Commentary: Post-Transplantation Cyclophosphamide Uniquely Restrains Alloreactive CD4+ T-Cell Proliferation and Differentiation After Murine MHC-Haploidentical Hematopoietic Cell Transplantation
 Ashley D. Hadjis1
Ashley D. Hadjis1 Natalia S. Nunes1
Natalia S. Nunes1 Shanzay M. Khan1
Shanzay M. Khan1 Rochelle E. Fletcher1
Rochelle E. Fletcher1 Alessandra de Paula Pohl1
Alessandra de Paula Pohl1 David J. Venzon2
David J. Venzon2 Michael A. Eckhaus3
Michael A. Eckhaus3 Christopher G. Kanakry1*
Christopher G. Kanakry1*- 1Experimental Transplantation and Immunotherapy Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 2Biostatistics and Data Management Section, Office of the Clinical Director, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 3Division of Veterinary Resources, Office of Research Services, National Institutes of Health, Bethesda, MD, United States
Post-transplantation cyclophosphamide (PTCy) reduces the incidence and severity of graft-versus-host disease (GVHD), thereby improving the safety and accessibility of allogeneic hematopoietic cell transplantation (HCT). We have shown that PTCy works by inducing functional impairment and suppression of alloreactive T cells. We also have identified that reduced proliferation of alloreactive CD4+ T cells at day +7 and preferential recovery of CD4+CD25+Foxp3+ regulatory T cells (Tregs) at day +21 are potential biomarkers associated with optimal PTCy dosing and timing in our B6C3F1→B6D2F1 MHC-haploidentical murine HCT model. To understand whether the effects of PTCy are unique and also to understand better the biology of GVHD prevention by PTCy, here we tested the relative impact of cyclophosphamide compared with five other optimally dosed chemotherapeutics (methotrexate, bendamustine, paclitaxel, vincristine, and cytarabine) that vary in mechanisms of action and drug resistance. Only cyclophosphamide, methotrexate, and cytarabine were effective in preventing fatal GVHD, but cyclophosphamide was superior in ameliorating both clinical and histopathological GVHD. Flow cytometric analyses of blood and spleens revealed that these three chemotherapeutics were distinct in constraining conventional T-cell numerical recovery and facilitating preferential Treg recovery at day +21. However, cyclophosphamide was unique in consistently reducing proliferation and expression of the activation marker CD25 by alloreactive CD4+Foxp3- conventional T cells at day +7. Furthermore, cyclophosphamide restrained the differentiation of alloreactive CD4+Foxp3- conventional T cells at both days +7 and +21, whereas methotrexate and cytarabine only restrained differentiation at day +7. No chemotherapeutic selectively eliminated alloreactive T cells. These data suggest that constrained alloreactive CD4+Foxp3- conventional T-cell numerical recovery and associated preferential CD4+CD25+Foxp3+ Treg reconstitution at day +21 may be potential biomarkers of effective GVHD prevention. Additionally, these results reveal that PTCy uniquely restrains alloreactive CD4+Foxp3- conventional T-cell proliferation and differentiation, which may explain the superior effects of PTCy in preventing GVHD. Further study is needed to determine whether these findings also hold true in clinical HCT.
Introduction
Allogeneic hematopoietic cell transplantation (HCT) is the only potentially curative therapy for many life-threatening hematologic diseases, but historically was not accessible to many patients for lack of a suitable human leukocyte antigen (HLA)-matched donor. HLA-haploidentical donors are available for nearly all patients, but early results of HLA-haploidentical HCT showed unacceptably high rates of graft failure, graft-versus-host disease (GVHD), and transplant-related mortality due to strong bi-directional alloreactivity (1). The administration of the chemotherapeutic cyclophosphamide on days +3 and +4 post-transplant (post-transplantation cyclophosphamide, PTCy) can greatly reduce the incidence and severity of acute and chronic graft-versus-host disease (GVHD) after HLA-haploidentical or HLA-partially mismatched unrelated donor HCT and consequently has been widely adopted (2). Despite very encouraging clinical outcomes, how PTCy works to prevent GVHD has not been well understood.
A better understanding of the immunological mechanisms by which PTCy works to prevent GVHD may allow for rational modifications of this platform in attempts to improve outcomes for patients. In murine HCT models, we have shown that PTCy works by inducing alloreactive T-cell functional impairment and subsequent suppression by CD4+Foxp3+ regulatory T cells (Tregs) (3–5). We also have shown in our B6C3F1→B6D2F1 MHC-haploidentical murine HCT model that optimal dosing and timing of PTCy are associated with reduced proliferation of alloreactive CD4+Foxp3- conventional T cells at day +7 and preferential recovery of CD4+CD25+Foxp3+ Tregs at day +21 (5, 6). We have proposed that together these T-cell endpoints may be potential biomarkers of effective GVHD control by PTCy (6).
The relative survival and recovery of T-cell subsets after optimally timed and dosed PTCy may be due to differential and dynamic expression of important drug resistance pathways, including aldehyde dehydrogenase (ALDH) and ATP-binding cassette (ABC) transporters (3, 4, 7). Human and mouse Tregs upregulate expression of ALDH, the main in vivo detoxifying pathway for cyclophosphamide (8), after alloantigen stimulation, contributing to resistance to cyclophosphamide-induced cell death in this specific context (3, 4, 9). These pathways also contribute to human CD8+ T-cell survival and recovery after cyclophosphamide (7). ALDH and ABC transporters are not only widely expressed throughout the hematopoietic system (10, 11), but also confer differential degrees of resistance to virtually all classes of chemotherapeutics (12–14). Each chemotherapeutic also may have additional specific mechanisms of resistance.
It is unclear whether the effects of cyclophosphamide (CY) given in the early post-transplant period are unique. In 1971, survival after murine MHC-haploidentical HCT was compared after treatment with cyclophosphamide, methotrexate, mercaptopurine, chlormethine, or cortisol, each administered on days +5, +8, +11, and +14 (15). Cyclophosphamide was the only drug found to be effective, while all other drugs had minimal impact (15). However, since 1971, not only are there more chemotherapeutics available, but we also have identified that PTCy is maximally effective in HCT when given between days +3 to +5 (6). Additionally, the relative effects of PTCy compared with other chemotherapeutics on T-cell subsets have not been examined. The only exception has been recent interest in post-transplantation bendamustine (BEN), which has been shown in pre-clinical studies to produce engraftment and GVHD prevention results similar to PTCy, while maintaining the graft-versus-leukemia effect (16–18); yet, phase I/II trials thus far have shown mixed results (17, 18).
To assess whether the biological effects of PTCy may be unique and provide further insights into our mechanistic understanding of the immunological mechanisms by which PTCy prevents GVHD, we investigated in our murine MHC-haploidentical HCT model (5) the relative efficacy of five other chemotherapeutics (methotrexate, bendamustine, paclitaxel, vincristine, and cytarabine). We specifically chose these drugs as they represent an array of mechanisms of action, metabolism, and drug resistance (Table 1) and include methotrexate, which has a long history of clinical use for GVHD prophylaxis, and bendamustine, which has recently been explored as an alternative to PTCy; topoisomerase inhibitors and other alkylators beyond bendamustine were intentionally excluded over theoretical concern for therapy-related myeloid neoplasms in any clinical application of these studies.
