 Bo Li2†
Bo Li2† Jian Jiao
Jian Jiao Wei Xu
Wei Xu- 1Department of Radiation Oncology, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 2Department of Orthopedic Oncology, Changzheng Hospital, Second Military Medical University, Shanghai, China
A substantial amount patients with cancer will develop bone metastases, with 70% of metastatic prostate and breast cancer patients harboring bone metastasis. Despite advancements in systemic therapies for advanced cancer, survival remains poor for those with bone metastases. The interaction between bone cells and the immune system contributes to a better understanding of the role that the immune system plays in the bone metastasis of cancer. The immune and bone systems share various molecules, including transcription factors, signaling molecules, and membrane receptors, which can stimulate the differentiation and activation of bone‐resorbing osteoclasts. The process of cancer metastasis to bone, which deregulates bone turnover and results in bone loss and skeletal-related events (SREs), is also controlled by primary cancer-related factors that modulate the intratumoral microenvironment as well as cellular immune process. The nuclear factor kappa B ligand (RANKL) and the receptor activator of nuclear factor kappa B (RANK) are key regulators of osteoclast development, bone metabolism, lymph node development, and T-cell/dendritic cell communication. RANKL is an osteoclastogenic cytokine that links the bone and the immune system. In this review, we highlight the role of RANKL and RANK in the immune microenvironment and bone metastases and review data on the role of the regulatory mechanism of immunity in bone metastases, which could be verified through clinical efficacy of RANKL inhibitors for cancer patients with bone metastases. With the discovery of the specific role of RANK signaling in osteoclastogenesis, the humanized monoclonal antibody against RANKL, such as denosumab, was available to prevent bone loss, SREs, and bone metastases, providing a unique opportunity to target RANKL/RANK as a future strategy to prevent bone metastases.
Introduction
Metastasis is a complicated, multistep process that is responsible for roughly 90% of cancer-related deaths (1). Bone metastases are common in patients with solid tumors (especially for prostate, breast, lung, and kidney cancers), and the spine is the most significant site of advanced disease (2), whereas lesions in flat and appendicular bones are infrequent. For example, up to 50% of patients diagnosed with lung cancer will develop spinal metastases (3). This is partially explained by the ease of access to vertebral bodies in the thoracic and lumbar spine through the plexus vertebral system (4, 5) and the high bone marrow flow of some skeletal elements (6). Bone metastatic cancer can be extremely debilitating and is associated with devastating clinical complications, such as severe pain and skeletal-related events (SREs) including pathological fractures, spinal cord and nerve compression, hypercalcemia, and bone marrow aplasia, which result in a poor prognosis (7). As the mechanisms and pharmacological treatment of bone metastases have been increasingly recognized, growing evidence suggests that the bone microenvironment and the immune system may contribute to cancer bone tropism.
Normal bone development and maintenance are controlled by a balance in normal bone remodeling through bone-forming osteoblasts and bone-resorbing osteoclasts (8–10). In patients with cancer metastatic to the bone, osteolysis bone lesions often develop because of the tumor-induced ability to exacerbate osteoclastic activity, leading to imbalanced osteoclast and osteoblast functions (11). It has been traditionally thought that certain types of solid tumors that metastasize to the bone are characterized by disruption in the homeostasis of osteoblasts or osteoclasts, being commonly both damaged in most solid tumors (2). Along with an increased understanding of the interplay among dormant tumor cells, the bone microenvironment, and the bone marrow, it has been shown that the complex interactions in the bone marrow microenvironment are crucial for the initiation and promotion of metastases in the bone (12–14). More recent work has revealed that the bone itself also has a unique local immune environment favoring bone metastasis, arising from diverse primary tumor types (15). Reports show that a tumor can systemically produce hormones such as parathyroid hormone-related protein (PTHrP) mediating osteoclastogenesis by enhancing the expression of the receptor activator of nuclear factor kappa B ligand (RANKL) (16). RANKL and RANK, a receptor–ligand pair of the TNF receptor superfamily, can regulate osteoclast development and bone metabolism (17, 18). In addition to the critical function of RANKL and RANK in normal bone resorption and remodeling, the RANKL/RANK pathway also controls many other physiological processes such as immunity, the proliferation and division of mammary epithelial cells, as well as mammary gland formation during lactation (19).
Osteoclasts and osteoclast precursors express RANK, whereas RANKL is mainly produced by osteoblastic lineage cells (20), bone-marrow stromal cells, immune cells (20–22), and some cancer cells (23). The binding of RANKL to RANK is an essential mediator of differentiation and osteoresorption function of the osteoclast, leading to bone resorption. Moreover, RANKL expression is crucial for the immune system, as it serves as an important molecule in optimal T-cell activation, mediates dendritic cell survival, and regulates the development of lymph nodes and Peyer’s patches (24, 25). The significance of the contribution of RANKL/RANK in the bone microenvironment and the immune system during bone metastasis process strengthens the notion that abrogation of RANK/RANKL signaling represents a key therapeutic target for cancer therapy. This review summarizes the current state and progress of the bone microenvironment and the immune system in the carcinogenesis process, which has propelled a deeper understanding of bone metastases. Unveiling the potential capacity of the RANKL/RANK axis to modulate the tumor immune microenvironment opens the door to the development of pharmacological inhibitors of RANKL as a potential therapy for bone disease in cancer patients.
Immune System in Cancer
Dating back to 1889, Stephen Paget postulated the idea of metastatic spread, that primary tumors induce changes to distant organs, due to the high degree of crosstalk between cancer cells and their microenvironment (26). The immune microenvironment, wherein the immune cells are primarily located in or around the cancer and the adjacent lymphoid tissues, plays a vital role in metastatic cascade in cancer (27, 28). Primary tumors can influence the systemic and local immune homeostasis, promoting chronic inflammation and consequently suppressing immune activity that facilitates immune evasion and metastasis formation. These immunosuppressive cell populations promoting metastasis include macrophages (29), conventional CD11b+Ly6G+ neutrophils (30, 31), platelets (32), as well as myeloid-derived suppressor cells (MDSCs) (33). Conversely, CD4+ T cells and CD8+ cytotoxic T lymphocytes (CTLs) exert antimetastatic effects by activating adaptive immunity to control primary tumors (34, 35). It is deemed that CD4+ T cells can either promote or prevent tumor progression. Regulatory T (Treg) and T helper 17 (Th17) CD4+ T-cell subsets have emerged as key elements facilitating a pro-tumor inflammation environment that could favor cancer initiation, progression, and metastasis (36). Several cytokines, such as RANKL, produced by CD4+ T cells, reportedly promote breast cancer progression and metastasis (37). The maturation and antigen presentation of dendritic cells (DCs) play diverse roles in the protective antitumor response (38, 39). CD8+ CTLs can recognize specific antigen bound to major histocompatibility complex (MHC)-I on DC, then they were activated and can produce interferon γ (IFNγ), perforin, and granzyme B that contribute to tumor cell cytolysis (40). Together, these results reveal that therapeutic strategies that can play roles in regulating immune responses may be a feasible immunotherapeutic action for patients with metastatic cancers.
Immune System in Bone Metastasis
Bone metastases have traditionally included osteolytic and osteoblastic metastases classified according to the predominance of lysis or sclerosis in the bone. Growing evidence has suggested the fact that mixed-type bone metastases containing both osteolytic and osteoblastic change were also observed in most patients (41). Bone metastases in breast cancer patients are dominantly mediated by osteoclast-induced osteolytic lesions; local bone forming and osteoblastic lesions are also observed (2). Bone metastases in many patients with lung cancer or multiple myeloma are preponderantly osteolytic, leading to the occurrence of focal bone destruction (42). Conversely, in the case of prostate cancer, the bone appears as dense osteosclerotic lesions and is characterized by increased osteoblast activity (43). The mechanism of bone metastases is complex; tumor cells, osteoblasts, osteoclasts, and the mineralized bone matrix cooperate to promote bone metastasis (Table 1) (44–58, 60, 63–65). In osteolytic bone metastases, multiple cytokines and growth factors secreted by tumor cells, such as PTHrP, RANKL, interleukins, prostaglandin E, tumor necrosis factor (TNF), and macrophage colony stimulating factor (MCSF), are responsible for hyperactive osteoclast activity, leading to osteoclast bone resorption and promoting osteolytic metastasis (59, 61, 62). In turn, osteoclastic bone resorption releases growth factors, such as TGFβ, IGFs, PDGFs, and BMPs, to promote cancer proliferation, sequentially further enhancing the secretion of osteolytic factors and driving a feedforward to fuel tumor growth in the bone (66, 67). In osteoblastic metastases, studies have established the scientific foundation for the vicious cycle of tumor cells and osteoblasts. Tumor cells produce factors, including FGFs, urokinase-type plasminogen activator (uPA), endothelin-1 (ET-1), prostate-specific antigen (PSA), insulin-like growth factors (IGFs), bone morphogenic proteins (BMPs), and vascular endothelial growth factor (VEGF), which stimulate osteoblast proliferation and differentiation (68, 69). This process, in turn, stimulates prostate cancer growth and invasion (70). Additionally, studies have also implicated the importance of several intracellular pathways, such as mitogen-activated protein kinase (MAPK), nitric oxide (NO), and Wnt signaling, in bone-forming osteoblasts (71, 72).

Table 1 Regulating factors associated with the bone metastasis process.
Owing to the close connection between the immune system and the bone in many diseases, such as inflammatory diseases and postmenopausal osteoporosis, we focus on how the two systems affect each other. Evidence is accumulating to support many shared regulatory factors, including cytokines, receptors, signaling molecules, and transcription factors, between the immune and bone systems (73, 74) (Figure 1). Soluble factors secreted from antigen-stimulated immune cells, such as interleukin-1 (IL-1) and RANKL, have a central role in activating osteoclast (75). It has been demonstrated that activated T lymphocytes might have the capacity to induce an osteoclastic phenotype by directly acting on osteoclast-precursor cells (76, 77). It was also observed that mice lacking cytotoxic T lymphocyte protein 4 (CTLA4) with systemically activated T lymphocytes trigger an increased osteoclast number response and induce an osteoporotic phenotype (78). However, not all T-cell responses associated with bone loss have such a deleterious outcome. The osteoprotegerin (OPG), IFNγ, interleukin (IL)-4, and transforming growth factor-β (TGF-β) produced by T cells appear to prevent T lymphocyte-mediated osteoclastogenesis (79, 80). In addition, Th1, Th2, and CD8+ T cells contribute to the repression of, other than the stimulation of, osteoclastogenesis in murine (81–85), and this blocking effect on murine and human osteoclast differentiation was observed in Treg cells (81). Arthritis was observed after the suppression of Treg cells, whereas bone destruction was ameliorated after the transfer of Treg cells into T-cell-deficient mice, suggesting that Treg cells are potentially beneficial in the bone protective effect (86, 87). The activated T cells showing pro-osteoclastogenic or anti-osteoclastogenic ability are usually thought to potentially depend on the local environment. New data have indicated the involvement of other immune cell types, such as DC, neutrophils, and natural killer cells, in increased osteoclastogenesis in diseases. For instance, DCs can undergo transdifferentiation into the osteoclast cell type under inflammatory conditions (88).
