
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Immunol., 01 February 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.819224
This article is part of the Research TopicReassessing the Immune System Contribution in Multiple Sclerosis: Therapeutic Target, Biomarkers of Disease and Immune PathogenesisView all 19 articles
 Lifei Hou1,2*
Lifei Hou1,2* Koichi Yuki1,2*
Koichi Yuki1,2*Due to the plasticity of IL-17-producing CD4 T cells (Th17 cells), a long-standing challenge in studying Th17-driven autoimmune is the lack of specific surface marker to identify the pathogenic Th17 cells in vivo. Recently, we discovered that pathogenic CD4 T cells were CXCR6 positive in experimental autoimmune encephalomyelitis (EAE), a commonly used Th17-driven autoimmune model. Herein, we further revealed that peripheral CXCR6+CD4 T cells contain a functionally distinct subpopulation, which is CCR6 positive and enriched for conventional Th17 molecules (IL-23R and RORγt) and cytotoxic signatures. Additionally, spinal cord-infiltrating CD4 T cells were highly cytotoxic by expressing Granzyme(s) along with IFNγ and GM-CSF. Collectively, this study suggested that peripheral CCR6+CXCR6+CD4 T cells were Th17 cells with cytotoxic property in EAE model, and highlighted the cytotoxic granzymes for EAE pathology.
Multiple sclerosis (MS) is a devastating autoimmune disease with progressive neurological dysfunction due to demyelination of the central nerve system (CNS) (1). EAE is the most commonly used murine MS model and driven by self-reactive CD4 T cells (CD4 cells thereafter) (1). The disease-inducing CD4 cells were initially thought to be Th1 cells, but later were described as Th17 cells (2, 3). Indeed, Th17 cells themselves are not pathogenic (4), but are converted, under the priming of myeloid cell-derived IL-1 and IL-23 (5–7), into pathogenic CD4 cells, which lose IL-17-producing ability and alternatively produce IFNγ and GM-CSF (8–12). Despite their cytokine profiles, little else is known about the pathogenic CD4 cells, mainly due to the lack of specific marker to precisely identify them in vivo.
We previously found SerpinB1 (sb1, Serine Protease Inhibitor B1), an endogenous protease inhibitor (13), to be a signature gene of IL-17-producing γδ T cells (14) and Th17 cells (15). Subsequent studies showed that sb1-ko mice were resistant to EAE with a paucity of CXCR6+CD4 cells (16), which were highly enriched in the spinal cord, secreted inflammatory cytokine IFNγ and GM-CSF, contained cytotoxic Granzyme-C, and proliferated rapidly (16). Depleting CXCR6+CD4 cells by anti-CXCR6 antibody dramatically ameliorated established EAE, confirming the CXCR6+CD4 cells as the driver of EAE pathology (16). In this study, we further characterized CXCR6+CD4 cells.
Wild type C57BL/6J mice (wild type, wt) were originally purchased from the Jackson laboratory and maintained in an animal facility at the Boston Children’s Hospital. Animal protocols were approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital. EAE model induction, flow cytometry, cell sorting, q-PCR analysis, cell counting, and RNA sequencing were performed as previously described (16). In brief, to induce EAE, wt mice in the C57BL/6J background were injected with MOG35–55 emulsified with complete Freund’s adjuvant followed by 200 ng pertussis toxin on days 0 and 2.
