 McKella Sylvester
McKella Sylvester Aran Son
Aran Son Daniella M. Schwartz
Daniella M. Schwartz- Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
Autoinflammatory diseases are a group of clinical syndromes characterized by constitutive overactivation of innate immune pathways. This results in increased production of or responses to monocyte- and neutrophil-derived cytokines such as interleukin-1β (IL-1β), Tumor Necrosis Factor-α (TNF-α), and Type 1 interferon (IFN). By contrast, clinical allergy is caused by dysregulated type 2 immunity, which is characterized by expansion of T helper 2 (Th2) cells and eosinophils, as well as overproduction of the associated cytokines IL-4, IL-5, IL-9, and IL-13. Traditionally, type 2 immune cells and autoinflammatory effectors were thought to counter-regulate each other. However, an expanding body of evidence suggests that, in some contexts, autoinflammatory pathways and cytokines may potentiate type 2 immune responses. Conversely, type 2 immune cells and cytokines can regulate autoinflammatory responses in complex and context-dependent manners. Here, we introduce the concepts of autoinflammation and type 2 immunity. We proceed to review the mechanisms by which autoinflammatory and type 2 immune responses can modulate each other. Finally, we discuss the epidemiology of type 2 immunity and clinical allergy in several monogenic and complex autoinflammatory diseases. In the future, these interactions between type 2 immunity and autoinflammation may help to expand the spectrum of autoinflammation and to guide the management of patients with various autoinflammatory and allergic diseases.
Introduction
Diseases of immune dysregulation affect up to 40% of the global population and can have devastating consequences including organ failure and death (1, 2). Conceptually, disorders of immune activation are divided into three major categories. Autoimmune diseases are caused by inappropriate antigen-specific immune responses to self-antigens, and inflammation is largely promoted by lymphocytes (3). Allergic diseases are also mediated by inappropriate activation of lymphocytes, but the immune responses are against foreign antigens, or allergens (4, 5). By contrast, autoinflammatory diseases are caused by activated myeloid cells that mediate antigen-independent innate immune pathology (3). Although this is a useful conceptual framework, many autoimmune diseases are driven by a combination of innate and adaptive immune dysregulation (6, 7). The role of autoinflammatory pathways in autoimmune diseases has become a major area of investigation, uncovering novel interactions between innate and adaptive immunity (6, 7).
While the boundaries between autoimmunity and autoinflammation have become less clear over time, less work has been done on the intersection of allergy and autoinflammation. In general, autoimmune and autoinflammatory responses have been thought to primarily repress allergic inflammation, and vice versa (8). This is largely due to the Th1-Th2 (T helper 1 – T helper 2) paradigm, where Th1 and Th2 cells have counterregulatory roles. Th1 cells are associated with Type 1 immune responses, which are also characterized by activated myeloid lineage cells, and which are associated with autoimmunity and autoinflammation (9). However, over the past several decades it has become clear that autoinflammatory-associated cytokines and pathways can promote allergy-associated type 2 immune responses (5, 10). In this review, we explore the interactions between autoinflammation and type 2, or allergy-associated, inflammation. We begin by providing a brief overview of autoinflammation and type 2 inflammation, including the human diseases associated with both immune responses. We then review the role of autoinflammation-associated cytokines and pathways in type 2 responses, and the role of type 2 immune factors in autoinflammation. Finally, we summarize results from studies exploring the prevalence of type 2 clinical and immunologic phenotypes in patients with monogenic and complex autoinflammatory diseases.
Part 1: An Overview of Autoinflammation and Type 2 Inflammation
Autoinflammation Results From Inappropriate Innate Immune Activation
The concept of autoinflammatory disease was coined in 1999 to describe a group of immune dysregulatory diseases characterized by recurrent episodes of fever and systemic inflammation. In contrast to autoimmune diseases, autoinflammatory disorders are typified by constitutive activation of myeloid cells rather than antigen-specific T cell or B cell responses (3). Given the central role of myeloid cells in the innate arm of immune responses, the concept of “autoinflammation” was subsequently broadened to characterize primary disorders of the innate immune system. This approach was further advanced by the discovery of monogenic autoinflammatory diseases caused by mutations in genes critical for innate immune function (11–16).
One useful framework for characterizing monogenic autoinflammatory diseases is by the innate immunologic pathways that are dysregulated by disease-causing mutations. Many autoinflammation-associated genes are critical to the inflammasome and IL-1β production pathway (Figure 1). This includes the MEFV gene, which causes the prototypical autoinflammatory disease Familial Mediterranean Fever (FMF). Other examples of inflammasome-regulating genes and associated autoinflammatory diseases include MVK (hyper-IgD syndrome; HIDS), NLRP3 (Cryopyrin-associated periodic fever syndrome; CAPS), PSTPIP1 (Pyogenic arthritis with pyoderma gangrenosum and acne; PAPA), WDR1 (periodic fever, immunodeficiency, and thrombocytopenia; PFIT), IL1RA (Deficiency of IL-1RA; DIRA), and NLRC4 (Macrophage activation syndrome; MAS). Inflammasomes are innate immune sensors; upon activation, they form multimeric complexes that cleave the protease caspase-1, which in turn cleaves and activates IL-1β and IL-18. Consequently, inflammasomopathies are characterized by overproduction of IL-1β, and affected patients respond clinically to inhibitors of IL-1β and its receptor (3).
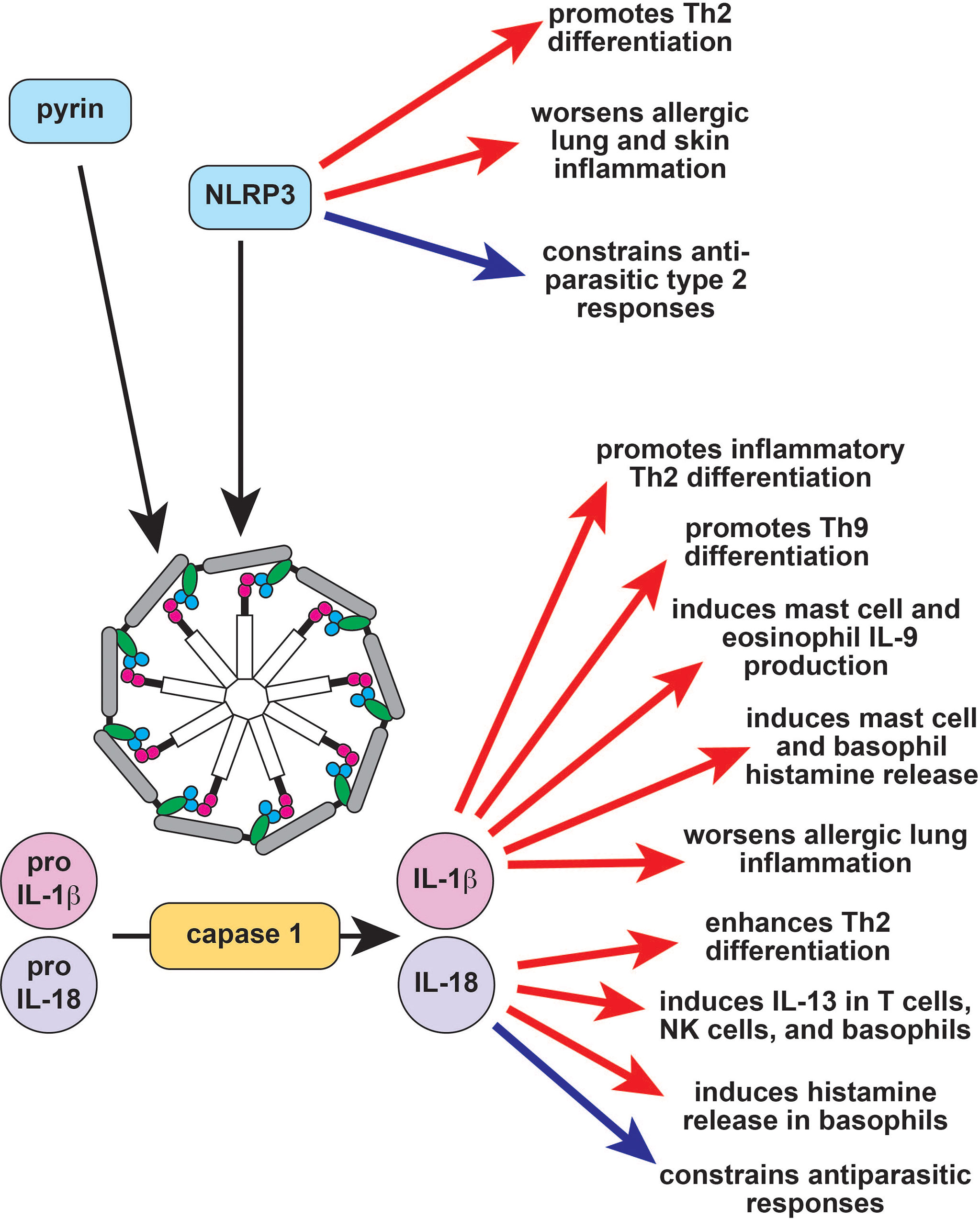
Figure 1 The role of the inflammasome in type 2 immune responses. Inflammasomes are large multimeric signaling molecules that process inactive pro-IL-1β and pro-IL-18 into their active forms. Constitutive activation of the pyrin inflammasome results in Familial Mediterranean Fever (FMF), while activation of NLRP3 causes the autoinflammatory disease cryopyrin-associated periodic fever syndrome (CAPS). NLRP3 induces Th2 differentiation through inflammasome-dependent and independent mechanisms (red arrows) but also acts as a brake on type 2 responses to parasites (blue arrow). IL-1b enhances allergic responses through a variety of effector cells (red arrows), while the effect of IL-18 is context-dependent (red and blue arrows). Th2, T helper 2; IL-9, interleukin 9; IL-13, interleukin 13; NK, natural killer.
Another group of diseases is caused by mutations in the tumor necrosis factor (TNF)/NF-κB signaling pathway, which modulates innate and adaptive immune responses (Figure 2) (17). The prototypical example of TNF-receptor associated periodic fever syndrome (TRAPS) is caused by mutations in TNFRSF1A, although the pathogenesis of TRAPS is complex and includes TNF-independent mechanisms (18). Downstream of the TNF receptor, ubiquitin-editing enzymes like OTULIN and A20 negatively regulate NF-κB signaling; inactivating mutations cause the autoinflammatory diseases Otulipenia and HA20, respectively (19, 20). Gain-of-function mutations in the NOD2 and CARD14 genes also cause autoinflammation due to constitutive activation of NF-κB signaling (21, 22). Although TNF inhibitors can be effective for this group of diseases, NF-κB can also be activated by TNF-independent agonists including IL-1β. Accordingly, some patients with NF-κB associated autoinflammatory diseases require treatment with other immunomodulators, including IL-1 pathway inhibitors (3, 18, 23).
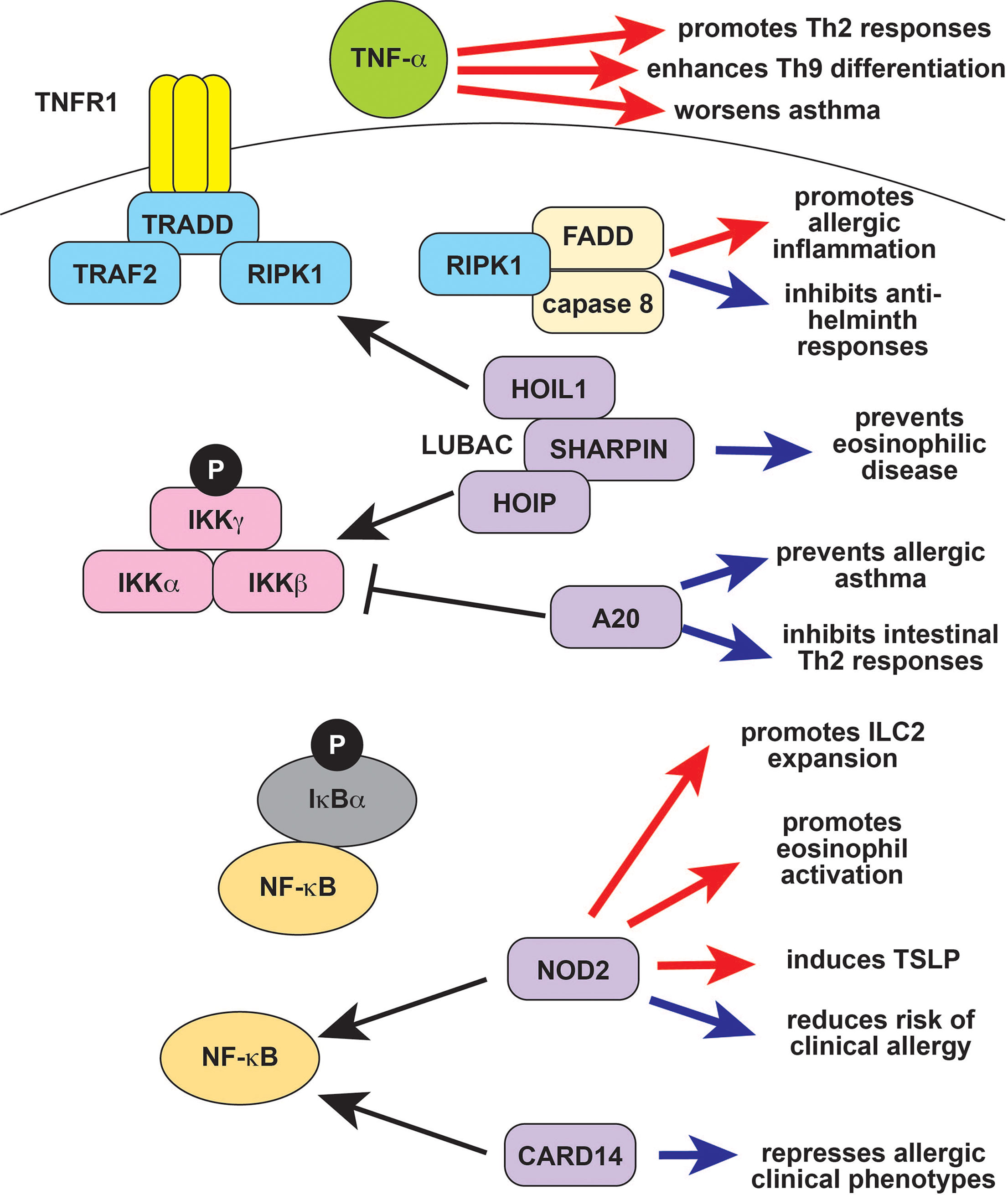
Figure 2 TNF-α and NF-κB signaling in type 2 immune responses. TNF-α exacerbates type 2 diseases like asthma in part by promoting Th9 and Th2 differentiation and function (red arrows). The protease caspase-8 forms the ripoptosome complex together with the TNF signaling molecule RIPK1 and FADD. The ripoptosome is regulates cell death, with caspase-8 and RIPK1 promoting apoptosis over necroptosis, so that defects in RIPK1 result in increased necroptosis and autoinflammation. The ripoptosome promotes type 2 responses in response to environmental allergens (red arrow) but can suppress type 2 responses to parasites (blue arrows). Ubiquitin editing proteins like A20 (TNFAIP3) and LUBAC (composed of HOIL-1, HOIP, and SHARPIN) modulate NF-κB signaling by targeting upstream molecules for activation and/or degradation. A20 negatively regulates NF-κB and also prevents allergic asthma as well as other type 2 responses (blue arrows). SHARPIN activates NF-κB, and deficiency results in eosinophilic tissue infiltration (blue arrow). The NF-κB signaling molecules CARD14 and CARD15/NOD2 also modulate type 2 responses. CARD14 prevents allergic disease, and deficiency results in clinical atopy (blue arrow). CARD15/NOD2 is reported to have both positive (red arrows) and negative (blue arrows) effects on type 2 immunity, and its role may be context-dependent. TNF-α, tumor necrosis factor alpha; RIPK1, Receptor Interacting Serine Threonine Kinase 1; FADD, Fas Associated via Death Domain; LUBAC, linear ubiquitin chain assembly complex; HOIL-1, Haem-Oxidized IRP2 Ubiquitin Ligase 1; HOIP, HOIL-1L Interacting Protein; SHARPIN, SHANK-associated RH-interacting protein; TNFAIP3, TNF-α induced protein 3; CARD14, caspase recruitment domain-containing protein 14; CARD15, caspase recruitment domain-containing protein 15; NOD2, nucleotide binding oligomerization domain-containing protein 2; Th2, T helper 2; IL-9, interleukin 9; IL-13, interleukin 13; IL-4, interleukin 4; NK, natural killer.
