 Mark R. Cronan
Mark R. Cronan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 07 March 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.820134
This article is part of the Research TopicImmunology of Infectious GranulomasView all 6 articles
The defining pathology of tuberculosis is the granuloma, an organized structure derived from host immune cells that surrounds infecting Mycobacterium tuberculosis. As the location of much of the bacteria in the infected host, the granuloma is a central point of interaction between the host and the infecting bacterium. This review describes the signals and cellular reprogramming that drive granuloma formation. Further, as a central point of host-bacterial interactions, the granuloma shapes disease outcome by altering host immune responses and bacterial susceptibility to antibiotic treatment, as discussed herein. This new understanding of granuloma biology and the signaling behind it highlights the potential for host-directed therapies targeting the granuloma to enhance antibiotic access and tuberculosis-specific immune responses.
The granuloma has been recognized by pathologists as the defining pathology of tuberculosis for more than 100 years (1–3). This structure is organized around infecting Mycobacterium tuberculosis, the etiologic agent of tuberculosis, from host immune cells (2, 4). As the location of much of the bacteria within infected individuals, the granuloma pivotally shapes the way that Mtb interacts with the host immune system (2). This structure also helps define our clinical approaches to Mtb treatment, serving as a substantial barrier to Mtb chemotherapeutics (5, 6). In this review, I will discuss the organization of the tuberculous granuloma and the host and bacterial signaling that shape its formation. I will also consider the ways in which this central structure of tuberculosis contributes to the pathogenicity and clinical outcomes of this disease. As a pivotal structure to mycobacterial pathogenesis and treatment, the potential of targeting this structure as an adjunctive therapy and potential pitfalls of this approach will be considered.
Mtb is an obligate human pathogen, passing from host to host in the aerosols generated by the cough of an Mtb-infected individual (7). Droplets containing Mtb bacilli are deposited into the lungs of newly infected individuals where the bacteria are rapidly phagocytosed by resident alveolar macrophages (7). These macrophages then bring the bacterium out of the airway, carrying the bacterium into the interstitium of the lung. Once out of the airway, the bacterium is able to use the concerted action of many virulence factors to evade immune-mediated killing and efficiently replicate within populations of macrophages and other recruited immune cells (7–10). As the infection progresses, continued recruitment and aggregation of macrophages and other immune populations to the site of Mtb infection leads to the formation of the granuloma, the defining pathology of tuberculosis infection (2, 4, 11).
The macrophage is central to the formation of the granuloma. Macrophages comprise much of the cell population of the granuloma and form the inner layers of the granuloma, serving as a central scaffold around which the other cell populations are nucleated (2, 4, 11). While macrophage populations are highly motile under normal physiological conditions or infection, during granuloma formation, these cells undergo a pronounced morphological differentiation termed ‘epithelioid differentiation’ where they tightly interdigitate and aggregate with their neighbors to form the granuloma (4, 12). These epithelioid macrophages are a central characteristic of the tuberculous granuloma, but they are joined by other macrophage populations, including conventional macrophages, lipid-laden foamy macrophages and multinucleate macrophage giant cells (2, 13). Beyond macrophage populations, the granuloma is characterized by a broader immune response that recruits many other cell types to the granuloma. Cell populations recruited to the granuloma include myeloid populations such as neutrophils, dendritic cells, eosinophils, and mast cells, lymphocyte populations including T cells, B cells, NK cells and ILCs and nonhematopoietic cells such as fibroblasts, endothelial and epithelial cells (Figure 1) (2, 14–20).
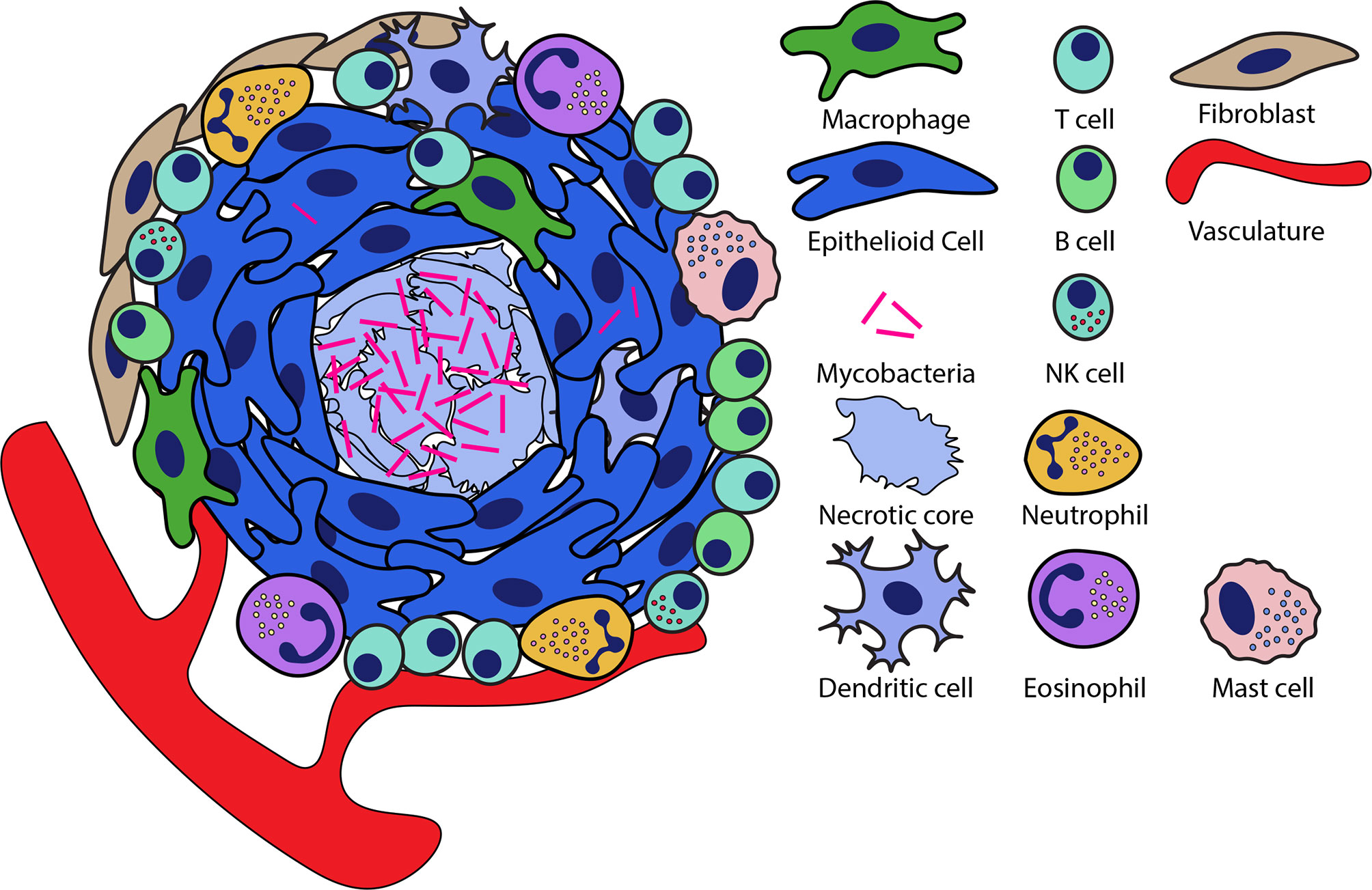
Figure 1 Organization of necrotic granulomas during mycobacterial infection. Necrotic granulomas are structured around a central core of necrotic cell debris in which much of the bacteria are concentrated. Layers of epithelioid macrophages surround the necrotic core interspersed with other macrophage populations. A diversity of other cell types are recruited to the granuloma and can be integrated into this structure at the periphery as well as within the epithelioid layers of the granuloma.
Granulomas in Mtb-infected patients take many shapes and forms. Tuberculosis is most classically associated with the formation of caseous necrotic granulomas, wherein the central region of the granuloma has undergone necrotic cell death, leading to the formation of a core of cell debris that frequently have a soft, cheese-like consistency termed caseum (4, 5, 21). These necrotic granulomas come in many forms including fibrotic granulomas (encapsulated by a fibrotic rim), calcified granulomas (necrotic core is mineralized), and suppurative granulomas (necrotic core infiltrated by neutrophils) (5, 18). Mtb infection also results in the formation of cellular or non-necrotic granulomas, where the central necrotic core is absent and the bacteria reside intracellularly (5, 18). Furthermore, the primary granulomas that arise from initial infection events are also morphologically distinct from the post-primary granulomas that form via encapsulation of caseus pneumonia from bronchogenic spread of Mtb in individuals previously exposed to the bacterium (22). This text will largely focus on the primary granulomas that have been the recent focus of the field, however, with the newfound re-recognition of post-primary tuberculosis and animal modeling of this process (23), future studies comparing these lesions will be of great interest.
Given the diversity of granuloma types and the on-demand nature of granuloma assembly, there is considerable heterogeneity in the organization of individual granulomas (5, 24, 25). However, a general pattern of spatial organization is observed in the granuloma, wherein the central regions of the granuloma are macrophage-rich with many of these macrophages having undergone epithelioid transition, whereas lymphocyte populations tend to be largely confined to the periphery where they comprise a lymphocyte-rich cuff that surrounds the granuloma (Figure 1) (2, 18, 26). At the edges of the granuloma, these lymphocytes can be organized along with antigen presenting cells into tertiary lymphoid follicles (also called iBALT in lungs) that allow for local antigen presentation at the site of infection (27, 28). In necrotic granulomas, bacteria are found largely in the necrotic core of granulomas, while smaller numbers of bacteria live intracellularly largely within the macrophage-rich regions surrounding the core (18, 26). Similarly, in cellular granulomas Mtb resides predominantly in the central regions of the granuloma, though the absence of necrosis in these granulomas means that Mtb resides intracellularly, predominantly within the macrophage populations that comprise the core of these granulomas (5, 18).
Granulomas are commonly presented as a number of distinct granuloma types, but it is important to recognize that all of these distinct granuloma types can be observed within a single infected host (21, 24, 29). Individual bacteria within the host can experience radically different granuloma environments and local inflammatory states depending on the type of granuloma they are in, the age of the granuloma and the regional immune response (24). Throughout the course of infection in a single infected individual, these local differences mean that while some granulomas progress, expand and consolidate with neighbors, other granulomas that are established subsequently regress (24). This is supported by both classical and recent findings that have demonstrated that sterile, frequently calcified, lesions freely coexist in Mtb infected individuals along with active lesions containing large numbers of Mtb (24). This considerable heterogeneity between individual lesions not only underlies differences in the infection outcomes within each lesion, but also likely complicates the clinical treatment of tuberculosis, as individual lesions respond heterogeneously to antibiotic treatment.
Not only is there substantial variation between granulomas, but the organization of individual granulomas is considerably more complex than is generally depicted. Granulomas are commonly represented as circular structures (including in the figures herein). While this circular depiction of granulomas is true to what is commonly observed in histological sections and is also representative of many smaller lesions, classical work as well as recent PET-CT and micro-CT analysis of granulomas in human patients has found that larger granulomas form substantially more complex structures (30–32). These larger granulomas are highly elaborated and branched, factors that make granuloma organization difficult to assess by histology (30–32). While researchers have worked to characterize the local environment of granulomas, these extended structures mean that a single granuloma may instead sample a much more diverse variety of environments within the lung than widely expected (30). In particular, many of the larger, elaborated granulomas, while lodged in lung tissue, were found to directly access the airways in these patients, potentially enabling the bronchogenic spread of Mtb from these granulomas through the airways (30). This complex, branched structure of the granuloma further suggests that even within a single granuloma, there may be substantial variation in local environment depending on where the bacteria are located within the granuloma, which may result in spatially distinct selective pressures even within a single granuloma.
