 Maximilian Haist1*
Maximilian Haist1* Frederic Ries2
Frederic Ries2 Matthias Gunzer3,4
Matthias Gunzer3,4 Monika Bednarczyk1Ekkehard Siegel5
Monika Bednarczyk1Ekkehard Siegel5 Michael Kuske1
Michael Kuske1 Stephan Grabbe1
Stephan Grabbe1 Markus Radsak2†
Markus Radsak2† Matthias Bros1†
Matthias Bros1† Daniel Teschner2,6†
Daniel Teschner2,6†- 1Department of Dermatology, University Medical Center of the Johannes Gutenberg University, Mainz, Germany
- 2Department of Hematology, Medical Oncology and Pneumology, University Medical Center of the Johannes Gutenberg University, Mainz, Germany
- 3Institute for Experimental Immunology and Imaging, University Hospital, University Duisburg-Essen, Essen, Germany
- 4Leibniz-Institut für Analytische Wissenschaften ISAS -e.V, Dortmund, Germany
- 5Institute for Medical Microbiology and Hygiene, University Medical Center of the Johannes Gutenberg University, Mainz, Germany
- 6Department of Internal Medicine II, University Hospital Würzburg, Würzburg, Germany
β2-integrins are heterodimeric surface receptors that are expressed specifically by leukocytes and consist of a variable α (CD11a-d) and a common β-subunit (CD18). Functional impairment of CD18, which causes leukocyte adhesion deficiency type-1 results in an immunocompromised state characterized by severe infections, such as invasive pulmonary aspergillosis (IPA). The underlying immune defects have largely been attributed to an impaired migratory and phagocytic activity of polymorphonuclear granulocytes (PMN). However, the exact contribution of β2-integrins for PMN functions in-vivo has not been elucidated yet, since the mouse models available so far display a constitutive CD18 knockout (CD18-/- or CD18hypo). To determine the PMN-specific role of β2-integrins for innate effector functions and pathogen control, we generated a mouse line with a Ly6G-specific knockdown of the common β-subunit (CD18Ly6G cKO). We characterized CD18Ly6G cKO mice in-vitro to confirm the PMN-specific knockdown of β2-integrins. Next, we investigated the clinical course of IPA in A. fumigatus infected CD18Ly6G cKO mice with regard to the fungal burden, pulmonary inflammation and PMN response towards A. fumigatus. Our results revealed that the β2-integrin knockdown was restricted to PMN and that CD18Ly6G cKO mice showed an aggravated course of IPA. In accordance, we observed a higher fungal burden and lower levels of proinflammatory innate cytokines, such as TNF-α, in lungs of IPA-infected CD18Ly6G cKO mice. Bronchoalveolar lavage revealed higher levels of CXCL1, a stronger PMN-infiltration, but concomitantly elevated apoptosis of PMN in lungs of CD18Ly6G cKO mice. Ex-vivo analysis further unveiled a strong impairment of PMN effector function, as reflected by an attenuated phagocytic activity, and a diminished generation of reactive oxygen species (ROS) and neutrophil-extracellular traps (NET) in CD18-deficient PMN. Overall, our study demonstrates that β2-integrins are required specifically for PMN effector functions and contribute to the clearance of A. fumigatus by infiltrating PMN, and the establishment of an inflammatory microenvironment in infected lungs.
1 Introduction
Humans are constantly exposed to spores of the ubiquitous environmental mould Aspergillus fumigatus (A. fumigatus) (1, 2). Although A. fumigatus is usually well controlled in healthy individuals, A. fumigatus can cause lethal invasive pulmonary aspergillosis (IPA) in immunocompromised patients, e.g., due to chemotherapeutic treatment of malignant diseases or immunosuppressive therapy after allogeneic hematopoietic stem cell transplantation, with mortality varying between 30% and 90% (1, 3). Commonly, disease follows the inhalation of airborne conidia, which germinate in the lung of immunocompromised hosts, sprouting there as hyphae (4). Despite the clinical application of potent antifungal drugs for prophylaxis and treatment of invasive fungal diseases in patients with severe immune deficiency, IPA continues to be a highly relevant health issue in daily clinical care (5).
The small size of A. fumigatus conidia (2-3µm) allows them to bypass the physiological epithelial defence of the nasal and bronchial cavities and to reach the lung alveoli without being cleared by the ciliated bronchial epithelium (6, 7). Although several in-vitro studies indicated that epithelial cells may internalize and subject conidia to phagolysosomal degradation (8), an engulfment of conidia by bronchial epithelium has not been observed in-vivo so far (9). Hence, the clearance of A. fumigatus conidia requires effective cellular and humoral immune responses.
The innate immune system is considered the key player in the clearance of conidia and the defence against the outgrowth of A. fumigatus conidia. Here, resident leukocytes present in the alveolar lung tissue, such as alveolar macrophages and dendritic cells (DC) initiate an early response against invasive aspergillosis (10, 11). However, the recruitment of polymorphonuclear neutrophils (PMN) to the lung tissue is essential for an efficient clearing of A. fumigatus (5, 6, 12). The importance of PMN for an effective protection against IPA was inferred from the observation, that quantitative [i.e., in neutropenic patients (13)] or qualitative [i.e., patients with chronic granulomatous disease (14)] defects of PMN are critical predisposing factors for IPA (13, 15). PMN mediate the killing of A. fumigatus via different effector mechanisms dependent on the size of conidia and hyphae:
Since the size of hyphae prevents phagocytosis, hyphal killing is mainly conferred by oxidative and non-oxidative PMN effector functions. These include the generation of reactive oxygen species (ROS), the formation of neutrophil extracellular traps (NET) and the release of neutrophil granular content (6, 16, 17). In the context of oxidative PMN functions, it has been observed, that the common beta subunit of β2 integrins (CD18) is critical for the recognition of A. fumigatus and the subsequent generation of ROS (18, 19).
By contrast, the small-size of A. fumigatus conidia allows for the phagocytosis by PMN, which is either mediated by direct recognition via complement receptor 3 (CR3, i.e., CD11b/CD18), Dectin-1 or indirectly via complement-dependant opsonization (6, 18, 19). The importance of β2 integrins in PMN-functions has been confirmed in more recent reports, which revealed that an antibody-mediated blockade of CD11b prevents the generation of ROS (20) and phagocytosis of A. fumigatus conidia by PMN (21).
The ß2 integrin-family consists of four members, which are formed by heterodimerization of the common beta subunit (CD18) with a variable alpha subunit (CD11a-CD11d) (22, 23). The integrin receptor CR-3 is primarily expressed by leukocytes of the myeloid lineage, which was name-giving (macrophage antigen 1, MAC-1) (22). MAC-1 serves as an adhesion receptor for various ligands, including intercellular adhesion molecule 1 (ICAM-1), which is necessary for the transendothelial migration of macrophages and PMN (5, 24). MAC1/CR3 also binds complement-opsonized pathogens, and immune complexes, non-opsonized pathogens, and numerous serum factors (25). In addition, MAC-1 serves as a coreceptor for the Fc-receptor-mediated uptake of antibody-opsonized pathogens (26). It has further been shown that MAC-1 acts as a regulator of LPS-induced signaling in macrophages and DC, and that the engagement of MAC-1 with yet unrecognized T cell receptors mediates T cell activation (27, 28). Last, MAC-1 is a modifier of various signaling pathways (29), such as TLR-induced inflammatory signaling (27), which is involved in the innate immune response to invasive aspergillosis (10).
In accordance with the importance of β2 integrins for immune responses, loss-of-function mutations of the CD18 gene in humans result in the so-called leukocyte adhesion deficiency type 1 (LAD1) syndrome, being characterized by severe, recurrent bacterial and fungal infections in patients, which require extensive treatment with anti-infective agents (30). Several studies have indicated that an impaired migration and phagocytic activity of CD18-deficient PMN might be largely causative for the spreading of pathogens in LAD1 patients (31).
However, the exact contribution of β2 integrins for PMN functions in-vivo has not been fully elucidated yet, since the mouse models available so far either display a constitutive CD18-knockdown (CD18hypo) or knockout (CD18-/-), which complicates to delineate the cell-type specific role of CD18. In order to reveal the PMN-specific role of β2-integrins for the control of infectious diseases such as IPA, we established a transgenic mouse with a floxed CD18 gene (CD18fl/fl Ly6GCre-). By crossing CD18fl/fl mice with transgenic mice expressing Cre recombinase under control of the PMN-specific (Ly6GCre+) promoter, offspring with a PMN-specific knockdown of CD18 have been generated, thus allowing to analyze the PMN-specific role in IPA.
In this study, we show that mice with a Ly6G-specific knockdown of CD18 (CD18Ly6G conditional knockout, in the following termed CD18Ly6G cKO) display an impaired survival during IPA as compared to control-mice (CD18fl/fl). The impaired survival of CD18Ly6G cKO mice is reflected by a higher fungal burden in the lung of these mice during the early phase of pulmonary infection and lower amounts of proinflammatory innate mediators, such as TNF-α in the bronchoalveolar lavage fluid (BALF). By contrast, we detected an enhanced bronchial infiltration of PMN and elevated levels of the PMN-chemoattractant CXCL-1 in BALF derived from infected CD18Ly6G cKO mice, which might reflect a compensatory mechanism. Moreover, we could observe that CD18-deficient PMN showed a strong attenuation of effector functions in-vitro, which might explain the higher fungal burden in the lungs of infected CD18Ly6G cKO mice. In particular, we observed an impaired phagocytic uptake of A. fumigatus conidia, and a diminished generation of ROS and NET in CD18-deficient PMN.
2 Materials and Methods
2.1 Fungal Strains and Cultivation Conditions
The wild type (WT; ATCC 46645) and the GFP-modified (AfS148) A. fumigatus strains (32) were cultured in Aspergillus minimal medium (AMM) with 1% (w/v) glucose, 1% Hutner´s trace element solution and 1M MgSO4 (Carl Roth, Karlsruhe, Germany) as described earlier (16). Briefly, conidia were incubated on AMM agar plates for 4 days at 37°C and 5% CO2. For preparation of spore suspensions, plates were washed with sterile water containing a small amount of glass pearls (Ø 4mm; Carl Roth, Karlsruhe, Germany) to detach conidia from agar plates. The obtained spore suspension was filtered twice through a sterile 40 μm nylon mesh and stored in sterile water at 4°C.
2.2 Mice
In order to allow for the assessment of the importance of β2-integrins specifically for PMN, we generated a transgenic mouse strain with a floxed CD18 gene (CD18fl/fl Ly6GCre- ; B6.Cg-Itgb2tm2.GrabS), which enabled a conditional knockout of β2 integrins in a cell-type specific manner (Supplementary Figure 1). The generation of mice with floxed exon 3 of the CD18 gene locus will be described in detail elsewhere. CD18fl/fl mice were bred with transgenic mice expressing Cre recombinase under control of the PMN-specific Ly6G promoter (33, 34) as described by Hasenberg and coworkers (Ly6GCre+, C57BL/6-Ly6g(tm2621(Cre-tdTomato)Arte mice) (35), yielding a mouse strain with diminished levels of CD18 on neutrophils (CD18Ly6G cKO). Resulting CD18wt/fl Ly6GCre- offspring were crossed back to CD18fl/fl background. Derived male CD18fl/fl Ly6GCre- mice were paired with CD18fl/fl Ly6GCre- females, yielding mice with diminished levels of CD18 on neutrophils (CD18fl/fl Ly6GCre-, in the following termed CD18Ly6G cKO) and CD18fl/fl Ly6GCre- mice at the same ratio.
The mouse strains (CD18fl/fl Ly6GCre- and CD18Ly6G cKO) were maintained in the Translational Animal Research Center of the University Medical Center Mainz under pathogen-free conditions on a standard diet. All animal procedures were performed in accordance with the institutional guidelines and approved by the responsible national authority (National Investigation Office Rhineland-Pfalz, Approval ID: 23177-07/G16-1-020). For the experiments, mice of both sexes were used, although most experiments were done with female mice. Mice used in the experiments were aged between 6-18 weeks unless stated otherwise.
2.3 Mouse Genotyping
Gene-targeted animals were verified by PCR (Supplementary Figure 1). To this end, ear biopsies of mice (2–6 weeks) were incubated with lysis buffer containing 100µl Direct PCR Ear Buffer (Viagen Biotec, Los Angeles, CA, USA) and 2µl proteinase K (ThermoFisher Scientific, Waltham, MA). Samples were incubated at 56°C for 1-3h under shaking. Subsequently the suspension was heated to 95°C for 5 min to inactivate proteinase K, and the lysate was put on ice until further processing. The typical PCR reaction contained a 25-μl volume containing 5µl PCR Reaction Mix (Sigma Aldrich, Merck, Darmstadt, Germany), 17,3µl H2O, 0,2µl of myTaq-Polymerase (Roche, Mannheim, Germany) and 1µl of the primers (10pmol/µl) for PCR 1 (Mix of 2 primers: CD18 ex3_s2, B2(s): 5´-GTGACACTTTAC TTGCGACCA-3´; CD18 loxp_as1,B3(as): 5´-TGCCAATAAAGAATTTCAGAGCC-3´, suspended 1:10 in H2O) or for PCR 2 (Mix of 3 primers: Ly6G [78]-s for 5´-CCTGCAACCTGGTCAGAGAG-3´, and 5064_61_rev for 5´-GAGGTCCAAGAGACTTTCTGG-3´, and 2240_31 for 5′-ACGTCCAGACACAGCATAGG-3′ suspended 1:10 in H2O). In PCR 2 we also included a control pair of primers for amplifying Actin as a wild-type allele (Actin FW: 5´-TGTTACCAACTGGGACGACA-3´ and Actin REV: 5´-GACATGCAAGGAGTGCAAGA. The following PCR conditions were applied for PCR 1:initial denaturation (3 min, 95°C), followed by 35 cycles (denaturation: 30 s, 95°C; annealing: 30 s, 58°C; elongation: 45 s, 72°C) and by a final elongation step (2 min 72°C). For PCR 2 the following PCR conditions were applied: initial denaturation (5 min, 95°C), followed by 35 cycles (denaturation: 30 s, 95°C; annealing: 30 s, 60°C; elongation: 1 min, 72°C) and by a final elongation step (10 min 72°C). PCR products were analyzed by agarose gel electrophoresis (Supplementary Figure 1).
2.4 Mouse Model of Invasive Aspergillosis
Mice were anesthetized with 14.5% Ketamin (50mg/ml)/5.7% Xylazin (0.2%) and were subsequently challenged with 107 A. fumigatus conidia (strain ATCC 46645) applied intratracheally as described (35, 36). In brief, a 22G indwelling venous catheter (Vasofix, B. Braun AG, Melsungen, Germany) was inserted into the trachea and 100 μl sterile fungal suspension was administered through the catheter. To enhance dispersion in the lungs, mice were ventilated mechanically with 250 breaths/min, 300 μl/breath for 2 min using an animal respirator (MiniVent, Hugo Sachs, March-Hugstetten, Germany) as previously described (16). In order to characterize the early immune response to fungal infection, 10 mice/group were sacrificed 24h after infection. In two additional groups (n=5-8 mice/group) the course of systemic infection was daily examined by evaluation of weight, activity, breathing, overall appearance (as assessed by posture, skin, and fur appearance), and survival was monitored for 14 days. Mice with severe symptoms as determined by clinical scoring were immediately euthanized as required by the institutional animal ethics guidelines. Where indicated, PMN depletion was induced by i.p. injection of anti-Gr-1 antibody (150 μg, clone RB6-8C5; BioXCell, Lebanon, NH) 1 day prior to inoculation with fungal suspension.
