
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 09 February 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.815936
This article is part of the Research Topic The Interaction between Apicomplexan Protozoan and the Host View all 7 articles
 Hong Zheng1,2,3†
Hong Zheng1,2,3† Xiao Lu1,3†
Xiao Lu1,3† Kai Li1Feng Zhu1Chenhao Zhao1
Kai Li1Feng Zhu1Chenhao Zhao1 Taiping Liu1Yan Ding1Yong Fu1Kun Zhang1Taoli Zhou1
Taiping Liu1Yan Ding1Yong Fu1Kun Zhang1Taoli Zhou1 Jigang Dai3*Yuzhang Wu2*
Jigang Dai3*Yuzhang Wu2* Wenyue Xu1,4*‡
Wenyue Xu1,4*‡Although exo-erythrocytic forms (EEFs) of liver stage malaria parasite in the parasitophorous vacuole (PV) are encountered with robust host innate immunity, EEFs can still survive and successfully complete the infection of hepatocytes, and the underlying mechanism is largely unknown. Here, we showed that sporozoite circumsporozoite protein (CSP) translocated from the parasitophorous vacuole into the hepatocyte cytoplasm significantly mediated the resistance to the killing of EEFs by interferon-gamma (IFN-γ). Attenuation of IFN-γ-mediated killing of EEFs by CSP was dependent on its ability to reduce the levels of autophagy-related genes (ATGs) in hepatocytes. The ATGs downregulation occurred through its enhanced ubiquitination mediated by E3 ligase NEDD4, an enzyme that was upregulated by CSP when it translocated from the cytoplasm into the nucleus of hepatocytes via its nuclear localization signal (NLS) domain. Thus, we have revealed an unrecognized role of CSP in subverting host innate immunity and shed new light for a prophylaxis strategy against liver-stage infection.
Malaria is still one of the most devastating diseases worldwide and is caused by infection with the genus Plasmodium that is initiated in the mammalian host by inoculation of sporozoites into the skin by Anopheles mosquitoes. From the skin, sporozoites travel to the liver via blood circulation, where they infect hepatocytes.
In the liver, sporozoites bind to highly sulfated heparin sulfate proteoglycans (HSPGs) on the hepatocyte surface by circumsporozoite protein (CSP), which triggers their invasion into hepatocytes (1). After invading hepatocytes, sporozoites inside a parasitophorous vacuole (PV) and transform into exo-erythrocytic forms (EEFs). To avoid fusion of the PV with lysosomes and to shield itself from the cytosol of the host, the PV membrane is modified by parasite-derived proteins, such as upregulated in infective sporozoite gene 4 (UIS4) and exported protein 1 (EXP-1) (2, 3). However, EEFs in the PV can be sensed by the Melanoma differentiation-associated gene 5 (MD-5) and elicit hepatocytes to generate a type-I IFN response (4). This recruits innate immune cells, such as natural killer (NK) and NK T cells, and eliminates the EEFs in the infected hepatocytes through the secretion of IFN-γ (5). The IFN-γ-mediated killing of EEFs was previously suggested to involve the production of nitric oxide (NO) (6, 7); it was recently reported to be regulated in an NO-independent manner (8). In addition, the infection of sporozoites could also induce Plasmodium-associated autophagy-related responses (PAAR) of hepatocytes to limit EEF development (9, 10). However, EEFs can still survive and successfully complete the infection of hepatocytes, and the underlying mechanism of parasite survival in this hostile environment remains largely unknown.
A previous study has shown that CSP, the major surface protein of sporozoites, translocating from the PV into the cytoplasm of hepatocytes, could promote liver stage development, which was thought to be associated with the reduced NF-κB activation in hepatocytes by CSP (11). However, in the present study we found that CSP enhanced the liver stage development through its resistance to the killing effect of IFN-γ on EEFs. Therefore, we uncovered a novel immune escape strategy for EEFs. Moreover, further study showed that the resistance mediated by CSP to the IFN-γ-killing of EEFs was dependent on its ability to downregulate autophagy-related genes (ATGs) through enhanced ubiquitination mediated by the E3 ligase NEDD4.
Plasmodium berghei ANKA (P.b ANKA) was maintained in our laboratory, and the P.b ANKA CSPmut parasite was constructed by mutation of pexel I-II of CSP using CRISPR-Cas9. All parasites were maintained by passage between Kunming mice (Swiss Webster strain) and Anopheles stephensi. ATG5fl/fl mice (B6.129S-Atg5<tm1Myok>) were obtained from the RIKEN BioResource Center (Ibaraki, Japan), and Alb-Cre mice (B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). ATG5fl/fl mice were crossed with Alb-Cre mice to conditionally delete the ATG5 cassette in hepatocytes. ATG5fl/fl mice were used as controls. IFN-γR1 knockout mice were gifts from Dr. Bo Guo (Army Medical University, Chongqing, China). Kunming mice were purchased from the Experimental Animal Center of the Army Medical University (Chongqing, China). Animals were kept in a specific pathogen-free laboratory at the Institute of Immunology of Army Medical University. All methods were carried out in accordance with the approved Guide for the Care and Use of Laboratory Animals of the Army Medical University (approval number AMUWEC20171828).
Anopheles stephensi (Hor strain) was maintained at 27°C and 70%–80% relative humidity and fed with a 5% sugar solution. For infection with the parasite, 3- to 5-day-old female adults were kept at 20–21°C and 70%–80% relative humidity and fed on P.b ANKA, P.b ANKA-RFP, or P.b ANKA-CSPmut-infected Kunming mice with gametocytemia ≥ 0.5%. Seven days after infection, the mosquitoes were dissected, and the oocysts on the midguts were examined under a light microscope.
Both HepG2 and HEK293T cells were purchased from the American Type Culture Collection (Manassas, VA, USA). HepG2 cells were maintained in DMEM (Hyclone) and HEK293T cells were maintained in RPMI-1640 medium (Hyclone), supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified atmosphere with 5% CO2. For transfection, 3 × 104 cells in 96-well, 1.2 × 105 cells in 24-well plates, 1 × 106 cells in 6-well plates, and 1 × 107 cells in 10-cm dishes were transfected with 0.2, 0.8, 5, or 24 μg recombinant plasmid using 0.5, 2, 7.5 or 37.5 μL Lipofectamine 3000 (Invitrogen, CA, USA), respectively, according to the manufacturer’s protocol. For virus infection, 1 × 105 HepG2 cells in 24-well plates were pretreated with 8 μg/mL polybrene (Santa Cruz, CA, USA) for 2 h, and then infected with Ad-mRFP-GRP-LC3 (Hanbio, Shanghai, China) at a multiplicity of infection (MOI) of 15. Four hours later, fresh cell culture containing 8 μg/mL polybrene was added. After 12 h of virus infection, the supernatant was discarded and replaced with fresh culture medium.
At 19 days after infection with P.b ANKA, P.b ANKA-RFP, or P.b ANKA-CSPmut, infected female mosquitoes were extensively washed in sterile PBS, and the salivary glands were dissected and collected in RPMI 1640 medium containing 2.5 μg/mL amphotericin B (Sangon Biotech, Shanghai, China), 100 units/mL penicillin, and 100 μg/mL streptomycin (Beyotime, Beijing, China). The sporozoites were released from salivary glands and counted, and then incubated with HepG2 cells at a ratio of 1:3. Three hours after infection, the supernatant was discarded and replaced with fresh culture medium with the three antibiotics as described above. For the in vivo assay, ATG5fl/fl and ATG5fl/fl-Alb-Cre were challenged with an intravenous injection of 1,000 sporozoites. Control and IFN-γ R1 knockout mice were challenged with an intravenous injection of 10,000 sporozoites.
The fragments of P.b ANKA full CSP (1020bp) were amplified from the genome of P.b ANKA using KOD-FX DNA polymerase (TOYOBO, Osaka, Japan). CSP-HA fragments were obtained by inserted HA-tag in C-terminal by PCR. CSPΔNLS (993 bp) and NLS (27 bp) fragments with initiation codons and HA-tag in C-terminal were synthesized by Sangon Biotech. CSP-HA and CSPΔNLS-HA with a 16-18 bp homologous sequence of the pcDNA3.1 vector at either the 5’ or 3’ end were obtained by PCR. The pcDNA3.1 vector was linearized using the restriction enzymes HindIII and BamHI. The fragments of CSP-HA and CSPΔNLS-HA were inserted into the pcDNA3.1 vector by seamless cloning according to the instructions of the In-fusion® HD Cloning Kit (Clontech, Palo Alto, CA, USA). NLS-HA was inserted into pcDNA3.1 using the High-Efficient Ligation Reagent Ligation High (TOYOBO). The pcDNA3.1 vector was linearized using the restriction enzymes BamHI and XholI (TAKARA, Otsu, Japan) for ATG5 and LC3B, and with KpnI and NotI for ATG7. The pFLAG-cmv8 vector was linearized using the restriction enzymes HindIII and EcoRI for ATG5 and ATG7. Human LC3B, ATG5, and ATG7 were inserted into the pcDNA3.1 or pFLAG-cmv8 vector, respectively, by seamless cloning according to the instructions of the In-fusion® HD Cloning Kit (Clontech). The pGL3-NEDD4 promoter reporter plasmid was constructed by cloning the NEDD4 promoter sequence (2000bp upstream form 1st exon) into the pGL3 vector through KpnI/HindIII. All recombinant plasmids were verified by DNA sequencing.
The CSP coding sequence of P.b ANKA was cloned downstream of the CMV-7 promoter in the Lenti-pCDH plasmid (a gift from Professor Ying Wan, Army Medical University, China). HEK293T cells were transfected with Lenti-pCDH-CSP or Lenti-pCDH control plasmids with psPAX2 and pMD2.G packaging plasmids at a ratio of 20:15:5 using Lipofectamine 3000 (Invitrogen) to obtain the lentivirus. Seventy-two hours after transfection, the supernatant was filtered through a 0.45-μm filter (Millipore), and the lentivirus was collected by ultracentrifugation (20,000 rpm) at 4°C for 2 h. HepG2 cells were infected with Lenti-pCDH-CSP or Lenti-pCDH control lentivirus at an MOI of 15 for 48 h. Then, the positively infected cells were screened using 5 μg/mL puromycin (Gibco). CSP-stably transfected and control cells were cloned by a limited-dilution method in a 96-well plate, and 3 μg/mL puromycin was used to maintain the resistance of stably transfected cells. CSP expression was confirmed using immunofluorescence.
The peptide containing three P.b ANKA CSP repeats, PAPPNANA-PAPPNANA- PAPPNANA, was synthesized by Wuhan GeneCreate Biological Engineering Co., Ltd. (Wuhan, China) and purified by high-performance liquid chromatography (HPLC) using a Waters XBridge C18 column (4.6 × 250 mm × 5 μm; Waters Corporation) on a Waters Corporation Prominence HPLC system (Massachusetts, USA).
The peptide was conjugated to bovine serum albumin (BSA). The resulting conjugated peptide was emulsified with Freund’s adjuvant (Sigma Aldrich) and immunized in a specific pathogen-free-grade rabbit at 0, 2, 4, and 6 weeks. Seven days after the final immunization, rabbit serum was collected, and the titer of the antibody against the epitope was detected using an enzyme-linked immunosorbent assay (cat. no. 44-2404-21, Nunc MaxiSorp flat bottom, Nalge Nunc International, Penfield, NY, USA). Protein G Sepharose (GE Healthcare) was used to purify the CSP antibody.
HepG2 cells (1 × 105) were placed on a 14-mm-diameter slide in a 24-well plate. To detect the distribution of CSP, HepG2 cells were infected with CSPwt or CSPmut sporozoites. Three hours after infection, the cells were washed with PBS to remove non-invaded sporozoites and incubated with fresh DMEM (Hyclone) containing 10% FBS (Gibco) and three antibiotics. Twenty-four hours later, the slides were immobilized with 4% paraformaldehyde (Sangon Biotech) for 10 min, permeabilized with 0.1% Triton-X 100 (Amersco, Albany, NY, USA) for 15 min, and then blocked with PBS containing 5% BSA and 0.02% Tween-20 for 1 h at room temperature. The cells were labeled with 1:500 goat anti-UIS4 (Sicgen, Cantanhede, Portugal) overnight at 4°C and 1:100 green donkey anti-goat secondary antibody (Abbkine, Wuhan, China) for 1 h at room temperature. After washing with PBS three times, the cells were labeled with 1:500 rabbit anti-CSP overnight at 4°C and 1:100 red donkey anti-rabbit secondary antibody (Abbkine) for 1 h at room temperature. Nuclei were counterstained with DAPI (Beyotime) for 5 min at room temperature.