While the spectrum of granuloma types has long been a hallmark of the human disease, granulomas are difficult to study in preclinical animal models due to discrepancies in granuloma morphology between models and human patients and species-specific limits in tool availability. The most widely used Mtb model are inbred mouse models such as C57BL/6 and BALB/c mice, which possess an expansive suite of immunological tools (33). However, these inbred mouse models fail to form the necrotic granulomas that define human disease (33, 34). Limitations of inbred mouse models have led to the development of new mouse models including C3HeB/FeJ mice (35), the C57BL/6 NOS2-/- ear infection model (36), CBA/J IL10-/- mice (37) and genetically diverse panels of mice, which develop necrotic lesions that more closely resemble the human disease (38, 39). However, the complex genetic backgrounds of these animals complicate the use of existing mouse genetic tools. Beyond mice, a number of other small mammal models have long been used as effective tuberculosis surrogate models, most notably rabbits and guinea pigs (33). These models phenocopy many features of human Mtb infection and granuloma formation, but also are restricted by tool availability and the limited use of genetic approaches in these models. The development of non-human primate models has allowed the interrogation of tuberculosis pathogenesis in a model that closely resemble human disease (29, 40), but cost, time, and ethical concerns limit the widespread adoption of this model. Outside of mammalian systems the zebrafish has emerged as a genetically tractable model to study mycobacterial granuloma formation (41–43). In this model, zebrafish are infected with Mycobacterium marinum, a close relative of Mtb that conserves many virulence factors with Mtb and is a natural pathogen of zebrafish (41). Infection of zebrafish can be performed within optically transparent zebrafish larvae to enable visualization of immune-mycobacterial interactions and early granuloma formation in live animals. Adult zebrafish can also be infected, resulting in a long-term infection and formation of necrotic granulomas that closely resemble the granulomas seen in human disease (42, 43). Findings in zebrafish have been predictive of human Mtb infection outcomes (44–48). However, as a surrogate model, there are several caveats to the zebrafish/M. marinum system including the use of M. marinum rather than Mtb itself, the degree of conservation of immune genes between humans and zebrafish and the absence of lungs in zebrafish. While many animal models exist for Mtb infection, each of these models have distinct strengths that allow researchers to match their question to an optimal model.
A central feature of mycobacterial granuloma formation in all of these models of tuberculosis is the epithelioid transformation of their macrophage populations. The term ‘Epithelioid cell’ was chosen by early pathologists due to the close resemblance of these macrophage populations to epithelial tissues (2, 4). Morphologically, these cells are characterized by a spread morphology, an elongated nucleus and tight interdigitation with surrounding macrophages (4, 11). The strikingly altered morphology and behavior of epithelioid cells compared to parental macrophages suggests that these cells undergo substantial reprogramming events during epithelioid transformation, but the nature of these reprogramming events remained unknown for over a century.
Using the zebrafish model, it was found that these granuloma macrophages undergo a bona fide epithelialization event during granuloma formation, broadly engaging epithelial-specific genes in a macrophage-epithelial transition that is analogous to mesenchymal-epithelial transition (46). Among the many characteristic rearrangements of epithelial tissues, these epithelioid cells formed functional E-cadherin-positive adherens junctions, desmosomes and tight junctions (Figure 2), alter their cytoskeletal structure and induce apical-basal polarity pathways (46). Engagement of this epithelialization program was essential to granuloma formation, as disruption of epithelialization by macrophage-specific expression of a dominant negative E-cadherin blocks granuloma formation and leads to the loss of epithelioid morphology (46). These rearrangements were observed not only in zebrafish, but also in samples from human patients by histological staining approaches (46, 47). Extensive engagement of epithelial markers could also be observed in mass spectrometry data sets from human granulomas, which further supports that epithelialization is an evolutionarily conserved feature of mycobacterial granulomas (49, 50). Beyond tuberculosis, E-cadherin induction has also been seen in schistosome granulomas as well as in dermal granulomas formed in sarcoidosis and foreign body reactions (51–55). The identification of epithelialization hallmarks within these diverse granuloma types also supports a wider involvement of epithelialization within granulomatous diseases generally.
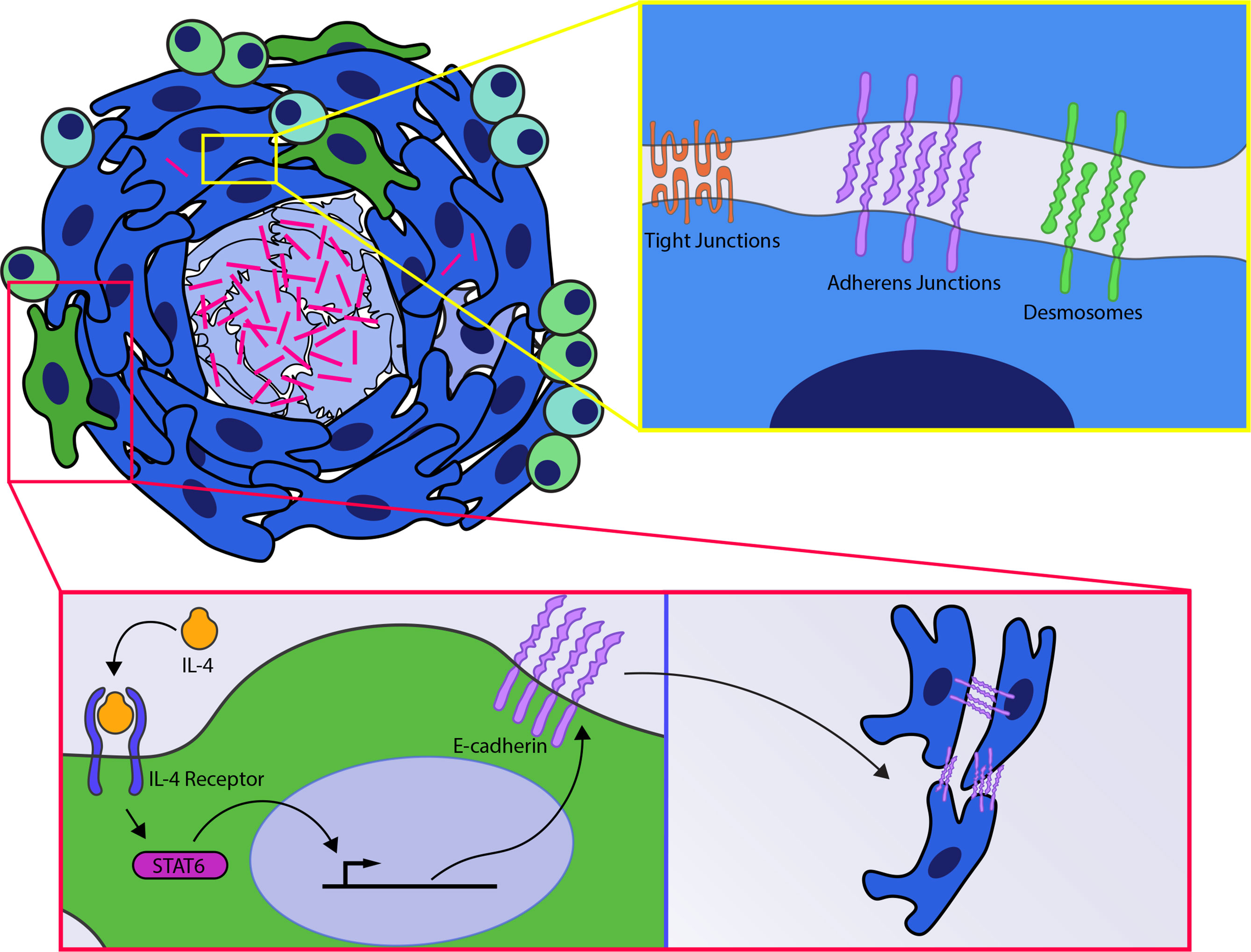
Figure 2 Host signals involved in granuloma formation. (Yellow box) Signaling events within epithelioid macrophages results in induction of epithelial cell-cell adhesion pathways including induction of adherens junctions, tight junctions and desmosomes within the macrophages of the granuloma. (Red box) IL4R signaling via stat6 is required for induction of E-cadherin within macrophages and necrotic granuloma formation.
That the macrophages of the granuloma undergo such profound cellular and molecular changes during granuloma formation raises the question of what factors could be driving these changes within macrophage populations. As central mediators of the immune response, cytokine signals are likely candidates for regulating granuloma formation. Classically, Mtb granulomas have been thought to be driven by type 1 immune responses (50, 56). Amongst type 1 cytokines, IFN-γ and TNF have been most closely associated with Mtb pathogenesis and granuloma formation. Both IFN-γ and TNF are crucial to the control of Mtb burden during infection in preclinical animal models and human patients (57–64). Loss of these cytokines has also been linked to altered granuloma architecture during Mtb infection, suggesting that signaling through these pathways may be required for ordered granuloma formation (61–64). However, in the case of TNF, subsequent longitudinal imaging experiments in zebrafish revealed that TNF deficient animals have normal granuloma formation early in infection, and that loss of granuloma morphology was instead due to the accelerated cell death caused by exuberant bacterial growth in these animals (65). Further evidence that TNF is required for bacterial restriction but not granuloma formation has been found in macaques, where neutralization of TNF with anti-TNF antibodies resulted in normal granuloma formation but markedly enhanced bacterial burden (66). Similar results have been observed for IFN-γ in mice infected with the attenuated vaccine strain M. bovis BCG, where IFN-γ-deficient animals infected with BCG still form similar numbers of morphologically normal granulomas compared to wildtype controls (67). Additionally, while C57BL/6 mice fail to form necrotic granulomas, disseminated infection in C57BL/6 mice deficient for the critical IFN-γ-induced factor iNOS resulted in the formation of sporadic necrotic granulomas that could be enhanced by neutralization of either IFN-γ or TNF (36). Therefore, while type 1 pathways are important for Mtb restriction, it is less clear that they are required for granuloma architecture, suggesting that other cytokine signaling pathways may be required for granuloma formation.
While Mtb pathology has commonly been thought to be driven by type 1 immunity, in Schistosome granulomas it has long been known that type 2 immunity is crucial to driving formation of the granuloma structure (12, 68). Type 1 and type 2 immunity are thought to drive substantially divergent transcriptional programs in macrophages (69). It seems unlikely that such disparate transcriptional programs could drive the formation of the same complex and highly organized granuloma structure. Instead, it is possible that the Mtb granuloma is driven by type 2 immunity and the degree of type 2 polarization within the tuberculous granuloma has been underestimated.
A growing number of studies have observed that there is a substantial type 2 inflammatory response during Mtb infection, seen in the lung, and within the granulomas in Mtb patients, as well as animal models that form necrotic granulomas (18, 47, 70–73). Owing to the identification of these type 2 responses in human disease, efforts have been made to model the involvement of these cytokines in infection and granulomatous responses. Due to the availability of genetic tools, these experiments were initially done in inbred mouse models deficient for type 2 signaling. However, in these models, loss of type 2 signaling had only mild effects on granuloma formation and modest to moderate effects on bacterial burden, suggesting that type 2 immunity was largely dispensable for granuloma formation during Mtb infection (74–76). One confounding point to these experiments is that the C57BL/6 and BALB/c mouse model used in these studies do not form the highly organized necrotic granulomas seen in human disease. Recognizing that the strong type 1 environment observed in many Mtb infected inbred mouse lines may limit the involvement of type 2 immune responses in these models, Heitmann et al. used transgenic mice overexpressing IL-13 to instead look at how enhanced type 2 responses within mice would alter the pathology and trajectory of Mtb infection (77). In contrast to non-transgenic C57BL/6 littermates, IL-13 overexpressing animals infected with Mtb formed organized necrotic granulomas that closely resembled human disease, suggesting that type 2 pathways contributed to necrotic granuloma formation (77). Necrotic granuloma formation was accompanied by increased bacterial burden in these animals, suggesting that either the enhanced type 2 signaling or granuloma formation itself was bacterial beneficial (77).
