
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 09 March 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.811551
 Xia Wu1,2*
Xia Wu1,2* Jessica B. Roberto1
Jessica B. Roberto1 Allison Knupp1
Allison Knupp1 Alexander L. Greninger1,3Camtu D. Truong1Nicole Hollingshead1Heidi L. Kenerson4Marianne Tuefferd5
Alexander L. Greninger1,3Camtu D. Truong1Nicole Hollingshead1Heidi L. Kenerson4Marianne Tuefferd5 Antony Chen5
Antony Chen5 David M. Koelle1,2,3,6,7Helen Horton5Keith R. Jerome1,3
David M. Koelle1,2,3,6,7Helen Horton5Keith R. Jerome1,3 Stephen J. Polyak1,7
Stephen J. Polyak1,7 Raymond S. Yeung4
Raymond S. Yeung4 Ian N. Crispe1
Ian N. Crispe1Precision-cut human liver slice cultures (PCLS) have become an important alternative immunological platform in preclinical testing. To further evaluate the capacity of PCLS, we investigated the innate immune response to TLR3 agonist (poly-I:C) and TLR4 agonist (LPS) using normal and diseased liver tissue. Pathological liver tissue was obtained from patients with active chronic HCV infection, and patients with former chronic HCV infection cured by recent Direct-Acting Antiviral (DAA) drug therapy. We found that hepatic innate immunity in response to TLR3 and TLR4 agonists was not suppressed but enhanced in the HCV-infected tissue, compared with the healthy controls. Furthermore, despite recent HCV elimination, DAA-cured liver tissue manifested ongoing abnormalities in liver immunity: sustained abnormal immune gene expression in DAA-cured samples was identified in direct ex vivo measurements and in TLR3 and TLR4 stimulation assays. Genes that were up-regulated in chronic HCV-infected liver tissue were mostly characteristic of the non-parenchymal cell compartment. These results demonstrated the utility of PCLS in studying both liver pathology and innate immunity.
We recently reported the development of PCLS as an immunological platform (1, 2). Compared with other approaches, the PCLS method has the advantage that hepatocytes and the major subsets of non-parenchymal cells (KC, LSEC and HSC) are cultured together in their normal anatomical relationships, enabling the analysis of liver cell function in the context of intercellular interaction. Furthermore, the patterns of liver disease that are present in fresh tissue are maintained over time in the slice cultures (1). For example, steatotic liver slices retained fat droplets, and cholestatic liver slices displayed yellow-green pigment in hepatocytes. However, it remains unclear whether PCLS has the capacity to reveal alternations in innate immunity that may occur in pathological liver tissue as compared with healthy controls. As a proof of principle, we used PCLS to compare the dynamic responses of liver tissue collected from three groups of patients which included non-infected subjects, chronic HCV-infected patients, and patients whose HCV infection was cured by DAA treatment.
HCV-infected liver tissue is of particular interest, because HCV infection is one of the most common liver diseases in the USA and globally. Approximately 71 million individuals are infected with HCV worldwide (3). The acute phase of infection is often subclinical, but 55–85% of infected individuals develop a chronic infection leading to progressive liver pathology (4). The World Health Organization (WHO) estimated that approximately 399,000 people died from HCV- related diseases in 2016, mostly from cirrhosis and hepatocellular carcinoma (HCC).
The recent introduction of highly effective direct-acting antiviral (DAA) drugs has presented a unique opportunity to investigate if the diseased liver returns to normal after HCV elimination. The extent to which such drugs revert the liver to normal is not fully resolved, with some studies suggesting that HCV clearance with DAA therapy leaves residual abnormalities in innate immunity in peripheral monocytes and NK cells (5, 6), adaptive immunity in peripheral CD4+ and CD8+ T cells (7–10), in γδ-T cells (11), in mucosal associated invariant T cells (12), and in the persisting risk to hepatocellular cancer (13–16). Other studies also found abnormal serum lipids (17), persistent epigenetic modifications (18–20), and sustained hepatic inflammation (21) in liver biopsies after DAA treatment. To our knowledge, no studies have evaluated hepatic innate immunity of untreated versus DAA-treated HCV infection in the context of intact liver tissue.
Fresh non-tumor liver tissues were obtained from patients undergoing liver resection at the University of Washington Medical Center (Seattle, WA, USA). All patients in this study prospectively consented to donate liver tissue for research under the Institutional Review Board protocol #00001852. Tissue from DAA-treated patients were collected from those who achieved sustained virologic response (SVR), with HCV viral load being non-detected after the completion of DAA therapy. 7 HCV-infected, 10 DAA-cured, and 11 non-infected controls were studied. The non-infected controls are subjects with no known history of HCV exposure, their HCV negative status verified by testing their liver tissue for HCV RNA. Clinical details of these patients are provided in Table S1.
PCLS was performed as previously described (1). Briefly, liver cores of 6 mm diameter were excised from the resected liver tissue using a biopsy punch (Integra Miltex, York, PA, USA), stored in BELZER-UW solution (Bridge to Life Ltd., Columbia, SC, USA), and transferred to the research laboratories typically within 1 h of tissue excision. Slices 250 μm thick were prepared using a vibrating microtome, Leica VT1200 S (Nussloch, Germany), using Dulbecco’s Modified Eagles Medium (DMEM) as the cutting medium. Liver slices were cultured individually on 0.4 µm millicell organotypic inserts in 24 well plates (Millipore Corporation, Billerica, MA, USA). The culturing medium comprised 1 × advanced-DMEM medium, 5% Fetal Bovine Serum (FBS), 1 × GlutaMAX, 0.5 × Penicillin-Streptomycin, 1 × Insulin-Transferrin-Selenium supplement and 15 mM HEPES (pH 7.2 - 7.5) (all from Gibco, Grand Island, NY, USA). The liver cultures were maintained on a rocking platform at 17 rpm in a humidified incubator at 5% CO2 and atmospheric concentration of O2 at 37°C. The medium was renewed every two to three days.
Liver slices were fixed with 10% neutral-buffered formalin at room temperature for 24 h. The fixed liver slices were embedded in paraffin and were sliced into 4 μm-thick sections. Trichrome stain, picrosirius red stain, and hematoxylin and eosin (H&E) stain were analyzed with the standard protocol in the Pathology Research Services Laboratory at Department of Laboratory Medicine and Pathology at UW. The fibrosis score of liver slices was analyzed by a pathologist blinded from the patient clinical diseases. The Scheuer/Batts-Ludwig method was used for fibrosis scoring, with 0: No fibrosis, 1: portal fibrosis, 2: peri-portal fibrosis, 3: bridging fibrosis, and 4: cirrhosis. The whole-slide images were recorded with Nanozoomer Whole Slide Scanner (Hamamatsu City, Shizuoka Pref., Japan) and visualized with NDP.view2 software (Hamamatsu).
Human liver cell isolation procedures were adapted from several sources (22, 23). Cell isolation was performed on perfused tissue wedges, rather than cores which do not maintain adequate vasculature to allow perfusion. Perfusion buffer contained 1 × Hank’s Balanced Salt Solution (HBSS, without Ca++, Mg++, or phenol red, from Gibco), 10 mM HEPES (pH 7.2-7.5) and 0.5 mM EDTA (pH 8.0). Collagenase buffer contained 1 × HBSS (Gibco), 5 mM MgCl2, 5 mM CaCl2, 5 mM HEPES (pH 7.2-7.5), 0.5% w/v Collagenase IV (Sigma-Aldrich, St. Louis, MO, USA), 0.25% w/v Protease (Sigma), 0.125% w/v Hyaluronidase (Sigma), 0.05% w/v DNase I (Sigma). Fresh aliquots of enzymes were added to the buffer on the day of the perfusion experiment. Forty mL of Perfusion buffer and 20 mL of Collagenase buffer were used for each 10 g of liver tissue. Buffers were pre-warmed to 37°C prior to the perfusion step.
Liver tissue wedges were sequentially perfused with Washing buffer (1 × HBSS and 10 mM HEPES, pH 7.2-7.5), Perfusion buffer, Washing buffer, and the recirculating Collagenase buffer at a flow rate of 12 mL/min (Gilson’s MINIPULS 3, Middleton, WI, USA). The perfused liver tissue samples were gently mashed with a sterile syringe plunger through a sterile mesh strainer in ice-cold DMEM medium. Cell extracts were filtered through a 100 μm sterile strainer and centrifuged at 50 × g at 4°C for 3 min to enrich for hepatocytes in the pellets and non-parenchymal cells in the supernatant. The supernatants were transferred to a new tube and kept on ice. The pellets were washed three times with ice-cold DMEM medium, and were pelleted each time at 50 × g at 4°C for 3 min. To further improve the purity of the isolated hepatocytes, cell pellets were resuspended with 5 mL of ice-cold PBS, overlaid with 10 mL of 25% Percoll gradient solution. The mixture was centrifuged at 1400 × g (no brake) at 4°C for 20 min. The pellet contained the purified live hepatocytes. The viability of the isolated hepatocytes was determined with the trypan blue exclusion assay (Thermo Fisher Scientific, Waltham, MA, USA). If the viability was greater than 50%, the isolated hepatocytes were stored for RNA extraction.
For the non-parenchymal cells (NPC), the 50 x g centrifuge supernatants were further centrifuged at 500 × g at 4°C for 7 min. The pellets were resuspended in 5 mL of ice-cold PBS, and overlaid on top with 50% and 25% Percoll gradients (10 mL layers each), and were centrifuged at 1400 × g (no brake) at 4°C for 20 min. Cell layers were collected into 20 mL PBS each, and centrifuged again at 500 × g at 4°C for 7 min. A fraction of the pellets were saved for RNA extraction, which were the total NPC. The rest of the pellets were resuspended with the ice-cold Flow buffer containing 1 × PBS, 2% FBS, and 1 mM EDTA. The experimental protocol is illustrated in Figure S1.
The isolated non-parenchymal cells were stained with an antibody mixture that included eBioscience (San Diego, CA, USA): anti-CD45 (Cat. No. 15-0459-42); BioLegend (San Diego, CA, USA): anti-CD3 (Cat. No. 317330), anti-CD11b (Cat. No. 553310), anti-CD14 (Cat. No. 301834), anti-CD31 (Cat. No. 303120), anti-CD32 (Cat. No. 303206), anti-CD68 (Cat. No. 333814), and anti-CD271 (Cat. No. 345110). In addition, cells were also stained with LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Cat. No. L10120, Life Technologies, Carlsbad, CA, USA). The incubation mixture was kept at 4°C for 30 min in the dark on a rocking platform. The mixture was centrifuged with 500 × g at 4°C for 7 min. Cells were washed once with Flow buffer.