The Type I interferon (IFN) pathway is important for antiviral immunity and for innate immune functions such as natural killer cell activation and antigen presentation (Figure 3) (24). Inborn errors of immunity that cause activation of Type I IFN signaling are termed interferonopathies. Proteasome-associated autoinflammatory syndromes (PRAAS) result from mutations in genes encoding proteasome subunits. Proteasome dysfunction induces the unfolded protein response (UPR), resulting in Type I IFN activation and autoinflammation (25). Several monogenic interferonopathies are caused by mutations in genes that modulate intracellular responses to nucleic acids. For example, mutations in the DNA sensor gene TMEM173 lead to STING-associated vasculopathy with onset in infancy (SAVI) (26). Mutations in interferon-response genes like STAT2 can also cause autoinflammation due to overactive signaling downstream of Type I IFN (27, 28).
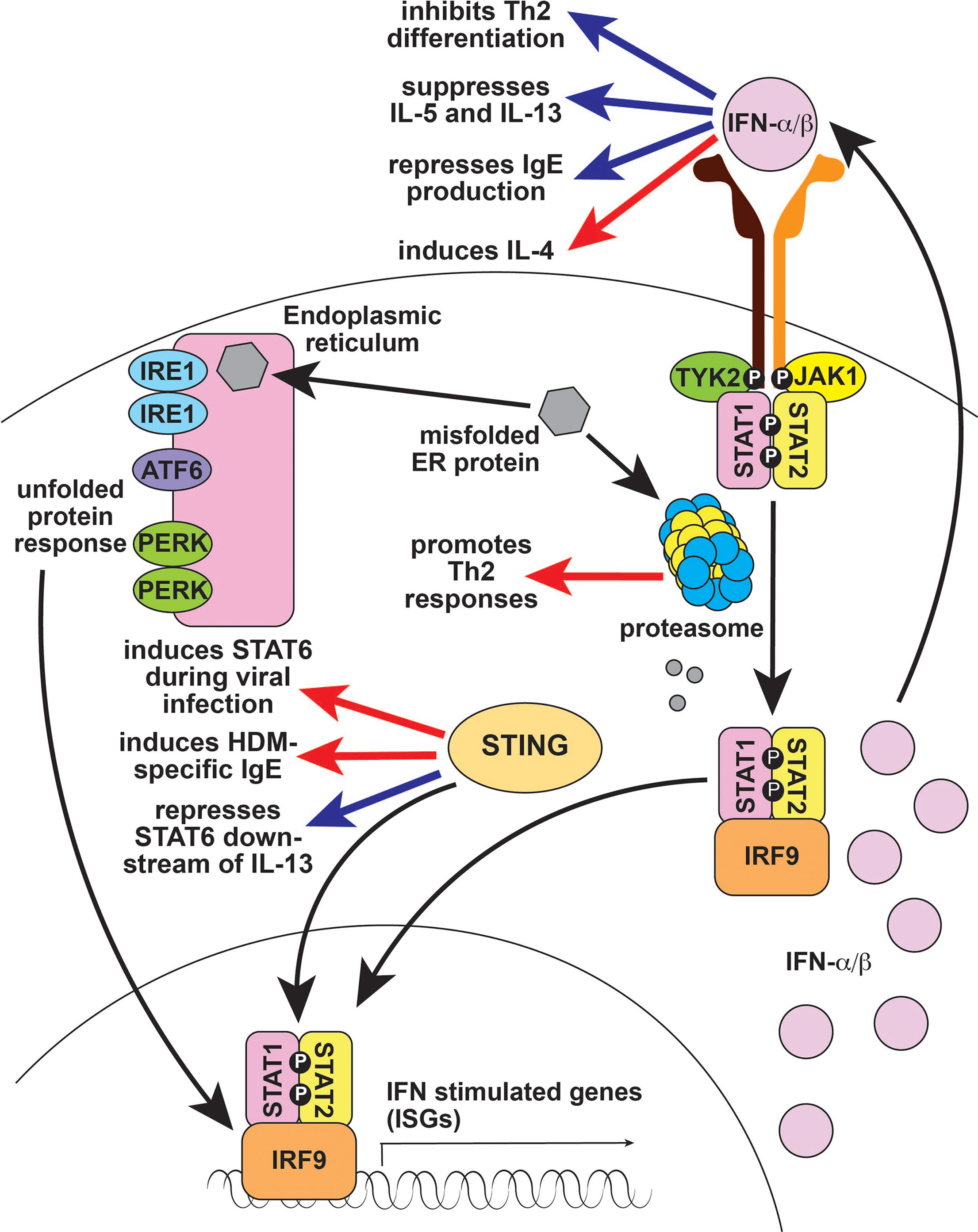
Figure 3 Type 1 interferon signaling in type 2 immune responses. Type 1 IFNs like IFN-α and IFN-β largely suppress type 2 responses (blue arrows), although they are reported to induce IL-4 (red arrow), which can enhance type 2 immunity. The proteasome is important for processing and degrading misfolded endoplasmic reticulum proteins; defects cause unfolded proteins to accumulate, resulting in Type 1 IFN production. The proteasome also regulates antigen processing and presentation which is critical for T cell immunity, including Th2 responses (red arrow). STING is a DNA sensor that activates Type 1 IFN. STING activates STAT6 in response to viral infection and promotes IgE production in response to HDM (red arrow), but also represses IL-13-induced STAT6 activation in subjects with rhinosinusitis (blue arrow). IFN, interferon; ER, endoplasmic reticulum; IRE1, Inositol Requiring Enzyme 1; ATF6, Activating Transcription Factor 6; PERK, PKR-like Endoplasmic Reticulum Kinase; STING, Stimulator of Interferon Genes; TYK2, Tyrosine Kinase 2; JAK1, Janus Kinase 1; STAT1, Signal Transducer and Activator of Transcription 1; IRF9, interferon regulatory factor 9; Th2, T helper 2; IL-4, interleukin 4; IL-5, interleukin 5; IL-13, interleukin 13; IgE, immunoglobulin E, NK, natural killer; HDM, house dust mite.
In addition to these canonical dysregulated pathways, autoinflammation can also be caused by mutations in genes important for other innate immune functions. Deficiency of ADA2 (DADA2) is caused by mutations in CERC1, which regulates monocyte differentiation (29). Mutations in complement pathway genes like CFH, C3, and CD46 can cause atypical hemolytic uremic syndrome (3, 30–32). Genes that regulate actin polymerization like WDR1 and CDC42 are also important for inflammasome assembly; mutations can therefore cause IL-1β and IL-18-dependent autoinflammation (3, 33, 34). The ripoptosome is a multimeric complex containing RIPK1, FADD, and caspase-8 that is important for regulating the balance between necroptotic and apoptotic cell death; inactivating mutations can therefore cause autoinflammation secondary to increased necroptosis (35–37). Somatic mutations in the ubiquitin-editing gene UBA1 lead to VEXAS, a treatment-refractory complex autoinflammatory syndrome characterized by activation of multiple immune pathways (38). Finally, a number of complex autoinflammatory diseases including systemic juvenile idiopathic arthritis (sJIA), Behcet’s disease, and periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome are linked to a combination of genetic polymorphisms and environmental factors (39, 40). As increased access to next-generation sequencing accelerates gene discovery, the spectrum of autoinflammatory diseases will likely broaden to comprise new mechanisms of innate immune dysregulation.
Type 2 Immunity Is Characterized by Allergy-Associated Effector Cytokines and Cells
Type 2 immunity was originally described as a counter-regulator of Th1-driven immune responses but was subsequently recognized as a distinct immune response with important roles in antihelminth defense, allergy, and wound repair (4, 5). Type 2 immunity is most commonly associated with Th2 cells and their hallmark effector cytokines IL-4, IL-5, and IL-13. However, type 2 inflammation is mediated by many other cell types including alternatively activated macrophages, type 2 innate lymphoid cells (ILC2), eosinophils, basophils, mast cells, and immunoglobulin E (IgE) secreting plasma cells (8). In addition to Th2-effector cytokines, type 2 immune cells secrete and respond to IL-9, IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) (Figure 4) (4).
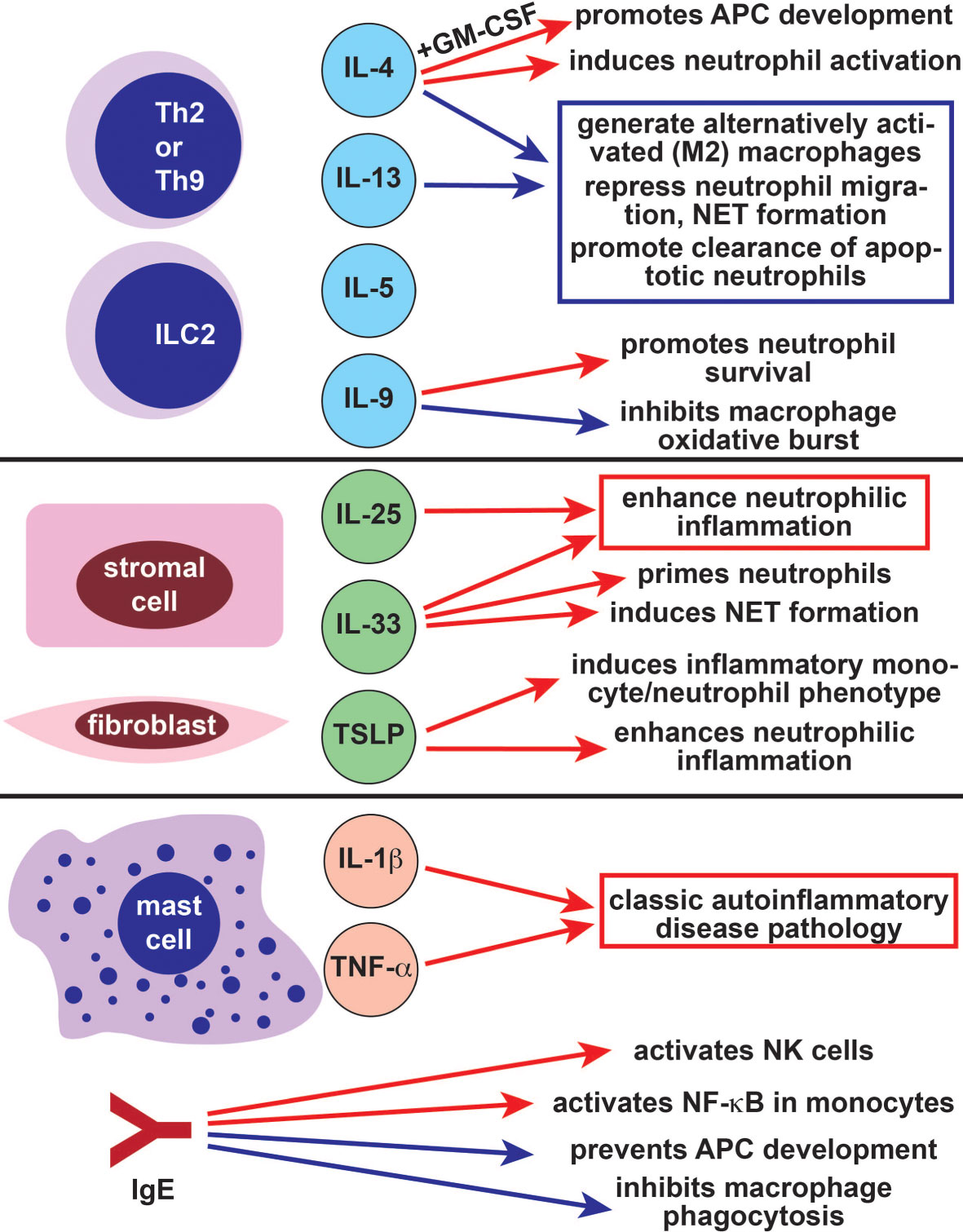
Figure 4 The role of type 2 immune cells and cytokines in autoinflammatory cells and pathways. The Th2- and ILC2-derived cytokines IL-4 and IL-13 largely suppress autoinflammatory pathology by inducing the differentiation of anti-inflammatory M2 alternatively activated macrophages and repressing neutrophil migration (blue arrows). IL-4 also has some positive effects on autoinflammatory cells (red arrows), particularly in combination with GM-CSF. Like IL-4, the Th2/Th9/ILC2-derived cytokine IL-9 has both positive (red arrow) and negative (blue arrow) effects on autoinflammatory cells. The alarmins IL-25, IL-33, and TSLP promote autoinflammatory pathology through their effects on neutrophils and monocytes (red arrows). Mast cells also promote classic autoinflammatory pathology by producing IL-1β and TNF-α. The allergy-associated immunoglobulin IgE can both promote (red arrows) and repress (blue arrows) autoinflammatory disease. Th2, T helper 2; ILC2, type 2 innate lymphoid cell, IL-, interleukin-; GM-CSF, granulocyte macrophage colony stimulating factor; NET, neutrophil extracellular trap; APC, antigen presenting cell; TSLP, thymic stromal lymphopoietin; TNF-α, tumor necrosis factor alpha; IgE, immunoglobulin E; NK, natural killer.
Immune responses have evolved to protect against discrete pathogens; in this context, type 2 immunity is critical to host defense against helminth infections. Accordingly, type 2 immune cells are found at barrier surfaces where they promote goblet cell hyperplasia, mucus secretion, and muscle contraction – all of which induce intestinal worm expulsion (4, 8). Many of these protective mechanisms can also promote tissue remodeling, making them important for wound repair after injury (4, 8). Type 2 dependent repair can ultimately result in tissue fibrosis, particularly when these pathways are chronically activated (4). Fibrosis is a highly pathological inflammatory endpoint that can result in significant morbidity and mortality secondary to organ failure. Thus, protective type 2 responses can easily become pathogenic when dysregulated or overactivated.
