
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 08 April 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.811525
 Nathan W. Zammit1,2*
Nathan W. Zammit1,2* Joseph McDowell1,2
Joseph McDowell1,2 Joanna Warren1,2Walter Muskovic2,3
Joanna Warren1,2Walter Muskovic2,3 Joanne Gamble4Yan-Chuan Shi2,5Dominik Kaczorowski2,3Chia-Ling Chan2,3Joseph Powell2,3
Joanne Gamble4Yan-Chuan Shi2,5Dominik Kaczorowski2,3Chia-Ling Chan2,3Joseph Powell2,3 Chris Ormandy2,6David Brown4Samantha R. Oakes2,6
Chris Ormandy2,6David Brown4Samantha R. Oakes2,6 Shane T. Grey1,2,6*
Shane T. Grey1,2,6*Women with autoimmune and inflammatory aetiologies can exhibit reduced fecundity. TNFAIP3 is a master negative regulator of inflammation, and has been linked to many inflammatory conditions by genome wide associations studies, however its role in fertility remains unknown. Here we show that mice harbouring a mild Tnfaip3 reduction-of-function coding variant (Tnfaip3I325N) that reduces the threshold for inflammatory NF-κB activation, exhibit reduced fecundity. Sub-fertility in Tnfaip3I325N mice is associated with irregular estrous cycling, low numbers of ovarian secondary follicles, impaired mammary gland development and insulin resistance. These pathological features are associated with infertility in human subjects. Transplantation of Tnfaip3I325N ovaries, mammary glands or pancreatic islets into wild-type recipients rescued estrous cycling, mammary branching and hyperinsulinemia respectively, pointing towards a cell-extrinsic hormonal mechanism. Examination of hypothalamic brain sections revealed increased levels of microglial activation with reduced levels of luteinizing hormone. TNFAIP3 coding variants may offer one contributing mechanism for the cause of sub-fertility observed across otherwise healthy populations as well as for the wide variety of auto-inflammatory conditions to which TNFAIP3 is associated. Further, TNFAIP3 represents a molecular mechanism that links heightened immunity with neuronal inflammatory homeostasis. These data also highlight that tuning-up immunity with TNFAIP3 comes with the potentially evolutionary significant trade-off of reduced fertility.
Autoimmune diseases are more frequent in women than men (1, 2), highlighting the potential impact of autoimmunity on female fecundity. Many autoimmune diseases can directly effect key endocrine and nervous systems (3–5), with thyroid autoimmunity being the most prevalent in women and impacting the hypothalamus. The hypothalamus participates in a hypothalamic–pituitary–gonadal axis essential for the normal menstrual cycle in preparation for fertilisation, egg implantation and embryo development (6). Further to this, women with rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) or multiple sclerosis (MS) can present with a number of fertility indications. These include a reduction in overall fecundity, increased time to pregnancy, as well as reduced birth weight for newborn (7–9).
Reproduction is a highly co-ordinated process that requires tight regulation of pro- and anti- inflammatory processes. Hypothalamic gonadotropin releasing hormone (GnRH) drives the production of luteinising hormone (LH) and follicle stimulating hormone (FSH) from the anterior pituitary which in turn triggers the release of ovarian estrogen and inhibin during the estrous cycle (6). Release of these hormones prepares the ovarian follicle for ovulation and induces decidualisation. This process involves release of pro-inflammatory cytokines and an increase of uterine natural killer cells, which populate the decidua (10). Ovulation itself causes damage to the ovarian surface, with vasculature and tissue remodelling being necessary to transform the follicle into a functional corpus luteum. Progesterone release, promotes uterine decidualisation, a prerequisite for successful embryo implantation (11). A wide variety of cytokines such as IL1, IL6, IL8 and TNF, the synthesis of which are controlled by nuclear factor kappa beta (NF-κB), are crucial mediators of this remodelling process and dysregulation of these inflammatory process can cause failure at any stage of follicle maturation, ovulation, implantation and placentation (11). Thus, the intersection of inflammatory disease with infertility reflects both the interplay between normal immune function and healthy reproductive physiology (3) but potentially a role for common genetic factors.
Human genome wide association scans (GWAS) have linked multiple immune genes with autoimmune disease raising the possibility that inflammatory genetic factors may also influence fertility in disease states. TNFAIP3 represents a possible inflammatory candidate gene that could influence fertility, as TNFAIP3 regulates inflammation through control of NF-κB which in turn influences many aspects of fertility, and GWAS data link TNFAIP3 SNPs with autoimmune diseases with female bias and associated infertility (i.e. with SLE, RA and MS). TNFAIP3, encoding A20, is an evolutionarily ancient controller of inflammation (12) – limiting over-zealous activation of innate or adaptive immune cells of the inflammatory response of non-hematopoietic cells through inhibition of NF-κB (12–14). A20 inhibits NF-κB and multiple downstream immune and danger sensing receptors via multiple ubiquitin-editing and interacting domains (15–18). A20 expression is regulated both at the level of gene transcription, whereby NF-κB activation induces TNFAIP3 gene transcription (19, 20) and by post-translational phosphorylation of a key residue Serine-381, which enhances the inhibitory activities of A20 enzymatic sites (21, 22). Thus, TNFAIP3 is a master negative regulator of NF-κB whose expression is induced during an inflammatory response to restore tissue and immune homeostasis.
The TNFAIP3 coding SNP F127C decreases TNFAIP3 anti-NF-κB function (23) and associates with RA, SLE, and MS (23–26). We tested the hypothesis that common TNFAIP3 genetic variants that subtly reduce the threshold for the proinflammatory NF-κB activation might also influence fertility. For our model of a TNFAIP3 hypomorphic variant we studied the mouse line carrying the I325N missense variant (Tnfaip3I325N) that diminishes A20’s anti-NF-κB inhibitory function (13). Tnfaip3I325N mice subsequently show reduced thresholds of immune cell NF-κB activation, heightened pathogen responses, and evidence of low-grade tissue inflammation that mirrors a subclinical autoimmune phenotype (13, 14). Here we show that carriers of the Tnfaip3I325N genotype exhibit impaired fertility, highlighting a molecular mechanism by which common variants in autoimmune risk alleles may link autoimmunity with infertility. These data also highlight that tuning-up immunity with TNFAIP3 comes with the potentially evolutionary significant trade-off of reduced fertility.
We have previously reported an A20-hypomorphic mouse line, which carries the I325N missense variant that impairs A20 phosphorylation at ser381 by ~ 50% (13). Reduced A20 phosphorylation subsequently results in a ~40% loss of NF-κB inhibitory function in Tnfaip3 (Tnfaip3I325N), resulting in reduced thresholds for both immune and non-immune cell intrinsic NF-κB activation (13, 14). Of interest, mice harbouring the Tnfaip3I325N allele exhibit evidence of a subclinical autoimmune phenotype characterised by low-grade pancreas and gut infiltrates that do not progress to overt disease (13). Thus, the I325N TNFAIP3 variant may be useful to model identified human coding variants for their impact on human reproductive physiology. Human coding variants include F127C, A125V and I207L, that reduce A20’s anti-NF-κB inhibitory activity and associate (GWAS) with human autoimmune and inflammatory disease (13, 23, 26). Furthermore, analysis of human genome data sets (e.g. GnomAD) reveal the presence of a human missense variant at the same I325 position as in the A20-hypomorphic mouse line, highlighting potential for direct relevance of the Tnfaip3I325N variant for human reproductive physiology.
When paired with wild-type males for 100 days, 30% of Tnfaip3I325N/I325N females failed to fall pregnant, with those successfully falling pregnant taking on average longer to do so compared to Tnfaip3+/+ and Tnfaip3+/I325N littermates (i.e. 39 versus 23 days) (Figures 1A, B). Despite a longer time to pregnancy, the average number of viable pups per litter was not different between genotypes (e.g. average number of pups per litter = ~4-7 for wild type versus ~4-8 for Tnfaip3I325N/I325N females). Together these data are indicative of a reduction in fecundity manifested as an increased time to pregnancy which is seen in women with autoimmune disease (7–9). Analysis of cervical swabs of cycling littermates between 5-8 weeks of age showed Tnfaip3I325N/I325N mice spent many more days in diestrous, or completely failed to enter estrous (Figures 1C, D and Supplementary Table 1). In addition, swabs of Tnfaip3I325N/I325N mice contained high numbers of neutrophils at all stages (Figure 1E). Because neutrophils are hormonally controlled to mediate the catabolic process of diestrous, these data are indicative of a failure to reach true estrous or exit diestrous and may point towards dysregulated hormonal signalling. In addition, Tnfaip3I325N/I325N mice exhibited a significantly reduced number of secondary follicles and a trend towards a decreased number of corpus luteum bodies. Ovarian area and primary follicle numbers were similar between genotype mice (Figures 1F–I). As the uterine estrous cycle is controlled by the temporal ovarian release of estrogen and progesterone stimulated by pituitary LH (6, 28), we investigated their serum levels at different stages of the estrous cycle. Subsequently, we found Tnfaip3I325N/I325N mice exhibited no difference in serum estradiol levels during bona fide proestrus and diestrus and a significantly increased level of progesterone during proestrus when compared to wild-type littermates. Serum samples taken during undefined cycle (UDC) in I325N homozygotes that typically exhibit features of estrus, proesteros and diestrous (Figure 1C) exhibited moderate levels of both hormones. Together these data suggest that ovaries of Tnfaip3I325N/I325N mice undergo a period of erroneous cycling characterised by a failure to enter full-esterous associated with dysregulated ovarian sex steroids and corpus luteal remodelling (Figures 1J, K). Therefore, these data show that Tnaip3I325N/I325N mice exhibit a sub-fertile phenotype. Reduced fecundity compared to their wild-type littermates was characterised by impaired ovarian follicle development, cyclic ovarian function, reduced estrous, and temporal dysregulated release or synthesis of primary sex steroid hormones.