2.5 Flow Cytometric Analysis
Blood samples, spleens and bone marrow were prepared from sacrificed mice, and lungs were flushed with 1 ml PBS. Spleen cell suspensions were generated via mechanical homogenization on a 40µm nylon mesh, washed twice with cold PBS, and red blood cells (RBC) were lysed with hypotonic Gey´s solution (155mM NH4Cl, 10mM KHCO3, 10µM EDTA at pH 7,4). RBC from blood samples were lysed in the same way. Cells derived from blood, spleen, bone marrow and bronchoalveolar lavage fluid (BALF) were analyzed by flow cytometry. To this end, cells were washed with staining buffer (PBS/2% FCS), and Fc receptors were blocked by incubation with rat anti-mouse CD16/CD32 antibody (clone 2.4G2) for 15 min at 4°C. Then, cells were incubated with FITC-conjugated anti-CD86 (GL-1), anti-CD45 (30F11), and anti-Annexin-V (Biolegend), PerCP-conjugated anti-Ly6C (HK1.4), APC-conjugated anti-CD18 (C71/16), anti-CD14 (Sa14-2), anti-Gr-1 (RB6-8C5) and anti-CD40 (1C10), APC-eFluor 780 conjugated anti-CD11c (N418), eFluor450-conjugated anti-MHCII (M5/114 15.2) and anti-F4/80 (BM8), eFluor506-conjugated anti-CD3 (500A2), Super Bright 600-conjugated anti-CD11b (M1/70), PE-conjugated anti-CD11a (M17/4), anti-CD80 (1610A1) and anti-Ly6G (1A8), PE-eFluor610-conjugated anti-Ly6G (1A8), PE-Cyanine7-conjugated anti-CD68 (FA11) and anti-CD62L (MEL-14). All antibodies were obtained from Biolegend (San Diego, CA) or Thermo Fisher (Waltham, MA). Viability was assessed using Fixable-viability-dye (FVD), conjugated either with APC eFluor 780, eFluor 450 or eFluor 506 (ThermoFisher). Samples were analyzed using a flow cytometer (Attune™ NxT Acoustic Focusing Cytometer, Thermo Fisher), and data were processed using FlowJo software V8.8.7 (Tree Star Inc., Ashland, OR, USA). The gating strategy is shown in Supplementary Figure 2.
2.6 Quantification of Fungal Burden
The right lungs of euthanized mice were removed, mechanically homogenized and serial dilutions were plated on Sabouraud-4% Glucose agar (Carl Roth, Karlsruhe, Germany), and cultivated at 37°C and 5% CO2. Colony-forming units (CFU) were counted after 24h and 48h.
Moreover, a D-Galactomannan assay based on the Platelia Aspergillus EIA (Bio-Rad Laboratories, Marne-La-Coquette, France) was employed to quantify the fungal load in BALF and serum derived from IPA-infected mice. This enzyme immunoassay is used in clinical routine and validated for the detection of A. fumigatus antigen. The test uses the rat monoclonal antibody EBA-2 directed against Aspergillus galactomannan. In brief, the antigen is first bound to the wells of the microplate coated with the EBA-2 antibody and then revealed by binding to the peroxidase-linked EBA-2 antibody resulting in a colorimetric reaction, which is measured via optical density on a Plate Reader as described previously (37).
2.7 Histopathologic Analysis
For histopathological analysis the left lungs of euthanized mice were filled with 10% formalin via the trachea. Paraffin-embedded blocks were prepared, and derived sections (5 μm) were stained with H&E to assess inflammatory responses. For this, H&E-stained sections were examined by microscopy in a blinded fashion for peribronchial, perivascular and tissue inflammation, using a scoring system (0–3). Furthermore, sections of lungs were stained with Grocott Gomori’s methenamine silver to assess the fungal burden of the lungs. Grocott stained sections were examined in a blinded fashion similar to H&E sections using a scoring system (0–3). In general, 3 randomly selected areas on each slide were analyzed with a BX40 microscope equipped with a CCD camera (Olympus, Hamburg, Germany).
2.8 Cytospin Analysis
For detection of lung infiltrating PMN, 100 μl of BALF containing 0.5-2x105 cells (see above) were cytospun onto microscope slides (3,500 rpm for 5 min; Cytospin 3, Thermo Fisher), treated with the Diff Quick Staining Set (Microptic, Barcelona, Spain), air-dried, and fixed as recommended. Samples were analyzed using a BX50WI microscope, equipped with a CCD camera (Olympus, Hamburg, Germany). PMN were identified based on their characteristic segmented nuclei.
2.9 Cytokine Detection
Serum and BALF were subjected to cytokine detection by Cytometric bead array (CBA) using the mouse CBA flex sets following the manufacturer’s instructions (BD Bioscience, San Jose, CA). Similarly, in-vitro cytokine generation by Ly6G+ PMN (105/100µl) immunomagnetically sorted from bone marrow of CD18fl/fl and CD18Ly6G cKO mice (see below) was quantified. Isolated PMN were incubated in Iscove’s medium (Thermo Fisher Scientific) supplemented with 5% (v/v) FCS, 2 mM l-glutamine, 50 μM ß-mercaptoethanol and 1 mM Na-pyruvate (SERVA Electrophoresis, Heidelberg, Germany) in 96-well plates (Greiner Bio One, Frickenhausen, Germany) and treated over-night with PBS, recombinant murine GM-CSF (100ng/ml; Miltenyi Biotec, Bergsich-Gladbach, Germany), LPS (1µg/ml, Merck-Millipore, Darmstadt, Germany), CpG (1µg/ml, In vivogen, Toulouse, France) or R8/48 (1µg/ml, In vivogen). Supernatants were taken 3h and 24h later from PMN aliquots generated in n=3 independent experiments.
2.10 Fungal Uptake by PMN
PMN were purified from bone marrow of CD18fl/fl and CD18Ly6G cKO mice by magnetic cell sorting (MACS) using biotin-labeled Ly6G-specific antibodies and streptavidin-conjugated beads (both from Miltenyi Biotec) according to the manufacturer’s protocol. The cell purity (Ly6G+) exceeded 90% as assessed by flow cytometry. Freshly isolated PMN were resuspended (106 cells/ml) in cell culture medium (see above), seeded into 96-well plates (100µl/well) and were incubated with GFP-fluorescent A. fumigatus conidia (5) at the indicated ratios in parallel at 4°C and 37°C to differentiate mere adhesion and energy-dependent uptake. After 1h of incubation PMN were washed twice with 500µl cold PBS and stained with anti-CD11b, anti-Ly6G, anti-MHCII and anti-CD62L specific antibodies, and FVD eFluor 506 to determine the uptake and activation status of GFP-labeled conidia by flow cytometry (Supplementary Figure 8 shows the gating strategy applied during the experiments).
2.11 Uptake of Inert Particles by PMN
To assess uptake of inert particles, we employed Cy5-labeled nanoparticles (Ø 50nm) and PE-labeled microBeads (Ø 2µm) (both Miltenyi Biotec). Immunomagnetically sorted PMN (106 cells/ml) were incubated in cell culture medium in 96-well plates (100µl) and treated over-night (12h) with GM-CSF (100ng/ml) or LPS (1µg/ml). Subsequently, PMN were washed once with 500µl cold PBS and were either left untreated, or incubated in parallel settings with particles, and particles pre-treated with native or heat-inactivated mouse serum (hiS; 56°C, 30min) at 4°C and 37°C for various periods of time (15-60 min). Pretreatment of particles with native versus heat-inactivated mouse serum served to elucidate the complement-dependent particle uptake. Subsequently PMN were washed twice with 500µl cold PBS and incubated with anti-CD11b, anti-Ly6G, anti-MHCII, anti-CD86, anti-Ly6C and anti-CD62L antibodies and FVD eFluor 506 to determine the PMN-specific uptake of inert particles by flow cytometry.
2.12 Assessment of Neutrophil Apoptosis
Freshly isolated PMN (1x106/ml) derived from bone marrow of either mouse strain were incubated in cell culture medium in 24-well plates and treated over-night (12h) in parallel w/o and with GM-CSF (100ng/ml), LPS (1µg/ml) and with GM-CSF plus LPS in order to differentiate spontaneous apoptosis (PBS-treated control), late-onset apoptosis (GM-CSF) and apoptosis upon LPS-treatment. Following over-night incubation, samples were washed twice with 1ml PBS and incubated with anti-Annexin V (FITC) and FVD (eFluor 506) according to the manufacturer´s protocol (ThermoFisher) to differentiate apoptosis and necrosis. Frequencies of apoptotic and necrotic PMN were determined by flow cytometry as described previously (38).
2.13 Analysis of ROS Production
To assess the rate of ROS production, PMN were isolated from bone marrow, were seeded into 96-well-plates (106/ml; 100µl/well) washed once with 200µl PBS and resuspended in 100 μl ROS-detection solution (2 μM 2´-7´Dichlorodihydrofluorescein [DCFDA] in PBS; Alexis Biochemicals, Lausen, CHE). After 20 min of incubation at 37°C the cells were washed with 200µl PBS, centrifuged, and the sedimented cells were dispersed in 200 μl PBS. Subsequently, PMN were stimulated with GM-CSF (100ng/ml), LPS (1µg/ml), A. fumigatus conidia (1:1), or 100 nM PMA (Sigma-Aldrich), respectively at 37°C, 5% CO2 in triplicates. Median fluorescence intensities (MFI) were measured using a SPARK multimode microplate-reader (TECAN Trading AG, CHE) at an excitation of 485nm and an emission of 530nm for 90min (intervals of 15min). After 90min cells were analyzed by flow cytometry for DCFDA-positive events.
2.14 Analysis of Neutrophil-Extracellular Traps Formation
To induce the release of neutrophil extracellular traps (NET) DNA, we isolated PMN from bone marrow as described previously and seeded PMN (105/100 µl) in 96-well plates with 100µl RPMI 1640 medium without phenol red (ThermoFisher, Waltham, CA). PMN were treated either with GM-CSF (100ng/ml), LPS (1µg/ml), A. fumigatus conidia (1:1), PMA (100 nM), or calcium ionophore (2,5µM; Sigma, Darmstadt, Germany), respectively. After incubation at 37°C for 3h, 5µM of Sytox orange nucleic stain (Invitrogen, Carlsbad, CA) was added and samples were incubated for 10min at room temperature in the dark. Subsequently, PMN were centrifugated and washed twice with 300µl cold PBS. MFI of Sytox orange was measured using a SPARK multimode microplate reader with an excitation of 547nm and an emission of 580nm. Then, cells were incubated with an anti-Ly6G antibody and analyzed by flow cytometry for Ly6G/Sytox orange double-positive cells.
2.15 RNA-Sequencing and Bioinformatical Analysis
First, PMN were isolated from bone marrow of CD18fl/fl and CD18Ly6G cKO mice (n=3). Each 106 PMN were either lysed directly after isolation or cultured overnight with GM-CSF (10ng/ml) plus LPS (1µg/ml). RNA was purified with the RNeasy Plus Micro Kit according to the manufacturer’s protocol (Qiagen). RNA was quantified with a Qubit 2.0 fluorometer (Invitrogen) and the quality was assessed on a Bioanalyzer 2100 (Agilent) using a RNA 6000 Pico chip (Agilent). Samples with an RNA integrity number (RIN) of > 8 were used for library preparation. Barcoded mRNA-seq cDNA libraries were prepared from 10ng of total RNA using NEBNext® Poly(A) mRNA Magnetic Isolation Module and NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® according to the manual with a final amplification of 15 PCR cycles. Quantity was assessed using Invitrogen’s Qubit HS assay kit and library size was determined using Agilent’s 2100 Bioanalyzer HS DNA assay. Barcoded RNA-Seq libraries were onboard clustered using HiSeq® Rapid SR Cluster Kit v2 using 8pM and 59bps were sequenced on the Illumina HiSeq2500 using HiSeq® Rapid SBS Kit v2 (59 Cycle). The raw output data of the HiSeq was preprocessed according to the Illumina standard protocol. Sequence reads were trimmed for adapter sequences and further processed using Qiagen’s software CLC Genomics Workbench (v20.0 with CLC’s default settings for RNA-Seq analysis). Reads were aligned to GRCm38 genome. Sequencing data were first analyzed with CLC Genomics Work Bench (Qiagen). Further processing was performed in R using the DESeq2 package for calling differential gene expression (39, 40). To determine the most up- or downregulated genes, genes were sorted on the basis of log2 [fold change] maximum-likelihood estimation, and the P-value cut-off was set to 0.05. Results were illustrated using the pheatmap package. Functional interaction networks were visualized using the STRING package in the open-source platform Cytoscape.
2.16 Statistical Analysis
Statistical analysis was conducted with GraphPad Prism (version 5.0a; GraphPad Software, San Diego, CA, USA). Comparison of two different parameters was performed using paired Student’s t-test. In case of comparison of more than two groups we employed one-way ANOVA and posthoc Tukey test. For survival analysis, Kaplan-Meier plots and hazard ratios have been calculated. For all analyses, p < 0.05 was considered as statistically significant. Abbreviations: *p<0.05, **p<0.005, ***p<0,001.
3 Results
3.1 Phenotype and Impairment of PMN Effector Functions of CD18Ly6G cKO Mice Assessed by In-Vitro Experiments
In murine leukocytes Ly6G is selectively expressed by PMN (41). To obtain mice with a diminished CD18 expression specifically on PMN (CD18Ly6G cKO), we crossed a mice with a floxed CD18 gene (CD18fl/fll) that was generated in our lab (will be described in detail elsewhere) with transgenic mice expressing the Cre recombinase under control of the Ly6G promoter (CD18wt/wt Ly6GCre+). Resulting offspring (CD18wt/fl Ly6GCre+ and CD18wt/fl Ly6GCre-) were fertile and showed no obvious phenotype. These mice were crossed back to CD18fl/fl background yielding CD18Ly6G cKO and CD18fl/fl Cre- mice at expected Mendelian ratios (not shown). All gene-targeted animals were verified by PCR (Supplementary Figure 1).