Consistent with the reciprocal inhibition seen for Th1 and Th2 cells, type 2 immune responses can also protect from autoimmune inflammation. This has largely been described in the context of murine inflammatory models, where Th2 cells and type 2 cytokines ameliorate autoimmune arthritis and encephalitis (8, 41, 42). However, the role of type 2 immunity in human autoimmune disease is complex: the type 2 effector cytokines IL-13 and IL-9, for example, are both thought to promote inflammation in patients with ulcerative colitis and psoriasis (43–46). Th2 cells and IgE can both promote kidney inflammation in patients with systemic lupus erythematosus (47, 48).
Allergic disorders make up the largest group of human diseases characterized by type 2 dysregulation and include asthma, atopic dermatitis, food allergy, and allergic rhinitis (2). Immunologically, allergy is caused by an exaggerated type 2 response to foreign antigens. However, many allergy-associated clinical syndromes have forms in which allergic sensitization cannot be demonstrated (49, 50). In some cases, this might be due to primary dysregulation of type 2 inflammatory cells and mediators. For example, some patients with late-onset eosinophilic asthma are thought to have primary dysregulation of ILC2, which produce type 2 cytokines independent of antigenic stimulation (51). Patients with NARES (nonallergic rhinitis with eosinophilia syndrome) are thought to have a primary eosinophilic disorder in at least some cases (50). In other cases, non-type 2 mediators can promote symptoms that are clinically indistinguishable from allergen-specific type 2 responses. For example, hormonal rhinitis can mimic allergic rhinitis but is caused by hormone-induced nasal vascular engorgement (50).
Part 2: The Role of Autoinflammation in Type 2 Immune Responses
Autoinflammation, Type 2 Immunity, and Clinical Allergy: A Complex Relationship
Type 1 cytokines have long been thought to primarily repress type 2 immunity based on the Th1-Th2 paradigm. Indeed, the type 1 cytokines IFN-γ and IL-12 inhibit Th2 differentiation and type 2 responses to helminth infection (8, 52, 53). However, other autoinflammatory and autoimmune cytokines can amplify type 2 inflammation, worsening type 2-driven pathology (54–56) Additionally, autoinflammatory and autoimmune cytokines can directly promote tissue inflammation, resulting in clinical phenotypes identical to type 2-driven allergic disease (57–59). The heterogeneity of inflammatory mechanisms driving common clinical phenotypes can present substantial barriers to understanding the crosstalk between type 2 inflammation and autoinflammation in human disease. To help address this complexity, one can approach the role of autoinflammation in type 2-mediated disease using the innate immunologic pathways that are used to categorize monogenic autoinflammatory diseases: inflammasomes, TNF-α, Type I IFN, and newer pathways including necroptosis.
Inflammasomes and Associated Cytokines in Type 2 Immunity
The pyrin inflammasome does not appear to have a major role in type 2 immune responses, and a recombinant pyrin domain was found to attenuate allergic inflammation in mice by suppressing NF-κB activation (60). By contrast, NLRP3 directly promotes Th2 differentiation independent of its inflammasome function by transcriptionally inducing Il4 in conjunction with IRF4 (Figure 1) (61). The NLRP3 inflammasome can also trigger a Th2-biased response in the context of both infection and allergic inflammation (Figure 1) (62–64). NLRP3 activation in bronchial epithelial cells promotes allergic lung inflammation, whereas activation in keratinocytes promotes eczema (63, 65). By contrast, Helicobacter pylori gastric infection protects from allergic asthma by activating NLRP3 in proximal dendritic cells (66). Similarly, helminths induce NLRP3, which then acts as a brake on type 2 responses via both inflammasome-independent and inflammasome-dependent mechanisms (Figure 1) (67–69). Taken together, these studies suggest that NLRP3 activation may primarily suppress type 2 responses to pathogens but promote dysregulated type 2 responses to environmental allergens.
The end products of inflammasome activation, IL-1α, IL-1β and IL-18, can also regulate type 2 immunity. Single nucleotide polymorphisms (SNPs) in IL1A, IL1B, and IL1R1 are all linked to asthma; accordingly, IL-1α and IL-1β both exacerbate murine allergic airway inflammation (Figure 1) (70–75). Type 2 immune cells like eosinophils and mast cells can release IL-1β, airway epithelial cells stimulated with the house dust mite (HDM) allergen can release IL-1α, and IL-1β can be found in allergic tissues, further suggesting that IL-1 has a role in type 2 responses (76–79). This hypothesis is supported by the observation that IL -1β enhances inflammatory Th2 differentiation and helps induce the differentiation of Th9 cells (Figure 1) (55, 80–82). IL-1β is also capable of regulating various type 2 innate effector cells to promote tissue inflammation. For example, IL-1β activates human ILC2s in the presence of IL-2, inducing proliferation and effector cytokine production (83, 84). IL-1β also induces histamine release from basophils and mast cells, and histamine enhances IL-1β release, which can induce a positive feedback loop (Figure 1) (85, 86). Eosinophils and mast cells stimulated with IL-1β produce IL-9, further supporting the hypothesis that IL-1β can enhance type 2 immune responses to promote allergic pathology (Figure 1) (87, 88). A pathogenic role for IL-1 signaling in allergy is further supported by a number of clinical studies demonstrating the efficacy of IL-1 pathway inhibitors in asthma and atopic dermatitis (89–91). Several larger randomized controlled clinical trials have been planned to follow up these encouraging observations but were halted early due to patient recruitment – particularly in light of the ongoing COVID-19 pandemic (NCT01122914, NCT04035109, NCT03513458).
Like IL-1β, IL-18 is reported to enhance Th2 differentiation and T-cell-derived IL-13 production (Figure 1) (54, 69). This effect is IL-4-dependent and may be because IL-18-induces IL-4 production or because it increases T cell sensitivity to IL-4 (54). IL-18 also induces IL-13 in natural killer (NK) cells and in basophils, suggesting that it may contribute to the innate arm of type 2 immune responses (Figure 1) (92, 93). In addition to IL-13, IL-18 also induces histamine from basophils and can promote eosinophil development and maturation in combination with IL-5 (94, 95). However, IL-18 can also repress type 2 responses in vivo. IL-18-deficient mice develop enhanced allergen-induced eosinophilia, and IL-18-deficient mice are protected from helminth infections (Figure 1) (96, 97). This suggests that the role of IL-18 in type 2 immunity may be context-dependent. Indeed, IL-18 can promote either Th1 or Th2 differentiation depending on genetic background and cytokine milieu (98). Similarly, IL-18 represses allergic pathology and IgE production in combination with IL-12 but induces both of these in the absence of IL-12 (95, 99, 100).
TNF-α and NF-κB Signaling in Type 2 Immunity
The inflammatory cytokine TNF-α has a role in both innate and adaptive immunity, underlying the efficacy of TNF inhibitors in patients with autoimmune conditions like rheumatoid arthritis (RA) and autoinflammatory conditions like Deficiency of ADA2 (DADA2) (101, 102). TNF-α and other TNF superfamily cytokines promote the differentiation of Th9 cells, suggesting that they may enhance type 2 immune responses (Figure 2) (103, 104). Many TNF superfamily cytokines are costimulatory molecules that more generally modulate division, survival, and activation in T cells. Several of these positively regulate of Th2 differentiation and function due to their role in costimulation (105). TNF-α also enhances the effect of IL-4 on eosinophils and enhances Th2- mediated responses at mucosal sites (Figure 2) (56, 106). This may be in part due to effects on non-immune cells that promote type 2 responses. For example, TNF-α and IL-1β synergize to promote airway hyperresponsiveness, which might partly underlie the role of TNF-α in asthma (Figure 2) (107, 108). The TNF-α inhibitor etanercept initially showed promise for severe refractory asthma, but a subsequent trial failed to show efficacy (107, 109). Clinical development was ultimately halted due to an increased rate of serious adverse effects, most notably respiratory infections, in a phase 2 trial of golimumab (110). Etanercept and infliximab are reported efficacious for the treatment of atopic dermatitis and have been used as an off-label treatment for severe disease (111, 112).
NF-κB signaling has long been known to play a role in type 2 immune responses, Th2 differentiation, IgE production, and the function of innate type 2 effectors like eosinophils, ILC2s, and mast cells (10, 113–115). Inactivating mutations in NF-κB pathway genes like CARD11 and CARD14 cause monogenic immune dysregulatory syndromes that include allergic phenotypes, indicating that physiologic NF-κB signaling can suppress type 2 pathology (Figure 2) (116, 117). The clinical phenotype of CARD14 loss-of-function (LOF) is particularly interesting in the context of autoinflammation, because activating CARD14 mutations cause a monogenic autoinflammatory disease (22). Similarly, NOD2 (CARD15) LOF polymorphisms are associated with an increased risk of clinical allergy and inflammatory bowel disease (IBD), whereas activating mutations cause the autoinflammatory disease Blau syndrome (Figure 2) (21, 118, 119). However, NOD2 also induces the type 2 cytokine TSLP, promotes ILC2 expansion, and induces eosinophil activation (Figure 2). These studies suggest that, in some cases, NF-kB signaling is primarily an inducer of type 2 immune responses.
The autoinflammation-associated NF-κB signaling repressor A20 (TNFAIP3) inhibits airway epithelial cytokine production in response to endotoxin, suppressing type 2 responses to HDM and preventing allergic asthma (Figure 2) (120). A20 also has a cell-intrinsic anti-inflammatory role in mast cells, inhibits intestinal Th2 responses, and prevents Th17 differentiation in response to HDM (Figure 2) (121–123). These observations may explain the negative associations of TNFAIP3 expression with allergic asthma, chronic rhinosinusitis, atopic dermatitis, and food allergy (124–127). The SHARPIN protein (Shank-interacting protein like 1) is a part of the LUBAC (linear ubiquitin chain assembly complex), which promotes NF-κB activation and is linked to autoinflammation and complex immune dysregulation. SHARPIN promotes regulatory T cell function, so deficiency promotes systemic inflammation (128). Additionally, SHARPIN deletion causes lymphocyte-independent eosinophilic esophagitis, and keratinocyte-specific deletion causes eosinophilic dermatitis (Figure 2) (129–131).
Type I IFN Signaling in Type 2 Immunity
Broadly, type I IFNs inhibit type 2 immune responses: they suppress IL-5 and IL-13 production, inhibit GATA3-dependent Th2 differentiation, and block B cell isotype switching to IgE (Figure 3) (24, 132–135). Accordingly, recombinant IFN-α is used to treat Idiopathic Hypereosinophilic Syndromes and Eosinophilic Granulomatosis with Polyangiitis (136, 137). Type I IFNs are also thought to play a role in asthma, where deficiency leads to increased viral infection and enhanced Th2 differentiation, worsening disease (138). However, type I IFNs are also reported to induce IL-4 production and to promote murine eosinophilic rhinosinusitis, possibly by increasing eosinophil recruitment (Figure 3) (134, 139). It remains to be determined whether these functions have any role in promoting type 2 immunity-related human diseases.
In addition to modulating type I IFNs through the unfolded protein response, the proteasome is important for antigen processing and presentation (140). Consequently, defects in the proteasome result in a general loss of T-cell-dependent immunity. Taken together with the antagonistic role of type I IFN on type 2 responses, it is not surprising that defects in the proteasome are associated with reduced Th2 responses (Figure 3) (141). By contrast, the DNA sensor STING activates the type 2 associated signaling molecule STAT6 in response to viral infection, although the result is enhanced antiviral immunity rather than a type 2 immune response (Figure 3) (142). STING also promotes HDM-induced IgE production by enhancing the function of T follicular helper cells (Figure 3) (143). However, STING represses IL-13-induced STAT6 phosphorylation in subjects with rhinosinusitis by increasing expression of the STAT6 inhibitor SOCS1 (suppressor of cytokine signaling 1) (Figure 3) (144). SOS1 induction may also underlie the observation that STING signaling in ILC2s promotes a phenotypic shift to Type 1 ILC (ILC1) during lung inflammation (145). Taken together, these studies suggest that the role of STING in type 2 immunity is complex and context dependent.
Other Autoinflammation-Associated Pathways in Type 2 Immunity
Cytoskeletal regulators that play a role in inflammasome activation, like CDC42 and WDR1, also play a role in adaptive immunity and nonhematopoietic cells. CDC42 is activated by the atypical guanine nucleotide exchange factor DOCK8, which is linked to autosomal recessive hyper-IgE syndrome (33). CDC42 signaling is also important for mast cell and eosinophil function, and CDC42-deficient invariant natural killer T cells have a defect in IL-4 secretion because CDC42 degradation induces IL-4 secretion in response to lipid antigens (146). Complement activation promotes Th1 differentiation and function, which can indirectly repress type 2 responses, but is not thought to directly regulate Th2-driven responses (147). The complement system can activate innate type 2 effectors like eosinophils and mast cells, however, and may therefore promote some type 2 associated pathology (148, 149). Environmental allergens can activate the ripoptosome to trigger type 2 inflammation through RIPK1 and caspase 8, which shunt cells away from necroptosis and towards apoptosis (Figure 2) (36, 150, 151). Caspase-8 can also promote allergic pathology by directly activating IL-1 cytokines (152). However, caspase 8 prevents type 2 immune responses to Trypanosoma cruzi infection, leading to increased parasitemia and chronic infection (150). Caspase-8 also promotes epithelial keratinocyte cohesion, so that epidermal-specific deficiency causes a spontaneous eczematoid dermatitis (153). Thus, the effect of the ripoptosome on type 2 immune responses may be context-dependent as for other autoinflammatory mediators.
Part 3: The Role of Type 2 Immunity in Autoinflammatory Cells and Pathways
Th2 and ILC2-Derived Cytokines in Autoinflammatory Cells and Pathways
The type 2 cytokines IL-4 and IL-13 have long been studied as modulators of innate immune function due to their role in the generation of alternatively activated macrophages (M2) (Figure 4) (154). In contrast to classical activation, which is induced by IFN-γ and characterized by type 1 cytokine production and microbial killing, alternative activation causes macrophages to develop an immunoregulatory function. M2 macrophages are not efficient killers of invading pathogens but produce growth factors and extracellular matrix components, making them important for wound healing (154). They also can generate or maintain type 2 immune responses. In the context of alternative activation, IL-4 promotes tissue resident macrophage activation and accumulation (155). Exposure to IL-4 in combination with GM-CSF (granulocyte-monocyte colony stimulating factor) causes peripheral monocytes to function as antigen presenting cells (Figure 4) (156). These monocyte-derived cells phenotypically resemble inflammatory dendritic cells rather than inflammatory macrophages (157). Indeed, the inflammatory macrophage phenotype is promoted by classical activation and inhibited by alternative activation (158). This may be because IL-4 inhibits NF-κB and inflammasome activation in macrophages, reducing responsiveness to lipopolysaccharide (158).
IL-4 also has a role in neutrophil biology and can even be produced by neutrophils (159). While IL-4 can induce neutrophil activation and phagocytosis, it also inhibits neutrophil migration (Figure 4) (160, 161). IL-4 also represses the formation of neutrophil extracellular traps (NETs), an important mechanism used for pathogen killing (162). Like IL-4, IL-13 inhibits neutrophil migration to inflamed tissues, although IL-13 also enhances production of several neutrophil effector proteins including IL-8 (163, 164). Finally, the IL-4 and IL-13 activated signaling molecule STAT6 is importance for clearance of apoptotic neutrophils, which promotes resolution of inflammatory responses (165). Taken together, these data suggest that type 2 cytokines primarily repress pathways associated with autoinflammation in macrophages and neutrophils.
