 Josephine A. Mapunda
Josephine A. Mapunda Houyam Tibar
Houyam Tibar Wafa Regragui
Wafa Regragui Britta Engelhardt
Britta Engelhardt- 1Theodor Kocher Institute, University of Bern, Bern, Switzerland
- 2Medical School of Rabat, Mohamed 5 University, Rabat, Morocco
- 3Hôpital des spécialités de Rabat, Ibn Sina University Hospital of Rabat, Rabat, Morocco
Multiple Sclerosis (MS) is considered the most frequent inflammatory demyelinating disease of the central nervous system (CNS). It occurs with a variable prevalence across the world. A rich armamentarium of disease modifying therapies selectively targeting specific actions of the immune system is available for the treatment of MS. Understanding how and where immune cells are primed, how they access the CNS in MS and how immunomodulatory treatments affect neuroinflammation requires a proper knowledge on the mechanisms regulating immune cell trafficking and the special anatomy of the CNS. The brain barriers divide the CNS into different compartments that differ with respect to their accessibility to cells of the innate and adaptive immune system. In steady state, the blood-brain barrier (BBB) limits immune cell trafficking to activated T cells, which can reach the cerebrospinal fluid (CSF) filled compartments to ensure CNS immune surveillance. In MS immune cells breach a second barrier, the glia limitans to reach the CNS parenchyma. Here we will summarize the role of the endothelial, epithelial and glial brain barriers in regulating immune cell entry into the CNS and which immunomodulatory treatments for MS target the brain barriers. Finally, we will explore current knowledge on genetic and environmental factors that may influence immune cell entry into the CNS during neuroinflammation in Africa.
Introduction
The human immune system has evolved to protect the body from microbial pathogens and trauma and thus ultimately to ensure host survival in a hostile environment (1). The skin as the outer body surface and the gut and respiratory tracts as the inner body surfaces are the most exposed sites for infection and injury. Their epithelial linings form highly specialized antimicrobial barriers towards the outside and are further fortified by site-specific immune defense mechanisms established by cells of the innate and adaptive immune system [summarized in (2)]. Melanization of the skin has been recognized as an essential component of skin innate immunity with melanocytes and melanin exerting antimicrobial functions [summarized in (3)]. Microbial or traumatic injury elicits a rapid stereotypic activation of tissue-resident innate immune mechanisms that allow for the killing of the microbes and the resolution of the inflammatory response. The innate immune response includes activation of tissue-resident dendritic cells (DCs), which will take up and process the antigens and travel via the afferent lymphatic vessels to the tissue-draining lymph nodes leading to activation of T and B lymphocytes and thus the adaptive immune response and immune memory against the specific microbes to provide an accelerated and amplified immune responses in case of a further encounter with the same antigen. During their priming, naïve lymphocytes are imprinted with navigation programs (expression of a combination of adhesion and chemoattractant receptors) that ensure their site-specific homing. In this context, DCs in gut and skin draining lymph nodes have been shown to play an essential role as they process food-derived vitamin A and ultraviolet-induced vitamin D3, respectively, to imprint gut homing and skin homing trafficking programs as well as site-specific effector functions in naïve lymphocytes summarized in (4)]. Skin complexion, sunlight exposure and dietary patterns will thus have a direct impact on immune cell priming. These site-specific effector functions i.e., production of cytokines, killing of infected tissue cells, and antibody production, ensure elimination of the injurious agent and reconstitution of tissue function and also establish a site-specific cellular immune memory with tissue-resident memory T (TRM) cells (5). Immune surveillance of a given tissue thus relies on drainage by lymphatic vessels to transport antigens and antigen-presenting DCs to the draining lymph nodes, as well as on blood vessels to allow for efficient immune cell trafficking to the respective tissues.
The anatomical location of the CNS within the skull and vertebral column provides robust protection from injury from the outside. Unless there is a penetrating injury, pathogens are thus unlikely to directly reach the CNS, unless they have escaped the innate and adaptive immune defense mechanisms at the outer surfaces of the body. However, the CNS resides behind blood-brain barriers that restrict pathogen and immune cell entry from the periphery into the CNS parenchyma and lacks lymphatic vessels. The CNS thus has a unique relationship with the immune system that differs from that of peripheral organs and is referred to as CNS immune privilege. The discovery of CNS immune privilege is based on the observation that foreign tissues, when grafted to peripheral sites like the skin, are readily rejected, but when grafted into the brain parenchyma, they survive for prolonged durations (6). These organs, in which experimentally implanted tissue grafts are incapable of provoking immunity leading to graft rejection, have since then been referred to as “immune privileged organs” (summarized in (7). CNS immune privilege also extends to innate immune responses as neither injection of bacterial products (8), nor experimental induction of cell death within the CNS parenchyma (9, 10) elicits a rapid infiltration of myelomonocytic cells as observed during the stereotypic innate immune response to such stimuli in peripheral organs (11).
Based on these observations, CNS immune privilege was originally thought to be based on “immune ignorance” where lack of lymphatic vessels and the endothelial blood-brain barrier (BBB) would inhibit the afferent and efferent arm of CNS immunity, respectively [summarized in (12)]. However, the observations that tissue grafts when transplanted into the cerebral ventricles were readily rejected (13, 14) and that foreign tissue grafts transplanted into the brain parenchyma of animals that had previously rejected a skin tissue graft of the same donor were readily destroyed (6) questioned this concept. Observations demonstrating that activated circulating T cells can cross the BBB in the absence of neuroinflammation [summarized in (15)] and that tracers injected into the cerebrospinal fluid (CSF) drain into the deep cervical lymph nodes (16) finally provided direct evidence for afferent and efferent connections of the CNS with the immune system and asked for revisiting the concept of CNS immune privilege. Recent advancements in the establishment of reporter mouse models combined with epifluorescence, near-infrared (NIR), and two-photon (2P) intravital microscopy (IVM) have led to the rediscovery of lymphatic vessels within the dura mater and their contribution to CSF drainage into the deep cervical lymph nodes and the proposal of a “glymphatic system” ensuring efficient mixing of CNS interstitial fluid (ISF) with the CSF (summarized by (17). These observations have led to questioning the existence of CNS immune privilege.
We have proposed that CNS immune privilege does exist but requires proper consideration of the special anatomy of the CNS and especially of the localization and function of the different brain barriers, which divide the CNS into compartments that differ with respect to their accessibility to mediators and cells of the innate and adaptive immune system (12). In this concept, the CNS parenchyma is immune privileged, allowing it to prioritize the proper function of neurons over eliciting an immune response, while the CNS ventricular spaces and border compartments (subarachnoid and perivascular spaces) are dedicated to CNS immunity and thus lack full CNS immune privilege.
The Brain Barriers
Under physiological conditions, the meningeal, endothelial, epithelial, and glial brain barriers maintain CNS homeostasis by protecting the CNS parenchyma from the constantly changing milieu of the bloodstream (Figure).
The Leptomeningeal Blood-Cerebrospinal Fluid (CSF) Barrier
The meningeal layers are the dura mater, the arachnoid mater, and the pia mater and cover the entire surface of the brain, and spinal cord (Figure 1A) and are mainly composed of fibroblasts (18–20). The dura mater is the outermost layer and is directly attached to the skull. Blood vessels in the dura mater lack a BBB and are thus different from those of the CNS proper (12). Along the superior and transversal sagittal sinuses, the dura mater also harbors lymphatic vessels suggested to drain antigens and immune cells from the CNS (21–23). This would require breaching the arachnoid mater below the dura mater, which establishes a bona fide blood– cerebrospinal fluid barrier (BCSFB) between the dura mater and the CSF filled subarachnoid space (SAS). The arachnoid fibroblasts are connected by tight junctions (24–28) prohibiting free diffusion of solutes and water-soluble molecules across this barrier and also express efflux pumps ensuring transport of toxic metabolites out of the CSF (29).
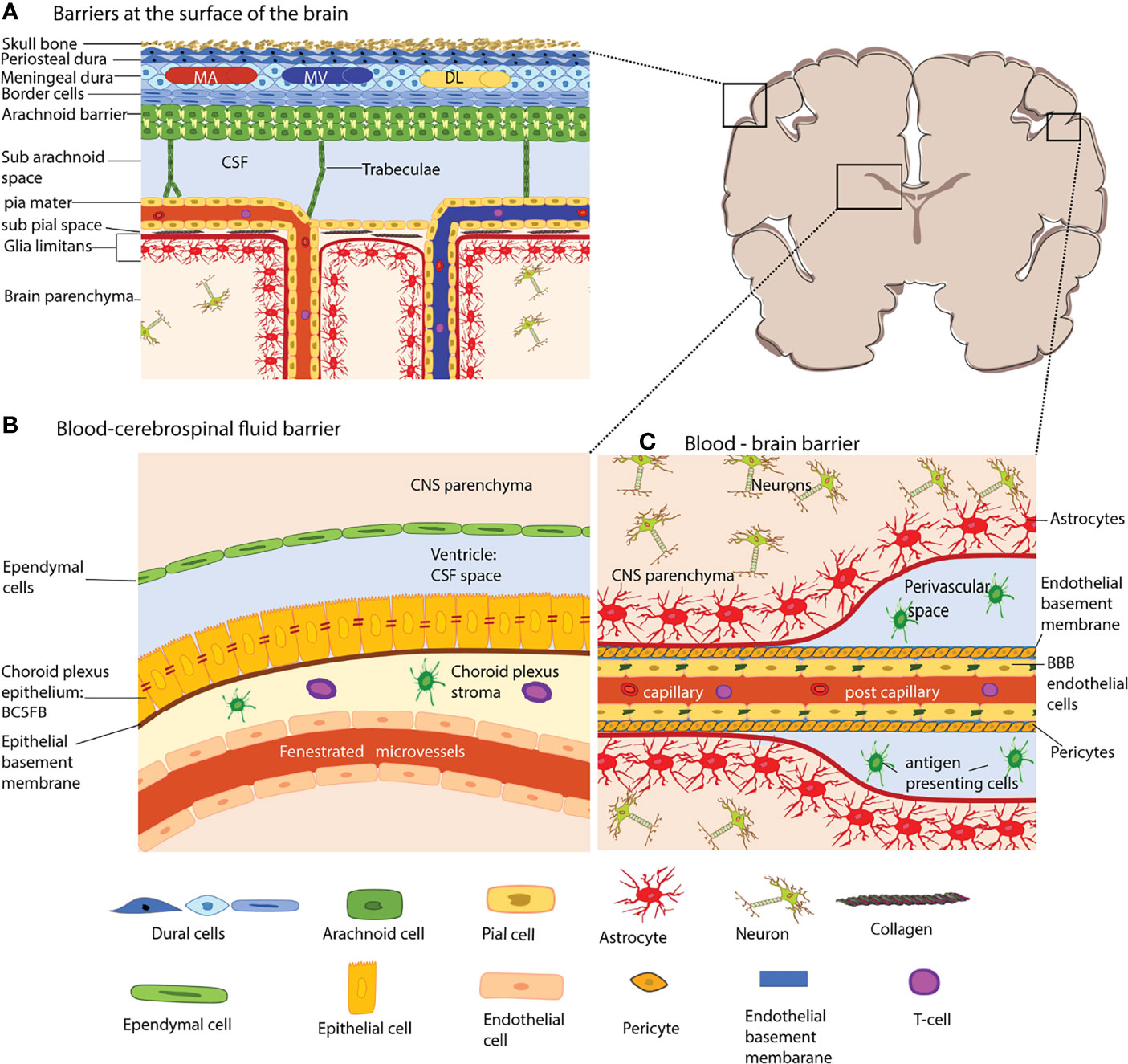
Figure 1 The brain barriers. The schematic coronal brain section depicts the localization of the different brain barriers shown in (A–C). (A) Barriers at the surface of the human brain. The meninges are composed of three layers, the dura mater, the arachnoid barrier, and the pia mater. The dura mater is directly connected to the skull bone. In humans, the dura mater is composed of three layers, the periosteal dura, the meningeal dura and the dural border cells. The dura mater has its own network of arteries (MA), veins (MV) and dural lymphatics (DL). The arachnoid barrier is formed by arachnoid fibroblasts which are connected by tight junctions and form a bona fide blood-cerebrospinal fluid barrier (BCSFB) – the arachnoid barrier – between the dura mater and the CSF filled subarachnoid space. Arachnoid trabeculae formed by a collagen core that is ensheathed by arachnoid and pial fibroblasts cross the SAS towards the pia mater and to the leptomeningeal blood vessels. The fibroblasts of the pia mater cover the veins and arteries in the SAS and separate the SAS from the subpial space filled with collagen bundles. The pia mater reflects of the surface where arteries dive into the brain parenchyma and at the same time ensheathes the arteries entering the brain. The glia limitans forms a barrier at all surfaces of the CNS parenchyma, this is the outer surface (glia limitians superficialis) and the perivascular surfaces (glia limitans perivascularis). (B) The blood–CSF barrier of the choroid plexus (ChP). The ChPs are localized in all four ventricles of the brain. The ChP epithelial cells are connected by unique parallel running tight junction stands and establish a BCSFB. The ChP stroma harbors dendritic cells and macrophages and the blood vessels of the ChP are fenestrated. (C) The blood–brain barrier (BBB) is formed by highly specialized microvascular endothelial cells connected by complex tight junctions. The endothelial basement membrane harbors a high number of pericytes. At the level of capillaries the endothelial basement membrane and the parenchymal basement membrane of the glia limitans merge. However that the post-capillary venule level they leave a small gap where single antigen-presenting cells can be found. The microvessels are surrounded by the glia limitans, which is composed of the parenchymal basement membrane and astrocyte end-feet. The extravasation of immune cells into the CNS parenchyma occurs at the level of postcapillary venules and thus involves crossing two barriers, the endothelial BBB and after reaching the perivascular space subsequent crossing of the glia limitans. The shapes of the cell types were adapted from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License.
