 Maria Karvouni
Maria Karvouni Marcos Vidal-Manrique
Marcos Vidal-Manrique Andreas Lundqvist
Andreas Lundqvist Evren Alici
Evren Alici
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 February 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.825979
This article is part of the Research TopicInnate Immune Cell Therapy of CancerView all 11 articles
Cell therapy is an innovative therapeutic concept where viable cells are implanted, infused, or grafted into a patient to treat impaired or malignant tissues. The term was first introduced circa the 19th century and has since resulted in multiple breakthroughs in different fields of medicine, such as neurology, cardiology, and oncology. Lately, cell and gene therapy are merging to provide cell products with additional or enhanced properties. In this context, adoptive transfer of genetically modified cytotoxic lymphocytes has emerged as a novel treatment option for cancer patients. To this day, five cell therapy products have been FDA approved, four of which for CD19-positive malignancies and one for B-cell maturation antigen (BCMA)-positive malignancies. These are personalized immunotherapies where patient T cells are engineered to express chimeric antigen receptors (CARs) with the aim to redirect the cells against tumor-specific antigens. CAR-T cell therapies show impressive objective response rates in clinical trials that, in certain instances, may reach up to 80%. However, the life-threatening side effects associated with T cell toxicity and the manufacturing difficulties of developing personalized therapies hamper their widespread use. Recent literature suggests that Natural Killer (NK) cells, may provide a safer alternative and an ‘off-the-shelf’ treatment option thanks to their potent antitumor properties and relatively short lifespan. Here, we will discuss the potential of NK cells in CAR-based therapies focusing on the applications of CAR-NK cells in cancer therapy and beyond.
Natural killer (NK) cells are characterized as cytotoxic lymphocytes of the innate immune system. They account for 5-15% of the circulating mononuclear lymphocytes and are phenotypically defined as CD3-CD56+NKp46+. NK cells develop in the bone marrow (BM) niche from hematopoietic stem cell (HSC) progenitors and undergo maturation in the BM or other secondary lymphoid organs, such as uterus, liver and tonsils (1). NK cells play important roles in host defense due to their ability to recognize and eradicate viral infected and malignant cells without the need for prior sensitization. They are equipped with a repertoire of receptors responsible for delivering activating or inhibitory signals, the relative balance of which dictates the cytotoxic activity and the canonical functions of the cell, such as proliferation and cytokine release (1, 2). Typical activating receptors are the Natural Cytotoxicity Receptors (NCRs) NKp44 (CD336), NKp46 (CD335), and NKp30 (CD337), as well as the Killer Immunoglobulin-like Receptors (KIRs) KIR2DS1, KIR2DS2, KIR2DS4, KIR2DS5, and KIR3DS1. Other important activating receptors are CD224 (2B4), CD226 (DNAM-1) and NKG2D. Several ligands to these activating receptors are upregulated upon cellular stress, infection, or malignant transformation. In cancer, commonly upregulated ligands include MICA and MICB (MHC-class I polypeptide-related sequence A and B), the UL16-binding proteins (ULBPs) and the adhesion molecules PVR (poliovirus receptor, also known as CD155) and Nectin-2. MICA/B and ULBPs are mediating activating signals via binding to NKG2D, whereas PVR and Nectin-2 ligate DNAM-1 (3). NCRs recognize a diverse set of ligands, such as heparan sulfate proteoglycans, cell surface proteins and proteins that reach the surface after their intracellular cleavage. These ligands are not exclusively activating but can also have an inhibitory effect depending on the splice variant of the receptor. The research on the identification of NCR ligands is ongoing. Some of the better studied ones include B7-H6 and HLA-B associated transcript 3 (BAT3) that bind to NKp30, and the proliferating cell nuclear antigen (PCNA) that binds to NKp44. The mechanism of upregulation of NK cell activating ligands is not fully elucidated, although increasing evidence suggests transcriptional and post-translational modifications taking place as a result of cell response to stressful stimuli and DNA damage (4, 5).
Besides the activating KIRs, many of the KIR group receptors are known to propagate inhibitory feedback upon interaction with their ligands. Such ligands are self-MHC (major histocompatibility complex) class I molecules that are expressed in all nucleated cell types and play a critical role in mitigating autoimmune reactions. The downregulation of surface MHC class I can occur under cellular stress conditions leading to increased targeting by NK cells. This is also known as ‘missing-self recognition’ and is a unique feature of NK cells. Inhibitory signals are also mediated by the receptors sialic acid-binding Ig-like lectin-7 (siglec-7) and 9 (siglec-9) that bind to sialic acid-containing carbohydrates (e.g. mucins) aberrantly expressed on tumor cells (6). Other inhibitory receptors are the complex NKG2A/CD94 and the receptors CD161 and KLRG1, that bind to HLA-E, lectin-like transcript 1, and cadherins, respectively.
In addition to the expression of activating and inhibitory receptors, mature NK cell subsets express the FcγRIIIa receptor CD16 that allows the recognition and elimination of antibody-coated cells through antibody-dependent cellular cytotoxicity (ADCC) (7). The mechanism of ADCC is being increasingly explored in cancer therapy. Such therapeutic approaches involve the use of monoclonal antibodies (mAbs) to specifically bind cell surface moieties. Subsequently, CD16- Fc region interaction triggers the antitumor effector immune response resulting in target cell killing. Today, a large number of mAbs have received regulatory approval for cancer treatment, including Rituximab (anti-CD20), Daratumumab (anti-CD38) and Elotuzumab (anti-SLAMF7) (8).
Upon target recognition and immunological synapse formation, NK cells induce target cell lysis via the secretion of lytic granules containing perforin, granzymes (mainly granzyme B) and granulysin (9). The process, also known as degranulation, involves the delivery of granzymes into the cytosol of the target cells, through pores formed by perforin. The granzymes are then cleaving several substrates including caspase-3, Bid and DNA-PKc, and initiate target cell death. A second NK cell killing mechanism is mediated by the engagement of the death receptors Fas and TNF-related apoptosis-inducing ligand (TRAIL)-R1/2 expressed on the surface of target cells, to their respective ligands FasL and TRAIL on NK cells (10). This interaction triggers cell death via the activation of caspase-8. The two pathways follow different kinetics, as granzyme B mediates killing in shorter time compared to death receptors (11).
Similar to CD8+ cytotoxic T cells, NK cells are ‘serial killers’ (12). Observations from time lapse video microscopy revealed that a single NK cell can eliminate up to six target cells. Moreover, it has been shown that NK cells switch from the ‘faster’ granzyme B to the ‘slower’ death receptor–mediated killing during serial target elimination (10). However, the exertion of cytotoxicity, especially when sequential, can lead to depletion of the cytotoxic granule payload, and consequently to NK cell anergy. Strategies to prevent NK cell exhaustion and restore cell fitness have been explored (12). Exposure to cytokines including IL-2, IL-15 and IFNα has been shown to prevent NK cell exhaustion.
Alongside the direct effector cell functions, activated NK cells release major inflammatory cytokines, such as IFNγ, TNFα, GM-CSF, and chemokines, like CCL1-5 and CXCL8 (13, 14). which play important immunomodulatory roles in cellular activation, differentiation, and migration.
Unlike T cells, the large-scale ex vivo expansion of autologous and donor-derived peripheral blood (PB) NK cells has been a challenge for many years. Today, NK cell expansion is performed either by a cytokine-based system or by a feeder cell-based one. The first method includes the use of IL-2, IL-12, IL-15, and IL-21, alone or in a combination of them, to provide the necessary activating and proliferating stimulus (15–17). Alternatively, the stimuli are provided by feeder cells. As feeder cells can be used autologous cells, recombinant human fibronectin fragment-stimulated T cells (18), Epstein-Barr virus-transformed lymphoblastoid cell lines (19), or genetically modified cells of the chronic myelogenous leukemia cell line K562 (20). The latter are engineered to express membrane bound IL-15 (mbIL15), mbIL21, MICA, and/or 4-1BB ligands (21, 22). Of note, feeder cells need to undergo an irradiation step prior to the initiation of the expansion to prevent cell division and obtain high purity of the final NK cell product.
Although an in-depth comparison between the different expansion strategies has yet to be done, it has been shown that the proliferative capacity, the cytotoxic potency (23), the metabolic function (24) and the receptor expression profile of the generated NK cells are heavily influenced by the expansion protocol (21). For instance, NK cell fold expansion can be negatively affected by telomere shortening; a process occurring due to the repeating replication cycles leading to NK cell senescence (25). The degree of telomere shortening is evidenced to be affected by the protocol used. Denman and colleagues showed that NK cell expansion with mbIL21-expressing feeder cells sustained or even increased telomere length while inducing a mean of 47,967-fold expansion (22). In comparison, the NK cell expansion with mbIL15-expressing feeder cells was limited to 825-fold and telomere shortening was observed (22, 26). Regarding the phenotype of the ex vivo expanded cells, studies have focused on the effect of the expansion in the upregulation of immune checkpoints and immunosuppressive molecules, as this can indicate limited efficacy. Markers associated with T cell exhaustion, such as PD-1 and Tim3, have been indeed found upregulated in healthy donor NK cells expanded with a clinically validated protocol of mbIL15-mb4-1BBL-K562 feeder cells + soluble IL-2 (27). Nevertheless, in vitro responsiveness assays verified the high cytotoxicity of the cells, suggesting that despite the upregulation of these markers, the cells were not functionally exhausted. Similar conclusions were drawn from cytokine-based NK cell expansions (28). Today, an increasing number of clinical trials is choosing feeder cell expansion systems. However, feeder-free expansion is still a feasible option as it is easier to adapt to GMP regulations and does not involve the hazard of infusing viable feeder cells to the patients (29).
In 2011, Parkhust and colleagues reported the infusion of ex vivo cytokine stimulated PB-derived autologous NK cells (30). Although the infusion was well-tolerated, none of the eight recruited patients responded to the treatment. This was hypothesized to be due to the inhibitory interactions between the NK cells and self MHC-class I molecules that are upregulated within the tumor microenvironment (TME). Moreover, the patients were heavily pre-treated, which by itself could have a negative impact on the function of NK cells. Nonetheless, the study provided valuable insight on the persistence of the cells in vivo reporting the presence of NK cells in peripheral blood of the patients between a week and several months post infusion. In an attempt to improve NK cell targeting of tumors, Lundqvist and colleagues investigated pre-treatment with the proteasome inhibitor Bortezomib (31, 32). Infusions of ex vivo expanded NK were well-tolerated with the exception of thyroiditis and constitutional symptoms related to IL-2 therapy. The study showed preliminary clinical evidence of antitumor immunity with best clinical response observed in 7/14 patients having stable disease (33).
In comparison to autologous NK cells, NK cells from haploidentical donors can elicit greater cytotoxicity due to the alloreactivity caused by the KIR-HLA mismatch (34, 35). This observation was made in an acute myeloid leukemia (AML) mouse model transplanted with HSC, and its translation to the clinic provided grounds to investigate haploidentical or HLA-mismatched NK cells in adoptive cell transfers (36). In 2005, Miller and colleagues conducted a phase I clinical trial in patients with poor-prognosis AML (37). Results showed that infusions of alloreactive NK cells derived from haploidentical donors are safe, have better in vivo persistence and resulted in remission of 5 out of 19 patients. Overall, the fact that allogeneic NK cells have low risk of causing graft-versus-host disease increased the applicability of NK cell therapy and encouraged discussions on off-the-shelf cell products (38).
To date, the majority of clinical studies on NK cell adoptive cell therapy (ACT) has utilized NK cells derived from peripheral blood. Through the years other NK cells sources have been explored (See Table 1). An example is the umbilical cord blood (UCB). UCB is a NK cell-rich source readily available as cryopreserved biobank material (39). The high proliferative capacity of the cells is particularly attractive, since it allows the generation of large amounts of clinical-grade NK cells (40). Furthermore, UCB-NK cells are suitable candidates for genetic manipulation strategies (41), as well as for combinational treatments with monoclonal antibodies (40), which allows their applicability in different immunotherapeutic strategies. A limitation of UCB-derived NK cells is the inevitable heterogeneity between the final NK cell products due to the use of different UCB donors among batches (42). In addition, comparative studies between PB NK and UCB-NK showed that the latter have some immature characteristics and phenotypic differences. More specifically, UCB-NK cells have increased expression of NKG2A and decreased expression of CD16, KIRs, target adhesion molecules (CD2, CD11a, CD18 and CD62L), perforin and granzyme B (43, 44). Strategies to overcome these issues include the culture of UCB-NK cells with the EBV-transformed HLA-I+ B lymphoblastoid cell line PLH, which provides the necessary inhibitory and activating signals to drive their maturation (45). Moreover, UCB-NK cells are often combined with cytokine support (e.g., IL-2 or IL-15) that enhances their in vivo activity and persistence. Nevertheless, it is worthy of mention that a direct comparison between genetically modified PB and UCB-NK led by Herrera and colleagues found both cell sources to induce similar levels of targeted in vitro cytotoxicity (46).
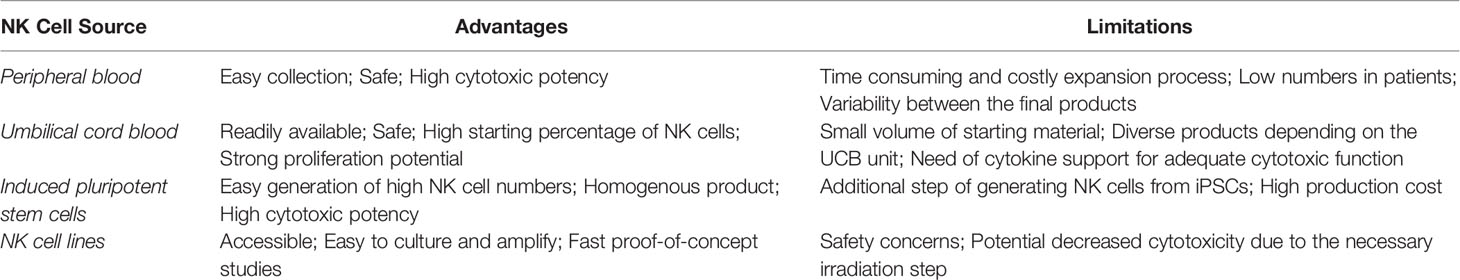
Table 1 Advantages and limitations of NK cell sources.
Stem cell-derived NK cells have been proposed as a viable alternative due to their suitability in standardized off-the-shelf settings. Different sources of stem cells have been explored so far, such as human embryonic stem cells (hESCs) (47), CD34+ HSCs (48) and induced pluripotent stem cells (iPSCs) (49). iPSCs have the advantage of being easier to generate and satisfy the clinical interest of providing greater donor diversity regarding KIR haplotypes (47). It has also been shown that irrespective of their KIR expression profiles, iPSC-NKs have similar killing capacity between them (50). iPSCs were firstly reported in 2006, when human somatic cells were reprogrammed by the simultaneous introduction of four factors: OCT3/4, SOX2, c-Myc and Klf4 (51, 52). In 2007, the combination of the factors OCT4, SOX2, NANOG and LIN28 was also proved effective (53). After successful reprogramming, iPSCs can undergo an essentially indefinite expansion in vitro and, subsequently, produce unlimited amounts of NK cells (54). Their production method is well described (42, 55, 56). Briefly, TrypLE-adapted iPSCs are cultured with human stem cell (SCF) and vascular endothelial growth (VEGF) factors for one week to induce their hematopoietic differentiation. Cells are then further differentiated into NK with the addition of IL-3, IL-15, IL-7, SCF and ftl3 ligand and expanded using feeder cell systems. Similar to UCB-NK cells, iPSC-NK are well susceptible to genetic manipulation and exhibit potent cytotoxicity (57). However, in comparison, the iPSC-derived NK cell population is reproducibly homogenous and consistent (58). Phenotypically, iPSC-NK cells have many similarities to the PB NK cells, with the exception of higher NKG2A and lower KIR expression, which indicates a degree of immaturity (49). The popularity of iPSC-NK cells has been increasing over the past years, with multiple preclinical studies and one clinical trial underway (See Table 2).
