 Xiaohui Wei
Xiaohui Wei Shen Li
Shen Li Suqiu Wang
Suqiu Wang Guojiao Feng
Guojiao Feng Xiaoli Xie
Xiaoli Xie Zhuolin Li
Zhuolin Li Nianzhi Zhang
Nianzhi Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 February 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.820881
This article is part of the Research Topic Crystal Clear: Visualizing the Immune Recognition for the Mechanism and Intervention View all 16 articles
Polymorphisms can affect MHC-I binding peptide length preferences, but the mechanism remains unclear. Using a random peptide library combined with LC-MS/MS and de novo sequencing (RPLD-MS) technique, we found that two swine MHC-I molecules with high sequence homology, SLA-1*04:01 and SLA-1*13:01, had significant differences in length preference of the binding peptides. Compared with SLA-1*04:01, SLA-1*13:01 binds fewer short peptides with 8-10 amino acids, but more long peptides. A dodecapeptide peptide (RW12) can bind to both SLA-1*04:01 and SLA-1*13:01, but their crystal structures indicate that the binding modes are significantly different: the entirety of RW12 is embedded in the peptide binding groove of SLA-1*04:01, but it obviously protrudes from the peptide binding groove of SLA-1*13:01. The structural comparative analysis showed that only five differential amino acids of SLA-1*13:01 and SLA-1*04:01 were involved in the binding of RW12, and they determine the different ways of long peptides binding, which makes SLA-1*04:01 more restrictive on long peptides than SLA-1*13:01, and thus binds fewer long peptides. In addition, we found that the N terminus of RW12 extends from the groove of SLA-1*13:01, which is similar to the case previously found in SLA-1*04:01. However, this unusual peptide binding does not affect their preferences of binding peptide length. Our study will be helpful to understand the effect of polymorphisms on the length distribution of MHC-I binding peptides, and to screen SLA-I-restricted epitopes of different lengths and to design effective epitope vaccines.
Highly polymorphic major histocompatibility complex class I (MHC-I) molecules present a variety of epitope peptides for specific TCR recognition, which is a key signal to start CTL (cytotoxic T lymphocyte) immunity (1). The MHC-I molecule presents the peptide through the closed peptide binding groove, and both ends of the peptide are anchored in the groove, so the length of the peptide is limited, generally 8-10 amino acids (2, 3). Long peptides (up to 15 peptides) can also be presented by MHC-I and cause a CTL response (4–6), but the ratio is relatively low. Long peptides often show a ‘bulge’ conformation, so both ends can be fixed in the peptide binding groove of MHC-I (7, 8). The conformational differences of peptides with different lengths will further affect the recognition of the TCR repertoire (6). However, some recent studies found that some long peptides can extend from the N- or C- end of the peptide binding groove, which reveals a new pattern of MHC-I presenting long peptides (9–13). However, compared with short peptides, the number of identified long peptides is much lower, which limits the in-depth study of the mechanism of MHC-I presenting long peptides. In the PDB library (https://www.rcsb.org/), the number of MHC-I complexes with short peptides is approximately 10 times that of long peptide complexes.
In recent years, the peptide length preference of MHC-I and its influence on the immune response have attracted increasing attention (5, 14, 15). The development of peptidomics provides powerful tools for accurately identifying the peptide binding characteristics of MHC-I, including peptide length preference (4, 13, 16–19). Mass spectrometry (MS) analysis of the human MHC-I (HLA-I) peptide ligand shows that some HLA-I alleles prefer to present longer peptides (5, 14, 15, 20). This indicates that the polymorphism of HLA-I can influence the length distribution of the presented peptides, but the mechanism behind it is still unclear.
Compared with humans and mice, there are fewer studies on the presentation of long peptides by MHC-I in other species. At present, only the complex structures of chicken and horse MHC-I and long peptides have been resolved (21, 22). However, due to the lack of high affinity monoclonal antibodies, cell lines and MHC-I haplotype experimental animals, the peptide length distributions of animal MHC-I revealed by cellular ligandome are relatively rare (9, 11, 22–27). Database-independent de novo peptide sequencing by MS can identify peptidomes without any database (28–30). We developed a new method in vitro to identify the peptide ligands of MHC-I through a random peptide library combined with LC-MS/MS and de novo sequencing (RPLD-MS) (17, 19). The random peptide library was refolded with MHC-I heavy chain and beta-2-microglobulin (β2-M) to form the peptide/MHC-I complex (pMHC-I). Then, pMHC-I was purified, and the bound peptides were eluted and sequenced by LC-MS/MS and de novo sequencing. Finally, the peptide-binding motif of MHC-I was determined. To date, the peptide binding properties of MHC-I molecules of five species, pig, bat, frog, lizard and shark, have been identified by RPLD-MS (17, 19, 31–33). This overcomes the limitations of laboratory animals, cell lines and antibodies, and lowers the threshold for the study of animal MHC-I.
Our studies on two highly homologous pig MHC-I (SLA-I) molecules, SLA-I*04:01 and SLA-1*13:01, showed that RPLD-MS can sensitively reflect the changes in peptide presentation caused by micropolymorphism. A key amino acid variation, Y99 in SLA-I*04:01 and F99 in SLA-1*13:01, makes SLA-1*04:01 present more nonapeptides than SLA-1*13:01. RPLD-MS can identify the length distribution of peptides presented by MHC-I in vitro, and it can focus on the influence of the polymorphism of MHC-I molecules, without considering the processes of peptide presentation in vivo (19). In addition, we also found a dodecapeptide RVEDVTNTAEYW (RW12 for short) which can bind to SLA-1*04:01. The complex structure (pSLA-1*04:01RW12) indicated that the first Arg of RW12 can extend from the N terminus of the peptide binding groove of SLA-1*04:01 (13). In this study, the length distributions of peptides presented by SLA-I*04:01 and SLA-1*13:01 were compared by RPLD-MS, and it was found that SLA-1*13:01 can bind more long peptides than SLA-1*04:01. Structural analysis revealed that SLA-1*04:01 and SLA-1*13:01 present RW12 in a markedly different manner, which is related to their different preferences for binding peptide lengths. This study will help us better understand how highly polymorphic MHC-I presents peptides of different lengths.
A random peptide repertoire was synthesized with a distribution of 19 amino acids other than cysteine at an equal molar ratio in every position, as previously reported (13, 29, 30). In brief, random peptide libraries of different lengths were synthesized separately according to this method and mixed in an equimolar ratio according to the average molecular mass before use.
Peptides with defined sequences used in this study, such as RVEDVTNTAEYW, were synthesized and purified to 99% by reverse-phase high performance liquid chromatography (HPLC) and mass spectrometry (SciLight Biotechnology).
All random peptide repertoires and peptides were stored in lyophilized aliquots at -80°C after synthesis and dissolved in dimethyl sulfoxide (DMSO) before use.
The DNA fragments encoding extracellular domains of SLA-1*13:01 (GenBank accession No. AB847437.1) and sβ2-M (GenBank accession No. BAG32341) were cloned into pET-21a (+) vectors (Novagen) and expressed as inclusion bodies in E. coli BL21 (DE3). The mutants at positions 99 of SLA-1*13:01 were cloned by overlap PCR and expressed as inclusion bodies in E. coli BL21 (DE3). The recombinant proteins were purified as described previously and dissolved in 6 M Gua-HCl (34).
The proteins were refolded by stepwise dilution using buffer containing 100 mM Tris-HCl (pH 8.0), 2 mM EDTA, 400 mM L-Arg HCl, 5 mM GSH, and 0.5 mM GSSH, and treated at 277 K for 12 hours. Before renaturation, a random peptide library containing 5.12 mg octapeptide, 5.76 mg nonapeptide, 6.4 mg decapeptide, 7.04 mg undecapeptide, 7.68 mg dodecapeptide, 8.32 mg thirteen peptide, 8.96 mg tetradecepeptide and 9.6 mg pentadecapeptide, or 10 mg of fixed peptide was dissolved and added to 1 L of refolding solution. Then, the solutions dissolved SLA-1 and sβ2-M inclusion bodies were individually added to refolding buffer at a 1:1 molar ratio. The refolded complexes were concentrated and purified with a Superdex 200 16/60 column in Tris buffer (20 mM Tris [pH 8] and 50 mM NaCl), followed by Resource Q anion-exchange chromatography (GE Healthcare) in Tris buffers (Buffer A: 20 mM Tris [pH 8] and 5 mM NaCl; Buffer B: 20 mM Tris [pH 8] and 500 mM NaCl).
The peptide-containing fraction in the complex was eluted according to a previous method (13, 19). In brief, the complex was treated with 10% acetic acid at 65°C for 15 minutes and the peptides were collected through a 3-kDa filter. Then, the peptide-containing fraction was desalted using a C18 tip and separated on the EasyNano LC 1000 system (San Jose, California, Thermo Fisher Scientific). Methods as follows: the peptide component was loaded into a trap column (5-μm pore size, 150-μm i.d. ×3-cm length, 100 Å) and separated by a custom C18 column (3-μm pore size, 75-μm i.d. 315-cm length, 100 Å), with a flow rate of 450 μL/minute. The 60-minute linear gradient was operated as follows: 3% B (0.1% formic acid in acetonitrile [v/v])/97% A (0.1% formic acid in H2O [v/v]) to 6% B in 8 min, 6% B to 22% B in 37 min, 22% B to 35% B in 8 min, 35% B to 100% B in 2 min, and 100% B for 5 min. The MS data were acquired by a Q Exactive HF (Bremen, Thermo Fisher Scientific) in data-dependent acquisition mode. The top 20 precursors by intensity in the mass range m/z 300 to 1800 were sequentially fragmented with higher-energy collisional dissociation and a normalized collision energy of 27. The dynamic exclusion time was 20 s. The automatic gain control for MS1 and MS2 was set to 3e6 and 1e, and the resolution for MS1 and MS2 was set to 120 and 30K.
