 Zeev Elkoshi
Zeev Elkoshi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 25 January 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.821598
The present article compares, side-by-side, cancer and autoimmune diseases in terms of innate and adaptive immune cells involvement, MHC Class I and Class II expression, TGFβ effect, immune modulating drugs effect and the effect of reactive oxygen species. The change in the inflammatory immune reaction during the progress of cancer and the effect of this change on the comorbidity of autoimmune diseases and cancer are discussed. The similar inflammatory properties of autoimmune diseases and early cancer, and the contrasting inflammatory properties of autoimmune diseases and advanced cancer elucidate the increased incidence of many types of cancer in patients with pre-existing autoimmune diseases and the decreased cancer-specific mortality of these patients. Stage-dependent effects of reactive oxygen-species on tumor proliferation are an additional probable cause for these epidemiological observations. The relationship: {standardized incidence ratio (SIR)} > {cancer-specific hazard ratio (HR)} for cancer patients with a history of autoimmune diseases is substantiated and rationalized.
Earlier work proposed a classification of chronic diseases into pro- and anti-inflammatory diseases based on the extent of regulatory T cells (Treg) activity observed (“high Treg” or “low Treg”) (1). This classification was found useful (a) in understanding the efficacy of drugs for the treatment of different chronic diseases, (b) in explaining the association of specific chronic diseases with specific pathogens, and (c) in interpreting the effects of different pathogens on cancer and autoimmune diseases (1–3).
The present work expands on the immunological properties of cancer and autoimmune diseases (AIDs) in relation to the binary classification proposed. During its course of growth, cancer progress from a pro-inflammatory, “low Treg” disease, to an anti-inflammatory, “high Treg” disease. This change is observed not only within the tumor microenvironment (TME) but also in the systemic circulation [Ref. 2 and references therein]. On the other hand, many autoimmune diseases are characterized by a decreased Tregs function or frequency (4), and may be regarded as proper pro-inflammatory “low Treg” chronic diseases. In addition, as will be described later, reactive oxygen species (ROS) exert opposite effects on the promotion of AIDs and advanced cancer. For these reasons, AIDs and advanced cancer can be considered immunological opposites. In contrast, AIDs and early cancer are triggered and promoted by a similar anti-inflammatory environment. As will be portrayed later, perspicacity of these time-related relationships helps in explaining the increased risk of cancer development in patients with AID history, a commonly reported observation which was referred to in the literature as “a paradox” (5). Noticing these unique relationships also helps in elucidating the longer survival of cancer patients with pre-existing AIDs compared to cancer patients without documented AIDs.
In the next two chapters, immune reactions to cancer and AIDs, and the effect of ROS on these immune reactions, including the roles of the major histocompatibility complex Class I (MHC-I) and Class II (MHC-II) and transforming growth factor β (TGFβ) will be reviewed and compared. The comorbidity of cancer and autoimmune diseases and the effect of ROS on this comorbidity will then be discussed.
CD8+ T cells attack cancer cells directly, and are of primary importance among several anti-cancer mechanisms reported in animals and man. By recognizing peptide-MHC-I complexes, CT8+ T cells identify cancer cells and destroy them through the release of perforin and the activation of the FAS apoptosis pathway (6).
Tregs suppress the function of CD8+ T cells (7, 8) and thus promote cancer. In addition, Tregs inhibit the proliferation of memory T cells (9) and of effector T cells such as Th1 (10) and Th2 (11). Th1 cytokine IL-2 demonstrated an anti-tumor activity in experimental models (12). A recombinant form of IL-2 is approved in several countries for the treatment of malignant melanoma and renal cell carcinoma. However another Th1 cytokine, IFNγ, exhibits pro- and anti-tumor effects (13).
In contrast to Th1 mediated immunity which is in general anti-cancer, Th2 mediated immunity present pro- and anti-tumor effects (14).
Th17 cells may display a pro- or anti-tumor role within the TME (15). Their frequency in the circulation of cancer patients increases compared to that in healthy controls. This was reported in many cancers, including renal cell carcinoma (16), ovarian cancer (17), breast cancer (18), cervical cancer (19, 20), endometrial cancer (21), hepatocellular carcinoma (22), and oral squamous cell carcinoma (23). However, low values of Th17 cells in the circulation of cancer patients have been reported in pancreatic cancer (24) and lung cancer (25). Th17 cells accumulate in the TME of cancers such as gastric cancer (26), breast cancer (18) and prostate cancer (27).
Regulatory T cells expansion in the peripheral blood of cancer-patients has been documented in ovarian cancer (17), hepatocellular carcinoma (22), oral squamous cell carcinoma (23), pancreatic cancer (24), gastric cancer (26), renal cell carcinoma (16) and cervical cancer (19, 20). High frequencies of Treg cells in the tumor microenvironment (compared to non-tumor tissues) were observed in breast cancer TME (18) and lung cancer TME (25). A correlation between Treg cells frequency and cancer progression was observed in breast cancer (18), lung cancer (25), hepatocellular carcinoma (22), oral squamous cell carcinoma (23), and gastric cancer (26),
Collectively, accumulation of Treg and Th17 cells in the tumor microenvironment along with increased levels of both cells in blood are observed in many cancers.
The effect of Th17 cells on cancer is usually limited. Inhibition of IL-23, a key cytokine for Th17 maintenance and expansion, by IL-23 inhibitor risankizumab, does not increase long-term cancer risk (28). Similarly, long-term treatment of psoriasis, psoriatic arthritis and ankylosing spondylitis patients with IL-17A inhibitor secukinumab, demonstrated a long-term low risk of malignancy (29). A pooled analysis of other long-term studies with secukinumab presented similar results (30). Another IL-17A inhibitor, ixekizumab, also demonstrated a long-term low risk of cancer (31). However other studies with these and other IL-23 inhibitors display conflicting results with pro-tumor or anti-tumor effects (32). It may be concluded that Th17 cells do not present significant anti-cancer or pro-cancer activities.
Tumor associated neutrophils (TANs) display plasticity in the TME. In the absence of TGFβ they attain a pro-inflammatory and anticancer mode of action (N1 phenotype) while in the presence of TGFβ they present an anti-inflammatory and pro-cancer response (N2 phenotype). The N1 phenotype which produces high levels of TNFα, NO and H2O2, prevails in the TME of early tumors when the level of TGFβ is low. The N2 phenotype predominates at later stages when TGFβ accumulates (33). Circulating neutrophils in tumor-bearing mice display two sub-populations classified by their densities in peripheral blood mononuclear cells (PBMC) fraction, after density gradient centrifugation of whole blood. High density neutrophils (HDN) prevail in tumor-free mice while low density neutrophils (LDN) are the dominant neutrophils in tumor-bearing mice. HDN were cytotoxic towards cancer cells in culture, whereas LDN were innocuous. Similar to TANs, high density neutrophils transform into the low density type in a TGFβ-dependent manner (33). It is not surprising therefore that circulating blood of patients with advanced-stage lung cancer was reported to be enriched with LDN, relative to early-stage patients, or healthy controls (34).
Macrophages can be classified as pro-inflammatory macrophages (M1) or anti-inflammatory macrophages (M2). M1 macrophages promote inflammation by the secretion of inflammatory cytokines such as TNFα, IL-1α, IL-1β, IL-6, IL-12, IL-18, and IL-23, and by the generation of ROS and reactive nitrogen species. They also express high levels of MHC that allow the activation of the adaptive immune arm. M2 macrophages secrete high levels of IL-10, PGE2 and TGFβ, cytokines that play an anti-inflammatory role. M2 macrophages express high levels of arginase-1, mannose receptor, and low levels of MHC class II complex (35).
Tumor associated macrophages (TAMs) include both phenotypes, M1 and M2. It has been proposed that macrophages display a pro-inflammatory antitumor effect (M1 phenotype) in early cancer and an anti-inflammatory pro-tumor effect (M2 phenotype) in established cancer (36).
The tumor microenvironment is rich in reactive oxygen species. As the tumor develops, ROS accumulate in the TME of many solid cancers, as a result of intensive production by tumor cells mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. In addition, ROS are released into the TME by cancer-associated fibroblasts (CAFs), TAMs, and myeloid-derived suppressor cells (MDSCs) (37). The effects of ROS on cancer proliferation are complex. They are partly mediated by the interaction of ROS with Tregs, but other pathways are possible. These interactions are presented below.
When mitochondrial oxidative phosphorylation in Tregs is impaired by the ablation of mitochondrial respiratory chain complex III in mice, the suppressive capacity of these Treg cells is lost without altering Treg cell proliferation and survival (38).
It has been demonstrated in vitro and in vivo that ROS generated by macrophages NADPH oxidase complex induce Treg frequency and function (39). In addition it was shown in psoriasis rat model that ROS prevent imiquimod-induced psoriatic dermatitis by enhancing Tregs function (40). On the other hand, TGFβ excreted by Treg cells induced the generation of ROS by NADPH oxidase (41, 42). In accordance with these observations, Treg-induced immunosuppression in the tumor microenvironment is mediated by Tregs-generated ROS (43).
MHC-I expression in tumor cells has been shown to increase following oxidative stress (44, 45). This increased expression of MHC-I facilitates the identification of cancer cells by CD8+ T cells and boosts tumor attack by these T cells.
Mitochondrial reactive oxygen species have a role in cross-presentation of MHC-I antigens by plasmacytoid dendritic cells. Reduction in mitochondrial generation of reactive oxygen species by plasmacytoid dendritic cells resulted in a substantial decrease in the ability of these cells to induce a CD8+ T cell reaction following cross-presentation (46). This behavior was also reported in normal dendritic cells, where NADPH oxidase isoform NOX2 (an enzyme with a prominent role in mitochondrial reactive oxygen species generation) was found essential for an efficient antigen cross-presentation to CD8+ T cells. Lack of NOX2 in DCs resulted in impaired cross-presentation (47). Dendritic cells from chronic granulomatous disease patients with an impaired catalytic subunit of NOX2 have shown defective cross-presentation of soluble antigens to CD8+ T cells (48). Since dendritic cells are major players in the control of cancer by adaptive immunity (49), a reduction in ROS production by dendritic cells is expected to promote cancer.