TABLE 1
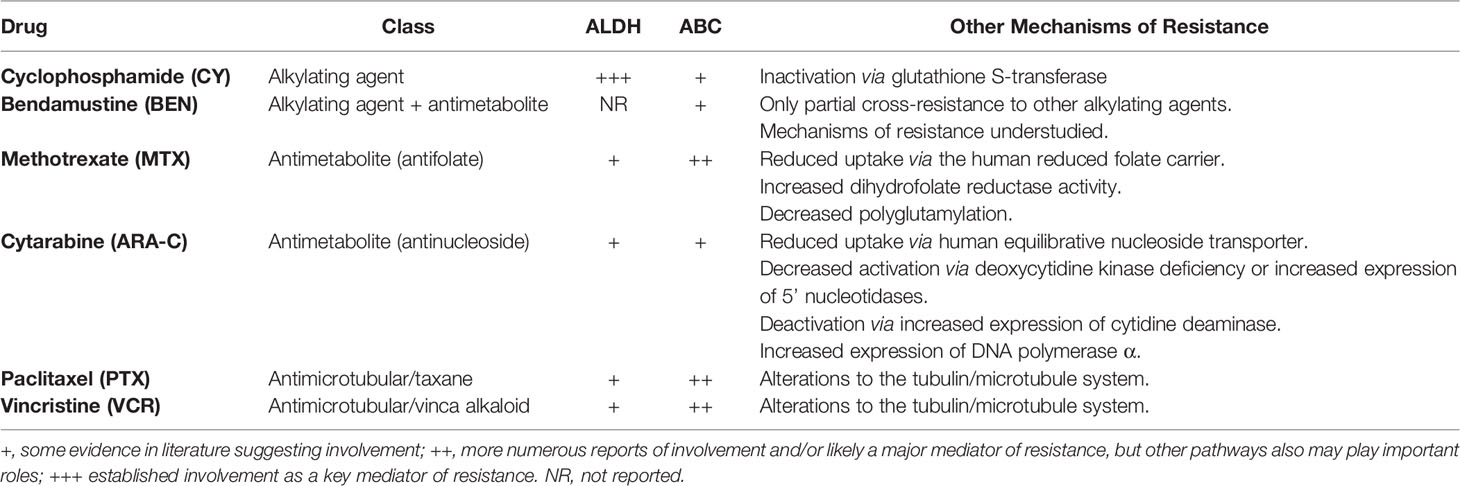
Table 1 Putative pathways of resistance to chemotherapeutics tested in this study as assessed by available literature (8, 12–14, 19–44).
Materials and Methods
Mice
B6C3F1/Crl (donor) and B6D2F1/Crl (recipient) female mice, 10-12 weeks old at the time of transplant, were obtained from the Charles River Laboratories. Mice were housed in specific-pathogen-free conditions at the NCI and were provided food and water ad libitum.
HCT
Spleens, tibias, and femurs were aseptically collected from donor B6C3F1 mice and processed as previously described, including red blood cell lysis of splenocytes and T-cell depletion of bone marrow (5). Recipient B6D2F1 mice were irradiated to 10.5 Gy in a single fraction and 6-8 hours later received 10x106 B6C3F1 T-cell-depleted bone marrow +/- 40x106 red blood cell-depleted B6C3F1 splenocytes via tail vein injection. Recipient mice received levofloxacin-treated water from days 0 to +14. Survival was followed daily, and blinded assessments of weights and clinical scores using a standardized rubric (5) were performed every three days. Tissue specimen preparation and blinded histopathologic assessments were performed as previously described (5).
Drug Preparation
On the day of administration, methotrexate (MTX, Intas Pharmaceuticals), paclitaxel (PTX, Athenex), vincristine (VCR, Hospira), and cytarabine (ARA-C, Hospira) were diluted with sterile PBS to appropriate concentrations, while bendamustine (BEN, TEVA Pharmaceutical Industries) was reconstituted using sterile water to 5 mg/ml per the manufacturer’s instructions and then further diluted to the appropriate concentrations with sterile PBS. Cyclophosphamide (CY, Baxter Oncology) was prepared as previously described (5) and diluted to a 1 mg/ml concentration with sterile PBS on the day of administration for injection. All drugs were diluted to concentrations allowing for administration of 300-500 µl per mouse and administered via intraperitoneal injection. Doses were based on the weight on the day of injection. Vehicle-treated mice received similar volumes of sterile PBS intraperitoneally.
Flow Cytometry
Blood and spleens were collected and processed as previously described (5). Viable cell counts were performed with dual-fluorescent imaging with a Cellometer Auto 2000 Cell Viability Counter (Nexcelom). 2x106 viable cells/sample were stained sequentially with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher), extracellular antibodies, fixation/permeabilization (eBioscience Foxp3/Transcription Factor Staining Kit), and intracellular antibodies. Single stains were used to generate compensations, and fluorescence-minus-one controls were prepared for CD25, Ki-67, and phospho-STAT5. Data were acquired on an LSRFortessa (BD Biosciences) and analyzed using FCS Express (De Novo Software). Cell subsets had to be at least 50 cells to allow for reliable further subsetting; subsets with denominators less than this threshold were excluded from further subsetting analyses.
Fluorochrome-conjugated monoclonal antibodies used for flow cytometry included BUV395 anti-CD3 (clone 145-2C11), BV786 anti-CD8a (clone 53-6.7), PE-CF594 anti-CD25 (clone PC61), AF700 anti-CD44 (clone IM7), BUV737 anti-CD62L (clone MEL-14), PE anti-H2kk (clone 36-7-5), and BV711 anti-H2kk (clone AF3-12.1) from BD Biosciences; PE-Cy5 anti-CD8 (clone 53-6.7), PE-Cy7 anti-H2kd (clone SF1-1.1), and BV605 anti-Ki67 (clone 16A8) from BioLegend; and APC-eFluor780 anti-CD4 (clone GK1.5), PerCP-eFluor710 anti-Vβ6 (clone RR4-7), eFluor450 anti-Foxp3 (clone FJK-16s), and PE anti-phospho-STAT5 (Tyr694) (clone SRBCZX) from Invitrogen.
Statistics
Survival distributions were compared using the exact log-rank test. Weight and clinical score area-under-the-curve (AUC) comparisons were performed using Wilcoxon’s rank sum test and were restricted to intervals in which ≥70% of vehicle-treated mice survived. Weight and clinical score data are shown as the mean +/- SEM. Due to strong serial correlations, weight and clinical score SEMs were not corrected for multiple measurements. Cell counts and median fluorescence intensities were natural logarithmically transformed and cell subset percentages were transformed using an arcsine transformation prior to one-way ANOVA. ANOVA results were followed with the Holm-Sidak post hoc correction for the multiple comparisons to the control group. Non-transformed data are displayed and are shown as box-and-whisker plots for ease of understanding, but transformed data were used for statistical testing. SAS/STAT software (SAS Institute Inc.), version 14.3, was used for analyses of survival, weight, and clinical score data. GraphPad Prism (GraphPad Software), version 8.4.3 was used for all other statistical analysis. P values <0.05 were considered statistically significant in an exploratory mode of analysis of repeated measurements of correlated immunologic outcomes.