The arachnoid trabeculae are mainly composed of collagen fibers and fibroblasts that add rigidity to the arachnoid barrier allowing to form a prominent SAS (30). The pia mater is formed by a single layer of flattened fibroblasts covering the surface of the brain and the spinal cord. The cells of the pia mater do not form tight junctions, thus making them permeable to solutes while however still limiting the passage of cellular elements like erythrocytes (31). Additionally, the pia mater sheathes all blood vessels in the SAS and does separate the SAS from the perivascular spaces by reflecting off the surface of the brain (31–33).
The Glia Limitans
The glia limitans envelops the brain and spinal cord parenchyma’s entire surface and the perivascular spaces. The glia limitans is composed of a parenchymal basement membrane produced by astrocytes and by astrocyte endfeet (12). In the healthy CNS, the polarized expression of the water channel aquaporin 4 (AQP4) in astrocyte endfeet regulates water transport at this barrier. In addition, astrocyte endfeet are joined together by gap junctions that allow for communication between the astrocytes (34). In the healthy CNS, the glia limitans provides a barrier for immune cells scanning the subarachnoid and perivascular spaces and prohibits their uncontrolled entry into the CNS parenchyma (35, 36).
The Endothelial Blood-Brain Barrier (BBB)
The endothelial BBB forms a barrier between the blood and the CNS. It is established by brain microvascular endothelial cells that are joined together by continuous and complex tight junctions which inhibit free paracellular diffusion of solutes and water-soluble molecules (37, 38) (Figure 1C). Combined with the low vesicular activity of BBB endothelial cells that prohibits uncontrolled transcellular diffusion, the BBB establishes a physical barrier for solutes and water-soluble molecules. Expression of specific enzymes, transporters, and efflux pumps make BBB endothelial cells biochemically unique and ensure the transport of nutrients into the CNS and toxic metabolites out of the CNS (38). BBB tight junctions are composed of the transmembrane proteins claudin-5, occludin, and members of the junctional adhesion molecules (JAM). While claudin-5 establishes a diffusion barrier for small molecules (39), occludin regulates calcium movement across the BBB and in addition to TJ stability and barrier function (40). JAM-A, JAM-B and JAM-C have been described in the brain microvascular endothelial cells and have been suggested to play a role in regulating the BBB stability by some but not others (41, 42). Members of the JAM family may however play a role in immune cell migration across the BBB (43, 44). Prerequisites for TJ formation are adherens junctions (AJs), and thus, in addition to their unique TJs, BBB endothelial cells display regular endothelial AJs [summarized in (37)]. VE-cadherin is the main transmembrane protein of the endothelial AJs and keeps neighboring cells attached by homophilic interactions (45). Additional transmembrane proteins localized to BBB cell-to-cell junctions are the platelet endothelial cell adhesion molecule-1 (PECAM-1) that contributes to vascular integrity (46) and CD99 which mediates leukocyte trafficking across the BBB (47).
The unique BBB phenotype in CNS microvascular endothelial cells is not intrinsic to the endothelial cells but relies on the continuous cross-talk with cellular and acellular elements surrounding the CNS microvascular endothelium forming the neurovascular unit (NVU). BBB endothelial cells produce the endothelial basement membrane composed of type IV collagen, α4 and α5 laminins (36). Additionally, a high number of pericytes is embedded in the endothelial basement membrane at the level of capillaries and possibly post-capillary venules, while smooth muscle cells form the mural cell population in arterioles and possibly venules (48). The CNS blood vessels are always separated from the CNS parenchyma proper by the glia limitans. The parenchymal basement membrane, which is secreted by the astrocytes, is with the expression of a1 and a2 laminin molecularly distinct from the endothelial basement membrane (36, 49, 50). At the capillary level, the parenchymal basement membrane fuses with the endothelial basement membrane bringing the astrocyte endfeet in close proximity to capillary pericytes and endothelial cells. At the level of the post-capillary venules, the two basement membranes detach to form a small perivascular space (51).
The Choroid Plexus and the Blood-Cerebrospinal Fluid Barrier (BCSFB)
The choroid plexus (ChP) extends into all four brain ventricles and is surrounded by epithelial cells that form a blood-cerebrospinal fluid barrier (BCSFB) (52) (Figure 1B). The ChP produces CSF and ChP epithelial cells are characterized by the expression of a particular combination of transporters (53). Paracellular diffusion across the ChP BCSFB is prohibited by unique tight junctions composed of claudin-1, -2, -3, and -11, occludin, and JAM-A and the scaffolding proteins ZO-1, -2, -3 (54). The capillaries in the choroid plexus stroma are fenestrated and thus allowing for free diffusion of blood-borne molecules into the ChP stroma. The ChP stroma harbors numerous cells of the innate but also the adaptive immune system (55). Furthermore, on the apical side of the ChP epithelial cells, epiplexus or Kolmer cells perform immune surveillance.
The Role of the Individual Brain Barriers in Regulating Immune Cell Entry Into the CNS
Immune Cell Trafficking Across the Endothelial Blood-Brain Barrier (BBB)
CNS immune surveillance has been shown to be ensured by peripherally activated circulating T cells that have the specific ability to cross the BBB to reach perivascular or subarachnoid spaces in the absence of neuroinflammation (12, 56). It should be noted that while immune cells trafficking occurs at the level of CNS post-capillary venules, transport of nutrients occurs at the level of CNS capillaries (57). This allows immune cells to reach perivascular or subarachnoid space, where they can encounter tissue resident antigen-presenting cells (APCs), like border associated macrophages (BAMs). Recognition of their cognate antigen on these CNS border associated APCs leads to local reactivation of T cells and is the prerequisite for subsequent T-cell migration across the glia limitans into the CNS parenchyma (58, 59).
Leucocyte extravasation is usually a multi-step process where after an initial tether or capture on the endothelium, selectins and their ligands allow immune cells to roll along the endothelium, reducing their speed and next recognize with their G-protein coupled receptors (GPCRs) chemotactic cues on the endothelium leading to their subsequent integrin-mediated arrest and crawling and finally their diapedesis across endothelial barrier (60). The unique barrier characteristics of the BBB extend to its characteristic immune quiescent phenotype. In contrast to peripheral endothelial cells, BBB endothelial cells lack storage of P-selectin protein in their endothelial Weibel Palade bodies [summarized in (61)] and constitutive expression of the atypical chemokine receptor 1 (ACKR1), which transports chemokines from the abluminal to the luminal surface of endothelial cells (62, 63). Thus, immune cell entry into the CNS is very low and limited to activated T cells that do not depend on these trafficking cues. Indeed, activated CD4 T cells were shown to be able to capture via α4-integrins on CNS endothelial VCAM-1 (64) and following LFA-1 dependent adhesive interactions to cross the BBB in the absence of neuroinflammation (65–67) (Figure 2).
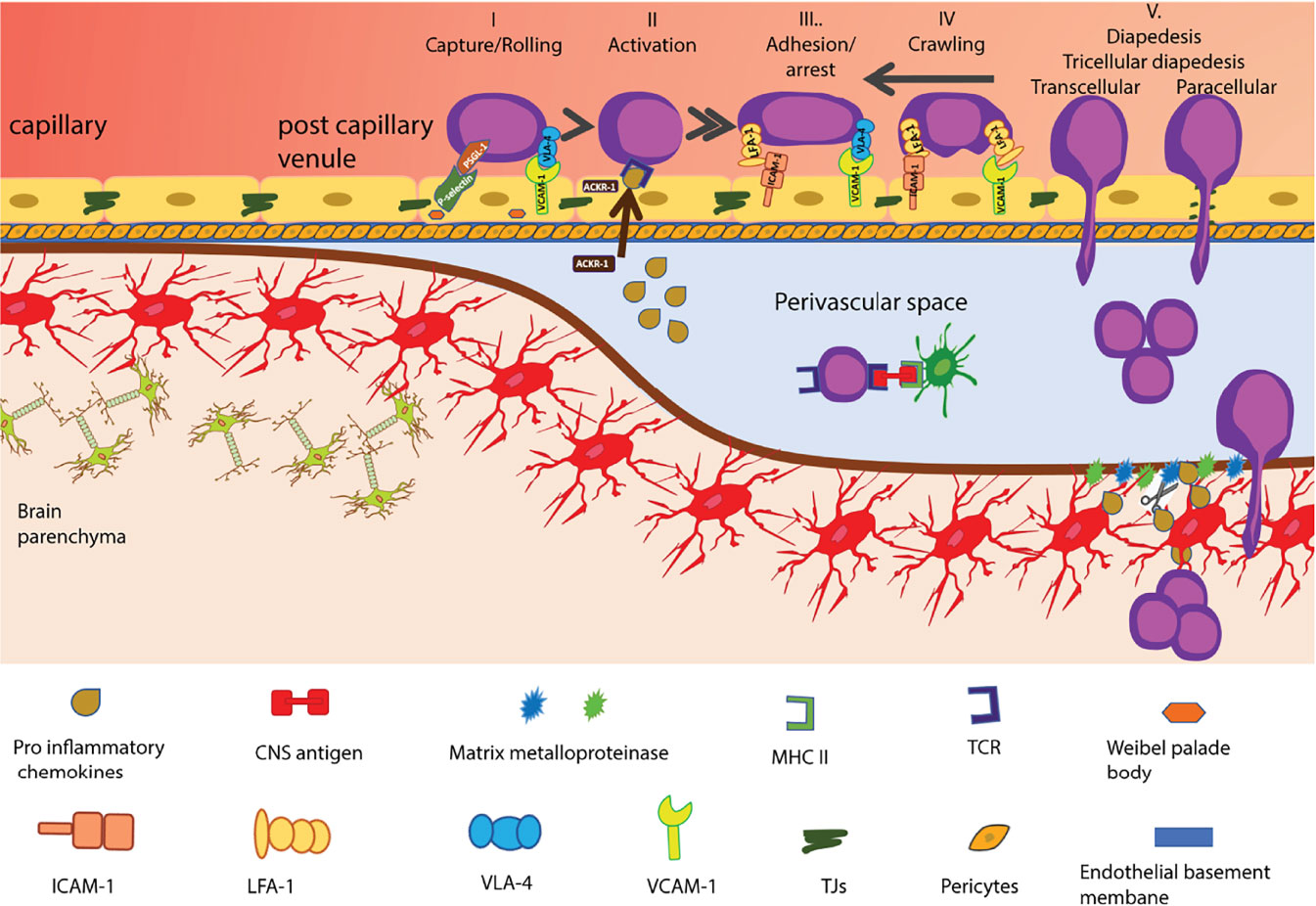
Figure 2 Multi-step T-cell extravasation across the BBB during neuroinflammation. Multi-step T cell extravasation across the BBB occurs at the level of CNS post capillary venules. During inflammation, the rolling of activated T-cells on the BBB endothelial cells is mediated by P-selectin and a4-integrins. After their GPCR-dependent arrest, T cells crawl on the BBB endothelium against the direction of blood flow. High levels of endothelial ICAM-1 and de novo expression of ACKR1 that can shuttle CNS chemokines across the BBB promote transcellular diapedesis of T cells while low levels of endothelial ICAM-1 direct T cells mainly to tricellular and bicellular junctions, i.e. paracellular sites of diapedesis. Once T cells have crossed the BBB endothelium they reach the perivascular space. The CNS-antigen-specific T cells may recognize their cognate antigens on perivascular APCs and become reactivated and start to proliferate. The change in local cytokine milieu leads to induction of matrix metalloproteinases -2 and -9 which cleave extracellular matrix receptors on astrocyte endfeet, allowing for T-cell passage across the glia limitans. Once in the CNS parenchyma, T cells induce tissue injury and clinical disease symptoms start to appear. The shapes of the cell types were adapted from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License.