Based on MS/MS information, peptides were resolved from each spectrum (FDR=1%) by de novo sequencing in Peaks Studio software. The parameters were set as follows: the enzyme was set to nonspecific, the variable modifications were adjusted oxidation (M)/deamidated (N,Q), the peptide mass tolerance was approximately ± 10 ppm, and the fragment mass tolerance was set to 0.02 Da. The identified peptides adjusted by the detection threshold (score ≥ 50) are listed in Table S1.
To determine the RXEDVTNTAEYW (X represents any of twenty amino acids) library, MaxQuant1.6 software was used to search peptides based on MS/MS information. Posttranslational modifications (PTMs) were set to false, and the MS/MS match tolerance was set to 20 ppm. No-specificity was selected in the digestion option. The protein FDR was set to 0.01.
The circular dichroism (CD) spectra of the peptide-SLA-1 (pSLA-1) complexes were measured on a CD instrument (Chirascan; Applied Photophysic, Ltd.) using a Jasco J-810 spectrometer equipped with a water-circulating cell holder. As the temperature rose from 20°C to 80°C at a rate of 1°C/minute. A 1-mm optical path length cell was used for monitoring at 218 nm. The instrument used a thermistor to detect the temperature of the solution. Finally, the ratio of unfolded protein to the mean residue ellipticity (θ) was calculated. The unfolded score is shown as (θ - θN)/(θU - θN), where θN and θU are the mean residual ellipticity values in fully folded and fully unfolded states, respectively. The midpoint transition temperature (Tm) is calculated from the denaturation curve data in the Origin 9.1 program (OriginLab).
The refolded and purified protein complex was concentrated and performed using the sitting-drop and hanging-drop vapor diffusion method at 277 K and 291K. Complex crystals of SLA-1*13:01 with peptide RVEDVTNTAEYW (pSLA-1*13:01RW12) were obtained under PEG/Ion solution No.3 (0.2 M ammonium fluoride, 20% w/v polyethylene glycol 3,350). Prior to X-ray diffraction, the crystals were immersed in a stock solution containing 17% glycerol as a cryoprotectant, and then flash-cooled in a 100 K gaseous nitrogen stream. Then, the diffraction data of pSLA-1*13:01RW12 were collected and the R-AXIS IV++ imaging plate detector was used to resolve the resolution of 2.5 Å on Beamline BL19U1 (wavelength, 0.97892 Å) of the Shanghai Synchrotron Radiation Facility (Shanghai, China). The HKL-3000 software package (HKL Research) was used to index, integrate, scale and merge data (35). The crystallographic statistics for the complexes are listed in Table 1.
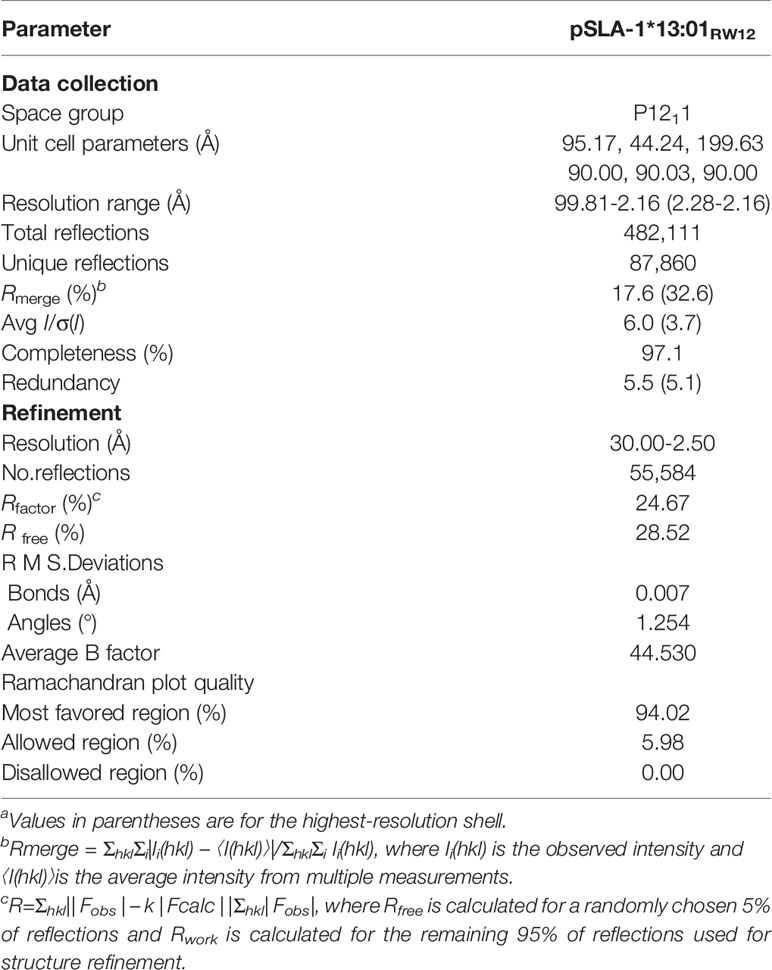
Table 1 X-ray diffraction data processing and refinement statistics.
Using SLA-1*13:01 (Protein Data Bank code 6KWO) as a search model, the structure of the pSLA-1*13:01RW12 complex was determined by molecular replacement using the Phaser program (36). The model was rebuilt using COOT (37) and refined by REFMACS5 (38). Refinement rounds were implemented using the refinement program in the PHENIX package with isotropic atomic displacement parameter (ADP) refinement and bulk solvent modeling (39). Finally, the stereochemical quality of the final model was assessed using the PROCHECK program (40). Structural illustrations and electron density-related figures were drawn using PyMOL software. Multiple-sequence alignment was performed with Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). The accessible surface area (ASA) and buried surface area (BSA) were calculated with PDB in Europe Proteins, Interfaces, Structures and Assemblies (PDBePISA, http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html), and the B factor was calculated with CCP4.
SLA-1*13:01 differs from SLA-1*04:01 by only eight residues, and they are all distributed on the peptide binding platform composed of α1 and α2 domains (Figures 1A, B). Our previous research showed that SLA-1*04:01 binds much more nonapeptides than SLA-1*13:01 (19). However, the in vitro refolding results of SLA-1*04:01 and SLA-1*13:01 with a mixed random peptide library of 7-15 amino acids showed that the peak of pSLA-1*13:01 is higher than the peak of pSLA-1*04:01, indicating SLA-1*13:01 can bind more random peptides of mixed length than SLA-1*04:01 (Figure 1C). RPLD-MS showed that compared with SLA-1*04:01 (13), SLA-1*13:01 could bind fewer short peptides of 8-10 amino acids, but more long peptides of 11-15 amino acids (Figure 1D and Table S1). It should be pointed out that among the peptides bound by SLA-1*13:01 and SLA-1*04:01, nonapeptide and decapeptide are the main peptides, which agrees with the consensus on the length distribution of MHC-I binding peptides. Compared with SLA-1*04:01, SLA-1*13:01 has a lower amount and proportion of binding nonapeptides (Figures 1D, E), which consists of the previous data (19).