We could observe a downregulation of CD18 and accordingly of the β2 integrin alpha subunits (CD11a and CD11b) on PMN of CD18Ly6G cKO mice. The extent of downregulation varied between 30-50% compared to CD18fl/fl mice depending on the investigated compartment (blood, spleen or bone marrow) (Figure 1A), which is in accordance with the extent of Ly6G Cre-mediated downregulation of targeted genes previously shown by Gunzer and coworkers (35). Notably, CD18-reduction was restricted to Ly6G+ PMN, and was not observed for CD3+ lymphocytes, F4/80+ macrophages and Ly6C+ monocytic cells, thus confirming the cell-type specific targeting of CD18 (Supplementary Figures 2, 3). Absolute PMN counts and relative amounts of PMN in both spleen and blood were found to be slightly higher, whereas PMN counts in the bone marrow did not show significant differences (Figure 1B). The percentages of monocytic and lymphocytic cells did not differ significantly between CD18Ly6G cKO mice and CD18fl/fl mice in spleen (Figure 1C, left panel) and blood (Figure 1C, right panel).
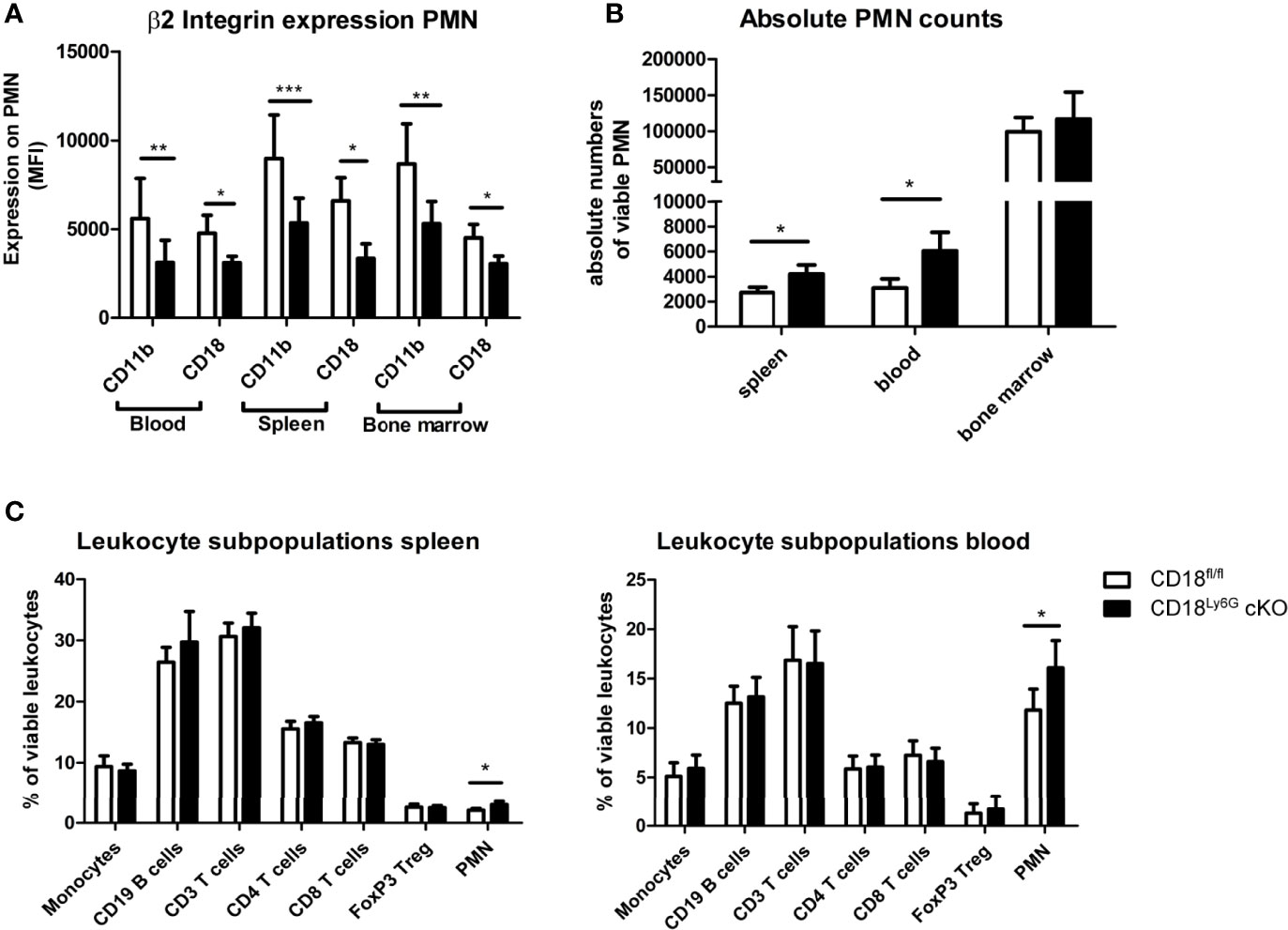
Figure 1 Phenotypical and functional characteristics of CD18Ly6G cKO mice compared to CD18fl/fl mice. We found a significant reduction of β2-integrin surface marker expression (CD11b, CD18) on PMN derived from blood, spleen, and bone marrow (A). Data depict the results of in-vitro experiments from n=7-17 mice/genotype. In the same set of experiments we further observed higher absolute and relative counts of PMN in CD18Ly6G cKO mice as compared to CD18fl/fl mice (B,C), whereas the proportions of other leukocyte subpopulations did not differ significantly (C) (n=10/genotype). Legend in (C) applies to all panels. Statistically significant differences between groups are indicated (*p<0.05, **p<0.005, ***p<0.001).
As β2 integrins have also been implicated in the differentiation and in survival signaling of myeloid cells (42), we next investigated whether the PMN-restricted CD18-knockdown affected PMN apoptosis in-vitro. Here, we did not find significant differences in the apoptosis of PMN after treatment with GM-CSF or LPS, as assessed by Annexin-V/FVD negative and Annexin-V positive/FVD negative PMN derived from spleens and bone marrow (not shown).
3.2 PMN-Specific Knockdown of β2-Integrins Results in an Aggravated Course of IPA
To assess the relevance of β2 integrins for PMN-specific clearance of pulmonary infection with A. fumigatus, we examined the course of disease in CD18Ly6G cKO and CD18fl/fl mice. In some mice an anti-Gr-1 antibody was applied prior to infection with A. fumigatus (d0) to deplete PMN as an internal control for the success of infection. As expected, all PMN-depleted mice died during the first days of infection (Figure 2), underlining the pivotal role of PMN to limit the spread of A. fumigatus. By contrast, all non-depleted CD18fl/fl mice survived infection monitored over 2 weeks, whereas 25% of CD18Ly6G cKO mice died within the first week of infection. This finding is consistent with the observation that clinical signs of IPA infection were more aggravated in case of CD18Ly6G cKO mice in the first days after inoculation. Furthermore, recovery of from clinical symptoms was delayed in CD18Ly6G cKO mice as compared to CD18fl/fl mice (Figure 2).
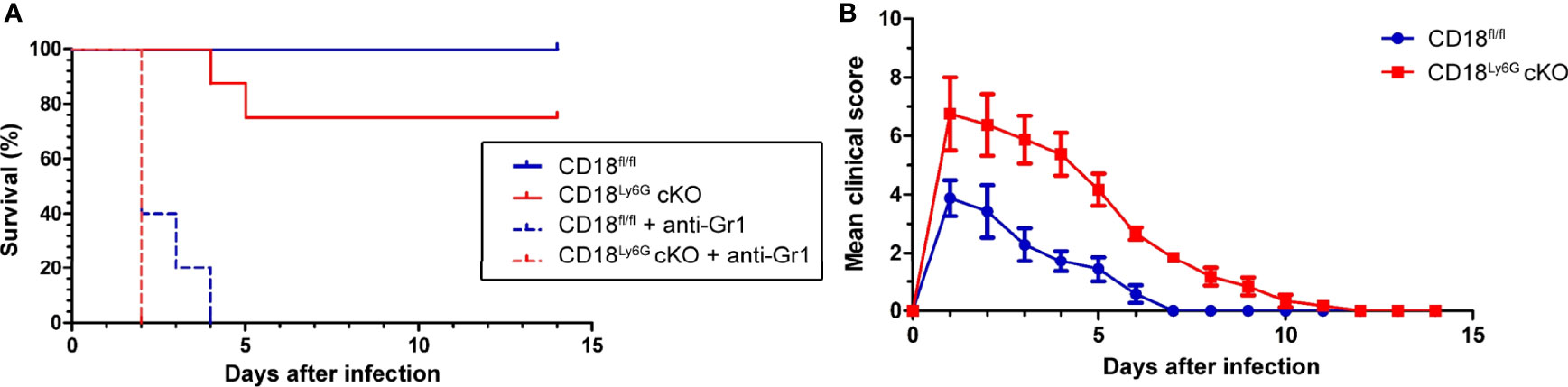
Figure 2 Infection with IPA caused an impaired survival (A) and an aggravated course of the disease (B) in CD18Ly6G cKO as compared to CD18fl/fl mice. CD18fl/fl and CD18Ly6G cKO mice were infected i.t. with A fumigatus (each 107 conidia/mouse) in 2 independent experiments. (A) Survival was monitored daily for 2 weeks and is presented in a Kaplan-Meier survival curve. In parallel settings PMN were depleted in some mice via injection of an anti-Gr-1 antibody one day before infection. Data show the cumulative results of two independent experiments with a total of 12 (CD18fl/fl) and 13 (CD18Ly6G cKO) mice/group. 5 mice/group received an anti-Gr-1 antibody in order to deplete PMN in these mice. All Gr-1 depleted mice died within the first days after IPA infection, whereas all non-depleted CD18fl/fl mice survived. By contrast, some non-depleted CD18Ly6G cKO mice (n=2) deceased within the first week after IPA infection. (B) The clinical course of IPA of monitoring was assessed in CD18fl/fl (n=7) and CD18Ly6G cKO mice (n=8) for 14 days. Parameters comprised breathing, reaction to pain overall appearance, hypothermia, strong weight loss, motoric disabilities and apathy (each 0-2).
3.3 CD18Ly6G cKO Mice Show a Higher Fungal Burden
Next, we focused on the course of the early innate immune response towards A. fumigatus infection, which is known to be driven by PMN (12). For this, lungs, BALF, and serum of infected mice were analyzed 24h after infection in more detail. Lung homogenates of CD18Ly6G cKO mice showed an enhanced amount of fungal conidia as compared to lungs from CD18fl/fl mice (Figure 3). Histopathological analysis confirmed a higher fungal burden and aggravated lung damage in in lungs of CD18Ly6G cKO mice as assessed by Grocott-silver and Hematoxylin & Eosin (H&E) staining. Notably, sprouting of hyphae has only been observed in CD18Ly6G cKO mice. Despite the strong differences in terms of fungal burden, H&E staining of lung tissues showed comparable levels of cellular inflammation, largely irrespective of the genotype (Figures 3A). D-Galactomannan-assays revealed that BALF derived from both mice strains contained A. fumigatus antigen above detection levels (>5.0), whereas serum analysis showed a higher fungal load in CD18Ly6G cKO mice (mean= 5.8 ± 0.14 vs. 4.7 ± 0.33, p=0.01).
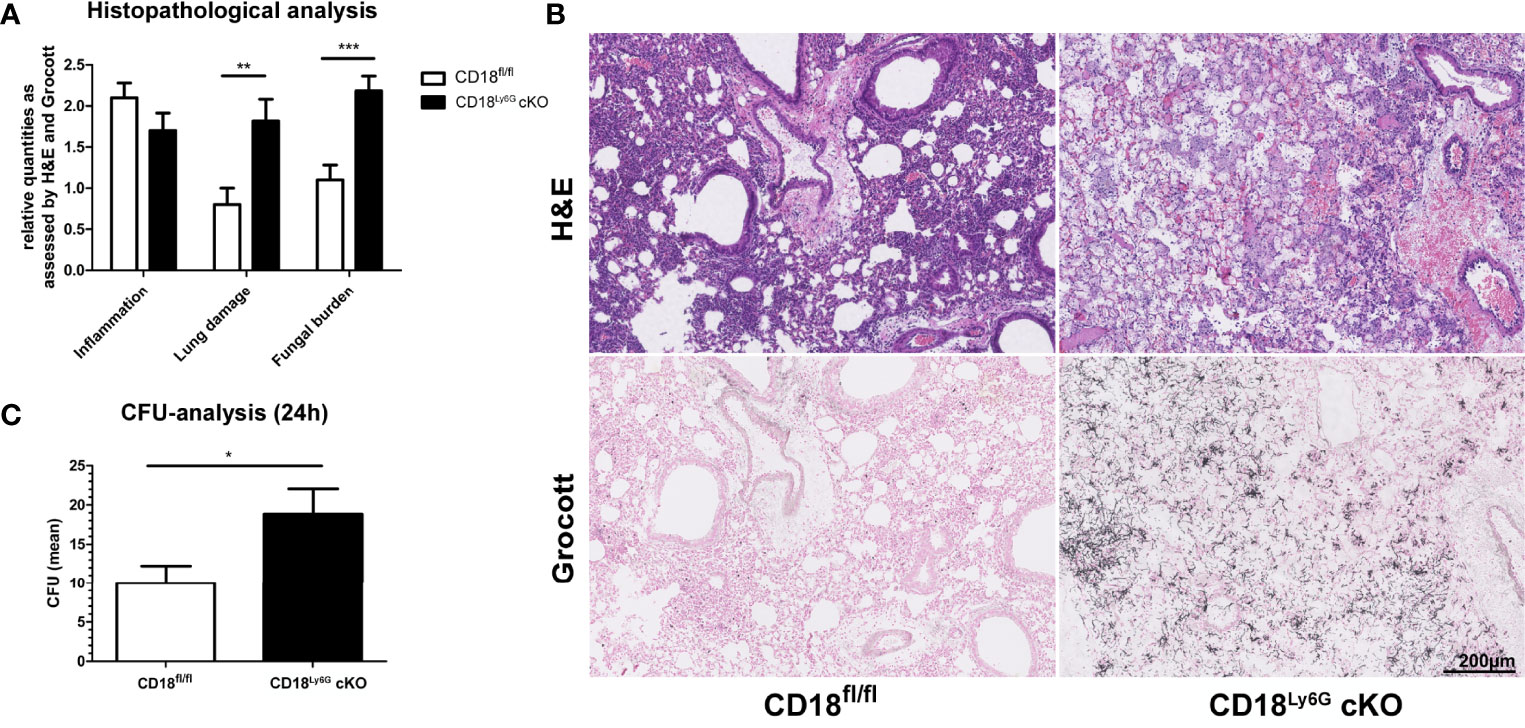
Figure 3 CD18Ly6G cKO mice show a higher pulmonary fungal burden. Histopathological analysis of H&E and Grocott-stained lungs derived from IPA-infected mice 24h upon A.fumigatus inoculation revealed a higher fungal burden and a stronger lung damage (i.e., hyaline membranes, fibrin-exudate within the alveoli) in CD18Ly6G cKO mice (A). Cellular inflammation did not show significant genotype-dependent differences. Representative examples of histological analysis are shown in (B) (Magnification 10x). Data in (A) denote results of histopathological analysis of n=9-10 mice/genotype. We further observed higher CFU counts in serial dilutions of lung homogenates (1:500) after incubation for 24h on Sabouraud-4% Glucose agar plates (C). Data show the mean ± SEM of 6 mice/group. Statistically significant differences between groups are indicated (*p<0.05, **p<0.005, ***p<0.001).