Study Approval
Mice were treated under a protocol approved by the NCI Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Results
MTX 5 mg/kg/day, BEN 10 mg/kg/day, PTX 1 mg/kg/day, VCR 0.05 mg/kg/day, and ARA-C 25 mg/kg/day Are Optimal Doses of Each Drug When Given on Days +3 and +4 in the B6C3F1→B6D2F1 MHC-Haploidentical HCT Model
We have previously shown that PTCy is maximally effective in our B6C3F1→B6D2F1 MHC-haploidentical HCT model when given between days +3 to +5 with dosing on days +3 and +4 being among the best dosing schedules and also is what is used clinically (2, 6). Therefore, we sought to identify the best dose of each alternative chemotherapeutic (MTX, BEN, PTX, VCR, ARA-C) when given on days +3 and +4 in this HCT model; we explored a wide range of doses, spanning what were thought to be below effective doses to near-lethal ranges based on prior studies (15, 16, 19, 45–56). MTX 5 mg/kg/day, BEN 10 mg/kg/day, PTX 1 mg/kg/day, VCR 0.05 mg/kg/day, and ARA-C 25 mg/kg/day were determined as best for each drug, although some had minimal to no efficacy in preventing fatal GVHD (Figure 1 and Supplementary Figure 1).
FIGURE 1
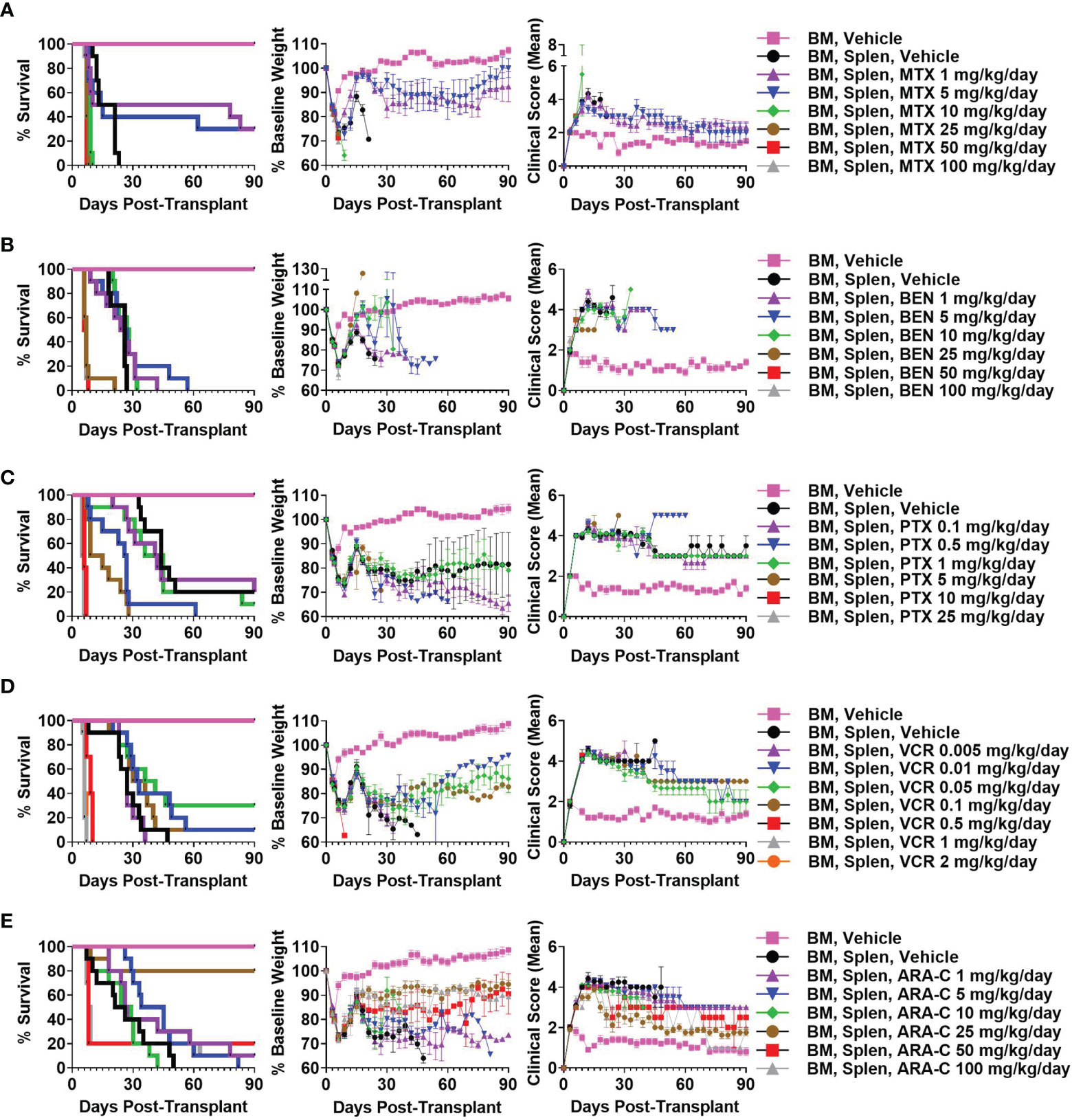
Figure 1 Methotrexate (MTX) 5 mg/kg/day, bendamustine (BEN) 10 mg/kg/day, paclitaxel (PTX) 1 mg/kg/day, vincristine (VCR) 0.05 mg/kg/day, and cytarabine (ARA-C) 25 mg/kg/day are optimal doses of each drug when given on days +3/+4 in the B6C3F1 → B6D2F1 MHC-haploidentical hematopoietic cell transplantation (HCT) model. On day 0, recipient 10-12-week-old female B6D2F1 mice were irradiated to 10.5 Gy in a single fraction and transplanted 6-8 hours later via intravenous injection with 10 x 106 T-cell-depleted bone marrow (BM) cells +/- 40 x 106 red-blood-cell-depleted splenocytes (Splen) from 10-12-week-old female B6C3F1 donors. Phosphate buffered saline (PBS) vehicle or the chemotherapeutic of interest was administered intraperitoneally on days +3 and +4. Although different chemotherapeutics had varying efficacy in mitigating fatal or severe graft-versus-host disease (GVHD), (A) MTX 5 mg/kg/day, (B) BEN 10 mg/kg/day, (C) PTX 1 mg/kg/day, (D) VCR 0.05 mg/kg/day, and (E) ARA-C 25 mg/kg/day were determined as optimal doses for each drug due to superior survival, weights, and/or clinical scores. Combined results from two independent experiments of n = 5 mice/group/experiment are shown.
CY 25 mg/kg/day Is Superior to All Other Chemotherapeutics in Preventing Severe GVHD
We next compared the optimal doses of these other chemotherapeutics with the previously established optimal dose of CY (25 mg/kg/day) in this MHC-haploidentical HCT model (5, 6), with all drugs being administered as daily doses on days +3 and +4. CY, MTX, and ARA-C all were partially effective in preventing fatal GVHD (Figure 2A). BEN slightly delayed survival in a subset of mice, while VCR and PTX were completely ineffective in preventing fatal GVHD (Figure 2A). In fact, PTX led to more rapid mortality (Figure 2A).