The antibody-labeled cells were sorted with a BD Aria III (BD Biosciences, San Jose, CA, USA). Analysis of cell populations was performed using FlowJo software, version 9.8.5 (FlowJo, LLC, Ashland, OR, USA). Kupffer cells were selected as the CD45+, CD3−, CD14+, CD68+, CD32+ populations (24, 25). LSECs were selected as CD45−, CD31+, CD11b− (26). HSCs were selected as CD45−, autofluorescence positive with the emission wavelength at 460 nm, SSC-H (high) (27, 28) (Figure S1). Cells were pelleted at 500 × g at 4°C for 7 min, and were stored in -80°C before RNA extraction.
The RNA of liver slices or the purified liver cells was isolated with TRIzol and the Direct-zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA). The RNA concentration was measured with nanoDrop (Thermo Fisher Scientific). The cDNA was synthesized with the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany).
A pre-amplification was performed before the multiplex qRT-PCR assays to improve the detection sensitivity (29, 30), which included PCR reactions of cDNA as templates, and the 0.2 fold- diluted primer mixture of TaqMan assays of interest (Thermo Fisher Scientific) and the BIO-X-ACT Short Mix reagents (Bioline USA Inc., Taunton, MA, USA). The pre-amplified samples were diluted five-fold with RNase- and DNase-free H2O. Samples were analyzed with a 48 × 48 dynamic array and a BioMark HD microfluidics system (Fluidigm, San Francisco, CA, USA). The Fluidigm Real-Time PCR Analysis software was used to calculate Ct thresholds, using the settings of quality threshold 0.65, baseline correction linear, Ct threshold method auto detection. Gene abundance of individual liver slices was normalized to the arithmetic mean of Ct values of ACTB, HPRT, and GAPDH. Typically, three liver slices were analyzed for each subject for each time point. The arithmetic mean delta Ct of three liver slices was used in the figures and for statistical analysis. The hierarchical clustering was analyzed with Cluster 3.0 (31). Heat map was visualized with Java Treeview (version 1.1.6r4) (32).
HCV RNA was measured with a Food and Drug Administration-approved Abbott Real-Time HCV assay (Abbott Molecular, Des Plaines, IL, USA) as previously described (21, 33). Fifty ng of total RNA for each sample was input in the assay, and HCV IU/ng total RNA was calculated from the calibration curve of positive controls. The reliable detection limit for HCV with this assay was 1.2 IU/50 ng liver total RNA.
Liver cell-type-specific genes for hepatocytes, Kupffer cells (KC), liver sinusoidal endothelial cells (LSEC) and hepatic stellate cells (HSC) were obtained from previous microarray analysis of liver FACS purified cells (1) and recent publications of liver single cell RNAseq (34, 35). Enrichment of the FACS purified liver cell types were confirmed by the selective expression of cell-type-specific genes.
Liver slices were cultured ex vivo for 7 days. Final concentrations of 15 μg/mL polyinosinic–polycytidylic acid (poly-I:C) (Sigma, Cat. No. P1530), 1.5 μg/mL lipopolysaccharide endotoxin (LPS) (Sigma, Cat. No. L2630) or 1 × PBS (control, matched by volume) were added to the growth medium. During the stimulation, liver slices were maintained on transwell inserts placed on a rocking platform at 37°C. Three liver slices were harvested at each time point, including 0 h (time zero control), 2 h, 4 h, 8 h, 12 h or 24 h. The concentrations of poly-I:C and LPS treatments were based on our previous studies with isolated liver leukocytes (36) and liver slices (1).
Some gene expression data from a subset of the non-infected control subjects (#1,3,4,5,6) in Figures 1, 5 and S2, S7 were previously published in a proof-of-concept study (1), and appear here in the pool of control data. Inclusion of these data expands the number of non-infected controls, allowing us better to resolve differences due to active or DAA-cured HCV infection. For genes that were evaluated in both data-sets, we include direct comparisons in Figure S9. The archived data were fully consistent with newer data, justifying their inclusion.
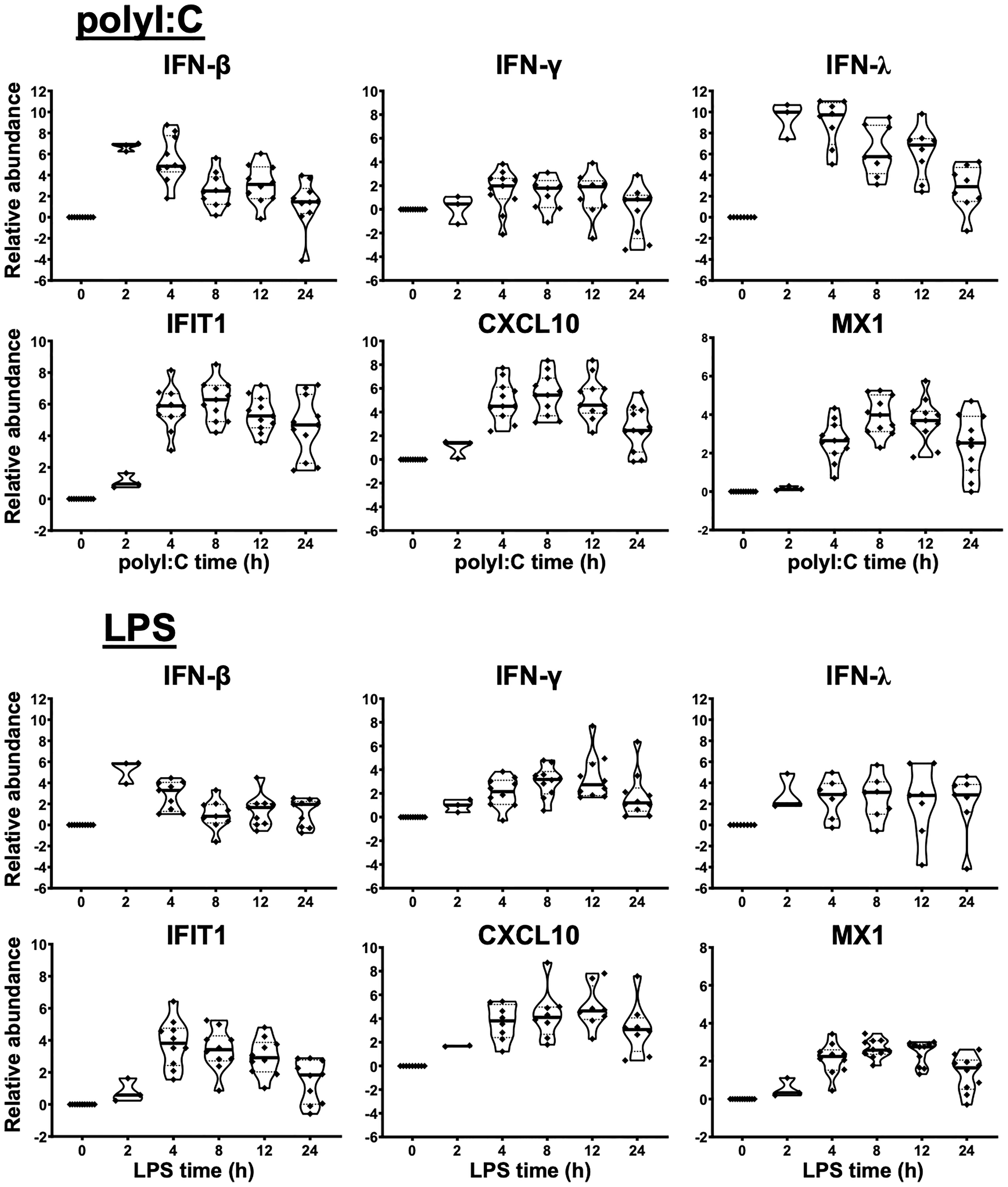
Figure 1 Innate immune response of human liver slices to poly-I:C or LPS treatment. Non-HCV-infected specimens are shown. IFN-β and IFN-λ expression peaked at 2-4 h, whereas interferon-stimulated gene expression for IFIT1, CXCL10 and MX1 peaked round 4-12 h. Each marker represents a patient time-point summary which consist of 2-3 biological replicates of liver slices. Relative abundance at the log2 scale is shown, with the median, 25% and 75% quantile values indicated with violin plots.
The non-parametric Mann Whitney test, Kruskal-Wallis test with Dunn’s post test with multiple test comparison correction, and Wilcoxon matched-pairs signed rank test were performed using Prism (version 9.1.0) (GraphPad Software Inc., CA, USA). Multiple comparison correction was not applied in the gene expression analysis, because a specific set of genes were targeted for analysis unlike the standard procedure in a data-driven functional genomics study. In this circumstance, correction for type I error is not advised, because the correction could lead to inflation of type II error (i.e. false negatives) (37). The issue of type II errors also arises because of the limited number of tissue samples available to us. For these reasons, we treated each gene we selected as an individual measurement.
As a first step, we verified the robust response of liver slices during poly-I:C and LPS exposure (Figures 1, S2). The 2 h time point confirmed that IFN-β and IFN-λ gene expression was stimulated prior to the elevation of interferon-stimulated genes IFIT1/2, CXCL10, MX1 and RSAD2 by poly-I:C. The induction of these antiviral genes was greater with poly-I:C compared with LPS treatment (Figures 1, S2). In contrast, IL-1B and IL-6 and IL-12B were more robustly stimulated by LPS. The response of IL-1B and IL-6 extended to 24 h post stimulation; in comparison, TNF peaked at 2 h, and gradually declined at 4, 8, 12, 24 h post-stimulation time points (Figures 1, S2). The temporal and gene specificity in poly-I:C and LPS response provided the basis for further dynamic testing of HCV-infected and DAA-cured liver tissue.
Liver tissue from eight chronic HCV-infected patients were studied. All eight patients were diagnosed with primary liver cancer, either hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC). Four HCV genotypes were identified including HCV G1a, G1b, G2, G6. Likewise, liver tissue from ten DAA-cured patients were studied. The DAA-cured patients completed 8-24 weeks of DAA therapy. All subjects achieve sustained virologic response (SVR) with HCV RNA below the limit of detection after completion of DAA treatment. However, these DAA-cured patients were diagnosed with primary liver cancer (HCC or ICC) post DAA therapy. The time from completion of DAA treatment to liver resection surgery (i.e. the time liver tissue was collected) ranged from less than 1 month to 20 months, with a median of 9 month post-DAA therapy completion (Table S1).