3.4 CD18Ly6G cKO Mice Reveal a Decreased Pulmonary Inflammation
In contrast to the increased fungal burden found in lung tissues of CD18Ly6G cKO mice, these mice displayed no significant differences in cellular inflammation as assessed by H&E staining (Figure 3). However, as depicted in Figure 4, BALF derived from infected CD18Ly6G cKO mice contained lower levels of pro-inflammatory cytokines (TNF-α), and chemokines (CCL2) compared to CD18fl/fl mice, albeit the reduction was below statistical significance in some cases (IL-1α, IL-1β and CCL5). Levels of IL-5, IL-6, IL-10, and GM-CSF were largely comparable. In contrast, BALF obtained from CD18Ly6G cKO mice contained higher levels of the chemokine CXCL-1 known as a relevant chemoattractant for PMN (43).
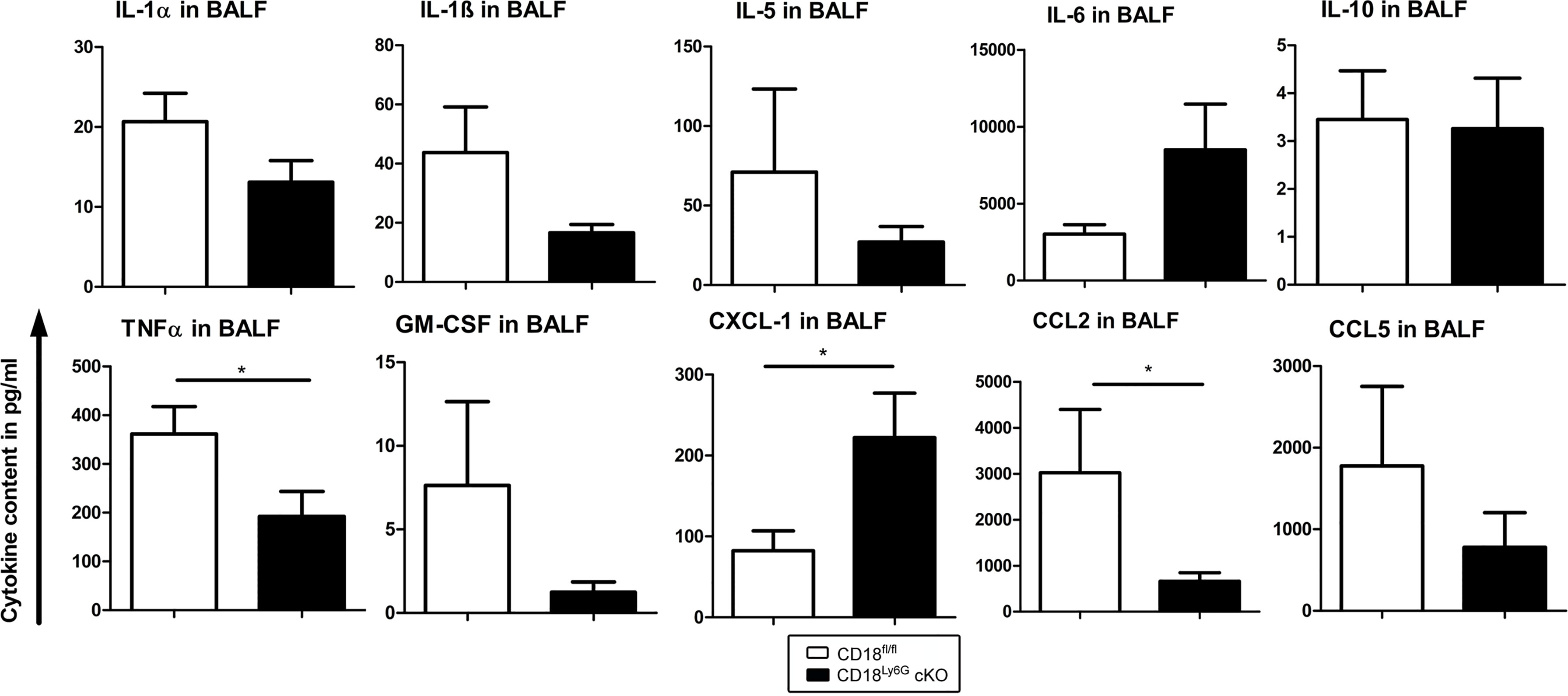
Figure 4 The BAL fluid of A. fumigatus infected CD18Ly6G cKO mice contains lower levels of proinflammatory cytokines. CD18fl/fl and CD18Ly6G cKO mice were infected i.t. with A. fumigatus. On the next day, mice were euthanized, and cytokines in BAL fluid were analyzed. Data denote the mean ± SEM of 6-10 samples analyzed per group. Statistically significant differences between groups are indicated (*p< 0.05).
In contrast, cytokine and chemokine levels in serum were largely comparable between A. fumigatus infected CD18fl/fl and CD18Ly6G cKO mice (Supplementary Figure 5).
3.5 Pulmonary PMN Infiltrates Are Increased in CD18Ly6G cKO Mice Upon IPA
In accordance with elevated CXCL-1 levels, we observed higher numbers of PMN in the BALF of infected CD18Ly6G cKO as compared to CD18fl/fl mice (Figure 5A). In contrast, PMN counts in spleen and blood remained comparable. Higher PMN numbers were also found in cytospin analysis (Figure 5B). Here, we could additionally observe lower counts of mononuclear cells in CD18Ly6G cKO mice. Consistent with this observation, results of FACS-analysis revealed lower macrophage counts in the BALF of CD18Ly6G cKO mice (Figure 5B).
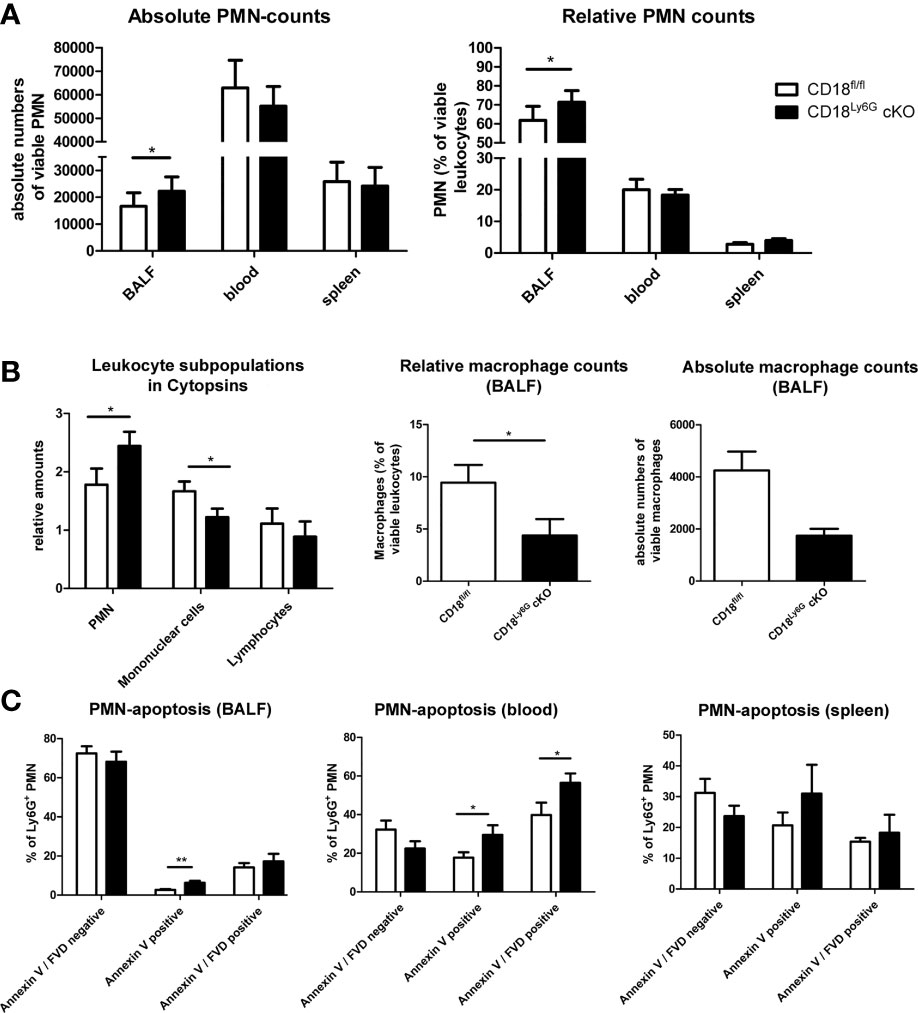
Figure 5 CD18Ly6G cKO mice infected with A. fumigatus are characterized by elevated lung infiltration of PMN, but not in spleens and blood (n=10/genotype) (A). Elevated PMN counts have also been found in Cytospins (bars depict the mean ± SEM of n=8 cytospins/genotype; cell infiltration has been assessed using a scoring system; 0=missing - 3=strongest infiltration) (B). Here, we could additionally observe higher numbers of mononuclear cells (B). This is consistent with the finding of higher macrophage counts in the BALF of CD18fl/fl mice as compared to CD18Ly6G cKO mice observed in FACS-analysis (B). Assessment of PMN apoptosis revealed a stronger expression of apoptosis marker Annexin V in PMN derived from BALF and blood of CD18Ly6G cKO mice (C). Bars depict the mean ± SEM of the relative cell counts found in n=8 cytospins/genotype. Legend in A applies to all panels. Statistically significant differences between groups are indicated (*p<0.05)
Notably, a higher frequency of PMN in BALF (Figure 5C, left panel) and blood (Figure 5C, center panel) obtained from IPA-infected CD18Ly6G cKO mice expressed the early apoptosis marker Annexin-V as compared to CD18fl/fl mice, indicating that CD18-deficient PMN might be more susceptible to apoptosis in response to A. fumigatus. In accordance, we observed a higher frequency of Annexin-V positive PMN in spleens of CD18Ly6G cKO mice, albeit the differences here were found to be below statistical significance (Figure 5C, right panel).
Besides, our data show that a smaller fraction of PMN derived from BALF of CD18Ly6G cKO mice expressed MHCII (1,6% vs. 4,6% of MHCIIhigh PMN), and CD80 (16.4% vs. 20.1% of CD80+ PMN) and showed a lower degree of degranulation as assessed by a low expression of CD62L (88.0% vs. 90.8% CD62Llow PMN) than observed for CD18fl/fl mice. BALF-derived PMN of both mice strains expressed the mouse DC marker CD11c at a moderate extent (Supplementary Figure 6). Infection-induced de novo expression of CD11c by PMN has been reported previously in different mouse infectious disease models (5).
Numbers of PMN, lymphocytes, and monocytes in the peripheral blood of A. fumigatus infected mice did not differ in a genotype-dependent manner (Supplementary Figure 7). In accordance with our in-vitro experiments, we could confirm that the knockdown of CD18 was restricted to Ly6G positive cells (Supplementary Figure 3). Similarly, we could observe a knockdown of the corresponding alpha subunits CD11a and CD11b on Ly6G positive PMN of IPA-infected mice (Supplementary Figure 4), which is consistent with the physiological role of CD18 as the rate-limiting subunit of β2-integrin surface expression.
3.6 Knockdown of CD18 Affects PMN Innate Effector Functions
3.6.1 Phagocytosis
Although PMN were able to infiltrate A. fumigatus infected lungs in CD18Ly6G cKO mice, we observed an impaired ability to limit fungal spreading. Hence, we analyzed whether the knockdown of CD18, and thereby β2 integrins, affected the commonly known pathogen-induced immune responses of PMN.
As phagocytosis is a major effector mechanism of PMN to clear A. fumigatus conidia, we analyzed purified bone marrow-derived Ly6G+ PMN to assess potential genotype-dependent differences in this regard. Here, we first investigated the uptake of inert nanoparticles (NP, Ø 50nm) and microBeads (Ø 2µm). In order to dichotomize mere adhesion and energy-dependent uptake we investigated the uptake in parallel settings at 4°C and 37°C. Since MAC-1 has been attributed to serve as a receptor to facilitate complement-opsonized phagocytosis of pathogens we further examined whether the addition of murine serum might enhance the uptake of particles. Heat-inactivated serum which lacks complement activity served as an internal negative control. We could observe for both kinds of particles that their uptake was strongly impaired in case of PMN with a β2 integrin knockdown. This effect was predominantly observed for serum-opsonized particles, indicating that the recognition of complement- opsonized particles might have been diminished in case of CD18 downregulation on PMN (Figures 6A). In accordance with the well-known role of MAC-1 (CD11b/CD18) for the binding and uptake of complement-opsonized material, we further observed a significant correlation between CD11b surface marker expression on PMN and the engagement of the aforementioned particles (Pearson´s r: 0.65; p= 0.0007).