For invasion capacity comparison, UIS4 staining was used to identify the CSPwt and CSPmut parasites in HepG2 cells, and images were obtained at 6 h after infection. To evaluate the co-localization of EEFs and LC3, control and CSP-stably transfected HepG2 cells were transfected with LC3B-RFP plasmid and infected with CSPwt or CSPmut sporozoites, and then labeled with goat anti-UIS4 and the secondary antibody as described above. To observe the effect of CSP on the IFN-γ-mediated killing of EEFs, control and CSP-stably transfected HepG2 cells were pre-treated with 1 U/mL recombinant human IFN-γ (Peprotech, Rocky Hill, NJ, USA) or equivoluminal medium for 6 h. The cells were then infected with CSPwt or CSPmut as stated above and co-treated with IFN-γ for 46 h.
For evaluating the effect of autophagy regulation on IFN-γ-mediated killing, HepG2 cells were pre-treated with 0.2 U/mL, 0.5 U/mL, and 1 U/mL IFN-γ for 6 h, and then infected with P.b ANKA-RFP sporozoites at a ratio of 1:3 and co-treated with IFN-γ and 0.25 μg/mL rapamycin, 10 μM LY294002 (Cell Signaling Technology, Danvers, MA, USA), 100nM wortmannin (Sigma-Aldrich), 1μM TAK-243 (MCE, NJ, USA) or equivoluminal medium for 24 or 46 h, respectively.
For autophagy flux observation, the control and CSP-stably transfected HepG2 cells were pre-treated with 8 μg/mL polybrene for 2 h, and then infected with adenovirus containing tandem GFP-RFP-LC3 (Hanbio, Shanghai, China). Since the cell culture was changed, the cells were treated with 0.25 μg/mL rapamycin (Cell Signaling Technology) or equivoluminal medium for 24 h.
For nuclear translocation of CSP observation, HepG2 cells were transfected with pcDNA3.1-CSP-HA and pcDNA3.1-CSPΔNLS-HA plasmids for 24 h. The cells were then labeled with rabbit anti-HA and the secondary antibody, as stated above.
The cells were immobilized, penetrated, and stained with DAPI. After washing with PBS, the slides were mounted on a microscope slide using DAKO Fluorescence Mounting Medium (Agilent). LSM 780 NLO microscope systems and ZEN Imaging Software (Zeiss) were used for image acquisition and export. Image J software (NIH) was used to analyze the number and size of the EEFs.
For parasite burden detection, the livers of mice were dissected at 46 h after infection with CSPwt or CSPmut sporozoites, and homogenized in 1.5 mL Trizol (Invitrogen), and HepG2 cells were collected 46 h after infection with CSPwt or CSPmut sporozoites and lysed with 1 mL Trizol (Invitrogen). Liver homogenate (100 μL) or 1 mL of cell lysis were used for total RNA extraction using TRIzol in accordance with the manufacturer’s instructions. cDNA was synthesized from equivalent total RNA using the PrimeScript™ RT reagent Kit with gDNA Eraser (TAKARA) in accordance with the manufacturer’s instructions. The parasite load in the livers and HepG2 cells was evaluated by Taqman PCR with primers and probes for 18S rRNA and GAPDH following the manufacturer’s instructions of Premix Ex Taq™ (Probe qPCR) (TAKARA). The mRNA levels of IFN-γ, iNOS, LC3B, ATG3, ATG5, ATG7, STUB1, SYVN1, CBL, SMURF1, PAFAH1B1, and NEDD4 were evaluated using TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) (TAKARA). CFX96 Touch™ Real-Time PCR Detection System and CFX ManagerTM Software (Bio-Rad, Hercules, CA, USA) were used for RT-PCR data collection and analysis. The annealing temperature used for the above two RT-qPCR experiments was 60°C.
To investigate the influence of CSP overexpression on the protein levels of ATGs, 1 × 106 control and CSP-stably transfected HepG2 cells were incubated in 6-well plates and treated with 0.25 μg/mL rapamycin for 0, 6, 12, and 24 h. To observe the influence of CSP overexpression on the half-life of ATG proteins, CSP-stably transfected HepG2 and control cells were treated with 20 μM cycloheximide (CHX, Cell Signaling Technology) or 10 μM MG132 (Cell Signaling Technology) for 0, 6, 12, and 24 h. To test whether the degeneration of ATGs was dependent on the ubiquitin-proteasome system, CSP-stably transfected HepG2 and control cells were treated with IFN-γ, 1μM TAK-243 or both for 24 h. Uniformly, the cells were lysed with 300 μL RIPA lysis buffer containing a protease and phosphatase inhibitor cocktail (Thermo Fisher) at various time points and protein concentrations were detected using the BCA Protein Assay Kit (Sangon Biotech). Total proteins (20 μg equivalent) were then separated by 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad), transferred using a 0.22-μm polyvinylidene fluoride (PVDF) filter (Millipore) and blocked with blocking solution (Sangon Biotech). The PVDF filter was incubated with 1:2000 rabbit anti-LC3B (Sigma-Aldrich), 1:2000 rabbit anti-ATG3 (Abcam, Cambridge, UK), 1:2000 rabbit anti-ATG5 (Invitrogen), 1:2000 rabbit anti-ATG7 (Sigma-Aldrich), 1:2000 rabbit anti-P62 (Sigma-Aldrich), 1:2000 rabbit anti-Beclin-1 (Cell Signaling Technology), and 1:2000 mouse anti-β-actin (Sigma-Aldrich) overnight at 4°C. Then, the PVDF filter was washed three times with TBS solution containing 1% Tween-20 (Amersco) and incubated with 1:20000 goat anti-rabbit or anti-mouse IgG-HRP secondary antibody (Santa Cruz) for 1 h at room temperature.
The ubiquitination of ATGs was detected by immunoprecipitation, as previously described (12). To detect the influence of CSP overexpression on the ubiquitination of ATGs, 2 × 106 CSP-stably transfected HepG2 and control cells in two 6-wells were transfected with 2μg His-UB, and 6 μg pFLAG-cmv8 or pFLAG-cmv8-human ATG5/7 plasmids, respectively. To confirm CSP-induced ubiquitination of ATGs via upregulation of NEDD4, 2 × 106 293FT cells in two 6-wells were co-transfected with 2 μg His-UB, 4 μg pFLAG-cmv8 or pFLAG-cmv8-human ATG5/7, 2 μg shNC or shNEDD4, and 2 μg pcDNA3.1 or pcDNA3.1-CSP-HA plasmids, respectively. To test the influence of the NLS domain on the CSP-induced ubiquitination of ATGs, 2 × 106 293FT cells in two 6-wells were co-transfected with 2μg His-UB plasmids, 4 μg pFLAG-cmv8-human ATG5/7, 4 μg pcDNA3.1, pcDNA3.1-CSP-HA, and pcDNA3.1-CSPΔNLS-HA, or pcDNA3.1-NLS-HA, respectively. Briefly, the cells were treated with 10 μM MG-132 for 24 h and lysed with 1 mL of western and IP cell lysis buffer. The protein concentrations of each group were determined and equalized as described above. Fusion FLAG-hATG5 and hATG7 proteins were captured using anti-FLAG ® M2 Magnetic Beads (Sigma-Aldrich) according to the manufacturer’s instructions. After washing five times with equilibrium buffer (50 mM Tris HCl, 150 mM NaCl, pH 7.4), the beads were boiled with SDS-PAGE loading buffer for 10 min. An equal volume of total cell lysates (input) was loaded and separated by 10% SDS-PAGE, as described above. The PVDF filter was incubated with 1:500 mouse anti-FLAG tag (Invitrogen), 1:1000 rabbit monoclonal anti-His-Tag (Cell Signaling Technology), 1:2000 mouse anti-β-actin (Sigma-Aldrich), or 1:500 rabbit anti-NEDD4 overnight at 4°C and incubated with secondary antibody (Santa Cruz) for 1 h at room temperature. The protein bands were visualized using the Western BLoT Hyper HRP Substrate (TAKARA) and exposed using a chemiluminescence imaging system (Fusion Solo S, Vilber, France). Image J software (NIH) was used for grey value analysis.
RNA interference for ATG5 and ATG7 in HepG2 cells was performed according to the protocol of the ATG5/7 human shRNA plasmid kit (Origene, Rockville, MD, USA). HepG2 cells (1 × 106) in 6-well plates were transfected with 4 μg ATG-interfering or scrambled non-effective plasmids using Lipofectamine 3000 following the manufacturer’s protocol. The plasmids containing the following sequences were used for ATG5 mRNA interference: 5-TCAGCTCTTCCTTGGAACATCACAGTACA-3, ATG7 mRNA interference, 5-CTTGGCTGCTACTTCTGCAATGATGTGGT-3, and shNC sequence, 5-GCACTACCAGAGCTAACTCAGATAGTACT-3’. For NEDD4 mRNA interference, 5-GTGAAATTGCACATAATGAGGTTCAAGAGACCTCATTATGTGCAATTTCAC-3, and shNC sequence, 5-GTTCTCCGAACGTGTCACGTCAAGAGATTACGTGACACGTTCGGAGAA-3′. Fresh culture medium containing 3 μg/mL puromycin (Gibco) was added to select puromycin-resistant cells. RNA interference efficiency was validated using quantitative real-time PCR and western blotting.
CSP-stably transfected or control cells (1 × 106) were lysed with 1 mL Trizol (Invitrogen), and total RNA was extracted as described above. A total of 2 μg RNA per sample was used as the input material for the RNA sample preparations. mRNA was purified from total RNA using poly T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperatures in the VAHTSTM First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using a random hexamer primer and M-MuLV reverse transcriptase (RNase H). Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3’ ends of the DNA fragments, an adaptor was ligated for library preparation. To select cDNA fragments of preferentially 150–200 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μL USER Enzyme (NEB, Ipswich, MA, UK) was incubated with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. Finally, PCR products were purified (AMPure XP system) and library quality was assessed using the Agilent Bioanalyzer 2100 system. Libraries were quantified and pooled. Paired-end sequencing of the library was performed using HiSeq XTen sequencers (Illumina, San Diego, CA). RAW data were submitted to the GEO database, and the series records were GSE129323. A heatmap was generated using R language version 4.0.2, and the pheatmap package v1.0.12.
Promoter activity assay was conducted by co-transfecting 100 ng pcDNA3.1 vector, pcDNA3.1-CSP-HA, pcDNA3.1-CSPΔNLS-HA or pcDNA3.1-NLS-HA, and 100ng pGL3-NEDD4 promoter and 1ng RL-TK into HEK293T cells in a 96-well 24 h later. The cells were then lysed and luciferase activity for each group was detected using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA).
SPSS software (version 19.0; IBM, Armonk, NY, USA) was used for statistical analysis. Student’s t-test for two groups or one-way ANOVA for multiple groups were used if the data were normally distributed to compare continuous variables, and if not, the Mann-Whitney U test was used for comparisons among groups. Pairwise differences in normally distributed variables were compared using the Tukey-Kramer statistic for multiple comparisons; if not, Dunnett’s test was used. Statistical significance was set at P < 0.05. Error bars represent standard deviations of the mean.
The pexel I-II domain of CSP has been demonstrated to mediate its translocation from the PV into the hepatocyte cytoplasm (11). To test whether CSP could facilitate EEF survival in the PV of hepatocytes, a CSP pexel I-II domain mutant Plasmodium berghei (P.b) ANKA, named after CSPmut, was constructed by replacing the wild-type (WT) CSP with a pexel I-II mutant CSP using CRISPR-Cas9 technology (Supplementary Figures S1A–C). As no mutations were found in any of the potential off-target regions screened by genome-wide comparison (Supplementary Table S1), the potential of genomic off-target mutations generated by the CRISPR-Cas9 strategy could be excluded. The growth of the blood-stage mutant parasite was normal as compared to that of the WT parasite (CSPwt), and the salivary gland sporozoites of CSPmut could be successfully generated after the mutant parasite-infected mosquitoes (Supplementary Figure S1D). Meanwhile, no difference of invasion ability into hepatocytes was found between CSPmut and CSPwt parasites (4.3 ± 0.2% vs 4.0 ± 0.1%) at 6 h post infection in vitro. Immunofluorescence assay with anti-UIS4, a parasitophorous vacuole membrane (PVM) specific marker and anti-CSP demonstrated that the CSP of mutant sporozoites was significantly inhibited to translocate from the PV into the hepatocyte cytoplasm when infecting HepG2 cells (Figure 1A).