At onset of neuroinflammation, T cells have been shown to cross venules in the subarachnoid space and crawl within the subarachnoid space, where they can also be washed off with the CSF (58).
During neuroinflammation, de novo expression of trafficking molecules like P-selectin and ACKR1 allow for increased immune cell entry into the CNS. The interaction between the P-selectin glycoprotein ligand (PSGL)-1 on T cells and E- and P-selectin on the BBB allows for tethering and rolling of activated CD4 T cells along the luminal side of inflamed spinal cord microvessels (68, 69). Rolling on the BBB allows T cells to interact with chemokines displayed on proteoglycans on the luminal surface of the endothelial cells via their specific GPCRs or possibly on ACKR1, leading to an inside-out-activation of integrins mediating the firm arrest of the immune cells on the luminal surface of the inflamed BBB endothelial cells (61). The interaction between the integrins LFA-1 and very late antigen-4 (VLA-4, α4β1 integrin) on the T cells and their endothelial ligands, ICAM-1 and VCAM-1, respectively mediates the firm adhesion of T cells to the BBB (70, 71). After their arrest, the T cells polarize and were observed to crawl over extended distances against the direction of the bloodstream on endothelial ICAM-1 and ICAM-2, obviously to find rare tricellular junctions as sites permissive for diapedesis across the BBB endothelium (58, 66, 67). Under neuroinflammatory conditions, high cell surface levels of endothelial ICAM-1 and de novo expression of ACKR-1 were shown to reduce T cell crawling distances and increase transcellular T-cell diapedesis across the BBB (62, 66). Importantly, although the BBB junctions become leaky under neuroinflammatory conditions and allow for uncontrolled diffusion of blood-borne molecules across the BBB, this is not accompanied by increased paracellular T cell diapedesis but rather leads to enhanced transcellular T cell diapedesis across the BBB. This underscores that the mechanisms that regulate the junctional integrity of the BBB are distinct from those regulating the cellular pathway of T cell diapedesis across the BBB.
In contrast to CD4 T cells, the molecular mechanisms involved in the multi-step migration of other immune cell subsets across the BBB are less well understood but are distinct from those of CD4 T cells. Although CD8 T cells also rely on α4-integrins to cross the BBB, they show enhanced dependence on LFA-1 to mediate shear resistant arrest and engage in addition endothelial JAM-B (72–76) which is not required for CD4 T cell diapedesis across the BBB (41). Also, although α4-integrins seem to be involved in the migration of most immune cell subsets across the BBB, the precise molecular mechanisms involved in every step of the multi-step extravasation of B cells (77, 78) or innate immune cells such as neutrophils (79), monocytes (75, 80–82) and dendritic cells (83–86) to cross the BBB, are not yet fully understood.
Additionally, several studies have proposed that other molecules such as the activated leukocyte cell adhesion molecule (ALCAM) (80, 87) and the melanoma cell adhesion molecule (MCAM) (88) as well as the nerve injury-induced protein (ninjurin-1) (89) might play a role in the migration of T-cells across the BBB during EAE and MS (76). Future studies to determine the precise role of these molecules in immune cell trafficking across the BBB still need to be done.
Immune Cell Trafficking Across the Glia Limitans
In experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis (MS), clinical symptoms start only upon immune cells crossing the glia limitans and reaching the CNS parenchyma (41, 90). This underscores that immune cell entry into the CNS is fundamentally different from that in peripheral tissues and involves two sequential and differentially regulated steps of crossing an outer brain barrier followed by progression across the glia limitans into the CNS parenchyma proper (91).
Under normal physiological conditions, the glia limitans act as a barrier for migrating immune cells by preventing their entry into the CNS parenchyma (52). During neuroinflammation, when BBB integrity is impaired, reactive astrocytes form tight junctions aiming to prohibit the parenchymal entry of humoral and cellular factors from the bloodstream (92). Nevertheless, it has also been observed that under neuroinflammatory conditions, such as during MS or its animal model EAE, immune cells first form a perivascular cuff around post-capillary venules and then can cross the glia limitans and infiltrate the CNS parenchyma initiating an onset of neurological symptoms (91). This process is mediated by local TNF-induced expression and activation of matrix metalloproteinase (MMP)-2 and MMP-9, which allow for cleavage of a-dystroglycan, an extracellular matrix receptor of astrocyte endfeet, and modulation of chemokines, thus enabling T-cell migration across the perivascular glia limitans into the CNS parenchyma (90, 93). In vivo imaging studies have provided ample evidence that T- cells can cross the walls of the leptomeningeal veins to reach the SAS (94). If this allows for their subsequent migration across the glia limitans on the surface of the brain and spinal cord into the CNS parenchyma is still a matter of debate.
Immune Cell Trafficking Across the Leptomeningeal Arachnoid Barrier
The role of the arachnoid barrier in regulating immune cell entry into the subarachnoid space is not well investigated. A recent study described the downregulation of claudin-11 in arachnoid barrier cells during EAE and MS. In EAE, the authors detected accumulation of T-cell infiltrates specifically in regions of the spinal cord associated with loss of claudin-11 immunostaining of arachnoid barrier fibroblast (95). This establishes a correlation with impairment of arachnoid barrier fibroblast TJs and CNS immune cell infiltration.
Recent studies have furthermore proposed that the dura mater harbors immune cells dedicated for CNS immune surveillance and directly sourced from nearby bone marrow cavities (96–98). Vice versa, it has been suggested that immune cells can readily reach the dural lymphatics from the subarachnoid space (22, 99).
Furthermore, the accumulation of B-cell follicles observed in the subarachnoid space of post mortem brain samples from progressive MS patients has ignited a discussion on the role played by the meninges in MS pathogenesis (100, 101). These meningeal B cell clusters have originally been described in EAE (102) and recent studies have suggested that these B-cells originate from the dura mater (97, 98) and may or may not migrate from the calvaria to the dura mater through specialized vascular channels traversing the inner skull bone. None of these studies has, however, integrated consideration of the arachnoid barrier which establishes a blood-CSF barrier between the dura mater and the subarachnoid space. Thus, it remains to be shown if the arachnoid barrier is a barrier for immune cell passage into the CNS during immunosurveillance and neuroinflammation.
Immune Cell Trafficking via the Choroid Plexus
The ChP has been proposed as an alternative CNS entry site for immune cells reaching the CSF-filled space during immunosurveillance and in neuroinflammation (55). The ChP microvessels do not form a BBB and have a phenotype rather resembling that of peripheral endothelial cells with e.g., constitutive storage of P-selectin in Weibel Palade bodies (103). To reach the CSF, immune cells would need to cross the BCSFB ensheathing the ChP stroma. Adhesion molecules such as ICAM-1 and VCAM-1 are expressed at the luminal surface of ChP epithelial cells, and in vitro studies have provided evidence that T cells can cross the monolayers of ChP epithelial cells from the abluminal to the luminal side with a contribution of epithelial ICAM-1 during the final step of diapedesis and release into the CSF space (104).
CSF from healthy individuals or individuals with non-neuroinflammatory disorders harbors tissue memory CD4+ T helper cells and CD8+ T cells (105, 106). It has been proposed that CSF T cells cross fenestrated capillaries of the ChP in a P-selectin-dependent manner to reach the ChP stroma (103). From there, at least Th17 cells expressing the chemokine receptor CCR6 were suggested to cross the BCSFB expressing the CCR6 ligand CCL20 in a CCR6/CCL20-dependent manner (107). Direct evidence for the migration of T cells from the ChP across the BCSFB in vivo awaits application of recently developed advanced imaging methodologies of the ChP (108). It has also been proposed that rather than crossing the BBB, immune cells exit the ChP stroma at the base of the ChP where it folds out from the ventricular wall. The BCSFB basement membrane was proposed to be in direct continuation with the parenchymal basement membrane of the glia limitans superficialis (53), allowing immune cells to crawl along the basement membranes reaching the SAS of the brain. Future studies on the precise anatomy of the base of the ChP are necessary to explore this potential immune cell entry route into the CNS.
Immune Cell Entry Into the CNS in Autoimmune Disease
MS is considered a prototypic organ-specific autoimmune disease targeting the CNS characterized by inflammatory lesions, brain barriers breakdown, demyelination, and axonal damage. The etiology of MS and its pathogenesis is not fully understood, and environmental and genetic factors have been shown to play a vital role in the development of MS. Many MS-associated genetic variants code for molecules related to the proper function of the immune system is consistent with the concept of MS as a T cell-mediated autoimmune disease of the CNS. Further support for a T-cell mediated autoimmunity in MS is derived from its animal model, experimental autoimmune encephalomyelitis (EAE), where neuro-antigen specific autoreactive CD4 T cells infiltrate the CNS and cause CNS pathology resembling that of MS (109).
Histopathologically, active lesions in early MS are characterized by focal white matter demyelination accompanied by perivenular immune cell infiltrates forming a typical perivascular cuff and consisting mainly of CD8 T cells, CD20 B cells, and plasmablasts as well as macrophages (110, 111). Immune cell trafficking to the CNS is thus central to MS pathogenesis and has been recognized as the therapeutic target for the treatment of MS.
MS Therapies Targeting Immune Cell Trafficking to the CNS
The options for the treatment of relapsing-remitting MS have significantly grown during the last years. These disease-modifying treatments (DMTs) have in common that they target specific actions of the immune system and come with different side effects. Only few DMTs directly target immune cell trafficking to the CNS. Natalizumab is a humanized function blocking monoclonal antibody binding to the α4-integrin subunit of α4β1-(VLA-4) and α4β1−integrin on the immune cell surface. In vivo imaging studies in experimental animals have shown that Natalizumab blocks α4- integrin mediated capture on CNS endothelial VCAM-1 in the absence of neuroinflammation (64) as well as sustained adhesion on inflamed BBB endothelium (112) and thus prohibits T cell migration across the BBB (113). This leads to the reduction of CNS inflammatory lesions with BBB breakdown as well as reduced numbers of CD4 and CD8 T cells detected in the CSF of MS patients (114).
The sphingosine phosphate 1 receptors (S1PR) S1PR1, S1PR3, S1PR4, and S1PR5 are expressed on many cell types including lymphocytes and the BBB and have been shown to be involved in the regulation of many biological processes including lymphocyte trafficking and vascular permeability. Four S1PR modulators, namely fingolimod, siponimod, ozanimod and ponesimod) are currently approved for the treatment of MS (summarized in (115). As S1P signaling is required for the egress of CCR7 expressing lymphocytes from lymph nodes, S1PR modulators trap naïve and central memory cells in lymph nodes while CCR7neg effector memory (Tem) and effector memory recently activated T cells (T EMRA) are not affected (116–118). The resulting lymphopenia and change in composition of circulating lymphocytes is thought to reduce immune cell trafficking into the CNS and is considered the main therapeutic effect of the S1PR modulators in MS. At the same time other effects including a direct effect on the BBB remains to be investigated (113).
In addition to their direct effects on immune cell trafficking glucocorticoids have been described to stabilize adherens and tight junctions of the BBB by upregulating expression of VE-cadherin and occludin and claudin-5 in brain endothelial cells (119). Similarly, interferon-beta has been proposed to restore barrier properties of the BBB which will eventually influence immune cell trafficking into the CNS (120).