As discussed above, ROS effect on tumor growth may be mediated by Tregs. However other routes for the effect of ROS on malignancy are possible. At the pre-cancerous stage, ROS may drive cancer initiation by inducing oxidative damage and base pair substitution mutations in tumor suppressor genes such as TP53 (50). As the tumor grows, the increased level of ROS in the TME induces tumor proliferation by enhancing Tregs activity. In addition ROS activate several canonical pathways involved in tumor propagation such as the NF-κB pathway, the MAPK pathway (including ERK1/2, JNK, MAPK-11 and MAPK1) and the PI3K/PTEN pathway (37). Reactive oxygen species also support the development of tumor metastases by driving epithelial–mesenchymal transition (EMT) (48, 51, 52) and by anoikis inhibition (anoikis is a type of programmed cell death induced by cell detachment from extracellular matrix) (53).
These and other pro-cancerous effects of increased ROS levels drive tumor progression. However, very high levels of oxidative agents may result in tumor tissue damage. Excessive ROS levels induce cell death by apoptosis, necrosis and ferroptosis (54). Hence, a moderately-high level of ROS within the TME would be optimal for tumor growth and sustainability (55–57). A tight control on ROS balance is therefore required for tumor progression. It may be stated that moderately-high intra- and extra-cellular ROS levels promotes solid cancer propagation, metastases and angiogenesis, whereas very high levels induce cancer cell death and low levels are cytostatic.
Using a mouse model, Mu et al. have shown that MHC-I transcription is enhanced by the transcription factor FoxP3 in T cells. This resulted in a higher expression of MHC-I in CD4(+)CD25(+) regulatory T cells than in conventional CD4(+)CD25(-) T cells. The authors also found that MHC-I expression by Tregs contributes to their regulatory function (58). High suppressive Treg activity drives advanced cancer and metastases (2). Mu et al. mention a study by Joetham et al. reporting that CD8+ T cells interaction with MHC-I on Tregs is required for Tregs activation (59). In relation to this observation, TCRs expressed in peripheral Treg cells can recognize foreign antigens with high affinity. Tregs induce antigen-specific suppression mainly by Treg-DCs interaction. A mechanism by which DCs present antigens to Treg cells as part of MHC-II, by means of Treg TCR, has been proposed. This process eventually results in the generation of antigen-specific tolerogenic DCs (60). One may speculate that antigens presented by DCs to Tregs may further bind to MHC-I to form protein-MHC-I complex. This complex may help CD8+ T cells in recognizing Tregs (the way CD8+ T cells recognize cancer cells), before they activate them by a direct contact.
The suppression of MHC-I expression by TGF-β1 has been demonstrated in the TGFβ1 null mouse where elevated mRNA levels of MHC-I (and MHC-II) were detected compared to normal or TGFβ1 heterozygous littermates (61). Incubation of two human uveal melanoma cell lines in the presence of TGF-β generated more than 50% decrease in MHC-I antigen expression (62).
As explained above, peptide-MHC-I complexes guide tumor attacks by CD8+ T cells. Many solid tumors demonstrate decreased expression of MHC-I antigen presentation (mediated through different pathways), and thus evade cancer immune control by CT8+ T cells (63).
MHC-II-mediated antigen presentation to CD4+ T cells supports immune response by both T helper cells and CD8+ T cells [CD4+ T cells are required for CD8+ priming and function (64)]. Even though MHC-II is expressed mainly by professional antigen presenting cells (macrophages, dendritic cells, and B cells) and by thymic epithelial cells, other cells may express MHC-II, including cancer cells. Indeed, in addition to changes in MHC-I expression by cancer cells, changes in the expression of MHC-II by cancer cells are also observed (65). The frequencies of MHC-II alleles in cancer patients are different from these observed in healthy subjects. These differences, which depend on the type of cancer, were found to correlate with the risk of developing the malignancy and with the response to treatment (66). It is tempting to believe that CD+4 T cells (and MHC-II expression) are important for anti-tumor immunity. In line with this, the response to PD-1 blockage in a synergistic murine model of melanoma required CD4+ T cells in addition to CD8+ T cells, and a costimulation by dendritic cells and macrophages (67).
Akkaya et al. have shown in a mouse model that one of the mechanisms by which regulatory T cells induce immune suppression is by the depletion of peptide-MHC-II complex from DCs (68). Since advanced cancer is a “high Treg” disease, this regulatory mode of action is expected to promote immune escape at advanced cancer stages.
Delvig et al. demonstrated that by reducing MHC-II expression on macrophages, TGFβ blocked antigen presentation of two T cell epitopes. In addition, TGFβ reduced the constitutive expression of MHC-II transactivator (CIITA), invariant chain, and HLA-DO mRNA (69). These findings are supported by another study, reporting of CIITA gene-attenuation mediated by Tregs (70). Since Treg cells are a major source of TGFβ, these results are consistence with the earlier observation of MHC-II depletion mediated by Tregs.
Treg cells activity (number and function) as well as TGFβ levels increase (in the TME and in the circulation) during the course of cancer development. Based on the findings aforementioned, MHC-II expression is expected to decline during the course of cancer progression. In line with this, MHC-II allele HLA-DRB1*07 expression was higher in lymph nodes of patients with early stage non-small cell lung cancer, compared to patients with an advanced stage of the disease (71).
Taken together, MHC-II expression is important for an effective anti-tumor immunity. Treg-mediated downregulation of MHC-II expression promotes tumor immune escape.
Considering the antitumor effects of CD8+ T cells and Th1 cells, it is not surprising that non-specific-action (but not target specific) immunosuppressive drugs increase the risk of cancer. For example, malignancy risk in transplant patients using immunosuppressive drugs such as cyclosporine or azathioprine increases 4 - 500 fold compared to age-matched controls in the general population (72).
Autoimmune diseases are a typical example of pro-inflammatory chronic disease (1). Many AIDs display impaired Tregs function (4). Moreover, the lack of Tregs induces AIDs. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare recessive disorder caused by mutations in the FoxP3 gene, considered to be the master regulator of Tregs. IPEX is associated with autoimmune enteropathy, Type 1 diabetes (T1D), autoimmune skin disorders and serious infections (73). Since Treg function is impaired in autoimmune diseases, CD4+ T cells activity intensifies. In particular, Th1 and Th17 cells are involved in the pathogenesis of many AIDs (74). Th17 cells frequency or function increases in multiple sclerosis (MS), psoriasis, RA, inflammatory bowel disease, and SLE (75).
Th2 cells mediate allergic immune responses (76) and also play a pathogenic role in systemic sclerosis and ulcerative colitis (74). Systemic frequency of Th2 in RA patients is similar to that in healthy controls (77). The data regarding Th2 cytokines concentration in SLE is conflicting (78, 79).
CD8+ T cells frequency (in affected organs or in the circulation) increases or their function is boosted in MS, systemic sclerosis, T1D, SLE, and severe aplastic anemia (80).
Neutrophils exhibit a pro-inflammatory function in AIDs. However their modus operandi may vary from one disease to another. The involvement of neutrophils in RA and SLE are described below as representative examples.
Neutrophils in RA patient’s joints are cytotoxic and neutrophils in the circulation of RA patients are primed for the production of ROS, in contrast to those in healthy subjects. Proteases released from lysosomal granules of neutrophils promote RA inflammation (81). Elevated levels of myeloperoxidase (MPO), a cytotoxic enzyme released from neutrophils granules, are found in blood, synovial fluid and tissues of RA patients. MPO increases vascular permeability and allows the penetration of pro-inflammatory immune cells. In addition, MPO attracts more neutrophils to the site of inflammation. Neutrophils in RA synovial fluid secrete inflammatory cytokines such as TNF, B cell-activating factor (BAFF) and receptor activator of nuclear factor kappa B ligand (RANKL). Neutrophil extracellular traps (NETs) are webs of histones and DNA fibers that participate in pathogens eradication. Citrullinated histones comprise around 70% of all NETs proteins. NETs can promote the inflammatory effects of fibroblast-like synoviocytes. NETs formation which is enhanced in RA, is believed to trigger autoimmunity to citrullinated proteins (81).
Similar to their role in RA, neutrophils in SLE display a pro-inflammatory function. They are mainly of the LDN phenotype and secrete type I IFN, IFN-γ, IL-6, IL-8 and TNFα. Like RA, SLE is characterized by enhanced NETs formation. NETs promote immune response in SLE by exposing antigens that are normally shielded by plasma membrane, by reducing T cells activation threshold, and by activation of autoreactive B-cells (82).
In many AIDs macrophages display the M1 pro-inflammatory phenotype (RA, SLE, primary biliary cholangitis, Sjögren’s syndrome, T1D). However, M2 phenotype has been detected in fibrotic AIDs such as systemic sclerosis and inflammatory bowel disease (83). In multiple sclerosis (MS), even though most macrophages in disease lesions display M1 characteristic, a large fraction of macrophages displays both M1 and M2 markers (84).
Chronic granulomatous disease (CGD) is a rare genetic disorder caused by defects in any of the five subunits of the NADPH oxidase complex. An increased frequency of several autoimmune diseases has been reported in CGD patients (85). Experimental evidence (observed in human and animals) supports a link between ROS deficiency and autoimmune diseases such as systemic lupus erythematosus (SLE) (86–88), rheumatoid arthritis (RA) (89–92), psoriasis (93) and Guillain–Barré syndrome (94)
Using a mouse model, Kim et al. have shown that imiquimod-induced psoriatic dermatitis was attenuated by elevated ROS levels, whereas lower ROS levels which induced Tregs dysfunction aggravated the disease (40). It is plausible that impaired Treg function under low ROS levels promoted this autoimmune disease.