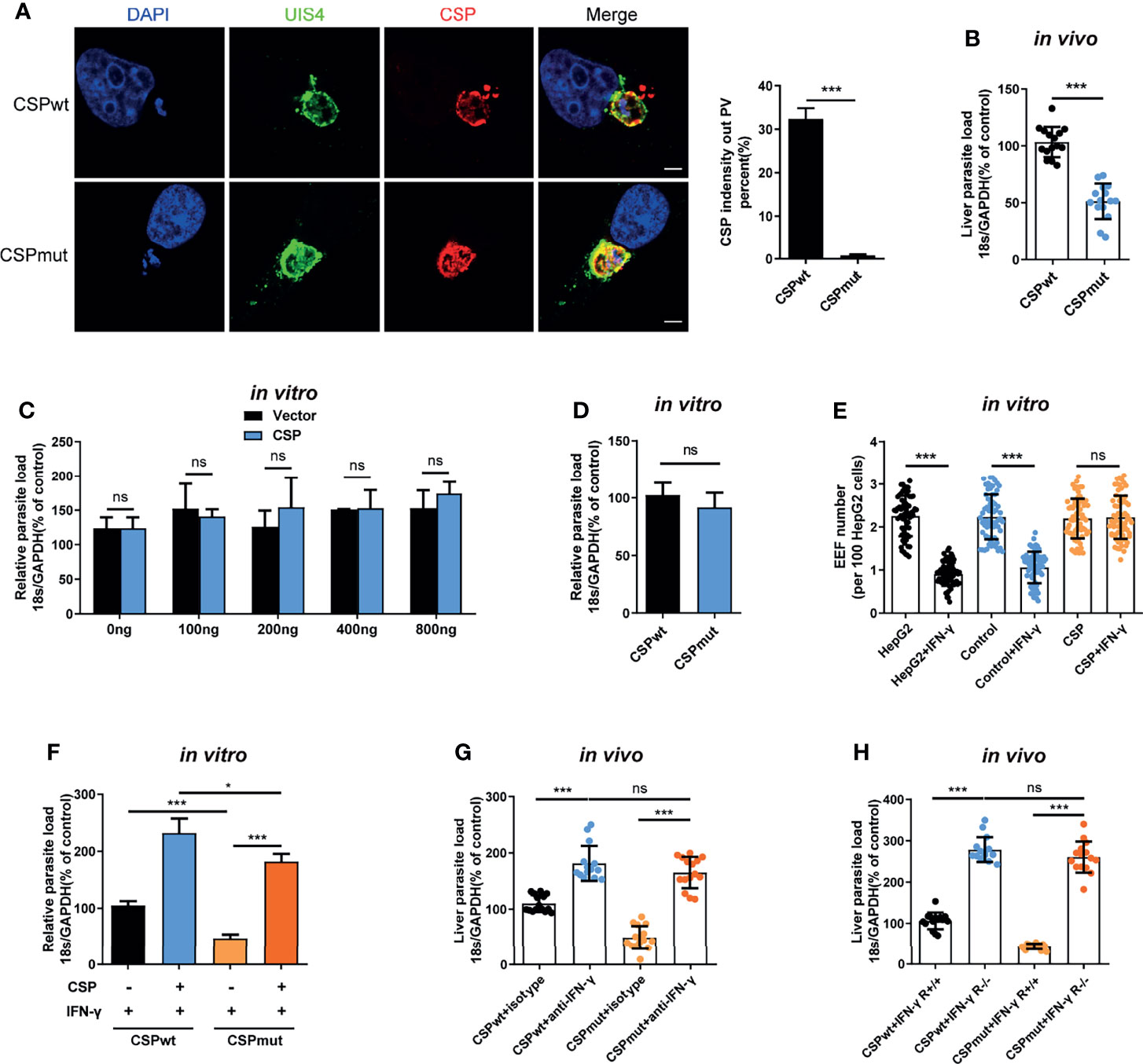
Figure 1 CSP translocated into cytoplasm of hepatocyte mediates the resistance to the killing of EEFs by IFN-γ. (A) After 1.2 × 105 HepG2 cells were incubated with 4 × 104 CSPwt and CSPmut P.b ANKA sporozoites for 24 h, then cells were stained with anti-UIS4, anti-CSP and DAPI. Translocation of CSP of EEFs (n=60) from the PV into HepG2 cytoplasm was observed under confocal microscopy (left), and quantified (right). Scale bar =5 μm. (B) After the mice were intravenously injected with 1,000 CSPwt or CSPmut parasite sporozoites for 46 h, the parasite burden in the liver was measured as the ratio of Plasmodium 18S rRNA to mouse GAPDH using Taqman real-time PCR. Each dot represents one mouse (n=15). (C) 1.2 × 105 HepG2 cells were transiently transfected with the indicated amount of CSP plasmid and then incubated with 4 × 104 CSPwt sporozoites for 46 h; the parasite burden was determined as the ratio of Plasmodium 18S rRNA to human GAPDH using Taqman real-time PCR. Three individual experiments have been performed. (D) After 1.2 × 105 HepG2 cells were incubated with 4 × 104 CSPwt or CSPmut sporozoites for 46 h, the parasite burden was determined as described in (C). The experiments have been performed for three times. (E) 1.2 × 105 HepG2 (without lentivirus infection), control (infected with Lenti-pCDH control lentivirus and selected by puromycin) and CSP-stably transfected HepG2 cells were pre-treated with or without 1 U/mL IFN-γ, and then incubated with 4 × 104 CSPwt sporozoites for 46 h. EEF number per 100 HepG2 cells was determined and statistically analyzed. Each dot represents one visual field (n=66, magnification, 630×). (F) 1.2 × 105 control and CSP-stably transfected HepG2 cells were treated with 1 U/mL IFN-γ, and then infected with CSPwt or CSPmut parasite as above; 46 h later, the parasite burden in hepatocytes was determined and compared as described in (C). Three individual experiments have been performed. (G) Mice pre-treated with anti-IFN-γ or isotype antibody, were infected with 1,000 CSPwt or CSPmut sporozoites. Then, the liver burden of parasite at 46h post infection was determined as described in (B). Each dot represents one mouse (n=15). (H) IFN-γR1 knockout and WT mice were infected with 1,000 CSPwt or CSPmut sporozoites, and liver burden was determined at 46 h after infection as described as above. Each dot represents one mouse (n=15). The pooled data of three repeated experiments was presented, data are represented as mean ± SD and analyzed by a student’s t-test or One-Way ANOVA. ns, not significant; *P < 0.05; ***P < 0.001.
Consistent with a previous study (11), we also found that the burden of CSPmut in the liver was greatly reduced compared to that of the CSPwt after the mice were infected (Figure 1B). However, transient transfection with full-length CSP plasmid did not enhance the development of EEFs in vitro, even when up to 800 ng of plasmids were transfected (Figure 1C). There was also no difference in parasite load in HepG2 cells infected with CSPwt or CSPmut parasites in vitro (Figure 1D). As sporozoite infection could trigger mice to generate immune responses against parasites (4), the different results between our in vivo and in vitro assays might be interpreted by the in vivo immune response that was not considered in our in vitro assay. Thus, we postulated that CSP might indirectly promote EEF development by suppressing host immune responses.
Plasmodium in the liver can trigger a type I interferon (IFN) response and recruit CD1d-restricted NKT cells (5). IFN-γ, predominately secreted by recruited NKT cells, is considered to be not only the major innate immune effector to suppress the development of the liver stage in vivo (5, 13), but also the critical effector to inhibit the EEFs in the PV of hepatocytes in vitro (14, 15). Altogether, these findings strongly suggest that IFN-γ should be included in our investigation of the effects of CSP on EEF development both in vitro and in vivo. Strikingly, we found that overexpression of CSP greatly inhibited the IFN-γ-mediated killing of EEFs, as both EEF number and parasite load in CSP-stably transfected HepG2 cells were much higher than those in control cells, when treated with IFN-γ (Figure 1E and Supplementary Figure S2). In addition, although the mutant parasite in HepG2 cells was susceptible to IFN-γ-mediated killing, overexpression of CSP remarkably reversed the killing effect of IFN-γ (Figure 1F). Subsequently, this finding was further confirmed in a physiologically relevant context, as no substantial difference in the liver parasite burden was found between the CSPwt and CSPmut-infected mice when IFN-γ was depleted (Figure 1G). Similar results were obtained when two parasite strains were infected with IFN-γR1 knockout mice (Figure 1H). The reduced parasite burden in CSPmut infected mouse livers was not due to the capacity of mutant parasites to induce the host to produce a much higher level of IFN-γ because a comparable mRNA level of IFN-γ was found in the livers of mice infected with CSPwt or CSPmut sporozoites at different doses (Supplementary Figure S3). Thus, our data demonstrates that translocated CSP from the PV into the hepatocyte cytoplasm confers resistance to IFN-γ-mediated killing of EEFs.
Previous studies have shown that IFN-γ prevents Plasmodium liver-stage development by inducing the expression of inducible nitric oxide synthase (iNOS), an enzyme required for the production of NO (6, 16, 17). Hence, we sought to investigate whether inhibition of the IFN-γ-mediated killing of EEFs by CSP was dependent on the downregulation of iNOS and NO. We found that transfection with CSP did not reduce the mRNA level of iNOS or the concentration of NO in the infected-HepG2 cells treated with IFN-γ (Supplementary Figure S4A). Similarly, both the levels of iNOS and NO were comparable between CSPwt and CSPmut parasite-infected HepG2 cells treated with IFN-γ (Supplementary Figure S4B). In addition, the resistance to the IFN-γ-mediated suppression of EEFs and the production of NO in CSP-transfected HepG2 cells treated with IFN-γ were not affected by treatment with two iNOS inhibitors, aminoguanidine (AG) and L-NAME. Although they could greatly inhibit the production of NO in lipopolysaccharide (LPS)-stimulated macrophages (Supplementary Figure S4C). Therefore, NO is not involved in the suppression of IFN-γ-mediated killing of EEFs by CSP.
Evidence has shown that autophagy and autophagy-related genes (ATGs) are essential for the IFN-γ-mediated killing of Toxoplasma gondii in macrophages (18–21), an apicomplexan parasite closely related to Plasmodium. Thus, we next sought to investigate whether autophagy was also involved in the regulation of IFN-γ-mediated killing of EEFs in HepG2 cells. Consistent with our previous study (22), autophagy alone, either enhanced by rapamycin or inhibited by LY294002, had no effect on liver stage development in HepG2 cells (Figures 2A, B). However, rapamycin-enhanced autophagy significantly reduced the EEF number, parasite load, and EEF size in HepG2 cells in the presence of 0.2 U/mL IFN-γ (Figure 2A and Supplementary Figure S5A). We found that rapamycin could not further reduce the parasite number and load, as well as the size, when treated with 0.5 U/mL or 1 U/mL IFN-γ (Figure 2A and Supplementary Figure S5A), possibly because most of the EEFs were already killed by IFN-γ at these higher concentrations (14), and enhanced autophagy could not further increase the effect of IFN-γ. Meanwhile, the autophagy inhibitor LY294002 considerably reverted the killing effect of 0.5 U/mL and 1 U/mL IFN-γ on EEFs (Figure 2B and Supplementary Figure S5B), and a similar result was also obtained using another autophagy inhibitor, wortmannin (Figure 2C). No toxicity effect on cells was found for any of the above three drugs and IFN-γ at the working concentrations (Supplementary Figure S5C), excluding the possible effect of these drugs on EEF development in HepG2 cells. Furthermore, knockdown of either ATG5 (key component of the ATG12-ATG5 conjugate) or ATG7(E1-like enzyme), by specific small hairpin RNA (shRNA) could significantly inhibit the IFN-γ-mediated suppression of EEF development (Figures 2D, E).