FIGURE 2
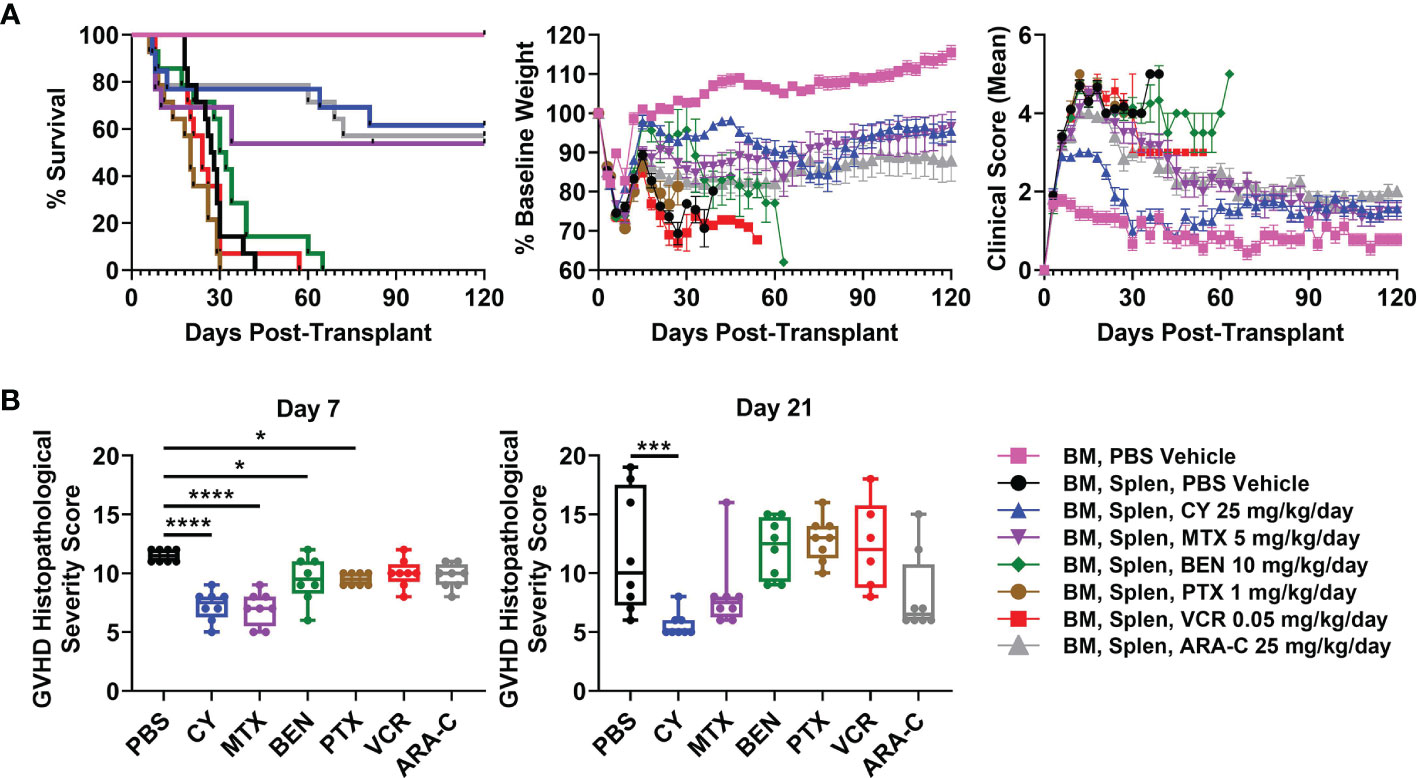
Figure 2 Post-transplantation cyclophosphamide (PTCy), MTX, and ARA-C all are partially effective in mitigating severe GVHD with PTCy having the most efficacy. Mice were transplanted as in Figure 1 and were given either PBS or the previously determined optimal dose of one of the tested chemotherapeutics. (A) CY (HR 0.12, p = 0.0008), MTX (HR 0.28, p = 0.012), and ARA-C (HR 0.11, p = 0.0004) all significantly prolonged survival compared with vehicle-treated mice, whereas PTX led to more rapid mortality (HR 2.42, p = 0.038). MTX and ARA-C had similar survival compared with CY, but BEN (HR 6.86, p = 0.0008), PTX (HR 8.41, p = 0.0005), and VCR (HR 5.86, p = 0.0012) had worse survival. However, of the three partially effective chemotherapeutics, only CY-treated mice had significantly higher weights (p = 0.015) compared with vehicle-treated mice (p > 0.10 for MTX and ARA-C). Conversely, VCR-treated mice had significantly lower weights (p = 0.011) and PTX trended towards lower weights (p = 0.075) compared with vehicle-treated mice. CY-treated mice had higher weights compared with PTX-treated (p = 0.0068), VCR-treated (p = 0.0011), and ARA-C-treated (p = 0.02) mice and also had marginally higher weights compared with MTX-treated mice (p = 0.052). CY (p < 0.0001), MTX (p = 0.03), and ARA-C (p < 0.0001) led to better clinical scores than vehicle-treated mice, and CY was significantly better than all other treatment groups including both MTX and ARA-C (p < 0.0001 for each). Statistical comparisons for clinical scores and weights are for the area-under-the-curve (AUC) calculations over the period of time in which ≥70% of vehicle-treated mice were alive. (B) Mice were taken for histopathology of GVHD target organs on day +7 or +21. Several chemotherapeutics (CY, MTX, BEN, PTX) significantly reduced histopathological GVHD at day +7, but only CY continued to significantly reduce histopathological GVHD at day +21. *p < 0.05, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant. Combined results from two independent experiments are shown; n = 5/group/experiment for the weights and clinical score assessments in A except TCD BM + PBS (n = 9 total), and n = 4/group/experiment for all groups in B except VCR at day +21 (n = 6 total) due to excess early deaths. Extra mice set up in B for both experiments were also followed and included in the survival graphs [n = 4 independent experiments with total n=14/group except CY (n = 13), MTX (n = 13), and TCD BM + PBS vehicle (n = 9)].
CY was superior to all other chemotherapeutics in ameliorating clinical GVHD (Figure 2A). CY-treated mice had significantly improved body weights and clinical scores compared with vehicle-treated mice as well as with MTX- and ARA-C-treated mice (Figure 2A). MTX and ARA-C had similar weights but significantly better clinical scores than vehicle-treated mice. BEN, VCR, and PTX did not significantly improve clinical scores compared with vehicle-treated mice (Figure 2A), while VCR had significantly lower body weights and PTX trended towards lower body weights compared with vehicle-treated mice (Figure 2A). Higher mean body weight in BEN-treated mice was a result of substantial weight gain from ascites, rather than clinical benefit from the chemotherapeutic (Figure 2A). Autopsy suggested that these mice were developing ascites secondary to GVHD-induced liver failure and protein-losing enteropathy, also evident in high average liver GVHD scores at day +21 (Supplementary Figure 2).
Histopathological scoring at day +7 revealed that mice receiving CY, MTX, BEN, and PTX all had significantly reduced total histopathological GVHD severity scores compared with vehicle-treated mice (Figure 2B and Supplementary Table 1). However, by day +21, only CY significantly reduced histopathological GVHD compared with vehicle-treated mice (Figure 2B and Supplementary Table 2). MTX and ARA-C both had lower median histopathological GVHD severity scores compared with vehicle-treated mice at day +21, but the lack of statistical significance was due to wider variability of scores between mice in those groups (Figure 2B).