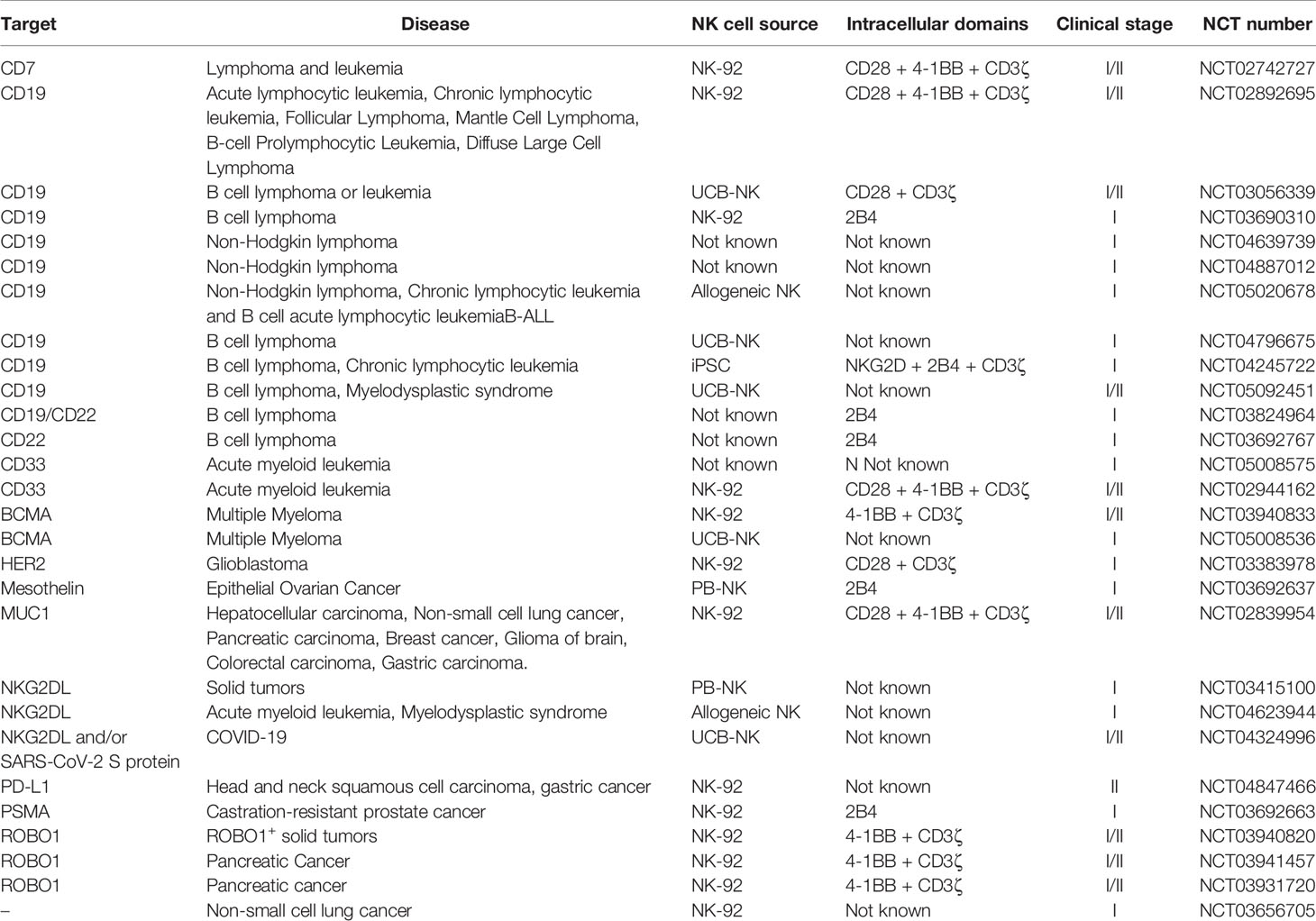
Table 2 Ongoing clinical trials using genetically modified NK cells.
A newer addition to the NK cell sources has been the memory-like (ML) NK cells. ML-NK cells are generated after viral infection (59), exposure to haptens (60) or cytokines, such as IL-12, IL-15 and IL-18 (61). These cells exhibit characteristics of adaptive immunity and have been reported to have higher anti-cancer reactivity in a clinical setting compared to conventional NK cells (62). The cells are well-susceptible to genetic manipulation and have been recently investigated in the context of chimeric antigen receptor (CAR)-NK therapy where introduction of an antiCD19-CAR further improved their targeting and cytotoxic activity (63).
In addition to primary NK cells, NK cell lines have also been used in ACT. Cell lines have the advantage of being easy to culture, expand and cryopreserve. Out of the available ones, NK-92, an IL-2 dependent non-Hodgkin lymphoma NK cell line, has been tested the most in proof-of-concept, preclinical and clinical settings. NK-92 cells have characteristics of activated NK cells, while lacking KIR expression (except KIR2DL4), which explains the potent cytotoxic responses they exhibit upon target cell recognition (64). The cell line NK-92, as well as its genetically modified IL-2- independent counterpart NK-92MI, have been considered for the use in ‘off-the-shelf’ NK cell-based immunotherapies. KHYG-1 and YT are two other NK cell lines with great potential (65). Nevertheless, the use of transformed cell lines in patients is in general met with safety concerns regarding the tumorigenic nature of the cell lines and the hazard of causing secondary NK lymphoma to the patients (66). These limitations can be overcome by implementing a high-dose (5-10 Gy) γ-irradiation step prior to the cell infusion (67, 68). While irradiation halts cell division, it can also negatively impact their long-term in vivo persistence which is necessary for better tumor control (69). Indeed, a study of irradiated and non-irradiated CAR-NK-92MI cells, demonstrated that irradiation with 5Gy reduced in vitro and in vivo proliferation of the cells and shortened their life span (69). Still, the cytotoxicity against target cell lines is not significantly compromised as shown by two different studies (69, 70). Another concern of γ-irradiation is the cellular damage that it causes, ranging from DNA breakage to radical formation and impairment of the cell membrane integrity (71, 72). An alternative method to γ-irradiation was proposed by Walcher and colleagues. Specifically, they demonstrated that low energy electron irradiation has considerable advantages, as it requires shorter treatment times, has more reproducible dose rates, is easier to implement in a laboratory or GMP setting and -importantly- maintains the high cytotoxic effector function of the NK-92 cells (73).
The unique advantages and weaknesses of each NK cell source are listed in Table 1.
The great potential of NK cells as off-the-shelf cellular treatments is often overshadowed by their sensitivity to cryopreservation. For instance, it has been shown that although cryopreserved NK cells can eliminate target cells in standard in vitro cytotoxicity assays, their efficacy against three-dimensional tumor models is reduced due to a 6-fold decrease in motility (74, 75). Moreover, in a mouse model where infusion of fresh or cryopreserved expanded NK cells was compared, the disadvantage of cryopreserved cells in homing, persistence and expansion was evident (76). Maintaining NK cell viability and cytotoxic function post-thaw is essential in a clinical setting, where high consistency and quality must be guaranteed. A number of studies are investigating the optimal cryopreservation conditions, assessing various freezing/thawing media, cooling rates, storing conditions, culture protocols and resting times. Although a gold standard has not been established yet, it is worth mentioning that substituting dimethyl sulfoxide (DMSO) with other small molecule sugar or protein-based cryoprotectants (e.g sucrose, proline, mannitol) in the freezing media formulation is a generally accepted alternative, associated with improved viability of immune cells post-thaw (77). Given the fact that cryostoring is a necessary step between the development of a cellular product and its infusion, intensifying the efforts to optimize the procedures will positively impact the universal application of immunotherapy.
In the clinical setting, the potency of ACT is dependent on the persistence of the infused NK cells. Accumulation of inhibitory factors, insufficient tumor targeting and failure to persist and expand in vivo are only a few of the challenges that need to be overcome. For most of these issues, genetic manipulation of the cell product provides a viable solution. NK cells, however, are typically less susceptible to such manipulation compared to other immune cell types (78). Therefore, although the genetic reprogramming of NK cells shares similar principles and methodologies to that of T cells, additional steps are often necessary.
Transfection is a method in which plasmid DNA, mRNA or proteins are introduced to a cell with the aim to initiate their expression. Depending on whether a short or a long-term expression is desired, different methods are applied. One of the most common transient transfection techniques is electroporation. The method uses electric pulses to permeabilize the cell membrane and create pores from where the genetic material is inserted. Electroporation is highly efficient in T cells, however, similar yields have yet to be observed in NK cells (79). Moreover, electroporation results in short-term expression of the transgene which can limit its applicability in the context of immunotherapy (80). Still, the cost-effectiveness of the technique and the ease of its application in large-scale clinical settings are attractive. Efforts to improve NK cell transfection efficiency focus on optimizing fundamental parameters, such as the number of cells, the voltage, and the concentration of material to be electroporated (81). Moreover, ways to minimize the considerable cell death that follows electroporation are being explored (82). Examples of the application of the method in NK cell therapy are mostly concerning CRISPR-mediated genome engineering (knock outs or edits) (83). If long-term expression is desired, the transposon-based technology may be an attractive method. Two such systems have been reported, namely the Sleeping Beauty (84) and the PiggyBac (85). These systems combine the efficiency of electroporation with the precise insertion of the genetic material into the host genome thanks to its integrating element. Nonetheless, to this day, the use of transposon-based systems is more commonly used in T cell rather than in NK cell studies.
Viral transduction is one of the most common methods for immune cell engineering. It results in stable transgene expression while maintaining higher cell viability compared to electroporation. Moreover, the method is well validated in preclinical and clinical studies, where it has repeatedly proven safe and efficient. The vast majority of the clinical studies are using either lentiviral (LV) or retroviral (RV) vectors, although some studies with adenoviral vectors have also been completed with mixed results (86). The first report of viral transduction of NK cells was made by Nagashima et al., in 1998, where he described the transduction of NK-92 cells using RVs (87). This study showed a transduction efficacy of about 2-3%, which although impressive for the time, it is considered low with today’s standards. Of note, recent publications of retroviral transductions of expanded NK cells resulted in about 70% transduction efficiency (88–90). RVs, and more specifically, self-inactivating γ-RVs were also the first viral vectors to enter clinical trials. Their application was further increased by the development of clinical grade RV-producing packaging cell lines. These cell lines, such as the murine cell line PG-13, are able to continuously generate large quantities of clinical-grade virus supernatant following their stable transduction by the vector of interest (91). A disadvantage of the RVs is the fact that they require active cell division in order to successfully integrate their vector to the host genome (92). Moreover, the integration itself can be at random sites, which rises concerns on tumorigenicity and the overall safety of the approach. Careful optimization of the transduction process is, therefore, necessary in order to limit the viral copy number per cell to the absolute minimum. Apart from the γ-RVs that have been monopolizing the transductions, α-RVs are gaining more attention, especially after studies showing superior transduction efficacies in primary NK cells of α-RVs compared to γ-RVs and LVs (93).
Unlike the RVs, LVs are able transduce cells irrespective of their cell cycle phase, leading to theoretically higher transduction efficacies. The vector of choice for T cell-based immunotherapies is the vesicular stomatitis virus (VSV-G) pseudetotyped vector, because of its broad tropism. However, these vectors have proved less effective in NK cells. Alternatively, LVs pseudotyped with a modified baboon envelope glycoprotein (BaEV-LVs) hold greater potential since their entry receptors sodium-dependent neutral amino acid transporter-1 and -2 (ASCT-1 and ASCT-2) are abundant in NK cells (94). Indeed, two independent investigations showed a 20-fold higher transduction efficacy with BaEV-LVs compared to VSV-G-LVs (94, 95). As far as safety is concerned, LVs, and more specifically the 3rd generation vectors, are considered safer viral vector options since the packaging genes gag/pol and rev are found in separate plasmids, thus making the generation of wild-type recombinant virus harder (96).
Due to their innate antiviral defense mechanisms, viral transduction of NK cells has proved to be challenging. A way to enhance the transduction efficacy is by reducing the NK cell virus repulsion, which in turn increases the internalization of viral particles into the cells independently of the viral receptors (97). Examples of such reagents are the cationic polymers polybrene and protamine sulfate. Although both reagents are routinely used in viral transduction protocols, they are also associated with decreased post-transduction cell viability. An alternative approach is to use recombinant human fibronectin fragment such as RetroNectin. RetroNectin is a 63kDa with adhesion sites for cells (integrin receptors VLA-4 and VLA-5) and viral particles (heparin domain) (98). Upon adhesion of both parties to the filament, transduction is facilitated thanks to the close proximity between them (99, 100). Transduction using RetroNectin is efficient and suitable for difficult to transduce or frail cells (98). Understanding the intracellular patterns of foreign genomic material recognition has contributed to finding additional strategies to increase transduction efficacy. More specifically, the inhibition of the TBK1/IKKϵ complex acting downstream of RIG-1, MDA-5 and TLR3, has significantly improved the viral transduction (101). Such reagent is the VyOz, which is reported to increase the efficacy of NK cell transduction by 4-fold (102). Of note, all the afore-mentioned reagents are available in GMP grade, further enabling the translation of the preclinical studies to the clinical setting.
Virus-like particles have also recently emerged as gene editing tools. One of these approaches uses engineered murine leukemia virus-like particles loaded with Cas9-sgRNA complexes (103). The system, commonly known as Nanoblade, is suitable for both in vitro and in vivo manipulations, providing high gene editing efficacy and precision in a cost-effective way. Although they have yet to be applied to NK cells, nanoblades have shown promising results in genome editing of human T, B and CD34+ cells (104).
Irrespective of the source of NK cells, the cytotoxic potential and the targeting capacity of the cells can be further increased with the expression of CARs. CARs are synthetic receptors comprising of three main regions; the antigen-binding single chain variable fragment (scFv), a short transmembrane region (TM) and one or more signal transduction domains (105).
The extracellular part, or ectodomain, derives from a tumor-specific antibody and consists of a heavy and a light chain that connect via a linker. Selecting the right targeted epitope, scFv and linker for each application is crucial. Indeed, a study comparing distinct scFvs targeting the same antigen demonstrated that the scFv domain can influence the expression of the CAR, as well as its functionality (106). A step further is the affinity optimization of the scFv. This is particularly relevant in the occasions where the targeted antigen is expressed on normal tissues (although in lower levels), and thus there are increased chances of on-target off-tumor toxicities. A strategy to reduce the recognition of antigenlow normal cells was described by Drent et al. (107). The researchers used ‘light-chain exchange technology’ to construct 124 new antibodies with 10 to >1000-fold less affinity to CD38, a target for multiple myeloma (MM). The selected scFvs were then assessed in both CAR-T (107) and CAR-NK (65) models, where effector cells expressing the affinity optimized CARs effectively discriminated between MM and normal cells. The selection of the scFv domain can be aided by modern computational methods of protein design (108).
The ectodomain is connected to the TM via the spacer or Hinge region. Similar to the linker, the type and length of the Hinge region can influence the binding capacity of the CAR, as it provides stability and flexibility to the receptor (109). The right selection of Hinge region can additionally protect the patient from off-target activation of the engineered cells and/or unintentional innate immune response; a phenomenon firstly observed in CARs with an immunoglobulin G (IgG) 1 Fc spacer domains interacting with IgG FcγRI receptors (110). It has also been reported that incorporation of a Hinge region to the CAR design enhances the expansion of CAR-transduced T cells (111). The most commonly used Hinge regions in CAR-NK cell therapy derive from the CD8a, CD28 and the IgG-based sequences (105). To our knowledge, there is no study comparing the Hinge regions in CAR-NK cells. Nonetheless, a study comparing the human CD28 and CD8a regions in anti-CD19 CAR-T cells showed that CD8a induced lower production of IFNγ and conferred better resistance to activation-induced cell death, while maintaining equal cytotoxicity to the CD28 counterpart (112). Apart from the functional purpose, Hinge regions act as a target for CAR-detection antibodies facilitating the confirmation of CAR expression (111).
After the Hinge region follows the TM. The TM region is responsible for anchoring the receptor on the cell membrane, as well as for transducing the signal from the extracellular to the intracellular domains of the CAR. Typical TMs used in T and NK cell studies are derived from CD3, CD8α and CD28 (112). In the context of NK cell therapy, TM domains of activating receptors, such as CD16, NKp44, NKp46, NKG2D, 2B4 and DNAM-1, have also been tested in an in vitro iPSC-NK based study (113). The comparison revealed that the combination of NKG2D-derived TM with 2B4 co-stimulatory domain and CD3ζ signaling domain confers strong antigen-specific cytotoxicity.
Regarding the intracellular region, the design of the CARs has advanced throughout the years, from having only a signal transduction domain (1st generation CAR) (114), to having one or two co-stimulatory domains additionally (2nd and 3rth generation respectively) (115, 116). The intracellular domains for the first CAR designs were inspired by the activating signaling pathways of T cells. To this day, the most broadly used co-stimulatory domains are CD28 and 4-1BB (CD137), while the most common signal transduction module is CD3ζ (117). Both CD28 and 4-1BB are shown to recapitulate natural co-stimulation and provide increased potency to the transduced effector cells. It is also known that CD28 confers a different set of advantages over 4-1BB (118). Studies comparing the two in a 2nd generation CAR setting showed that 4-1BB promotes survival and proliferation, whereas CD28 attributes a stronger cytotoxic potential. Depending on the clinical application, the use of either or both domains is preferred.