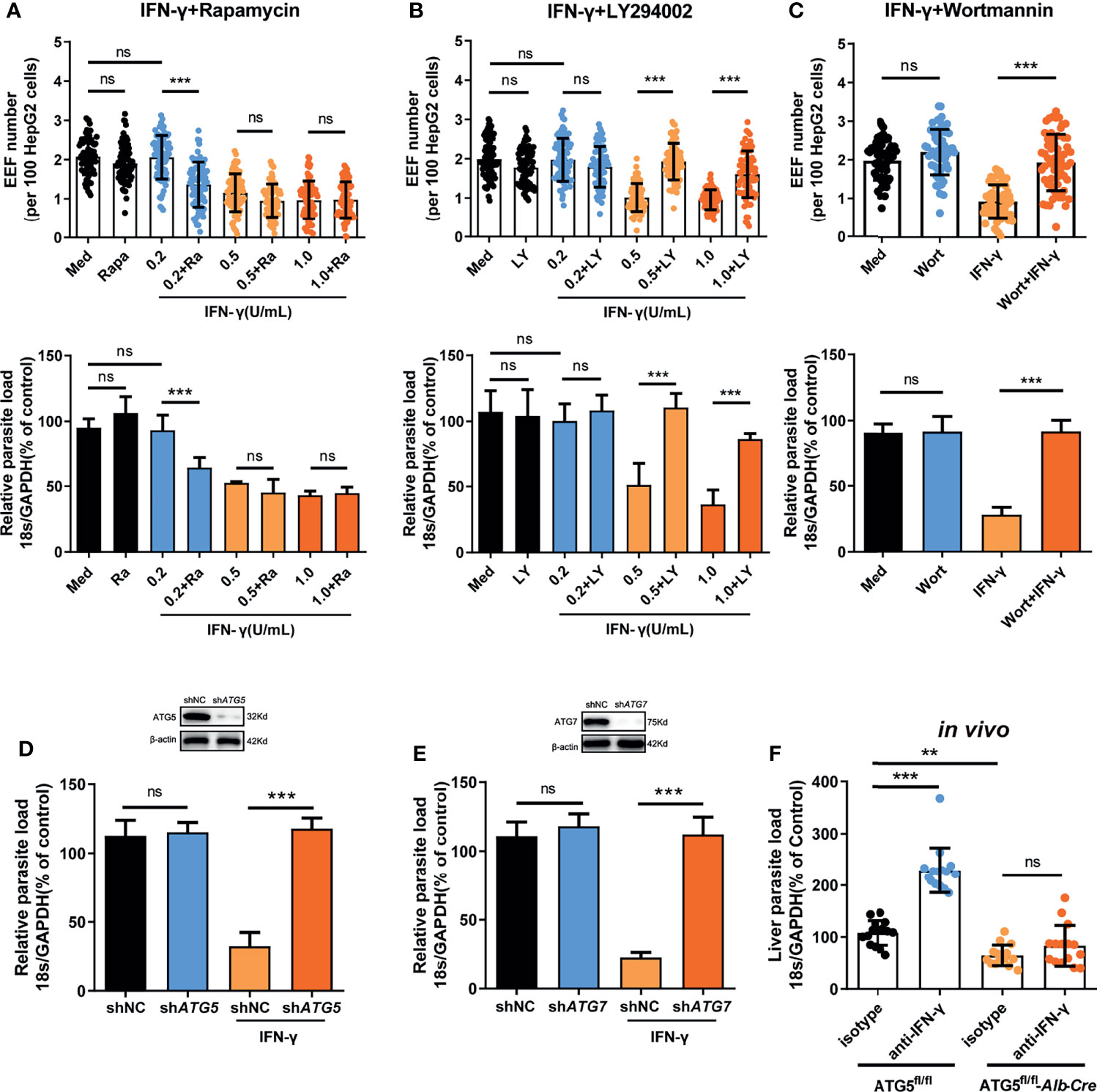
Figure 2 Autophagy and ATGs are essential for the IFN-γ-mediated killing of EEFs. (A) 1.2 × 105 HepG2 cells were pre-treated with or without the autophagy inducer rapamycin (Ra) and indicated concentrations of IFN-γ and then incubated with 4 × 104 CSPwt sporozoites. The EEF number (up, each dot represents one visual field, n=75, magnification, 630×) and the parasite load (bottom) at 46 h post-infection were determined and compared. Three individual experiments have been performed. Med, medium. (B) HepG2 cells were pre-treated with or without the autophagy inhibitor LY294002 (LY) and IFN-γ at the indicated concentrations and then incubated with CSPwt sporozoites as above. The EEF number (up, each dot represents one visual field, n=75, magnification, 630×) and the parasite load (bottom) at 46 h post-infection were determined and compared. The experiment has been repeated for three times. Med, medium. (C) HepG2 cells were pre-treated with or without the autophagy inhibitor wortmannin (Wort) at 100 mM and 1 U/mL IFN-γ, and then incubated with CSPwt sporozoites as above for 46 h. The EEF number (up, each dot represents one visual field, n=60, magnification, 630×) and the parasite burden (bottom) at 46 h post-infection were determined and compared. Three individual experiments have been performed. Med, medium. (D) HepG2 cells were transiently transfected with shNC or shATG5 plasmids, and then treated with 1 U/mL IFN-γ and infected with CSPwt sporozoites as above for 46 h. The knockdown of ATG5 was verified by western blot (up), and the parasite burden (down) in HepG2 cells was determined and compared. Three individual experiments have been performed. (E) HepG2 cells were transiently transfected with shNC or shATG7 plasmids and then treated with 1 U/mL IFN-γ and infected with CSPwt sporozoites as above for 46 h. The knockdown of ATG7 was verified by western blotting (up), and the parasite burden (down) in HepG2 cells was determined and compared. The experiment has been performed for three time. (F) After IFN-γ was depleted with or without anti-IFN-γ, both control (ATG5fl/fl) and ATG5fl/fl-Alb-Cre mice were intravenously injected with 1,000 CSPwt sporozoites, 46h later, the liver parasite burdens were measured and compared. Each dot represents one mouse (n=15). The pooled data of three repeated experiments was presented, data are represented as mean ± SD and analyzed by One-Way ANOVA and analyzed by One-Way ANOVA. ns, not significant; **P < 0.01; ***P < 0.001.
In addition, the depletion of IFN-γ greatly increased the liver parasite load in control ATG5fl/fl mice but had no effect on parasite burden in ATG5fl/fl-Alb-Cre mice, in which ATG5 in hepatocytes was specifically knocked out (Figure 2F), indicating the essential role of ATG5 in mediating the IFN-γ-killing of EEFs. However, we also noted that the level of liver burden in ATG5fl/fl-Alb-Cre mice was even lower than that in control ATG5fl/fl mice in the presence of IFN-γ (Figure 2F). The reduced liver burden in ATG5fl/fl-Alb-Cre mice may be caused by completely abolishing of canonical autophagy after ATG5 knockout. As canonical autophagy could promote parasite development by supplying essential nutrients (23), and the lack of canonical autophagy greatly limited the replication of EEF in hepatocyte, even in the absence of IFN-γ. Thus, our findings show that both autophagy and ATGs are pivotal for the killing and suppression of EEF development in hepatocytes by IFN-γ, which is consistent with the report that an ATGs-mediated LAP-like process was also involved in the IFN-γ-mediated killing of the malaria liver stage (8).
We then investigated whether CSP could inhibit autophagy and the expression of ATGs. Immunofluorescence assay showed that rapamycin-induced autophagy was significantly inhibited in CSP-stably transfected HepG2 cells as either the fluorescence intensity of the key autophagy marker, microtubule-associated protein 1 light chain 3 (LC3)-RFP or LC3-GFP puncta, induced by rapamycin, was reduced by more than 3 folds in CSP-stably transfected HepG2 cells as compared to that in control cells (Figure 3A). In addition, the intensity of LC3 fluorescence around the CSPmut EEFs was much stronger than that of CSPwt EEFs in HepG2 cells, and the level of LC3 fluorescence intensity around CSPmut EEFs was comparable to that around CSPwt EEFs in CSP-overexpressing HepG2 cells (Figure 3B). Next, the effect of CSP on the expression of ATGs was investigated. LC3 levels were significantly reduced in CSP-stably transfected HepG2 cells treated with rapamycin at all test time points when compared with control group, and the levels of autophagy-related genes, including Beclin-1, ATG3, ATG5, and ATG7, were also significantly decreased at 24 h. In contrast, the autophagy adaptor protein SQSTM1 (p62) significantly accumulated in CSP-stably transfected HepG2 cells, indicating the suppression of autophagy (Figure 3C and Supplementary Figure S6). Overall, these findings demonstrate that CSP could inhibit autophagy and the expression of ATGs.
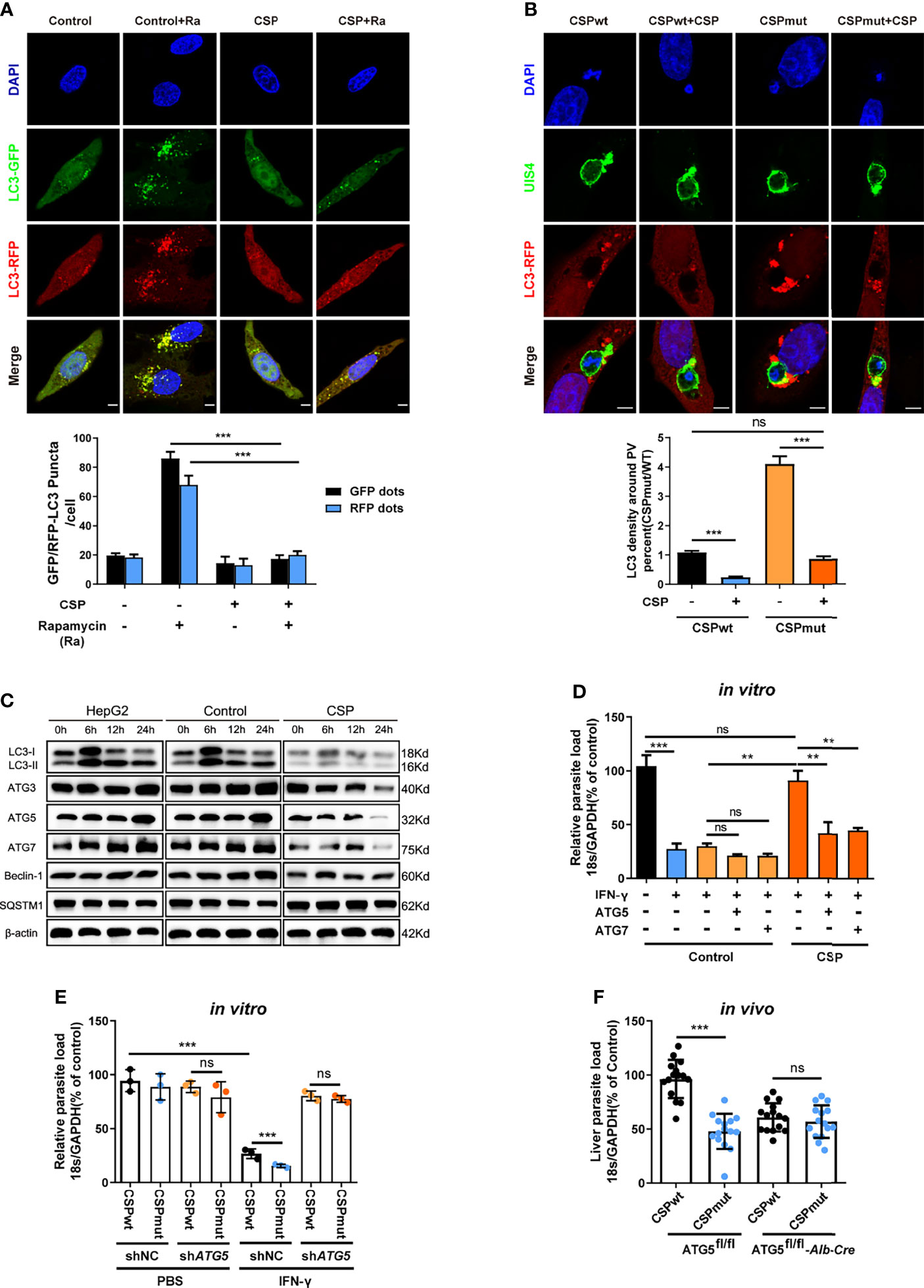
Figure 3 CSP inhibits the IFN-γ-mediated suppression of EEFs by the downregulation of ATGs. (A) 1.2 × 105 control and CSP-stably transfected HepG2 cells were infected with Ad-mRFP-GFP-LC3 virus, and then treated with the autophagy inducer rapamycin for 24 h. Both LC3-RFP or LC3-GFP puncta in HepG2 cells were imaged (up) and qualified (down). Scale bar = 5 μm, n=60. (B) 1.2 × 105 control and CSP-stably transfected HepG2 cells were transiently transfected with LC3-RFP plasmid, and then incubated with 4 × 104 CSPwt or CSPmut sporozoites for 24 h. Cells were fixed and stained with anti-UIS4, and LC3 surrounding the EEFs was observed under confocal microscopy (up), and the intensity of LC3 around PV was quantified (down), Scale bar = 5 μm, n (EEFs) =60. (C) HepG2 (without lentivirus infection), control (infected with Lenti-pCDH control lentivirus and selected by puromycin) and CSP-stably transfected HepG2 cells were treated with rapamycin for the indicated times. Protein levels of ATGs, including LC3I/LC3II, ATG3, ATG5, ATG7, Beclin-1, and p62 (SQSTM1), was determined by western blotting. (D) Control and CSP-stably transfected HepG2 cells transfected with or without ATG5, or ATG7 plasmid, and then treated with 1 U/mL IFN-γ and infected with CSPwt sporozoites as above for 46 h. The parasite load in HepG2 cells was determined and compared. Three individual experiments have been performed. (E) HepG2 cells were transiently transfected with Scramble or ATG5 shRNA plasmids, and then treated with 1 U/mL IFN-γ and incubated with CSPwt or CSPmut sporozoites as above. 46 h later, the parasite load in the HepaG2 cells was determined and compared. The experiment has been performed for three times. (F) Both control (ATG5fl/fl) and ATG5fl/fl-Alb-Cre mice were infected by intravenous injection with 1,000 CSPwt or CSPmut sporozoites, 46 h later, the liver burden of the two parasites in control and ATG5fl/fl-Alb-Cre mice were determined and compared. Each dot represents one mouse, n=15. The pooled data of three repeated experiments was presented, data are represented as mean ± SD, and analyzed by a student’s t-test, Mann-Whitney U test or One-Way ANOVA; ns, not significant; **P < 0.01; ***P < 0.001.
