 Yunlong Liu†
Yunlong Liu† Yan Sun†
Yan Sun† Juening KangZiqi HeQuan LiuJihua WuDerong LiXiang WangZhiwei TaoXiaofeng GuanWusheng SheHua XuYaoliang Deng*
Juening KangZiqi HeQuan LiuJihua WuDerong LiXiang WangZhiwei TaoXiaofeng GuanWusheng SheHua XuYaoliang Deng*- Department of Urology, First Affiliated Hospital of Guangxi Medical University, Nanning, China
Calcium oxalate nephrolithiasis is a common and highly recurrent disease in urology; however, its precise pathogenesis is still unknown. Recent research has shown that renal inflammatory injury as a result of the cell-crystal reaction plays a crucial role in the development of calcium oxalate kidney stones. An increasing amount of research have confirmed that inflammation mediated by the cell-crystal reaction can lead to inflammatory injury of renal cells, promote the intracellular expression of NADPH oxidase, induce extensive production of reactive oxygen species, activate NLRP3 inflammasome, discharge a great number of inflammatory factors, trigger inflammatory cascading reactions, promote the aggregation, nucleation and growth process of calcium salt crystals, and ultimately lead to the development of intrarenal crystals and even stones. The renal tubular epithelial cells (RTECs)-crystal reaction, macrophage-crystal reaction, calcifying nanoparticles, endoplasmic reticulum stress, autophagy activation, and other regulatory factors and mechanisms are involved in this process.
Introduction
One of the most frequently encountered diseases in urology is kidney stones, which are closely related to multiple factors such as environment, genetics, and metabolic abnormalities; however, its exact pathogenesis is still unclear (1). Over the past few years, the incidence of kidney stones has been increasing year by year, with the incidence rate reaching 8.8% in the United States and 3.5−7.4% in Asia (2, 3). In addition, the recurrence rate of kidney stones is as high as 50% within 5−10 years following drug therapy or surgical treatment, and the recurrence rate within 20 years is even higher at 75% (4). Because of the high incidence and recurrence rate, kidney stone disease does not only greatly affects human life and health, but also significantly increases the financial burden of the healthcare system (5). Therefore, exploring the exact pathogenesis of kidney stones and finding new therapeutic targets has important theoretical significance and application value in the treatment and prevention of stones.
It is known that kidney stone formation involves a series of complicated processes, including urine supersaturation, calcium salt crystal adhesion, accumulation, nucleation, growth, and retention in the kidney (6). Common crystals in the kidney mainly include oxalate (Ox), calcium oxalate (CaOx), calcium phosphate, and urate, among which CaOx crystals are the most common (7). The retention and subsequent growth of crystals within the kidney are inseparable from kidney stone formation. A large amount of research has shown that the cell-crystal-induced inflammatory response plays a crucial role in the formation of CaOx kidney stones (8–10). In this process, the massive generation of reactive oxygen species (ROS) and the activation of NLRP3 inflammasomes promote the maturation and discharge of inflammatory factors, leading to inflammation in the kidneys and renal tubular epithelial cells (RTECs) (11, 12). In this review, we will describe in detail the role of inflammation arising from ROS-induced NLRP3 inflammasome stimulation in the formation of CaOx kidney stones.
ROS-Induced Activation of NLRP3 in RTECs-Crystal Reaction
ROS refers to a series of active oxygen clusters and their metabolites produced by organisms in the process of aerobic metabolism, and includes superoxide anion (O2-), hydrogen peroxide (H2O2), hydroxyl radical (OH-), etc., which are mainly produced on the electron respiration chain of mitochondria under the catalysis of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (13, 14). Due to its high biological activity, ROS have a crucial role in the pathophysiological process of many diseases (15–17). Many studies have shown that the origination of CaOx kidney stones is intricately associated with the excessive production of ROS mediated by oxidative stress in RTECs (18–20). Clinical studies have confirmed that the levels of malondialdehyde (MDA), thiobarbituric acid reactive substances (TBARS), α-glutathione-s-transferase (α-GST), β-galactosidase (GAL), and N-acetyl-β-D-glucosaminidase (NAG) in the urine of CaOx nephrolithiasis patients are significantly higher than those of healthy patients, suggesting that the development of renal calculi is related to RTEC injury induced by ROS (20). In our previous study, we found that when taurine, N-acetylcysteine (NAC), catalase, or superoxide dismutase (SOD) were added to the rat CaOx kidney stone model, the activity of NADPH oxidase and the production of ROS and LDH decreased, the renal tissue and cell damage and inflammatory reaction triggered by Ox and/or CaOx crystals were alleviated, the deposition of crystal in the kidney was significantly decreased, and the formation rate of CaOx kidney stones was reduced (21). In further experimental studies, we found that oxalic acid or CaOx crystals could prompt oxidative stress in RTECs. The production of a large number of ROS initiates the NF-κB signal pathway, promotes the secretion of a series of cytokines, and induces inflammation, resulting in RTEC degeneration, injury, exfoliation, and basement membrane exposure, which creates conditions favorable for the conservation of crystals in the lumen of renal tubules, and is ultimately conducive to the development of CaOx kidney stones (22, 23).
Inflammasome is a multiprotein complex that is triggered and activated after cells recognize dangerous-associated molecular patterns (DAMPs) for instance, monosodium urate and silica, and pathogen-associated molecular patterns (PAMPs) such as viruses and bacteria (24). The main members of the inflammasome family are NOD-like receptors (NLRs), AIM2-like receptors, and pyrin inflammasome. Of these, NLRP3 inflammasome is the most studied multiprotein complex, and mainly consists of NLRP3, caspase-1, and apoptosis-associated spot-like protein (ASC) (25, 26). After endogenous or exogenous stimulation, activated NLRP3 can recruit ASC protein and activate caspase-1, subsequently induce the maturation and release of inflammatory factors such as interleukin (IL)-1β and IL-18, and participate in a variety of inflammatory responses in the body, which has become a hot topic in the study of inflammatory mechanisms (27). Previous research has indicated that CaOx crystals can directly stimulate the secretion of IL-1β by renal dendritic cells via the NLRP3/ASC/caspase-1 axis, in addition to damaging RTECs to release ATP, indirectly activating the NLRP3 inflammasome, and then stimulating the secretion of IL-1β by dendritic cells, resulting in renal inflammatory response and inflammatory injury of RTECs (28). Studies have also shown that after knockout of NLRP3, the rate of renal stone formation in mice, who were fed a high oxalic acid diet, as well as caspase-1 expression and IL-1β secretion in RTECs all decreased significantly (8, 10, 29). Furthermore, the induction of NLRP3 inflammasome can participate in the formation of renal stones by changing the adhesion of cells to crystals. Khan and other researchers have found that ROS can up-regulate the expression of hyaluronic acid (HA), osteopontin (OPN), and CD44 via the p38MAPK pathway, and change the adhesion of RTECs to CaOx crystals as well as stimulate the formation of CaOx stones (30). Qi et al. (31) found that the induction of the NLRP3 inflammasome plays a connective role in the process of ROS changing the cell adhesion to crystals and promoting stone formation through the p38MAPK signaling pathway, and related proteins such as phosphorylated p38 and c-Jun play an important regulatory role. Research by Joshi et al. (10) has indicated that in NLRP3 gene-deficient mice, who received a high oxalate diet, the expression levels of OPN, HA, and CD44 on the surface of RTECs were significantly decreased, and the formation speed of CaOx stones was significantly lower compared to that of normal mice.