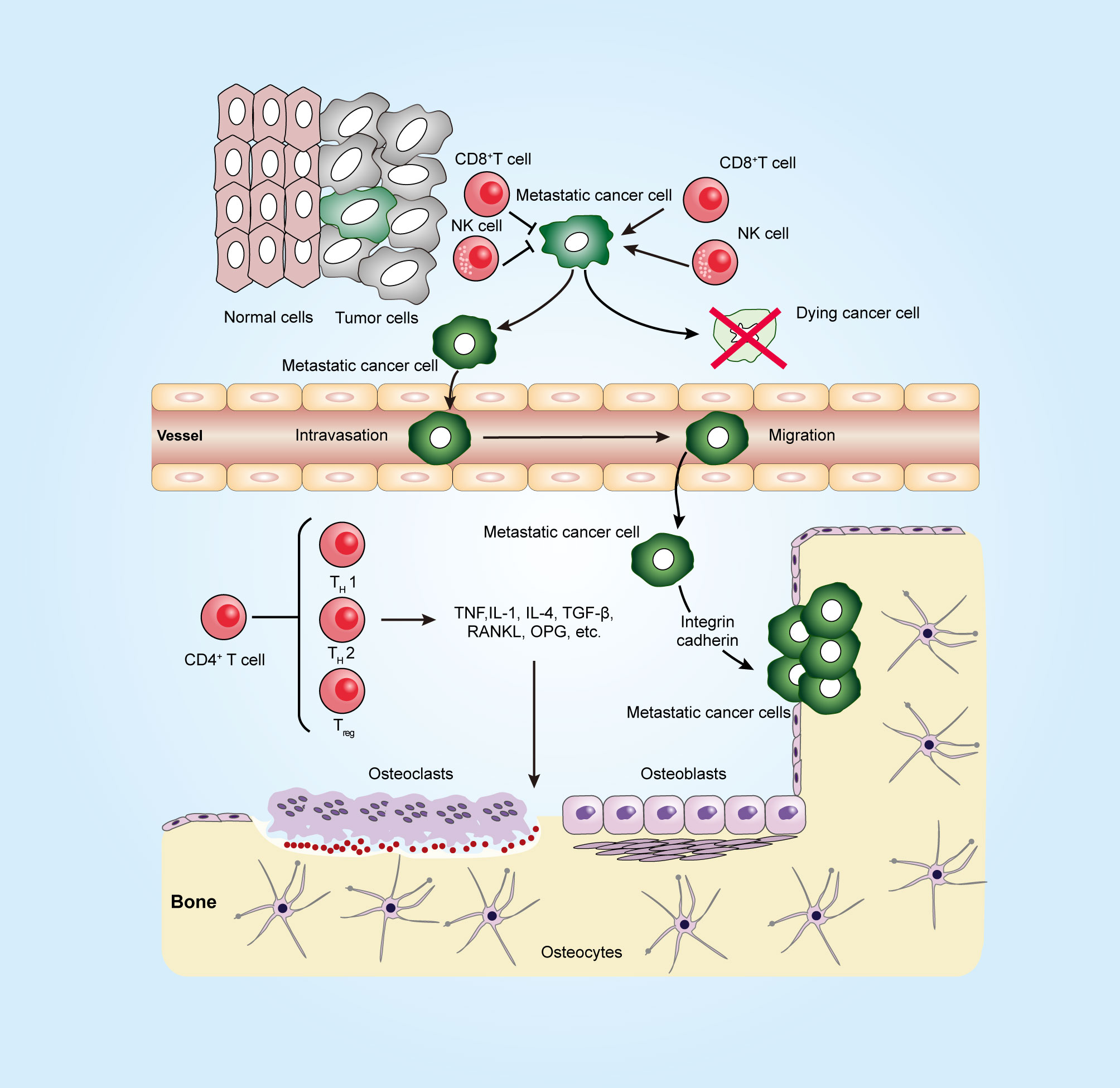
Figure 1 Immune system in a long journey to develop bone metastasis. To establish the metastatic tumor, cancer cells escape from the tumoricidal immune response that is mediated by killer cells, such as CD8+ T cells and natural killer (NK) cells, and then invade through the surrounding stroma and intrude into blood vessels (intravasation). At the metastatic site, the arrested tumor cells in microvessels escape from the blood vessel (extravasation). Metastatic tumor cells can interact with both osteoclasts and osteoblasts in the bone microenvironment and release factors, such as TNF, TGF-β, and RANKL, to promote osteoclastogenesis and osteoblastogenesis. CD4+ T cells can produce not only osteoclastogenic cytokines such as IL17 and TNF but also anti-osteoclastogenic cytokines such as IFNγ and IL4. The figure was designed using Adobe Illustrator CC.
More findings provide evidence that the immune system performs a critical role in the progression and the treatment resistance of tumors. It is known that a complex interplay of immunity in primary tumors can have a profound impact on the development of tumors (89, 90). Here, we focus on the contributions of the immune microenvironment to the bone metastasis of primary cancer. In this contribution, the multiple inhibitory and stimulatory effects of the immune system on host cells within the bone microenvironment were systematically reviewed, emphasizing the critical roles of immunity in the modulation of bone metastases (91). Partially the regression of tumor bone metastasis is due to the antagonistic effect of several programs, such as natural killer (NK) cell- or effector T cell-mediated lysis. However the majority of immune cells, such as regulatory T cells (Treg), dendritic cells (DCs), MDSCs, and macrophages, serve as a phenotype promoting cancer bone metastasis. Immunosuppressive myeloid-derived suppressor cells infiltrate in tumors driving tumor growth, sensed by inhibiting the host antitumor immune responses in the tumor. In bone metastasis, signals derived from the tumor and the microenvironment allow myeloid-derived suppressor cells to act as progenitors of the osteoclast to enhance tumor-induced osteolysis (92, 93). The impact of T cells on bone metastasis has also been recognized. Treg cells, which maintain immune homeostasis through impeding aberrant immune response against self-antigens, also suppress antitumor immune response (94–96). A study found that a dose-dependent increase in the recruitment of Treg cells in a mouse model of breast cancer treated with PGE2 derived from tumor leads to increases in the cellular apoptosis of CD8+ T cells and promotes bone metastasis (97).
The Crucial Role of RANKL/Rank Signaling in Bone Remodeling
RANK was first identified in 1997 via expression cloning techniques in the course of studying a novel tumor necrosis factor receptor (TNFR) superfamily member. RANKL, also known as TNFSF11 (tumor necrosis factor receptor super family 11), TRANCE (TNF-related activation-induced cytokine), OPGL (osteoprotegerin ligand), and ODF (osteoclast differentiation factor), was characterized as the cognate ligand of TNFR superfamily member (21). Over 20 years, much has been researched about the important biology of TNFSF, their receptors and the intracellular signaling pathways in patients with autoimmune or inflammatory disorders. TNFSF and TNFR members are broadly expressed in immune cells including macrophages and brain glial cells and are usually related to the expression of immune system cells (98). TNFSF proteins perform immunoregulatory functions through displaying a lymphoid tissue organizer and lymphocyte stimulation activity and increasing the survival and function of lymphocytes (99–103). Moreover, TNFSF is closely associated with neuronal activity, such as CNS injury and neurodegenerative diseases (104). Recent evidence indicates that TNFSF is one of the major mechanisms of the development and survival of osteoclasts (103). These scientific discoveries have led to the identification of novel therapeutic options, through inhibiting or activating TNFSF signaling pathways, to control diseases.
The TNF superfamily of cytokine-like molecules included a superfamily of 19 ligands, while the TNFR superfamily consisted of 29 proteins acting as receptors of these ligands (105). One important example is the system consisting of RANKL and its signaling receptor RANK. In 2016, a study reported that leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4, also called GPR48) is another receptor for RANKL. The findings suggest that LGR4 competes with RANK for RANKL binding in osteoclasts, and a negative regulatory feedback loop of LGR4 has been unveiled to restrict RANKL osteoclastogenesis through multiple mechanisms (106). On the other hand, further research about the molecular mechanisms for the crucial roles of LGR4 mediation of RANKL signaling in many other processes has not appeared. The gene TNFSF11, located on human chromosome 13q14 and a conserved syntenic region on mouse chromosome 14, encoded the RANKL protein (107, 108). RANKL was initially identified on murine thymoma cell line EL40.5, and then in 1997, functional genomics and biochemical analyses identified RANKL, expressed by activated T cells, as a stimulator of the DCs (109), and RANKL played an essential role in regulating the DCs–T cells interaction (110). RANKL is a molecule acting as a three-part complex, containing a small N-terminal intracellular domain, a transmembrane segment, and a c-terminal extracellular domain consisting of a stalk and a receptor-binding region (108).On the other hand, RANKL exists in soluble forms upon proteolytic cleavage of the extracellular stalk region by matrix metalloproteinases (MMPs) such as MMP-14 (111, 112). However, RANKL as a membrane-anchored molecule functions significantly more efficiently (113). RANKL mediated oligomerization of the receptor RANK on cells, and it activated RANKL/RANK signaling and downstream responses. The activation of the RANKL/RANK pathway initiates a cascade of intracellular signaling molecules such as tumor necrosis factor receptor-associated factor (TRAF) family members, which can culminate in nuclear factor κB (NFκB), AP-1, and NFATc1 signaling activation that are responsible for osteoclast differentiation and function (114, 115). By contrast, it is reported that RANK can activate osteoclasts independent of ligand through receptor self-association (116). However, most physiological functions of RANK are generally deemed to be ligand-dependent to be the starting point of the RANKL/RANK signaling cascade (117).
The bone is a dynamic tissue that consists of osteoblasts and is resorbed by osteoclasts in the bone remodeling cycle, which is responsive to the skeleton for optimal function. Once a bone matrix of osteoclasts, most osteoblasts will undergo apoptosis, then a proportion of cells will differentiate into osteocytes, which make up the majority of bone cells in the adult skeleton (118). Of note, osteocytes are reprogrammed from differentiated osteoblast cells, and the functions of osteocytes are still being investigated. Although it appears that osteocytes do not express RANKL mRNA (119), data from previous study suggest that osteocytes could regulate RANKL expression through other bone cells. Osteocytes, based on the location within the bone matrix and cellular morphology, could regulate bone remodeling through modulating the production of RANKL (112, 120). The resorptive effect reflects the important role of RANKL on osteoclasts and/or their precursors (121). Studies in mice lacking TNFSF11 or TNFRSF11a gene have illuminated the cellular and molecular mechanisms by which RANKL controls tooth eruption owing to the function of RANKL in osteoclast development and bone metabolism (122, 123). A RANKL transgenic mice model greatly reduced bone mass and increased the number and enhanced the activity of osteoclasts, demonstrating the systemic nature of the increased bone turnover (124). Binding of RANKL to its receptor RANK, located on the surface of pre-osteoclasts and mature osteoclast cells, triggers pre-osteoclast differentiation into multinucleated, fully functional osteoclasts (125). A fine balance of osteoclasts and osteoblasts is tightly related to the RANKL–RANK system because mice deficient in either RANK or RANKL show a complete absence of osteoclasts and develop severe osteopetrosis (126, 127). It has also been reported that RANKL inhibition in osteocyte-specific RANKL-deficient mice was effective to protect against bone loss and deletion of osteocyte RANKL, conferring an increased cancellous bone mass in osteogenesis imperfecta mice (128, 129). It was also suggested that vesicular RANK, which is secreted from the maturing osteoclasts, binds osteoblastic RANKL and promotes bone formation by triggering RANKL reverse signaling (130). These results suggest that osteocyte RANKL, whether in healthy or diseased individuals, greatly affects bone resorption, and targeting RANKL can be classified as appropriate for the development of drugs.
RANKL–Rank Axis in Immunity
It has become clear that cells of both innate and adaptive immunity are able to respond to tumors and promote the effective elimination of cancer cells, which ultimately help fuel tumor progression. With the crucial role for driving either co-stimulation or co-inhibition of the immune response through various mechanisms, TNFSF members provide unique biophysical and biochemical cues to control both innate and adaptive immunity (105). Efforts on study about the RANKL–RANK pathway showed that RANKL provided by T cells following antigen receptor stimulation can significantly stimulate the survival and function of DCs, chief inducers of adaptive immune response across the body (107, 131, 132). The RANKL/RANK pathway not only stimulates immune the system (e.g., lymph-node, B-, and T-cell development) but also inhibits the immune system (e.g., generation of regulatory T cells and induction of T-cell tolerance) (133, 134).
Although a rare population of DCs exists among the immune cells within tumors and lymphoid organs, DCs are central to adaptive immune response, acting to initiate the antigen-specific immunity and tolerance during invasive microorganisms (135, 136). Consequently, multiple clinical strategies, including harnessing the activity of DCs and the generation of DC-based vaccines, are being detected to target DCs for cancer treatment. Findings reveal the mechanisms of DCs in the activation of immunity that rely not only on capturing antigens and presenting them to T cells in the lymph nodes but also on providing immunomodulatory signals through cell–cell contacts and cytokines (137, 138). DCs initiate CD8+ or CD4+ T-cell activation by antigen-loaded MHC class I or II molecules, respectively expressed on the DC surface, and by providing potent co-stimulatory signals expressed by the T cells (139, 140). In fact, differentiation of naive CD4+ T cells into each T-cell subset, such as Th1, Th2, and Th17 cells, is dictated by DCs providing the additional activation and developmental signals (140). Importantly, the maintenance, homeostasis, maturation, and activation of DCs during an immune response are supported by TNFRSF members, such as RANK, CD40, and LTβR, that are strongly expressed on DCs (21, 141). The study explored that the role of the interaction between RANKL on the activated T cell and RANK on the activated DC in enhancing the survival of DC may be through the induction of Bcl-xL and the upregulation of CD40 (105). Transforming growth factor β-activated kinase 1 (TAK1), a mitogen-activated protein kinase, mediates signal transduction from CD40 and RANK. The result that reduced survival of TAK1-deficient DCs cannot be rescued with neither RANKL nor an anti-CD40 agonistic antibody clarified that RANKL plays essential roles in promoting DC survival in a TAK1-dependent manner (142).