Like IL-4 and IL-13, the type 2 cytokines IL-5 and IL-9 are derived primarily from T helper cells and ILC2s. While neither IL-5 nor IL-9 is implicated in alternative activation of macrophages, both cytokines can modulate the function of monocytes and neutrophils. IL-5 receptor is expressed on neutrophils, including airway-resident neutrophils from asthma patients, although its function in neutrophils is not well characterized (166, 167). IL-5 indirectly regulates dendritic cells by inducing eosinophils, which repress plasmacytoid dendritic cell derived type I IFN production (168). IL-9 represses autoinflammation-associated responses by inhibiting oxidative burst and TNFα release in LPS-stimulated human monocytes and alveolar macrophages (Figure 4) (169, 170). However, IL-9 can also promote neutrophil survival and neutrophil-derived IL-8 release, enhancing type 1 inflammatory responses (171, 172). This suggests that the role of IL-9 in autoinflammation is complex and context-dependent.
Alarmins in Autoinflammatory Cells and Pathways
Type 2 innate cytokines, or alarmins, are produced by epithelial cells, endothelial cells, stromal cells, and fibroblasts in response to injury. These alarmins include IL-25, IL-33 and TSLP; they activate ILC2, Th2, eosinophils, mast cells, and other type 2 effectors (5, 10, 173, 174). Because activated ILC2 and Th2 cells produce large amounts of IL-4 and IL-13, alarmins can indirectly promote ILC2-dependent immunosuppressive functions in neutrophils (175, 176). In some cases, alarmins can also directly regulate neutrophils and monocytes. For example, IL-33 primes neutrophils so that they are rapidly recruited to sites of infection and inflammation, whereas IL-25 promotes neutrophilic airway infiltration (Figure 4) (177–180). IL-33 overexpression causes spontaneous neutrophilic arthritis and sterile inflammation possibly due to increased NET formation (181). The alarmin TSLP enhances neutrophilic inflammation and induces a proinflammatory phenotype in circulating monocytes and neutrophils (Figure 4) (182, 183). Further supporting its role in neutrophil-mediated host defense, TSLP enhances neutrophilic microbicidal activity against methicillin-resistant Staphylococcus Aureus (184). Together, these data suggest that type 2 alarmins can promote autoinflammatory pathology in some contexts.
Mast Cells and High Affinity IgE Receptor in Autoinflammatory Cells and Pathways
Mast cells produce IL-1β; which is cleaved and activated by caspase 1, caspase 8, and serine proteases (Figure 4) (185). Mast cell IL-1β production is NLRP3-dependent, suggesting that mast cells may have a role in NLRP3-associated autoinflammatory processes. Accordingly, patients with NLRP3 mutations develop cold-induced histamine-independent urticariform lesions, and mast cells are a major source of IL-1β in affected skin (77, 79). Mast cells also produce IL-1β in patients with the adult-onset autoinflammatory disease Schnitzler’s syndrome, in subjects with chronic recurrent multifocal osteomyelitis, and have been found in inflamed joints from patients with FMF (186–188). In mice, mast cells promote sterile joint and central nervous system inflammation (185, 189). Mast cell derived TNF-α induces urticariform rashes in patients with NLRP3 mutations, although the role of mast cell derived TNF-α in other autoinflammatory diseases is not known (Figure 4) (190).
IgE is a critical inducer of many type 2 effector cells, including mast cells, through its high affinity receptor Fc epsilon RI. Fc epsilon RI is also expressed and functional in several type 1 innate effector cells. IgE crosslinking suppresses monocyte function by blocking phagocytosis and preventing differentiation into dendritic cells (Figure 4) (191, 192). Simultaneously, engagement of Fc epsilon RI activates NF-κB in monocytes and dendritic cells, which promotes secretion of IL-6, IL-10, and TNF-α (192, 193). Macrophage Fc epsilon RI engagement also reprograms alternatively activated tumor-resident macrophages to be more proinflammatory, enhancing their antitumoral functions (194). The functions of IgE and its receptor are not as well characterized in other type 1 innate cells. However, Fc epsilon RI is expressed in both dendritic cells and neutrophils, where it delays neutrophil apoptosis (195–197). IgE can also activate NK cells through the lower affinity Fc gamma RIII receptor (198). Future studies will be needed to further characterize the roles of IgE and its receptors in autoinflammation-associated innate immune cells.
Part 4: The Epidemiology of Allergy in Autoinflammatory Diseases
The Epidemiology of Allergy in Monogenic Autoinflammatory Diseases
One way to investigate the interaction between autoinflammatory pathways and type 2 immunity is to investigate the prevalence of allergic clinical and immunological phenotypes in subjects with monogenic autoinflammatory diseases (Table 1). Because single gene mutations promote activation of discrete innate pathways, this approach can assess the in vivo roles of dysregulated autoinflammatory pathways in regulating human type 2 immune responses (199). This question has been most extensively studied in FMF, perhaps because it was the first autoinflammatory disease to be linked to a causative gene (16) (Table 1). Several studies have suggested that FMF protected against asthma and atopy, potentially due to protective linkage of MEFV with asthma associated genes like IL4RA (199–201). Although one study suggested that Turkish FMF patients may have elevated total serum IgE relative to healthy volunteers, this result was not seen in other cohorts, where there was a trend towards reduced serum IgE (199, 201, 202). Taken together, these results suggest that activation of the pyrin inflammasome attenuates human type 2 immune responses.
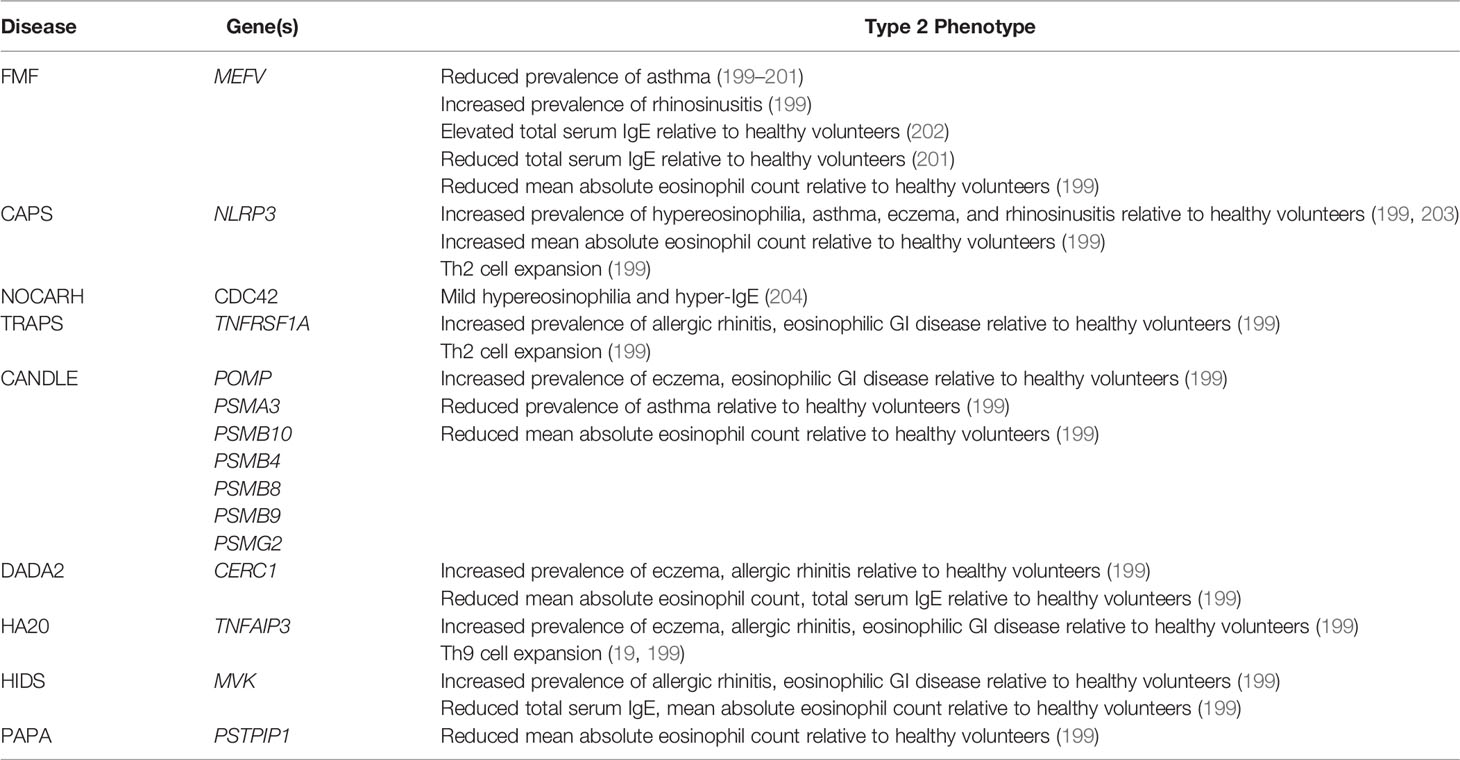
Table 1 Associations of monogenic autoinflammatory diseases with type 2 clinical and immunological phenotypes.
By contrast, the autoinflammatory disease CAPS, caused by NLRP3 mutations, is associated with peripheral eosinophilia and eosinophilic skin infiltration (199, 203) (Table 1). Eosinophilia correlates with CAPS disease activity, suggesting that NLRP3 activation promotes eosinophilia (199). This is consistent with the role of NLRP3 and IL-1β in promoting the differentiation and function of type 2 effectors like Th2 cells, mast cells, and eosinophils. CAPS is also characterized by an increased prevalence of eczema, asthma, and allergic rhinitis relative to both the general population and FMF (199). This is consistent with the observation that NLRP3 activation exacerbates murine models of asthma and eczematous dermatitis (63, 65). Finally, the urticariform lesions of CAPS are characterized by IL-1β and TNF-α producing mast cell infiltration, once again linking the NLRP3 inflammasome to type 2 effector activation in humans (77, 79, 190). Overall, these results suggest that in humans, constitutive activation of the NLRP3 inflammasome promotes type 2 immune responses. Because helminth infections are extremely uncommon in countries with highly developed CAPS cohorts, it remains to be determined whether NLRP3 activation suppresses type 2 responses to pathogens in humans, as it does in murine models (67–69).
CDC42 is a plasma membrane associated GTPase involved in diverse processes including cell division, phagocytosis, and epithelial cell morphology (204). Mutations are linked to NOCARH (neonatal onset of pancytopenia, autoinflammation, rash, and episodes of HLH) an IL-1-responsive autoinflammatory disease with features of macrophage activation syndrome (MAS) (33, 205). CDC42 alternates between an inactive cytosolic form and an active plasma membrane bound form; mutations affecting trafficking alter the subcellular localization independent of the protein’s activation state (33). This, in turn, alters the partners that bind to CDC42, ultimately leading to NF-κB overactivation and autoinflammation (204). In addition to autoinflammation, one patient with NOCARH also developed mild hypereosinophilia and hyper-IgE, although no clinical allergic diagnoses were reported (204) (Table 1). As additional patients are identified, careful phenotyping will be needed to determine whether type 2 immune activation is a common feature of NOCARH.
Far less is known about the prevalence of type 2 immune activation in other autoinflammatory diseases. In one systematic population study, clinical diagnoses of allergic rhinitis were highly prevalent in almost all autoinflammatory diseases, including FMF (199). This included diseases with reduced clinical laboratory markers of type 2 inflammation relative to the general population, like HIDS and DADA2. This might be because autoinflammation-associated cytokines like IL-1β and TNF-α can promote sinus mucosal thickening independent of type 2 immune activation (206). Thus, in some cases, autoinflammatory pathology may mimic type 2 associated disease, and this may be a potential confounder in epidemiologic studies.
The Epidemiology of Allergy in Complex Autoinflammatory Diseases
Unlike their monogenic counterparts, complex autoinflammatory diseases are linked to multiple genetic and environmental factors that contribute to their pathogenesis. Behcet’s disease is a heterogeneous and complex autoinflammatory disease that manifests with orogenital ulcers, pustular skin disease, arthritis, eye disease, gastrointestinal inflammation, and vascular complications (207). Genetic studies have identified a number of risk alleles that overlap with both recurrent aphthous stomatitis and PFAPA syndrome, allowing the three syndromes to be grouped together as Behcet’s spectrum disorders (39). Amongst the susceptibility loci shared by Behcet’s spectrum disorders are multiple genes associated with Th1-driven immunity, such as STAT4 and IL12A (39). Th1 cells are thought to repress Th2 cells, and perhaps for this reason patients with Behcet’s disease were found in several studies to have lower rates of allergic sensitization and lower IgE levels than the general population (201, 208) (Table 2). However, a separate group of studies reported increased rates of atopy and elevated levels of type 2 cytokines in subjects with Behcet’s spectrum disorders, particularly children with PFAPA (209–212). Moreover, the IgE-blocking monoclonal antibody has been reported to alleviate symptoms and reduce autoinflammation in one subject with concurrent Behcet’s disease and asthma (213) (Table 2). These disparate findings may be partly due to the genetic and phenotypic heterogeneity of patients with Behcet’s spectrum disorders, which can vary substantially between cohorts with different ancestries (207).
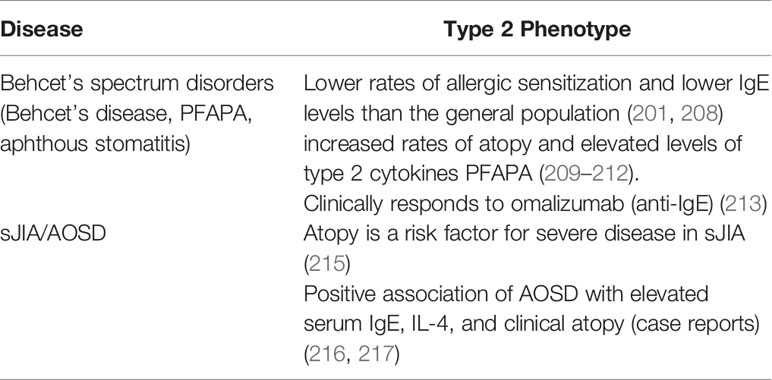
Table 2 Associations of complex autoinflammatory diseases with type 2 clinical and immunological phenotypes.
Systemic juvenile idiopathic arthritis (sJIA) is another complex autoinflammatory disease with genetic and phenotypic links to both autoinflammation and autoimmune inflammation (40). Children with allergic disease were found to be at a higher risk of developing JIA in a Taiwanese cohort, although sJIA was not differentiated from other forms (214). Atopy may also be a risk factor for increased disease severity in sJIA, although this has only been investigated in one small prospective study (215). Adult-onset Still’s disease (AOSD) is an adult-onset clinical syndrome that phenotypically resembles sJIA (216). Cases of AOSD have been reported in association with elevated serum IgE, IL-4, and clinical atopy, but the prevalence of these features has not yet been systematically investigated (216, 217).