Genetic and Environmental Factors Influencing MS in Africa
MS prevalence is increasing worldwide and shows a heterogeneous distribution globally, with the highest prevalence in Europe and North America (121, 122). In Africa, although MS has not been widely studied, epidemiological reports have shown a diverse distribution of the disease with a higher occurrence in North Africa as compared to Sub-Saharan Africa (122, 123). With the unknown etiology of MS and the complex interplay between genetic and environmental factors involved in disease pathogenesis has been proposed to be necessary for MS development. Most epidemiological studies focusing on the impact of genetic and environmental risk factors on MS development were performed in Caucasian populations with a representation of 85-99% of the population, while with a representation of 56% the African population is under represented (121). Very little information is available on the African populations, which have great genotypic and phenotypic variability, but several studies have shown that being a member of the African population is itself a risk factor in developing a severe course of the disease. This was proven throughout many studies based on different parameters of severity evaluation of the disease ranging from disability scores, radiological activity or even atrophy (124–127).
Genetic Factors
Genome-wide association studies (GWAS) identified many single nucleotide polymorphisms (SNPs) in genes coding for molecules regulating functions of the immune system (128), which is consistent with the concept of MS being a T-cell mediated autoimmune disease targeting the CNS. There is, however, an overrepresentation of immune cells in the transcriptional, epigenetic and pathway analysis datasets used in the GWAS studies to interpret the relevance of SNPs to MS susceptibility, which naturally favors identification of MS risk factors associated with immune cells. Inclusion of CNS datasets in GWAS is just emerging (129). which may allow to discover additional risk factors outside of the immune system. To this end in Caucasian populations, the most vital genetic link to MS has been found in MHC haplotypes, especially those containing HLA-DRB1*15.01, HLA DQB1*06.02, and DQA1*01.02 (130). The few studies in black Africans have revealed a diverse distribution in the HLA-DRB1 and -DQB1 loci expression. For instance, a study in Morocco showed a positive association between the HLA-DRB1-15 and the genetic predisposition to MS in a Moroccan population of MS patients (131). This gene has been reported to play a role in immunity, a study showed that in HLA-DR1-15 positive patients, Th1 lymphocytes auto-proliferate in an elevated way and leading to the binding and presentation of CNS antigens to T cells (132). African ethnic groups that have a higher distribution of these alleles are protected against parasitic infections like malaria but are at higher risks of developing autoimmune diseases like MS (133–135). Individuals lacking expression of the atypical chemokine receptor 1 (ACKR1), formally referred to as DARC (Duffy blood group antigen receptor for chemokines) are for example resistant to malaria. ACKR1 mediates inflammatory chemokine shuttling across the BBB and enhances transcellular T-cell diapedesis across the BBB during EAE (62, 63). Lack of ACKR1 ameliorates development of EAE and it remains to be shown if individuals lacking functional ACKR1 are protected from MS. Alternatively, also different ACKR1 haplotypes could affect susceptibility to MS. To this end over 900 ACKR1 haplotypes were identified (136). There is in fact evidence that a strong selective pressure for malaria resistance in the Ethiopian population correlates with the development and maintenance of certain ACKR1 haplotypes (137). A correlation of ACKR1 haplotypes with susceptibility to MS has not yet been investigated.
There is also first studies highlighting polymorphisms in adhesion molecules, e.g. for ICAM4, among African ethnicities (138). It will be interesting to see if polymorphisms in adhesion molecules involved in MS pathogenesis may influence susceptibility to MS in the African population.
Moreover, studies have shown that color tones of the skin influence MS pathogenesis. In two population-based case control studies done in Australia, the researchers assessed the skin phenotype spectrophotometrically by measuring the melanin density of the skin at the upper inner arm and buttock aiming for body sites that are usually not exposed to sunlight. Both studies assessed the association between the skin phenotype and likelihood of developing MS. They concluded that people with a pale skin had a 32.4% increase of developing first demyelinating events. Additionally, low melanin density at the buttock and fair skin were associated with earlier onset of disease. Suggesting that pale-skinned people have a higher risk of developing MS and show earlier MS symptoms as compared to people with black skin (139, 140).
Ultraviolet Radiation and Vitamin D Levels
The prevalence of MS increases directly proportional to an increase in distance from the equator. Several studies have confirmed the association between lower sun exposure with lower Vitamin D levels and the increased risk of developing MS. Considering its geographical location, African countries experience more sun exposure during the year compared to other continents (141). Although there are no studies assessing the impact of sun exposure on MS development in African countries, the observed low prevalence of MS in African countries might be due to increased sun exposure.
There is ample epidemiological evidence implicating lack of Vitamin D as a risk for the development of MS. Vitamin D interacts with its specific receptor expressed by all immune cells that influence the transcription rate of Vitamin D responsive genes resulting in strong immunoregulatory effects (142). In addition, in skin-draining lymph nodes DCs metabolize Vitamin D to imprint trafficking and effector programs in naïve T cells (143) (Figure 3).
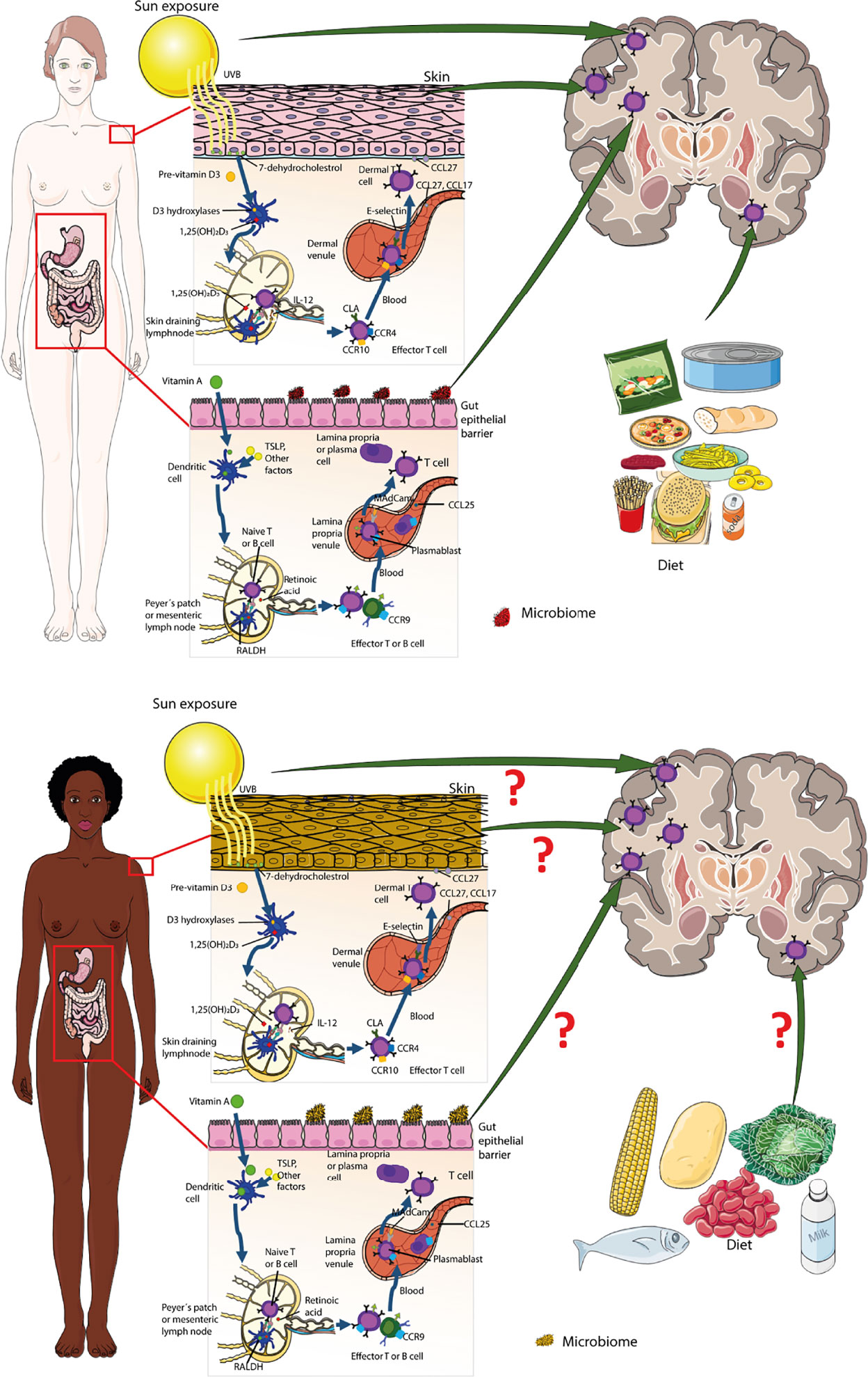
Figure 3 Genetic and environmental factors influencing immune cell entry into the brain. Encounter of microbes takes place at the inner and outer surfaces of the body equipped with special barrier forming epithelia and innate immune cells residing behind these barriers. Priming of T cells in skin and gut-draining lymph nodes imprints their effector function, i.e. expression of trafficking molecules. Pale-skinned people have a higher risk of developing MS as compared to people with black skin. The schematic representation shows imprinting of trafficking properties in T-cells primed in the skin and the gut (adapted from (2), chapter 14). Experimental animal studies have shown that autoagressive T cells primed in skin-draining lymph nodes express CXCR6 and can enter the CNS white and grey matter, while when these T cells are primed in gut-draining lymph odes they express P2rx7 and only infiltrate CNS white matter. How skin color and the gut microbiome of the African population affects T cell priming and their CNS homing properties remains to be shown. The shapes of the cell types were adapted from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License.
Seasonal differences in MS activity have also been reported. The predicted correlation between sun exposure and increased levels of vitamin D would suggest higher disease activity during low sun exposure seasons such as fall and winter (144). However, recent studies have rather suggested the opposite, namely that disease activity increases during spring and summer (145). Studies from Africa have shown that the majority of the African population have low levels of Vitamin D (146–148). These findings contradict that lower prevalence of MS in Africa is correlated to Vitamin D and rather suggests that immunomodulatory effects of Vitamin D and their potential impact on immune cell trafficking need further investigation (149). Furthermore, many of the reported therapeutic essays of vitamin D supplementation for MS patients still do not prove any efficacy on the EDSS score or annual relapses rate (150). Also studies from us in African MS patients supplemented with high dosage Vitamin D did not now show a significant association between Vitamin D levels and MS status (148).
Infections
In genetically MS predisposed individuals, studies have shown that microbial infections can act as environmental triggers in inducing or promoting the onset of clinical signs of MS. This has ignited an active debate as to whether infections prevent or precipitate autoimmune diseases [summarized in (151)]. Furthermore, studies conducted in the developed countries have shown that people who were exposed to a higher level of sanitation during childhood had a higher risk of developing MS in adulthood (152) therefore supporting the hypothesis that infections early in live protect rather than induce or accelerate autoimmune diseases like MS.
Further protective evidence of infections in autoimmune diseases was demonstrated by interventional studies where it was reported that individuals treated with anti-helminth drugs showed an increased MS activity (153).
In most African countries, soil-transmitted helminths (STH) affect primarily the people living in rural areas or urban settings with a lack of clean water and poor sanitation (154). STH is still a considerable burden in children aged 5-14 years in Sub Saharan Africa, although a recent study has shown a vast decline in the prevalence of STH in the last decade (155). Even though there are no studies investigating the correlation between STH infections and the risk of developing MS in Africa, we can speculate that exposure to STH during childhood might contribute to the observed low MS prevalence in Africa. Nevertheless, the question remains if we should consider helminths as beneficial commensals or harmful pathogens. Furthermore, if they are beneficial commensals, will deworming the population with anti-helminth drugs cause an increase in autoimmune disorders?
Moreover, there are some viral infections that have been reported to increase the risk of developing MS later in life. A recent study has suggested a causal role of the Epstein–Barr virus (EBV) in MS, where MS patients seropositive to EBV had high levels of HLA1-B*07+ genes. They suggested that these HLA-class I molecules present antigens to T lymphocytes and initiate immune response against viruses, and thus supporting the potential role of EBV in MS pathology (156). Prior exposure to EBV has been shown to increase the risk of developing MS in both white and black individuals (157).
Furthermore, a recent meta-analysis has shown a strong association between infection with human herpes viruses (HHV) and MS, suggesting that infection with HHS increases the risk of developing MS although the precise mechanisms remain unclear (158). In addition, few studies have shown a MS protective role of prior Cytomegalovirus (CMV) infection (132).