As CSP could significantly suppress autophagy and its related proteins, we next investigated whether the resistance of CSP to IFN-γ-mediated suppression of EEFs was closely associated with its ability to suppress ATGs expression. Although the CSP-stably transfected HepG2 cells had a significant resistance to the IFN-γ-mediated suppression of EEFs, the overexpression of either ATG5 or ATG7 significantly reduced this inhibitory effect (Figure 3D). Similarly, the CSPwt and CSPmut parasite burdens in ATG5-knockdown HepG2 cells were comparable when the cells were treated with IFN-γ (Figure 3E). To further confirm this finding, both control (ATG5fl/fl) and ATG5fl/fl-Alb-Cre mice were infected with either CSPwt or CSPmut sporozoites, and as expected, there was no difference in the liver burdens in ATG5fl/fl-Alb-Cre mice between the two parasites (Figure 3F). Taken together, these findings demonstrate the essential role of ATGs in the resistance to IFN-γ-mediated suppression by CSP.
To explore the underlying mechanism by which CSP downregulates the expression of ATGs, mRNAs from CSP-stably transfected and control HepG2 cells treated with rapamycin were sequenced and compared. Unexpectedly, there was no significant downregulation in the mRNA levels of most ATGs in CSP-stably transfected HepG2 cells as compared to that of the control (Figure 4A), which was further confirmed by real-time PCR (Figure 4B). As CSP downregulated the protein levels of ATGs, this finding suggests that CSP does not regulate the expression of ATGs at the transcriptional level.
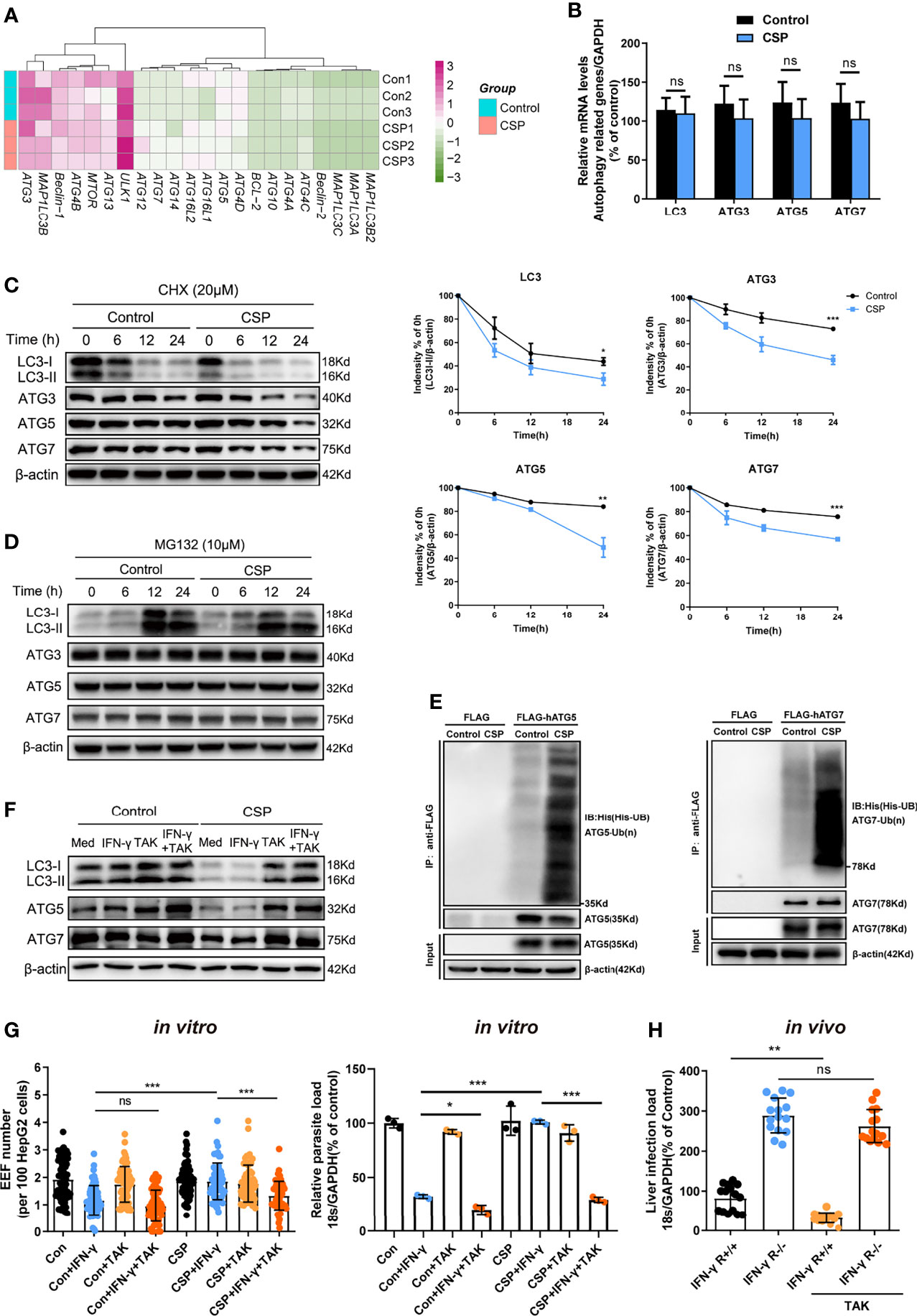
Figure 4 The CSP-mediated resistance to IFN-γ- killing of EEFs through the enhance of ATGs ubiquitination. (A) The mRNA levels of ATGs between control and CSP-stably transfected HepG2 cells in the heatmap were compared, and three biological repeats were performed. (B) The different mRNA levels of ATGs (ATG3, ATG5, ATG7, and LC3) between control and CSP-stably transfected HepG2 cells were compared using real-time PCR. Three individual experiments have been performed. (C) Control and CSP-stably transfected HepG2 cells were treated with CHX for the indicated times, and the protein levels of LC3I/LC3II, ATG3, ATG5 and ATG7 were detected by western blotting (left); the relative expression levels of LC3I/LC3II, ATG3, ATG5, and ATG7 to β-actin were quantified (right). Three individual experiments have been performed. (D) Control and CSP-stably transfected HepG2 cells were treated with the proteasome inhibitor MG132 for the indicated times, and the protein levels of LC3I/LC3II, ATG3, ATG5, and ATG7 were detected by western blot. The experiment have been performed for three times. (E) Ubiquitination assay showed that CSP promoted the ubiquitination of ATG5 (left) and ATG7 (right). Control and CSP-stably transfected HepG2 cells were transfected with His-UB, and FLAG-hATG5 or FLAG-hATG7 plasmids, and treated with MG-132 for 24 h. Then, cells were collected for immunoprecipitation of FLAG-hATG5 and FLAG-hATG7 followed by immunoblot analyses of ubiquitin (His-UB). Cells transfected with FLAG vector was used as negative controls. Total cell lysates (input) were also immunoblotted with antibodies against FLAG and β-actin. The experiment has been repeated for three times. (F) The levels of protein LC3, ATG5 and ATG7 were determined by western blotting in both control and CSP-stably transfected HepG2 treated with or without 1μM TAK (TAK-243) and 1 U/mL IFN-γ for 24 h. (G) 1.2 × 105 CSP-stably transfected HepG2 or control cells were treated with or without 1μM TAK (TAK-243), and then were treated with 1 U/mL IFN-γ and infected with 4 × 104 CSPwt sporozoites for 46 h. The EEF number (left, each dot represents one visual field, n=57-73, magnification, 630×) and parasite load (right) were determined and compared as above. Three individual experiments have been performed (H) IFN-γ R1 knockout and WT mice were per-treated with or without 20mg/kg TAK-243 and infected with 1,000 CSPwt sporozoites in the next day, then liver parasite burden was determined at 46 h after infection as described as above. Each dot represents one mouse (n=15). The pooled data of three repeated experiments was presented, data are represented as mean ± SD and analyzed by a student’s t-test or One-way ANOVA; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Eukaryotic cells use the autophagy-lysosome and ubiquitin-proteasome system as their major intracellular protein degradation pathways (24). However, autophagy level was significantly reduced in the CSP-stably transfected HepG2 cells (Figure 3A), indicating that the autophagy-lysosome system may not be responsible for the downregulation of the ATGs protein levels. Therefore, we next investigated whether CSP could downregulate the expression of ATGs at the post-translational level through the ubiquitin-proteasome pathway, which is a major intracellular degradation pathway and a widespread post-translational modification (25). The half-lives of LC3, ATG3, ATG5, and ATG7 in the CSP-stably transfected HepG2 cells were significantly reduced compared to those of the control, when cells were treated with the protein synthesis inhibitor cycloheximide (CHX) (Figure 4C). In addition, the levels of LC3, ATG3, ATG5, and ATG7 in CSP-stably transfected and control HepG2 cells were comparable when the cells were treated with the specific proteasome inhibitor MG132 (Figure 4D and Supplementary Figure S7). These findings indicate that CSP could regulate the degradation of ATGs in a proteasome-dependent manner. To investigate whether the degradation of ATGs in the proteasome was dependent on ubiquitination, the CSP-stably transfected and control cells were transfected with His-UB, FLAG-hATG5 (human ATG5), or FLAG-hATG7 (human ATG7) plasmids, and co-immunoprecipitation was performed using anti-FLAG beads to detect ubiquitin bound to ATGs. The ubiquitination assay showed that CSP promoted the ubiquitination of both ATG5 and ATG7 in HepG2 cells (Figure 4E).
To verify that the resistance of CSP to IFN-γ-mediated suppression of EEF development was due to the enhancement of ATGs ubiquitination, a ubiquitination-specific inhibitor TAK-243 (26), was used at non-toxic concentrations in vitro and in vivo (Supplementary Figure S8) to suppress ubiquitination. We found that treatment with TAK-243 significantly elevated the protein levels of ATGs (Figure 4F) and enhanced the IFN-γ-mediated killing of EEFs in CSP-stably transfected HepG2 cells, as compared to those in control cells (Figure 4G). Furthermore, the administration of TAK-243 greatly reduced the liver parasite burden in WT mice but had no significant effect on liver parasite burden in IFN-γ R1 knockout mice (Figure 4H). Thus, we demonstrate that CSP mediated resistance to IFN-γ- killing of EEFs through the upregulation of specific ubiquitination of ATGs.