Conclusions
Although autoinflammation and type 2 immunity have traditionally thought to counter-regulate each other, a growing body of literature demonstrates that the relationship between type 1 and type 2 immune responses is more nuanced than this canonical view would suggest. Some autoinflammatory cytokines, like IL-1β and TNF-α; enhance the differentiation and function of type 2 effector cells and exacerbate allergic pathology. Others, like type 1 IFN, largely repress type 2 inflammation but can promote type 2 cytokine production in certain contexts. And some autoinflammatory signaling molecules like NLRP3 may constrain type 2 responses in the context of parasitic infection, while inducing type 2 immunity in the setting of allergic inflammation. These observations suggest that the role of autoinflammation in type 2 immunity may rely on a broad array of genetic and environmental factors involved in driving the immune response.
Similarly, the role of type 2 immunity in the pathogenesis of autoinflammation is complex and context-dependent. While Th2- and ILC2-derived type 2 cytokines like IL-4 and IL-13 generally repress type 1 inflammation, they can promote neutrophil activation in certain context. Moreover, alarmins like IL-33 and TSLP clearly induce autoinflammatory effectors like neutrophils and monocytes, causing local and systemic inflammation. Mast cells, which are generally considered type 2 effectors, have a clear role in NLRP3-associated autoinflammatory diseases and may play a role in diseases linked to other genes like MEFV. Future investigation will be required to determine the roles played by type 2 cytokines and effectors in modulating pathology in subjects with monogenic and complex autoinflammatory diseases.
Clinical epidemiology studies in patients with autoinflammatory diseases paint a similarly nuanced picture. Given the role of NLRP3 in promoting allergic pathology, for example, it is not surprising that the phenotypic spectrum of CAPS comprises eosinophilia and clinical allergy in addition to systemic autoinflammation. In other syndromes, autoinflammation appears to have a negative effect on type 2 immunity – most notably for FMF – although the mechanisms are not well-defined. Finally, in some cases, it appears that autoinflammatory pathology can mimic allergic disease, causing a phenotype that is indistinguishable from clinical allergy but that is not mediated by type 2 effectors. These observations have clinical implications for subjects with autoinflammatory diseases, where type 2 directed therapies have been reported effective in some cases. They may also have repercussions for subjects with clinical allergy-associated diagnoses like asthma, where non-allergic endotypes are unlikely to respond to type 2 directed therapies. The ability of autoinflammatory cytokines and mediators to both potentiate and clinically phenocopy type 2 pathology suggests that some of these patients might benefit from autoinflammation-directed treatments. In the future, dissecting the interactions between these two not-so-separate arms of the immune response should help to refine our understanding of – and improve treatments for – monogenic and complex immune dysregulatory disorders.
Author Contributions
MS, AS, and DS organized and composed the manuscript. MS created tables. DS created figures and provided supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH/NIAID intramural research program (grant no. 1ZIAAI001251).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dinse GE, Parks CG, Weinberg CR, Co CA, Wilkerson J, Zeldin DC, et al. Increasing Prevalence of Antinuclear Antibodies in the United States. Arthritis Rheumatol (2020) 72:1026–35. doi: 10.1002/art.41214
3. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The Monogenic Autoinflammatory Diseases Define New Pathways in Human Innate Immunity and Inflammation. Nat Immunol (2017) 18:832–42. doi: 10.1038/ni.3777
4. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 Immunity in Tissue Repair and Fibrosis. Nat Rev Immunol (2018) 18:62–76. doi: 10.1038/nri.2017.90
5. Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 Cells in Health and Disease. Annu Rev Immunol (2017) 35:53–84. doi: 10.1146/annurev-immunol-051116-052350
6. Gupta S, Kaplan MJ. Bite of the Wolf: Innate Immune Responses Propagate Autoimmunity in Lupus. J Clin Invest (2021) 131:e144918. doi: 10.1172/JCI144918
7. Weyand CM, Goronzy JJ. The Immunology of Rheumatoid Arthritis. Nat Immunol (2021) 22:10–8. doi: 10.1038/s41590-020-00816-x
8. Wynn TA. Type 2 Cytokines: Mechanisms and Therapeutic Strategies. Nat Rev Immunol (2015) 15:271–82. doi: 10.1038/nri3831
9. Annunziato F, Romagnani C, Romagnani S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J Allergy Clin Immunol (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
10. Rodriguez-Rodriguez N, Gogoi M, McKenzie ANJ. Group 2 Innate Lymphoid Cells: Team Players in Regulating Asthma. Annu Rev Immunol (2021) 39:167–98. doi: 10.1146/annurev-immunol-110119-091711
11. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 Forms an IL-1β-Processing Inflammasome With Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity (2004) 20:319–25. doi: 10.1016/S1074-7613(04)00046-9
12. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a New Gene Encoding a Putative Pyrin-Like Protein Causes Familial Cold Autoinflammatory Syndrome and Muckle-Wells Syndrome. Nat Genet (2001) 29:301–5. doi: 10.1038/ng756
13. Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the Gene Encoding Mevalonate Kinase Cause Hyper-IgD and Periodic Fever Syndrome. International Hyper-IgD Study Group. Nat Genet (1999) 22:178–81. doi: 10.1038/9696
14. Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, Encoding Mevalonate Kinase, Cause Hyperimmunoglobulinaemia D and Periodic Fever Syndrome. Nat Genet (1999) 22:175–7. doi: 10.1038/9691
15. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
16. Pras E, Aksentijevich I, Gruberg L, Balow JE Jr, Prosen L, Dean M, et al. Mapping of a Gene Causing Familial Mediterranean Fever to the Short Arm of Chromosome 16. N Engl J Med (1992) 326:1509–13. doi: 10.1056/NEJM199206043262301
17. Bazzoni F, Beutler B. The Tumor Necrosis Factor Ligand and Receptor Families. N Engl J Med (1996) 334:1717–25. doi: 10.1056/NEJM199606273342607
18. Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF Receptor-Associated Periodic Syndrome (TRAPS): Current Perspectives. Int J Mol Sci (2020) 21:3263. doi: 10.3390/ijms21093263
19. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-Of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48:67–73. doi: 10.1038/ng.3459
20. Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell (2016) 166:1215–1230 e20. doi: 10.1016/j.cell.2016.07.019
21. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-Onset Sarcoidosis and CARD15 Mutations With Constitutive Nuclear factor-kappaB Activation: Common Genetic Etiology With Blau Syndrome. Blood (2005) 105:1195–7. doi: 10.1182/blood-2004-07-2972
22. Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, et al. PSORS2 Is Due to Mutations in CARD14. Am J Hum Genet (2012) 90:784–95. doi: 10.1016/j.ajhg.2012.03.012
23. Aeschlimann FA, Batu ED, Canna SW, Go E, Gul A, Hoffmann P, et al. A20 Haploinsufficiency (HA20): Clinical Phenotypes and Disease Course of Patients With a Newly Recognised NF-kB-Mediated Autoinflammatory Disease. Ann Rheum Dis (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
24. Gonzales-van Horn SR, Farrar JD. Interferon at the Crossroads of Allergy and Viral Infections. J Leukoc Biol (2015) 98:185–94. doi: 10.1189/jlb.3RU0315-099R
25. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive Loss-of-Function Proteasome Subunit Mutations in CANDLE/PRAAS Patients Promote Type I IFN Production. J Clin Invest (2015) 125:4196–211. doi: 10.1172/JCI81260
26. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
27. Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe Type I Interferonopathy and Unrestrained Interferon Signaling Due to a Homozygous Germline Mutation in STAT2. Sci Immunol (2019) 4:eaav7501. doi: 10.1126/sciimmunol.aav7501
28. Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 Gain-of-Function Mutation by Loss of USP18 Activity in a Patient With Type I Interferonopathy. J Exp Med (2020) 217:e20192319. doi: 10.1084/jem.20192319
29. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset Stroke and Vasculopathy Associated With Mutations in ADA2. N Engl J Med (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
30. Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, et al. Familial Haemolytic Uraemic Syndrome and an MCP Mutation. Lancet (2003) 362:1542–7. doi: 10.1016/S0140-6736(03)14742-3
31. Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, Vera M, Lopez-Trascasa M, Rodriguez de Cordoba S, et al. Clustering of Missense Mutations in the C-Terminal Region of Factor H in Atypical Hemolytic Uremic Syndrome. Am J Hum Genet (2001) 68:478–84. doi: 10.1086/318201
32. Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, et al. Factor H Mutations in Hemolytic Uremic Syndrome Cluster in Exons 18-20, a Domain Important for Host Cell Recognition. Am J Hum Genet (2001) 68:485–90. doi: 10.1086/318203
33. Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A Novel Disorder Involving Dyshematopoiesis, Inflammation, and HLH Due to Aberrant CDC42 Function. J Exp Med (2019) 216:2778–99. doi: 10.1084/jem.20190147
34. Standing AS, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory Periodic Fever, Immunodeficiency, and Thrombocytopenia (PFIT) Caused by Mutation in Actin-Regulatory Gene WDR1. J Exp Med (2017) 214:59–71. doi: 10.1084/jem.20161228
35. Zinngrebe J, Rieser E, Taraborrelli L, Peltzer N, Hartwig T, Ren H, et al. –LUBAC Deficiency Perturbs TLR3 Signaling to Cause Immunodeficiency and Autoinflammation. J Exp Med (2016) 213:2671–89. doi: 10.1084/jem.20160041
36. Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, et al. Mutations That Prevent Caspase Cleavage of RIPK1 Cause Autoinflammatory Disease. Nature (2020) 577:103–8. doi: 10.1038/s41586-019-1828-5
37. Cuchet-Lourenco D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 Mutations in Humans Cause Severe Immunodeficiency, Arthritis, and Intestinal Inflammation. Science (2018) 361:810–3. doi: 10.1126/science.aar2641
38. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
39. Manthiram K, Preite S, Dedeoglu F, Demir S, Ozen S, Edwards KM, et al. Common Genetic Susceptibility Loci Link PFAPA Syndrome, Behcet’s Disease, and Recurrent Aphthous Stomatitis. Proc Natl Acad Sci USA (2020) 117:14405–11. doi: 10.1073/pnas.2002051117
40. Ombrello MJ, Arthur VL, Remmers EF, Hinks A, Tachmazidou I, Grom AA, et al. Genetic Architecture Distinguishes Systemic Juvenile Idiopathic Arthritis From Other Forms of Juvenile Idiopathic Arthritis: Clinical and Therapeutic Implications. Ann Rheum Dis (2017) 76:906–13. doi: 10.1136/annrheumdis-2016-210324
41. Shaw MK, Lorens JB, Dhawan A, DalCanto R, Tse HY, Tran AB, et al. Local Delivery of Interleukin 4 by Retrovirus-Transduced T Lymphocytes Ameliorates Experimental Autoimmune Encephalomyelitis. J Exp Med (1997) 185:1711–4. doi: 10.1084/jem.185.9.1711
42. Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous Gammaglobulin Suppresses Inflammation Through a Novel T(H)2 Pathway. Nature (2011) 475:110–3. doi: 10.1038/nature10134
43. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 Cells That Express the Transcription Factor PU.1 Drive T Cell-Mediated Colitis via IL-9 Receptor Signaling in Intestinal Epithelial Cells. Nat Immunol (2014) 15:676–86. doi: 10.1038/ni.2920
44. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 Cells Are Skin-Tropic and Have Autocrine and Paracrine Proinflammatory Capacity. Sci Transl Med (2014) 6:219ra8. doi: 10.1126/scitranslmed.3007828
45. Spadaro A, Rinaldi T, Riccieri V, Valesini G, Taccari E. Interleukin 13 in Synovial Fluid and Serum of Patients With Psoriatic Arthritis. Ann Rheum Dis (2002) 61:174–6. doi: 10.1136/ard.61.2.174
46. Karmele EP, Pasricha TS, Ramalingam TR, Thompson RW, Gieseck RL 3rd, Knilans KJ, et al. Anti-IL-13Ralpha2 Therapy Promotes Recovery in a Murine Model of Inflammatory Bowel Disease. Mucosal Immunol (2019) 12:1174–86. doi: 10.1038/s41385-019-0189-6
47. Charles N, Hardwick D, Daugas E, Illei GG, Rivera J. Basophils and the T Helper 2 Environment can Promote the Development of Lupus Nephritis. Nat Med (2010) 16:701–7. doi: 10.1038/nm.2159