Gut Microbiota
The gut microbiota plays a vital role in maintaining the host’s homeostasis and preventing inflammatory diseases. Diet is considered as the major driving factor in shaping the gut microbiota across the lifetime (159). Mice raised in the germ-free environment are protected from developing clinical EAE (160). These mice developed EAE only when they were exposed to feces from mice that were colonized with gut microbiota, and the subsequent disease was observed to be very mild, suggesting that the gut microbiota participates in the activation of adaptive immune cells (161). This has been further supported by the observation that transplantation of MS twin-derived microbiota to a transgenic mouse model of spontaneous brain autoimmunity induced a significantly higher incidence of disease when compared to transplantation of the healthy twin -derived microbiota (162). These findings provide evidence for pathogenic microbial components in human MS. Considering the geographical and cultural differences between Europe, America, Asia, Australia and Africa, there is a diverse difference in the gut microbiota, which might impact on MS prevalence (51). Studies comparing protective and pathogenic microbial components in MS in different continents will thus be of fundamental importance to understand if the low prevalence of MS in Africa is also due to a specific gut microbiome affecting the priming and trafficking of immune cell subsets.
Lifestyle Risk Factors
Both active and passive tobacco smoking has been highly associated with MS onset with a clear dose-dependent relationship. The prevalence rate of tobacco smoking in Africa is low as compared to the Americas and Eastern Mediterranean. However, it is currently increasing at a very high speed when compared to other parts of the world (163). In 2010, the Lancet survey published that Mozambique has seen a 220% growth in cigarette consumption over the past 16 years (164). The increase in the number of smokers is yet to determine if it will increase the prevalence of MS in Africa in the coming years.
Lately, there has been much discussion regarding the contribution of dietary intake to MS incidence and severity. As we know, the diet has a significant influence on the gut microbiome, leading to altered immune function. High salt diet food has been described to promote CD4 T-cell differentiation to Th17 cells, thus leading to earlier disease onset with severe clinical manifestations (165). Furthermore, a high-fat diet has been associated with the development of obesity which puts an individual at a high risk of developing MS (166). The available statistics show increasing trends of body mass index and obesity in Africa (167) which heralds an increase in the MS incidence in the coming years.
Conclusions
CNS autoimmunity is suggested to be either triggered by molecular mimicry where the adaptive immune response is raised against microbial antigens resembling those of the host or by inflammatory cytokine induced bystander activation, where APCs upregulate, co-stimulatory molecules leading to loss of self-tolerance. The initial activation of these autoaggressive immune cells most likely takes place at outer and inner body surfaces, aka the skin and mucosal surfaces, respectively (Figure 3). This has relevance to their CNS trafficking properties. In an EAE model autoaggressive T cells primed in skin draining lymph nodes were shown to infiltrate in addition to the CNS white matter also CNS grey matter using CXCR6 (168). In contrast, autoaggressive T cells primed in gut draining lymph nodes solely infiltrated CNS white matter. Thus, the site of immune cell priming will have a significant impact on T cell effector functions that may not be adequately described with the current immune cell classifications. How skin color and the gut microbiome of the African population affects T cell priming and their CNS homing properties remains to be shown. There is thus an unmet need to compare the specific characteristics of the barrier associated lymphoid tissues in the African population and their impact on immune cell priming during infections to understand the molecular underpinnings of the lower prevalence of MS in Africa and to prevent a future increase of MS in Africa.
Author Contributions
JM wrote the first draft and compiled all figures. HT and WG wrote part of the document. BE designed the overall layout and edited the entire document. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge funding of our research by the Swiss National Science Foundation grants CRSII3_154483, 310030E_189312 and 310030_189080 and the Fidelity Bermuda Foundation to BE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Alyssa Baert for her immense help in assembling the images.
Abbreviations
aEAE, Active EAE; 2P-IVM, Two-photon intravital microscopy; AJs, Adherens junctions; APC, Antigen presenting cell; AQP4, Aquaporin 4; BBB, Blood-brain barrier; BCSFB, Blood-cerebrospinal-fluid barrier; ChP, Choroid plexus; CLDN, Claudin; CNS, Central nervous system; CSF, Cerebrospinal fluid; E-cadherin, Epithelial cadherin; ICAM-1, Intercellular adhesion molecule 1; JAMs, Junctional adhesion molecules; LFA-1, Lymphocyte function-associated antigen 1; NVU, Neurovascular unit; PECAM-1, Platelet endothelial cell adhesion molecule 1; PSGL, P-selectin glycoprotein ligand; STH, soil-transmitted helminths; TJs, Tight junctions; VCAM-1, Vascular cell adhesion molecule 1; VE-cadherin, Vascular endothelial cadherin; ZO, Zona occludens.
References
1. Flajnik MF, Du Pasquier L. Evolution of Innate and Adaptive Immunity: Can We Draw a Line? Trends Immunol (2004) 25(12):640–4. doi: 10.1016/j.it.2004.10.001
2. Abbas A, Lichtman A, Pillai S. Cellular and Molecular Immunology. 9th ed. Oxford: Elsevier (2018).
3. Mackintosh JA. The Antimicrobial Properties of Melanocytes, Melanosomes and Melanin and the Evolution of Black Skin. J Theor Biol (2001) 211(2):101–13. doi: 10.1006/jtbi.2001.2331
4. Sigmundsdottir H, Butcher EC. Environmental Cues, Dendritic Cells and the Programming of Tissue-Selective Lymphocyte Trafficking. Nat Immunol (2008) 9(9):981–7. doi: 10.1038/ni.f.208
5. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-Resident Memory T Cells in Tissue Homeostasis, Persistent Infection, and Cancer Surveillance. Immunol Rev (2018) 283(1):54–76. doi: 10.1111/imr.12650
6. Medawar PB. Immunity to Homologous Grafted Skin; the Fate of Skin Homografts Transplanted to the Brain, to Subcutaneous Tissue, and to the Anterior Chamber of the Eye. Br J Exp Pathol (1948) 29(1):58–69.
7. Billingham RE, Brent L, Medawar PB. ‘Actively Acquired Tolerance’ of Foreign Cells. Nature (1953) 172(4379):603–6. doi: 10.1038/172603a0
8. Perry VH, Andersson PB. The Inflammatory Response in the CNS. Neuropathol Appl Neurobiol (1992) 18(5):454–9. doi: 10.1111/j.1365-2990.1992.tb00811.x
9. Locatelli G, Wörtge S, Buch T, Ingold B, Frommer F, Sobottka B, et al. Primary Oligodendrocyte Death Does Not Elicit Anti-CNS Immunity. Nat Neurosci (2012) 15(4):543–50. doi: 10.1038/nn.3062
10. Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B. Oligodendrocyte Death Results in Immune-Mediated CNS Demyelination. Nat Neurosci (2016) 19(1):65–74. doi: 10.1038/nn.4193
11. Phillipson M, Kubes P. The Neutrophil in Vascular Inflammation. Nat Med (2011) 17(11):1381–90. doi: 10.1038/nm.2514
12. Engelhardt B, Vajkoczy P, Weller RO. The Movers and Shapers in Immune Privilege of the CNS. Nat Immunol (2017) 18(2):123–31. doi: 10.1038/ni.3666
13. Murphy JB, Sturm E. Conditions Determining the Transplantability Of Tissues In The Brain. J Exp Med (1923) 38(2):183–97. doi: 10.1084/jem.38.2.183
14. Mason DW, Charlton HM, Jones AJ, Lavy CB, Puklavec M, Simmonds SJ. The Fate of Allogeneic and Xenogeneic Neuronal Tissue Transplanted Into the Third Ventricle of Rodents. Neuroscience (1986) 19(3):685–94. doi: 10.1016/0306-4522(86)90292-7
15. Zamvil SS, Steinman L. The T Lymphocyte in Experimental Allergic Encephalomyelitis. Annu Rev Immunol (1990) 8:579–621. doi: 10.1146/annurev.iy.08.040190.003051
16. Bradbury MW, Cole DF. The Role of the Lymphatic System in Drainage of Cerebrospinal Fluid and Aqueous Humour. J Physiol (1980) 299:353–65. doi: 10.1113/jphysiol.1980.sp013129
17. Proulx ST. Cerebrospinal Fluid Outflow: A Review of the Historical and Contemporary Evidence for Arachnoid Villi, Perineural Routes, and Dural Lymphatics. Cell Mol Life Sci (2021) 78(6):2429–57. doi: 10.1007/s00018-020-03706-5
18. Dorrier CE, Jones HE, Pintarić L, Siegenthaler JA, Daneman R. Emerging Roles for CNS Fibroblasts in Health, Injury and Disease. Nat Rev Neurosci (2021) 23:23–34. doi: 10.1038/s41583-021-00525-w
19. Siegenthaler JA, Pleasure SJ. We Have Got You ‘Covered’: How the Meninges Control Brain Development. Curr Opin Genet Dev (2011) 21(3):249–55. doi: 10.1016/j.gde.2010.12.005
20. Dasgupta K, Jeong J. Developmental Biology of the Meninges. J Genet Dev (2019) 57(5):e23288. doi: 10.1002/dvg.23288
21. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A Dural Lymphatic Vascular System That Drains Brain Interstitial Fluid and Macromolecules. J Exp Med (2015) 212(7):991–9. doi: 10.1084/jem.20142290
22. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
23. Da Mesquita S, Fu Z, Kipnis J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron (2018) 100(2):375–88. doi: 10.1016/j.neuron.2018.09.022
24. Nabeshima S, Reese TS, Landis DMD, Brightman MW. Junctions in the Meninges and Marginal Glia. J Comp Neurol (1975) 164(2):127–69. doi: 10.1002/cne.901640202
25. Coles JA, Myburgh E, Brewer JM, McMenamin PG. Where Are We? The Anatomy of the Murine Cortical Meninges Revisited for Intravital Imaging, Immunology, and Clearance of Waste From the Brain. Prog Neurobiol (2017) 156:107–48. doi: 10.1016/j.pneurobio.2017.05.002
26. Saunders N, Habgood M, Møllgård K, Dziegielewska K. The Biological Significance of Brain Barrier Mechanisms: Help or Hindrance in Drug Delivery to the Central Nervous System? [Version 1; Peer Review: 2 Approved]. (2016) 5:F1000 FacultyRev-313. doi: 10.12688/f1000research.7378.1
27. Yasuda K, Cline C, Vogel P, Onciu M, Fatima S, Sorrentino BP, et al. Drug Transporters on Arachnoid Barrier Cells Contribute to the Blood-Cerebrospinal Fluid Barrier. Drug Metab Disposition (2013) 41(4):923–31. doi: 10.1124/dmd.112.050344
28. Figarella-Branger D, Pellissier JF, Bouillot P, Bianco N, Mayan M, Grisoli F, et al. Expression of Neural Cell-Adhesion Molecule Isoforms and Epithelial Cadherin Adhesion Molecules in 47 Human Meningiomas: Correlation With Clinical and Morphological Data. Modern Pathol (1994) 7(7):752–61.
29. Alcolado R, Weller RO, Parrish EP, Garrod D. The Cranial Arachnoid and Pia Mater in Man: Anatomical and Ultrastructural Observations. Neuropathol Appl Neurobiol (1988) 14(1):1–17. doi: 10.1111/j.1365-2990.1988.tb00862.x
30. Saboori P, Sadegh A. Histology and Morphology of the Brain Subarachnoid Trabeculae. Anat Res Int (2015) 2015:279814–. doi: 10.1155/2015/279814
31. Hutchings M, Weller RO. Anatomical Relationships of the Pia Mater to Cerebral Blood Vessels in Man. J Neurosurg (1986) 65(3):316–25. doi: 10.3171/jns.1986.65.3.0316
32. Weller RO. Microscopic Morphology and Histology of the Human Meninges. Morphologie (2005) 89(284):22–34. doi: 10.1016/S1286-0115(05)83235-7
33. Decimo I, Fumagalli G, Berton V, Krampera M, Bifari F. Meninges: From Protective Membrane to Stem Cell Niche. Am J Stem Cells (2012) 1(2):92–105.