The Partially Effective Chemotherapeutics (CY, MTX, and ARA-C) All Constrain T-Cell Recovery at Day +21
Some have contended that PTCy prevents GVHD via in vivo T-cell depletion, but our recent work showed that day +7 total numbers of T cells in blood, spleens, peripheral lymph nodes, and liver after CY 25 mg/kg/day, the optimal dose in this model, were similar to or in the same log range as vehicle-treated mice (5). Similar to these findings, total T-cell numbers in blood and spleen from CY and most other tested chemotherapeutics were not significantly reduced at day +7 when compared with vehicle-treated mice (Figure 3A). The only exception was that MTX reduced total T-cell concentrations in blood at day +7, which was attributable to a decrease in total CD8+ T-cell concentrations at that timepoint (Figure 3A). CY did not significantly reduce total T-cell numbers in either blood or spleen, but did result in a significant reduction of CD4+ T cells in the spleen at day +7 (Figure 3A). By contrast, CY, MTX, and ARA-C all significantly reduced total T-cell, including CD4+ and CD8+ T-cell subset, numbers at day +21, whereas the ineffective chemotherapeutics had total T-cell numbers similar to vehicle-treated mice (Figure 3B). This suggests that effective GVHD control may be associated with constrained T-cell recovery at day +21.
FIGURE 3
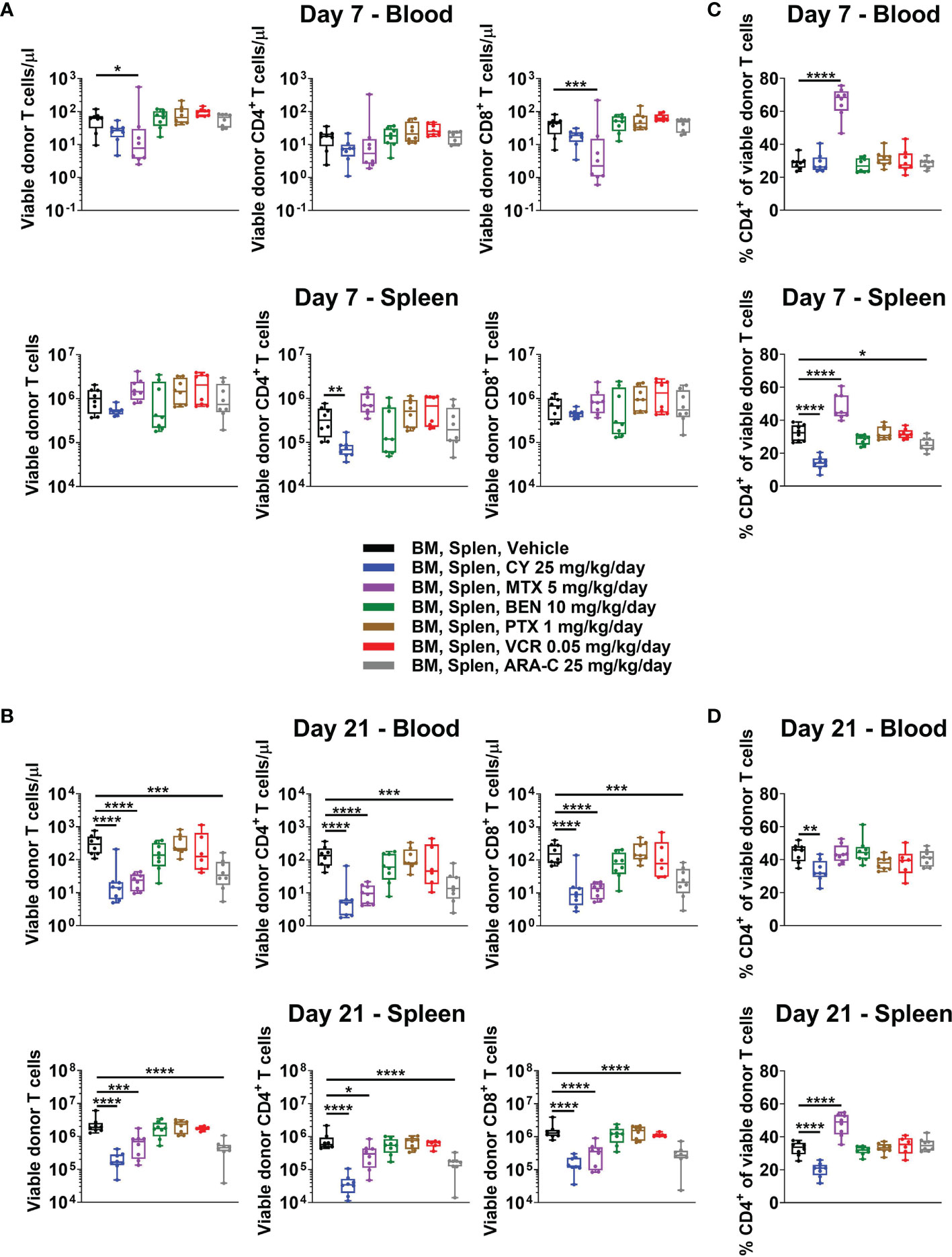
Figure 3 All partially effective chemotherapeutics constrain T-cell recovery at day +21, but MTX and CY differentially affect the balance of CD4+ versus CD8+ T cells, distinct from all other chemotherapeutics. Mice were transplanted as in Figure 1 and received intraperitoneal injections on days +3 and +4 with either PBS vehicle or the optimal dose of one of the chemotherapeutics of interest. At day +7 or +21, mice were euthanized, and their blood and spleens were assessed by flow cytometry. (A) Total numbers of CD3+ T cells and CD4+ and CD8+ T-cell subsets were not significantly reduced in BEN-, PTX-, VCR-, or ARA-C-treated mice at day +7. However, MTX significantly reduced total number of CD3+ and CD8+ T cells in the blood, while CY significantly reduced total numbers of CD4+ T cells in spleens at day +7. (B) At day +21, CY, MTX, and ARA-C all constrained recovery of CD4+ and CD8+ T-cell subsets. (C, D) MTX and CY had opposite effects at both (C) day +7 and (D) day +21 on the balance of CD4+ and CD8+ T cells, divergent from effects seen in vehicle-treated mice and mice treated with other chemotherapeutics. Combined results from two independent experiments are shown with n = 4/group/experiment except for VCR at day +21 (n = 6 total) due to excess early deaths in one experiment prior to day +21. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant.
The CD4+/CD8+ T-Cell Ratio Is Not Affected by Chemotherapeutics Other Than MTX, Which Increases It, and CY, Which Decreases It
MTX administration facilitated a markedly distinct recovery of CD4+ versus CD8+ T cells compared with all other treatment groups and opposite that of CY-treated mice (Figures 3C, D and Supplementary Figure 3). At day +7, MTX demonstrated higher percentages of CD4+ T cells in both blood and spleen compared with vehicle-treated mice, while CY-treated mice had similar percentages of CD4+ T cells in the blood and reduced percentages in the spleen (Figure 3C). At day +21, splenic CD4+ T-cell percentages remained high in MTX-treated mice, while CY again reduced percentages in both blood and spleens (Figure 3D). The ineffective chemotherapeutics all had similar percentages of CD4+ T cells compared with vehicle-treated mice across tissues at both days +7 and +21 (Figures 3C, D). This unique, contrasting T-cell recovery after MTX compared with that observed after PTCy is especially interesting considering that MTX was found to be effective in this model (Figure 2), albeit less so than PTCy, suggesting that MTX and PTCy may have distinct mechanisms of GVHD prevention.