CD4 effector cells (CD44+CD4) were sort-purified from pooled draining lymph node (dLN) cells and spinal cords of MOG-immunized wt mice by FACSAria system at the disease onset. The cells were stimulated for 4 h with PMA (50 ng/ml) and ionomycin (750 ng/ml) (Sigma-Aldrich), and RNA was purified using QIAGEN RNeasy Plus Mini kits and quantified by optical density at 260/280/230 nm. TruSeq RNA V2 kits were used to construct transcript-specific libraries that were sequenced on Illumina HiSeq2500. The resulting 4.5 Gb/genotype of raw data was trimmed, and 20 million reads were mapped. Genes that had expression levels (FPKM) ≥1.0 were analyzed for differential expression (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192878)
CCR6+CXCR6-CD44+CD4, CCR6+CXCR6+CD44+CD4, CCR6-CXCR6+CD44+CD4 were sort-purified from pooled lymph node cells of MOG-immunized wt mice by FACSAria system at disease onset. RNA was isolated using RNeasy Plus kits (74134, Qiagen) according to the manufacturer’s protocol and reverse-transcribed using the iScript™ cDNA Synthesis kit (Bio-Rad). The qPCR assays were performed on the CFX96™ Real-Time System (Bio-Rad) with the iTaq™ Universal SYBR Green Supermix (Bio-Rad) using 30 s denaturation at 95°C and 40 cycles of 5 s at 95°C and 30 s at 61°C using the primers. Relative expression level for each gene was calculated by using the ΔΔCt method and normalizing to Actb.
Cells were stained with fluorochrome-conjugated antibodies to surface markers. Fluorochrome-conjugated antibodies were from Biolegend: FITC- or PE-Cy7-anti-mCD3 (145-2C11), Pacific blue- or PE-anti-mCD45 (30-F11), Pacific blue- or PE-Cy7- or APC- anti-mCD4 (GK1.5), PE-anti-mIL1R1 (JAMA-147), FITC-anti-mCD44 (IM7), APC-anti-mCXCR6 (SA051D1), APC- or PE-Cy7-anti-mCCR6 (29-2L17). From R&D system: PE-anti-mIL-23R (753317). Data were acquired on a Canto II cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star). Cell counting was achieved by using AccuCount beads (Spherotech).
After red blood cell lysis, both draining lymph node cells and peripheral blood cells were stimulated for 4 h with PMA (50 ng/ml) and ionomycin (750 ng/ml) (Sigma-Aldrich) in the presence of Brefeldin A. Cells were then collected and resuspended in cell staining buffer (Fluidigm; San Francisco, CA). After centrifugation, Fc receptor blocking reagent (clone 93, Biolegend) was used at a 1:100 dilution in for 10 min, followed by incubation with metal conjugated surface antibodies for 30 min. Then the cells were fixed and permeabilized by using fixation/permeabilization reagents (BD Bioscience). The permeabilization buffer was made by diluting the 10× stock solution (51-2091, BD Biosciences) in UltraPure distilled water (Invitrogen). All antibodies were purchased from the CyTOF core facility at Brigham and Women’s Hospital. DNA was labeled with iridium intercalator solution (Fluidigm). Samples were subsequently washed and reconstituted in Milli-Q filtered distilled water in the presence of EQ Four Element Calibration beads (Fluidigm, catalog 201078). Samples were acquired on a Helios CyTOF Mass Cytometer (Fluidigm) at Cellular Profiling Core Facility (School of Public Health, Harvard Medical School, Boston, MA, USA); Data were analyzed by using spanning-tree progression analysis of density-normalized events (SPADE) and vi stochastic neighbor embedding (SNE) on Cytobank software.
Statistical analyses were performed using Prism 4 (Graphpad Software). Student’s t-test was used. P-values <0.05 were considered significant.
We immunized the wild type (wt) mice with MOG to induce EAE and monitored the CXCR6+CD4 cells in the blood. Results showed that CXCR6+CD4 cells did not exist in naïve wt mice, but were dramatically induced in the blood with EAE induction (Figures 1A, B). Although CXCR6+CD4 cells dramatically accumulated in the peripheral blood of EAE mice, Cxcr6-ko and wt mice developed comparable EAE symptom (17), suggesting that CXCR6 might only serve as a marker and be dispensable for CD4 cell chemotaxis in EAE. Since CCR6 has been well established as a chemokine receptor preferentially expressed on conventional Th17 cells (18), especially the one generated in vitro, we then co-stained the CCR6 and CXCR6 on draining lymph node (dLN)-derived CD4 cells at EAE onset. Strikingly, we found CXCR6+CD4 cells could be divided into two subpopulations: CCR6 positive and CCR6 negative ones (Figure 1C). We then sorted out the CCR6+CXCR6- (as control), CCR6+CXCR6+, and CCR6-CXCR6+ effector CD4 cells from dLN at EAE onset. Q-PCR experiment showed that CCR6+CXCR6+ effector CD4 cells majorly expressed conventional Th17 signatures including Il23r, Il17a and Rorc, while CCR6−CXCR6+ ones majorly expressed Ifng and Csf2 (encoding GM-CSF), a typical ex-Th17 phenotype (Figure 1D). Flow cytometry analysis further confirmed that IL-23R and IL-1R were exclusively expressed on CCR6+CXCR6+CD4 cells (Figure 1E).
