 Aaron H. S. Heng
Aaron H. S. Heng Caleb W. Han
Caleb W. Han Caitlin Abbott
Caitlin Abbott Shaun R. McColl
Shaun R. McColl Iain Comerford
Iain Comerford- The Chemokine Biology Laboratory, Department of Molecular and Biomedical Science, School of Biological Sciences, Faculty of Science, The University of Adelaide, Adelaide, SA, Australia
Pro-inflammatory CD4+ T helper (Th) cells drive the pathogenesis of many autoimmune conditions. Recent advances have modified views of the phenotype of pro-inflammatory Th cells in autoimmunity, extending the breadth of known Th cell subsets that operate as drivers of these responses. Heterogeneity and plasticity within Th1 and Th17 cells, and the discovery of subsets of Th cells dedicated to production of other pro-inflammatory cytokines such as GM-CSF have led to these advances. Here, we review recent progress in this area and focus specifically upon evidence for chemokine receptors that drive recruitment of these various pro-inflammatory Th cell subsets to sites of autoimmune inflammation in the CNS. We discuss expression of specific chemokine receptors by subsets of pro-inflammatory Th cells and highlight which receptors may be tractable targets of therapeutic interventions to limit pathogenic Th cell recruitment in autoimmunity.
Introduction
Th cells play pivotal roles in tissue inflammation in a variety of human autoimmune conditions such as multiple sclerosis (MS), psoriasis and rheumatoid arthritis (RA). Various subsets of Th cells that produce distinct arrays of cytokines can either promote or inhibit autoimmune inflammation (1). Th subsets are well-recognised for their functional specialisation in polarisation of immune responses towards effector responses tailored for destruction/elimination of specific types of microbial assault. For example, intracellular microbes are combated by the actions of IFNγ-secreting Th1 cells; responses suited for expulsion of helminths and nematodes are triggered following activation of interleukin (IL)-4/5/13-secreting Th2 cells; and extracellular bacteria and fungus are controlled by phagocyte responses amplified by IL-17A/F-secreting Th17 cells. An underlying pathological driver of numerous chronic autoimmune diseases has been attributed to the actions of misdirected Th1 and/or Th17 responses. These subsets have been implicated to be drivers of tissue damage through their local production of pro-inflammatory cytokines in the local microenvironment that facilitates the persistent recruitment and activation of tissue-damaging myeloid cells, thus culminating in a chronic inflammatory response. When considering the pathogenesis of Th cell mediated tissue inflammation, an essential step is their recruitment from the peripheral circulation into sites of inflammation (2). Thus, it is of importance to elucidate and understand the molecular mechanisms pro-inflammatory Th cells use to traffic into tissues where they drive inflammatory disease. In doing so, molecular targets amenable to therapeutic intervention in autoimmunity may be realized.
Pro-Inflammatory Th Cells
Prior to 2005, IFNγ-producing Th1 cells were considered the main CD4+ T cell subset responsible for driving autoimmune inflammation in the central nervous system (CNS). These cells were abundantly recruited to sites of autoimmune inflammation where they supported monocyte recruitment, macrophage activation and other cell-mediated effector responses. Th1 cells rely on IFNγ and IL-12 for their differentiation and are characterized by expression of the transcription factor T-Bet. Supporting their pro-inflammatory function in autoimmunity, studies using a mouse model of MS, experimental autoimmune encephalomyelitis (EAE), demonstrated that deletion of the p40 subunit of IL-12 or the IL-12Rβ1 chain rendered mice resistant to neuroinflammation with impaired Th1 responses (3). However, the discovery of the cytokine IL-23 and its receptor fundamentally altered the Th1-driven view of autoimmunity. IL-23 shares the p40 subunit of IL-12 and the IL-23 receptor also uses the IL-12Rβ1 chain (4, 5). Deletion of the IL-12p35 subunit or the IL-12Rβ2 chain, which are specific for IL-12, had no effect on EAE pathogenesis while loss of the IL-23p19 subunit or IL-23R chain resulted in resistance to EAE (3, 5, 6). Subsequent studies into the IL-23-dependent cells in EAE revealed a population of CD4+ T cells in the inflamed CNS that were predominantly IL-17A-producers (7). IL-23 signalling was also demonstrated to promote the emergence and expansion of IL-17A-secreting CD4+ T cells in both recently activated and memory CD4+ T cell compartments, and IL-23-driven MOG-reactive CD4+ T cells were shown to be sufficient in driving pathogenesis in adoptive transfer of EAE. Th17 cells quickly became established as a distinct lineage of Th cells (8, 9), with a wide variety of roles in adaptive immunity to extracellular microbes and strongly linked to pro-inflammatory responses in autoimmunity.
Th17 cells are characterised by secretion of IL-17A/F and the expression of the transcription factor retinoic acid receptor-related orphan receptor C (RORγt) (8, 10). These cells are primarily involved in the maintenance of mucosal barrier function and defence against extracellular pathogens (11, 12). During homeostasis, Th17 cells are present at mucosal surfaces to maintain tissue homeostasis and prevent microbial colonisation (13). Antigen-specific activation in the presence of IL-6 triggers differentiation of naïve T cells into Th17 cells via JAK1 and JAK2 signalling, which triggers activation of the STAT3 pathway (14, 15). Th17 cells produce their signature cytokines IL-17A, IL-17F, IL-21 and IL-22, which are involved in host mucosal defence against extracellular pathogens by inducing chemokine production for increased neutrophil recruitment and enhancing expression of antimicrobial peptides. IL-17A has been implicated in an array of autoimmune diseases such as in MS, psoriasis, SLE, and RA. Engagement of IL-17A with the IL-17 receptor activates the NFκB pathway, which stimulates production of proinflammatory cytokines including IL-1β, GM-CSF and chemokines such as CCL2 (16), CXCL1 (17) and CCL20 (18). These cytokines and chemokines act in concert to accelerate the inflammatory response and to direct neutrophils and monocytes to the site of inflammation. An interesting characteristic of Th17 cells is their context-dependant plasticity, allowing Th17 cells to transdifferentiate and reprogram for production of a different array of cytokines in response to signals from antigen-presenting cells (APCs) (19–25). For example, studies have shown that Th17 cells transdifferentiate into IFNγ-producing Th1-like cells, IL-4-producing Th2-like cells, type 1 regulatory cell (Tr1)-like cells and IgA -promoting T follicular helper-like cells (26–28). The reverse has also been shown to be possible to a certain extent. Studies have shown that Th1-like cells that emerge from Th17 cells are able to transdifferentiate back into Th17 cells, and that FOXP3+ T cells are able to transdifferentiate into Th17 cells in a model of RA (29, 30). Th17 cells can also turn off production of IL-17A, transdifferentiating into so called ‘exTh17’ cells (31). This plasticity appears to give Th17 cells a degree of flexibility that allows them to appropriately respond to an evolving antigenic threat by either amplifying, attenuating, or modulating the inflammatory response.
In EAE, neutralisation of IL-17A was reported to delay the onset of clinical disease (32), however multiple studies have also concluded that IL-17A is not essential for EAE pathogenesis (33–35). This has prompted further study of the Th17-derived cytokines that drive EAE. These studies have identified GM-CSF as being a critical T cell-derived cytokine for the pathogenesis of classical EAE (35–37). GM-CSF-deficient mice are highly resistant to EAE induction (38), and GM-CSF-deficient myelin-specific T cells are unable to transfer disease (35). Furthermore, dysregulated overexpression of GM-CSF in polyclonal Th cells was sufficient to induce neuroinflammation by promoting an influx of phagocytes into the CNS (39). T cell-derived GM-CSF was more specifically shown to act on a variety of myeloid subsets including inflammatory CCR2+Ly6Chi monocytes, monocyte-derived dendritic cells (moDCs) and microglia, all of which have been previously ascribed pathogenic roles in EAE via GM-CSF-dependent signalling (40). Moreover, loss of the GM-CSF receptor on myeloid cells rendered mice resistant to EAE induction (40, 41). While most studies indicate that GM-CSF is essential for inducing EAE pathogenesis, two studies have disputed this claim by showing that EAE can be induced via both direct and indirect immunisation of GM-CSF-deficient mice (42, 43). Despite these findings, both studies showed that GM-CSF is a potent driver of inflammation during EAE.
Mechanisms driving the conversion of IL-17-secreting Th17 cells to proinflammatory GM-CSF-secreting Th17 or exTh17 cells in vivo have been elucidated to some extent. IL-23 and IL-1β have been demonstrated to induce a more inflammatory GM-CSF/IFNγ cytokine production profile in Th17 cells in vitro and have also been shown to have indispensable roles in the pathogenesis of EAE in knock-out studies. Studies have also shown that Th17 cells generated in the presence of TGFβ3 acquired inflammatory capacity after prolonged exposure to IL-23, upregulating IFNγ and adopting a Th1-like phenotype (44, 45). TGFβ3 restrains IL-10 production and supports the production of proinflammatory cytokines by Th17 cells through induction of Smad1 and Smad5 (46). Additionally, IL-7, which is expressed highly in the CNS of mice with EAE, has also been implicated in the induction of GM-CSF production and acquisition of a more Th1-like phenotype by Th17 cells (47, 48). Although it has been shown that IL-7Ra expression is induced by IL-23 (49), IL-7R signalling requires IL-23 responsiveness in Th17 cells to promote their pathogenicity in EAE (47). However, recent studies suggests that IL-7 may be involved in the generation or maintenance of memory Th17 cells at the site of inflammation (50). Interestingly, a recent study using a reporter and fate mapper of GM-CSF showed that Th cells that had produced GM-CSF have a distinct epigenetic profile indicative of long-term stability (51). It is possible that IL-7 contributes to stability of GM-CSF-secreting cells, but this remains to be tested. Furthermore, Th17 cells in the CNS have the highest expression of IL-7R followed by Th1-like Th17 cells and then Th1 cells, which express the lowest levels of this receptor (47). This further implicates a role for IL-7 signalling in the expansion or maintenance of exTh17 cells in the CNS.
Recent studies have identified another subset of Th cells that abundantly produces GM-CSF and low amounts of cytokines associated with other Th subsets (52). These cells, dubbed ThGM cells, are believed to be a distinct Th subset from Th1 or Th17 cells as they do not require the expression of RORγt or T-Bet to support high levels of GM-CSF production, and unlike Th1 and Th17 cells, which develop via a STAT4 and STAT3 pathway respectively, they have been shown to derive from naïve CD4+ precursors that activate the STAT5 pathway following IL-7 and/or IL-2 stimulation (48). In support, evidence implicates STAT5 as being an important factor in EAE. Mice harbouring STAT5-deficient CD4+ T cells are less susceptible to EAE compared to WT, and this was associated with reduced GM-CSF production without impacting IFNγ or IL-17. This is further supported by studies that demonstrated that GM-CSF-producing CD4+ T cells generated through an IL-7/STAT5 pathway were more encephalitogenic in an adoptive EAE model compared to Th17 or Th1 cells (48).
Migratory Mechanisms of Autoreactive Th Cells in EAE/MS
The CNS has several unique barriers in place to maintain homeostasis and limit leukocyte infiltration. To initiate CNS autoimmunity, autoantigen-specific T cells are activated in the periphery and travel to the CNS, where they are reactivated by APCs presenting the autoantigen. This triggers the release of cytokines to activate and recruit other inflammatory cells to cause inflammation. However, the CNS employs various obstacles such as the blood brain barrier (BBB) and the blood cerebral spinal fluid (CSF) barrier (BCSFB) to limit access of T cells [reviewed in (53)]. In order to breech these barriers, the coordinated use of selectins, integrins and chemokines are required for efficient T cell migration into the CNS (depicted in Figure 1).

Figure 1 T cell recruitment into the CNS during neuroinflammation. (1) After activation in the periphery, CD4+ T cells roll across the surface of the blood vessel in a process mediated by P-selectin and PSGL-1 interactions. (2) Binding of chemokine receptors to chemokines expressed on the surface of the endothelial cells activate G-proteins which trigger conformational changes of the integrins VLA-4 and LFA-1, allowing for arrest (3) of the T cells to VCAM-1 and ICAM-1 respectively. (4) T cells crawl across the endothelial cells, (5) undergo diapedesis and eventually enter the perivascular space. (6) Perivascular macrophages and dendritic cells reactivate the T cells by presenting antigen, (7) which allows T cells to migrate into the CNS parenchyma. (8-10) Once in the parenchyma, T cells can be reactivated by antigen presenting cells and release inflammatory cytokines such as GM-CSF which activates and recruits Ly6C+ monocytes and monocyte-derived dendritic cells that drive demyelination. Figure was created using Biorender.
Selectins
Selectins are a family of transmembrane C-type lectins expressed by bone marrow-derived cells and endothelial cells (54). The selectin family consists of three members: L-selectin, P-selectin, and E-selectin. While selectins and their ligands are clearly involved in T cell rolling on cerebrovascular endothelial cells, a clear picture of the essential selectin-mediated signals required for T cell entry to the CNS has yet to emerge and differences in selectin usage by distinct subsets of Th cells remains to be determined. L-selectin is expressed on many subsets of T cells, including naïve and some memory T cells where it is required for homing to lymph nodes. While studies have also implicated L-selectin in the pathogenesis of EAE/MS (55, 56), this appears to be mainly due to the role of this adhesion molecule in T cell priming rather than their effector functions (57). PSGL-1 and its ligand P-Selectin have clearly been shown to mediate T cell rolling on BBB microvasculature (58, 59), however this interaction between P-selectin and PSGL-1 is evidently dispensable to T cell recruitment to the CNS and for EAE pathogenesis (60). More recently the P-selectin ligand TIM-1 was implicated as the key adhesive signal that mediated Th1 and Th17 rolling on brain microvasculature (61). Another recent study has implicated the E-selectin ligand CD43 on T cells as a key mediator of Th17 adhesion to the brain endothelial cells, but this appears to be independent of interactions with E-selectin (62). Taken together, there is still much to learn about the essential early steps in pathogenic T cell rolling on brain vasculature and which selectin molecules, or other interactions, mediate this process in distinct subsets of these cells.