34. Nualart-Marti A, Solsona C, Fields RD. Gap Junction Communication in Myelinating Glia. Biochim Biophys Acta (2013) 1828(1):69–78. doi: 10.1016/j.bbamem.2012.01.024
35. Howell OW, Schulz-Trieglaff EK, Carassiti D, Gentleman SM, Nicholas R, Roncaroli F, et al. Extensive Grey Matter Pathology in the Cerebellum in Multiple Sclerosis Is Linked to Inflammation in the Subarachnoid Space. Neuropathol Appl Neurobiol (2015) 41(6):798–813. doi: 10.1111/nan.12199
36. Sorokin L. The Impact of the Extracellular Matrix on Inflammation. Nat Rev Immunol (2010) 10(10):712–23. doi: 10.1038/nri2852
37. Tietz S, Engelhardt B. Brain Barriers: Crosstalk Between Complex Tight Junctions and Adherens Junctions. J Cell Biol (2015) 209(4):493–506. doi: 10.1083/jcb.201412147
38. Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional Morphology of the Blood-Brain Barrier in Health and Disease. Acta Neuropathol (2018) 135(3):311–36. doi: 10.1007/s00401-018-1815-1
39. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-Selective Loosening of the Blood-Brain Barrier in Claudin-5-Deficient Mice. J Cell Biol (2003) 161(3):653–60. doi: 10.1083/jcb.200302070
40. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex Phenotype of Mice Lacking Occludin, a Component of Tight Junction Strands. Mol Biol Cell (2000) 11(12):4131–42. doi: 10.1091/mbc.11.12.4131
41. Tietz S, Périnat T, Greene G, Enzmann G, Deutsch U, Adams R, et al. Lack of Junctional Adhesion Molecule (JAM)-B Ameliorates Experimental Autoimmune Encephalomyelitis. Brain Behav Immun (2018) 73:3–20. doi: 10.1016/j.bbi.2018.06.014
42. Wyss L, Schäfer J, Liebner S, Mittelbronn M, Deutsch U, Enzmann G, et al. Junctional Adhesion Molecule (JAM)-C Deficient C57BL/6 Mice Develop a Severe Hydrocephalus. PLoS One (2012) 7(9):e45619. doi: 10.1371/journal.pone.0045619
43. Cunningham SA, Rodriguez JM, Arrate MP, Tran TM, Brock TA. JAM2 Interacts With Alpha4beta1. Facilitation by JAM3. J Biol Chem (2002) 277(31):27589–92. doi: 10.1074/jbc.C200331200
44. Ludwig RJ, Hardt K, Hatting M, Bistrian R, Diehl S, Radeke HH, et al. Junctional Adhesion Molecule (JAM)-B Supports Lymphocyte Rolling and Adhesion Through Interaction With Alpha4beta1 Integrin. Immunology (2009) 128(2):196–205. doi: 10.1111/j.1365-2567.2009.03100.x
45. Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, Dejana E, et al. VE-Cadherin Is Not Required for the Formation of Nascent Blood Vessels But Acts to Prevent Their Disassembly. Blood (2005) 105(7):2771–6. doi: 10.1182/blood-2004-06-2244
46. Wimmer I, Tietz S, Nishihara H, Deutsch U, Sallusto F, Gosselet F, et al. PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier During Neuroinflammation. Front Immunol (2019) 10:711. doi: 10.3389/fimmu.2019.00711
47. Bixel G, Kloep S, Butz S, Br P, Engelhardt B, Vestweber D. Mouse CD99 Participates in T-Cell Recruitment Into Inflamed Skin. Blood (2004) 104(10):3205–13. doi: 10.1182/blood-2004-03-1184
48. Hartmann D, Underly R, Grant R, Watson A, Lindner V, Shih A. Pericyte Structure and Distribution in the Cerebral Cortex Revealed by High-Resolution Imaging of Transgenic Mice. J Neurophotonics (2015) 4):041402. doi: 10.1117/1.NPh.2.4.041402
49. Pizzo ME, Wolak DJ, Kumar NN, Brunette E, Brunnquell CL, Hannocks M-J, et al. Intrathecal Antibody Distribution in the Rat Brain: Surface Diffusion, Perivascular Transport and Osmotic Enhancement of Delivery. J Physiol (2018) 596(3):445–75. doi: 10.1113/JP275105
50. Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and Function of Laminins in the Embryonic and Mature Vasculature. Physiol Rev (2005) 85(3):979–1000. doi: 10.1152/physrev.00014.2004
51. Hansen MEB, Rubel MA, Bailey AG, Ranciaro A, Thompson SR, Campbell MC, et al. Population Structure of Human Gut Bacteria in a Diverse Cohort From Rural Tanzania and Botswana. Genome Biol (2019) 20(1):16. doi: 10.1186/s13059-018-1616-9
52. Engelhardt B, Sorokin L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin Immunopathol (2009) 31(4):497–511. doi: 10.1007/s00281-009-0177-0
53. Wolburg H, Paulus W. Choroid Plexus: Biology and Pathology. Acta Neuropathol (2010) 119(1):75–88. doi: 10.1007/s00401-009-0627-8
54. Castro Dias M, Mapunda JA, Vladymyrov M, Engelhardt B. Structure and Junctional Complexes of Endothelial, Epithelial and Glial Brain Barriers. Int J Mol Sci (2019) 20(21):5372. doi: 10.3390/ijms20215372
55. Ghersi-Egea J-F, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B. Molecular Anatomy and Functions of the Choroidal Blood-Cerebrospinal Fluid Barrier in Health and Disease. Acta Neuropathol (2018) 135(3):337–61. doi: 10.1007/s00401-018-1807-1
56. Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT, et al. Phenotypic and Functional Analysis of T Cells Homing Into the CSF of Subjects With Inflammatory Diseases of the CNS. J Leukoc Biol (2003) 73(5):584–90. doi: 10.1189/jlb.1202598
57. Ransohoff RM, Engelhardt B. The Anatomical and Cellular Basis of Immune Surveillance in the Central Nervous System. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
58. Bartholomäus I, Kawakami N, Odoardi F, Schläger C, Miljkovic D, Ellwart JW, et al. Effector T Cell Interactions With Meningeal Vascular Structures in Nascent Autoimmune CNS Lesions. Nature (2009) 462(7269):94–8. doi: 10.1038/nature08478
59. Kawakami N, Flügel A. Knocking at the Brain's Door: Intravital Two-Photon Imaging of Autoreactive T Cell Interactions With CNS Structures. Semin Immunopathol (2010) 32(3):275–87. doi: 10.1007/s00281-010-0216-x
60. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
61. Marchetti L, Engelhardt B. Immune Cell Trafficking Across the Blood-Brain Barrier in the Absence and Presence of Neuroinflammation. Vasc Biol (2020) 2(1):H1–H18. doi: 10.1530/VB-19-0033
62. Marchetti L, Francisco D, Soldati S, Haghayegh Jahromi N, Barcos S, Gruber I, et al. ACKR1 Favors Transcellular Over Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier in Neuroinflammation In Vitro. Eur J Immunol (2021) 52(1):161–77. doi: 10.1002/eji.202149238
63. Minten C, Alt C, Gentner M, Frei E, Deutsch U, Lyck R, et al. DARC Shuttles Inflammatory Chemokines Across the Blood-Brain Barrier During Autoimmune Central Nervous System Inflammation. Brain (2014) 137(Pt 5):1454–69. doi: 10.1093/brain/awu045
64. Vajkoczy P, Laschinger M, Engelhardt B. Alpha4-Integrin-VCAM-1 Binding Mediates G Protein-Independent Capture of Encephalitogenic T Cell Blasts to CNS White Matter Microvessels. J Clin Invest (2001) 108(4):557–65. doi: 10.1172/JCI12440
65. Laschinger M, Vajkoczy P, Engelhardt B. Encephalitogenic T cells Use LFA-1 for Transendothelial Migration But Not During Capture and Initial Adhesion Strengthening in Healthy Spinal Cord Microvessels In Vivo. Eur J Immunol (2002) 32: (12):3598–606. doi: 10.1002/1521-4141(200212)32:12<3598::AIDIMMU3598>3.0.CO;2-6
66. Abadier M, Haghayegh Jahromi N, Cardoso Alves L, Boscacci R, Vestweber D, Barnum S, et al. Cell Surface Levels of Endothelial ICAM-1 Influence the Transcellular or Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier. Eur J Immunol (2015) 45(4):1043–58. doi: 10.1002/eji.201445125
67. Castro Dias M, Odriozola Quesada A, Soldati S, Bösch F, Gruber I, Hildbrand T, et al. Brain Endothelial Tricellular Junctions as Novel Sites for T Cell Diapedesis Across the Blood-Brain Barrier. J Cell Sci (2021) 134(8):jcs253880. doi: 10.1242/jcs.253880
68. Sathiyanadan K, Coisne C, Enzmann G, Deutsch U, Engelhardt B. PSGL-1 and E/P-Selectins Are Essential for T-Cell Rolling in Inflamed CNS Microvessels But Dispensable for Initiation of EAE. Eur J Immunol (2014) 44(8):2287–94. doi: 10.1002/eji.201344214
69. Angiari S, Rossi B, Piccio L, Zinselmeyer BH, Budui S, Zenaro E, et al. Regulatory T Cells Suppress the Late Phase of the Immune Response in Lymph Nodes Through P-Selectin Glycoprotein Ligand-1. J Immunol (Baltimore Md 1950) (2013) 191(11):5489–500. doi: 10.4049/jimmunol.1301235
70. Man S, Tucky B, Bagheri N, Li X, Kochar R, Ransohoff RM. Alpha4 Integrin/FN-CS1 Mediated Leukocyte Adhesion to Brain Microvascular Endothelial Cells Under Flow Conditions. J Neuroimmunol (2009) 210(1-2):92–9. doi: 10.1016/j.jneuroim.2009.03.008
71. Laschinger M, Engelhardt B. Interaction of Alpha4-Integrin With VCAM-1 Is Involved in Adhesion of Encephalitogenic T Cell Blasts to Brain Endothelium But Not in Their Transendothelial Migration In Vitro. J Neuroimmunol (2000) 102(1):32–43. doi: 10.1016/S0165-5728(99)00156-3
72. Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T Cells From Patients With Acute Multiple Sclerosis Display Selective Increase of Adhesiveness in Brain Venules: A Critical Role for P-Selectin Glycoprotein Ligand-1. Blood (2003) 101(12):4775–82. doi: 10.1182/blood-2002-10-3309
73. Martin-Blondel G, Pignolet B, Tietz S, Yshii L, Gebauer C, Perinat T, et al. Migration of Encephalitogenic CD8 T Cells Into the Central Nervous System Is Dependent on the α4β1-Integrin. Eur J Immunol (2015) 45(12):3302–12. doi: 10.1002/eji.201545632
74. Rudolph H, Klopstein A, Gruber I, Blatti C, Lyck R, Engelhardt B. Postarrest Stalling Rather Than Crawling Favors CD8(+) Over CD4(+) T-Cell Migration Across the Blood-Brain Barrier Under Flow In Vitro. Eur J Immunol (2016) 46(9):2187–203. doi: 10.1002/eji.201546251
75. Alvarez JI, Kébir H, Cheslow L, Chabarati M, Larochelle C, Prat A. JAML Mediates Monocyte and CD8 T Cell Migration Across the Brain Endothelium. Ann Clin Transl Neurol (2015) 2(11):1032–7. doi: 10.1002/acn3.255
76. Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonnière L, et al. Central Nervous System Recruitment of Effector Memory CD8+ T Lymphocytes During Neuroinflammation Is Dependent on α4 Integrin. Brain (2011) 134(Pt 12):3560–77. doi: 10.1093/brain/awr268
77. Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, et al. Determinants of Human B Cell Migration Across Brain Endothelial Cells. J Immunol (Baltimore Md 1950) (2003) 170(9):4497–505. doi: 10.4049/jimmunol.170.9.4497
78. Michel L, Grasmuck C, Charabati M, Lécuyer MA, Zandee S, Dhaeze T, et al. Activated Leukocyte Cell Adhesion Molecule Regulates B Lymphocyte Migration Across Central Nervous System Barriers. Sci Trans Med (2019) 11(518):eaaw0475. doi: 10.1126/scitranslmed.aaw0475