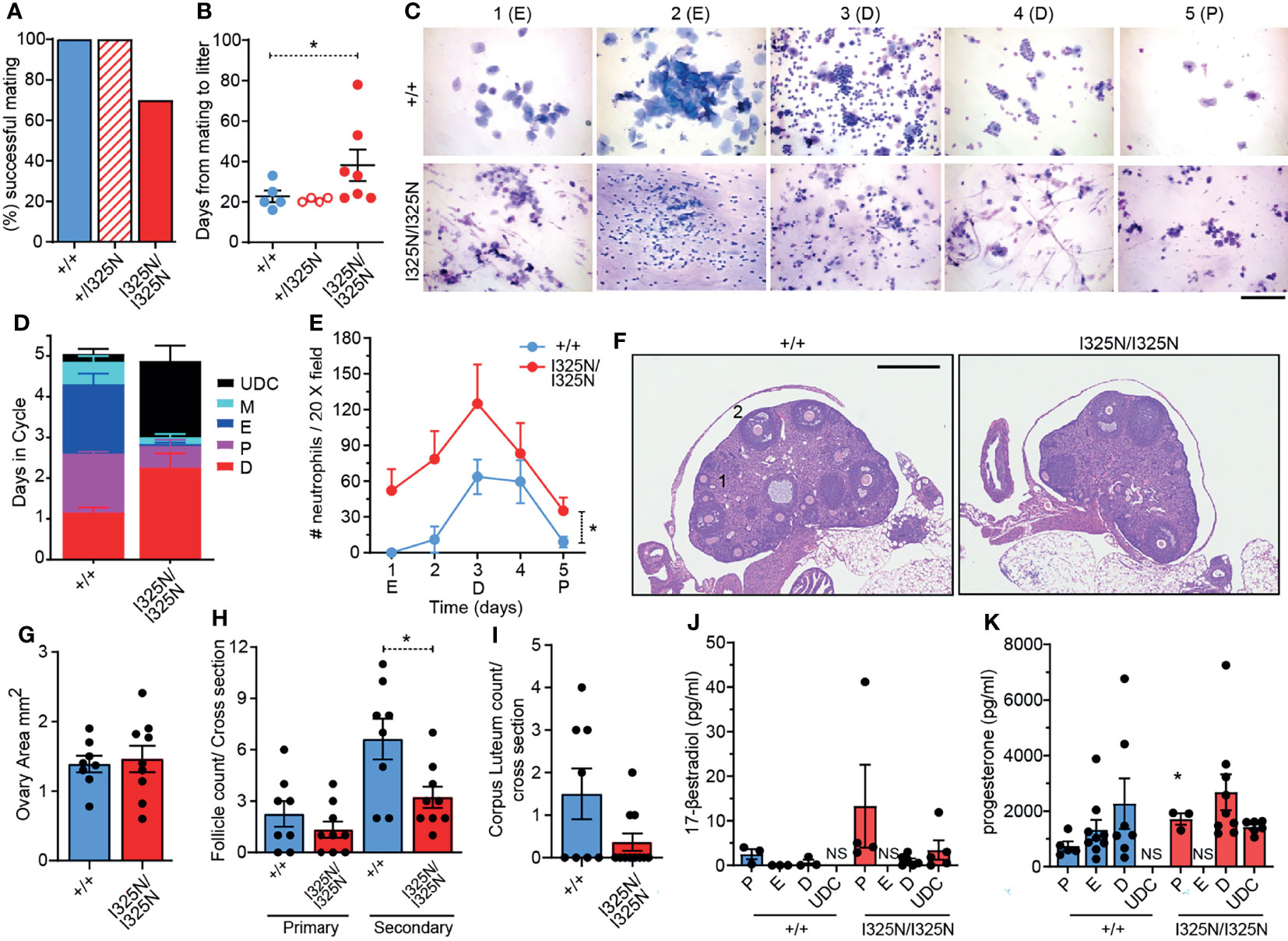
Figure 1 Tnfaip3I325N drives ovarian dysfunction in homozygous mice. Adult (8-12 weeks) female mice of the indicated genotypes were paired with a similar aged wild-type males and (A) successful mating and (B) time to litter recorded over a 100 day period. (n = 5 Tnfaip3+/+, 4 Tnfaip3+/I325N and 10 Tnfaip3I325N/I325N). (C) Diff-Quick stain of vaginal smears over 5 days (D1-D5) from female mice induced to synchronous estrous at 5 weeks of age [Whitten effect (27)]. Data is representative of 1 box of 8 week old mice from 3 independently housed groups (n = 5 Tnfaip3+/+ and 6 Tnfaip3I325N/I325N). Determined cycle stage for wild-type smears is indicated in parenthesis; E = estrous, D = diesterous, P = proesterous. (D) Days spent in each cycle-stage cumulated from all groups in (C). (E) The average number of neutrophils identified in a 20X field view of vaginal smears taken in (C) Neutrophil count was determined for each slide by numerating and averaging neutrophil number from five random 20X field of views. Counts for 1 full cycle were averaged against counts from 4 additional cycles from the same mouse. (F) Representative H&E sections of ovaries from cycling Tnfaip3 I325N mice of the indicated genotypes. Primary follicles and secondary follicles indicated by 1 and 2 respectively. Scale = 500 µm. (G) Ovarian area, (H) primary and secondary follicle and (I) corpus luteum counts per cross sectional area for Tnfaip3+/+ and Tnfaip3I325N/I325N ovaries. Each point represents an individual ovary. (J) 17-β estradiol and (K) progesterone ELISA from serum of cycling mice (n ≥ 3). Error bars represent s.e.m and One-Way Anova (B), (D) Area under the curve or Student’s T test (G–K) used for significance analysis * = P < 0.05. UDC = undefined cycle; NS = no sample.
Ovarian steroid hormones, estrogen and progesterone are crucial for mammary gland development by promoting post-pubertal ductal elongation and side branching, respectively, and for establishment of the lobuloalveoli during pregnancy (29). In mice post-pubertal ductal morphogenesis is complete by 8-14 weeks of age depending on strain. As Tnfaip3I325N/I325N mice exhibited dysregulated ovarian cycling, we investigated mammary morphology of adult 14-week old virgin mice. We observed a marked reduction in ductal elongation in the 3rd (axial) and 4th (inguinal) mammary glands of Tnfaip3I325N/I325N mice accompanied by grossly enlarged lymph nodes compared to wild-type littermates (Figure 2A). Ductal morphogenesis inTnfaip3I325N/I325N was partially rescued by pregnancy where the mammary epithelium completely penetrated the mammary fat pad, but lobuloalveolar development was impaired, characterized by smaller and more compacted lobuloalveoli indicative of attenuated secretory activation or slowed milk secretion into the alveolar luminal space (Figure 2B). Despite the impaired lobuloalveolar development, Tnfaip3I325N/I325N mice were able to support pup weight gain post-partum (Figure 2C). In addition, no significant difference in the proportion of mammary epithelial cells with either estrogen or progesterone receptor expression was detected (Figures 2D, E and Supplementary Figure 1). Lastly, we next examined for the presence of mammary paracrine hormones RANKL and OPG in 10-week old developing mammary glands. Estrogen stimulates OPG expression (30, 31), and progesterone, RANKL expression, critical for ductal side branching and alveologenesis (32–35). We found Tnfaip3I325N/I325N mammary glands to exhibit increased levels of both OPG and RANKL paracrine hormones, compared to Tnfaip3+/+ littermates (Figure 2F), which may fit with the serum presence of both estrogen and progesterone hormones during the prolonged UDC stage of Tnfaip3I325N/I325N mice. Together, the above data support a sub-fertility phenotype driven by dysregulated ovarian cycling.
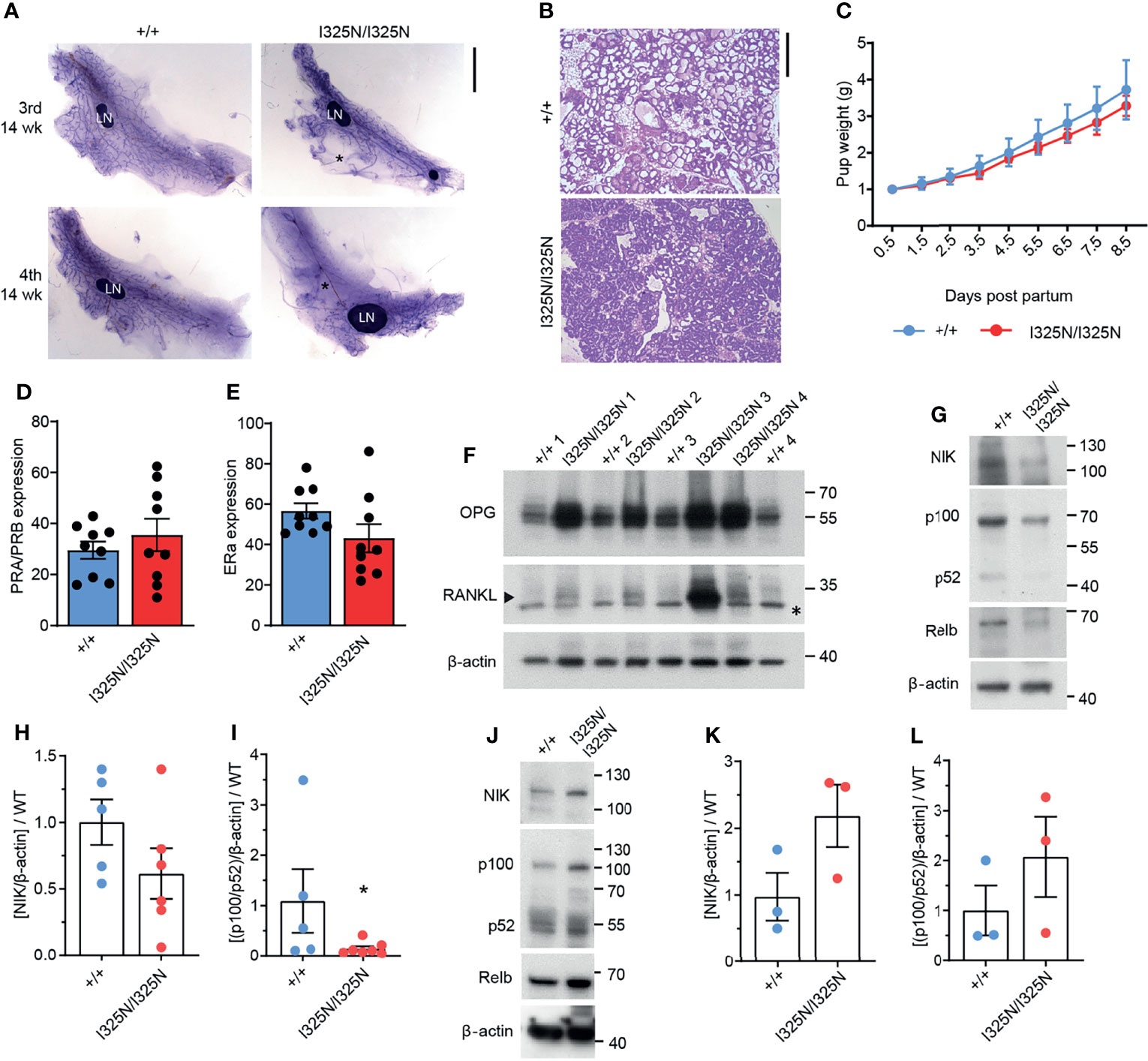
Figure 2 Tnfaip3 I325N mice exhibit impaired mammary development, elevated non-canonical RankL signalling and dampened non-canonical NF-κB signalling. (A) Mammary gland whole mounts of the 3rd and 4th mammary gland from 14-week virgin Tnfaip3I325N mice of indicated genotype. * denotes empty fat pad space. LN = lymph node. (B) Representative H&E of mammary gland from does 8.5 days post-partum, and (C) pup weight of resulting pairs, normalised to litter size. (D, E) Quantified positive immunohistochemistry staining for (D) progesterone receptor (PR) or (E) estrogen receptor (ER) (n = 9 Tnfaip3+/+ and 9 Tnfaip3I325N/I325N). (F) 4th Mammary gland from 10-week-old mice were collected and lysed for immunoblot assessment of paracrine hormones OPG and RANKL with Beta-actin used as the loading control.* denotes non-specific band and arrow head denotes specific band. Molecular weight in Kilodaltons is shown to the right of each blot. (G–I) 4th Mammary gland and its lymph node (J–L) from 10-week-old mice were collected for immunoblot (G, J) and densitometry analysis (H, I, K, L) for non-canonical NF-κB components NIK, p100/p52 and RelB. Immunoblots are representative of 2 independent experiments with n=6 (H, I) or n = 3 (K, L) biological replicates for both WT and HOM donors. Densitometry values were normalised to the average WT value of each blot to allow cumulative quantification. Error bars represent s.e.m and Student’s T test used for significance analysis * = P < 0.05.