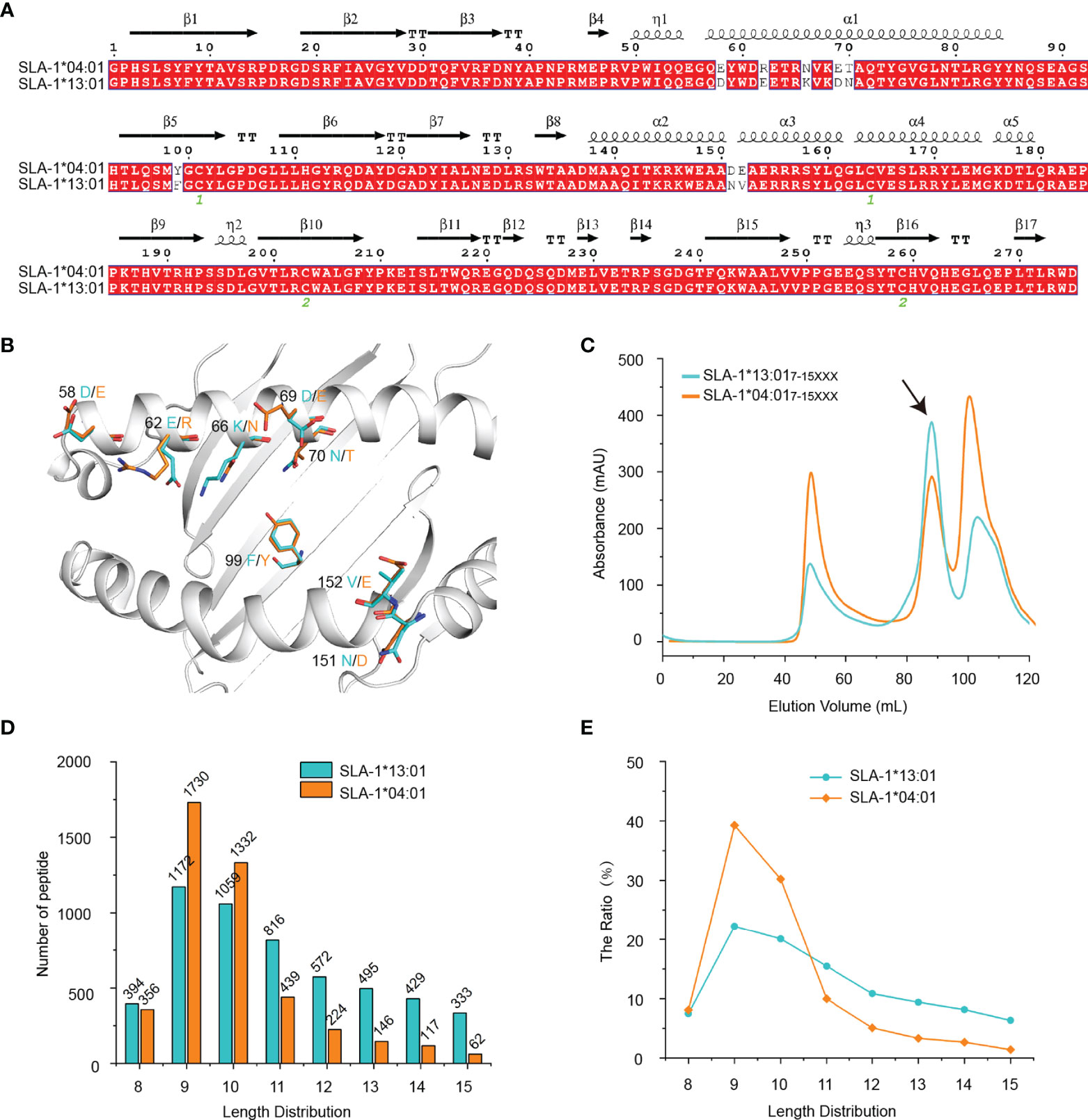
Figure 1 Analysis of the refolding of SLA-1*13:01 and SLA-1*04:01 bound peptide libraries and the determination of the eluted peptide length and quantity. (A) Structure-based sequence alignment of SLA-1*04:01 and SLA-1*13:01. (B) The structural location of the different residues between SLA-1*13:01 and SLA-1*04:01. (C) Gel filtration chromatograms of the in vitro refolding test of SLA-1*13:01 and SLA-1*04:01 with random peptides of mixed length. The black arrows point to the peak of the compound. (D, E) The length and quantity comparison between SLA-1*13:01 and SLA-1*04:01 eluted peptides.
Structural analysis of SLA-1*13:01 and SLA-1*04:01 complexed with long peptides is helpful to clarify their different preferences for peptide length. We have previously identified a dodecapeptide (RW12) that can bind to SLA-1*04:01 and solved the structure of their complex pSLA-1*04:01RW12 (13). Here, an in vitro refolding experiment showed that RW12 can also combine with SLA-1*13:01 to form the stable complex pSLA-1*13:01RW12 (Figure 2A). After purification and crystal screening of pSLA-1*13:01RW12, we successfully obtained the crystal and finally solved its structure at 2.5 Å, the same resolution of pSLA-1*04:01RW12 (Table 1). Structural analysis confirms that bound peptide conformation is not affected by crystal-crystal packing. The structural comparison between pSLA-1*04:01RW12 and pSLA-1*13:01RW12 showed that their overall structural similarity was high (RMSD = 0.9561), but the conformations of the presented RW12 peptide were obviously different (RMSD = 2.089) (Figure 2B).
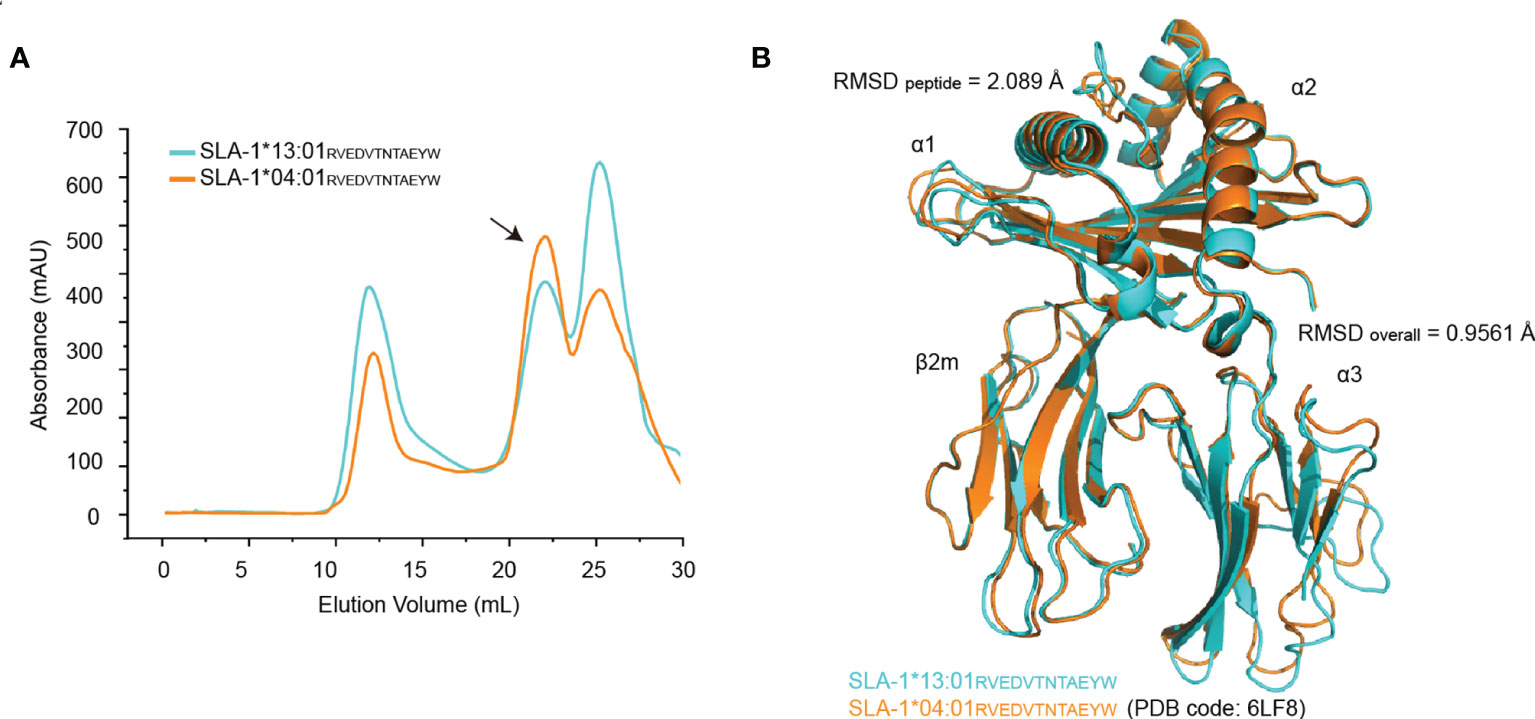
Figure 2 Structural determination of the pSLA-1*13:01RVEDVTNTAEYW complex. (A) Visual display of SLA-1*13:01 and SLA-1*04:01 in vitro refolding efficiency with the peptide RVEDVTNTAEYW by gel filtration chromatograms. The black arrows point to the peak of the compound. (B) The overall structural comparison between pSLA-1*13:01RW12 (cyan) and pSLA-1*04:01RW12 (orange) presented in cartoon form.
In the structures of pSLA-1*04:01RW12 and pSLA-1*13:01RW12, the electronic density maps of the RW12 peptide are clear and can be used for credible comparative analysis (Figure 3A). Our previous study showed that the binding mode of RW12 is different from the classical MHC-I peptide binding mode. The first residue Arg (P-1-R) at the N terminus of RW12 extends out of the peptide binding groove of SLA-1*04:01, the second residue Val (P1-V) is turned over and its side chain is accommodated by the A pocket (13). The same is true of the interaction between the N terminus of RW12 and SLA-1*13:01 (Figure 3B). Except for the N terminus, the conformations of the rest of RW12 are significantly different in SLA-1*13:01 and SLA-1*04:01. RW12 obviously protrudes from the peptide binding groove of SLA-1*13:01 but is deeply embedded in the groove of SLA-1*04:01 (Figure 3B), and the side chain orientation of each residue is also different (Figure 3C).