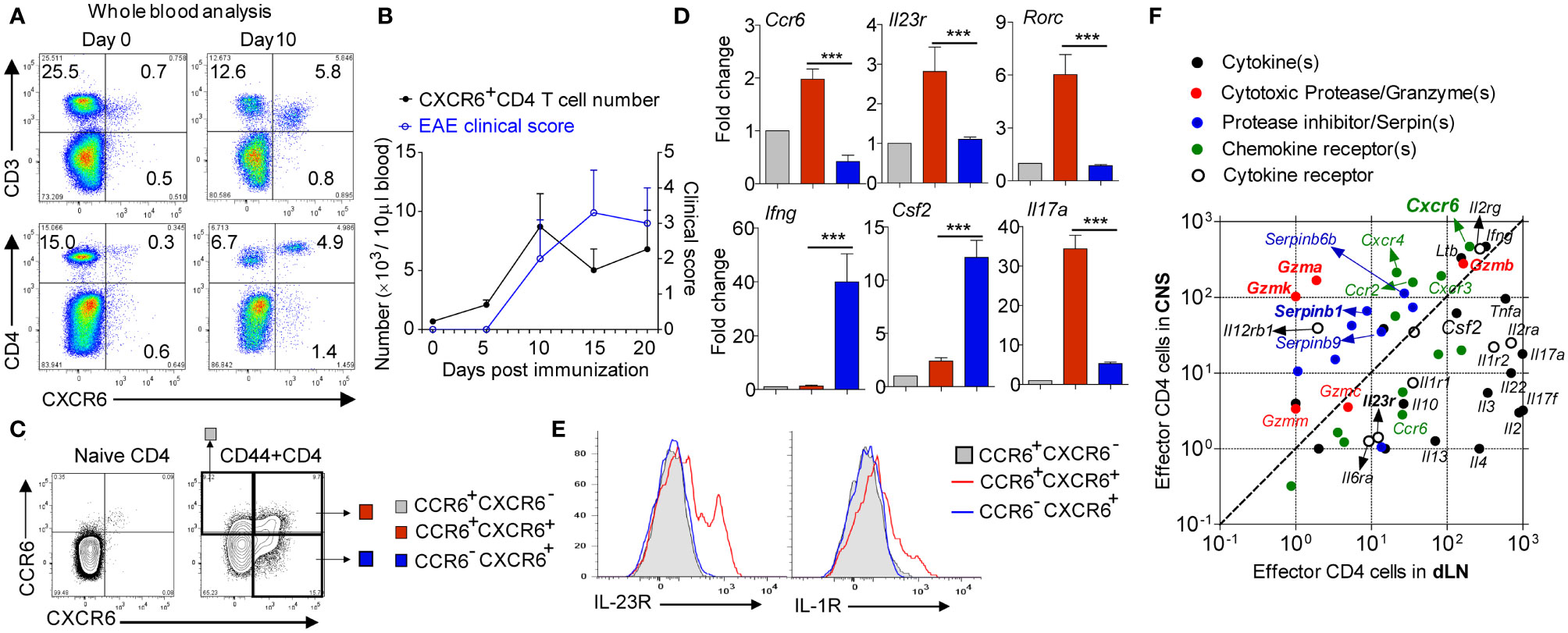
Figure 1 CXCR6+CD4 cells in EAE. (A) Representative FACS data gating on blood leukocyte. (B) Cell number. N = 4 in each time points. (C) Gating strategy for FACS sorting or analysis of CD4 cells in dLN at EAE onset. (D) Relative gene expression of CCR6+CXCR6−, CCR6+CXCR6+and CCR6−CXCR6+ effector CD4 cells; CCR6+CXCR6− was used as control. Data are mean ± SEM of 3 independent experiments. ***p <0.001. (E) Representative FACS data showing IL23R and IL1R expression on CD4 cells in dLN at EAE onset. (F) Effector CD4 cells were isolated from both dLN and spinal cord at EAE onset, and subjected to bulk RNAseq. Data are presented by FMPK.