It has been established that ROS are a crucial component in the activation of NLRP3 inflammasomes (32, 33). The application of ROS inhibitors can impede the activation of NLRP3 inflammasomes, alleviate cell inflammatory damage, and delay progression of the disease (34, 35). In vivo as well as in vitro studies have shown that CaOx crystals are capable of inducing the generation of ROS in the kidney, and then activating NLRP3 inflammasome to cause inflammatory damage to RTECs and renal tissue, thus promoting the formation of CaOx stones (10). We found in our previous research that atorvastatin treatment downregulated ROS production, suppressed the induction of NLRP3 inflammasome pathways, decreased the release of IL-1β, IL-6, IL-18, and tumor necrosis factor-α (TNF-α), and ameliorated inflammatory injury and crystal deposition induced by CaOx crystals in kidney tissues of rats and HK-2 cells (36). Therefore, we believe that oxalic acid or CaOx crystals induce ROS production in RTECs during the formation of CaOx renal calculi and mediate the activation of NLRP3 inflammatory corpuscles. This process leads to inflammatory cell infiltration and RTEC degeneration and necrosis, and causes a renal inflammatory cascade effect, promotes the adhesion, accumulation, nucleation, and secondary growth of CaOx crystals, ultimately resulting in the formation of renal stones.
ROS-Induced Activation of NLRP3 in Macrophage-Crystal Reaction
In the cell-crystal inflammatory response, macrophages, as an important inflammatory cell with phagocytic function, participate in CaOx crystals formation in the kidney (37). The expression of macrophage-related genes in renal papillary calcified plaque tissues was found to be significantly higher in patients with kidney stones than in patients without (38). The results that have been reported in in vitro studies show that under the stimulation of CaOx crystals, kidney cells produce a variety of inflammatory factors, which induce monocytes or macrophages to migrate to the place where the stone crystals are deposited and transported through pinocytosis. At the same time, the crystals can promote the expression of NADPH oxidase in the cell to generate a large amount of ROS, induce the nuclear factor κB signal transduction pathway, and produce a great amount of inflammatory factors including TNF-α, IL -6, and IL-1β. This further causes inflammatory damage to kidney cells, leading to cell degeneration, necrosis, exposure of basement membrane, etc., which is conducive to the formation of renal calcification plaques (39, 40). In the hyperoxaluria rat model, hyperoxaluria can activate the NF-κB signal transduction pathway through ROS, induce the kidney to express a great amount of inflammatory factors including MCP-1, IL-6, and IL-1β. Following these processes, inflammatory cell infiltration (such as macrophages) and renal interstitial damage occur, creating conditions for CaOx crystal formation in the kidneys (41). This indicates that the ROS-mediated macrophage-crystal inflammatory response plays a key role in the formation and development of CaOx kidney stones.
In the resting state, the expression level of NLRP3 in dendritic cells, macrophages, and other innate immune cells is relatively low, but after endogenous and exogenous stimulation, activated NLRP3 can recruit the ASC protein, activate caspase-1, and then induce the maturation and release of inflammatory factors including IL-18 and IL-1β (27). It has been reported that macrophages can cause intracellular lysosomes to rupture after they internalize pathogens or crystals. After the lysosome ruptures, the protease (such as cathepsin B) in the lysosome is released into the cytoplasm (42), leading to the opening of effector cell membrane ion channels, causing changes in intracellular ion concentration, which promotes the activation of NLRP3 inflammasomes. Hydroxyapatite crystals can activate the NLRP3 receptor through ROS to stimulate human monocyte macrophages and rat macrophages to produce the inflammatory factor IL-1β (43). Macrophages activate the NADPH enzyme to produce ROS after engulfing CaOx crystals and accumulate in the kidneys, leading to aggravation of OS; after which ROS can activate inflammatory pathways such as NLRP3 and TLR4 (44). Blocking NLRP3 may protect macrophages from oxalate damage (45). Therefore, we speculate that after macrophages engulf CaOx crystals, up-regulation of ROS may mediate the activation of NLRP3 inflammasomes, induce the secretion of inflammatory factors, and cause aggravation of the inflammatory damage in kidney tissues and RTECs, thereby promoting the formation process of CaOx kidney stones.
High mobility group box 1 (HMGB1) is an important inflammatory mediator and inflammatory cytokine and is widely present in various cells. It can be actively or passively released by activated mononuclear macrophages and damaged or necrotic cells to trigger the biological effect of initiating, maintaining, and enhancing the inflammatory response (46). Studies have found that the application of anti-HMGB1 antibodies can significantly reduce the infiltration of renal tubular interstitial neutrophils and monocytes, alleviate the inflammatory damage of RTECs, and inhibit the release of inflammatory factors in kidney tissues such as MCP-1, TNF-α, and IL-6. By blocking the inflammatory cascade mediated by HMGB1, the renal function can be improved and renal ischemia-reperfusion injury reduced (47). Wang et al. (48) reported that in patients with calcium-containing kidney stones, an increased expression of HMGB1 and MCP-1 was found in their urine. The results of in vitro studies show that high calcium ions stimulate RTECs to activate the HMGB1-RAGE/TLR4-NF-κB signaling pathway and stimulate the secretion of inflammatory factors TNF-α, IL-1β, and IL-6 (49). Moreover, previous studies have also indicated that there is a close relationship between the secretion of HMGB1 and the activation of NLRP3 inflammasomes (50, 51). Lamkanfi et al. (46) found that lipopolysaccharide-induced secretion of HMGB1 by activated macrophages requires the participation of the NLRP3, ASC, and caspase-1 inflammatory complex.
In summary, we speculate that after macrophages engulf CaOx crystals, ROS generated by oxidative stress in the cells stimulate macrophages by activating NLRP3 inflammasomes, inducing increased secretion of HMGB1, and activating the NF-κB signal transduction pathways to stimulate macrophages to secrete inflammatory factors including TNF-α and IL-1β. On the other hand, HMGB1 itself can in turn activate the NLRP3 inflammasome, forming a positive feedback effect of inflammation, and thereby maintaining or even amplifying the inflammatory cascade, which causes severe inflammatory damage to the kidney. This promotes the adhesion, accumulation, nucleation, and subsequent growth of CaOx crystals, and ultimately leads to crystals and even stones being formed in the kidney.
ROS-Induced Activation of NLRP3 in Calcifying Nanoparticles
Randall plaque is a kind of calcified tissue located under the mucosa of the renal papilla (9). It originates from the basement membrane of the tubular epithelial cells in the slender part of the medullary loop, gradually extends to the interstitium of the renal medulla, and finally deposits in the renal papilla’s interstitial tissue (52). Matlaga et al. (53) used endoscopic techniques to observe renal papillary plaques in 23 patients who had idiopathic CaOx stones and found that there were Randall plaques in 24 kidneys (24/46) and 156 renal papillae (156/172) indicating that most patients with CaOx stones have Randall plaques in their kidneys. Near the tubule basement membrane, the main constituent of Randall’s spots is calcium phosphate. As it gradually migrates to the lumen of renal tubules, the components of the plaque shows an obvious migration and transformation phenomenon in the form of “calcium phosphate→calcium phosphate and CaOx→CaOx” (1). Scholars believe that the secretion of hydrogen in the distal renal tubules can acidify the urine, which causes the ion activity of calcium phosphate to decrease significantly, and promotes the dissolution of calcium phosphate in the tubules to release calcium and phosphorus ions (54). In the final urine at the end of the collecting tubule, calcium ions integrate with oxalate ions to form CaOx (55).