To confirm HCV infection in the samples from subjects with chronic HCV infection, HCV copy number and genotype were analyzed by UW Virology using CLIA-approved HCV assays. Liver slices originated from the chronic HCV patients were all confirmed with detectable HCV RNA, ranged from 1.2 to 9760.1 with a median value of 3325.4 IU/50 ng total RNA. In comparison, none of the non-infected or DAA-treated subjects were positive for HCV RNA ex vivo based on the limit of detection of 1.2 IU/50 ng total RNA (Figure 2A). Moreover, chronic HCV-infected liver slices continued to be HCV positive after 7 days of ex vivo culture (Figure 2B), with a range of 1.8-14365.8, and median of 405 IU/50 ng total RNA. The viral load on day 7 did not significantly differ from day 0 (Wilcoxon matched-pairs signed rank test, two-tailed, P=0.4688).
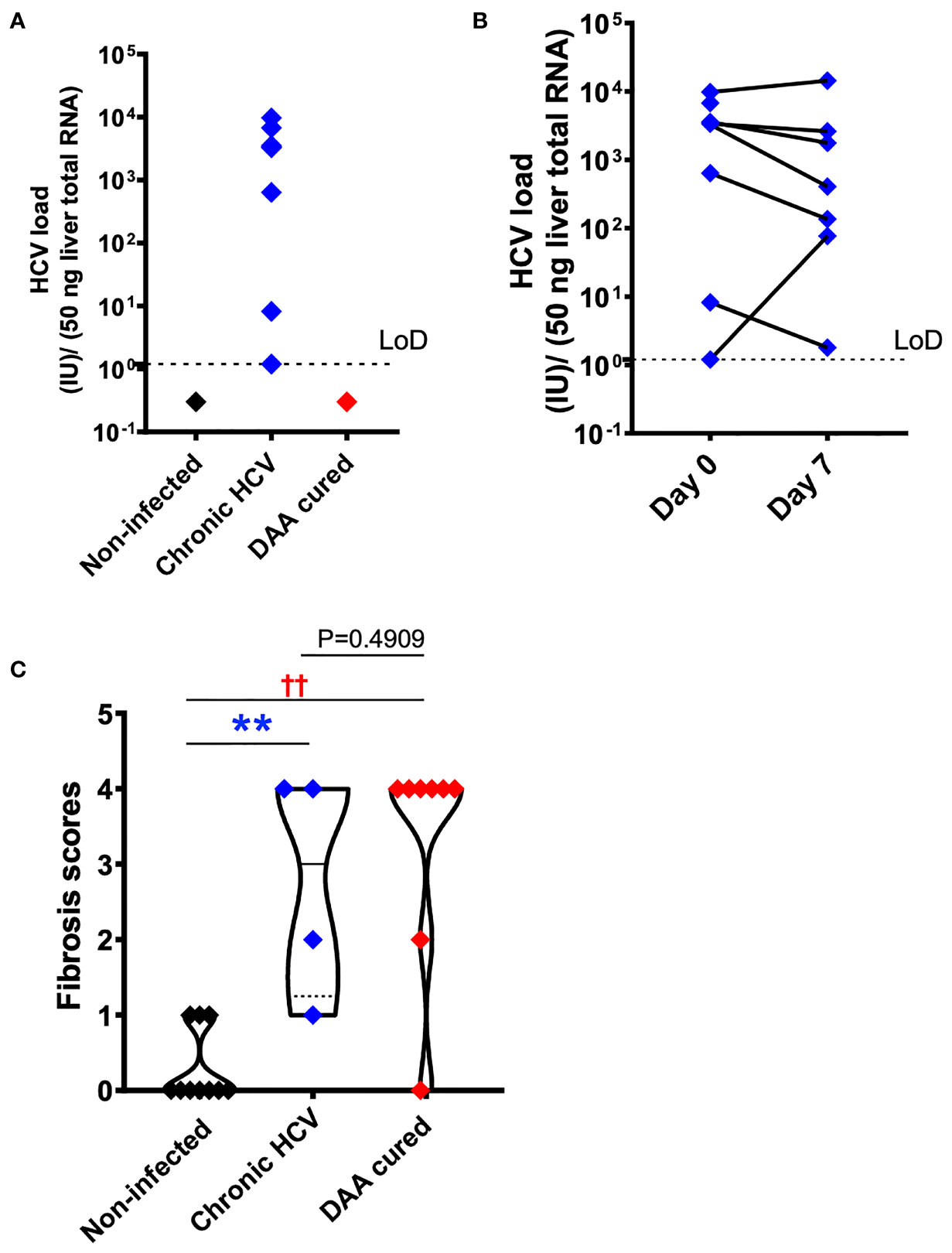
Figure 2 HCV and fibrosis analysis with human liver slices. (A) All eight chronic HCV samples (blue color) were confirmed with positive HCV RNA detection. None of the DAA cured subjects (red color) or the non-infected controls (black color) was positive for HCV RNA. The limit of reliable detection (LoD) was 1.2 IU/50 ng liver total RNA. (B) HCV RNA remained robustly detected in the day 7 liver slices cultured from chronically HCV-infected patients. The viral load between day 0 and day 7 liver slices was not statistically significant (P = 0.4688, Wilcoxon matched-pairs signed rank test). (C) Fibrosis analysis of the day 0 ex vivo liver specimens with trichrome staining and picrosirius red staining indicated that in DAA-treated, and now HCV-negative patients in this study there was persistent fibrosis, similar to untreated HCV and different from tissue without a history of HCV infection. The scoring system was based on the Scheuer/Batts-Ludwig method. The fibrosis score included 10 non-infected patients, 4 chronic HCV patients, and 8 DAA-treated patients. In other words, 4 out of the 7 HCV+ subjects, and 8 out of the 10 DAA-cured subjects were analyzed. Due to tissue availability, not all subjects could be analyzed in this way. The minimum and maximum data points for each subgroup are shown. Statistical significance was based on Mann-Whitney test. **P < 0.01. ††P < 0.01.
Because chronic HCV infection induced liver cirrhosis, we examined the liver tissue for fibrosis using picrosirius red and Masson’s trichrome staining (Figure 2C). Previous indirect measurements with transient elastography and noninvasive fibrosis indices reported regression of liver fibrosis after DAA treatment (38, 39). Nevertheless, direct analysis of the liver tissue revealed that six out of eight DAA-cured patients met the definition of cirrhosis (i.e., fibrosis score of 4, Scheuer/Batts-Ludwig method) in livers despite the absence of HCV RNA, even after 20 months after DAA therapy (Figures 2C, 3). Likewise, the untreated chronic HCV livers were fibrotic as expected (40). None of the non-infected subjects had advanced liver fibrosis. Since the duration between the liver tissue collection and the time point of SVR ranged from less than 1 month to 20 months, it remains plausible that the fibrosis will be slowly resolved over a greater time span (41, 42). Since only the HCV+ and previously HCV+ liver tissue samples were fibrotic, an important caveat relating to our study is that the abnormal immune gene expression in both the HCV-infected and the DAA-cured liver slices could result from HCV infection, as a consequence of fibrosis, or from cross-talk between these two pathological processes.
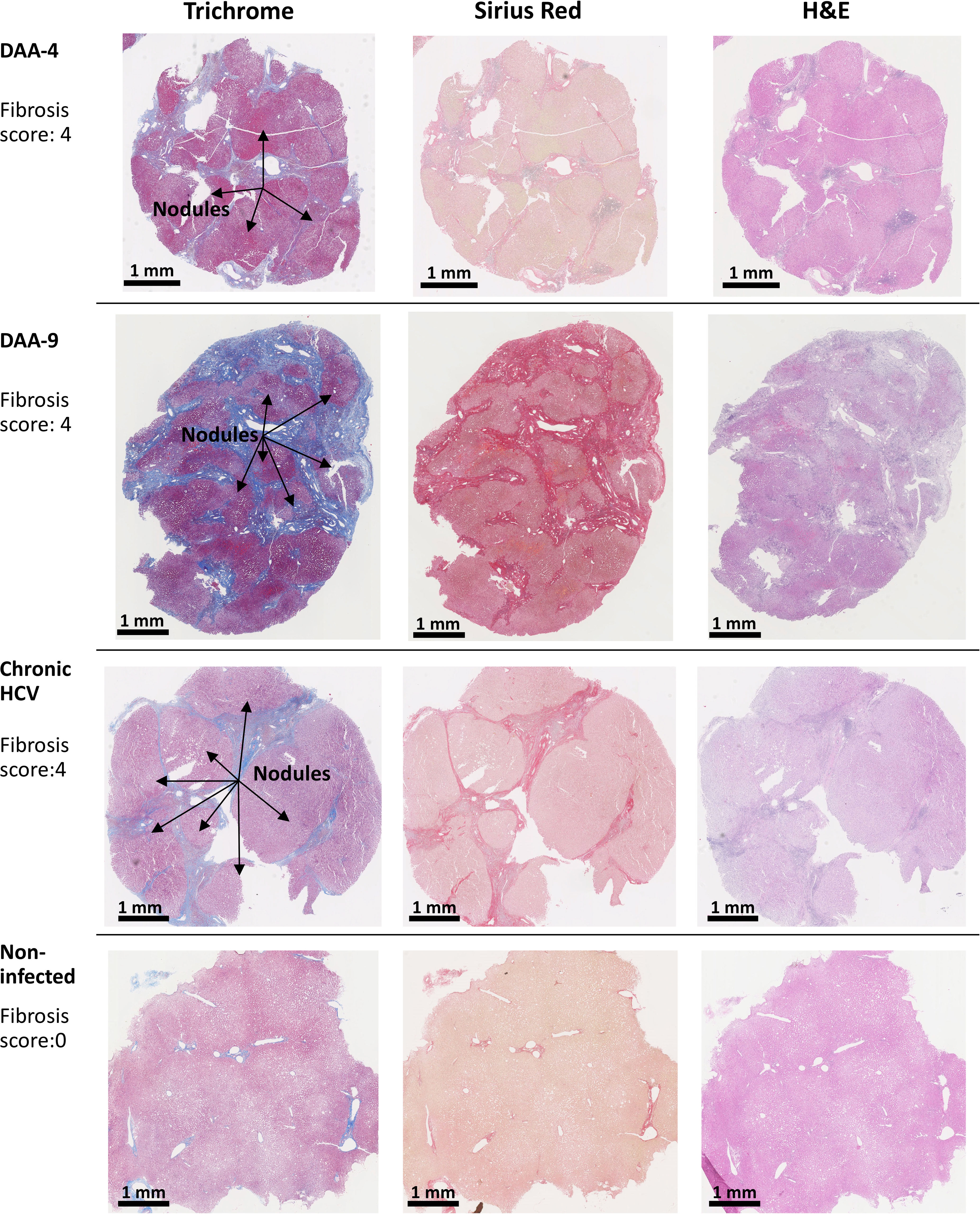
Figure 3 Examples of trichrome stain, picrosirius red stain, and H&E stain analysis of human liver slices. Arrow indicates the nodule formation in the cirrhosis livers. The Scheuer/Batts-Ludwig method was used for fibrosis scoring, with 0, No fibrosis; 1, portal fibrosis; 2, peri-portal fibrosis; 3, bridging fibrosis; and 4, cirrhosis. The DAA-4 liver tissue was collected 12 months after the completion of 24-week DAA therapy. DAA-9 liver tissue was collected 20 months after the completion of 12-week DAA therapy.