A long-standing difficulty in treating autoimmune disease, such as MS, is the lack of understanding of inflammatory tissue-infiltrating CD4 cells. Given that most of CNS-infiltrating CD4 cells are CXCR6 positive, we compared the transcriptome profile between CNS-infiltrating CD4 cells and peripheral effector CD4 cells in EAE. We sorted out the spinal cord-infiltrated CD4 cells, and compared them with effector CD4 from dLN at EAE onset and discovered that all of Il17a, Ccr6, and Il23r were downregulated in spinal cord-infiltrated CD4 cells, suggesting a terminal differentiated effector status. Interestingly, we found that CD4 cells in the spinal cord were cytotoxic by highly expressing Granzyme(s) and cognate serine protease inhibitor Serpin(s) along with IFNγ and GM-CSF (Figure 1F). Indeed, this finding matches with the human MS study that CD4 cells from human secondary progressive MS autopsy brain samples are cytotoxic by co-expression of Eomes and Granzyme-B (19).
To further confirm the existence of distinct subpopulation within CXCR6+CD4 cells in the periphery at EAE onset, cytometry by time of flight (CyTOF) was applied (20), which is a powerful discovery tool in identifying rare cell population and cluster. A CyTOF study of 22 markers for effector CD4 cells showed that all of IFNγ (IL-2 negative portion), GM-CSF, IL-17, IL-23R, CD107a, Granzyme-B, IL-1R1 and CCR6 and partial of T-bet and RORγt overlapped with CXCR6 expression, suggesting CXCR6 as a marker for pathogenic CD4 cells (Figure 2A). We then focused on CXCR6+CD44+CD4 cells, and finally confirmed that they contained a distinct CCR6 positive subpopulation, which was enriched with conventional Th17 molecules (IL-1R, IL-17, IL-23R, and RORγt), and also highly cytotoxic by expressing Perforin, CD107a and Granzyme-B (Figure 2B). We observed partial overlap among IFNγ, IL-17 and GM-CSF, which might suggest the co-existence of conventional Th17 and ex-Th17 cells. Interestingly, IFNγ staining did not match with T-bet well, deemed as Th1 transcriptional factor responsible for IFNγ expression. Similarly, IL-17 staining did not match with RORγt well. This discrepancy between cytokines and corresponding transcriptional factors might be due to that PMA and ionomycin stimulation will preferentially accumulate the cytokine, not the transcriptional factors, for detection. It also could be due to the complex roles of transcriptional factors involved in the CD4 cell plasticity in autoimmunity. Purified PBMC from the peripheral blood were subjected to CyTOF analysis in parallel and showed identical profile as dLN (data not shown).