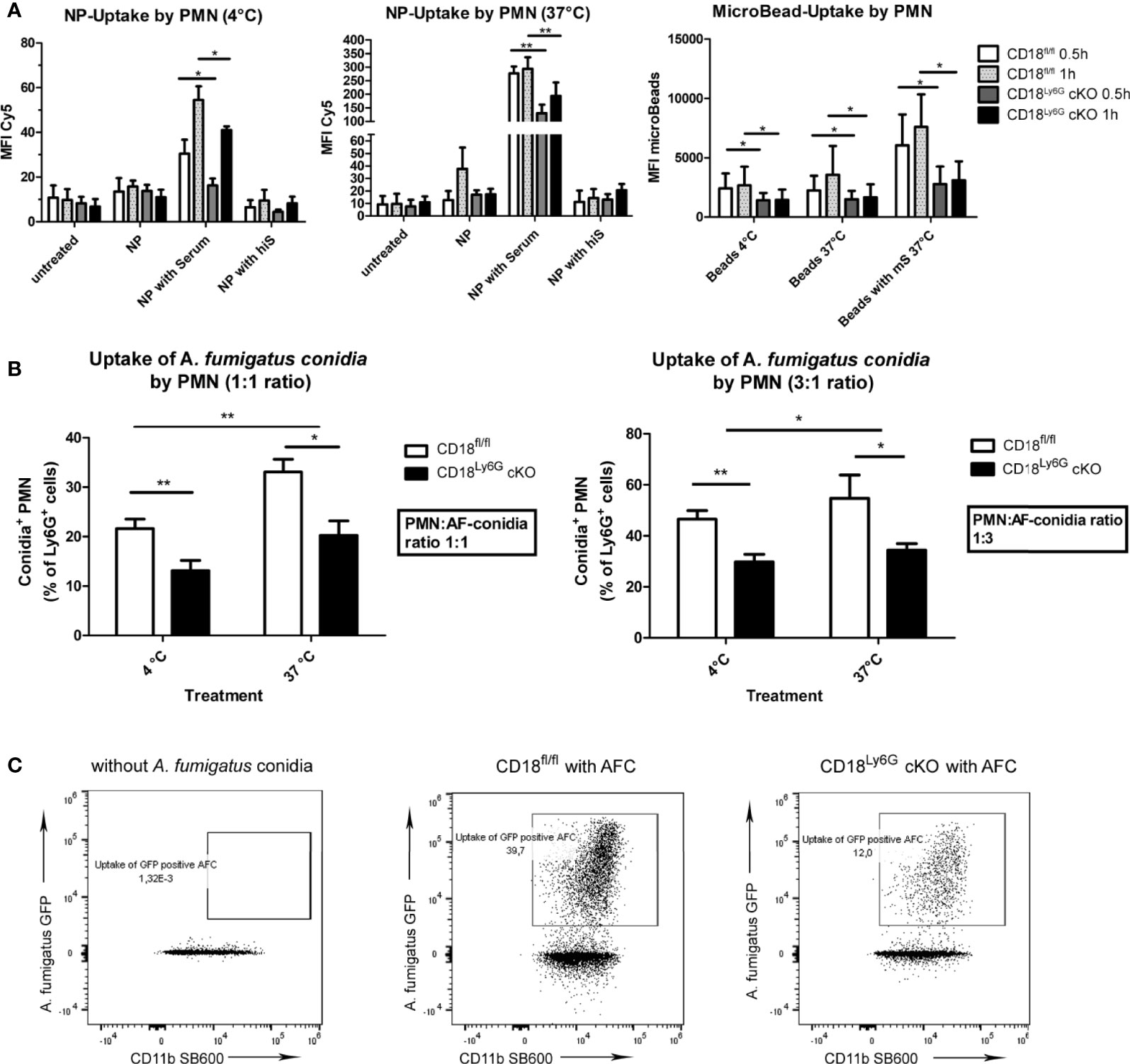
Figure 6 Phagocytosis of inert particles and fungal conidia is less effective in PMN of CD18Ly6G cKO mice. Freshly isolated PMN were co-incubated either with nanoparticles (NP), microBeads (A) or with GFP-fluorescent A. fumigatus conidia (AFC) at 37°C with indicated ratios (B). Simultaneous co-incubation at 4°C served to differentiate mere adhesion from temperature-dependent binding. After 30 min and 60 min the frequency of either Cy5 positive NP, PE-positive microBeads (A), or GFP-positive PMN (B) was determined by flow cytometry. Data represent the mean ± SEM of 3 samples analyzed/group. Exemplary flow cytometry data depicting the diminished uptake of GFP-fluorescent conidia by PMN from CD18Ly6G cKO mice are shown in (C). Statistically significant differences between groups are indicated (*p<0.05, **p<0.005).
Subsequently, we analyzed the phagocytic capacity of PMN after incubation with A. fumigatus conidia. Similar to previous experiments with inert particles, we observed a significantly lower phagocytic uptake of A. fumigatus conidia by PMN derived from CD18Ly6G cKO mice (Figure 6B, C).
3.6.2 NETosis
We also investigated the rate of NET-formation of freshly isolated PMN after differential stimulation. Here, we could observe that the formation of NET by PMN derived from CD18Ly6G cKO mice was significantly impaired after treatment with PMA or A. fumigatus conidia, as assessed by Sytox orange staining. After treatment with GM-CSF or LPS the differences in the formation of NET by PMN derived from CD18Ly6G cKO vs CD18fl/fl mice were below statistical significance (Figure 7A).
3.6.3 ROS-Production
Next, we analyzed the generation of ROS as another important effector mechanism in the innate pathogen defense of PMN. To this end, we incubated freshly isolated PMN with GM-CSF, LPS, PMA or A. fumigatus conidia and assessed the generation of ROS via DCFDA staining in time intervals of 15min for a total period of 90min. Our results revealed that PMN isolated from CD18Ly6G cKO mice generated significantly lower amounts of ROS after incubation with A. fumigatus conidia, suggesting that a CD18 knockdown might impair the ability of PMN to exercise this important effector mechanisms in pathogen-defense (Figure 7B). Referring particularly to the time kinetics of ROS-generation we could further observe that the ability to generate ROS was mainly impaired in the course of the first 60min, which indicates that β2 integrins might be implicated in the early generation of ROS (not shown).
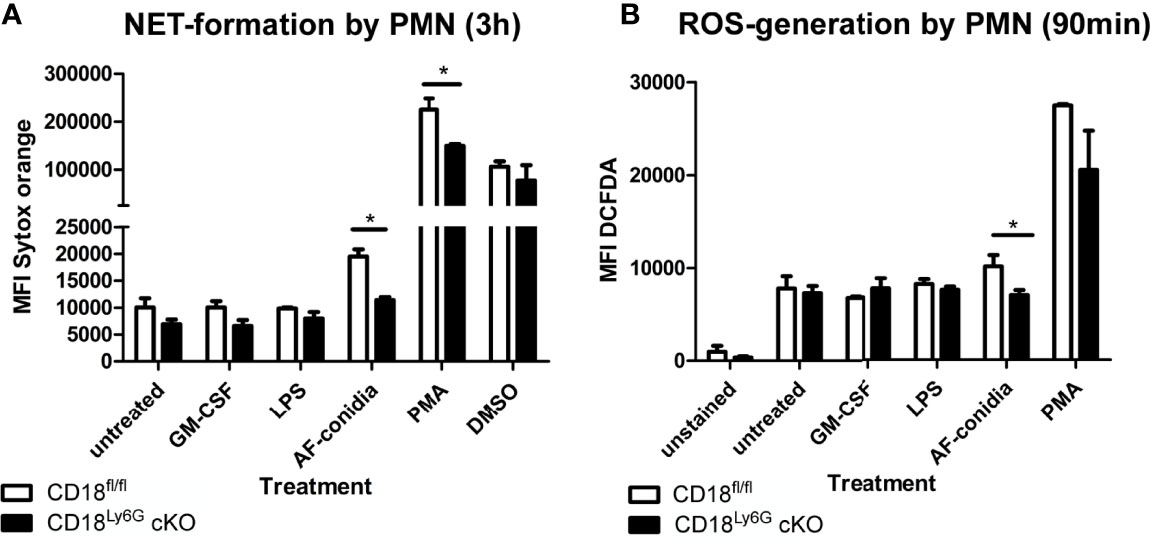
Figure 7 Impaired oxidative and non-oxidative effector functions of CD18-deficient PMN. We could observe that the formation of NET after 3h of incubation (A) and ROS-generation (B) were significantly lower in PMN derived from CD18Ly6G cKO mice, particularly after stimulation with A. fumigatus conidia. Data represent the mean ± SEM of 3 samples analyzed/group. Statistically significant differences between groups are indicated (*p<0.05)
3.6.4 Cytokine Secretion
β2 integrins have been found to regulate various signaling pathways in myeloid cells, which modulate the secretion of inflammatory cytokines (27). Hence, we have investigated the generation of cytokines by PMN after in-vitro stimulation with GM-CSF, LPS (TLR4 agonist), CpG (TLR9 agonist), and R8/48 (TLR7/8 agonist): Here, we could observe that PMN derived from CD18Ly6G cKO mice generated significantly less amounts of TNF-α upon treatment with LPS (Figure 8). On the other hand, we detected significant concentrations of IL-1β, IL-6 and IL-10 upon PMN stimulation, although genotype-dependent differences were largely below statistical significance (Supplementary Figure 9). Other cytokines (IL-12, IL-23 or IFN-γ) showed very low concentrations (not shown), suggesting that these cytokines might not be secreted by PMN under the conditions applied. These in-vitro data are consistent with our observations from in-vivo analysis, showing that BALF and blood derived from CD18Ly6G cKO mice contained lower amounts of TNF-α or IL-1.
Figure 8 PMN derived from CD18Ly6G cKO mice generate lower amounts of TNF-α after previous stimulation with LPS, CpG or R848. We have purified PMN from CD18fl/fl (n=3) mice and CD18Ly6G cKO (n=3) mice and incubated them for 24h at the indicated conditions. After 3h and 24h supernatants have been taken and were analyzed using a CBA. Results show significantly lower levels of TNF-α in supernatants derived from CD18Ly6G cKO mice. *p<0.05, #p<0.05 when comparing cytokine concentrations from supernatants at 3 vs 24h.
3.6.5 RNA-Sequencing Analysis
Last, we have analyzed the impact of the β2 integrin knockdown on the transcriptome of PMN. To this end, we performed RNA-sequencing analysis of either freshly isolated PMN from CD18Ly6G cKO and CD18fl/fl mice or treated aliquots of isolated PMN over-night with LPS (1µg/ml). This genome-wide gene expression analysis confirmed that both, freshly isolated and LPS-treated CD18-deficient PMN showed a significant downregulation of Itgb2 and Ly6G. Referring to the expression of other integrin genes, we could further observe a downregulation Itgb3 and Itgb7, whereas the CD11c encoding gene Itgax, and Itgb2l were found to be upregulated in CD18Ly6G cKO PMN. Moreover, RNA-sequencing data revealed that CD18-deficient PMN showed a higher expression of genes implicated in NFκB signaling, such as CD180, Ly86, CD14, Bach2 or the LPS antagonistic neutrophilic granule protein Ngp (44). On the other hand, we could observe a downregulation of genes involved in the inhibition of oxidative effector functions in PMN, such as S100a9 (45), and a downregulation of genes being implicated in PMN chemotaxis and microbicidal functions (i.e., Defb40) in CD18Ly6G cKO PMN (Figure 9).
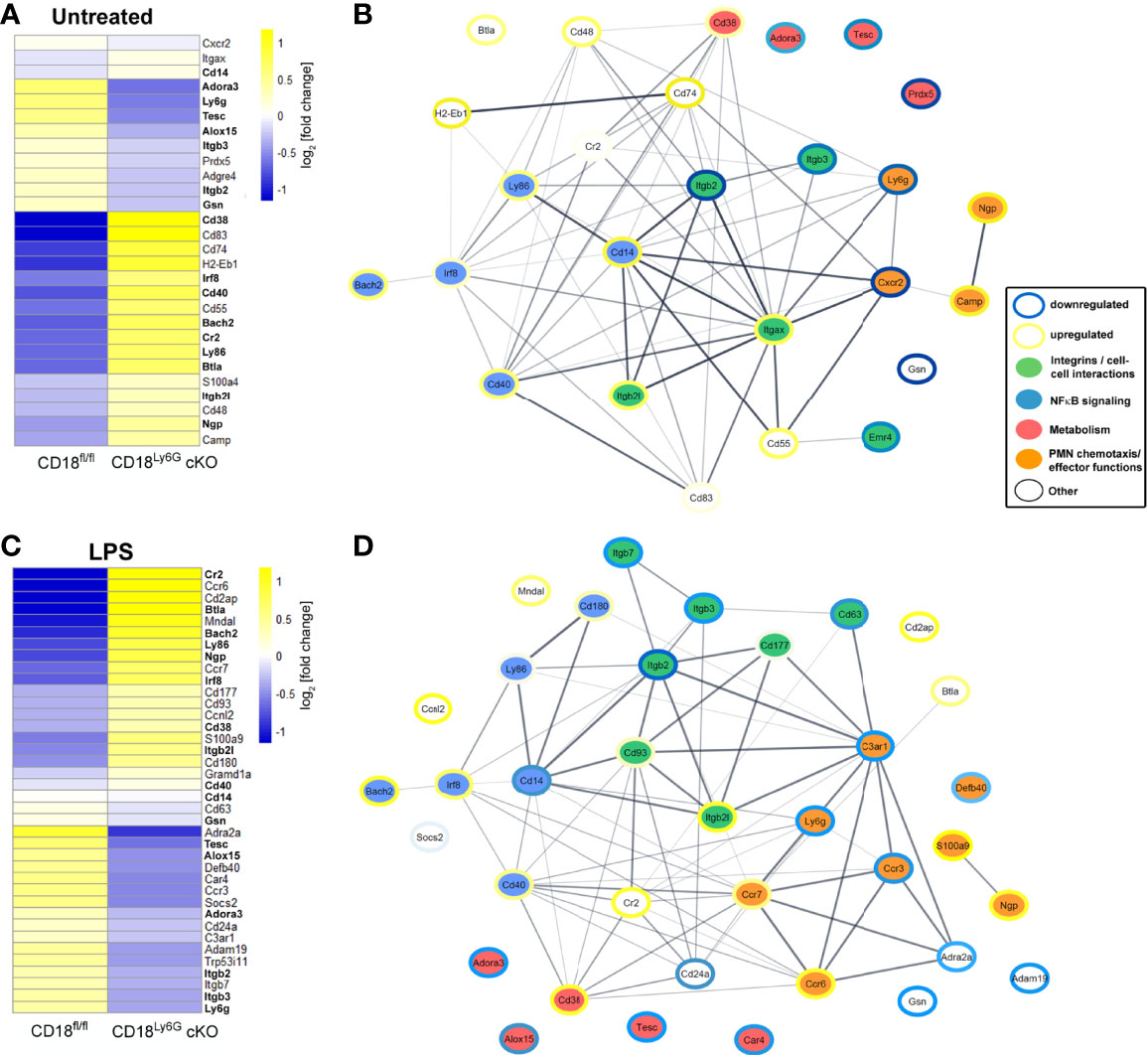
Figure 9 Transcriptomes and functional interaction networks of PMN-associated genes directly after isolation or upon over-night treatment with LPS. PMN were sorted from CD18fl/fl and CD18Ly6G cKO mice (each n=3) and RNA-seq was performed from untreated PMN (A) or LPS-treated PMN (C). Expression of indicated PMN-associated genes was analyzed using CLC Genomics Workbench. Genes being differentially regulated both in LPS-treated and freshly isolated PMN are shown in bold (A, C). Predicted interaction networks of the encoded proteins were being visualized using the STRING package in Cytoscape. Genes shown in the interaction networks of untreated PMN (B) or LPS-treated PMN (D) were categorized into 4 groups affecting either PMN cell-cell interactions, NFκB singaling, PMN metabolism or PMN chemotaxis and PMN effector functions. Colored borders illustrate the degree of the up- or downregulation (log fold change) found for the genes of PMN isolated from CD18Ly6G cKO mice as compared to PMN isolated from CD18fl/fl mice. Legend in (B, D).
4 Discussion
The critical role of β2 integrins for immunological functions is confirmed by the severe immunocompromised state of LAD1 patients, which regularly results in reoccurring invasive bacterial and fungal infections (22, 30). PMN are considered the first line of defense to prevent the spread of inhaled pathogens in the lung (46), and were shown to require β2-integrins for transendothelial migration (24), phagocytosis of opsonized pathogens (21), as well as oxidative, and non-oxidative effector mechanisms (16). Due to the importance of β2-integrins for PMN effector functions and the frequent observation of IPA in LAD1 patients, we aimed to investigate the cell-type specific role of β2 integrins for PMN antifungal effector functions in the early innate immune response to IPA.
Here, we have obtained several key findings that corroborate previous concepts of the pathophysiological role of β2-integrins in the context of severe infections. Our results put these observations into a cell-type specific context and allow insights into the role of CD18 for antifungal effector mechanisms of PMN in the course of IPA, which have not been shown previously.