Integrins
Integrins have been long implicated in recruitment of CD4+ T cells to the CNS during autoimmunity. For example, the interaction between the α4β1 integrin (CD29/CD49d, VLA-4) on effector T cells and its ligand VCAM-1 on BBB endothelial cells has been implicated in early entry of T cells into the CNS. Neutralisation of the α4 integrin inhibits both EAE pathogenesis and prevents encephalitogenic T cell recruitment into the CNS parenchyma (63–65). These discoveries led to the development of Natalizumab, an anti-α4 integrin blocking monoclonal antibody, now used for the treatment of MS. Natalizumab displays significant clinical efficacy in reducing relapses and disease progression in relapsing-remitting MS patients (66–69). While blocking of α4 integrin clearly inhibits EAE and MS pathogenesis, the specific role of this integrin in the recruitment of pro-inflammatory T cell subsets to the CNS is more complex. VLA-4 is strongly implicated in monocyte and Th1 cell adhesion to the inflamed BBB, but Th17 recruitment to the brain during EAE appears to be entirely independent of this integrin (70, 71). Instead, Th17 cells appear to be more reliant on the αLβ2 integrin (LFA-1) for entry into the CNS (71), a characteristic they also appear to share with Tregs (72). In addition to mediating recruitment to the inflamed CNS, recent 2-photon imaging studies have also implicated the αLβ2 integrin in regulating both Th1 and Th17 motility within the CNS, and blocking this integrin using intrathecal anti-LFA-1 antibodies substantially attenuated disease pathogenesis in EAE mice (73).
Other T cell expressed integrins have also been implicated in recruitment to the inflamed CNS. The integrin ανβ3 was found to be expressed specifically by Th17 cells, and was upregulated through IL-23 signalling (74). This identified that ανβ3-expressing Th17/exTh17 cells increased in number during EAE, and deficiency of this integrin using KO mice or using an inhibitor severely dampened Th17-mediated EAE, while not affecting Th1-mediated EAE. Transfer of ανβ3-deficient Th17 cells also resulted in reduced severity of Th17-mediated EAE. Interestingly, this study suggested ανβ3-deficient Th17 cells were unable to infiltrate the CNS, pointing to a role of ανβ3 in allowing for pathogenic Th17 cell migration to the CNS during EAE (74). In support of this finding, another study using an ανβ3 binding peptide also showed efficacy in reducing clinical signs of EAE in rats (75).
Together, while α4 integrins have been shown to be involved in T cell recruitment to the CNS EAE, other integrins such as ανβ3 and αLβ2 likely also contribute to the migration of pathogenic Th cells into the CNS. It is probable that these integrins play a coordinated role in influencing the trafficking of Th cells and is therefore likely to be able to compensate for the loss of either integrin. Therefore, future research into elucidating the exact roles these integrins play during EAE, as well as the location these integrins can allow Th cells to enter the CNS should be investigated. Doing so will undoubtably uncover new insights into the properties and process of pathogenic T cell migration during CNS autoimmunity. The remainder of this review will now focus on the chemokine receptors that control the mobility of autoinflammatory Th cells, as well as their impact in the pathogenesis of EAE.
Chemokine Receptor-Driven Migration of Th Cells to the CNS in EAE/MS
Chemokines and their associated receptors are key modulators of T cell migration. Chemokines are a large group of small structurally related cytokines that function as chemoattractants and play a fundamental role in T cell biology by directing cell migration through interactions with chemokine receptors [reviewed in (76, 77)]. Chemokine receptors are G-protein coupled receptors that upon ligation trigger intracellular signalling cascades resulting in leading edge polarisation, integrin activation and ultimately directed cell migration towards extracellular sources of chemokines (78). This allows for T cell traffic to specific microanatomical sites and/or areas of inflammation. Differential chemokine receptor expression profiles of Th subsets functionally distinguish them according to their differing circulation patterns and migratory potential, and chemokine receptor profiling has been used to characterise and identify specific populations of Th cells (79, 80). For example, the expression of the chemokine receptor CCR7 is typically present on naïve CD4 T cells and some memory Th cells to allow them to home towards the chemokines CCL19 and CCL21 within T cell areas of the secondary lymphoid organs, where they encounter mature APCs (81). In contrast, activated Th cells downregulate CCR7 and express an array of inflammatory chemokine receptors (such as CXCR3, CXCR6, CCR2, CCR4, CCR5, CCR6) that act in concert to drive recruitment to inflamed tissues where ligands for these receptors are produced in response to tissue damage or infection (80, 82, 83). Th1 cells are well known to express CXCR3 that directs these cells to sites of production of CXCL9/10/11, which are induced by type-I interferon signalling (84, 85). Th17 cells are known to express CCR6 (21, 86, 87), which along with its sole cognate chemokine ligand CCL20, plays a role in both homeostasis and inflammation (88). Under resting conditions, constitutive CCL20 expression in the intestinal mucosa attracts CCR6-expressing cells such as Th17 cells, B cells, dendritic cells (DC) and regulatory T cells (Treg), whereas under inflammatory conditions CCR6 recruits activated T cells, DCs and macrophages to areas of acute inflammation at epithelial sites (89, 90).
In EAE/MS, autoreactive T cells gain access to the CNS in spite of significant physical barriers in place to impede this process. The two main barriers that impede immune cell entry into the CNS are the BBB and BCSFB. Bypassing the BBB and BCSFB allows access to the perivascular space and the CSF subarachnoid space (SAS) respectively, whereby the cells can then access the CNS by bypassing the glia limitans from the perivascular space or through the CSF [reviewed in (91)]. The chemokines expressed in the CNS under non-inflammatory conditions to mediate initial recruitment of encephalitogenic Th cells in EAE are not yet fully understood, if indeed a chemokine-dependent signal is required. Candidate chemokines expressed in the non-inflamed CNS are CX3CL1, CXCL12, CCL20 and CCL21 (92–98). While the CX3CL1 receptor CX3CR1 clearly impacts on CNS autoimmunity through effects on myeloid cells, its role in T cell recruitment to the brain during neuroinflammation is controversial with one study indicating no T cell intrinsic role for CX3CR1 in EAE and another study showing CX3CR1-dependent recruitment of T cells to the brain during neuroinflammation (99, 100). CXCL12 is expressed along the basolateral surfaces of CNS endothelial cells (101). Here, it appears retain T cells, which almost ubiquitously express CXCR4, that have crossed the BBB to the perivascular space and CXCR4 limits their infiltration of the CNS parenchyma. The role of CCL21/CCR7 in T cell entry to the uninflamed CNS has not been fully explored but clinical EAE does not appear to be affected by whole animal deletion of CCR7 (102); and a study where CCR7-deficiency was limited to T cells imply that this receptor is involved in T cell priming during EAE but not entry to the CNS (103). However, a recent study administration of a dietary flavonoid downregulated CCR7 on CD4+ T cells in the CNS, thus reducing infiltration of pathogenic T cells into the CNS and thus reducing EAE severity (104). CCL20 is constitutively expressed on the epithelial cells of the choroid plexus, and may be the chemokine signal that allows CCR6+ T cells to bypass the BCSFB (95). Another study by Murakami and colleagues indicated that CCL20 was expressed by dorsal blood vessels in the 5th lumbar spinal cord where it is induced by IL-6 and that this provides a site of pathogenic T cell recruitment into the CNS in EAE (105).
The molecular mechanisms governing how GM-CSF-secreting exTh17 cells or ThGM cells infiltrate the CNS have yet to be revealed. While it has been shown that CCR6+ Th17 cells infiltrate the choroid plexus early during the initial stages of EAE, where they are reactivated by local APCs (95, 106), not much is known about the mechanisms used by pathogenic Th subsets to enter the CNS during the later phases of EAE. It is possible that cytokines released by early infiltrating Th17 cells (such as TNFα or IL-17A) disrupt BBB integrity and then allow cells expressing chemokine receptors such as CCR2 or CXCR6 to enter the CNS directly from brain capillaries (107, 108). Alternatively, pathogenic T cells could manoeuvre into the CNS via expression of other chemokine receptors that may allow entry through the BBB or BCSFB. Expression of chemokines such as CXCL16 and CXCL12 are upregulated in the inflamed CNS (109, 110), and whether these chemokines are involved in recruiting T cells such as ThGM cells into the CNS remains an interesting area for future research.
It has been shown that various chemokine receptors are involved during the pathogenesis of EAE/MS. These receptors are expressed on various T cell subsets involved in EAE such as Th17 cells and ThGM cells and their expression is regulated temporally during EAE. Th17 cells initially express CCR6 but can switch chemokine receptor expression and upregulate other receptors such as CCR2, which reflects an increase in their pathogenic signature (111). GM-CSF-producing cells, including Th17, exTh17 cells, and ThGM cells also potentially utilise these receptors to infiltrate the CNS. The chemokine receptor expression profile of the various subsets of Th cells implicated in EAE and the relationship between these cell types is depicted in Figure 2. We will now discuss each of the main candidate chemokine receptors that appear likely to contribute to pathogenic T cell recruitment to the CNS in detail.
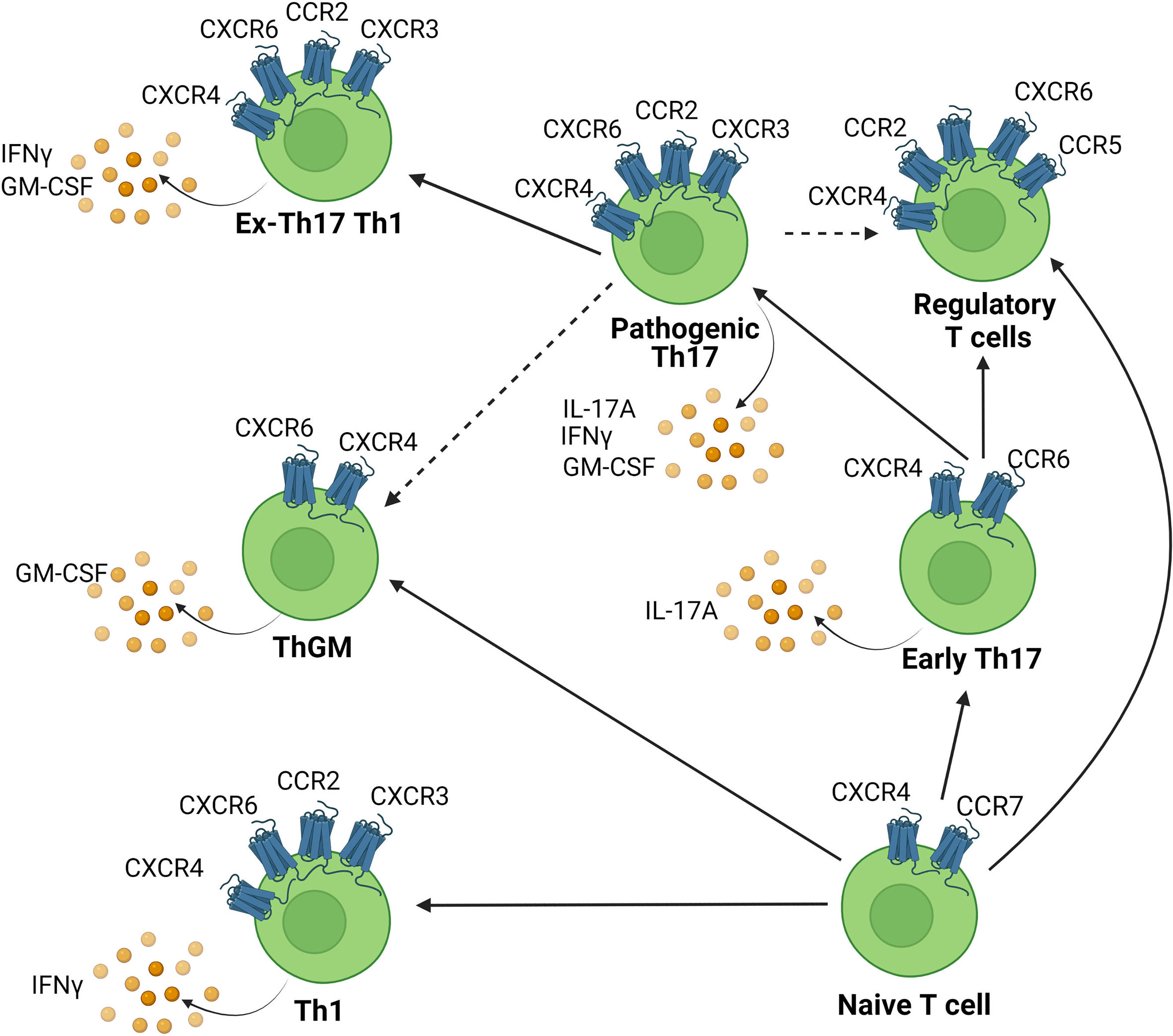
Figure 2 Chemokine receptor expression on CNS infiltrating Th cell subsets. Chemokines attract chemokine receptor expressing Th cells to infiltrate the CNS. Various Th subsets are capable of migration into the CNS to cause inflammation. This includes Th1, Th17, ThGM cells and each are associated with distinct chemokine receptor signatures. Solid lines indicate pathways of cellular transdifferentiation. Dotted lines indicate potential pathways that remain to be elucidated. Figure was created using Biorender.
CCR6
Th17 cells have been extensively shown to express CCR6 (21, 86, 87), the sole receptor for the chemokine CCL20 (88, 112). Studies have also reported increased expression of CCL20 in the draining lymph nodes (dLN) and spinal cord of mice following immunisation for EAE (113, 114). CCL20 is also expressed in the CNS by astrocytes (115), and decreased production by astrocytes is correlated with a reduced clinical disease in EAE (116). Furthermore, CCL20 also appears to be involved in Th17 cell egress from the LN, prior to migration into the CNS (114). Neutralizing either CCL20 or CCR6 in EAE mice decreased disease severity in various studies, highlighting the role of the CCL20/CCR6 axis in EAE (113, 114, 117). A seminal study by Reboldi et al., demonstrated that Th17 cells migrate through the blood-CSF barrier into the uninflamed brain through the interaction of CCR6 with CCL20 expressed on the epithelial cells of the choroid plexus (95). This study also showed that Ccr6-/- mice were resistant to the induction of EAE and that transfer of CCR6-sufficient T cells restored susceptibility of these mice to EAE. It was noted that the subsequent recruitment of Th17 cells into the CNS was largely CCR6-independent, which established a model that CCR6 coordinates the first wave of Th17 cells required to initiate inflammation in EAE but is dispensable for the subsequent infiltration of autoreactive cells into the inflamed CNS.