We used multiplex qRT-PCR to examine the expression of 140 cellular genes with functions in immune activation and suppression, and tissue repair. Forty immune genes differed in expression in chronic HCV-infected livers versus non-infected patients (Mann Whitney test, two-tailed, P < 0.05) (Figure 4A). This included antiviral interferon (IFN)-stimulated genes (ISGs) including IFIT1/2/3, ISG15, RSAD2, MX1, CXCL9, CXCL10, consistent with previous reports (43–46). In addition, immune activation genes including MHC genes HLA-A, HLA-DRA, CIITA, CD80, CD86, tumor necrosis factor (TNF) superfamily genes OX40, 4-1BB, TNFSF9, TNFRSF18 and inflammation genes including CASP1, IL-12A, IL-12B, TNF, IL-18, and NFKB were also significantly up-regulated in HCV-infected livers (Figure 4A).
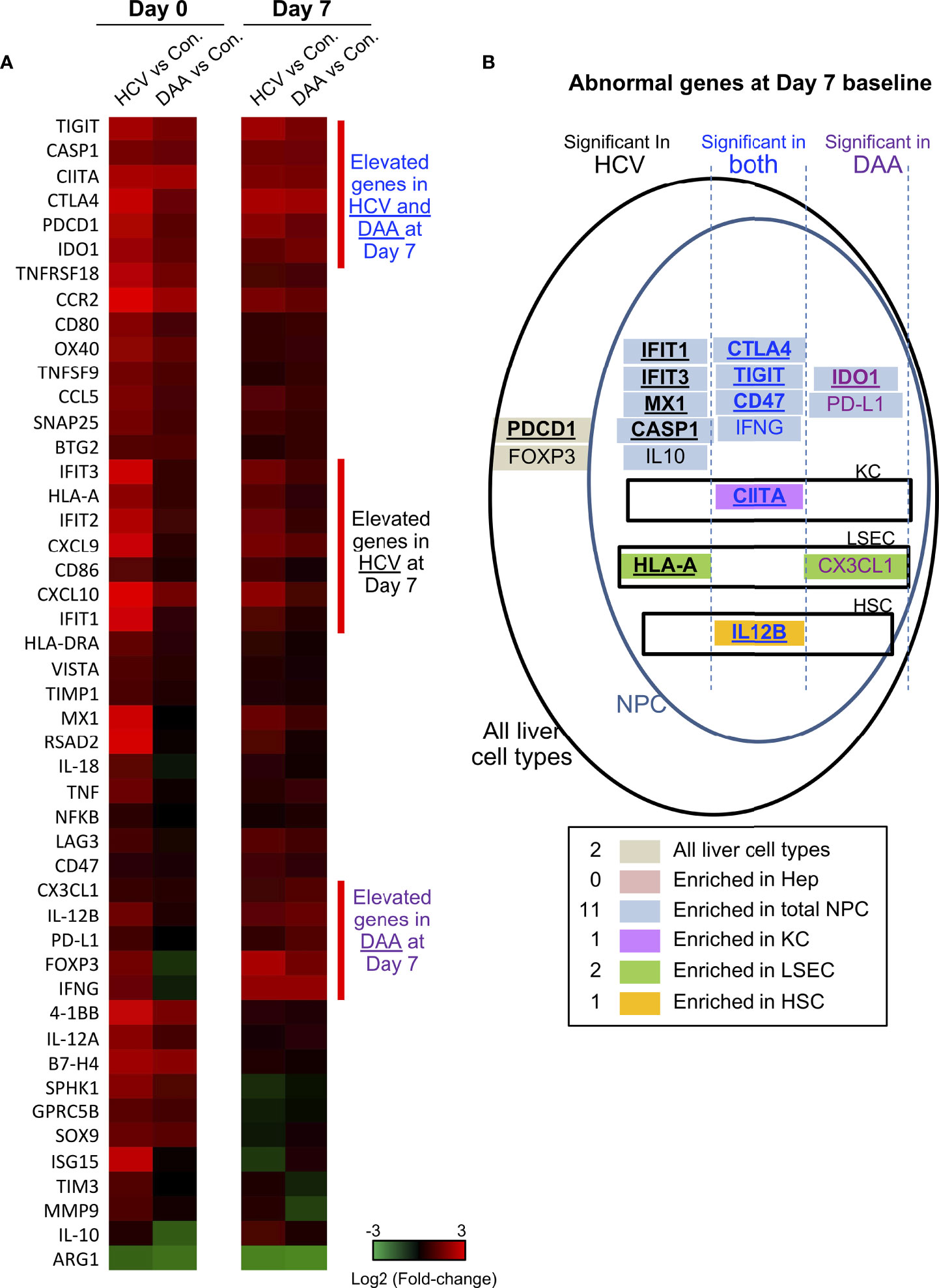
Figure 4 Immune gene changes at the day 7 baseline in chronic HCV-infected and DAA-cured livers. (A) Hierarchical clustering of immune genes with statistically significant elevation in HCV or DAA-cured subjects at Day 0 or Day 7 time points. The group-level fold changes are shown, which were calculated with the mean Ct value of each gene within each patient group. Decreased fold changes are colored in green, and increased fold changes are colored in red. (B) The up-regulated genes in HCV and DAA-cured at day 7 time points. Genes were mapped to liver cell types according to the cell type analysis with day 0 non-infected livers. Genes are grouped by expression patterns in cell compartments and in specimen type. The overlapped genes up-regulated at both day 0 and day 7 time points are bolded and underscored.
DAA treatment (i.e., HCV cure) restored 28/40 of the antiviral genes to baseline, as reported previously (47), but 11 immune genes remained elevated after cure of HCV infection. These included immune activation genes (HLA-A, CIITA, 4-1BB, CCL5, CCR2), inflammatory genes (CASP1, IL-12A), and immune suppression-associated genes (CTLA4, TIGIT, IDO1). Another immune suppression-related gene ARG1 was significantly decreased in HCV+ and DAA-cured livers, compared with non-infected controls (Figure 4A and Table 1).
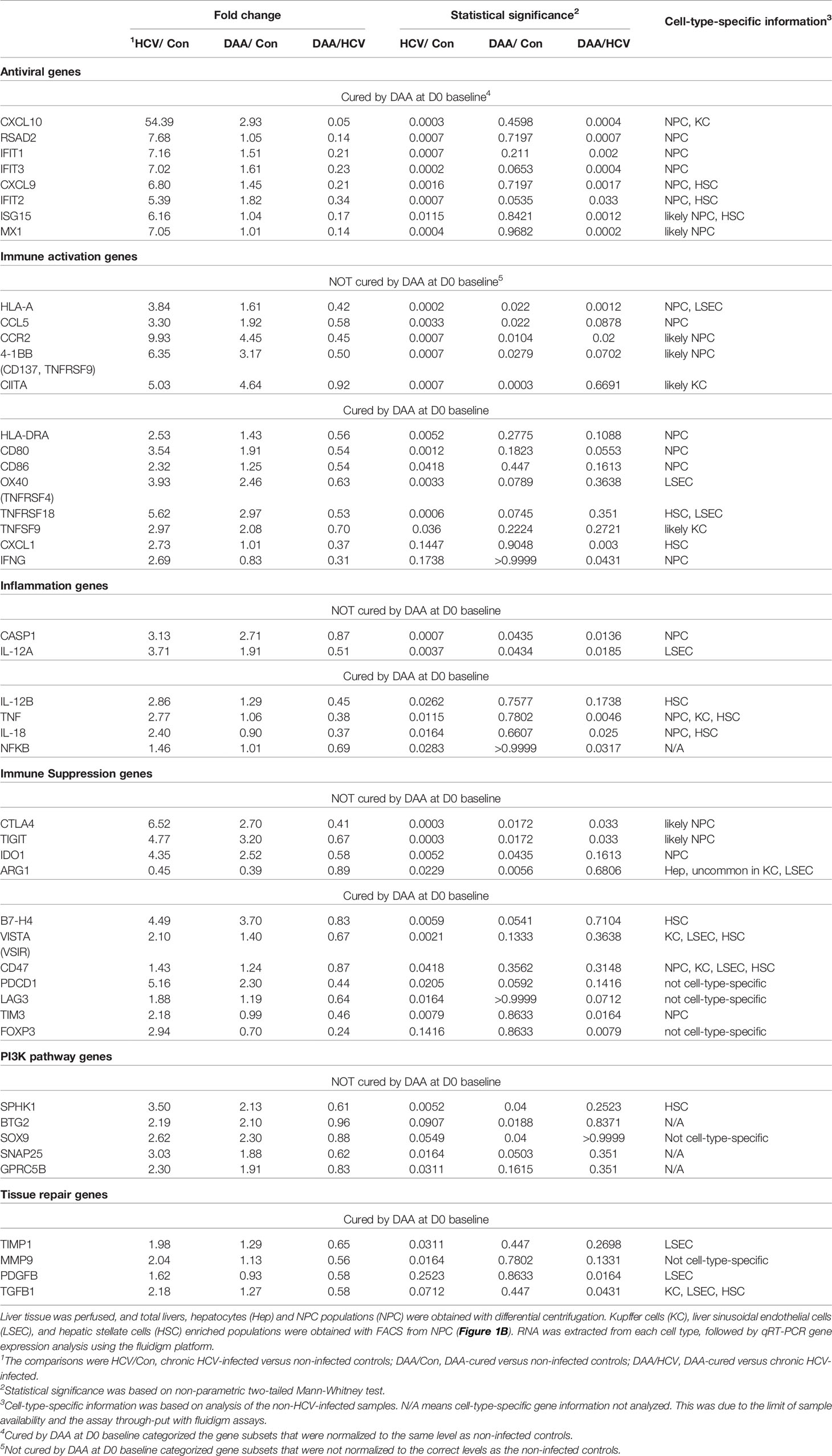
Table 1 Immune gene changes ex vivo (Day 0).
The phosphoinositol-3-kinase (PI3K) pathway-linked genes are susceptible to modification by HCV infection (18, 20, 48) and liver fibrosis (49, 50). We found that SPHK1, BTG2, SOX9, SNAP25, and GPRC5B were significantly up-regulated in chronic HCV infected livers. Furthermore, SPHK1, BTG2, and SOX9 remained significantly up-regulated after HCV clearance with DAA treatment (Figure 4A).