First, we could observe that the fungal clearance and the early innate immune response in CD18Ly6G cKO mice are significantly impaired. In particular, we found that 24 hours after infection, lungs derived from CD18Ly6G cKO mice showed an enhanced fungal burden and a lower bronchial inflammation as compared to those of CD18fl/fl mice. When PMN are activated upon contact with pathogens and by various danger signals (i.e., the A. fumigatus cell wall component ß-glucan), they contribute to the inflammatory immune response in infected tissues by secreting proinflammatory cytokines and chemokines (47). We observed lower levels of innate proinflammatory mediators, such as TNF-α, IL-1α, IL-1ß, and chemokines, like CCL2 and CCL5 in BALF derived from CD18Ly6G cKO mice, suggesting that the knockdown of β2-integrins might have impaired the ability of PMN to generate these inflammatory mediators. Moreover, we observed lower expression levels of markers for PMN degranulation (CD62L) and activation (MHCII, CD80) in PMN derived from CD18Ly6G cKO mice, indicating that the inflammatory signaling pathways in PMN might have also been impaired by CD18 deficiency. In agreement, we found that A. fumigatus infected CD18Ly6G cKO mice showed an aggravated course of IPA.
Despite, the significant impairment of the early innate immune response mediated by PMN, the overall survival of A. fumigatus infected CD18Ly6G cKO mice was not significantly impaired, suggesting that CD18 despite its pivotal immunoregulatory function might not be critical for the long-term control of IPA or that the residual β2-integrin expression found on PMN of CD18Ly6G cKO mice was sufficient for PMN-mediated pathogen clearance in some mice. Additionally, our results revealed several mechanisms, which may serve to compensate for the impaired effector functions of PMN in CD18Ly6G cKO mice upon infection: Particularly, we could observe a higher level of the PMN-attracting chemokine CXCL-1 in BALF obtained from CD18Ly6G cKO mice. Accordingly, a significantly higher bronchial infiltration by PMN has been found in these mice. These findings were unexpected as β2 integrins were reported to be necessary for the firm adhesion of PMN to vessel endothelium as a prerequisite of PMN migration into the extravascular space (48). In this regard, it has been suggested, that the requirement of CD18 for PMN infiltration might depend on the type of pathogen used in CD18−/− mice (49) and the disease specific context investigated (50). Also, it has been suggested by Mackarel and coworkers, that PMN migration into inflamed lungs might occur either via a CD18-dependent or CD18-independent route, which is selected depending on whether inflammation is acute or chronic (51). In particular, Mizgerd and coworkers reported that intratracheal instillation with E. coli or Ps. aeruginosa resulted in a limited pulmonary PMN-infiltration, whereas infection with S. pneumonia yielded a stronger PMN-infiltration in a CD18-independent manner (49). These observations are consistent with previous reports, which have demonstrated that CD11b−/−mice infected with either S. pneumoniae (52) or A. fumigatus (5), showed an elevated PMN infiltration 24 hours upon infection. However, also in these disease models a higher pulmonary burden and a diminished cellular inflammation have been reported. Similarly, a stronger pulmonary infiltration by PMN has been observed in LAD1 patients suffering from pneumonia (53), suggesting that MAC-1 might not be essential for PMN migration. Rather β2 integrin deficiency may be compensated by other adhesion receptors in a disease specific manner (22). In this context, some studies have reported that LFA-1 (CD11a/CD18) may play a dominant role for transendothelial migration of PMN (51, 54). Our results could however not reveal an upregulation of CD11a on PMN, but rather showed a significant downregulation of CD11a in the context of CD18-deficiency. This is in line with the physiological regulation of β2-integrins on PMN. In particular, the downregulation of CD18 in our knock-out mouse model limits the amount of intracellular available CD18 protein and thus heterodimerization with the corresponding alpha subunits on the cell surface is also being restricted, resulting in lower expression levels of LFA-1 (CD11a/CD18) and MAC-1 (CD11b/CD18) on PMN. On the other hand, RNA-sequencing results indicated that other β2 integrin-associated genes (such as the CD11c coding Itgax or Itgb2l) and genes coding for chemokine receptors (i.e., Ccr7) might be upregulated in PMN isolated from CD18Ly6G cKO mice potentially revealing another compensatory mechanism (Figure 9). Altogether, our results suggest that the knockdown of β2 integrins (LFA-1, MAC-1) might not significantly impair the pulmonary migration of PMN. However, when interpreting the results of our analysis, it has to be taken into account that a significant residual expression of CD18 was still being observed on PMN, which might allow for the CD18-dependent migration of PMN into inflamed pulmonary tissue.
Furthermore, we could demonstrate that PMN isolated from CD18Ly6G cKO mice showed an impaired phagocytic activity towards opsonized A. fumigatus conidia and inert particles as compared to CD18fl/fl PMN, which is consistent with the enhanced pulmonary fungal burden found in A. fumigatus infected CD18Ly6G cKO mice. This finding is in agreement with previous observations that MAC-1 is required in human PMN to recognize ß-glucan containing structures (55), such as A. fumigatus conidia, and thus to kill conidia by phagocytic uptake (21, 56).
In contrast to small-sized conidia, recognition of A. fumigatus hyphae has been largely attributed to IgG and Fcγ receptors (21). However, cross-linking of MAC-1 upon pathogen-recognition, also results in an NADPH-oxidase-dependent oxidative burst by PMN, which is required for an efficient fungal clearance of both A. fumigatus conidia and hyphae (19, 21, 57–59). Oxidative burst protects against invasive fungal infections, because it induces apoptosis-like cell death in fungal conidia (60) and contributes to the formation of NET (61, 62). The latter is considered a mechanism of extracellular killing of hyphae, being too large to be phagocytosed (63). The proposed role of MAC-1 for the induction of ROS and the formation of NET by PMN upon incubation with A. fumigatus conidia indicates that both antifungal killing-mechanisms might be impaired in CD18Ly6G cKO mice. These findings are again consistent with previous reports, which demonstrated that CD11b−/− mice displayed an attenuated PMN killing activity and increased fungal burdens in a mouse model of candidiasis, thus underpinning the pivotal role of β2 integrins for antifungal effector mechanisms (64), such as CR3-mediated phagocytosis, NETosis (65) and ROS-generation (20, 66). Interestingly, Yakubenko and coworkers have more recently observed that neutrophil oxidative burst might further contribute to a positive feedback loop with β2 integrins by enhancing the affinity of MAC-1 ligands to MAC-1 on macrophages, thus stimulating their migratory activity (67).
Next to the direct cytotoxic effects exerted by PMN, some studies reported that the engagement of MAC-1 with extracellular pathogens also promotes proinflammatory signaling pathways in PMN via activation of members of the NF-κB transcription factor family, thus yielding an elevated production of proinflammatory cytokines such as IL-1 and TNF-α (68, 69). In agreement we observed that a knockdown of β2 integrins impaired the secretion of TNF-α. Moreover, it has been found that CD11b facilitates TLR-4 mediated proinflammatory immune responses by promoting MyD88 signaling pathways (27). Hence, the impaired induction of an inflammatory milieu in the lungs of CD18Ly6G cKO mice might be a consequence of the attenuated PMN activation, resulting from a reduced activity of CD18-deficient PMN to recognize and phagocytose A. fumigatus conidia and to promote TLR-4-induced signaling pathways.
Besides the diminished levels of proinflammatory cytokines found in BALF obtained from CD18Ly6G cKO mice, we could also observe lower levels of macrophage attracting chemokines CCL2 and CCL5 therein. CCL5 is known to attract many leukocyte populations, such as macrophages and PMN (70–72). Early in the course of inhalative inflammation, CCL5 is generated by various activated cell types, including airway epithelial cells (73) or lung fibroblasts (74). Moreover, A. fumigatus was reported to induce CCL5 in platelets (75), and activated PMN were demonstrated to produce CCL5 when incubated with Toxoplasma gondii (76). Therefore, it is conceivable that a reduced level of β2 integrins on PMN might impair their ability to generate CCL5.
CCL2, also known as monocyte chemoattractant protein (MCP)-1 is an important regulator of monocyte and macrophage trafficking during infection and in the presence of inflammation (77–79). CCL2 is generated by pulmonary epithelium (80), endothelial cells (81), fibroblasts and T cells upon induction with inflammatory stimuli such as LPS or IFN-γ (82). Notably, also PMN contribute to CCL2 generation, which can be induced upon TLR2-/TLR4-activation (82, 83). CCL2 mainly serves as a chemoattractant for monocytes and macrophages (82, 84–86). Beyond its role as a monocyte chemoattractant CCL2 has been implicated in various molecular and cellular processes impacting myeloid cell functions and their response to pathogens. In particular, it has been shown that CCL2 induces β2 integrin expression on monocytes, thus promoting their migration into inflamed tissues (82, 87) Moreover an enhanced survival and an augmented generation of proinflammatory cytokines by CD11b+ cells has been demonstrated upon CCL2 treatment (88). CCL2 treatment has further been shown to induce respiratory burst in monocytes, thus contributing to myeloid cell effector functions in response to pathogens (82, 89). In agreement, increased CCL2 levels have been reported to improve the clearance of pathogens and the survival of S. pneumonia infected mice (90). These studies are consistent with our observations that CD18fl/fl mice show higher levels of CCL2, a lower fungal burden and a stronger pulmonary infiltration with macrophages, which might exert critical antifungal effector mechanisms in the early innate response to A. fumigatus infection (90). Due to impaired signaling in CD18-deficient PMN it also seems conceivable, that PMN might generate less CCL2 and CCL5 in CD18Ly6G cKO mice. However, as for the multiple sources of these chemokines, further studies are required to elucidate which cell types are responsible for the different concentrations of CCL2 and CCL5 in the lungs of A. fumigatus infected CD18Ly6G cKO mice and which cells are most likely to be attracted in response to these chemokines.
In addition to migration, pathogen recognition/phagocytosis, and the regulation of cell signaling, MAC-1 has also been implicated in myeloid cell survival. Referring particularly to PMN apoptosis, we could observe that PMN derived from A. fumigatus infected CD18Ly6G cKO mice showed a stronger expression of apoptosis marker Annexin-V, suggesting that a knockdown of β2 integrins might impair PMN survival. This is in contrast to previous in-vitro experiments from Coxon and coworkers, which suggested that CD11b contributes to PMN survival, as CD11b−/− PMN isolated from the peritoneum after injection of thioglycollate were characterized by lower apoptosis than their wild-type counterparts (42). However, the contribution of MAC-1 signaling to apoptosis of activated PMN is still subject to controversial discussion. For example, another report by Zhang et al. showed that phagocytosis of pathogens by PMN promoted apoptosis of the latter, which was associated with the induction of reactive oxygen species and was enhanced by TNF-α (91). In contrast, CD11b−/− PMN were not found to undergo phagocytosis-induced apoptosis. Similar findings were reported for human PMN (92). On the contrary, Yan and coworkers showed that antibody-mediated blockade of β2 integrins on human PMN elevated apoptosis after their activation by TNF-α or microbial stimuli (93). Since CD18Ly6G cKO mice only showed a moderate, PMN-restricted, LAD1 phenotype with a residual β2-integrin expression on PMN it seems conceivable that apoptosis may not have been significantly impaired, whereas the same moderate reduction of CD18 on PMN might yet affect other PMN effector functions, as well as the overall course of the disease. Hence, further studies are warranted to elucidate the exact role of MAC-1 on PMN viability during pathogen control.
Although our study focused on the role of β2 integrins for PMN effector mechanisms during early innate immune responses towards inhalative infection with A. fumigatus, it is likely that a knockdown of β2 integrins might not only impair PMN functions but may also modulate their interaction with other immune cells implicated in IPA-resolution, such as DC (94), macrophages, lymphocytes or eosinophils (95, 96). Here, a report by Park and coworkers could show that PMN contribute to pulmonary infiltration of CD11b+ conventional DC in IPA by activating CD11b+ DC via DC-SIGN (94). This C-type lectin receptor expressed by DC and macrophages mediates the phagocytic uptake of A. fumigatus conidia (97) and engages with PMN-bound MAC-1 upon DC-PMN interaction (98). Hence, MAC-1 on PMN may further contribute to the activation of infiltrating DC, which produce IL-12 and IL-23, thus inducing Th1 immunity in IPA (99). Notably, IL-23 has also been reported to stimulate IL-17 production in PMN, and IL-17 induced ROS production by PMN (100), contributing to the killing of A. fumigatus conidia and hyphae. Thus, the diminished expression of CD18 on PMN might further impair their interaction with DC, contributing to an impaired antifungal immune response in CD18Ly6G cKO mice. However, we could not find significant differences neither in IL-17 nor IL-23 secretion in BALF and serum. An important limitation of our experiments is the residual expression of β2-integrins on PMN derived from CD18dLy6G mice which may only result in a moderate impairment of PMN effector. On first sight, an adoptive transfer of PMN from CD11b-/- mice into infected WT mice after depletion of WT PMN may be suitable to give more comprehensive insights into the PMN-specific role of β2-integrins during invasive A. fumigatus infections and exclude compensatory effects that might result from an intermediate PMN phenotype. In this context it would also be interesting to evaluate whether the addition of WT PMN into CD18Ly6G cKO mice might reverse a severe course of the disease. However, such adoptive transfer studies might be subject to methodological bias, including the rather short life span of PMN in general, the influence of β2 integrins on PMN viability and the possibility of artificial PMN activation during adoptive transfer procedures. In conclusion, our results demonstrate, that the PMN-specific downregulation of CD18 allows for a distinct cell-type specific analysis of the role of β2 integrins for PMN effector functions, PMN signaling, survival and the role of β2 integrins as regulators within the immune cell network (47). We could further show that CD18 deficiency on PMN particularly affects the early course of IPA, which might be attributed to the critical role of MAC−1 for PMN antifungal effector mechanisms, such as phagocytosis and ROS-generation (5). However, we cannot rule out that the CD18-knockdown might cause additional unrecognized effects in PMN effector functions, such as the release of primary granules or MPO-activity contributing to the clearance of A. fumigatus or that residual CD18 expression might compensate some impaired effector functions. Taking into account that previous PMN-specific knock-out models, such as the Sykfl/- MRP8CreTg mice reported by van Ziffle and Lowell (101), also showed a residual expression of the targeted proteins on PMN, further work is necessary to generate knock-out models which might allow for a complete knock-out of β2-integrins on PMN. Also, additional studies will be necessary to elucidate the long-term course of IPA in CD18Ly6G cKO mice with regard to the interplay of PMN with DC, the efficacy of adaptive immune responses and the contribution of chemokines such as CCL2 and CCL5.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Gene Expression Omnibus, GSE195444.
Ethics Statement
The animal study was reviewed and approved by the National State Investigation Office Rhineland-Palatinate, Approval ID: 23177-07/G16-1-020.