Further evidence for the role of type 2 immunity in mycobacterial granuloma formation has come from recent studies on macaques and zebrafish. Using histological staining of macaque granulomas and scRNA-seq analysis of the organized necrotic granulomas that form in zebrafish it was found that the epithelialized macrophage populations in the granuloma had increased type 2 inflammatory signaling (47). These type 2 responses were required for necrotic granuloma formation and epithelialization (Figure 2), as zebrafish deficient for type 2 signaling by deletion of either the IL-4 receptor or the downstream transcription factor stat6 failed to undergo macrophage-epithelial transformation and no longer formed necrotic granulomas (47). Loss of type 2 signaling was associated with increased bacterial burden in stat6-deficient animals, raising the possibility that elevated bacterial burden could be driving necrotic granuloma breakdown (47). To test this hypothesis, mixed marrow chimeras possessing both wildtype and stat6-deficient cell populations were used to assess whether stat6 functioned cell autonomously. Infection of these mixed marrow chimera animals resulted in the formation of necrotic granulomas. However, these necrotic granulomas were predominantly composed from the wildtype donor tissue (47). Despite the presence of organized necrotic granulomas in these animals, stat6-deficient populations failed to undergo epithelialization and were largely excluded from these granulomas, localizing instead to the periphery of these structures (47). Further supporting that type 2 signaling is required for granuloma organization independent of its burden effects, an ex vivo culture model (78) was used to test the effects of stat6 inhibitors on established granulomas ex vivo. Addition of a stat6 inhibitor to isolated granulomas reversed granuloma epithelialization and drove disaggregation of these structures, suggesting that continued stat6 signaling was required to maintain granuloma organization (47). Thus type 2 responses appear to play a critical role in the organization of necrotic granulomatous structures. At later stages of infection, it is thought that erosion of these lesions into the airway can drive transmission in the process of cavitation (5, 21). Interestingly, work in human patients has indicated that elevated type 2 signaling is also associated with cavitation and transmission, suggesting that type 2 signaling may play crucial roles in multiple phases of the Mtb life cycle (71, 79).
While these studies have implicated type 2 immune responses driven by IL-4 and IL-13 in granuloma pathology, there are still a number of outstanding questions about how, when and where these cytokines are produced. In zebrafish, scRNA-seq demonstrated that IL-4 and IL-13 were produced by T cell and eosinophil populations within the granuloma (47). By contrast, in macaques, scRNA-seq found that IL-4 and IL-13 were particularly highly produced by recruited mast cells, indicating that there may be some species specific variation in the cell types that are producing type 2 cytokines (15). Beyond IL-4 and IL-13, it seems likely that other cytokines and signals are also required to drive necrotic granuloma formation, as elevated levels of IL-4 and IL-13 are observed in many inflammatory responses that do not lead to granuloma formation (80). The identity of these potential accessory factors is unknown, but experiments characterizing the induction of the epithelial cell-cell adhesion molecule E-cadherin in macrophages in culture found that E-cadherin induction by IL-4 and IL-13 could be enhanced by costimulation with IL-10 and TGFβ (52), suggesting that these two cytokines may act as accessory factors, facilitating granuloma formation through potentiation of the macrophage-epithelial transitions that underlie granuloma formation (46). Similarly, cell-cell interactions during infection and granuloma formation may also contribute to type 2 polarization and epithelioid transformation. Coculture of macrophages with platelets has found that platelets drive these macrophages into an M2 phenotype that is accompanied by the acquisition of epithelioid-like characteristics (81).
The granuloma has frequently been thought of as a host driven structure. However, studies of bacterial growth in mycobacterial infected hosts have found that bacterial expansion actually coincides with the formation of this structure (82, 83). The connection between bacterial growth and granuloma formation suggests that the mycobacteria themselves benefit from driving this structure (82, 83). The attenuated vaccine strain M. bovis BCG has previously been used to identify regions of the mycobacterial genome that are potentially involved in virulence (84–86). The ΔRD1 region of M. bovis was identified as a particularly crucial region that was lost in BCG strains and necessary for virulence (84, 85, 87). This region is broadly conserved in pathogenic mycobacterial species; mycobacterial species lacking the ΔRD1 region are strongly attenuated in vitro and in vivo (82, 87, 88). Studies of ΔRD1 M. marinum have found that this region is necessary for effective granuloma formation either by innate immunity alone in larval zebrafish or in the context of both innate and adaptive arms in adult zebrafish (42, 82, 83). These defects were associated in part with decreased recruitment of macrophages and led to the formation of smaller, less well-organized granulomas (83, 89). In larval zebrafish, the crucial role for the ΔRD1 region in mycobacteria was found to involve the secreted protein ESAT6, which is encoded within this region (89). ESAT6 was released from infected macrophages and was subsequenty taken up by the surrounding epithelium, where it drives expression of the chemotactic metalloprotease MMP9 within these epithelial populations (89). Elevated MMP9 levels were found in turn to lead to the recruitment of surrounding macrophages to the granuloma, facilitating granuloma growth and expansion (89).
Beyond secreted bacterial effectors, mycobacteria are known to produce a number of lipid species that are critically important to the ability of mycobacteria to influence host immune responses (90). This was demonstrated in classical experiments which found that injections of killed mycobacteria, particularly when emulsified within Freund’s adjuvant (so called Complete Freund’s Adjuvant) could organize a robust granulomatous response, suggesting that lipids or other heat-stable mycobacterial fractions were important mediators of the granulomatous response (91, 92). Early efforts to identify crucial lipids by Sabin identified three distinct lipid fractions, all which could induce varying levels of epithelioid and giant cell differentiation and granuloma formation within rabbits (91, 93). Subsequent work in mice identified the mycolic acids, particularly Trehalose 6, 6’-Dimycolate (TDM), as a major mediator of epithelioid transformation and granuloma formation (94–96). Specifically these studies found that TDM when emulsified in incomplete Freund’s Adjuvant or TDM coated beads was sufficient to induce epithelioid granuloma formation in vivo (94, 95, 97). Interestingly, experiments in mice found that the effects of TDM depend on the particle size of emulsions and the density of TDM on the surface of beads, which together with studies on TDM organization in vitro, suggested that TDM geometry on the surface of mycobacteria is crucial to the granulomatous response (95, 98, 99). Importantly, removal of cell surface TDM and other mycolic acids and lipid species by lipid extraction abrogates granuloma formation in mice, instead leading to a predominantly neutrophilic response with minimal macrophage involvement (94, 100). However, these findings with delipidated bacteria are complicated as these bacteria are rapidly removed by host immune responses (94, 100).
Taken together with the previous section, orderly formation of granulomas requires the concerted action of both host and bacterial responses. The granuloma has long been presumed to be a host-protective response, containing and walling off the mycobacteria within this structure. This effective, host beneficial response can be observed in the sterile calcified granulomas seen in human patients and animal models (24). However, given the long-term coevolution between Mtb and humans, the granuloma likely represents a stalemate, affording benefits for not only the host but also the pathogen. This view is further supported by the recognition that bacteria also drive this structure with conserved mediators to facilitate their expansion (82, 89, 94, 96). While aspects of the granuloma may benefit both host the bacterium, as we discuss in the following sections, this structure critically shapes the immune responses to mycobacteria and the clinical treatment of infection.
The hallmark of the granuloma is its complex three-dimensional organization of immune cells into the granulomatous structure. Despite the ad hoc assembly of granulomas by the host immune system, this process results in a generally reproducible spatial organization, with bacterial populations largely constrained to the central region of the structure, while distinct immune populations are distributed in the surrounding cellular layers (18). With the presence of so many bacteria in the core regions of this structure, effective immune responses must be mounted through the complex and polarized cellular and inflammatory milieu of this structure (18, 26, 49). Many immune populations require close contact with infecting mycobacteria to restrict these pathogens; the spatial distribution of bacteria and immune populations within the granuloma suggests that the highly structured granuloma could serve as a barrier to mycobacterial recognition, critically shaping the interaction of bacteria with the host immune system. Recent work described below, has supported this notion, finding the cellular environment of the granuloma plays a crucial role in shaping host immune responses.
While CD4+ T cells are clearly critical to control of Mtb infection, the persistent survival of Mtb in actively infected individuals demonstrates that these responses are ultimately unable to clear the infection in many individuals (101). One possible explanation for this disconnect is that the complex environment of the granuloma is able to shape and limit effective T cell responses. In mouse models, it was found that direct interaction between CD4 T cells and infected macrophages is critical to effective T cell help in Mtb infection (102). However, T cells are predominantly located at the periphery of the granuloma, distant from the bacteria in the myeloid-rich core regions of the granuloma (18, 26). Thus, the inability for T cell responses to completely eliminate Mtb in infected patients appears to be, at least in part, due to the spatial restriction of T cells to sites distant from infected macrophages (26). Similarly, both NK cells and CD8 T cells are known to be recruited to granulomas in humans and animal models of Mtb infection (15, 103, 104). CD8 cells, have been found to be crucial to control of Mtb burden in the mouse model in vivo (105, 106). NK cells by contrast have been demonstrated to be important for Mtb control in T cell-deficient animals, suggesting that these cells can also participate in functional responses to Mtb (107). Aspects of the responses of both of these cell types require close contact between the immune cells and Mtb-infected cells (108, 109), suggesting that granuloma architecture may also play a critical role in the Mtb-specific responses of these populations as well. In the case of T cells, the importance of spatial separation between these cells and bacterial populations is also supported by computational modeling of tuberculous granulomas, which indicated that the spatial organization of the granuloma was one of the major drivers limiting T cell responsiveness in the granuloma (110).
The factors driving the pronounced spatial separation between T cells and macrophages are likely complex and multifactorial, but some potential mechanisms contributing to this asymmetry have been identified. Intravital microscopy of Mtb-specific T cells in mouse granulomas demonstrated that these cells largely fail to arrest within the granuloma as Mtb antigens appear to be poorly recognized within the granuloma (111). The limited recognition of mycobacterial antigens within the granuloma is not due to failure by the macrophages to sense these antigens as administration of exogenous antigens led to rapid arrest of T cell populations (111). Lack of recognition of Mtb antigens within the granuloma may be due to repression of antigen presentation by Mtb within infected macrophages (112, 113) or by the failure of T cells to recognize specific Mtb antigens within infected macrophages (114). In mice and cell culture models, infected macrophages have also been found to export Mtb antigens to surrounding cells, which limits the ability of T cells to directly recognize infected cells within granuloma and which could also potentially lead to arrest of T cells at sites distant to infected cells (115).