RANKL can modulate immune responses, balancing inflammatory processes and immunosuppression. The DC survival promoted by RANKL raises the immune response and increases inflammation. It was found that RANKL signaling in DCs significantly upregulated in Fas-deficient strain MRL/lpr mice leads to a rapidly progressing autoimmunity, suggesting that the activation of RANKL signaling may be regulated by Fas-induced signaling (143). On the other hand, RANKL can regulate DC-mediated immunosuppression. The data demonstrate that RANKL expression induced on keratinocytes can stimulate a molecular pathway that couples the epidermis to local and systemic immunosuppression by regulating the function of epidermal DCs, which is crucial for the peripheral homeostasis of regulatory T cells (144). Thus, the RANKL–RANK interaction could selectively either promote or suppress immunity determined by the specific phase of the immunity cycle at which this pathway is activated (145).
Activated CD4+ and CD8+ T cells can produce RANKL, which is also expressed in lymph nodes, spleen, thymus, and immature CD4 CD8 thymocytes (146, 147). Emerging insights into how RANKL expression is influenced provide the view that calcineurin, extracellular regulated protein kinases (ERK1/ERK2), and protein kinase C (PKC)-regulated signaling pathways can regulate RANKL expression in T cells (148, 149). In addition to the role of T cells in RANKL, RANKL/RANK signaling plays a key role in T-cell physiology. RANKL and RANK knockout mice, which failed to develop lymph nodes, have been shown to have intact splenic architecture and Peyer’s patches (150, 151). In adjuvant arthritis, a T-cell-driven experimental arthritis, the immune response is potentially influenced by RANKL blockade (152). On the other hand, specific primary T-cell responses were increased following RANKL treatment. Interestingly, crucial memory responses were observed only in mice injected with RANKL-treated dendritic cells (132). RANKL inhibition in vivo, including the use of a soluble RANK-Fc molecule, does not prevent the priming of LCMV-specific T cells, but it has damaged effects on proliferation of CD4+ T cells to the viral antigen after a period of infection, reaching a point at which RANKL seems to play a role in memory T-cell responses (153). At the same time, data in lymphocytes suggest that decreasing RANKL expression does not help regulate the generation of cytotoxic T cells in normal lymph node condition (153).
The development of B cells relies on the production and regulation of chemical species, such as RANKL, OPG, IL-7, and C-X-C Motif Chemokine Ligand 12 (CXCL12) that are produced by bone marrow stromal cells and osteoblasts (154). RANKL or RANK deficiencies created an interference of B-cell development due to the loss of the B-cell development environment. However, a study revealed that neither obvious loss of B-cell development in the bone marrow nor functions such as immunoglobulin secretion were observed in RANK-depleted mice, suggesting that the normal development and homeostasis of B cells do not require B-cell intrinsic RANK expression (155). Thus, these results showed that the effect of RANKL in B-cell development was caused by the interaction between RANKL and an alternative receptor.
RANKL–Rank Axis in Bone Metastasis
Although RANK expression was primarily found in osteoclasts and their progenitors, recent data also indicate that RANK is expressed on tumor cells, appearing to regulate metastases from establishing tumors (156). The role of RANKL/RANK in immune and bones is as essential for autoimmune diseases affecting the bone as the development of bone metastasis from tumors. Causes of RANKL/RANK in bone metastasis could be divided into those that enhance osteolysis and those that have an effect on promoting metastasis (Figure 2).
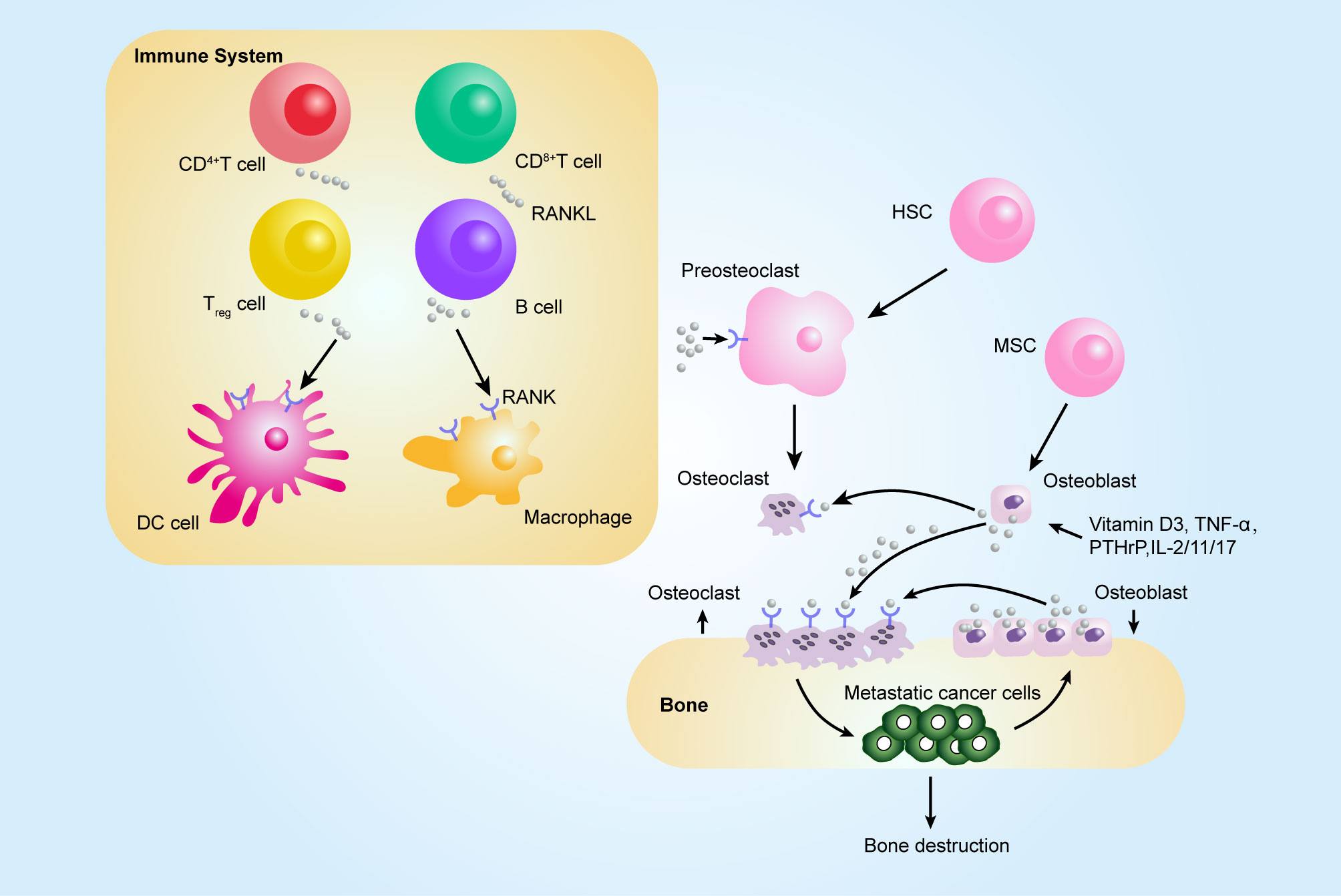
Figure 2 Contribution of the RANKL–RANK axis in immune system and bone metastasis. Among tumor-infiltrating immune cells, the expression of RANKL has been observed on all immune cell types, such CD8+ T, CD4+ T, and Treg cells, and RANKL can act on dendritic cells (DCs) to promote their survival and to prolong T–DC interactions. Bone-resorbing factors, such as vitamin D3, PTHrP, IL-1, IL-11, IL-17, and TNF-α, act on osteoblasts to induce RANKL, which binds to RANK present at the surface of osteoclast progenitors (pre-osteoclasts), which results in bone resorption by mature osteoclasts. The figure was designed using Adobe Illustrator CC.
RANKL was found to exert promigratory effects in a breast cancer model and to promote their metastasis to the bone via PTHrP produced by breast cancer cells (157). Recent investigations have shown that osteolytic bone metastases through osteoclast activation may be the important function of RANKL/RANK in tumor bone metastasis. Moreover, this pathway also contributes to tumor growth and metastasis in a bone-independent manner. The RANKL/RANK pathway is an important checkpoint that influences the antitumor immune response or directly affects bone metastasis (158). It was demonstrated that hormone receptor-negative breast cancer patients with higher RANK expression are at a higher risk of bone metastasis and have worse recurrence-free survival (159). It was also suggested that RANKL, with chemokine function for recruiting cancer cells to the bone, can stimulate the migration of breast cancer cells to the bone (156). With respect to the role of RANKL outside of the bone in triggering metastasis, one study showed that infiltrating Tregs expression of RANKL can drive the metastasis of breast cancer through altering the secretion of inflammatory factors in metastasis (37). With the increased knowledge regarding the molecular pathways downstream of RANKL/RANK signaling in bone metastasis, it is expected that RANK signaling has a close connection with metastatic potential through the activation of IKK-a (160).
Pro-osteoclastogenic factors, including parathyroid hormone (PTH), vitamin D3, TNF-α, Wnt 5A, and Sclerostin (SOST), can promote osteoclastogenesis through inducing RANKL expression in osteoblasts, stromal cells, or T cells (161–163). There are many additional osteotropic factors, including IL-1, IL-6, and IL-11, exerting osteoclastogenic activity by leading to RANKL expression in osteoblasts (164). Upon the phosphorylation of JNK, Wnt5a-Ror2 upregulates the expression of RANK in osteoclast precursors, c-Jun is recruited to the promoter of the RANK gene, and RANKL-induced osteoclastogenesis is activated (162, 165). RANKL levels can also influence the activation of osteoclast-specific genes (TRAP, cathepsin K, and MMP-9) (166).
Metalloprotease–disintegrin TNF-a convertase (TACE) (98), including MMP-7 and MMP-14, can convert the membrane-bound form of RANKL to a soluble form. In a MMP-7-deficient prostate cancer model, it is observed that MMP-7-deficient treatment reduces the risk of osteolytic bone metastasis as a result of aberrations in RANKL processing (167). However, the impact of circulating sRANKL in cancer biology is now in dispute. Increased serum sRANKL levels were observed in a cohort of 61 patients with lung or breast cancers with bone metastasis compared with healthy volunteers (168). In other studies, serum sRANKL levels between bone metastasis patients from prostate, lung, or breast cancer and those without bone metastasis did not discriminate (169–171).
Development of RANKL Inhibitors
Building on these foundations about the roles of the RANKL/RANK pathway in the immune system, bone metastasis, and many other physiological processes such as the proliferation and division of mammary epithelial cells as well as the mammary gland formation during lactation (Table 2) (37, 172–179, 182, 183), researchers have exploited the inhibitors targeting this axis to control the primary tumor development, reduce bone metastasis, and even exert a direct antitumor effect via regulating antitumor immune responses (180, 181). Antibodies targeting RANKL were preferably used; this behavior might be attributable to an unintended receptor agonist with those targeting the receptor. Denosumab, a humanized monoclonal RANKL antibody, is one such candidate, which can play an important role in different physiological activity including the immune system, bone, or cancer (108). On target cells, denosumab can bind to the DE loop region of both soluble and membrane-bound RANKL to inhibit RANKL/RANK intracellular signaling. The DE loop region is one of the surface loop structures of human RANKL binding to and oligomerizing its receptor RANK on responding cells (184). In 2001, Amgen started studies about significant therapeutic and clinical safety of denosumab, which was applied for osteoporosis patients, bone destruction caused by advanced cancer, and bone loss generated by cancer treatment in early-stage cancer.