Transitioning into NK cells, the expression of already validated T cell-based CAR designs was initially assessed. The first CAR-NK cell was reported by Tran et al. (119). In this in vitro study NK cells successfully expressed a functional CD4-CD3ζ CAR, which redirected the cells against HIV-infected CD4+ T cells and NK cell-resistant tumor cells. Soon after, applying the CAR know-how from T to NK cells without adjustments was deemed sub-optimal. This is because besides some common signaling moieties that the two cell types share, such as CD3ζ and 4-1BB, most of the frequently used co-stimulatory domains are absent in NK cells. Therefore, questions were raised on whether the true potential of CAR-NK therapy was harnessed with the early CAR designs. Novel constructs for NK cell therapy are substituting the signaling domains of the CARs with those of activating NK cell moieties. The domains that NK cells typically use for downstream signaling are the CD3ζ, DAP10, DAP12 and FcRγ chains (120, 121). In comparison to CD3ζ that has three ITAM domains, the rest of the molecules have a single ITAM. It is also worth mentioning that DAP10 is the adapter protein of NKG2D, whereas DAP12 mediates signaling of activating KIRs, NKG2C and NKp44. In experiments of primary NK cells, DAP10 was found to be functional only when NKG2D was used as the ectodomain, whereas when NKG2D was utilized as the TM region, DAP10 decreased the functionality of the CAR (122, 123). On the contrary, DAP12, as a signaling domain, was met with greater success outperforming CD3ζ-based CARs in two independent in vitro and in vivo studies of primary NK cells (124, 125). Another co-stimulatory domain under investigation is the NK cell specific receptor 2B4. A couple of studies comparing NK-92 and PB-NK cells expressing 2nd generation antiCD5 or antiCD123 CAR constructs with either 2B4 or 4-1BB co-stimulatory domains, showed that 2B4-CD3ζ CARs provided superior antitumor efficacy compared to 4-1BB-based CAR constructs (121, 126). A similar effect was observed in a study comparing NK-like CARs (NKG2D TM and 2B4 + CD3ζ intracellular domains) and T-like CARs (CD28 TM and 4-1BB + CD3ζ intracellular domains), which showed that iPSC-derived NK cells expressing NK-like CARs induced tumor regression and prolonged mice survival (113). NK cell-like stimulatory domains are being increasingly used in the design of CAR constructs for NK cell immunotherapies, especially for the treatment of solid tumors (113, 125, 127, 128). Regarding 3rd generation CARs, the investigation on finding the optimal combination of signaling domains is currently of high interest. Overall, the findings suggest that NK cell-like co-stimulatory domains unleash superior antitumor responses by CAR-NK cell products.
The first CAR designs were comprising of scFv domains deriving from mouse antibodies. It has been shown, however, that murine scFvs can be immunogenic and that the triggering of the host immune response often results in early elimination of the CAR products (129). In an effort to prevent that, fully human CARs are being developed, showing comparable targeting capacity to the originally reported scFvs (130). Besides scFvs, which are typically used as the antigen recognition domain, alternatives for improving the CAR features or facilitating their design process have emerged throughout the years. For instance, when target cells express a cell-specific surface antigen whose ligand is known, CAR constructs can incorporate the ligand itself as the extracellular antigen-recognition domain, instead of an scFv. Of note, Zhuang and colleagues demonstrated that CD28H-based CAR-NK cells could be used to recognize and kill target cells expressing B7H7, its natural ligand (131).
A more recent advancement is the use of nanobody-based constructs. Nanobodies, also known as VHH antibodies, derive from the variable domain of heavy chain-only antibodies naturally existing in Camelidae and shark species (132, 133). Nanobodies have several advantages over traditional antibodies, such as increased capacity of reaching inaccessible epitopes thanks to their long CDR3 sequence, ability to maintain their physiochemical properties in extreme conditions, easier humanization process and less probable folding and assembly issues (134–136). A study of generating CD7-nanobody based CAR-NK-92MI cells demonstrated potent antitumor effect against T-cell leukemia cell lines and primary cells (137). Similarly, nanobody-based CAR-NK-92 cells targeting CD38 in multiple myeloma showed high specificity and cytotoxic activity in primary human bone marrow samples (138). A comparison between scFv- and nanobody- based CARs was done in clinical studies of CAR-T cells, where comparable efficacy and safety were reported (136). A further step is the design of affinity optimized nanobodies, using a recently developed algorithm that helps predict the residues whose modulation would confer specific binding characteristics (139).
As previously mentioned, the long-term persistence of NK cells after adoptive cell transfer can be a concern without cytokine support (21). Due to the association of cytokine administration with serious adverse effects, recent efforts are incorporating an ectopic cytokine support system into the CAR plasmid, developing thus a so-called ‘armored’ CAR. In a study of Liu et al, CB-NK cells were transduced with a viral vector encoding for an anti-CD19 CAR and the IL-15 gene (41). The results were promising as the generated NK cells exhibited increased cytotoxicity against CD19-positive targets in vitro and led to prolonged survival in vivo (41). In a phase I/II clinical trial, the same approach showed response to 8/11 patients, without the cause of serious adverse effects (140). In a different study, Wang et al. coupled IL-15 transgene expression to an inducible MyD88/CD40 system and achieved increase of the in vivo CAR-NK cell persistence for a minimal of 40-50 days (141).
Ensuring the safety of the adoptive cell transfer is pivotal, especially after the reports of neurotoxicity, on target-off tumor effects and cytokine storm in CAR-T cell clinical studies (142). Although NK cells have in general a safer profile than T cells, additional safety measures can be lifesaving during an emergency situation. A method to rapidly terminate a cell-based treatment is by incorporating a suicide gene into the therapeutic transgene. In NK cells, the strategy has been tested using the inducible safety switch caspase-9 (iCasp9) suicide gene that expresses a modified caspase-9 fused to the human FK605 binding protein (41). The system is pharmacologically activated, meaning that upon administration of AP1903 (a chemical inducer of dimerization) a caspase-mediated apoptosis is induced (143). Similar drug-induced CAR-NK cell elimination was achieved by an orthogonal rapamycin-regulated caspase-9 switch (141). Notably, cell-cycle dependent suicide genes, such as HSV-TK, are not recommended for use in CAR therapy, as they require active cell division in order to function and can, additionally, induce immunogenicity (144). An alternative strategy for increasing safety and minimizing on-target off-tumor effects was applied in an anti-CD147 CAR-NK study for hepatocellular carcinoma (70). Tseng and colleagues designed a GPC3-synNotch-inducible anti-CD147 CAR, in which the CAR-mediated antitumor responses were unleashed only when GPC3 and CD147 were co-expressed on the surface of the target cell.
Antigen escape, the partial or total loss of the targeted antigen expression from the surface of malignant cells, is a known mechanism that impedes the efficacy of CAR therapy in cancer. A study addressed this issue by designing a CAR construct with dual specificity for the tumor associated antigens (TAA) EGFR and its mutant form EGFRvIII for the treatment of glioblastoma (145). Dual-specific NK cells eliminated cells positive for both or either of the antigens, in contrast to the CAR-NK cells targeting a single epitope, which resulted in the significant extension of survival of glioblastoma bearing mice. A different approach was proposed by Li and colleagues, who engineered a plasmid encoding for NKG2D and an antiPD-1-CAR, with the intracellular domain of DAP10, aiming to induce synergistic activation of NK-92 cells by parallel recognition of PD-1 and NKG2D ligands (146). Functional assays showed increased cytotoxic activity of the engineered cells and underlined the potential of the method. Notably, dual targeting can be also achieved with the use of tandem CAR constructs. Tandem CARs are constructed by consecutively linking two different antigen binding domains (either scFv or nanobody-based) to a single intracellular domain. Although this approach has been assessed in CAR-T studies, tandem CARs have not yet been evaluated in the NK cell setting (147).
Grote S. et al. further increased the versatility of CAR-targeting by proposing modular CARs (148). Modular or ‘adapter’ CARs (AdCARs) recognize biotin-labeled antibodies specifically targeted against TAAs. This would mean that the CAR-mediated cytotoxicity is fully dependent on the selection of the biotin-labeled antibodies and can be easily modified if needed. The novel AdCAR-NK-92 cell product demonstrated superior cytotoxic responses against CD19+ and/or CD20+ primary cell targets and gave ground for discussions on off-the-shelf universal CAR-NK cell therapy.
Cell therapy is traditionally administrated via intravenous infusion. Therefore, trafficking of CAR-NK cells to the targeted tissue is critical for the exhibition of a therapeutic effect. To aid this process, the incorporation of a chemokine receptor transgene into the CAR design was assessed. In a model of AML, Jamali and colleagues, generated transgenically augmented anti-CD19 CAR-NK cells (TRACKs) expressing the chemokine receptor CXCR4, which is implicated in the retention of NK cells in the bone marrow niche (149). Improved migration and superior lysis of target cells was demonstrated in vitro. The strategy was also applied in a solid tumor setting, where expression of CXCR4 by EGFRvIII CAR-NK cells induced specific chemotaxis towards the CXCL12/SDF-1α positive glioblastoma cell line U87-MG (150).
In the occasion that one of the tumor-associated antigens overexpressed in the tumor is a death receptor, transduction of CAR-NK cells with its respective apoptosis-inducing ligand could effectively redirect CAR-mediated antitumor responses towards the cancer cells. The approach was assessed by Lee Ye and collaborators in a pancreatic ductal adenocarcinoma model, where TRAIL transgene was cloned in the FRα-specific CAR vector (151). The generated CAR-NK-92 cells demonstrated enhanced and targeted cytotoxicity.
CRISPR technology is a powerful gene editing tool that has been increasingly used in cellular immunotherapy. CRISPR/Cas9 is successfully used to insert the CAR transgene into specific loci of the effector cell genome with high precision (152). Apart from that, the technology is extensively studied in the context of increasing the functionality and persistence of CAR-modified cells. Such strategy involves the knocking in of genes associated with effector cell activation, and/or the knocking out (KO) of inhibitory genes. In a study on EGFRvIII CAR-T cells, the authors used the CRISPR/Cas9 system to specifically disrupt the PD-1 gene without causing further alterations to the CAR-T phenotype (153). This resulted in the in vitro inhibition of the glioblastoma cell growth. Similarly, CRISPR-mediated KO of the endogenous TGF-β receptor II (TGFBR2) in CAR-T cells unleased immunosuppressive breaks and reduced the CAR-T exhaustion (154). In NK cells, blockade of NKG2A expression resulted in highly functional NK cells that overcame NKG2A-mediated inhibition (155). Although the last study used specifically designed protein expression blockers, rather than the CRISPR-technology, it is believed that both methods could be applied for the generation of NKG2Anull NK cells.
An alternative way of using the CRIPSR/Cas9 system is enabling the application of an approach in a particular setting. For example, CD38 is a validated target for MM therapy, as it is overexpressed on the malignant cells. However, CD38 is also highly expressed by effector immune cell types, such as the NK cells (156). Because of that, antiCD38 CAR-NK therapy is characterized by effector cell fratricide shortly after the CAR is expressed. In a study of antiCD38 CAR-NK cells, CRISPR/Cas9 KO of CD38 on the NK cells provided a practical solution to avoid fratricide while maintaining their functionality and cytotoxic potential (65).
The metabolic state of the CAR-NK cells in the hostile environment of the TME affects their functional fate to a great extent. For example, limiting glycolysis or oxidative phosphorylation (OXPHOS) is known to impair the production of cytokines, such as IFNγ and Granzyme B in NK cells (157, 158). Efforts are made to elevate NK cell metabolism in the TME using gene editing techniques. Mammalian target or rapamycin complex 1 (mTORC1) is a key regulator of NK cell development and effector responses, activated upon cytokine stimulation (e.g IL-15). A method to maintain mTORC1 within the TME is via deletion of the cytokine-inducible SH2-containing protein (CIS) (159). This results in increased JAK/STAT and mTORC1 signaling after IL-15 stimulation, and consequently improves metabolic fitness, cytotoxicity and in vivo persistence (160, 161). However, caution should be taken during long exposure of the edited NK cell to IL-15, as it may lead to opposite results, such as NK cell exhaustion and reduced cytotoxicity (24). Another pathway that could be targeted is the cMyc signaling. Adequate levels of cMyc protein are of vital importance for sustaining elevated rates of glycolysis and OXPHOS in NK cells (162). Nevertheless, within TME it is observed a rapid loss of cMyc expression. A strategy to sustain the levels of cMyc is by targeting its degradation pathway mediated by the kinase glycogen synthase kinase-3 (GSK3). Indeed, GSK3 inhibitors are found to restore NK cell cytotoxicity and enhance IFNγ and TNFα production in in vivo models of AML and ovarian cancer (163, 164). Alternatively, expression of cMyc protein is rescued by increasing the availability of glutamine. Suppression of glutamine metabolism in tumors can be achieved by treatment with the compound JHU083 (165). Last but not least, the potential benefit of the hypoxia-inducible factor 1α (HIF1α) deletion is being discussed, although the complexity of its regulation requires further investigation (166).
With the advances of CAR technology and the discovery of alternative NK cell sources, many of the initial obstacles that CAR-NK therapy faces were addressed. However, there are still opportunities for further improvements. As we have previously discussed, NK cell homing is crucial for the success of immunotherapy. Reaching the appropriate effector to target cell ratio within the malignant site has proven a challenge, especially with regards to solid tumors (167). Independent studies have provided evidence that 1) pre-conditioning of patient with lymphodepletion (168), 2) complement cytokine support (76), 3) pharmacological intervention (169, 170), 4) reduction of immunosuppression in TME (171–173) and 5) expression of chemokine receptors by effector cells (174), among others, have a positive effect. Combining different strategies into a multi-dimensional novel approach appears to be the best chance of improving the baseline trafficking and infiltration.
Pharmaceutical intervention can counteract antigen escape by increasing or maintaining the expression of the targeted antigen. The drugs used for this application are mediating epigenetic modulations, post-translational modulations or inhibit antigen cleavage from the cell surface (175). An example is the use of all-trans retinoic acid (ATRA), an anti-leukemic agent known to enhance the expression of CD38 and folate receptor β (FRβ), to increase the potency of anti-CD38 (176) or anti-FRβ (177) CAR-T therapy respectively. Similarly, treatment with histone deacetylase inhibitors, such as valproic acid, causes upregulation of the NKG2DL MICA/B and ULBP2 from the tumor cells (178). This effect was successfully harnessed in a study of NK cell-mediated lysis of hepatocellular carcinoma cells (179). It could additionally be exploited, however, by the NKG2D-based CAR modified T (180) and NK cells (124, 181). Other examples of drug CAR therapy combinations reported in literature are: 1) adenosine 2a receptor antagonists and mesothelin CARs (182), 2) DNA methyltransferase inhibitors (e.g decitabine) and mucin 1 CARs (183), 3) enhancer of Zeste homolog 2 (e.g GSK126 and tazemetostat) and GD2 CARs (184), protein kinase C modulators (e.g bryostatin-1) and CD22 CARs (185), and 4) γ-secretase inhibitors and BCMA CARs (186).
The efficacy of CAR-NK therapy against solid tumors is limited. To tackle problems associated with insufficient migration of the modified cells to the solid tumor, several studies have investigated the synergistic effect of anti-angiogenic agents with CAR-NK cell infusion. Zhang and colleagues investigated the combination of regorafenib and EpCAM-specific CAR-NK-92 cells in a mouse model of human colorectal cancer xenografts and found the combination to have superior antitumor response compared to each monotherapy (169). Another study by Wu et al. combined apatinib with anti-HER-2 CAR-NK-92 cells and assessed the efficacy against gastric cancer xenografts (170). The strategy achieved improved CAR-NK cell infiltration into the larger tumor xenografts and resulted in better tumor growth suppression (169, 170).
Checkpoint inhibition and adoptive cell transfer have revolutionized modern cancer treatment. The combination of these individually successful immunotherapeutic approaches has been assessed. Specifically, blockade of the immune checkpoint CD73 enhanced the cytotoxicity of NKG2D-targeting CAR-NK-92 cells in CD73+ human lung cancer xenograft models (128). Furthermore, the administration of an anti-PD-1 monoclonal antibody together with anti-HER2 CAR-NK-92 cells for the treatment of glioblastoma has been reported in an abstract by Strassheimer et al. (187).
The combination of adoptive NK cell transfer with therapeutic monoclonal antibodies (mAbs) is a promising therapeutic approach, due to the innate ability of NK cells to induce ADCC via its FcγRIII receptor CD16 (188). The efficacy of this combination is dependent on the CD16 polymorphisms, as well as the affinity of the mAb to the CD16 receptor (189). Engineered Fc receptors, with optimized affinity and enhanced durability within the in vivo environment have emerged the last years in an effort to increase the efficacy of cell and antibody therapy combination. Indeed, iPSC-derived NK cells expressing a high affinity non-cleavable CD16-construct (hnCD16-iNK cells) showed enhanced ADCC-mediated effector functions against target cells coated with an anti-CD20 mAb (190). To our knowledge, CAR-NK cells have not been combined with mAbs to this day in a peer-reviewed journal. However, a relevant approach was proposed by Goodridge and colleagues in an abstract form, where iPSC-NK cells co-transduced with hnCD16 and antiCD19-CAR constructs showed promising results in combination with Rituximab (191).
A similar approach was described in a study reporting the combination of anti-TF CAR-NK cell therapy with chimeric antibody-like homodimer immunoconjugates that also target TF, called ICON and L-ICON, in triple-negative breast cancer (192). The combination was assessed in vitro showing enhanced cytotoxicity deriving both from the CAR and the ADCC response, compared to the individual treatments.