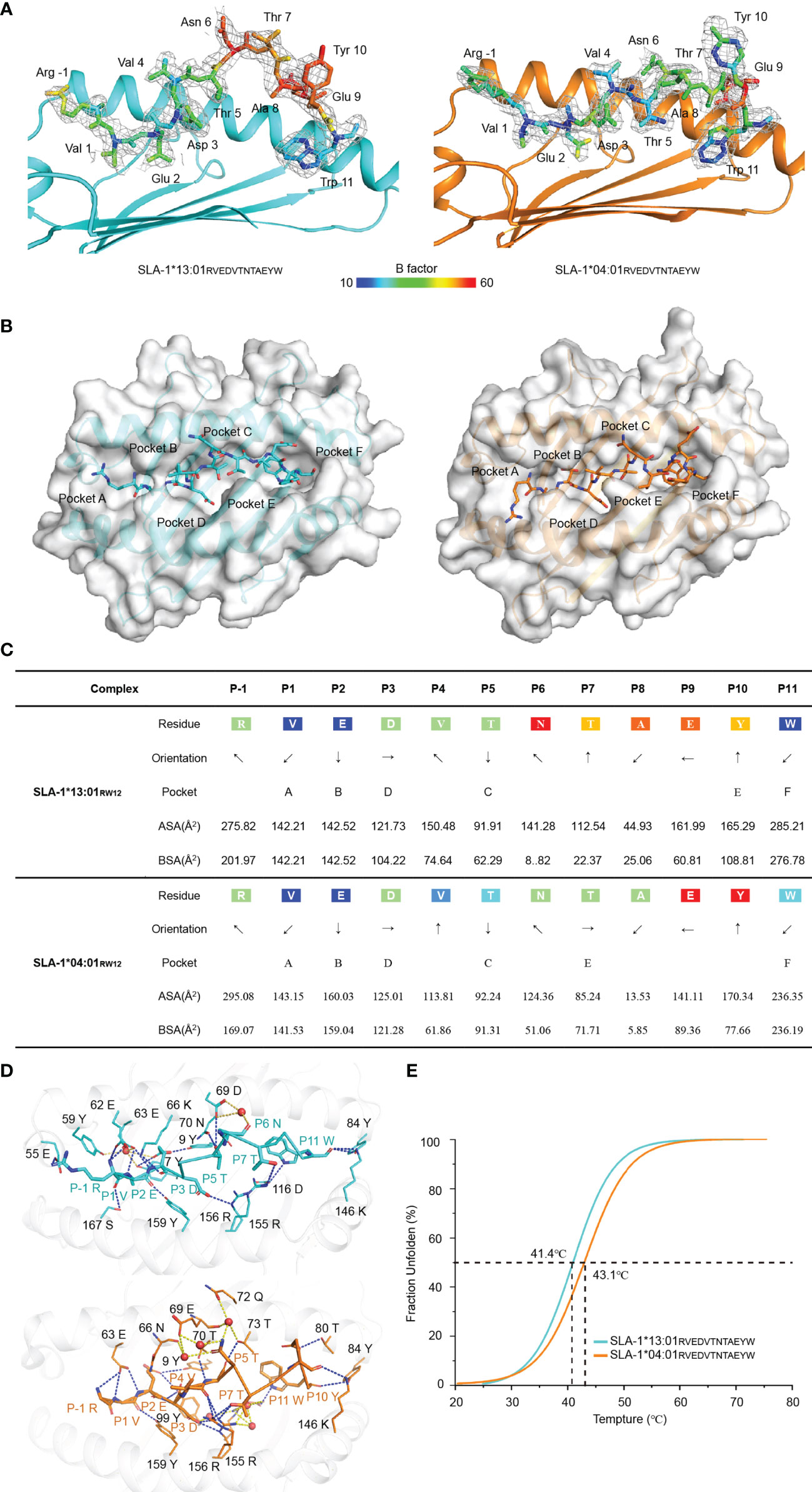
Figure 3 Electron density and overall conformation of the structurally defined peptides. (A) Electron densities and overall conformations of the peptide RVEDVTNTAEYW from the solved pSLA-1*13:01RW12 and pSLA-1*04:01RW12 complexes. Simulated CNS annealing omit maps calculated for the peptides are shown in blue at a contour of 1.0. (B) The structural location of the peptide RVEDVTNTAEYW residues inserted into the pocket. (C) General side chain orientations and the different interfacing areas of peptides presented in a table, as viewed in profile from the peptide N-terminus toward the C-terminus. Black arrows indicate the directions in which the residues point: up is toward the TCR, down is toward the floor of the ABG, left is toward the α1 helix domain, and right is toward the α2 helix domain. Pockets accommodating each residue are listed under the corresponding anchors within the ABG. ASA, accessible surface area of each residue; BSA, buried surface area of the residues. (D) Comparison of the forces that mediate peptide stabilization between pSLA-1*13:01RW12 and pSLA-1*04:01RW12. The red sphere is the solvent molecule. (E) Thermal stabilities of pSLA-1*13:01RW12 and pSLA-1*04:01RW12 analyzed by the CD spectroscopy. The stabilities can be measured by the Tm value. The Tm values of the complexes are labeled.
The hydrogen bonds between RW12 and SLA-1*13:01 are significantly less than that between RW12 and SLA-1*04:01, including the hydrogen bonds mediated by water molecules, especially in the middle region of the peptide binding groove (Figure 3D and Table 2). Compared with pSLA-1*04:01RW12, the residues from P6 to P10 of RW12 in pSLA-1*13:01RW12 have higher B factors, which indicates that these residues protruding from the peptide binding groove are less constrained (Figure 3A). The results of circular dichroism showed that the thermal stability of pSLA-1*04:01RW12 is slightly higher than that of pSLA-1*13:01RW12, consisting with the numbers of hydrogen bonds with RW12, although both of them can keep stable and crystallize (Figure 3E).
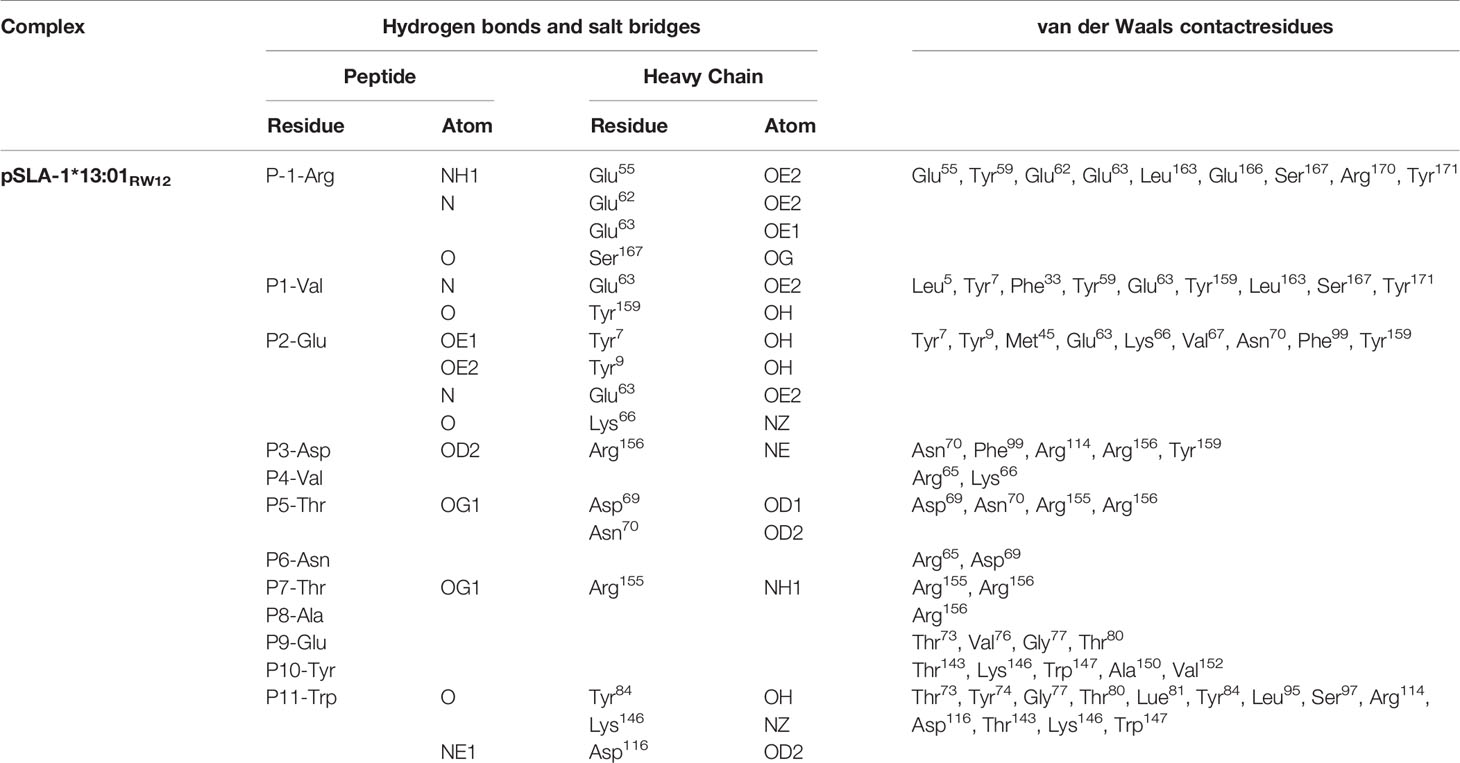
Table 2 The interactions between the peptide and the PBG of pSLA-1*13:01RW12.
The conformational difference between RW12 presented by SLA-1*13:01 and SLA-1*04:01 is caused by the different amino acids between these two SLA-I alleles. Structural analysis showed that only five of the eight differential amino acids directly interact with RW12, and the variable residues at positions 57, 151 and 152 do not contact RW12. 62E, 66K and 69D in SLA-1*13:01 can form hydrogen bonds with P-1-R, P2-E and P5-T of RW12 (Figure 4A), and 70T and 99Y in SLA-1*04:01 can form hydrogen bonds with P3-D and P5-T of RW12 (Figure 4B).