Since then, various studies have shown that CCR6 signalling does not inherently drive pathogenic T cell recruitment in EAE, as Ccr6-/- mice showed delayed disease onset but eventually develop disease with increased severity compared to WT controls (118, 119). This highlights the essential role of other chemokine receptors or migratory factors that can compensate for the loss of CCR6 for recruitment of inflammatory T cells to the CNS. Furthermore, Treg cells also express CCR6, and have been shown to migrate to the CNS via CCR6 to control inflammation during EAE (117). In addition, exTh17 cells that have transitioned to a Th1-like phenotype were found to downregulate CCR6, suggesting that pathogenic cells associated with driving EAE utilise a different mode of migration (31). In support, a study from our laboratory (111) found that Th17 cells undergo an IL-23-dependent switch of chemokine receptor expression during EAE, which involves downregulation of CCR6 as they increase pro-inflammatory GM-CSF and IFNγ production. In humans, CCR6 is commonly used in combination with CXCR3 to identify Th17 cells (120, 121). While a CCR6+CXCR3- profile enriches for Th17 cells, Th17 cells that co-express IFNγ have been found to also express CXCR3 (122). These CCR6/CXCR3 double expressing Th17 cells (dubbed Th17.1 or non-classic Th1) have also been associated with MS disease severity (123, 124).
From the discussion above, while CCR6/CCL20 clearly play a role in EAE/MS, there are other factors that must be considered with regards to their potential as therapeutic targets to treat MS. These includes the likely effectiveness of disrupting CCR6 activity, which could affect the trafficking of Tregs or other regulatory leukocyte populations into the CNS, as well as having no effect on inflammatory T cell subsets that have downregulated CCR6 or do not express this receptor.
CCR2
CCR2 is involved largely in monocyte and macrophage mobilization (125–127) but is also expressed by activated T cells, astrocytes, microglia cells, and has been shown to be expressed by infiltrating autoinflammatory Th cells in the CNS during MS/EAE (111, 128–130). CCR2 is potently activated by the chemokine CCL2, which has been shown to be expressed by astroglial cells in EAE (131, 132). Multiple studies have demonstrated significant protective effects of CCR2-deficiency in EAE, as CCR2-deficient mice are resistant to EAE induction, and neutralising antibodies against CCL2 reduced EAE severity (133–135). This has been proposed to be due to the effects on CCR2+ Ly6Chi monocytes, which have been shown to be important in mediating tissue inflammation during neuronal autoimmunity through a GM-CSF-dependent pathway (40, 127). A study which transferred primed MOG-specific T cells from WT or CCR2-deficient mice into WT or CCR2-deficient hosts showed that while Ccr2-/- T cells were able to transfer disease to a WT mice, primed T cells from WT mice were unable to do so when transferred into Ccr2-/- mice (134). This suggest that the major role for CCR2 in EAE was extrinsic to antigen-specific T cells. Similar results were found in Ccl2-/- mice, which had delayed onset of EAE, but were able to generate Th cells that could transfer disease into WT mice (136). Another study looking at depletion of CCL2 from astrocytes in EAE mice reported delayed onset and severity of disease, but also discovered an increase in Th17 cell infiltration in the CNS along with increased levels of Il6 mRNA (132). This suggests that in the absence of astroglial CCL2, either an increase in other chemotactic factors allows for an increase in Th17 cells migrating in, or that astrocytes increased production of IL-6 which resulted in an increased amount of Th17 differentiation in the CNS. A recent study that silenced activity of excitatory neurons in EAE resulted in decreased expression of Ccl2 and Ccr2 mRNA in EAE lesions, suppressing migration of Th cells and alleviating motor deficits of EAE (137). Interestingly, a study has shown that blocking CCR2 can lead to an increase in severity of experimental arthritis, which is in contrast to the proinflammatory nature of CCR2 (138). This has been explained to be due to the inability of CCR2-expressing Tregs to migrate to the affected areas to regulate the inflammation.
CCR2 and CCL2 have also been implicated with MS disease severity. CCR2 has been found to be highly expressed by infiltrating T cells within and around active MS lesions, and CCL2 is expressed by astrocytes within lesions (115, 129, 139, 140). As such, CCR2/CCL2 targeting drugs have been explored as treatments for MS and entered as candidates in clinical trials. While early trials reported a good safety profile of the drug candidates, they failed to induce significant therapeutic benefits and the trials were abandoned (141, 142). The lack of efficacy of CCR2 antagonists in MS patients may stem from complex compensatory mechanisms T cells have to traffic into the CNS during MS. Thus, inhibition of CCR2 function alone would thus be unable to deter T cell infiltration. In support of this, a recent study demonstrated that Clozapine, a commonly used drug in MS patient treatment, reduced disease onset and severity in EAE by attenuating migration of immune cells including T cells by specifically reducing both CCL2 and CCL5 levels (143). Our chemokine receptor expression profiling of Th17 cell indicated that Th17 cells express both CCR6 and CCR2 in EAE mice. Over the course of the model, Th17 cells exhibited a temporal transition from CCR6 to CCR2 expression, which was associated with a more inflammatory IFNγ/GM-CSF-producing state (111). Moreover, mice lacking CCR2 specifically in the T cell compartment and transfer of Ccr2-deficient T cells showed reduced EAE and fewer GM-CSF-secreting Th17 cells in the CNS compared to controls. However, mice with a T cell compartment deficient in CCR2 were still able to induce EAE after a delay, suggesting that inhibiting CCR2 is not essential for all autoreactive T cell entry into the CNS. Therefore, other migratory factors aside from CCR2 on T cells should be explored as potential targets.
CXCR3
CXCR3 has been extensively studied with regards to its role in inflammatory T cell recruitment during neuroinflammation. This receptor is abundantly expressed on CNS-infiltrating T lymphocytes from MS patients (144) and co-ordinates migration in response to its three ligands, CXCL9, CXCL10 and CXCL11 (145–147). Commonly associated with IFNγ-producing Th1 cells and CTLs, CXCR3 is expressed on activated and memory T cells and is induced after exposure to IFNγ (148, 149). Some Th17 cells also express CXCR3, and it has been shown to mediate migration of these cells in models of autoimmunity (150–152). Studies of the role of CXCR3 in EAE studies have yielded divergent results. Contrary to the expectation that CXCR3 would play a role in mediating migration of pathogenic Th subsets into the CNS, disrupting CXCR3 using antibodies or using Cxcr3-/- mice revealed that lack of CXCR3 signalling renders mice more susceptible to EAE (153–156). A study using anti-CXCR3 neutralising antibodies in rats showed that while there are high levels of CXCR3+ infiltrating T cells in the CNS of EAE rats, CXCR3 was redundant for T cell infiltration of the CNS (153). This study also showed that while anti-CXCR3 treatment was strongly effective against adoptive transfer of MBP-specific T cells, it did not affect active immunisation. This could be explained by active immunisation eliciting multiple arms of the immune response, which triggers activation of APCs leading to production of additional CXCR3-independent migratory factors in the CNS, thus allowing T cells recruitment to the CNS without CXCR3 function in this setting. Studies using Cxcr3-/- mice also showed that these mice did not differ from WT in terms of the amount or quality of infiltrating leukocytes during peak EAE. Instead, Cxcr3-/- mice exhibited severe BBB disruption, decreased levels of IFNγ, increased demyelination and axonal damage (155, 156). While the number of CD4+ T cells infiltrating the CNS was similar between Cxcr3-/- and WT mice, FOXP3+ regulatory T cells were significantly reduced in Cxcr3-/- mice, which may contribute to exacerbated disease. To add to the complexity regarding the role of CXCR3 in EAE, a study by Chung et al., showed that Cxcr3-/- mice were more susceptible to both actively and passively induced EAE compared to WT mice (154). This study highlighted that activated glial cell numbers are increased in Cxcr3-/- mice, and these Cxcr3-/- glial cells produce cytokines that promote Th17 cells, as mRNA levels of IL-6, IL-23p19 and IL-17A were all increased in glial cells in the absence of CXCR3. These findings correlated with an increased number and proliferative capabilities of Th17 cells in the CNS. This points to an indirect role of glial cell-derived CXCR3 in controlling Th17 levels in the CNS and hence restraining EAE pathogenesis in CXCR3-sufficient mice.
Contrary to studies that show that CXCR3 inhibits EAE pathogenesis, our previous studies have shown that administration of a truncated CXCL11 antagonist of CXCR3 prevented infiltration of CD4+ T cells into the CNS of mice with relapsing-remitting EAE and resulted in reduced disease severity (157). We found that CXCR3 antagonism blocked adoptive transfer of EAE but did not impact on T cell priming. These findings are consistent with the findings from Sporici and Issekutz in rat models of EAE where CXCR3 antibody blockade affects passively transferred EAE (153). In further support of this, Fife et al. reported that CXCL10 administration induced infiltration of CXCR3+ T cells into the CNS (158). A study by Schmitz et al. also reported a reduction in EAE clinical scores on Cxcr3-/- mice, which was associated with a reduction in IL-17A levels as compared to WT mice (159). Altogether, these studies point towards a complex role for CXCR3 in EAE. While CXCR3 signalling clearly can have suppressive effects on EAE pathogenesis, there is strong evidence that it is indeed used by subsets of CD4+ T cells to invade the CNS and this includes in some settings inflammatory CD4+ T cells that drive the disease. This requires future investigation with more targeted analysis of the known inflammatory subsets of T cells that have been identified in recent years. Of note, adoptive transfer of WT and Cxcr3-/- highly polarised encephalitogenic Th1 cells has clearly shown that recruitment of these cells to the CNS is independent of CXCR3 (160), and this also seems to be the case with adoptively transferred polarised Th17 cells (154).
In humans, CXCR3 levels are increased in the CSF and brain lesions of MS patients (161, 162). The levels of CXCL10 was also found to increase during relapses, and is expressed by macrophages and astrocytes in demyelinating lesions (162, 163). Moreover, treatment with IFNβ, which is an approved therapy for treatment of MS (164), specifically decreased CXCR3 expression levels on CD4+ T cells (165). As such, there seems to be compelling evidence that CXCR3 is involved in MS development and hence therapeutics targeting it may be beneficial for managing MS. However, there has only been one CXCR3 antagonist that has entered clinical trial for the treatment of psoriasis, and has been withdawn due to a lack of efficacy (166). However, as discussed below, there may still be utility in targeting CXCR3, particularly if this is in combination with other key migratory receptors and in contexts where this system is known to be driving pathogenesis.
CCR5
Another chemokine receptor associated with T cell recruitment and MS/EAE pathogenesis is CCR5. Widely known for its role as an HIV-1 co-receptor, CCR5 has been implicated in T cell trafficking in various diseases including MS (167, 168). The ligands for CCR5, (in humans CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL8 and CCL16) have been shown to be produced by cells in the CNS including microglia and astrocytes and may possibly play a role in attracting T cells into the CNS (169–171).
CCR5-expressing T cells were increased in the peripheral blood and demyelinating lesions of MS patients as compared to healthy patients and were also found in the brains of MS patient cadavers (168). CCR5+ T cells were also reported to produce more IFNγ than other T cells, consistent with a Th1-like phenotype. A study by Sato and colleagues reported that a subset of CCR2+CCR5+CCR6- CD4+ T cells were enriched in the CSF of patients with severe MS compared to patients with other neurologic diseases (129). Furthermore, these cells produced various molecules that influence neuroinflammation, such as IFNγ and IL-17A, matrix metalloproteinase-9 (MMP9), and osteopontin. MMP9 has been shown to play a role in BBB disruption during EAE/MS (172, 173), while osteopontin is a phosphoprotein which has been associated with MS/EAE severity (174–176). This study also showed an increased potential of CCR2+CCR5+CCR6- CD4+ T cells to traffic into the brain parenchyma, possibly due to their increased production of MMP9 and osteopontin (129).
In EAE, a clear picture of the role of CCR5 has yet to emerge. A study using mice deficient in either CCR5 or CCL3 reported that these mice were susceptible to EAE induction and were very similar to WT mice (177). However, another study recently reported that Ccr5-/- mice were resistant to EAE induction, displaying lowered disease severity than WT mice (178). Lower clinical disease was linked to several factors in that study, including impaired infiltration of leukocytes into the CNS, attenuated activation of astrocytes and microglial cells, and decreased levels of cytokines such as IL-1β in the spinal cord of Ccr5-/- mice. In support of a role of CCR5 in driving EAE, several studies have shown that CCR5 antagonists reduce migration of inflammatory cells into the CNS, resulting in reduced disease incidence, delayed onset, and severity in EAE mice (179–182). Furthermore, a study that neutralised the ligands of CCR5 ameliorated ongoing EAE (183). However, it should be noted that CCL3 and CCL5 are also ligands for CCR1 and CCR3, and hence the effect shown by this study was not necessarily a result of impairment of CCR5 activity. A recent study determining the effect of the Let-7 microRNAs determined that expression of Let-7 microRNA inhibited expression of CCR5 and CCR2 in pathogenic Th17 cells, leading to fewer numbers of infiltrating CD4 T cells in the CNS and reduced EAE pathogenesis (184). Intriguingly, CCR5, together with CXCR3, appear to contribute to adhesion to leptomeningeal structures and thus influence migration of T cells from the CSF into the CNS (185).Together, the evidence strongly suggests that CCR5 has a role in mediating the migration of autoinflammatory cells into the CNS during EAE, but whether CCR5 plays a prominent role in migration of recently identified inflammatory GM-CSF-secreting Th subsets will require further studies and use of experimental systems that can reveal the cell-intrinsic role of this receptor in pathogenic Th17 cells.
CXCR6
Initially classified as a co-receptor for allowing the viruses SIV and HIV to enter cells (186), CXCR6 has been shown to be expressed on CD4+ T cells and is involved in the homing of CD4+ T cells to tissue sites of inflammation such as joints of RA patients (187, 188). Interestingly, CXCR6 has been reported to be highly expressed on IFNγ-producing and GM-CSF-producing CD4+ T cells (51, 189). The ligand for CXCR6, CXCL16, is a unique chemokine that acts both as an adhesion molecule to bind CXCR6-expressing cells and is cleaved as a soluble protein that mediates chemotaxis (190, 191). Membrane-tethered CXCL16 is also a scavenger receptor for oxidized low-density lipoprotein (192). CXCR6/CXCL16 have been implicated in various diseases, such as cancer, liver diseases and atherosclerosis (188, 193, 194). Expression of CXCL16, while low in the uninflamed brain, is drastically increased during inflammation as astrocytes and glial cells upregulate CXCL16 during neuroinflammation, and mRNA levels of Cxcl16 have been shown to greatly increase in the brain and spinal cord during EAE (195, 196). Being selectively expressed on APCs such as DCs, CXCL16 has been shown to play a role in mediating the interactions between T cells and APCs in the splenic red pulp, allowing for the recruitment and activation of T cells and NKT cells (189, 197, 198). CXCR6 is used as a marker of T cell activation, as CXCR6 is drastically upregulated following priming by APCs (189, 199, 200).