To identify the cellular origin of the dysregulated immune gene subsets, we investigated gene expression with liver cells subsets purified using FACS-sorting (1). Six liver wedge samples of non-infected liver tissue were large enough in size and anatomically ideal for perfusion through the vasculature, which allowed them to be dissociated and analyzed for gene expression. We could not do this with samples from HCV-infected or DAA-cured patients due to limitations in the amount of liver tissue obtained. The purity of isolated liver cell types was confirmed based on the expression of cell marker genes (1, 34, 51), including FCN1, NLRP3, ITGA4 for KC, EMCN, VWF, CD34 for LSEC, FGFR2, PDGFRB, COL1A1, LRAT for HSC, and FBP1, RBP4, ALB for hepatocytes (Figure S3).
Many immune genes that were up-regulated in HCV-infected or DAA-cured fibrotic liver tissue were significantly enriched in the NPC compartment (Table 1 and Figure 4B). For a smaller subset of immune genes, we were able to map them to specific immune cells with greatest expression: for example CIITA mapped to Kupffer cells (KC), HLA-A and CX3CL1 mapped to liver sinusoidal endothelial cells (LSEC), and IL-12B mapped to Hepatic Stellate Cells (HSC) (Figures 4B, S4). Even though the NPC fraction may not include the cell types targeted by HCV infection, previous studies demonstrate that NPC can be activated through paracrine signaling from the neighboring HCV-infected hepatocytes (44, 52, 53).
We stimulated liver slices with poly-I:C and LPS for varying times using the method established in our previous study (1). We verified the immune genes activated by tissue slicing had broadly stabilized after 4 days of ex vivo culture for HCV-infected and DAA-cured slices (Figures S5A, B), similar to the non-infected slices described previously (1). Day 7 was thus an ideal time-point to add exogenous stimuli, and the expression of immune genes was recorded at the day 7 baseline (T0) prior to the treatment. Twelve of the immune genes remained at elevated baseline in HCV-infected or DAA-cured samples compared with non-infected control (Figures 4A, B). The PBS mock stimulation did not significantly induce IFNs and ISGs in liver slices (1). Notably, expression of TLR3, TLR4, and NF-κB were not significantly different at day 7 among chronic HCV-infected, DAA-treated and non-infected liver slices (Figure S6A). We used the ΔCt method with internal normalization to ACTB, GAPDH and HPRT to evaluate the gene expression level post stimulation (Figure S7). We also used the ΔΔCt method against the day 7 T0 to assess the enhanced net increase of genes during TLR3/TLR4 response (Figure 5). We confirmed that TLR3 and TLR4 are primarily expressed in the NPC compartment (Figure S6B), consistent with the expectation that NPCs in the liver are the primary responders to these triggers (54).
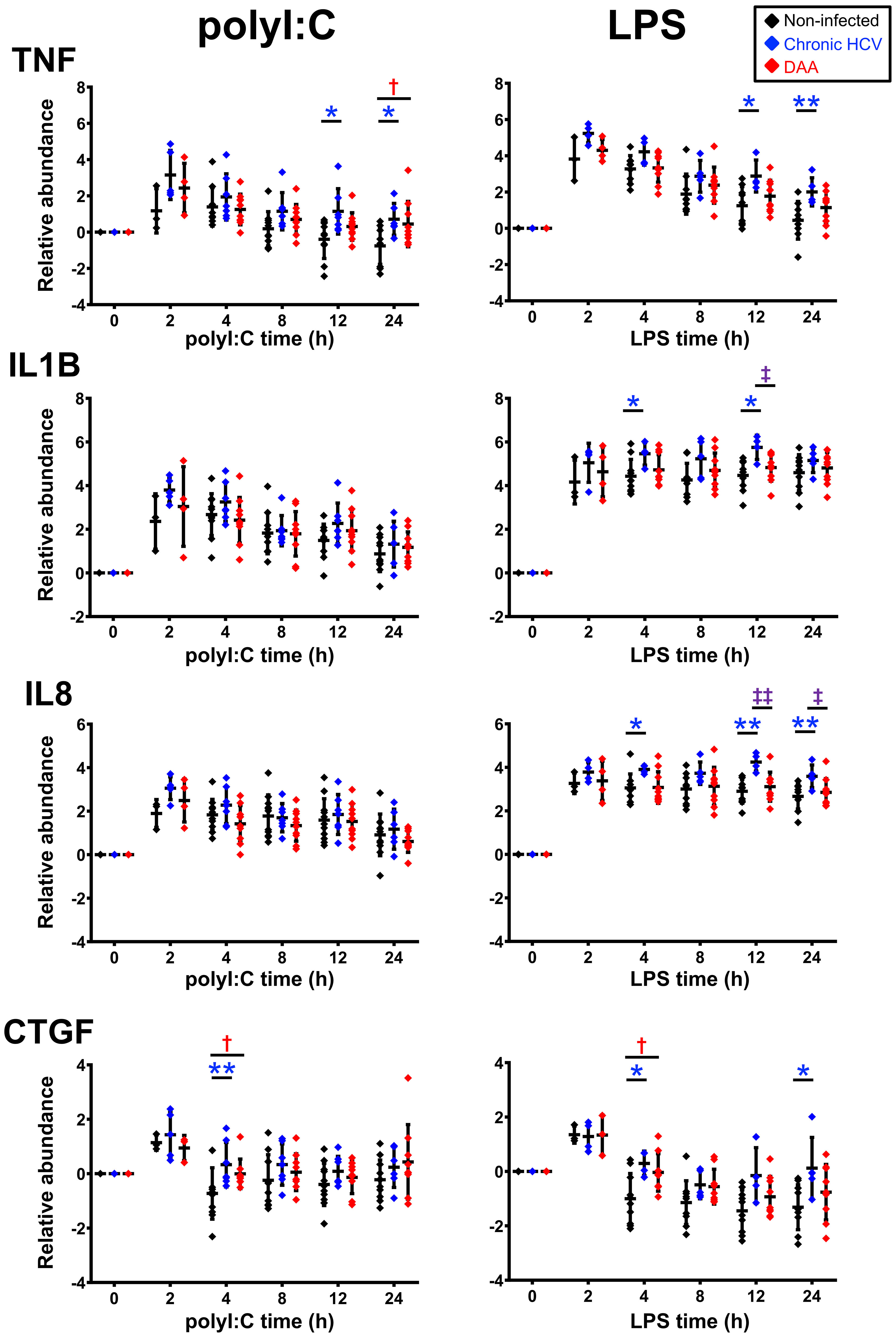
Figure 5 Delta-delta Ct analysis showing up-regulated genes post- poly-I:C or LPS stimulation in chronic HCV-infected and DAA-cured liver slices. The relative gene abundance was normalized to to the arithmetic mean of Ct values of ACTB, GAPDH and HPRT1. The relative abundance was further normalized to the day 7 time zero measurements. Statistical significance was based on two-tailed Mann-Whitney test. *, statistical significantly different between non-infected versus chronic HCV liver slices. †, statistical significantly different between non-infected versus DAA-cured liver slices. ‡, statistical significantly different between chronic HCV versus DAA-cured liver slices. Levels of statistical significance are indicated thus: *P < 0.05 **P < 0.01. The mean and standard deviation within each group are plotted.
These studies revealed the following findings. First, robust induction of IFNs and ISGs by both TLR3 and TLR4 agonists was observed in liver slices from all three groups of liver specimens (i.e., non-infected, HCV-infected, and DAA-cured). The sequential signaling cascade induced by TLR3 and TLR4 pathways described in Figures 1, S1 for non-infected liver tissue, were also pronounced in the HCV-infected and DAA-cured liver tissue. For example, IFNB1 and IFNL3 (IL28B) peaked at 2-4 h, prior to the maximum abundance of ISGs (IFIT 1/2/3, RSAD2 and MX1) which occurred at 4-8 (Figures 5, S6).
Second, some immune genes were not suppressed but induced more strongly by TLR3 agonist in liver slices of chronic HCV-infected liver at least at one time point (Mann-Whitney test, two tailed, P < 0.05). These included antiviral genes, MX1, RSAD2, IFNG, chemokine genes CCL7, CX3CL1, and inflammatory genes TNF and CASP1 (Figure S8). Other common activation markers for TLR3 signaling, including IFNB1, IL28B, IFIT1/2/3, ISG15, CXCL10, IL-1B and CCL-5, were also responsive in chronic HCV-infected slices with elevation of greater than 10-fold, as strong as changes in the non-infected liver slices. Thus, TLR3 sensing and signaling did not appear to be impaired in chronic HCV-infected liver slices.
TLR4 sensing and signaling were similarly robust in chronic HCV-infected livers. IFIT1/2/3 and CCL7 were identified at greater abundance post LPS stimulation in chronic HCV-infected liver slices on at least one time point (Mann-Whitney test, two tailed, P < 0.05) (Figures 6A, S7).