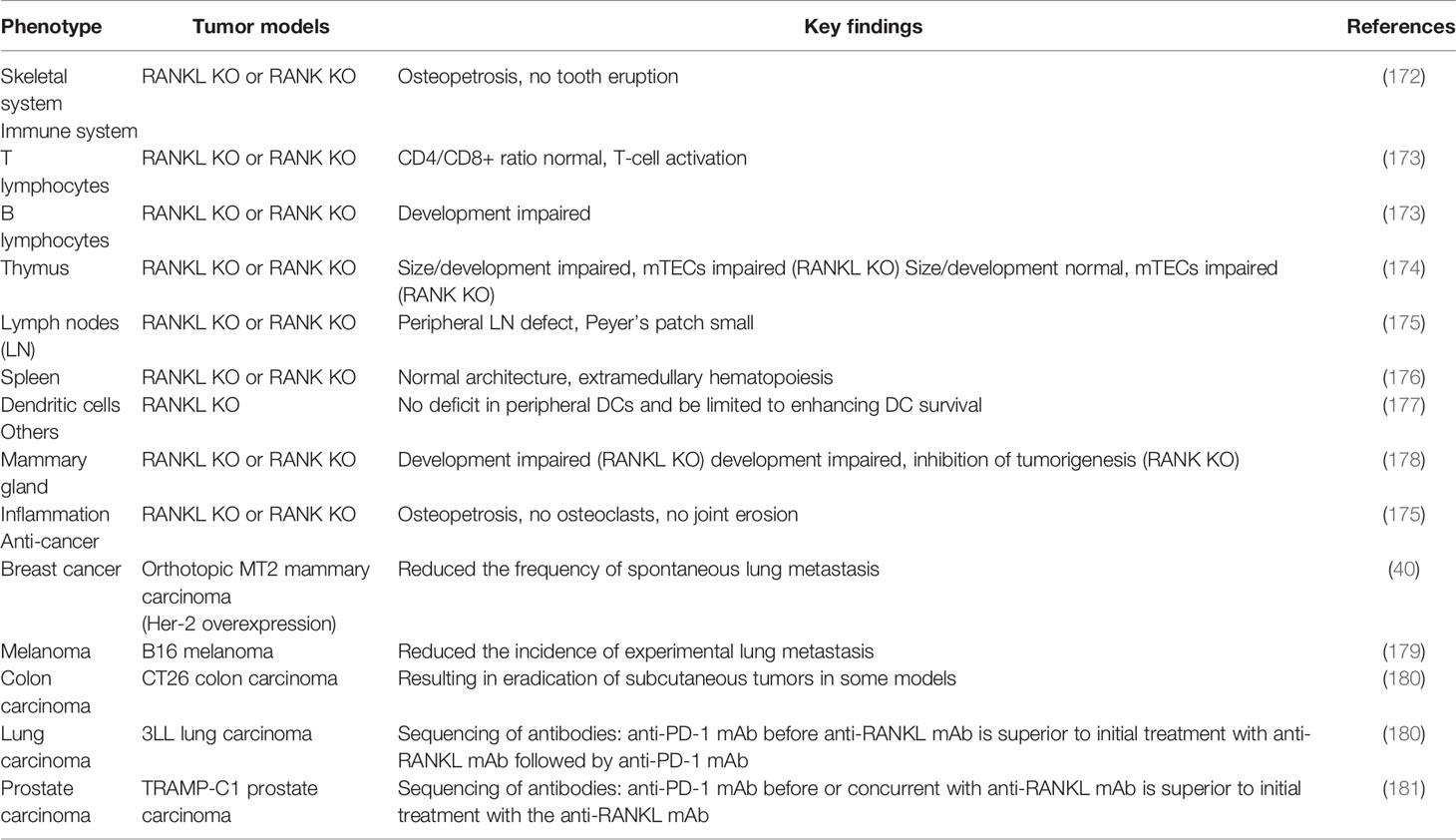
Table 2 Mice genetic phenotypes on the RANKL/RANK axis.
Osteoporosis is an increasingly common medical and socioeconomic matter characterized by decreased bone mass and strength, resulting in the propensity of fractures and substantial bone-associated morbidities. Denosumab, due to its role in osteoclastogenesis, is one of the most promising novel treatments of osteoporosis and has recently been approved in Europe and the US (185). The optimal denosumab dosage used in the treatment of osteoporosis turned out to be 60 mg at 6-month intervals subcutaneously. The antiresorptive effects of denosumab inhibiting the recruitment and activity of osteoclasts decline rapidly >6 months after each injection, and rapid decreases in bone mineral density (BMD) and bone resorption occur, losing fracture protection (186). Therefore, denosumab administration every 6 months is recommended after the last denosumab use to prevent a potential rebound effect (187). Very rare cases of osteonecrosis of the jaw and atypical femoral fractures occurred in osteoporosis cases under long-term denosumab exposure, but these possible side effects remain to be confirmed in long-term follow-up (188).
In a study including 252 postmenopausal women with early-stage breast cancer who receive endocrinotherapy with an aromatase inhibitor, after 24 months of follow-up, the result suggested that lumbar spine BMD in the denosumab group is 7.6% above the placebo group, and in the total hip, BMD is 4.7% above in denosumab individuals (189). Initiation of androgen-deprivation therapy (ADT) in localized prostate cancer patients is associated with rapidly decreased BMD and increased risk of fracture (190, 191). In a placebo-controlled trial in 1,468 nonmetastatic prostate cancer patients receiving ADT, a significant reduced incidence of new vertebral fractures was observed in the denosumab group and an increased BMD occurred, leading to significant differences in the denosumab and placebo groups (192).
Bone metastases from tumors result in RANKL upregulation, inducing the stimulation of excessive bone resorption, which can lead to SREs (193). Denosumab has been approved on the basis of a high proportion of patients with solid tumor bone metastases or multiple myeloma achieving almost complete osteoclast inhibition with denosumab (121). The encouraging preclinical trials have provided the evidence for the role of denosumab in affecting the SREs of patients with bone metastases from tumors. The endpoints in clinical trials have included SRE incidence, time to first SRE, or bone marker changes (194, 195). In these trials, those who were treated with denosumab had lower biochemical markers of bone turnover (such as urinary Ntelopeptide, uNTX) and received a significant improvement in median time to first SRE in breast cancer and castration-resistant prostate cancer patients with metastatic bone disease (196–198). Other studies indicated the result that when breast cancer and prostate cancer patients were excluded, denosumab used in solid tumor and multiple myeloma was also superior to zoledronic acid (194). Denosumab, which can determine the course of a malignancy and potentially prevent metastatic outgrowth, provides one potential strategy for metastasis prevention. In men with nonmetastatic castration-resistant prostate cancer, denosumab increased the median bone metastasis-free survival by 4.2 months, and the occurrence of the first bone metastasis event was delayed using denosumab compared with placebo (199). However, no statistically superior bone metastasis-free survival was observed in high-risk early breast cancer with denosumab in the D-CARE study, which was an international, double-blind, randomized, placebo-controlled, phase 3 study conducted at 389 centers in 39 countries in women with early-stage breast cancer at moderate to high risk of disease recurrence (181). On the other hand, in the ABCSG-18 study including 3,425 postmenopausal women with estrogen receptor-positive or progesterone receptor-positive early breast cancer, adjuvant denosumab therapy delayed the disease recurrence in postmenopausal patients with early-stage lower-risk breast cancer (180). These results suggested that the role of denosumab to delay or inhibit the occurrence of bone metastasis in solid tumors requires further investigation to provide better information and more details about its biological effect (Figure 3).
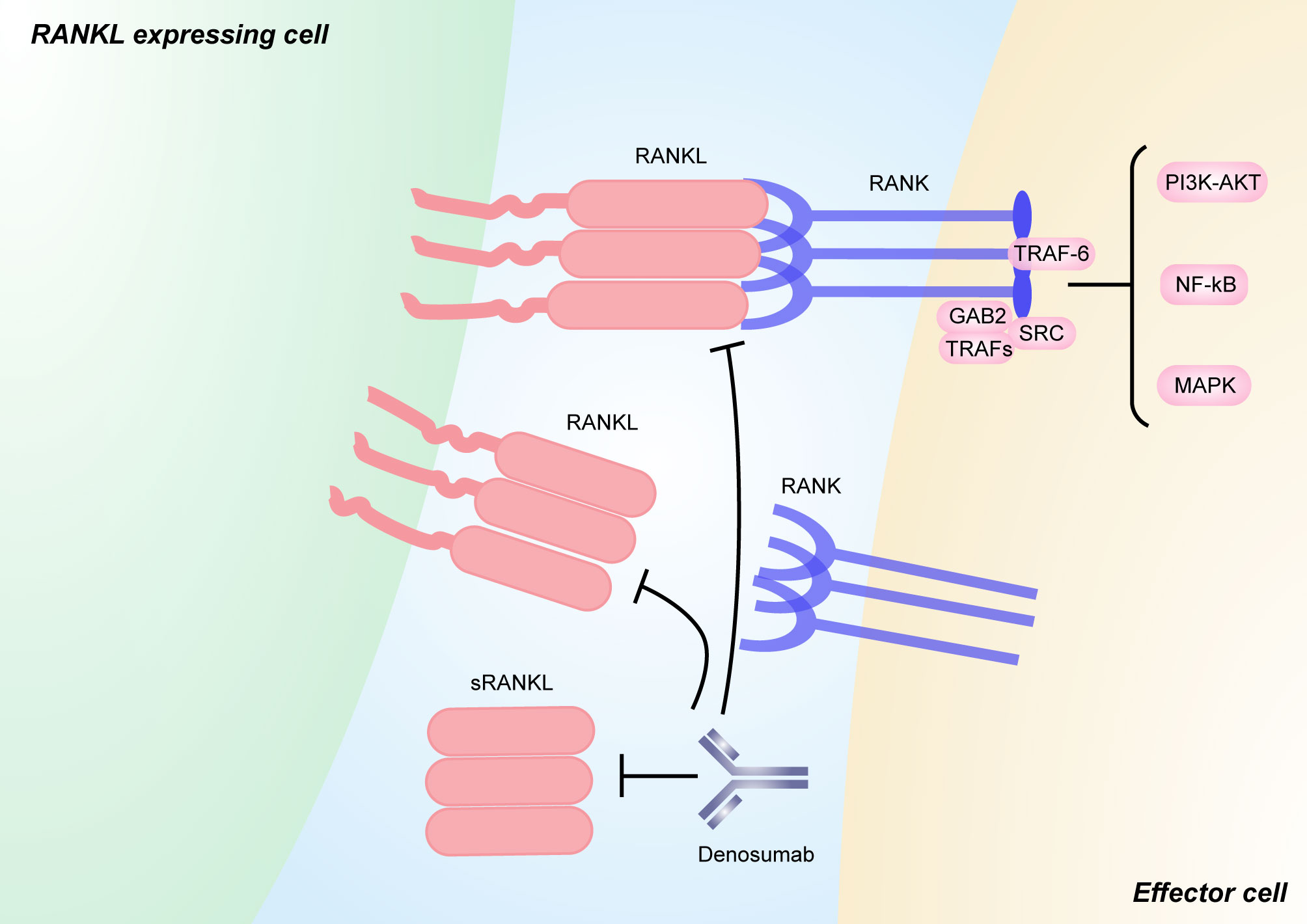
Figure 3 RANKL/RANK signaling and its inhibitors. RANKL is a type II transmembrane molecule that contains a small N-terminal intracellular domain, a transmembrane region, and a c-terminal extracellular domain consisting of a stalk and a receptor-binding region [111]. sRANKL is derived from the membrane-bound form through alternative splicing or proteolytic cleavage. Binding between RANKL and RANK induces the recruitment of TNF receptor associated (TRAF) proteins (including TRAF6), GRB-associated-binding protein 2 (GAB2), and SRC, which activates downstream signaling pathways, such as NF-κB, MAPK, and PI3K–AKT pathways. Pharmacological inhibitors targeting both soluble and membrane-bound forms of RANKL that have been used in humans include the anti-RANKL monoclonal antibody denosumab. The figure was designed using Adobe Illustrator CC.
Denosumab in Combination With ICI
The randomized clinical trials about the efficacy of therapy using combinations of denosumab and immune checkpoint inhibitors (ICIs) targeting CTLA4, programmed cell death protein 1 (PD1), or programmed cell death 1 ligand 1 (PDL1) have not been prospectively unfolded in tumor bone metastasis patients. A few retrospective case series and case reports in bone metastatic melanoma have reported the synergism of ICI with denosumab. In case series including 10 patients with metastatic melanoma, the objective response rate was 60% and the disease control rate was 80% treated with denosumab and ICI (predominantly antiPD1 mAbs with or without antiCTLA4 mAbs), with the average treatment time of 9.8 months (200). Following these case studies, combination therapy targeting CTLA4 and RANKL provides more effective treatment inhibiting metastatic activities than either one of the agents alone in various mouse models (158). In 2016, a case of a melanoma patient, with aggressive and symptomatic bone metastases, was concomitantly treated with denosumab and anti-CTLA-4 antibody ipilimumab and was demonstrated to have a dramatic response and was alive at 62 weeks (177). Whether the combined effect of anti-CTLA-4 and anti-RANKL is successful is dependent on the lymphocytes’ action, as treatment was completely ineffective in mice lacking all lymphocytes and in mice depleted of natural killer cells (177). In another study, inhibition of RANKL has been shown to augment the antimetastatic efficacy of anti-PD-1/PD-L1 monoclonal antibodies in the prostate, colorectal cancer, and melanoma cell lines in mouse models (178). Mechanisms underlying the synergistic effect of anti-RANKL and ICIs remain unclear. Ahern et al. provide interesting possibilities that the optimal combination depended on the presence of activating Fc receptors and lymphocytes (particularly natural killer and CD8+ T cells) (201). The data on anti-RANKL/anti-PD1/anti-CTLA4 combination therapy showed promising results in a higher proportion of tumor-infiltrating CD4+ and CD8+ T cells that can produce both IFNγ and TNFα after anti-RANKL treatment. Such TME changes can increase ICB response through the attenuation of PD1 expression (178). Dougall et al. further underline the role of CD8+ T cells and IFNγ in enhancing PD-1 antibody efficacy. RANKL/RANK blockade can reverse the role of PD-1 blockade in CD8+ T-cell dysfunction and improve the antitumor efficacy of PD-1 antibody. However, the effect was abrogated by the depletion of CD8+ T cells or neutralization of IFNγ, suggesting major roles for RANKL/RANK blockade in an immune cell-dependent manner in the ICI response (202).