Unlike CY, MTX and ARA-C Do Not Control CD25 Expression by CD4+Foxp3- Conventional T Cells at Day +7
Consistent with increased percentages of CD4+ T cells in MTX-treated mice (Figure 3C), total numbers of CD4+Foxp3- conventional T cells were similar to slightly higher in MTX-treated compared with vehicle-treated mice at day +7 (Figure 4A). Furthermore, at day +7, much higher percentages of CD4+Foxp3- conventional T cells appeared to have an activated phenotype (CD25+Foxp3-) in MTX-treated mice and, to a lesser extent, ARA-C-treated mice, whereas this percentage was reduced in CY-treated mice (Figures 4B, C). Interestingly, this difference in CD25 expression of CD4+Foxp3- conventional T cells did not equate to differences in STAT5 phosphorylation, which was not decreased in CY-treated mice (Supplementary Figure 4). Corresponding to the higher CD25 expression within CD4+Foxp3- conventional T cells after MTX and ARA-C at day +7, MTX- and ARA-C-treated mice had worse weights and clinical scores than CY-treated mice (Figure 2A). This clinical effect was seen despite increased percentages of CD4+CD25+Foxp3+ Tregs at day +7 in MTX-treated mice, whereas these percentages were reduced in the spleens of CY-treated mice at this timepoint (Figure 4B).
FIGURE 4
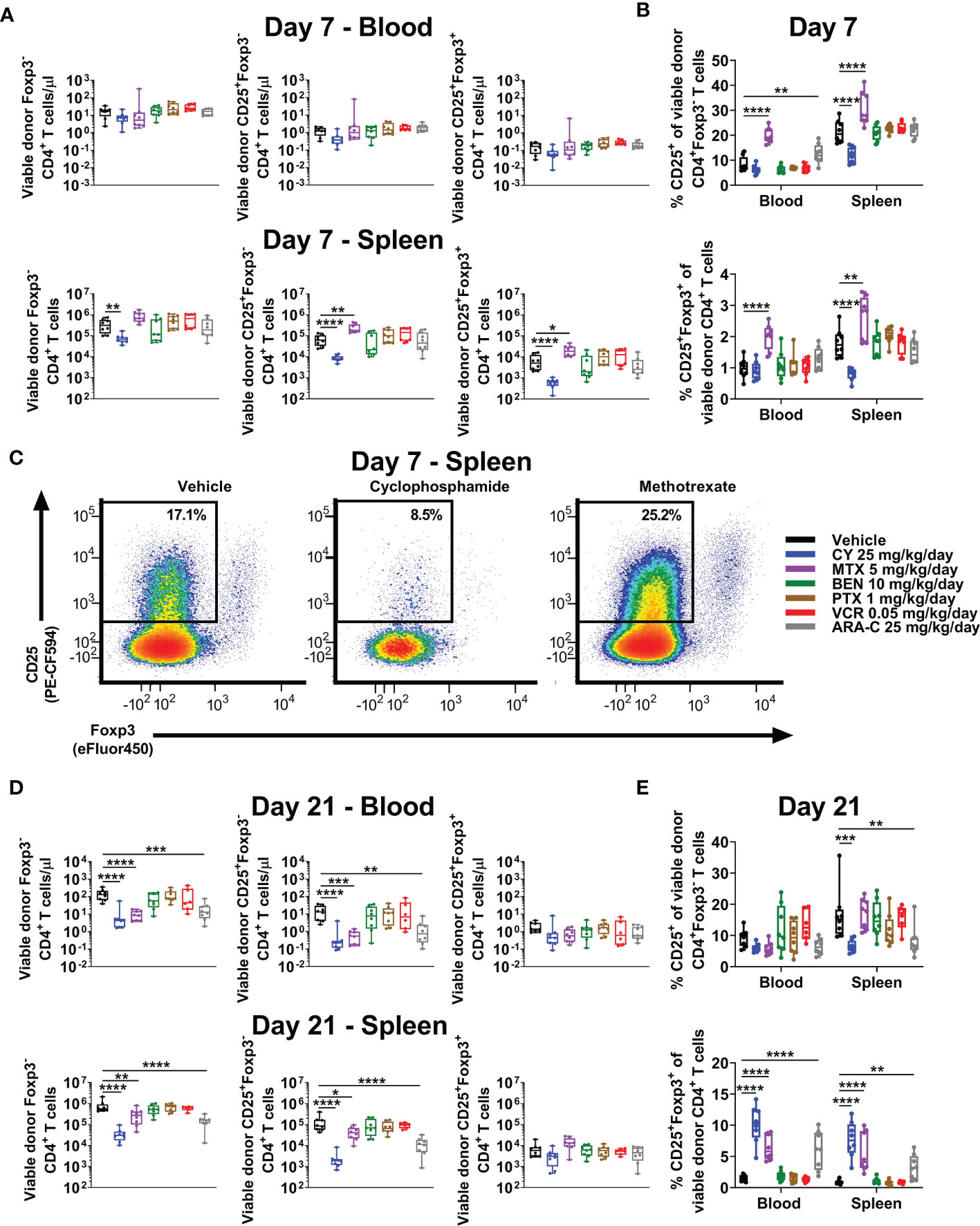
Figure 4 MTX and ARA-C do not control activated conventional CD4+ T cells at day +7, but at day +21 CY, MTX, and ARA-C (all partially effective chemotherapeutics) constrain conventional CD4+ T-cell recovery and facilitate preferential CD4+CD25+Foxp3+ regulatory T-cell recovery. Mice were transplanted, treated with PBS or a chemotherapeutic on days +3 and +4, and euthanized for flow cytometric assessment at day +7 or +21 as in Figure 3. (A) At day +7, total numbers of CD4+ T-cell subsets in the blood were not significantly different across treatment groups, but CY significantly reduced and MTX significantly increased total numbers of CD4+ T-cell subsets in the spleen. (B) Although MTX increased percentages of CD25+Foxp3+ regulatory T cells (Tregs) at day +7 in both the blood and spleen, MTX also significantly increased percentages of conventional (Foxp3-) CD4+ T cells with an activated (CD25+) phenotype. This was distinct from CY, which reduced percentages of both. All other chemotherapeutics did not significantly alter these percentages in comparison with vehicle-treated mice except for ARA-C, which increased percentages of activated (CD25+) conventional CD4+ T cells only in the blood. (C) Representative flow cytometric plots are shown of CD4+ T cells gated on CD25 versus Foxp3 expression, showing percentages of CD25+Foxp3- CD4+ T cells at day +7 that were decreased after CY but increased after MTX. CD25-positivity was gated based on the use of a fluorescence-minus-one (FMO) control. (D) At day +21, total numbers of Foxp3- and CD25+Foxp3- conventional CD4+ T cells were decreased in mice treated with the partially effective chemotherapeutics (CY, MTX, and ARA-C), whereas total numbers of CD4+CD25+Foxp3+ Tregs were similar across treatment groups. (E) Due to this balance, CY, MTX, and ARA-C all were associated with increased percentages of CD4+CD25+Foxp3+ Tregs at day +21, while CY and ARA-C also reduced the percentages of activated (CD25+) conventional CD4+ T cells. Combined results from two independent experiments are shown with n = 4/group/experiment for (A–E) except for VCR (n = 6 total) in (D, E) due to excess early deaths. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant.