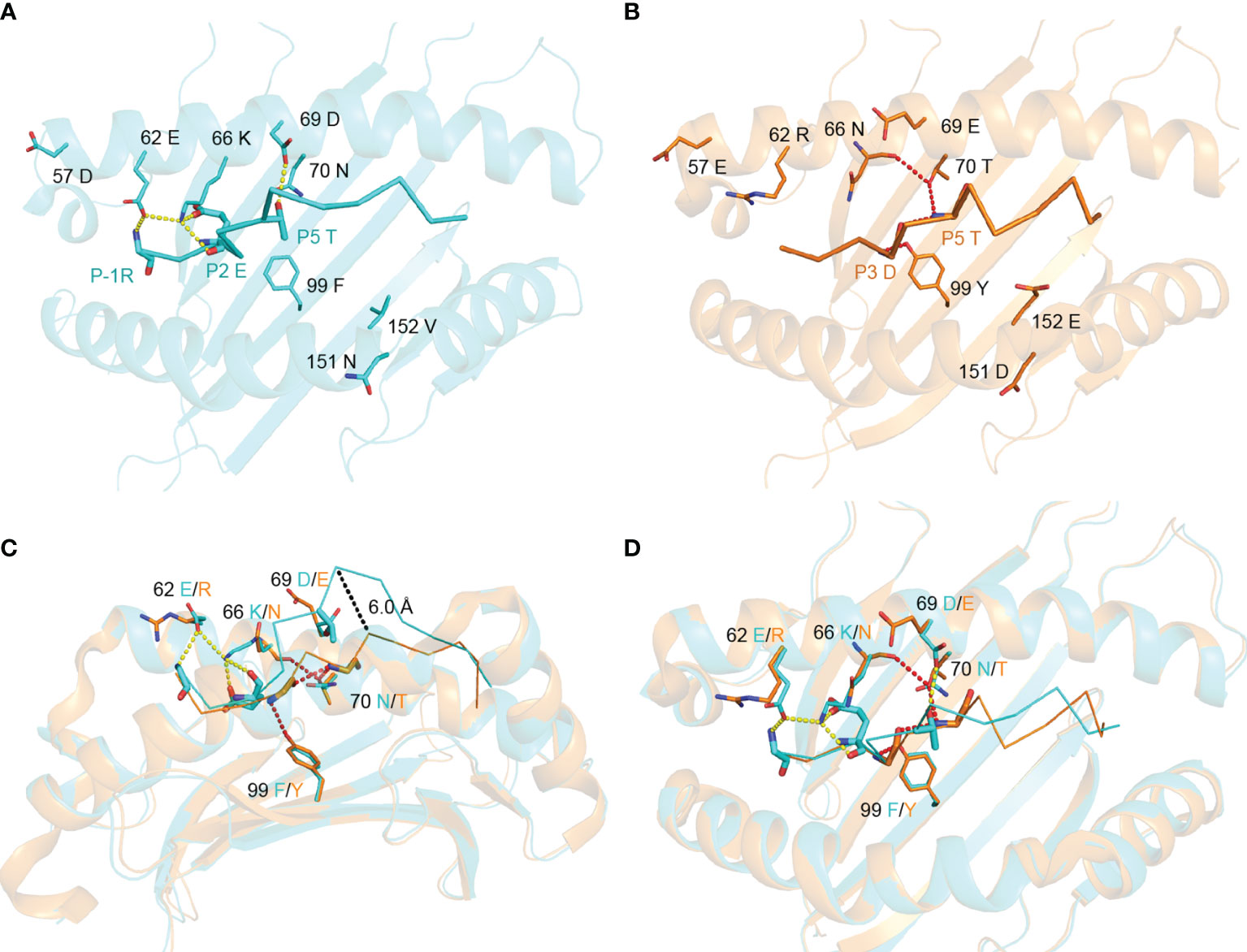
Figure 4 Contributions of variant amino acids between SLA-1*13:01 and SLA-1*04:01 to the conformation of peptide RVEDVTNTAEYW. (A) Analysis of the interaction between different amino acids and peptide in pSLA-1*13:01RW12. (B) Analysis of the interaction between variant amino acids and peptide in pSLA-1*04:01 RW12. (C, D) Comparison of the effect of amino acid differences between SLA-1*13:01 and SLA-1*04:01 on peptide conformation.
62E/R and 66K/N at the N terminus of the peptide binding groove did significantly alter the conformation of the P-1 to P2 residues of RW12 (Figures 4C, D). The conformational difference of RW12 starts from P3-D, the P3-D position in pSLA-1*13:01RW12 is relatively higher (Figure 4C), the positions of the subsequent residues are more elevated, and their side chain orientations are more variable (Figure 3C). These results indicated that the difference between SLA-1*04:01 and SLA-1*13:01 in presenting RW12 peptide may start from the interaction with P3-D and be consolidated later, while 69D/E, 70N/T and 99F/Y are just located in this region (Figures 4C, D).
Our previous study showed that 99Y/F is the key factor that leads to a significant difference between SLA-1*04:01 and SLA-1*13:01 when binding nonapeptides (19). 99Y in SLA-1*04:01 can form a hydrogen bond with the main chain of the P3 residue, but in SLA-1*13:01, 99F cannot form a similar hydrogen bond and leads to a decrease in its ability to bind nonapeptides. If the 99Y/F of SLA-1*04:01 and SLA-1*13:01 are replaced by mutations, the ability to bind nonapeptides between them will also be exchanged. Similar to the binding of nonapeptides, the main chain of P3-D of RW12 forms a hydrogen bond with 99Y of SLA-1*04:01, but not with 99F of SLA-1*13:01. The conformational difference of RW12 also starts from P3-D, 99Y of SLA-1*04:01 makes P3-D of RW12 tied to the bottom of the groove but 99F of SLA-1*13:01 cannot (Figure 4). To further analyze the role of 99Y/F, we compared the other solved structures of SLA-1*04:01, SLA-1*13:01 and its mutant SLA-1*13:01(F99Y). 99Y can form a conserved hydrogen bond with the P3 main chain in all structures (Figure 5A), so we believe that 99Y/F has an important influence on peptide binding, regardless of peptide length.
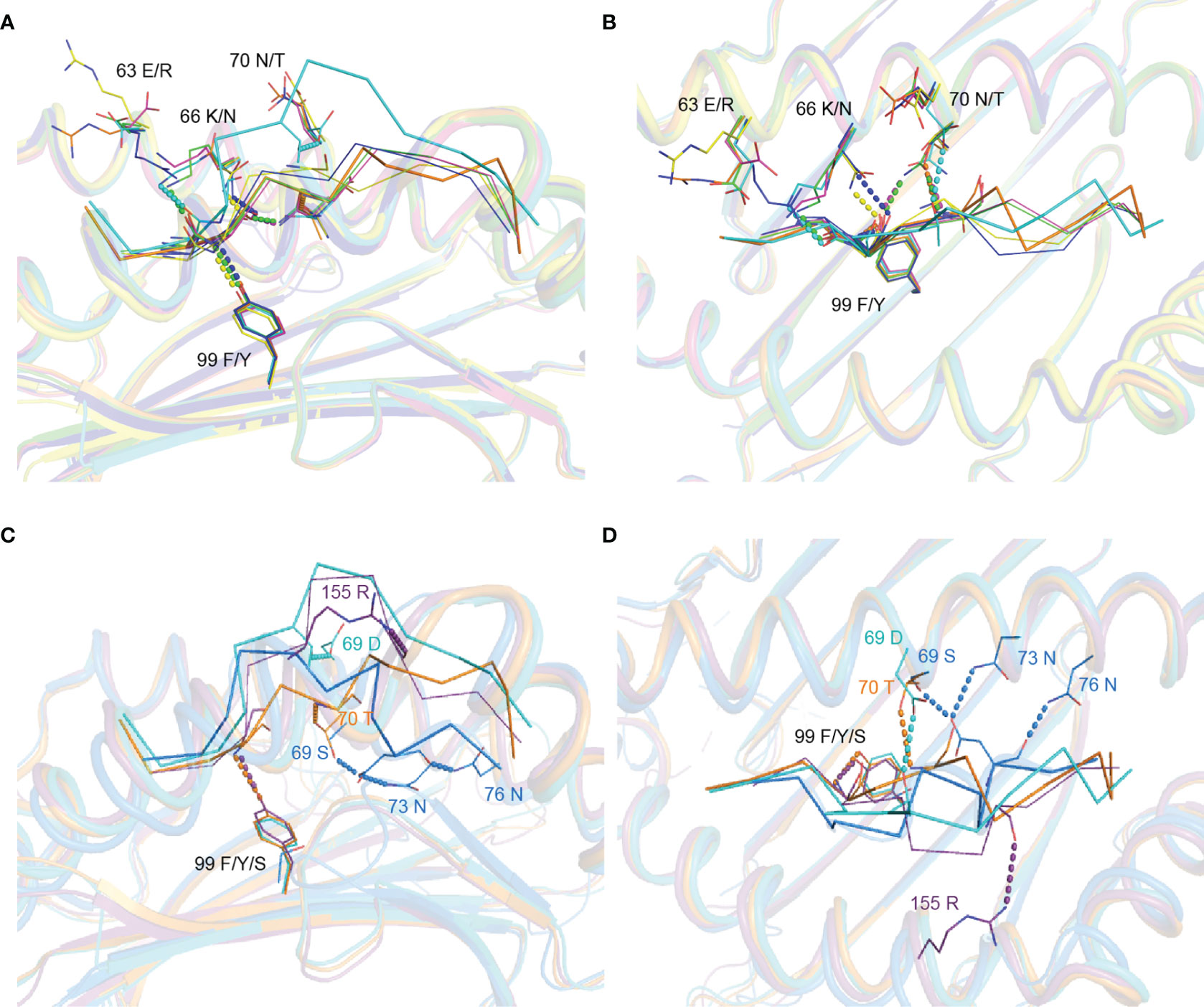
Figure 5 Analysis of the effect of variant residues between SLA-1*13:01 and SLA-1*04:01 on MHC-I binding peptides. (A, B) Structural comparison between determined pSLA-1*13:01RW12 (cyan), pSLA-1*04:01RW12 (orange, PDB code 6LF8), pSLA-1*04:01MY9 (blue, PDB code 6KWK), pSLA-1*13:01(F99Y)NW9 (green, PDB code 6KWN) and pSLA-1*13:01EW9 (magenta, PDB code 6KWO). The dotted lines of different colors indicate the interaction between the peptides and the residues of the antigen binding groove in different structures. (C, D) Structural comparison between determined pSLA-1*13:01RW12 (cyan), pSLA-1*04:01RW12 (orange, PDB code 6LF8), pMamu*B17IW11 (purple, PDB code 3RWD) and pBF2*21:01GL11 (purple blue, PDB code 2YF1). The dotted lines of different colors indicate the interaction between the peptides and the residues of the antigen binding groove in different structures.