In EAE, a study of a fate mapper and reporter model for GM-CSF production discovered that cells that are or had previously produced GM-CSF are marked with a unique gene expression profile, and that CXCR6 is highly correlated with the encephalitogenic GM-CSF-producers in the CNS (51). This finding is supported by another recent study by Hou et al. (201), which also identifies CXCR6 to be a marker of GM-CSF- and IFNγ-expressing Th cells which may also secrete IL-17A, lending support to the potential of CXCR6 as a marker for the pathogenic Th17/exTh17 subsets. Furthermore, a recent study demonstrated that SLAMF6+ Th17 cells in secondary lymphoid organs and intestines represent a stem-like Th17 population that convert to CXCR6+ GM-CSF-secreting pathogenic Th cells that invade the CNS during EAE (202). The specific role of CXCR6 in recruiting Th17 cells into the CNS during EAE is not currently clear. Cxcr6-/- mice exhibited similar clinical EAE scores to WT mice, seemingly indicating that CXCR6 is not a critical factor for EAE despite being expressed on pathogenic GM-CSF-producing Th17 cells (196). This finding contrasted somewhat with two studies which used neutralising antibodies against CXCL16 and CXCR6, which both reported impairment in the generation and recruitment of MOG35-55-primed T cells into the CNS and thus resulted in a decrease in severity of EAE (201, 203). Future studies that specifically address the cell-intrinsic role of CXCR6 in Th17 trafficking to the CNS in EAE are required to resolve these issues. Interestingly, some studies have questioned the potency of CXCR6 in mediating chemotaxis to CXCL16, possibly indicating that CXCR6 is not involved in migration of pathogenic cells into the CNS (196, 200), which raises the possibility that other activities of CXCL16, such as its LDL scavenging function, may instead contribute to EAE pathogenesis. Future studies that specifically address these questions will be required to elucidate the function of CXCR6 on inflammatory Th cells during EAE.
CXCR4 and ACKR3
CXCR4 and its sole ligand, CXCL12, also play complex roles in EAE and have been implicated in inflammatory and regulatory T cell trafficking to the CNS (204). CXCR4 is expressed on a broad range of cells including T cells, monocytes, and endothelial cells (205, 206), while CXCL12 is also widely produced in multiple tissues including the brain and the bone marrow (109). Interestingly, CXCR4 has recently been identified to be part of a core Th signature, co-expressed with GM-CSF, that is associated with MS (207), which strongly implicates this receptor in pathogenic T cell homing. In addition to CXCR4, CXCL12 also binds an atypical chemokine receptor, ACKR3 (208). ACKR3 functions as a scavenger of CXCL12, creating CXCL12 gradients to modulate migration, and can form heterodimers with CXCR4 to modulate its activity (209, 210). The CXCR4-CXCL12-ACKR3 axis have been implicated in a variety of diseases including inflammatory diseases and cancer (206, 211).
In non-inflamed brains, the CXCR4-CXCL12-ACKR3 axis is involved in inducing the survival, differentiation, and migration of neuronal cells (212–214). This has led to theories that this axis could possibly be involved in remyelination (215). CXCL12 is increased in the CSF of MS patients compared to healthy controls and produced by astrocytes in response to IL-1β and MBP (216). Using immunohistochemical analysis, studies have shown that CXCL12 is constitutively expressed by endothelial cells on the abluminal surface of the BBB, but that CXCL12 production during MS shifts to the luminal site of the endothelium and this change is linked to increased MS severity (101, 217, 218). In EAE studies, this has been shown to be due to increased ACKR3 expression at abluminal sites, which caused an increased scavenging of CXCL12, although a recent study attributes this redistribution of CXCL12 to the actions of IL-20Rβ signalling in the CNS (219, 220). Recently, a study using neural network-based computer learning algorithms on MS patients identified CXCR4 expression to be a signature of the GM-CSF-expressing T cell population (207). These studies thus propose an important involvement of the CXCR4/CXCL12/ACKR3 axis during EAE/MS.
Absence of CXCR4, CXCL12 or ACKR3 are fatal to mice due to defects in embryonic development, which has limited analysis of the role of these molecules in EAE using knock-out models. Instead, antagonists of CXCR4/CXCL12/ACKR3 have been widely used. Administration of CXCR4 antagonists reduced disease severity and delayed onset of EAE (157, 221). In contrast, a CXCL12-globulin fusion protein, which acts as a CXCR4 agonist, reduced EAE severity associated with increased regulatory T cell recruitment (222). Furthermore, administration of a small molecule CXCR4 antagonist, AMD3100, resulted in an exacerbation of EAE scores, which was attributed to a loss of CXCL12 function in retaining CXCR4+ mononuclear cells to the perivascular space and limiting their entry into the CNS parenchyma (101). Interestingly, AMD3100 is also an agonist for ACKR3, and thus serve as both an antagonist and an agonist for two molecules of the same pathway (223). As such, the contributions of ACKR3 to pathogenic T cell trafficking EAE may also be significant. Specific blockade of ACKR3 function using a small molecule antagonist attenuated EAE due to reduced parenchymal leukocyte infiltration (219). Recently, a study using a CXCR7 antagonist resulted in a significant decrease in pathogenic Th17 levels in the CNS, and a reduced disease severity. The study postulates that CXCR7 blockade increased circulating CXCL12 levels, hence sequestering CXCR4+ T cell in the CNS (224). Taken together, the decrease in CXCL12 due to the increased scavenging role of ACKR3 may be important for leukocyte infiltration during EAE as CXCL12 constrains leukocytes from entering the parenchyma. This could thus explain why AMD3100 exacerbated EAE unlike other CXCR4 antagonists which do not activate ACKR3 signalling. The CXCR4/CXCL12/ACKR3 axis is thus an attractive target to investigate during EAE and may significantly impact infiltration of pathogenic T cells. Higher levels of CXCL12 functionality brought about by disrupting ACKR3 may reduce inflammatory T cell trafficking in EAE, although this remains to be formally demonstrated.
One challenge in targeting chemokine receptors as a treatment for inflammatory disease is that these same receptors are commonly used by regulatory components of the immune response such as Tregs. Indeed, Treg function often necessitates co-localisation with effector T cells (225). To do this Tregs will often express the same chemokine receptors as effector T cells. Tregs also express majority of the chemokine receptors mentioned in this review due to their requirement of entering areas of inflammation to suppress excessive inflammation. Targeting these chemokine receptors would also thus potentially inhibit Tregs from being able to enter the CNS and thus be unable to alleviate the inflammatory response (117, 138). Our laboratory has shown that specific deletion of CCR6 in T cells caused a delayed in EAE but ultimately led to a more severe course of disease, whereas deletion of CCR2 in T cells reduced overall disease severity without affecting onset of disease (111). It is therefore likely that different chemokine receptors differentially affect trafficking of Tregs and inflammatory T cells into the inflamed CNS and the evidence suggests that Treg migration relies more on CCR6 than CCR2. Therefore, if migratory receptors can be identified that are highly constrained to the pathogenic T cell subsets, this may minimise the impact on Treg migration and thus allow for a therapeutic avenue for EAE/MS. Further, more detailed profiling of multiple migratory receptors expressed by pathogenic T cells and Tregs in distinct compartments and at different time-points in CNS autoimmunity is therefore critical to determining these targets.
CCR9
Recently, CCR9 has been shown to direct trafficking of Th17 during EAE. A study using Tlr4-/- mice showed that CCR9 and its ligand CCL25 were specifically decreased and the migration of Th17 cells to the CNS of Tlr4-/- mice was also negatively affected (226). In support of these findings, Kadowaki et al. reported that most of the CD4+ T memory cells in the CNS at early timepoints are indeed CCR9+ (227). However, these cells also expressed the co-inhibitory molecule LAG3 during the later phase of EAE, and an increase in these cells ultimately reduced severity of EAE, indicating that CCR9+ Th cells may have regulatory function during EAE. This would be consistent with studies of Th17 cells in murine colitis models (228). Therefore, it appears that CCR9 may have multifaceted roles during EAE and may be involved in trafficking pathogenic Th subsets early during EAE, and either Tregs or exTh17/Tregs enter during the late stages of EAE, however the definitive cell-intrinsic studies remain to be done to determine this definitively.
Discussion and Future Perspectives
Much has been discovered over the years of research into how Th cells are able to traffic into the CNS parenchyma during MS. Several key questions regarding the mechanisms guiding T cell recruitment into the inflamed CNS remain unanswered. Impairment of a single chemokine-chemokine receptor interaction or adhesion molecule interaction is demonstrably not sufficient to completely inhibit CNS infiltration of autoreactive Th cells. This is likely due to the partially redundant expression of multiple migratory and adhesion molecules that can compensate for the lack of a single axis. These same migratory mechanisms are also used by regulatory cells to enter the CNS to some extent too. This highlights the importance of selecting receptors that are predominately required by pathogenic T cells and sparingly used by Tregs to infiltrate the CNS. The nature of the pathogenic T cell subsets that drive autoimmune inflammation and how they can be discriminated from regulatory cells based on their migratory requirements is therefore a key issue. Th17 cells and exTh17 cells have been implicated as the T cell subsets that are predominantly responsible for driving EAE through the production of GM-CSF (35, 229). However, the discovery of T cells dedicated to GM-CSF production that do not appear to emerge from Th17 or Th17 cells raises the possibility that multiple distinct GM-CSF-secreting T cell subsets contribute to autoimmune inflammation (51, 207).
Currently, the ThGM subset is associated with two chemokine receptors: CXCR6 and CXCR4 (51, 207). CXCR6 expression could conceivably promote recruitment of these cells into the CNS as astrocytes and glial cells upregulate CXCL16 during EAE. However, studies have shown that CXCR6 is not required for T cell entry into the CNS (196, 200). It is therefore likely that CXCR6 acts either in combination with other migratory receptors or does not directly contribute to recruitment to the CNS. CXCR4 is known to promote infiltration of CXCR4-expressing cells into the perivascular space of the CNS but functions to restrict entry into the CNS parenchyma. This could mean that CXCR4 activity on GM-CSF-secreting T cells prevents neuroinflammation rather than promoting infiltration of these cells. However, this remains to be elucidated. Therefore, there is a clear need for better understanding of the specific role of CXCR6 and CXCR4 with respect to recruitment of pathogenic T cells to CNS and for more detailed understanding of additional migratory receptors expressed by these cells.
Concluding Remarks
The numerous mechanisms used by pathogenic T cells to enter the CNS to initiate and perpetuate inflammation are currently not well understood. Chemokine receptors, integrins and selectins are all key players in T cell motility, and combinatory targeting of specific chemokine receptors, integrins and selectins might be necessary to prevent inflammatory T cell entry into the CNS during EAE/MS. Whether ThGM cells and pathogenic Th17/exTh17 cells use similar mechanisms to enter the CNS is an area of great interest, as targeting the molecules involved in the mechanisms may thus yield a therapeutic that minimises side effects of targeting other cells. Future investigation is required to identify the key molecules that affect Th cell motility during neuroinflammation, and ultimately define the best molecular targets to limit pathogenic T cell trafficking in human autoimmune diseases such as MS.
Author Contributions
AH wrote the first draft of the manuscript. CWH and CA contributed to writing of the manuscript and preparation of figures. SM and IC wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
IC and SM hold NHMRC project grant funding (1163327 and 1163335). IC is supported by a senior fellowship from MS Australia.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
IC is supported by a senior research fellowship from MS Australia.