With the development of modern molecular biology and cell biology technology, Kumar et al. (56) discovered that there are many round or quasi-circular electronic high-density small bodies that are very similar to calcifying nanoparticles (CNPs) in the Randall plaque. Although it remains to be confirmed whether these high-density bodies are CNPs, some scholars have shown that the characteristics of the thin apatite shell on the surface of CNPs are highly consistent with the Randall patch structure at the initial stage (57). CNPs can be cultured in the tissues around Randall’s spots, stone specimens, and renal pelvic urine of patients with kidney stones (58). García et al. (59) found that the rat kidney stone model can be successfully constructed by intravenous injection or direct intra-renal injection of CNPs. Furthermore, Ciftcioglu et al. (60) verified the expression of CNPs in renal calcification plaques in human kidney tissue specimens through a variety of methods, and believed that CNPs are one of the main causes of renal calcification plaques. Therefore, we can infer that there is a close correlation between Randall’s spot and CNPs.
CNPs-mediated inflammation is closely related to kidney stone formation. Our previous research found that in the co-culture system of CNPs and RTECs, RTECs can be observed to adhere to and phagocytize CNPs, resulting in an increased NADPH oxidase activity, stimulating cells to produce a large amount of ROS, leading to RTEC inflammatory damage, which in turn can cause cell death in severe cases (61). Wu et al. (58) reported that CNPs can cause mitochondrial damage after entering the cell, and then generate ROS, which mediates the damage of RTECs and the formation of stones through the ROS-JNk signaling pathway. After Shiekh et al. intravenously injected CNPs into Wistar rats, calcium deposits were observed in the pathological sections of kidneys, while inflammatory cell infiltration and aggregation were seen in the renal medulla and cortex of rats (62). After the aggregated CNPs are swallowed by macrophages, they can induce mitochondrial damage and generate ROS, which leads to the activation of caspase-1 to mediate the secretion of IL-1β, resulting in an inflammatory response (63). As the effector protein of NLRP3 inflammasome, caspase-1 is responsible for cutting the inactive pro-inflammatory cytokine pro-IL-1β into mature IL-1β (64). We speculate that CNPs may activate NLRP3 inflammasomes through the production of ROS, thereby mediating the cell-crystal inflammatory response, leading to renal tissue inflammatory damage, inducing calcium phosphate-CaOx heterogeneous nucleation, and ultimately leading to the formation of intra-renal crystals.
ROS-Induced Activation of NLRP3 in Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is an important organelle widely present in mammalian cells. It is the main intracellular site for protein manufacture and processing, and the main calcium storehouse for maintaining calcium homeostasis (65). When the body is stimulated by glucose deficiency, oxidative stress, and Ca2+ metabolic disorders, the imbalance of ER protein dynamics leads to ER stress, and as a result, initiates the unfolded protein response (UPR), and participates in the pathophysiological process of many diseases (66). During observations under the electron microscope, our team accidentally discovered that a sequence of abnormal morphological changes in the ER, for instance, enlargement and malformation, appeared in the kidney cells of CaOx kidney stone rats (67). Further research has shown that ER stress is related to the development of CaOx kidney stones (67, 68), which is consistent with the results as reported by Yang (69) and other research teams. Much evidence shows that the regulation of CaOx kidney stone formation by ER stress is inextricably connected to the accumulation of intracellular ROS and the activation of NLRP3 inflammasome (10, 70).
ROS are viewed as a sign of oxidative stress. Studies have reported that ROS have twofold functions in signaling ER stress. During ER stress, NADPH oxidase (NOX), present in the ER, can stimulate the production of ROS, which in turn can regulate the UPR and reinstate ER homeostasis (71). Nevertheless, if the strong stimulus continues or is not removed timely, the ER pressure cannot be relieved, and ER oxidase 1 (ERO1) will partially trigger an elevation in ROS (72). Excessive ROS production in the ER leads to the deposition of calcium in mitochondria and increases the damage in mitochondria (73). We found in previous studies that SOD can alleviate the stress of ROS, and inhibiting SOD will increase the accumulation of ROS, which in turn aggravates ER stress and promotes the formation of kidney stones (74). It is currently known that the stimulation of ROS in the presence of oxidative and ER stress is crucial for the induction of NLRP3 inflammasomes in macrophages. Aside from NOX4 activating NF-κB via ROS, which mediates the activation of NLRP3, it can also activate mitogen-activated protein kinase (MAPK) to stimulate the release of pro-inflammatory factors (75). Furthermore, NOX2 can regulate the expression of dsRNA-activated protein kinase R (PKR) during ER stress (76). The autophosphorylation of PKR results in the de novo binding of NLRP3, caspase-1, and ASC, which increases the activation of inflammasomes. The lack of protein kinase receptors significantly suppresses the release of HMGB1, IL-1β, and IL-18 (50).
In addition, Ca2+ released by the ER may be a general stimulus that results in activation of the NLRP3 inflammasome (77). The ER is the primary organelle where Ca2+ is stored, and Ca2+ mobilization plays a key role in the activation of NLRP3 inflammasomes. Excessive Ca2+ release results in an overload of mitochondrial calcium and mitochondrial damage. The accumulation of mitochondrial ROS increases, which leads to additional activation of inflammasomes and IL-1β production. Blocking Ca2+ mobilization can suppress the generation and activation of the NLRP3 inflammasome complex (78, 79). The C/EPB homologous protein (CHOP), a transcription factor considered to be a regulator of ER Ca2+ release during ER stress, can regulate the release of ER Ca2+ through the inositol 1,4,5-trisphosphate receptor. Under ER stress, loss of CHOP leads to a weakened Ca2+ release from the ER, which reduces ROS and improves cell survival (80–82). Therefore, CHOP and ER stress are thought to be a potential mechanism for amplifying the activity of NLRP3 inflammasomes to increase the inflammatory response (83).
In summary, we believe that ER stress may be an upstream or intermediate mediation mechanism of the ROS-NLRP3 signaling pathway that induces CaOx nephrolithiasis. This mechanism is not directly impacted by the IRE1, PERK, and ATF6 pathways in the classic UPR pathway (84). However, it may directly impact the expression of the terminal signal in the UPR and play a role by generating ROS or mediating Ca2+ mobilization.