Conclusion and Perspectives
Advances in research about the early events of tumor seeding, dormancy, and local niche remodeling have provided new insights into the role of the bone microenvironment to facilitate the growth of metastatic tumors in the bone. The studies discussed above illustrate how the changes of the immune microenvironment contribute to bone metastases, providing a highly modular platform potentially applicable to a broad range of cancer bone metastases. New molecules targeting bone metastases should combine with immune modulation in order to achieve effective eradication of metastatic lesions with minimal side effects.
In the past decades, many studies have uncovered the biological network associated with RANKL–RANK in immune systems, the development of lymphoid organs, bone metastasis regulation, and the initiation and progression of sex hormone-driven mammary cancer. Now that RANKL inhibitors to block osteoclast differentiation could be effectively used in treatments of osteoporosis, bone loss, and bone metastasis, the knowledge about how to modulate RANK signaling is needed to be further excavated and long-term clinical studies are needed to estimate whether the combined use of inhibitors with conventional therapies or ICIs leads to optimal treatment for bone metastasis patients.
Author Contributions
BL and PW provided direction and guidance throughout the preparation of this manuscript. JJ and HW conducted the literature review and drafted the manuscript. PZ and WX reviewed the manuscript and made significant revisions on the drafts. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81902733), the Shanghai Science and Technology Committee (Grant No. 17411950300, 17411950301), and the Sailing Plan Project of Shanghai Municipal Commission of Science and Technology (19YF1448100).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chaffer CL, Weinberg RA. A Perspective on Cancer Cell Metastasis. Science (2011) 331:1559–64. doi: 10.1126/science.1203543
2. Mundy GR. Metastasis to Bone: Causes, Consequences and Therapeutic Opportunities. Nat Rev Cancer (2002) 2:584–93. doi: 10.1038/nrc867
3. Sugiura H, Yamada K, Sugiura T, Hida T, Mitsudomi T. Predictors of Survival in Patients With Bone Metastasis of Lung Cancer. Clin Orthop Relat Res (2008) 466:729–36. doi: 10.1007/s11999-007-0051-0
4. Batson OV. The Function of the Vertebral Veins and Their Role in the Spread of Metastases. Ann Surg (1940) 112:138–49. doi: 10.1097/00000658-194007000-00016
5. Aydinli U, Ozturk C, Bayram S, Sarihan S, Evrensel T, Yilmaz HS. Evaluation of Lung Cancer Metastases to the Spine. Acta Orthop Belg (2006) 72:592–7.
6. Kahn D, Weiner GJ, Ben-Haim S, Ponto LL, Madsen MT, Bushnell DL, et al. Positron Emission Tomographic Measurement of Bone Marrow Blood Flow to the Pelvis and Lumbar Vertebrae in Young Normal Adults. Blood (1994) 83:958–63. doi: 10.1182/blood.V83.4.958.958
7. Coleman RE, Rubens RD. The Clinical Course of Bone Metastases From Breast Cancer. Br J Cancer (1987) 55:61–6. doi: 10.1038/bjc.1987.13
8. Guise TA. Breaking Down Bone: New Insight Into Site-Specific Mechanisms of Breast Cancer Osteolysis Mediated by Metalloproteinases. Genes Dev (2009) 23:2117–23. doi: 10.1101/gad.1854909
9. Roodman GD. Mechanisms of Bone Metastasis. N Engl J Med (2004) 350:1655–64. doi: 10.1056/NEJMra030831
10. Blonder J, Xiao Z, Veenstra TD. Proteomic Profiling of Differentiating Osteoblasts. Expert Rev Proteomics (2006) 3:483–96. doi: 10.1586/14789450.3.5.483
11. Vicent S, Perurena N, Govindan R, Lecanda F. Bone Metastases in Lung Cancer. Potential Novel Approaches to Therapy. Am J Respir Crit Care Med (2015) 192:799–809. doi: 10.1164/rccm.201503-0440SO
12. Aft R, Naughton M, Trinkaus K, Watson M, Ylagan L, Chavez-MacGregor M, et al. Effect of Zoledronic Acid on Disseminated Tumour Cells in Women With Locally Advanced Breast Cancer: An Open Label, Randomised, Phase 2 Trial. Lancet Oncol (2010) 11:421–8. doi: 10.1016/S1470-2045(10)70054-1
13. Pantel K, Muller V, Auer M, Nusser N, Harbeck N, Braun S, et al. Detection and Clinical Implications of Early Systemic Tumor Cell Dissemination in Breast Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2003) 9:6326–34.
14. Braun S, Kentenich C, Janni W, Hepp F, de Waal J, Willgeroth F, et al. Lack of Effect of Adjuvant Chemotherapy on the Elimination of Single Dormant Tumor Cells in Bone Marrow of High-Risk Breast Cancer Patients. J Clin Oncol Off J Am Soc Clin Oncol (2000) 18:80–6. doi: 10.1200/JCO.2000.18.1.80
15. Fornetti J, Welm AL, Stewart SA. Understanding the Bone in Cancer Metastasis. J Bone Miner Res (2018) 33:2099–113. doi: 10.1002/jbmr.3618
16. Boabaid F, Berry JE, Koh AJ, Somerman MJ, McCcauley LK. The Role of Parathyroid Hormone-Related Protein in the Regulation of Osteoclastogenesis by Cementoblasts. J Periodontol (2004) 75:1247–54. doi: 10.1902/jop.2004.75.9.1247
17. Leibbrandt A, Penninger JM. RANK/RANKL: Regulators of Immune Responses and Bone Physiology. Ann N Y Acad Sci (2008) 1143:123–50. doi: 10.1196/annals.1443.016
18. Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK Signaling in Osteoclastogenesis and Bone Disease. Trends Mol Med (2006) 12:17–25. doi: 10.1016/j.molmed.2005.11.007
19. Wittrant Y, Theoleyre S, Chipoy C, Padrines M, Blanchard F, Heymann D, et al. RANKL/RANK/OPG: New Therapeutic Targets in Bone Tumours and Associated Osteolysis. Biochim Biophys Acta (2004) 1704:49–57. doi: 10.1016/j.bbcan.2004.05.002
20. Gori F, Hofbauer LC, Dunstan CR, Spelsberg TC, Khosla S, Riggs BL, et al. The Expression of Osteoprotegerin and RANK Ligand and the Support of Osteoclast Formation by Stromal-Osteoblast Lineage Cells is Developmentally Regulated. Endocrinology (2000) 141:4768–76. doi: 10.1210/endo.141.12.7840
21. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A Homologue of the TNF Receptor and Its Ligand Enhance T-Cell Growth and Dendritic-Cell Function. Nature (1997) 390:175–9. doi: 10.1038/36593
22. Khosla S. Minireview: The OPG/RANKL/RANK System. Endocrinology (2001) 142:5050–5. doi: 10.1210/endo.142.12.8536
23. Nosaka K, Miyamoto T, Sakai T, Mitsuya H, Suda T, Matsuoka M. Mechanism of Hypercalcemia in Adult T-Cell Leukemia: Overexpression of Receptor Activator of Nuclear Factor kappaB Ligand on Adult T-Cell Leukemia Cells. Blood (2002) 99:634–40. doi: 10.1182/blood.v99.2.634
24. Schoppet M, Preissner KT, Hofbauer LC. RANK Ligand and Osteoprotegerin: Paracrine Regulators of Bone Metabolism and Vascular Function. Arterioscler Thromb Vasc Biol (2002) 22:549–53. doi: 10.1161/01.atv.0000012303.37971.da
25. Yeung RS. The Osteoprotegerin/Osteoprotegerin Ligand Family: Role in Inflammation and Bone Loss. J Rheumatol (2004) 31:844–6.
26. Maman S, Witz IP. A History of Exploring Cancer in Context. Nat Rev Cancer (2018) 18:359–76. doi: 10.1038/s41568-018-0006-7
27. Valiente M, Ahluwalia MS, Boire A, Brastianos PK, Goldberg SB, Lee EQ, et al. The Evolving Landscape of Brain Metastasis. Trends Cancer (2018) 4:176–96. doi: 10.1016/j.trecan.2018.01.003
28. Ceelen W, Ramsay RG, Narasimhan V, Heriot AG, De Wever O. Targeting the Tumor Microenvironment in Colorectal Peritoneal Metastases. Trends Cancer (2020) 6:236–46. doi: 10.1016/j.trecan.2019.12.008
29. Georgoudaki AM, Prokopec KE, Boura V, Hellqvist F, Sohn E, Ostling S, et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
30. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor Exosomal RNAs Promote Lung Pre-Metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell (2016) 30:243–56. doi: 10.1016/j.ccell.2016.06.021
31. Wculek SK, Malanchi I. Neutrophils Support Lung Colonization of Metastasis-Initiating Breast Cancer Cells. Nature (2015) 528:413–7. doi: 10.1038/nature16140
32. Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell (2015) 28:666–76. doi: 10.1016/j.ccell.2015.09.018
33. Vences-Catalan F, Rajapaksa R, Srivastava MK, Marabelle A, Kuo CC, Levy R, et al. Tetraspanin CD81 Promotes Tumor Growth and Metastasis by Modulating the Functions of T Regulatory and Myeloid-Derived Suppressor Cells. Cancer Res (2015) 75:4517–26. doi: 10.1158/0008-5472.CAN-15-1021
34. Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial Watch: Prognostic and Predictive Value of the Immune Infiltrate in Cancer. Oncoimmunology (2012) 1:1323–43. doi: 10.4161/onci.22009
35. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature (2017) 544:250–4. doi: 10.1038/nature21724
36. Marshall EA, Ng KW, Kung SH, Conway EM, Martinez VD, Halvorsen EC, et al. Emerging Roles of T Helper 17 and Regulatory T Cells in Lung Cancer Progression and Metastasis. Mol Cancer (2016) 15:67. doi: 10.1186/s12943-016-0551-1
37. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-Infiltrating Regulatory T Cells Stimulate Mammary Cancer Metastasis Through RANKL-RANK Signalling. Nature (2011) 470:548–53. doi: 10.1038/nature09707
38. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host Type I IFN Signals are Required for Antitumor CD8+ T Cell Responses Through CD8{alpha}+ Dendritic Cells. J Exp Med (2011) 208:2005–16. doi: 10.1084/jem.20101159
39. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26:938. doi: 10.1016/j.ccell.2014.11.010
40. Lakshmi Narendra B, Eshvendar Reddy K, Shantikumar S, Ramakrishna S. Immune System: A Double-Edged Sword in Cancer. Inflamm Res (2013) 62:823–34. doi: 10.1007/s00011-013-0645-9
41. Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, et al. Basic Mechanisms Responsible for Osteolytic and Osteoblastic Bone Metastases. Clin Cancer Res an Off J Am Assoc Cancer Res (2006) 12:6213s–6s. doi: 10.1158/1078-0432.CCR-06-1007
42. Roato I. Bone Metastases: When and How Lung Cancer Interacts With Bone. World J Clin Oncol (2014) 5:149–55. doi: 10.5306/wjco.v5.i2.149
43. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging Alpha4beta1-Positive Osteoclast Progenitors. Cancer Cell (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
44. Yao Z, Lei W, Duan R, Li Y, Luo L, Boyce BF, et al. RANKL Cytokine Enhances TNF-Induced Osteoclastogenesis Independently of TNF Receptor Associated Factor (TRAF) 6 by Degrading TRAF3 in Osteoclast Precursors. J Biol Chem (2017) 292:10169–79. doi: 10.1074/jbc.M116.771816
45. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A Multigenic Program Mediating Breast Cancer Metastasis to Bone. Cancer Cell (2003) 3:537–49. doi: 10.1016/s1535-6108(03)00132-6
46. Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 Directly Inhibits Osteoclast Differentiation by Suppressing Receptor Activator of NF-kappaB Signaling Pathways. J Biol Chem (2008) 283:11535–40. doi: 10.1074/jbc.M607999200
47. Kim JH, Sim JH, Lee S, Seol MA, Ye SK, Shin HM, et al. Interleukin-7 Induces Osteoclast Formation via STAT5, Independent of Receptor Activator of NF-kappaB Ligand. Front Immunol (2017) 8:1376. doi: 10.3389/fimmu.2017.01376
48. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and T-Cell Responses: What We do and Don't Know. Cell Res (2006) 16:126–33. doi: 10.1038/sj.cr.7310017
49. Hernandez I, Moreno JL, Zandueta C, Montuenga L, Lecanda F. Novel Alternatively Spliced ADAM8 Isoforms Contribute to the Aggressive Bone Metastatic Phenotype of Lung Cancer. Oncogene (2010) 29:3758–69. doi: 10.1038/onc.2010.130
50. Romas E, Udagawa N, Zhou H, Tamura T, Saito M, Taga T, et al. The Role of Gp130-Mediated Signals in Osteoclast Development: Regulation of Interleukin 11 Production by Osteoblasts and Distribution of Its Receptor in Bone Marrow Cultures. J Exp Med (1996) 183:2581–91. doi: 10.1084/jem.183.6.2581
51. Sethi N, Dai X, Winter CG, Kang Y. Tumor-Derived JAGGED1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell (2011) 19:192–205. doi: 10.1016/j.ccr.2010.12.022
52. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2010) 16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329
53. Chen WG, Sun J, Shen WW, Yang SZ, Zhang Y, Hu X, et al. Sema4D Expression and Secretion Are Increased by HIF-1alpha and Inhibit Osteogenesis in Bone Metastases of Lung Cancer. Clin Exp Metastasis (2019) 36:39–56. doi: 10.1007/s10585-018-9951-5
54. Van Sant C, Wang G, Anderson MG, Trask OJ, Lesniewski R, Semizarov D. Endothelin Signaling in Osteoblasts: Global Genome View and Implication of the Calcineurin/NFAT Pathway. Mol Cancer Ther (2007) 6:253–61. doi: 10.1158/1535-7163.MCT-06-0574
55. Fielder PJ, Rosenfeld RG, Graves HC, Grandbois K, Maack CA, Sawamura S, et al. Biochemical Analysis of Prostate Specific Antigen-Proteolyzed Insulin-Like Growth Factor Binding Protein-3. Growth Regul (1994) 4:164–72.