References
1. Zhu J, Paul WE. Peripheral CD4+ T-Cell Differentiation Regulated by Networks of Cytokines and Transcription Factors. Immunol Rev (2010) 238:247–62. doi: 10.1111/j.1600-065X.2010.00951.x
2. Norman MU, Hickey MJ. Mechanisms of Lymphocyte Migration in Autoimmune Disease. Tissue Antigens (2005) 66:163–72. doi: 10.1111/j.1399-0039.2005.00434.x
3. Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, et al. IL-12p35-Deficient Mice are Susceptible to Experimental Autoimmune Encephalomyelitis: Evidence for Redundancy in the IL-12 System in the Induction of Central Nervous System Autoimmune Demyelination. J Immunol (2002) 169:7104–10. doi: 10.4049/jimmunol.169.12.7104
4. Becher B, Durell BG, Noelle RJ. Experimental Autoimmune Encephalitis and Inflammation in the Absence of Interleukin-12. J Clin Invest (2002) 110:493–7. doi: 10.1172/JCI0215751
5. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce C, Seymour B, et al. Interleukin-23 Rather Than Interleukin-12 Is the Critical Cytokine for Autoimmune Inflammation of the Brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
6. Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of Experimental Autoimmune Encephalomyelitis in IL-12 Receptor-Beta 2-Deficient Mice: IL-12 Responsiveness Is Not Required in the Pathogenesis of Inflammatory Demyelination in the Central Nervous System. J Immunol (2003) 170:2153–60. doi: 10.4049/jimmunol.170.4.2153
7. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation. J Exp Med (2005) 201:233–40. doi: 10.1084/jem.20041257
8. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6:1123–32. doi: 10.1038/ni1254
9. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
10. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
11. Stockinger B, Omenetti S. The Dichotomous Nature of T Helper 17 Cells. Nat Rev Immunol (2017) 17:535–44. doi: 10.1038/nri.2017.50
12. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-Induced Human TH17 Cells Produce IFN-Gamma or IL-10 and Are Regulated by IL-1beta. Nature (2012) 484:514–8. doi: 10.1038/nature10957
13. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
14. Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O'Malley JT, et al. Stat3 and Stat4 Direct Development of IL-17-Secreting Th Cells. J Immunol (2007) 178:4901–7. doi: 10.4049/jimmunol.178.8.4901
15. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 Regulates Cytokine-Mediated Generation of Inflammatory Helper T Cells. J Biol Chem (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
16. Shahrara S, Pickens SR, Mandelin AM 2nd, Karpus WJ, Huang Q, Kolls JK, et al. IL-17-Mediated Monocyte Migration Occurs Partially Through CC Chemokine Ligand 2/Monocyte Chemoattractant Protein-1 Induction. J Immunol (Baltimore Md 1950) (2010) 184:4479–87. doi: 10.4049/jimmunol.0901942
17. Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17a Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release From Brain Endothelial Cells. Int J Mol Sci (2017) 18:1000. doi: 10.3390/ijms18051000
18. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential Recruitment of CCR6-Expressing Th17 Cells to Inflamed Joints via CCL20 in Rheumatoid Arthritis and Its Animal Model. J Exp Med (2007) 204:2803–12. doi: 10.1084/jem.20071397
19. Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, et al. IL-9 as a Mediator of Th17-Driven Inflammatory Disease. J Exp Med (2009) 206:1653–60. doi: 10.1084/jem.20090246
20. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a TH 17 Cytokine, Mediates IL-23-Induced Dermal Inflammation and Acanthosis. Nature (2007) 445:648–51. doi: 10.1038/nature05505
21. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and Functional Features of Human Th17 Cells. J Exp Med (2007) 204:1849–61. doi: 10.1084/jem.20070663
22. Guo B. IL-10 Modulates Th17 Pathogenicity During Autoimmune Diseases. J Clin Cell Immunol (2016) 7:400. doi: 10.4172/2155-9899.1000400
23. Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E, et al. Cutting Edge: The Pathogenicity of IFN-γ-Producing Th17 Cells Is Independent of T-Bet. J Immunol (Baltimore Md 1950) (2013) 190:4478–82. doi: 10.4049/jimmunol.1203172
24. Martin F, Apetoh L, Ghiringhelli F. Controversies on the Role of Th17 in Cancer: A TGF-β-Dependent Immunosuppressive Activity? Trends Mol Med (2012) 18:742–9. doi: 10.1016/j.molmed.2012.09.007
25. Ye J, Su X, Hsueh EC, Zhang Y, Koenig JM, Hoft DF, et al. Human Tumor-Infiltrating Th17 Cells Have the Capacity to Differentiate Into IFN-γ+ and FOXP3+ T Cells With Potent Suppressive Function. Eur J Immunol (2011) 41:936–51. doi: 10.1002/eji.201040682
26. Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a Novel Subset of Human Circulating Memory CD4+ T Cells That Produce Both IL-17A and IL-4. J Allergy Clin Immunol (2010) 125:222–230. e224. doi: 10.1016/j.jaci.2009.10.012
27. Hirota K, Turner J-E, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of TH 17 Cells in Peyer's Patches Is Responsible for the Induction of T Cell–Dependent IgA Responses. Nat Immunol (2013) 14:372–9. doi: 10.1038/ni.2552
28. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 Cells Transdifferentiate Into Regulatory T Cells During Resolution of Inflammation. Nature (2015) 523:221–5. doi: 10.1038/nature14452
29. Leipe J, Pirronello F, Klose A, Schulze-Koops H, Skapenko A. Increased Plasticity of Non-Classic Th1 Cells Toward the Th17 Phenotype. Mod Rheumatol (2020) 30(5):930–6. doi: 10.1080/14397595.2019.1667473
30. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic Conversion of Foxp3+ T Cells Into TH17 Cells in Autoimmune Arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
31. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate Mapping of IL-17-Producing T Cells in Inflammatory Responses. Nat Immunol (2011) 12:255–63. doi: 10.1038/ni.1993
32. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 Plays an Important Role in the Development of Experimental Autoimmune Encephalomyelitis. J Immunol (2006) 177:566–73. doi: 10.4049/jimmunol.177.1.566
33. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do Not Contribute Vitally to Autoimmune Neuro-Inflammation in Mice. J Clin Invest (2009) 119:61–9. doi: 10.1172/JCI35997
34. Gonzalez-Garcia I, Zhao Y, Ju S, Gu Q, Liu L, Kolls JK, et al. IL-17 Signaling-Independent Central Nervous System Autoimmunity Is Negatively Regulated by TGF-Beta. J Immunol (2009) 182:2665–71. doi: 10.4049/jimmunol.0802221
35. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat Drives Production of the Cytokine GM-CSF in Helper T Cells, Which Is Essential for the Effector Phase of Autoimmune Neuroinflammation. Nat Immunol (2011) 12:560–7. doi: 10.1038/ni.2027
36. Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, et al. Targeting the GM-CSF Receptor for the Treatment of CNS Autoimmunity. J Autoimmun (2017) 84:1–11. doi: 10.1016/j.jaut.2017.06.005
37. Kroenke MA, Chensue SW, Segal BM. EAE Mediated by a Non-IFN-Gamma/Non-IL-17 Pathway. Eur J Immunol (2010) 40:2340–8. doi: 10.1002/eji.201040489
38. McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte Macrophage Colony-Stimulating Factor: A New Putative Therapeutic Target in Multiple Sclerosis. J Exp Med (2001) 194:873–82. doi: 10.1084/jem.194.7.873
39. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity (2017) 46:245–60. doi: 10.1016/j.immuni.2017.01.007
40. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
41. Ko HJ, Brady JL, Ryg-Cornejo V, Hansen DS, Vremec D, Shortman K, et al. GM-CSF-Responsive Monocyte-Derived Dendritic Cells are Pivotal in Th17 Pathogenesis. J Immunol (2014) 192:2202–9. doi: 10.4049/jimmunol.1302040
42. Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF Promotes Chronic Disability in Experimental Autoimmune Encephalomyelitis by Altering the Composition of Central Nervous System–Infiltrating Cells, But Is Dispensable for Disease Induction. J Immunol (2018) 200:966–73. doi: 10.4049/jimmunol.1701484
43. Pierson ER, Goverman JM. GM-CSF Is Not Essential for Experimental Autoimmune Encephalomyelitis But Promotes Brain-Targeted Disease. JCI Insight (2017) 2(7):e92362. doi: 10.1172/jci.insight.92362
44. Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 Effector Cells Induce Experimental Autoimmune Encephalomyelitis With Different Pathological Phenotypes. J Immunol (2009) 183:7169–77. doi: 10.4049/jimmunol.0901906
45. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, et al. Late Developmental Plasticity in the T Helper 17 Lineage. Immunity (2009) 30:92–107. doi: 10.1016/j.immuni.2008.11.005
46. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and Molecular Signature of Pathogenic TH17 Cells. Nat Immunol (2012) 13:991–9. doi: 10.1038/ni.2416
47. Arbelaez CA, Glatigny S, Duhen R, Eberl G, Oukka M, Bettelli E. IL-7/IL-7 Receptor Signaling Differentially Affects Effector CD4+ T Cell Subsets Involved in Experimental Autoimmune Encephalomyelitis. J Immunol (2015) 195:1974–83. doi: 10.4049/jimmunol.1403135
48. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 Programs a Distinct Subset of GM-CSF-Producing T Helper Cells That Is Essential for Autoimmune Neuroinflammation. Cell Res (2014) 24:1387–402. doi: 10.1038/cr.2014.154
49. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The Interleukin 23 Receptor Is Essential for the Terminal Differentiation of Interleukin 17-Producing Effector T Helper Cells In Vivo. Nat Immunol (2009) 10:314–24. doi: 10.1038/ni.1698
50. Chen Y, Chauhan SK, Tan X, Dana R. Interleukin-7 and -15 Maintain Pathogenic Memory Th17 Cells in Autoimmunity. J Autoimmun (2017) 77:96–103. doi: 10.1016/j.jaut.2016.11.003
51. Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, et al. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1&X3b2. Immunity (2019) 50:1289–304.e1286. doi: 10.1016/j.immuni.2019.04.006
52. Noster R, Riedel R, Mashreghi M-F, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF Expression Are Antagonistically Regulated by Human T Helper Cells. Sci Trans Med (2014) 6:241ra280–241ra280. doi: 10.1126/scitranslmed.3008706
53. Engelhardt B, Sorokin L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin Immunopathol (2009) 31:497–511. doi: 10.1007/s00281-009-0177-0
54. Barthel SR, Gavino JD, Descheny L, Dimitroff CJ. Targeting Selectins and Selectin Ligands in Inflammation and Cancer. Expert Opin Ther Targets (2007) 11:1473–91. doi: 10.1517/14728222.11.11.1473
55. Möβner R, Fassbender K, Kühnen J, Schwarte A, Hennend M. Circulating L-Selectin in Multiple Sclerosis Patients With Active, Gadolinium-Enhancing Brain Plaques. J Neuroimmunol (1996) 65:61–5. doi: 10.1016/0165-5728(96)00003-3
56. Archelos JJ, Jung S, Rinner W, Lassmann H, Miyasaka M, Hartung H-P. Role of the Leukocyte-Adhesion Molecule L-Selectin in Experimental Autoimmune Encephalomyelitis. J Neurol Sci (1998) 159:127–34. doi: 10.1016/S0022-510X(98)00154-3
57. Li O, Liu J-Q, Zhang H, Zheng P, Liu Y, Bai X-F. CD62L Is Required for the Priming of Encephalitogenic T Cells But Does Not Play a Major Role in the Effector Phase of Experimental Autoimmune Encephalomyelitis. Scand J Immunol (2006) 64:117–24. doi: 10.1111/j.1365-3083.2006.01783.x
58. Kerfoot SM, Kubes P. Overlapping Roles of P-Selectin and Alpha 4 Integrin to Recruit Leukocytes to the Central Nervous System in Experimental Autoimmune Encephalomyelitis. J Immunol (2002) 169:1000–6. doi: 10.4049/jimmunol.169.2.1000
59. Piccio L, Rossi B, Scarpini E, Laudanna C, Giagulli C, Issekutz AC, et al. Molecular Mechanisms Involved in Lymphocyte Recruitment in Inflamed Brain Microvessels: Critical Roles for P-Selectin Glycoprotein Ligand-1 and Heterotrimeric G(i)-Linked Receptors. J Immunol (2002) 168:1940–9. doi: 10.4049/jimmunol.168.4.1940
60. Kerfoot SM, Norman MU, Lapointe BM, Bonder CS, Zbytnuik L, Kubes P. Reevaluation of P-Selectin and Alpha 4 Integrin as Targets for the Treatment of Experimental Autoimmune Encephalomyelitis. J Immunol (2006) 176:6225–34. doi: 10.4049/jimmunol.176.10.6225
61. Angiari S, Donnarumma T, Rossi B, Dusi S, Pietronigro E, Zenaro E, et al. TIM-1 Glycoprotein Binds the Adhesion Receptor P-Selectin and Mediates T Cell Trafficking During Inflammation and Autoimmunity. Immunity (2014) 40:542–53. doi: 10.1016/j.immuni.2014.03.004
62. Velazquez FE, Anastasiou M, Carrillo-Salinas FJ, Ngwenyama N, Salvador AM, Nevers T, et al. Sialomucin CD43 Regulates T Helper Type 17 Cell Intercellular Adhesion Molecule 1 Dependent Adhesion, Apical Migration and Transendothelial Migration. Immunology (2019) 157:52–69. doi: 10.1111/imm.13047
63. Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA Jr. Surface Expression of Alpha 4 Integrin by CD4 T Cells is Required for Their Entry Into Brain Parenchyma. J Exp Med (1993) 177:57–68. doi: 10.1084/jem.177.1.57
64. Kent SJ, Karlik SJ, Cannon C, Hines DK, Yednock TA, Fritz LC, et al. A Monoclonal Antibody to Alpha 4 Integrin Suppresses and Reverses Active Experimental Allergic Encephalomyelitis. J Neuroimmunol (1995) 58:1–10. doi: 10.1016/0165-5728(94)00165-K
65. Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of Experimental Autoimmune Encephalomyelitis by Antibodies Against Alpha 4 Beta 1 Integrin. Nature (1992) 356:63–6. doi: 10.1038/356063a0
66. Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N Engl J Med (2003) 348:15–23. doi: 10.1056/NEJMoa020696
67. Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N Engl J Med (2006) 354:899–910. doi: 10.1056/NEJMoa044397
68. Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab Plus Interferon Beta-1a for Relapsing Multiple Sclerosis. N Engl J Med (2006) 354:911–23. doi: 10.1056/NEJMoa044396
69. Tubridy N, Behan PO, Capildeo R, Chaudhuri A, Forbes R, Hawkins CP, et al. The Effect of Anti-Alpha4 Integrin Antibody on Brain Lesion Activity in MS. The UK Antegren Study Group. Neurology (1999) 53:466–72. doi: 10.1212/WNL.53.3.466
70. Glatigny S, Duhen R, Oukka M, Bettelli E. Cutting Edge: Loss of Alpha4 Integrin Expression Differentially Affects the Homing of Th1 and Th17 Cells. J Immunol (2011) 187:6176–9. doi: 10.4049/jimmunol.1102515
71. Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B, et al. Th17 Lymphocytes Traffic to the Central Nervous System Independently of Alpha4 Integrin Expression During EAE. J Exp Med (2011) 208:2465–76. doi: 10.1084/jem.20110434
72. Glatigny S, Duhen R, Arbelaez C, Kumari S, Bettelli E. Integrin Alpha L Controls the Homing of Regulatory T Cells During CNS Autoimmunity in the Absence of Integrin Alpha 4. Sci Rep (2015) 5:7834. doi: 10.1038/srep07834
73. Dusi S, Angiari S, Pietronigro EC, Lopez N, Angelini G, Zenaro E, et al. LFA-1 Controls Th1 and Th17 Motility Behavior in the Inflamed Central Nervous System. Front Immunol (2019) 10:2436. doi: 10.3389/fimmu.2019.02436
74. Du F, Garg AV, Kosar K, Majumder S, Kugler DG, Mir GH, et al. Inflammatory Th17 Cells Express Integrin αvβ3 for Pathogenic Function. Cell Rep (2016) 16:1339–51. doi: 10.1016/j.celrep.2016.06.065
75. Han S, Zhang F, Hu Z, Sun Y, Yang J, Davies H, et al. Dose-Dependent Anti-Inflammatory and Neuroprotective Effects of an ανβ3 Integrin-Binding Peptide. Mediators Inflammation (2013) 2013:268486–6. doi: 10.1155/2013/268486
76. Griffith JW, Sokol CL, Luster AD. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
77. Zlotnik A, Yoshie O. The Chemokine Superfamily Revisited. Immunity (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
78. Melchers F, Rolink AG, Schaniel C. The Role of Chemokines in Regulating Cell Migration During Humoral Immune Responses. Cell (1999) 99:351–4. doi: 10.1016/S0092-8674(00)81521-4
79. Lintermans LL, Rutgers A, Stegeman CA, Heeringa P, Abdulahad WH. Chemokine Receptor Co-Expression Reveals Aberrantly Distributed TH Effector Memory Cells in GPA Patients. Arthritis Res Ther (2017) 19:136. doi: 10.1186/s13075-017-1343-8
80. Strazza M, Mor A. Consider the Chemokines: A Review of the Interplay Between Chemokines and T Cell Subset Function. Discovery Med (2017) 24:31–9.