The RANKL/RANK/OPG axis directs ductal morphogenesis and pregnancy in response to estrogen and progesterone by fine tuning non-canonical NF-κB mediated transcription of cell cycle mediators such as Cyclin D1 to drive proliferation of the mammary gland (36). RANKL binds RANK receptor (RANK) to activate non-canonical NF-κB, and OPG acts to limit RANKL binding to RANK by binding to RANKL with higher avidity (31, 36). Therefore, we next probed for components of the non-canonical pathway to determine whether observed upregulation of OPG and RANKL in I325N allelic mice resulted in changes in downstream signalling. We observed reduced NIK stabilisation, and decreased levels of P100 to P52 processing (Figures 2G–I), indicative of decreased non-canonical NF-κB signalling in Tnfaip3I325N/I325N mammary glands. These data conflicts with previous observations in pancreatic islets, where we have reported Tnfaip3I325N/I325N islets to exhibit increased non-canonical NF-κB activation (13), which associates with metabolic dysregulation (37). When we examined non-canonical NF-κB signalling in mammary lymph-nodes, we found a trend increase in non-canonical NF-κB activation, highlighting a potential for tissue specific effects (Figures 2J–L). Indeed, these data suggest that dysregulated paracrine signalling characterised by high OPG, is the major driver of dampened non-canonical NF-κB activation in the mammary glands of Tnfaip3I325N/I325N mice. These data are consistent with previous observations demonstrating that loss of RANKL signalling causes impaired mammary lobuloalveolar development and lactation (38). Together, these data show that the I325N Tnfaip3 allele drives an impairment in mammary gland development through dysregulated paracrine signalling.
The Tnfaip3I325N/I325N allele drives a sub-clinical, low-grade inflammation in the colon, liver and kidneys under SPF conditions, however these mice do not spontaneously develop overt autoimmune or inflammatory disease, (13). As A20 is expressed in ovaries (i.e. https://www.proteinatlas.org/ENSG00000118503-TNFAIP3/tissue) we next assessed the uteri of allelic mice. We observed sub-clinical inflammation evidenced by thickening of the uterine wall and presence of an immune infiltrate along the uterine wall but without overt signs of tissue damage (Supplementary Figure 2A). Further to this, we also observed an increased frequency of macrophages (CD45+CD11b+F4/80+ cells), and an increased frequency of ICAM-1+ macrophages compared to allelic null mice (Supplementary Figures 2B, C). These data are consistent with a heightened inflammatory state of I325N uteri. As local inflammation represents a barrier to fertility, we next tested whether the effects of Tnfaip3I325N/I325N on ovarian function and mammary gland development were cell autonomous. To test this question, we transplanted Tnfaip3I325N/I325N mammary epithelial tissue or ovaries into wild-type mice. Transplantation of Tnfaip3I325N/I325N mammary epithelial tissue into wild-type hosts rescued post-pubertal mammary ductal elongation and mammary branching morphogenesis during pregnancy (Figure 3A; right panel). Tnfaip3I325N/I325N mutant mice bearing Tnfaip3+/+ ovaries showed a similar failed estrous cycle to Tnfaip3I325N/I325N mutant mice, but transplantation of Tnfaip3I325N/I325N ovaries into ovariectomized wild-type recipients rescued the estrous cycle (Figure 3B). Futhermore, wild-type mice bearing Tnfaip3I325N/I325N ovaries exhibited fully developed mammary glands 12 days following pregnancy (Figure 3C; bottom panel). In contrast, Tnfaip3I325N/I325N mice bearing wild-type ovaries exhibited impaired development 12 days following pregnancy (Figure 3C; top panel). Together, these data support a cell-extrinsic mechanism for infertility in Tnfaip3I325N/I325N mice.
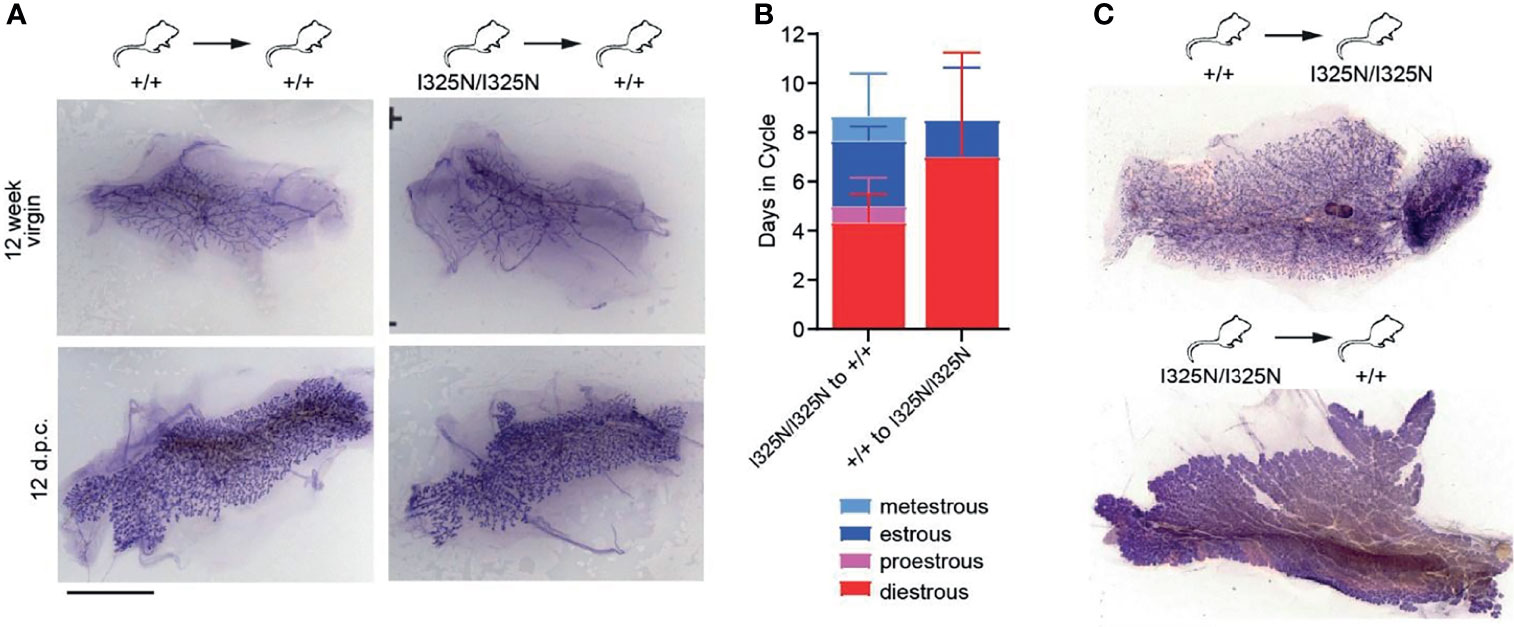
Figure 3 Transplantation of I325N mammary tissue or ovaries into WT recipients rescues mammary development and esterous cycling. (A) Representative whole mounts of mammary gland at 8 weeks following mammary epithelial transplants from wild-type (left column) or I325N homozygous (right column) donors into wild-type recipients. Whole mounts were assessed from virgin mice (top row – 12-week virgin) or following pairing with a wild-type male and 12 days post coitum (d.p.c.). Scale bar = 5mm. (B) Average days spent in each cycle-stage from wild type mice receiving a Tnfaip3I325N/I325N ovary transplant or Tnfaip3I325N/I325N mice receiving a wild-type ovary. Diff-Quick stain of cervical smears was to determine cycle-stage. (C) Representative whole mount of mammary glands 8 days post-partum of transplanted female7s in (B). Scale bar = 5mm. Error bars represent s.d..
Infertility is commonly associated with insulin resistance (39). Following a 16-hour fast Tnfaip3I325N/I325N mice exhibited lower blood glucose levels compared to wild-type littermates, but were not hyperinsulinemic (Supplementary Figures 3A, B). Similar to previously reported (13), Tnfaip3I325N/I325N mice are glucose tolerant following administration of an intraperitoneal or intravenous bolus of glucose (Supplementary Figures 3C, D). However, Tnfaip3I325N/I325N mice were found to release up to 3 times more insulin in the blood compared to wild-type litter mates in response to the glucose bolus (Supplementary Figure 3E), which increased with age (Supplementary Figures 3F, G). Hyperinsulinemia is a hallmark of peripheral insulin resistance, typically caused by chronic and systemic inflammation (40–42). Indeed, we previously reported Tnfaip3I325N/I325N mice to exhibit marked mononuclear infiltrate within peripheral tissues such as the liver, kidney, colon and pancreas (13), as well as, increased levels of circulating and splenic activated/memory T cells and B cells (13). In addition, macrophages from Tnfaip3I325N/I325N mice are more sensitive to activating stimuli (13). Consistent with these data Tnfaip3I325N/I325N mice harbour higher frequencies of mononuclear immune cells expressing NF-κB regulated activation markers, including an increased frequency of ICAM1+ splenic macrophages, and an increased frequency of CD44+ T cells in both the spleen and lymph nodes (Supplementary Figure 4) (43–45).
To further test the idea of peripheral insulin resistance as the primary mechanism for hyperinsulinemia following glucose challenge in Tnfaip3I325N/I325N mice, we isolated and transplanted wild-type islets into Tnfaip3+/+ or Tnfaip3I325N/I325N recipients. Next, we measured blood glucose and insulin output following a glucose bolus in euglycemic mice 14-days post-transplant (Supplementary Figure 3H). Tnfaip3I325N/I325N recipients harbouring wild-type islets exhibited 3 times higher blood insulin levels compared to wild-type islets transplanted into wild-type recipients (Supplementary Figure 3J). These data point to systemic inflammation, extrinsic to the pancreatic beta cell, as the cause for high insulin output following a glucose challenge in Tnfaip3I325N/I325N mice. Together, these data illustrate that Tnfaip3I325N mice exhibit features for peripheral insulin resistance, however they do not alone explain the observed systemic hormone dysregulation and infertility.