48. Dema B, Charles N, Pellefigues C, Ricks TK, Suzuki R, Jiang C, et al. Immunoglobulin E Plays an Immunoregulatory Role in Lupus. J Exp Med (2014) 211:2159–68. doi: 10.1084/jem.20140066
49. Peters SP. Asthma Phenotypes: Nonallergic (Intrinsic) Asthma. J Allergy Clin Immunol Pract (2014) 2:650–2. doi: 10.1016/j.jaip.2014.09.006
50. Dykewicz MS, Wallace DV, Amrol DJ, Baroody FM, Bernstein JA, Craig TJ, et al. Rhinitis 2020: A Practice Parameter Update. J Allergy Clin Immunol (2020) 146:721–67. doi: 10.1016/j.jaci.2020.07.007
51. Khalaf K, Paoletti G, Puggioni F, Racca F, De Luca F, Giorgis V, et al. Asthma From Immune Pathogenesis to Precision Medicine. Semin Immunol (2019) 46:101294. doi: 10.1016/j.smim.2019.101294
52. Finkelman FD, Madden KB, Cheever AW, Katona IM, Morris SC, Gately MK, et al. Effects of Interleukin 12 on Immune Responses and Host Protection in Mice Infected With Intestinal Nematode Parasites. J Exp Med (1994) 179:1563–72. doi: 10.1084/jem.179.5.1563
53. Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental Commitment to the Th2 Lineage by Extinction of IL-12 Signaling. Immunity (1995) 2:665–75. doi: 10.1016/1074-7613(95)90011-X
54. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 Regulates Both Th1 and Th2 Responses. Annu Rev Immunol (2001) 19:423–74. doi: 10.1146/annurev.immunol.19.1.423
55. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 Acts Directly on CD4 T Cells to Enhance Their Antigen-Driven Expansion and Differentiation. Proc Natl Acad Sci USA (2009) 106:7119–24. doi: 10.1073/pnas.0902745106
56. Artis D, Humphreys NE, Bancroft AJ, Rothwell NJ, Potten CS, Grencis RK. Tumor Necrosis Factor Alpha Is a Critical Component of Interleukin 13-Mediated Protective T Helper Cell Type 2 Responses During Helminth Infection. J Exp Med (1999) 190:953–62. doi: 10.1084/jem.190.7.953
57. Terada M, Tsutsui H, Imai Y, Yasuda K, Mizutani H, Yamanishi K, et al. Contribution of IL-18 to Atopic-Dermatitis-Like Skin Inflammation Induced by Staphylococcus Aureus Product in Mice. Proc Natl Acad Sci USA (2006) 103:8816–21. doi: 10.1073/pnas.0602900103
58. Konishi H, Tsutsui H, Murakami T, Yumikura-Futatsugi S, Yamanaka K, Tanaka M, et al. IL-18 Contributes to the Spontaneous Development of Atopic Dermatitis-Like Inflammatory Skin Lesion Independently of IgE/stat6 Under Specific Pathogen-Free Conditions. Proc Natl Acad Sci USA (2002) 99:11340–5. doi: 10.1073/pnas.152337799
59. Whitehead GS, Thomas SY, Shalaby KH, Nakano K, Moran TP, Ward JM, et al. TNF Is Required for TLR Ligand-Mediated But Not Protease-Mediated Allergic Airway Inflammation. J Clin Invest (2017) 127:3313–26. doi: 10.1172/JCI90890
60. Piao H, Choi YH, Li H, Wang C, Xian Z, Ogasawara M, et al. Recombinant Pyrin Domain Protein Attenuates Allergic Inflammation by Suppressing NF-KappaB Pathway in Asthmatic Mice. Scand J Immunol (2019) 89:e12720. doi: 10.1111/sji.12720
61. Bruchard M, Rebe C, Derangere V, Togbe D, Ryffel B, Boidot R, et al. The Receptor NLRP3 Is a Transcriptional Regulator of TH2 Differentiation. Nat Immunol (2015) 16:859–70. doi: 10.1038/ni.3202
62. Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, et al. An NLRP3 Inflammasome-Triggered Th2-Biased Adaptive Immune Response Promotes Leishmaniasis. J Clin Invest (2015) 125:1329–38. doi: 10.1172/JCI79526
63. Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, et al. NLRP3 Inflammasome Activation by Mitochondrial ROS in Bronchial Epithelial Cells Is Required for Allergic Inflammation. Cell Death Dis (2014) 5:e1498. doi: 10.1038/cddis.2014.460
64. Wu HM, Zhao CC, Xie QM, Xu J, Fei GH. TLR2-Melatonin Feedback Loop Regulates the Activation of NLRP3 Inflammasome in Murine Allergic Airway Inflammation. Front Immunol (2020) 11:172. doi: 10.3389/fimmu.2020.00172
65. Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, et al. Activation of the IL-1beta-Processing Inflammasome Is Involved in Contact Hypersensitivity. J Invest Dermatol (2007) 127:1956–63. doi: 10.1038/sj.jid.5700819
66. Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, et al. Helicobacter Urease-Induced Activation of the TLR2/NLRP3/IL-18 Axis Protects Against Asthma. J Clin Invest (2015) 125:3297–302. doi: 10.1172/JCI79337
67. Chenery AL, Alhallaf R, Agha Z, Ajendra J, Parkinson JE, Cooper MM, et al. Inflammasome-Independent Role for NLRP3 in Controlling Innate Antihelminth Immunity and Tissue Repair in the Lung. J Immunol (2019) 203:2724–34. doi: 10.4049/jimmunol.1900640
68. Alhallaf R, Agha Z, Miller CM, Robertson AAB, Sotillo J, Croese J, et al. The NLRP3 Inflammasome Suppresses Protective Immunity to Gastrointestinal Helminth Infection. Cell Rep (2018) 23:1085–98. doi: 10.1016/j.celrep.2018.03.097
69. Zaiss MM, Maslowski KM, Mosconi I, Guenat N, Marsland BJ, Harris NL. IL-1beta Suppresses Innate IL-25 and IL-33 Production and Maintains Helminth Chronicity. PloS Pathog (2013) 9:e1003531. doi: 10.1371/journal.ppat.1003531
70. Karjalainen J, Hulkkonen J, Pessi T, Huhtala H, Nieminen MM, Aromaa A, et al. The IL1A Genotype Associates With Atopy in Nonasthmatic Adults. J Allergy Clin Immunol (2002) 110:429–34. doi: 10.1067/mai.2002.126784
71. Karjalainen J, Nieminen MM, Aromaa A, Klaukka T, Hurme M. The IL-1beta Genotype Carries Asthma Susceptibility Only in Men. J Allergy Clin Immunol (2002) 109:514–6. doi: 10.1067/mai.2002.121948
72. Rossios C, Pavlidis S, Hoda U, Kuo CH, Wiegman C, Russell K, et al. Unbiased Biomarkers for the Prediction of Respiratory Diseases Outcomes Consortia Project, Sputum Transcriptomics Reveal Upregulation of IL-1 Receptor Family Members in Patients With Severe Asthma. J Allergy Clin Immunol (2018) 141:560–70. doi: 10.1016/j.jaci.2017.02.045
73. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha Controls Allergic Sensitization to Inhaled House Dust Mite via the Epithelial Release of GM-CSF and IL-33. J Exp Med (2012) 209:1505–17. doi: 10.1084/jem.20112691
74. Qian X, Aboushousha R, van de Wetering C, Chia SB, Amiel E, Schneider RW, et al. IL-1/Inhibitory kappaB Kinase Epsilon-Induced Glycolysis Augment Epithelial Effector Function and Promote Allergic Airways Disease. J Allergy Clin Immunol (2018) 142:435–450 e10. doi: 10.1016/j.jaci.2017.08.043
75. Agoro R, Piotet-Morin J, Palomo J, Michaudel C, Vigne S, Maillet I, et al. IL-1r1-MyD88 Axis Elicits Papain-Induced Lung Inflammation. Eur J Immunol (2016) 46:2531–41. doi: 10.1002/eji.201646366
76. Esnault S, Kelly EA, Nettenstrom LM, Cook EB, Seroogy CM, Jarjour NN. Human Eosinophils Release IL-1ss and Increase Expression of IL-17A in Activated CD4+ T Lymphocytes. Clin Exp Allergy (2012) 42:1756–64. doi: 10.1111/j.1365-2222.2012.04060.x
77. Nakamura Y, Kambe N, Saito M, Nishikomori R, Kim YG, Murakami M, et al. Mast Cells Mediate Neutrophil Recruitment and Vascular Leakage Through the NLRP3 Inflammasome in Histamine-Independent Urticaria. J Exp Med (2009) 206:1037–46. doi: 10.1084/jem.20082179
78. Nutan FN, Kanwar AJ, Parsad D. The Effect of Topically Applied Corticosteroids on Interleukin 1beta Levels in Patients With Atopic Dermatitis. J Eur Acad Dermatol Venereol (2012) 26:1020–2. doi: 10.1111/j.1468-3083.2011.04133.x
79. Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Nunez G. Critical Role for Mast Cells in Interleukin-1beta-Driven Skin Inflammation Associated With an Activating Mutation in the Nlrp3 Protein. Immunity (2012) 37:85–95. doi: 10.1016/j.immuni.2012.04.013
80. Caucheteux SM, Hu-Li J, Guo L, Bhattacharyya N, Crank M, Collins MT, et al. IL-1beta Enhances Inflammatory TH2 Differentiation. J Allergy Clin Immunol (2016) 138:898–901.e4. doi: 10.1016/j.jaci.2016.02.033
81. Helmby H, Grencis RK. Interleukin 1 Plays a Major Role in the Development of Th2-Mediated Immunity. Eur J Immunol (2004) 34:3674–81. doi: 10.1002/eji.200425452
82. Xue G, Jin G, Fang J, Lu Y. IL-4 Together With IL-1beta Induces Antitumor Th9 Cell Differentiation in the Absence of TGF-Beta Signaling. Nat Commun (2019) 10:1376. doi: 10.1038/s41467-019-09401-9
83. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17:636–45. doi: 10.1038/ni.3444
84. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 Is a Critical Regulator of Group 2 Innate Lymphoid Cell Function and Plasticity. Nat Immunol (2016) 17:646–55. doi: 10.1038/ni.3447
85. Haakfrendscho M, Dinarello C, Kaplan A. Recombinant Human Interleukin-1 Beta Causes Histamine Release From Human Basophils. J Allergy Clin Immunol (1988) 82:218–23. doi: 10.1016/0091-6749(88)91002-0
86. Conti P, Caraffa A, Tete G, Gallenga CE, Ross R, Kritas SK, et al. Mast Cells Activated by SARS-CoV-2 Release Histamine Which Increases IL-1 Levels Causing Cytokine Storm and Inflammatory Reaction in COVID-19. J Biol Regul Homeost Agents (2020) 34:1629–32. doi: 10.23812/20-2EDIT
87. Gounni AS, Nutku E, Koussih L, Aris F, Louahed J, Levitt RC, et al. IL-9 Expression by Human Eosinophils: Regulation by IL-1beta and TNF-Alpha. J Allergy Clin Immunol (2000) 106:460–6. doi: 10.1067/mai.2000.109172
88. Hultner L, Kolsch S, Stassen M, Kaspers U, Kremer JP, Mailhammer R, et al. In Activated Mast Cells, IL-1 Up-Regulates the Production of Several Th2-Related Cytokines Including IL-9. J Immunol (2000) 164:5556–63. doi: 10.4049/jimmunol.164.11.5556
89. Hernandez ML, Mills K, Almond M, Todoric K, Aleman MM, Zhang H, et al. IL-1 Receptor Antagonist Reduces Endotoxin-Induced Airway Inflammation in Healthy Volunteers. J Allergy Clin Immunol (2015) 135:379–85. doi: 10.1016/j.jaci.2014.07.039
90. Pascoe S, Kanniess F, Bonner J, Lloyd P, Lowe P, Beier J, et al. A Monoclonal Antibody to IL-1B Attenuates the Late Asthmatic Response to Antigen Challenge in Patients With Mild Asthma. Eur Respir J (2006) 28:752.
91. Simpson E. Abstract 11191. In: AAD Annual Meeting. Washington DC, USA (2019). Available at: https://www.aad.org/member/meetings/archive/handouts.
92. Hoshino T, Wiltrout RH, Young HA. IL-18 Is a Potent Coinducer of IL-13 in NK and T Cells: A New Potential Role for IL-18 in Modulating the Immune Response. J Immunol (1999) 162:5070–7.
93. Kroeger KM, Sullivan BM, Locksley RM. IL-18 and IL-33 Elicit Th2 Cytokines From Basophils via a MyD88- and P38alpha-Dependent Pathway. J Leukoc Biol (2009) 86:769–78. doi: 10.1189/jlb.0708452
94. Kandikattu HK, Upparahalli Venkateshaiah S, Mishra A. Synergy of Interleukin (IL)-5 and IL-18 in Eosinophil Mediated Pathogenesis of Allergic Diseases. Cytokine Growth Factor Rev (2019) 47:83–98. doi: 10.1016/j.cytogfr.2019.05.003
95. Yoshimoto T, Tsutsui H, Tominaga K, Hoshino K, Okamura H, Akira S, et al. IL-18, Although Antiallergic When Administered With IL-12, Stimulates IL-4 and Histamine Release by Basophils. Proc Natl Acad Sci USA (1999) 96:13962–6. doi: 10.1073/pnas.96.24.13962
96. Helmby H, Takeda K, Akira S, Grencis RK. Interleukin (IL)-18 Promotes the Development of Chronic Gastrointestinal Helminth Infection by Downregulating IL-13. J Exp Med (2001) 194:355–64. doi: 10.1084/jem.194.3.355
97. Kodama T, Matsuyama T, Kuribayashi K, Nishioka Y, Sugita M, Akira S, et al. IL-18 Deficiency Selectively Enhances Allergen-Induced Eosinophilia in Mice. J Allergy Clin Immunol (2000) 105:45–53. doi: 10.1016/S0091-6749(00)90176-3
98. Xu D, Trajkovic V, Hunter D, Leung BP, Schulz K, Gracie JA, et al. IL-18 Induces the Differentiation of Th1 or Th2 Cells Depending Upon Cytokine Milieu and Genetic Background. Eur J Immunol (2000) 30:3147–56. doi: 10.1002/1521-4141(200011)30:11<3147::AID-IMMU3147>3.0.CO;2-J
99. Yoshimoto T, Okamura H, Tagawa YI, Iwakura Y, Nakanishi K. Interleukin 18 Together With Interleukin 12 Inhibits IgE Production by Induction of Interferon-Gamma Production From Activated B Cells. Proc Natl Acad Sci USA (1997) 94:3948–53. doi: 10.1073/pnas.94.8.3948
100. Yoshimoto T, Mizutani H, Tsutsui H, Noben-Trauth N, Yamanaka K, Tanaka M, et al. IL-18 Induction of IgE: Dependence on CD4+ T Cells, IL-4 and STAT6. Nat Immunol (2000) 1:132–7. doi: 10.1038/77811
101. Croft M, Siegel RM, Beyond TNF. TNF Superfamily Cytokines as Targets for the Treatment of Rheumatic Diseases. Nat Rev Rheumatol (2017) 13:217–33. doi: 10.1038/nrrheum.2017.22
102. Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment Strategies for Deficiency of Adenosine Deaminase 2. N Engl J Med (2019) 380:1582–4. doi: 10.1056/NEJMc1801927