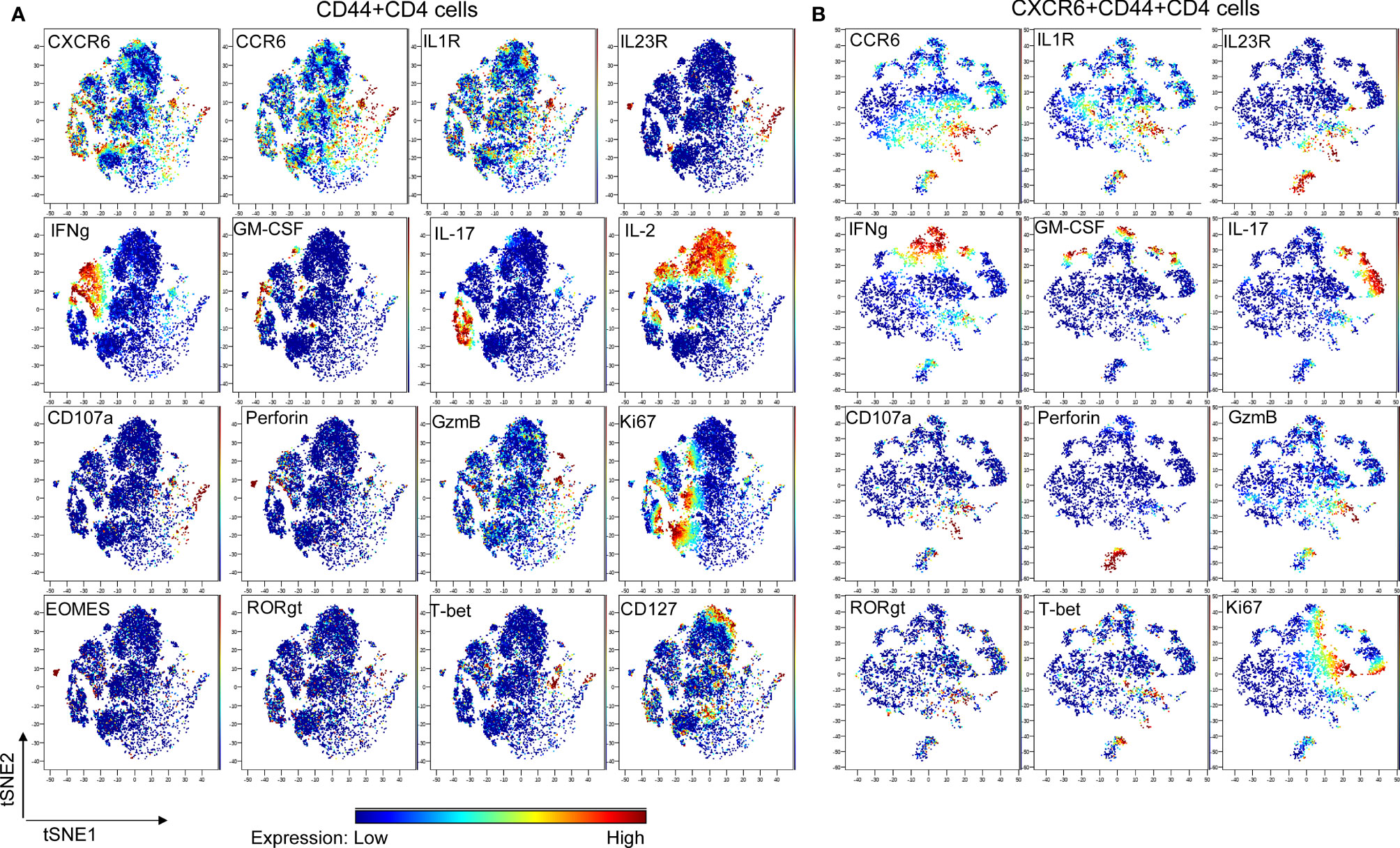
Figure 2 CyTOF study. Wt mice were induced to develop EAE and sacrificed at the disease onset (N = 9; dLN from every 3 mice were pooled and deemed as one biological sample). Lymphocytes were stimulated with PMA & Ionomycin for 4 h in the presence of Brefeldin A, then were stained with metal-conjugated mAbs and subjected to CyTOF analysis. CXCR6 mAb and CD107a mAb were added during the stimulation. Shown are viSNE plots of CyTOF of effector CD4 (A) and CXCR6+ effector CD4 (B) of one of 3 biological samples with the same pattern. The color indicates expression level of labeled marker.
