 Katharina Eva Ruppel
Katharina Eva Ruppel Stephan Fricke
Stephan Fricke Ulrike Köhl
Ulrike Köhl Dominik Schmiedel
Dominik Schmiedel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 March 2022
Sec. NK and Innate Lymphoid Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.822298
This article is part of the Research TopicTranslating NK Cell Scientific Research to Clinical Product ManufacturingView all 10 articles
Cancer immunotherapies utilize the capabilities of the immune system to efficiently target malignant cells. In recent years, chimeric antigen receptor (CAR) equipped T cells showed promising results against B cell lymphomas. Autologous CAR-T cells require patient-specific manufacturing and thus extensive production facilities, resulting in high priced therapies. Along with potentially severe side effects, these are the major drawbacks of CAR-T cells therapies. Natural Killer (NK) cells pose an alternative for CAR equipped immune cells. Since NK cells can be safely transferred from healthy donors to cancer patients, they present a suitable platform for an allogeneic “off-the-shelf” immunotherapy. However, administration of activated NK cells in cancer therapy has until now shown poor anti-cancer responses, especially in solid tumors. Genetic modifications such as CARs promise to enhance recognition of tumor cells, thereby increasing anti-tumor effects and improving clinical efficacy. Although the cell biology of T and NK cells deviates in many aspects, the development of CAR-NK cells frequently follows within the footsteps of CAR-T cells, meaning that T cell technologies are simply adopted to NK cells. In this review, we underline the unique properties of NK cells and their potential in CAR therapies. First, we summarize the characteristics of NK cell biology with a focus on signaling, a fine-tuned interaction of activating and inhibitory receptors. We then discuss why tailored NK cell-specific CAR designs promise superior efficacy compared to designs developed for T cells. We summarize current findings and developments in the CAR-NK landscape: different CAR formats and modifications to optimize signaling, to target a broader pool of antigens or to increase in vivo persistence. Finally, we address challenges beyond NK cell engineering, including expansion and manufacturing, that need to be addressed to pave the way for CAR-NK therapies from the bench to the clinics.
Cancer immunotherapies utilize the capabilities of the immune system to efficiently target tumor cells. In recent years, T cells equipped with a chimeric antigen receptor (CAR) showed promising results against B cell malignancies, proving to be a possibly curative treatment option (1, 2). CARs are artificial proteins that are composed of an antibody-derived extracellular target-binding domain, predominantly of a single chain variable fragment (scFv), that is connected to an intracellular signaling domain by a hinge region and a transmembrane domain. Upon binding, e.g., to tumor-specific or -associated antigens, intracellular CAR signaling is activated, resulting in cytotoxicity of the CAR-modified immune cell towards the target cell as well as cytokine release (3).
As of 2021, five CAR-T cell products have been approved by FDA and EMA. With BCMA (B cell maturation antigen), a second target antigen in addition to CD19 has been granted market access (1). Therefore, CAR immunotherapies are now available for the treatment of multiple myeloma and specific B cell malignancies (2). The steadily growing number of clinical CAR-T cell trials enables the treatment of additional tumor indications. All currently approved CAR-T cell products are autologous medications, which require patient-individual manufacturing. This process is complex and requires suitable production facilities, leading to high prices. Additionally, the quality of autologous CAR-T cell products is often affected by the heavy pre-treatment of patients, leading to production failure. Especially in solid tumors, CAR-T cells frequently lack clinical efficacy, caused by cellular exhaustion that can result in therapy failure (4). These factors pose major hurdles for the implementation of future widely applicable CAR therapies. Other drawbacks of CAR-T cell therapies are possibly severe side effects, e.g. cytokine release syndrome (CRS) or neurotoxicities (1). An additional risk when modifying cancer patient’s immune cells is described in a case report: During the manufacturing process of autologous, CD19-specific CAR-T cells, the CAR transgene was mistakenly introduced into a single leukemic B cell resulting in relapse with a B cell lymphoma clone resistant to CAR-T cell therapy (5).
To overcome the above stated limitations and to generate more standardized, cost-effective immunotherapies against cancer, strategies to generate CAR-T cells that are allogeneically applicable are currently developed (6). In parallel, several approaches are explored to use immune cells of healthy, allogeneic donors as a source for immunotherapies. To this end, different immune cell types are tested, especially focusing on Natural Killer (NK) cells, as these immune cells, unlike T cells, do not cause graft versus host disease (GvHD), even if donor and recipient have an HLA mismatch (7).
NK cells pose an interesting alternative to T cells for the generation of CAR equipped cells as an “off the shelf” cell therapy due to their potential to be safe and adoptively transferred from healthy donors to cancer patients. For over twenty years, NK cells have been used as adoptive therapy both in tumors of hematological and solid origin (8, 9). NK cells, collected either from patients or healthy donors, are predominantly activated and expanded ex vivo using cytokines like IL-2, IL-15, IL-12, IL-18, IL-21 or combinations thereof, and then infused to the patient (10–12). Additionally, in some clinical settings, patients were subjected to cytokines like IL-2 after DLI (donor lymphocyte infusion) in order to extend NK cell life-span and cellular activity in vivo. Several studies showed that NK cells can be safely applied in virtually unlimited numbers without the appearance of major side effects (8, 9). In some studies, NK cell infusions to patients recovering after hematopoietic stem cell transplantation showed beneficial effects (13). The overall clinical benefit for patients suffering from tumors of detectable burden, however, is fairly limited in most studies, likely due to poor recognition of tumor cells and quick exhaustion of NK cells in the immuno-suppressive tumor microenvironment (TME) (9, 14).
The great clinical success of CAR-T cells is a stepping stone for the development of next generation NK cell therapies. Using artificial receptors to recognize tumor cells, the limited efficacy of NK cells can be overcome, while retaining the favorable safety properties of NK cells when used as an allogeneic “off-the-shelf” therapy. Therefore, CAR-NK cells have been generated and assessed for efficacy in many pre-clinical and clinical studies, suggesting similar clinical benefits using CAR-NK cells as compared to CAR-T cells (8).
NK and T cells are both lymphocytes with cytotoxic or regulatory functions. When encountering and killing target cells, both exhibit similar mechanisms of action (15). NK cells can induce cell death by activating death receptors like FAS or TRAIL, but the predominant mode of action is the release of perforin and granzyme B to induce cellular apoptosis (16, 17), also utilized in CAR-mediated cytotoxicity.
In T cells, TCR signaling is the only genuine activating signal and an ultimate requirement to trigger cytotoxicity. Additionally, co-stimulatory signals exist that significantly modify the T cell response when triggered simultaneously with the TCR, such as CD28, 4-1BB, OX-40 or NKG2D (18, 19). These costimulatory signals prevent cellular exhaustion, or stimulate persistence, proliferation, differentiation or cytokine secretion, respectively. In consequence, clinically used CAR-T designs consist of a signaling domain assembled from CD3ζ signaling motifs (that mimic TCR signaling) and a single co-stimulatory domain (CD28 in Yescarta® and Tecartus™; 4-1BB in Kymriah®, Breyanzi® and Abecma®) (2). In preclinical studies, additional formats that enhance functionality or safety are being tested, e.g. the combination of several co-stimulatory domains, the effects of artificial, CAR signaling-dependent promoter elements or logic gated CAR expression (20–24).
NK cells encode a vast array of both activating and inhibitory receptors which are, compared to the clonotypic TCR, germline encoded (Figure 1). There are several activating receptors that activate NK cells upon recognition of ligands on target cells, e.g. virus infected cells or tumor cells. Diverse co-stimulatory receptors distinctively guide the NK cell gene expression profile and fine-tune their activation (25). Thereby, the extent of signaling of each individual receptor is taken into account and the multitude of integrated signals determines NK cell behavior (25, 26). On top, the receptor expression of both activating and inhibitory depends on NK cell residency in tissues, is dynamic and surprisingly diverse on a single cell level (27–29). Depending on cellular activation status, exposure to ligands or cytokines, different receptors are being expressed. The fine-tuned interaction of the receptors with inhibitory and activating signals determines whether NK cells initiate cytotoxicity, cytokine secretion or proliferation, or remain tolerant. Molecularly, upon ligand binding, both activating and inhibitory signaling motifs are phosphorylated by Src family kinases (30). Subsequently, activating receptors recruit kinases which trigger phosphorylation cascades when activated, whereas inhibitory receptors recruit and activate phosphatases which counteract activating signaling by removing phosphorylations, thereby in turn interrupting these cascades. Therefore, the balance of kinase and phosphatase activities determines NK cell tolerance and activation.