Organized granuloma formation may also diminish immune cell recognition of Mtb by physical means. This could potentially result from macrophage reprogramming and induction of epithelial modules within the granuloma which may in turn limit the ability of immune populations to recognize or migrate to bacteria within the central regions of the granuloma. Supporting this idea in zebrafish, partially disrupted granulomas formed by mosaic expression of dominant negative E-cadherin, neutrophils are preferentially recruited to the areas of the granuloma deficient for epithelial transformation, suggesting that epithelialization limits recruitment of these cells (46). Similarly, the physical packing of the individual cells in the granuloma may also constitute a barrier to immune recognition in this context. In intravital imaging experiments of BCG liver granulomas in mice, it was found that T cell recruitment into the granuloma required morphological rearrangement of granuloma macrophages to accommodate incoming T cells (116). While the degree to which these physical constraints serve to limit mycobacterial recognition is still to be fully investigated, at least in the case of epithelialization-dependent neutrophil exclusion, altered immune access confers a bacterial beneficial role, as epithelialization-deficient animals had increased bacterial burden (46). This suggests that altering granuloma epithelialization or otherwise loosening the tight apposition of immune cell populations within the granuloma could potentially improve antimycobacterial immune responses. Illustrating the potential of therapies targeted at granuloma structure, work in macaques has found that IDO inhibition can reorganize granulomas, resulting in increased numbers of T cells at the core of the granuloma and decreased bacterial burden (117). Outside of cellular response, it could also be expected that loosening of the granuloma would facilitate the access of other immune mediators like antibodies. B cells are known to be recruited to granulomas in humans and animal models (28, 118, 119), although the effects of B cells and antibodies on infection have been varied despite the effective recognition of Mtb by B cell produced antibodies (120).
Beyond physical separation of immune cell and bacterial populations, the highly organized granuloma environment alters host immune responsiveness through the generation of local inflammatory environments that shape the cell signaling and responses of cells within the granuloma. Histological and mass spectrometry characterization of human and macaque granulomas found that there is a pronounced type 1 inflammatory signature in the central regions of the granuloma, presumably due to recognition of adjacent bacteria within the necrotic core while more distal regions expressed an anti-inflammatory signature (18, 47, 49). Immune markers were found to correlate with the local cytokine environment, as mass spectrometry studies indicated that the caseum is specifically enriched for the type 1 inflammatory cytokine TNF, eicosanoids and downstream components of IFN signaling (49). Studies in cell culture have indicated that TNF is induced by macrophages in response to mycobacteria and mycobacterial products (121, 122), suggesting that this local TNF signal could result from interaction of macrophages with mycobacterial products at the edge of the caseum. Local expression of type 2 immune mediators can also shape the granuloma environment. TGFβ, a potent type 2 cytokine, has long been known to alter immune responses by driving anti-inflammatory programs in diverse immune populations (123). Elevated levels of TGFβ are found in the plasma of individuals with active Mtb infection (124). Interrogation of the local inflammatory environment within granulomas in Mtb infected mice and macaques also found that there was a pronounced TGFβ signature within the granuloma (125). Similarly, using histological techniques and mass spectrometry, elevated TGFβ signaling and induction of downstream genes have recently been described in human patient samples as well (103). Using TGFβR-deficient mice, TGFβ was found to act in a T cell intrinsic fashion to repress Th1 differentiation within the granuloma (125). Loss of TGFβ signaling led to increased IFN-γ production by granuloma T cells and reduced bacterial burden in these animals (125).
The anti-inflammatory cytokine IL-10 has also been studied during Mtb infection, particularly within the granuloma. Computational studies have suggested that IL-10 may play a crucial role in maintaining a host-detrimental anti-inflammatory environment within the granulomas (126). Short-term studies in macaques indicated that while neutralization of IL-10 had no effect on bacterial burden, it altered the cytokine environment within the granuloma and led to increased fibrosis throughout the structure (127). While ongoing studies will continue to identify the specific cytokines that shape the granuloma, it is clear that the granuloma creates a constrained space in which cytokines can generate a local inflammatory environment to shape the host response to Mtb.
That tuberculosis persists in the face of effective antibiotic therapies is a testament to the difficulty in clinically treating tuberculosis. As a slow growing bacterium, treatment necessitates the use of combination therapies composed of multiple antibiotics over extended time frames of 6 months or longer (128). In particular, the use of combination therapies is essential to prevent the acquisition of antibiotic resistance by Mtb. Despite these safeguards, antibiotic resistance is a growing problem, particularly due to difficulties in maintaining these complex drug regimens in resource-limited settings. This has led to the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains, which in turn have substantially longer and more complicated treatment regimens and greatly increased mortality (129). While many factors contribute to the complexities of treating tuberculosis in the clinic, the granuloma has been found to be a crucial structure that shapes antibiotic access to the bacterium and the responsiveness of the bacteria contained within.
Antimycobacterial antibiotics are first transported to their site of action via the host vasculature (6). Mtb granulomas, while largely composed from host immune populations, also recruit host vasculature to the nascent granuloma as observed in patient samples as well as animal models of infection (130, 131). The vascularization of granulomatous tissue is the result of angiogenic signals produced by the granuloma itself (19, 130, 132, 133). While multiple angiogenic factors are produced by the immune populations of the granuloma, the growth factor VEGF, predominantly produced by the macrophages of the granuloma, is crucial for efficient granuloma vascularization in both zebrafish and rabbits (130, 132, 134). In zebrafish, VEGF production by granuloma macrophages and angiogenesis is driven at least in part by recognition of bacterial TDM by these macrophage populations (134). However, there is considerable heterogeneity in the degree of vascularization between individual granulomas, which may restrict drug delivery to certain lesions (5, 30, 131). In addition, despite the strong vascularization of certain lesions, the improvised nature of the vasculature formed around the granuloma may limit the degree to which this vasculature can deliver drugs to the granuloma. Using anti-VEGF therapies to limit vascularization at the granuloma in rabbits, it was found that anti-VEGF despite reducing the degree of granuloma vascularization, enhanced delivery of small molecules to the granuloma by stabilizing the residual vasculature (132). In agreement with VEGF findings, the broad spectrum MMP inhibitor Marimastat was also found to stabilize the blood vessels of the granuloma in mice, leading to increased delivery of Mtb chemotherapeutics to the granuloma (135). Thus, variations in the degree vascularization and the organization and stabilization of granuloma vascularization are expected to influence accessibility of the lesion to mycobacterial chemotherapies.
After delivery to the granuloma via the vasculature, antimycobacterial therapies must cross the cellular layers that compose the granuloma and access Mtb within its niche in the granuloma. There are two distinct populations of bacteria within the granuloma, intracellular bacteria within macrophages and other immune cells and extracellular populations generally residing within the necrotic core of the granuloma. Antibiotic access to these disparate bacterial locations is driven by different considerations. In intracellular mycobacterial populations there is considerable variability in the degree to which individual antibiotics are able to enter and be retained by immune populations such as macrophages (136–139). Additionally, the metabolic changes driven in Mtb by the intracellular environment also alter the antibiotic susceptibility profile of Mtb (140–142). By contrast, for bacteria located within the central necrotic regions, antibiotics must not only penetrate the cellular layers of the granuloma but must also be able to diffuse efficiently into the caseum within the center of the granuloma (6, 143). Caseum in particular comprises a substantial barrier to antibiotic therapies, as many antibiotics either diffuse poorly into caseum or irreversibly bind to caseous material limiting deep penetration of antibiotics into the granuloma (136, 143, 144). The necrotic core of the granuloma and the composition of caseum itself also drives broad metabolic reprogramming of Mtb that make it resistant to many antibiotics (143, 145). Finally the reduced oxygen availability within the granuloma can also drive mycobacterial populations into a metabolically shifted, non-replicating state that can lead to altered susceptibility to antibiotics (146, 147).
The use of animal models with and without necrosis (148–150) as well as identification of spatial distribution of antibiotics with imaging mass spectrometry (136, 150–152) has shed light on the degree to which the complex structure of the tuberculous granuloma controls antibiotic access to Mtb. Consistent with their strong efficacy in the clinic, the frontline chemotherapeutics isoniazid and pyrazinamide effectively penetrate the outer cellular layers of the granuloma as well as the necrotic core in human patients and granuloma forming C3HeB/FeJ mice (136, 150). The other frontline therapies, rifampicin and ethambutol, were initially excluded from necrotic regions but concentrated within the necrotic core at later time points in either human lesions (rifampicin) or rabbit lesions (ethambutol) (136, 152). Despite the effective penetration of these antibiotics into the granuloma, experiments in granuloma-forming mouse models found that organized granuloma formation still limited the effectiveness of frontline antibiotics (148, 150), suggesting that the granuloma complicates treatment even for antibiotics with favorable distributions. Consistent with this, modeling of pharmacokinetic and pharmacodynamic data from humans found that many frontline and second-line therapies failed to reach desired concentrations in a subset of lesion types, with cavitary and caseous lesions being most difficult to treat (153).
Many second-line therapies also have limited diffusion into the core of necrotic granulomas (136, 137, 139, 150). Imaging of moxifloxacin in human and rabbit granulomas demonstrated that moxifloxacin penetrated the necrotic core far less efficiently than the cellular layers surrounding the core (136, 139). This asymmetric distribution of moxifloxacin and other fluoroquinolones was driven in part through strong uptake of this antibiotic by macrophage populations within the granuloma (139). While the clinical efficacy of moxifloxacin and pharmacodynamic studies demonstrate that moxifloxacin reaches effective concentrations within the granuloma (154), experiments in Nos2-deficient mice that form necrotic granulomas have suggested that the granuloma still serves as a barrier to effective therapy (148). Moxifloxacin treatment of Nos2-deficient animals prior to necrotic granuloma formation was approximately a log more effective in reducing bacterial burden than in animals that have already formed necrotic granulomas, consistent with granuloma necrosis limiting moxifloxacin efficacy (148). Similarly, while clinically equivalent moxifloxacin doses in rabbits were able to effectively clear bacterial burden in all types of granulomas, the use of suboptimal doses of moxifloxacin that mimic the case of patients with poor pharmacokinetics led to diminished clearance of bacteria within necrotic granulomas (154). By contrast, bacterial killing within cellular granulomas in these rabbits was equivalent for clinical and suboptimal dosing (154).
The complex structure of the granuloma also limits the use of other second-line therapies. Clofazimine is widely used to treat leprosy in clinical settings and has also been used against drug resistant Mtb (155, 156). However, Imaging of clofazimine localization in human patient samples revealed that although the drug penetrated the cellular layers of the granuloma very effectively, clofazimine was almost entirely excluded from the necrotic regions of the granuloma (136). Experiments in mouse models using the necrotic granuloma forming C3HeB/FeJ mice as well as BALB/c mouse models, which fail to form necrotic granulomas, found that while clofazimine was highly active in BALB/c mice, it was almost completely inactive in the necrotic granulomas that form in the lungs of C3HeB/FeJ mice (149). Further confirming that these differences in clofazimine effectiveness were likely related to granuloma structure, Mtb was found to be highly responsive to clofazimine in the spleens of C3HeB/FeJ mice, which lack necrotic granuloma formation (149). Taken together, these studies demonstrate that in Mtb infected individuals, the disparate features of the granuloma are a substantial barrier to therapy.
The central role of the granuloma in shaping host immune responses suggests that targeting the formation of granulomas could be used as a potential adjunctive therapy to facilitate productive immune responses within Mtb infected individuals. The disruption of the granuloma may serve to improve the ability of crucial cell types to recognize and eliminate mycobacteria within the granuloma either directly or through helper responses. Loss of granuloma integrity may enhance diffusion and recognition of mycobacterial products by the host immune system, potentially facilitating a more productive response within the host immune system. Similarly, loss of granuloma integrity may heighten the ability of host cytokines or immune molecules such as antibodies to access the bacteria and cells at the center of the granuloma. Beyond its effects on the host immune response, the granuloma also serves as a substantial barrier to the therapeutic efficacy of antibiotic treatments. Disruption of granulomas may increase the effectiveness of existing antimycobacterial therapies. Granuloma disruption could also enhance the spectrum of therapies that are effective against Mtb. Many chemotherapeutics are known to be effective against Mtb in vitro but have failed to translate into new clinical antibiotics. While the failure modes of these antibiotics are in many cases not fully understood, given the strong partitioning of some clinically effective anti-Mtb therapies, it is possible that at least some of these therapies that are effective in vitro fail due to limited penetration of these therapies in the context of organized granulomas. Thus, adjunctive therapeutic approaches targeting the granuloma may also facilitate the development of new antibiotics that are effective within disrupted granulomas.