In addition to ovary-derived sex steroids, cyclic ovarian function is also under the control of CNS hormones, namely luteinizing hormone (LH) emanating from the anterior pituitary (6). We found Tnfaip3I325N/I325N mice exhibited a ~ 50% reduction in serum LH compared to Tnfaip3+/+ littermates (Figure 4A). A reduction in serum LH was associated with a decrease in LH-β subunit RNA and a trend towards elevated levels of Gndh RNA in hypothalamic-pituitary lysates of Tnfaip3 allelic mice (Figures 4B, C). As LH synthesis and secretion is sensitive to inflammation (46), we assessed the preoptic area that contains neurons responsible for LH release from the pituitary, for the presence of inflammatory factors. We subsequently found only a small subset of genes to be significantly upregulated compared to Tnfaip3 wild-type littermates (Figure 4D). The upregulated genes associated strongly with microglial activation and included antigen presentation genes (Tap1, B2m) (47–49) and activation genes (Ccl12, IL-34 and Icam1) (50–52) that occur in neurodegenerative disease, brain injury and viral infection, and are NF-κB regulated. We next used RTPCR as a more sensitive approach to probe for the presence of other inflammatory mediators within the hypothalamus of Tnfaip3 littermates, which revealed a significant increase for Il1β, ccl7, cxcl2 and cxcl1 (Figure 4E), genes associated with microglial-mediated chronic neurodegeneration (53). Indeed, Tnfaip3 critically restricts CNS microglia activation and microglia-dependent inflammation (54, 55). RTPCR analysis of Tnfaip3I325N/I325N brains revealed an increase in Aif1 mRNA that encodes the microglial marker Iba-1 (Figure 4F). Morphological analysis revealed an increase in activated microglia in the hypothalamus of Tnfaip3 I325N homozygotes exemplified by increased ramification compared to Iba-1+ cells in the brains of wild-type littermates (Figures 4G–I). In contrast, Tnfaip3I325N/I325N mice did not exhibit evidence of chronic systemic inflammation as determined by an absence of IL-6 in either the serum or hypothalamus of unstimulated brains (i.e. for Tnfaip3I325N/I325N mice versus wild type mice serum levels below detection limit of an IL-6 ELISA; n = ≥ 4 mice per group; data not shown). Together, these data show that the Tnfaip3 I325N hypomorph causes spontaneous inflammation in the CNS of homozygous mice, associated with a decrease in LH, likely explaining the cell-extrinsic effects (Figure 3) on ovarian function, fertility (Figure 1) and subsequently mammary gland development (Figure 2).
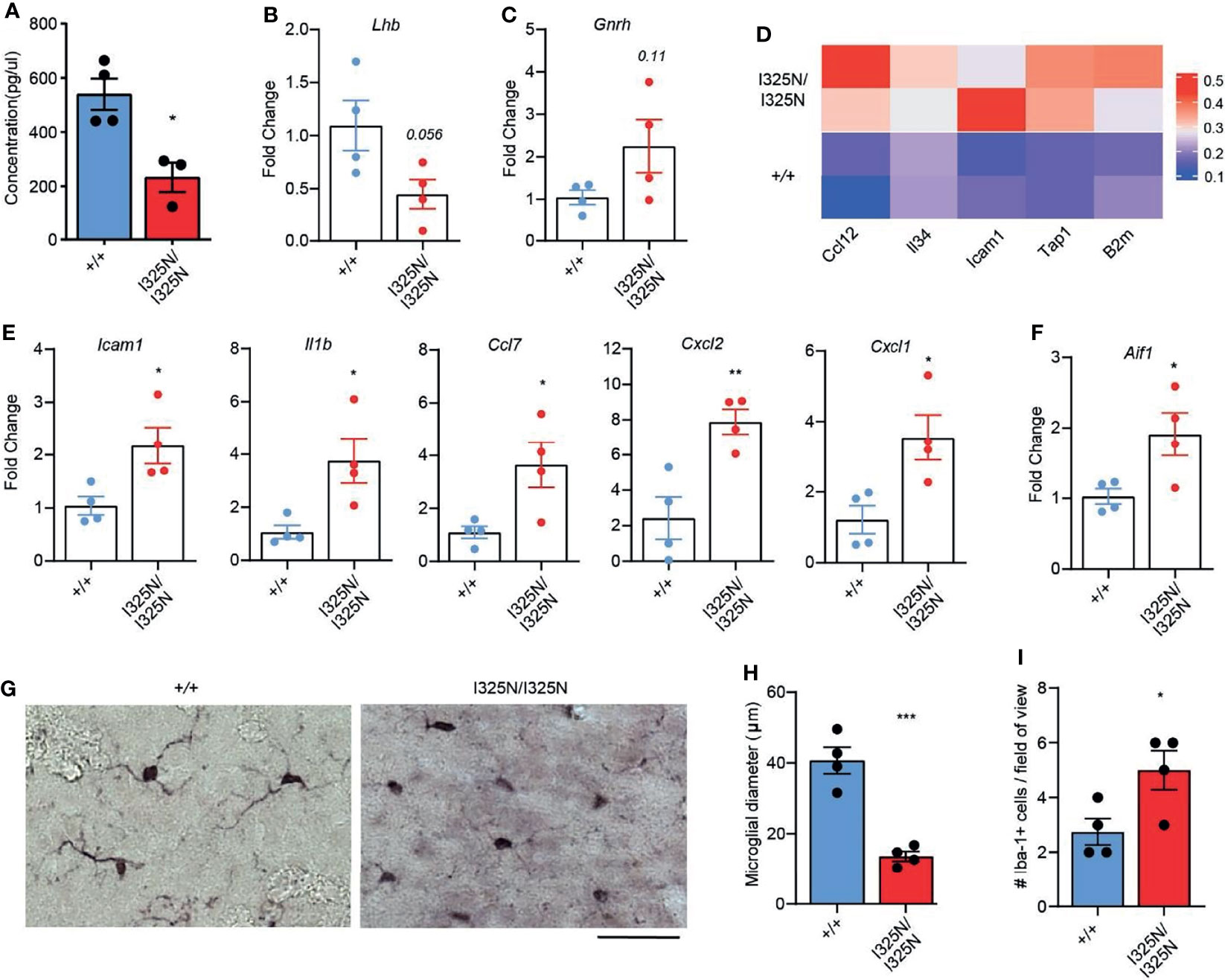
Figure 4 Tnfaip3I325N mice exhibit reduced serum luteinizing hormone and spontaneous CNS inflammation. (A) ELISA for luteinizing hormone from serum plasma from Tnfap3I325N littermates of indicated genotypes, and (B) RTPCR analysis of brain lysates obtained from dissected pituitary and hypothalamus for luteinizing hormone or (C) gonadotropin-releasing hormone mRNA. (D) Visium transcriptomic analysis of preoptic area (PO) of Tnfaip3I325N littermates of indicated genotypes revealed an upregulation of microglial associated genes at steady state. Each row represents data from an individual mouse. (E) RTPCR analysis of brain lysates from dissected pituitary and hypothalamus for microglial-associated inflammatory markers from Tnfaip3I325N littermates, and for (F) the microglial marker AIF1 that encodes Iba-1. (G) Representative immunohistochemistry images for Iba-1 from Tnfaip3I325N littermates of indicated genotypes showing a more ramified phenotype in Tnfaip3 homozygotes. Scale bar = 20 µm. (H) Quantification of microglial diameter and (I) number of Iba-1+ cells per field of view from (F). Error bars represent s.e.m and Student’s T test used for significance analysis * = P < 0.05; ** = P < 0.01; *** = P < 0.005.
Mammalian reproduction is energetically demanding and to ensure enough energy is available for puberty and the maintenance of reproductive capacity a tight feed-back control network exists, regulated by paracrine, autocrine and neuronal signals (56). The concentration of adipocyte-derived hormone leptin provides information about the amount of available stored energy in the body (57). While the concentration of metabolic hormones insulin and ghrelin, provide ‘real-time’ updates on nutrient availability, absorption and use (56, 58, 59). These signals act at multiple levels, namely the gonads and CNS, were signals are integrated through the complex interactions of the hypothalamic-pituitary-gonadal (HPG) axis (56–59). Together, these systems ensure reproductive success by allowing reproduction to proceed only when conditions are favourable. For example, caloric deficit, stress and insulin resistance can abrogate the HPG axis to promote hypothalamic amenorrhea (60) or polycystic ovary syndrome (PCOS) (61) respectively, and thus down-tune fecundity. In this way finite resources can be temporarily diverted to energetically demanding catabolic processes such as immunity and tissue repair to enhance future reproductive success (62–64). However, how the body genetically tunes these cross-tissue adaptations is still largely unknown.
Here we show that the anti-inflammatory enzyme TNFAIP3 can contribute to the tuning of fecundity. The Tnfaip3 I325N reduction-of-function allele limits fecundity by driving systemic hormonal dysregulation characterised by reduced serum LH. These changes were associated with increased levels of basal CNS inflammation – as may occur during stress or infection (65, 66). We have previously shown Tnfaip3 I325N to increase host immunity by lowering NF-κB activating thresholds of T cells, B cells and dendritic cells, achieving superior protective immunity to Coxsackie virus at the expense of microbial tolerance (13). In addition to a loss of microbial tolerance, here we illustrate that fertility is an additional trade-off for increased protective immunity (13, 64).
The Tnfaip3 I325N allele allows for increased cellular NF-κB activation by impairing A20 function via the down-tuning of A20-phosphorylation at ser-381, which enforces A20’s enzymatic activities (21, 22). The severity of the Tnfaip3 I325N reduction-of-function effect biochemically lies between two additional Tnfaip3 alleles found in humans that also impair A20 phosphorylation at ser-381 (13). The most severely impaired allele, C243Y, is causative for autosomal Bechets disease (67), while the subtlest, I207L, has been found to beneficially up-tune immunity (13). The I207L allele exhibits a specific population distribution, having been adaptively introgressed into modern peoples of Oceania from ancient Denisovan hominins (13, 68). Other potentially functional TNFAIP3 coding variants with specific population distributions also exist, namely F127C, A125V and I325V (as identified in GnomAD), raising the potential for subtle genetic tuning of A20 activity to have wide-spread impact on female fertility. In addition, our data would predict that disease associated non-coding variants, as well as more penetrant A20 haploinsufficieny (12, 13, 69) may have significant implications for fertility in inflammatory disease. Thus, by altering immune activation thresholds human TNFAIP3 genetic variants may contribute to the fertility impairment observed in females with autoimmune disease (7–9). None-the-less, these findings widen the scope of clinical phenotypes impacted by Tnfaip3 to include fertility.
The transplant studies showing rescue of the I325N ovarian phenotype when placed in wild type hosts (Figure 3) support an extrinsic mechanism whereby loss of LH secretion associating with CNS inflammation is sufficient for driving infertility in Tnfaip3 I325N mice. We also observed a trend to decreased in Gndh mRNA in hypothalamic-pituitary lysates of Tnfaip3 allelic mice, and future studies should investigate this result further by protein analysis. However, TNFAIP3 is expressed by ovaries, suggesting the potential for an ovary intrinsic effect contributing to impaired fertility, indeed intrinsic ovarian inflammation due to the reduced anti-inflammatory activity of the I325N variant may act as an important susceptibility factor following an initial extrinsic trigger (14). Further to this, the transplant data do not rule out an additional contribution by bone marrow derived immune cells that reside within the ovary. Myeloid cells and lymphocytes are activated upon the LH surge to assist proper ovulation. As the Tnfaip3 I325N variant increases the activation state of host immunity (13) (Supplementary Figures 2, 4) it is probable that hyper-activated and resident ovary immune cells could contribute to improper ovulation and therefore altered ovarian sex-steroid production (70, 71).