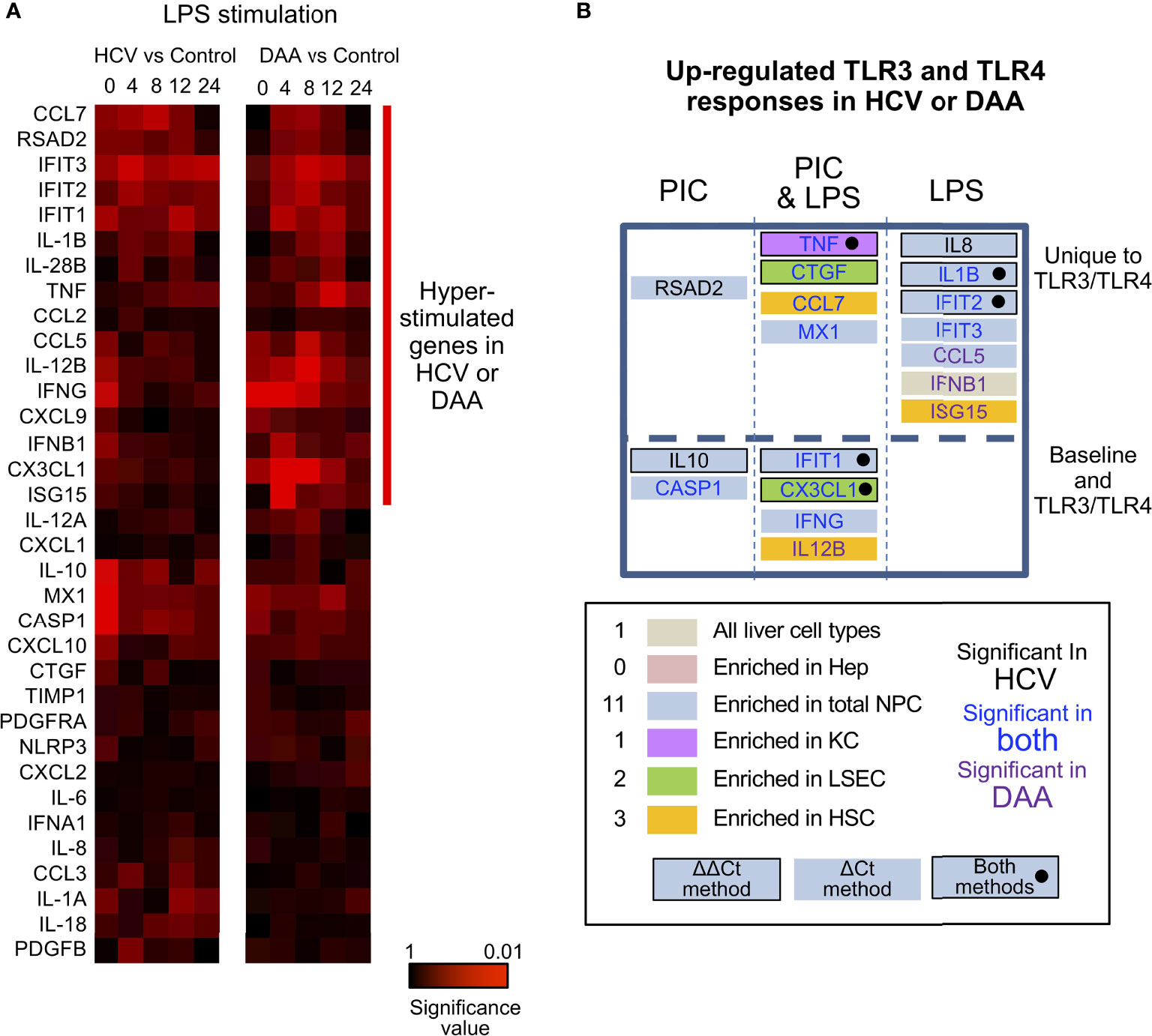
Figure 6 Altered TLR3 and TLR4 response in chronic HCV-infected liver slices not restored in DAA-cured liver slices. (A) Hierarchical clustering of immune responsive genes with 2-fold or greater induction during LPS stimulation. The statistical significance P value with ΔCt method is shown. Gene abundance of individual liver slices was normalized to the arithmetic mean of Ct values of ACTB, GAPDH and HPRT. Two-tailed Mann-Whitney test was used. Greater significance P value is colored with darker red. (B) Genes up-regulated with P < 0.05 at one time point in either poly-I:C or LPS treatment were included as up-regulated genes. Genes boxes were colored with the enriched expression in cell compartment of day 0 information. Using the ΔΔCt method, genes that increased in expression during TLR3/TLR4 response are highlighted with a black bordering; genes shown to increase based on both ΔΔCt and ΔCt methods are highlighted with black bordering and black solid circles. Genes names are colored by in HCV- or DAA- samples or both. These genes were further grouped by whether they were differentially expressed in TLR3 or TLR4 treatment not at the baseline level, and whether poly-I: C or LPS treatment revealed differential responses.
Third, most of the hyper-induction of the genes in HCV-infected tissue (9/11 genes in TLR3 response, 10/11 genes in TLR4 response) was not reversed in the tissue from formerly HCV+, but now successfully treated donors. IFNB1, CCL5, ISG15 and IL-12B were more strongly induced by LPS or poly-I:C in DAA-cured tissue, previously not identified with HCV-infected tissue (Figures 6A, B).
In terms of the ΔΔCt net increase, TNF, IL-1B, IL-8, CTGF, and CX3CL1 were more strongly induced in HCV-infected liver tissue during LPS or poly-I:C stimulation. Likewise, TNF, CTGF and IFIT2 were abnormally induced in DAA-cured liver tissue (Figures 5, 6B). The abnormal induction of TNF, IL-1B, IL-8, and CTGF at 12 h and 24 h in chronic HCV liver slices, indicated persistent inflammation (Figure 5). Importantly, a subset of these genes TNF in KC, CTGF in LSEC, CCL7, ISG15 for HSC, and IL-8, IL-1B, IFIT2/3, RSAD2, MX1, CCL5 for NPCs, and IFNB1 were only revealed through the TLR3 and TLR4 stimulation assays, but not with the ex vivo day 0 measurements. These results highlighted the unique value to measure dynamic changes with liver tissue to reveal immune signaling defects (Figures 6B, S9).
In this study, we pioneered the PCLS technology to study innate immune response within normal and pathological liver tissue. We showed that immune abnormalities persist after the elimination of HCV by anti-viral therapy, and the persisting inflammatory and immune signatures featured genes characteristic of hepatic non-parenchymal cells. Innate immunity in response to TLR3 and TLR4 agonists was not suppressed but enhanced in HCV-infected tissue, and these abnormalities were not corrected after effective DAA treatment.
In human hepatocyte cultures (55, 56) HCV infection triggered cleavage of MAVS and TRIF host proteins by HCV NS3/4A protease, disrupting TLR3-antiviral signaling. Reduction of protein levels of MAVS and TRIF was also observed previously in chronic HCV-infected livers (57, 58). The novel aspect of PCLS is to study whole liver slices consisting of hepatocytes (infected cells) and non-parenchymal cells (NPC, non-infected cells). We observed a robust response to TLR3 signaling in the HCV-infected liver, which supports the idea that ISG induction may be weak in HCV infected hepatocytes, but is strong in HCV infected liver due to paracrine activation of NPC. The data on the direct effect of HCV on the TLR3 response in liver tissue are novel and distinct from previous work focused on the IFN-induced gene expression in chronically HCV-infected liver by pegylated IFN therapy (59, 60), since such direct activation of IFN signaling bypasses MAVS and TRIF.
A previous study reported that HBV does not interfere with innate immune response based on chronic HBV-infected liver biopsy tissues (61). However, in chronic HCV-infected tissue, we found innate immunity via TLR3 and TLR4 was not suppressed but enhanced. The different outcome may reflect the differences in immune evasion mechanisms between DNA and RNA viruses. Furthermore, the tissue status and treatment method were not identical between the two studies. In chronic HCV-infected tissue, advanced fibrosis was observed, and this study also used the day 7 as treatment time zero when immune activation by tissue slicing was broadly stabilized.
From the days of IFN-based therapy, we know that HCV-induced liver disease can regress when the virus is eradicated from the liver (62). However, halting or reversing liver disease upon IFN cure of HCV has not been universal; some patients with advanced disease still go on to develop HCC (63–65). DAA drugs are now able to cure the majority of HCV infections, even in subjects with advanced liver disease. Nevertheless, clearance of HCV with DAA treatment may only partially restore immune cell function. In this study, we have identified immune genes that were restored to normal by DAA treatment, and more importantly immune genes that were not restored ex vivo (i.e., at day 0 time point), including a subset of immune activation genes, inflammation genes, immune suppression, and PI3K pathway genes.
In North America, the era during which some, but not all HCV patients were treated with DAA was brief and has passed. Within this narrow time frame, we worked with tissue from patients who were treated for surgical resection of a liver lesion. Accordingly, there are inherent limitations to this study. First, due to the small sample size, we did not further classify patient subgroups based on HCV genotypes. Likewise, other subject characteristics including gender, age, genetic polymorphisms (such as IL28B), and liver fibrosis status, all of which may affect immunity and responses to HCV infection (13, 66, 67) were not used as factors to subdivide patient groups in the downstream analysis. Nonetheless, we observed significant effects that cut across these hidden variables, with consistent identification of aberrant immune gene expression at baseline day 0 and day 7, as well as those with altered TLR3 and TLR4 responses. Second, we cannot determine whether the sustained abnormality is due to the host response to hepatic fibrosis or HCC rather than HCV-mediated. The DAA-cured subjects we studied were based only on those who developed HCC or ICC post DAA therapy, since these were the patients who underwent liver resection. Thus, our DAA-cured samples only covered a unique sub-group of DAA-cured patients. Third, only KC, LSEC, HSC enriched genes could be assigned to specific liver cell type. Many other differentially expressed genes were mapped to the NPC compartment, but the analysis of isolated cells did not give us sufficient resolution to allow us to assign any genes to other liver cell types, such as CD8+ T cells, CD4+ T cells, NK cells, dendritic cells and monocytes.
Previous analysis with DAA-cured PBMC suggest both the functional phenotypes (8, 11, 68, 69) and the frequency/composition (70, 71) of immune cells are relevant aspects not fully restored by DAA therapy. Since we did not interrogate the expression of these genes in NPCs from samples other than the non-infected controls, it is unclear whether the gene expression changes were contributed mainly by the NPC cell phenotypes or by the overall greater abundance of immune cells in HCV+ and DAA-cured livers. Hence, IHC analysis of overall abundance of NPC populations in liver slices between groups would be impactful to address this question. Future study may extend the conclusion through analysis of a more diverse group of clinical situations, including chronic HCV patients and DAAs-SVR cases with or without fibrosis or HCC, as well as non-HCV related liver tissues with fibrosis or HCC. Liver tissue with longer time span post DAA therapy (>20 months) will also help clarify the resolution of liver cirrhosis by DAA therapy.
We made use of the PCLS approach opportunistically to obtain data from the group of untreated HCV-infected subjects, a group that has now disappeared from the surgical patient pool, at least in North America. Despite the heterogeneity of both subjects and controls, this study demonstrates the power of PCLS to document functional responses of intact human liver tissue.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Board, University of Washington. The patients/participants provided their written informed consent to participate in this study.
XW, JR, AC, HH, RY, and IC designed research; XW, JR, AK, AG, NH, HK, MT, and AC performed research; DK, KJ, SP, and RY contributed reagents/analytic tools; XW, AG, CT, and IC analyzed data; XW, SP, and IC wrote the manuscript. All authors reviewed the manuscript and provided suggestions.
This work was supported by NIH NIAID R56 grant AI143683, US Department of Defense grant CA150370, Janssen Research and Development, the Seattle Foundation, and the University of Washington.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Pathology Research Services Laboratory, and Histology and Imaging Core (HIC) at UW for their help in data and image acquisition.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.811551/full#supplementary-material
PCLS, precision-cut liver slice; TLR, toll-like receptor; NPC, non-parenchymal cell; KC, Kupffer cells; LSEC, Liver sinusoidal endothelial cells; HSC, hepatic stellate cells; HCV, hepatitis C virus; DAA, direct-acting antiviral; ISGs, interferon-stimulated genes; HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; SVR, sustained virologic response.