ROS-Induced Activation of NLRP3 in Autophagy
Autophagy is a very well conserved intracellular degradation pathway. In the process of starvation, hypoxia, or oxidative stress, cells use lysosomes to degrade damaged macromolecular proteins or organelles, which can maintain intracellular environmental homeostasis and adapt to microenvironmental changes (85, 86). Studies have shown that an intimate association exists between autophagy and inflammatory responses (87). Autophagy can mitigate the inflammatory response by clearing inflammatory protein aggregates and down-regulating the release of pro-inflammatory cytokines. Conversely, excessive activation of autophagy can stimulate the inflammasomes to release a great number of inflammatory factors and accelerate the progression in inflammatory response. It has been reported that in a renal ischemia-reperfusion injury rat model, autophagy activation can downregulate the expression of pro-inflammatory factors HMGB1, TNF-α, and IL-6, increase the release of anti-inflammatory factor IL-10, and reduce renal inflammatory injury (88). Kirkland et al. (89) revealed that excessive accumulation of intracellular ROS can directly cause a certain degree of inflammatory damage to cells. In addition, a high concentration of ROS can induce excessive activation of autophagy and directly cause cell death (90).
ROS and inflammatory responses can not only activate autophagy, but also play an important regulatory role in the formation of CaOx kidney stones. The results of our previous studies have indicated that a significantly higher level of renal autophagy can be observed in patients with CaOx kidney stones than those with normal kidneys. CaOx crystals can induce the production of ROS in tubular epithelial cells, which mediates autophagy overactivation, whereas suppression of autophagy can effectively ameliorate CaOx crystal-induced tubular epithelial cell damage and decrease renal injury and CaOx crystal deposition caused by ethylene glycol, thereby reducing the rate of kidney stone formation (90, 91). Duan et al. (92) also found that chloroquine, an autophagy inhibitor, may reduce oxidative stress damage, mitochondrial damage, and lower excretion of urinary oxalate and kidney crystal deposition in rats by suppressing the activation of the p38 signaling pathways and the expression of the renal oxalate transporter SLC26A6, ultimately suppressing the formation of CaOx crystals in rat kidneys. Sun et al. (93) performed in vitro and in vivo experiments and revealed that application of the antioxidant taurine can alleviate the oxidative stress injury of RTECs caused by CaOx crystals. The responsible mechanism is inhibition of the excessive activation of autophagy mediated by ROS through up-regulation of the Akt/mTOR signal transduction pathway by taurine. Therefore, ROS mediated autophagy also plays an important role in the formation of CaOx kidney stones.
Autophagy plays a bidirectional regulatory role in diseases associated with activation of the NLRP3 inflammasome. Saitoh (94) et al. reported for the first time in 2008 that autophagy can regulate inflammasome activation, and that LPS could induce activation of inflammasome in macrophages after knockout of the autophagy regulation gene Atg16L1, suggesting that inhibition of autophagy can stimulate the maturation and release of inflammatory factors IL-18 and IL-1β. Ko et al. (95) reported that applying the autophagy activator rapamycin to increase the level of autophagy can result in inhibition of NLRP3 inflammasome activation and its mediated inflammatory response by eliminating ROS in mitochondria. However, other studies have revealed that the excessive activation of ROS mediated autophagy can promote NLRP3 inflammasome activation and IL-1β production. Zhang (96) has shown that mechanical ventilation can mediate excessive activation of autophagy after stimulating the generation of mitochondrial ROS in pulmonary macrophages, and the inflammatory injury in the lung as a result of mechanical ventilation is caused by the activation of NLRP3 inflammasomes, which is mediated by autophagy signals and the secretion of pro-inflammatory cytokines such as IL-18 and IL-1β in pulmonary macrophages. A study by Qiu et al. (97) has indicated that arsenic trioxide (As2O3) can induce excessive autophagy in hepatocytes and stimulate the activation of NLRP3 inflammasome, while the antioxidant taurine can ameliorate the inflammatory response of hepatocytes induced by As2O3 by inhibiting the autophagy-inflammasome pathway. Moreover, inducing the NLRP3 inflammasome can also inhibit or promote autophagy. By activating inflammasomes, especially NLRP3 inflammasomes, the level of mitochondrial autophagy can be inhibited and the self-clearance of mitochondria affected, thus promoting the occurrence and development of diseases (98). Allaeys (99) et al. reported that sodium urate crystals can positively manage the formation of autophagosomes in cells by up-regulating NLRP3 inflammasome activation.
Therefore, we speculate that the excessive activation of autophagy mediated by mitochondria-derived ROS could trigger the NLRP3 inflammasome pathway, up-regulate the secretion of inflammatory factors IL-18 and IL-1β, which leads to inflammatory cell invasion and renal interstitial inflammatory injury in the course of CaOx kidney stone formation. The activation of NLRP3 inflammasome may further increase the level of autophagy, resulting in an inflammatory chain reaction in the kidney, and accelerating the formation process of nephrolithiasis.
Summary and Perspectives
In recent years, with the development of modern biology technology, the role of the cell-crystal inflammatory response theory in the formation of CaOx kidney stones has been increasingly valued. A growing body of literature suggests that the RTECs-crystal reaction, macrophage-crystal reaction, calcifying nanoparticles, ER stress, autophagy activation, and other regulatory factors can induce ROS production during the course of CaOx kidney stones formation. These processes can also mediate the activation of the NLRP3 inflammasome, promote the release of inflammatory factors such as IL-1β andIL-18, cause inflammatory cell infiltration and renal degeneration of tubular epithelial cells, necrosis, the kidney inflammatory cascade effect, hence stimulating the adhesion, aggregation, nucleation, and subsequent growth of CaOx crystals, and ultimately the formation of CaOx nephrolithiasis (Figure 1). In conclusion, effective intervention for ROS-induced activation of NLRP3 inflammasome may be a potential therapeutic target in the prevention of CaOx kidney stones formation and recurrence, which has important theoretical significance and practical value.