Inflammation plays a central role in the metabolic syndrome (40), and neuroinflammation is now considered a central driver for a wide range of neurodegenerative diseases (72, 73) and psychiatric illness’s (74–76). Neuroinflammation may also explain clinical links between autoimmune disorders and neuropsychiatric disease (77). Indeed, Tnfaip3 a central immune regulator, is linked to increased susceptibility to many inflammatory and autoimmune diseases (12, 69), and has recently been found to play a central protective role for the maintenance of CNS homeostasis. Tnfaip3 deficiency causes spontaneous CNS inflammation characterised by microglial activation and induction of inflammatory mRNAs in the brain (54). A20 is highly expressed in microglia (78), the resident immune cell of the brain (79), and its deficiency within microglia drives a potent inflammatory response, triggering the infiltration of CD8 T-cells and neurodegenerative disease (80, 81). Here we observe increased Aif1 mRNA and Iba-1 positive microglia with an ‘activated’ morphometry in the brains of Tnfaip3 I325N mice. These data are consistent with the hyper-inflammatory state of peritoneal macrophages harbouring the same Tnfaip3 allele (13) or with A20 deficiency (24). Given microglia’s important role in synapse formation, synapse pruning as well as neural myelination (82–84) all important for CNS signal transmission and function, loss of A20 function may impact or predispose to a wide range of neurodegenerative disease including neuropsychiatric conditions (85), as well as fertility as described here.
Neuroinflammation has been suggested to drive infertility by disrupting the hypothalamic-pituitary-gonadal axis, through yet completely understood mechanisms. One potential mechanism is through driving insensitivity of CNS insulin receptors to insulin, termed insulin resistance. Similar to our study, female mice with CNS-specific KO of insulin receptor exhibited ovarian dysfunction and reduced serum and RNA levels of LH (58). Indeed, mice with the Tnfaip3 I355N allele exhibit peripheral insulin resistance and CNS inflammation, thus identifying a shared mechanism of infertility. A role for inflammatory driven insulin resistance that is independent to obesity induced hormonal dysregulation, such as of leptin, was demonstrated in a transgenic line of lean normoglycemic mice with insulin resistance that exhibited altered duration of estrous cycle and dysfunctional ovarian follicles (86). Lastly, conditions such as Type A syndrome and lipoatrophic diabetes that are characterised by severe insulin resistance, exhibit perturbed hypothalamic-pituitary-gonadal function and ovarian dysfunction (87, 88).
These data underline a novel role for Tnfaip3 in the maintenance of fertility. Future studies would focus on determining whether neuronal inflammation is causal to the sub-fertility impact of the Tnfaip3 I355N allele, and the full nature of the fecundity defect including whether it involves a delay in puberty. As NF-κB controls male mouse sperm counts (89) these data also highlight a mechanism for TNFAIP3 to regulate male fertility. Finally, TNFAIP3 coding variants have been identified with population distinct distributions. It has been hypothesised that common genetic variants with medium to low penetrance may play an important role in influencing infertility, which is globally wide-spread and common (~5-25% of the female population), yet not well explained (90–92). TNFAIP3 coding variants may offer one mechanism for the cause of sub-fertility observed across otherwise healthy populations as well as for the wide variety of auto-inflammatory conditions to which TNFAIP3 is associated. Therapies targeting pathways and inflammatory products normally controlled by TNFAIP3 may serve as promising treatments to restore the functioning of hypothalamic-pituitary axis and fertility.
The Tnfaip3 lasvegas strain (Tnfaip3I325N) was generated by N-ethyl-N-nitrosourea (ENU) mutagenesis in C57BL/6 mice, and propagated by backcrossing to C57BL/6 (13, 14). The strain was maintained as heterozygote breeding pairs so that wild type littermates could be used for controls. C57BL/6 and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice were purchased from Australian Bioresources. Animal studies were conducted in compliance with guidelines of the Garvan Institute Animal Ethics Committee.
Adult female wild type and Tnfaip3I325N variant mice (8-12 weeks of age) were paired with wild type male mice, and pregnancy was confirmed by the observation of a cervical plug. For fecundity studies n = 5 wild type, n = 4 Tnfaip3I325N/+ and n = 6 Tnfaip3I325N/I325N mice were examined. The onset to pregnancy for Tnfaip3I325N variant mice was significantly delayed as shown in Results, but the average number of viable pups per litter was not significantly different between genotypes (average 4-5 pups per litter).
5-week-old Tnfaip3 mice were monitored for the stage of the estrous cycle by gentle daily vaginal smearing and staining with Diff-Quik (ProSciTech) Romanowsky stain as per the manufacturers instructions.
Mammary whole-mounts were performed using the Carmine alum technique as described before (93). Briefly, at experimental time points, mice were euthanised with CO2 asphyxiation and cervical dislocation and mammary glands harvested and spread onto Superfrost plus glass slides and placed into 10% neutral buffered formalin overnight at room temperature. Mammary wholemounts were defatted with at least 4 changes of acetone and then rehydrated before staining in a 0.2 w/v solution of Carmine alum containing potassium aluminium sulphate and thymol. Mammary wholemounts were then dehydrated with graded alcohols before clearing in Slightbright for 1hr at RT and storage in methylsalycilate. Wholemounts were photographed on a Leica DMRB light microscope and imaged using a Leica DC200 camera.
Mammary epithelial transplants were performed exactly as previously described (94). Note that to determine whether the mammary phenotype was cell autonomous, either Tnfaip3I325N tissue or wild type tissue was transplanted into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ recipients. Ovarian transplants were performed on 3-4-week-old donor and recipients. Mice were paired and anaesthetized simultaneously with 75mg/kg ketamine and 0.75mg/kg medetomidine IP. A small incision was made in the left and right dorsal flanks of each mouse perpendicular to the spine and across the abdomen just caudal to the last rib and the abdomen was opened. The ovarian fat pad was grasped and externalised onto a sterile gauze so that a small incision could be made in the ovarian bursa and the ovary dissected free. Pressure was placed on the ovarian stalk for a 20-30 secs using forceps to stop any bleeding. Each donor ovary was then placed in in the left and right empty ovarian bursas of recipient mice and the ovary and uterus re-internalised into the abdomen. The abdominal wall and skin closed with silk sutures and recipient mice monitored for estrous cycling from 5 weeks of age.
Islets were isolated as previously described (95), and counted for islet transplantation using a Leica MZ9.5 stereomicroscope. Islets were transplanted under the kidney capsule of diabetic C57BL/6 littermates as described (96, 97). Diabetes was induced by intraperitoneal injection of 180 mg/kg streptozotocin (Sigma-Aldrich) dissolved in 0.1 M citrate buffer (pH 4.2) at a concentration of 20 mg/ml. Diabetes was determined as [blood glucose] ≥16 mM on two consecutive days measured by FreeStyle Lite® glucometer and Abbott Diabetes Care test strips following tail tipping.
Flow cytometric staining was performed as described (13). For mouse lymphocytes, fluorochrome-conjugated antibody clones against the following surface antigens were: CD4 (RM4-5), CD44 (IM7), CD62L (MEL-14), CD45.2 (104), CD11b (M1/70), F4/80 (RUO), ICAM1 (HCD54). Antibodies were purchased from BD, eBioscience or BioLegend. Data were acquired with LSR II and FORTESSA flow cytometers (BD) and analysed using FlowJo software (Tree Star).
Blood glucose levels were determined using a FreeStyle Lite® glucometer and blood glucose test strips (Abbott Diabetes Care) via tail tipping. Intraperitoneal glucose tolerance tests (IP-GTT) were conducted following an overnight fast (16 h) with access to water. The following day mice were weighed and fasting blood glucose measurements taken. Subsequently, mice were injected intraperitoneally with 2 g/kg 20% dextrose (Sigma Aldrich). Blood glucose levels were measured from the tail vein at 15, 30, 60 and 120 min post-dextrose administration. Intravenous (IV) GTT was conducted in a similar manner; however, glucose (1 g/kg) was administered intravenously into the tail vein and blood glucose measurements taken at 0, 5, 10, 15, 20, 30, and 60 min post-injection. During the IV-GTT blood samples were also taken for the determination of insulin content via ELISA, conducted as per the manufacture’s instructions (Cayman Chemical). Serum ELISA for 17β-estradiol (Cayman Chemical; 501890), progesterone (Cayman Chemical; 582601) and Luteinizing hormone (Elabscience; E-EL-M3053) were performed as per manufacturers’ instructions
Ovarian tissues were fixed in 10% neutral buffered formalin (Sigma-Aldrich), paraffin embedded and parallel sections (5-10 µm) prepared. Sections were stained with hematoxylin and eosin (H&E Sigma-Aldrich). Images were captured using a Leica DM 4000 or Leica DM 6000 Power Mosaic microscope (Leica Microsystems). Quantification of the number of follicles per cross sectional area was conducted by taking the average from 5 serial sections separated by 500 µm each. Progesterone and estrogen receptor staining was performed as previously described (98). Briefly, formalin fixed paraffin embedded sections were dewaxed, dehydrated and antigen retrieval performed with pH6 retrieval solution (Dako S1699) in a pressure cooker at 125°C for 30 sec. Endogenous peroxidase was blocked by 3% H2O2 and sections were stained with an antibody to ERα (Santa Cruz MC20 SC-542) or PRA and PRB (Dako A0098) for 30 mins at RT. The sections were then incubated with Envision rabbit HRP secondary (Dako K4002) for 30 min at RT prior to application of 3,3’Diaminobenzidine plus tertiary substrate (Dako K3468) for 10 mins at RT. Image quantification of the percentage positive epithelial nuclei was performed using macros designed for the FIJI image analysis software (http://fiji.sc/Fiji). The percentage positive of 3 representative images from each animal (5 per group) is shown in Figures 2D, E.
CNS tissue was examined by IHC. Briefly, mice were deeply anesthetized (350mg/kg pentobarbitone sodium) and perfused through the left ventricle with 100 ml 0.9% saline, followed by 150 ml ice-cold 4% paraformaldehyde in sodium borate buffer at pH 9.5 using a peristaltic pump (Gilson). Brains were harvested, postfixed in 4% paraformaldehyde for 5 hours, and cryoprotected for 16 hours in 20% sucrose/50 mM potassium PBS (KPBS) at 4°C. Next, 30-μm-thick frozen brain sections were obtained with a sliding microtome (SM2010R; Leica Biosystem). Sections were stored in cryoprotectant solution (30% ethylene glycol, 20% glycol in 50 mM sodium phosphate buffer, pH 7.4) at –20°C. Next, 30-μm-thick floating tissue sections were treated with 0.3% hydrogen peroxide for 10 minutes to inhibit endogenous peroxidase, followed by 7 minutes of 1% sodium borohydride.
Tissue sections were then immunostained with monoclonal antibody against Iba-1 (rabbit anti-mouse Iba-1:1,000; abcam AB108539) in 2% donkey serum and 0.3% Triton X-100. Tissue sections were incubated in primary antibody for 48 hours at 4°C on an orbital rocker (50 rpm; Ratek Instrument), followed by biotinylated secondary staining (donkey anti-rat 1:200; Jackson ImmunoResearch) in the same buffer as above for 1 hour at room temperature. After secondary immunostaining, sections were labelled with avidin using the Vecta kit (Vector Laboratories) per the manufacturer’s protocol. Sections were then washed twice (10 minutes per wash) with KPBS, and staining was developed in DAB without nickel enhancement (0.5 mg/ml DAB, 2 mg/ml D β-glucose, 0.4 mg/ml ammonium chloride, 1 U/ml glucose oxidase; Sigma-Aldrich) in KPBS to give brown labelling. Sections were mounted onto slides in an aqueous mounting buffer and coverslipped. Iba-1 positive microglia where quantified using random fields of view from brain sections across four levels. Microglial diameter was assessed by measuring the diameter of the boundary circle using Image J.