1. Wu X, Roberto JB, Knupp A, Kenerson HL, Truong CD, Yuen SY, et al. Precision-Cut Human Liver Slice Cultures as an Immunological Platform. J Immunol Methods (2018) 455:71–9. doi: 10.1016/j.jim.2018.01.012
2. Kenerson HL, Sullivan KM, Labadie KP, Pillarisetty VG, Yeung RS. Protocol for Tissue Slice Cultures From Human Solid Tumors to Study Therapeutic Response. STAR Protoc (2021) 2:100574. doi: 10.1016/j.xpro.2021.100574
3. Polaris Observatory, H.C.V.C. Global Prevalence and Genotype Distribution of Hepatitis C Virus Infection in 2015: A Modelling Study. Lancet Gastroenterol Hepatol (2017) 2:161–76. doi: 10.1016/S2468-1253(16)30181-9
4. Ringelhan M, Mckeating JA, Protzer U. Viral Hepatitis and Liver Cancer. Philos Trans R Soc Lond B Biol Sci (2017) 372:1–11. doi: 10.1098/rstb.2016.0274
5. Spaan M, Van Oord G, Kreefft K, Hou J, Hansen BE, Janssen HL, et al. Immunological Analysis During Interferon-Free Therapy for Chronic Hepatitis C Virus Infection Reveals Modulation of the Natural Killer Cell Compartment. J Infect Dis (2016) 213:216–23. doi: 10.1093/infdis/jiv391
6. Zhang Q, Wang Y, Zhai N, Song H, Li H, Yang Y, et al. HCV Core Protein Inhibits Polarization and Activity of Both M1 and M2 Macrophages Through the TLR2 Signaling Pathway. Sci Rep (2016) 6:36160. doi: 10.1038/srep36160
7. Najafi Fard S, Schietroma I, Corano Scheri G, Giustini N, Serafino S, Cavallari EN, et al. Direct-Acting Antiviral Therapy Enhances Total CD4+ and CD8+ T-Cells Responses, But Does Not Alter T-Cells Activation Among HCV Mono-Infected, and HCV/HIV-1 Co-Infected Patients. Clin Res Hepatol Gastroenterol (2018) 42:319–29. doi: 10.1016/j.clinre.2017.11.006
8. Aregay A, Sekyere SO, Deterding K, Port K, Dietz J, Berkowski C, et al. Elimination of Hepatitis C Virus has Limited Impact on the Functional and Mitochondrial Impairment of HCV-Specific CD8+ T Cell Responses. J Hepatol (2019) 71(5):889–99. doi: 10.1016/j.jhep.2019.06.025
9. Telatin V, Nicoli F, Frasson C, Menegotto N, Barbaro F, Castelli E, et al. In Chronic Hepatitis C Infection, Myeloid-Derived Suppressor Cell Accumulation and T Cell Dysfunctions Revert Partially and Late After Successful Direct-Acting Antiviral Treatment. Front Cell Infect Microbiol (2019) 9:190. doi: 10.3389/fcimb.2019.00190
10. Vranjkovic A, Deonarine F, Kaka S, Angel JB, Cooper CL, Crawley AM. Direct-Acting Antiviral Treatment of HCV Infection Does Not Resolve the Dysfunction of Circulating CD8(+) T-Cells in Advanced Liver Disease. Front Immunol (2019) 10:1926. doi: 10.3389/fimmu.2019.01926
11. Ghosh A, Mondal RK, Romani S, Bagchi S, Cairo C, Pauza CD, et al. Persistent Gamma Delta T-Cell Dysfunction in Chronic HCV Infection Despite Direct-Acting Antiviral Therapy Induced Cure. J Viral Hepat (2019) 26:1105–16. doi: 10.1111/jvh.13121
12. Hengst J, Strunz B, Deterding K, Ljunggren HG, Leeansyah E, Manns MP, et al. Nonreversible MAIT Cell-Dysfunction in Chronic Hepatitis C Virus Infection Despite Successful Interferon-Free Therapy. Eur J Immunol (2016) 46:2204–10. doi: 10.1002/eji.201646447
13. Conti F, Buonfiglioli F, Scuteri A, Crespi C, Bolondi L, Caraceni P, et al. Early Occurrence and Recurrence of Hepatocellular Carcinoma in HCV-Related Cirrhosis Treated With Direct-Acting Antivirals. J Hepatol (2016) 65:727–33. doi: 10.1016/j.jhep.2016.06.015
14. Reig M, Marino Z, Perello C, Inarrairaegui M, Ribeiro A, Lens S, et al. Unexpected High Rate of Early Tumor Recurrence in Patients With HCV-Related HCC Undergoing Interferon-Free Therapy. J Hepatol (2016) 65:719–26. doi: 10.1016/j.jhep.2016.04.008
15. Kuftinec G, Loehfelm T, Corwin M, Durbin-Johnson B, Candido M, Hluhanich R, et al. De Novo Hepatocellular Carcinoma Occurrence in Hepatitis C Cirrhotics Treated With Direct-Acting Antiviral Agents. Hepat Oncol (2018) 5:HEP06. doi: 10.2217/hep-2018-0003
16. Dong Ji EA. Increase in Incidence of Hepatocellular Carcinoma in Chronic Hepatitis C Chinese After Sustained Virologic Response With Direct Acting Antiviral Agents as Compared to Pegylated Interferon Plus Ribavirin Therapy: A Prospective Open-Label Study. Metabol Open (2021) 10:100090. doi: 10.1016/j.metop.2021.100090
17. Gitto S, Cicero AFG, Loggi E, Giovannini M, Conti F, Grandini E, et al. Worsening of Serum Lipid Profile After Direct Acting Antiviral Treatment. Ann Hepatol (2018) 17:64–75. doi: 10.5604/01.3001.0010.7536
18. Hamdane N, Juhling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology (2019) 156:2313–2329 e2317. doi: 10.1053/j.gastro.2019.02.038
19. Perez S, Kaspi A, Domovitz T, Davidovich A, Lavi-Itzkovitz A, Meirson T, et al. Hepatitis C Virus Leaves an Epigenetic Signature Post Cure of Infection by Direct-Acting Antivirals. PloS Genet (2019) 15:e1008181. doi: 10.1371/journal.pgen.1008181
20. Juhling F, Hamdane N, Crouchet E, Li S, El Saghire H, Mukherji A, et al. Targeting Clinical Epigenetic Reprogramming for Chemoprevention of Metabolic and Viral Hepatocellular Carcinoma. Gut (2021) 70:157–69. doi: 10.1136/gutjnl-2019-318918
21. Whitcomb E, Choi WT, Jerome KR, Cook L, Landis C, Ahn J, et al. Biopsy Specimens From Allograft Liver Contain Histologic Features of Hepatitis C Virus Infection After Virus Eradication. Clin Gastroenterol Hepatol (2017) 15:1279–85. doi: 10.1016/j.cgh.2017.04.041
22. Bhogal RH, Hodson J, Bartlett DC, Weston CJ, Curbishley SM, Haughton E, et al. Isolation of Primary Human Hepatocytes From Normal and Diseased Liver Tissue: A One Hundred Liver Experience. PloS One (2011) 6:e18222. doi: 10.1371/journal.pone.0018222
23. Mohar I, Brempelis KJ, Murray SA, Ebrahimkhani MR, Crispe IN. Isolation of Non-Parenchymal Cells From the Mouse Liver. Methods Mol Biol (2015) 1325:3–17. doi: 10.1007/978-1-4939-2815-6_1
24. Alabraba EB, Curbishley SM, Lai WK, Wigmore SJ, Adams DH, Afford SC. A New Approach to Isolation and Culture of Human Kupffer Cells. J Immunol Methods (2007) 326:139–44. doi: 10.1016/j.jim.2007.06.014
25. Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, et al. Distinct Development and Functions of Resident and Recruited Liver Kupffer Cells/Macrophages. J Leukoc Biol (2013) 94:1325–36. doi: 10.1189/jlb.0313144
26. Elvevold K, Smedsrod B, Martinez I. The Liver Sinusoidal Endothelial Cell: A Cell Type of Controversial and Confusing Identity. Am J Physiol Gastrointest Liver Physiol (2008) 294:G391–400. doi: 10.1152/ajpgi.00167.2007
27. Buhring HJ, Battula VL, Treml S, Schewe B, Kanz L, Vogel W. Novel Markers for the Prospective Isolation of Human MSC. Ann N Y Acad Sci (2007) 1106:262–71. doi: 10.1196/annals.1392.000
28. D'ambrosio DN, Walewski JL, Clugston RD, Berk PD, Rippe RA, Blaner WS. Distinct Populations of Hepatic Stellate Cells in the Mouse Liver Have Different Capacities for Retinoid and Lipid Storage. PloS One (2011) 6:e24993. doi: 10.1371/journal.pone.0024993
29. Jang JS, Simon VA, Feddersen RM, Rakhshan F, Schultz DA, Zschunke MA, et al. Quantitative miRNA Expression Analysis Using Fluidigm Microfluidics Dynamic Arrays. BMC Genomics (2011) 12:144. doi: 10.1186/1471-2164-12-144
30. Brempelis KJ, Yuen SY, Schwarz N, Mohar I, Crispe IN. Central Role of the TIR-Domain-Containing Adaptor-Inducing Interferon-Beta (TRIF) Adaptor Protein in Murine Sterile Liver Injury. Hepatology (2017) 65:1336–51. doi: 10.1002/hep.29078
31. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster Analysis and Display of Genome-Wide Expression Patterns. Proc Natl Acad Sci USA (1998) 95:14863–8. doi: 10.1073/pnas.95.25.14863
32. Saldanha AJ. Java Treeview–extensible Visualization of Microarray Data. Bioinformatics (2004) 20:3246–8. doi: 10.1093/bioinformatics/bth349
33. Elmasry S, Wadhwa S, Bang BR, Cook L, Chopra S, Kanel G, et al. Detection of Occult Hepatitis C Virus Infection in Patients Who Achieved a Sustained Virologic Response to Direct-Acting Antiviral Agents for Recurrent Infection After Liver Transplantation. Gastroenterology (2017) 152:550–553 e558. doi: 10.1053/j.gastro.2016.11.002
34. Macparland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat Commun (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
35. Aizarani N, Saviano A, Sagar ML, Durand S, Herman JS, Pessaux P, et al. A Human Liver Cell Atlas Reveals Heterogeneity and Epithelial Progenitors. Nature (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
36. Tu Z, Bozorgzadeh A, Pierce RH, Kurtis J, Crispe IN, Orloff MS. TLR-Dependent Cross Talk Between Human Kupffer Cells and NK Cells. J Exp Med (2008) 205:233–44. doi: 10.1084/jem.20072195
37. Armstrong RA. When to Use the Bonferroni Correction. Ophthalmic Physiol Opt (2014) 34:502–8. doi: 10.1111/opo.12131