Recently, we identified that chemokine receptor CXCR6 was preferentially expressed on pathogenic CD4 cells in EAE model (16), which i) secreted inflammatory cytokine IFNγ and GM-CSF, ii) contained cytotoxic granules, and iii) proliferated rapidly. In the current study, we further revealed that CXCR6+CD4 cells in EAE mice were heterogeneous, divided into two distinct subpopulations. One was CCR6 positive, enriched for cytotoxic signatures (Perforin, CD107a and GzmB) and conventional Th17 molecules (IL-17, IL-23R and RORγt); while the other was CCR6 negative, enriched solely for cytokine IFNγ and GM-CSF. Of note, although CCR6 has been established as a chemokine receptor of conventional Th17 cells, its role in EAE is still controversial since EAE disease varies from total absence (21, 22) to be delayed but more severe or totally normal (23, 24) in ccr6-deficient mice.
Regarding the EAE pathology, the current perception is that CD4 cells mainly interact with myeloid cells to indirectly induce the myelin sheath damage in EAE. It is unknown whether CD4 cells directly mediate the CNS damage and which molecule(s) mediate the process when that is the case. Although CD4 cell-derived GM-CSF is thought to be essential for EAE development, GM-CSF-deficient mice develop comparable EAE disease as wild type (wt) mice when regulatory T cells are depleted (25), suggesting that GM-CSF might function through peripheral priming step to mediate EAE. Indeed, the clinical trial of anti-GM-CSF antibody hasn’t been successful to date for MS. Thus, it is possible that EAE pathology is induced by GM-CSF-producing CD4 cells, rather than GM-CSF itself, and there might be other factors that are responsible for CD4 cell pathogenicity in EAE and human MS.
In this study, we discovered that CNS-infiltrated CD4 cells were highly cytotoxic by highly expressing various cytotoxic granzymes and cognate inhibitor Serpin(s). Granzyme(s) possesses broad pathological functions, such as triggering apoptosis of target cells (26), degrading the extracellular matrix (27), activating the myeloid cells (28, 29), and inducing self-inflicted cell death (30, 31). The finding that CNS-infiltrated CD4 cells highly expressed all of Gzm-A, -B, -K and endogenous inhibitor Serpin(s) strongly suggests that the pathogenic CD4 cells may use the Granzyme(s) to directly mediate CNS damage or to induce activation-induced cell death to maintain the homeostasis.
Overall, in the current study, we identified two distinct subpopulations in CXCR6+CD4 cells in EAE: CCR6+ cells (enriched for conventional Th17 signature with unexpected cytotoxic signature) and CCR6- cells (enriched for cytokine IFNγ and GM-CSF, an ex-Th17 signature). The discovery of these two subpopulations makes in vivo generated pathogenic CD4 cells amenable for direct investigation. Our study also highlights the existence of cytotoxic protease(s) of encephalitogenic CD4 cells, which might be the mediator to directly mediate the CNS damage in MS. The relationship between Th17 programming and acquisition of cytotoxicity is not understood; the co-existence of these two functionally distinct subpopulations is elusive and intriguing, which requires the further investigations.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI GEO, GSE192878 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192878.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital.
Both authors, LH and KY, designed the research, did the experiment, analyzed the data and wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Author LH is a cofounder and shareholder of Edelweiss Immune Inc. Both authors declare that the research was conducted in the absence of any other commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Eileen Remold-O’Donnell for providing the essential reagents and discussion, and XiaoDong Wang for assisting the CyTOF. This study is partially funded by the Anesthesia Research Distinguished Ignition Award (LH).