Radiotherapy is a commonly used regime for the treatment of cancer, particularly in solid tumor malignancies. Radiation introduces cell damage to the tumor and the adjacent cells which generates neoantigens or induces stress ligand upregulation (193). The accumulation of activating ligands triggers the immune system and facilitates cancer cell recognition. For this reason, radiotherapy and immunotherapy have been assessed in a combinational approach with promising results (194). In 2020, Kim et al. reported their findings from the synergy of radiotherapy and ACT of ex vivo activated NK cells in a human triple-negative breast cancer xenograft model (195). The combination treatment showed enhanced NK cell tumor infiltration, reduced tumor burden, prolonged NK cell retention to the tumor site and suppression of metastasis. CAR-based therapy and radiotherapy have been assessed together only in the context of CAR-T therapy for glioblastoma treatment. In this study, NKG2D-specific CAR-T cells were combined with radiation therapy due to the upregulation of NKG2DL occurring post-radiation (196). The study showed increased CAR-T cell activation and improved outcomes in terms of survival and tumor control. Taken together, the combination of CAR-NK cells and radiotherapy is worth exploring.
Oncolytic virotherapy is a fast-developing field within the cancer immunotherapy, attracting particular interest the last two decades. The field is based on the notion of utilizing viruses to selectively replicate within malignant cells, leading to target cell lysis while normal cells remain unaffected (197). Indeed, oncolytic viruses (OVs) have shown impressive cancer cell elimination in murine models, without causing severe side effects in multiple studies (198–200). There are currently three OVs that have received governmental regulatory approval: 1) the herpes simplex virus T-VEC (Imlygic), approved in USA and EU, 2) the adenovirus H101 (Oncocrine), approved in China, and 3) the Rigvir enterovirus, approved in Latvia, Armenia, Georgia and Uzbekistan (201). OVs and CAR therapy have been successfully combined, although mainly in the context of CAR-T cells. The combinational approaches have the form of either sequential treatment courses of OV and CAR therapy, or CAR cells are used as the vessels to deliver OVs to the tumor site (202). There are many advantages of combining two immunotherapies with different mechanism of action. These include better tumor infiltration of the CAR cells following the initial direct tumor cell lysis by the OVs, upregulation of stress markers from the OV-infected cells leading to enhanced effector cell persistence, proliferation and functionality and additional antitumor activity in the case the CAR cells become anergic in the TME (203). CAR-NK cells and OVs have also been studied together. More specifically, in a mouse model of breast cancer brain metastasis, sequential intratumoral administration of OVs (herpes simplex virus) and anti-EGFR CAR-NK-92 cells resulted in better tumor control and prolonged survival, compared to the effects of the individual treatments (204). Moreover, in the in vitro experiments of the same study, CAR-NK cells displayed higher cytolytic activity and cytokine release after co-culture with breast cancer cell lines. Additional evidence on the potential of such combinational approach was given by a recently published study on glioblastoma, where OVs expressing the IL-15/IL-15Rα complex (OV-IL15C) were combined with off-the-shelf EGFR CAR-NK cells demonstrating strong antitumor response (205).
The combination of recombinant viruses with CAR-NK cells has been reported in a preliminary abstract format. Specifically, HER2-specific AAV-mediated gene transfer of a PD-1 inhibitor together with local administration of anti-HER2 CAR-NK-92 cells has been suggested for the treatment glioblastoma (206).
An interesting approach on limiting the insufficient delivery of nanoparticle-based drug formulations was suggested by Siegler et al, which involved the use of CAR-NK cells as carriers (207). The researchers used Abraxane, an FDA-approved nanoparticle-based formulation of the chemotherapeutic agent paclitaxel, to load multilamellar liposomal vesicles, which they then cross-linked to the CAR-NK cell surface. The combinatorial approach was assessed in vitro and in vivo against HER2+ and CD19+ cancers where enhanced targeted cytotoxicity was observed.
Although less of a combinational approach and more of an unexplored field, CAR-NK cell-derived EVs are worth mentioning. EVs are membranous vesicles secreted by multiple cell types. They enclose proteins, lipids, nucleic acids and other cytoplasmic components of the parent cell and, upon uptake, they mediate biological effects, phenotypic changes and cell-to-cell communications (208). EVs can be categorized into microvesicles, exosomes and apoptotic bodies, depending on their generation mechanism and size. Of particular interest are the exosomes. These are typically 30-150nm in diameter and originate from the endosomal cell compartment. Studies on the content of the NK-cell derived exosomes (NK-exos) revealed that they are loaded with perforin, granzymes, DNAM-1, IFNγ, and other functional molecules, which provides reason for their exploration in cancer therapy (209, 210). Indeed, exosomes derived from the NK cell line NK-92 were found to exert potent antitumor activity in in vitro and in vivo studies of aggressive melanoma (211). Importantly, NK-exos can be modified, either directly [e.g via electroporation (212)] or by the genetic manipulation of the parent cells (213). The latter approach has been extended into the CAR-T field, offering a new and potentially ‘off-the-shelf’ treatment option. More specifically, primary T cells were lentivirally transduced to express a 2nd generation CAR construct (214). The deriving exosomes were analyzed for their content, where it was found that apart from the cytotoxic molecules, the CAR protein was also present. In contrast, the PD-1 receptor was not detected. The cytolytic potential of the exosomes was assessed against relevant cancer cell lines, showing targeted cancer cell death in a concentration-dependent manner. Taking everything into consideration, we believe that the CAR-NK derived exosomes is also an immunotherapeutic platform worthy of exploring.
CAR-NK-based therapies have been extensively studied for the treatment of hematological malignancies, showing a clear in vitro and in vivo advantage of CAR expressing NK cells over control NK (41, 46, 63, 149, 160, 190, 215–217). CD19 targeting has been in the epicenter of this research, following the FDA approval of the antiCD19 CAR-T products Yescarta and Kymriah. Overall, multiple studies demonstrated that CAR-NK cells were efficient in eradiating CD19+ targets (41, 46, 63, 149, 160, 215–217). Regarding the nature of the assessed CAR constructs, it was shown that 2nd generation anti-CD19 CAR-NK cells containing CD3ζ and CD28 costimulatory domains outperformed 1st generation CARs, whereas 2nd and 3rd generation CAR constructs did not seem to confer substantial differences in NK cell functionality (215, 217). Investigations on the optimal NK cell source were also conducted, where it was suggested that primary NK sources might be a better option than antiCD19 CAR-NK-92 cells (218, 219). In a comparison of CAR PB-NK versus CAR UCB-NK expressing the same antiCD19 CAR construct, it was shown that the former was better in eliminating CD19+ target cells in an effector to target ratio of 1:1 (46). However, the latter could be obtained at higher numbers with less inter-donor variability and stimulation with IL-2 and IL-15 improved more their functionality compared to CAR PB-NK cells. Naturally, the question of whether anti-CD19 CAR-T or CAR-NK cells had better efficacy was addressed. A study comparing CD19-targeting CAR T cell and CAR PB-NK cell anti-leukemia responses in vivo demonstrated prolonged survival and reduced adverse effects in mice treated with the CAR-NK cell product, highlighting the potential of CAR-NK therapy in CD19+ malignancies (216).
Other studies have evaluated the preclinical efficacy of CAR-NK cell products for the treatment of hematological malignancies targeting CD5, CD20, CD38, FLT3 or B7H7. All these CAR-NK cell products showed enhanced antitumor responses against target cells expressing the respective antigen (65, 125, 131, 138, 190, 220).
CAR-NK therapy has been evaluated as a therapeutic option of various solid tumors, targeting different antigens for each application. More specifically, CAR-NK cell therapy has been assessed in ovarian cancer (NKG2D ligands (NKG2DL), PSMA, FRα, CD24, HER2 or mesothelin) (113, 124, 221–224), glioblastoma (NKG2DL, EGFRvIII and ErbB2) (128, 225, 226), colorectal cancer (NKG2DL and EpCAM)124,187, prostate cancer (PSMA and NKG2DL) (128, 227), hepatocellular carcinomas (c-MET, GPC3 or CD147) (70, 127, 228), pancreatic cancer (mesothelin and FRα) (151, 229, 230), high-risk myosarcoma (ErbB2) (231), gastric cancer (HER2) (170), breast cancer (ErbB2, EGFR and TF) (192, 232, 233), head and neck cancer (PD-L1) (234, 235), neuroblastomas and melanoma (GD2) (236) and lung cancer (NKG2DL and EGFR) (128, 190). Overall, these preclinical studies showed superior antitumor responses in vitro and/or in vivo compared to non-transduced or control NK cells. However, solid tumors pose additional challenges for CAR-NK cell efficacy, namely intra-tumor infiltration, tumor trafficking, immunosuppressive microenvironment, among others (237). Strategies to overcome these issues and enhance CAR-NK cell functionality against solid tumors have been previously discussed.
A number of studies have explored the potential of CAR-NK therapy for the treatment of infectious diseases, such as AIDS (acquired immunodeficiency syndrome) and COVID-19 (coronavirus disease). Regarding AIDS, a universal CAR-NK cell product was designed to recognize 2,4-dinitrophenyl (DNP)-tagged antibodies that target the gp160 glycoprotein expressed on the HIV-infected cells (238). The study demonstrated effective degranulation against gp160+ cells, as well as killing of HIV-infected primary CD4+ T cells. Although a comparison between DNP CAR-NK cells and anti-gp-160 CAR-NK cells showed the first to be less cytotoxicity, the versatility of the approach and the ability to target multiple variants/isoforms of the HIV gp160 glycoprotein depending on the DNP-tagged antibodies used is an advantage. Therefore, considering the high mutational rate of HIV, the universal DNP CAR-NK cell product poses a very attractive and potentially effective strategy to treat AIDS.
COVID-19 is an infectious disease caused by the recently emerged SARS-CoV-2 virus (239). The virus can cause severe acute respiratory syndrome, for which no effective treatment exists at the moment. Independent studies have explored the potential of ‘off-the-shelf’ CAR-NK cell therapy in this setting, although only one of them is published in a peer-reviewed paper. In that study, Ma and colleagues generated a CAR-NK-92 cell product using the scFv domain of the neutralizing antibody S309, which recognizes a highly conserved region of the virus’ spike glycoprotein (240). S309 CAR-NK-92 cells showed increased degranulation and cytotoxicity in vitro, while targeting four different variants of the spike protein. Other studies generated similar spike protein-targeting constructs displaying promising results (241).
The incapability of follicular helper CD4+ T cells (TFH) to prevent aberrant immune responses is associated with the development of several autoimmune diseases (242–244). Current therapeutic strategies are insufficient in providing a permanent solution and are additionally causing serious side effects. CAR-NK therapy holds potential, as it can confer targeted elimination of the pathological immune cells in the autoimmune milieu. For their approach, Reighard and colleagues targeted PD-1, a marker moderately expressed on physiological cells, but overexpressed on TFH cells (245). They generated antiPD-1 CAR-NK-92 cells and reported cytotoxicity against PD-1high but not PD-1low cells in vitro studies. The results were further validated in an NSG lupus-like mouse model. Autoantibodies have been described in many other autoimmune diseases, such as inflammatory bowel disease and rheumatoid arthritis, where the potential of CAR-NK cell therapy could be investigated.
To our knowledge, only a few studies evaluating the clinical efficacy of CAR-NK cell for the treatment of hematological malignancies and solid tumors have been published to date. The clinical studies evaluated NK-92, PB-NK and UCB-based CAR-NK cell products.
A phase I clinical study in patients with AML evaluated the safety of (60) Co-irradiated antiCD33 CAR-NK-92 cells. The cells were transduced with a lentiviral vector encoding for a 3rd generation CAR with CD28, 4-1BB and CD3ζ co-stimulatory domains (246). All three of the enrolled patients were recruited after experiencing relapse from at least one chemotherapeutic regimen. They displayed up to 37,5% blasts in the BM, of which 20.4 to 99.9% were CD33+. Overall, the treatment was found well-tolerated by all patients and the maximum tolerable dose was not reached even with the dose of 50 billion cells. The first two patients were diagnosed with grade I cytokine release syndrome (CRS). To evaluate the response to the treatment, bone marrow aspirates were collected 1-4 months post-infusion. Only one of the patients achieved objective response (OR), but shortly relapsed and all three patients eventually reached a concentration of blasts of at least 75%. Analysis of CD33 positivity in two of the patients revealed 49% and 94,6% CD33+ blasts. The limited efficacy was hypothesized to be primarily due to the decreased cytotoxic potency that the CAR-NK-92 cells had after the irradiation step, as well as due to the insufficient phenotypic evaluation of the CD33+/high AML populations. As a solution, the researchers proposed treatment with non-irradiated CAR-NK-92 cells engineered with a suicide gene. This could allow a better control of the lifespan and proliferation of the NK-92 lymphoma cell line in the patients whilst maintaining high viability and cytotoxicity. Further improvements would be the optimization of the CAR construct towards recognizing different CD33 isoforms present in AML patients, and/or the targeting of alternative antigens. Finally, the authors acknowledge the need to elucidate the factors that determine CAR-NK-92 responsiveness in AML in order to better predict the response in patients.
Another study published the results of the clinical evaluation of anti-Robo1 CAR-NK-92 to a patient with pancreatic ductal adenocarcinoma and liver metastasis (NCT03941457) (247). Here, cells were generated with a lentiviral vector carrying a 2nd generation CAR with 4-1BB and CD3ζ co-stimulatory domains. The patient was refractory to chemotherapy and had Robo1+ non-operative tumor. The patient was infused with 10 billion anti-Robo1 CAR-NK-92 cells on days 1 and 3, and liver metastasis was treated with percutaneous administration of this CAR product on days 2 and 4. Overall, no substantial treatment-related adverse events were reported. The patient progressed two months after the final infusion, with an overall survival of 8 months.
CAR-transduced UCB- and PB-NK cells have also been investigated in clinical trials. The promising preclinical results obtained with CAR UCB-NK cells co-transduced with IL-15 and iCasp9 genes encouraged the investigation of the clinical efficacy of this therapy (41). Therefore, a phase I/II study was published 2 years later using UCB-NK cells retrovirally transduced with a 2nd generation antiCD19 CAR construct endowed with CD28 and CD3ζ intracellular signaling domains (NCT03056339) (140). In addition, these UCB-NK cells were also transduced with IL-15 gene and iCasp9. Briefly, a single dose of HLA-mismatched antiCD19 CAR UCB-NK cells was administered to 11 relapsed/refractory patients with CD19+ lymphomas after undergoing lymphodepleting chemotherapy. These patients already received a median of 4 lines of therapy before. The administered doses ranged from 1 to 100 x 10 (5) cells per kg. The maximum tolerated dose was not reached, and no CRS, neurotoxicity or GvHD was detected. Despite the HLA-mismatch, CAR-NK cells were found at least 12 months after infusion, probably due to the inclusion of IL-15 gene in the engineered cells. With a median follow-up of 13.8 months, the ORR is 73% (8 patients), with 7 patients showing CR and 1 PR.
Last, colorectal cancer patients were treated with anti-NKG2DL CAR-expressing autologous or allogeneic PB-NK cells in an haploidentical setting (125). The product was generated by mRNA electroporation of the CAR gene which contained a single DAP12 co-stimulatory domain. Intraperitoneal infusion reduced EpCAM+ cancer cells in two patients, while a third patient showed tumor size reduction four days after the first injection with allogeneic CAR-NK cells. In addition, the third patient showed almost no uptake of fludeoxyglucose by PET/CT imaging after completion of the treatment, an indication of tumor regression. Tumor sites injected with CAR-NK cells demonstrated necrotic lesions, which were not apparent in non-injected tumor sites. Moreover, CAR-NK cell injected tumor regions showed loss of expression of the NKG2D ligands MICA/B, Villin and CDX2 (markers of adenocarcinoma of intestinal origin), supporting the argument of local antitumor effect in the patients.
The current ongoing clinical studies evaluating the safety and efficacy of CAR-NK cell therapy in various indications are summarized in Table 2. We found that about half of the listed trials are based on the NK-92 cell line. These much-anticipated results are believed to shed light on the potential of NK cell lines as the source of off-the-shelf CAR-NK cell products. The rest of the trials with disclosed information concern mostly PB- and UCB-based CAR-NK cells, while there is also one iPSC-based CAR-NK cell trial (NCT04245722). Being the first registered trial investigating the clinical efficacy and tolerability of this approach, the insights gained from this study could boost further investigations of iPSC-derived CAR-NK cells in the clinical setting.
Even though the applicability of CAR-NK cell therapy in various indications has been proved preclinically, it is clear that the vast majority of the registered clinical trials is focusing on cancer. CD19 remains the most commonly targeted antigen (34% of the trials), while ROBO1 and NKG2DL are being increasingly investigated, counting for about 10% of the listed trials each. It is also worth mentioning that although cell therapy is traditionally used to treat hematological malignancies, there are currently 10 registered trials focusing on solid tumors. Moreover, for the first time, CAR-NK cells are under clinical investigation for the treatment of the pandemic-causing infectious disease COVID-19 (NCT04324996).
As previously mentioned, NK cell-based co-stimulatory domains may induce more potent CAR-NK cell-mediated antitumor responses compared to T cell-based CARs (113, 122–126). The potential of NK cell-based CARs is being increasingly investigated in the clinical setting, counting for 37% of the trials with relative disclosed information. The results of these studies are believed to influence the next generation of CAR-NK therapies.
Lastly, it is worth mentioning that non-CAR genetically engineered NK cell products are also under clinical investigation (e.g., NCT03656705 or NCT04023071). The reports of these results are highly anticipated.