79. Gorina R, Lyck R, Vestweber D, Engelhardt B. β2 Integrin-Mediated Crawling on Endothelial ICAM-1 and ICAM-2 Is a Prerequisite for Transcellular Neutrophil Diapedesis Across the Inflamed Blood-Brain Barrier. J Immunol (Baltimore Md 1950) (2014) 192(1):324–37. doi: 10.4049/jimmunol.1300858
80. Lyck R, Lécuyer M-A, Abadier M, Wyss CB, Matti C, Rosito M, et al. ALCAM (CD166) Is Involved in Extravasation of Monocytes Rather Than T Cells Across the Blood-Brain Barrier. J Cereb Blood Flow Metab (2017) 37(8):2894–909. doi: 10.1177/0271678X16678639
81. Séguin R, Biernacki K, Rotondo RL, Prat A, Antel JP. Regulation and Functional Effects of Monocyte Migration Across Human Brain-Derived Endothelial Cells. J Neuropathol Exp Neurol (2003) 62(4):412–9. doi: 10.1093/jnen/62.4.412
82. Veenstra M, Williams DW, Calderon TM, Anastos K, Morgello S, Berman JW. Frontline Science: CXCR7 Mediates CD14(+)CD16(+) Monocyte Transmigration Across the Blood Brain Barrier: A Potential Therapeutic Target for NeuroAIDS. J Leukoc Biol (2017) 102(5):1173–85. doi: 10.1189/jlb.3HI0517-167R
83. Jain P, Coisne C, Enzmann G, Rottapel R, Engelhardt B. Alpha4beta1 Integrin Mediates the Recruitment of Immature Dendritic Cells Across the Blood-Brain Barrier During Experimental Autoimmune Encephalomyelitis. J Immunol (Baltimore Md 1950) (2010) 184(12):7196–206. doi: 10.4049/jimmunol.0901404
84. Sagar D, Foss C, El Baz R, Pomper MG, Khan ZK, Jain P. Mechanisms of Dendritic Cell Trafficking Across the Blood-Brain Barrier. J Neuroimmune Pharmacol (2012) 7(1):74–94. doi: 10.1007/s11481-011-9302-7
85. Garnier A, Laffont S, Garnier L, Kaba E, Deutsch U, Engelhardt B, et al. CD49d/CD29-Integrin Controls the Accumulation of Plasmacytoid Dendritic Cells Into the CNS During Neuroinflammation. Eur J Immunol (2019) 49(11):2030–43. doi: 10.1002/eji.201948086
86. Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic Cell Transmigration Through Brain Microvessel Endothelium Is Regulated by MIP-1alpha Chemokine and Matrix Metalloproteinases. J Immunol (Baltimore Md 1950) (2007) 178(1):520–9. doi: 10.4049/jimmunol.178.1.520
87. Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, et al. Activated Leukocyte Cell Adhesion Molecule Promotes Leukocyte Trafficking Into the Central Nervous System. Nat Immunol (2008) 9(2):137–45. doi: 10.1038/ni1551
88. Larochelle C, Cayrol R, Kebir H, Alvarez JI, Lécuyer MA, Ifergan I, et al. Melanoma Cell Adhesion Molecule Identifies Encephalitogenic T Lymphocytes and Promotes Their Recruitment to the Central Nervous System. Brain (2012) 135(Pt 10):2906–24. doi: 10.1093/brain/aws212
89. Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, et al. T Cells Become Licensed in the Lung to Enter the Central Nervous System. Nature (2012) 488(7413):675–9. doi: 10.1038/nature11337
90. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan Is Selectively Cleaved at the Parenchymal Basement Membrane at Sites of Leukocyte Extravasation in Experimental Autoimmune Encephalomyelitis. J Exp Med (2006) 203(4):1007–19. doi: 10.1084/jem.20051342
91. Owens T, Bechmann I, Engelhardt B. Perivascular Spaces and the Two Steps to Neuroinflammation. J Neuropathol Exp Neurol (2008) 67(12):1113–21. doi: 10.1097/NEN.0b013e31818f9ca8
92. Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, et al. Astrocytic Tight Junctions Control Inflammatory CNS Lesion Pathogenesis. J Clin Invest (2017) 127(8):3136–51. doi: 10.1172/JCI91301
93. Song J, Wu C, Korpos E, Zhang X, Agrawal Smriti M, Wang Y, et al. Focal MMP-2 and MMP-9 Activity at the Blood-Brain Barrier Promotes Chemokine-Induced Leukocyte Migration. Cell Rep (2015) 10(7):1040–54. doi: 10.1016/j.celrep.2015.01.037
94. Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-Cell Trafficking Between the Leptomeninges and the Cerebrospinal Fluid. Nature (2016) 530(7590):349–53. doi: 10.1038/nature16939
95. Uchida Y, Sumiya T, Tachikawa M, Yamakawa T, Murata S, Yagi Y, et al. Involvement of Claudin-11 in Disruption of Blood-Brain, -Spinal Cord, and -Arachnoid Barriers in Multiple Sclerosis. Mol Neurobiol (2019) 56(3):2039–56. doi: 10.1007/s12035-018-1207-5
96. Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and Vertebral Bone Marrow Are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science (2021) 373(6553):eabf7844. doi: 10.1126/science.abf7844
97. Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Börsch A-L, et al. Single-Cell Profiling of CNS Border Compartment Leukocytes Reveals That B Cells and Their Progenitors Reside in Non-Diseased Meninges. Nat Neurosci (2021) 24(9):1225–34. doi: 10.1038/s41593-021-00880-y
98. Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of Meningeal B Cells Reveals a Lymphopoietic Niche at the CNS Borders. Science (New York NY) (2021) 373(6553):eabf9277. doi: 10.1126/science.abf9277
99. Rustenhoven J, Drieu A, Mamuladze T, de Lima KA, Dykstra T, Wall M, et al. Functional Characterization of the Dural Sinuses as a Neuroimmune Interface. Cell (2021) 184(4):1000–16.e27. doi: 10.1016/j.cell.2020.12.040
100. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate With Early Onset of Disease and Severe Cortical Pathology. Brain (2007) 130(4):1089–104. doi: 10.1093/brain/awm038
101. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of Neuronal Loss and Meningeal Inflammation in Multiple Sclerosis. Ann Neurol (2010) 68(4):477–93. doi: 10.1002/ana.22230
102. Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral Expression of CXCL13 and BAFF Is Accompanied by Formation of Lymphoid Follicle-Like Structures in the Meninges of Mice With Relapsing Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2004) 148(1-2):11–23. doi: 10.1016/j.jneuroim.2003.10.056
103. Kivisäkk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human Cerebrospinal Fluid Central Memory CD4+ T Cells: Evidence for Trafficking Through Choroid Plexus and Meninges via P-Selectin. PNAS (2003) 100: (14):8389–94. doi: 10.1073/pnas.1433000100
104. Nishihara H, Soldati S, Mossu A, Rosito M, Rudolph H, Muller WA, et al. Human CD4+ T Cell Subsets Differ in Their Abilities to Cross Endothelial and Epithelial Brain Barriers In Vitro. Fluids Barriers CNS (2020) 17(1):3. doi: 10.1186/s12987-019-0165-2
105. Enose-Akahata Y, Azodi S, Smith BR, Billioux BJ, Vellucci A, Ngouth N, et al. Immunophenotypic Characterization of CSF B Cells in Virus-Associated Neuroinflammatory Diseases. PLoS Pathog (2018) 14(4):e1007042. doi: 10.1371/journal.ppat.1007042
106. Hrastelj J, Andrews R, Loveless S, Morgan J, Bishop SM, Bray NJ, et al. CSF-Resident CD4+ T-Cells Display a Distinct Gene Expression Profile With Relevance to Immune Surveillance and Multiple Sclerosis. Brain Commun (2021) 3(3):fcab155. doi: 10.1093/braincomms/fcab155
107. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C Chemokine Receptor 6-Regulated Entry of TH-17 Cells Into the CNS Through the Choroid Plexus Is Required for the Initiation of EAE. Nat Immunol (2009) 10(5):514–23. doi: 10.1038/ni.1716
108. Shipley FB, Dani N, Xu H, Deister C, Cui J, Head JP, et al. Tracking Calcium Dynamics and Immune Surveillance at the Choroid Plexus Blood-Cerebrospinal Fluid Interface. Neuron (2020) 108(4):623–39.e10. doi: 10.1016/j.neuron.2020.08.024
109. Krishnamoorthy G, Wekerle H. EAE: An Immunologist's Magic Eye. Eur J Immunol (2009) 39(8):2031–5. doi: 10.1002/eji.200939568
110. Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The Relation Between Inflammation and Neurodegeneration in Multiple Sclerosis Brains. Brain (2009) 132(Pt 5):1175–89. doi: 10.1093/brain/awp070
111. Lassmann H. Multiple Sclerosis Pathology. Cold Spring Harb Perspect Med (2018) 8(3):a028936. doi: 10.1101/cshperspect.a028936
112. Coisne C, Mao W, Engelhardt B. Cutting Edge: Natalizumab Blocks Adhesion But Not Initial Contact of Human T Cells to the Blood-Brain Barrier In Vivo in an Animal Model of Multiple Sclerosis. J Immunol (Baltimore Md 1950) (2009) 182(10):5909–13. doi: 10.4049/jimmunol.0803418
113. Martin R, Sospedra M, Rosito M, Engelhardt B. Current Multiple Sclerosis Treatments Have Improved Our Understanding of MS Autoimmune Pathogenesis. Eur J Immunol (2016) 46(9):2078–90. doi: 10.1002/eji.201646485
114. Stüve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, et al. Altered CD4+/CD8+ T-Cell Ratios in Cerebrospinal Fluid of Natalizumab-Treated Patients With Multiple Sclerosis. Arch Neurol (2006) 63(10):1383–7. doi: 10.1001/archneur.63.10.1383
115. McGinley MP, Cohen JA. Sphingosine 1-Phosphate Receptor Modulators in Multiple Sclerosis and Other Conditions. Lancet (Lond Engl) (2021) 398(10306):1184–94. doi: 10.1016/S0140-6736(21)00244-0
116. Grebenciucova E, Pruitt A. Infections in Patients Receiving Multiple Sclerosis Disease-Modifying Therapies. Curr Neurol Neurosci Rep (2017) 17(11):88. doi: 10.1007/s11910-017-0800-8
117. Chaudhry BZ, Cohen JA, Conway DS. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics (2017) 14(4):859–73. doi: 10.1007/s13311-017-0565-4
118. Hunter SF, Bowen JD, Reder AT. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs (2016) 30(2):135–47. doi: 10.1007/s40263-015-0297-0
119. Blecharz KG, Drenckhahn D, Förster CY. Glucocorticoids Increase VE-Cadherin Expression and Cause Cytoskeletal Rearrangements in Murine Brain Endothelial cEND Cells. J Cereb Blood Flow Metab (2008) 28(6):1139–49. doi: 10.1038/jcbfm.2008.2
120. Kraus J, Voigt K, Schuller AM, Scholz M, Kim KS, Schilling M, et al. Interferon-Beta Stabilizes Barrier Characteristics of the Blood-Brain Barrier in Four Different Species In Vitro. Multiple Scler (Houndmills Basingstoke Engl) (2008) 14(6):843–52. doi: 10.1177/1352458508088940
121. Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights From the Atlas of MS, Third Edition. Multiple Scler (Houndmills Basingstoke Engl) (2020) 26(14):1816–21. doi: 10.1177/1352458520970841
122. Leray E, Moreau T, Fromont A, Edan G. Epidemiology of Multiple Sclerosis. Rev Neurol (2016) 172(1):3–13. doi: 10.1016/j.neurol.2015.10.006
123. Yamout BI, Assaad W, Tamim H, Mrabet S, Goueider R. Epidemiology and Phenotypes of Multiple Sclerosis in the Middle East North Africa (MENA) Region. Mult Scler J Exp Transl Clin (2020) 6(1):2055217319841881. doi: 10.1177/2055217319841881