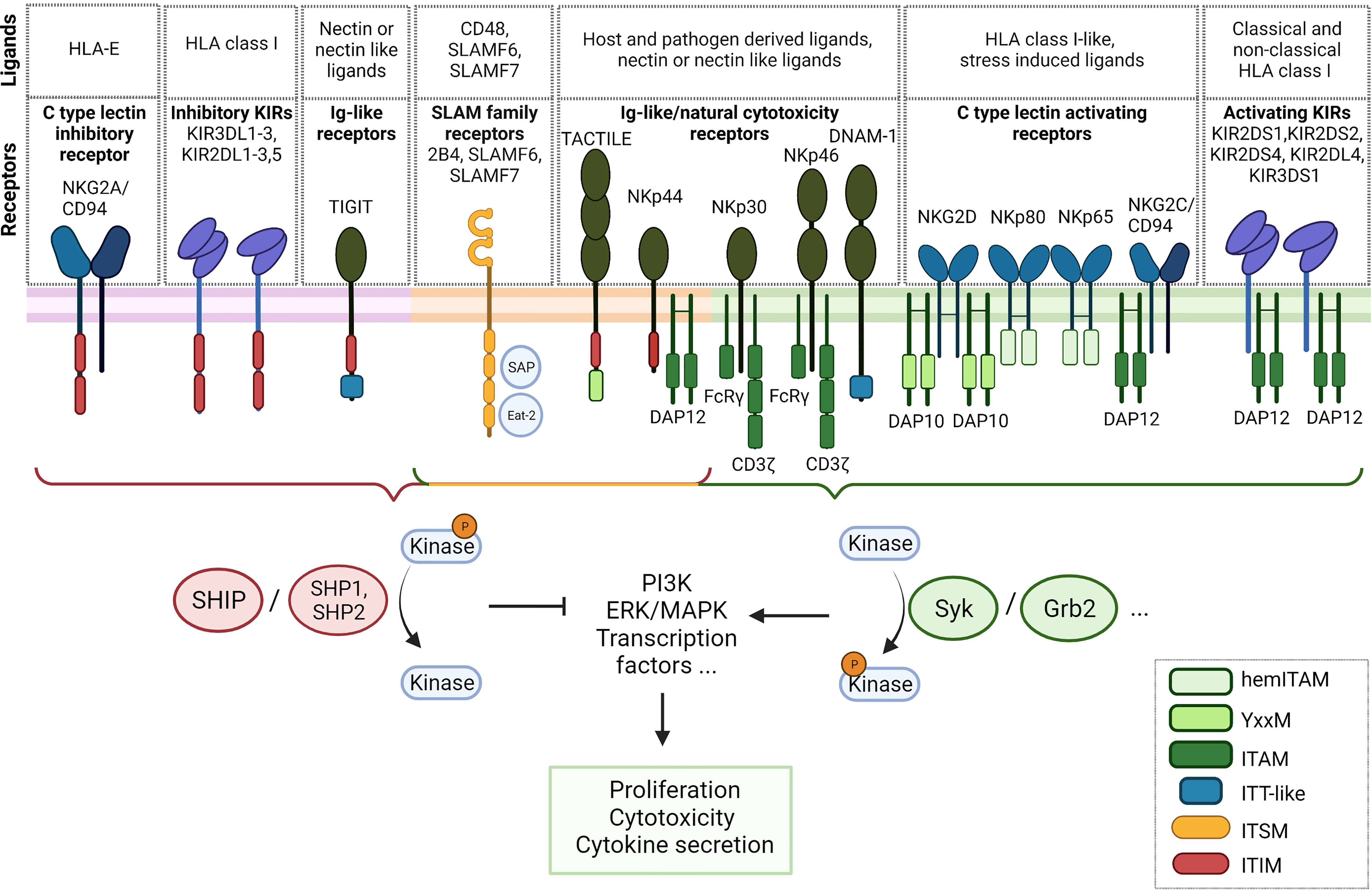
Figure 1 Overview of NK cell receptors and their signaling adapters. Inhibitory receptors are situated on the left (violet membrane), receptors with activating and inhibitory signaling motifs central (orange membrane) and activating receptors on the right (green membrane). ITAM, immunoreceptor tyrosine-based activation motif; hemITAM, hemi-immunoreceptor tyrosine-based activation motif; ITSM, immunoreceptor tyrosine based switch motifs; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITT, immunoglobulin tail tyrosine.
The repertoire of NK cell receptors can be clustered into different families. The killer cell immunoglobulin-like receptors (KIRs) comprise both activating and inhibitory receptors which predominantly bind to HLA-A, -B and -C (26, 31). The nomenclature of KIRs is derived from the length of the cytoplasmic tail. Inhibitory KIRs have a long cytoplasmic tail with an immunoreceptor tyrosine-based inhibitory motif (ITIM) (31). Upon binding to their respective ligands, phosphorylation of the ITIM motif leads to the recruitment of the Src homology (SH) 2 domain-containing protein tyrosine phosphatases SHP-1 and SHP-2 and the inositol phosphatase SHIP. These downstream phosphatases dephosphorylate phosphorylated members of activating signaling cascades. SHIP, e.g., dephosphorylates the second messenger PIP3 within the PI3K pathway, a central signaling pathway that is crucial for NK cell cytotoxicity (31–33). In contrast, activating KIRs generally have a short cytoplasmic domain and lack a signaling domain. However, by means of a positively charged amino acid residue in their transmembrane domain, they recruit the activating signaling adapter DAP12 containing immunoreceptor tyrosine-based activation motifs (ITAM) that leads to the subsequent activation of Syk and PI3K (31, 34). Inhibitory and activating KIRs can share the same ligand, e.g. activating KIR2DS1 and inhibitory KIR2DL1 both bind to HLA-C2 (31). Additionally, some KIRs recognize non-classical HLA class I molecules, such as HLA-G, recognized by KIR2DL4 (35), or HLA-F, recognized by KIR3DS1 (36). Recently, HHLA2, a non-HLA ligand, has been described to interact with the inhibitory receptor KIR3DL3, an interaction suggested as a potential target for immune checkpoint inhibition (37, 38).
Inhibitory KIRs are considered the major mediator to maintain tolerance towards healthy tissues by sensing HLA class I, along with the inhibitory receptor NKG2A (39). NKG2A belongs to the C-type lectin superfamily that also comprises inhibitory and activating receptors (40). NKG2A, as well as its activating counterpart NKG2C, forms heterodimers with CD94 and recognizes the non-classical HLA class I molecule HLA-E. The intracellular tail of NKG2A contains two ITIM motifs to recruit phosphatases that inhibit NK cell signaling. Interestingly, in the sequence of NKG2C, both relevant tyrosine residues are mutated, therefore the ITIM motifs are dysfunctional. Instead, the NKG2C receptor recruits DAP12 to induce an activating signal via Syk (41). The homodimeric activating receptor NKG2D can recruit the DAP10 signaling adapter that contains a YxxM motif which signals via PI3K (42). This receptor recognizes a family of HLA class I-like, stress-induced ligands, named MICA, MICB and ULBP1-6, that frequently appear on the surface of tumor cells, and is critically involved in immune-surveillance (43). Two additional lectin-like NK cell receptors are NKp80, which recognizes AICL, and NKp65 which binds KACL. Both receptors encode a modified ITAM motif with reduced signaling capacity termed hemi-immunoreceptor tyrosine-based activation motif (hemITAM) (44, 45).
A group of activating Ig-like receptors termed natural cytotoxicity receptors (NCR) plays an important role in the recognition of both tumor cells and virus infected cells. The family comprises the three type I transmembrane receptors NKp30, NKp44 and NKp46. NCRs recognize heterogenous host- and pathogen-derived ligands. NKp46 and NKp30 recruit the activating downstream adaptors CD3ζ or FcRγ. Interestingly, different isoforms of the NKp44 have shown to transduce either inhibitory signals via a cytoplasmic ITIM motiv, or activating signals by associating with the downstream adaptor protein DAP12 (46, 47). Of note, NK cells encode multiple so-called paired receptors (48). These receptors are either activating or inhibitory but share the same ligands, usually with the inhibitory receptor possessing higher affinity and therefore being dominant when expressed at the same time. An example for these paired receptors pose the Ig superfamily receptors DNAM-1, TIGIT, CD112R/PVRIG and CD96/TACTILE which bind to tumor associated ligands of the Nectin or Nectin-like (Necl) family. Binding of DNAM-1 to its ligands such as CD155 promotes the cytotoxicity of NK cells against a range of tumor cells, mediated by an immunoglobulin tail tyrosine (ITT)-like motif with downstream Grb2 signaling (49). The inhibitory receptors TIGIT, CD112R and TACTILE act suppressive when binding to the same family of ligands (50). CD112R contains an inhibitory ITIM consensus sequence. TIGIT possesses both an inhibitory ITIM and an ITT-like motif that directly interacts with downstream regulators, e.g. SHIP1 and Grb2 (51, 52). The role of TACTILE remains unclear as it contains both an inhibitory ITIM motif and an YxxM motif that is found on various activating receptors (53, 54).