38. Dolmazashvili E, Abutidze A, Chkhartishvili N, Karchava M, Sharvadze L, Tsertsvadze T. Regression of Liver Fibrosis Over a 24-Week Period After Completing Direct-Acting Antiviral Therapy in Patients With Chronic Hepatitis C Receiving Care Within the National Hepatitis C Elimination Program in Georgia: Results of Hepatology Clinic HEPA Experience. Eur J Gastroenterol Hepatol (2017) 29:1223–30. doi: 10.1097/MEG.0000000000000964
39. Hsu WF, Lai HC, Su WP, Lin CH, Chuang PH, Chen SH, et al. Rapid Decline of Noninvasive Fibrosis Index Values in Patients With Hepatitis C Receiving Treatment With Direct-Acting Antiviral Agents. BMC Gastroenterol (2019) 19:63. doi: 10.1186/s12876-019-0973-5
40. Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol (2016) 14:124–131 e121. doi: 10.1016/j.cgh.2015.07.019
41. Casado JL, Quereda C, Moreno A, Perez-Elias MJ, Marti-Belda P, Moreno S. Regression of Liver Fibrosis is Progressive After Sustained Virological Response to HCV Therapy in Patients With Hepatitis C and HIV Coinfection. J Viral Hepat (2013) 20:829–37. doi: 10.1111/jvh.12108
42. Rockey DC, Friedman SL. Fibrosis Regression After Eradication of Hepatitis C Virus: From Bench to Bedside. Gastroenterology (2021) 160:1502–1520 e1501. doi: 10.1053/j.gastro.2020.09.065
43. Lau DT, Fish PM, Sinha M, Owen DM, Lemon SM, Gale M Jr. Interferon Regulatory Factor-3 Activation, Hepatic Interferon-Stimulated Gene Expression, and Immune Cell Infiltration in Hepatitis C Virus Patients. Hepatology (2008) 47:799–809. doi: 10.1002/hep.22076
44. Brownell J, Wagoner J, Lovelace ES, Thirstrup D, Mohar I, Smith W, et al. Independent, Parallel Pathways to CXCL10 Induction in HCV-Infected Hepatocytes. J Hepatol (2013) 59:701–8. doi: 10.1016/j.jhep.2013.06.001
45. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1beta Production Through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection With Liver Inflammation and Disease. PloS Pathog (2013) 9:e1003330. doi: 10.1371/journal.ppat.1003330
46. Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M Jr., et al. Direct, Interferon-Independent Activation of the CXCL10 Promoter by NF-kappaB and Interferon Regulatory Factor 3 During Hepatitis C Virus Infection. J Virol (2014) 88:1582–90. doi: 10.1128/JVI.02007-13
47. Meissner EG, Wu D, Osinusi A, Bon D, Virtaneva K, Sturdevant D, et al. Endogenous Intrahepatic IFNs and Association With IFN-Free HCV Treatment Outcome. J Clin Invest (2014) 124:3352–63. doi: 10.1172/JCI75938
48. Polyak SJ, Crispe IN, Baumert TF. Liver Abnormalities After Elimination of HCV Infection: Persistent Epigenetic and Immunological Perturbations Post-Cure. Pathogens (2021) 10:1–11. doi: 10.3390/pathogens10010044
49. Cai CX, Buddha H, Castelino-Prabhu S, Zhang Z, Britton RS, Bacon BR, et al. Activation of Insulin-PI3K/Akt-P70s6k Pathway in Hepatic Stellate Cells Contributes to Fibrosis in Nonalcoholic Steatohepatitis. Dig Dis Sci (2017) 62:968–78. doi: 10.1007/s10620-017-4470-9
50. Wang L, Feng Y, Xie X, Wu H, Su XN, Qi J, et al. Neuropilin-1 Aggravates Liver Cirrhosis by Promoting Angiogenesis via VEGFR2-Dependent PI3K/Akt Pathway in Hepatic Sinusoidal Endothelial Cells. EBioMedicine (2019) 43:525–36. doi: 10.1016/j.ebiom.2019.04.050
51. Wu X, Hollingshead N, Roberto J, Knupp A, Kenerson H, Chen A, et al. Human Liver Macrophage Subsets Defined by CD32. Front Immunol (2020) 11:2108. doi: 10.3389/fimmu.2020.02108
52. Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C, et al. Short-Range Exosomal Transfer of Viral RNA From Infected Cells to Plasmacytoid Dendritic Cells Triggers Innate Immunity. Cell Host Microbe (2012) 12:558–70. doi: 10.1016/j.chom.2012.08.010
53. Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, et al. Simultaneous Detection of Hepatitis C Virus and Interferon Stimulated Gene Expression in Infected Human Liver. Hepatology (2014) 59:2121–30. doi: 10.1002/hep.26770
54. Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, et al. Toll-Like Receptor-Induced Innate Immune Responses in Non-Parenchymal Liver Cells Are Cell Type-Specific. Immunology (2010) 129:363–74. doi: 10.1111/j.1365-2567.2009.03179.x
55. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune Evasion by Hepatitis C Virus NS3/4A Protease-Mediated Cleavage of the Toll-Like Receptor 3 Adaptor Protein TRIF. Proc Natl Acad Sci USA (2005) 102:2992–7. doi: 10.1073/pnas.0408824102
56. Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, et al. Viral and Therapeutic Control of IFN-Beta Promoter Stimulator 1 During Hepatitis C Virus Infection. Proc Natl Acad Sci USA (2006) 103:6001–6. doi: 10.1073/pnas.0601523103
57. Bellecave P, Sarasin-Filipowicz M, Donze O, Kennel A, Gouttenoire J, Meylan E, et al. Cleavage of Mitochondrial Antiviral Signaling Protein in the Liver of Patients With Chronic Hepatitis C Correlates With a Reduced Activation of the Endogenous Interferon System. Hepatology (2010) 51:1127–36. doi: 10.1002/hep.23426
58. Kar P, Kumar D, Gumma PK, Chowdhury SJ, Karra VK. Down Regulation of TRIF, TLR3, and MAVS in HCV Infected Liver Correlates With the Outcome of Infection. J Med Virol (2017) 89:2165–72. doi: 10.1002/jmv.24849
59. Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, et al. Interferon Signaling and Treatment Outcome in Chronic Hepatitis C. Proc Natl Acad Sci USA (2008) 105:7034–9. doi: 10.1073/pnas.0707882105
60. Mcgilvray I, Feld JJ, Chen L, Pattullo V, Guindi M, Fischer S, et al. Hepatic Cell-Type Specific Gene Expression Better Predicts HCV Treatment Outcome Than IL28B Genotype. Gastroenterology (2012) 142:1122–31.e1121. doi: 10.1053/j.gastro.2012.01.028
61. Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology (2018) 154:1778–90. doi: 10.1053/j.gastro.2018.01.034
62. D'ambrosio R, Colombo M. Should Surveillance for Liver Cancer be Modified in Hepatitis C Patients After Treatment-Related Cirrhosis Regression? Liver Int (2016) 36:783–90. doi: 10.1111/liv.13106
63. Makiyama A, Itoh Y, Kasahara A, Imai Y, Kawata S, Yoshioka K, et al. Characteristics of Patients With Chronic Hepatitis C Who Develop Hepatocellular Carcinoma After a Sustained Response to Interferon Therapy. Cancer (2004) 101:1616–22. doi: 10.1002/cncr.20537
64. Van Der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, et al. Association Between Sustained Virological Response and All-Cause Mortality Among Patients With Chronic Hepatitis C and Advanced Hepatic Fibrosis. JAMA (2012) 308:2584–93. doi: 10.1001/jama.2012.144878
65. Van Der Meer AJ, Feld JJ, Hofer H, Almasio PL, Calvaruso V, Fernandez-Rodriguez CM, et al. Risk of Cirrhosis-Related Complications in Patients With Advanced Fibrosis Following Hepatitis C Virus Eradication. J Hepatol (2017) 66:485–93. doi: 10.1016/j.jhep.2016.10.017
66. Schott E, Witt H, Hinrichsen H, Neumann K, Weich V, Bergk A, et al. Gender-Dependent Association of CTLA4 Polymorphisms With Resolution of Hepatitis C Virus Infection. J Hepatol (2007) 46:372–80. doi: 10.1016/j.jhep.2006.09.011
67. Yan Z, Wang Y. Viral and Host Factors Associated With Outcomes of Hepatitis C Virus Infection (Review). Mol Med Rep (2017) 15:2909–24. doi: 10.3892/mmr.2017.6351
68. Langhans B, Nischalke HD, Kramer B, Hausen A, Dold L, Van Heteren P, et al. Increased Peripheral CD4(+) Regulatory T Cells Persist After Successful Direct-Acting Antiviral Treatment of Chronic Hepatitis C. J Hepatol (2017) 66:888–96. doi: 10.1016/j.jhep.2016.12.019
69. Ning G, Li YT, Chen YM, Zhang Y, Zeng YF, Lin CS. Dynamic Changes of the Frequency of Classic and Inflammatory Monocytes Subsets and Natural Killer Cells in Chronic Hepatitis C Patients Treated by Direct-Acting Antiviral Agents. Can J Gastroenterol Hepatol (2017) 2017:3612403. doi: 10.1155/2017/3612403
70. Ravens S, Hengst J, Schlapphoff V, Deterding K, Dhingra A, Schultze-Florey C, et al. Human Gammadelta T Cell Receptor Repertoires in Peripheral Blood Remain Stable Despite Clearance of Persistent Hepatitis C Virus Infection by Direct-Acting Antiviral Drug Therapy. Front Immunol (2018) 9:510. doi: 10.3389/fimmu.2018.00510
Keywords: liver slice culture, innate immunity, hepatitis C virus, direct-acting antiviral treatment, inflammation, fibrosis
Citation: Wu X, Roberto JB, Knupp A, Greninger AL, Truong CD, Hollingshead N, Kenerson HL, Tuefferd M, Chen A, Koelle DM, Horton H, Jerome KR, Polyak SJ, Yeung RS and Crispe IN (2022) Response of Human Liver Tissue to Innate Immune Stimuli. Front. Immunol. 13:811551. doi: 10.3389/fimmu.2022.811551
Received: 09 November 2021; Accepted: 08 February 2022;
Published: 09 March 2022.
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Matthew Alan Burchill, University of Colorado Denver, United StatesCopyright © 2022 Wu, Roberto, Knupp, Greninger, Truong, Hollingshead, Kenerson, Tuefferd, Chen, Koelle, Horton, Jerome, Polyak, Yeung and Crispe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Wu, eGlhd3UyQHV3LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.