E3 ubiquitin ligase, which transfers the ubiquitin of the E2 enzyme to its attached substrate, is the key enzyme in the ubiquitination process (25). Therefore, we hypothesized that E3 ubiquitin ligases may be involved in the ubiquitination of ATGs (27). Six candidate E3 ligases, including STIP1 homology and U box-containing protein 1 (STUB1), Synovial apoptosis inhibitor 1 (SYVN1), Cbl proto-oncogene (CBL), SMAD specific E3 ubiquitin protein ligase 1 (SMURF1), Platelet activating factor acetylhydrolase 1b regulatory subunit 1 (PAFAH1B1), and neural precursor cell expressed, developmentally down-regulated 4 (NEDD4), were predicted to regulate the ubiquitination of at least two ATGs using an online prediction tool (Supplementary Figure S9 and Table S2) (27). However, the RNA-seq data showed that among these ligases, only NEDD4 was significantly upregulated in CSP-stably transfected HepG2 cells (Figure 5A), and the increase in both the mRNA and protein levels of NEDD4 was confirmed by real-time PCR and western blotting, respectively (Figures 5B, C). Furthermore, infection with the CSPwt parasite, but not the CSPmut parasite, significantly elevated the mRNA level of NEDD4 in HepG2 cells (Figure 5D), strongly suggesting that the translocation of CSP from the PV into the cytoplasm could upregulate the expression of E3 ubiquitin ligase NEDD4. Consistently, the upregulation of NEDD4 in sporozoite-infected hepatocyte cell lines was also found in the P. b ANKA infection model (data from GSE78931 and GSE72049) (28) (Figure 5E). Moreover, knockdown of NEDD4 by shRNA significantly reduced CSP-mediated ubiquitination of ATG5 and ATG7 (Figure 5F). In addition, the resistance of CSP-overexpression to IFN-γ-mediated killing of CSPwt parasites was significantly reduced after NEDD4 was silenced (Figure 5G). This finding strongly suggests that NEDD4 was essential for CSP-mediated resistance to IFN-γ suppression of EEFs.
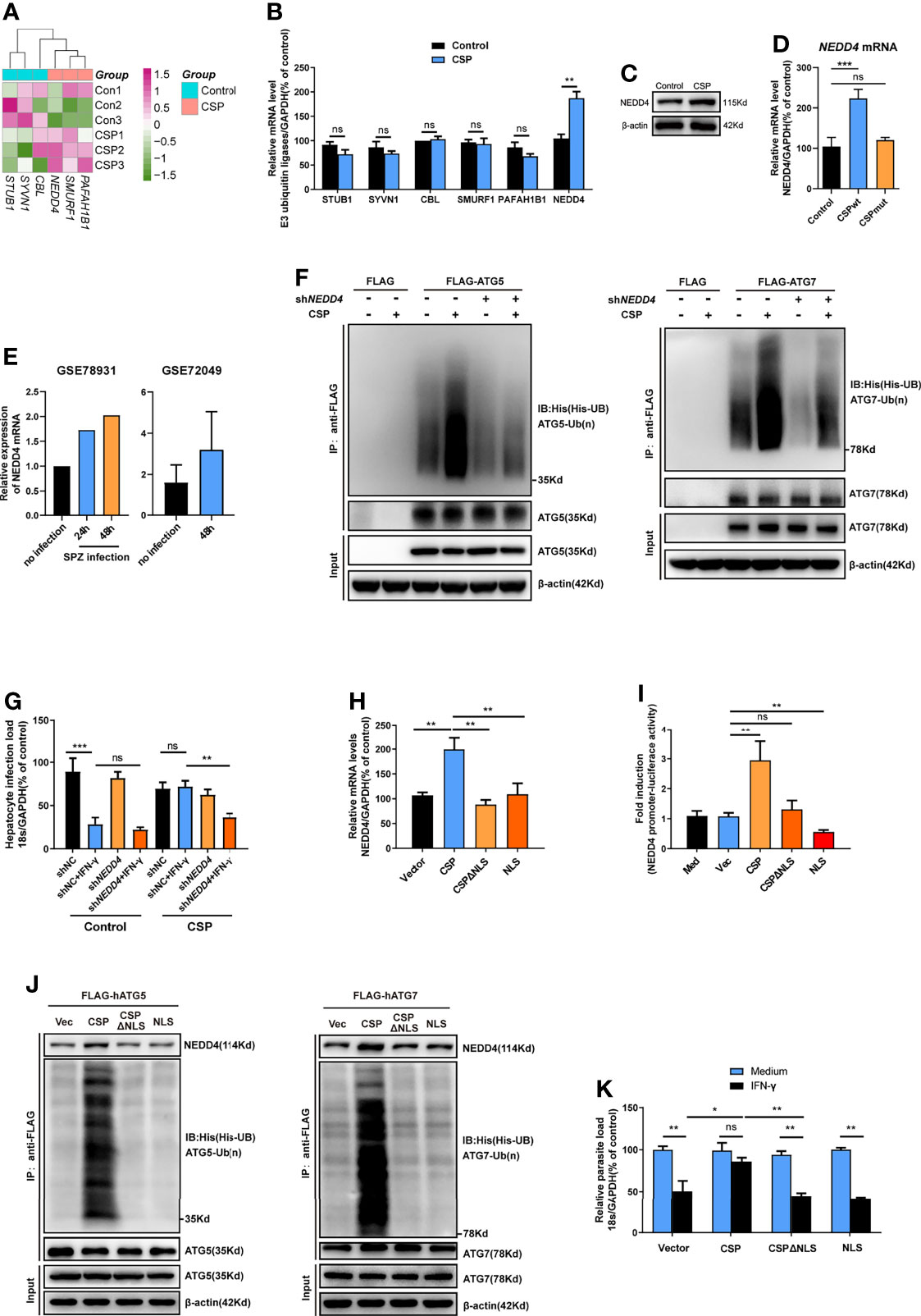
Figure 5 CSP upregulates the E3 ubiquitin ligase NEDD4 to promote the ubiquitination of ATGs. (A) The mRNA levels of six E3 ubiquitin ligases between control and CSP-stably transfected HepG2 cells in the heatmap were compared, and three biological replicates were performed. (B) The different mRNA levels of six E3 ubiquitin ligases in both control and CSP-stably transfected HepG2 cells at 24 after culture were confirmed by real-time PCR. Three individual experiments have been performed. (C) The protein levels of NEDD4 in both control and CSP-stably transfected HepG2 cells at 24 after culture were confirmed by western blotting. Three individual experiments have been performed. (D) After HepG2 cells were infected with CSPwt or CSPmut sporozoites as described as before for 24 h, the mRNA level of NEDD4 was determined by real-time PCR, uninfected HepG2 cells were used as control. The experiment has been repeated for three times. (E) Relative level of NEDD4 mRNA in P.b ANKA sporozoite infected Huh7 and HepG2 cells were analyzed based on data extracted from GSE78931 and GSE72049. (F) Ubiquitination assay showed that shNEDD4 deceased CSP-induced hATG5 (left) and hATG7 (right) ubiquitination in 293FT cells. 293FT cells were transfected with His-UB, pcDNA3.1 vector or pcDNA3.1-CSP, shNC or shNEDD4, and FLAG vector, FLAG-hATG5 or FLAG-hATG7 plasmids, and treated with MG-132 for 24 h. Then, cells were collected for immunoprecipitation of FLAG-hATG5 and FLAG-hATG7 followed by immunoblot analyses of ubiquitin (His-UB). Cells transfected with FLAG vector was used as negative controls. Total cell lysates (input) were also immunoblotted with antibodies against FLAG and β-actin. Three individual experiments have been performed. (G) 1.2 × 105 control and CSP-stably transfected HepG2 cells were transfected with shNC or shNEDD4 and treated with 1 U/mL IFN-γ or not, then infected with 4 × 104 CSPwt sporozoites, for 46 h. The parasite load in HepG2 cells was determined as above. The experiment has been performed for three times. (H) HepG2 cells were transfected with vector (Vec), CSP, CSPΔNLS, or NLS plasmids for 24 h, and the transcription of NEDD4 was determined by real-time PCR. Three individual experiments have been performed. (I) HEK293T cells were transfected with RL-TK, NEDD4 promoter reporter plasmid, and with or without Vec, CSP, CSPΔNLS, or NLS plasmids for 24 h, and the ratio of firefly luciferase to Renilla luciferase were determined using Dual Luciferase Assay kit. Three individual experiments have been performed. Med, medium. (J) The ubiquitination assay showed that deletion of NLS domain (ΔNLS) deceased CSP-induced hATG5 (left) and hATG7 (right) ubiquitination in 293FT cells. 293FT cells were transfected His-UB, and Vec, CSP, CSPΔNLS or NLS, and FLAG-hATG5 or FLAG-hATG7 plasmids, and treated with MG-132 for 24 h. Then, cells were collected for immunoprecipitation of FLAG-hATG5 and FLAG-hATG7 followed by immunoblot analyses of ubiquitin (His-UB). Cells transfected with pcDNA3.1 vector was used as negative controls. Total cell lysates (input) were also immunoblotted with antibodies against FLAG and β-actin. The experiment has been repeated for three times. (K) 1.2 × 105 HepG2 cells were transfected with Vec, CSP, CSPΔNLS, or NLS plasmids and treated with or without 1 U/mL IFN-γ, followed by the infection with 4 × 104 CSPwt sporozoites for 46 h. The parasite load in HepG2 cells was determined as above. Three individual experiments have been performed. The pooled data of three repeated experiments was presented (except E), data are represented as mean ± SD and analyzed by Mann-Whitney U test or One-way ANOVA. UB, ubiquitin; ns, not significant; *P < 0.05; **P < 0.01, ***P < 0.001.
A previous study showed that CSP contained a nuclear location signal (NLS) domain, which could import the CSP into the nucleus (11), and we also confirmed that the transfection of CSP, but not CSPΔNLS plasmid (CSP without NLS domain), could import into the nucleus of HepG2 cells (Supplementary Figure S10). To better elucidate the mechanism of the upregulation of NEDD4 by CSP, the effects of CSP, CSPΔNLS, and NLS (only NLS domain) expression on the transcription of NEDD4 in HepG2 cells were determined. Although the transfection of CSP upregulated NEDD4 expression, neither the transfection of CSPΔNLS nor NLS influenced the transcription of NEDD4 in HepG2 cells (Figure 5H). The dual luciferase reporter assay confirmed that only CSP significantly activated the NEDD4 promoter (Figure 5I). These data indicate that translocation into the nucleus was essential for CSP to modulate the transcription of NEDD4, but the modulatory motif might not exist in the sequence of CSPΔNLS. In addition, co-immunoprecipitation results showed that more NEDD4 proteins could directly bind to both ATG5 and ATG7 and increased the ubiquitination of these two proteins in CSP, but not in the vector, CSPΔNLS or NLS-transfected 293FT cells (Figure 5J), and only the transfection of CSP remarkably inhibited the IFN-γ-mediated suppression of EEFs in HepG2 cells (Figure 5K). Overall, these data suggest that NEDD4 was an E3 ubiquitin ligase that specifically mediated the ubiquitination of ATG5 and ATG7, and that CSP enhanced the ubiquitination of ATGs through the upregulation of NEDD4.
Considerable progress has been made in identifying the molecules of sporozoites involved in their selective attachment to hepatocytes and the formation of the PV after their invasion (1, 29–31); however, the underlying mechanism of the survival of parasites in the PV has thus far remained unknown. Here, we found that nuclear translocation of cytoplasmic CSP could mediate the resistance to IFN-γ-killing of EEFs and facilitate the survival of parasites in the PV.
CSP is a multifunctional protein of the malaria parasite that is not only critical for the invasion of sporozoites into hepatocytes but is also essential for sporozoite development in mosquitoes (32–38). Therefore, CSP is regarded as a leading candidate protective antigen (39), which is included in several subunit vaccines to induce CSP-specific antibody and/or CD8+T cell responses against sporozoites (40–46). Our finding of this new aspect of CSP to subvert the host innate immune response not only supports the important role of CSP as the dominant protective antigen but also sheds new light on a novel prophylactic strategy against the liver stage.