Although the other variant residues do not interact with peptides in the stable way (Figures 5A, B), but they also have their own unique role in influencing peptide conformation. 70T of SLA-1*04:01forms a hydrogen bond with the main chain of P5-T, further limiting the subsequent residues of RW12 in the peptide binding groove (Figures 4B–D). 69D of SLA-1*13:01 forms a hydrogen bond with P5-T, which fixes the middle of RW12 outside the peptide binding groove, jointly determine the conformation of RW12 protruding out of the SLA-I*13:01 groove (Figures 4A, C, D). Overall, RW12 presents a typical ‘bulge’ conformation in pSLA-1*13:01RW12, while it is embedded in the groove in pSLA-1*04:01RW12. The maximum position deviation between them can reach 6.0 Å (Figure 4C).
By comparing the structures of other resolved MHC-I and undecapeptide complexes, we also found that the 99th residue cannot independently determine the conformation of the peptide. For example, 99S of the chicken MHC-I molecule (BF2*2101) does not form hydrogen bonds with the P3 residue, similar to SLA-1*13:01 and RW12, but its 69S, 73N and 76N residues are bound to peptide, which limits the whole peptide in the groove, similar to SLA-1*04:01 and RW12. 99Y of the monkey MHC-I molecule (Mamu*B17) forms a hydrogen bond with the P3 residue, similar to SLA-1*04:01 and RW12, but there is no strong restriction on the peptide, similar to SLA-1*13:01 and RW12 (Figures 5C, D). Therefore, for SLA-1*04:01 and SLA-1*13:01, the difference of 99Y/F should be critical factor for the binding of peptide, but it needs to cooperate with other different amino acids to jointly determine the conformation of peptide.
The N terminus of RW12 can extend out of the peptide binding groove of SLA-1*04:01 and SLA-1*13:01, while 62E and 66K in SLA-1*13:01 can form hydrogen bonds with P-1-R and P2-E (Figure 4A). This may result in SLA-1*13:01 having a stronger ability to present N-terminal extended peptides than SLA-1*04:01, thus affecting their preference for long peptides. Therefore, it is necessary to compare the capabilities of SLA-1*04:01 and SLA-1*13:01 in presenting N-terminal extended peptides.
At present, there are only four pMHC-I complexes with N-terminal extending peptides, pSLA-1*04:01RW12, pSLA-1*13:01RW12, pHLA-B*57:01TSTLQEQIGW (pHLA-B*57:01TW10) and pHLA-B*58:01TW10 (9, 12, 13). SLA-I and HLA-I have similar patterns of binding N-terminal extension peptide, but they also clearly show distinguishable species characteristics (Figure 6A). The change in the binding mode of the P1 residue of the peptide is the key to the extension of the peptide N terminus. Compared with other peptides in classical presentation mode, the P1 residues of RW12 and TW10 are reversed, and their side chains are inserted into the A pocket, which leads to the elevation of the Cα position of the P1 residue. Then, the main chain N atom and O atom of the P1 residue form hydrogen bonds with 63E on the α1 helix and 159Y on the α2 helix, respectively. The 63rd residue is polymorphic, and our previous mutation studies show that 63E is necessary for this binding pattern (13). The angle between P-1 and P1 residues in RW12 is obviously larger than that in TW10 (Figure 6B) because the species characteristic of 167S in SLA-I alleles makes the N terminus of the peptide binding groove of SLA-I open and is beneficial to the extension of the P-1 residue (13).
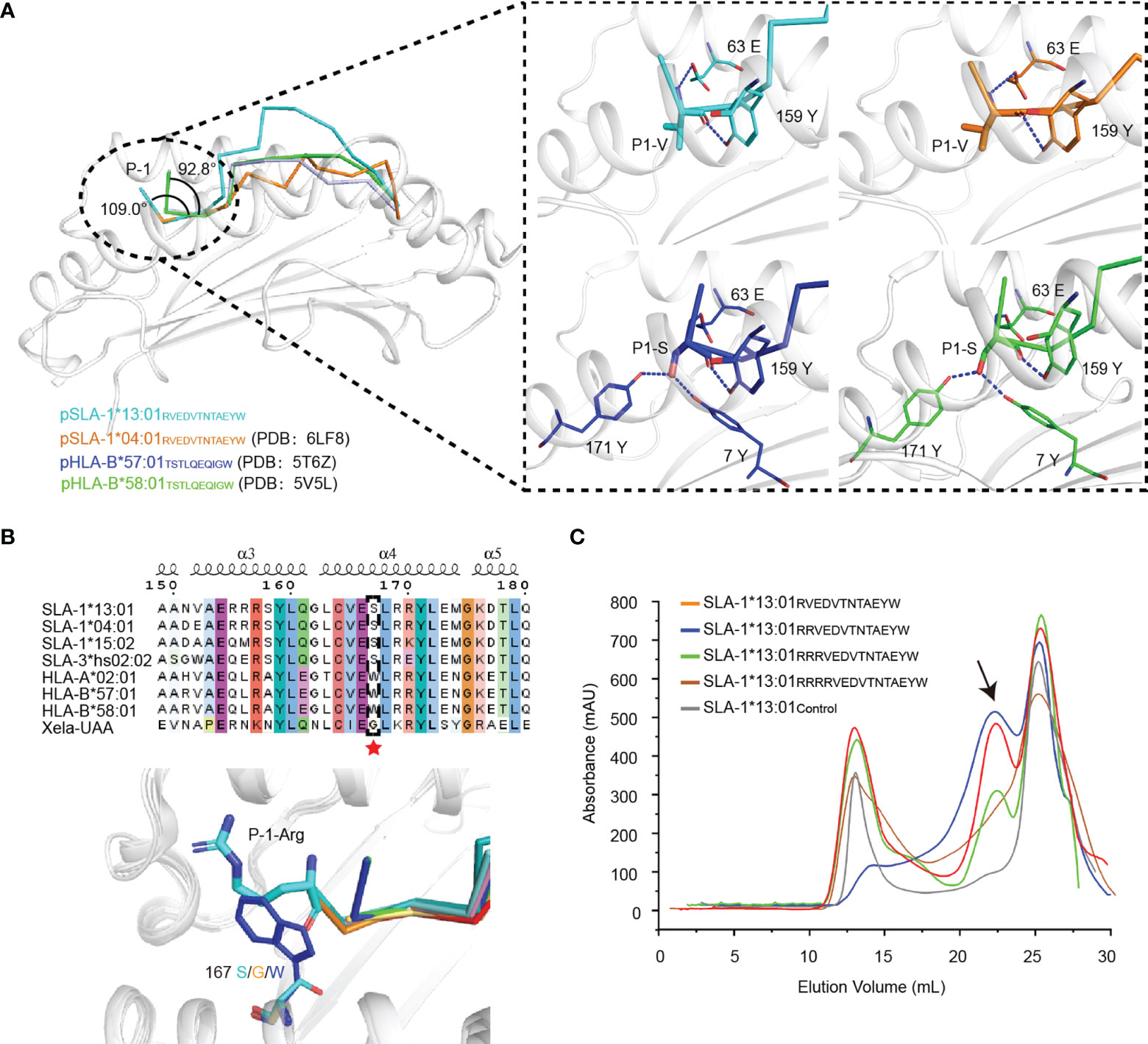
Figure 6 The peptide N-terminal extension mode of the MHC-I family. (A) The consistent mode of the N-terminally extended peptide presented by MHC-I. Comparison of the forces between the A pocket and peptides in SLA-I and HLA-I. (B) Sequence alignment and structural comparison of residue at position 167 in SLA-I, HLA-I and Xela-UAA. The amino acids at position 167 are highlighted by a red pentacle. (C) The in vitro refolding efficiency of SLA-1*13:01 was measured with peptides with different numbers of arginines added to the N-terminus of RW12. The black arrow indicates the peak of the complex.