NK cells have three signaling lymphocytic activation molecule (SLAM) receptors, namely 2B4/SLAMF4, NTB-A/SLAMF6 and CRACC/SLAMF7 (55, 56). These Ig-like receptors play an important role in fine tuning cytotoxic responses of NK cells. Whereas SLAMF4 binds to CD48, SLAMF6 and SLAMF7 bind themselves in a homophilic manner in cis and trans (55, 56). They act activating, upon phosphorylation of the cytoplasmic immunoreceptor tyrosine based switch motifs (ITSMs) and the downstream adaptors SLAM-associated protein (SAP) and Ewing sarcoma-associated transcript (EAT-2). SLAMF6 can additionally act inhibitory upon downstream recruitment of SHP-1, SHP-2 and SHIP1 phosphatases that compete with SAP for the same, phosphorylated ITSMs (55, 57–60).
There are additional NK cell specific receptors like KLRG1 (61) and Siglec family of receptors (62) which are not described here. The repertoire of NK cell receptors is very complex and few synergistic effects between receptors have been described (63–65). However, research on (novel) receptors, ligands and functional mechanisms to better understand NK biology, but also their potential for immunotherapy, is still ongoing. The balance of activation or inhibition of NK cells via phosphorylation by kinases and dephosphorylation by phosphatases is a fine-tuned process with some variables still under investigation (26, 66–68). Therefore, the exact transcriptional changes induced in NK cells when encountering a target cell are barely predictable using our current knowledge about potential additive, synergistic or counteracting signaling pathways. A systematic assessment of the signaling pathways with a particular focus on the interplay of activating signals is still lacking, and CAR technology may provide a suitable means of addressing such open questions in NK cell biology.
Although the cell biology of T and NK cells deviates dramatically in terms of signaling acquisition and integration, the development of CAR-NK cells follows frequently within the footsteps of CAR-T cells, and T cell technologies are simply adopted for NK cells. Accordingly, in the first published clinical trial of CAR-NK cells, the CAR signaling domains are derived from the TCR and 4-1BB like used for CAR-T cells. These T cell based CAR constructs showed promising efficiency against B cell malignancies (69). The large repertoire of NK cell activating receptors and adaptor proteins, however, provides a pool of signaling domains that might improve CAR signaling responses for NK cells (Figure 2).
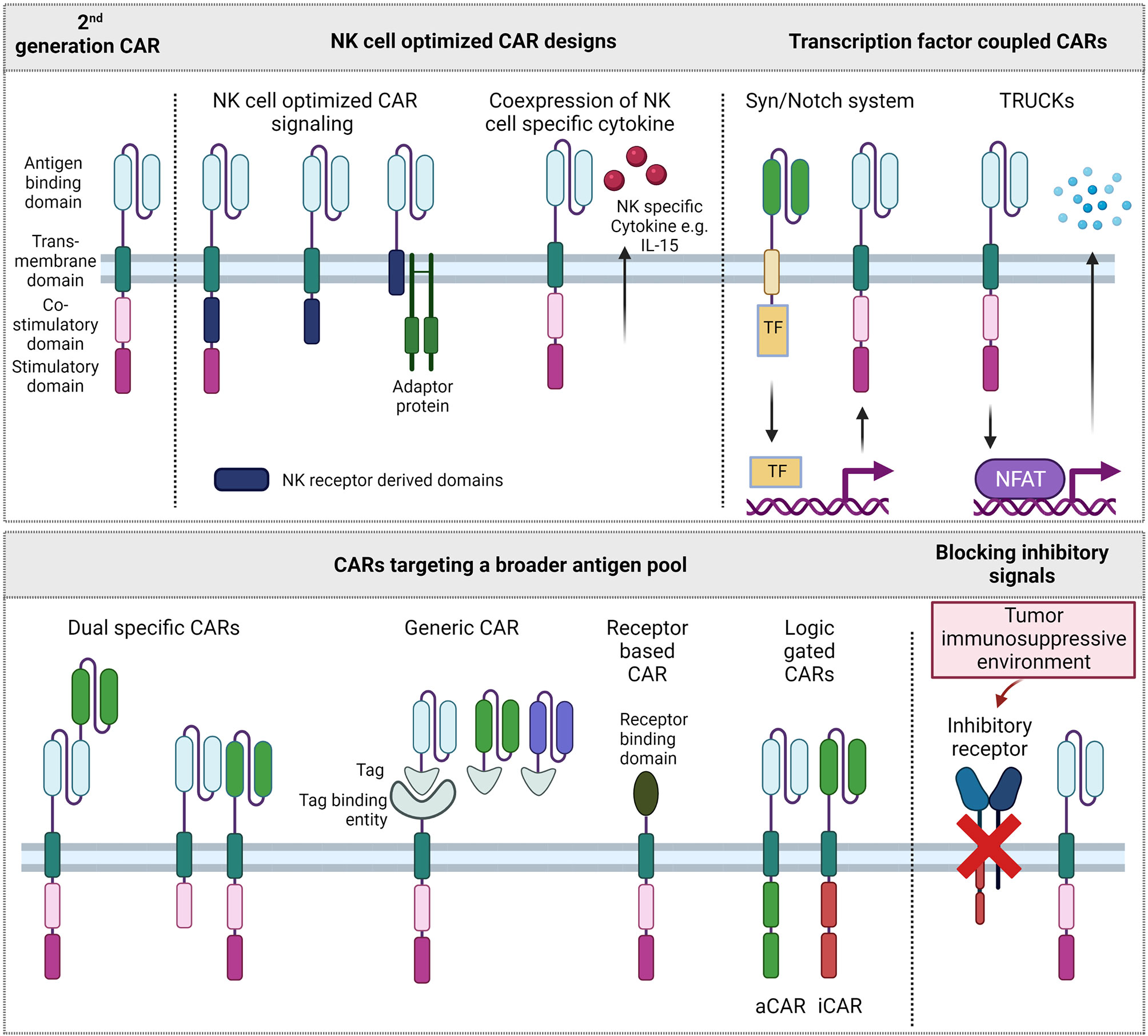
Figure 2 Different CAR formats. Upper left: 2nd generation CAR format that is approved on the market for T cells. Other CAR designs improve signaling for NK cells, couple CAR expression and activation to transcription factor systems, target a broader pool of antigens or aim to overcome inhibitory signals of the tumor immunosuppressive environment. TF, Transcription factor; aCAR, activating CAR; iCAR, inhibitory CAR.
Among the many NK cell activating receptor and adaptor protein domains that could be explored as CAR signaling domains, the signaling domains of the activating receptor 2B4, and the adaptor proteins DAP10 and DAP12 have shown to induce superior cytotoxic activity of CAR-NK cells towards their respective targets. Two early studies explored the CAR constructs with a DAP12 derived signaling domain in the NK cell line YTS and showed potent cytotoxicity towards target cells (70, 71). In a study targeting Mesothelin, combinations of costimulatory domains upstream of a CD3ζ domain were screened. Notably, in addition to intracellular signaling parts, NK cell receptor derived transmembrane domains CD16, NKp46, NKp44 and NKG2D were included in the screening. As transmembrane domains of several activating NK cell receptors, like KIR2DS1, NKp46, NKG2D, possess a charged amino acid in their transmembrane region capable of directly interacting with downstream signaling adapters (72–74), CAR constructs including an NKG2D transmembrane domain as well as DAP10 and 2B4 costimulatory domain were identified as the most promising candidate to increase CAR-NK cell mediated cytotoxicity (75). A recent study explored DNAM1- and 2B4-derived costimulatory domains in CAR constructs in a combination with CD3ζ for NK-92 cells, a NK cell line with cytotoxic abilities, showing that the NK cell-specific costimulatory signal increased the persistency, proliferation and cytotoxicity of CAR-NK92 cells compared to CD3ζ alone or combined with CD28 signaling (76).
In an alternative approach, the design of the CAR construct is solely based on the NK cell specific receptor KIR2DS4 as transmembrane and intracellular domain, without a fused signaling adapter (77). Instead, KIR2DS4 adaptor protein DAP12 is co-expressed with the CAR, separated by a P2A site. Although DAP12 in this case is expressed independent of CAR target binding, the CAR-NK cells show a specific cytotoxicity against target tumor cells. The CAR in this study is directed against HLA-G, an antigen that is expressed on numerous solid tumors but restricted on healthy tissues, mostly in immune-privileged sites (77). Generally, finding suitable antigen targets for CAR therapies is crucial as targets need to be specific for the tumor while not being expressed on healthy tissue. Multiple novel targets are tested for CAR therapies, mostly on T cells, results however might be applied to NK cells (78). As single and tumor specific targets are hard to find, binding domains of receptors targeting ligands on diseased cells can be used to target a broader spectrum of antigens. Therefore, some CAR constructs exploit and enhance the natural tumor recognition of NK cells. NKG2D can bind up to eight stress induced ligands that are frequently upregulated in tumor cells and less expressed on healthy tissues. NKG2D-based CAR-NK cells have been shown effective against multiple myeloma (MM) cells in a preclinical setting (79), in a clinical study with colorectal cancer patients (80) and in various other studies using a NKG2D construct in T cells. Similarly, multiple tumors were targeted with NK cell receptor based NKp44 or NKp46 -CAR in T cells (81, 82).
By using two or more antigen directed scFvs within CAR cells, the spectrum of targets can be broadened further. These tandem or bispecific CARs combine two scFv domains within the same CAR binding domain such as in two phase I CAR-T cell studies targeting CD19 and CD22, CD19 and CD20 or BCMA and CD38. Targeting more than one antigen might thereby reduce the risk of antigen negative relapse (83–85). Alternatively, two CARs targeting different antigens can be coexpressed in the same cell. This approach was followed in a CAR-T cell study targeting CD19 and CD123 and prevented antigen negative relapses in xenograft models (86). The intracellular costimulatory and stimulatory domains of the two individually expressed CARs can either be identical or different to each other. Given the large variety of NK cell activating receptors and adaptor proteins, only few combinations of intracellular signaling adapters or domains were tested as CAR-NK signaling domains. There might be a hidden potential in assessing synergistic but also inhibiting effects. This potential has been recently tapped in NK cells by developing a logic-gated approach of an inhibitory CAR (iCAR) which will inhibit the cytotoxic signaling of an activating CAR (aCAR) upon target binding. In this approach, healthy tissue is shielded while more antigens can be explored on tumor cells (87).
In a modular approach, CAR modified cells are directed against multiple and or varying targets. An antigen targeting element, mostly scFv derived, is fused to a tag or another CAR binding property. The CAR consists of a tag or adaptor binding region and the usual signaling module. Although mostly applied in the T cell setting (88, 89), this setting has been successfully translated to NK cells (90) and, in terms of targets, this approach is rather flexible and multiple targets can be addressed at once.
A special interest should be taken to T cell malignancies with currently poor clinical outcomes. As in B cell settings, T cells express unique targets which cannot be addressed with the common autologous CAR-T cell setting due to self-targeting leading to fratricide (91). NK cells can overcome this limitation as they are negative for the two mostly used T cell markers CD3 and CD4. In a preclinical study, anti CD3-CAR-NK-92 cells significantly prolonged survival of mice challenged with Jurkat cells line (92).
CAR therapies are restricted to surface antigens. In contrast, T cell receptor (TCR) recognizes degraded intracellular proteins presented by MHC. In TCR based cell therapies, effector cells express an artificially designed high-affinity TCR (93). Although TCR-based cell therapies are also mostly explored on the T cell platform, the feasibility of TCR NK cells in the NK-92 model was previously shown (94, 95).
Besides addressing more targets, CAR design can also focus on enhancing immune responses of NK cells. A limitation of NK cell therapies is the short lifetime and persistence in vivo (96). The integration of autocrine growth factor IL-15 as a downstream IL-15 cassette has shown increased life span and persistence in the first published clinical CAR-NK study (69, 97). An inducible MyD88/CD40 protein switch in CAR-NK cells, which lead to increased cytokine secretion upon activation and further synergistic effects with transgenic IL-15 was previously reported. A different approach to activate NK cells with IL-15 is taken in the clinical trial NKX101 where a membrane bound IL-15 is tested to increase persistence of NKG2D-OX40-CD3ζ CAR-NK cells (97). To safeguard from potential toxicity of engineered CAR-NK cells, an inducible caspase was included in addition to the CAR in several studies (69, 98).
Especially in solid tumors, the TME is immunosuppressive, due to limited supply of nutrients, decreased levels of oxygen and an accumulation of inhibitory molecules and cells. Overcoming but also exploiting and redirecting this suppressive environment is key to combating solid malignancies. Based on the success of checkpoint inhibitor therapies, making CAR immune cells more resistant to inhibitory signals is also strived for. A reduced surface expression of the immune checkpoint receptor NKG2A showed increased cytotoxicity against HLA-E-expressing tumor cells (99). The blockage of another immune checkpoint in NK cells, TIGIT, prevented NK cell exhaustion (99, 100). Another negative regulator of IL-15 signaling is the cytokine-inducible Src homology 2–containing (CIS) protein. Genetic knockout of its gene, CISH, via CRISPR-Cas9 showed enhanced fitness of NK cells that additionally expressed IL-15 and a CD19 specific CAR (101). Blocking of inhibitory cytokine sensing receptors such as the of the TGF-β receptor TGF-βR2 has increased the resistance of NK cells under TGF-β inhibitory conditions (102). In a different approach, the immune inhibition is reversed by fusing the exodomain of inhibitory cytokine sensing receptors to an activating endodomain or CAR signaling domain. These chimeric cytokine receptors rather enhance than inhibit the antitumoral response of effector cells within an inhibitory tumor microenvironment (103, 104).
Immune response modulating capacities are further exploited in T cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs) (105). In the often so called 4th generation of CAR-T cells, CARs are modified to express transgenic cytokines upon CAR stimulation: Antigen binding and resulting downstream phosphorylation cascades via the intracellular CAR signaling domain also phosphorylate NFAT transcription factor which can subsequently bind to NFAT response elements. In TRUCK design, stimulatory cytokines such as IL-7, IL-12, IL-15, IL-18, IL-23 are genetically introduced after NFAT response elements and their induced expression at the tumor site promotes on site effect, e.g. activating other immune cells (105, 106). Another transcription factor driven system is the SynNotch System. In a CAR setting this system can be applied to as in TRUCKs to induce expression of cytokines like IL-12 (107). However, the expression of various other (effector) proteins is imaginable and currently explored, possibly overcoming some issues of finding tumor specific antigens. A system in which a SynNotch receptor first specifically binds to neoantigens EGFRvIII which induces the expression of tandem α-EphA2/IL13Rα2 CAR was previously described. This system shows high antitumor activity with restricted off-target effects and the opportunity to target cancer-associated but not completely tumor specific antigens (22).
In summary, there are many recent innovations in CAR design, with emphasis to broaden the pool of addressable antigens, and to enhance safety or efficiency of CAR cells. Many methods initially developed in the CAR-T cell context are now utilized in NK cells. However, to further enhance CAR-NK cell tumor cell killing and in vivo persistence, the latest CAR-based technologies developed in the T cell context (e.g. TRUCKS) need to be translated to CAR-NK cells as well, and by acknowledging NK cell characteristics in terms of activation, or by selecting the most suitable cytokines for overexpression, the chances are best to succeed. The application and development of CAR innovations on the NK cell platform is still a large playground with a lot of potential to overcome the limitations of the CAR-T cell market.
Signaling enhancement and tailored adaptation of the CAR construct for NK cells constitutes just one step to success for NK-cell based immunotherapies. Deciphering critical parameters that affect in vivo persistence and anti-cancer efficacy of NK cells beyond the CAR-mediated effects will be critical for a clinical “off-the-shelf” product, and additional genetic modifications or combination therapies with chemo-, radio-, or immune checkpoint inhibitor therapies may be required to yield optimal results of immunotherapeutic approaches. On top, beyond biological properties and in vivo behavior of engineered NK cells, there are several additional technological hurdles to overcome to get NK cell technologies market-ready.
Currently, several sources for NK cells are under investigation. NK cells collected from peripheral blood (PB-NK) are easily accessible from healthy donors, but show high donor variation in terms of expansion rate, transgene-expression and efficacy, and possess overall a heterogenous and mature phenotype following expansion. NK cells derived from cord blood (CB-NK) are less mature before and after expansion, easier to engineer and easily accessible as well (108). Additionally, NK cells can be differentiated from hematopoietic stem cells (HSCs) (109, 110), induced pluripotent stem cells (iPSCs) (75) as well as from other immune cells using direct reprogramming (drNK) within a few days (111). These cells are more difficult to manufacture in research facilities due to the added complexity of differentiating NK cells in a first step, however, stem cells pose a virtually unlimited source of homogenous, highly active NK cell populations. However, if signaling and immune-suppressive factors of the TME affect NK cells of all sources in the same manner is still an open question, as most studies utilize just a single source of cells, with few exceptions (111), and side-by-side comparisons, especially under challenging conditions, are scarce. Possibly, genetic engineering has to be adjusted both to source and the anticipated tumor environment that needs to be overcome, therefore, differential CAR signaling, metabolic reprogramming or deletion of different inhibitory receptors need to be functionally assessed and predictive markers for NK cell in vivo efficacy established. On top, it is yet unclear if the heterogeneous, blood-derived NK cell products may perform better in patients compared to uniform, homogeneous stem cell-derived NK cells as they may react more plastic to immune suppression and possibly elicit a more varied anti-tumor response (112). Next to primary NK cells, also the cell line NK-92 is under clinical investigation (113).
Next to biological obstacles, technical hurdles still exist. NK cells are considered hard-to-engineer and hard-to-expand compared to T cells. Recently, lentiviral transductions were significantly enhanced by introducing new transduction enhancers that facilitate viral entry (114) or suppress anti-viral cellular signaling (115). On top, a novel viral envelope derived from the baboon endogenous virus (BaEV) showed superior efficacy as compared to other lentiviral envelope proteins to successfully gene-edit NK cells (116, 117) that outperforms also alpha-retroviruses previously tested (118). There is a high donor-to-donor-variation in terms of expansion rates, transduction efficiency and in vitro anti-tumor efficacy, however, thinking of an “off-the-shelf” CAR-NK cell therapy, a thorough donor-selection for the most suitable NK cell starting material is likely to be performed. Transposon-based DNA delivery systems are currently assessed in clinical trials for the generation of CAR-T cells as an alternative approach for long-term transgene expression (119–122). Although the occurrence of T cell lymphomas in few patients using PiggyBac transposon system mediated gene transfer poses a setback for this technology (123, 124), the transposon technology will likely be further developed and be assessed for the generation of CAR-NK cells.
To expand NK cells efficiently, feeder cell-free and feeder cell-based protocols were established. On the one hand, feeder cell-based approaches, mostly performed with genetically optimized K562 feeder cells, show considerably superior expansion rates and were successfully used for the generation of CAR-NK cells used in the only clinical phase I trial published (69, 125). On the other hand, feeder cell-free systems are still considered and widely favored for their anticipated ease in the regulatory approval processes. Additionally, feeder free processes can be performed in existing, closed (semi-)automated production lines without major adaptations required for feeder cell line cultivation and irradiation (126–129).
Lastly, storage of an NK cell based cell product is essential to serve as an “off-the-shelf” therapy which is produced in large batches for multiple patients. However, NK cells are very sensitive to cryo-induced damages. Although cytokine activated cells exhibit higher survival rates and cytotoxic capabilities than unactivated NK cells, still, important parameters that determine efficacy in vivo, like proliferation or migration (130), are majorly impaired following a freeze-thaw cycle. Importantly, such parameters are frequently not addressed in quality control protocols to assess quality of NK cell products (131), therefore, reasons for potential treatment failures may also be due to undetected, limited NK cell fitness. Efficient cryopreservation protocols, preferentially without the use of toxic cryoprotectant DMSO, are still under investigation (132), and need to ensure long-term product stability, safe means of transport, and enable direct infusion of the cell product into patients from cryobags.
It is widely accepted that allogeneic products will be the next step in the development of anti-cancer cell therapies, as they promise high quality standards, immediate availability, significant cost reductions and a reduced utilization of manufacturing capacities.
However, CAR-NK cell based therapies need to prove their abilities in additional clinical trials so as to show that they are at least as effective as CAR-T cell therapies which currently monopolize the market of CAR therapy. Postulated advantages of NK cells, like innate anti-cancer immunity to possibly overcome antigen escape and cancer heterogeneity (133) or a different cytokine profile that may direct other immune cells like dendritic cells (134) into the tumor site, need to prove clinical relevance. The limited efficacy in the treatment of solid tumors observed in CAR-T cells in vivo may pose a similar challenge for CAR-NK cells as studies on allogeneic, activated NK cells imply that they face cellular exhaustion and poor infiltration into tumor sites (14, 135–137). To overcome immune suppression in the TME, additional arming of immune cells through genetic engineering is pushed for, e.g., by genetic ablations (138, 139) or metabolic reprogramming of NK cells (140, 141).
The generation and clinical administration of CAR-T cells is widely performed, and clinical manufacturing pipelines are established and approved. In contrast, technologies required for the optimization of cellular behavior and manufacturing processes of engineered NK cells still need refinement.
Both NK cell and T cell therapies require further research to overcome their respective limitations and to turn them into efficient and affordable living anti-cancer drugs applicable beyond B cell malignancies. Therefore, the question if T cells or NK cells are to become the dominating platform for allogeneic, “off-the-shelf” CAR cell therapies remains unanswered.
KR and DS outlined, wrote, and referenced the manuscript. KR prepared the figures. SF and UK carefully edited the work. All authors contributed to the article and approved the submitted version.
This work was supported by the Fraunhofer Internal Programs under Grant No. Attract 131-600004 (to DS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figures were generated using BioRender.com.
1. Fischer JW, Bhattarai N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front Immunol (2021) 12:693016. doi: 10.3389/fimmu.2021.693016
2. Vucinic V, Quaiser A, Lückemeier P, Fricke S, Platzbecker U, Koehl U. Production and Application of CAR T Cells: Current and Future Role of Europe. Front Med (2021) 8:713401. doi: 10.3389/fmed.2021.713401
3. Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev (2019) 12:145–56. doi: 10.1016/j.omtm.2018.12.009
4. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res Ther (2021) 12:81. doi: 10.1186/s13287-020-02128-1
5. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat Med (2018) 24:1499–503. doi: 10.1038/s41591-018-0201-9
6. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. “Off-The-Shelf” Allogeneic CAR T Cells: Development and Challenges. Nat Rev Drug Discovery (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
7. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295:2097–100. doi: 10.1126/science.1068440
8. Myers JA, Miller JS. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat Rev Clin Oncol (2021) 18:85–100. doi: 10.1038/s41571-020-0426-7
9. Geller MA, Miller JS. Use of Allogeneic NK Cells for Cancer Immunotherapy. Immunotherapy (2011) 3:1445–59. doi: 10.2217/imt.11.131
10. Granzin M, Wagner J, Köhl U, Cerwenka A, Huppert V, Ullrich E. Shaping of Natural Killer Cell Antitumor Activity by Cultivation. Front Immunol (2017) 8:458. doi: 10.3389/fimmu.2017.00458
11. Brehm C, Huenecke S, Quaiser A, Esser R, Bremm M, Kloess S, et al. IL-2 Stimulated But Not Unstimulated NK Cells Induce Selective Disappearance of Peripheral Blood Cells: Concomitant Results to a Phase I/II Study. PloS One (2011) 6:e27351. doi: 10.1371/journal.pone.0027351
12. Koehl U, Kalberer C, Spanholtz J, Lee DA, Miller JS, Cooley S, et al. Advances in Clinical NK Cell Studies: Donor Selection, Manufacturing and Quality Control. Oncoimmunology (2016) 5:e1115178. doi: 10.1080/2162402X.2015.1115178
13. Sivori S, Meazza R, Quintarelli C, Carlomagno S, Della Chiesa M, Falco M, et al. NK Cell-Based Immunotherapy for Hematological Malignancies. J Clin Med Res (2019) 8:e84. doi: 10.3390/jcm8101702
14. Merino AM, Kim H, Miller JS, Cichocki F. Unraveling Exhaustion in Adaptive and Conventional NK Cells. J Leukoc Biol (2020) 108:1361–8. doi: 10.1002/JLB.4MR0620-091R
15. Chiang SCC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of Primary Human Cytotoxic T-Cell and Natural Killer Cell Responses Reveal Similar Molecular Requirements for Lytic Granule Exocytosis But Differences in Cytokine Production. Blood (2013) 121:1345–56. doi: 10.1182/blood-2012-07-442558
16. Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK Cells Switch From Granzyme B to Death Receptor-Mediated Cytotoxicity During Serial Killing. J Exp Med (2019) 216:2113–27. doi: 10.1084/jem.20181454
17. Prager I, Watzl C. Mechanisms of Natural Killer Cell-Mediated Cellular Cytotoxicity. J Leukoc Biol (2019) 105:1319–29. doi: 10.1002/JLB.MR0718-269R
18. Mascarelli DE, Rosa RSM, Toscaro JM, Semionatto IF, Ruas LP, Fogagnolo CT, et al. Boosting Antitumor Response by Costimulatory Strategies Driven to 4-1BB and OX40 T-Cell Receptors. Front Cell Dev Biol (2021) 9:692982. doi: 10.3389/fcell.2021.692982
19. Perez C, Prajapati K, Burke B, Plaza-Rojas L, Zeleznik-Le NJ, Guevara-Patino JA. NKG2D Signaling Certifies Effector CD8 T Cells for Memory Formation. J Immunother Cancer (2019) 7:48. doi: 10.1186/s40425-019-0531-2
20. Chmielewski M, Abken H. TRUCKS, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv Cell Gene Ther (2020) 3. doi: 10.1002/acg2.84
21. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell (2019) 35:489–503.e8. doi: 10.1016/j.ccell.2019.02.003
22. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T Cells Overcome Challenges of Specificity, Heterogeneity, and Persistence in Treating Glioblastoma. Sci Transl Med (2021) 13(591):eabe7378. doi: 10.1126/scitranslmed.abe7378
23. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR Circuits Enhance Solid Tumor Recognition and Promote Persistent Antitumor Activity in Mouse Models. Sci Transl Med (2021) 13(591):eabd8836. doi: 10.1126/scitranslmed.abd8836
24. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and Characterization of an “All-In-One” Lentiviral Vector System Combining Constitutive Anti-G CAR Expression and Inducible Cytokines. Cancers (2020) 12:375. doi: 10.3390/cancers12020375
25. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell Mol Immunol (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
26. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu Rev Immunol (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005
27. Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell (2020) 180:749–763.e13. doi: 10.1016/j.cell.2020.01.022
28. Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity (2018) 49:971–986.e5. doi: 10.1016/j.immuni.2018.09.009
29. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
30. Rajasekaran K, Riese MJ, Rao S, Wang L, Thakar MS, Sentman CL, et al. Signaling in Effector Lymphocytes: Insights Toward Safer Immunotherapy. Front Immunol (2016) 7:176. doi: 10.3389/fimmu.2016.00176
31. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front Immunol (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
32. Mace EM. Phosphoinositide-3-Kinase Signaling in Human Natural Killer Cells: New Insights From Primary Immunodeficiency. Front Immunol (2018) 9:445. doi: 10.3389/fimmu.2018.00445
33. Dempke WCM, Uciechowski P, Fenchel K, Chevassut T. Targeting SHP-1, 2 and SHIP Pathways: A Novel Strategy for Cancer Treatment? Oncology (2018) 95:257–69. doi: 10.1159/000490106
34. Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 Bearing a Tyrosine-Based Activation Motif is Involved in Activating NK Cells. Nature (1998) 391:703–7. doi: 10.1038/35642
35. Rajagopalan S, Long EO. KIR2DL4 (CD158d): An Activation Receptor for HLA-G. Front Immunol (2012) 3:258. doi: 10.3389/fimmu.2012.00258
36. Garcia-Beltran WF, Hölzemer A, Martrus G, Chung AW, Pacheco Y, Simoneau CR, et al. Open Conformers of HLA-F Are High-Affinity Ligands of the Activating NK-Cell Receptor KIR3DS1. Nat Immunol (2016) 17:1067–74. doi: 10.1038/ni.3513
37. Bhatt RS, Berjis A, Konge JC, Mahoney KM, Klee AN, Freeman SS, et al. KIR3DL3 Is an Inhibitory Receptor for HHLA2 That Mediates an Alternative Immunoinhibitory Pathway to PD1. Cancer Immunol Res (2021) 9:156–69. doi: 10.1158/2326-6066.CIR-20-0315
38. Wei Y, Ren X, Galbo PM Jr, Moerdler S, Wang H, Sica RA, et al. KIR3DL3-HHLA2 Is a Human Immunosuppressive Pathway and a Therapeutic Target. Sci Immunol (2021) 6(61):eabf9792. doi: 10.1126/sciimmunol.abf9792
39. Orr MT, Lanier LL. Natural Killer Cell Education and Tolerance. Cell (2010) 142:847–56. doi: 10.1016/j.cell.2010.08.031
40. Brown GD, Willment JA, Whitehead L. C-Type Lectins in Immunity and Homeostasis. Nat Rev Immunol (2018) 18:374–89. doi: 10.1038/s41577-018-0004-8
41. Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 With Activating CD94/NKG2C NK Cell Receptors. Immunity (1998) 8:693–701. doi: 10.1016/s1074-7613(00)80574-9
42. Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 Triggers Human NK Cell-Mediated Killing via a Syk-Independent Regulatory Pathway. Nat Immunol (2003) 4:557–64. doi: 10.1038/ni929
43. Schmiedel D, Mandelboim O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front Immunol (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
44. Rückrich T, Steinle A. Attenuated Natural Killer (NK) Cell Activation Through C-Type Lectin-Like Receptor NKp80 Is Due to an Anomalous Hemi-Immunoreceptor Tyrosine-Based Activation Motif (HemITAM) With Impaired Syk Kinase Recruitment Capacity. J Biol Chem (2013) 288:17725–33. doi: 10.1074/jbc.M113.453548
45. Bauer B, Wotapek T, Zöller T, Rutkowski E, Steinle A. The Activating C-Type Lectin-Like Receptor NKp65 Signals Through a Hemi-Immunoreceptor Tyrosine-Based Activation Motif (hemITAM) and Spleen Tyrosine Kinase (Syk). J Biol Chem (2017) 292:3213–23. doi: 10.1074/jbc.M116.759977
46. Hecht M-L, Rosental B, Horlacher T, Hershkovitz O, De Paz JL, Noti C, et al. Natural Cytotoxicity Receptors NKp30, NKp44 and NKp46 Bind to Different Heparan Sulfate/Heparin Sequences. J Proteome Res (2009) 8:712–20. doi: 10.1021/pr800747c
47. Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol (2019) 10:909. doi: 10.3389/fimmu.2019.00909
48. Martinet L, Smyth MJ. Balancing Natural Killer Cell Activation Through Paired Receptors. Nat Rev Immunol (2015) 15:243–54. doi: 10.1038/nri3799
49. Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM-1 Controls NK Cell Activation via an ITT-Like Motif. J Exp Med (2015) 212:2165–82. doi: 10.1084/jem.20150792
50. Jin H-S, Park Y. Hitting the Complexity of the TIGIT-CD96-CD112R-CD226 Axis for Next-Generation Cancer Immunotherapy. BMB Rep (2021) 54:2–11. doi: 10.5483/bmbrep.2021.54.1.229
51. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-Like Motif of TIGIT Suppresses Granule Polarization and Cytotoxicity of NK Cells. Cell Death Differ (2013) 20:456–64. doi: 10.1038/cdd.2012.141
52. Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E. SHP-1-Mediated Inhibitory Signals Promote Responsiveness and Anti-Tumour Functions of Natural Killer Cells. Nat Commun (2014) 5:5108. doi: 10.1038/ncomms6108
53. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers (2019) 11:877. doi: 10.3390/cancers11060877
54. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The Receptors CD96 and CD226 Oppose Each Other in the Regulation of Natural Killer Cell Functions. Nat Immunol (2014) 15:431–8. doi: 10.1038/ni.2850
55. Claus M, Meinke S, Bhat R, Watzl C. Regulation of NK Cell Activity by 2B4, NTB-A and CRACC. Front Biosci (2008) 13:956–65. doi: 10.2741/2735
56. Claus M, Urlaub D, Fasbender F, Watzl C. SLAM Family Receptors in Natural Killer Cells – Mediators of Adhesion, Activation and Inhibition via Cis and Trans Interactions. Clin Immunol (2019) 204:37–42. doi: 10.1016/j.clim.2018.10.011
57. Eissmann P, Beauchamp L, Wooters J, Tilton JC, Long EO, Watzl C. Molecular Basis for Positive and Negative Signaling by the Natural Killer Cell Receptor 2B4 (CD244). Blood (2005) 105:4722–9. doi: 10.1182/blood-2004-09-3796
58. Tassi I, Colonna M. The Cytotoxicity Receptor CRACC (CS-1) Recruits EAT-2 and Activates the PI3K and Phospholipase Cgamma Signaling Pathways in Human NK Cells. J Immunol (2005) 175:7996–8002. doi: 10.4049/jimmunol.175.12.7996
59. Cruz-Munoz M-E, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM Family Receptor Coupled to the Adaptor EAT-2, on Natural Killer Cell Function. Nat Immunol (2009) 10:297–305. doi: 10.1038/ni.1693
60. Meinke S, Watzl C. NK Cell Cytotoxicity Mediated by 2B4 and NTB-A Is Dependent on SAP Acting Downstream of Receptor Phosphorylation. Front Immunol (2013) 4:3. doi: 10.3389/fimmu.2013.00003
61. Müller-Durovic B, Lanna A, Covre LP, Mills RS, Henson SM, Akbar AN. Killer Cell Lectin-Like Receptor G1 Inhibits NK Cell Function Through Activation of Adenosine 5’-Monophosphate-Activated Protein Kinase. J Immunol (2016) 197:2891–9. doi: 10.4049/jimmunol.1600590
62. Daly J, Carlsten M, O’Dwyer M. Sugar Free: Novel Immunotherapeutic Approaches Targeting Siglecs and Sialic Acids to Enhance Natural Killer Cell Cytotoxicity Against Cancer. Front Immunol (2019) 10:1047. doi: 10.3389/fimmu.2019.01047
63. Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy Among Receptors on Resting NK Cells for the Activation of Natural Cytotoxicity and Cytokine Secretion. Blood (2006) 107:159–66. doi: 10.1182/blood-2005-04-1351
64. Zamai L, Del Zotto G, Buccella F, Gabrielli S, Canonico B, Artico M, et al. Understanding the Synergy of NKp46 and Co-Activating Signals in Various NK Cell Subpopulations: Paving the Way for More Successful NK-Cell-Based Immunotherapy. Cells (2020) 9:877. doi: 10.3390/cells9030753
65. Kim HS, Long EO. Complementary Phosphorylation Sites in the Adaptor Protein SLP-76 Promote Synergistic Activation of Natural Killer Cells. Sci Signal (2012) 5:ra49. doi: 10.1126/scisignal.2002754
66. Chen Y, Lu D, Churov A, Fu R. Research Progress on NK Cell Receptors and Their Signaling Pathways. Mediators Inflammation (2020) 2020:6437057. doi: 10.1155/2020/6437057
67. Kwon H-J, Kim N, Kim HS. Molecular Checkpoints Controlling Natural Killer Cell Activation and Their Modulation for Cancer Immunotherapy. Exp Mol Med (2017) 49:e311. doi: 10.1038/emm.2017.42
68. Paul S, Lal G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front Immunol (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
69. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
70. Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J Immunol (2015) 194:3201–12. doi: 10.4049/jimmunol.1400330
71. Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK Cells Modified With an EGFRvIII-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-Secreting Glioblastoma. J Immunother (2015) 38:197–210. doi: 10.1097/CJI.0000000000000082
72. Chapel A, Garcia-Beltran WF, Hölzemer A, Ziegler M, Lunemann S, Martrus G, et al. Peptide-Specific Engagement of the Activating NK Cell Receptor KIR2DS1. Sci Rep (2017) 7:2414. doi: 10.1038/s41598-017-02449-x
73. Westgaard IH, Berg SF, Vaage JT, Wang LL, Yokoyama WM, Dissen E, et al. Rat NKp46 Activates Natural Killer Cell Cytotoxicity and is Associated With FcepsilonRIgamma and CD3zeta. J Leukoc Biol (2004) 76:1200–6. doi: 10.1189/jlb.0903428
74. Garrity D, Call ME, Feng J, Wucherpfennig KW. The Activating NKG2D Receptor Assembles in the Membrane With Two Signaling Dimers Into a Hexameric Structure. Proc Natl Acad Sci USA (2005) 102:7641–6. doi: 10.1073/pnas.0502439102
75. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell (2018) 23:181–192.e5. doi: 10.1016/j.stem.2018.06.002
76. Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 Costimulatory Domains Enhance the Cytotoxicity of Anti-GPC3 Chimeric Antigen Receptor-Modified Natural Killer Cells Against Hepatocellular Cancer Cells In Vitro. Cancer Manag Res (2020) 12:3247–55. doi: 10.2147/CMAR.S253565
77. Jan C-I, Huang S-W, Canoll P, Bruce JN, Lin Y-C, Pan C-M, et al. Targeting Human Leukocyte Antigen G With Chimeric Antigen Receptors of Natural Killer Cells Convert Immunosuppression to Ablate Solid Tumors. J Immunother Cancer (2021) 9:e003050. doi: 10.1136/jitc-2021-003050
78. Liu B, Yan L, Zhou M. Target Selection of CAR T Cell Therapy in Accordance With the TME for Solid Tumors. Am J Cancer Res (2019) 9:228–41.
79. Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. NKG2D-CAR-Transduced Natural Killer Cells Efficiently Target Multiple Myeloma. Blood Cancer J (2021) 11:146. doi: 10.1038/s41408-021-00537-w
80. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
81. Eisenberg V, Shamalov K, Meir S, Hoogi S, Sarkar R, Pinker S, et al. Targeting Multiple Tumors Using T-Cells Engineered to Express a Natural Cytotoxicity Receptor 2-Based Chimeric Receptor. Front Immunol (2017) 8:1212. doi: 10.3389/fimmu.2017.01212
82. Tal Y, Yaakobi S, Horovitz-Fried M, Safyon E, Rosental B, Porgador A, et al. An NCR1-Based Chimeric Receptor Endows T-Cells With Multiple Anti-Tumor Specificities. Oncotarget (2014) 5:10949–58. doi: 10.18632/oncotarget.1919
83. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S, et al. CAR T Cells With Dual Targeting of CD19 and CD22 in Pediatric and Young Adult Patients With Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia: A Phase 1 Trial. Nat Med (2021) 27:1797–805. doi: 10.1038/s41591-021-01497-1
84. Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: A Phase 1 Dose Escalation and Expansion Trial. Nat Med (2020) 26:1569–75. doi: 10.1038/s41591-020-1081-3
85. Mei H, Li C, Jiang H, Zhao X, Huang Z, Jin D, et al. A Bispecific CAR-T Cell Therapy Targeting BCMA and CD38 in Relapsed or Refractory Multiple Myeloma. J Hematol Oncol (2021) 14:161. doi: 10.1186/s13045-021-01170-7
86. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 Targeting Prevents Antigen-Loss Relapses After CD19-Directed Immunotherapies. J Clin Invest (2016) 126:3814–26. doi: 10.1172/JCI87366
87. Gonzalez A, Roguev A, Frankel NW, Garrison BS, Lee D, Gainer M, et al. Abstract LB028: Development of Logic-Gated CAR-NK Cells to Reduce Target-Mediated Healthy Tissue Toxicities. Clin Res (Excluding Clin Trials) Am Assoc Cancer Res (2021) 81(13_Suppl):Abstract nr LB028. doi: 10.1158/1538-7445.am2021-lb028
88. Sutherland AR, Owens MN, Geyer CR. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21197222
89. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A Universal Strategy for Adoptive Immunotherapy of Cancer Through Use of a Novel T-Cell Antigen Receptor. Cancer Res (2012) 72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890
90. Lim RM, Rong L, Zhen A, Xie J. A Universal CAR-NK Cell Targeting Various Epitopes of HIV-1 Gp160. ACS Chem Biol (2020) 15:2299–310. doi: 10.1021/acschembio.0c00537
91. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR-T Cell Therapy in T-Cell Malignancies: Is Success a Low-Hanging Fruit? Stem Cell Res Ther (2021) 12:527. doi: 10.1186/s13287-021-02595-0
92. Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel Anti-CD3 Chimeric Antigen Receptor Targeting of Aggressive T Cell Malignancies. Oncotarget (2016) 7:56219–32. doi: 10.18632/oncotarget.11019
93. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol (2019) 0:2250. doi: 10.3389/fimmu.2019.02250
94. Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Myhre MR, et al. NK Cells Specifically TCR-Dressed to Kill Cancer Cells. EBioMedicine (2019) 40:106–17. doi: 10.1016/j.ebiom.2019.01.031
95. Walseng E, Köksal H, Sektioglu IM, Fåne A, Skorstad G, Kvalheim G, et al. Wälchli S. A TCR-Based Chimeric Antigen Receptor. Sci Rep (2017) 7:10713. doi: 10.1038/s41598-017-11126-y
96. Mehta RS, Rezvani K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front Immunol (2018) 9:283. doi: 10.3389/fimmu.2018.00283
97. Bachier C, Borthakur G, Hosing C, Blum W, Rotta M, Ojeras P, et al. NA Phase 1 Study of NKX101, an Allogeneic CAR Natural Killer (NK) Cell Therapy, in Subjects With Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) or Higher-Risk Myelodysplastic Syndrome (MDS). Blood (2020) 136:42–3. doi: 10.1182/blood-2020-134625
98. Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible MyD88/CD40 Synergizes With IL-15 to Enhance Antitumor Efficacy of CAR-NK Cells. Blood Adv (2020) 4:1950–64. doi: 10.1182/bloodadvances.2020001510
99. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking Expression of Inhibitory Receptor NKG2A Overcomes Tumor Resistance to NK Cells. J Clin Invest (2019) 129:2094–106. doi: 10.1172/JCI123955
100. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
101. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a Cytokine Checkpoint Enhances the Fitness of Armored Cord Blood CAR-NK Cells. Blood (2021) 137:624–36. doi: 10.1182/blood.2020007748
102. Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, et al. The TGF-β/SMAD Pathway is an Important Mechanism for NK Cell Immune Evasion in Childhood B-Acute Lymphoblastic Leukemia. Leukemia (2016) 30:800–11. doi: 10.1038/leu.2015.327
103. Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol Ther (2014) 22:1211–20. doi: 10.1038/mt.2014.47
104. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther (2017) 25:249–58. doi: 10.1016/j.ymthe.2016.10.016