1. Compston A, Coles A. Multiple Sclerosis. Lancet (2008) 372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7
2. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 Rather Than Interleukin-12 is the Critical Cytokine for Autoimmune Inflammation of the Brain. Nature (2003) 421(6924):744–8. doi: 10.1038/nature01355
3. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
4. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do Not Contribute Vitally to Autoimmune Neuro-Inflammation in Mice. J Clin Invest (2009) 119(1):61–9. doi: 10.1172/JCI35997
5. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A Crucial Role for Interleukin (IL)-1 in the Induction of IL-17-Producing T Cells That Mediate Autoimmune Encephalomyelitis. J Exp Med (2006) 203(7):1685–91. doi: 10.1084/jem.20060285
6. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The Interleukin 23 Receptor Is Essential for the Terminal Differentiation of Interleukin 17-Producing Effector T Helper Cells. vivo Nat Immunol (2009) 10(3):314–24. doi: 10.1038/ni.1698
7. Ronchi F, Basso C, Preite S, Reboldi A, Baumjohann D, Perlini L, et al. Experimental Priming of Encephalitogenic Th1/Th17 Cells Requires Pertussis Toxin-Driven IL-1beta Production by Myeloid Cells. Nat Commun (2016) 7:11541. doi: 10.1038/ncomms11541
8. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat Drives Production of the Cytokine GM-CSF in Helper T Cells, Which Is Essential for the Effector Phase of Autoimmune Neuroinflammation. Nat Immunol (2011) 12(6):560–7. doi: 10.1038/ni.2027
9. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The Encephalitogenicity of T(H)17 Cells Is Dependent on IL-1- and IL-23-Induced Production of the Cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi: 10.1038/ni.2031
10. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte Macrophage Colony-Stimulating Factor: A New Putative Therapeutic Target in Multiple Sclerosis. J Exp Med (2001) 194(7):873–82. doi: 10.1084/jem.194.7.873
11. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of Pathogenic T(H)17 Cells in the Absence of TGF-Beta Signalling. Nature (2010) 467(7318):967–71. doi: 10.1038/nature09447
12. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate Mapping of IL-17-Producing T Cells in Inflammatory Responses. Nat Immunol (2011) 12(3):255–63. doi: 10.1038/ni.1993
13. Remold-O'Donnell E, Chin J, Alberts M. Sequence and Molecular Characterization of Human Monocyte/Neutrophil Elastase Inhibitor. Proc Natl Acad Sci USA (1992) 89(12):5635–9. doi: 10.1073/pnas.89.12.5635
14. Zhao P, Hou L, Farley K, Sundrud MS, Remold-O'Donnell E. SerpinB1 Regulates Homeostatic Expansion of IL-17+ Gammadelta and CD4+ Th17 Cells. J Leukoc Biol (2014) 95(3):521–30. doi: 10.1189/jlb.0613331
15. Hou L, Cooley J, Swanson R, Ong PC, Pike RN, Bogyo M, et al. The Protease Cathepsin L Regulates Th17 Cell Differentiation. J Autoimmun (2015) 65:56–63. doi: 10.1016/j.jaut.2015.08.006
16. Hou L, Rao DA, Yuki K, Cooley J, Henderson LA, Jonsson AH, et al. SerpinB1 Controls Encephalitogenic T Helper Cells in Neuroinflammation. Proc Natl Acad Sci U S A (2019) 116(41):20635–43. doi: 10.1073/pnas.1905762116
17. Kim JV, Jiang N, Tadokoro CE, Liu L, Ransohoff RM, Lafaille JJ, et al. Two-Photon Laser Scanning Microscopy Imaging of Intact Spinal Cord and Cerebral Cortex Reveals Requirement for CXCR6 and Neuroinflammation in Immune Cell Infiltration of Cortical Injury Sites. J Immunol Methods (2010) 352(1-2):89–100. doi: 10.1016/j.jim.2009.09.007
18. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and Functional Features of Human Th17 Cells. J Exp Med (2007) 204(8):1849–61. doi: 10.1084/jem.20070663
19. Raveney BJE, Sato W, Takewaki D, Zhang C, Kanazawa T, Lin Y, et al. Involvement of Cytotoxic Eomes-Expressing CD4(+) T Cells in Secondary Progressive Multiple Sclerosis. Proc Natl Acad Sci U S A (2021) 118(11):e2021818118. doi: 10.1073/pnas.2021818118
20. Kimball AK, Oko LM, Bullock BL, Nemenoff RA, van Dyk LF, Clambey ET. A Beginner's Guide to Analyzing and Visualizing Mass Cytometry Data. J Immunol (2018) 200(1):3–22. doi: 10.4049/jimmunol.1701494
21. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C Chemokine Receptor 6-Regulated Entry of TH-17 Cells Into the CNS Through the Choroid Plexus is Required for the Initiation of EAE. Nat Immunol (2009) 10(5):514–23. doi: 10.1038/ni.1716
22. Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, et al. Inhibition of CCR6 Function Reduces the Severity of Experimental Autoimmune Encephalomyelitis via Effects on the Priming Phase of the Immune Response. J Immunol (2009) 182(5):3121–30. doi: 10.4049/jimmunol.0713169
23. Villares R, Cadenas V, Lozano M, Almonacid L, Zaballos A, Martínez AC, et al. CCR6 Regulates EAE Pathogenesis by Controlling Regulatory CD4+ T-Cell Recruitment to Target Tissues. Eur J Immunol (2009) 39(6):1671–81. doi: 10.1002/eji.200839123
24. Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ. Mice Deficient for CCR6 Fail to Control Chronic Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2009) 213(1-2):91–9. doi: 10.1016/j.jneuroim.2009.05.011
25. Ghosh D, Curtis AD 2nd, Wilkinson DS, Mannie MD. Depletion of CD4+ CD25+ Regulatory T Cells Confers Susceptibility to Experimental Autoimmune Encephalomyelitis (EAE) in GM-CSF-Deficient Csf2-/- Mice. J Leukoc Biol (2016) 100(4):747–60. doi: 10.1189/jlb.3A0815-359R
26. Bird CH, Christensen ME, Mangan MS, Prakash MD, Sedelies KA, Smyth MJ, et al. The Granzyme B-Serpinb9 Axis Controls the Fate of Lymphocytes After Lysosomal Stress. Cell Death Differ (2014) 21(6):876–87. doi: 10.1038/cdd.2014.7
27. Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, et al. Extracellular Matrix Remodeling by Human Granzyme B via Cleavage of Vitronectin, Fibronectin, and Laminin. J Biol Chem (2005) 280(25):23549–58. doi: 10.1074/jbc.M412001200
28. Metkar SS, Menaa C, Pardo J, Wang B, Wallich R, Freudenberg M, et al. Human and Mouse Granzyme A Induce a Proinflammatory Cytokine Response. Immunity (2008) 29(5):720–33. doi: 10.1016/j.immuni.2008.08.014
29. Wensink AC, Hack CE, Bovenschen N. Granzymes Regulate Proinflammatory Cytokine Responses. J Immunol (2015) 194(2):491–7. doi: 10.4049/jimmunol.1401214
30. Phillips T, Opferman JT, Shah R, Liu N, Froelich CJ, Ashton-Rickardt PG. A Role for the Granzyme B Inhibitor Serine Protease Inhibitor 6 in CD8+ Memory Cell Homeostasis. J Immunol (2004) 173(6):3801–9. doi: 10.4049/jimmunol.173.6.3801
Keywords: CXCR6, CCR6, Th17, cytotoxicity, EAE, Multiple Sclerosis
Citation: Hou L and Yuki K (2022) CCR6 and CXCR6 Identify the Th17 Cells With Cytotoxicity in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 13:819224. doi: 10.3389/fimmu.2022.819224
Received: 21 November 2021; Accepted: 11 January 2022;
Published: 01 February 2022.
Edited by:
Luisa María Villar, Ramón y Cajal University Hospital, SpainReviewed by:
Luc Van Kaer, Vanderbilt University, United StatesCopyright © 2022 Hou and Yuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifei Hou, TGlmZWkuSG91QGNoaWxkcmVucy5oYXJ2YXJkLmVkdQ==; Koichi Yuki, S29pY2hpLll1a2lAY2hpbGRyZW5zLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.