Author Contributions
MH designed methods for in-vitro and ex-vivo experiments, carried out experiments, performed image analysis, carried out data analysis, calculated statistics, designed and generated figures, compiled tables, and wrote the manuscript. MBro designed experiments, designed methods for in-vitro experiments, performed image analyses, edited and designed figures and tables, and helped writing the manuscript. FR, DT and MK helped to design and carry out the in-vivo experiments, helped writing the manuscript and edited the manuscript. MK and ES helped carrying out the experiments. SG and MR helped designing the experiments, as well as writing and editing the manuscript. MBed (Monika Bednarczyk) generated the CD18fl/fl and CD18Ly6G cKO mouse. MG helped generating the CD18Ly6G cKO mouse strain, provided C57BL/6-Ly6g(tm2621(Cre-tdTomato)Arte mice and AF strains (ATCC 46645 and Afs148) and helped editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MH was supported by the Clinician Scientist Fellowship “TransMed Jumpstart Program: 2019_A72” supported by the Else Kröner Fresenius Foundation and by an intramural research funding of the UMC Mainz. MR is funded by the TRR156 KS01 and SFB1066 TPB10. SG is funded by the DFG (TRR156, B11; SFB1066, B04).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank C. Braun, F. Zarate, I. Tubbe, S. Hamdi and E. Montermann for excellent technical assistance. Also, we gratefully acknowledge the support by Matthias Klein, the Forschungszentrum Immunologie (FZI) and the FZI sequencing core facility at the University Medical Center Mainz for the generation and analysis of RNA-sequencing data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.823121/full#supplementary-material
References
1. Gresnigt MS, Rekiki A, Rasid O, Savers A, Jouvion G, Dannaoui E, et al. Reducing Hypoxia and Inflammation During Invasive Pulmonary Aspergillosis by Targeting the Interleukin-1 Receptor. Sci Rep (2016) 6(1):26490. doi: 10.1038/srep26490
2. O’Gorman CM, Fuller HT. Prevalence of Culturable Airborne Spores of Selected Allergenic and Pathogenic Fungi in Outdoor Air. Atmos Environ (2008) 42(18):4355–68. doi: 10.1016/j.atmosenv.2008.01.009
3. Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. Hidden Killers: Human Fungal Infections. Sci Transl Med (2012) 4(165):165rv13. doi: 10.1126/scitranslmed.3004404
4. Feldmesser M. Role of Neutrophils in Invasive Aspergillosis. Infect Immun (2006) 74(12):6514–6. doi: 10.1128/IAI.01551-06
5. Teschner D, Cholaszczynska A, Ries F, Beckert H, Theobald M, Grabbe S, et al. CD11b Regulates Fungal Outgrowth But Not Neutrophil Recruitment in a Mouse Model of Invasive Pulmonary Aspergillosis. Front Immunol (2019) 10:123. doi: 10.3389/fimmu.2019.00123
6. Braem SG, Rooijakkers SH, van Kessel KP, de Cock H, Wosten HA, van Strijp JA, et al. Effective Neutrophil Phagocytosis of Aspergillus Fumigatus Is Mediated by Classical Pathway Complement Activation. J Innate Immun (2015) 7(4):364–74. doi: 10.1159/000369493
7. Latge JP. The Pathobiology of Aspergillus Fumigatus. Trends Microbiol (2001) 9(8):382–9. doi: 10.1016/S0966-842X(01)02104-7
8. Botterel F, Gross K, Ibrahim-Granet O, Khoufache K, Escabasse V, Coste A, et al. Phagocytosis of Aspergillus Fumigatus Conidia by Primary Nasal Epithelial Cells In Vitro. BMC Microbiol (2008) 8(1):97. doi: 10.1186/1471-2180-8-97
9. Rammaert B, Jouvion G, de Chaumont F, Garcia-Hermoso D, Szczepaniak C, Renaudat C, et al. Absence of Fungal Spore Internalization by Bronchial Epithelium in Mouse Models Evidenced by a New Bioimaging Approach and Transmission Electronic Microscopy. Am J Pathol (2015) 185(9):2421–30. doi: 10.1016/j.ajpath.2015.04.027
10. Luther K, Torosantucci A, Brakhage AA, Heesemann J, Ebel F. Phagocytosis of Aspergillus Fumigatus Conidia by Murine Macrophages Involves Recognition by the Dectin-1 Beta-Glucan Receptor and Toll-Like Receptor 2. Cell Microbiol (2007) 9(2):368–81. doi: 10.1111/j.1462-5822.2006.00796.x
11. Braedel S, Radsak M, Einsele H, Latge JP, Michan A, Loeffler J, et al. Aspergillus Fumigatus Antigens Activate Innate Immune Cells via Toll-Like Receptors 2 and 4. Br J Haematol (2004) 125(3):392–9. doi: 10.1111/j.1365-2141.2004.04922.x
12. Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM. Essential Role for Neutrophils But Not Alveolar Macrophages at Early Time Points Following Aspergillus Fumigatus Infection. J Infect Dis (2009) 200(4):647–56. doi: 10.1086/600380
13. Post MJ, Lass-Floerl C, Gastl G, Nachbaur D. Invasive Fungal Infections in Allogeneic and Autologous Stem Cell Transplant Recipients: A Single-Center Study of 166 Transplanted Patients. Transpl Infect Dis (2007) 9(3):189–95. doi: 10.1111/j.1399-3062.2007.00219.x
14. Cohen MS, Isturiz RE, Malech HL, Root RK, Wilfert CM, Gutman L, et al. Fungal Infection in Chronic Granulomatous Disease. The Importance of the Phagocyte in Defense Against Fungi. Am J Med (1981) 71(1):59–66. doi: 10.1016/0002-9343(81)90259-X
15. Gerson SL, Talbot GH, Hurwitz S, Strom BL, Lusk EJ, Cassileth PA. Prolonged Granulocytopenia: The Major Risk Factor for Invasive Pulmonary Aspergillosis in Patients With Acute Leukemia. Ann Intern Med (1984) 100(3):345–51. doi: 10.7326/0003-4819-100-3-345
16. Prufer S, Weber M, Stein P, Bosmann M, Stassen M, Kreft A, et al. Oxidative Burst and Neutrophil Elastase Contribute to Clearance of Aspergillus Fumigatus Pneumonia in Mice. Immunobiology (2014) 219(2):87–96. doi: 10.1016/j.imbio.2013.08.010
17. Diamond RD, Clark RA. Damage to Aspergillus Fumigatus and Rhizopus Oryzae Hyphae by Oxidative and Nonoxidative Microbicidal Products of Human Neutrophils In Vitro. Infect Immun (1982) 38(2):487–95. doi: 10.1128/iai.38.2.487-495.1982
18. Leal SM Jr., Vareechon C, Cowden S, Cobb BA, Latge JP, Momany M, et al. Fungal Antioxidant Pathways Promote Survival Against Neutrophils During Infection. J Clin Invest (2012) 122(7):2482–98. doi: 10.1172/JCI63239
19. Boyle KB, Gyori D, Sindrilaru A, Scharffetter-Kochanek K, Taylor PR, Mocsai A, et al. Class IA Phosphoinositide 3-Kinase Beta and Delta Regulate Neutrophil Oxidase Activation in Response to Aspergillus Fumigatus Hyphae. J Immunol (2011) 186(5):2978–89. doi: 10.4049/jimmunol.1002268
20. Aarts CEM, Hiemstra IH, Beguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated Neutrophils Exert Myeloid-Derived Suppressor Cell Activity Damaging T Cells Beyond Repair. Blood Adv (2019) 3(22):3562–74. doi: 10.1182/bloodadvances.2019031609
21. Gazendam RP, van Hamme JL, Tool AT, Hoogenboezem M, van den Berg JM, Prins JM, et al. Human Neutrophils Use Different Mechanisms To Kill Aspergillus Fumigatus Conidia and Hyphae: Evidence From Phagocyte Defects. J Immunol (2016) 196(3):1272–83. doi: 10.4049/jimmunol.1501811
22. Bednarczyk M, Stege H, Grabbe S, Bros M. Beta2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci (2020) 21(4):6. doi: 10.3390/ijms21041402
23. Arnaout MA. Biology and Structure of Leukocyte Beta 2 Integrins and Their Role in Inflammation. F1000Res (2016) 5(2433):1. doi: 10.12688/f1000research.9415.1
24. Basoni C, Nobles M, Grimshaw A, Desgranges C, Davies D, Perretti M, et al. Inhibitory Control of TGF-Beta1 on the Activation of Rap1, CD11b, and Transendothelial Migration of Leukocytes. FASEB J (2005) 19(7):822–4. doi: 10.1096/fj.04-3085fje
25. Ehlers MR. CR3: A General Purpose Adhesion-Recognition Receptor Essential for Innate Immunity. Microbes Infect (2000) 2(3):289–94. doi: 10.1016/S1286-4579(00)00299-9
26. Huang ZY, Hunter S, Chien P, Kim MK, Han-Kim TH, Indik ZK, et al. Interaction of Two Phagocytic Host Defense Systems: Fcgamma Receptors and Complement Receptor 3. J Biol Chem (2011) 286(1):160–8. doi: 10.1074/jbc.M110.163030
27. Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, et al. Integrin CD11b Positively Regulates TLR4-Induced Signalling Pathways in Dendritic Cells But Not in Macrophages. Nat Commun (2014) 5(1):3039. doi: 10.1038/ncomms4039
28. Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, et al. Active MAC-1 (CD11b/CD18) on DCs Inhibits Full T-Cell Activation. Blood (2007) 109(2):661–9. doi: 10.1182/blood-2005-12-023044
29. Lim J, Hotchin NA. Signalling Mechanisms of the Leukocyte Integrin Alphambeta2: Current and Future Perspectives. Biol Cell (2012) 104(11):631–40. doi: 10.1111/boc.201200013
30. Harris ES, Weyrich AS, Zimmerman GA. Lessons From Rare Maladies: Leukocyte Adhesion Deficiency Syndromes. Curr Opin Hematol (2013) 20(1):16–25. doi: 10.1097/MOH.0b013e32835a0091
31. Anderson DC, Schmalsteig FC, Finegold MJ, Hughes BJ, Rothlein R, Miller LJ, et al. The Severe and Moderate Phenotypes of Heritable Mac-1, LFA-1 Deficiency: Their Quantitative Definition and Relation to Leukocyte Dysfunction and Clinical Features. J Infect Dis (1985) 152(4):668–89. doi: 10.1093/infdis/152.4.668
32. Lother J, Breitschopf T, Krappmann S, Morton CO, Bouzani M, Kurzai O, et al. Human Dendritic Cell Subsets Display Distinct Interactions With the Pathogenic Mould Aspergillus Fumigatus. Int J Med Microbiol (2014) 304(8):1160–8. doi: 10.1016/j.ijmm.2014.08.009
33. Lee PY, Wang JX, Parisini E, Dascher CC, Nigrovic PA. Ly6 Family Proteins in Neutrophil Biology. J Leukoc Biol (2013) 94(4):585–94. doi: 10.1189/jlb.0113014
34. Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW, et al. High-Dimensional Analysis of the Murine Myeloid Cell System. Nat Immunol (2014) 15(12):1181–9. doi: 10.1038/ni.3006
35. Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, et al. Catchup: A Mouse Model for Imaging-Based Tracking and Modulation of Neutrophil Granulocytes. Nat Methods (2015) 12(5):445–52. doi: 10.1038/nmeth.3322
36. Alflen A, Prufer S, Ebner K, Reuter S, Aranda Lopez P, Scharrer I, et al. ADAMTS-13 Regulates Neutrophil Recruitment in a Mouse Model of Invasive Pulmonary Aspergillosis. Sci Rep (2017) 7(1):7184. doi: 10.1038/s41598-017-07340-3
37. Lamoth F. Galactomannan and 1,3-Beta-D-Glucan Testing for the Diagnosis of Invasive Aspergillosis. J Fungi (Basel) (2016) 2(3):22. doi: 10.3390/jof2030022
38. Majewska E, Sulowska Z, Baj Z. Spontaneous Apoptosis of Neutrophils in Whole Blood and Its Relation to Apoptosis Gene Proteins. Scand J Immunol (2000) 52(5):496–501. doi: 10.1046/j.1365-3083.2000.00802.x
39. Team RC. R Core Team R: A Language and Environment for Statistical Computing, Foundation for Statistical Computing, 2020.
40. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
41. Wang JX, Bair AM, King SL, Shnayder R, Huang YF, Shieh CC, et al. Ly6G Ligation Blocks Recruitment of Neutrophils via a Beta2-Integrin-Dependent Mechanism. Blood (2012) 120(7):1489–98. doi: 10.1182/blood-2012-01-404046
42. Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, et al. A Novel Role for the β2 Integrin CD11b/CD18 in Neutrophil Apoptosis: A Homeostatic Mechanism in Inflammation. Immunity (1996) 5(6):653–66. doi: 10.1016/S1074-7613(00)80278-2
43. Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, et al. Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity (2018) 49(6):1062–76.e6. doi: 10.1016/j.immuni.2018.09.018
44. Hong J, Qu P, Wuest T, Lin PC. Neutrophilic Granule Protein Is a Novel Murine LPS Antagonist. J Immunol (2020) 204(1):60.21–1. doi: 10.4110/in.2019.19.e34
45. Sroussi HY, Lu Y, Zhang QL, Villines D, Marucha PT. S100A8 and S100A9 Inhibit Neutrophil Oxidative Metabolism In-Vitro: Involvement of Adenosine Metabolites. Free Radic Res (2010) 44(4):389–96. doi: 10.3109/10715760903431434
46. Baddley JW, Andes DR, Marr KA, Kontoyiannis DP, Alexander BD, Kauffman CA, et al. Factors Associated With Mortality in Transplant Patients With Invasive Aspergillosis. Clin Infect Dis (2010) 50(12):1559–67. doi: 10.1086/652768
47. Yang F, Feng C, Zhang X, Lu J, Zhao Y. The Diverse Biological Functions of Neutrophils, Beyond the Defense Against Infections. Inflammation (2017) 40(1):311–23. doi: 10.1007/s10753-016-0458-4
48. Langer HF, Chavakis T. Leukocyte-Endothelial Interactions in Inflammation. J Cell Mol Med (2009) 13(7):1211–20. doi: 10.1111/j.1582-4934.2009.00811.x
49. Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM. Effects of CD18 Deficiency on the Emigration of Murine Neutrophils During Pneumonia. J Immunol (1999) 163(2):995–9.
50. Cunin P, Lee PY, Kim E, Schmider AB, Cloutier N, Pare A, et al. Differential Attenuation of Beta2 Integrin-Dependent and -Independent Neutrophil Migration by Ly6G Ligation. Blood Adv (2019) 3(3):256–67. doi: 10.1182/bloodadvances.2018026732
51. Mackarel AJ, Russell KJ, Ryan CM, Hislip SJ, Rendall JC, FitzGerald MX, et al. CD18 Dependency of Transendothelial Neutrophil Migration Differs During Acute Pulmonary Inflammation. J Immunol (2001) 167(5):2839–46. doi: 10.4049/jimmunol.167.5.2839
52. Rijneveld AW, de Vos AF, Florquin S, Verbeek JS, van der Poll T. CD11b Limits Bacterial Outgrowth and Dissemination During Murine Pneumococcal Pneumonia. J Infect Dis (2005) 191(10):1755–60. doi: 10.1086/429633
53. Hawkins HK, Heffelfinger SC, Anderson DC. Leukocyte Adhesion Deficiency: Clinical and Postmortem Observations. Pediatr Pathol (1992) 12(1):119–30. doi: 10.3109/15513819209023288
54. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative Contribution of LFA-1 and Mac-1 to Neutrophil Adhesion and Migration. J Immunol (1999) 163(9):5029–38.