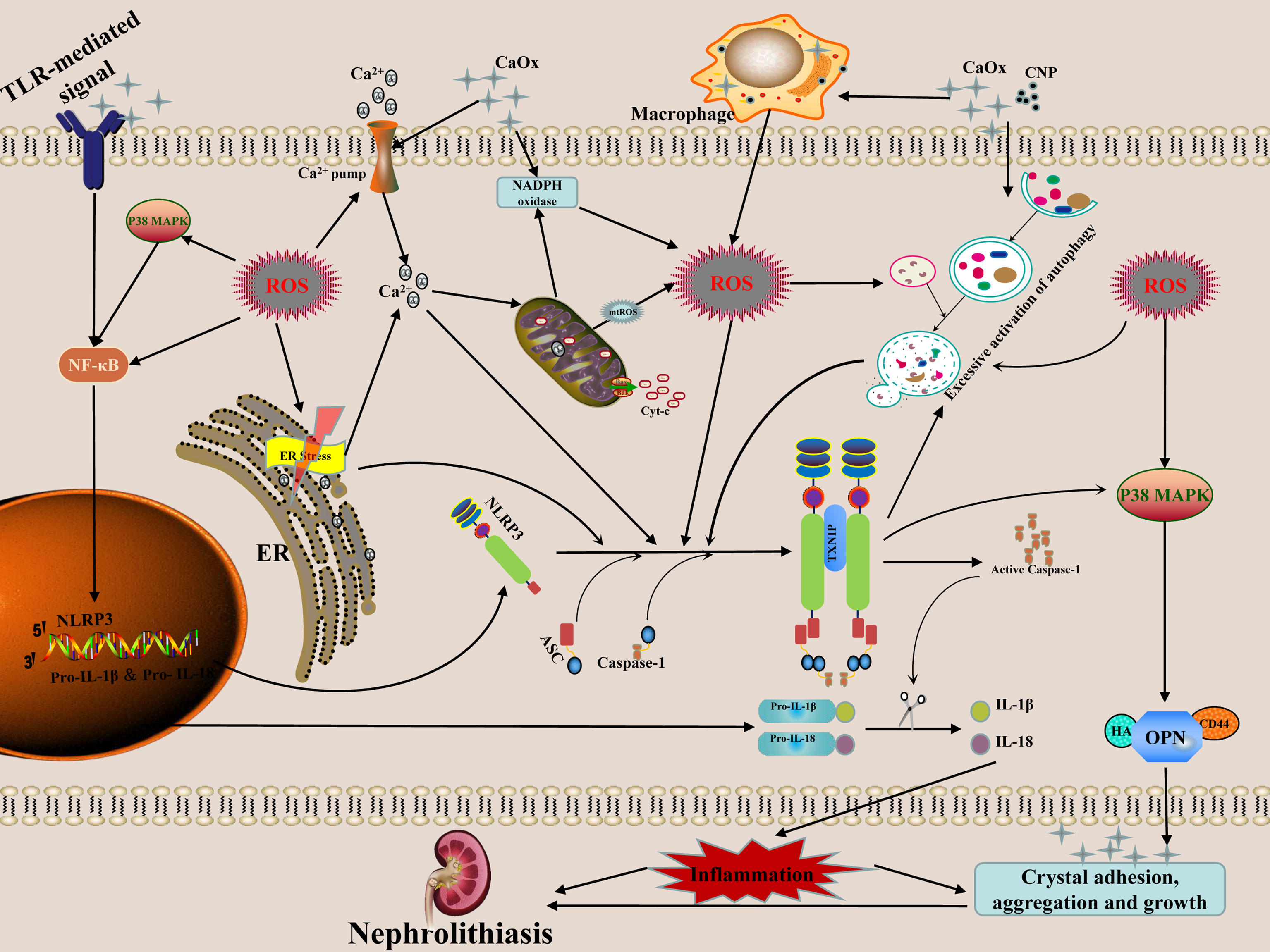
Figure 1 Possible mechanism and regulation of ROS-induced NLRP3 inflammasome activation in the formation of calcium oxalate nephrolithiasis. The secretion of mature forms of IL-1β and IL-18 is the result of NLRP3 inflammasome activation. These mediators have the properties of pro-inflammatory activation, which in turn promote the adhesion, aggregation and growth of crystals. The interaction of crystals or nanoparticles with cells can cause mitochondrial damage and increased NADPH oxidase activity, and then generate ROS, which mediates NLRP3 inflammasome transcription and activation through the ROS-dependent of NF-κB and autophagy signaling pathway. ER induces ROS production via NOX4 and ERO1 during stress. The release of Ca2+in the ER causes mitochondrial damage which further aggravates the release of ROS. High concentration of ROS can induce excessive activation of autophagy, thereby stimulate the inflammasomes to release a great number of inflammatory factors. The activation of NLRP3 inflammasome may further increase the level of autophagy, resulting in an inflammatory chain reaction.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
HX, YD, and JK contributed to conception and design of the study. YL wrote the first draft of the manuscript. YS, ZH, QL, JW, DL, XW, ZT, XG, and WS wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by National Natural Science Foundations of China (81960138, 81760127, 81360113, 30960455 and 30860280) and the Scientific Research and Technology Development Program of Guangxi (AB16380225).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RTECs, The renal tubular epithelial cells; Ox, oxalate; CaOx, calcium oxalate; ROS, reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; MDA, malondialdehyde; TBARS, thiobarbituric acid reactive substances; α-GST, α-glutathione-s-transferase; GAL, β-galactosidase; NAG, N-acetyl-β-D-glucosaminidase; NAC, N-acetylcysteine; SOD, superoxide dismutase; DAMPs, dangerous-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; NLRs, NOD-like receptors;ASC, apoptosis-associated spot-like protein; HA, hyaluronic acid; OPN, osteopontin; TNF-α, tumor necrosis factor-α; CNPs, nanoparticles; HMGB1, High mobility group box-1; ER, endoplasmic reticulum; UPR, unfolded protein response; NOX, NADPH oxidase; ERO1, ER oxidase 1; MAPK, mitogen-activated protein kinase; PKR, protein kinase R; CHOP, C/EPB homologous protein; As2O3, arsenic trioxide.
References
1. Deng YL, Liu YL, Tao ZW, Wang X. The Role of Cell-Crystal Reaction Mediated Inflammation in the Formation of Intrarenal Calcium Oxalate Crystals. Zhonghua Wai Ke Za Zhi (2018) 56:733–6. doi: 10.3760/cma.j.issn.0529-5815.2018.10.004
2. Shoag J, Tasian GE, Goldfarb DS, Eisner BH. The New Epidemiology of Nephrolithiasis. Adv Chronic Kidney Dis (2015) 22:273–8. doi: 10.1053/j.ackd.2015.04.004
3. Ziemba JB, Matlaga BR. Epidemiology and Economics of Nephrolithiasis. Investig Clin Urol (2017) 58:299. doi: 10.4111/icu.2017.58.5.299
5. Antonelli JA, Maalouf NM, Pearle MS, Lotan Y. Use of the National Health and Nutrition Examination Survey to Calculate the Impact of Obesity and Diabetes on Cost and Prevalence of Urolithiasis in 2030. Eur Urol (2014) 66:724–9. doi: 10.1016/j.eururo.2014.06.036
6. Tsujihata M. Mechanism of Calcium Oxalate Renal Stone Formation and Renal Tubular Cell Injury. Int J Urol (2008) 15:115–20. doi: 10.1111/j.1442-2042.2007.01953.x
7. Chung MJ. Urolithiasis and Nephrolithiasis. JAAPA (2017) 30:49–50. doi: 10.1097/01.JAA.0000522145.52305.aa
8. Anders H-J, Suarez-Alvarez B, Grigorescu M, Foresto-Neto O, Steiger S, Desai J, et al. The Macrophage Phenotype and Inflammasome Component NLRP3 Contributes to Nephrocalcinosis-Related Chronic Kidney Disease Independent From IL-1-Mediated Tissue Injury. Kidney Int (2018) 93:656–69. doi: 10.1016/j.kint.2017.09.022
9. Khan SR, Canales BK, Dominguez-Gutierrez PR. Randall’s Plaque and Calcium Oxalate Stone Formation: Role for Immunity and Inflammation. Nat Rev Nephrol (2021) 17:417–33. doi: 10.1038/s41581-020-00392-1
10. Joshi S, Wang W, Peck AB, Khan SR. Activation of the NLRP3 Inflammasome in Association With Calcium Oxalate Crystal Induced Reactive Oxygen Species in Kidneys. J Urol (2015) 193:1684–91. doi: 10.1016/j.juro.2014.11.093
11. Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-Parkin Pathway of Mitophagy Protects Against Contrast-Induced Acute Kidney Injury via Decreasing Mitochondrial ROS and NLRP3 Inflammasome Activation. Redox Biol (2019) 26:101254. doi: 10.1016/j.redox.2019.101254
12. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive Oxygen Species Promote Tubular Injury in Diabetic Nephropathy: The Role of the Mitochondrial Ros-Txnip-Nlrp3 Biological Axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
13. Lu M, Gong X. Upstream Reactive Oxidative Species (ROS) Signals in Exogenous Oxidative Stress-Induced Mitochondrial Dysfunction. Cell Biol Int (2009) 33:658–64. doi: 10.1016/j.cellbi.2009.03.009
14. Joshi S, Peck AB, Khan SR. NADPH Oxidase as a Therapeutic Target for Oxalate Induced Injury in Kidneys. Oxid Med Cell Longev (2013) 2013:462361. doi: 10.1155/2013/462361
15. Sies H, Jones DP. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat Rev Mol Cell Biol (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
16. Harris IS, DeNicola GM. The Complex Interplay Between Antioxidants and ROS in Cancer. Trends Cell Biol (2020) 30:440–51. doi: 10.1016/j.tcb.2020.03.002
17. Farmer EE, Mueller MJ. ROS-Mediated Lipid Peroxidation and RES-Activated Signaling. Annu Rev Plant Biol (2013) 64:429–50. doi: 10.1146/annurev-arplant-050312-120132