103. Meylan F, Siegel RM. TNF Superfamily Cytokines in the Promotion of Th9 Differentiation and Immunopathology. Semin Immunopathol (2017) 39:21–8. doi: 10.1007/s00281-016-0612-y
104. Jiang Y, Chen J, Bi E, Zhao Y, Qin T, Wang Y, et al. TNF-Alpha Enhances Th9 Cell Differentiation and Antitumor Immunity via TNFR2-Dependent Pathways. J Immunother Cancer (2019) 7:28. doi: 10.1186/s40425-018-0494-8
105. Aggarwal BB. Signalling Pathways of the TNF Superfamily: A Double-Edged Sword. Nat Rev Immunol (2003) 3:745–56. doi: 10.1038/nri1184
106. Liu LY, Bates ME, Jarjour NN, Busse WW, Bertics PJ, Kelly EA. Generation of Th1 and Th2 Chemokines by Human Eosinophils: Evidence for a Critical Role of TNF-Alpha. J Immunol (2007) 179:4840–8. doi: 10.4049/jimmunol.179.7.4840
107. Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a Role of Tumor Necrosis Factor Alpha in Refractory Asthma. N Engl J Med (2006) 354:697–708. doi: 10.1056/NEJMoa050580
108. Horiba M, Qutna N, Gendapodi P, Agrawal S, Sapkota K, Abel P, et al. Effect of IL-1beta and TNF-Alpha vs IL-13 on Bronchial Hyperresponsiveness, Beta2-Adrenergic Responses and Cellularity of Bronchial Alveolar Lavage Fluid. Auton Autacoid Pharmacol (2011) 31:37–49. doi: 10.1111/j.1474-8673.2011.00465.x
109. Holgate ST, Noonan M, Chanez P, Busse W, Dupont L, Pavord I, et al. Efficacy and Safety of Etanercept in Moderate-to-Severe Asthma: A Randomised, Controlled Trial. Eur Respir J (2011) 37:1352–9. doi: 10.1183/09031936.00063510
110. Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, et al. A Randomized, Double-Blind, Placebo-Controlled Study of Tumor Necrosis Factor-Alpha Blockade in Severe Persistent Asthma. Am J Respir Crit Care Med (2009) 179:549–58. doi: 10.1164/rccm.200809-1512OC
111. Jacobi A, Antoni C, Manger B, Schuler G, Hertl M. Infliximab in the Treatment of Moderate to Severe Atopic Dermatitis. J Am Acad Dermatol (2005) 52:522–6. doi: 10.1016/j.jaad.2004.11.022
112. Buka RL, Resh B, Roberts B, Cunningham BB, Friedlander S. Etanercept Is Minimally Effective in 2 Children With Atopic Dermatitis. J Am Acad Dermatol (2005) 53:358–9. doi: 10.1016/j.jaad.2005.03.013
113. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB Signaling in Inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
114. Schwartz C, Willebrand R, Huber S, Rupec RA, Wu D, Locksley R, et al. Eosinophil-Specific Deletion of IkappaBalpha in Mice Reveals a Critical Role of NF-kappaB-Induced Bcl-xL for Inhibition of Apoptosis. Blood (2015) 125:3896–904. doi: 10.1182/blood-2014-10-607788
115. Marquardt DL, Walker LL. Dependence of Mast Cell IgE-Mediated Cytokine Production on Nuclear factor-kappaB Activity. J Allergy Clin Immunol (2000) 105:500–5. doi: 10.1067/mai.2000.104942
116. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
117. Peled A, Sarig O, Sun G, Samuelov L, Ma CA, Zhang Y, et al. Loss-Of-Function Mutations in Caspase Recruitment Domain-Containing Protein 14 (CARD14) Are Associated With a Severe Variant of Atopic Dermatitis. J Allergy Clin Immunol (2019) 143:173–81.e10. doi: 10.1016/j.jaci.2018.09.002
118. Daley D, Lemire M, Akhabir L, Chan-Yeung M, He JQ, McDonald T, et al. Analyses of Associations With Asthma in Four Asthma Population Samples From Canada and Australia. Hum Genet (2009) 125:445–59. doi: 10.1007/s00439-009-0643-8
119. Kabesch M, Peters W, Carr D, Leupold W, Weiland SK, von Mutius E. Association Between Polymorphisms in Caspase Recruitment Domain Containing Protein 15 and Allergy in Two German Populations. J Allergy Clin Immunol (2003) 111:813–7. doi: 10.1067/mai.2003.1336
120. Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm Dust and Endotoxin Protect Against Allergy Through A20 Induction in Lung Epithelial Cells. Science (2015) 349:1106–10. doi: 10.1126/science.aac6623
121. Heger K, Fierens K, Vahl JC, Aszodi A, Peschke K, Schenten D, et al. A20-Deficient Mast Cells Exacerbate Inflammatory Responses In Vivo. PloS Biol (2014) 12:e1001762. doi: 10.1371/journal.pbio.1001762
122. Chen D, Ma L, Hu T, Liu J, Chen B, Yang P, et al. A20 Restores Impaired Intestinal Permeability and Inhibits Th2 Response in Mice With Colitis. Dig Dis Sci (2020) 65:1340–7. doi: 10.1007/s10620-019-05860-8
123. Vroman H, Bergen IM, van Hulst JAC, van Nimwegen M, van Uden D, Schuijs MJ, et al. TNF-Alpha-Induced Protein 3 Levels in Lung Dendritic Cells Instruct TH2 or TH17 Cell Differentiation in Eosinophilic or Neutrophilic Asthma. J Allergy Clin Immunol (2018) 141:1620–33.e12. doi: 10.1016/j.jaci.2017.08.012
124. Hrusch CL, Stein MM, Gozdz J, Holbreich M, von Mutius E, Vercelli D, et al. T-Cell Phenotypes Are Associated With Serum IgE Levels in Amish and Hutterite Children. J Allergy Clin Immunol (2019) 144:1391–401.e10. doi: 10.1016/j.jaci.2019.07.034
125. Krusche J, Twardziok M, Rehbach K, Bock A, Tsang MS, Schroder PC, et al. TNF-Alpha-Induced Protein 3 Is a Key Player in Childhood Asthma Development and Environment-Mediated Protection. J Allergy Clin Immunol (2019) 144:1684–96.e12. doi: 10.1016/j.jaci.2019.07.029
126. Devos M, Mogilenko DA, Fleury S, Gilbert B, Becquart C, Quemener S, et al. Keratinocyte Expression of A20/TNFAIP3 Controls Skin Inflammation Associated With Atopic Dermatitis and Psoriasis. J Invest Dermatol (2019) 139:135–45. doi: 10.1016/j.jid.2018.06.191
127. Li MY, Zhu M, Linghu EQ, Feng F, Zhu B, Wu C, et al. Interleukin-13 Suppresses Interleukin-10 via Inhibiting A20 in Peripheral B Cells of Patients With Food Allergy. Oncotarget (2016) 7:79914–24. doi: 10.18632/oncotarget.13107
128. Park Y, Jin HS, Lopez J, Lee J, Liao L, Elly C, et al. SHARPIN Controls Regulatory T Cells by Negatively Modulating the T Cell Antigen Receptor Complex. Nat Immunol (2016) 17:286–96. doi: 10.1038/ni.3352
129. Chien SJ, Silva KA, Kennedy VE, HogenEsch H, Sundberg JP. The Pathogenesis of Chronic Eosinophilic Esophagitis in SHARPIN-Deficient Mice. Exp Mol Pathol (2015) 99:460–7. doi: 10.1016/j.yexmp.2015.08.012
130. Liang Y, Seymour RE, Sundberg JP. Inhibition of NF-kappaB Signaling Retards Eosinophilic Dermatitis in SHARPIN-Deficient Mice. J Invest Dermatol (2011) 131:141–9. doi: 10.1038/jid.2010.259
131. Sundberg JP, Pratt CH, Goodwin LP, Silva KA, Kennedy VE, Potter CS, et al. Keratinocyte-Specific Deletion of SHARPIN Induces Atopic Dermatitis-Like Inflammation in Mice. PloS One (2020) 15:e0235295. doi: 10.1371/journal.pone.0235295
132. Huber JP, Ramos HJ, Gill MA, Farrar JD. Cutting Edge: Type I IFN Reverses Human Th2 Commitment and Stability by Suppressing GATA3. J Immunol (2010) 185:813–7. doi: 10.4049/jimmunol.1000469
133. Schandene L, Del Prete GF, Cogan E, Stordeur P, Crusiaux A, Kennes B, et al. Recombinant Interferon-Alpha Selectively Inhibits the Production of Interleukin-5 by Human CD4+ T Cells. J Clin Invest (1996) 97:309–15. doi: 10.1172/JCI118417
134. Shibuya H, Hirohata S. Differential Effects of IFN-Alpha on the Expression of Various TH2 Cytokines in Human CD4+ T Cells. J Allergy Clin Immunol (2005) 116:205–12. doi: 10.1016/j.jaci.2005.03.016
135. Pritchard AL, Carroll ML, Burel JG, White OJ, Phipps S, Upham JW. Innate IFNs and Plasmacytoid Dendritic Cells Constrain Th2 Cytokine Responses to Rhinovirus: A Regulatory Mechanism With Relevance to Asthma. J Immunol (2012) 188:5898–905. doi: 10.4049/jimmunol.1103507
136. Seeliger B, Forster M, Happe J, Forberg T, Moeser A, Neumann T, et al. Interferon-Alpha for Induction and Maintenance of Remission in Eosinophilic Granulomatosis With Polyangiitis: A Single-Center Retrospective Observational Cohort Study. J Rheumatol (2017) 44:806–14. doi: 10.3899/jrheum.160907
137. Butterfield JH, Gleich GJ. Interferon-Alpha Treatment of Six Patients With the Idiopathic Hypereosinophilic Syndrome. Ann Intern Med (1994) 121:648–53. doi: 10.7326/0003-4819-121-9-199411010-00003
138. Rich HE, Antos D, Melton NR, Alcorn JF, Manni ML, Insights Into Type I. And III Interferons in Asthma and Exacerbations. Front Immunol (2020) 11:574027. doi: 10.3389/fimmu.2020.574027
139. Jang YJ, Lim JY, Kim S, Lee Y, Kweon MN, Kim JH. Enhanced Interferon-Beta Response Contributes to Eosinophilic Chronic Rhinosinusitis. Front Immunol (2018) 9:2330. doi: 10.3389/fimmu.2018.02330
140. Loureiro J, Ploegh HL. Antigen Presentation and the Ubiquitin-Proteasome System in Host-Pathogen Interactions. Adv Immunol (2006) 92:225–305. doi: 10.1016/S0065-2776(06)92006-9
141. Volkov A, Hagner S, Loser S, Alnahas S, Raifer H, Hellhund A, et al. Beta5i Subunit Deficiency of the Immunoproteasome Leads to Reduced Th2 Response in OVA Induced Acute Asthma. PloS One (2013) 8:e60565. doi: 10.1371/journal.pone.0060565
142. Chen H, Sun H, You F, Sun W, Zhou X, Chen L, et al. Activation of STAT6 by STING Is Critical for Antiviral Innate Immunity. Cell (2011) 147:436–46. doi: 10.1016/j.cell.2011.09.022
143. Nunokawa H, Murakami Y, Ishii T, Narita T, Ishii H, Takizawa H, et al. Crucial Role of Stimulator of Interferon Genes-Dependent Signaling in House Dust Mite Extract-Induced IgE Production. Sci Rep (2021) 11:13157. doi: 10.1038/s41598-021-92561-w
144. Wang H, Hu DQ, Xiao Q, Liu YB, Song J, Liang Y, et al. Defective STING Expression Potentiates IL-13 Signaling in Epithelial Cells in Eosinophilic Chronic Rhinosinusitis With Nasal Polyps. J Allergy Clin Immunol (2021) 147:1692–703. doi: 10.1016/j.jaci.2020.12.623
145. Cavagnero KJ, Badrani JH, Naji LH, Amadeo MB, Leng AS, Lacasa LD, et al. Cyclic-Di-GMP Induces STING-Dependent ILC2 to ILC1 Shift During Innate Type 2 Lung Inflammation. Front Immunol (2021) 12:618807. doi: 10.3389/fimmu.2021.618807
146. Wang L, Liu Z, Wang L, Wu Q, Li X, Xie D, et al. Spatial Distribution of IL4 Controls iNKT Cell-DC Crosstalk in Tumors. Cell Mol Immunol (2020) 17:496–506. doi: 10.1038/s41423-019-0243-z
147. West EE, Kolev M, Kemper C. Complement and the Regulation of T Cell Responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
148. McBrien CN, Menzies-Gow A. The Biology of Eosinophils and Their Role in Asthma. Front Med (Lausanne) (2017) 4:93. doi: 10.3389/fmed.2017.00093
149. Elieh Ali Komi D, Shafaghat F, Kovanen PT, Meri S. Mast Cells and Complement System: Ancient Interactions Between Components of Innate Immunity. Allergy (2020) 75:2818–28. doi: 10.1111/all.14413
150. Oikonomou N, Schuijs MJ, Chatzigiagkos A, Androulidaki A, Aidinis V, Hammad H, et al. Airway Epithelial Cell Necroptosis Contributes to Asthma Exacerbation in a Mouse Model of House Dust Mite-Induced Allergic Inflammation. Mucosal Immunol (2021) 14:1160–71. doi: 10.1038/s41385-021-00415-5
151. Brusilovsky M, Rochman M, Rochman Y, Caldwell JM, Mack LE, Felton JM, et al. Environmental Allergens Trigger Type 2 Inflammation Through Ripoptosome Activation. Nat Immunol (2021) 22:1316–26. doi: 10.1038/s41590-021-01011-2
152. Qi X, Gurung P, Malireddi RK, Karmaus PW, Sharma D, Vogel P, et al. Critical Role of Caspase-8-Mediated IL-1 Signaling in Promoting Th2 Responses During Asthma Pathogenesis. Mucosal Immunol (2017) 10:128–38. doi: 10.1038/mi.2016.25
153. Li C, Lasse S, Lee P, Nakasaki M, Chen SW, Yamasaki K, et al. Development of Atopic Dermatitis-Like Skin Disease From the Chronic Loss of Epidermal Caspase-8. Proc Natl Acad Sci USA (2010) 107:22249–54. doi: 10.1073/pnas.1009751108
154. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
155. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local Macrophage Proliferation, Rather Than Recruitment From the Blood, Is a Signature of TH2 Inflammation. Science (2011) 332:1284–8. doi: 10.1126/science.1204351
156. Sallusto F, Lanzavecchia A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells Is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor Plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J Exp Med (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
157. Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt AL, et al. Cellular Differentiation of Human Monocytes Is Regulated by Time-Dependent Interleukin-4 Signaling and the Transcriptional Regulator Ncor2. Immunity (2017) 47:1051–1066 e12. doi: 10.1016/j.immuni.2017.11.024
158. Czimmerer Z, Daniel B, Horvath A, Ruckerl D, Nagy G, Kiss M, et al. The Transcription Factor STAT6 Mediates Direct Repression of Inflammatory Enhancers and Limits Activation of Alternatively Polarized Macrophages. Immunity (2018) 48:75–90.e6. doi: 10.1016/j.immuni.2017.12.010
159. Brandt E, Woerly G, Younes AB, Loiseau S, Capron M. IL-4 Production by Human Polymorphonuclear Neutrophils. J Leukoc Biol (2000) 68:125–30.