55. van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, Chapel H, et al. Complement Receptor 3, Not Dectin-1, Is the Major Receptor on Human Neutrophils for Beta-Glucan-Bearing Particles. Mol Immunol (2009) 47(2-3):575–81. doi: 10.1016/j.molimm.2009.09.018
56. Moalli F, Doni A, Deban L, Zelante T, Zagarella S, Bottazzi B, et al. Role of Complement and Fc{gamma} Receptors in the Protective Activity of the Long Pentraxin PTX3 Against Aspergillus Fumigatus. Blood (2010) 116(24):5170–80. doi: 10.1182/blood-2009-12-258376
57. Nilsson M, Weineisen M, Andersson T, Truedsson L, Sjobring U. Critical Role for Complement Receptor 3 (CD11b/CD18), But Not for Fc Receptors, in Killing of Streptococcus Pyogenes by Neutrophils in Human Immune Serum. Eur J Immunol (2005) 35(5):1472–81. doi: 10.1002/eji.200424850
58. Futosi K, Fodor S, Mocsai A. Neutrophil Cell Surface Receptors and Their Intracellular Signal Transduction Pathways. Int Immunopharmacol (2013) 17(3):638–50. doi: 10.1016/j.intimp.2013.06.034
59. Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, et al. Protein Kinase C-Beta Contributes to NADPH Oxidase Activation in Neutrophils. Biochem J (2000) 347 Pt 1(Pt 1):285–9. doi: 10.1042/bj3470285
60. Shlezinger N, Irmer H, Dhingra S, Beattie SR, Cramer RA, Braus GH, et al. Sterilizing Immunity in the Lung Relies on Targeting Fungal Apoptosis-Like Programmed Cell Death. Science (2017) 357(6355):1037–41. doi: 10.1126/science.aan0365
61. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
62. Clark HL, Abbondante S, Minns MS, Greenberg EN, Sun Y, Pearlman E. Protein Deiminase 4 and CR3 Regulate Aspergillus Fumigatus and Beta-Glucan-Induced Neutrophil Extracellular Trap Formation, But Hyphal Killing Is Dependent Only on CR3. Front Immunol (2018) 9:1182. doi: 10.3389/fimmu.2018.01182
63. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense Against Candida Albicans. PloS Pathog (2009) 5(10):e1000639. doi: 10.1371/journal.ppat.1000639
64. Soloviev DA, Jawhara S, Fonzi WA. Regulation of Innate Immune Response to Candida Albicans Infections by Alphambeta2-Pra1p Interaction. Infect Immun (2011) 79(4):1546–58. doi: 10.1128/IAI.00650-10
65. O'Brien XM, Reichner JS. Neutrophil Integrins and Matrix Ligands and NET Release. Front Immunol (2016) 7:363. doi: 10.3389/fimmu.2016.00363
66. Gazendam RP, van Hamme JL, Tool AT, van Houdt M, Verkuijlen PJ, Herbst M, et al. Two Independent Killing Mechanisms of Candida Albicans by Human Neutrophils: Evidence From Innate Immunity Defects. Blood (2014) 124(4):590–7. doi: 10.1182/blood-2014-01-551473
67. Yakubenko VP, Cui K, Ardell CL, Brown KE, West XZ, Gao D, et al. Oxidative Modifications of Extracellular Matrix Promote the Second Wave of Inflammation via Beta2 Integrins. Blood (2018) 132(1):78–88. doi: 10.1182/blood-2017-10-810176
68. Kim CH, Lee K-H, Lee C-T, Kim YW, Han SK, Shim Y-S, et al. Aggregation of β2integrins Activates Human Neutrophils Through the Iκb/NF-κb Pathway. J Leukoc Biol (2004) 75(2):286–92. doi: 10.1189/jlb.0103038
69. Kettritz R, Choi M, Rolle S, Wellner M, Luft FC. Integrins and Cytokines Activate Nuclear Transcription factor-kappaB in Human Neutrophils. J Biol Chem (2004) 279(4):2657–65. doi: 10.1074/jbc.M309778200
70. Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. CCL5 Promotes Macrophage Recruitment and Survival in Human Adipose Tissue. Arterioscler Thromb Vasc Biol (2010) 30(1):39–45. doi: 10.1161/ATVBAHA.109.197442
71. Culley FJ, Pennycook AM, Tregoning JS, Dodd JS, Walzl G, Wells TN, et al. Role of CCL5 (RANTES) in Viral Lung Disease. J Virol (2006) 80(16):8151–7. doi: 10.1128/JVI.00496-06
72. Yu C, Zhang S, Wang Y, Zhang S, Luo L, Thorlacius H. Platelet-Derived CCL5 Regulates CXC Chemokine Formation and Neutrophil Recruitment in Acute Experimental Colitis. J Cell Physiol (2016) 231(2):370–6. doi: 10.1002/jcp.25081
73. Propst SM, Denson R, Rothstein E, Estell K, Schwiebert LM. Proinflammatory and Th2-Derived Cytokines Modulate CD40-Mediated Expression of Inflammatory Mediators in Airway Epithelia: Implications for the Role of Epithelial CD40 in Airway Inflammation. J Immunol (2000) 165(4):2214–21. doi: 10.4049/jimmunol.165.4.2214
74. Teran LM, Mochizuki M, Bartels J, Valencia EL, Nakajima T, Hirai K, et al. Th1- and Th2-Type Cytokines Regulate the Expression and Production of Eotaxin and RANTES by Human Lung Fibroblasts. Am J Respir Cell Mol Biol (1999) 20(4):777–86. doi: 10.1165/ajrcmb.20.4.3508
75. Rodland EK, Ueland T, Pedersen TM, Halvorsen B, Muller F, Aukrust P, et al. Activation of Platelets by Aspergillus Fumigatus and Potential Role of Platelets in the Immunopathogenesis of Aspergillosis. Infect Immun (2010) 78(3):1269–75. doi: 10.1128/IAI.01091-09
76. Bennouna S, Bliss SK, Curiel TJ, Denkers EY. Cross-Talk in the Innate Immune System: Neutrophils Instruct Recruitment and Activation of Dendritic Cells During Microbial Infection. J Immunol (2003) 171(11):6052–8. doi: 10.4049/jimmunol.171.11.6052
77. Yadav A, Saini V, Arora S. MCP-1: Chemoattractant With a Role Beyond Immunity: A Review. Clin Chim Acta (2010) 411(21-22):1570–9. doi: 10.1016/j.cca.2010.07.006
78. Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, et al. Alveolar Epithelial Cells Direct Monocyte Transepithelial Migration Upon Influenza Virus Infection: Impact of Chemokines and Adhesion Molecules. J Immunol (2006) 177(3):1817–24. doi: 10.4049/jimmunol.177.3.1817
79. Lai C, Wang K, Zhao Z, Zhang L, Gu H, Yang P, et al. C-C Motif Chemokine Ligand 2 (CCL2) Mediates Acute Lung Injury Induced by Lethal Influenza H7N9 Virus. Front Microbiol (2017) 8:587. doi: 10.3389/fmicb.2017.00587
80. Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Konigshoff M, Howell DC, et al. Pulmonary Epithelium Is a Prominent Source of Proteinase-Activated Receptor-1-Inducible CCL2 in Pulmonary Fibrosis. Am J Respir Crit Care Med (2009) 179(5):414–25. doi: 10.1164/rccm.200712-1827OC
81. Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, et al. Minimally Modified Low Density Lipoprotein Induces Monocyte Chemotactic Protein 1 in Human Endothelial Cells and Smooth Muscle Cells. Proc Natl Acad Sci USA (1990) 87(13):5134–8. doi: 10.1073/pnas.87.13.5134
82. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol (2019) 10(2759):2759. doi: 10.3389/fimmu.2019.02759
83. Yoshimura T, Takahashi M. IFN-Gamma-Mediated Survival Enables Human Neutrophils to Produce MCP-1/CCL2 in Response to Activation by TLR Ligands. J Immunol (2007) 179(3):1942–9. doi: 10.4049/jimmunol.179.3.1942
84. Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, et al. Monocytes Are Potent Facilitators of Alveolar Neutrophil Emigration During Lung Inflammation: Role of the CCL2-CCR2 Axis. J Immunol (2003) 170(6):3273–8. doi: 10.4049/jimmunol.170.6.3273
85. Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and Characterization of a Novel Monocyte Chemotactic and Activating Factor Produced by a Human Myelomonocytic Cell Line. J Exp Med (1989) 169(4):1485–90. doi: 10.1084/jem.169.4.1485
86. Reichel CA, Rehberg M, Lerchenberger M, Berberich N, Bihari P, Khandoga AG, et al. Ccl2 and Ccl3 Mediate Neutrophil Recruitment via Induction of Protein Synthesis and Generation of Lipid Mediators. Arterioscler Thromb Vasc Biol (2009) 29(11):1787–93. doi: 10.1161/ATVBAHA.109.193268
87. Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte Chemoattractant Protein-1 Regulates Adhesion Molecule Expression and Cytokine Production in Human Monocytes. J Immunol (1992) 148(8):2423–8.
88. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and Interleukin-6 Promote Survival of Human CD11b+ Peripheral Blood Mononuclear Cells and Induce M2-Type Macrophage Polarization. J Biol Chem (2009) 284(49):34342–54. doi: 10.1074/jbc.M109.042671
89. Rollins BJ, Walz A, Baggiolini M. Recombinant Human MCP-1/JE Induces Chemotaxis, Calcium Flux, and the Respiratory Burst in Human Monocytes. Blood (1991) 78(4):1112–6. doi: 10.1182/blood.V78.4.1112.1112
90. Winter C, Taut K, Srivastava M, Langer F, Mack M, Briles DE, et al. Lung-Specific Overexpression of CC Chemokine Ligand (CCL) 2 Enhances the Host Defense to Streptococcus Pneumoniae Infection in Mice: Role of the CCL2-CCR2 Axis. J Immunol (2007) 178(9):5828–38. doi: 10.4049/jimmunol.178.9.5828
91. Zhang B, Hirahashi J, Cullere X, Mayadas TN. Elucidation of Molecular Events Leading to Neutrophil Apoptosis Following Phagocytosis: Cross-Talk Between Caspase 8, Reactive Oxygen Species, and MAPK/ERK Activation. J Biol Chem (2003) 278(31):28443–54. doi: 10.1074/jbc.M210727200
92. Walzog B, Jeblonski F, Zakrzewicz A, Gaehtgens P. Beta2 Integrins (CD11/CD18) Promote Apoptosis of Human Neutrophils. FASEB J (1997) 11(13):1177–86. doi: 10.1096/fasebj.11.13.9367353
93. Yan SR, Sapru K, Issekutz AC. The CD11/CD18 (Beta2) Integrins Modulate Neutrophil Caspase Activation and Survival Following TNF-Alpha or Endotoxin Induced Transendothelial Migration. Immunol Cell Biol (2004) 82(4):435–46. doi: 10.1111/j.0818-9641.2004.01268.x
94. Park SJ, Burdick MD, Mehrad B. Neutrophils Mediate Maturation and Efflux of Lung Dendritic Cells in Response to Aspergillus Fumigatus Germ Tubes. Infect Immun (2012) 80(5):1759–65. doi: 10.1128/IAI.00097-12
95. Guerra ES, Lee CK, Specht CA, Yadav B, Huang H, Akalin A, et al. Central Role of IL-23 and IL-17 Producing Eosinophils as Immunomodulatory Effector Cells in Acute Pulmonary Aspergillosis and Allergic Asthma. PloS Pathog (2017) 13(1):e1006175. doi: 10.1371/journal.ppat.1006175
96. Lilly LM, Scopel M, Nelson MP, Burg AR, Dunaway CW, Steele C. Eosinophil Deficiency Compromises Lung Defense Against Aspergillus Fumigatus. Infect Immun (2014) 82(3):1315–25. doi: 10.1128/IAI.01172-13
97. Serrano-Gomez D, Dominguez-Soto A, Ancochea J, Jimenez-Heffernan JA, Leal JA, Corbi AL. Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Nonintegrin Mediates Binding and Internalization of Aspergillus Fumigatus Conidia by Dendritic Cells and Macrophages. J Immunol (2004) 173(9):5635–43. doi: 10.4049/jimmunol.173.9.5635
98. van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y. Neutrophils Mediate Immune Modulation of Dendritic Cells Through Glycosylation-Dependent Interactions Between Mac-1 and DC-SIGN. J Exp Med (2005) 201(8):1281–92. doi: 10.1084/jem.20041276
99. Gafa V, Lande R, Gagliardi MC, Severa M, Giacomini E, Remoli ME, et al. Human Dendritic Cells Following Aspergillus Fumigatus Infection Express the CCR7 Receptor and a Differential Pattern of Interleukin-12 (IL-12), IL-23, and IL-27 Cytokines, Which Lead to a Th1 Response. Infect Immun (2006) 74(3):1480–9. doi: 10.1128/IAI.74.3.1480-1489.2006
100. Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, Cobb BA, et al. Activation of Neutrophils by Autocrine IL-17a-IL-17RC Interactions During Fungal Infection is Regulated by IL-6, IL-23, RORgammat and Dectin-2. Nat Immunol (2014) 15(2):143–51. doi: 10.1038/ni.2797
Keywords: β2 integrins, CD18, CD11b, polymorphonuclear neutrophils, Aspergillus fumigatus, pneumonia, complement receptor 3, phagocytosis
Citation: Haist M, Ries F, Gunzer M, Bednarczyk M, Siegel E, Kuske M, Grabbe S, Radsak M, Bros M and Teschner D (2022) Neutrophil-Specific Knockdown of β2 Integrins Impairs Antifungal Effector Functions and Aggravates the Course of Invasive Pulmonal Aspergillosis. Front. Immunol. 13:823121. doi: 10.3389/fimmu.2022.823121
Received: 26 November 2021; Accepted: 28 April 2022;
Published: 06 June 2022.
Edited by:
Marko Radic, University of Tennessee College of Medicine, United StatesReviewed by:
Rodrigo Tinoco Figueiredo, Federal University of Rio de Janeiro, BrazilMeiqing Shi, University of Maryland, College Park, United States
Copyright © 2022 Haist, Ries, Gunzer, Bednarczyk, Siegel, Kuske, Grabbe, Radsak, Bros and Teschner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maximilian Haist, TWF4aW1pbGlhbi5IYWlzdEB1bmltZWRpemluLW1haW56LmRl
†These authors have contributed equally to this work