Mammary and lymphoid tissue were collected in Cell Lysis Buffer (Cell Signalling Technology), supplemented with EDTA-free protease inhibitor 1045 [Roche]). Protein concentration was measured using the Bradford assay (Bio-Rad) and total protein (20-25 µg) resolved on a 7 – 10% SDS PAGE gel and then transferred to a nitrocellulose membrane, Immobilon-P® (Merck Millipore). Non-cannonical NFκB pathway activation was assessed as described (37). Briefly, membranes were incubated anti-OPG (SANTA CRUZ E-10; Sc-390518), anti RANKL (R&D AF462), anti NIK (4994), anti-NF-kB2 P100/P52 (4882), anti-Relb (C1E4, 4922) (Cell signalling Technology), and anti-beta-actin (AC15) (Sigma-Aldrich), followed by horseradish peroxidase (HRP)-labelled secondary antibody goat-anti mouse IgG Fc (Pierce Antibodies) or donkey-anti-rabbit IgG (GE Life Sciences). HRP conjugates bound to antigen were detected and visualized by using an ECL detection kit (GE Life Sciences). Densitometry analysis was conducted using ImageJ software (https://imagej.nih.gov/ij/).
Brains from euthanised mice were set in OCT blocks and snap frozen in liquid nitrogen-chilled isopentane. Individual brains were then and stored at minus 80 until use. For cryosectioning, both the tissue block and the Visium slide were equilibrated inside the cryostat for 15-30 min. Next using an Allen Brain atlas 10 um coronal sections made to capture the medial preoptic area (MPO; Figure 29). Array slides containing sections were stored at minus 80 for a maximum of 1 week before use. Samples were processed according to the Visium Spatial Gene Expression User Guide (10× Genomics) and all reagents were from the Visium Spatial Gene Expression Kit (10× Genomics). Sections were first methanol fixed, stained with H&E and imaged at 10X using a Leica DM6000 microscope. Slides were then processed using an optimised permeabilization time of 9 min (determined using the Visium Spatial Tissue Optimization Kit). The resulting cDNA library was checked for both quality and quantity, and sequenced using an Illumina NovaSeq6000 system using the recommended parameters at the recommended depth, based on slide area occupancy.
Reads were demultiplexed and mapped to the mouse reference genome GRCm38 using the Space Ranger software v.1.1.0 (10x Genomics). Count matrices were analysed with Seurat v.4.0.0 for all subsequent data filtering, normalization, dimensional reduction and visualization. Data normalization was performed on independent tissue sections using the variance-stabilizing transformation method implemented in the Seurat function SCTransform. Voxels corresponding to the medial preoptic area (MPO) were manually defined following expert annotation. Mean normalised expression values of microglial associated genes were quantified across the MPO of each section and visualised as a heatmap.
Total RNA was extracted from brain tissue using the RNeasy Plus Mini Kit (Qiagen) and reverse transcribed using Quantitect Reverse Transcription Kit (Qiagen). PCR reactions were performed on the LightCycler® 480 Real Time PCR System (Roche) using the TaqMan system (Thermofisher; Supplementary Table 2). Cyclophilin (CPH2) and ACTB were used as housekeeping genes and data analysed using the 2ΔΔCT method. Data were normalized to average WT value. Initial denaturation was = performed at 95°C for 10 sec, followed by a three-step cycle consisting of 95°C for 15 sec = (4.8°C/s, denaturation), 63°C for 30 sec (2.5°C/sec, annealing), and 72°C for 30 sec (4.8°C/s, elongation). A melt-curve was performed after finalization of 45 cycles at 95°C for 2 min, 40°C for 3 min and gradual increase to 95°C with 25 acquisitions/°C.
All data are presented as mean ± s.e.m. Student’s t-test or 1- or 2- way ANOVA analysis were performed, depending on experimental design, to determine statistical difference between groups. A P value less than 0.05 was considered significant. Tests were conducted on Prism (v8) software (GraphPad Software).
All procedures involving animals were carried out according to the guidelines established by the Australian Institutional Animal Ethics Committee guidelines. Animal studies were approved by the Garvan/St Vincent’s Animal Ethics Committee. All procedures performed complied with the Australian Code of Practice for Care and Use of Animals for Scientific Purposes.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Garvan Institute Animal Ethics Committee.
Experimental analysis of ovarian dysfunction in Tnfaip3I355N mice conducted by NZ, SO, CO, and JM. Mammary gland development studies conducted by NZ, CO, and SO. Ovary transplantation studies performed by CO and SO. Islet transplant and metabolic studies performed by NZ. ELISA studies conducted by JW. Brain gene expression analysis by NZ and JW. Brain pathology analysis JG and DB. Visium sample preparation conducted by JW, YS, and NZ. Visium analysis conducted by WM, DK, CC, JP and NZ. Experimental design and analysis performed by NZ, SO, and SG. Manuscript preparation by NZ, SO, and SG. SG led the study and is the study guarantor. All authors contributed to the article and approved the submitted version.
The research was supported by grants to SG from the NIH (DK076169) and NHMRC (GNT1130222; GNT1189235), a NHMRC Fellowship to CJO (481310), a National Breast Cancer Foundation Fellowship to SRO (ECF-13-08; ECF-16-022), and an Australian Postgraduate Award and International Pancreas and Islet Transplant Association (IPITA) Derek Gray Scholarship to NZ. SG is supported by an NHMRC Senior Research Fellowship Level B (GNT1140691).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Biological Testing Facility at the Garvan Institute of Medical Research for animal care. The authors declare they have no conflicting financial interests.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.811525/full#supplementary-material
Supplementary Figure 1 | Representative photomicrograph of estrogen receptor (ERa) or progesterone receptor (PRA/PRB) stained sections of mammary glands from mice of the indicated genotypes (n = 5 Tnfaip3+/+ and 5 Tnfaip3I325N/I325N).
Supplementary Figure 2 | (A) Representative H&E of uterine walls of mice with or without the I325N allele. Scale bar for paired low (left column) and high power (right column) images = 500 and 200 µm respectively. (B, C) Flow cytometric analysis of digested uteri showing the frequency of infiltrating cells that are (B), CD45+ CD11b+ F4/80+ cells, and which are defined as macrophages and (C), ICAM1+ macrophages (CD45+ CD11b+ F4/80+ cells) (B). * = area of high immune infiltrate.
Supplementary Figure 3 | A20 I325N mice produce high levels of insulin following a glucose challenge and exhibit peripheral insulin resistance. (A) Fasting glucose and (B) insulin levels of chow fed 16-week-old female mice with the indicated genotypes. (C, D) Blood glucose levels (BGL) were monitored following an (C) intraperitoneal (IP; 2 g/kg) (+/+ n = 10; I325N/+ n = 14; I325N/I325N n = 8) or (D) intravenous injection (IV; 1 g/kg) of glucose (+/+ n = 6; I325N/+ n = 3; I325N/I325N n = 5). (E) Blood insulin levels (ng/ml) were measured following intravenous injection in (D). Blood insulin levels following IVGTT was also measured in 32 (F) and 52 (G) week old female mice. (H–J) To test if A20 I325N mice exhibit peripheral insulin resistance, wild-type (+/+) islets were isolated and transplanted into age and weight-matched streptozotocin-induced diabetic syngeneic recipients that were homozygous, or null for the I325N variant (i.e. WT -> WT n=4; WT -> HOM n = 4 [donor -> recipient]). (I) Following transplantation blood glucose levels (BGLs) were monitored for 13 days. (I) At post-operative day (POD) 14 glucose tolerance was tested via IV injection of a glucose bolus (1g/kg). (J) Blood insulin levels (ng/ml) were measured via ELISA from blood samples taken. Statistical significance calculated by area under the curve analysis followed by student’s t test; * = P < 0.05; error bars ± SEM.
Supplementary Figure 4 | The frequency of CD4+ effector memory cells in the (A), Spleen; (B), LN; and (C), peripheral blood. Frequency of macrophages (CD45+ CD11b+ F4/80+ cells) in the (D) spleen; and, frequency of ICAM1+ macrophages in the spleen (E). Error bars represent S.E.M and Student’s T test used for significance analysis * = P < 0.05; ** = P < 0.01.
Supplementary Table 1 | Ovarian cycling was assessed from 5-8 weeks of age and monitored weekly. Note the homozygous I325N mice did not cycle in early stages, then ultimately became pregnant but also with a delay i.e. time to pregnancy for I325N mice = 38.7 days versus 22.8 days for wild type mice.