In Figure 1 we have provided a schematic representation of the different aspects involved in the development of a CAR-NK cell therapy, as well as recent advances of the field. The success story of CAR therapy, as well as the advances in receptor engineering, inspired the development of other constructs for T and NK cell therapy of cancer. Some of the ones that have been applied to NK cells are listed below.
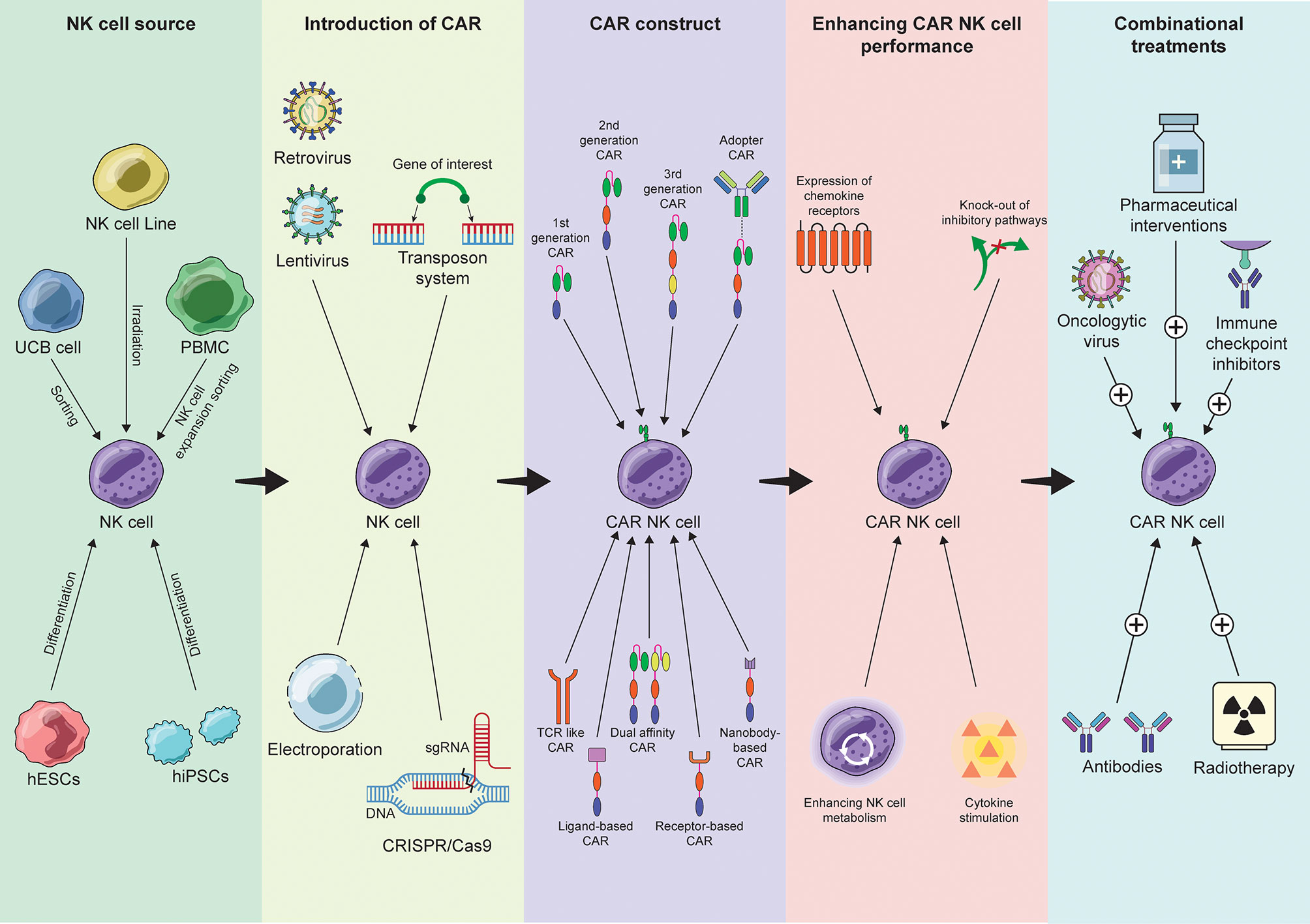
Figure 1 Summary of the recent advances in the process of CAR-NK cell therapy development from NK cell source selection to combinational approaches.
Dimeric antigen receptors, or DARs, are a novel category of artificial receptors that share many of the transmembrane and the signal transduction compartments of the CARs (248). While the antigen-targeting domain of the CARs is a scFv domain, however, DARs utilize the whole Fab part of the antibody. This is presumed to increase the stability of the synapse, as well as the targeting specificity. To this day, evidence on the preclinical efficacy of the DARs has only been provided by T cell-based approaches for the treatment of relapsed/refractory multiple myeloma. Nevertheless, the preclinical evaluation of anti-CD38 iPSC-derived DAR-NK cells for the same indication has also been announced.
As TME is a major factor dictating the success of an immunotherapy, strategies have been developed to switch the negative effects of immune suppression into positive, using chimeric switch receptors (CSRs). CSRs are cleverly designed to bind to inhibitory ligands on the malignant cells and transmit activating signal instead, thanks to their intracellular signaling domain. The ectodomains of the checkpoint inhibitors PD1 (249), TIGIT (250) and CTLA-4 (251) are particularly attractive and have shown promising results in T cell studies with regards to resistance to immunosuppression and restored effector function (252). Application of the approach to NK cells is gaining popularity the last years. Notably, a study on NK-92 expressing a novel PD1-NKG2D-41BB receptor demonstrated rapid elimination of PD1+ lung cancer target cells in an in vitro setting (253). A similar re-targeting approach is the chimeric chemokine receptors (CCRs). Although the potential of CCR-NK cells has yet to be explored, co-expression of chemokine receptors, such as CCR2b, has shown increased migration of CAR-T cells to the site of the malignancy (254).
The genetic modification of NK cells to express tumor specific T cell receptors (TCRs) has recently been attempted in vitro. In contrast to CARs that bind to cell surface antigens, TCRs can recognize antigenic peptides from degraded protein presented on the MHC, and therefore, are theoretically less restricted by the localization of the targeted molecule. An obstacle to the TCR expressing NK cells, however, is the lack of the accessory TCR signaling components that are present in T cells. Taking this into consideration, the engineering of TCR-NK cells becomes more challenging as the presence of the CD3 complex on the cell surface is necessary for the TCR to be functional. Mensali et al, in 2019, provided evidence that TCRs can be successfully expressed on NK-92 cells and that are able to mediate pMHC-specific cytotoxicity (255).
CAR-NK therapy has given new hope to the patients battling ‘incurable’ diseases and a new platform for researchers to explore the potential of CAR-based cell therapy. Although the challenges regarding the ex vivo expansion of the cells, in vivo persistence and insufficient cell trafficking remain, recent advances in cell and molecular biology provide viable solutions. Furthermore, CAR-NK cells are proven versatile and customizable, which expands their applicability to diseases beyond cancer. Looking into the future, next generation CAR-NK therapy is incorporating more and more state-of-the-art technology, adapting from the discoveries of CAR-T research, but also harnessing the unique features of NK cells. Taken together, CAR-NK therapy is believed to play an even greater role in the clinics in the forthcoming years, by providing efficient and safe off-the-shelf products.
MK, MV-M, AL, and EA contributed equally for this review. All authors contributed to the article and approved the submitted version.
EA is a founder of Vycellix and XNK and he is a consultant for Vycellix.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yoon SR, TD K, Choi I. Understanding of Molecular Mechanisms in Natural Killer Cell Therapy. Exp Mol Med (2015) 47(2). doi: 10.1038/emm.2014.114
2. Bryceson YT, March ME, Barber DF, Ljunggren HG, Long EO. Cytolytic Granule Polarization and Degranulation Controlled by Different Receptors in Resting NK Cells. J Exp Med (2005) 202(7):1001–12. doi: 10.1084/jem.20051143
3. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as Cell Surface Ligands for the Human DNAM-1 (CD226) Activating Molecule. J Exp Med (2003) 198(4):557–67. doi: 10.1084/jem.20030788
4. Pazina T, Shemesh A, Brusilovsky M, Porgador A, Campbell KS. Regulation of the Functions of Natural Cytotoxicity Receptors by Interactions With Diverse Ligands and Alterations in Splice Variant Expression. Front Immunol (2017) 8:369. doi: 10.3389/fimmu.2017.00369
5. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA Damage Pathway Regulates Innate Immune System Ligands of the NKG2D Receptor. Nature (2005) 436(7054):1186–90. doi: 10.1038/nature03884
6. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, et al. Interactions Between Siglec-7/9 Receptors and Ligands Influence NK Cell-Dependent Tumor Immunosurveillance. J Clin Invest (2014) 124(4):1810–20. doi: 10.1172/JCI65899
7. Amand M, Iserentant G, Poli A, Sleiman M, Fievez V, Sanchez IP, et al. Human CD56dimCD16dimCells As an Individualized Natural Killer Cell Subset. Front Immunol (2017) 8:699. doi: 10.3389/fimmu.2017.00699
8. Cai HH. Monoclonal Antibodies for Cancer Therapy Approved by FDA. MOJ Immunol (2016) 4(2):2–4. doi: 10.15406/moji.2016.04.00120
9. Li Y, Orange JS. Degranulation Enhances Presynaptic Membrane Packing, Which Protects NK Cells From Perforin-Mediated Autolysis. PloS Biol (2021) 19(8):1–29. doi: 10.1371/journal.pbio.3001328
10. Prager I, Liesche C, Van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK Cells Switch From Granzyme B to Death Receptor-Mediated Cytotoxicity During Serial Killing. J Exp Med (2019) 216(9):2113–27. doi: 10.1084/jem.20181454
11. Zhu Y, Huang B, Shi J. Fas Ligand and Lytic Granule Differentially Control Cytotoxic Dynamics of Natural Killer Cell Against Cancer Target. Oncotarget (2016) 7(30):47163–72. doi: 10.18632/oncotarget.9980
12. Bhat R, Watzl C. Serial Killing of Tumor Cells by Human Natural Killer Cells - Enhancement by Therapeutic Antibodies. PloS One (2007) 2(3). doi: 10.1371/journal.pone.0000326
13. Jewett A, Gan XH, Lebow LT, Bonavida B. Differential Secretion of TNF-α and IFN-γ by Human Peripheral Blood-Derived NK Subsets and Association With Functional Maturation. J Clin Immunol (1996) 16(1):46–54. doi: 10.1007/BF01540972
14. Blaser B, Caligiuri M. Natural Killer Cells: Development, Receptor Biology, and Their Role in Cancer Immunotherapy. Hematologist (2008) 5(1). doi: 10.1182/hem.V5.1.6144
15. Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-Grade, Large-Scale, Feeder-Free Expansion of Highly Active Human Natural Killer Cells for Adoptive Immunotherapy Using an Automated Bioreactor. Cytotherapy (2010) 12(8):1044–55. doi: 10.3109/14653249.2010.504770
16. Saito S, Harada Y, Morodomi Y, Onimaru M, Yoshida K, Kyuragi R, et al. Ex Vivo Generation of Highly Purified and Activated Natural Killer Cells From Human Peripheral Blood. Hum Gene Ther Methods (2013) 24(4):241–52. doi: 10.1089/hgtb.2012.183
17. Gao C, Li X, Ma J, Wu X, Wang F, Li M, et al. Ex Vivo Expansion of Highly Purified Human NK Cells. Blood (2009) 114(22):2157. doi: 10.1182/blood.V114.22.2157.2157
18. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I Clinical Trial of Autologous NK Cell Therapy Using Novel Expansion Method in Patients With Advanced Digestive Cancer. J Trans Med (2015) 13(1):1–13. doi: 10.1186/s12967-015-0632-8
19. Granzin M, Soltenborn S, Müller S, Kollet J, Berg M, Cerwenka A, et al. Fully Automated Expansion and Activation of Clinical-Grade Natural Killer Cells for Adoptive Immunotherapy. Cytotherapy (2015) 17(5):621–32. doi: 10.1016/j.jcyt.2015.03.611
20. Gong W, Xiao W, Hu M, Weng X, Qian L, Pan X, et al. Ex Vivo Expansion of Natural Killer Cells With High Cytotoxicity by K562 Cells Modified to Co-Express Major Histocompatibility Complex Class I Chain-Related Protein A, 4-1BB Ligand, and Interleukin-15. Tissue Antigens (2010) 76(6):467–75. doi: 10.1111/j.1399-0039.2010.01535.x
21. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of Highly Cytotoxic Human Natural Killer Cells for Cancer Cell Therapy. Cancer Res (2009) 69(9):4010–7. doi: 10.1158/0008-5472.CAN-08-3712
22. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar Pv, Kopp LM, Johnson JL, et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PloS One (2012) 7(1). doi: 10.1371/journal.pone.0030264
23. Wagner J, Pfannenstiel V, Waldmann A, Bergs JWJ, Brill B, Huenecke S, et al. A Two-Phase Expansion Protocol Combining Interleukin (IL)-15 and IL-21 Improves Natural Killer Cell Proliferation and Cytotoxicity Against Rhabdomyosarcoma. Front Immunol (2017) 8:676. doi: 10.3389/fimmu.2017.00676
24. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous Treatment With IL-15 Exhausts Human NK Cells via a Metabolic Defect. JCI Insight (2018) 3(3). doi: 10.1172/jci.insight.96219
25. Xu L, Li S, Stohr BA. The Role of Telomere Biology in Cancer. Annu Rev Pathology: Mech Dis (2013) 8:49–78. doi: 10.1146/annurev-pathol-020712-164030
26. Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative Potential of Human Natural Killer Cells. Br J Haematol (2009) 145(5):606–13. doi: 10.1111/j.1365-2141.2009.07667.x
27. Lieberman NAP, DeGolier K, Haberthur K, Chinn H, Moyes KW, Bouchlaka MN, et al. An Uncoupling of Canonical Phenotypic Markers and Functional Potency of Ex Vivo -Expanded Natural Killer Cells. Front Immunol (2018) 9:150(FEB). doi: 10.3389/fimmu.2018.00150
28. Alici E, Sutlu T, Björkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous Antitumor Activity by NK Cells Expanded From Myeloma Patients Using GMP-Compliant Components. Blood (2008) 111(6):3155–62. doi: 10.1182/blood-2007-09-110312
29. Kundu S, Gurney M, O’Dwyer M. Generating Natural Killer Cells for Adoptive Transfer: Expanding Horizons. Cytotherapy (2021) 23(7):559–66. doi: 10.1016/j.jcyt.2020.12.002
30. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells But Does Not Mediate Tumor Regression. Clin Cancer Res (2011) 17(19):6287–97. doi: 10.1158/1078-0432.CCR-11-1347
31. Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, et al. Bortezomib and Depsipeptide Sensitize Tumors to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand: A Novel Method to Potentiate Natural Killer Cell Tumor Cytotoxicity. Cancer Res (2006) 66(14):7317–25. doi: 10.1158/0008-5472.CAN-06-0680
32. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib Treatment and Regulatory T-Cell Depletion Enhance the Antitumor Effects of Adoptively Infused NK Cells. Blood (2009) 113(24):6120–7. doi: 10.1182/blood-2008-11-190421
33. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib Treatment to Potentiate the Anti-Tumor Immunity of Ex-Vivo Expanded Adoptively Infused Autologous Natural Killer Cells. J Cancer (2011) 2(1):383–5. doi: 10.7150/jca.2.383
34. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
35. Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural Killer Cell-Directed Therapies: Moving From Unexpected Results to Successful Strategies. Nat Immunol (2008) 9(5):486–94. doi: 10.1038/ni1580
36. Davis ZB, Felices M, Verneris MR, Miller JS. Natural Killer Cell Adoptive Transfer Therapy: Exploiting the First Line of Defense Against Cancer. Cancer J (United States) (2015) 21(6):486–91. doi: 10.1097/PPO.0000000000000156
37. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful Adoptive Transfer and In Vivo Expansion of Human Haploidentical NK Cells in Patients With Cancer. Blood (2005) 105(8):3051–7. doi: 10.1182/blood-2004-07-2974
38. Locatelli F, Moretta F, Brescia L, Merli P. Natural Killer Cells in the Treatment of High-Risk Acute Leukaemia. Semin Immunol (2014) 26(2):173–9. doi: 10.1016/j.smim.2014.02.004
39. Lin SJ, Kuo ML. Cytotoxic Function of Umbilical Cord Blood Natural Killer Cells: Relevance to Adoptive Immunotherapy. Pediatr Hematol Oncol (2011) 28(8):640–6. doi: 10.3109/08880018.2011.613092
40. Reina-Ortiz C, Constantinides M, Fayd-Herbe-de-Maudave A, Présumey J, Hernandez J, Cartron G, et al. Expanded NK Cells From Umbilical Cord Blood and Adult Peripheral Blood Combined With Daratumumab are Effective Against Tumor Cells From Multiple Myeloma Patients. OncoImmunology (2021) 10(1). doi: 10.1080/2162402X.2020.1853314
41. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
42. Zhu H, Kaufman DS. An Improved Method to Produce Clinical-Scale Natural Killer Cells From Human Pluripotent Stem Cells. Methods Mol Biol (2019) 2048:107–19. doi: 10.1007/978-1-4939-9728-2_12
43. Tanaka H, Kai S, Yamaguchi M, Misawa M, Fujimori Y, Yamamoto M, et al. Analysis of Natural Killer (NK) Cell Activity and Adhesion Molecules on NK Cells From Umbilical Cord Blood. Eur J Haematol (2003) 71(1):29–38. doi: 10.1034/j.1600-0609.2003.00081.x
44. Dalle JH, Menezes J, Wagner É, Blagdon M, Champagne J, Champagne MA, et al. Characterization of Cord Blood Natural Killer Cells: Implications for Transplantation and Neonatal Infections. Pediatr Res (2005) 57(5 I):649–55. doi: 10.1203/01.PDR.0000156501.55431.20
45. Sanchez-Martinez D, Allende-Vega N, Orecchioni S, Talarico G, Cornillon A, Vo DN, et al. Expansion of Allogeneic NK Cells With Efficient Antibody-Dependent Cell Cytotoxicity Against Multiple Tumors. Theranostics (2018) 8(14):3856–69. doi: 10.7150/thno.25149
46. Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult Peripheral Blood and Umbilical Cord Blood NK Cells are Good Sources for Effective CAR Therapy Against CD19 Positive Leukemic Cells. Sci Rep (2019) 9(1):1–10. doi: 10.1038/s41598-019-55239-y
47. Woll PS, Martin CH, Miller JS, Kaufman DS. Human Embryonic Stem Cell-Derived NK Cells Acquire Functional Receptors and Cytolytic Activity. J Immunol (2005) 175(8):5095–103. doi: 10.4049/jimmunol.175.8.5095