All Partially Effective Chemotherapeutics Facilitate Preferential Treg Recovery at Day +21
We have shown that Tregs are necessary for GVHD prevention by PTCy and that this role is increasingly important as time progresses post-transplant in suppressing surviving alloreactive T cells (3–5). PTCy also allows preferential recovery of Tregs in mice and patients (3–6); indeed, increased percentages of Tregs at day +21 are associated in our MHC-haploidentical HCT model with more effective dosing schedules of PTCy (5, 6). Consistent with our previous work, CY facilitated increased percentages of Tregs at day +21 in both blood and spleens (Figures 4D, E), an effect that also was seen with the two other partially effective chemotherapeutics, MTX and ARA-C (Figures 4D, E). Conversely, the ineffective drugs, BEN, PTX, and VCR, were not associated with increased percentages of Tregs at either timepoint (Figures 4B, E). These data further support preferential recovery of Tregs at day +21 as a potential biomarker of successful GVHD prevention, as this T-cell endpoint has been consistent between all effective chemotherapeutics here and also for maximally effective dosing schedules of PTCy (5, 6).
Alloreactive T Cells Persist After All Chemotherapeutics, but Alloantigen-Specific Tregs Are Increased at Day +7 After CY and MTX
Previously PTCy was thought to work via selective elimination of alloreactive T cells, since these cells would be proliferating rapidly in the early post-transplant setting. However, our recently published work showed that PTCy does not selectively eliminate alloreactive T cells (5, 6). Indeed, neither after PTCy, nor after any of the other chemotherapeutics tested, were alloreactive T cells eliminated (Figures 5A, B and Supplementary Figure 5). At day +7, the percentages of CD4+Foxp3- T cells that were Vβ6+ were slightly reduced after PTCy (Figure 5A). By contrast, these percentages, and those of CD8+ T cells that were Vβ6+ actually were increased in the blood after MTX (Figure 5A). Interestingly, both PTCy and MTX were associated with increased percentages of alloantigen-specific (Vβ6+) Tregs at day +7 (Figure 5A). At day +21, across all T-cell subsets and treatment groups, there were no significant differences in percentages of alloreactive T cells compared with vehicle-treated mice (Figure 5B).
FIGURE 5
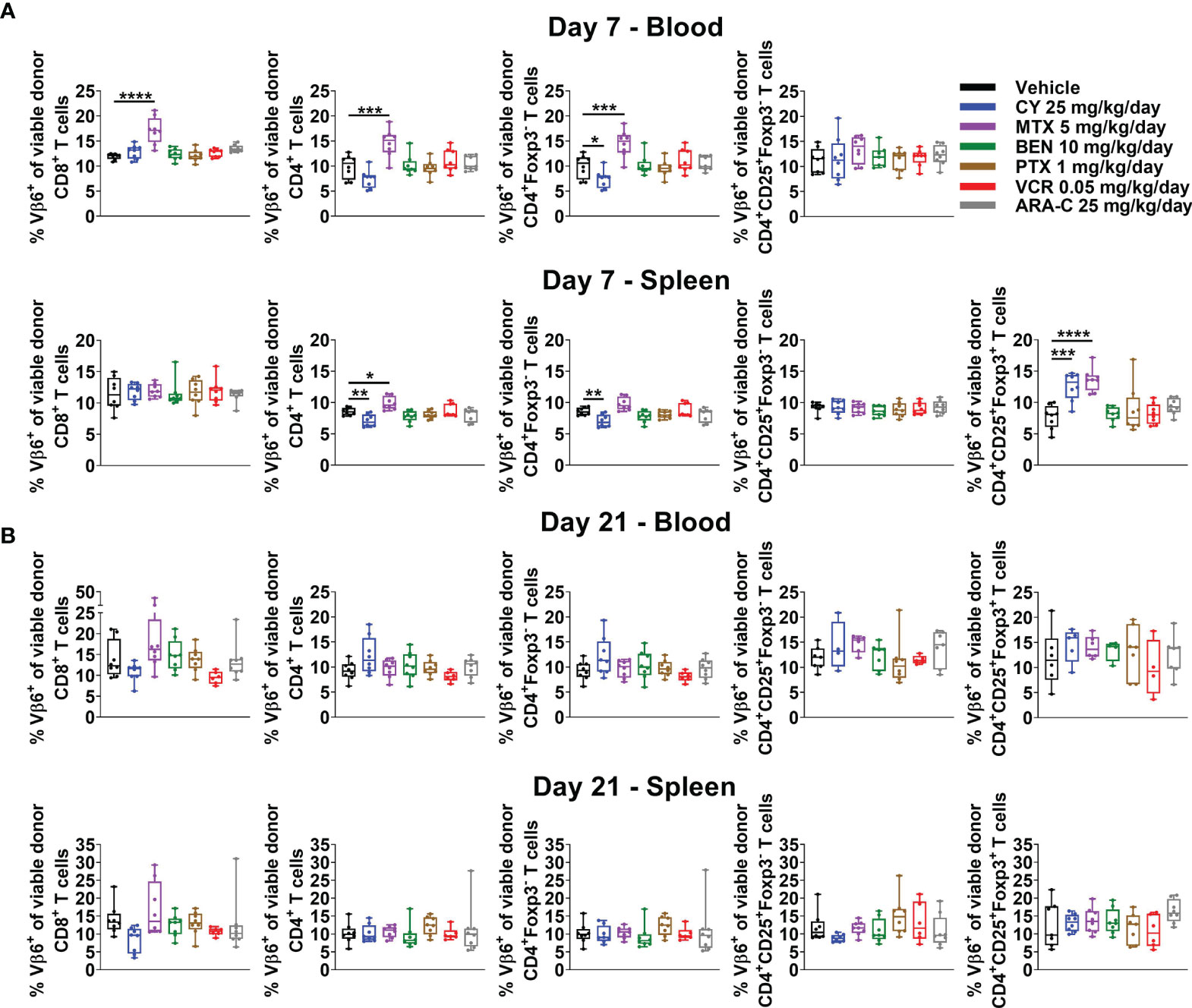
Figure 5 Alloreactive T cells persist after administration of all chemotherapeutics, but alloantigen-specific Tregs are increased at day +7 after CY and MTX. Mice were transplanted, treated with PBS or a chemotherapeutic on days +3 and +4, and euthanized for flow cytometric assessment at day +7 or +21 as in Figure 3. Alloreactive Vβ6+ T cells were not eliminated by any chemotherapeutic at either (A) day +7 or (B) day +21. (A) In fact, at day +7, percentages of CD8+, CD4+, and CD4+Foxp3- T cells that were Vβ6+ were increased in MTX-treated mice. The percentages of CD4+Foxp3- T cells that were Vβ6+ were slightly reduced after CY although other T-cell subsets, including percentages of CD4+CD25+Foxp3- that were Vβ6+, were not affected. Interestingly, alloantigen-specific CD4+CD25+Foxp3+ cells were increased in CY- and MTX-treated mice at day +7 in the spleen; percentages in the blood were not included due to low total numbers of CD4+CD25+Foxp3+ cells at day +7 across treatment groups that did not permit reliable determination of further subsetting. (B) At day +21, there were no significant differences in percentages of Vβ6+ T cells across T-cell subsets or treatment groups. Combined results from two independent experiments are shown with n = 4/group/experiment except VCR (n = 6 total) in B due to excess early deaths. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant.