81. Yan Y, Chen R, Wang X, Hu K, Huang L, Lu M, et al. CCL19 and CCR7 Expression, Signaling Pathways, and Adjuvant Functions in Viral Infection and Prevention. Front Cell Dev Biol (2019) 7. doi: 10.3389/fcell.2019.00212
82. Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, et al. Differential Expression of Chemokine Receptors and Chemotactic Responsiveness of Type 1 T Helper Cells (Th1s) and Th2s. J Exp Med (1998) 187:129–34. doi: 10.1084/jem.187.1.129
83. Lian J, Luster AD. Chemokine-Guided Cell Positioning in the Lymph Node Orchestrates the Generation of Adaptive Immune Responses. Curr Opin Cell Biol (2015) 36:1–6. doi: 10.1016/j.ceb.2015.05.003
84. Campanella GSV, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, et al. Chemokine Receptor CXCR3 and its Ligands CXCL9 and CXCL10 are Required for the Development of Murine Cerebral Malaria. Proc Natl Acad Sci USA (2008) 105:4814–9. doi: 10.1073/pnas.0801544105
85. Kristensen NN, Gad M, Thomsen AR, Lu B, Gerard C, Claesson MH. CXC Chemokine Receptor 3 Expression Increases the Disease-Inducing Potential of CD4+ CD25- T Cells in Adoptive Transfer Colitis. Inflammation Bowel Dis (2006) 12:374–81. doi: 10.1097/01.MIB.0000217337.15442.e1
86. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface Phenotype and Antigenic Specificity of Human Interleukin 17-Producing T Helper Memory Cells. Nat Immunol (2007) 8:639–46. doi: 10.1038/ni1467
87. Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T Cells That are Able to Produce IL-17 Express the Chemokine Receptor CCR6. J Immunol (2008) 180:214–21. doi: 10.4049/jimmunol.180.1.214
88. Comerford I, Bunting M, Fenix K, Haylock-Jacobs S, Litchfield W, Harata-Lee Y, et al. An Immune Paradox: How can the Same Chemokine Axis Regulate Both Immune Tolerance and Activation?: CCR6/CCL20: A Chemokine Axis Balancing Immunological Tolerance and Inflammation in Autoimmune Disease. Bioessays (2010) 32:1067–76. doi: 10.1002/bies.201000063
89. Paradis TJ, Cole SH, Nelson RT, Gladue RP. Essential Role of CCR6 in Directing Activated T Cells to the Skin During Contact Hypersensitivity. J Invest Dermatol (2008) 128:628–33. doi: 10.1038/sj.jid.5701055
90. Varona R, Villares R, Carramolino L, Goya Í, Zaballos Á, Gutiérrez J, et al. CCR6-Deficient Mice Have Impaired Leukocyte Homeostasis and Altered Contact Hypersensitivity and Delayed-Type Hypersensitivity Responses. J Clin Invest (2001) 107:R37–45. doi: 10.1172/JCI11297
91. Goverman J. Autoimmune T Cell Responses in the Central Nervous System. Nat Rev Immunol (2009) 9:393–407. doi: 10.1038/nri2550
92. Alt C, Laschinger M, Engelhardt B. Functional Expression of the Lymphoid Chemokines CCL19 (ELC) and CCL 21 (SLC) at the Blood-Brain Barrier Suggests Their Involvement in G-Protein-Dependent Lymphocyte Recruitment Into the Central Nervous System During Experimental Autoimmune Encephalomyelitis. Eur J Immunol (2002) 32:2133–44. doi: 10.1002/1521-4141(200208)32:8<2133::AID-IMMU2133>3.0.CO;2-W
93. Columba-Cabezas S, Serafini B, Ambrosini E, Aloisi F. Lymphoid Chemokines CCL19 and CCL21 are Expressed in the Central Nervous System During Experimental Autoimmune Encephalomyelitis: Implications for the Maintenance of Chronic Neuroinflammation. Brain Pathol (2003) 13:38–51. doi: 10.1111/j.1750-3639.2003.tb00005.x
94. Comerford I, Nibbs RJ, Litchfield W, Bunting M, Harata-Lee Y, Haylock-Jacobs S, et al. The Atypical Chemokine Receptor CCX-CKR Scavenges Homeostatic Chemokines in Circulation and Tissues and Suppresses Th17 Responses. Blood (2010) 116:4130–40. doi: 10.1182/blood-2010-01-264390
95. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C Chemokine Receptor 6-Regulated Entry of TH-17 Cells Into the CNS Through the Choroid Plexus is Required for the Initiation of EAE. Nat Immunol (2009) 10:514–23. doi: 10.1038/ni.1716
96. Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Chemokines and Their Receptors in the Central Nervous System. Front Neuroendocrinol (2001) 22:147–84. doi: 10.1006/frne.2001.0214
97. Bajetto A, Bonavia R, Barbero S, Piccioli P, Costa A, Florio T, et al. Glial and Neuronal Cells Express Functional Chemokine Receptor CXCR4 and its Natural Ligand Stromal Cell-Derived Factor 1. J Neurochem (1999) 73:2348–57. doi: 10.1046/j.1471-4159.1999.0732348.x
98. Schwaeble WJ, Stover CM, Schall TJ, Dairaghi DJ, Trinder PK, Linington C, et al. Neuronal Expression of Fractalkine in the Presence and Absence of Inflammation. FEBS Lett (1998) 439:203–7. doi: 10.1016/S0014-5793(98)01384-2
99. Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, et al. Regulation of Adaptive Immunity by the Fractalkine Receptor During Autoimmune Inflammation. J Immunol (2013) 191:1063–72. doi: 10.4049/jimmunol.1300040
100. Rayasam A, Kijak JA, Dallmann M, Hsu M, Zindl N, Lindstedt A, et al. Regional Distribution of CNS Antigens Differentially Determines T-Cell Mediated Neuroinflammation in a CX3CR1-Dependent Manner. J Neurosci (2018) 38:7058–71. doi: 10.1523/JNEUROSCI.0366-18.2018
101. McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. CXCL12 Limits Inflammation by Localizing Mononuclear Infiltrates to the Perivascular Space During Experimental Autoimmune Encephalomyelitis. J Immunol (2006) 177:8053. doi: 10.4049/jimmunol.177.11.8053
102. Pahuja A, Maki RA, Hevezi PA, Chen A, Verge GM, Lechner SM, et al. Experimental Autoimmune Encephalomyelitis Develops in CC Chemokine Receptor 7-Deficient Mice With Altered T-Cell Responses. Scand J Immunol (2006) 64:361–9. doi: 10.1111/j.1365-3083.2006.01787.x
103. Belikan P, Buhler U, Wolf C, Pramanik GK, Gollan R, Zipp F, et al. CCR7 on CD4(+) T Cells Plays a Crucial Role in the Induction of Experimental Autoimmune Encephalomyelitis. J Immunol (2018) 200:2554–62. doi: 10.4049/jimmunol.1701419
104. Niu X, Sang H, Wang J. Naringenin Attenuates Experimental Autoimmune Encephalomyelitis by Protecting the Intact of Blood-Brain Barrier and Controlling Inflammatory Cell Migration. J Nutr Biochem (2021) 89:108560. doi: 10.1016/j.jnutbio.2020.108560
105. Arima Y, Harada M, Kamimura D, Park JH, Kawano F, Yull FE, et al. Regional Neural Activation Defines a Gateway for Autoreactive T Cells to Cross the Blood-Brain Barrier. Cell (2012) 148:447–57. doi: 10.1016/j.cell.2012.01.022
106. Kivisäkk P, Imitola J, Rasmussen S, Elyaman W, Zhu B, Ransohoff RM, et al. Localizing Central Nervous System Immune Surveillance: Meningeal Antigen-Presenting Cells Activate T Cells During Experimental Autoimmune Encephalomyelitis. Ann Neurol (2009) 65:457–69. doi: 10.1002/ana.21379
107. Steinman L. Inflammatory Cytokines at the Summits of Pathological Signal Cascades in Brain Diseases. Sci Signal (2013) 6:pe3. doi: 10.1126/scisignal.2003898
108. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular Mechanisms of IL-17-Induced Blood-Brain Barrier Disruption. FASEB J (2010) 24:1023–34. doi: 10.1096/fj.09-141978
109. Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, et al. SDF-1 (CXCL12) is Upregulated in the Ischemic Penumbra Following Stroke: Association With Bone Marrow Cell Homing to Injury. J Neuropathol Exp Neurol (2004) 63:84–96. doi: 10.1093/jnen/63.1.84
110. Kim JV, Tadokoro CE, Shen S, Jiang N, Lafaille JJ, Dustin ML. CXCR6 Is Required for T Cell Recruitment Into Injured Gray Matter in EAE. FASEB J (2008) 22:424–4. doi: 10.1096/fasebj.22.2_supplement.424
111. Kara EE, McKenzie DR, Bastow CR, Gregor CE, Fenix KA, Ogunniyi AD, et al. CCR2 Defines In Vivo Development and Homing of IL-23-Driven GM-CSF-Producing Th17 Cells. Nat Commun (2015) 6:8644. doi: 10.1038/ncomms9644
112. Schutyser E, Struyf S, Van Damme J. The CC Chemokine CCL20 and its Receptor CCR6. Cytokine Growth Factor Rev (2003) 14:409–26. doi: 10.1016/S1359-6101(03)00049-2
113. Kohler RE, Caon AC, Willenborg DO, Clark-Lewis I, McColl SR. A Role for Macrophage Inflammatory Protein-3α/CC Chemokine Ligand 20 in Immune Priming During T Cell-Mediated Inflammation of the Central Nervous System. J Immunol (2003) 170:6298–306. doi: 10.4049/jimmunol.170.12.6298
114. Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, et al. Inhibition of CCR6 Function Reduces the Severity of Experimental Autoimmune Encephalomyelitis via Effects on the Priming Phase of the Immune Response. J Immunol (2009) 182:3121–30. doi: 10.4049/jimmunol.0713169
115. Ambrosini E, Columba-Cabezas S, Serafini B, Muscella A, Aloisi F. Astrocytes are the Major Intracerebral Source of Macrophage Inflammatory Protein-3alpha/CCL20 in Relapsing Experimental Autoimmune Encephalomyelitis and In Vitro. Glia (2003) 41:290–300. doi: 10.1002/glia.10193
116. Li R, Xu W, Chen Y, Qiu W, Shu Y, Wu A, et al. Raloxifene Suppresses Experimental Autoimmune Encephalomyelitis and NF-κb-Dependent CCL20 Expression in Reactive Astrocytes. PloS One (2014) 9:e94320. doi: 10.1371/journal.pone.0094320
117. Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. CCR6 Regulates the Migration of Inflammatory and Regulatory T Cells. J Immunol (2008) 181:8391–401. doi: 10.4049/jimmunol.181.12.8391
118. Elhofy A, DePaolo RW, Lira SA, Lukacs NW, Karpus WJ. Mice Deficient for CCR6 Fail to Control Chronic Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2009) 213:91–9. doi: 10.1016/j.jneuroim.2009.05.011
119. Villares R, Cadenas V, Lozano M, Almonacid L, Zaballos A, Martínez AC, et al. CCR6 Regulates EAE Pathogenesis by Controlling Regulatory CD4+ T-Cell Recruitment to Target Tissues. Eur J Immunol (2009) 39:1671–81. doi: 10.1002/eji.200839123
120. Mahnke YD, Beddall MH, Roederer M. OMIP-017: Human CD4(+) Helper T-Cell Subsets Including Follicular Helper Cells. Cytometry A (2013) 83:439–40. doi: 10.1002/cyto.a.22269
121. Pandya JM, Lundell AC, Hallström M, Andersson K, Nordström I, Rudin A. Circulating T Helper and T Regulatory Subsets in Untreated Early Rheumatoid Arthritis and Healthy Control Subjects. J Leukoc Biol (2016) 100:823–33. doi: 10.1189/jlb.5A0116-025R
122. Ramstein J, Broos CE, Simpson LJ, Ansel KM, Sun SA, Ho ME, et al. IFN-γ-Producing T-Helper 17.1 Cells Are Increased in Sarcoidosis and Are More Prevalent Than T-Helper Type 1 Cells. Am J Respir Crit Care Med (2016) 193:1281–91. doi: 10.1164/rccm.201507-1499OC
123. van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T Helper 17.1 Cells Associate With Multiple Sclerosis Disease Activity: Perspectives for Early Intervention. Brain (2018) 141:1334–49. doi: 10.1093/brain/awy069
124. Restorick SM, Durant L, Kalra S, Hassan-Smith G, Rathbone E, Douglas MR, et al. CCR6(+) Th Cells in the Cerebrospinal Fluid of Persons With Multiple Sclerosis are Dominated by Pathogenic non-Classic Th1 Cells and GM-CSF-Only-Secreting Th Cells. Brain Behav Immun (2017) 64:71–9. doi: 10.1016/j.bbi.2017.03.008
125. Kurihara T, Warr G, Loy J, Bravo R. Defects in Macrophage Recruitment and Host Defense in Mice Lacking the CCR2 Chemokine Receptor. J Exp Med (1997) 186:1757–62. doi: 10.1084/jem.186.10.1757
126. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe Reduction in Leukocyte Adhesion and Monocyte Extravasation in Mice Deficient in CC Chemokine Receptor 2. Proc Natl Acad Sci (1997) 94:12053–8. doi: 10.1073/pnas.94.22.12053
127. Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, et al. CCR2+Ly-6Chi Monocytes are Crucial for the Effector Phase of Autoimmunity in the Central Nervous System. Brain (2009) 132:2487–500. doi: 10.1093/brain/awp144
128. Komiya H, Takeuchi H, Ogawa Y, Hatooka Y, Takahashi K, Katsumoto A, et al. CCR2 is Localized in Microglia and Neurons, as Well as Infiltrating Monocytes, in the Lumbar Spinal Cord of ALS Mice. Mol Brain (2020) 13:64. doi: 10.1186/s13041-020-00607-3
129. Sato W, Tomita A, Ichikawa D, Lin Y, Kishida H, Miyake S, et al. CCR2(+)CCR5(+) T Cells Produce Matrix Metalloproteinase-9 and Osteopontin in the Pathogenesis of Multiple Sclerosis. J Immunol (2012) 189:5057–65. doi: 10.4049/jimmunol.1202026
130. Simpson J, Newcombe J, Cuzner M, Woodroofe M. Expression of Monocyte Chemoattractant Protein-1 and Other β-Chemokines by Resident Glia and Inflammatory Cells in Multiple Sclerosis Lesions. J Neuroimmunol (1998) 84:238–49. doi: 10.1016/S0165-5728(97)00208-7
131. Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG. Role of CCR2 in Inflammatory Conditions of the Central Nervous System. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2014) 34:1425–9. doi: 10.1038/jcbfm.2014.120
132. Moreno M, Bannerman P, Ma J, Guo F, Miers L, Soulika AM, et al. Conditional Ablation of Astroglial CCL2 Suppresses CNS Accumulation of M1 Macrophages and Preserves Axons in Mice With MOG Peptide EAE. J Neurosci (2014) 34:8175–85. doi: 10.1523/JNEUROSCI.1137-14.2014
133. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to Experimental Autoimmune Encephalomyelitis in Mice Lacking the CC Chemokine Receptor (CCR)2. J Exp Med (2000) 192:1075–80. doi: 10.1084/jem.192.7.1075
134. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC Chemokine Receptor 2 Is Critical for Induction of Experimental Autoimmune Encephalomyelitis. J Exp Med (2000) 192:899–906. doi: 10.1084/jem.192.6.899
135. dos Santos AC, Barsante MM, Arantes RME, Bernard CC, Teixeira MM, Carvalho-Tavares J. CCL2 and CCL5 Mediate Leukocyte Adhesion in Experimental Autoimmune Encephalomyelitis—an Intravital Microscopy Study. J Neuroimmunol (2005) 162:122–9. doi: 10.1016/j.jneuroim.2005.01.020
136. Huang D, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of Monocyte Chemoattractant Protein 1 in Mice Leads to Decreased Local Macrophage Recruitment and Antigen-Specific T Helper Cell Type 1 Immune Response in Experimental Autoimmune Encephalomyelitis. J Exp Med (2001) 193:713–26. doi: 10.1084/jem.193.6.713
137. Nakazato Y, Fujita Y, Nakazato M, Yamashita T. Neurons Promote Encephalitogenic CD4(+) Lymphocyte Infiltration in Experimental Autoimmune Encephalomyelitis. Sci Rep (2020) 10:7354–4. doi: 10.1038/s41598-020-64363-z
138. Brühl H, Cihak J, Schneider MA, Plachý J, Rupp T, Wenzel I, et al. Dual Role of CCR2 During Initiation and Progression of Collagen-Induced Arthritis: Evidence for Regulatory Activity of CCR2+ T Cells. J Immunol (2004) 172:890–8. doi: 10.4049/jimmunol.172.2.890
139. Szczuciński A, Losy J. Chemokines and Chemokine Receptors in Multiple Sclerosis. Potential Targets for New Therapies. Acta Neurol Scand (2007) 115:137–46. doi: 10.1111/j.1600-0404.2006.00749.x
140. Kim RY, Hoffman AS, Itoh N, Ao Y, Spence R, Sofroniew MW, et al. Astrocyte CCL2 Sustains Immune Cell Infiltration in Chronic Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2014) 274:53–61. doi: 10.1016/j.jneuroim.2014.06.009
141. Horuk R. Chemokine Receptor Antagonists: Overcoming Developmental Hurdles. Nat Rev Drug Discovery (2009) 8:23–33. doi: 10.1038/nrd2734
142. Pease J, Horuk R. Chemokine Receptor Antagonists. J Medicinal Chem (2012) 55:9363–92. doi: 10.1021/jm300682j
143. Robichon K, Patel V, Connor B, La Flamme AC. Clozapine Reduces Infiltration Into the CNS by Targeting Migration in Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2020) 17:53. doi: 10.1186/s12974-020-01733-4
144. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of Specific Chemokines and Chemokine Receptors in the Central Nervous System of Multiple Sclerosis Patients. J Clin Invest (1999) 103:807–15. doi: 10.1172/JCI5150
145. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine Receptor Specific for IP10 and Mig: Structure, Function, and Expression in Activated T-Lymphocytes. J Exp Med (1996) 184:963–9. doi: 10.1084/jem.184.3.963
146. Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, et al. Interferon–inducible T Cell Alpha Chemoattractant (I-TAC): A Novel Non-ELR CXC Chemokine With Potent Activity on Activated T Cells Through Selective High Affinity Binding to CXCR3. J Exp Med (1998) 187:2009–21. doi: 10.1084/jem.187.12.2009
147. Piali L, Weber C, LaRosa G, Mackay CR, Springer TA, Clark-Lewis I, et al. The Chemokine Receptor CXCR3 Mediates Rapid and Shear-Resistant Adhesion-Induction of Effector T Lymphocytes by the Chemokines IP10 and Mig. Eur J Immunol (1998) 28:961–72. doi: 10.1002/(SICI)1521-4141(199803)28:03<961::AID-IMMU961>3.0.CO;2-4
148. Nakajima C, Mukai T, Yamaguchi N, Morimoto Y, Park WR, Iwasaki M, et al. Induction of the Chemokine Receptor CXCR3 on TCR-Stimulated T Cells: Dependence on the Release From Persistent TCR-Triggering and Requirement for IFN-γ Stimulation. Eur J Immunol (2002) 32:1792–801. doi: 10.1002/1521-4141(200206)32:6<1792::AID-IMMU1792>3.0.CO;2-0
149. Xie JH, Nomura N, Lu M, Chen SL, Koch GE, Weng Y, et al. Antibody-Mediated Blockade of the CXCR3 Chemokine Receptor Results in Diminished Recruitment of T Helper 1 Cells Into Sites of Inflammation. J Leukoc Biol (2003) 73:771–80. doi: 10.1189/jlb.1102573
150. Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, et al. CXCR3-Dependent Recruitment and CCR6-Mediated Positioning of Th-17 Cells in the Inflamed Liver. J Hepatol (2012) 57:1044–51. doi: 10.1016/j.jhep.2012.07.008
151. Panzer U, Steinmetz OM, Paust H-J, Meyer-Schwesinger C, Peters A, Turner J-E, et al. Chemokine Receptor CXCR3 Mediates T Cell Recruitment and Tissue Injury in Nephrotoxic Nephritis in Mice. J Am Soc Nephrol (2007) 18:2071. doi: 10.1681/ASN.2006111237
152. Steinmetz OM, Turner J-E, Paust H-J, Lindner M, Peters A, Heiss K, et al. CXCR3 Mediates Renal Th1 and Th17 Immune Response in Murine Lupus Nephritis. J Immunol (2009) 183:4693. doi: 10.4049/jimmunol.0802626
153. Sporici R, Issekutz TB. CXCR3 Blockade Inhibits T-Cell Migration Into the CNS During EAE and Prevents Development of Adoptively Transferred, But Not Actively Induced, Disease. Eur J Immunol (2010) 40:2751–61. doi: 10.1002/eji.200939975
154. Chung C-Y, Liao F. CXCR3 Signaling in Glial Cells Ameliorates Experimental Autoimmune Encephalomyelitis by Restraining the Generation of a Pro-Th17 Cytokine Milieu and Reducing CNS-Infiltrating Th17 Cells. J Neuroinflamm (2016) 13:76–6. doi: 10.1186/s12974-016-0536-4
155. Liu L, Huang D, Matsui M, He TT, Hu T, DeMartino J, et al. Severe Disease, Unaltered Leukocyte Migration, and Reduced IFN-γ Production in CXCR3–/– Mice With Experimental Autoimmune Encephalomyelitis. J Immunol (2006) 176:4399–409. doi: 10.4049/jimmunol.176.7.4399
156. Müller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, et al. CXCR3 Signaling Reduces the Severity of Experimental Autoimmune Encephalomyelitis by Controlling the Parenchymal Distribution of Effector and Regulatory T Cells in the Central Nervous System. J Immunol (2007) 179:2774–86. doi: 10.4049/jimmunol.179.5.2774
157. Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR. Antagonism of the Chemokine Receptors CXCR3 and CXCR4 Reduces the Pathology of Experimental Autoimmune Encephalomyelitis. Brain Pathol (2008) 18:504–16. doi: 10.1111/j.1750-3639.2008.00154.x
158. Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD, et al. CXCL10 (IFN-γ-Inducible Protein-10) Control of Encephalitogenic CD4+ T Cell Accumulation in the Central Nervous System During Experimental Autoimmune Encephalomyelitis. J Immunol (2001) 166:7617–24. doi: 10.4049/jimmunol.166.12.7617
159. Schmitz K, Pickert G, Wijnvoord N, Häussler A, Tegeder I. Dichotomy of CCL21 and CXCR3 in Nerve Injury-Evoked and Autoimmunity-Evoked Hyperalgesia. Brain Behav Immun (2013) 32:186–200. doi: 10.1016/j.bbi.2013.04.011
160. Lalor SJ, Segal BM. Th1-Mediated Experimental Autoimmune Encephalomyelitis Is CXCR3 Independent. Eur J Immunol (2013) 43:2866–74. doi: 10.1002/eji.201343499
161. Sindern E, Patzold T, Ossege LM, Gisevius A, Malin J-P. Expression of Chemokine Receptor CXCR3 on Cerebrospinal Fluid T-Cells Is Related to Active MRI Lesion Appearance in Patients With Relapsing–Remitting Multiple Sclerosis. J Neuroimmunol (2002) 131:186–90. doi: 10.1016/S0165-5728(02)00263-1
162. Mahad DJ, Howell SJ, Woodroofe MN. Expression of Chemokines in the CSF and Correlation With Clinical Disease Activity in Patients With Multiple Sclerosis. J Neurol Neurosurg Psychiatry (2002) 72:498–502. doi: 10.1136/jnnp.72.4.498
163. Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of the Interferon-Gamma-Inducible Chemokines IP-10 and Mig and Their Receptor, CXCR3, in Multiple Sclerosis Lesions. Neuropathol Appl Neurobiol (2000) 26:133–42. doi: 10.1046/j.1365-2990.2000.026002133.x
164. Tur C, Montalban X, Tintoré M, Nos C, Río J, Aymerich FX, et al. Interferon β-1b for the Treatment of Primary Progressive Multiple Sclerosis: Five-Year Clinical Trial Follow-Up. Arch Neurol (2011) 68:1421–7. doi: 10.1001/archneurol.2011.241
165. Sørensen TL, Sellebjerg F. Selective Suppression of Chemokine Receptor CXCR3 Expression by Interferon-Beta1a in Multiple Sclerosis. Mult Scler (2002) 8:104–7. doi: 10.1191/1352458502ms781oa
166. Johnson M, Li AR, Liu J, Fu Z, Zhu L, Miao S, et al. Discovery and Optimization of a Series of Quinazolinone-Derived Antagonists of CXCR3. Bioorg Med Chem Lett (2007) 17:3339–43. doi: 10.1016/j.bmcl.2007.03.106
167. Subileau EA, Rezaie P, Davies HA, Colyer FM, Greenwood J, Male DK, et al. Expression of Chemokines and Their Receptors by Human Brain Endothelium: Implications for Multiple Sclerosis. J Neuropathol Exp Neurol (2009) 68:227–40. doi: 10.1097/NEN.0b013e318197eca7
168. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T Cells Are Increased in Multiple Sclerosis and Their Ligands MIP-1α and IP-10 are Expressed in Demyelinating Brain Lesions. Proc Natl Acad Sci (1999) 96:6873–8. doi: 10.1073/pnas.96.12.6873
169. Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular Localization of the Chemokine Receptor CCR5. Correlation to Cellular Targets of HIV-1 Infection. Am J Pathol (1997) 151:1341.
170. Westmoreland S, Alvarez X, Debakker C, Aye P, Wilson M, Williams K, et al. Developmental Expression Patterns of CCR5 and CXCR4 in the Rhesus Macaque Brain. J Neuroimmunol (2002) 122:146–58. doi: 10.1016/S0165-5728(01)00457-X
171. Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM. Synchronous Synthesis of Alpha-and Beta-Chemokines by Cells of Diverse Lineage in the Central Nervous System of Mice With Relapses of Chronic Experimental Autoimmune Encephalomyelitis. Am J Pathol (1997) 150:617.