18. Khan SR. Reactive Oxygen Species, Inflammation and Calcium Oxalate Nephrolithiasis. Transl Androl Urol (2014) 3:256–76. doi: 10.3978/j.issn.2223-4683.2014.06.04
19. Khan SR. Crystal-Induced Inflammation of the Kidneys: Results From Human Studies, Animal Models, and Tissue-Culture Studies. Clin Exp Nephrol (2004) 8:75–88. doi: 10.1007/s10157-004-0292-0
20. Khan SR. Reactive Oxygen Species as the Molecular Modulators of Calcium Oxalate Kidney Stone Formation: Evidence From Clinical and Experimental Investigations. J Urol (2013) 189:803–11. doi: 10.1016/j.juro.2012.05.078
21. Li CY, Deng YL, Sun BH. Taurine Protected Kidney From Oxidative Injury Through Mitochondrial-Linked Pathway in a Rat Model of Nephrolithiasis. Urol Res (2009) 37:211–20. doi: 10.1007/s00240-009-0197-1
22. Huang H-S, Ma M-C, Chen J. Chronic L-Arginine Administration Increases Oxidative and Nitrosative Stress in Rat Hyperoxaluric Kidneys and Excessive Crystal Deposition. Am J Physiol Renal Physiol (2008) 295:F388–396. doi: 10.1152/ajprenal.00405.2007
23. Khan SR, Thamilselvan S. Nephrolithiasis: A Consequence of Renal Epithelial Cell Exposure to Oxalate and Calcium Oxalate Crystals. Mol Urol (2000) 4:305–12.
24. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
25. Swanson KV, Deng M, Ting JP-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
26. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat Rev Drug Discovery (2018) 17:688. doi: 10.1038/nrd.2018.149
27. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 Ubiquitin Ligase TRIM31 Attenuates NLRP3 Inflammasome Activation by Promoting Proteasomal Degradation of NLRP3. Nat Commun (2016) 7:13727. doi: 10.1038/ncomms13727
28. Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium Oxalate Crystals Induce Renal Inflammation by NLRP3-Mediated IL-1β Secretion. J Clin Invest (2013) 123:236–46. doi: 10.1172/JCI63679
29. Knauf F, Asplin JR, Granja I, Schmidt IM, Moeckel GW, David RJ, et al. NALP3-Mediated Inflammation Is a Principal Cause of Progressive Renal Failure in Oxalate Nephropathy. Kidney Int (2013) 84:895–901. doi: 10.1038/ki.2013.207
30. Khan SR. Role of Renal Epithelial Cells in the Initiation of Calcium Oxalate Stones. Nephron Exp Nephrol (2004) 98:e55–60. doi: 10.1159/000080257
31. Qi S, Wang Q, Xie B, Chen Y, Zhang Z, Xu Y. P38 MAPK Signaling Pathway Mediates COM Crystal-Induced Crystal Adhesion Change in Rat Renal Tubular Epithelial Cells. Urolithiasis (2020) 48:9–18. doi: 10.1007/s00240-019-01143-z
32. Tschopp J, Schroder K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat Rev Immunol (2010) 10:210–5. doi: 10.1038/nri2725
33. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1β Production via Suppressing the NF-κb Pathway and ROS Production. J Hepatol (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
34. Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting Edge: Reactive Oxygen Species Inhibitors Block Priming, But Not Activation, of the NLRP3 Inflammasome. J Immunol (2011) 187:613–7. doi: 10.4049/jimmunol.1100613
35. Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li P-L. Endothelial NLRP3 Inflammasome Activation and Enhanced Neointima Formation in Mice by Adipokine Visfatin. Am J Pathol (2014) 184:1617–28. doi: 10.1016/j.ajpath.2014.01.032
36. Sun Y, Liu Y, Guan X, Kang J, Wang X, Liu Q, et al. Atorvastatin Inhibits Renal Inflammatory Response Induced by Calcium Oxalate Crystals via Inhibiting the Activation of TLR4/NF-κb and NLRP3 Inflammasome. IUBMB Life (2020) 72:1065–74. doi: 10.1002/iub.2250
37. de Water R, Leenen PJ, Noordermeer C, Nigg AL, Houtsmuller AB, Kok DJ, et al. Cytokine Production Induced by Binding and Processing of Calcium Oxalate Crystals in Cultured Macrophages. Am J Kidney Dis (2001) 38:331–8. doi: 10.1053/ajkd.2001.26098
38. Taguchi K, Hamamoto S, Okada A, Unno R, Kamisawa H, Naiki T, et al. Genome-Wide Gene Expression Profiling of Randall’s Plaques in Calcium Oxalate Stone Formers. J Am Soc Nephrol (2017) 28:333–47. doi: 10.1681/ASN.2015111271
39. Umekawa T, Chegini N, Khan SR. Increased Expression of Monocyte Chemoattractant Protein-1 (MCP-1) by Renal Epithelial Cells in Culture on Exposure to Calcium Oxalate, Phosphate and Uric Acid Crystals. Nephrol Dial Transplant (2003) 18:664–9. doi: 10.1093/ndt/gfg140
40. Deng Y, Sun B, Li C. UP-3.135: COM Crystals Stimulate the Expression and Activity of NADPH Oxidase in Macrophage. Urology (2009) 74:S337–7. doi: 10.1016/j.urology.2009.07.140
41. Toblli JE, Cao G, Casas G, Stella I, Inserra F, Angerosa M. NF-kappaB and Chemokine-Cytokine Expression in Renal Tubulointerstitium in Experimental Hyperoxaluria. Role of the Renin-Angiotensin System. Urol Res (2005) 33:358–67. doi: 10.1007/s00240-005-0484-4
42. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome Through Phagosomal Destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
43. Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, et al. NLRP3 Inflammasome Plays a Critical Role in the Pathogenesis of Hydroxyapatite-Associated Arthropathy. Proc Natl Acad Sci USA (2011) 108:14867–72. doi: 10.1073/pnas.1111101108
44. Li Y, Yan G, Zhang J, Chen W, Ding T, Yin Y, et al. LncRNA HOXA11-AS Regulates Calcium Oxalate Crystal-Induced Renal Inflammation via miR-124-3p/MCP-1. J Cell Mol Med (2020) 24:238–49. doi: 10.1111/jcmm.14706
45. Dominguez-Gutierrez PR, Kwenda EP, Khan SR, Canales BK. Immunotherapy for Stone Disease. Curr Opin Urol (2020) 30:183–9. doi: 10.1097/MOU.0000000000000729
46. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia. J Immunol (2010) 185:4385–92. doi: 10.4049/jimmunol.1000803