56. Bhattacharyya RS, Stern PH. IGF-I and MAP Kinase Involvement in the Stimulatory Effects of LNCaP Prostate Cancer Cell Conditioned Media on Cell Proliferation and Protein Synthesis in MC3T3-E1 Osteoblastic Cells. J Cell Biochem (2003) 90:925–37. doi: 10.1002/jcb.10726
57. Jung Y, Song J, Shiozawa Y, Wang J, Wang Z, Williams B, et al. Hematopoietic Stem Cells Regulate Mesenchymal Stromal Cell Induction Into Osteoblasts Thereby Participating in the Formation of the Stem Cell Niche. Stem Cells (2008) 26:2042–51. doi: 10.1634/stemcells.2008-0149
58. Hall CL, Keller ET. The Role of Wnts in Bone Metastases. Cancer Metastasis Rev (2006) 25:551–8. doi: 10.1007/s10555-006-9022-2
59. Weilbaecher KN, Guise TA, McCauley LK. Cancer to Bone: A Fatal Attraction. Nat Rev Cancer (2011) 11:411–25. doi: 10.1038/nrc3055
60. Liao J, Guise TA, McCauley LK. Tumor Expressed PTHrP Facilitates Prostate Cancer-Induced Osteoblastic Lesions. Int J Cancer (2008) 123:2267–78. doi: 10.1002/ijc.23602
61. Ell B, Kang Y. SnapShot: Bone Metastasis. Cell (2012) 151:690.e691. doi: 10.1016/j.cell.2012.10.005
62. Raje N, Terpos E, Willenbacher W, Shimizu K, Garcia-Sanz R, Durie B, et al. Denosumab Versus Zoledronic Acid in Bone Disease Treatment of Newly Diagnosed Multiple Myeloma: An International, Double-Blind, Double-Dummy, Randomised, Controlled, Phase 3 Study. Lancet Oncol (2018) 19:370–81. doi: 10.1016/S1470-2045(18)30072-X
63. Krzeszinski JY, Wan Y. New Therapeutic Targets for Cancer Bone Metastasis. Trends Pharmacol Sci (2015) 36:360–73. doi: 10.1016/j.tips.2015.04.006
64. Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, et al. Evidence for a Causal Role of Parathyroid Hormone-Related Protein in the Pathogenesis of Human Breast Cancer-Mediated Osteolysis. J Clin Invest (1996) 98:1544–9. doi: 10.1172/JCI118947
65. Roodman GD. Biology of Osteoclast Activation in Cancer. J Clin Oncol Off J Am Soc Clin Oncol (2001) 19:3562–71. doi: 10.1200/JCO.2001.19.15.3562
66. Guise TA. The Vicious Cycle of Bone Metastases. J Musculoskelet Neuronal Interact (2002) 2:570–2.
67. Adamik J, Galson DL, Roodman GD. Osteoblast Suppression in Multiple Myeloma Bone Disease. J Bone Oncol (2018) 13:62–70. doi: 10.1016/j.jbo.2018.09.001
68. Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P, Amadori D, et al. Pathogenesis of Osteoblastic Bone Metastases From Prostate Cancer. Cancer (2010) 116:1406–18. doi: 10.1002/cncr.24896
69. Lin SC, Yu-Lee LY, Lin SH. Osteoblastic Factors in Prostate Cancer Bone Metastasis. Curr Osteoporos Rep (2018) 16:642–7. doi: 10.1007/s11914-018-0480-6
70. Wang W, Yang X, Dai J, Lu Y, Zhang J, Keller ET. Prostate Cancer Promotes a Vicious Cycle of Bone Metastasis Progression Through Inducing Osteocytes to Secrete GDF15 That Stimulates Prostate Cancer Growth and Invasion. Oncogene (2019) 38:4540–59. doi: 10.1038/s41388-019-0736-3
71. Robling AG, Turner CH. Mechanical Signaling for Bone Modeling and Remodeling. Crit Rev Eukaryot Gene Expr (2009) 19:319–38. doi: 10.1615/critreveukargeneexpr.v19.i4.50
72. Rubin J, Rubin C, Jacobs CR. Molecular Pathways Mediating Mechanical Signaling in Bone. Gene (2006) 367:1–16. doi: 10.1016/j.gene.2005.10.028
73. Takayanagi H. Mechanistic Insight Into Osteoclast Differentiation in Osteoimmunology. J Mol Med (Berl) (2005) 83:170–9. doi: 10.1007/s00109-004-0612-6
74. Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, et al. Osteoimmunology: Interplay Between the Immune System and Bone Metabolism. Annu Rev Immunol (2006) 24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646
75. Horton JE, Raisz LG, Simmons HA, Oppenheim JJ, Mergenhagen SE. Bone Resorbing Activity in Supernatant Fluid From Cultured Human Peripheral Blood Leukocytes. Science (1972) 177:793–5. doi: 10.1126/science.177.4051.793
76. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T Cells Regulate Bone Loss and Joint Destruction in Adjuvant Arthritis Through Osteoprotegerin Ligand. Nature (1999) 402:304–9. doi: 10.1038/46303
77. Takayanagi H. Osteoimmunology: Shared Mechanisms and Crosstalk Between the Immune and Bone Systems. Nat Rev Immunol (2007) 7:292–304. doi: 10.1038/nri2062
78. Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: Interactions of the Bone and Immune System. Endocr Rev (2008) 29:403–40. doi: 10.1210/er.2007-0038
79. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-Cell-Mediated Regulation of Osteoclastogenesis by Signalling Cross-Talk Between RANKL and IFN-Gamma. Nature (2000) 408:600–5. doi: 10.1038/35046102
80. Quinn JM, Itoh K, Udagawa N, Hausler K, Yasuda H, Shima N, et al. Transforming Growth Factor Beta Affects Osteoclast Differentiation via Direct and Indirect Actions. J Bone Miner Res (2001) 16:1787–94. doi: 10.1359/jbmr.2001.16.10.1787
81. Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, et al. Treg Cells Suppress Osteoclast Formation: A New Link Between the Immune System and Bone. Arthritis Rheum (2007) 56:4104–12. doi: 10.1002/art.23138
82. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J Exp Med (2006) 203:2673–82. doi: 10.1084/jem.20061775
83. Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ, Kim HM, et al. Osteoclastogenesis is Enhanced by Activated B Cells But Suppressed by Activated CD8(+) T Cells. Eur J Immunol (2001) 31:2179–88. doi: 10.1002/1521-4141(200107)31:7<2179::aid-immu2179>3.0.co;2-x
84. Grcevic D, Lukic IK, Kovacic N, Ivcevic S, Katavic V, Marusic A, et al. Activated T Lymphocytes Suppress Osteoclastogenesis by Diverting Early Monocyte/Macrophage Progenitor Lineage Commitment Towards Dendritic Cell Differentiation Through Down-Regulation of Receptor Activator of Nuclear factor-kappaB and C-Fos. Clin Exp Immunol (2006) 146:146–58. doi: 10.1111/j.1365-2249.2006.03181.x
85. Schett G, David JP. The Multiple Faces of Autoimmune-Mediated Bone Loss. Nat Rev Endocrinol (2010) 6:698–706. doi: 10.1038/nrendo.2010.190
86. Frey O, Petrow PK, Gajda M, Siegmund K, Huehn J, Scheffold A, et al. The Role of Regulatory T Cells in Antigen-Induced Arthritis: Aggravation of Arthritis After Depletion and Amelioration After Transfer of CD4+CD25+ T Cells. Arthritis Res Ther (2005) 7:R291–301. doi: 10.1186/ar1484
87. Takayanagi H. Osteoimmunology and the Effects of the Immune System on Bone. Nat Rev Rheumatol (2009) 5:667–76. doi: 10.1038/nrrheum.2009.217
88. Wakkach A, Mansour A, Dacquin R, Coste E, Jurdic P, Carle GF, et al. Bone Marrow Microenvironment Controls the In Vivo Differentiation of Murine Dendritic Cells Into Osteoclasts. Blood (2008) 112:5074–83. doi: 10.1182/blood-2008-01-132787
89. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune Microenvironments in Solid Tumors: New Targets for Therapy. Genes Dev (2011) 25:2559–72. doi: 10.1101/gad.169029.111
90. Smith HA, Kang Y. The Metastasis-Promoting Roles of Tumor-Associated Immune Cells. J Mol Med (Berl) (2013) 91:411–29. doi: 10.1007/s00109-013-1021-5
91. Brylka LJ, Schinke T. Chemokines in Physiological and Pathological Bone Remodeling. Front Immunol (2019) 10:2182. doi: 10.3389/fimmu.2019.02182
92. Sawant A, Ponnazhagan S. Myeloid-Derived Suppressor Cells as Osteoclast Progenitors: A Novel Target for Controlling Osteolytic Bone Metastasis. Cancer Res (2013) 73:4606–10. doi: 10.1158/0008-5472.CAN-13-0305
93. Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling J, et al. Myeloid-Derived Suppressor Cells Expand During Breast Cancer Progression and Promote Tumor-Induced Bone Destruction. Oncoimmunology (2012) 1:1484–94. doi: 10.4161/onci.21990
94. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
95. Tanaka A, Sakaguchi S. Regulatory T Cells in Cancer Immunotherapy. Cell Res (2017) 27:109–18. doi: 10.1038/cr.2016.151
96. Garner H, de Visser KE. Immune Crosstalk in Cancer Progression and Metastatic Spread: A Complex Conversation. Nat Rev Immunol (2020) 20:483–97. doi: 10.1038/s41577-019-0271-z
97. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
98. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front Immunol (2014) 5:511. doi: 10.3389/fimmu.2014.00511
99. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell (2001) 104:487–501. doi: 10.1016/s0092-8674(01)00237-9
100. Croft M. Co-Stimulatory Members of the TNFR Family: Keys to Effective T-Cell Immunity? Nat Rev Immunol (2003) 3:609–20. doi: 10.1038/nri1148
101. Croft M. The Role of TNF Superfamily Members in T-Cell Function and Diseases. Nat Rev Immunol (2009) 9:271–85. doi: 10.1038/nri2526
102. Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF Superfamily in Inflammatory Disease: Translating Basic Insights. Trends Immunol (2012) 33:144–52. doi: 10.1016/j.it.2011.10.004
103. Croft M, Siegel RM. Beyond TNF: TNF Superfamily Cytokines as Targets for the Treatment of Rheumatic Diseases. Nat Rev Rheumatol (2017) 13:217–33. doi: 10.1038/nrrheum.2017.22
104. Yang QQ, Zhou JW. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia (2019) 67:1017–35. doi: 10.1002/glia.23571
105. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol Rev (2019) 99:115–60. doi: 10.1152/physrev.00045.2017
106. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. LGR4 Is a Receptor for RANKL and Negatively Regulates Osteoclast Differentiation and Bone Resorption. Nat Med (2016) 22:539–46. doi: 10.1038/nm.4076
107. Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, et al. TRANCE (Tumor Necrosis Factor [TNF]-Related Activation-Induced Cytokine), a New TNF Family Member Predominantly Expressed in T Cells, Is a Dendritic Cell-Specific Survival Factor. J Exp Med (1997) 186:2075–80. doi: 10.1084/jem.186.12.2075
108. Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, et al. Bench to Bedside: Elucidation of the OPG-RANK-RANKL Pathway and the Development of Denosumab. Nat Rev Drug Discov (2012) 11:401–19. doi: 10.1038/nrd3705
109. Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a Novel Ligand of the Tumor Necrosis Factor Receptor Family That Activates C-Jun N-Terminal Kinase in T Cells. J Biol Chem (1997) 272:25190–4. doi: 10.1074/jbc.272.40.25190
110. Han Y, You X, Xing W, Zhang Z, Zou W. Paracrine and Endocrine Actions of Bone-the Functions of Secretory Proteins From Osteoblasts, Osteocytes, and Osteoclasts. Bone Res (2018) 6:16. doi: 10.1038/s41413-018-0019-6
111. Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, et al. Negative Regulation of Osteoclastogenesis by Ectodomain Shedding of Receptor Activator of NF-kappaB Ligand. J Biol Chem (2006) 281:36846–55. doi: 10.1074/jbc.M606656200
112. Nakashima T, Hayashi M, Takayanagi H. New Insights Into Osteoclastogenic Signaling Mechanisms. Trends Endocrinol Metab (2012) 23:582–90. doi: 10.1016/j.tem.2012.05.005
113. Miyamoto T, Arai F, Ohneda O, Takagi K, Anderson DM, Suda T, et al. An Adherent Condition is Required for Formation of Multinuclear Osteoclasts in the Presence of Macrophage Colony-Stimulating Factor and Receptor Activator of Nuclear Factor Kappa B Ligand. Blood (2000) 96:4335–43. doi: 10.1182/blood.V96.13.4335
114. Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-Mediated Activation of NF-Kappa B by TNF Receptor 2 and CD40. Science (1995) 269:1424–7. doi: 10.1126/science.7544915
115. Edwards JR, Mundy GR. Advances in Osteoclast Biology: Old Findings and New Insights From Mouse Models. Nat Rev Rheumatol (2011) 7:235–43. doi: 10.1038/nrrheum.2011.23
116. Kanazawa K, Kudo A. Self-Assembled RANK Induces Osteoclastogenesis Ligand-Independently. J Bone Miner Res (2005) 20:2053–60. doi: 10.1359/JBMR.050706
117. Crockett JC, Mellis DJ, Shennan KI, Duthie A, Greenhorn J, Wilkinson DI, et al. Signal Peptide Mutations in RANK Prevent Downstream Activation of NF-Kappab. J Bone Miner Res (2011) 26:1926–38. doi: 10.1002/jbmr.399
119. Kearns AE, Khosla S, Kostenuik PJ. Receptor Activator of Nuclear Factor kappaB Ligand and Osteoprotegerin Regulation of Bone Remodeling in Health and Disease. Endocr Rev (2008) 29:155–92. doi: 10.1210/er.2007-0014
120. Seeman E, Delmas PD. Bone Quality–the Material and Structural Basis of Bone Strength and Fragility. N Engl J Med (2006) 354:2250–61. doi: 10.1056/NEJMra053077
121. Boyle WJ, Simonet WS, Lacey DL. Osteoclast Differentiation and Activation. Nature (2003) 423:337–42. doi: 10.1038/nature01658
122. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a Key Regulator of Osteoclastogenesis, Lymphocyte Development and Lymph-Node Organogenesis. Nature (1999) 397:315–23. doi: 10.1038/16852
123. Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the Intrinsic Hematopoietic Cell Surface Receptor That Controls Osteoclastogenesis and Regulation of Bone Mass and Calcium Metabolism. Proc Natl Acad Sci USA (2000) 97:1566–71. doi: 10.1073/pnas.97.4.1566
124. Mizuno A, Kanno T, Hoshi M, Shibata O, Yano K, Fujise N, et al. Transgenic Mice Overexpressing Soluble Osteoclast Differentiation Factor (sODF) Exhibit Severe Osteoporosis. J Bone Miner Metab (2002) 20:337–44. doi: 10.1007/s007740200049
125. Ming J, Cronin SJF, Penninger JM. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front Oncol (2020) 10:1283. doi: 10.3389/fonc.2020.01283
126. Reddy SV, Menaa C, Singer FR, Demulder A, Roodman GD. Cell Biology of Paget's Disease. J Bone Miner Res (1999) 14 Suppl 2:3–8. doi: 10.1002/jbmr.5650140203
127. Adebanjo OA, Moonga BS, Yamate T, Sun L, Minkin C, Abe E, et al. Mode of Action of Interleukin-6 on Mature Osteoclasts. Novel Interactions With Extracellular Ca2+ Sensing in the Regulation of Osteoclastic Bone Resorption. J Cell Biol (1998) 142:1347–56. doi: 10.1083/jcb.142.5.1347
128. Graves DT, Alshabab A, Albiero ML, Mattos M, Correa JD, Chen S, et al. Osteocytes Play an Important Role in Experimental Periodontitis in Healthy and Diabetic Mice Through Expression of RANKL. J Clin Periodontol (2018) 45:285–92. doi: 10.1111/jcpe.12851
129. Zimmerman SM, Heard-Lipsmeyer ME, Dimori M, Thostenson JD, Mannen EM, O'Brien CA, et al. Loss of RANKL in Osteocytes Dramatically Increases Cancellous Bone Mass in the Osteogenesis Imperfecta Mouse (Oim). Bone Rep (2018) 9:61–73. doi: 10.1016/j.bonr.2018.06.008
130. Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, et al. Coupling of Bone Resorption and Formation by RANKL Reverse Signalling. Nature (2018) 561:195–200. doi: 10.1038/s41586-018-0482-7
131. Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, et al. TRANCE, a TNF Family Member, Activates Akt/PKB Through a Signaling Complex Involving TRAF6 and C-Src. Mol Cell (1999) 4:1041–9. doi: 10.1016/s1097-2765(00)80232-4
132. Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, et al. TRANCE, a Tumor Necrosis Factor Family Member, Enhances the Longevity and Adjuvant Properties of Dendritic Cells In Vivo. J Exp Med (2000) 191:495–502. doi: 10.1084/jem.191.3.495
133. Cheng ML, Fong L. Effects of RANKL-Targeted Therapy in Immunity and Cancer. Front Oncol (2014) 3:329. doi: 10.3389/fonc.2013.00329
134. Ferrari-Lacraz S, Ferrari S. Do RANKL Inhibitors (Denosumab) Affect Inflammation and Immunity? Osteoporos Int (2011) 22:435–46. doi: 10.1007/s00198-010-1326-y
135. Steinman RM. Decisions About Dendritic Cells: Past, Present, and Future. Annu Rev Immunol (2012) 30:1–22. doi: 10.1146/annurev-immunol-100311-102839
136. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
137. Collin M, Bigley V. Human Dendritic Cell Subsets: An Update. Immunology (2018) 154:3–20. doi: 10.1111/imm.12888
138. Merad M, Sathe P, Helft J, Miller J, Mortha A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu Rev Immunol (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
139. Mildner A, Jung S. Development and Function of Dendritic Cell Subsets. Immunity (2014) 40:642–56. doi: 10.1016/j.immuni.2014.04.016
140. Kapsenberg ML. Dendritic-Cell Control of Pathogen-Driven T-Cell Polarization. Nat Rev Immunol (2003) 3:984–93. doi: 10.1038/nri1246
141. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol Rev (2017) 97:1295–349. doi: 10.1152/physrev.00036.2016
142. Wang Y, Huang G, Vogel P, Neale G, Reizis B, Chi H. Transforming Growth Factor Beta-Activated Kinase 1 (TAK1)-Dependent Checkpoint in the Survival of Dendritic Cells Promotes Immune Homeostasis and Function. Proc Natl Acad Sci USA (2012) 109:E343–352. doi: 10.1073/pnas.1115635109
143. Izawa T, Ishimaru N, Moriyama K, Kohashi M, Arakaki R, Hayashi Y. Crosstalk Between RANKL and Fas Signaling in Dendritic Cells Controls Immune Tolerance. Blood (2007) 110:242–50. doi: 10.1182/blood-2006-11-059980
144. Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL Controls Regulatory T-Cell Numbers via Activation Dendritic Cells. Nat Med (2006) 12:1372–9. doi: 10.1038/nm1518
145. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
146. American Society for B. Mineral Research President's Committee on, N. Proposed Standard Nomenclature for New Tumor Necrosis Factor Family Members Involved in the Regulation of Bone Resorption. The American Society for Bone and Mineral Research President's Committee on Nomenclature. J Bone Miner Res (2000) 15:2293–6. doi: 10.1359/jbmr.2000.15.12.2293
147. Iberg CA, Jones A, Hawiger D. Dendritic Cells As Inducers of Peripheral Tolerance. Trends Immunol (2017) 38:793–804. doi: 10.1016/j.it.2017.07.007
148. Proposed Standard Nomenclature for New Tumor Necrosis Factor Members Involved in the Regulation of Bone Resorption. The American Society for Bone and Mineral Research President's Committee on Nomenclature. Bone (2000) 27:761–4. doi: 10.1016/s8756-3282(00)00420-8
149. Miyamoto N, Higuchi Y, Mori K, Ito M, Tsurudome M, Nishio M, et al. Human Osteosarcoma-Derived Cell Lines Produce Soluble Factor(s) That Induces Differentiation of Blood Monocytes to Osteoclast-Like Cells. Int Immunopharmacol (2002) 2:25–38. doi: 10.1016/s1567-5769(01)00134-5
150. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is Essential for Osteoclast and Lymph Node Development. Genes Dev (1999) 13:2412–24. doi: 10.1101/gad.13.18.2412
151. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The Osteoclast Differentiation Factor Osteoprotegerin-Ligand is Essential for Mammary Gland Development. Cell (2000) 103:41–50. doi: 10.1016/s0092-8674(00)00103-3
152. Gravallese EM. Bone Destruction in Arthritis. Ann Rheum Dis (2002) 61(Suppl 2):ii84–86. doi: 10.1136/ard.61.suppl_2.ii84
153. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T Cells, Bone Loss, and Mammalian Evolution. Annu Rev Immunol (2002) 20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753
154. Nagasawa T. Microenvironmental Niches in the Bone Marrow Required for B-Cell Development. Nat Rev Immunol (2006) 6:107–16. doi: 10.1038/nri1780
155. Perlot T, Penninger JM. Development and Function of Murine B Cells Lacking RANK. J Immunol (2012) 188:1201–5. doi: 10.4049/jimmunol.1102063
156. Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of Cancer Cell Migration and Bone Metastasis by RANKL. Nature (2006) 440:692–6. doi: 10.1038/nature04524
157. Campbell JP, Karolak MR, Ma Y, Perrien DS, Masood-Campbell SK, Penner N, et al. Stimulation of Host Bone Marrow Stromal Cells by Sympathetic Nerves Promotes Breast Cancer Bone Metastasis in Mice. PloS Biol (2012) 10:e1001363. doi: 10.1371/journal.pbio.1001363
158. de Groot AF, Appelman-Dijkstra NM, van der Burg SH, Kroep JR. The Anti-Tumor Effect of RANKL Inhibition in Malignant Solid Tumors - A Systematic Review. Cancer Treat Rev (2018) 62:18–28. doi: 10.1016/j.ctrv.2017.10.010
159. Vidula N, Yau C, Li J, Esserman LJ, Rugo HS. Receptor Activator of Nuclear Factor Kappa B (RANK) Expression in Primary Breast Cancer Correlates With Recurrence-Free Survival and Development of Bone Metastases in I-SPY1 (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat (2017) 165:129–38. doi: 10.1007/s10549-017-4318-1
160. Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, et al. Nuclear Cytokine-Activated IKKalpha Controls Prostate Cancer Metastasis by Repressing Maspin. Nature (2007) 446:690–4. doi: 10.1038/nature05656
161. Takahashi N, Udagawa N, Suda T. A New Member of Tumor Necrosis Factor Ligand Family, ODF/OPGL/TRANCE/RANKL, Regulates Osteoclast Differentiation and Function. Biochem Biophys Res Commun (1999) 256:449–55. doi: 10.1006/bbrc.1999.0252
162. Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, et al. Wnt5a-Ror2 Signaling Between Osteoblast-Lineage Cells and Osteoclast Precursors Enhances Osteoclastogenesis. Nat Med (2012) 18:405–12. doi: 10.1038/nm.2653
163. Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic Niche Supports the Growth of Quiescent Multiple Myeloma Cells. Blood (2014) 123:2204–8. doi: 10.1182/blood-2013-07-517136
164. Roodman GD. Cell Biology of the Osteoclast. Exp Hematol (1999) 27:1229–41. doi: 10.1016/s0301-472x(99)00061-2