48. Luevano M, Madrigal A, Saudemont A. Generation of Natural Killer Cells From Hematopoietic Stem Cells In Vitro for Immunotherapy. Cell Mol Immunol (2012) 9(4):310–20. doi: 10.1038/cmi.2012.17
49. Knorr. PROTOCOLS AND MANUFACTURING FOR CELL-BASED THERAPIES Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Trans Med (2013) 274–83. Published online. doi: 10.5966/sctm.2012-0084
50. Goldenson BH, Zhu H, Wang YZM, Heragu N, Bernareggi D, Ruiz-Cisneros A, et al. Umbilical Cord Blood and iPSC-Derived Natural Killer Cells Demonstrate Key Differences in Cytotoxic Activity and KIR Profiles. Front Immunol (2020) 11:561553(October). doi: 10.3389/fimmu.2020.561553
51. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells From Adult Human Fibroblasts by Defined Factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
52. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
53. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced Pluripotent Stem Cell Lines Derived From Human Somatic Cells. Science (2007) 318(5858):1917–20. doi: 10.1126/science.1151526
54. Valamehr B, Robinson M, Abujarour R, Rezner B, Vranceanu F, Le T, et al. Platform for Induction and Maintenance of Transgene-Free hiPSCs Resembling Ground State Pluripotent Stem Cells. Stem Cell Rep (2014) 2(3):366–81. doi: 10.1016/j.stemcr.2014.01.014
55. Kaufman D. Human Pluripotent Stem Cell-Derived Natural Killer Cells for Anti-Cancer Therapy. Exp Hematol (2016) 44:S31. doi: 10.1016/j.exphem.2016.06.023
56. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJN, et al. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Trans Med (2013) 2(4):274–83. doi: 10.5966/sctm.2012-0084
57. Maali A, Atashi A, Ghaffari S, Kouchaki R, Abdolmaleki F, Azad M. A Review on Leukemia and iPSC Technology: Application in Novel Treatment and Future. Curr Stem Cell Res Ther (2018) 13(8). doi: 10.2174/1574888X13666180731155038
58. Zhu H, Kaufman DS. Engineered Human Pluripotent Stem Cell-Derived Natural Killer Cells: The Next Frontier for Cancer Immunotherapy. Blood Sci (2019) 1(1):4–11. doi: 10.1097/bs9.0000000000000023
59. Sun JC, Beilke JN, Lanier LL. Adaptive Immune Features of Natural Killer Cells. Nature (2009) 457(7229):557–61. doi: 10.1038/nature07665
60. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical Role for the Chemokine Receptor CXCR6 in NK Cell-Mediated Antigen-Specific Memory of Haptens and Viruses. Nat Immunol (2010) 11(12):1127–35. doi: 10.1038/ni.1953
61. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine Activation Induces Human Memory-Like NK Cells. Blood (2012) 120(24):4751–60. doi: 10.1182/blood-2012-04-419283
62. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-Induced Memory-Like Natural Killer Cells Exhibit Enhanced Responses Against Myeloid Leukemia. Sci Trans Med (2016) 8(357). doi: 10.1126/scitranslmed.aaf2341
63. Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-Modified Memory-Like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood (2020) 136(20):2308–18. doi: 10.1182/BLOOD.2020006619
64. Liu H, Yang B, Sun T, Lin L, Hu Y, Deng M, et al. Specific Growth Inhibition of ErbB2-Expressing Human Breast Cancer Cells by Genetically Modified NK-92 Cells. Oncol Rep (2015) 33(1):95–102. doi: 10.3892/or.2014.3548
65. Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 Knockout Natural Killer Cells Expressing an Affinity Optimized CD38 Chimeric Antigen Receptor Successfully Target Acute Myeloid Leukemia With Reduced Effector Cell Fratricide. Haematologica (2020). doi: 10.3324/haematol.2020.271908 Online ahe.
66. Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK Cell-Based Cancer Immunotherapy: From Basic Biology to Clinical Development. J Hematol Oncol (2021) 14(1). doi: 10.1186/s13045-020-01014-w
67. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the Allogeneic Cell Line NK-92 in Patients With Advanced Renal Cell Cancer or Melanoma: A Phase I Trial. Cytotherapy (2008) 10(6):625–32. doi: 10.1080/14653240802301872
68. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of Patients With Advanced Cancer With the Natural Killer Cell Line NK-92. Cytotherapy (2013) 15(12):1563–70. doi: 10.1016/j.jcyt.2013.06.017
69. Liu Q, Xu Y, Mou J, Tang K, Fu X, Li Y, et al. Irradiated Chimeric Antigen Receptor Engineered NK-92MI Cells Show Effective Cytotoxicity Against CD19+ Malignancy in a Mouse Model. Cytotherapy (2020) 22(10):552–62. doi: 10.1016/j.jcyt.2020.06.003
70. Tseng HC, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of Anti-CD147 Chimeric Antigen Receptors Targeting Hepatocellular Carcinoma. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-18444-2
71. Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, et al. Ionizing Radiation Induces Mitochondrial Reactive Oxygen Species Production Accompanied by Upregulation of Mitochondrial Electron Transport Chain Function and Mitochondrial Content Under Control of the Cell Cycle Checkpoint. Free Radical Biol Med (2012) 53(2):260–70. doi: 10.1016/j.freeradbiomed.2012.04.033
72. Smith TA, Kirkpatrick DR, Smith S, Smith TK, Pearson T, Kailasam A, et al. Radioprotective Agents to Prevent Cellular Damage Due to Ionizing Radiation. J Trans Med (2017) 15(1):1–18. doi: 10.1186/s12967-017-1338-x
73. Walcher L, Kistenmacher AK, Sommer C, Böhlen S, Ziemann C, Dehmel S, et al. Low Energy Electron Irradiation Is a Potent Alternative to Gamma Irradiation for the Inactivation of (CAR-)NK-92 Cells in ATMP Manufacturing. Front Immunol (2021) 12:684052. doi: 10.3389/fimmu.2021.684052
74. Mark C, Czerwinski T, Roessner S, Mainka A, Hörsch F, Heublein L, et al. Cryopreservation Impairs 3-D Migration and Cytotoxicity of Natural Killer Cells. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-19094-0
75. Domogala A, Alejandro Madrigal J, Saudemont A. Cryopreservation has No Effect on Function of Natural Killer Cells Differentiated In Vitro From Umbilical Cord Blood CD34+ Cells. Cytotherapy (2016) 18(6):754–9. doi: 10.1016/j.jcyt.2016.02.008
76. Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and Homing of Adoptively Transferred Human Natural Killer Cells in Immunodeficient Mice Varies With Product Preparation and InVivo Cytokine Administration: Implications for Clinical Therapy. Biol Blood Marrow Transplant (2014) 20(8):1252–7. doi: 10.1016/j.bbmt.2014.05.004
77. Rui L, Johnson R, Yu G, McKenna DH, Hubel A. Preservation of Cell-Based Immunotherapies for Clinical Trials. Cytotherapy (2019) 21(9):943–57. doi: 10.1016/j.jcyt.2019.07.004
78. Alici E, Sutlu T, Sirac Dilber M. Retroviral Gene Transfer Into Primary Human Natural Killer Cells. Methods Mol Biol (2009) 506:127–37. doi: 10.1007/978-1-59745-409-4_10
79. Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol Ther (2017) 25(8):1769–81. doi: 10.1016/j.ymthe.2017.06.012
80. Matosevic S. Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. J Immunol Res (2018) 2018. doi: 10.1155/2018/4054815
81. Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human CAR NK Cells: A New non-Viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front Immunol (2019) 10:957(APR). doi: 10.3389/fimmu.2019.00957
82. Rols MP. Parameters Affecting Cell Viability Following Electroporation In Vitro. Handb Electroporation (2017) 1-4:1–2998. doi: 10.1007/978-3-319-32886-7
83. Lambert M, Leijonhufvud C, Segerberg F, Melenhorst JJ, Carlsten M. CRISPR/Cas9-Based Gene Engineering of Human Natural Killer Cells: Protocols for Knockout and Readouts to Evaluate Their Efficacy. Methods Mol Biol (2020) 2121:213–39. doi: 10.1007/978-1-0716-0338-3_18
84. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I Trials Using Sleeping Beauty to Generate CD19-Specific CAR T Cells. J Clin Invest (2016) 126(9):3363–76. doi: 10.1172/JCI86721
85. Gregory T, Cohen AD, Costello CL, Ali SA, Berdeja JG, Ostertag EM, et al. Efficacy and Safety of P-Bcma-101 CAR-T Cells in Patients With Relapsed/Refractory (R/R) Multiple Myeloma (Mm). Blood (2018) 132(1):1012. doi: 10.1182/blood-2018-99-111419
86. Thaci B, Ulasov IV, Wainwright DA, Lesniak MS. The Challenge for Gene Therapy: Innate Immune Response to Adenoviruses. Oncotarget (2011) 2(3):113–21. doi: 10.18632/oncotarget.231
87. Nagashima S, Mailliard R, Kashii Y, Reichert TE, Herberman RB, Robbins P, et al. Stable Transduction of the Interleukin-2 Gene Into Human Natural Killer Cell Lines and Their Phenotypic and Functional Characterization In Vitro and In Vivo. Blood (1998) 91(10):3850–61. doi: 10.1182/blood.v91.10.3850
88. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SMH, Coustan-Smith E, et al. Autonomous Growth and Increased Cytotoxicity of Natural Killer Cells Expressing Membrane-Bound Interleukin-15. Blood (2014) 124(7):1081–8. doi: 10.1182/blood-2014-02-556837
89. Imai C, Iwamoto S, Campana D. Genetic Modification of Primary Natural Killer Cells Overcomes Inhibitory Signals and Induces Specific Killing of Leukemic Cells. Blood (2005) 106(1):376–83. doi: 10.1182/blood-2004-12-4797
90. Guven H, Konstantinidis KV, Alici E, Aints A, Abedi-Valugerdi M, Christensson B, et al. Efficient Gene Transfer Into Primary Human Natural Killer Cells by Retroviral Transduction. Exp Hematol (2005) 33(11):1320–8. doi: 10.1016/j.exphem.2005.07.006
91. Loew R, Meyer Y, Kuehlcke K, Gama-Norton L, Wirth D, Hauser H, et al. A New PG13-Based Packaging Cell Line for Stable Production of Clinical-Grade Self-Inactivating γ-Retroviral Vectors Using Targeted Integration. Gene Ther (2010) 17(2):272–80. doi: 10.1038/gt.2009.134
92. Poletti V, Mavilio F. Interactions Between Retroviruses and the Host Cell Genome. Mol Ther - Methods Clin Dev (2018) 8(March):31–41. doi: 10.1016/j.omtm.2017.10.001
93. Suerth JD, Morgan MA, Kloess S, Heckl D, Neudörfl C, Falk CS, et al. Efficient Generation of Gene-Modified Human Natural Killer Cells via Alpharetroviral Vectors. J Mol Med (2016) 94(1):83–93. doi: 10.1007/s00109-015-1327-6
94. Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front Immunol (2019) 10:2873. doi: 10.3389/fimmu.2019.02873
95. Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front Immunol (2019) 10:2001(AUG). doi: 10.3389/fimmu.2019.02001
96. Milone MC, O’Doherty U. Clinical Use of Lentiviral Vectors. Leukemia (2018) 32(7):1529–41. doi: 10.1038/s41375-018-0106-0
97. Davis HE, Morgan JR, Yarmush ML. Polybrene Increases Retrovirus Gene Transfer Efficiency by Enhancing Receptor-Independent Virus Adsorption on Target Cell Membranes. Biophys Chem (2002) 97(2-3):159–72. doi: 10.1016/S0301-4622(02)00057-1
98. Takarabio.com. RetroNectin Reagent (2021). Available at: https://www.takarabio.com/products/gene-function/t-cell-transduction-and-culture/retronectin-reagent (Accessed September 24, 2021).
99. Hanenberg H, Li Xiao X, Dilloo D, Hashino K, Kato I, Williams DA. Colocalization of Retrovirus and Target Cells on Specific Fibronectin Fragments Increases Genetic Transduction of Mammalian Cells. Nature (1996) 2(5):601–3. doi: 10.1038/nm0896-876
100. Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front Immunol (2020) 10:3123. doi: 10.3389/fimmu.2019.03123
101. Sutlu T, Nyström S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of Intracellular Antiviral Defense Mechanisms Augments Lentiviral Transduction of Human Natural Killer Cells: Implications for Gene Therapy. Hum Gene Ther (2012) 23(10):1090–100. doi: 10.1089/hum.2012.080
102. Alici E, Duru A, Sutlu T. Enhanced Gene Delivery to Natural Killer Cells, Hematopoietic Stem Cells and Macrophages. (2019). Published online.
103. Mangeot PE, Risson V, Fusil F, Marnef A, Laurent E, Blin J, et al. Genome Editing in Primary Cells and In Vivo Using Viral-Derived Nanoblades Loaded With Cas9-sgRNA Ribonucleoproteins. Nat Commun (2019) 10(1):1–15. doi: 10.1038/s41467-018-07845-z
104. Gutierrez-Guerrero A, Abrey Recalde MJ, Mangeot PE, Costa C, Bernadin O, Périan S, et al. Baboon Envelope Pseudotyped “Nanoblades” Carrying Cas9/gRNA Complexes Allow Efficient Genome Editing in Human T, B, and CD34+ Cells and Knock-In of AAV6-Encoded Donor DNA in CD34+ Cells. Front Genome Editing (2021) 3:604371(February). doi: 10.3389/fgeed.2021.604371
105. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther - Methods Clin Dev (2019) 12(March):145–56. doi: 10.1016/j.omtm.2018.12.009
106. Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv Structure in Chimeric Antigen Receptor on Receptor Expression Efficiency and Antigen Recognition Properties. Biochem Biophys Res Commun (2020) 527(2):350–7. doi: 10.1016/j.bbrc.2020.03.071
107. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther (2017) 25(8):1946–58. doi: 10.1016/j.ymthe.2017.04.024
108. Krokhotin A, Du H, Hirabayashi K, Popov K, Kurokawa T, Wan X, et al. Computationally Guided Design of Single-Chain Variable Fragment Improves Specificity of Chimeric Antigen Receptors. Mol Ther - Oncolytics (2019) 15(December):30–7. doi: 10.1016/j.omto.2019.08.008
109. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of Human T Cells to Tumor-Associated MUC1: The Evolution of a Chimeric Antigen Receptor. J Immunol (2008) 180(7):4901–9. doi: 10.4049/jimmunol.180.7.4901
110. Hombach A, Hombach AA, Abken H. Adoptive Immunotherapy With Genetically Engineered T Cells: Modification of the IgG1 Fc Spacer Domain in the Extracellular Moiety of Chimeric Antigen Receptors Avoids Off-Target Activation and Unintended Initiation of an Innate Immune Response. Gene Ther (2010) 17(10):1206–13. doi: 10.1038/gt.2010.91
111. Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, et al. Incorporation of a Hinge Domain Improves the Expansion of Chimeric Antigen Receptor T Cells. J Hematol Oncol (2017) 10(1):1–11. doi: 10.1186/s13045-017-0437-8
112. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of Novel Anti-CD19 Chimeric Antigen Receptors With Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol Ther (2017) 25(11):2452–65. doi: 10.1016/j.ymthe.2017.07.013
113. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell (2018) 23(2):181–192.e5. doi: 10.1016/j.stem.2018.06.002
114. Eshhar Z, Waks T, Gross G, Schindler DG. Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody-Binding Domains and the γ or ζ Subunits of the Immunoglobulin and T-Cell Receptors. Proc Natl Acad Sci USA (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
115. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric Receptors Providing Both Primary and Costimulatory Signaling in T Cells From a Single Gene Product. J Immunol (Baltimore Md: 1950) (1998) 161(6):2791–7.
116. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of Large, Established Tumor Xenografts With Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc Natl Acad Sci USA (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
117. Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
118. Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel Comparison of 4-1BB or CD28 Co-Stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol Ther - Oncolytics (2019) 15(52):60–8. doi: 10.1016/j.omto.2019.08.002
119. Tran AC, Zhang D, Byrn R, Roberts MR. Chimeric Zeta-Receptors Direct Human Natural (1995). Available at: http://www.jimmunol.org/content/155/.
120. Lanier LL. NK Cell Recognition. Annu Rev Immunol (2005) 23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526
121. Christodoulou I, Ho WJ, Marple A, Ravich JW, Tam A, Rahnama R, et al. Engineering CAR-NK Cells to Secrete IL-15 Sustains Their Anti-AML Functionality But is Associated With Systemic Toxicities. J ImmunoTherapy Cancer (2021) 9(12):e003894. doi: 10.1136/jitc-2021-003894
122. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A Chimeric Receptor With NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res (2013) 73(6):1777–86. doi: 10.1158/0008-5472.CAN-12-3558
123. Guo C, Wang X, Zhang H, Zhi L, Lv T, Li M, et al. Structure-Based Rational Design of a Novel Chimeric PD1-NKG2D Receptor for Natural Killer Cells. Mol Immunol (2019) 114(May):108–13. doi: 10.1016/j.molimm.2019.07.009
124. Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J Immunol (2015) 194(7):3201–12. doi: 10.4049/jimmunol.1400330
125. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27(6):1114–25. doi: 10.1016/j.ymthe.2019.03.011
126. Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 Costimulatory Domain Enhancing Cytotoxic Ability of Anti-CD5 Chimeric Antigen Receptor Engineered Natural Killer Cells Against T Cell Malignancies. J Hematol Oncol (2019) 12(1):1–13. doi: 10.1186/s13045-019-0732-7
127. Liu B, Liu ZZ, Zhou ML, Lin JW, Chen XM, Li Z, et al. Development of C-MET-Specific Chimeric Antigen Receptor-Engineered Natural Killer Cells With Cytotoxic Effects on Human Liver Cancer HepG2 Cells. Mol Med Rep (2019) 20(3):2823–31. doi: 10.3892/mmr.2019.10529
128. Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic Targeting Enhances Immunotherapy of CD73+ Solid Tumors With Piggybac-Engineered Chimeric Antigen Receptor Natural Killer Cells. J ImmunoTherapy Cancer (2018) 6(1):1–14. doi: 10.1186/s40425-018-0441-8
129. Tan Y, Cai H, Li C, Deng B, Song W, Ling Z, et al. A Novel Full-Human CD22-CAR T Cell Therapy With Potent Activity Against CD22low B-ALL. Blood Cancer J (2021) 11(4). doi: 10.1038/s41408-021-00465-9
130. Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, et al. Fully Human CD19-Specific Chimeric Antigen Receptors for T-Cell Therapy. Leukemia (2017) 31(10):2191–9. doi: 10.1038/leu.2017.57
131. Zhuang X, Long EO. CD28 Homolog is a Strong Activator of Natural Killer Cells for Lysis of B7H7+ Tumor Cells. Cancer Immunol Res (2019) 7(6):939–51. doi: 10.1158/2326-6066.CIR-18-0733
132. Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annu Rev Biochem (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
133. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF. A New Antigen Receptor Gene Family That Undergoes Rearrangement and. Nature (1995) 374(9):168–73. doi: 10.1038/374168a0
134. Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody-Based Chimeric Antigen Receptor T Cells Designed by CRISPR/Cas9 Technology for Solid Tumor Immunotherapy. Signal Transduct Target Ther (2021) 6(1). doi: 10.1038/s41392-021-00462-1
135. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies as Antitumor Therapeutics. Front Immunol (2017) 8:1603(NOV). doi: 10.3389/fimmu.2017.01603
136. Bao C, Gao Q, Li LL, Han L, Zhang B, Ding Y, et al. The Application of Nanobody in CAR-T Therapy. Biomolecules (2021) 11(2):1–18. doi: 10.3390/biom11020238
137. You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A Novel CD7 Chimeric Antigen Receptor-Modified NK-92MI Cell Line Targeting T-Cell Acute Lymphoblastic Leukemia. Am J Cancer Res (2019) 9(1):64–78.
138. Hambach J, Riecken K, Cichutek S, Schütze K, Albrecht B, Petry K, et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro With Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells (2020) 9(321):1–14. doi: 10.3390/cells9020321
139. Hacisuleyman A, Erman B. ModiBodies: A Computational Method for Modifying Nanobodies in Nanobody-Antigen Complexes to Improve Binding Affinity and Specificity. J Biol Phys (2020) 46(2):189–208. doi: 10.1007/s10867-020-09548-3
140. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. New Engl J Med (2020) 382(6):545–53. doi: 10.1056/nejmoa1910607
141. Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Henri Bayle J. Inducible MyD88/CD40 Synergizes With IL-15 to Enhance Antitumor Efficacy of CAR-NK Cells. Blood Adv (2020) 4(9):1950–64. doi: 10.1182/bloodadvances.2020001510
142. Yu S, Yi M, Qin S, Wu K. Next Generation Chimeric Antigen Receptor T Cells: Safety Strategies to Overcome Toxicity. Mol Cancer (2019) 18(1). doi: 10.1186/s12943-019-1057-4
143. Gargett T, Brown MP. The Inducible Caspase-9 Suicide Gene System as a “Safety Switch” to Limit on-Target, Off-Tumor Toxicities of Chimeric Antigen Receptor T-Cells. Front Pharmacol (2014) 5:235. doi: 10.3389/fphar.2014.00235
144. Andrea AE, Chiron A, Bessoles S, Hacein-Bey-abina S. Engineering Next-Generation Car-T Cells for Better Toxicity Management. Int J Mol Sci (2020) 21(22):1–25. doi: 10.3390/ijms21228620
145. Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual Targeting of Glioblastoma With Chimeric Antigen Receptor-Engineered Natural Killer Cells Overcomes Heterogeneity of Target Antigen Expression and Enhances Antitumor Activity and Survival. OncoImmunology (2016) 5(4):1–12. doi: 10.1080/2162402X.2015.1119354
146. Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A Novel Bispecific Chimeric PD1-DAP10/NKG2D Receptor Augments NK92-Cell Therapy Efficacy for Human Gastric Cancer SGC-7901 Cell. Biochem Biophys Res Commun (2020) 523(3):745–52. doi: 10.1016/j.bbrc.2020.01.005
147. Rafei H, Daher M, Rezvani K. Chimeric Antigen Receptor (CAR) Natural Killer (NK)-Cell Therapy: Leveraging the Power of Innate Immunity. Br J Haematol (2021) 193(2):216–30. doi: 10.1111/bjh.17186
148. Grote S, Mittelstaet J, Baden C, Chan KCH, Seitz C, Schlegel P, et al. Adapter Chimeric Antigen Receptor (AdCAR)-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Universal Tumor Targeting. OncoImmunology (2020) 9(1):1–11. doi: 10.1080/2162402X.2020.1825177
149. Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR4. Front Immunol (2020) 11:2028. doi: 10.3389/fimmu.2020.02028
150. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK Cells Modified With an EGFRvIII-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-Secreting Glioblastoma. J Immunother (2015) 38(5):197–210. doi: 10.1097/CJI.0000000000000082
151. Lee YE, Ju A, Choi HW, Kim JC, Kim EEK, Kim TS, et al. Rationally Designed Redirection of Natural Killer Cells Anchoring a Cytotoxic Ligand for Pancreatic Cancer Treatment. J Controlled Rel (2020) 326(April):310–23. doi: 10.1016/j.jconrel.2020.07.016
152. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC Locus With CRISPR/Cas9 Enhances Tumour Rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
153. Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted Primary Human Third-Generation CAR-T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells (2020) 9(4):1–20. doi: 10.3390/cells9040998
154. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β Inhibition via CRISPR Promotes the Long-Term Efficacy of CAR T Cells Against Solid Tumors. JCI Insight (2020) 5(4). doi: 10.1172/jci.insight.133977
155. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking Expression of Inhibitory Receptor NKG2A Overcomes Tumor Resistance to NK Cells. J Clin Invest (2019) 129(5):2094–106. doi: 10.1172/JCI123955
156. Malona R, Funaro A, Zubiar M, Baj G, Ausiello CM, Tacchetti C, et al. Signaling Through CD38 Induces NK Cell Activation. Int Immunol (2001) 13(4):397–409. doi: 10.1093/intimm/13.4.397
157. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. Mtorc1-Dependent Metabolic Reprogramming Is a Prerequisite for NK Cell Effector Function. J Immunol (2014) 193(9):4477–84. doi: 10.4049/jimmunol.1401558
158. Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic Reprogramming Supports IFN-γ Production by CD56 Bright NK Cells. J Immunol (2016) 196(6):2552–60. doi: 10.4049/jimmunol.1501783
159. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-Tumor Activity. Cell Stem Cell (2020) 27(2):224–37.e6. doi: 10.1016/j.stem.2020.05.008
160. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a Cytokine Checkpoint Enhances the Fitness of Armored Cord Blood CAR-NK Cells. Blood (2021) 137(5):624–36. doi: 10.1182/blood.2020007748
161. Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a Potent Checkpoint in NK Cell-Mediated Tumor Immunity. Nat Immunol (2016) 17(7):816–24. doi: 10.1038/ni.3470
162. Loftus RM, Assmann N, Kedia-Mehta N, O’Brien KL, Garcia A, Gillespie C, et al. Amino Acid-Dependent Cmyc Expression is Essential for NK Cell Metabolic and Functional Responses in Mice. Nat Commun (2018) 9(1):152–60. doi: 10.1038/s41467-018-04719-2
163. Parameswaran R, Ramakrishnan P, Moreton SA, Xia Z, Hou Y, Lee DA, et al. Repression of GSK3 Restores NK Cell Cytotoxicity in AML Patients. Nat Commun (2016) 7:1–11. doi: 10.1038/ncomms11154
164. Cichocki F, Valamehr B, Bjordahl R, Zhang B, Rezner B, Rogers P, et al. GSK3 Inhibition Drives Maturation of NK Cells and Enhances Their Antitumor Activity. Cancer Res (2017) 77(20):5664–75. doi: 10.1158/0008-5472.CAN-17-0799
165. Leone RD, Zhao L, Englert JM, Min-Hee Oh IS, Matthew L, Arwood I-HS, et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science (2019) 366(6468):1012–21. doi: 10.1126/science.aav2588
166. Choi C, Finlay DK. Optimising NK Cell Metabolism to Increase the Efficacy of Cancer Immunotherapy. Stem Cell Res Ther (2021) 12(1):1–10. doi: 10.1186/s13287-021-02377-8
167. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-Cell Infiltration Into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov (2014) 4(5):522–6. doi: 10.1158/2159-8290.CD-13-0985
168. Mihara K, Bhattacharyya J, Takihara Y, Kimura A. All-Trans Retinoic Acid Enhances the Cytotoxic Effect of T Cells With Anti-CD38 Chimeric Receptor in Acute Myeloid Leukemia. Blood (2012) 120(21):1901–1. doi: 10.1182/blood.v120.21.1901.1901
169. Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination Therapy With EpCAM-CAR-NK-92 Cells and Regorafenib Against Human Colorectal Cancer Models. J Immunol Res (2019) 2019:2070562. doi: 10.1155/2019/2070562
170. Wu X, Huang S. HER2-Specific Chimeric Antigen Receptor-Engineered Natural Killer Cells Combined With Apatinib for the Treatment of Gastric Cancer. Bull du Cancer (2019) 106(11):946–58. doi: 10.1016/j.bulcan.2019.03.012
171. Ponzetta A, Benigni G, Antonangeli F, Sciumè G, Sanseviero E, Zingoni A, et al. Multiple Myeloma Impairs Bone Marrow Localization of Effector Natural Killer Cells by Altering the Chemokine Microenvironment. Cancer Res (2015) 75(22):4766–77. doi: 10.1158/0008-5472.CAN-15-1320
172. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-Derived TGF-β1 Modulates the Chemokine Receptor Repertoire of Human Resting NK Cells. J Immunol (2013) 190(10):5321–8. doi: 10.4049/jimmunol.1202693
173. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab Plus Ipilimumab in Patients With Metastatic Melanoma. Cancer Immunol Res (2014) 2(7):632–42. doi: 10.1158/2326-6066.CIR-14-0053
174. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy Against Several Solid Malignancies. Cancers (2019) 11(5). doi: 10.3390/cancers11050674
175. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming Heterogeneity of Antigen Expression for Effective Car T Cell Targeting of Cancers. Cancers (2020) 12(5). doi: 10.3390/cancers12051075
176. Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of Folate Receptor β on Acute Myeloid Leukemia Blasts With Chimeric Antigen Receptor-Expressing T Cells. Blood (2015) 125(22):3466–76. doi: 10.1182/blood-2014-11-612721
177. Bhat J, Dubin S, Dananberg A, Quabius ES, Fritsch J, Marie Dowds C, et al. Histone Deacetylase Inhibitor Modulates NKG2D Receptor Expression and Memory Phenotype of Human Gamma/Delta T Cells Upon Interaction With Tumor Cells. Front Immunol (2019) 10:569. doi: 10.3389/fimmu.2019.00569
178. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural Killer Cell-Mediated Lysis of Hepatoma Cells via Specific Induction of NKG2D Ligands by the Histone Deacetylase Inhibitor Sodium Valproate. Cancer Res (2005) 65(14):6321–9. doi: 10.1158/0008-5472.CAN-04-4252
179. Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ. Chimeric NKG2D CAR-Expressing T Cell-Mediated Attack of Human Ovarian Cancer is Enhanced by Histone Deacetylase Inhibition. Hum Gene Ther (2013) 24(3):295–305. doi: 10.1089/hum.2012.143
180. Masoumi E, Jafarzadeh L, Mirzaei HR, Alishah K, Fallah-Mehrjardi K, Rostamian H, et al. Genetic and Pharmacological Targeting of A2a Receptor Improves Function of Anti-Mesothelin CAR T Cells. J Exp Clin Cancer Res (2020) 39(1):1–12. doi: 10.1186/s13046-020-01546-6
181. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of Tumor Destruction by Chimeric Antigen Receptor-Modified T Cells. Mol Ther (2014) 22(3):623–33. doi: 10.1038/mt.2013.262
182. Kailayangiri S, Altvater B, Lesch S, Balbach S, Göttlich C, Kühnemundt J, et al. EZH2 Inhibition in Ewing Sarcoma Upregulates G D2 Expression for Targeting With Gene-Modified T Cells. Mol Ther (2019) 27(5):933–46. doi: 10.1016/j.ymthe.2019.02.014
183. Hardman C, Ho S, Shimizu A, Luu-Nguyen Q, Sloane JL, Soliman MSA, et al. Synthesis and Evaluation of Designed PKC Modulators for Enhanced Cancer Immunotherapy. Nat Commun (2020) 11(1):1–11. doi: 10.1038/s41467-020-15742-7
184. Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. G-Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma. Blood (2019) 134(19):1585–97. doi: 10.1182/blood.2019000050
185. Strassheimer F, Strecker M, Alekseeva T, Macas J, Demes M, Mildenberger I, et al. Combination Therapy of CAR-NK-Cells and Anti-PD-1 Antibody Results in High Efficacy Against Advanced-Stage Glioblastoma in a Syngeneic Mouse Model and Induces Protective Anti-Tumor Immunity In Vivo. J ImmunoTherapy Cancer (2020) 8(Suppl 2):1–2. doi: 10.1136/jitc-2020-ITOC7.91
186. Deng X, Terunuma H, Nieda M, Xiao W, Nicol A. Synergistic Cytotoxicity of Ex Vivo Expanded Natural Killer Cells in Combination With Monoclonal Antibody Drugs Against Cancer Cells. Int Immunopharmacol (2012) 14(4):593–605. doi: 10.1016/j.intimp.2012.09.014
187. Bowles JA, Wang SY, Link BK, Allan B, Beuerlein G, Campbell MA, et al. Anti-CD20 Monoclonal Antibody With Enhanced Affinity for CD16 Activates NK Cells at Lower Concentrations and More Effectively Than Rituximab. Blood (2006) 108(8):2648–54. doi: 10.1182/blood-2006-04-020057
188. Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent Stem Cell-Derived NK Cells With High-Affinity Noncleavable CD16a Mediate Improved Antitumor Activity. Blood (2020) 135(6):399–410. doi: 10.1182/blood.2019000621
189. Goodridge JP, Mahmood S, Zhu H, Gaidarova S, Blum R, Bjordahl R, et al. FT596: Translation of First-Of-Kind Multi-Antigen Targeted Off-The-Shelf CAR-NK Cell With Engineered Persistence for the Treatment of B Cell Malignancies. Blood (2019) 134(Supplement_1):301–1. doi: 10.1182/blood-2019-129319
190. Hu Z. Tissue Factor as a New Target for CAR-NK Cell Immunotherapy of Triple-Negative Breast Cancer. Sci Rep (2020) 10(1):1–13. doi: 10.1038/s41598-020-59736-3
191. Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature (2017) 552:116–20. doi: 10.1038/nature24673
192. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front Pharmacol (2018) 9:185(MAR). doi: 10.3389/fphar.2018.00185
193. Kim KW, Jeong JUK, Lee KH, Uong TNT, Rhee JH, Ahn SJ, et al. Combined NK Cell Therapy and Radiation Therapy Exhibit Long-Term Therapeutic and Antimetastatic Effects in a Human Triple Negative Breast Cancer Model. Int J Radiat Oncol Biol Phys (2020) 108(1):115–25. doi: 10.1016/j.ijrobp.2019.09.041
194. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res (2018) 78(4):1031–43. doi: 10.1158/0008-5472.CAN-17-1788
195. Davis JJ, Fang B. Oncolytic Virotherapy for Cancer Treatment: Challenges and Solutions. J Gene Med (2005) 7(11):1380–9. doi: 10.1002/jgm.800
196. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol Ther (2017) 25(12):2620–34. doi: 10.1016/j.ymthe.2017.08.016
197. Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, et al. Phase 1 Trial of Vocimagene Amiretrorepvec and 5-Fluorocytosine for Recurrent High-Grade Glioma. Sci Trans Med (2016) 8(341). doi: 10.1126/scitranslmed.aad9784
198. Lang FF, Conrad C, Gomez-Manzano C, Alfred Yung WK, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol (2018) 36(14):1419–27. doi: 10.1200/JCO.2017.75.8219
199. Group R. Rigvir - Rigvir Group (2021). Available at: https://www.rigvir.com/products/rigvir.php (Accessed October 14, 2021).