Recent studies have shown that parasites can shed host autophagic proteins from the PVM to escape the deleterious effects of the PAAR response (47) and UIS3 sequestered LC3 to suppress autophagy in EEFs (48). These findings support the existence of subversive strategies for EEFs against hepatocyte autonomous immunity. In this study, we found that CSP translocated from the PV into the cytoplasm of hepatocytes inhibited the IFN-γ-mediated killing of EEFs by downregulating the expression of ATGs by ubiquitination, revealing a novel strategy for the liver stage of malaria to subvert host innate immunity. However, a previous study suggested that liver-stage development enhanced by CSP was associated with its ability to suppress NF-κB activation in infected hepatocytes. Although the real reason is definitively unknown for the different mechanisms of CSP to promote EEF development between our study and previous studies, the possible interpretation might be the missed consideration of IFN-γ in the natural malaria infection in a previous study.
It is well known that IFN-γ-mediated killing of intracellular T. gondii is regulated by IFN-γ-inducible GTPase Irg6 and guanylate-binding protein (GBP)1 (49–51), whereas Irg6 is not involved in the IFN-γ-mediated killing of EEFs (49). Indeed, we found that the knockout of GBP-1 also had no effect on EEF development in HepG2 cells treated with IFN-γ (Supplementary Figure S11). Thus, the roles of GTPase and GBPs in the IFN-γ-mediated killing of EEFs might differ from those in T. gondii and should be investigated in further detail.
Recent studies have demonstrated that infection with sporozoites could induce both canonical autophagy and the PAAR response of hepatocytes to EEFs (9, 22). Although canonical autophagy promotes parasite development by supplying essential nutrients (23), the role of PAAR response in liver stage development is conflicting (9, 52). One possible reason is that conditional ATG knockout of non-permissive mouse embryonic fibroblasts (MEFs) has been used in previous studies (9, 52). In the present study, we found that knockdown of either ATG5 or ATG7 in HepG2 cells, the permissive cells of sporozoites, had no significant effect on EEF development (Figures 2D, E). In contrast, the induction of autophagy by rapamycin enhanced the killing effect of IFN-γ (Figure 2A), and autophagy inhibitors or knockdown of ATGs attenuated the killing of EEFs by IFN-γ (Figures 2B–E). This demonstrates the essential role of autophagy in the regulation of the IFN-γ-mediated killing of EEFs. Therefore, we showed that autophagy alone has no significant effect on EEF development in vitro, but it plays a pivotal role in the regulation of IFN-γ-mediated killing of EEFs.
Taken together, we found that CSP translocated from the PV into the cytoplasm of hepatocytes could be imported into the nucleus and upregulated the transcription of E3 ligase NEDD4, depending on its NLS domain. The E3 ligase NEDD4 enhances the ubiquitination of ATGs, leading to a decrease in the protein level of ATGs in hepatocytes. As ATGs are essential for IFN-γ-mediated killing of EEFs, CSP resists the IFN-γ-mediated killing of EEFs and facilitates the survival of EEFs in hepatocytes (Figure 6). Thus, our findings explain why CSP translocated from the PV into the hepatocyte cytoplasm could promote liver stage development, and provide novel prophylaxis strategies to eliminate liver stage infection by chemically targeting CSP pexel, NLS domain, or ubiquitination in hepatocytes.
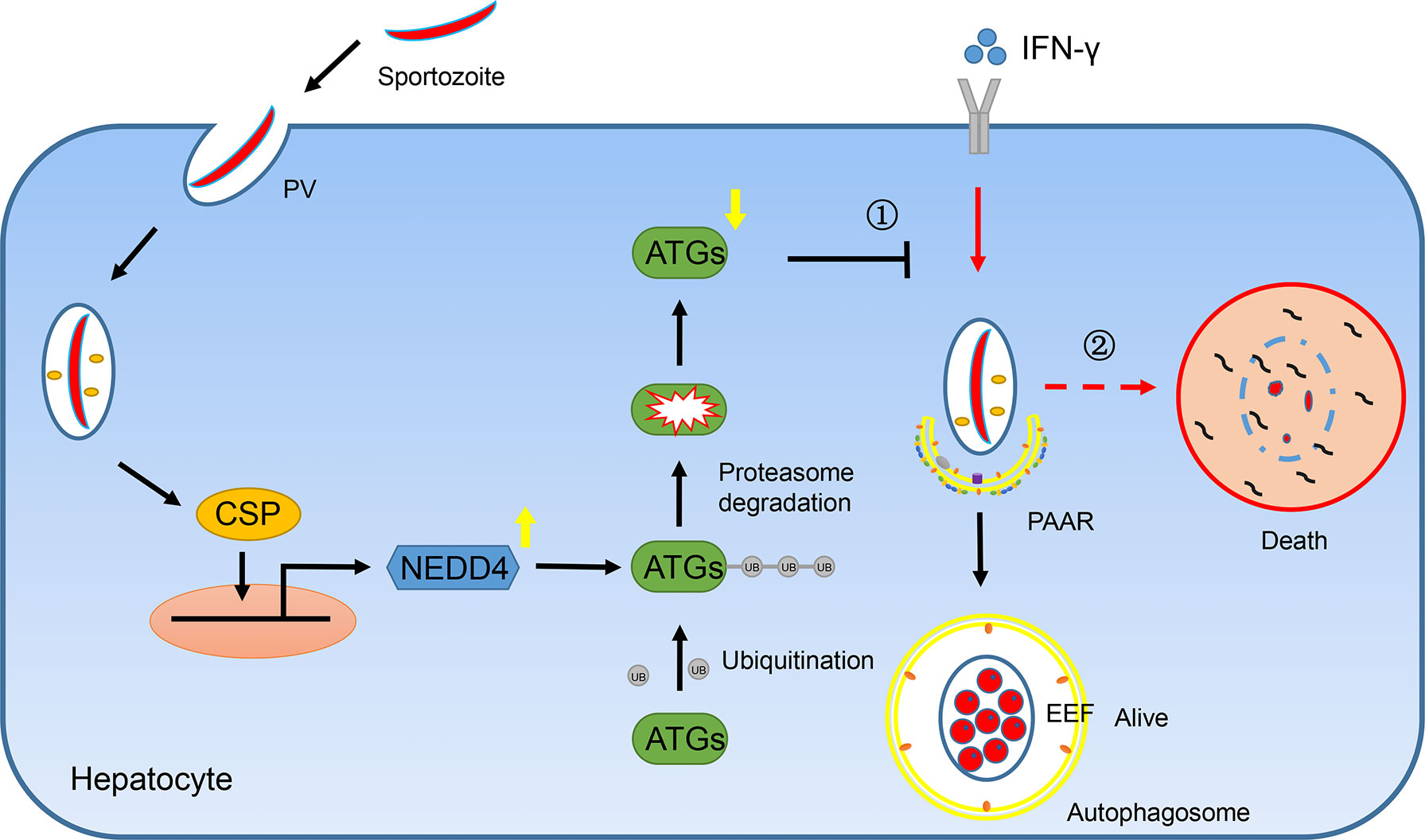
Figure 6 A mechanistic pathway of CSP-mediated resistance to IFN-γ- killing of EEFs in hepatocytes. ①After sporozoite invaded into hepatocytes, CSP on its surface would be translocated from PV into the cytoplasm of hepatocytes and is then introduced into nucleus dependent on its NLS domain. As a result, CSP activates the promoter of ligase3 NEDD4 and enhances its transcription. The upregulated NEDD4 promotes the ubiquitination of ATGs, leading to the degradation of ATGs. As ATGs are essential for IFN-γ-mediated killing of EEFs, the CSP-mediated resistance to IFN-γ, which is dependent on its ability to downregulate ATGs through enhancing NEDD4-mediated ubiquitination, facilitates the survival of EEFs. ②Otherwise, EEFs are destroyed by IFN-γ.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE129323.
The animal study was reviewed and approved by Animal Ethics Committee of Army Medical University.
Conceptualization, WX, YW, HZ, and JD. Writing – Original Draft, WX and HZ. Writing–Review & Editing, WX, YW, JD, and HZ. Methodology, HZ and XL. Investigation, HZ, XL, KL, KZ, CZ, TL, FZ, TZ, YD, and YF. Funding Acquisition, WX. Resources, WX, YW, and JD. Supervision, WX, YW, and JD. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 81672053 and 81702247), the State Key Program of the National Natural Science Foundation of China (No. 81830067), and the Miaopu Talent Grant from Army Medical University (2019R057).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.815936/full#supplementary-material
1. Coppi A, Tewari R, Bishop JR, Bennett BL, Lawrence R, Esko JD, et al. Heparan Sulfate Proteoglycans Provide a Signal to Plasmodium Sporozoites to Stop Migrating and Productively Invade Host Cells. Cell Host Microbe (2007) 2(5):316–27. doi: 10.1016/j.chom.2007.10.002
2. Mueller AK, Camargo N, Kaiser K, Andorfer C, Frevert U, Matuschewski K, et al. Plasmodium Liver Stage Developmental Arrest by Depletion of a Protein at the Parasite-Host Interface. Proc Natl Acad Sci USA (2005) 102(8):3022–7. doi: 10.1073/pnas.0408442102
3. Sa ECC, Nyboer B, Heiss K, Sanches-Vaz M, Fontinha D, Wiedtke E, et al. Plasmodium Berghei EXP-1 Interacts With Host Apolipoprotein H During Plasmodium Liver-Stage Development. Proc Natl Acad Sci USA (2017) 114(7):E1138–47. doi: 10.1073/pnas.1606419114
4. Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-Cell Sensors for Plasmodium Activate Innate Immunity Against Liver-Stage Infection. Nat Med (2014) 20(1):47–53. doi: 10.1038/nm.3424
5. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe S. Interferon-Mediated Innate Immune Responses Against Malaria Parasite Liver Stages. Cell Rep (2014) 7(2):436–47. doi: 10.1016/j.celrep.2014.03.018
6. Klotz FW, Scheller LF, Seguin MC, Kumar N, Marletta MA, Green SJ, et al. Co-Localization of Inducible-Nitric Oxide Synthase and Plasmodium Berghei in Hepatocytes From Rats Immunized With Irradiated Sporozoites. J Immunol (1995) 154(7):3391–5.
7. Nahrevanian H, Dascombe MJ. Nitric Oxide and Reactive Nitrogen Intermediates During Lethal and Nonlethal Strains of Murine Malaria. Parasite Immunol (2001) 23(9):491–501. doi: 10.1046/j.1365-3024.2001.00406.x
8. Boonhok R, Rachaphaew N, Duangmanee A, Chobson P, Pattaradilokrat S, Utaisincharoen P, et al. LAP-Like Process as an Immune Mechanism Downstream of IFN-Gamma in Control of the Human Malaria Plasmodium Vivax Liver Stage. Proc Natl Acad Sci USA (2016) 113(25):E3519–28. doi: 10.1073/pnas.1525606113
9. Prado M, Eickel N, De Niz M, Heitmann A, Agop-Nersesian C, Wacker R, et al. Long-Term Live Imaging Reveals Cytosolic Immune Responses of Host Hepatocytes Against Plasmodium Infection and Parasite Escape Mechanisms. Autophagy (2015) 11(9):1561–79. doi: 10.1080/15548627.2015.1067361
10. Agop-Nersesian C, Niklaus L, Wacker R, Theo HV. Host Cell Cytosolic Immune Response During Plasmodium Liver Stage Development. FEMS Microbiol Rev (2018) 42(3):324–34. doi: 10.1093/femsre/fuy007
11. Singh AP, Buscaglia CA, Wang Q, Levay A, Nussenzweig DR, Walker JR, et al. Plasmodium Circumsporozoite Protein Promotes the Development of the Liver Stages of the Parasite. CELL (2007) 131(3):492–504. doi: 10.1016/j.cell.2007.09.013
12. Huang Z, Wu Q, Guryanova OA, Cheng L, Shou W, Rich JN, et al. Deubiquitylase HAUSP Stabilizes REST and Promotes Maintenance of Neural Progenitor Cells. Nat Cell Biol (2011) 13(2):142–52. doi: 10.1038/ncb2153
13. Liehl P, Meireles P, Albuquerque IS, Pinkevych M, Baptista F, Mota MM, et al. Innate Immunity Induced by Plasmodium Liver Infection Inhibits Malaria Reinfections. Infect Immun (2015) 83(3):1172–80. doi: 10.1128/IAI.02796-14
14. Schofield L, Ferreira A, Altszuler R, Nussenzweig V, Nussenzweig RS. Interferon-Gamma Inhibits the Intrahepatocytic Development of Malaria Parasites In Vitro. J Immunol (1987) 139(6):2020–5.