47. Li J, Gong Q, Zhong S, Wang L, Guo H, Xiang Y, et al. Neutralization of the Extracellular HMGB1 Released by Ischaemic Damaged Renal Cells Protects Against Renal Ischaemia-Reperfusion Injury. Nephrol Dial Transplant (2011) 26:469–78. doi: 10.1093/ndt/gfq466
48. Wang Y, Sun C, Li C, Deng Y, Zeng G, Tao Z, et al. Urinary MCP-1、HMGB1 Increased in Calcium Nephrolithiasis Patients and the Influence of Hypercalciuria on the Production of the Two Cytokines. Urolithiasis (2017) 45:159–75. doi: 10.1007/s00240-016-0902-9
49. Zhao J, Cheng J, Li C, Xu M, Ma C, Qin L, et al. Ethyl Pyruvate Attenuates CaCl2-Induced Tubular Epithelial Cell Injury by Inhibiting Autophagy and Inflammatory Responses. Kidney Blood Press Res (2018) 43:1585–95. doi: 10.1159/000494445
50. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature (2012) 488:670–4. doi: 10.1038/nature11290
51. Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 Release by Inflammasomes. Protein Cell (2013) 4:163–7. doi: 10.1007/s13238-012-2118-2
52. Chung H-J. The Role of Randall Plaques on Kidney Stone Formation. Transl Androl Urol (2014) 3:251–4. doi: 10.3978/j.issn.2223-4683.2014.07.03
53. Matlaga BR, Williams JC, Kim SC, Kuo RL, Evan AP, Bledsoe SB, et al. Endoscopic Evidence of Calculus Attachment to Randall’s Plaque. J Urol (2006) 175:1720–1724; discussion 1724. doi: 10.1016/S0022-5347(05)01017-7
54. Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, et al. The B1-Subunit of the H(+) ATPase Is Required for Maximal Urinary Acidification. Proc Natl Acad Sci USA (2005) 102:13616–21. doi: 10.1073/pnas.0506769102
55. Tiselius H-G, Lindbäck B, Fornander A-M, Nilsson M-A. Studies on the Role of Calcium Phosphate in the Process of Calcium Oxalate Crystal Formation. Urol Res (2009) 37:181–92. doi: 10.1007/s00240-009-0191-7
56. Kumar V, Farell G, Yu S, Harrington S, Fitzpatrick L, Rzewuska E, et al. Cell Biology of Pathologic Renal Calcification: Contribution of Crystal Transcytosis, Cell-Mediated Calcification, and Nanoparticles. J Investig Med (2006) 54:412–24. doi: 10.2310/6650.2006.06021
57. Kajander EO, Ciftcioglu N, Aho K, Garcia-Cuerpo E. Characteristics of Nanobacteria and Their Possible Role in Stone Formation. Urol Res (2003) 31:47–54. doi: 10.1007/s00240-003-0304-7
58. Wu J, Tao Z, Deng Y, Liu Q, Liu Y, Guan X, et al. Calcifying Nanoparticles Induce Cytotoxicity Mediated by ROS-JNK Signaling Pathways. Urolithiasis (2019) 47:125–35. doi: 10.1007/s00240-018-1048-8
59. García Cuerpo E, Olavi Kajander E, Ciftçioglu N, Lovaco Castellano F, Correa C, González J, et al. Nanobacteria. An Experimental Neo-Lithogenesis Model. Arch Esp Urol (2000) 53:291–303.
60. Ciftçioğlu N, Vejdani K, Lee O, Mathew G, Aho KM, Kajander EO, et al. Association Between Randall’s Plaque and Calcifying Nanoparticles. Int J Nanomed (2008) 3:105–15. doi: 10.2147/ijn.s2553
61. Deng YL, Meng DD, Li CY. E18 The Interaction of the Calcifying Nanoparticles (CNP) With Human Renal Tubular Epithelial Cells (HK-2) In Vitro. Eur Urol Suppl (2011) 10(7):468. doi: 10.1016/S1569-9056(11)61167-6
62. Shiekh FA, Khullar M, Singh SK. Lithogenesis: Induction of Renal Calcifications by Nanobacteria. Urol Res (2006) 34:53–7. doi: 10.1007/s00240-005-0034-0
63. Peng H-H, Wu C-Y, Young D, Martel J, Young A, Ojcius DM, et al. Physicochemical and Biological Properties of Biomimetic Mineralo-Protein Nanoparticles Formed Spontaneously in Biological Fluids. Small (2013) 9:2297–307. doi: 10.1002/smll.201202270
64. Haneklaus M, O’Neill LAJ. NLRP3 at the Interface of Metabolism and Inflammation. Immunol Rev (2015) 265:53–62. doi: 10.1111/imr.12285
65. Anelli T, Sitia R. Protein Quality Control in the Early Secretory Pathway. EMBO J (2008) 27:315–27. doi: 10.1038/sj.emboj.7601974
66. Oakes SA, Papa FR. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu Rev Pathol (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
67. Sun Y, Kang J, Guan X, Xu H, Wang X, Deng Y. Regulation of Endoplasmic Reticulum Stress on the Damage and Apoptosis of Renal Tubular Epithelial Cells Induced by Calcium Oxalate Crystals. Urolithiasis (2021) 49:291–9. doi: 10.1007/s00240-021-01261-7
68. Sun Y, Kang J, Tao Z, Wang X, Liu Q, Li D, et al. Effect of Endoplasmic Reticulum Stress-Mediated Excessive Autophagy on Apoptosis and Formation of Kidney Stones. Life Sci (2020) 244:117232. doi: 10.1016/j.lfs.2019.117232
69. Yang B, Lu X, Li Y, Li Y, Yu D, Zhang W, et al. A Proteomic Network Approach Across the Kidney Stone Disease Reveals Endoplasmic Reticulum Stress and Crystal-Cell Interaction in the Kidney. Oxid Med Cell Longev (2019) 2019:9307256. doi: 10.1155/2019/9307256
70. Lv P, Liu H, Ye T, Yang X, Duan C, Yao X, et al. XIST Inhibition Attenuates Calcium Oxalate Nephrocalcinosis-Induced Renal Inflammation and Oxidative Injury via the miR-223/NLRP3 Pathway. Oxid Med Cell Longev (2021) 2021:1676152. doi: 10.1155/2021/1676152
71. Ochoa CD, Wu RF, Terada LS. ROS Signaling and ER Stress in Cardiovascular Disease. Mol Aspects Med (2018) 63:18–29. doi: 10.1016/j.mam.2018.03.002
72. Görlach A, Klappa P, Kietzmann T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxid Redox Signal (2006) 8:1391–418. doi: 10.1089/ars.2006.8.1391
73. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER Tubules Mark Sites of Mitochondrial Division. Science (2011) 334:358–62. doi: 10.1126/science.1207385
74. Kang J, Sun Y, Deng Y, Liu Q, Li D, Liu Y, et al. Autophagy-Endoplasmic Reticulum Stress Inhibition Mechanism of Superoxide Dismutase in the Formation of Calcium Oxalate Kidney Stones. Biomed Pharmacother (2020) 121:109649. doi: 10.1016/j.biopha.2019.109649