165. Takada I, Mihara M, Suzawa M, Ohtake F, Kobayashi S, Igarashi M, et al. A Histone Lysine Methyltransferase Activated by Non-Canonical Wnt Signalling Suppresses PPAR-Gamma Transactivation. Nat Cell Biol (2007) 9:1273–85. doi: 10.1038/ncb1647
166. Jules J, Zhang P, Ashley JW, Wei S, Shi Z, Liu J, et al. Molecular Basis of Requirement of Receptor Activator of Nuclear Factor kappaB Signaling for Interleukin 1-Mediated Osteoclastogenesis. J Biol Chem (2012) 287:15728–38. doi: 10.1074/jbc.M111.296228
167. Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, et al. MMP-7 Promotes Prostate Cancer-Induced Osteolysis via Solubilization RANKL. Cancer Cell (2005) 7:485–96. doi: 10.1016/j.ccr.2005.04.013
168. Mountzios G, Dimopoulos MA, Bamias A, Papadopoulos G, Kastritis E, Syrigos K, et al. Abnormal Bone Remodeling Process is Due to an Imbalance in the Receptor Activator of Nuclear factor-kappaB Ligand (RANKL)/osteoprotegerin (OPG) Axis in Patients With Solid Tumors Metastatic to the Skeleton. Acta Oncol (2007) 46:221–9. doi: 10.1080/02841860600635870
169. Jung K, Lein M, Stephan C, Von Hosslin K, Semjonow A, Sinha P, et al. Comparison of 10 Serum Bone Turnover Markers in Prostate Carcinoma Patients With Bone Metastatic Spread: Diagnostic and Prognostic Implications. Int J Cancer (2004) 111:783–91. doi: 10.1002/ijc.20314
170. Leeming DJ, Koizumi M, Byrjalsen I, Li B, Qvist P, Tanko LB. The Relative Use of Eight Collagenous and Noncollagenous Markers for Diagnosis of Skeletal Metastases in Breast, Prostate, or Lung Cancer Patients. Cancer Epidemiol Biomark Prev (2006) 15:32–8. doi: 10.1158/1055-9965.EPI-05-0492
171. Jung K, Stephan C, Semjonow A, Lein M, Schnorr D, Loening SA, et al. Serum Osteoprotegerin and Receptor Activator of Nuclear Factor-Kappa B Ligand as Indicators of Disturbed Osteoclastogenesis in Patients With Prostate Cancer. J Urol (2003) 170:2302–5. doi: 10.1097/01.ju.0000094191.39574.cb
172. Rossi SW, Kim MY, Leibbrandt A, Parnell SM, Jenkinson WE, Glanville SH, et al. RANK Signals From CD4(+)3(-) Inducer Cells Regulate Development of Aire-Expressing Epithelial Cells in the Thymic Medulla. J Exp Med (2007) 204:1267–72. doi: 10.1084/jem.20062497
173. Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, et al. The Tumor Necrosis Factor Family Receptors RANK and CD40 Cooperatively Establish the Thymic Medullary Microenvironment and Self-Tolerance. Immunity (2008) 29:423–37. doi: 10.1016/j.immuni.2008.06.015
174. Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, et al. A Chemokine-Driven Positive Feedback Loop Organizes Lymphoid Follicles. Nature (2000) 406:309–14. doi: 10.1038/35018581
175. Wu Q, Wang Y, Wang J, Hedgeman EO, Browning JL, Fu YX, et al. The Requirement of Membrane Lymphotoxin for the Presence of Dendritic Cells in Lymphoid Tissues. J Exp Med (1999) 190:629–38. doi: 10.1084/jem.190.5.629
176. Whyte MP. Paget's Disease of Bone and Genetic Disorders of RANKL/OPG/RANK/NF-kappaB Signaling. Ann N Y Acad Sci (2006) 1068:143–64. doi: 10.1196/annals.1346.016
177. Smyth MJ, Yagita H, McArthur GA. Combination Anti-CTLA-4 and Anti-RANKL in Metastatic Melanoma. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34:e104–106. doi: 10.1200/JCO.2013.51.3572
178. Ahern E, Harjunpaa H, O'Donnell JS, Allen S, Dougall WC, Teng MWL, et al. RANKL Blockade Improves Efficacy of PD1-PD-L1 Blockade or Dual PD1-PD-L1 and CTLA4 Blockade in Mouse Models of Cancer. Oncoimmunology (2018) 7:e1431088. doi: 10.1080/2162402X.2018.1431088
179. Liede A, Hernandez RK, Wade SW, Bo R, Nussbaum NC, Ahern E, et al. An Observational Study of Concomitant Immunotherapies and Denosumab in Patients With Advanced Melanoma or Lung Cancer. Oncoimmunology (2018) 7:e1480301. doi: 10.1080/2162402X.2018.1480301
180. Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, et al. Adjuvant Denosumab in Postmenopausal Patients With Hormone Receptor-Positive Breast Cancer (ABCSG-18): Disease-Free Survival Results From a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2019) 20:339–51. doi: 10.1016/S1470-2045(18)30862-3
181. Coleman R, Finkelstein DM, Barrios C, Martin M, Iwata H, Hegg R, et al. Adjuvant Denosumab in Early Breast Cancer (D-CARE): An International, Multicentre, Randomised, Controlled, Phase 3 Trial. Lancet Oncol (2020) 21:60–72. doi: 10.1016/S1470-2045(19)30687-4
182. Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res (2016) 76:1089–100. doi: 10.1158/0008-5472.CAN-15-1703
183. Morris MJ, Molina A, Small EJ, de Bono JS, Logothetis CJ, Fizazi K, et al. Radiographic Progression-Free Survival as a Response Biomarker in Metastatic Castration-Resistant Prostate Cancer: COU-AA-302 Results. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33:1356–63. doi: 10.1200/JCO.2014.55.3875
184. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin Ligand Is a Cytokine That Regulates Osteoclast Differentiation and Activation. Cell (1998) 93:165–76. doi: 10.1016/s0092-8674(00)81569-x
185. Harslof T, Langdahl BL. New Horizons in Osteoporosis Therapies. Curr Opin Pharmacol (2016) 28:38–42. doi: 10.1016/j.coph.2016.02.012
186. Miller PD, Bolognese MA, Lewiecki EM, McClung MR, Ding B, Austin M, et al. Effect of Denosumab on Bone Density and Turnover in Postmenopausal Women With Low Bone Mass After Long-Term Continued, Discontinued, and Restarting of Therapy: A Randomized Blinded Phase 2 Clinical Trial. Bone (2008) 43:222–9. doi: 10.1016/j.bone.2008.04.007
187. Terpos E, Zamagni E, Lentzsch S, Drake MT, Garcia-Sanz R, Abildgaard N, et al. Treatment of Multiple Myeloma-Related Bone Disease: Recommendations From the Bone Working Group of the International Myeloma Working Group. Lancet Oncol (2021) 22:e119–30. doi: 10.1016/S1470-2045(20)30559-3
188. Papapoulos S, Lippuner K, Roux C, Lin CJ, Kendler DL, Lewiecki EM, et al. The Effect of 8 or 5 Years of Denosumab Treatment in Postmenopausal Women With Osteoporosis: Results From the FREEDOM Extension Study. Osteoporos Int (2015) 26:2773–83. doi: 10.1007/s00198-015-3234-7
189. Ellis GK, Bone HG, Chlebowski R, Paul D, Spadafora S, Smith J, et al. Randomized Trial of Denosumab in Patients Receiving Adjuvant Aromatase Inhibitors for Nonmetastatic Breast Cancer. J Clin Oncol Off J Am Soc Clin Oncol (2008) 26:4875–82. doi: 10.1200/JCO.2008.16.3832
190. Shahinian VB, Kuo YF, Freeman JL, Goodwin JS. Risk of Fracture After Androgen Deprivation for Prostate Cancer. N Engl J Med (2005) 352:154–64. doi: 10.1056/NEJMoa041943
191. Smith MR, Boyce SP, Moyneur E, Duh M, Raut S, Brandman MK, et al. Risk of Clinical Fractures After Gonadotropin-Releasing Hormone Agonist Therapy for Prostate Cancer. J Urol (2006) 175:136–139; discussion 139. doi: 10.1016/S0022-5347(05)00033-9
192. Smith MR, Egerdie B, Hernandez Toriz N, Feldman R, Tammela TL, Saad F, et al. Denosumab in Men Receiving Androgen-Deprivation Therapy for Prostate Cancer. N Engl J Med (2009) 361:745–55. doi: 10.1056/NEJMoa0809003
193. Dougall WC. Molecular Pathways: Osteoclast-Dependent and Osteoclast-Independent Roles of the RANKL/RANK/OPG Pathway in Tumorigenesis and Metastasis. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18:326–35. doi: 10.1158/1078-0432.CCR-10-2507
194. Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, et al. Randomized, Double-Blind Study of Denosumab Versus Zoledronic Acid in the Treatment of Bone Metastases in Patients With Advanced Cancer (Excluding Breast and Prostate Cancer) or Multiple Myeloma. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29:1125–32. doi: 10.1200/JCO.2010.31.3304
195. Hirsh V, Major PP, Lipton A, Cook RJ, Langer CJ, Smith MR, et al. Zoledronic Acid and Survival in Patients With Metastatic Bone Disease From Lung Cancer and Elevated Markers of Osteoclast Activity. J Thorac Oncol (2008) 3:228–36. doi: 10.1097/JTO.0b013e3181651c0e
196. Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, et al. Denosumab Compared With Zoledronic Acid for the Treatment of Bone Metastases in Patients With Advanced Breast Cancer: A Randomized, Double-Blind Study. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28:5132–9. doi: 10.1200/JCO.2010.29.7101
197. Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, et al. Denosumab Versus Zoledronic Acid for Treatment of Bone Metastases in Men With Castration-Resistant Prostate Cancer: A Randomised, Double-Blind Study. Lancet (2011) 377:813–22. doi: 10.1016/S0140-6736(10)62344-6
198. Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, et al. Randomized Phase II Trial of Denosumab in Patients With Bone Metastases From Prostate Cancer, Breast Cancer, or Other Neoplasms After Intravenous Bisphosphonates. J Clin Oncol Off J Am Soc Clin Oncol (2009) 27:1564–71. doi: 10.1200/JCO.2008.19.2146
199. Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. Denosumab and Bone-Metastasis-Free Survival in Men With Castration-Resistant Prostate Cancer: Results of a Phase 3, Randomised, Placebo-Controlled Trial. Lancet (2012) 379:39–46. doi: 10.1016/S0140-6736(11)61226-9
200. Ahern E, Smyth MJ, Dougall WC, Teng MWL. Roles of the RANKL-RANK Axis in Antitumour Immunity - Implications for Therapy. Nat Rev Clin Oncol (2018) 15:676–93. doi: 10.1038/s41571-018-0095-y
201. Ahern E, Harjunpaa H, Barkauskas D, Allen S, Takeda K, Yagita H, et al. Co-Administration of RANKL and CTLA4 Antibodies Enhances Lymphocyte-Mediated Antitumor Immunity in Mice. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23:5789–801. doi: 10.1158/1078-0432.CCR-17-0606
Keywords: bone metastasis, osteoclasts, osteoblast, RANKL/RANK, immune cells, denosumab
Citation: Li B, Wang P, Jiao J, Wei H, Xu W and Zhou P (2022) Roles of the RANKL–RANK Axis in Immunity—Implications for Pathogenesis and Treatment of Bone Metastasis. Front. Immunol. 13:824117. doi: 10.3389/fimmu.2022.824117
Received: 28 November 2021; Accepted: 16 February 2022;
Published: 21 March 2022.
Edited by:
Antonio Maurizi, University of L’Aquila, ItalyReviewed by:
Marco Ponzetti, University of L’Aquila, ItalyShunji Takahashi, Japanese Foundation For Cancer Research, Japan
Allan Lipton, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2022 Li, Wang, Jiao, Wei, Xu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingting Zhou, cHR6aG91Xzlob3NwaXRhbEAxNjMuY29t; Wei Xu, eHV3ZWljaGFuZ3poZW5nQGhvdG1haWwuY29t
†These authors have contributed equally to this work