1. Ngo ST, Steyn FJ, McCombe PA. Gender Differences in Autoimmune Disease. Front Neuroendocrinol (2014) 35(3):347–69. doi: 10.1016/j.yfrne.2014.04.004
2. Greer JM, McCombe PA. Role of Gender in Multiple Sclerosis: Clinical Effects and Potential Molecular Mechanisms. J Neuroimmunol (2011) 234(1-2):7–18. doi: 10.1016/j.jneuroim.2011.03.003
3. Sen A, Kushnir VA, Barad DH, Gleicher N. Endocrine Autoimmune Diseases and Female Infertility. Nat Rev Endocrinol (2014) 10(1):37–50. doi: 10.1038/nrendo.2013.212
4. Poppe K, Velkeniers B, Glinoer D. Thyroid Disease and Female Reproduction. Clin Endocrinol (Oxf) (2007) 66(3):309–21. doi: 10.1111/j.1365-2265.2007.02752.x
5. Deligeoroglou E, Vrachnis N, Athanasopoulos N, Iliodromiti Z, Sifakis S, Iliodromiti S, et al. Mediators of Chronic Inflammation in Polycystic Ovarian Syndrome. Gynecol Endocrinol (2012) 28(12):974–8. doi: 10.3109/09513590.2012.683082
6. Lindholm J, Nielsen EH. Pituitary-Gonadal Axis: Historical Notes. Pituitary (2009) 12(3):226–35. doi: 10.1007/s11102-008-0128-0
7. Clowse ME, Chakravarty E, Costenbader KH, Chambers C, Michaud K. Effects of Infertility, Pregnancy Loss, and Patient Concerns on Family Size of Women With Rheumatoid Arthritis and Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken) (2012) 64(5):668–74. doi: 10.1002/acr.21593
8. Ostensen M. Sexual and Reproductive Health in Rheumatic Disease. Nat Rev Rheumatol (2017) 13(8):485–93. doi: 10.1038/nrrheum.2017.102
9. Khizroeva J, Nalli C, Bitsadze V, Lojacono A, Zatti S, Andreoli L, et al. Infertility in Women With Systemic Autoimmune Diseases. Best Pract Res Clin Endocrinol Metab (2019) 33(6):101369. doi: 10.1016/j.beem.2019.101369
10. King AE, Critchley HO. Oestrogen and Progesterone Regulation of Inflammatory Processes in the Human Endometrium. J Steroid Biochem Mol Biol (2010) 120(2-3):116–26. doi: 10.1016/j.jsbmb.2010.01.003
11. Jabbour HN, Sales KJ, Catalano RD, Norman JE. Inflammatory Pathways in Female Reproductive Health and Disease. Reproduction (2009) 138(6):903–19. doi: 10.1530/REP-09-0247
12. Ma A, Malynn BA. A20: Linking a Complex Regulator of Ubiquitylation to Immunity and Human Disease. Nat Rev Immunol (2012) 12(11):774–85. doi: 10.1038/nri3313
13. Zammit NW, Siggs OM, Gray PE, Horikawa K, Langley DB, Walters SN, et al. Denisovan, Modern Human and Mouse TNFAIP3 Alleles Tune A20 Phosphorylation and Immunity. Nat Immunol (2019) 20(10):1299–310. doi: 10.1038/s41590-019-0492-0
14. Zammit NW, Walters SN, Seeberger KL, O'Connell PJ, Korbutt GS, Grey ST. A20 as an Immune Tolerance Factor Can Determine Islet Transplant Outcomes. JCI Insight (2019) 4(21):e131028. doi: 10.1172/jci.insight.131028
15. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-Ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-kappaB Signalling. Nature (2004) 430(7000):694–9. doi: 10.1038/nature02794
16. Lu TT, Onizawa M, Hammer GE, Turer EE, Yin Q, Damko E, et al. Dimerization and Ubiquitin Mediated Recruitment of A20, A Complex Deubiquitinating Enzyme. Immunity (2013) 38(5):896–905. doi: 10.1016/j.immuni.2013.03.008
17. Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, et al. Zinc-Finger Protein A20, a Regulator of Inflammation and Cell Survival, has De-Ubiquitinating Activity. Biochem J (2004) 378(Pt 3):727–34. doi: 10.1042/bj20031377
18. Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, et al. Ubiquitin Binding to A20 ZnF4 Is Required for Modulation of NF-KappaB Signaling. Mol Cell (2010) 40(4):548–57. doi: 10.1016/j.molcel.2010.10.009
19. Krikos A, Laherty CD, Dixit VM. Transcriptional Activation of the Tumor Necrosis Factor Alpha-Inducible Zinc Finger Protein, A20, Is Mediated by Kappa B Elements. J Biol Chem (1992) 267:17971–6. doi: 10.1016/S0021-9258(19)37138-8
20. Liuwantara D, Elliot M, Smith MW, Yam AO, Walters SN, Marino E, et al. Nuclear factor-KappaB Regulates Beta-Cell Death: A Critical Role for A20 in Beta-Cell Protection. Diabetes (2006) 55(9):2491–501. doi: 10.2337/db06-0142
21. Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. IkappaB Kinase Beta Phosphorylates the K63 Deubiquitinase A20 to Cause Feedback Inhibition of the NF-KappaB Pathway. Mol Cell Biol (2007) 27(21):7451–61. doi: 10.1128/MCB.01101-07
22. Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, et al. Phosphorylation and Linear Ubiquitin Direct A20 Inhibition of Inflammation. Nature (2015) 528(7582):370–5. doi: 10.1038/nature16165
23. Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, et al. Multiple Polymorphisms in the TNFAIP3 Region Are Independently Associated With Systemic Lupus Erythematosus. Nat Genet (2008) 40(9):1062–4. doi: 10.1038/ng.202
24. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M, et al. A20 (TNFAIP3) Deficiency in Myeloid Cells Triggers Erosive Polyarthritis Resembling Rheumatoid Arthritis. Nat Genet (2011) 43(9):908–12. doi: 10.1038/ng.874
25. Westra HJ, Martinez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, et al. Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes. Nat Genet (2018) 50(10):1366–74. doi: 10.1038/s41588-018-0216-7
26. Lodolce JP, Kolodziej LE, Rhee L, Kariuki SN, Franek BS, McGreal NM, et al. African-Derived Genetic Polymorphisms in TNFAIP3 Mediate Risk for Autoimmunity. J Immunol (2010) 184(12):7001–9. doi: 10.4049/jimmunol.1000324
27. Whitten MK. Effect of Exteroceptive Factors on the Oestrous Cycle of Mice. Nature (1957) 180(4599):1436. doi: 10.1038/1801436a0
28. Plant TM. 60 YEARS OF NEUROENDOCRINOLOGY: The Hypothalamo-Pituitary-Gonadal Axis. J Endocrinol (2015) 226(2):T41–54. doi: 10.1530/JOE-15-0113
29. Oakes SR, Hilton HN, Ormandy CJ. The Alveolar Switch: Coordinating the Proliferative Cues and Cell Fate Decisions That Drive the Formation of Lobuloalveoli From Ductal Epithelium. Breast Cancer Res (2006) 8(2):207. doi: 10.1186/bcr1411
30. Saika M, Inoue D, Kido S, Matsumoto T. 17beta-Estradiol Stimulates Expression of Osteoprotegerin by a Mouse Stromal Cell Line, ST-2, via Estrogen Receptor-Alpha. Endocrinology (2001) 142(6):2205–12. doi: 10.1210/endo.142.6.8220
31. Hanada R, Hanada T, Sigl V, Schramek D, Penninger JM. RANKL/RANK-Beyond Bones. J Mol Med (Berl) (2011) 89(7):647–56. doi: 10.1007/s00109-011-0749-z
32. Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y, et al. Two Distinct Mechanisms Underlie Progesterone-Induced Proliferation in the Mammary Gland. Proc Natl Acad Sci U S A (2010) 107(7):2989–94. doi: 10.1073/pnas.0915148107
33. Fernandez-Valdivia R, Mukherjee A, Ying Y, Li J, Paquet M, DeMayo FJ, et al. The RANKL Signaling Axis Is Sufficient to Elicit Ductal Side-Branching and Alveologenesis in the Mammary Gland of the Virgin Mouse. Dev Biol (2009) 328(1):127–39. doi: 10.1016/j.ydbio.2009.01.019
34. Lee HJ, Gallego-Ortega D, Ledger A, Schramek D, Joshi P, Szwarc MM, et al. Progesterone Drives Mammary Secretory Differentiation via RankL-Mediated Induction of Elf5 in Luminal Progenitor Cells. Development (2013) 140(7):1397–401. doi: 10.1242/dev.088948
35. Oakes SR, Rogers RL, Naylor MJ, Ormandy CJ. Prolactin Regulation of Mammary Gland Development. J Mammary Gland Biol Neoplasia (2008) 13(1):13–28. doi: 10.1007/s10911-008-9069-5
36. Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, et al. IKKalpha Provides an Essential Link Between RANK Signaling and Cyclin D1 Expression During Mammary Gland Development. Cell (2001) 107(6):763–75. doi: 10.1016/S0092-8674(01)00599-2
37. Malle EK, Zammit NW, Walters SN, Koay YC, Wu J, Tan BM, et al. Nuclear Factor KappaB-Inducing Kinase Activation as a Mechanism of Pancreatic Beta Cell Failure in Obesity. J Exp Med (2015) 212(8):1239–54. doi: 10.1084/jem.20150218
38. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The Osteoclast Differentiation Factor Osteoprotegerin-Ligand Is Essential for Mammary Gland Development. Cell (2000) 103(1):41–50. doi: 10.1016/S0092-8674(00)00103-3
39. Nestler JE. Metformin for the Treatment of the Polycystic Ovary Syndrome. N Engl J Med (2008) 358(1):47–54. doi: 10.1056/NEJMct0707092
40. Donath MY, Shoelson SE. Type 2 Diabetes as an Inflammatory Disease. Nat Rev Immunol (2011) 11(2):98–107. doi: 10.1038/nri2925
41. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of Obesity- and Diet-Induced Insulin Resistance With Salicylates or Targeted Disruption of Ikkbeta. Science (2001) 293(5535):1673–7. doi: 10.1126/science.1061620
42. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-Beta Links Inflammation to Obesity-Induced Insulin Resistance. Nat Med (2005) 11(2):191–8. doi: 10.1038/nm1185
43. Oh H, Ghosh S. NF-Kappab: Roles and Regulation in Different CD4(+) T-Cell Subsets. Immunol Rev (2013) 252(1):41–51. doi: 10.1111/imr.12033
44. Villanueva JE, Walters SN, Saito M, Malle EK, Zammit NW, Watson KA, et al. Targeted Deletion of Traf2 Allows Immunosuppression-Free Islet Allograft Survival in Mice. Diabetologia (2017) 60(4):679–89. doi: 10.1007/s00125-016-4198-7
45. Hubbard AK, Giardina C. Regulation of ICAM-1 Expression in Mouse Macrophages. Inflammation (2000) 24(2):115–25. doi: 10.1023/A:1007029409521
46. Wojtulewicz K, Krawczynska A, Tomaszewska-Zaremba D, Wojcik M, Herman AP. Effect of Acute and Prolonged Inflammation on the Gene Expression of Proinflammatory Cytokines and Their Receptors in the Anterior Pituitary Gland of Ewes. Int J Mol Sci (2020) 21(18):6939. doi: 10.3390/ijms21186939
47. Malone KE, Stohlman SA, Ramakrishna C, Macklin W, Bergmann CC. Induction of Class I Antigen Processing Components in Oligodendroglia and Microglia During Viral Encephalomyelitis. Glia (2008) 56(4):426–35. doi: 10.1002/glia.20625
48. Izzy S, Liu Q, Fang Z, Lule S, Wu L, Chung JY, et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front Cell Neurosci (2019) 13:307. doi: 10.3389/fncel.2019.00307
49. Sarlus H, Heneka MT. Microglia in Alzheimer’s Disease. J Clin Invest (2017) 127(9):3240–9. doi: 10.1172/JCI90606
50. Popiolek-Barczyk K, Ciechanowska A, Ciapala K, Pawlik K, Oggioni M, Mercurio D, et al. The CCL2/CCL7/CCL12/CCR2 Pathway Is Substantially and Persistently Upregulated in Mice After Traumatic Brain Injury, and CCL2 Modulates the Complement System in Microglia. Mol Cell Probes (2020) 54:101671. doi: 10.1016/j.mcp.2020.101671