While there is considerable promise in the development of adjunctive therapies targeting granuloma organization, there are also potential concerns of how granuloma disruption could alter disease outcomes during Mtb infection. In particular, the act of dissociating the granuloma may lead to the development of disseminated disease, a manifestation associated with poor disease outcome. Support for this idea comes from findings in HIV-positive individuals, where the degree of CD4 depletion is associated with disseminated disease and disruption of granuloma structure (157, 158). However, in contrast to findings in HIV-infected individuals, the granuloma has also been associated with dissemination in early infection (82, 83). A second potential limitation in targeting granuloma formation is that disruption of the granuloma and liberation of a large number of bacteria from the central core of the granuloma may lead to the host mounting an exuberant, host-detrimental response again Mtb in granuloma disrupted individuals. Excessive and problematic inflammatory responses are seen in Mtb infected, HIV-positive individuals, after beginning anti-retroviral therapy in a syndrome termed TB-immune reconstitution syndrome (TB-IRIS) (159). Finally, disruption of granulomas may enhance bacterial growth or allow the access to distinct niches within the infected host, potentially outweighing any advantages gained in antibiotic penetration and altered immune recognition.
While it remains to be seen whether there will be detrimental effects to therapies targeting granuloma structure, it may be possible to manage any potential drawbacks by timing the dissociation of granulomas. For instance, reduction of bacterial numbers through an initial period of antibiotic treatment may ultimately enable the safe disassembly of granulomas to improve subsequent therapies. Regardless, the ongoing difficulties in treating tuberculosis suggest that novel approaches to treating tuberculosis are worth pursuing. Adjunctively targeting the granuloma may enhance the effectiveness of existing treatments and shorten the duration of therapy required for these complex therapies. Granuloma dissociation could also enhance antimycobacterial immune responses, potentially boosting existing therapeutic approaches for both drug-sensitive and drug-resistant tuberculosis which could be further combined with immunomodulatory and antibiotic therapies. Lastly, targeting granuloma integrity may allow for the development of a wider palette of chemotherapeutics including drugs that fail to penetrate complex millieu of the organized granuloma. With the continued burden of tuberculosis worldwide and the increasing threat of drug-resistant Mtb, targeting of granuloma structure is a distinct approach to enhance the treatment of Mtb and is complementary to ongoing efforts to improve existing therapies and the development of new therapeutics. With the recent advances made in understanding granuloma formation and the molecular changes that underlie it, potential new points of intervention for therapies altering granuloma formation have been identified. Targeting these pathways may enable the rational development of new granuloma-targeted interventions.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by the Max Planck Society.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
I would like to thank David Tobin for critical reading of the manuscript and Molly Matty and Erika Hughes for original artwork that provided inspiration for the figures herein.
1. Metchnikoff E. Lectures on the Comparative Pathology of Inflammation: Delivered at the Pasteur Institute in 1891. London: Kegan Paul, Trench, Trübner (1893).
2. Ramakrishnan L. Revisiting the Role of the Granuloma in Tuberculosis. Nat Rev Immunol (2012) 12(5):352–66. doi: 10.1038/nri3211
3. Schüppel O. Untersuchungen Uber Lymphdrüsen-Tuberkulose Sowie Über Die Damit Verwandten Und Verwechselten Drüsenkrankheiten. Tübingen: Lauppsch (1871).
5. Lenaerts A, Barry CE 3rd, Dartois V. Heterogeneity in Tuberculosis Pathology, Microenvironments and Therapeutic Responses. Immunol Rev (2015) 264(1):288–307. doi: 10.1111/imr.12252
6. Dartois V. The Path of Anti-Tuberculosis Drugs: From Blood to Lesions to Mycobacterial Cells. Nat Rev Microbiol (2014) 12(3):159–67. doi: 10.1038/nrmicro3200
7. Ernst JD. The Immunological Life Cycle of Tuberculosis. Nat Rev Immunol (2012) 12(8):581–91. doi: 10.1038/nri3259
8. Saelens JW, Viswanathan G, Tobin DM. Mycobacterial Evolution Intersects With Host Tolerance. Front Immunol (2019) 10:528. doi: 10.3389/fimmu.2019.00528
9. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium Tuberculosis Infects Dendritic Cells With High Frequency and Impairs Their Function in vivo. J Immunol (2007) 179(4):2509–19. doi: 10.4049/jimmunol.179.4.2509
10. Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, et al. Intracellular Bacillary Burden Reflects a Burst Size for Mycobacterium Tuberculosis in vivo. PloS Pathog (2013) 9(2):e1003190. doi: 10.1371/journal.ppat.1003190
11. Adams DO. The Structure of Mononuclear Phagocytes Differentiating In Vivo. I. Sequential Fine and Histologic Studies of the Effect of Bacillus Calmette-Guerin (BCG). Am J Pathol (1974) 76(1):17–48.
12. Pagan AJ, Ramakrishnan L. The Formation and Function of Granulomas. Annu Rev Immunol (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
13. Laval T, Chaumont L, Demangel C. Not Too Fat to Fight: The Emerging Role of Macrophage Fatty Acid Metabolism in Immunity to Mycobacterium Tuberculosis. Immunol Rev (2021) 301(1):84–97. doi: 10.1111/imr.12952
14. Garcia-Rodriguez KM, Bini EI, Gamboa-Dominguez A, Espitia-Pinzon CI, Huerta-Yepez S, Bulfone-Paus S, et al. Differential Mast Cell Numbers and Characteristics in Human Tuberculosis Pulmonary Lesions. Sci Rep (2021) 11(1):10687. doi: 10.1038/s41598-021-89659-6
15. Gideon HP, Hughes TK, Wadsworth MH, Tu AA, Gierahn TM, Peters JM, et al. Multimodal Profiling of Lung Granulomas Reveals Cellular Correlates of Tuberculosis Control. bioRxiv (2021) 2020.10.24.352492. doi: 10.1101/2020.10.24.352492
16. Lasco TM, Turner OC, Cassone L, Sugawara I, Yamada H, McMurray DN, et al. Rapid Accumulation of Eosinophils in Lung Lesions in Guinea Pigs Infected With Mycobacterium Tuberculosis. Infect Immun (2004) 72(2):1147–9. doi: 10.1128/IAI.72.2.1147-1149.2004
17. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, et al. Initiation of the Adaptive Immune Response to Mycobacterium Tuberculosis Depends on Antigen Production in the Local Lymph Node, Not the Lungs. J Exp Med (2008) 205(1):105–15. doi: 10.1084/jem.20071367
18. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in Tuberculous Granulomas are Delineated by Distinct Populations of Macrophage Subsets and Expression of Nitric Oxide Synthase and Arginase Isoforms. J Immunol (2013) 191(2):773–84. doi: 10.4049/jimmunol.1300113
19. Hortle E, Oehlers SH. Host-Directed Therapies Targeting the Tuberculosis Granuloma Stroma. Pathog Dis (2020) 78(2):ftaa015. doi: 10.1093/femspd/ftaa015
20. Bohrer AC, Castro E, Hu Z, Queiroz ATL, Tocheny CE, Assmann M, et al. Eosinophils are Part of the Granulocyte Response in Tuberculosis and Promote Host Resistance in Mice. J Exp Med (2021) 218(10):e20210469. doi: 10.1084/jem.20210469
21. Canetti G. The Tubercle Bacillus in the Pulmonary Lesion of Man: Histobacteriology and Its Bearing on the Therapy of Pulmonary Tuberculosis. New York: Springer Publishing Company (1955).
22. Hunter RL. Tuberculosis as a Three-Act Play: A New Paradigm for the Pathogenesis of Pulmonary Tuberculosis. Tuberc (Edinb) (2016) 97:8–17. doi: 10.1016/j.tube.2015.11.010
23. Hunter RL, Actor JK, Hwang SA, Khan A, Urbanowski ME, Kaushal D, et al. Pathogenesis and Animal Models of Post-Primary (Bronchogenic) Tuberculosis, A Review. Pathogens (2018) 7(1):19. doi: 10.3390/pathogens7010019
24. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of Granulomas is Common in Active and Latent Tuberculosis Despite Within-Host Variability in Bacterial Killing. Nat Med (2014) 20(1):75–9. doi: 10.1038/nm.3412
25. Lin PL, Flynn JL. The End of the Binary Era: Revisiting the Spectrum of Tuberculosis. J Immunol (2018) 201(9):2541–8. doi: 10.4049/jimmunol.1800993
26. Kauffman KD, Sallin MA, Sakai S, Kamenyeva O, Kabat J, Weiner D, et al. Defective Positioning in Granulomas But Not Lung-Homing Limits CD4 T-Cell Interactions With Mycobacterium Tuberculosis-Infected Macrophages in Rhesus Macaques. Mucosal Immunol (2018) 11(2):462–73. doi: 10.1038/mi.2017.60
27. Marin ND, Dunlap MD, Kaushal D, Khader SA. Friend or Foe: The Protective and Pathological Roles of Inducible Bronchus-Associated Lymphoid Tissue in Pulmonary Diseases. J Immunol (2019) 202(9):2519–26. doi: 10.4049/jimmunol.1801135
28. Ulrichs T, Kosmiadi GA, Trusov V, Jorg S, Pradl L, Titukhina M, et al. Human Tuberculous Granulomas Induce Peripheral Lymphoid Follicle-Like Structures to Orchestrate Local Host Defence in the Lung. J Pathol (2004) 204(2):217–28. doi: 10.1002/path.1628
29. Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, et al. Quantitative Comparison of Active and Latent Tuberculosis in the Cynomolgus Macaque Model. Infect Immun (2009) 77(10):4631–42. doi: 10.1128/IAI.00592-09
30. Wells G, Glasgow JN, Nargan K, Lumamba K, Madansein R, Maharaj K, et al. microCT Analysis of the Human Tuberculous Lung Reveals Remarkable Heterogeneity in 3D Granuloma Morphology. Am J Respir Crit Care Med (2021) 204(5):583–95. doi: 10.1164/rccm.202101-0032OC
31. Coleman MT, Chen RY, Lee M, Lin PL, Dodd LE, Maiello P, et al. PET/CT Imaging Reveals a Therapeutic Response to Oxazolidinones in Macaques and Humans With Tuberculosis. Sci Transl Med (2014) 6(265):265ra167. doi: 10.1126/scitranslmed.3009500
32. Chen RY, Dodd LE, Lee M, Paripati P, Hammoud DA, Mountz JM, et al. PET/CT Imaging Correlates With Treatment Outcome in Patients With Multidrug-Resistant Tuberculosis. Sci Transl Med (2014) 6(265):265ra166. doi: 10.1126/scitranslmed.3009501
33. Flynn JL. Lessons From Experimental Mycobacterium Tuberculosis Infections. Microbes Infect (2006) 8(4):1179–88. doi: 10.1016/j.micinf.2005.10.033
34. Kramnik I, Beamer G. Mouse Models of Human TB Pathology: Roles in the Analysis of Necrosis and the Development of Host-Directed Therapies. Semin Immunopathol (2016) 38(2):221–37. doi: 10.1007/s00281-015-0538-9
35. Kramnik I, Dietrich WF, Demant P, Bloom BR. Genetic Control of Resistance to Experimental Infection With Virulent Mycobacterium Tuberculosis. Proc Natl Acad Sci USA (2000) 97(15):8560–5. doi: 10.1073/pnas.150227197
36. Reece ST, Loddenkemper C, Askew DJ, Zedler U, Schommer-Leitner S, Stein M, et al. Serine Protease Activity Contributes to Control of Mycobacterium Tuberculosis in Hypoxic Lung Granulomas in Mice. J Clin Invest (2010) 120(9):3365–76. doi: 10.1172/JCI42796
37. Cyktor JC, Carruthers B, Kominsky RA, Beamer GL, Stromberg P, Turner J. IL-10 Inhibits Mature Fibrotic Granuloma Formation During Mycobacterium Tuberculosis Infection. J Immunol (2013) 190(6):2778–90. doi: 10.4049/jimmunol.1202722
38. Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, et al. Lung Necrosis and Neutrophils Reflect Common Pathways of Susceptibility to Mycobacterium Tuberculosis in Genetically Diverse, Immune-Competent Mice. Dis Model Mech (2015) 8(9):1141–53. doi: 10.1242/dmm.020867
39. Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, et al. Tuberculosis Susceptibility and Vaccine Protection Are Independently Controlled by Host Genotype. mBio (2016) 7(5):e01516–16. doi: 10.1128/mBio.01516-16
40. Capuano SV 3rd, Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, et al. Experimental Mycobacterium Tuberculosis Infection of Cynomolgus Macaques Closely Resembles the Various Manifestations of Human M. Tuberculosis Infection. Infect Immun (2003) 71(10):5831–44. doi: 10.1128/IAI.71.10.5831-5844.2003
41. Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-Time Visualization of Mycobacterium-Macrophage Interactions Leading to Initiation of Granuloma Formation in Zebrafish Embryos. Immunity (2002) 17(6):693–702. doi: 10.1016/S1074-7613(02)00475-2
42. Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. Mycobacterium Marinum Infection of Adult Zebrafish Causes Caseating Granulomatous Tuberculosis and is Moderated by Adaptive Immunity. Infect Immun (2006) 74(11):6108–17. doi: 10.1128/IAI.00887-06
43. Parikka M, Hammaren MM, Harjula SK, Halfpenny NJ, Oksanen KE, Lahtinen MJ, et al. Mycobacterium Marinum Causes a Latent Infection That can be Reactivated by Gamma Irradiation in Adult Zebrafish. PloS Pathog (2012) 8(9):e1002944. doi: 10.1371/journal.ppat.1002944
44. Tobin DM, Vary JC Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The Lta4h Locus Modulates Susceptibility to Mycobacterial Infection in Zebrafish and Humans. Cell (2010) 140(5):717–30. doi: 10.1016/j.cell.2010.02.013
45. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host Genotype-Specific Therapies can Optimize the Inflammatory Response to Mycobacterial Infections. Cell (2012) 148(3):434–46. doi: 10.1016/j.cell.2011.12.023
46. Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity (2016) 45(4):861–76. doi: 10.1016/j.immuni.2016.09.014
47. Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, et al. A non-Canonical Type 2 Immune Response Coordinates Tuberculous Granuloma Formation and Epithelialization. Cell (2021) 184(7):1757–74 e14. doi: 10.1016/j.cell.2021.02.046
48. Ramakrishnan L. Mycobacterium Tuberculosis Pathogenicity Viewed Through the Lens of Molecular Koch's Postulates. Curr Opin Microbiol (2020) 54:103–10. doi: 10.1016/j.mib.2020.01.011
49. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory Signaling in Human Tuberculosis Granulomas is Spatially Organized. Nat Med (2016) 22(5):531–8. doi: 10.1038/nm.4073
50. Ndlovu H, Marakalala MJ. Granulomas and Inflammation: Host-Directed Therapies for Tuberculosis. Front Immunol (2016) 7:434. doi: 10.3389/fimmu.2016.00434
51. Wanat KA, Rosenbach M, Zoiber AF, Zhang PJ. Schaffer A. E-Cadherin is Expressed by Mono- and Multinucleated Histiocytes in Cutaneous Sarcoidal and Foreign Body Granulomas. Am J Dermatopathol (2014) 36(8):651–4. doi: 10.1097/DAD.0b013e31828de7e0
52. Van den Bossche J, Bogaert P, van Hengel J, Guerin CJ, Berx G, Movahedi K, et al. Alternatively Activated Macrophages Engage in Homotypic and Heterotypic Interactions Through IL-4 and Polyamine-Induced E-Cadherin/Catenin Complexes. Blood (2009) 114(21):4664–74. doi: 10.1182/blood-2009-05-221598
53. Van den Bossche J, Laoui D, Naessens T, Smits HH, Hokke CH, Stijlemans B, et al. E-Cadherin Expression in Macrophages Dampens Their Inflammatory Responsiveness In Vitro, But Does Not Modulate M2-Regulated Pathologies In Vivo. Sci Rep (2015) 5:12599. doi: 10.1038/srep12599
54. Takaki KK, Rinaldi G, Berriman M, Pagan AJ, Ramakrishnan L. Schistosoma Mansoni Eggs Modulate the Timing of Granuloma Formation to Promote Transmission. Cell Host Microbe (2021) 29(1):58–67.e5. doi: 10.1016/j.chom.2020.10.002
55. Caputo R, Gianotti F. Junctions Between Histiocytes: Role of Coated Vesicles. J Ultrastruct Res (1979) 68(3):256–64. doi: 10.1016/s0022-5320(79)90158-8
56. Chensue SW. Chemokines in Innate and Adaptive Granuloma Formation. Front Immunol (2013) 4:43:43. doi: 10.3389/fimmu.2013.00043
57. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An Essential Role for Interferon Gamma in Resistance to Mycobacterium Tuberculosis Infection. J Exp Med (1993) 178(6):2249–54. doi: 10.1084/jem.178.6.2249
58. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated Tuberculosis in Interferon Gamma Gene-Disrupted Mice. J Exp Med (1993) 178(6):2243–7. doi: 10.1084/jem.178.6.2243
59. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor Necrosis Factor-Alpha is Required in the Protective Immune Response Against Mycobacterium Tuberculosis in Mice. Immunity (1995) 2(6):561–72. doi: 10.1016/1074-7613(95)90001-2
60. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis Associated With Infliximab, a Tumor Necrosis Factor Alpha-Neutralizing Agent. N Engl J Med (2001) 345(15):1098–104. doi: 10.1056/NEJMoa011110
61. Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural Deficiencies in Granuloma Formation in TNF Gene-Targeted Mice Underlie the Heightened Susceptibility to Aerosol Mycobacterium Tuberculosis Infection, Which is Not Compensated for by Lymphotoxin. J Immunol (1999) 162(6):3504–11.
62. Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF Regulates Chemokine Induction Essential for Cell Recruitment, Granuloma Formation, and Clearance of Mycobacterial Infection. J Immunol (2002) 168(9):4620–7. doi: 10.4049/jimmunol.168.9.4620
63. Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, et al. Effects of Tumor Necrosis Factor Alpha on Host Immune Response in Chronic Persistent Tuberculosis: Possible Role for Limiting Pathology. Infect Immun (2001) 69(3):1847–55. doi: 10.1128/IAI.69.3.1847-1855.2001
64. Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, et al. Interferon-Gamma-Receptor Deficiency in an Infant With Fatal Bacille Calmette-Guerin Infection. N Engl J Med (1996) 335(26):1956–61. doi: 10.1056/NEJM199612263352604
65. Clay H, Volkman HE, Ramakrishnan L. Tumor Necrosis Factor Signaling Mediates Resistance to Mycobacteria by Inhibiting Bacterial Growth and Macrophage Death. Immunity (2008) 29(2):283–94. doi: 10.1016/j.immuni.2008.06.011
66. Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, et al. Tumor Necrosis Factor Neutralization Results in Disseminated Disease in Acute and Latent Mycobacterium Tuberculosis Infection With Normal Granuloma Structure in a Cynomolgus Macaque Model. Arthritis Rheum (2010) 62(2):340–50. doi: 10.1002/art.27271
67. Hogan LH, Markofski W, Bock A, Barger B, Morrissey JD, Sandor M. Mycobacterium Bovis BCG-Induced Granuloma Formation Depends on Gamma Interferon and CD40 Ligand But Does Not Require CD28. Infect Immun (2001) 69(4):2596–603. doi: 10.1128/IAI.69.4.2596-2603.2001
68. Jankovic D, Kullberg MC, Noben-Trauth N, Caspar P, Ward JM, Cheever AW, et al. Schistosome-Infected IL-4 Receptor Knockout (KO) Mice, in Contrast to IL-4 KO Mice, Fail to Develop Granulomatous Pathology While Maintaining the Same Lymphokine Expression Profile. J Immunol (1999) 163(1):337–42.
69. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
70. Subbian S, Tsenova L, Yang G, O'Brien P, Parsons S, Peixoto B, et al. Chronic Pulmonary Cavitary Tuberculosis in Rabbits: A Failed Host Immune Response. Open Biol (2011) 1(4):110016. doi: 10.1098/rsob.110016
71. van Crevel R, Karyadi E, Preyers F, Leenders M, Kullberg BJ, Nelwan RH, et al. Increased Production of Interleukin 4 by CD4+ and CD8+ T Cells From Patients With Tuberculosis is Related to the Presence of Pulmonary Cavities. J Infect Dis (2000) 181(3):1194–7. doi: 10.1086/315325
72. Ashenafi S, Aderaye G, Bekele A, Zewdie M, Aseffa G, Hoang AT, et al. Progression of Clinical Tuberculosis is Associated With a Th2 Immune Response Signature in Combination With Elevated Levels of SOCS3. Clin Immunol (2014) 151(2):84–99. doi: 10.1016/j.clim.2014.01.010
73. Hammaren MM, Oksanen KE, Nisula HM, Luukinen BV, Pesu M, Ramet M, et al. Adequate Th2-Type Response Associates With Restricted Bacterial Growth in Latent Mycobacterial Infection of Zebrafish. PloS Pathog (2014) 10(6):e1004190. doi: 10.1371/journal.ppat.1004190
74. Guler R, Parihar SP, Savvi S, Logan E, Schwegmann A, Roy S, et al. IL-4Ralpha-Dependent Alternative Activation of Macrophages is Not Decisive for Mycobacterium Tuberculosis Pathology and Bacterial Burden in Mice. PloS One (2015) 10(3):e0121070. doi: 10.1371/journal.pone.0121070
75. Hernandez-Pando R, Aguilar D, Hernandez ML, Orozco H, Rook G. Pulmonary Tuberculosis in BALB/c Mice With non-Functional IL-4 Genes: Changes in the Inflammatory Effects of TNF-Alpha and in the Regulation of Fibrosis. Eur J Immunol (2004) 34(1):174–83. doi: 10.1002/eji.200324253
76. Jung YJ, LaCourse R, Ryan L, North RJ. Evidence Inconsistent With a Negative Influence of T Helper 2 Cells on Protection Afforded by a Dominant T Helper 1 Response Against Mycobacterium Tuberculosis Lung Infection in Mice. Infect Immun (2002) 70(11):6436–43. doi: 10.1128/IAI.70.11.6436-6443.2002
77. Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, McKenzie AN, et al. The IL-13/IL-4Ralpha Axis is Involved in Tuberculosis-Associated Pathology. J Pathol (2014) 234(3):338–50. doi: 10.1002/path.4399
78. Cronan MR, Matty MA, Rosenberg AF, Blanc L, Pyle CJ, Espenschied ST, et al. An Explant Technique for High-Resolution Imaging and Manipulation of Mycobacterial Granulomas. Nat Methods (2018) 15(12):1098–107. doi: 10.1038/s41592-018-0215-8
79. Holscher C, Heitmann L, Owusu-Dabo E, Horstmann RD, Meyer CG, Ehlers S, et al. A Mutation in IL4RA Is Associated With the Degree of Pathology in Human TB Patients. Mediators Inflammation (2016) 2016:4245028. doi: 10.1155/2016/4245028
80. Hassoun D, Malard O, Barbarot S, Magnan A, Colas L. Type 2 Immunity-Driven Diseases: Towards a Multidisciplinary Approach. Clin Exp Allergy (2021) 51(12):1538–52. doi: 10.1111/cea.14029
81. Feng Y, Dorhoi A, Mollenkopf HJ, Yin H, Dong Z, Mao L, et al. Platelets Direct Monocyte Differentiation Into Epithelioid-Like Multinucleated Giant Foam Cells With Suppressive Capacity Upon Mycobacterial Stimulation. J Infect Dis (2014) 210(11):1700–10. doi: 10.1093/infdis/jiu355
82. Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. Tuberculous Granuloma Formation is Enhanced by a Mycobacterium Virulence Determinant. PloS Biol (2004) 2(11):e367. doi: 10.1371/journal.pbio.0020367
83. Davis JM, Ramakrishnan L. The Role of the Granuloma in Expansion and Dissemination of Early Tuberculous Infection. Cell (2009) 136(1):37–49. doi: 10.1016/j.cell.2008.11.014
84. Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, et al. Comparative Genomics of BCG Vaccines by Whole-Genome DNA Microarray. Science (1999) 284(5419):1520–3. doi: 10.1126/science.284.5419.1520
85. Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular Analysis of Genetic Differences Between Mycobacterium Bovis BCG and Virulent M. Bovis. J Bacteriol (1996) 178(5):1274–82. doi: 10.1128/jb.178.5.1274-1282.1996
86. Philipp WJ, Nair S, Guglielmi G, Lagranderie M, Gicquel B, Cole ST. Physical Mapping of Mycobacterium Bovis BCG Pasteur Reveals Differences From the Genome Map of Mycobacterium Tuberculosis H37Rv and From M. Bovis. Microbiol (Reading) (1996) 142(Pt 11):3135–45. doi: 10.1099/13500872-142-11-3135
87. Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, et al. Deletion of RD1 From Mycobacterium Tuberculosis Mimics Bacille Calmette-Guerin Attenuation. J Infect Dis (2003) 187(1):117–23. doi: 10.1086/345862
88. Sherman DR, Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Smith S. Mycobacterium Tuberculosis H37Rv: Delta RD1 is More Virulent Than M. Bovis Bacille Calmette-Guerin in Long-Term Murine Infection. J Infect Dis (2004) 190(1):123–6. doi: 10.1086/421472
89. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous Granuloma Induction via Interaction of a Bacterial Secreted Protein With Host Epithelium. Science (2010) 327(5964):466–9. doi: 10.1126/science.1179663
90. Jackson M. The Mycobacterial Cell Envelope-Lipids. Cold Spring Harb Perspect Med (2014) 4(10):a021105. doi: 10.1101/cshperspect.a021105
91. Sabin FR. Cellular Reactions to Fractions Isolated From Tubercle Bacilli. Physiol Rev (1932) 12(2):141–65. doi: 10.1152/physrev.1932.12.2.141
92. Chang JC, Jagirdar J, Lesser M. Long-Term Evolution of BCG- and CFA-Induced Granulomas in Rat Lungs. Correlation of Histologic Features With Cells in Bronchoalveolar Lavage Samples. Am J Pathol (1986) 125(1):16–27.
93. Sabin F, Doan C, Forkner C. Reaction of the Connective Tissues of the Normal Rabbit to Lipoids From the Tubercle Bacillus, Strain H-37. J Exptl Med Suppl (1930) 3(52):1–88.
94. Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6'-Dimycolate and Lipid in the Pathogenesis of Caseating Granulomas of Tuberculosis in Mice. Am J Pathol (2006) 168(4):1249–61. doi: 10.2353/ajpath.2006.050848
95. Retzinger GS, Meredith SC, Hunter RL, Takayama K, Kezdy FJ. Identification of the Physiologically Active State of the Mycobacterial Glycolipid Trehalose 6,6'-Dimycolate and the Role of Fibrinogen in the Biologic Activities of Trehalose 6,6'-Dimycolate Monolayers. J Immunol (1982) 129(2):735–44.
96. Bekierkunst A. Acute Granulomatous Response Produced in Mice by Trehalose-6,6-Dimycolate. J Bacteriol (1968) 96(4):958–61. doi: 10.1128/jb.96.4.958-961.1968
97. Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In Vivo Activity of Released Cell Wall Lipids of Mycobacterium Bovis Bacillus Calmette-Guerin is Due Principally to Trehalose Mycolates. J Immunol (2005) 174(8):5007–15. doi: 10.4049/jimmunol.174.8.5007
98. Yarkoni E, Rapp HJ. Granuloma Formation in Lungs of Mice After Intravenous Administration of Emulsified Trehalose-6,6'-Dimycolate (Cord Factor): Reaction Intensity Depends on Size Distribution of the Oil Droplets. Infect Immun (1977) 18(2):552–4. doi: 10.1128/iai.18.2.552-554.1977
99. Behling CA, Bennett B, Takayama K, Hunter RL. Development of a Trehalose 6,6'-Dimycolate Model Which Explains Cord Formation by Mycobacterium Tuberculosis. Infect Immun (1993) 61(6):2296–303. doi: 10.1128/iai.61.6.2296-2303.1993
100. Silva CL, Ekizlerian SM, Fazioli RA. Role of Cord Factor in the Modulation of Infection Caused by Mycobacteria. Am J Pathol (1985) 118(2):238–47.
101. Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T Cells and Adaptive Immunity to Mycobacterium Tuberculosis in Humans. Immunol Rev (2015) 264(1):74–87. doi: 10.1111/imr.12274
102. Srivastava S, Ernst JD. Cutting Edge: Direct Recognition of Infected Cells by CD4 T Cells is Required for Control of Intracellular Mycobacterium Tuberculosis In Vivo. J Immunol (2013) 191(3):1016–20. doi: 10.4049/jimmunol.1301236
103. McCaffrey EF, Donato M, Keren L, Chen Z, Delmastro A, Fitzpatrick MB, et al. The Immunoregulatory Landscape of Human Tuberculosis Granulomas. Nat Immunol (2022) 23(2):318–29. doi: 10.1038/s41590-021-01121-x
104. Portevin D, Via LE, Eum S, Young D. Natural Killer Cells are Recruited During Pulmonary Tuberculosis and Their Ex Vivo Responses to Mycobacteria Vary Between Healthy Human Donors in Association With KIR Haplotype. Cell Microbiol (2012) 14(11):1734–44. doi: 10.1111/j.1462-5822.2012.01834.x
105. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The Relative Importance of T Cell Subsets in Immunity and Immunopathology of Airborne Mycobacterium Tuberculosis Infection in Mice. J Exp Med (2001) 193(3):271–80. doi: 10.1084/jem.193.3.271
106. van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P. Control of Latent Mycobacterium Tuberculosis Infection is Dependent on CD8 T Cells. Eur J Immunol (2000) 30(12):3689–98. doi: 10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4
107. Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, et al. NK Cell-Derived IFN-Gamma Differentially Regulates Innate Resistance and Neutrophil Response in T Cell-Deficient Hosts Infected With Mycobacterium Tuberculosis. J Immunol (2006) 177(10):7086–93. doi: 10.4049/jimmunol.177.10.7086
108. Lin PL, Flynn JL. CD8 T Cells and Mycobacterium Tuberculosis Infection. Semin Immunopathol (2015) 37(3):239–49. doi: 10.1007/s00281-015-0490-8
109. Allen M, Bailey C, Cahatol I, Dodge L, Yim J, Kassissa C, et al. Mechanisms of Control of Mycobacterium Tuberculosis by NK Cells: Role of Glutathione. Front Immunol (2015) 6:508:508. doi: 10.3389/fimmu.2015.00508
110. Millar JA, Butler JR, Evans S, Mattila JT, Linderman JJ, Flynn JL, et al. Spatial Organization and Recruitment of Non-Specific T Cells May Limit T Cell-Macrophage Interactions Within Mycobacterium Tuberculosis Granulomas. Front Immunol (2021) 11:613638. doi: 10.3389/fimmu.2020.613638
111. Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital Imaging Reveals Limited Antigen Presentation and T Cell Effector Function in Mycobacterial Granulomas. Immunity (2011) 34(5):807–19. doi: 10.1016/j.immuni.2011.03.022
112. Noss EH, Harding CV, Boom WH. Mycobacterium Tuberculosis Inhibits MHC Class II Antigen Processing in Murine Bone Marrow Macrophages. Cell Immunol (2000) 201(1):63–74. doi: 10.1006/cimm.2000.1633
113. Kincaid EZ, Ernst JD. Mycobacterium Tuberculosis Exerts Gene-Selective Inhibition of Transcriptional Responses to IFN-Gamma Without Inhibiting STAT1 Function. J Immunol (2003) 171(4):2042–9. doi: 10.4049/jimmunol.171.4.2042
114. Yang JD, Mott D, Sutiwisesak R, Lu YJ, Raso F, Stowell B, et al. Mycobacterium Tuberculosis-Specific CD4+ and CD8+ T Cells Differ in Their Capacity to Recognize Infected Macrophages. PloS Pathog (2018) 14(5):e1007060. doi: 10.1371/journal.ppat.1007060
115. Srivastava S, Grace PS, Ernst JD. Antigen Export Reduces Antigen Presentation and Limits T Cell Control of M. Tuberculosis. Cell Host Microbe (2016) 19(1):44–54. doi: 10.1016/j.chom.2015.12.003
116. Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T Cell Dynamics During the Development and Disintegration of Mycobacterial Granulomas. Immunity (2008) 28(2):271–84. doi: 10.1016/j.immuni.2007.12.010
117. Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, et al. In Vivo Inhibition of Tryptophan Catabolism Reorganizes the Tuberculoma and Augments Immune-Mediated Control of Mycobacterium Tuberculosis. Proc Natl Acad Sci USA (2018) 115(1):E62–71. doi: 10.1073/pnas.1711373114
118. Phuah JY, Mattila JT, Lin PL, Flynn JL. Activated B Cells in the Granulomas of Nonhuman Primates Infected With Mycobacterium Tuberculosis. Am J Pathol (2012) 181(2):508–14. doi: 10.1016/j.ajpath.2012.05.009
119. Maglione PJ, Xu J, Chan J. B Cells Moderate Inflammatory Progression and Enhance Bacterial Containment Upon Pulmonary Challenge With Mycobacterium Tuberculosis. J Immunol (2007) 178(11):7222–34. doi: 10.4049/jimmunol.178.11.7222
120. Rijnink WF, Ottenhoff THM. Joosten SA. B-Cells and Antibodies as Contributors to Effector Immune Responses in Tuberculosis. Front Immunol (2021) 12:640168. doi: 10.3389/fimmu.2021.640168
121. Valone SE, Rich EA, Wallis RS, Ellner JJ. Expression of Tumor Necrosis Factor In Vitro by Human Mononuclear Phagocytes Stimulated With Whole Mycobacterium Bovis BCG and Mycobacterial Antigens. Infect Immun (1988) 56(12):3313–5. doi: 10.1128/iai.56.12.3313-3315.1988
122. Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. TNF Influences Chemokine Expression of Macrophages In Vitro and That of CD11b+ Cells In Vivo During Mycobacterium Tuberculosis Infection. J Immunol (2004) 172(11):6846–57. doi: 10.4049/jimmunol.172.11.6846
123. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-Beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol (2017) 9(6):a022236. doi: 10.1101/cshperspect.a022236
124. Olobo JO, Geletu M, Demissie A, Eguale T, Hiwot K, Aderaye G, et al. Circulating TNF-Alpha, TGF-Beta, and IL-10 in Tuberculosis Patients and Healthy Contacts. Scand J Immunol (2001) 53(1):85–91. doi: 10.1046/j.1365-3083.2001.00844.x
125. Gern BH, Adams KN, Plumlee CR, Stoltzfus CR, Shehata L, Moguche AO, et al. TGFbeta Restricts Expansion, Survival, and Function of T Cells Within the Tuberculous Granuloma. Cell Host Microbe (2021) 29(4):594–606.e6. doi: 10.1016/j.chom.2021.02.005
126. Cilfone NA, Ford CB, Marino S, Mattila JT, Gideon HP, Flynn JL, et al. Computational Modeling Predicts IL-10 Control of Lesion Sterilization by Balancing Early Host Immunity-Mediated Antimicrobial Responses With Caseation During Mycobacterium Tuberculosis Infection. J Immunol (2015) 194(2):664–77. doi: 10.4049/jimmunol.1400734
127. Wong EA, Evans S, Kraus CR, Engelman KD, Maiello P, Flores WJ, et al. IL-10 Impairs Local Immune Response in Lung Granulomas and Lymph Nodes During Early Mycobacterium Tuberculosis Infection. J Immunol (2020) 204(3):644–59. doi: 10.4049/jimmunol.1901211
128. Lee A, Xie YL, Barry CE, Chen RY. Current and Future Treatments for Tuberculosis. BMJ (2020) 368:m216. doi: 10.1136/bmj.m216
129. Allue-Guardia A, Garcia JI, Torrelles JB. Evolution of Drug-Resistant Mycobacterium Tuberculosis Strains and Their Adaptation to the Human Lung Environment. Front Microbiol (2021) 12:612675. doi: 10.3389/fmicb.2021.612675
130. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of Host Angiogenic Signalling Limits Mycobacterial Growth. Nature (2015) 517(7536):612–5. doi: 10.1038/nature13967
131. Ulrichs T, Kosmiadi GA, Jorg S, Pradl L, Titukhina M, Mishenko V, et al. Differential Organization of the Local Immune Response in Patients With Active Cavitary Tuberculosis or With Nonprogressive Tuberculoma. J Infect Dis (2005) 192(1):89–97. doi: 10.1086/430621
132. Datta M, Via LE, Kamoun WS, Liu C, Chen W, Seano G, et al. Anti-Vascular Endothelial Growth Factor Treatment Normalizes Tuberculosis Granuloma Vasculature and Improves Small Molecule Delivery. Proc Natl Acad Sci USA (2015) 112(6):1827–32. doi: 10.1073/pnas.1424563112
133. Oehlers SH, Cronan MR, Beerman RW, Johnson MG, Huang J, Kontos CD, et al. Infection-Induced Vascular Permeability Aids Mycobacterial Growth. J Infect Dis (2017) 215(5):813–7. doi: 10.1093/infdis/jiw355
134. Walton EM, Cronan MR, Cambier CJ, Rossi A, Marass M, Foglia MD, et al. Cyclopropane Modification of Trehalose Dimycolate Drives Granuloma Angiogenesis and Mycobacterial Growth Through Vegf Signaling. Cell Host Microbe (2018) 24(4):514–25.e6. doi: 10.1016/j.chom.2018.09.004
135. Xu Y, Wang L, Zimmerman MD, Chen KY, Huang L, Fu DJ, et al. Matrix Metalloproteinase Inhibitors Enhance the Efficacy of Frontline Drugs Against Mycobacterium Tuberculosis. PloS Pathog (2018) 14(4):e1006974. doi: 10.1371/journal.ppat.1006974
136. Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O'Brien P, et al. The Association Between Sterilizing Activity and Drug Distribution Into Tuberculosis Lesions. Nat Med (2015) 21(10):1223–7. doi: 10.1038/nm.3937
137. Ernest JP, Sarathy J, Wang N, Kaya F, Zimmerman MD, Strydom N, et al. Lesion Penetration and Activity Limit the Utility of Second-Line Injectable Agents in Pulmonary Tuberculosis. Antimicrob Agents Chemother (2021) 65(10):AAC0050621. doi: 10.1128/AAC.00506-21
138. Tulkens PM. Intracellular Distribution and Activity of Antibiotics. Eur J Clin Microbiol Infect Dis (1991) 10(2):100–6. doi: 10.1007/BF01964420
139. Blanc L, Daudelin IB, Podell BK, Chen PY, Zimmerman M, Martinot AJ, et al. High-Resolution Mapping of Fluoroquinolones in TB Rabbit Lesions Reveals Specific Distribution in Immune Cell Types. Elife (2018) 7:e41115. doi: 10.7554/eLife.41115
140. VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, et al. Novel Inhibitors of Cholesterol Degradation in Mycobacterium Tuberculosis Reveal How the Bacterium's Metabolism is Constrained by the Intracellular Environment. PloS Pathog (2015) 11(2):e1004679. doi: 10.1371/journal.ppat.1004679
141. Bellerose MM, Proulx MK, Smith CM, Baker RE, Ioerger TR, Sassetti CM. Distinct Bacterial Pathways Influence the Efficacy of Antibiotics Against Mycobacterium Tuberculosis. mSystems (2020) 5(4):e00396–20. doi: 10.1128/mSystems.00396-20
142. Pisu D, Huang L, Narang V, Theriault M, Le-Bury G, Lee B, et al. Single Cell Analysis of M. Tuberculosis Phenotype and Macrophage Lineages in the Infected Lung. J Exp Med (2021) 218(9):e20210615. doi: 10.1084/jem.20210615
143. Sarathy JP, Dartois V. Caseum: A Niche for Mycobacterium Tuberculosis Drug-Tolerant Persisters. Clin Microbiol Rev (2020) 33(3):e00159–19. doi: 10.1128/CMR.00159-19
144. Sarathy JP, Zuccotto F, Hsinpin H, Sandberg L, Via LE, Marriner GA, et al. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect Dis (2016) 2(8):552–63. doi: 10.1021/acsinfecdis.6b00051
145. Sarathy JP, Via LE, Weiner D, Blanc L, Boshoff H, Eugenin EA, et al. Extreme Drug Tolerance of Mycobacterium Tuberculosis in Caseum. Antimicrob Agents Chemother (2018) 62(2):e02266–17. doi: 10.1128/AAC.02266-17
146. Prosser G, Brandenburg J, Reiling N, Barry CE 3rd, Wilkinson RJ, Wilkinson KA. The Bacillary and Macrophage Response to Hypoxia in Tuberculosis and the Consequences for T Cell Antigen Recognition. Microbes Infect (2017) 19(3):177–92. doi: 10.1016/j.micinf.2016.10.001
147. Hudock TA, Foreman TW, Bandyopadhyay N, Gautam US, Veatch AV, LoBato DN, et al. Hypoxia Sensing and Persistence Genes Are Expressed During the Intragranulomatous Survival of Mycobacterium Tuberculosis. Am J Respir Cell Mol Biol (2017) 56(5):637–47. doi: 10.1165/rcmb.2016-0239OC
148. Gengenbacher M, Duque-Correa MA, Kaiser P, Schuerer S, Lazar D, Zedler U, et al. NOS2-Deficient Mice With Hypoxic Necrotizing Lung Lesions Predict Outcomes of Tuberculosis Chemotherapy in Humans. Sci Rep (2017) 7(1):8853. doi: 10.1038/s41598-017-09177-2
149. Irwin SM, Gruppo V, Brooks E, Gilliland J, Scherman M, Reichlen MJ, et al. Limited Activity of Clofazimine as a Single Drug in a Mouse Model of Tuberculosis Exhibiting Caseous Necrotic Granulomas. Antimicrob Agents Chemother (2014) 58(7):4026–34. doi: 10.1128/AAC.02565-14
150. Irwin SM, Prideaux B, Lyon ER, Zimmerman MD, Brooks EJ, Schrupp CA, et al. Bedaquiline and Pyrazinamide Treatment Responses Are Affected by Pulmonary Lesion Heterogeneity in Mycobacterium Tuberculosis Infected C3HeB/FeJ Mice. ACS Infect Dis (2016) 2(4):251–67. doi: 10.1021/acsinfecdis.5b00127
151. Prideaux B, Dartois V, Staab D, Weiner DM, Goh A, Via LE, et al. High-Sensitivity MALDI-MRM-MS Imaging of Moxifloxacin Distribution in Tuberculosis-Infected Rabbit Lungs and Granulomatous Lesions. Anal Chem (2011) 83(6):2112–8. doi: 10.1021/ac1029049
152. Zimmerman M, Lestner J, Prideaux B, O'Brien P, Dias-Freedman I, Chen C, et al. Ethambutol Partitioning in Tuberculous Pulmonary Lesions Explains Its Clinical Efficacy. Antimicrob Agents Chemother (2017) 61(9):e00924–17. doi: 10.1128/AAC.00924-17
153. Strydom N, Gupta SV, Fox WS, Via LE, Bang H, Lee M, et al. Tuberculosis Drugs' Distribution and Emergence of Resistance in Patient's Lung Lesions: A Mechanistic Model and Tool for Regimen and Dose Optimization. PloS Med (2019) 16(4):e1002773. doi: 10.1371/journal.pmed.1002773
154. Sarathy J, Blanc L, Alvarez-Cabrera N, O'Brien P, Dias-Freedman I, Mina M, et al. Fluoroquinolone Efficacy Against Tuberculosis Is Driven by Penetration Into Lesions and Activity Against Resident Bacterial Populations. Antimicrob Agents Chemother (2019) 63(5):e02516–18. doi: 10.1128/AAC.02516-18
155. Tang S, Yao L, Hao X, Liu Y, Zeng L, Liu G, et al. Clofazimine for the Treatment of Multidrug-Resistant Tuberculosis: Prospective, Multicenter, Randomized Controlled Study in China. Clin Infect Dis (2015) 60(9):1361–7. doi: 10.1093/cid/civ027
156. Van Deun A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, et al. Short, Highly Effective, and Inexpensive Standardized Treatment of Multidrug-Resistant Tuberculosis. Am J Respir Crit Care Med (2010) 182(5):684–92. doi: 10.1164/rccm.201001-0077OC
157. Diedrich CR, O'Hern J, Wilkinson RJ. HIV-1 and the Mycobacterium Tuberculosis Granuloma: A Systematic Review and Meta-Analysis. Tuberc (Edinb) (2016) 98:62–76. doi: 10.1016/j.tube.2016.02.010
158. Esmail H, Riou C, Bruyn ED, Lai RP, Harley YXR, Meintjes G, et al. The Immune Response to Mycobacterium Tuberculosis in HIV-1-Coinfected Persons. Annu Rev Immunol (2018) 36:603–38. doi: 10.1146/annurev-immunol-042617-053420
Keywords: tuberculosis, granuloma, granuloma organization, host-directed therapies, macrophage reprogramming, macrophage
Citation: Cronan MR (2022) In the Thick of It: Formation of the Tuberculous Granuloma and Its Effects on Host and Therapeutic Responses. Front. Immunol. 13:820134. doi: 10.3389/fimmu.2022.820134
Received: 22 November 2021; Accepted: 15 February 2022;
Published: 07 March 2022.
Edited by:
Antonio Pagán, University of Cambridge, United KingdomReviewed by:
Alan Sher, National Institutes of Health (NIH), United StatesCopyright © 2022 Cronan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark R. Cronan, Y3JvbmFuQG1waWliLWJlcmxpbi5tcGcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.