15. Ferreira A, Schofield L, Enea V, Schellekens H, van der Meide P, Collins WE, et al. Inhibition of Development of Exoerythrocytic Forms of Malaria Parasites by Gamma-Interferon. Science (1986) 232(4752):881–4. doi: 10.1126/science.3085218
16. Sedegah M, Finkelman F, Hoffman SL. Interleukin 12 Induction of Interferon Gamma-Dependent Protection Against Malaria. Proc Natl Acad Sci USA (1994) 91(22):10700–2. doi: 10.1073/pnas.91.22.10700
17. Mellouk S, Green SJ, Nacy CA, Hoffman SL. IFN-Gamma Inhibits Development of Plasmodium Berghei Exoerythrocytic Stages in Hepatocytes by an L-Arginine-Dependent Effector Mechanism. J Immunol (1991) 146(11):3971-3976.
18. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome-Independent Essential Function for the Autophagy Protein Atg5 in Cellular Immunity to Intracellular Pathogens. Cell Host Microbe (2008) 4(5):458–69. doi: 10.1016/j.chom.2008.10.003
19. Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, et al. the Parasitophorous Vacuole Membrane of Toxoplasma Gondii Is Targeted for Disruption by Ubiquitin-Like Conjugation Systems of Autophagy. Immunity (2014) 40(6):924–35. doi: 10.1016/j.immuni.2014.05.006
20. Ohshima J, Lee Y, Sasai M, Saitoh T, Su MJ, Kamiyama N, et al. Role of Mouse and Human Autophagy Proteins in IFN-Gamma-Induced Cell-Autonomous Responses Against Toxoplasma Gondii. J Immunol (2014) 192(7):3328–35. doi: 10.4049/jimmunol.1302822
21. Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, et al. Vacuolar and Plasma Membrane Stripping and Autophagic Elimination of Toxoplasma Gondii in Primed Effector Macrophages. J Exp Med (2006) 203(9):2063–71. doi: 10.1084/jem.20061318
22. Zhao C, Liu T, Zhou T, Fu Y, Zheng H, Ding Y, et al. the Rodent Malaria Liver Stage Survives in the Rapamycin-Induced Autophagosome of Infected Hepa1-6 Cells. Sci Rep (2016) 6:38170. doi: 10.1038/srep38170
23. Grutzke J, Rindte K, Goosmann C, Silvie O, Rauch C, Heuer D, et al. the Spatiotemporal Dynamics and Membranous Features of the Plasmodium Liver Stage Tubovesicular Network. Traffic (2014) 15(4):362–82. doi: 10.1111/tra.12151
24. Varshavsky A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu Rev Biochem (2017) 86:123–8. doi: 10.1146/annurev-biochem-061516-044859
25. Hershko A, Ciechanover A. The Ubiquitin System. Annu Rev Biochem (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
26. Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A Small-Molecule Inhibitor of the Ubiquitin Activating Enzyme for Cancer Treatment. Nat Med (2018) 24(2):186–93. doi: 10.1038/nm.4474
27. Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z, et al. an Integrated Bioinformatics Platform for Investigating the Human E3 Ubiquitin Ligase-Substrate Interaction Network. Nat Commun (2017) 8(1):347. doi: 10.1038/s41467-017-00299-9
28. Maruthi M, Singh D, Reddy SR, Mastan BS, Mishra S, Kumar KA. Modulation of Host Cell Sumoylation Facilitates Efficient Development of Plasmodium Berghei and Toxoplasma Gondii. Cell Microbiol (2017) 19(7):e12723. doi: 10.1111/cmi.12723
29. Silvie O, Rubinstein E, Franetich JF, Prenant M, Belnoue E, Renia L, et al. Hepatocyte CD81 Is Required for Plasmodium Falciparum and Plasmodium Yoelii Sporozoite Infectivity. Nat Med (2003) 9(1):93–6. doi: 10.1038/nm808
30. Yalaoui S, Zougbede S, Charrin S, Silvie O, Arduise C, Farhati K, et al. Hepatocyte Permissiveness to Plasmodium Infection Is Conveyed by a Short and Structurally Conserved Region of the CD81 Large Extracellular Domain. PloS Pathog (2008) 4(2):e1000010. doi: 10.1371/journal.ppat.1000010
31. Kaushansky A, Douglass AN, Arang N, Vigdorovich V, Dambrauskas N, Kain HS, et al. Malaria Parasites Target the Hepatocyte Receptor Epha2 for Successful Host Infection. Science (2015) 350(6264):1089–92. doi: 10.1126/science.aad3318
32. Menard R, Sultan AA, Cortes C, Altszuler R, van Dijk MR, Janse CJ, et al. Circumsporozoite Protein Is Required for Development of Malaria Sporozoites in Mosquitoes. Nature (1997) 385(6614):336–40. doi: 10.1038/385336a0
33. Cerami C, Frevert U, Sinnis P, Takacs B, Clavijo P, Santos MJ, et al. the Basolateral Domain of the Hepatocyte Plasma Membrane Bears Receptors for the Circumsporozoite Protein of Plasmodium Falciparum Sporozoites. Cell (1992) 70(6):1021–33. doi: 10.1016/0092-8674(92)90251-7
34. Rathore D, Sacci JB, de la Vega P, McCutchan TF. Binding and Invasion of Liver Cells by Plasmodium Falciparum Sporozoites. Essential Involvement of the Amino Terminus of Circumsporozoite Protein. J Biol Chem (2002) 277(9):7092–8. doi: 10.1074/jbc.M106862200
35. Pinzon-Ortiz C, Friedman J, Esko J, Sinnis P. The Binding of the Circumsporozoite Protein to Cell Surface Heparan Sulfate Proteoglycans Is Required for Plasmodium Sporozoite Attachment to Target Cells. J Biol Chem (2001) 276(29):26784–91. doi: 10.1074/jbc.M104038200
36. Frevert U, Sinnis P, Cerami C, Shreffler W, Takacs B, Nussenzweig V. Malaria Circumsporozoite Protein Binds to Heparan Sulfate Proteoglycans Associated With the Surface Membrane of Hepatocytes. J Exp Med (1993) 177(5):1287–98. doi: 10.1084/jem.177.5.1287
37. Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, et al. the Malaria Circumsporozoite Protein Has Two Functional Domains, Each With Distinct Roles as Sporozoites Journey From Mosquito to Mammalian Host. J Exp Med (2011) 208(2):341–56. doi: 10.1084/jem.20101488
38. Wang Q, Fujioka H, Nussenzweig V. Exit of Plasmodium Sporozoites From Oocysts Is an Active Process That Involves the Circumsporozoite Protein. PloS Pathog (2005) 1(1):e9. doi: 10.1371/journal.ppat.0010009
39. Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, et al. the Circumsporozoite Protein Is an Immunodominant Protective Antigen in Irradiated Sporozoites. Nature (2006) 444(7121):937–40. doi: 10.1038/nature05361
40. Gordon DM, McGovern TW, Krzych U, Cohen JC, Schneider I, LaChance R, et al. Safety, Immunogenicity, and Efficacy of a Recombinantly Produced Plasmodium Falciparum Circumsporozoite Protein-Hepatitis B Surface Antigen Subunit Vaccine. J Infect Dis (1995) 171(6):1576–85. doi: 10.1093/infdis/171.6.1576
41. Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. a Preliminary Evaluation of a Recombinant Circumsporozoite Protein Vaccine Against Plasmodium Falciparum Malaria. RTS,s Malaria Vaccine Evaluation Group. N Engl J Med (1997) 336(2):86–91. doi: 10.1056/NEJM199701093360202
42. Kubler-Kielb J, Majadly F, Biesova Z, Mocca CP, Guo C, Nussenzweig R, et al. a Bicomponent Plasmodium Falciparum Investigational Vaccine Composed of Protein-Peptide Conjugates. Proc Natl Acad Sci USA (2010) 107(3):1172–7. doi: 10.1073/pnas.0913374107
43. Foquet L, Hermsen CC, van Gemert GJ, Van Braeckel E, Weening KE, Sauerwein R, et al. Vaccine-Induced Monoclonal Antibodies Targeting Circumsporozoite Protein Prevent Plasmodium Falciparum Infection. J Clin Invest (2014) 124(1):140–4. doi: 10.1172/JCI70349
44. Aliprandini E, Tavares J, Panatieri RH, Thiberge S, Yamamoto MM, Silvie O, et al. Cytotoxic Anti-Circumsporozoite Antibodies Target Malaria Sporozoites in the Host Skin. Nat Microbiol (2018) 3(11):1224–33. doi: 10.1038/s41564-018-0254-z
45. Kisalu NK, Idris AH, Weidle C, Flores-Garcia Y, Flynn BJ, Sack BK, et al. a Human Monoclonal Antibody Prevents Malaria Infection by Targeting a New Site of Vulnerability on the Parasite. Nat Med (2018) 24(4):408–16. doi: 10.1038/nm.4512
46. Tan J, Sack BK, Oyen D, Zenklusen I, Piccoli L, Barbieri S, et al. a Public Antibody Lineage That Potently Inhibits Malaria Infection Through Dual Binding to the Circumsporozoite Protein. Nat Med (2018) 24(4):401–7. doi: 10.1038/nm.4513
47. Agop-Nersesian C, De Niz M, Niklaus L, Prado M, Eickel N, Heussler VT. Shedding of Host Autophagic Proteins From the Parasitophorous Vacuolar Membrane of Plasmodium Berghei. Sci Rep (2017) 7(1):2191. doi: 10.1038/s41598-017-02156-7
48. Real E, Rodrigues L, Cabal GG, Enguita FJ, Mancio-Silva L, Mello-Vieira J, et al. Plasmodium UIS3 Sequesters Host LC3 to Avoid Elimination by Autophagy in Hepatocytes. Nat Microbiol (2018) 3(1):17–25. doi: 10.1038/s41564-017-0054-x
49. Liesenfeld O, Parvanova I, Zerrahn J, Han SJ, Heinrich F, Munoz M, et al. the IFN-Gamma-Inducible Gtpase, Irga6, Protects Mice Against Toxoplasma Gondii But Not Against Plasmodium Berghei and Some Other Intracellular Pathogens. PloS One (2011) 6(6):e20568. doi: 10.1371/journal.pone.0020568
50. Virreira WS, Niedelman W, Jensen KD, Rosowski EE, Julien L, Spooner E, et al. Determinants of GBP Recruitment to Toxoplasma Gondii Vacuoles and the Parasitic Factors That Control It. PloS One (2011) 6(9):e24434. doi: 10.1371/journal.pone.0024434
51. Selleck EM, Fentress SJ, Beatty WL, Degrandi D, Pfeffer K, Virgin HT, et al. Guanylate-Binding Protein 1 (Gbp1) Contributes to Cell-Autonomous Immunity Against Toxoplasma Gondii. PloS Pathog (2013) 9(4):e1003320. doi: 10.1371/journal.ppat.1003320
Keywords: Plasmodium, malaria, sporozoites, exo-erythrocytic forms, circumsporozoite protein, IFN-γ, ATGs, ubiquitination
Citation: Zheng H, Lu X, Li K, Zhu F, Zhao C, Liu T, Ding Y, Fu Y, Zhang K, Zhou T, Dai J, Wu Y and Xu W (2022) ATG Ubiquitination Is Required for Circumsporozoite Protein to Subvert Host Innate Immunity Against Rodent Malaria Liver Stage. Front. Immunol. 13:815936. doi: 10.3389/fimmu.2022.815936
Received: 16 November 2021; Accepted: 20 January 2022;
Published: 09 February 2022.
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Katsuyuki Yui, Nagasaki University, JapanCopyright © 2022 Zheng, Lu, Li, Zhu, Zhao, Liu, Ding, Fu, Zhang, Zhou, Dai, Wu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenyue Xu, eHV3ZW55dWVAdG1tdS5lZHUuY24=; Yuzhang Wu, d3V5dXpoYW5nQHRtbXUuZWR1LmNu; Jigang Dai, ZGFpamlnYW5nQHRtbXUuZWR1LmNu
†These authors have contributed equally to this work
‡Lead contact
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.