200. Vanseggelen H, Tantalo DGM, Afsahi A, Hammill JA, Bramson JL. Chimeric Antigen Receptor-Engineered T Cells as Oncolytic Virus Carriers. Mol Ther - Oncolytics (2015) 2(August):15014. doi: 10.1038/mto.2015.14
201. Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L, et al. Tumor-Targeted, Systemic Delivery of Therapeutic Viral Vectors Using Hitchhiking on Antigen-Specific T Cells. Nat Med (2005) 11(10):1073–81. doi: 10.1038/nm1297
202. Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A Combinational Therapy of EGFR-CAR NK Cells and Oncolytic Herpes Simplex Virus 1 for Breast Cancer Brain Metastases. Oncotarget (2016) 7(19):27764–77. doi: 10.18632/oncotarget.8526
203. Ma R, Lu T, Li Z, Teng KY, Mansour AG, Yu M, et al. An Oncolytic Virus Expressing Il15/Il15ra Combined With Off-the-Shelf Egfr-Car Nk Cells Targets Glioblastoma. Cancer Res (2021) 81(13):3635–48. doi: 10.1158/0008-5472.CAN-21-0035
204. Strecker M, Strassheimer F, Reul J, Harter PN, Tonn T, Steinbach JP, et al. A Novel Local Treatment Approach? Targeted Immunotherapy of Glioblastoma via AAV-Mediated Gene Transfer of Checkpoint Inhibitors Through Locally Administered HER2-AAVs in Combination With CAR-NK Cells. J ImmunoTherapy Cancer (2020) 8(Suppl 2):47. doi: 10.1136/jitc-2020-ITOC7.92
205. Siegler EL, Kim YJ, Chen X, Siriwon N, Mac J, Rohrs JA, et al. Combination Cancer Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells as Drug Carriers. Mol Ther (2017) 25(12):2607–19. doi: 10.1016/j.ymthe.2017.08.010
206. Van Niel G, D’Angelo G, Raposo G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
207. Laura A, Pace D, Tumino N, Besi F, Alicata C, Conti LA, et al. Characterization of Human NK Cell-Derived Exosomes: Role of DNAM1 Receptor in Exosome-Mediated Cytotoxicity Against Tumor. Cancers (2020) 1:1–16. doi: 10.3390/cancers12030661
208. Enomoto Y, Li P, Jenkins LM, Anastasakis D, Lyons GC, Hafner M, et al. Cytokine-Enhanced Cytolytic Activity of Exosomes From NK Cells. Cancer Gene Ther (2021) 3:17–9. doi: 10.1038/s41417-021-00352-2
209. Zhu L, Kalimuthu S, Gangadaran P, Oh JM, Lee HW, Baek SH, et al. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics (2017) 7(10):2732–45. doi: 10.7150/thno.18752
210. Han D, Wang K, Zhang T, Gao GC, Xu H. Natural Killer Cell-Derived Exosome-Entrapped Paclitaxel can Enhance its Anti-Tumor Effect. Eur Rev Med Pharmacol Sci (2020) 24(10):5703–13. doi: 10.26355/eurrev_202005_21362
211. Neviani P, Wise PM, Murtadha M, Liu CW, Wu CH, Jong AY, et al. Natural Killer–Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res (2019) 79(6):1151–64. doi: 10.1158/0008-5472.CAN-18-0779
212. Fu W, Lei C, Liu S, Cui Y, Wang C, Qian K, et al. CAR Exosomes Derived From Effector CAR-T Cells Have Potent Antitumour Effects and Low Toxicity. Nat Commun (2019) 10(1). doi: 10.1038/s41467-019-12321-3
213. De Oliveira SN, Ryan C, Giannoni F, Hardee CL, Tremcinska I, Katebian B, et al. Modification of Hematopoietic Stem/Progenitor Cells With CD19-Specific Chimeric Antigen Receptors as a Novel Approach for Cancer Immunotherapy. Hum Gene Ther (2013) 24(10):824–39. doi: 10.1089/hum.2012.202
214. Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of Third-Party Chimeric Antigen Receptor Modified Peripheral Blood Natural Killer Cells for Adoptive Cell Therapy of B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia (2020) 34(4):1102–15. doi: 10.1038/s41375-019-0613-7
215. Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol Res (2018) 6(4):467–80. doi: 10.1158/2326-6066.CIR-17-0207
216. Bergman H, Sissala N, Hägerstrand H, Lindqvist C. Human NK-92 Cells Function as Target Cells for Human NK Cells – Implications for CAR NK-92 Therapies. Anticancer Res (2020) 40(10):5355–9. doi: 10.21873/anticanres.14543
217. Kloess S, Oberschmidt O, Dahlke J, Vu XK, Neudoerfl C, Kloos A, et al. Preclinical Assessment of Suitable Natural Killer Cell Sources for Chimeric Antigen Receptor Natural Killer-Based “Off-the-Shelf” Acute Myeloid Leukemia Immunotherapies. Hum Gene Ther (2019) 30(4):381–401. doi: 10.1089/hum.2018.247
218. Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, et al. Expression of miR-17-92 Enhances Anti-Tumor Activity of T-Cells Transduced With the Anti-EGFRvIII Chimeric Antigen Receptor in Mice Bearing Human GBM Xenografts. J ImmunoTherapy Cancer (2013) 1:1–12. doi: 10.1186/2051-1426-1-21
219. Nakazawa T, Murakami T, Natsume A, Nishimura F, Morimoto T, Matsuda R, et al. KHYG-1 Cells With EGFRvIII-Specific CAR Induced a Pseudoprogression-Like Feature in Subcutaneous Tumours Derived From Glioblastoma-Like Cells. Anticancer Res (2020) 40(6):3231–7. doi: 10.21873/anticanres.14304
220. Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pietà A, Barbieri V, et al. Anti-PSMA CAR-Engineered NK-92 Cells: An Off-The-Shelf Cell Therapy for Prostate Cancer. Cells (2020) 9(6). doi: 10.3390/cells9061382
221. Yu M, Luo H, Fan M, Wu X, Shi B, Di S, et al. Development of GPC3-Specific Chimeric Antigen Receptor-Engineered Natural Killer Cells for the Treatment of Hepatocellular Carcinoma. Mol Ther (2018) 26(2):366–78. doi: 10.1016/j.ymthe.2017.12.012
222. Jin Z, Maiti S, Huls H, Singh H, Olivares S, Mátés L, et al. The Hyperactive Sleeping Beauty Transposase SB100X Improves the Genetic Modification of T Cells to Express a Chimeric Antigen Receptor. Gene Ther (2011) 18(9):849–56. doi: 10.1038/gt.2011.40
223. Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, et al. Engraftment of Mesothelin Chimeric Antigen Receptor Using a Hybrid Sleeping Beauty/minicircle Vector Into NK-92MI Cells for Treatment of Pancreatic Cancer. Surg (United States) (2019) 166(4):503–8. doi: 10.1016/j.surg.2019.05.047
224. Merker M, Wagner J, Kreyenberg H, Heim C, Moser LM, Wels WS, et al. ERBB2-CAR-Engineered Cytokine-Induced Killer Cells Exhibit Both CAR-Mediated and Innate Immunity Against High-Risk Rhabdomyosarcoma. Front Immunol (2020) 11:581468. doi: 10.3389/fimmu.2020.581468
225. Liu Y, Zhou Y, Huang KH, Fang X, Li Y, Wang F, et al. Targeting Epidermal Growth Factor-Overexpressing Triple-Negative Breast Cancer by Natural Killer Cells Expressing a Specific Chimeric Antigen Receptor. Cell Prolif (2020) 53(8):1–12. doi: 10.1111/cpr.12858
226. Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical Grade Manufacturing of Genetically Modified, CAR-Expressing NK-92 Cells for the Treatment of ErbB2-Positive Malignancies. Cancer Immunol Immunother (2018) 67(1):25–38. doi: 10.1007/s00262-017-2055-2
227. Robbins Y, Greene S, Friedman J, Clavijo PE, Van Waes C, Fabian KP, et al. Tumor Control via Targeting Pd-L1 With Chimeric Antigen Receptor Modified Nk Cells. eLife (2020) 9:1–18. doi: 10.7554/eLife.54854
228. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM, Greiner JW, et al. An NK Cell Line (haNK) Expressing High Levels of Granzyme and Engineered to Express the High Affinity CD16 Allele. Oncotarget (2016) 7(52):86359–73. doi: 10.18632/oncotarget.13411
229. Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, et al. “UniCAR”-Modified Off-the-Shelf NK-92 Cells for Targeting of GD2-Expressing Tumour Cells. Sci Rep (2020) 10(1):1–16. doi: 10.1038/s41598-020-59082-4
230. Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent Stem Cell-Derived NK Cells With High-Affinity Noncleavable CD16a Mediate Improved Antitumor Activity (2020). Available at: http://ashpublications.org/blood/article-pdf/135/6/399/1633322/bloodbld2019000621.pdf.
231. Wrona E, Borowiec M, Potemski P. CAR-NK Cells in the Treatment of Solid Tumors. Int J Mol Sci (2021) 22(11):1–19. doi: 10.3390/ijms22115899
232. Lim RM, Rong L, Zhen A, Xie J. A Universal CAR-NK Cell Targeting Various Epitopes of HIV-1 Gp160. ACS Chem Biol (2020) 15(8):2299–310. doi: 10.1021/acschembio.0c00537
233. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol Sin (2020) 41(9):1141–9. doi: 10.1038/s41401-020-0485-4
234. Ma MT, Badeti S, Chen CH, Kim J, Choudhary A, Honnen B, et al. CAR-NK Cells Effectively Target SARS-CoV-2-Spike-Expressing Cell Lines In Vitro. Front Immunol (2021) 12:652223. doi: 10.3389/fimmu.2021.652223
235. Ma M, Badeti S, Geng K, Liu D. Efficacy of Targeting SARS-CoV-2 by CAR-NK Cells. bioRxiv (2020). Published online. doi: 10.1101/2020.08.11.247320
236. Tangye SG, Ma CS, Brink R, Deenick EK. The Good, the Bad and the Ugly-T FH Cells in Human Health and Disease. Nat Rev Immunol (2013) 13(6):412–26. doi: 10.1038/nri3447
237. Vinuesa CG, Sanz Ĩ, Cook MC. Dysregulation of Germinal Centres in Autoimmune Disease. Nat Rev Immunol (2009) 9(12):845–57. doi: 10.1038/nri2637
238. Craft JE. Follicular Helper T Cells in Immunity and Systemic Autoimmunity. Nat Rev Rheumatol (2012) 8(6):337–47. doi: 10.1038/nrrheum.2012.58
239. Reighard SD, Cranert SA, Rangel KM, Ali A, Gyurova IE, de la Cruz-Lynch AT, et al. Therapeutic Targeting of Follicular T Cells With Chimeric Antigen Receptor-Expressing Natural Killer Cells. Cell Rep Med (2020) 1(1):100003. doi: 10.1016/j.xcrm.2020.100003
240. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-In-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients With Relapsed and Refractory Acute Myeloid Leukemia. Am J Cancer Res (2018) 8(9):1899.
241. Li C, Yang N, Li H, Wang Z. Robo1−specific Chimeric Antigen Receptor Natural Killer Cell Therapy for Pancreatic Ductal Adenocarcinoma With Liver Metastasis. J Cancer Res Ther (2020) 16:393–6. doi: 10.4103/jcrt.JCRT
242. Martin U. The Biologics News and Reports Portal. Pipelinereview (2021). Available at: https://pipelinereview.com/index.php/2021080278884/DNA-RNA-and-Cells/Sorrento-Receives-FDA-Authorization-to-Start-Phase-1-Clinical-Trial-of-Proprietary-Off-the-Shelf-Allogeneic-anti-CD38-DAR-T-Dimeric-Antigen-Receptor-T-Cell-Therapy-to-T.html (Accessed October 9, 2021).
243. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res (2016) 76(6):1578–90. doi: 10.1158/0008-5472.CAN-15-2524
244. Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT-Based Chimeric Co-Stimulatory Switch Receptor Improves T-Cell Anti-Tumor Function. J Immunother Cancer (2019) 7(1):243. doi: 10.1186/s40425-019-0721-y
245. Lin S, Cheng L, Ye W, Li S, Zheng D, Qin L, et al. Chimeric CTLA4-CD28-CD3z T Cells Potentiate Antitumor Activity Against CD80/CD86–Positive B Cell Malignancies. Front Immunol (2021) 12:642528. doi: 10.3389/fimmu.2021.642528
246. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126(8):3130–44. doi: 10.1172/JCI83092
247. Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A Novel Chimeric PD1-NKG2D-41BB Receptor Enhances Antitumor Activity of NK92 Cells Against Human Lung Cancer H1299 Cells by Triggering Pyroptosis. Mol Immunol (2020) 122(May):200–6. doi: 10.1016/j.molimm.2020.04.016
248. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother (2010) 33(8):780–8. doi: 10.1097/CJI.0b013e3181ee6675
249. Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Myhre MR, et al. NK Cells Specifically TCR-Dressed to Kill Cancer Cells. EBioMedicine (2019) 40:106–17. doi: 10.1016/j.ebiom.2019.01.031
250. Woan KV, Miller JS. Harnessing Natural Killer Cell Antitumor Immunity: From the Bench to Bedside. Cancer Immunol Res (2019) 7(11):1742–7. doi: 10.1158/2326-6066.CIR-19-0404
251. Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. NKG2D-CAR-Transduced Natural Killer Cells Efficiently Target Multiple Myeloma. Blood Cancer J (2021) 11(8):1–11. doi: 10.1038/s41408-021-00537-w
252. Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically Engineered CAR NK Cells Display Selective Cytotoxicity Against FLT3-Positive B-ALL and Inhibit In Vivo Leukemia Growth. Int J Cancer (2019) 145(7):1935–45. doi: 10.1002/ijc.32269
253. Ao X, Yang Y, Li W, Tan Y, Guo W. Anti-αfr CAR-Engineered NK-92 Cells Display Potent Cytotoxicity Against αfr-Positive Ovarian Cancer. J Immunother (2019) 42(08):284–96. doi: 10.1097/CJI.0000000000000286
254. Klapdor R, Wang S, Morgan M, Dörk T, Hacker U, Hillemanns P, et al. Characterization of a Novel Third-Generation Anti-CD24-CAR Against Ovarian Cancer. Int J Mol Sci (2019) 20(3):1–15. doi: 10.3390/ijms20030660
255. Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, et al. Use of Chimeric Antigen Receptor NK-92 Cells to Target Mesothelin in Ovarian Cancer. Biochem Biophys Res Commun (2020) 524(1):96–102. doi: 10.1016/j.bbrc.2020.01.053
Keywords: natural killer cells, chimeric antigen receptors, cancer, autoimmunity, clinical trials, CAR-NK, Preclinical studies
Citation: Karvouni M, Vidal-Manrique M, Lundqvist A and Alici E (2022) Engineered NK Cells Against Cancer and Their Potential Applications Beyond. Front. Immunol. 13:825979. doi: 10.3389/fimmu.2022.825979
Received: 30 November 2021; Accepted: 13 January 2022;
Published: 15 February 2022.
Edited by:
Rizwan Romee, Dana–Farber Cancer Institute, United StatesReviewed by:
Congcong Zhang, Miltenyi Biotec, GermanyCopyright © 2022 Karvouni, Vidal-Manrique, Lundqvist and Alici. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evren Alici, ZXZyZW4uYWxpY2lAa2kuc2U=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.