160. Woytschak J, Keller N, Krieg C, Impellizzieri D, Thompson RW, Wynn TA, et al. Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration During Infection and Inflammation. Immunity (2016) 45:172–84. doi: 10.1016/j.immuni.2016.06.025
161. Boey H, Rosenbaum R, Castracane J, Borish L. Interleukin-4 Is a Neutrophil Activator. J Allergy Clin Immunol (1989) 83:978–84. doi: 10.1016/0091-6749(89)90115-2
162. Impellizzieri D, Ridder F, Raeber ME, Egholm C, Woytschak J, Kolios AGA, et al. IL-4 Receptor Engagement in Human Neutrophils Impairs Their Migration and Extracellular Trap Formation. J Allergy Clin Immunol (2019) 144:267–279 e4. doi: 10.1016/j.jaci.2019.01.042
163. Panda SK, Wigerblad G, Jiang L, Jimenez-Andrade Y, Iyer VS, Shen Y, et al. IL-4 Controls Activated Neutrophil FcgammaR2b Expression and Migration Into Inflamed Joints. Proc Natl Acad Sci USA (2020) 117:3103–13. doi: 10.1073/pnas.1914186117
164. Girard D, Paquin R, Naccache PH, Beaulieu AD. Effects of Interleukin-13 on Human Neutrophil Functions. J Leukoc Biol (1996) 59:412–9. doi: 10.1002/jlb.59.3.412
165. Nepal S, Tiruppathi C, Tsukasaki Y, Farahany J, Mittal M, Rehman J, et al. STAT6 Induces Expression of Gas6 in Macrophages to Clear Apoptotic Neutrophils and Resolve Inflammation. Proc Natl Acad Sci USA (2019) 116:16513–8. doi: 10.1073/pnas.1821601116
166. Gorski SA, Lawrence MG, Hinkelman A, Spano MM, Steinke JW, Borish L, et al. Expression of IL-5 Receptor Alpha by Murine and Human Lung Neutrophils. PloS One (2019) 14:e0221113. doi: 10.1371/journal.pone.0221113
167. Welch C, Lawrence MG, Spano MS, Steinke JW, Braciale T, Teague WG, et al. Il-5r Expression on Airway Neutrophils in Treatment-Refractory Asthma. Am J Respir Crit Care Med (2019) 199:A3808. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A3808
168. Skrzeczynska-Moncznik J, Zabieglo K, Bossowski JP, Osiecka O, Wlodarczyk A, Kapinska-Mrowiecka M, et al. Eosinophils Regulate Interferon Alpha Production in Plasmacytoid Dendritic Cells Stimulated With Components of Neutrophil Extracellular Traps. J Interferon Cytokine Res (2017) 37:119–28. doi: 10.1089/jir.2016.0036
169. Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, et al. Oxidative Burst in Lipopolysaccharide-Activated Human Alveolar Macrophages Is Inhibited by Interleukin-9. Eur Respir J (2002) 20:1198–205. doi: 10.1183/09031936.02.00005402
170. Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, et al. IL-9 Inhibits Oxidative Burst and TNF-Alpha Release in Lipopolysaccharide-Stimulated Human Monocytes Through TGF-Beta. J Immunol (2002) 168:4103–11. doi: 10.4049/jimmunol.168.8.4103
171. Chowdhury K, Kumar U, Das S, Chaudhuri J, Kumar P, Kanjilal M, et al. Synovial IL-9 Facilitates Neutrophil Survival, Function and Differentiation of Th17 Cells in Rheumatoid Arthritis. Arthritis Res Ther (2018) 20:18. doi: 10.1186/s13075-017-1505-8
172. Abdelilah S, Latifa K, Esra N, Cameron L, Bouchaib L, Nicolaides N, et al. Functional Expression of IL-9 Receptor by Human Neutrophils From Asthmatic Donors: Role in IL-8 Release. J Immunol (2001) 166:2768–74. doi: 10.4049/jimmunol.166.4.2768
173. Bouffi C, Rochman M, Zust CB, Stucke EM, Kartashov A, Fulkerson PC, et al. IL-33 Markedly Activates Murine Eosinophils by an NF-KappaB-Dependent Mechanism Differentially Dependent Upon an IL-4-Driven Autoinflammatory Loop. J Immunol (2013) 191:4317–25. doi: 10.4049/jimmunol.1301465
174. Xu J, Guardado J, Hoffman R, Xu H, Namas R, Vodovotz Y, et al. IL33-Mediated ILC2 Activation and Neutrophil IL5 Production in the Lung Response After Severe Trauma: A Reverse Translation Study From a Human Cohort to a Mouse Trauma Model. PloS Med (2017) 14:e1002365. doi: 10.1371/journal.pmed.1002365
175. Liang Y, Yi P, Yuan DMK, Jie Z, Kwota Z, Soong L, et al. IL-33 Induces Immunosuppressive Neutrophils via a Type 2 Innate Lymphoid Cell/IL-13/STAT6 Axis and Protects the Liver Against Injury in LCMV Infection-Induced Viral Hepatitis. Cell Mol Immunol (2019) 16:126–37. doi: 10.1038/cmi.2017.147
176. Talabot-Ayer D, Martin P, Vesin C, Seemayer CA, Vigne S, Gabay C, et al. Severe Neutrophil-Dominated Inflammation and Enhanced Myelopoiesis in IL-33-Overexpressing CMV/IL33 Mice. J Immunol (2015) 194:750–60. doi: 10.4049/jimmunol.1402057
177. Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA Jr., Auxiliadora-Martins M, et al. Interleukin-33 Attenuates Sepsis by Enhancing Neutrophil Influx to the Site of Infection. Nat Med (2010) 16:708–12. doi: 10.1038/nm.2156
178. Le HT, Tran VG, Kim W, Kim J, Cho HR, Kwon B. IL-33 Priming Regulates Multiple Steps of the Neutrophil-Mediated Anti-Candida Albicans Response by Modulating TLR and Dectin-1 Signals. J Immunol (2012) 189:287–95. doi: 10.4049/jimmunol.1103564
179. Hueber AJ, Alves-Filho JC, Asquith DL, Michels C, Millar NL, Reilly JH, et al. IL-33 Induces Skin Inflammation With Mast Cell and Neutrophil Activation. Eur J Immunol (2011) 41:2229–37. doi: 10.1002/eji.201041360
180. Panova V, Gogoi M, Rodriguez-Rodriguez N, Sivasubramaniam M, Jolin HE, Heycock MWD, et al. Group-2 Innate Lymphoid Cell-Dependent Regulation of Tissue Neutrophil Migration by Alternatively Activated Macrophage-Secreted Ear11. Mucosal Immunol (2021) 14:26–37. doi: 10.1038/s41385-020-0298-2
181. Yazdani HO, Chen HW, Tohme S, Tai S, van der Windt DJ, Loughran P, et al. IL-33 Exacerbates Liver Sterile Inflammation by Amplifying Neutrophil Extracellular Trap Formation. J Hepatol (2017) S0168-8278(17):32291–2. doi: 10.1016/j.jhep.2017.09.010
182. Yu Q, Li Y, Wang H, Xiong H. TSLP Induces a Proinflammatory Phenotype in Circulating Innate Cells and Predicts Prognosis in Sepsis Patients. FEBS Open Bio (2019) 9:2137–48. doi: 10.1002/2211-5463.12746
183. Tanaka J, Watanabe N, Kido M, Saga K, Akamatsu T, Nishio A, et al. And TLR3 Ligands Promote Differentiation of Th17 Cells With a Central Memory Phenotype Under Th2-Polarizing Conditions. Clin Exp Allergy (2009) 39:89–100. doi: 10.1111/j.1365-2222.2008.03151.x
184. West EE, Spolski R, Kazemian M, Yu ZX, Kemper C, Leonard WJ. A TSLP-Complement Axis Mediates Neutrophil Killing of Methicillin-Resistant Staphylococcus Aureus. Sci Immunol (2016) 1:eaaf8471. doi: 10.1126/sciimmunol.aaf8471
185. Bonnekoh H, Scheffel J, Kambe N, Krause K. The Role of Mast Cells in Autoinflammation. Immunol Rev (2018) 282:265–75. doi: 10.1111/imr.12633
186. de Koning HD, van Vlijmen-Willems IM, Rodijk-Olthuis D, van der Meer JW, Zeeuwen PL, Simon A, et al. Mast-Cell Interleukin-1beta, Neutrophil Interleukin-17 and Epidermal Antimicrobial Proteins in the Neutrophilic Urticarial Dermatosis in Schnitzler’s Syndrome. Br J Dermatol (2015) 173:448–56. doi: 10.1111/bjd.13857
187. Young S, Sharma N, Lee JH, Chitu V, Neumeister V, Sohr E, et al. Mast Cells Enhance Sterile Inflammation in Chronic Nonbacterial Osteomyelitis. Dis Model Mech (2019) 12:dmm040097. doi: 10.1242/dmm.040097
188. Al-Salam S, Conca W. Novel Protagonists in Autoinflammatory Arthritis of Familial Mediterranean Fever. Pediatrics (2011) 128:e464–70. doi: 10.1542/peds.2010-2998
189. Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, et al. Mast Cells Contribute to Initiation of Autoantibody-Mediated Arthritis via IL-1. Proc Natl Acad Sci USA (2007) 104:2325–30. doi: 10.1073/pnas.0610852103
190. Nakamura Y, Kambe N. Linkage of Bacterial Colonization of Skin and the Urticaria-Like Rash of NLRP3-Mediated Autoinflammatory Syndromes Through Mast Cell-Derived TNF-Alpha. J Dermatol Sci (2013) 71:83–8. doi: 10.1016/j.jdermsci.2013.04.009
191. Pyle DM, Yang VS, Gruchalla RS, Farrar JD, Gill MA. IgE Cross-Linking Critically Impairs Human Monocyte Function by Blocking Phagocytosis. J Allergy Clin Immunol (2013) 131:491–500.e1-5. doi: 10.1016/j.jaci.2012.11.037
192. Novak N, Bieber T, Katoh N. Engagement of Fc Epsilon RI on Human Monocytes Induces the Production of IL-10 and Prevents Their Differentiation in Dendritic Cells. J Immunol (2001) 167:797–804. doi: 10.4049/jimmunol.167.2.797
193. Kraft S, Novak N, Katoh N, Bieber T, Rupec RA. Aggregation of the High-Affinity IgE Receptor Fc(epsilon)RI on Human Monocytes and Dendritic Cells Induces NF-kappaB Activation. J Invest Dermatol (2002) 118:830–7. doi: 10.1046/j.1523-1747.2002.01757.x
194. Pellizzari G, Hoskin C, Crescioli S, Mele S, Gotovina J, Chiaruttini G, et al. IgE Re-Programs Alternatively-Activated Human Macrophages Towards Pro-Inflammatory Anti-Tumoural States. EBioMedicine (2019) 43:67–81. doi: 10.1016/j.ebiom.2019.03.080
195. Mora J, Riggs EK, Fu J, MacGlashan DW Jr., Fox SA, Yu B, et al. Expression of the High Affinity IgE Receptor by Neutrophils of Individuals With Allergic Asthma Is Both Minimal and Insensitive to Regulation by Serum IgE. Clin Immunol (2009) 132:132–40. doi: 10.1016/j.clim.2009.03.513
196. Saffar AS, Alphonse MP, Shan L, Hayglass KT, Simons FE, Gounni AS. IgE Modulates Neutrophil Survival in Asthma: Role of Mitochondrial Pathway. J Immunol (2007) 178:2535–41. doi: 10.4049/jimmunol.178.4.2535
197. Novak N, Tepel C, Koch S, Brix K, Bieber T, Kraft S. Evidence for a Differential Expression of the FcepsilonRIgamma Chain in Dendritic Cells of Atopic and Nonatopic Donors. J Clin Invest (2003) 111:1047–56. doi: 10.1172/JCI200315932
198. Arase N, Arase H, Hirano S, Yokosuka T, Sakurai D, Saito T. IgE-Mediated Activation of NK Cells Through Fc Gamma RIII. J Immunol (2003) 170:3054–8. doi: 10.4049/jimmunol.170.6.3054
199. Schwartz DM, Kitakule MM, Dizon BL, Gutierrez-Huerta C, Blackstone SA, Burma AM, et al. Systematic Evaluation of Nine Monogenic Autoinflammatory Diseases Reveals Common and Disease-Specific Correlations With Allergy-Associated Features. Ann Rheum Dis (2021). doi: 10.1136/annrheumdis-2020-219137
200. Brenner-Ullman A, Melzer-Ofir H, Daniels M, Shohat M. Possible Protection Against Asthma in Heterozygotes for Familial Mediterranean Fever. Am J Med Genet (1994) 53:172–5. doi: 10.1002/ajmg.1320530210
201. Yazici A, Orge Gonullu E, Kardes B, Cefle A. The Prevalence of Atopy in Patients With Familial Mediterranean Fever and Behcet’s Disease. Clin Exp Rheumatol (2013) 31:68–70.
202. Aydogmus C, Ayaz NA, Cakan M, Cipe F, Topal N, Oner OB, et al. Is There Any Difference Regarding Atopy Between Children With Familial Mediterranean Fever and Healthy Controls? Allergol Immunopathol (Madr) (2017) 45:549–52. doi: 10.1016/j.aller.2016.12.006
203. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De Novo CIAS1 Mutations, Cytokine Activation, and Evidence for Genetic Heterogeneity in Patients With Neonatal-Onset Multisystem Inflammatory Disease (NOMID): A New Member of the Expanding Family of Pyrin-Associated Autoinflammatory Diseases. Arthritis Rheum (2002) 46:3340–8. doi: 10.1002/art.10688
204. Bekhouche B, Tourville A, Ravichandran Y, Tacine R, Abrami L, Dussiot M, et al. A Toxic Palmitoylation of Cdc42 Enhances NF-kappaB Signaling and Drives a Severe Autoinflammatory Syndrome. J Allergy Clin Immunol (2020) 146:1201–1204 e8. doi: 10.1016/j.jaci.2020.03.020
205. Gernez Y, de Jesus AA, Alsaleem H, Macaubas C, Roy A, Lovell D, et al. Severe Autoinflammation in 4 Patients With C-Terminal Variants in Cell Division Control Protein 42 Homolog (CDC42) Successfully Treated With IL-1beta Inhibition. J Allergy Clin Immunol (2019) 144:1122–5.e6. doi: 10.1016/j.jaci.2019.06.017
206. Ahmadi N, Brewer CC, Zalewski C, King KA, Butman JA, Plass N, et al. Cryopyrin-Associated Periodic Syndromes: Otolaryngologic and Audiologic Manifestations. Otolaryngol Head Neck Surg (2011) 145:295–302. doi: 10.1177/0194599811402296
207. Yazici H, Seyahi E, Hatemi G, Yazici Y. Behcet Syndrome: A Contemporary View. Nat Rev Rheumatol (2018) 14:107–19. doi: 10.1038/nrrheum.2017.208
208. Chang HK, Lee SS, Kim JW, Jee YK, Kim JU, Lee YW, et al. The Prevalence of Atopy and Atopic Diseases in Behcet’s Disease. Clin Exp Rheumatol (2003) 21:S31–4.
209. Ozyurt K, Celik A, Sayarlioglu M, Colgecen E, Inci R, Karakas T, et al. Th2 and Th17 Cytokine Profiles and Alpha-Enolase Levels in Recurrent Aphthous Stomatitis. J Oral Pathol Med (2014) 43:691–5. doi: 10.1111/jop.12182
210. Broderick L, Carvalho D, Magit A, Jiang W, Bothwell M, Kearns D, et al. Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA) Syndrome: Evaluation of Patients in San Diego, California. J Allergy Clin Immunol (2012) 129:AB214. doi: 10.1016/j.jaci.2011.12.084
211. Tasher D, Somekh E, Dalal I. PFAPA Syndrome: New Clinical Aspects Disclosed. Arch Dis Child (2006) 91:981–4. doi: 10.1136/adc.2005.084731
212. Gul U, Gonul M, Cakmak SK, Kilic A, Olcay I. Atopy in Behcet’s Disease. Int J Dermatol (2006) 45:1011–3. doi: 10.1111/j.1365-4632.2006.02773.x
213. Yalcin AD, Yalcin AN. A Case of Asthma With Behcet’s Disease: Successful Treatment With Omalizumab and Its Effects on Recurrent Aphthous Lesions. Immunopharmacol Immunotoxicol (2020) 42:379–82. doi: 10.1080/08923973.2020.1789656
214. Lin CH, Lin CL, Shen TC, Wei CC. Epidemiology and Risk of Juvenile Idiopathic Arthritis Among Children With Allergic Diseases: A Nationwide Population-Based Study. Pediatr Rheumatol Online J (2016) 14:15. doi: 10.1186/s12969-016-0074-8
215. Zhang WM, Xu LY, Lu YM, Cao LF. Atopy in Children With Systemic Onset Juvenile Idiopathic Arthritis (SoJIA) Is Associated With a Worse Outcome. Immunopharmacol Immunotoxicol (2014) 36:176–81. doi: 10.3109/08923973.2014.898068
216. Arlet JB, Le TH, Marinho A, Amoura Z, Wechsler B, Papo T, et al. Reactive Haemophagocytic Syndrome in Adult-Onset Still’s Disease: A Report of Six Patients and a Review of the Literature. Ann Rheum Dis (2006) 65:1596–601. doi: 10.1136/ard.2005.046904
Keywords: autoinflammation, autoinflammatory diseases (AID), allergy, type 2 immune response, type 2 immunity
Citation: Sylvester M, Son A and Schwartz DM (2022) The Interactions Between Autoinflammation and Type 2 Immunity: From Mechanistic Studies to Epidemiologic Associations. Front. Immunol. 13:818039. doi: 10.3389/fimmu.2022.818039
Received: 18 November 2021; Accepted: 02 February 2022;
Published: 24 February 2022.
Edited by:
Qingjun Pan, Affiliated Hospital of Guangdong Medical University, ChinaReviewed by:
Raif Geha, Boston Children’s Hospital and Harvard Medical School, United StatesHirohito Kita, Mayo Clinic, United States
Copyright © 2022 Sylvester, Son and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniella M. Schwartz, RGFuaWVsbGEuU2Nod2FydHpAbmloLmdvdg==