Although 62E and 66K in SLA-1*13:01 can form hydrogen bonds with P-1-R and P2-E, there is no strong interaction with the P1 residue (Figure 4A), nor does it change the RW12 N-terminal extension (Figure 6). By adding excess residues at the N terminus, we previously proved that SLA-I * 04:01 can allow the extension of up to three residues (13). SLA-1*13:01 could only tolerate the extension of 1-3 residues from the N terminus of RW12 (Figure 6C). Therefore, we believe that SLA-1*13:01 does not have a stronger binding ability for the N-terminal extended peptide than SLA-I*04:01, and the N-terminal extended peptide presentation mode has no obvious influence on the difference in peptide length preferences between SLA-1*13:01 and SLA-1*04:01.
The length of the peptide can affect the recognition of MHC-I restricted epitopes by TCR, so the identification of T cell epitopes should consider the length of epitopes (6). Different HLA-I alleles have various preferences for the length of binding peptides (5). However, naturally presented peptides are not only influenced by MHC allele-specific binding preference, but also depend on many processes such as digestion and transportation of peptides in cells (41–45). Therefore, there are certain limitations in studying the peptide length preference of MHC-I alleles by MHC-I ligandomes in vivo, which further affects the prediction, screening and identification of long peptide epitopes.
RPLD-MS can determine the MHC-I random peptide ligandome in vitro and quickly identify MHC-I restricted peptide epitopes, which is especially suitable for studying unknown animal MHC-I molecules lacking research background and conditions (17, 19, 31–33). Our previous studies proved that RPLD-MS can sensitively reflect the different nonapeptide binding capabilities in vitro between SLA-1*04:01 and SLA-1*13:01 caused by a single residue variation (99Y/F) (19). In this study, we quantitatively mapped the length distribution of SLA-1*04:01 and SLA-1*13:01 ligand peptidomes with RPLD-MS. These results showed that RPLD-MS is suitable for studying the effect of polymorphisms on peptide length preferences of MHC-I alleles: first, it eliminates the interference of peptide processing on the peptide length distribution in vivo, and focuses on the binding preference of the MHC-I molecule itself; second, its sensitivity and accuracy are enough to reflect the influence of micropolymorphism on peptide length preference. Ultimately, of course, identified natural presented peptidomes will be required to determine whether the preference for peptide length by MHC-I itself is sufficiently pronounced.
Compared with SLA-1*04:01, why is SLA-1*13:01 weak in binding short peptides but strong in binding long peptides? This is due to the difference in amino acids between them, which leads to different binding ways of short and long peptides. For short peptides, regardless of SLA-1*04:01 or SLA-I*13:01, the whole peptide should be accommodated in the peptide binding groove to form enough affinity with limited length. The differential amino acids of SLA-1*04:01 and SLA-1*13:01 are not located in the pockets that restrict the side chains of peptides, so the short peptides that they can bind to have similar motifs. However, 99F leads to the lack of hydrogen bond between SLA-1*13:01 and the mainchain of P3 residue, which plays a key role in stabilizing peptide binding. Therefore, only short peptides that strongly bind to SLA-1*04:01 can bear the loss of affinity caused by 99F and stably bind to SLA-1*13:01. This caused the capability of SLA-1*13:01 to bind short peptides to be weaker than SLA-1*04:01 (19). For the binding of long peptides with more than 10 amino acids, these two SLA-I molecules are different. 99Y and 70T of SLA-1*04:01 are bound to the main chain of RW12, which limits RW12 in the peptide binding groove. This binding does not involve the sidechain of the peptide, indicating that SLA-1*04:01 should bind other long peptides in a similar way. The whole long peptide needs to be completely contained by its groove, so SLA-1*04:01 is restrictive for long peptides. However, 99F and 70N of SLA-1*13:01 have no restriction on long peptides, and long peptides can protrude out of the binding groove if they can bind stably. Therefore, SLA-1*13:01 reduces the restriction on long peptides and increases the types and quantities of long peptides that may be bound.
It is a cross-species phenomenon that both ends of the peptide extend from the peptide binding groove of MHC-I (9–13). At present, extension of the N terminus of peptides has been found in HLA-I (HLA-B*57:01, HLA-B*58:01) and SLA-I (SLA-1*04:01, SLA-1*13:01), and all the residues contained in the A pocket have turned over in a similar way. The species characteristic of S167 opens the N terminus of the SLA-I peptide binding groove, which is helpful for the N-terminal extension of the peptide. However, both SLA-1*04:01 and SLA-1*13:01 can only bear 1-3 redundant N-terminal residues, so this novel peptide binding mode does not affect their preferences for peptide length.
At present, the identification of MHC-I restricted CTL epitopes focuses on short peptides with 8-10 amino acids and relatively ignores long peptide epitopes. However, there is increasing evidence that long peptide epitopes also play an important role in the CTL response (4–6). Our study showed that long peptides accounted for a considerable proportion of the SLA-I restricted epitopes. First, SLA-I species-specific S167 facilitates N-terminal nested long peptide binding; Second, some alleles, such as SLA-1*13:01, bind to a higher proportion of long peptides. Therefore, long peptides should not be ignored in the identification of SLA-I limiting epitopes. Because the sequences of SLA-1*04:01 and SLA-1*13:01 are highly homologous, it is helpful to understand how the MHC-I polymorphism affects peptide length preference by analyzing their peptide length distribution and basis. Further exploration of the relationship between SLA-I polymorphism and the length of binding peptide will help us to screen SLA-I restricted T cell epitopes more comprehensively, avoid the omission of long peptide epitopes, and develop effective epitope vaccines.
The coordinate and structure factor for the pSLA-1*13:01RW12 complex have been deposited in the Protein Data Bank (https://www.rcsb.org/) with the following accession number: 6LF9. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/) partner repository with the dataset identifier PXD020818.
NZ and XW conceived and designed the study. XW and SL performed the experiments. SW and GF collected and analyzed the crystal data. XX and ZL analyzed the LC-MS/MS data. XW and NZ wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported financially by grants from the National Key Research and Development Program of China (2021YFD1800100), the National Natural Science Foundation of China (NSFC) (31830097, http://www.nsfc.gov.cn), the National Natural Science Foundation of China (NSFC) (31201887, http://www.nsfc.gov.cn), the Natural Science Foundation of Beijing Municipality (6182029, http://www.bjkw.gov.cn/), Major Science and Technology Project of Liaoning Province (2020JH1/10200003) and the 2115 Talent Development Program of China Agricultural University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the staff at the Shanghai Synchrotron Radiation Facility of China (SSRF) for their technical assistance during the data collection.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.820881/full#supplementary-material
1. Kaufman J. Unfinished Business: Evolution of the MHC and the Adaptive Immune System of Jawed Vertebrates. Annu Rev Immunol (2018) 36:383–409. doi: 10.1146/annurev-immunol-051116-052450
2. Josephs TM, Grant EJ, Gras S. Molecular Challenges Imposed by MHC-I Restricted Long Epitopes on T Cell Immunity. Biol Chem (2017) 398:1027–36. doi: 10.1515/hsz-2016-0305
3. Saper MA, Bjorkman PJ, Wiley DC. Refined Structure of the Human Histocompatibility Antigen HLA-A2 at 2.6 A Resolution. J Mol Biol (1991) 219:277–319. doi: 10.1016/0022-2836(91)90567-p
4. Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-Allelic Cells Enables More Accurate Epitope Prediction. Immunity (2017) 46:315–26. doi: 10.1016/j.immuni.2017.02.007
5. Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, et al. The Length Distribution of Class I-Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele-Specific Binding Preference. J Immunol (2016) 196:1480–7. doi: 10.4049/jimmunol.1501721
6. Ekeruche-Makinde J, Miles JJ, van den Berg HA, Skowera A, Cole DK, Dolton G, et al. Peptide Length Determines the Outcome of TCR/peptide-MHCI Engagement. Blood (2013) 121:1112–23. doi: 10.1182/blood-2012-06-437202
7. Speir JA, Stevens J, Joly E, Butcher GW, Wilson IA. Two Different, Highly Exposed, Bulged Structures for an Unusually Long Peptide Bound to Rat MHC Class I RT1-Aa. Immunity (2001) 14:81–92. doi: 10.1016/s1074-7613(01)00091-7