51. Obst J, Simon E, Martin-Estebane M, Pipi E, Barkwill LM, Gonzalez-Rivera I, et al. Inhibition of IL-34 Unveils Tissue-Selectivity and Is Sufficient to Reduce Microglial Proliferation in a Model of Chronic Neurodegeneration. Front Immunol (2020) 11:579000. doi: 10.3389/fimmu.2020.579000
52. Lutton EM, Razmpour R, Andrews AM, Cannella LA, Son YJ, Shuvaev VV, et al. Acute Administration of Catalase Targeted to ICAM-1 Attenuates Neuropathology in Experimental Traumatic Brain Injury. Sci Rep (2017) 7(1):3846. doi: 10.1038/s41598-017-03309-4
53. Song WM, Colonna M. The Identity and Function of Microglia in Neurodegeneration. Nat Immunol (2018) 19(10):1048–58. doi: 10.1038/s41590-018-0212-1
54. Guedes RP, Csizmadia E, Moll HP, Ma A, Ferran C, da Silva CG. A20 Deficiency Causes Spontaneous Neuroinflammation in Mice. J Neuroinflamm (2014) 11:122. doi: 10.1186/1742-2094-11-122
55. Herman AP, Krawczynska A, Bochenek J, Dobek E, Herman A, Tomaszewska-Zaremba D. LPS-Induced Inflammation Potentiates the IL-1beta-Mediated Reduction of LH Secretion From the Anterior Pituitary Explants. Clin Dev Immunol (2013) 2013:926937. doi: 10.1155/2013/926937
56. Fernandez-Fernandez R, Martini AC, Navarro VM, Castellano JM, Dieguez C, Aguilar E, et al. Novel Signals for the Integration of Energy Balance and Reproduction. Mol Cell Endocrinol (2006) 254-255:127–32. doi: 10.1016/j.mce.2006.04.026
57. Moschos S, Chan JL, Mantzoros CS. Leptin and Reproduction: A Review. Fertil Steril (2002) 77(3):433–44. doi: 10.1016/S0015-0282(01)03010-2
58. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science (2000) 289(5487):2122–5. doi: 10.1126/science.289.5487.2122
59. Sliwowska JH, Fergani C, Gawalek M, Skowronska B, Fichna P, Lehman MN. Insulin: Its Role in the Central Control of Reproduction. Physiol Behav (2014) 133:197–206. doi: 10.1016/j.physbeh.2014.05.021
60. Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, et al. A Genetic Basis for Functional Hypothalamic Amenorrhea. N Engl J Med (2011) 364(3):215–25. doi: 10.1056/NEJMoa0911064
61. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, et al. Polycystic Ovary Syndrome. Nat Rev Dis Primers (2016) 2:16057. doi: 10.1038/nrdp.2016.57
62. Wang A, Luan HH, Medzhitov R. An Evolutionary Perspective on Immunometabolism. Science (2019) 363(6423):eaar3932. doi: 10.1126/science.aar3932
63. Odegaard JI, Chawla A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science (2013) 339(6116):172–7. doi: 10.1126/science.1230721
64. Schwenke RA, Lazzaro BP, Wolfner MF. Reproduction-Immunity Trade-Offs in Insects. Annu Rev Entomol (2016) 61:239–56. doi: 10.1146/annurev-ento-010715-023924
65. Liu YZ, Wang YX, Jiang CL. Inflammation: The Common Pathway of Stress-Related Diseases. Front Hum Neurosci (2017) 11:316. doi: 10.3389/fnhum.2017.00316
66. Klein RS, Garber C, Howard N. Infectious Immunity in the Central Nervous System and Brain Function. Nat Immunol (2017) 18(2):132–41. doi: 10.1038/ni.3656
67. Shigemura T, Kaneko N, Kobayashi N, Kobayashi K, Takeuchi Y, Nakano N, et al. Novel Heterozygous C243Y A20/TNFAIP3 Gene Mutation Is Responsible for Chronic Inflammation in Autosomal-Dominant Behcet’s Disease. RMD Open (2016) 2(1):e000223. doi: 10.1136/rmdopen-2015-000223
68. Gittelman RM, Schraiber JG, Vernot B, Mikacenic C, Wurfel MM, Akey JM. Archaic Hominin Admixture Facilitated Adaptation to Out-Of-Africa Environments. Curr Biol (2016) 26(24):3375–82. doi: 10.1016/j.cub.2016.10.041
69. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-Of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48(1):67–73. doi: 10.1038/ng.3459
70. Duffy DM, Ko C, Jo M, Brannstrom M, Curry TE. Ovulation: Parallels With Inflammatory Processes. Endocr Rev (2019) 40(2):369–416. doi: 10.1210/er.2018-00075
71. Oakley OR, Frazer ML, Ko C. Pituitary-Ovary-Spleen Axis in Ovulation. Trends Endocrinol Metab (2011) 22(9):345–52. doi: 10.1016/j.tem.2011.04.005
72. Brown DA, Sawchenko PE. Time Course and Distribution of Inflammatory and Neurodegenerative Events Suggest Structural Bases for the Pathogenesis of Experimental Autoimmune Encephalomyelitis. J Comp Neurol (2007) 502(2):236–60. doi: 10.1002/cne.21307
73. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms Underlying Inflammation in Neurodegeneration. Cell (2010) 140(6):918–34. doi: 10.1016/j.cell.2010.02.016
74. Depino AM. Peripheral and Central Inflammation in Autism Spectrum Disorders. Mol Cell Neurosci (2013) 53:69–76. doi: 10.1016/j.mcn.2012.10.003
75. Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and Psychiatric Illness. J Neuroinflamm (2013) 10:43. doi: 10.1186/1742-2094-10-43
76. Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased Inflammatory Markers Identified in the Dorsolateral Prefrontal Cortex of Individuals With Schizophrenia. Mol Psychiatry (2013) 18(2):206–14. doi: 10.1038/mp.2012.110
77. Kayser MS, Dalmau J. The Emerging Link Between Autoimmune Disorders and Neuropsychiatric Disease. J Neuropsychiatry Clin Neurosci (2011) 23(1):90–7. doi: 10.1176/appi.neuropsych.23.1.90
78. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J Neurosci (2014) 34(36):11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
79. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
80. Voet S, McGuire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 Critically Controls Microglia Activation and Inhibits Inflammasome-Dependent Neuroinflammation. Nat Commun (2018) 9(1):2036. doi: 10.1038/s41467-018-04376-5
81. Mohebiany AN, Ramphal NS, Karram K, Di Liberto G, Novkovic T, Klein M, et al. Microglial A20 Protects the Brain From CD8 T-Cell-Mediated Immunopathology. Cell Rep (2020) 30(5):1585–97.e6. doi: 10.1016/j.celrep.2019.12.097
82. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (2011) 333(6048):1456–8. doi: 10.1126/science.1202529
83. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat Commun (2016) 7:12540. doi: 10.1038/ncomms12540
84. Kyrargyri V, Vega-Flores G, Gruart A, Delgado-Garcia JM, Probert L. Differential Contributions of Microglial and Neuronal IKKbeta to Synaptic Plasticity and Associative Learning in Alert Behaving Mice. Glia (2015) 63(4):549–66. doi: 10.1002/glia.22756
85. Daems C, Sékulic MV, Vulsteke V, van Loo G, D’Hooge R, Callaerts-Végh Z, et al. A20/TNFAIP3 Heterozygosity Predisposes to Behavioral Symptoms in a Mouse Model for Neuropsychiatric Lupus. Brain Behav Immun - Health (2020) 2:100018. doi: 10.1016/j.bbih.2019.100018
86. Nandi A, Wang X, Accili D, Wolgemuth DJ. The Effect of Insulin Signaling on Female Reproductive Function Independent of Adiposity and Hyperglycemia. Endocrinology (2010) 151(4):1863–71. doi: 10.1210/en.2009-0788
87. Dunaif A. Insulin Resistance and the Polycystic Ovary Syndrome: Mechanism and Implications for Pathogenesis. Endocrinol Rev (1997) 18(6):774–800. doi: 10.1210/edrv.18.6.0318
88. Nakamura F, Taira M, Hashimoto N, Makino H, Sasaki N. Familial Type C Syndrome of Insulin Resistance and Short Stature With Possible Autosomal Dominant Transmission. Endocrinol Jpn (1989) 36(3):349–58. doi: 10.1507/endocrj1954.36.349
89. Blomberg Jensen M, Andreassen CH, Jorgensen A, Nielsen JE, Juel Mortensen L, Boisen IM, et al. RANKL Regulates Male Reproductive Function. Nat Commun (2021) 12(1):2450. doi: 10.1038/s41467-021-22734-8
90. Larsen U. Primary and Secondary Infertility in Sub-Saharan Africa. Int J Epidemiol (2000) 29(2):285–91. doi: 10.1093/ije/29.2.285
91. Stephen EH, Chandra A. Declining Estimates of Infertility in the United States: 1982-2002. Fertil Steril (2006) 86(3):516–23. doi: 10.1016/j.fertnstert.2006.02.129
92. Liang S, Chen Y, Wang Q, Chen H, Cui C, Xu X, et al. Prevalence and Associated Factors of Infertility Among 20-49 Year Old Women in Henan Province, China. Reprod Health (2021) 18(1):254. doi: 10.1186/s12978-021-01298-2
93. Bradbury JM, Edwards PA, Niemeyer CC, Dale TC. Wnt-4 Expression Induces a Pregnancy-Like Growth Pattern in Reconstituted Mammary Glands in Virgin Mice. Dev Biol (1995) 170(2):553–63. doi: 10.1006/dbio.1995.1236
94. Deome KB, Faulkin LJ Jr., Bern HA, Blair PB. Development of Mammary Tumors From Hyperplastic Alveolar Nodules Transplanted Into Gland-Free Mammary Fat Pads of Female C3H Mice. Cancer Res (1959) 19(5):515–20.
95. Grey ST, Lock J, Bach FH, Ferran C. Adenovirus-Mediated Gene Transfer of A20 in Murine Islets Inhibits Fas-Induced Apoptosis. Transplant Proc (2001) 33(1-2):577–8. doi: 10.1016/S0041-1345(00)02149-7
96. Grey ST, Longo C, Shukri T, Patel VI, Csizmadia E, Daniel S, et al. Genetic Engineering of a Suboptimal Islet Graft With A20 Preserves Beta Cell Mass and Function. J Immunol (2003) 170(12):6250–6. doi: 10.4049/jimmunol.170.12.6250
97. Zammit NW, Tan BM, Walters SN, Liuwantara D, Villanueva JE, Malle K, et al. Low-Dose Rapamycin Unmasks the Protective Potential of Targeting Intragraft NF-KappaB for Islet Transplants. Cell Transplant (2013) 22(12):2355–66. doi: 10.3727/096368912X658737
98. Hilton HN, Doan TB, Graham JD, Oakes SR, Silvestri A, Santucci N, et al. Acquired Convergence of Hormone Signaling in Breast Cancer: ER and PR Transition From Functionally Distinct in Normal Breast to Predictors of Metastatic Disease. Oncotarget (2014) 5(18):8651–64. doi: 10.18632/oncotarget.2354
Keywords: TNFAIP3, A20, inflammation, reproduction, fertility, neuroinflammation, life-history, evolutionary medicine
Citation: Zammit NW, McDowell J, Warren J, Muskovic W, Gamble J, Shi Y, Kaczorowski D, Chan C-L, Powell J, Ormandy C, Brown D, Oakes SR and Grey ST (2022) TNFAIP3 Reduction-of-Function Drives Female Infertility and CNS Inflammation. Front. Immunol. 13:811525. doi: 10.3389/fimmu.2022.811525
Received: 08 November 2021; Accepted: 21 February 2022;
Published: 08 April 2022.
Edited by:
Trine N. Jorgensen, Case Western Reserve University, United StatesReviewed by:
Katsuhiko Ishihara, Kawasaki Medical School, JapanCopyright © 2022 Zammit, McDowell, Warren, Muskovic, Gamble, Shi, Kaczorowski, Chan, Powell, Ormandy, Brown, Oakes and Grey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan W. Zammit, bi56YW1taXRAZ2FydmFuLm9yZy5hdQ==; Shane T. Grey, cy5ncmV5QGdhcnZhbi5vcmcuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.