75. Feng C, Zhang Y, Yang M, Lan M, Liu H, Huang B, et al. Oxygen-Sensing Nox4 Generates Genotoxic ROS to Induce Premature Senescence of Nucleus Pulposus Cells Through MAPK and NF-κb Pathways. Oxid Med Cell Longev (2017) 2017:7426458. doi: 10.1155/2017/7426458
76. Li G, Scull C, Ozcan L, Tabas I. NADPH Oxidase Links Endoplasmic Reticulum Stress, Oxidative Stress, and PKR Activation to Induce Apoptosis. J Cell Biol (2010) 191:1113–25. doi: 10.1083/jcb.201006121
77. Krebs J, Agellon LB, Michalak M. Ca(2+) Homeostasis and Endoplasmic Reticulum (ER) Stress: An Integrated View of Calcium Signaling. Biochem Biophys Res Commun (2015) 460:114–21. doi: 10.1016/j.bbrc.2015.02.004
78. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
79. Csordás G, Hajnóczky G. SR/ER-Mitochondrial Local Communication: Calcium and ROS. Biochim Biophys Acta (2009) 1787:1352–62. doi: 10.1016/j.bbabio.2009.06.004
80. Li G, Mongillo M, Chin K-T, Harding H, Ron D, Marks AR, et al. Role of ERO1-Alpha-Mediated Stimulation of Inositol 1,4,5-Triphosphate Receptor Activity in Endoplasmic Reticulum Stress-Induced Apoptosis. J Cell Biol (2009) 186:783–92. doi: 10.1083/jcb.200904060
81. Oyadomari S, Mori M. Roles of CHOP/GADD153 in Endoplasmic Reticulum Stress. Cell Death Differ (2004) 11:381–9. doi: 10.1038/sj.cdd.4401373
82. Tabas I, Ron D. Integrating the Mechanisms of Apoptosis Induced by Endoplasmic Reticulum Stress. Nat Cell Biol (2011) 13:184–90. doi: 10.1038/ncb0311-184
83. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc Natl Acad Sci USA (2012) 109:11282–7. doi: 10.1073/pnas.1117765109
84. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER Stress Activates the NLRP3 Inflammasome via an UPR-Independent Pathway. Cell Death Dis (2012) 3:e261. doi: 10.1038/cddis.2011.132
85. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy (2021) 17:1–382. doi: 10.1080/15548627.2020.1797280
86. Awan MUF, Deng Y. Role of Autophagy and its Significance in Cellular Homeostasis. Appl Microbiol Biotechnol (2014) 98:5319–28. doi: 10.1007/s00253-014-5721-8
87. Jones SA, Mills KHG, Harris J. Autophagy and Inflammatory Diseases. Immunol Cell Biol (2013) 91:250–8. doi: 10.1038/icb.2012.82
88. Ling H, Chen H, Wei M, Meng X, Yu Y, Xie K. The Effect of Autophagy on Inflammation Cytokines in Renal Ischemia/Reperfusion Injury. Inflammation (2016) 39:347–56. doi: 10.1007/s10753-015-0255-5
89. Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of Cardiolipin and Mitochondria During Programmed Neuronal Death: Evidence of a Role for Lipid Peroxidation and Autophagy. Neuroscience (2002) 115:587–602. doi: 10.1016/s0306-4522(02)00512-2
90. Liu Y, Liu Q, Wang X, He Z, Li D, Guan X, et al. Inhibition of Autophagy Attenuated Ethylene Glycol Induced Crystals Deposition and Renal Injury in a Rat Model of Nephrolithiasis. Kidney Blood Pressure Res (2018) 43:246–55. doi: 10.1159/000487678
91. Liu Y, Li D, He Z, Liu Q, Wu J, Guan X, et al. Inhibition of Autophagy-Attenuated Calcium Oxalate Crystal-Induced Renal Tubular Epithelial Cell Injury In Vivo and In Vitro. Oncotarget (2017) 9(4):4571–82. doi: 10.18632/oncotarget.23383
92. Duan X, Kong Z, Mai X, Lan Y, Liu Y, Yang Z, et al. Autophagy Inhibition Attenuates Hyperoxaluria-Induced Renal Tubular Oxidative Injury and Calcium Oxalate Crystal Depositions in the Rat Kidney. Redox Biol (2018) 16:414–25. doi: 10.1016/j.redox.2018.03.019
93. Sun Y, Dai S, Tao J, Li Y, He Z, Liu Q, et al. Taurine Suppresses ROS-Dependent Autophagy via Activating Akt/mTOR Signaling Pathway in Calcium Oxalate Crystals-Induced Renal Tubular Epithelial Cell Injury. Aging (Albany NY) (2020) 12:17353–66. doi: 10.18632/aging.103730
94. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature (2008) 456:264–8. doi: 10.1038/nature07383
95. Ko JH, Yoon S-O, Lee HJ, Oh JY. Rapamycin Regulates Macrophage Activation by Inhibiting NLRP3 Inflammasome-P38 MAPK-Nfκb Pathways in Autophagy- and P62-Dependent Manners. Oncotarget (2017) 8:40817–31. doi: 10.18632/oncotarget.17256
96. Zhang Y, Liu G, Dull RO, Schwartz DE, Hu G. Autophagy in Pulmonary Macrophages Mediates Lung Inflammatory Injury via NLRP3 Inflammasome Activation During Mechanical Ventilation. Am J Physiol Lung Cell Mol Physiol (2014) 307:L173–185. doi: 10.1152/ajplung.00083.2014
97. Qiu T, Pei P, Yao X, Jiang L, Wei S, Wang Z, et al. Taurine Attenuates Arsenic-Induced Pyroptosis and Nonalcoholic Steatohepatitis by Inhibiting the Autophagic-Inflammasomal Pathway. Cell Death Dis (2018) 9:946. doi: 10.1038/s41419-018-1004-0
98. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: Disturbed Interplay Between Autophagy and Inflammasomes. Aging (Albany NY) (2012) 4:166–75. doi: 10.18632/aging.100444
Keywords: reactive oxygen species, inflammasome, macrophage, nanoparticles, endoplasmic reticulum stress, autophagy
Citation: Liu Y, Sun Y, Kang J, He Z, Liu Q, Wu J, Li D, Wang X, Tao Z, Guan X, She W, Xu H and Deng Y (2022) Role of ROS-Induced NLRP3 Inflammasome Activation in the Formation of Calcium Oxalate Nephrolithiasis. Front. Immunol. 13:818625. doi: 10.3389/fimmu.2022.818625
Received: 19 November 2021; Accepted: 11 January 2022;
Published: 27 January 2022.
Edited by:
Quan Hong, Chinese PLA General Hospital, ChinaReviewed by:
Chengguo Wei, Mount Sinai Hospital, United StatesLu Zhang, First Affiliated Hospital of Xiamen University, China
Copyright © 2022 Liu, Sun, Kang, He, Liu, Wu, Li, Wang, Tao, Guan, She, Xu and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaoliang Deng, ZHlsa2YzMTdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship