GSH) levels and γ-glutamyl-transpeptidase activity are reduced in neutrophils of patients with active SLE leading to reduced redox capacity (100). NETs are induced by ribonucleoprotein immune complexes (RNP-ICs) found in SLE through augmented production of mitochondrial ROS production (24). Low-density granulocytes (LDGs), a distinct subset of pro-inflammatory neutrophils, have also been described in SLE patients (24), and are associated with enhanced disease activity (196). LDGs exhibit increased mitochondrial ROS (24) associated with increased NET formation (99). Furthermore, NET formation by LDGs is shown to be responsible for endothelial damage (96, 98). Lupus symptoms are rescued through treatment with 2-DG, metformin and rapamycin in mice models, confirming a potential role of metabolic alterations in SLE (197) (Table 1). It remains unclear why NET formation is increased in SLE neutrophils in spite of reduced expression of glucose transporters on these cells. Perhaps, other factors, such as increased mitochondrial ROS activity, can compensate reduced glycolytic activity.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovitis and joint destruction through extensive activation of the immune system (3). Infiltrated neutrophils in joint synovial fluid (SF) release cytotoxic proteases (198) and show high ROS production (198) and increased expression of cytokines (103, 199). The hypoxic conditions in joints of RA patients increase the survival of SF neutrophils (103). Impaired beta cell function and insulin resistance in RA patients leads to a higher risk for diabetes (200). Interestingly, enhanced and accelerated NET formation is observed in RA patients (104). In RA, the NETs are involved in the degradation of cartilage as well as in the activation of fibroblast-like synoviocytes and macrophages, resulting ultimately in joint damage (106). Within RA joint milieu, due to low oxygen availability, HIF-1α is upregulated in neutrophils leading to increased glycolytic activity by upregulating the expression of G3PDH and triosephosphate isomerse-1 to provide ATP energy supply (102) (Table 1).
Atherosclerosis
Plaques containing LDL and cholesterol, as well as hyperlipidemia and inflammation, are the hallmarks of atherosclerosis (3). Neutrophil functions modulated by metabolic changes are reported to play a role in the pathogenesis of atherosclerosis (201, 202). Accordingly, hyperlipidemia and hypercholesterolemia trigger atherosclerosis through induction of neutrophilia (201). NET formation is induced by oxidized LDL and cholesterol crystals (131). Furthermore, the decrease of CXCL1 levels, resulting in inhibition of NAMPT and NAD+ biosynthesis, leads to diminished neutrophil infiltration in atherosclerotic plaques (203). NETs were also seen in atherosclerotic lesions in mice, together with an elevation in the pro-inflammatory markers IL-1 and IL-6 (204). Moreover, luminally adherent neutrophils releasing DNA in apolipoprotein-deficient (Apoe-/-) mice fed a high-fat diet for 4–6 weeks were identified using a two-photon microscopy intravital technique, but no neutrophil adhesion and thus no NET release were observed in mice receiving chow diet (130, 132, 182) (Table 1).
COVID-19
COVID-19 is a proinflammatory condition with a disproportionately high incidence of developing acute respiratory distress syndrome (ARDS), acute renal failure, arrhythmia, and shock (205, 206). A number of NET-associated indicators, such as cell-free DNA (cfDNA), MPO/NE-DNA complexes, and citrullinated histone H3 (CitH3) are considerably elevated in the circulation and/or tracheal aspirates of COVID-19 patients (127, 129, 207–215). In vitro treatment of neutrophils isolated from healthy controls with serum/plasma from COVID-19 patients led to formation of extracellular DNA traps, confirming the presence of NET-inducing substances in the circulation of COVID-19 patients (127, 129, 215–218). Furthermore, neutrophils obtained from COVID-19 patients also exhibited increased spontaneous NET formation in vitro (127, 207, 209, 210).
Several metabolic pathways were reported to be dysregulated in neutrophils from COVID-19 patients such as the tryptophan metabolism, TCA cycle, polyunsaturated fatty acid mobilization, and eicosanoid biosynthesis. In addition to these metabolic changes, it has also been reported that COVID-19 viral infection itself can cause metabolic reprograming in both neutrophils and PBMC (219, 220). Recent studies confirmed elevated levels of inflammatory cytokines, such as IL-1β and IL-6, as well as insufficient IL-10 responses in individuals with severe COVID-19. Furthermore, soluble tumor necrosis factor receptor 1 (sTNFR1), a surrogate receptor for circulating TNF-α, and pyruvate kinase M2 (PKM2) were significantly increased as well in COVID-19 patients. PKM2, an enzyme that catalyze the final step of glycolysis is a coactivator of HIF-1α (221), which induces the expression of proglycolytic enzymes, such as LDH, PDKs (pyruvate dehydrogenase kinases), and GLUT1. Additionally, enhanced cytosolic lactate and succinate levels have been detected in COVID-19 neutrophils compared to healthy control neutrophils.
Interestingly, accumulation of cytosolic succinate resulted in decreased breakdown of HIF-1α (222) and therefore enhanced the cytosolic levels of HIF-1α in COVID-19 neutrophils (124). The increased cytosolic lactate: pyruvate ratio has been revealed as fundamental metabolic change, suggesting a shift towards glycolysis in COVID-19 neutrophils (124). Glycolysis and lactate production are crucial processes in the development of neutrophil antimicrobial responses (43, 176). The increased glycolytic activity of COVID-19 neutrophils might provide a mechanistic explanation for the increased spontaneous NET formation by neutrophils isolated from patients with severe COVID-19 (209, 210). However, contrasting results have also been reported that suggested an impaired glycolytic flow and oxidative metabolism in neutrophils isolated from COVID-19 patients with ARDS symptoms (223). Currently, the reasons for these different findings remain unclear.
Sepsis
Sepsis is a systemic inflammatory syndrome characterized by impaired neutrophil migration and functions (3). Reduced expression of chemokine receptor CXCR2, GPCR receptor kinase and impaired actin polymerization cause defective neutrophil chemotaxis in sepsis (224, 225). Furthermore, impaired mitochondrial activity, largely complex V (ATP synthesis) and ETC complex III and IV were linked to dysfunction of neutrophil chemotaxis driven by LPS in septic conditions (113). Increased autophagy in association with augmented NET formation has been reported to improve the survival of sepsis patients (116). The induction of autophagy during sepsis leads to functional rejuvenation of neutrophil population and alteration of neutrophil metabolism through production of intermediate products (116). On the other hand, enhanced NET levels are associated with increased sepsis severity and subsequent organ damage (111, 117). NETs are accompanied with intestinal barrier dysfunction, ER stress activation and ROS production in sepsis patients, leading to increased serum intestinal fatty-acid binding protein (I-FABP) and D-lactate that positively correlates with NET formation, indicating that fatty acid and lipid metabolism might be required for neutrophil maturation during sepsis (Figure 5) (63, 119). Moreover, peroxisomal lipid synthesis regulated inflammation by sustaining neutrophil membrane phospholipid composition and viability (110). Increased expression of G6PD and phosphogluconate dehydrogenase suggest an induced PPP pathway as well (Figure 2) (114, 118). Additionally, plasma ATP is required for neutrophil activation during sepsis (226), while systemic ATP reduces neutrophil activation and chemotaxis through the disruption of endogenous purinergic signaling mechanisms (115). Accordingly, blockage of endogenous ATP signaling by suramin, a P2-receptor antagonist, increases bacterial growth and mortality whereas apyrase, an ATP diphosphatase, improves survival in sepsis through improved bacterial clearance and removal of systemic ATP (115) (Table 1).
Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
G6PD deficiency is an inherited enzymatic disorder characterized by a decrease in NADPH production, resulting in glutathione depletion and hemolytic anemia (3, 227, 228). G6PD is required for the switch of the cellular metabolism from glycolysis to PPP (229). Moreover, G6PD activity is increased after PMA stimulation in neutrophils and switches the metabolism toward the PPP pathway, resulting in increased NET formation. On the other hand, in the absence of G6PD activity, the PPP metabolism and NET formation is diminished (58). Neutrophils exhibiting G6PD deficiency, similar to mild CGD, showed impaired microbicidal and metabolic activity (107). Typically, bactericidal activity and neutrophil function are not impaired in mild G6PD-deficient neutrophils (108, 109). However, in patients with severe enzymatic deficiency reduced G6PD activity, increased susceptibility to infections due to reduced NADPH generation and subsequently impaired NET formation have been observed (109). Interestingly, rapid neutrophil turnover and diurnal fluctuating levels of G6PD activity are sufficient to induce an effective respiratory burst (230). Furthermore, nicotinamide nucleotide biosynthesis is suggested to be sufficient to compensate the G6PD deficiency, resulting in comparable rates of NET formation in G6PD patients and healthy controls (231) (Table 1).
Conclusions
Neutrophils are capable of adapting to the tissue environment under pathological conditions by modifying their metabolic activity utilizing several metabolic pathways. An increased mitochondrial activity and its contribution within a network of neutrophil metabolic pathways has been shown to be critical for anti-bacterial functions mediated by neutrophils. Pathological changes of these pathways contribute to disease, resulting in either immunodeficiency or accelerated inflammatory responses. The molecular mechanism of NET formation heavily relies on an intact cellular metabolism in neutrophils.
Author Contributions
Conceptualization: DS, SY, and H-US. Methodology: DS, LG, and SP. Investigation: DS, LG, and SP. Visualization: DS, LG, SP, and SY. Funding acquisition: SY and H-US. Project administration: DS, SY, and H-US. Supervision: SY and H-US. Writing – original draft: DS, SY, and H-US. Writing – review & editing: DS, RL, PR, AK, AR, NB, SY, and H-US. All authors contributed to the article and approved the submitted version.
Funding
Swiss National Science Foundation, grant No. 31003A_173215 (SY); Swiss National Science Foundation, grant No. 310030_184816 (H-US); Russian Government Program “Recruitment of the Leading Scientists into the Russian Institutions of Higher Education”, grant No. 075-15-2021-600 (H-US). AR was partially supported by Kazan Federal University (KFU) Strategic Academic Leadership Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Bouma G, Ancliff PJ, Thrasher AJ, Burns SO. Recent Advances in the Understanding of Genetic Defects of Neutrophil Number and Function. Br J Haematol (2010) 151:312–26. doi: 10.1111/j.1365-2141.2010.08361.x
2. Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J. The Role of Neutrophils in Immune Dysfunction During Severe Inflammation. Crit Care (2016) 20:73. doi: 10.1186/s13054-016-1250-4
3. Kumar S, Dikshit M. Metabolic Insight of Neutrophils in Health and Disease. Front Immunol (2019) 10:2099. doi: 10.3389/fimmu.2019.02099
4. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What’s Your Age Again? Determination of Human Neutrophil Half-Lives Revisited. J Leukoc Biol (2013) 94:595–601. doi: 10.1189/jlb.1112571
5. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human Neutrophil Kinetics: Modeling of Stable Isotope Labeling Data Supports Short Blood Neutrophil Half-Lives. Blood (2016) 127:3431–8. doi: 10.1182/blood-2016-03-700336
6. Hidalgo A, Chilvers ER, Summers C, Koenderman L. The Neutrophil Life Cycle. Trends Immunol (2019) 40:584–97. doi: 10.1016/j.it.2019.04.013
7. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
8. Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and Glutathionylation Balance Cytoskeletal Dynamics in Neutrophil Extracellular Trap Formation. J Cell Biol (2017) 216:4073–90. doi: 10.1083/jcb.201611168
9. Amini P, Stojkov D, Felser A, Jackson CB, Courage C, Schaller A, et al. Neutrophil Extracellular Trap Formation Requires OPA1-Dependent Glycolytic ATP Production. Nat Commun (2018) 9:2958. doi: 10.1038/s41467-018-05387-y
10. Dahlgren C, Karlsson A, Bylund J. Intracellular Neutrophil Oxidants: From Laboratory Curiosity to Clinical Reality. J Immunol (2019) 202:3127–34. doi: 10.4049/jimmunol.1900235
11. Faurschou M, Sorensen OE, Johnsen AH, Askaa J, Borregaard N. Defensin-Rich Granules of Human Neutrophils: Characterization of Secretory Properties. Biochim Biophys Acta (2002) 1591:29–35. doi: 10.1016/S0167-4889(02)00243-4
12. Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil Granules: A Library of Innate Immunity Proteins. Trends Immunol (2007) 8:340–5. doi: 10.1016/j.it.2007.06.002
13. Lawrence SM, Corriden R, Nizet V. The Ontogeny of a Neutrophil: Mechanisms of Granulopoiesis and Homeostasis. Microbiol Mol Biol Rev (2018) 82:e00057–00017. doi: 10.1128/MMBR.00057-17
14. Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: Molecules,Ffunctions and Pathophysiological Aspects. Lab Invest (2000) 5:617–53. doi: 10.1038/labinvest.3780067
15. Hatanaka E, Shimomi FM, Curi R, Campa A. Sodium Chloride Inhibits Cytokine Production by Lipopolysaccharide-Stimulated Human Neutrophils and Mononuclear Cells. Shock (2007) 1:32–5. doi: 10.1097/01.shk.0000238061.69579.a5
16. Selvatici R, Falzarano S, Mollica A, Spisani S. Signal Transduction Pathways Triggered by Selective Formylpeptide Analogues in Human Neutrophils. Eur J Pharmacol (2006) 534:1–11. doi: 10.1016/j.ejphar.2006.01.034
17. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
18. Clark SR, Ma AC, Tavener SA, Mcdonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
19. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable Neutrophils Release Mitochondrial DNA to Form Neutrophil Extracellular Traps. Cell Death Differ (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
20. Mcilroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA Neutrophil Extracellular Traps are Formed After Trauma and Subsequent Surgery. J Crit Care (2014) 29:1133.e1131–1135. doi: 10.1016/j.jcrc.2014.07.013
21. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol (2015) 67:3190–200. doi: 10.1002/art.39296
22. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, et al. NET Formation Can Occur Independently of RIPK3 and MLKL Signaling. Eur J Immunol (2016) 46:178–84. doi: 10.1002/eji.201545615
23. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized Mitochondrial Nucleoids Released by Neutrophils Drive Type I Interferon Production in Human Lupus. J Exp Med (2016) 213:697–713. doi: 10.1084/jem.20151876
24. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
25. Ingelsson B, Soderberg D, Strid T, Soderberg A, Bergh AC, Loitto V, et al. Lymphocytes Eject Interferogenic Mitochondrial DNA Webs in Response to Cpg and Non-Cpg Oligodeoxynucleotides of Class C. Proc Natl Acad Sci USA (2018) 115:E478–87. doi: 10.1073/pnas.1711950115
26. Cristinziano L, Modestino L, Loffredo S, Varricchi G, Braile M, Ferrara AL, et al. Anaplastic Thyroid Cancer Cells Induce the Release of Mitochondrial Extracellular DNA Traps by Viable Neutrophils. J Immunol (2020) 204:1362–72. doi: 10.4049/jimmunol.1900543
27. Klopf J, Brostjan C, Eilenberg W, Neumayer C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int J Mol Sci (2021) 22:559. doi: 10.3390/ijms22020559
28. Von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-Independent Antimicrobial Activity of Mast Cells by Means of Extracellular Trap Formation. Blood (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
29. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-Like Release of Mitochondrial DNA by Eosinophils Contributes to Antibacterial Defense. Nat Med (2008) 14:949–53. doi: 10.1038/nm.1855
30. Chow OA, Von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe (2010) 8:445–54. doi: 10.1016/j.chom.2010.10.005
31. Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon HU. Eosinophil Extracellular DNA Traps in Skin Diseases. J Allergy Clin Immunol (2011) 127:194–9. doi: 10.1016/j.jaci.2010.11.002
32. Morshed M, Yousefi S, Stockle C, Simon HU, Simon D. Thymic Stromal Lymphopoietin Stimulates the Formation of Eosinophil Extracellular Traps. Allergy (2012) 67:1127–37. doi: 10.1111/j.1398-9995.2012.02868.x
33. Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H, et al. NADPH Oxidase-Independent Formation of Extracellular DNA Traps by Basophils. J Immunol (2014) 192:5314–23. doi: 10.4049/jimmunol.1303418
34. Yousefi S, Morshed M, Amini P, Stojkov D, Simon D, Von Gunten S, et al. Basophils Exhibit Antibacterial Activity Through Extracellular Trap Formation. Allergy (2015) 70:1184–8. doi: 10.1111/all.12662
35. Germic N, Fettrelet T, Stojkov D, Hosseini A, Horn MP, Karaulov A, et al. The Release Kinetics of Eosinophil Peroxidase and Mitochondrial DNA Is Different in Association With Eosinophil Extracellular Trap Formation. Cells (2021) 10:306. doi: 10.3390/cells10020306
36. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J Thromb Haemost (2014) 12:2074–88. doi: 10.1111/jth.12710
37. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular Mechanisms of NET Formation and Degradation Revealed by Intravital Imaging in the Liver Vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
38. Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse Stimuli Engage Different Neutrophil Extracellular Trap Pathways. Elife (2017) 6:e24437. doi: 10.7554/eLife.24437
39. Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, et al. Neutrophil Extracellular Traps Drive Inflammatory Pathogenesis in Malaria. Sci Immunol (2019) 4:eaaw0336. doi: 10.1126/sciimmunol.aaw0336
40. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
41. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH Oxidase and Myeloperoxidase in Neutrophil Extracellular Trap Formation Differ Depending on the Stimulus. J Leukoc Biol (2012) 92:841–9. doi: 10.1189/jlb.1211601
42. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights Into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol (2016) 7:302. doi: 10.3389/fimmu.2016.00302
43. Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology (2015) 145:213–24. doi: 10.1111/imm.12437
44. Gibellini L, De Biasi S, Paolini A, Borella R, Boraldi F, Mattioli M, et al. Altered Bioenergetics and Mitochondrial Dysfunction of Monocytes in Patients With COVID-19 Pneumonia. EMBO Mol Med (2020) 12:e13001. doi: 10.15252/emmm.202013001
45. Fox CJ, Hammerman PS, Thompson CB. Fuel Feeds Function: Energy Metabolism and the T-Cell Response. Nat Rev Immunol (2005) 5:844–52. doi: 10.1038/nri1710
46. Bordbar A, Mo ML, Nakayasu ES, Schrimpe-Rutledge AC, Kim YM, Metz TO, et al. Model-Driven Multi-Omic Data Analysis Elucidates Metabolic Immunomodulators of Macrophage Activation. Mol Syst Biol (2012) 8:558. doi: 10.1038/msb.2012.21
47. O’Neill LA, Hardie DG. Metabolism of Inflammation Limited by AMPK and Pseudo-Starvation. Nature (2013) 493:346–55. doi: 10.1038/nature11862
48. Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
49. Borregaard N, Herlin T. Energy Metabolism of Human Neutrophils During Phagocytosis. J Clin Invest (1982) 70:550–7. doi: 10.1172/JCI110647
50. Van Raam BJ, Sluiter W, De Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial Membrane Potential in Human Neutrophils is Maintained by Complex III Activity in the Absence of Supercomplex Organisation. PloS One (2008) 3:e2013. doi: 10.1371/journal.pone.0002013
51. Zucker-Franklin D. Electron Microscopic Studies of Human Granulocytes: Structural Variations Related to Function. Semin Hematol (1968) 5:109–33.
52. Bainton DF, Ullyot JL, Farquhar MG. The Development of Neutrophilic Polymorphonuclear Leukocytes in Human Bone Marrow. J Exp Med (1971) 134:907–34. doi: 10.1084/jem.134.4.907
53. Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The Mitochondrial Network of Human Neutrophils: Role in Chemotaxis, Phagocytosis, Respiratory Burst Activation, and Commitment to Apoptosis. J Immunol (2003) 170:1964–72. doi: 10.4049/jimmunol.170.4.1964
54. Karinch AM, Pan M, Lin CM, Strange R, Souba WW. Glutamine Metabolism in Sepsis and Infection. J Nutr (2001) 131:2535S–8S. discussion 2550S-2531S. doi: 10.1093/jn/131.9.2535S
55. Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, et al. Mitochondrial Rhomboid PARL Regulates Cytochrome C Release During Apoptosis via OPA1-Dependent Cristae Remodeling. Cell (2006) 126:163–75. doi: 10.1016/j.cell.2006.06.021
56. Frezza C, Cipolat S, Martins De Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 Controls Apoptotic Cristae Remodeling Independently From Mitochondrial Fusion. Cell (2006) 126:177–89. doi: 10.1016/j.cell.2006.06.025
57. Han J, Thompson-Lowrey AJ, Reiss A, Mayorov V, Jia H, Biousse V, et al. OPA1 Mutations and Mitochondrial DNA Haplotypes in Autosomal Dominant Optic Atrophy. Genet Med (2006) 8:217–25. doi: 10.1097/01.gim.0000214299.61930.c0
58. Azevedo EP, Rochael NC, Guimaraes-Costa AB, De Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A Metabolic Shift Toward Pentose Phosphate Pathway is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-Induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
59. Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, Sigrist N, Sadek H, et al. AK2 Deficiency Compromises the Mitochondrial Energy Metabolism Required for Differentiation of Human Neutrophil and Lymphoid Lineages. Cell Death Dis (2015) 6:e1856. doi: 10.1038/cddis.2015.211
60. Jones R, Mcdonald KE, Willson JA, Ghesquiere B, Sammut D, Daniel E, et al. Mutations in Succinate Dehydrogenase B (SDHB) Enhance Neutrophil Survival Independent of HIF-1alpha Expression. Blood (2016) 127:2641–4. doi: 10.1182/blood-2016-02-696922
61. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity (2017) 47:466–80.e465. doi: 10.1016/j.immuni.2017.08.005
62. Richer BC, Salei N, Laskay T, Seeger K. Changes in Neutrophil Metabolism Upon Activation and Aging. Inflammation (2018) 41:710–21. doi: 10.1007/s10753-017-0725-z
63. Sbarra AJ, Karnovsky ML. The Biochemical Basis of Phagocytosis. I. Metabolic Changes During the Ingestion of Particles by Polymorphonuclear Leukocytes. J Biol Chem (1959) 234:1355–62. doi: 10.1016/S0021-9258(18)70011-2
64. Jeon JH, Hong CW, Kim EY, Lee JM. Current Understanding on the Metabolism of Neutrophils. Immune Netw (2020) 20:e46. doi: 10.4110/in.2020.20.e46
65. Weisdorf DJ, Craddock PR, Jacob HS. Granulocytes Utilize Different Energy Sources for Movement and Phagocytosis. Inflammation (1982) 6:245–56. doi: 10.1007/BF00916406
66. Alba-Loureiro TC, Hirabara SM, Mendonca JR, Curi R, Pithon-Curi TC. Diabetes Causes Marked Changes in Function and Metabolism of Rat Neutrophils. J Endocrinol (2006) 188:295–303. doi: 10.1677/joe.1.06438
67. Gaber T, Strehl C, Buttgereit F. Metabolic Regulation of Inflammation. Nat Rev Rheumatol (2017) 13:267–79. doi: 10.1038/nrrheum.2017.37
68. Curi TC, De Melo MP, De Azevedo RB, Curi R. Glutamine Utilization by Rat Neutrophils. Biochem Soc Trans (1997) 25:249S. doi: 10.1042/bst025249s
69. Curi R, Newsholme P, Pithon-Curi TC, Pires-De-Melo M, Garcia C, Homem-De-Bittencourt Junior PI, et al. Metabolic Fate of Glutamine in Lymphocytes, Macrophages and Neutrophils. Braz J Med Biol Res (1999) 32:15–21. doi: 10.1590/S0100-879X1999000100002
70. Newsholme P. Why is L-Glutamine Metabolism Important to Cells of the Immune System in Health, Postinjury, Surgery or Infection? J Nutr (2001) 131:2515S–22S. discussion 2523S-2514S. doi: 10.1093/jn/131.9.2515S
71. Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, et al. Involvement of a Ferroprotein Sensor in Hypoxia-Mediated Inhibition of Neutrophil Apoptosis. Blood (2002) 100:3008–16. doi: 10.1182/blood-2002-02-0454
72. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-Induced Neutrophil Survival is Mediated by HIF-1alpha-Dependent NF-Kappab Activity. J Exp Med (2005) 201:105–15. doi: 10.1084/jem.20040624
73. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-Mediated Selection of Cells With Diminished Apoptotic Potential in Solid Tumours. Nature (1996) 379:88–91. doi: 10.1038/379088a0
74. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is Essential for Myeloid Cell-Mediated Inflammation. Cell (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
75. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. Mtor- and HIF-1alpha-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
76. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J Biol Chem (1994) 269:23757–63. doi: 10.1016/S0021-9258(17)31580-6
77. Halligan DN, Murphy SJ, Taylor CT. The Hypoxia-Inducible Factor (HIF) Couples Immunity With Metabolism. Semin Immunol (2016) 28:469–77. doi: 10.1016/j.smim.2016.09.004
78. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian Target of Rapamycin Regulates Neutrophil Extracellular Trap Formation via Induction of Hypoxia-Inducible Factor 1 Alpha. Blood (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
79. Vollger L, Akong-Moore K, Cox L, Goldmann O, Wang Y, Schafer ST, et al. Iron-Chelating Agent Desferrioxamine Stimulates Formation of Neutrophil Extracellular Traps (Nets) in Human Blood-Derived Neutrophils. Biosci Rep (2016) 36:e00333. doi: 10.1042/BSR20160031
80. Branitzki-Heinemann K, Mollerherm H, Vollger L, Husein DM, De Buhr N, Blodkamp S, et al. Formation of Neutrophil Extracellular Traps Under Low Oxygen Level. Front Immunol (2016) 7:518. doi: 10.3389/fimmu.2016.00518
81. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-Inducible Factor 2alpha Regulates Key Neutrophil Functions in Humans, Mice, and Zebrafish. Blood (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
82. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The Return of Metabolism: Biochemistry and Physiology of the Pentose Phosphate Pathway. Biol Rev Camb Philos Soc (2015) 90:927–63. doi: 10.1111/brv.12140
83. Tibrewal S, Ivanir Y, Sarkar J, Nayeb-Hashemi N, Bouchard CS, Kim E, et al. Hyperosmolar Stress Induces Neutrophil Extracellular Trap Formation: Implications for Dry Eye Disease. Invest Ophthalmol Vis Sci (2014) 55:7961–9. doi: 10.1167/iovs.14-15332
84. Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, Bonora B, et al. Netosis Is Induced by High Glucose and Associated With Type 2 Diabetes. Acta Diabetol (2015) 52:497–503. doi: 10.1007/s00592-014-0676-x
85. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo Netosis, Which Impairs Wound Healing. Nat Med (2015) 21:815–9. doi: 10.1038/nm.3887
86. Park JH, Kim JE, Gu JY, Yoo HJ, Park SH, Kim YI, et al. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp Clin Endocrinol Diabetes (2016) 124:557–61. doi: 10.1055/s-0042-101792
87. Barliya T, Dardik R, Nisgav Y, Dachbash M, Gaton D, Kenet G, et al. Possible Involvement of Netosis in Inflammatory Processes in the Eye: Evidence From a Small Cohort of Patients. Mol Vis (2017) 23:922–32.
88. Wang L, Zhou X, Yin Y, Mai Y, Wang D, Zhang X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front Immunol (2018) 9:3076. doi: 10.3389/fimmu.2018.03076
89. D’Abbondanza M, Martorelli EE, Ricci MA, De Vuono S, Migliola EN, Godino C, et al. Increased Plasmatic Nets by-Products in Patients in Severe Obesity. Sci Rep (2019) 9:14678. doi: 10.1038/s41598-019-51220-x
90. Cichon I, Ortmann W, Kolaczkowska E. Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (Nets) Differ in Obesity and Systemic Inflammation. Int J Mol Sci (2021) 22:7718. doi: 10.3390/ijms22147718
91. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-Elicited Neutrophils Engage Mitochondrial Metabolism to Circumvent Nutrient Limitations and Maintain Immune Suppression. Nat Commun (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
92. Hsu BE, Tabaries S, Johnson RM, Andrzejewski S, Senecal J, Lehuede C, et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility That Facilitates Breast Cancer Liver Metastasis. Cell Rep (2019) 27:3902–15.e3906. doi: 10.1016/j.celrep.2019.05.091
93. Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res (2019) 79:5626–39. doi: 10.1158/0008-5472.CAN-19-0800
94. Ancey PB, Contat C, Boivin G, Sabatino S, Pascual J, Zangger N, et al. GLUT1 Expression in Tumor-Associated Neutrophils Promotes Lung Cancer Growth and Resistance to Radiotherapy. Cancer Res (2021) 81:2345–57. doi: 10.1158/0008-5472.CAN-20-2870
95. Rogers T, DeBerardinis RJ. Metabolic Plasticity of Neutrophils: Relevance to Pathogen Responses and Cancer. Trends Cancer (2021) 7:700–13. doi: 10.1016/j.trecan.2021.04.007
96. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated From Patients With Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I Ifns. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
97. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of Neutrophil Extracellular Trap Degradation Is Associated With Lupus Nephritis. Proc Natl Acad Sci USA (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
98. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
99. Knight JS, Kaplan MJ. Lupus Neutrophils: ‘NET’ Gain in Understanding Lupus Pathogenesis. Curr Opin Rheumatol (2012) 24:441–50. doi: 10.1097/BOR.0b013e3283546703
100. Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged Bioenergetics and Defective Redox Capacity in T Lymphocytes and Neutrophils are Related to Cellular Dysfunction and Increased Oxidative Stress in Patients With Active Systemic Lupus Erythematosus. Clin Dev Immunol (2012) 2012:548516. doi: 10.1155/2012/548516
101. Mahajan A, Herrmann M, Munoz LE. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front Immunol (2016) 7:35. doi: 10.3389/fimmu.2016.00035
102. Fresneda Alarcon M, Mclaren Z, Wright HL. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front Immunol (2021) 12:649693. doi: 10.3389/fimmu.2021.649693
103. Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial Fluid Neutrophils Transcribe and Express Class II Major Histocompatibility Complex Molecules in Rheumatoid Arthritis. Arthritis Rheum (2003) 48:2796–806. doi: 10.1002/art.11253
104. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. Nets are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci Transl Med (2013) 5:178ra140. doi: 10.1126/scitranslmed.3005580
105. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced Neutrophil Extracellular Trap Generation in Rheumatoid Arthritis: Analysis of Underlying Signal Transduction Pathways and Potential Diagnostic Utility. Arthritis Res Ther (2014) 16:R122. doi: 10.1186/ar4579
106. Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil Extracellular Traps Mediate Articular Cartilage Damage and Enhance Cartilage Component Immunogenicity in Rheumatoid Arthritis. JCI Insight (2020) 5:e139388. doi: 10.1172/jci.insight.139388
107. Gray GR, Stamatoyannopoulos G, Naiman SC, Kliman MR, Klebanoff SJ, Austin T, et al. Neutrophil Dysfunction, Chronic Granulomatous Disease, and non-Spherocytic Haemolytic Anaemia Caused by Complete Deficiency of Glucose-6-Phosphate Dehydrogenase. Lancet (1973) 2:530–4. doi: 10.1016/S0140-6736(73)92350-7
108. Ardati KO, Bajakian KM, Tabbara KS. Effect of Glucose-6-Phosphate Dehydrogenase Deficiency on Neutrophil Function. Acta Haematol (1997) 97:211–5. doi: 10.1159/000203685
109. Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, et al. Severe Glucose-6-Phosphate Dehydrogenase Deficiency Leads to Susceptibility to Infection and Absent Netosis. J Allergy Clin Immunol (2017) 139:212–219 e213. doi: 10.1016/j.jaci.2016.04.041
110. Lodhi IJ, Wei X, Yin L, Feng C, Adak S, Abou-Ezzi G, et al. Peroxisomal Lipid Synthesis Regulates Inflammation by Sustaining Neutrophil Membrane Phospholipid Composition and Viability. Cell Metab (2015) 21:51–64. doi: 10.1016/j.cmet.2014.12.002
111. Czaikoski PG, Mota JM, Nascimento DC, Sonego F, Castanheira FV, Melo PH, et al. Neutrophil Extracellular Traps Induce Organ Damage During Experimental and Clinical Sepsis. PloS One (2016) 11:e0148142. doi: 10.1371/journal.pone.0148142
112. Liu Z, Bone N, Jiang S, Park DW, Tadie JM, Deshane J, et al. AMP-Activated Protein Kinase and Glycogen Synthase Kinase 3β Modulate the Severity of Sepsis-Induced Lung Injury. Mol Med (2016) 21:937–50. doi: 10.2119/molmed.2015.00198
113. Liu Z, Bone N, Jiang S, Park DW, Tadie JM, Deshane J, et al. AMP-Activated Protein Kinase and Glycogen Synthase Kinase 3beta Modulate the Severity of Sepsis-Induced Lung Injury. Mol Med (2016) 21:937–50. doi: 10.2119/molmed.2015.00198
114. Nalos M, Parnell G, Robergs R, Booth D, Mclean AS, Tang BM. Transcriptional Reprogramming of Metabolic Pathways in Critically Ill Patients. Intensive Care Med Exp (2016) 4:21. doi: 10.1186/s40635-016-0094-1
115. Li X, Kondo Y, Bao Y, Staudenmaier L, Lee A, Zhang J, et al. Systemic Adenosine Triphosphate Impairs Neutrophil Chemotaxis and Host Defense in Sepsis. Crit Care Med (2017) 45:e97–e104. doi: 10.1097/CCM.0000000000002052
116. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation During Sepsis. Am J Respir Crit Care Med (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
117. Colon DF, Wanderley CW, Franchin M, Silva CM, Hiroki CH, Castanheira FVS, et al. Neutrophil Extracellular Traps (Nets) Exacerbate Severity of Infant Sepsis. Crit Care (2019) 23:113. doi: 10.1186/s13054-019-2407-8
118. Kumar V. Immunometabolism: Another Road to Sepsis and its Therapeutic Targeting. Inflammation (2019) 42:765–88. doi: 10.1007/s10753-018-0939-8
119. Sun S, Duan Z, Wang X, Chu C, Yang C, Chen F, et al. Neutrophil Extracellular Traps Impair Intestinal Barrier Functions in Sepsis by Regulating TLR9-Mediated Endoplasmic Reticulum Stress Pathway. Cell Death Dis (2021) 12:606. doi: 10.1038/s41419-021-03896-1
120. Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, et al. CXCR2 Mediates NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation in Cystic Fibrosis Airway Inflammation. Nat Med (2010) 16:1018–23. doi: 10.1038/nm.2209
121. Cifani N, Pompili B, Anile M, Patella M, Diso D, Venuta F, et al. Reactive-Oxygen-Species-Mediated P. Aeruginosa Killing Is Functional in Human Cystic Fibrosis Macrophages. PloS One (2013) 8:e71717. doi: 10.1371/journal.pone.0071717
122. Laval J, Touhami J, Herzenberg LA, Conrad C, Taylor N, Battini JL, et al. Metabolic Adaptation of Neutrophils in Cystic Fibrosis Airways Involves Distinct Shifts in Nutrient Transporter Expression. J Immunol (2013) 190:6043–50. doi: 10.4049/jimmunol.1201755
123. Gray RD, Hardisty G, Regan KH, Smith M, Robb CT, Duffin R, et al. Delayed Neutrophil Apoptosis Enhances NET Formation in Cystic Fibrosis. Thorax (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
124. McElvaney OJ, Mcevoy NL, Mcelvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med (2020) 202:812–21. doi: 10.1164/rccm.202005-1583OC
125. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
126. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue Damage From Neutrophil-Induced Oxidative Stress in COVID-19. Nat Rev Immunol (2020) 20:515–6. doi: 10.1038/s41577-020-0407-1
127. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136:1169–79. doi: 10.1182/blood.2020007008
128. Schonrich G, Raftery MJ, Samstag Y. Devilishly Radical Network in COVID-19: Oxidative Stress, Neutrophil Extracellular Traps (Nets), and T Cell Suppression. Adv Biol Regul (2020) 77:100741. doi: 10.1016/j.jbior.2020.100741
129. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5:e138999. doi: 10.1172/jci.insight.138999
130. Megens RT, Vijayan S, Lievens D, Doring Y, Van Zandvoort MA, Grommes J, et al. Presence of Luminal Neutrophil Extracellular Traps in Atherosclerosis. Thromb Haemost (2012) 107:597–8. doi: 10.1160/TH11-09-0650
131. Awasthi D, Nagarkoti S, Kumar A, Dubey M, Singh AK, Pathak P, et al. Oxidized LDL Induced Extracellular Trap Formation in Human Neutrophils via TLR-PKC-IRAK-MAPK and NADPH-Oxidase Activation. Free Radic Biol Med (2016) 93:190–203. doi: 10.1016/j.freeradbiomed.2016.01.004
132. Mostafa MN, Osama M. The Implications of Neutrophil Extracellular Traps in the Pathophysiology of Atherosclerosis and Atherothrombosis. Exp Biol Med (Maywood) (2020) 245:1376–84. doi: 10.1177/1535370220945989
133. Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional Characterization of Mitochondria in Neutrophils: A Role Restricted to Apoptosis. Cell Death Differ (2004) 11:143–53. doi: 10.1038/sj.cdd.4401320
134. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil Metabolic Shift During Their Lifecycle: Impact on Their Survival and Activation. Int J Mol Sci (2019) 21:287. doi: 10.3390/ijms21010287
135. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, et al. Glucose Transporter Expression on the Plasma Membrane of Resting and Activated White Blood Cells. Eur J Clin Invest (2007) 37:282–90. doi: 10.1111/j.1365-2362.2007.01786.x
136. Huang JB, Kindzelskii AL, Petty HR. Hexokinase Translocation During Neutrophil Activation, Chemotaxis, and Phagocytosis: Disruption by Cytochalasin D, Dexamethasone, and Indomethacin. Cell Immunol (2002) 218:95–106. doi: 10.1016/S0008-8749(02)00582-8
137. Cooper MR, Dechatelet LR, Mccall CE, Lavia MF, Spurr CL, Baehner RL. Complete Deficiency of Leukocyte Glucose-6-Phosphate Dehydrogenase With Defective Bactericidal Activity. J Clin Invest (1972) 51:769–78. doi: 10.1172/JCI106871
138. Perner A, Nielsen SE, Rask-Madsen J. High Glucose Impairs Superoxide Production From Isolated Blood Neutrophils. Intensive Care Med (2003) 29:642–5. doi: 10.1007/s00134-002-1628-4
139. Robinson JM, Karnovsky ML, Karnovsky MJ. Glycogen Accumulation in Polymorphonuclear Leukocytes, and Other Intracellular Alterations That Occur During Inflammation. J Cell Biol (1982) 95:933–42. doi: 10.1083/jcb.95.3.933
140. Sadiku P, Willson JA, Ryan EM, Sammut D, Coelho P, Watts ER, et al. Neutrophils Fuel Effective Immune Responses Through Gluconeogenesis and Glycogenesis. Cell Metab (2021) 33:411–23.e414. doi: 10.1016/j.cmet.2020.11.016
141. Jun HS, Lee YM, Cheung YY, Mcdermott DH, Murphy PM, De Ravin SS, et al. Lack of Glucose Recycling Between Endoplasmic Reticulum and Cytoplasm Underlies Cellular Dysfunction in Glucose-6-Phosphatase-Beta-Deficient Neutrophils in a Congenital Neutropenia Syndrome. Blood (2010) 116:2783–92. doi: 10.1182/blood-2009-12-258491
142. Wachstein M. The Distribution of Histochemically Demonstrable Glycogen in Human Blood and Bone Marrow Cells. Blood (1949) 4:54–9. doi: 10.1182/blood.V4.1.54.54
143. Scott RB. Glycogen in Human Peripheral Blood Leukocytes. I. Characteristics of the Synthesis and Turnover of Glycogen in Vitro. J Clin Invest (1968) 47:344–52. doi: 10.1172/JCI105730
144. Curi R, Levada-Pires AC, Silva EBD, Poma SO, Zambonatto RF, Domenech P, et al. The Critical Role of Cell Metabolism for Essential Neutrophil Functions. Cell Physiol Biochem (2020) 54:629–47. doi: 10.33594/000000245
145. Borregaard N, Juhl H. Activation of the Glycogenolytic Cascade in Human Polymorphonuclear Leucocytes by Different Phagocytic Stimuli. Eur J Clin Invest (1981) 11:257–63. doi: 10.1111/j.1365-2362.1981.tb02114.x
146. Rath L, Bui MV, Ellis J, Carey J, Baker J, Taylor L, et al. Fast and Safe: Optimising Multiple Sclerosis Infusions During COVID-19 Pandemic. Mult Scler Relat Disord (2021) 47:102642. doi: 10.1016/j.msard.2020.102642
147. Veiga-Da-Cunha M, Gerin I, Chen YT, De Barsy T, De Lonlay P, Dionisi-Vici C, et al. A Gene on Chromosome 11q23 Coding for a Putative Glucose- 6-Phosphate Translocase Is Mutated in Glycogen-Storage Disease Types Ib and Ic. Am J Hum Genet (1998) 63:976–83. doi: 10.1086/302068
148. Hayee B, Antonopoulos A, Murphy EJ, Rahman FZ, Sewell G, Smith BN, et al. G6PC3 Mutations Are Associated With a Major Defect of Glycosylation: A Novel Mechanism for Neutrophil Dysfunction. Glycobiology (2011) 21:914–24. doi: 10.1093/glycob/cwr023
149. Ambruso DR, Mccabe ER, Anderson D, Beaudet A, Ballas LM, Brandt IK, et al. Infectious and Bleeding Complications in Patients With Glycogenosis Ib. Am J Dis Child (1985) 139:691–7. doi: 10.1001/archpedi.1985.02140090053027
150. Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY. Molecular Mechanisms of Neutrophil Dysfunction in Glycogen Storage Disease Type Ib. Blood (2014) 123:2843–53. doi: 10.1182/blood-2013-05-502435
151. Kuijpers TW, Maianski NA, Tool AT, Smit GP, Rake JP, Roos D, et al. Apoptotic Neutrophils in the Circulation of Patients With Glycogen Storage Disease Type 1b (GSD1b). Blood (2003) 101:5021–4. doi: 10.1182/blood-2002-10-3128
152. Wortmann SB, Van Hove JLK, Derks TGJ, Chevalier N, Knight V, Koller A, et al. Treating Neutropenia and Neutrophil Dysfunction in Glycogen Storage Disease Type Ib With an SGLT2 Inhibitor. Blood (2020) 136:1033–43. doi: 10.1182/blood.2019004465
153. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients (2018) 10:1564. doi: 10.3390/nu10111564
154. Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras Decouples Glucose and Glutamine Metabolism to Support Cancer Cell Growth. Mol Syst Biol (2011) 7:523. doi: 10.1038/msb.2011.56
155. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab (2012) 15:110–21. doi: 10.1016/j.cmet.2011.12.009
156. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-Driven Oxidative Phosphorylation Is a Major ATP Source in Transformed Mammalian Cells in Both Normoxia and Hypoxia. Mol Syst Biol (2013) 9:712. doi: 10.1038/msb.2013.65
157. Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, et al. Nicotinamide Adenine Dinucleotide Is Transported Into Mammalian Mitochondria. Elife (2018) 7:e33246. doi: 10.7554/eLife.33246
158. Pithon-Curi TC, Levada AC, Lopes LR, Doi SQ, Curi R. Glutamine Plays a Role in Superoxide Production and the Expression of P47phox, P22phox and Gp91phox in Rat Neutrophils. Clin Sci (Lond) (2002) 103:403–8. doi: 10.1042/cs1030403
159. Furukawa S, Saito H, Fukatsu K, Hashiguchi Y, Inaba T, Lin MT, et al. Glutamine-Enhanced Bacterial Killing by Neutrophils From Postoperative Patients. Nutrition (1997) 13:863–9. doi: 10.1016/S0899-9007(97)00271-2
160. Furukawa S, Saito H, Inoue T, Matsuda T, Fukatsu K, Han I, et al. Supplemental Glutamine Augments Phagocytosis and Reactive Oxygen Intermediate Production by Neutrophils and Monocytes From Postoperative Patients in Vitro. Nutrition (2000) 16:323–9. doi: 10.1016/S0899-9007(00)00228-8
161. Khaw KT, Friesen MD, Riboli E, Luben R, Wareham N. Plasma Phospholipid Fatty Acid Concentration and Incident Coronary Heart Disease in Men and Women: The EPIC-Norfolk Prospective Study. PloS Med (2012) 9:e1001255. doi: 10.1371/journal.pmed.1001255
162. Rodrigues HG, Takeo Sato F, Curi R, Vinolo MAR. Fatty Acids as Modulators of Neutrophil Recruitment, Function and Survival. Eur J Pharmacol (2016) 785:50–8. doi: 10.1016/j.ejphar.2015.03.098
163. Burdge GC, Calder PC. Introduction to Fatty Acids and Lipids. World Rev Nutr Diet (2015) 112:1–16. doi: 10.1159/000365423
164. Martins De Lima T, Gorjao R, Hatanaka E, Cury-Boaventura MF, Portioli Silva EP, Procopio J, et al. Mechanisms by Which Fatty Acids Regulate Leucocyte Function. Clin Sci (Lond) (2007) 113:65–77. doi: 10.1042/CS20070006
165. Fan Z, Ley K. Developing Neutrophils Must Eat … Themselves! Immunity (2017) 47:393–5. doi: 10.1016/j.immuni.2017.08.013
166. Randle PJ, Garland PB, Hales CN, Newsholme EA. The Glucose Fatty-Acid Cycle. Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet (1963) 1:785–9. doi: 10.1016/S0140-6736(63)91500-9
167. Longo N, Amat Di San Filippo C, Pasquali M. Disorders of Carnitine Transport and the Carnitine Cycle. Am J Med Genet C Semin Med Genet (2006) 142C:77–85. doi: 10.1002/ajmg.c.30087
168. Houten SM, Wanders RJA, Ranea-Robles P. Metabolic Interactions Between Peroxisomes and Mitochondria With a Special Focus on Acylcarnitine Metabolism. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165720. doi: 10.1016/j.bbadis.2020.165720
169. Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, et al. Untangling “Netosis” From Nets. Eur J Immunol (2019) 49:221–7. doi: 10.1002/eji.201747053
170. Kirchner T, Moller S, Klinger M, Solbach W, Laskay T, Behnen M. The Impact of Various Reactive Oxygen Species on the Formation of Neutrophil Extracellular Traps. Mediators Inflamm (2012) 2012:849136. doi: 10.1155/2012/849136
171. Stoiber W, Obermayer A, Steinbacher P, Krautgartner WD. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (Ets) in Humans. Biomolecules (2015) 5:702–23. doi: 10.3390/biom5020702
172. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET Formation by Gene Therapy in CGD Controls Aspergillosis. Blood (2009) 114:2619–22. doi: 10.1182/blood-2009-05-221606
173. Insuela DBR, Azevedo CT, Coutinho DS, Magalhaes NS, Ferrero MR, Ferreira TPT, et al. Glucagon Reduces Airway Hyperreactivity, Inflammation, and Remodeling Induced by Ovalbumin. Sci Rep (2019) 9:6478. doi: 10.1038/s41598-019-42981-6
174. Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, Satyamoorthy K. High Glucose Modulates IL-6 Mediated Immune Homeostasis Through Impeding Neutrophil Extracellular Trap Formation. FEBS Lett (2013) 587:2241–6. doi: 10.1016/j.febslet.2013.05.053
175. Arampatzioglou A, Papazoglou D, Konstantinidis T, Chrysanthopoulou A, Mitsios A, Angelidou I, et al. Clarithromycin Enhances the Antibacterial Activity and Wound Healing Capacity in Type 2 Diabetes Mellitus by Increasing LL-37 Load on Neutrophil Extracellular Traps. Front Immunol (2018) 9:2064. doi: 10.3389/fimmu.2018.02064
176. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis Dependent Lactate Formation in Neutrophils: A Metabolic Link Between NOX-Dependent and Independent Netosis. Biochim Biophys Acta Mol Basis Dis (2019) 1865:165542. doi: 10.1016/j.bbadis.2019.165542
177. Riyapa D, Rinchai D, Muangsombut V, Wuttinontananchai C, Toufiq M, Chaussabel D, et al. Transketolase and Vitamin B1 Influence on ROS-Dependent Neutrophil Extracellular Traps (Nets) Formation. PloS One (2019) 14:e0221016. doi: 10.1371/journal.pone.0221016
178. Alarcon P, Manosalva C, Conejeros I, Carretta MD, Munoz-Caro T, Silva LMR, et al. D(-) Lactic Acid-Induced Adhesion of Bovine Neutrophils Onto Endothelial Cells is Dependent on Neutrophils Extracellular Traps Formation and CD11b Expression. Front Immunol (2017) 8:975. doi: 10.3389/fimmu.2017.00975
179. Sofoluwe A, Bacchetta M, Badaoui M, Kwak BR, Chanson M. ATP Amplifies NADPH-Dependent and -Independent Neutrophil Extracellular Trap Formation. Sci Rep (2019) 9:16556. doi: 10.1038/s41598-019-53058-9
180. Quiroga J, Alarcon P, Manosalva C, Taubert A, Hermosilla C, Hidalgo MA, et al. Mitochondria-Derived ATP Participates in the Formation of Neutrophil Extracellular Traps Induced by Platelet-Activating Factor Through Purinergic Signaling in Cows. Dev Comp Immunol (2020) 113:103768. doi: 10.1016/j.dci.2020.103768
181. Yousefi S, Simon HU. Netosis - Does it Really Represent Nature’s “Suicide Bomber”? Front Immunol (2016) 7:328. doi: 10.3389/fimmu.2016.00328
182. Yousefi S, Simon D, Stojkov D, Karsonova A, Karaulov A, Simon HU. In Vivo Evidence for Extracellular DNA Trap Formation. Cell Death Dis (2020) 11:300. doi: 10.1038/s41419-020-2497-x
183. Tackenberg H, Moller S, Filippi MD, Laskay T. The Small Gtpase Cdc42 Negatively Regulates the Formation of Neutrophil Extracellular Traps by Engaging Mitochondria. Front Immunol (2021) 12:564720. doi: 10.3389/fimmu.2021.564720
184. Naffah De Souza C, Breda LCD, Khan MA, De Almeida SR, Camara NOS, Sweezey N, et al. Alkaline Ph Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front Immunol (2017) 8:1849. doi: 10.3389/fimmu.2017.01849
185. Khan MA, Philip LM, Cheung G, Vadakepeedika S, Grasemann H, Sweezey N, et al. Regulating Netosis: Increasing Ph Promotes NADPH Oxidase-Dependent Netosis. Front Med (Lausanne) (2018) 5:19. doi: 10.3389/fmed.2018.00019
186. Alarcon P, Manosalva C, Quiroga J, Belmar I, Alvarez K, Diaz G, et al. Oleic and Linoleic Acids Induce the Release of Neutrophil Extracellular Traps via Pannexin 1-Dependent ATP Release and P2X1 Receptor Activation. Front Vet Sci (2020) 7:260. doi: 10.3389/fvets.2020.00260
187. Khan MA, Pace-Asciak C, Al-Hassan JM, Afzal M, Liu YF, Oommen S, et al. Furanoid F-Acid F6 Uniquely Induces Netosis Compared to C16 and C18 Fatty Acids in Human Neutrophils. Biomolecules (2018) 8:144. doi: 10.3390/biom8040144
188. Palladino END, Katunga LA, Kolar GR, Ford DA. 2-Chlorofatty Acids: Lipid Mediators of Neutrophil Extracellular Trap Formation. J Lipid Res (2018) 59:1424–32. doi: 10.1194/jlr.M084731
189. Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, Curi R, et al. Neutrophil Function and Metabolism in Individuals With Diabetes Mellitus. Braz J Med Biol Res (2007) 40:1037–44. doi: 10.1590/S0100-879X2006005000143
190. Kuwabara WM, Curi R, Alba-Loureiro TC. Autophagy is Impaired in Neutrophils From Streptozotocin-Induced Diabetic Rats. Front Immunol (2017) 8:24. doi: 10.3389/fimmu.2017.00024
191. Bonaventura A, Vecchie A, Abbate A, Montecucco F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells (2020) 9:9010231. doi: 10.3390/cells9010231
192. Roberts HM, Grant MM, Hubber N, Super P, Singhal R, Chapple ILC. Impact of Bariatric Surgical Intervention on Peripheral Blood Neutrophil (PBN) Function in Obesity. Obes Surg (2018) 28:1611–21. doi: 10.1007/s11695-017-3063-1
193. Khan MA, Ali ZS, Sweezey N, Grasemann H, Palaniyar N. Progression of Cystic Fibrosis Lung Disease From Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes (Basel) (2019) 10:183. doi: 10.3390/genes10030183
194. Cools-Lartigue J, Spicer J, Mcdonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells and Promote Metastasis. J Clin Invest (2013) 123:3446–58. doi: 10.1172/JCI67484
195. Cristinziano L, Modestino L, Antonelli A, Marone G, Simon HU, Varricchi G, et al. Neutrophil Extracellular Traps in Cancer. Semin Cancer Biol (2021) S1044-1579X(1021)00206. doi: 10.1016/j.semcancer.2021.07.011
196. Midgley A, Beresford MW. Increased Expression of Low Density Granulocytes in Juvenile-Onset Systemic Lupus Erythematosus Patients Correlates With Disease Activity. Lupus (2016) 25:407–11. doi: 10.1177/0961203315608959
197. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci Transl Med (2015) 7:274ra218. doi: 10.1126/scitranslmed.aaa0835
198. Wipke BT, Allen PM. Essential Role of Neutrophils in the Initiation and Progression of a Murine Model of Rheumatoid Arthritis. J Immunol (2001) 167:1601–8. doi: 10.4049/jimmunol.167.3.1601
199. Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in Expression of Membrane TNF, NF-Kb Activation and Neutrophil Apoptosis During Active and Resolved Inflammation. Ann Rheum Dis (2011) 70:537–43. doi: 10.1136/ard.2010.138065
200. Pi H, Zhou H, Jin H, Ning Y, Wang Y. Abnormal Glucose Metabolism in Rheumatoid Arthritis. BioMed Res Int (2017) 2017:9670434. doi: 10.1155/2017/9670434
201. Drechsler M, Megens RT, Van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-Triggered Neutrophilia Promotes Early Atherosclerosis. Circulation (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
202. Soehnlein O. Multiple Roles for Neutrophils in Atherosclerosis. Circ Res (2012) 110:875–88. doi: 10.1161/CIRCRESAHA.111.257535
203. Nencioni A, Da Silva RF, Fraga-Silva RA, Steffens S, Fabre M, Bauer I, et al. Nicotinamide Phosphoribosyltransferase Inhibition Reduces Intraplaque CXCL1 Production and Associated Neutrophil Infiltration in Atherosclerotic Mice. Thromb Haemost (2014) 111:308–22. doi: 10.1160/TH13-07-0531
204. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil Extracellular Traps License Macrophages for Cytokine Production in Atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
205. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
206. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
207. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
208. Park HH, Park W, Lee YY, Kim H, Seo HS, Choi DW, et al. Bioinspired Dnase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated Netosis Dysregulation. Adv Sci (2020) 7:2001940. doi: 10.1002/advs.202001940
209. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
210. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, De Lima M, Nascimento DC, et al. SARS-Cov-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
211. Fernandez-Perez MP, Aguila S, Reguilon-Gallego L, De Los Reyes-Garcia AM, Minano A, Bravo-Perez C, et al. Neutrophil Extracellular Traps and Von Willebrand Factor are Allies That Negatively Influence COVID-19 Outcomes. Clin Transl Med (2021) 11:e268. doi: 10.1002/ctm2.268
212. Gueant JL, Gueant-Rodriguez RM, Fromonot J, Oussalah A, Louis H, Chery C, et al. Elastase and Exacerbation of Neutrophil Innate Immunity are Involved in Multi-Visceral Manifestations of COVID-19. Allergy (2021) 76:1846–58. doi: 10.1111/all.14746
213. Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-Acting Nanoparticulate Dnase-1 for Effective Suppression of SARS-Cov-2-Mediated Neutrophil Activities and Cytokine Storm. Biomaterials (2021) 267:120389. doi: 10.1016/j.biomaterials.2020.120389
214. Ouwendijk WJD, Raadsen MP, Van Kampen JJA, Verdijk RM, Von Der Thusen JH, Guo L, et al. High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients With Coronavirus Disease 2019. J Infect Dis (2021) 223:1512–21. doi: 10.1093/infdis/jiab050
215. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of Neutrophil Activation, More Than Platelet Activation, With Thrombotic Complications in Coronavirus Disease 2019. J Infect Dis (2021) 223:933–44. doi: 10.1093/infdis/jiaa756
216. Arcanjo A, Logullo J, Menezes CCB, De Souza Carvalho Giangiarulo TC, Dos Reis MC, De Castro GMM, et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci Rep (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
217. Busch MH, Timmermans S, Nagy M, Visser M, Huckriede J, Aendekerk JP, et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation (2020) 142:1787–90. doi: 10.1161/CIRCULATIONAHA.120.050656
218. Strich JR, Ramos-Benitez MJ, Randazzo D, Stein SR, Babyak A, Davey RT, et al. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J Infect Dis (2021) 223:981–4. doi: 10.1093/infdis/jiaa789
219. Gardinassi LG, Souza COS, Sales-Campos H, Fonseca SG. Immune and Metabolic Signatures of COVID-19 Revealed by Transcriptomics Data Reuse. Front Immunol (2020) 11:1636. doi: 10.3389/fimmu.2020.01636
220. Hazeldine J, Lord JM. Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front Immunol (2021) 12:680134. doi: 10.3389/fimmu.2021.680134
221. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’meally R, et al. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
222. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate Is an Inflammatory Signal That Induces IL-1β Through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
223. Reyes L, M AS-G, Morrison T, Howden AJM, Watts ER, Arienti S, et al. A Type I IFN, Prothrombotic Hyperinflammatory Neutrophil Signature Is Distinct for COVID-19 ARDS. Wellcome Open Res (2021) 6:38. doi: 10.12688/wellcomeopenres.16584.2
224. Arraes SM, Freitas MS, Da Silva SV, De Paula Neto HA, Alves-Filho JC, Auxiliadora Martins M, et al. Impaired Neutrophil Chemotaxis in Sepsis Associates With GRK Expression and Inhibition of Actin Assembly and Tyrosine Phosphorylation. Blood (2006) 108:2906–13. doi: 10.1182/blood-2006-05-024638
225. Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil Dysregulation During Sepsis: An Overview and Update. J Cell Mol Med (2017) 21:1687–97. doi: 10.1111/jcmm.13112
226. Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, et al. Plasma ATP Is Required for Neutrophil Activation in a Mouse Sepsis Model. Shock (2014) 42:142–7. doi: 10.1097/SHK.0000000000000180
227. Arese P, De Flora A. Pathophysiology of Hemolysis in Glucose-6-Phosphate Dehydrogenase Deficiency. Semin Hematol (1990) 27:1–40.
228. Cappellini MD, Fiorelli G. Glucose-6-Phosphate Dehydrogenase Deficiency. Lancet (2008) 371:64–74. doi: 10.1016/S0140-6736(08)60073-2
229. Stanton RC. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life (2012) 64:362–9. doi: 10.1002/iub.1017
230. Wolach B, Ashkenazi M, Grossmann R, Gavrieli R, Friedman Z, Bashan N, et al. Diurnal Fluctuation of Leukocyte G6PD Activity. A Possible Explanation for the Normal Neutrophil Bactericidal Activity and the Low Incidence of Pyogenic Infections in Patients With Severe G6PD Deficiency in Israel. Pediatr Res (2004) 55:807–13. doi: 10.1203/01.PDR.0000120680.47846.47
Keywords: neutrophil, metabolism, neutrophil extracellular traps, metabolic switch, glycolysis
Citation: Stojkov D, Gigon L, Peng S, Lukowski R, Ruth P, Karaulov A, Rizvanov A, Barlev NA, Yousefi S and Simon H-U (2022) Physiological and Pathophysiological Roles of Metabolic Pathways for NET Formation and Other Neutrophil Functions. Front. Immunol. 13:826515. doi: 10.3389/fimmu.2022.826515
Received: 30 November 2021; Accepted: 20 January 2022;
Published: 09 February 2022.
Edited by:
Carlos Rodrigo Hermosilla, University of Giessen, GermanyReviewed by:
Ioannis Mitroulis, Democritus University of Thrace, GreeceAnderson Guimarães-Costa, Federal University of Rio de Janeiro, Brazil
Copyright © 2022 Stojkov, Gigon, Peng, Lukowski, Ruth, Karaulov, Rizvanov, Barlev, Yousefi and Simon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Uwe Simon, aHVzQHBraS51bmliZS5jaA==