124. Aurenção JC, Vasconcelos CC, Thuler LC, Alvarenga RM. Disability and Progression in Afro-Descendant Patients With Multiple Sclerosis. Arquivos Neuro-psiquiatria (2016) 74(10):836–41. doi: 10.1590/0004-282X20160118
125. Seyman E, Jones A, Guenette M, Vosoughi R, Selchen D, Amezcua L, et al. Clinical and MRI Characteristics of Multiple Sclerosis in Patients of Middle Eastern and North African Ancestry Residing in Ontario, Canada. Multiple Scler (Houndmills Basingstoke Engl) (2021) 27(7):1027–36. doi: 10.1177/1352458520948212
126. Jeannin S, Bourg V, Berthier F, Lebrun C. [Phenotypical Aspects and Clinical Course of Multiple Sclerosis in 76 Patients With a North African Ethnic Background Followed at the Nice University Hospital]. Rev Neurol (2007) 163(4):440–7. doi: 10.1016/S0035-3787(07)90419-1
127. Caldito NG, Saidha S, Sotirchos ES, Dewey BE, Cowley NJ, Glaister J, et al. Brain and Retinal Atrophy in African-Americans Versus Caucasian-Americans With Multiple Sclerosis: A Longitudinal Study. Brain (2018) 141(11):3115–29. doi: 10.1093/brain/awy245
128. Axisa PP, Hafler DA. Multiple Sclerosis: Genetics, Biomarkers, Treatments. Curr Opin Neurol (2016) 29(3):345–53. doi: 10.1097/WCO.0000000000000319
129. International Multiple Sclerosis Genetics C. Multiple Sclerosis Genomic Map Implicates Peripheral Immune Cells and Microglia in Susceptibility. Science (New York NY) (2019) 365(6460):eaav7188. doi: 10.1126/science.aav7188
130. Gourraud P-A, Harbo HF, Hauser SL, Baranzini SE. The Genetics of Multiple Sclerosis: An Up-to-Date Review. Immunol Rev (2012) 248(1):87–103. doi: 10.1111/j.1600-065X.2012.01134.x
131. Ouadghiri S, El Alaoui Toussi K, Brick C, Ait Benhaddou EH, Benseffaj N, Benomar A, et al. Genetic Factors and Multiple Sclerosis in the Moroccan Population: A Role for HLA Class II. Pathologie-biologie (2013) 61(6):259–63. doi: 10.1016/j.patbio.2013.05.002
132. Waubant E, Lucas R, Mowry E, Graves J, Olsson T, Alfredsson L, et al. Environmental and Genetic Risk Factors for MS: An Integrated Review. Ann Clin Transl Neurol (2019) 6(9):1905–22. doi: 10.1002/acn3.50862
133. Paximadis M, Mathebula TY, Gentle NL, Vardas E, Colvin M, Gray CM, et al. Human Leukocyte Antigen Class I (A, B, C) and II (DRB1) Diversity in the Black and Caucasian South African Population. Hum Immunol (2012) 73(1):80–92. doi: 10.1016/j.humimm.2011.10.013
134. Lulli P, Mangano VD, Onori A, Batini C, Luoni G, Sirima BS, et al. HLA-DRB1 and -DQB1 Loci in Three West African Ethnic Groups: Genetic Relationship With Sub-Saharan African and European Populations. Hum Immunol (2009) 70(11):903–9. doi: 10.1016/j.humimm.2009.07.025
135. Torcia MG, Santarlasci V, Cosmi L, Clemente A, Maggi L, Mangano VD, et al. Functional Deficit of T Regulatory Cells in Fulani, an Ethnic Group With Low Susceptibility to Plasmodium Falciparum Malaria. PNAS (2008) 105(2):646–51. doi: 10.1073/pnas.0709969105
136. Srivastava K, Fratzscher A-S, Lan B, Flegel WA. Cataloguing Experimentally Confirmed 80.7 Kb-Long ACKR1 Haplotypes From the 1000 Genomes Project Database. BMC Bioinf (2021) 22(1):273. doi: 10.1186/s12859-021-04169-6
137. Yin Q, Srivastava K, Gebremedhin A, Makuria AT, Flegel WA. Long-Range Haplotype Analysis of the Malaria Parasite Receptor Gene ACKR1 in an East-African Population. Hum Genome Variation (2018) 5(1):26. doi: 10.1038/s41439-018-0024-8
138. Yin Q, Srivastava K, Schneider JB, Gebremedhin A, Makuria AT, Flegel WA. Molecular Analysis of the ICAM4 Gene in an Autochthonous East African Population. Transfusion (2019) 59(5):1880–1. doi: 10.1111/trf.15217
139. Lucas RM, Ponsonby AL, Dear K, Valery PC, Pender MP, Taylor BV, et al. Sun Exposure and Vitamin D Are Independent Risk Factors for CNS Demyelination. Neurology (2011) 76(6):540–8. doi: 10.1212/WNL.0b013e31820af93d
140. van der Mei IAF, Ponsonby AL, Dwyer T, Blizzard L, Simmons R, Taylor BV, et al. Past Exposure to Sun, Skin Phenotype, and Risk of Multiple Sclerosis: Case-Control Study. BMJ (2003) 327(7410):316–. doi: 10.1136/bmj.327.7410.316
141. Wright CY, Reddy T, Mathee A, Street RA. Sun Exposure, Sun-Related Symptoms, and Sun Protection Practices in an African Informal Traditional Medicines Market. Int J Environ Res Public Health (2017) 14(10):1142. doi: 10.3390/ijerph14101142
142. Ramagopalan SV, Maugeri NJ, Handunnetthi L, Lincoln MR, Orton S-M, Dyment DA, et al. Expression of the Multiple Sclerosis-Associated MHC Class II Allele HLA-DRB1*1501 Is Regulated by Vitamin D. PLoS Genet (2009) 5(2):e1000369–e. doi: 10.1371/journal.pgen.1000369
143. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs Metabolize Sunlight-Induced Vitamin D3 to 'Program' T Cell Attraction to the Epidermal Chemokine CCL27. Nat Immunol (2007) 8(3):285–93. doi: 10.1038/ni1433
144. Rosecrans R, Dohnal JC. Seasonal Vitamin D Changes and the Impact on Health Risk Assessment. Clin Biochem (2014) 47(7):670–2. doi: 10.1016/j.clinbiochem.2014.02.004
145. Spelman T, Gray O, Trojano M, Petersen T, Izquierdo G, Lugaresi A, et al. Seasonal Variation of Relapse Rate in Multiple Sclerosis Is Latitude Dependent. Ann Neurol (2014) 76(6):880–90. doi: 10.1002/ana.24287
146. O'Connor MY, Thoreson CK, Ramsey NLM, Ricks M, Sumner AE. The Uncertain Significance of Low Vitamin D Levels in African Descent Populations: A Review of the Bone and Cardiometabolic Literature. Prog Cardiovasc Dis (2013) 56(3):261–9. doi: 10.1016/j.pcad.2013.10.015
147. Bouillon R. Vitamin D Status in Africa Is Worse Than in Other Continents. Lancet Global Health (2020) 8(1):20–1. doi: 10.1016/S2214-109X(19)30492-9
148. Skalli A, Ait Ben Haddou EH, El Jaoudi R, Razine R, Mpandzou GA, Tibar H, et al. Association of Vitamin D Status With Multiple Sclerosis in a Case-Control Study From Morocco. Rev Neurol (2018) 174(3):150–6. doi: 10.1016/j.neurol.2017.06.030
149. Bscheider M, Butcher EC. Vitamin D Immunoregulation Through Dendritic Cells. Immunology (2016) 148(3):227–36. doi: 10.1111/imm.12610
150. Feige J, Moser T, Bieler L, Schwenker K, Hauer L, Sellner J. Vitamin D Supplementation in Multiple Sclerosis: A Critical Analysis of Potentials and Threats. Nutrients (2020) 12(3):783. doi: 10.3390/nu12030783
151. Christen U, von Herrath MG. Infections and Autoimmunity–Good or Bad? J Immunol (Baltimore Md 1950) (2005) 174(12):7481–6. doi: 10.4049/jimmunol.174.12.7481
152. Bach J-F. The Effect of Infections on Susceptibility to Autoimmune and Allergic Diseases. N Engl J Med (2002) 347(12):911–20. doi: 10.1056/NEJMra020100
153. Correale J, Farez MF. The Impact of Parasite Infections on the Course of Multiple Sclerosis. J Neuroimmunol (2011) 233(1):6–11. doi: 10.1016/j.jneuroim.2011.01.002
154. Hotez PJ, Bundy DAP, Beegle K, Brooker S, Drake L, de Silva N, et al. Helminth Infections: Soil-Transmitted Helminth Infections and Schistosomiasis. In: Jamison DT, Breman JG, Measham AR, et al, editors. Disease Control Priorities in Developing Countries, vol. 2. New York: Oxford University Press (2006).
155. Sartorius B, Cano J, Simpson H, Tusting LS, Marczak LB, Miller-Petrie MK, et al. Prevalence and Intensity of Soil-Transmitted Helminth Infections of Children in Sub-Saharan Africa, 2000-18: A Geospatial Analysis. Lancet Glob Health (2021) 9(1):e52–60. doi: 10.1016/S2214-109X(20)30398-3
156. Agostini S, Mancuso R, Guerini FR, D'Alfonso S, Agliardi C, Hernis A, et al. HLA Alleles Modulate EBV Viral Load in Multiple Sclerosis. J Trans Med (2018) 16(1):80. doi: 10.1186/s12967-018-1450-6
157. Xiao D, Ye X, Zhang N, Ou M, Guo C, Zhang B, et al. A Meta-Analysis of Interaction Between Epstein-Barr Virus and HLA-DRB1*1501 on Risk of Multiple Sclerosis. Sci Rep (2015) 5:18083. doi: 10.1038/srep18083
158. Pormohammad A, Azimi T, Falah F, Faghihloo E. Relationship of Human Herpes Virus 6 and Multiple Sclerosis: A Systematic Review and Meta-Analysis. J Cell Physiol (2018) 233(4):2850–62. doi: 10.1002/jcp.26000
159. Thursby E, Juge N. Introduction to the Human Gut Microbiota. Biochem J (2017) 474(11):1823–36. doi: 10.1042/BCJ20160510
160. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal Microbiota and Myelin Autoantigen Cooperate to Trigger Autoimmune Demyelination. Nature (2011) 479(7374):538–41. doi: 10.1038/nature10554
161. Mor F, Cohen IR. Beta-Lactam Antibiotics Modulate T-Cell Functions and Gene Expression via Covalent Binding to Cellular Albumin. Proc Natl Acad Sci U S A (2013) 110(8):2981–6. doi: 10.1073/pnas.1215722110
162. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut Microbiota From Multiple Sclerosis Patients Enables Spontaneous Autoimmune Encephalomyelitis in Mice. PNAS (2017) 114(40):10719–24. doi: 10.1073/pnas.1711233114
163. Bertollini R, Ribeiro S, Mauer-Stender K, Galea G. Tobacco Control in Europe: A Policy Review. Eur Respir Rev (2016) 25(140):151–7. doi: 10.1183/16000617.0021-2016
164. Baleta A. Africa's Struggle to be Smoke Free. Lancet (2010) 375(9709):107–8. doi: 10.1016/S0140-6736(10)60032-3
165. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium Chloride Drives Autoimmune Disease by the Induction of Pathogenic TH17 Cells. Nature (2013) 496(7446):518–22. doi: 10.1038/nature11868
166. Hedström AK, Olsson T, Alfredsson L. High Body Mass Index Before Age 20 Is Associated With Increased Risk for Multiple Sclerosis in Both Men and Women. Multiple Scler (Houndmills Basingstoke Engl) (2012) 18(9):1334–6. doi: 10.1177/1352458512436596
167. Group NCDRFCAW. Trends in Obesity and Diabetes Across Africa From 1980 to 2014: An Analysis of Pooled Population-Based Studies. Int J Epidemiol (2017) 46(5):1421–32. doi: 10.1093/ije/dyx078
Keywords: blood-brain barrier, blood-cerebrospinal fluid barrier, immune cell trafficking, arachnoid barrier, multiple sclerosis
Citation: Mapunda JA, Tibar H, Regragui W and Engelhardt B (2022) How Does the Immune System Enter the Brain? Front. Immunol. 13:805657. doi: 10.3389/fimmu.2022.805657
Received: 30 October 2021; Accepted: 31 January 2022;
Published: 22 February 2022.
Edited by:
Willias Masocha, Kuwait University, KuwaitReviewed by:
Jorge Ivan Alvarez, University of Pennsylvania, United StatesHartmut Wekerle, Max Planck Society, Germany
Copyright © 2022 Mapunda, Tibar, Regragui and Engelhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Britta Engelhardt, YmVuZ2VsQHRraS51bmliZS5jaA==