105. Chmielewski M, Abken H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
106. Chmielewski M, Abken H. TRUCKS, the Fourth Generation CAR T Cells: Current Developments and Clinical Translation. Adv Cell Gene Ther (2020) 3:e84. doi: 10.5283/EPUB.44684
107. Luo H, Wu X, Sun R, Su J, Wang Y, Dong Y, et al. Target-Dependent Expression of IL12 by Synnotch Receptor-Engineered NK92 Cells Increases the Antitumor Activities of CAR-T Cells. Front Oncol (2019) 9:1448. doi: 10.3389/fonc.2019.01448
108. Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult Peripheral Blood and Umbilical Cord Blood NK Cells are Good Sources for Effective CAR Therapy Against CD19 Positive Leukemic Cells. Sci Rep (2019) 9:18729. doi: 10.1038/s41598-019-55239-y
109. Matsubara H, Niwa A, Nakahata T, Saito MK. Induction of Human Pluripotent Stem Cell-Derived Natural Killer Cells for Immunotherapy Under Chemically Defined Conditions. Biochem Biophys Res Commun (2019) 515:1–8. doi: 10.1016/j.bbrc.2019.03.085
110. Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I, et al. High Log-Scale Expansion of Functional Human Natural Killer Cells From Umbilical Cord Blood CD34-Positive Cells for Adoptive Cancer Immunotherapy. PloS One (2010) 5:e9221. doi: 10.1371/journal.pone.0009221
111. Kim H-S, Kim JY, Seol B, Song CL, Jeong JE, Cho YS. Directly Reprogrammed Natural Killer Cells for Cancer Immunotherapy. Nat BioMed Eng (2021) 147:3725–34. doi: 10.1038/s41551-021-00768-z
112. Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol Res (2018) 6:467–80. doi: 10.1158/2326-6066.CIR-17-0207
113. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-The-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front Immunol (2017) 8:533. doi: 10.3389/fimmu.2017.00533
114. Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front Immunol (2019) 10:3123. doi: 10.3389/fimmu.2019.03123
115. Chockley P, Patil SL, Gottschalk S. Transient Blockade of TBK1/Ikkϵ Allows Efficient Transduction of Primary Human Natural Killer Cells With Vesicular Stomatitis Virus G-Pseudotyped Lentiviral Vectors. Cytotherapy (2021) 23:787–92. doi: 10.1016/j.jcyt.2021.04.010
116. Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, et al. Corrigendum: A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front Immunol (2019) 10:2784. doi: 10.3389/fimmu.2019.02784
117. Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front Immunol (2019) 10:2873. doi: 10.3389/fimmu.2019.02873
118. Suerth JD, Morgan MA, Kloess S, Heckl D, Neudörfl C, Falk CS, et al. Efficient Generation of Gene-Modified Human Natural Killer Cells via alpharetroviral vectors. J Mol Med (2016) 94:83–93. doi: 10.1007/s00109-015-1327-6
119. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-Cell Engineering Using non-Viral Sleeping Beauty Transposition From Minicircle Vectors. Leukemia (2017) 31:186–94. doi: 10.1038/leu.2016.180
120. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I Clinical Trial of EGFR-Specific CAR-T Cells Generated by the Piggybac Transposon System in Advanced Relapsed/Refractory non-Small Cell Lung Cancer Patients. J Cancer Res Clin Oncol (2021). doi: 10.1007/s00432-021-03613-7
121. Manuri PVR, Wilson MH, Maiti SN, Mi T, Singh H, Olivares S, et al. Piggybac Transposon/Transposase System to Generate CD19-Specific T Cells for the Treatment of B-Lineage Malignancies. Hum Gene Ther (2010) 21:427–37. doi: 10.1089/hum.2009.114
122. Prommersberger S, Reiser M, Beckmann J, Danhof S, Amberger M, Quade-Lyssy P, et al. CARAMBA: A First-in-Human Clinical Trial With SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther (2021) 28:560–71. doi: 10.1038/s41434-021-00254-w
123. Micklethwaite KP, Gowrishankar K, Gloss BS, Li Z, Street JA, Moezzi L, et al. Investigation of Product-Derived Lymphoma Following Infusion of Piggybac-Modified CD19 Chimeric Antigen Receptor T Cells. Blood (2021) 138:1391–405. doi: 10.1182/blood.2021010858
124. Bishop DC, Clancy LE, Simms R, Burgess J, Mathew G, Moezzi L, et al. Development of CAR T-Cell Lymphoma in 2 of 10 Patients Effectively Treated With Piggybac-Modified CD19 CAR T Cells. Blood (2021) 138:1504–9. doi: 10.1182/blood.2021010813
125. Liu E, Ang SOT, Kerbauy L, Basar R, Kaur I, Kaplan M, et al. GMP-Compliant Universal Antigen Presenting Cells (uAPC) Promote the Metabolic Fitness and Antitumor Activity of Armored Cord Blood CAR-NK Cells. Front Immunol (2021) 12:626098. doi: 10.3389/fimmu.2021.626098
126. Gratch YS, Bowles P, Krishnan R, Loo-Yong-Kee S, Iyer RK. Dulgar-Tulloch A. A Semi-Automated, High-Purity Process for Natural Killer (NK) Cell Manufacturing in a Rocking Bioreactor. Cytotherapy (2018) 20:e8–9. doi: 10.1016/j.jcyt.2018.03.029
127. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-Grade Generation of Active NK Cells From Cord Blood Hematopoietic Progenitor Cells for Immunotherapy Using a Closed-System Culture Process. PloS One (2011) 6:e20740. doi: 10.1371/journal.pone.0020740
128. Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 With Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum Gene Ther (2017) 28:897–913. doi: 10.1089/hum.2017.157
129. Granzin M, Soltenborn S, Müller S, Kollet J, Berg M, Cerwenka A, et al. Fully Automated Expansion and Activation of Clinical-Grade Natural Killer Cells for Adoptive Immunotherapy. Cytotherapy (2015) 17:621–32. doi: 10.1016/j.jcyt.2015.03.611
130. Mark C, Czerwinski T, Roessner S, Mainka A, Hörsch F, Heublein L, et al. Cryopreservation Impairs 3-D Migration and Cytotoxicity of Natural Killer Cells. Nat Commun (2020) 11:5224. doi: 10.1038/s41467-020-19094-0
131. Oberschmidt O, Morgan M, Huppert V, Kessler J, Gardlowski T, Matthies N, et al. Development of Automated Separation, Expansion, and Quality Control Protocols for Clinical-Scale Manufacturing of Primary Human NK Cells and Alpharetroviral Chimeric Antigen Receptor Engineering. Hum Gene Ther Methods (2019) 30:102–20. doi: 10.1089/hgtb.2019.039
132. Yao X, Matosevic S. Cryopreservation of NK and T Cells Without DMSO for Adoptive Cell-Based Immunotherapy. BioDrugs (2021) 35:529–45. doi: 10.1007/s40259-021-00494-7
133. Majzner RG, Mackall CL. Tumor Antigen Escape From CAR T-Cell Therapy. Cancer Discovery (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
134. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of Cdc1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004
135. Judge SJ, Murphy WJ, Canter RJ. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front Cell Infect Microbiol (2020) 10:49. doi: 10.3389/fcimb.2020.00049
136. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
137. Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting Natural Killer Cells in Solid Tumors. Cell Mol Immunol (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
138. Zhu H, Blum R, Wu Z, Bahena A, Hoel HJ, Ask EH, et al. Notch Activation Rescues Exhaustion in CISH-Deleted Human iPSC-Derived Natural Killer Cells to Promote In Vivo Persistence and Enhance Anti-Tumor Activity. Blood (2018) 132:1279–9. doi: 10.1182/blood-2018-99-112791
139. Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Bühler L, et al. Single-Cell RNA Sequencing of Tumor-Infiltrating NK Cells Reveals That Inhibition of Transcription Factor HIF-1α Unleashes NK Cell Activity. Immunity (2020) 52:1075–1087.e8. doi: 10.1016/j.immuni.2020.05.001
140. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-Tumor Activity. Cell Stem Cell (2020) 27:224–237.e6. doi: 10.1016/j.stem.2020.05.008
Keywords: chimeric antigen receptor (CAR), CAR-NK cell, cell signaling, gene therapy, CAR-T cells, allogenic cell therapy, NK cell receptors and ligands, immunotherapy
Citation: Ruppel KE, Fricke S, Köhl U and Schmiedel D (2022) Taking Lessons from CAR-T Cells and Going Beyond: Tailoring Design and Signaling for CAR-NK Cells in Cancer Therapy. Front. Immunol. 13:822298. doi: 10.3389/fimmu.2022.822298
Received: 25 November 2021; Accepted: 18 February 2022;
Published: 18 March 2022.
Edited by:
Volker Huppert, Glycostem, NetherlandsReviewed by:
Frank M. Cichocki, University of Minnesota Twin Cities, United StatesCopyright © 2022 Ruppel, Fricke, Köhl and Schmiedel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominik Schmiedel, ZG9taW5pay5zY2htaWVkZWxAaXppLmZyYXVuaG9mZXIuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.