172. Dubois B, Masure S, Hurtenbach U, Paemen L, Heremans H, Van Den Oord J, et al. Resistance of Young Gelatinase B–deficient Mice to Experimental Autoimmune Encephalomyelitis and Necrotizing Tail Lesions. J Clin Invest (1999) 104:1507–15. doi: 10.1172/JCI6886
173. Lindberg RL, De Groot CJ, Montagne L, Freitag P, van der Valk P, Kappos L, et al. The Expression Profile of Matrix Metalloproteinases (MMPs) and Their Inhibitors (TIMPs) in Lesions and Normal Appearing White Matter of Multiple Sclerosis. Brain (2001) 124:1743–53. doi: 10.1093/brain/124.9.1743
174. Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, et al. The Influence of the Proinflammatory Cytokine, Osteopontin, on Autoimmune Demyelinating Disease. Science (2001) 294:1731–5. doi: 10.1126/science.1062960
175. Jansson M, Panoutsakopoulou V, Baker J, Klein L, Cantor H. Cutting Edge: Attenuated Experimental Autoimmune Encephalomyelitis in Eta-1/Osteopontin-Deficient Mice. J Immunol (2002) 168:2096–9. doi: 10.4049/jimmunol.168.5.2096
176. Chiocchetti A, Comi C, Indelicato M, Castelli L, Mesturini R, Bensi T, et al. Osteopontin Gene Haplotypes Correlate With Multiple Sclerosis Development and Progression. J Neuroimmunol (2005) 163:172–8. doi: 10.1016/j.jneuroim.2005.02.020
177. Tran EH, Kuziel WA, Owens T. Induction of Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice Deficient in Either the Chemokine Macrophage Inflammatory Protein-1α or its CCR5 Receptor. Eur J Immunol (2000) 30:1410–5. doi: 10.1002/(SICI)1521-4141(200005)30:5<1410::AID-IMMU1410>3.0.CO;2-L
178. Gu SM, Park MH, Yun HM, Han SB, Oh KW, Son DJ, et al. CCR5 Knockout Suppresses Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice. Oncotarget (2016) 7:15382–93. doi: 10.18632/oncotarget.8097
179. Ni J, Zhu YN, Zhong XG, Ding Y, Hou LF, Tong XK, et al. The Chemokine Receptor Antagonist, TAK-779, Decreased Experimental Autoimmune Encephalomyelitis by Reducing Inflammatory Cell Migration Into the Central Nervous System, Without Affecting T Cell Function. Br J Pharmacol (2009) 158:2046–56. doi: 10.1111/j.1476-5381.2009.00528.x
180. Zheng H-M, Jiang Y, Wang J-R, Gong X-L, Guo B-Y. Mimic Peptides Bonding Specifically With the First and Second Extracellular Loops of the CC Chemokine Receptor 5 Derived From a Phage Display Peptide Library are Potent Inhibitors of Experimental Autoimmune Encephalomyelitis. Inflammation Res (2011) 60:759–67. doi: 10.1007/s00011-011-0331-8
181. Glass WG, Hickey MJ, Hardison JL, Liu MT, Manning JE, Lane TE. Antibody Targeting of the CC Chemokine Ligand 5 Results in Diminished Leukocyte Infiltration Into the Central Nervous System and Reduced Neurologic Disease in a Viral Model of Multiple Sclerosis. J Immunol (2004) 172:4018–25. doi: 10.4049/jimmunol.172.7.4018
182. Karampoor S, Zahednasab H, Amini R, Esghaei M, Sholeh M, Keyvani H. Maraviroc Attenuates the Pathogenesis of Experimental Autoimmune Encephalitis. Int Immunopharmacol (2020) 80:106138. doi: 10.1016/j.intimp.2019.106138
183. Sapir Y, Vitenshtein A, Barsheshet Y, Zohar Y, Wildbaum G, Karin N. A Fusion Protein Encoding the Second Extracellular Domain of CCR5 Arrests Chemokine-Induced Cosignaling and Effectively Suppresses Ongoing Experimental Autoimmune Encephalomyelitis. J Immunol (2010) 185:2589–99. doi: 10.4049/jimmunol.1000666
184. Angelou CC, Wells AC, Vijayaraghavan J, Dougan CE, Lawlor R, Iverson E, et al. Differentiation of Pathogenic Th17 Cells Is Negatively Regulated by Let-7 MicroRNAs in a Mouse Model of Multiple Sclerosis. Front Immunol (2019) 10:3125. doi: 10.3389/fimmu.2019.03125
185. Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-Cell Trafficking Between the Leptomeninges and the Cerebrospinal Fluid. Nature (2016) 530:349–53. doi: 10.1038/nature16939
186. Loetscher M, Amara A, Oberlin E, Brass N, Legler D, Loetscher P, et al. TYMSTR, a Putative Chemokine Receptor Selectively Expressed in Activated T Cells, Exhibits HIV-1 Coreceptor Function. Curr Biol (1997) 7:652–60. doi: 10.1016/S0960-9822(06)00292-2
187. Slauenwhite D, Gebremeskel S, Doucette CD, Hoskin DW, Johnston B. Regulation of Cytokine Polarization and T Cell Recruitment to Inflamed Paws in Mouse Collagen-Induced Arthritis by the Chemokine Receptor Cxcr6. Arthritis Rheumatol (2014) 66:3001–12. doi: 10.1002/art.38816
188. Butcher MJ, Wu C-I, Waseem T, Galkina EV. CXCR6 Regulates the Recruitment of Pro-Inflammatory IL-17A-Producing T Cells Into Atherosclerotic Aortas. Int Immunol (2016) 28:255–61. doi: 10.1093/intimm/dxv068
189. Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A Transmembrane CXC Chemokine Is a Ligand for HIV-Coreceptor Bonzo. Nat Immunol (2000) 1:298–304. doi: 10.1038/79738
190. Shimaoka T, Nakayama T, Fukumoto N, Kume N, Takahashi S, Yamaguchi J, et al. Cell Surface-Anchored SR-PSOX/CXC Chemokine Ligand 16 Mediates Firm Adhesion of CXC Chemokine Receptor 6-Expressing Cells. J Leukoc Biol (2004) 75:267–74. doi: 10.1189/jlb.1003465
191. Wilbanks A, Zondlo SC, Murphy K, Mak S, Soler D, Langdon P, et al. Expression Cloning of the STRL33/BONZO/TYMSTRligand Reveals Elements of CC, CXC, and CX3C Chemokines. J Immunol (2001) 166:5145–54. doi: 10.4049/jimmunol.166.8.5145
192. Minami M, Kume N, Shimaoka T, Kataoka H, Hayashida K, Akiyama Y, et al. Expression of SR-PSOX, a Novel Cell-Surface Scavenger Receptor for Phosphatidylserine and Oxidized LDL in Human Atherosclerotic Lesions. Arterioscler Thromb Vasc Biol (2001) 21:1796–800. doi: 10.1161/hq1001.096652
193. Ke C, Ren Y, Lv L, Hu W, Zhou W. Association Between CXCL16/CXCR6 Expression and the Clinicopathological Features of Patients With Non-Small Cell Lung Cancer. Oncol Lett (2017) 13:4661–8. doi: 10.3892/ol.2017.6088
194. Wehr A, Baeck C, Ulmer F, Gassler N, Hittatiya K, Luedde T, et al. Pharmacological Inhibition of the Chemokine CXCL16 Diminishes Liver Macrophage Infiltration and Steatohepatitis in Chronic Hepatic Injury. PloS One (2014) 9:e112327. doi: 10.1371/journal.pone.0112327
195. Ludwig A, Schulte A, Schnack C, Hundhausen C, Reiss K, Brodway N, et al. Enhanced Expression and Shedding of the Transmembrane Chemokine CXCL16 by Reactive Astrocytes and Glioma Cells. J Neurochem (2005) 93:1293–303. doi: 10.1111/j.1471-4159.2005.03123.x
196. Kim JV, Jiang N, Tadokoro CE, Liu L, Ransohoff RM, Lafaille JJ, et al. Two-Photon Laser Scanning Microscopy Imaging of Intact Spinal Cord and Cerebral Cortex Reveals Requirement for CXCR6 and Neuroinflammation in Immune Cell Infiltration of Cortical Injury Sites. J Immunol Methods (2010) 352:89–100. doi: 10.1016/j.jim.2009.09.007
197. Jiang X, Shimaoka T, Kojo S, Harada M, Watarai H, Wakao H, et al. Cutting Edge: Critical Role of CXCL16/CXCR6 in NKT Cell Trafficking in Allograft Tolerance. J Immunol (2005) 175:2051. doi: 10.4049/jimmunol.175.4.2051
198. Veinotte L, Gebremeskel S, Johnston B. CXCL16-Positive Dendritic Cells Enhance Invariant Natural Killer T Cell-Dependent IFNgamma Production and Tumor Control. Oncoimmunology (2016) 5:e1160979. doi: 10.1080/2162402X.2016.1160979
199. Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, et al. Bonzo/CXCR6 Expression Defines Type 1-Polarized T-Cell Subsets With Extralymphoid Tissue Homing Potential. J Clin Invest (2001) 107:595–601. doi: 10.1172/JCI11902
200. Markus L, Karkada M, Issekutz TB. CXCR6 is Expressed on T Cells in Both T Helper Type 1 (Th1) Inflammation and Allergen-Induced Th2 Lung Inflammation But Is Only a Weak Mediator of Chemotaxis. Immunology (2007) 121:555–64. doi: 10.1111/j.1365-2567.2007.02603.x
201. Hou L, Rao DA, Yuki K, Cooley J, Henderson LA, Jonsson AH, et al. SerpinB1 Controls Encephalitogenic T Helper Cells in Neuroinflammation. Proc Natl Acad Sci (2019) 116:20635–43. doi: 10.1073/pnas.1905762116
202. Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BML, et al. Stem-Like Intestinal Th17 Cells Give Rise to Pathogenic Effector T Cells During Autoimmunity. Cell (2021) 184:6281–6298 e6223. doi: 10.1016/j.cell.2021.11.018
203. Fukumoto N, Shimaoka T, Fujimura H, Sakoda S, Tanaka M, Kita T, et al. Critical Roles of CXC Chemokine Ligand 16/Scavenger Receptor That Binds Phosphatidylserine and Oxidized Lipoprotein in the Pathogenesis of Both Acute and Adoptive Transfer Experimental Autoimmune Encephalomyelitis. J Immunol (2004) 173:1620–7. doi: 10.4049/jimmunol.173.3.1620
204. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science (1996) 272:872. doi: 10.1126/science.272.5263.872
205. Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, et al. CXCR4–CCR5: A Couple Modulating T Cell Functions. Proc Natl Acad Sci (2008) 105:10101. doi: 10.1073/pnas.0804286105
206. Meng Y-M, Liang J, Wu C, Xu J, Zeng D-N, Yu X-J, et al. Monocytes/Macrophages Promote Vascular CXCR4 Expression via the ERK pathway in hepatocellular carcinoma. Oncoimmunology (2018) 7:e1408745. doi: 10.1080/2162402X.2017.1408745
207. Galli E, Hartmann FJ, Schreiner B, Ingelfinger F, Arvaniti E, Diebold M, et al. GM-CSF and CXCR4 Define a T Helper Cell Signature in Multiple Sclerosis. Nat Med (2019) 25:1290–300. doi: 10.1038/s41591-019-0521-4
208. Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, et al. The Chemokine SDF-1/CXCL12 Binds to and Signals Through the Orphan Receptor RDC1 in T Lymphocytes. J Biol Chem (2005) 280:35760–6. doi: 10.1074/jbc.M508234200
209. Luker KE, Lewin SA, Mihalko LA, Schmidt BT, Winkler JS, Coggins NL, et al. Scavenging of CXCL12 by CXCR7 Promotes Tumor Growth and Metastasis of CXCR4-Positive Breast Cancer Cells. Oncogene (2012) 31:4750–8. doi: 10.1038/onc.2011.633
210. Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 Heterodimerizes With CXCR4 and Regulates CXCL12-Mediated G Protein Signaling. Blood (2009) 113:6085–93. doi: 10.1182/blood-2008-12-196618
211. Dotan I, Werner L, Vigodman S, Weiss S, Brazowski E, Maharshak N, et al. CXCL12 Is a Constitutive and Inflammatory Chemokine in the Intestinal Immune System. Inflamm Bowel Dis (2010) 16:583–92. doi: 10.1002/ibd.21106
212. Patel JR, McCandless EE, Dorsey D, Klein RS. CXCR4 Promotes Differentiation of Oligodendrocyte Progenitors and Remyelination. Proc Natl Acad Sci (2010) 107:11062. doi: 10.1073/pnas.1006301107
213. Dziembowska M, Tham TN, Lau P, Vitry S, Lazarini F, Dubois-Dalcq M. A Role for CXCR4 Signaling in Survival and Migration of Neural and Oligodendrocyte Precursors. Glia (2005) 50:258–69. doi: 10.1002/glia.20170
214. Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE. Migration of Engrafted Neural Stem Cells is Mediated by CXCL12 Signaling Through CXCR4 in a Viral Model of Multiple Sclerosis. Proc Natl Acad Sci (2010) 107:11068–73. doi: 10.1073/pnas.1006375107
215. Williams JL, Patel JR, Daniels BP, Klein RS. Targeting CXCR7/ACKR3 as a Therapeutic Strategy to Promote Remyelination in the Adult Central Nervous System. J Exp Med (2014) 211:791–9. doi: 10.1084/jem.20131224
216. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, Ransohoff RM, et al. Chemokines in Multiple Sclerosis: CXCL12 and CXCL13 Up-Regulation is Differentially Linked to CNS Immune Cell Recruitment. Brain (2006) 129:200–11. doi: 10.1093/brain/awh680
217. Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, et al. A Role for CXCL12 (SDF-1alpha) in the Pathogenesis of Multiple Sclerosis: Regulation of CXCL12 Expression in Astrocytes by Soluble Myelin Basic Protein. J Neuroimmunol (2006) 177:27–39. doi: 10.1016/j.jneuroim.2006.05.003
218. McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, et al. Pathological Expression of CXCL12 at the Blood-Brain Barrier Correlates With Severity of Multiple Sclerosis. Am J Pathol (2008) 172:799–808. doi: 10.2353/ajpath.2008.070918
219. Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, et al. CXCR7 Influences Leukocyte Entry Into the CNS Parenchyma by Controlling Abluminal CXCL12 Abundance During Autoimmunity. J Exp Med (2011) 208:327–39. doi: 10.1084/jem.20102010
220. Dayton JR, Yuan Y, Pacumio LP, Dorflinger BG, Yoo SC, Olson MJ, et al. Expression of IL-20 Receptor Subunit β Is Linked to EAE Neuropathology and CNS Neuroinflammation. Front Cell Neurosci (2021) 15:683687. doi: 10.3389/fncel.2021.683687
221. Hanes MS, Salanga CL, Chowdry AB, Comerford I, McColl SR, Kufareva I, et al. Dual Targeting of the Chemokine Receptors CXCR4 and ACKR3 With Novel Engineered Chemokines *. J Biol Chem (2015) 290:22385–97. doi: 10.1074/jbc.M115.675108
222. Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1alpha) Suppresses Ongoing Experimental Autoimmune Encephalomyelitis by Selecting Antigen-Specific Regulatory T Cells. J Exp Med (2008) 205:2643–55. doi: 10.1084/jem.20080730
223. Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 Is a CXCR7 Ligand With Allosteric Agonist Properties. Mol Pharmacol (2009) 75:1240. doi: 10.1124/mol.108.053389
224. Pouzol L, Baumlin N, Sassi A, Tunis M, Marrie J, Vezzali E, et al. ACT-1004-1239, a First-in-Class CXCR7 Antagonist With Both Immunomodulatory and Promyelinating Effects for the Treatment of Inflammatory Demyelinating Diseases. FASEB J (2021) 35:e21431. doi: 10.1096/fj.202002465R
225. Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune Homeostasis Enforced by Co-Localized Effector and Regulatory T Cells. Nature (2015) 528:225–30. doi: 10.1038/nature16169
226. Zhang Y, Han J, Wu M, Xu L, Wang Y, Yuan W, et al. Toll-Like Receptor 4 Promotes Th17 Lymphocyte Infiltration Via CCL25/CCR9 in Pathogenesis of Experimental Autoimmune Encephalomyelitis. J Neuroimmune Pharmacol (2019) 14:493–502. doi: 10.1007/s11481-019-09854-1
227. Kadowaki A, Saga R, Lin Y, Sato W, Yamamura T. Gut Microbiota-Dependent CCR9+CD4+ T Cells Are Altered in Secondary Progressive Multiple Sclerosis. Brain (2019) 142:916–31. doi: 10.1093/brain/awz012
228. Evans-Marin HL, Cao AT, Yao S, Chen F, He C, Liu H, et al. Unexpected Regulatory Role of CCR9 in Regulatory T Cell Development. PloS One (2015) 10:e0134100. doi: 10.1371/journal.pone.0134100
Keywords: chemokine, migration, EAE (experimental autoimmune encephalomyelitis), multiple sclerosis, Th subsets
Citation: Heng AHS, Han CW, Abbott C, McColl SR and Comerford I (2022) Chemokine-Driven Migration of Pro-Inflammatory CD4+ T Cells in CNS Autoimmune Disease. Front. Immunol. 13:817473. doi: 10.3389/fimmu.2022.817473
Received: 18 November 2021; Accepted: 25 January 2022;
Published: 16 February 2022.
Edited by:
Gordana Leposavić, University of Belgrade, SerbiaReviewed by:
Cheng-Chih Hsiao, Amsterdam University Medical Center, NetherlandsBarbara Rossi, University of Verona, Italy
Makoto Inoue, University of Illinois at Urbana-Champaign, United States
Copyright © 2022 Heng, Han, Abbott, McColl and Comerford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iain Comerford, aWFpbi5jb21lcmZvcmRAYWRlbGFpZGUuZWR1LmF1