8. Tynan FE, Borg NA, Miles JJ, Beddoe T, El-Hassen D, Silins SL, et al. High Resolution Structures of Highly Bulged Viral Epitopes Bound to Major Histocompatibility Complex Class I. Implications for T-Cell Receptor Engagement and T-Cell Immunodominance. J Biol Chem (2005) 280:23900–9. doi: 10.1074/jbc.M503060200
9. Pymm P, Illing PT, Ramarathinam SH, O’Connor GM, Hughes VA, Hitchen C, et al. MHC-I Peptides Get Out of the Groove and Enable a Novel Mechanism of HIV-1 Escape. Nat Struct Mol Biol (2017) 24:387–94. doi: 10.1038/nsmb.3381
10. Xiao J, Xiang W, Zhang Y, Peng W, Zhao M, Niu L, et al. An Invariant Arginine in Common With MHC Class II Allows Extension at the C-Terminal End of Peptides Bound to Chicken MHC Class I. J Immunol (2018) 201:3084–95. doi: 10.4049/jimmunol.1800611
11. McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, Bardet W, et al. Toxoplasma Gondii Peptide Ligands Open the Gate of the HLA Class I Binding Groove. Elife (2016) 5:e12556. doi: 10.7554/eLife.12556
12. Li X, Lamothe PA, Walker BD, Wang JH. Crystal Structure of HLA-B*5801 With a TW10 HIV Gag Epitope Reveals a Novel Mode of Peptide Presentation. Cell Mol Immunol (2017) 14:631–4. doi: 10.1038/cmi.2017.24
13. Wei X, Wang S, Wang S, Xie X, Zhang N. Structure and Peptidomes of Swine MHC Class I With Long Peptides Reveal the Cross-Species Characteristics of the Novel N-Terminal Extension Presentation Mode. J Immunol (2022) 208(2):480–91. doi: 10.4049/jimmunol.2001207
14. Gfeller D, Guillaume P, Michaux J, Pak HS, Daniel RT, Racle J, et al. The Length Distribution and Multiple Specificity of Naturally Presented HLA-I Ligands. J Immunol (2018) 201:3705–16. doi: 10.4049/jimmunol.1800914
15. Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, et al. HLA Peptide Length Preferences Control CD8+ T Cell Responses. J Immunol (2013) 191:561–71. doi: 10.4049/jimmunol.1300292
16. Illing PT, Pymm P, Croft NP, Hilton HG, Jojic V, Han AS, et al. HLA-B57 Micropolymorphism Defines the Sequence and Conformational Breadth of the Immunopeptidome. Nat Commun (2018) 9:4693. doi: 10.1038/s41467-018-07109-w
17. Qu Z, Li Z, Ma L, Wei X, Zhang L, Liang R, et al. Structure and Peptidome of the Bat MHC Class I Molecule Reveal a Novel Mechanism Leading to High-Affinity Peptide Binding. J Immunol (2019) 202:3493–506. doi: 10.4049/jimmunol.1900001
18. Gfeller D, Bassani-Sternberg M. Predicting Antigen Presentation-What Could We Learn From a Million Peptides? Front Immunol (2018) 9:1716. doi: 10.3389/fimmu.2018.01716
19. Wei X, Wang S, Li Z, Li Z, Qu Z, Wang S, et al. Peptidomes and Structures Illustrate Two Distinguishing Mechanisms of Alternating the Peptide Plasticity Caused by Swine MHC Class I Micropolymorphism. Front Immunol (2021) 12:592447. doi: 10.3389/fimmu.2021.592447
20. Stevens J, Wiesmüller KH, Walden P, Joly E. Peptide Length Preferences for Rat and Mouse MHC Class I Molecules Using Random Peptide Libraries. Eur J Immunol (1998) 28:1272–9. doi: 10.1002/(sici)1521-4141(199804)28:04<1272::aid-immu1272>3.0.co;2-e
21. Yao S, Liu J, Qi J, Chen R, Zhang N, Liu Y, et al. Structural Illumination of Equine MHC Class I Molecules Highlights Unconventional Epitope Presentation Manner That Is Evolved in Equine Leukocyte Antigen Alleles. J Immunol (2016) 196:1943–54. doi: 10.4049/jimmunol.1501352
22. Koch M, Camp S, Collen T, Avila D, Salomonsen J, Wallny HJ, et al. Structures of an MHC Class I Molecule From B21 Chickens Illustrate Promiscuous Peptide Binding. Immunity (2007) 27:885–99. doi: 10.1016/j.immuni.2007.11.007
23. Nielsen M, Connelley T, Ternette N. Improved Prediction of Bovine Leucocyte Antigens (BoLA) Presented Ligands by Use of Mass-Spectrometry-Determined Ligand and In Vitro Binding Data. J Proteome Res (2018) 17:559–67. doi: 10.1021/acs.jproteome.7b00675
24. Wallny HJ, Avila D, Hunt LG, Powell TJ, Riegert P, Salomonsen J, et al. Peptide Motifs of the Single Dominantly Expressed Class I Molecule Explain the Striking MHC-Determined Response to Rous Sarcoma Virus in Chickens. Proc Natl Acad Sci USA (2006) 103:1434–9. doi: 10.1073/pnas.0507386103
25. Wynne JW, Woon AP, Dudek NL, Croft NP, Ng JH, Baker ML, et al. Characterization of the Antigen Processing Machinery and Endogenous Peptide Presentation of a Bat MHC Class I Molecule. J Immunol (2016) 196:4468–76. doi: 10.4049/jimmunol.1502062
26. Pavlovich SS, Lovett SP, Koroleva G, Guito JC, Arnold CE, Nagle ER, et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell (2018) 173:1098–110.e18. doi: 10.1016/j.cell.2018.03.070
27. Cumberbatch JA, Brewer D, Vidavsky I, Sharif S. Chicken Major Histocompatibility Complex Class II Molecules of the B Haplotype Present Self and Foreign Peptides. Anim Genet (2006) 37:393–6. doi: 10.1111/j.1365-2052.2006.01459.x
28. Alvarez B, Barra C, Nielsen M, Andreatta M. Computational Tools for the Identification and Interpretation of Sequence Motifs in Immunopeptidomes. Proteomics (2018) 18:e1700252. doi: 10.1002/pmic.201700252
29. Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, et al. Deep Learning Enables De Novo Peptide Sequencing From Data-Independent-Acquisition Mass Spectrometry. Nat Methods (2019) 16:63–6. doi: 10.1038/s41592-018-0260-3
30. Tran NH, Zhang X, Xin L, Shan B, Li M. De Novo Peptide Sequencing by Deep Learning. Proc Natl Acad Sci USA (2017) 114:8247–52. doi: 10.1073/pnas.1705691114
31. Wu Y, Zhang N, Wei X, Lu S, Li S, Hashimoto K, et al. The Structure of a Peptide-Loaded Shark MHC Class I Molecule Reveals Features of the Binding Between β(2)-Microglobulin and H Chain Conserved in Evolution. J Immunol (2021) 207:308–21. doi: 10.4049/jimmunol.2001165
32. Ma L, Zhang N, Qu Z, Liang R, Zhang L, Zhang B, et al. A Glimpse of the Peptide Profile Presentation by Xenopus laevis MHC Class I: Crystal Structure of Pxela-UAA Reveals a Distinct Peptide-Binding Groove. J Immunol (2020) 204:147–58. doi: 10.4049/jimmunol.1900865
33. Wang Y, Qu Z, Ma L, Wei X, Zhang N, Zhang B, et al. The Crystal Structure of the MHC Class I (MHC-I) Molecule in the Green Anole Lizard Demonstrates the Unique MHC-I System in Reptiles. J Immunol (2021) 206:1653–67. doi: 10.4049/jimmunol.2000992
34. Zhang N, Qi J, Feng S, Gao F, Liu J, Pan X, et al. Crystal Structure of Swine Major Histocompatibility Complex Class I SLA-1 0401 and Identification of 2009 Pandemic Swine-Origin Influenza A H1N1 Virus Cytotoxic T Lymphocyte Epitope Peptides. J Virol (2011) 85:11709–24. doi: 10.1128/JVI.05040-11
35. Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol (1997) 276:307–26. doi: 10.1016/S0076-6879(97)76066-X
36. McCoy AJ. Solving Structures of Protein Complexes by Molecular Replacement With Phaser. Acta Crystallogr D Biol Crystallogr (2007) 63:32–41. doi: 10.1107/s0907444906045975
37. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr D Biol Crystallogr (2010) 66:486–501. doi: 10.1107/S0907444910007493
38. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr D Biol Crystallogr (2011) 67:355–67. doi: 10.1107/S0907444911001314
39. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr D Biol Crystallogr (2010) 66:213–21. doi: 10.1107/s0907444909052925
40. Laskowski RA, Moss DS, Thornton JM. Main-Chain Bond Lengths and Bond Angles in Protein Structures. J Mol Biol (1993) 231:1049–67. doi: 10.1006/jmbi.1993.1351
41. Olson E, Geng J, Raghavan M. Polymorphisms of HLA-B: Influences on Assembly and Immunity. Curr Opin Immunol (2020) 64:137–45. doi: 10.1016/j.coi.2020.05.008
42. Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA Class I Supertypes: A Revised and Updated Classification. BMC Immunol (2008) 9:1. doi: 10.1186/1471-2172-9-1
43. Uebel S, Kraas W, Kienle S, Wiesmuller KH, Jung G, Tampe R. Recognition Principle of the TAP Transporter Disclosed by Combinatorial Peptide Libraries. Proc Natl Acad Sci USA (1997) 94:8976–81. doi: 10.1073/pnas.94.17.8976
44. Purcell AW, Gorman JJ, Garcia-Peydro M, Paradela A, Burrows SR, Talbo GH, et al. Quantitative and Qualitative Influences of Tapasin on the Class I Peptide Repertoire. J Immunol (2001) 166:1016–27. doi: 10.4049/jimmunol.166.2.1016
Keywords: MHC class I, micropolymorphism, binding peptide length, crystal structure, peptidomes
Citation: Wei X, Li S, Wang S, Feng G, Xie X, Li Z and Zhang N (2022) Peptidomes and Structures Illustrate How SLA-I Micropolymorphism Influences the Preference of Binding Peptide Length. Front. Immunol. 13:820881. doi: 10.3389/fimmu.2022.820881
Received: 23 November 2021; Accepted: 10 February 2022;
Published: 28 February 2022.
Edited by:
William J. Liu, National Institute for Viral Disease Control and Prevention (China CDC), ChinaCopyright © 2022 Wei, Li, Wang, Feng, Xie, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nianzhi Zhang, emhhbmduaWFuemhpQGNhdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.