Strickland et al. demonstrated that oxidant-treated T cells induced lupus-like disease in mice. In addition, genes known to be associated with lupus in SLE patients and animal models of SLE were upregulated by ROS. This effect was most pronounced with peroxynitrite (ONOO-) as the oxidant (95). Peroynitrite is at equilibrium with peroxynitrous acid which rapidly decays in non-alkaline media to •NO2 and •OH free radicals (~ 30% yield) and the rest of peroxynitrous acid isomerizes quickly [k = 1.2 sec-1 (96)] to the nitrate ion (97). For the treatment of T cells, Strickland et al. have used 20 µM peroynitrite solution (95). This highly oxidative medium triggered the disease.
T cell mitochondrial dysfunction has been proposed as the generator of oxidative stress in SLE (98). Mitochondrial oxidative stress and DNA damage was reported in Treg cells extracted from patients with different AIDs. This mitochondrial oxidative stress and DNA damage which resulted in Treg cell death, was also observed in the EAE mouse model (99). Oxidative stress has been shown to affect the Treg/Th17 balance in SLE (100).
It is evident that oxidative stress (which is synonymous with very high ROS levels) promotes autoimmunity.
Administration of several immunosuppressive agents to renal transplant recipients (with an uneventful postoperative course and stable renal function) before transplantation, indicated a statistically significant increase in ROS levels (101).
Blocking TGFβ signaling in T cells induces autoimmunity in mice (102, 103).
BXSB-Yaa is a murine-strain that develops spontaneous SLE-like disease. It was demonstrated that β2-microglobulin-deficient BXSB-Yaa mice (BXSB-Yaa mice with a defective MHC-I function) develop more aggressive and lethal SLE-like disease than BXSB-Yaa controls (104). MRL/lpr mouse-strain is another model for spontaneous lupus. β2m-deficient MRL/lpr mice showed accelerated lupus skin lesions accompanied by attenuated nephritis (105). However, other works presented contradicting data: MHC-I deficiency diminished SLE symptoms in a mouse-model of lupus (106) and evoked resistance to experimental induction of lupus in another mouse model (107). The effect of MHC-I on SLE is therefore unclear. It was also reported that MHC-I alleles are associated with only few AIDs (ankylosing spondylitis, psoriasis) whereas MHC-II alleles are associated with more AIDs (RA, T1D, MS, celiac disease and chronic beryllium disease) (108).
It can be concluded that MHC-I is not involved with the pathogenesis of all AIDs and its effect is controversial.
Genetic studies have confirmed that associations between MHC and autoimmune diseases are mostly related to MHC-II alleles. Certain MHC-II alleles increase the probability of developing specific AIDs (a positive association) while other alleles reduce this probability (a negative association). Grave’s disease, narcolepsy, autoimmune thyroiditis, RA, MS, T1D, SLE, ulcerative colitis and Crohn’s disease are all associated with MHC-II polymorphism (109, 110). Ulcerative colitis and Crohn’s disease are associated (but less consistently) with Class I alleles as well (110).
Strong induction of MHC -II expression in mice retina was observed in an experimental mouse-model of autoimmune uveitis (111). Upregulated endothelial MHC-II expression has been reported in dilated cardiomyopathy, RA, SLE, MS and Crohn’s disease patients (112).
Pathogen-stimulated PBMC from subjects homozygous for autoimmune vitiligo high-risk SNP haplotype, demonstrated increased production of IFN-γ and IL-1β than cells from subjects homozygous for a low-risk haplotype (113).
Collectively, AIDs are consistently associated with MHC-II polymorphism. MHC-II expression is upregulated in AIDs and induces a pro-inflammatory effect.
Autoimmune disease patients treated with checkpoint inhibitors for cancer are at increased risk of AID relapses. More than 30% of these patients develop flares of their pre-existing AIDs and some develop new autoimmune manifestations (114).
All the aforementioned data concerning cancer and AIDs is summarized in Table 1 and presented comparatively.
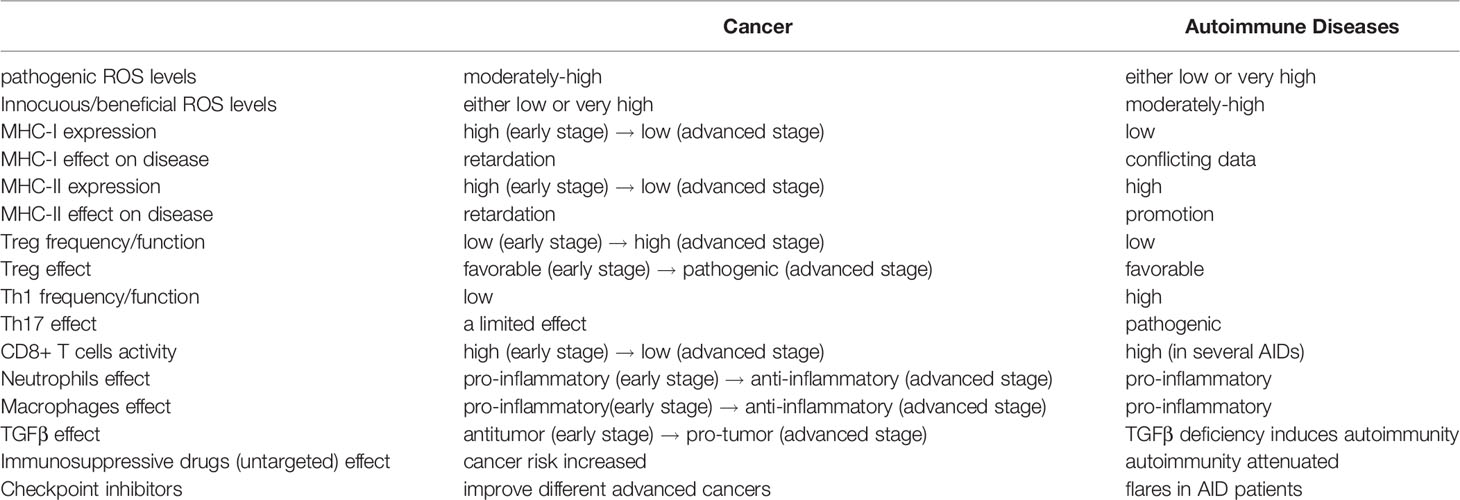
Table 1 A comparison of cancer and autoimmune diseases in terms of immunological properties, ROS effect, TGFβ effect and drug effect.
Discussing the comorbidity of cancer and autoimmune diseases, the difference between AIDs developed in the context of pre-existing cancer (paraneoplastic AIDs) and cancer developed in the context of pre-existing AIDs should be acknowledged. Paraneoplastic AIDs are typically alleviated or even resolved by reducing the tumor burden following a surgical removal of the growth or by the use of anti-cancer drugs (115). Paraneoplastic AIDs are generally rare. For example, paraneoplastic neurological syndromes affect one in every 334 patients with cancer (116). There are case reports describing paraneoplastic AIDs such as paraneoplastic polymyalgia rheumatica associated with esophageal carcinoma (117), and paraneoplastic SLE associated with colorectal cancer (118). As an exception, paraneoplastic AIDs associated with lymphoma are not rare and display a 7.6% incidence rate of AIDs in non-Hodgkin’s lymphoma (NHL) and 8.6% in Hodgkin’s lymphoma (HL) patients. The primary AIDs associated with lymphoma are Sjogren’s syndrome in NHL and autoimmune thyroiditis in HL. In most cases, autoimmunity develops after the diagnosis of HL but before the diagnosis of NHL (119).
Paraneoplastic AIDs are often a result of genetic alterations in tumor cells. For example, gene gain-of-function was demonstrated in paraneoplastic cerebellar degenerations with anti-Yo antibodies (Yo-PCD) (rare syndromes caused by an auto-immune response against neuronal antigens expressed by tumor cells). In Yo-PCD ovarian carcinomas, a high percentage of the tumors were found to harbor at least one genetic alteration of Yo antigens which was not observed in ovarian carcinoma samples extracted from patients without Yo-PCD (120).
Systemic sclerosis is another example where genetic alterations trigger an autoimmune response. It was found that tumors from systemic sclerosis patients concurrently diagnosed with cancer, carry mutations in the polymerase III polypeptide A (POLR3A) gene, and the presence of certain anti-nuclear antibodies (anti-Scl70, anti PM/Scl-100, anti RNA Polymerase III) in systemic sclerosis patients increases the risk of cancer (121). It was also found that in a subgroup of scleroderma patients with anti-RNA polymerase I/III autoantibodies, scleroderma developed within 2 years of cancer diagnosis, and nucleolar RNA polymerase III expression was enhanced in tumors of patients in this subgroup only (122). The authors hypothesized that tumors expressing high concentrations of RNA polymerase III initiate an immune response to these autoantigens which triggers scleroderma. The authors also raised the possibility that in some scleroderma patients without cancer pre-diagnosis who are tested positive for anti-RNA polymerase antibodies, scleroderma-specific (originally anti-tumor) immune response aborted the underlying malignancy (122).
Similar mechanisms are reported in cancer developed in patients with pre-existing autoimmune diseases. If tumor cells express antigens to AID autoantibodies, these antibodies may inhibit the antigen related effects. Autoantibodies binding to an antigen with an anti-tumor effect will promote cancer. For example, anti p155/140 antibodies have been detected in sera of adult patients with cancer-associated polymyositis/dermatomyositis (PM/DM) (123). Anti p155/140 antibodies target the transcriptional intermediary factor 1-γ (TIF1-γ). This transcription factor plays an ambivalent role in cancer. TIF1-γ promotes cancer by inhibiting the anti-tumor p-53 gene and by activating estrogen-dependent genes. Accordingly, it displays a pro-tumor effect in breast cancer. However by inhibiting the retinoic acid pathway, TIF1-γ has been shown to suppress liver cancer in a mouse model [Ref. 123 and references therein]. PM and DM are highly associated with the risk of cancer: the SIR value reported for malignancy in PM/DM (averaged over 20 studies) is 4.07 (3.02-5.12) (124).
Even though genetic alterations associated with cancer may elicit immune response that explains the strong associations of certain cancers with autoimmune diseases, this mechanism is either limited to a small number of AIDs or to a small number of patients. The cancer-modulating effect of autoantibodies is also restricted to a few AIDs.
The present work proposes a mechanism that explains the relationship between cancer and AIDs in most AIDs. It is suggested that in the majority of AIDs, the risks of cancer and of cancer-specific mortality are mainly affected by the temporal change of immune response along the course of cancer development.
Table 1 reveals that several immune reactions, triggers, cytokines and drug effects associated with advanced cancer are reciprocal to those associated with autoimmune diseases. The immune reaction to advanced cancer may be regarded as a “high Treg” and anti-inflammatory response while immune reaction to autoimmune diseases is a “low Treg” and pro-inflammatory response. On the other hand, the immune reaction to early cancer (prior to immune evasion) is pro-inflammatory, just like the immune reaction to AIDs. The pro-inflammatory environment that underlies AIDs supports the initiation and growth of early malignancy and increases the risk of cancer in AID patients. Indeed, higher incidence of cancer development is reported in many autoimmune diseases (125). In agreement, tumor infiltrating regulatory T cells (which release high amounts of TGFβ in the TME) improve survival in cancers with a long pre-metastatic periods like lymphoma, but have a negative effect on survival in cancers with an early immune evasion like breast cancer, lung cancer, or aggressive squamous cell carcinoma (2).
Table 2 presents published values of mean standardized incidence ratios (SIR) for different types of cancer, averaged over a large number of AIDs (including 95% confidence intervals). An increased SIR value in cancer patients with a history of AIDs was reported in lung cancer (126), kidney cancer, bladder cancer, prostate cancer (127), hepatobiliary cancer, primary liver cancer, gallbladder cancer, extrahepatic bile duct cancer (128) and multiple myeloma (129). Similarly, SIR for the development of lymphoma, increased in patients with SLE or RA (133). Cancer SIR (averaged over several types of cancer) increased in SLE, Sjogren’s syndrome, systemic sclerosis, and marginally in rheumatoid arthritis (134).
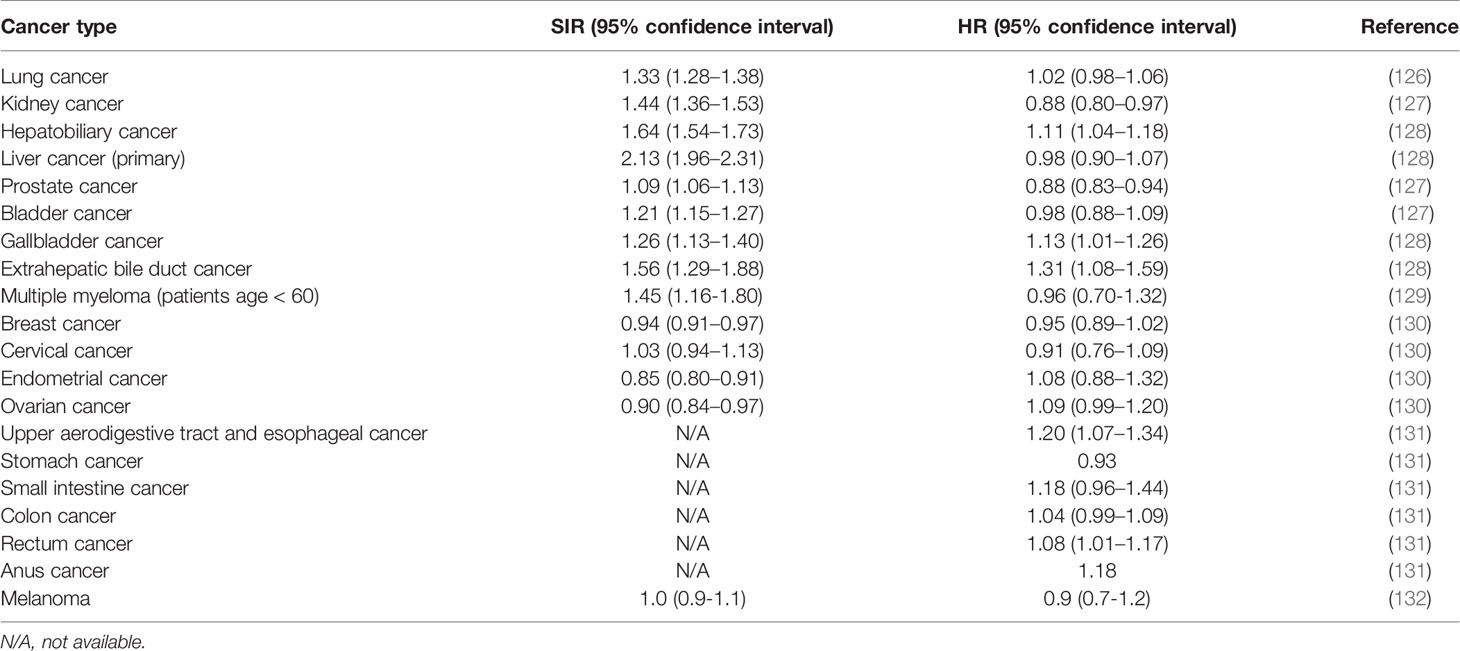
Table 2 Standardized incidence ratios (SIR) and cancer-specific hazard ratios (HR) (averaged over a wide-range of autoimmune diseases) for different types of cancer.
Turning to the survival of cancer patients with AIDs, the pro-inflammatory nature of AIDs in advanced cancer is expected to slow down cancer propagation and to correct the negative effect of AIDs in early cancer. Since cancer-related mortality occurs almost always at advanced stages of the disease, the hazard ratio for cancer-specific mortality is expected to be lower than the cancer standardized incidence ratio. Inspection of Table 2 reveal that in 11 out of 14 types of cancer the relation {SIR > HR} (where HR refers to cancer-specific mortality) holds true, and in 9 types of cancer, cancer-specific mortality in patients with AIDs is even lower compared to patients without an autoimmune background (i.e. HR < 1), even though the effect may be statistically non-significant in several cancers. In addition, a decrease in mortality was reported in breast cancer patients where a statistically significant 54% reduced risk of breast cancer mortality was observed in women with Th1 dominant autoimmune diseases (135).
In this respect, the distinction between cancer-specific mortality and overall mortality is important. Overall mortality is affected by comorbidities other than cancer (mainly by comorbidities related to AIDs) and is higher than cancer-specific mortality. This was recorded, for example, in kidney cancer, bladder cancer and prostate cancer (127). Therefore, the relationship {SIR > HR} may not be valid for the hazard ratio of overall mortality.
It must be delineated that the results presented in Table 2, are averages over a wide range of autoimmune diseases. The effect of a specific autoimmune disease may strongly deviate from the average.
It should also be realized that factors other than immune reaction, may affect the survival of cancer patients with AIDs differently from those cancer patients without AIDs.
For instance, cancer patients with formerly diagnosed AIDs are often undertreated with respect to their malignancy. The major reason for this insufficient treatment is “poor initial performance status or frailty” (136). An increased frequency of leukopenia due to the simultaneous use of immunosuppressive and chemotherapeutic drugs in these patients is a possible reason for shorter anti-cancer treatment periods or lower doses of anti-cancer drugs. In line with this, higher cancer-specific hazard ratios were reported in breast, cervical, and endometrial cancer in a subgroup of patients with severe AIDs (patients with at least 3 hospitalizations) compared to the mean values over the entire group (that included patients with less severe autoimmune disease) (130). The subgroup with a more severe disease was possibly undertreated.
If insufficient treatment of cancer patients with AIDs is considered as a confounding factor in mortality hazard assessment, the beneficial effect of AIDs on cancer-specific mortality may be even larger, and the “true” HR values should be lower than the values presented in Table 2.
To summarize, the incidence of cancer increases in many autoimmune diseases (digestive tract malignancies are exceptions) while cancer-specific mortality of most solid cancers either decreases or is unaffected by pre-existing autoimmune disease. The inequality {SIR > HR} (where HR refers to cancer-specific mortality) is valid in the overwhelming majority of cancer types in patients with formerly diagnosed AID (including digestive tract cancers).
It is noted that ROS levels that promote or suppress cancer are a mirror image of the levels that promote or suppress AIDs (see Figure 1). Whereas cancer proliferate and metastasize best under moderately-high ROS levels, AIDs develop and thrive under either low or very high ROS levels. Even though the high-low borders are not well defined, it is clear that very high ROS levels promote AIDs (98). Reactive oxygen species that accumulate in the circulation of AID patients act as pro-cancer agents at early cancer stages but as anti-cancer agents at advanced stages (50). This stage-related effect is another factor that contributes to the pathogenic effect of AIDs in early cancer and to their beneficial effect in advanced cancer, leading to the relationship {SIR > HR} discussed before.
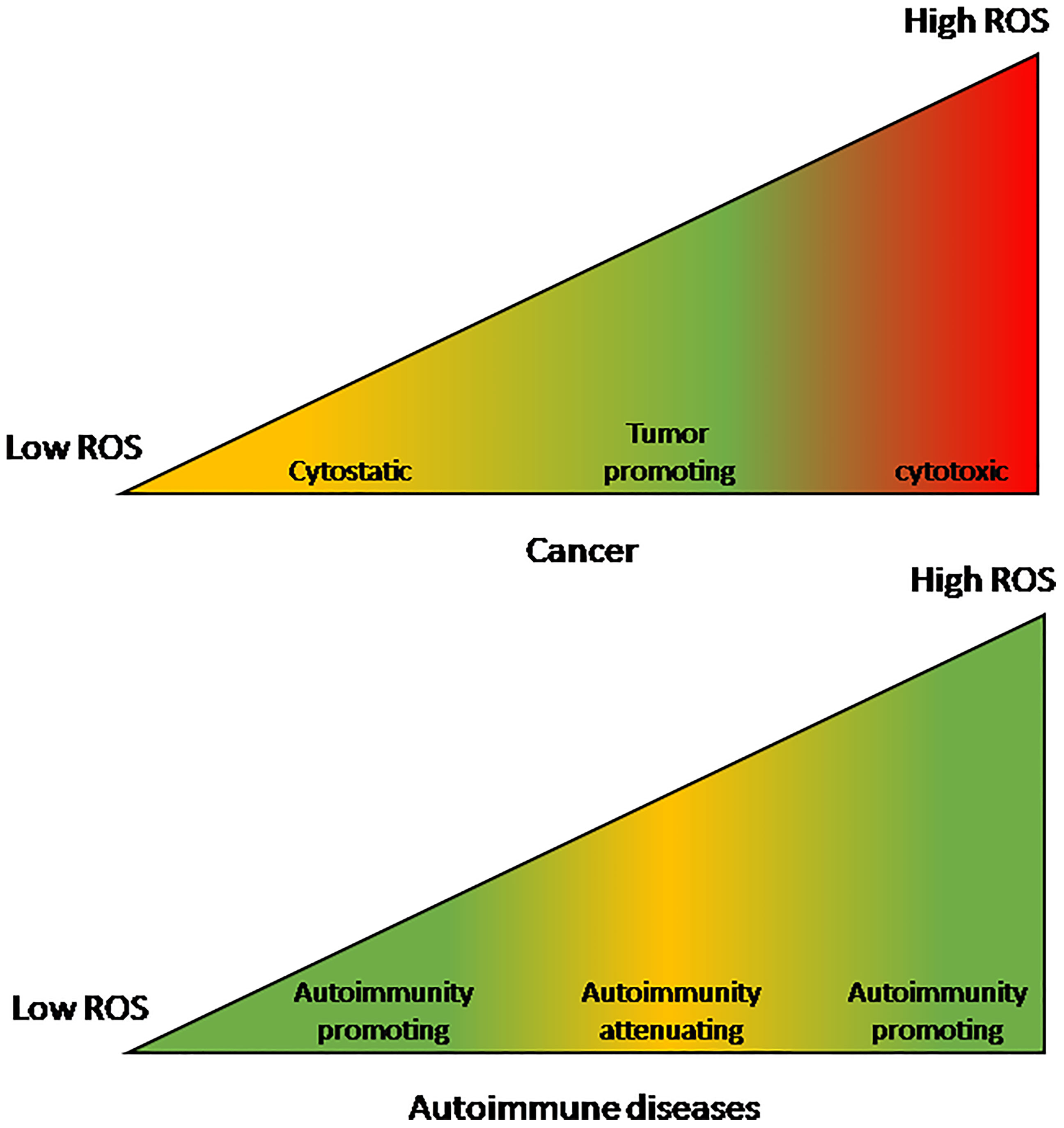
Figure 1 The effect of reactive oxygen species levels on cancers and autoimmune diseases. The effect on cancers is a mirror-image of the effect on autoimmune diseases. Cancers are optimally promoted by moderately-high ROS levels while autoimmune diseases are promoted by either low or very high ROS levels. On the other hand, at moderately-high ROS levels autoimmune diseases are attenuated while very high ROS levels are cancer-cytotoxic and low levels are cancer-cytostatic.
Inflammation drives both autoimmune diseases and early stage cancer while it slows down advanced cancer. Low or very high levels of reactive oxygen species induce autoimmunity while cancer propagates optimally at moderately-high levels of reactive oxygen species.
Due to both effects, initiation of cancer and their early growth are promoted by pre-existing autoimmune diseases leading to an increased risk of cancer, while advanced cancer growth and spread are hindered in patients with pre-existing autoimmune diseases, resulting in an improved survival.
The author confirms being the sole contributor of this work and has approved it for publication.
The views and opinions expressed, and/or conclusions drawn, in this article are those of the author and do not necessarily reflect those of Taro Pharmaceutical Industries Ltd., its affiliates, directors or employees.
ZE is employed by Taro Pharmaceutical Industries Ltd.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Elkoshi Z. The Binary Classification of Chronic Diseases. J Inflamm Res (2019) 12:319–33. doi: 10.2147/JIR.S227279
2. Elkoshi Z. "High Treg" Inflammations Promote (Most) Non-Hematologic Cancers While "Low Treg" Inflammations Promote Lymphoid Cancers. J Inflamm Res (2020) 13:209–21. doi: 10.2147/JIR.S249384
3. Elkoshi Z. The Binary Model of Chronic Diseases Applied to COVID-19. Front Immunol (2021) 12:716084. doi: 10.3389/fimmu.2021.716084
4. Buckner JH. Mechanisms of Impaired Regulation by CD4(+)CD25(+)FOXP3(+) Regulatory T Cells in Human Autoimmune Diseases. Nat Rev Immunol (2010) 10(12):849–59. doi: 10.1038/nri2889
5. Valencia JC, Egbukichi N, Erwin-Cohen RA. Autoimmunity and Cancer, the Paradox Comorbidities Challenging Therapy in the Context of Preexisting Autoimmunity. J Interferon Cytokine Res (2019) 39(1):72–84. doi: 10.1089/jir.2018.0060
6. van den Broek ME, Kägi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, et al. Decreased Tumor Surveillance in Perforin-Deficient Mice. J Exp Med (1996) 1184(5):1781–90. doi: 10.1084/jem.184.5.1781
7. Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, et al. Regulatory T Cells Reversibly Suppress Cytotoxic T Cell Function Independent of Effector Differentiation. Immunity (2006) 25(1):129–41. doi: 10.1016/j.immuni.2006.04.015
8. Hasenkrug KJ, Myers LM. In Vitro and In Vivo Analyses of Regulatory T Cell Suppression of CD8+ T Cells. Methods Mol Biol (2011) 707:45–54. doi: 10.1007/978-1-61737-979-6_4
9. Levings MK, Sangregorio R, Roncarolo MG. Human Cd25(+)Cd4(+) T Regulatory Cells Suppress Naive and Memory T Cell Proliferation and can be Expanded In Vitro Without Loss of Function. J Exp Med (2001) 193(11):1295–302. doi: 10.1084/jem.193.11.1295
10. Shen E, Zhao K, Wu C, Yang B. The Suppressive Effect of CD25+Treg Cells on Th1 Differentiation Requires Cell-Cell Contact Partially via TGF-β Production. Cell Biol Int (2011) 35(7):705–12. doi: 10.1042/CBI20100528
11. Bellinghausen I, König B, Böttcher I, Knop J, Saloga J. Inhibition of Human Allergic T-Helper Type 2 Immune Responses by Induced Regulatory T Cells Requires the Combination of Interleukin-10-Treated Dendritic Cells and Transforming Growth Factor-Beta for Their Induction. Clin Exp Allergy (2006) 36(12):1546–55. doi: 10.1111/j.1365-2222.2006.02601.x
12. Xu HM. Th1 Cytokine-Based Immunotherapy for Cancer. Hepatobiliary Pancreat Dis Int (2014) 13(5):482–94. doi: 10.1016/s1499-3872(14)60305-2
13. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. Biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
14. Ellyard JI, Simson L, Parish CR. Th2-Mediated Anti-Tumour Immunity: Friend or Foe? Tissue Antigens (2007) 70(1):1–11. doi: 10.1111/j.1399-0039.2007.00869.x
15. Ye J, Livergood RS, Peng G. The Role and Regulation of Human Th17 Cells in Tumor Immunity. Am J Pathol (2013) 182(1):10–20. doi: 10.1016/j.ajpath.2012.08.041
16. Li L, Yang C, Zhao Z, Xu B, Zheng M, Zhang C, et al. Skewed T-Helper (Th)1/2- and Th17/T Regulatory−Cell Balances in Patients With Renal Cell Carcinoma. Mol Med Rep (2015) 11(2):947–53. doi: 10.3892/mmr.2014.2778
17. Wang LH, Wang LL, Zhang J, Zhang P, Li SZ. [Th1/Th2 and Treg/Th17 Cell Balance in Peripheral Blood of Patients With Ovarian Cancer]. Nan Fang Yi Ke Da Xue Xue Bao (2017) 37(8):1066–70. doi: 10.3969/j.issn.1673-4254.2017.08.11
18. Wang J, Cai D, Ma B, Wu G, Wu J. Skewing the Balance of Regulatory T-Cells and T-Helper 17 Cells in Breast Cancer Patients. J Int Med Res (2011) 39(3):691–701. doi: 10.1177/147323001103900301
19. Chen Z, Ding J, Pang N, Du R, Meng W, Zhu Y, et al. The Th17/Treg Balance and the Expression of Related Cytokines in Uygur Cervical Cancer Patients. Diagn Pathol (2013) 8:61. doi: 10.1186/1746-1596-8-61
20. Zhang Y, Ma D, Zhang Y, Tian Y, Wang X, Qiao Y, et al. The Imbalance of Th17/Treg in Patients With Uterine Cervical Cancer. Clin Chim Acta (2011) 412(11-12):894–900. doi: 10.1016/j.cca.2011.01.015
21. Zhang W, Hou F, Zhang Y, Tian Y, Jiao J, Ma D, et al. Changes of Th17/Tc17 and Th17/Treg Cells in Endometrial Carcinoma. Gynecol Oncol (2014) 132(3):599–605. doi: 10.1016/j.ygyno.2013.12.036
22. Lan YT, Fan XP, Fan YC, Zhao J, Wang K. Change in the Treg/Th17 Cell Imbalance in Hepatocellular Carcinoma Patients and Its Clinical Value. Med (Baltimore) (2017) 96(32):e7704. doi: 10.1097/MD.0000000000007704
23. Wang L, Zhang Y, Xie F. T-Regulatory Cell/T Helper 17 Cell Imbalance Functions as Prognostic Biomarker of Oral Squamous Cell Carcinoma - CONSORT. Med (Baltimore) (2020) 99(49):e23145. doi: 10.1097/MD.0000000000023145
24. Wang X, Wang L, Mo Q, Dong Y, Wang G, Ji A. Changes of Th17/Treg Cell and Related Cytokines in Pancreatic Cancer Patients. Int J Clin Exp Pathol (2015) 8(5):5702–8.
25. Duan MC, Zhong XN, Liu GN, Wei JR. The Treg/Th17 Paradigm in Lung Cancer. J Immunol Res (2014) 2014:730380. doi: 10.1155/2014/730380
26. Li Q, Li Q, Chen J, Liu Y, Zhao X, Tan B, et al. Prevalence of Th17 and Treg Cells in Gastric Cancer Patients and Its Correlation With Clinical Parameters. Oncol Rep (2013) 30(3):1215–22. doi: 10.3892/or.2013.2570
27. Gong Y, Wang L, Yu H, Alpert N, Cohen MD, Prophete C, et al. Prostate Cancer in World Trade Center Responders Demonstrates Evidence of an Inflammatory Cascade. Mol Cancer Res (2019) 17(8):1605–12. doi: 10.1158/1541-7786.MCR-19-0115
28. Gordon KB, Lebwohl M, Papp KA, Bachelez H, Wu JJ, Langley RG, et al. Long-Term Safety of Risankizumab From 17 Clinical Trials in Patients With Moderate-To-Severe Plaque Psoriasis. Br J Dermatol (2021). doi: 10.1111/bjd.20818
29. Lebwohl M, Deodhar A, Griffiths CEM, Menter MA, Poddubnyy D, Bao W, et al. The Risk of Malignancy in Patients With Secukinumab-Treated Psoriasis, Psoriatic Arthritis and Ankylosing Spondylitis: Analysis of Clinical Trial and Postmarketing Surveillance Data With Up to Five Years of Follow-Up. Br J Dermatol (2021) 185(5):935–44. doi: 10.1111/bjd.20136
30. Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long-Term Safety of Secukinumab in Patients With Moderate-to-Severe Plaque Psoriasis, Psoriatic Arthritis, and Ankylosing Spondylitis: Integrated Pooled Clinical Trial and Post-Marketing Surveillance Data. Arthritis Res Ther (2019) 21(1):111. doi: 10.1186/s13075-019-1882-2
31. Armstrong A, Paul C, Puig L, Boehncke WH, Freeman M, Torii H, et al. Safety of Ixekizumab Treatment for Up to 5 Years in Adult Patients With Moderate-To-Severe Psoriasis: Results From Greater Than 17,000 Patient-Years of Exposure. Dermatol Ther (Heidelb) (2020) 10(1):133–50. doi: 10.1007/s13555-019-00340-3
32. Ergen EN, Yusuf N. Inhibition of Interleukin-12 and/or Interleukin-23 for the Treatment of Psoriasis: What is the Evidence for an Effect on Malignancy? Exp Dermatol (2018) 27(7):737–47. doi: 10.1111/exd.13676
33. Uribe-Querol E, Rosales C. Neutrophils in Cancer: Two Sides of the Same Coin. J Immunol Res (2015) 2015:983698. doi: 10.1155/2015/983698
34. Shaul ME, Eyal O, Guglietta S, Aloni P, Zlotnik A, Forkosh E, et al. Circulating Neutrophil Subsets in Advanced Lung Cancer Patients Exhibit Unique Immune Signature and Relate to Prognosis. FASEB J (2020) 34(3):4204–18. doi: 10.1096/fj.201902467R
35. Duan Z, Luo Y. Targeting Macrophages in Cancer Immunotherapy. Signal Transduct Target Ther (2021) 6(1):127. doi: 10.1038/s41392-021-00506-6
36. Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The Role of Macrophages in Cancer Development and Therapy. Cancers (Basel) (2021) 13(8):1946. doi: 10.3390/cancers13081946
37. Liao Z, Chua D, Tan NS. Reactive Oxygen Species: A Volatile Driver of Field Cancerization and Metastasis. Mol Cancer (2019) 18(1):65. doi: 10.1186/s12943-019-0961-y
38. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
39. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of Regulatory T Cells by Macrophages is Dependent on Production of Reactive Oxygen Species. Proc Natl Acad Sci USA (2010) 107(41):17686–91. doi: 10.1073/pnas.1012016107
40. Kim HR, Lee A, Choi EJ, Hong MP, Kie JH, Lim W, et al. Reactive Oxygen Species Prevent Imiquimod-Induced Psoriatic Dermatitis Through Enhancing Regulatory T Cell Function. PloS One (2014) 9(3):e91146. doi: 10.1371/journal.pone.0091146
41. Chen X, Song M, Zhang B, Zhang Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med Cell Longev (2016) 2016:1580967. doi: 10.1155/2016/1580967
42. Chang CH, Pauklin S. ROS and Tgfβ: From Pancreatic Tumour Growth to Metastasis. J Exp Clin Cancer Res (2021) 40(1):152. doi: 10.1186/s13046-021-01960-4
43. Kotsafti A, Scarpa M, Castagliuolo I, Scarpa M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers (Basel) (2020) 12(7):1748. doi: 10.3390/cancers12071748
44. Oliva MR, Iradi A, Garrido F, Ramos M, Oltra AM, Muñiz P, et al. Oxidative Stress Induces the Expression of the Major Histocompatibility Complex in Murine Tumor Cells. Free Radic Res (2001) 35(2):119–28. doi: 10.1080/10715760100300661
45. Charni S, de Bettignies G, Rathore MG, Aguiló JI, van den Elsen PJ, Haouzi D, et al. Oxidative Phosphorylation Induces De Novo Expression of the MHC Class I in Tumor Cells Through the ERK5 Pathway. J Immunol (2010) 185(6):3498–503. doi: 10.4049/jimmunol.1001250
46. Oberkampf M, Guillerey C, Mouriès J, Rosenbaum P, Fayolle C, Bobard A, et al. Mitochondrial Reactive Oxygen Species Regulate the Induction of CD8+ T Cells by Plasmacytoid Dendritic Cells. Nat Commun (2018) 9(1):2241. doi: 10.1038/s41467-018-04686-8
47. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 Controls Phagosomal pH to Regulate Antigen Processing During Crosspresentation by Dendritic Cells. Cell (2006) 126(1):205–18. doi: 10.1016/j.cell.2006.05.035
48. Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, et al. NADPH Oxidase Controls Phagosomal pH and Antigen Cross-Presentation in Human Dendritic Cells. Blood (2008) 112(12):4712–22. doi: 10.1182/blood-2008-01-134791
49. Hansen M, Andersen MH. The Role of Dendritic Cells in Cancer. Semin Immunopathol (2017) 39(3):307–16. doi: 10.1007/s00281-016-0592-y
50. Assi M. The Differential Role of Reactive Oxygen Species in Early and Late Stages of Cancer. Am J Physiol Regul Integr Comp Physiol (2017) 313(6):R646–53. doi: 10.1152/ajpregu.00247.2017
51. Jiang J, Wang K, Chen Y, Chen H, Nice EC, Huang C. Redox Regulation in Tumor Cell Epithelial-Mesenchymal Transition: Molecular Basis and Therapeutic Strategy. Signal Transduct Target Ther (2017) 2:17036. doi: 10.1038/sigtrans.2017.36
52. Lee SY, Ju MK, Jeon HM, Lee YJ, Kim CH, Park HG, et al. Reactive Oxygen Species Induce Epithelial−Mesenchymal Transition, Glycolytic Switch, and Mitochondrial Repression Through the Dlx−2/Snail Signaling Pathways in MCF−7 Cells. Mol Med Rep (2019) 20(3):2339–46. doi: 10.3892/mmr.2019.10466
53. Du S, Miao J, Zhu Z, Xu E, Shi L, Ai S, et al. NADPH Oxidase 4 Regulates Anoikis Resistance of Gastric Cancer Cells Through the Generation of Reactive Oxygen Species and the Induction of EGFR. Cell Death Dis (2018) 9(10):948. doi: 10.1038/s41419-018-0953-7
54. Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, et al. The Double-Edged Roles of ROS in Cancer Prevention and Therapy. Theranostics (2021) 11(10):4839–57. doi: 10.7150/thno.56747
55. Weinberg F, Ramnath N, Nagrath D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers (Basel) (2019) 11(8):1191. doi: 10.3390/cancers11081191
56. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in Cancer Therapy: The Bright Side of the Moon. Exp Mol Med (2020) 52(2):192–203. doi: 10.1038/s12276-020-0384-2
58. Mu J, Tai X, Iyer SS, Weissman JD, Singer A, Singer DS. Regulation of MHC Class I Expression by Foxp3 and its Effect on Regulatory T Cell Function. J Immunol (2014) 192(6):2892–903. doi: 10.4049/jimmunol.1302847
59. Joetham A, Takeda K, Miyahara N, Matsubara S, Ohnishi H, Koya T, et al. Activation of Naturally Occurring Lung CD4(+)CD25(+) Regulatory T Cells Requires CD8 and MHC I Interaction. Proc Natl Acad Sci USA (2007) 104(38):15057–62. doi: 10.1073/pnas.0706765104
60. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
61. Geiser AG, Letterio JJ, Kulkarni AB, Karlsson S, Roberts AB, Sporn MB. Transforming Growth Factor Beta 1 (TGF-Beta 1) Controls Expression of Major Histocompatibility Genes in the Postnatal Mouse: Aberrant Histocompatibility Antigen Expression in the Pathogenesis of the TGF-Beta 1 Null Mouse Phenotype. Proc Natl Acad Sci USA (1993) 90(21):9944–8. doi: 10.1073/pnas.90.21.9944
62. Ma D, Niederkorn JY. Transforming Growth Factor-Beta Down-Regulates Major Histocompatibility Complex Class I Antigen Expression and Increases the Susceptibility of Uveal Melanoma Cells to Natural Killer Cell-Mediated Cytolysis. Immunology (1995) 86(2):263–9.
63. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
64. Phares TW, Stohlman SA, Hwang M, Min B, Hinton DR, Bergmann CC. CD4 T Cells Promote CD8 T Cell Immunity at the Priming and Effector Site During Viral Encephalitis. J Virol (2012) 86(5):2416–27. doi: 10.1128/JVI.06797-11
65. Seliger B, Kloor M, Ferrone S. HLA Class II Antigen-Processing Pathway in Tumors: Molecular Defects and Clinical Relevance. Oncoimmunology (2017) 6(2):e1171447. doi: 10.1080/2162402X.2016.1171447
66. Wang H, Zhao S, Zhang X, Jia K, Deng J, Zhou C, et al. Major Histocompatibility Complex Class II Molecule in non-Small Cell Lung Cancer Diagnosis, Prognosis and Treatment. Onco Targets Ther (2019) 12:7281–8. doi: 10.2147/OTT.S214231
67. Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol Res (2016) 4(10):845–57. doi: 10.1158/2326-6066.CIR-16-0060
68. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T Cells Mediate Specific Suppression by Depleting Peptide-MHC Class II From Dendritic Cells. Nat Immunol (2019) 20(2):218–31. doi: 10.1038/s41590-018-0280-2
69. Delvig AA, Lee JJ, Chrzanowska-Lightowlers ZM, Robinson JH. TGF-Beta1 and IFN-Gamma Cross-Regulate Antigen Presentation to CD4 T Cells by Macrophages. J Leukoc Biol (2002) 72(1):163–6.
70. Nandan D, Reiner NE. TGF-Beta Attenuates the Class II Transactivator and Reveals an Accessory Pathway of IFN-Gamma Action. J Immunol (1997) 158(3):1095–101.
71. Bulut I, Meral M, Kaynar H, Pirim I, Bilici M, Gorguner M. Analysis of HLA Class I and II Alleles Regarding to Lymph Node and Distant Metastasis in Patients With non-Small Cell Lung Cancer. Lung Cancer (2009) 66(2):231–6. doi: 10.1016/j.lungcan.2009.01.012
72. Vial T, Descotes J. Immunosuppressive Drugs and Cancer. Toxicology (2003) 185(3):229–40. doi: 10.1016/s0300-483x(02)00612-1
73. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
74. Zhang XM, Liu CY, Shao ZH. Advances in the Role of Helper T Cells in Autoimmune Diseases. Chin Med J (Engl) (2020) 133(8):968–74. doi: 10.1097/CM9.0000000000000748
75. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When Worlds Collide: Th17 and Treg Cells in Cancer and Autoimmunity. Cell Mol Immunol (2018) 15(5):458–69. doi: 10.1038/s41423-018-0004-4
76. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The Role of the T Cell in Autoimmune Inflammation. Arthritis Res Ther (2005) 7 Suppl 2(Suppl 2):S4–14. doi: 10.1186/ar1703
77. Chen J, Li J, Gao H, Wang C, Luo J, Lv Z, et al. Comprehensive Evaluation of Different T-Helper Cell Subsets Differentiation and Function in Rheumatoid Arthritis. J BioMed Biotechnol (2012) 2012:535361. doi: 10.1155/2012/535361
78. Wong CK, Ho CY, Li EK, Lam CW. Elevation of Proinflammatory Cytokine (IL-18, IL-17, IL-12) and Th2 Cytokine (IL-4) Concentrations in Patients With Systemic Lupus Erythematosus. Lupus (2000) 9(8):589–93. doi: 10.1191/096120300678828703
79. Csiszár A, Nagy G, Gergely P, Pozsonyi T, Pócsik E. Increased Interferon-Gamma (IFN-Gamma), IL-10 and Decreased IL-4 mRNA Expression in Peripheral Blood Mononuclear Cells (PBMC) From Patients With Systemic Lupus Erythematosus (SLE). Clin Exp Immunol (2000) 122(3):464–70. doi: 10.1046/j.1365-2249.2000.01369.x
80. Deng Q, Luo Y, Chang C, Wu H, Ding Y, Xiao R. The Emerging Epigenetic Role of CD8+T Cells in Autoimmune Diseases: A Systematic Review. Front Immunol (2019) 10:856. doi: 10.3389/fimmu.2019.00856
81. Zhang L, Yuan Y, Xu Q, Jiang Z, Chu CQ. Contribution of Neutrophils in the Pathogenesis of Rheumatoid Arthritis. J BioMed Res (2019) 34(2):86–93. doi: 10.7555/JBR.33.20190075
82. Wirestam L, Arve S, Linge P, Bengtsson AA. Neutrophils-Important Communicators in Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front Immunol (2019) 10:2734. doi: 10.3389/fimmu.2019.02734
83. Ma WT, Gao F, Gu K, Chen DK. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front Immunol (2019) 10:1140. doi: 10.3389/fimmu.2019.01140
84. Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, et al. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. J Neuroinflamm (2013) 10:35. doi: 10.1186/1742-2094-10-35
85. Gardiner GJ, Deffit SN, McLetchie S, Pérez L, Walline CC, Blum JS. A Role for NADPH Oxidase in Antigen Presentation. Front Immunol (2013) 4:295. doi: 10.3389/fimmu.2013.00295
86. Urbonaviciute V, Luo H, Sjöwall C, Bengtsson A, Holmdahl R. Low Production of Reactive Oxygen Species Drives Systemic Lupus Erythematosus. Trends Mol Med (2019) 25(10):826–35. doi: 10.1016/j.molmed.2019.06.001
87. Kelkka T, Kienhöfer D, Hoffmann M, Linja M, Wing K, Sareila O, et al. Reactive Oxygen Species Deficiency Induces Autoimmunity With Type 1 Interferon Signature. Antioxid Redox Signal (2014) 21(16):2231–45. doi: 10.1089/ars.2013.5828
88. Kienhöfer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental Lupus is Aggravated in Mouse Strains With Impaired Induction of Neutrophil Extracellular Traps. JCI Insight (2017) 2(10):e92920. doi: 10.1172/jci.insight.92920
89. Olofsson P, Holmberg J, Tordsson J, Lu S, Akerström B, Holmdahl R. Positional Identification of Ncf1 as a Gene That Regulates Arthritis Severity in Rats. Nat Genet (2003) 33(1):25–32. doi: 10.1038/ng1058
90. Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages Suppress T Cell Responses and Arthritis Development in Mice by Producing Reactive Oxygen Species. J Clin Invest (2007) 117:3020–8. doi: 10.1172/JCI31935
91. Hultqvist M, Bäcklund J, Bauer K, Gelderman KA, Holmdahl R. Lack of Reactive Oxygen Species Breaks T Cell Tolerance to Collagen Type II and Allows Development of Arthritis in Mice. J Immunol (2007) 179(3):1431–7. doi: 10.4049/jimmunol.179.3.1431
92. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring Oxidant Signaling Suppresses Proarthritogenic T Cell Effector Functions in Rheumatoid Arthritis. Sci Transl Med (2016) 8(331):331ra38. doi: 10.1126/scitranslmed.aad7151
93. Khmaladze IA, Kelkka T, Guerard S, Wing K, Pizzolla A, Saxena A, et al. Mannan Induces ROS-Regulated, IL-17A-Dependent Psoriasis Arthritis-Like Disease in Mice. Proc Natl Acad Sci USA (2014) 111(35):E3669–78. doi: 10.1073/pnas.1405798111
94. Huberle A, Beyeen AD, Ockinger J, Ayturan M, Jagodic M, de Graaf KL, et al. Advanced Intercross Line Mapping Suggests That Ncf1 (Ean6) Regulates Severity in an Animal Model of Guillain-Barre Syndrome. J Immunol (2009) 182(7):4432–8. doi: 10.4049/jimmunol.0803847
95. Strickland FM, Li Y, Johnson K, Sun Z, Richardson BC. CD4(+) T Cells Epigenetically Modified by Oxidative Stress Cause Lupus-Like Autoimmunity in Mice. J Autoimmun (2015) 62:75–80. doi: 10.1016/j.jaut.2015.06.004
96. Koppenol W. The Chemistry of Peroxynitrite, a Biological Toxin. Química Nova (1998) 21:1998. doi: 10.1590/S0100-40421998000300014
97. Carballal S, Bartesaghi S, Radi R. Kinetic and Mechanistic Considerations to Assess the Biological Fate of Peroxynitrite. Biochim Biophys Acta (2014) 1840(2):768–80. doi: 10.1016/j.bbagen.2013.07.005
98. Perl A. Oxidative Stress in the Pathology and Treatment of Systemic Lupus Erythematosus. Nat Rev Rheumatol (2013) 9(11):674–86. doi: 10.1038/nrrheum.2013.147
99. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial Oxidative Damage Underlies Regulatory T Cell Defects in Autoimmunity. Cell Metab (2020) 32(4):591–604.e7. doi: 10.1016/j.cmet.2020.07.001
100. Yang J, Yang X, Zou H, Li M. Oxidative Stress and Treg and Th17 Dysfunction in Systemic Lupus Erythematosus. Oxid Med Cell Longev (2016) 2016:2526174. doi: 10.1155/2016/2526174
101. Perrea DN, Moulakakis KG, Poulakou MV, Vlachos IS, Papachristodoulou A, Kostakis AI. Correlation Between Oxidative Stress and Immunosuppressive Therapy in Renal Transplant Recipients With an Uneventful Postoperative Course and Stable Renal Function. Int Urol Nephrol (2006) 38(2):343–8. doi: 10.1007/s11255-006-0054-x
102. Aoki CA, Borchers AT, Li M, Flavell RA, Bowlus CL, Ansari AA, et al. Transforming Growth Factor Beta (TGF-Beta) and Autoimmunity. Autoimmun Rev (2005) 4(7):450–9. doi: 10.1016/j.autrev.2005.03.006
103. Gorelik L, Flavell RA. Abrogation of TGFbeta Signaling in T Cells Leads to Spontaneous T Cell Differentiation and Autoimmune Disease. Immunity (2000) 12(2):171–81. doi: 10.1016/s1074-7613(00)80170-3
104. McPhee CG, Sproule TJ, Shin DM, Bubier JA, Schott WH, Steinbuck MP, et al. MHC Class I Family Proteins Retard Systemic Lupus Erythematosus Autoimmunity and B Cell Lymphomagenesis. J Immunol (2011) 187(9):4695–704. doi: 10.4049/jimmunol.1101776
105. Chan OT, Paliwal V, McNiff JM, Park SH, Bendelac A, Shlomchik MJ. Deficiency in Beta(2)-Microglobulin, But Not CD1, Accelerates Spontaneous Lupus Skin Disease While Inhibiting Nephritis in MRL-Fas(lpr) Nice: An Example of Disease Regulation at the Organ Level. J Immunol (2001) 167(5):2985–90. doi: 10.4049/jimmunol.167.5.2985
106. Christianson GJ, Blankenburg RL, Duffy TM, Panka D, Roths JB, Marshak-Rothstein A, et al. Beta2-Microglobulin Dependence of the Lupus-Like Autoimmune Syndrome of MRL-Lpr Mice. J Immunol (1996) 156(12):4932–9.
107. Mozes E, Kohn LD, Hakim F, Singer DS. Resistance of MHC Class I-Deficient Mice to Experimental Systemic Lupus Erythematosus. Science (1993) 261(5117):91–3. doi: 10.1126/science.8316860
108. Fiorillo MT, Paladini F, Tedeschi V, Sorrentino R. HLA Class I or Class II and Disease Association: Catch the Difference If You Can. Front Immunol (2017) 8:1475. doi: 10.3389/fimmu.2017.01475
109. Zakharova MY, Belyanina TA, Sokolov AV, Kiselev IS, Mamedov AE. The Contribution of Major Histocompatibility Complex Class II Genes to an Association With Autoimmune Diseases. Acta Naturae (2019) 11(4):4–12. doi: 10.32607/20758251-2019-11-4-4-12
110. Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, et al. Defining the Role of the MHC in Autoimmunity: A Review and Pooled Analysis. PloS Genet (2008) 4(4):e1000024. doi: 10.1371/journal.pgen.1000024
111. Lipski DA, Dewispelaere R, Foucart V, Caspers LE, Defrance M, Bruyns C, et al. MHC Class II Expression and Potential Antigen-Presenting Cells in the Retina During Experimental Autoimmune Uveitis. J Neuroinflamm (2017) 14(1):136. doi: 10.1186/s12974-017-0915-5
112. Turesson C. Endothelial Expression of MHC Class II Molecules in Autoimmune Disease. Curr Pharm Des (2004) 10(2):129–43. doi: 10.2174/1381612043453414
113. Cavalli G, Hayashi M, Jin Y, Yorgov D, Santorico SA, Holcomb C, et al. MHC Class II Super-Enhancer Increases Surface Expression of HLA-DR and HLA-DQ and Affects Cytokine Production in Autoimmune Vitiligo. Proc Natl Acad Sci USA (2016) 113(5):1363–8. doi: 10.1073/pnas.1523482113
114. Coureau M, Meert AP, Berghmans T, Grigoriu B. Efficacy and Toxicity of Immune -Checkpoint Inhibitors in Patients With Preexisting Autoimmune Disorders. Front Med (Lausanne) (2020) 7:137. doi: 10.3389/fmed.2020.00137
115. Racanelli V, Prete M, Minoia C, Favoino E, Perosa F. Rheumatic Disorders as Paraneoplastic Syndromes. Autoimmun Rev (2008) 7(5):352–8. doi: 10.1016/j.autrev.2008.02.001
116. Vogrig A, Muñiz-Castrillo S, Desestret V, Joubert B, Honnorat J. Pathophysiology of Paraneoplastic and Autoimmune Encephalitis: Genes, Infections, and Checkpoint Inhibitors. Ther Adv Neurol Disord (2020) 13:1756286420932797. doi: 10.1177/1756286420932797
117. Umetsu A, Shimizu T, Iwamoto N, Hashiguchi K, Eguchi M, Okamoto M, et al. Paraneoplastic Syndrome Presenting With Polymyalgia Rheumatica-Like Accumulations on 18F-Fluorodeoxyglucose-Positron Emission Tomography/Computed Tomography. Intern Med (2019) 58(6):861–4. doi: 10.2169/internalmedicine.1847-18
118. Rees MJ, Steinberg A, Romas E, Ford S, Roberts V, Ierino FL. Paraneoplastic Systemic Lupus Erythematosus Associated With Colorectal Cancer. Oxf Med Case Rep (2019) 2019(2):omy131. doi: 10.1093/omcr/omy131
119. Hagler KT, Lynch JW Jr. Paraneoplastic Manifestations of Lymphoma. Clin Lymphoma (2004) 5(1):29–36. doi: 10.3816/clm.2004.n.007
120. Small M, Treilleux I, Couillault C, Pissaloux D, Picard G, Paindavoine S, et al. Genetic Alterations and Tumor Immune Attack in Yo Paraneoplastic Cerebellar Degeneration. Acta Neuropathol (2018) 135(4):569–79. doi: 10.1007/s00401-017-1802-y
121. Giat E, Ehrenfeld M, Shoenfeld Y. Cancer and Autoimmune Diseases. Autoimmun Rev (2017) 16(10):1049–57. doi: 10.1016/j.autrev.2017.07.022
122. Shah AA, Rosen A, Hummers L, Wigley F, Casciola-Rosen L. Close Temporal Relationship Between Onset of Cancer and Scleroderma in Patients With RNA Polymerase I/III Antibodies. Arthritis Rheum (2010) 62(9):2787–95. doi: 10.1002/art.27549
123. Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al. Myositis-Specific Anti-155/140 Autoantibodies Target Transcription Intermediary Factor 1 Family Proteins. Arthritis Rheum (2012) 64(2):513–22. doi: 10.1002/art.33403
124. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and Malignancy Risk: A Meta Analysis Study. J Rheumatol (2015) 42(2):282–91. doi: 10.3899/jrheum.140566
125. Franks AL, Slansky JE. Multiple Associations Between a Broad Spectrum of Autoimmune Diseases, Chronic Inflammatory Diseases and Cancer. Anticancer Res (2012) 32(4):1119–36.
126. Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Effect of Autoimmune Diseases on Risk and Survival in Histology-Specific Lung Cancer. Eur Respir J (2012) 40(6):1489–95. doi: 10.1183/09031936.00222911
127. Liu X, Ji J, Forsti A, Sundquist K, Sundquist J, Hemminki K. Autoimmune Disease and Subsequent Urological Cancer. J Urol (2013) 189(6):2262–8. doi: 10.1016/j.juro.2012.12.014
128. Castro FA, Liu X, Försti A, Ji J, Sundquist J, Sundquist K, et al. Increased Risk of Hepatobiliary Cancers After Hospitalization for Autoimmune Disease. Clin Gastroenterol Hepatol (2014) 12(6):1038–45.e7. doi: 10.1016/j.cgh.2013.11.007
129. Hemminki K, Liu X, Försti A, Ji J, Sundquist J, Sundquist K. Effect of Autoimmune Diseases on Incidence and Survival in Subsequent Multiple Myeloma. J Hematol Oncol (2012) 5:59. doi: 10.1186/1756-8722-5-59
130. Hemminki K, Liu X, Ji J, Försti A, Sundquist J, Sundquist K. Effect of Autoimmune Diseases on Risk and Survival in Female Cancers. Gynecol Oncol (2012) 127(1):180–5. doi: 10.1016/j.ygyno.2012.07.100
131. Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Effect of Autoimmune Diseases on Mortality and Survival in Subsequent Digestive Tract Cancers. Ann Oncol (2012) 23(8):2179–84. doi: 10.1093/annonc/mdr590
132. Kaae J, Wohlfahrt J, Boyd HA, Wulf HC, Biggar RJ, Melbye M. The Impact of Autoimmune Diseases on the Incidence and Prognosis of Cutaneous Malignant Melanoma. Cancer Epidemiol Biomarkers Prev (2007) 16(9):1840–4. doi: 10.1158/1055-9965.EPI-07-0459
133. Zintzaras E, Voulgarelis M, Moutsopoulos HM. The Risk of Lymphoma Development in Autoimmune Diseases: A Meta-Analysis. Arch Intern Med (2005) 165(20):2337–44. doi: 10.1001/archinte.165.20.2337
134. Machado RI, Braz Ade S, Freire EA. Incidence of Neoplasms in the Most Prevalent Autoimmune Rheumatic Diseases: A Systematic Review. Rev Bras Reumatol (2014) 54(2):131–9.
135. Truman S. The Impact of Autoimmune Disease on Breast Cancer Survival. An MSc Thesis (2017). Available at: https://scholarcommons.sc.edu/cgi/viewcontent.cgi?article=5395&context=etd.
Keywords: cancer, autoimmune diseases, reactive oxygen species, standardized incidence ratio, hazard ratio, regulatory T cells, inflammation, major histocompability complex
Citation: Elkoshi Z (2022) Cancer and Autoimmune Diseases: A Tale of Two Immunological Opposites? Front. Immunol. 13:821598. doi: 10.3389/fimmu.2022.821598
Received: 24 November 2021; Accepted: 06 January 2022;
Published: 25 January 2022.
Edited by:
Sinisa Savic, University of Leeds, United KingdomReviewed by:
Michael Francis McDermott, University of Leeds, United KingdomCopyright © 2022 Elkoshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zeev Elkoshi, emVldi5lbGtvc2hpQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.