CY Is the Only Effective Chemotherapeutic That Consistently Reduces Alloreactive CD4+ T-Cell Proliferation at Day +7
Although PTCy does not selectively eliminate alloreactive T cells, effective GVHD control by PTCy is associated with a decrease in percentages of CD4+, including alloreactive CD4+ T cells, that are proliferating at day +7 (5, 6). Consistent with previous results, CY reduced percentages of proliferating CD4+ T cells, including alloreactive (Vβ6+) conventional T-cell subsets, in both blood and spleens (Figures 6A, C). Conversely, neither MTX nor ARA-C significantly reduced proliferation of alloreactive T cells at day +7, except within CD4+CD25+Foxp3-Vβ6+ T cells after ARA-C (Figure 6C). By contrast, proliferation of alloreactive T cells had normalized and was similar across treatment groups and T-cell subsets at day +21 (Figures 6B, D).
FIGURE 6
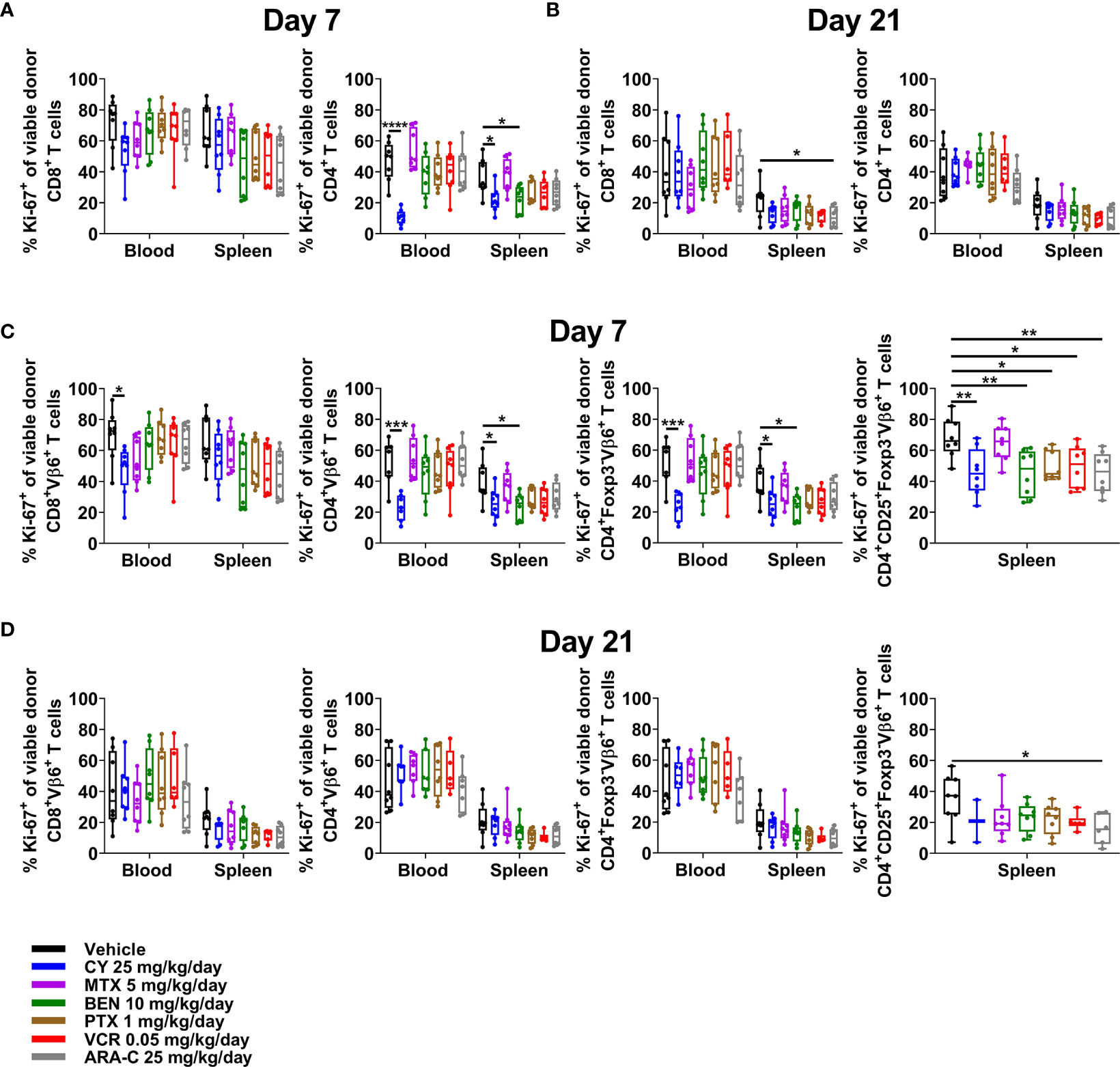
Figure 6 Only CY consistently reduces alloreactive CD4+ T-cell proliferation at day +7. Mice were transplanted, treated with PBS or a chemotherapeutic on days +3 and +4, and euthanized for flow cytometric assessment at day +7 or +21 as in Figure 3. (A) In both the blood and spleens at day +7, CY reduced proliferation (Ki-67+) of CD4+ T cells. (B) At day +21, proliferation was generally similar across all groups. (C) CY also reduced proliferation of alloreactive (Vβ6+) conventional CD4+ T cells at day +7. Interestingly, BEN, PTX, VCR, and ARA-C significantly reduced percentages of proliferating alloreactive CD4+CD25+Foxp3- T cells at day +7, but did not affect proliferation of other alloreactive CD4+ T cells. (D) At day +21, proliferation of alloreactive subsets again was generally similar across all groups. Percentages of proliferating alloreactive CD4+CD25+Foxp3- T cells in the blood are not shown in (C, D) due to low total numbers of CD4+CD25+Foxp3-Vβ6+ cells across treatment groups that did not permit reliable determination of further subsetting. Combined results from two independent experiments are shown with n = 4/group/experiment for (A–D) except for VCR (n = 6 total) in (B, D) due to excess early deaths. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant.
All Partially Effective Chemotherapeutics Restrain Alloreactive CD4+ Conventional T-Cell Differentiation at Day +7, but CY Has the Greatest Effect and Is the Only Drug That Maintains This Effect at Day +21
CY, MTX, and ARA-C, the three chemotherapeutics that were partially effective in ameliorating GVHD, all were distinct in restraining alloreactive CD4+Foxp3- T-cell differentiation at day +7, with less effector/effector memory and more naïve/central memory phenotypes (Figure 7A). This effect was most pronounced for CY and even more evident when looking at all CD4+Foxp3- T cells (Supplementary Figure 6), which also would include alloreactive T cells beyond those that are Vβ6+. Furthermore, the restraint of alloreactive CD4+ T-cell differentiation continued after CY at day +21, whereas MTX-treated mice actually had slightly more highly differentiated alloreactive CD4+ T cells at that time point (Figure 7B and Supplementary Figure 6).
FIGURE 7
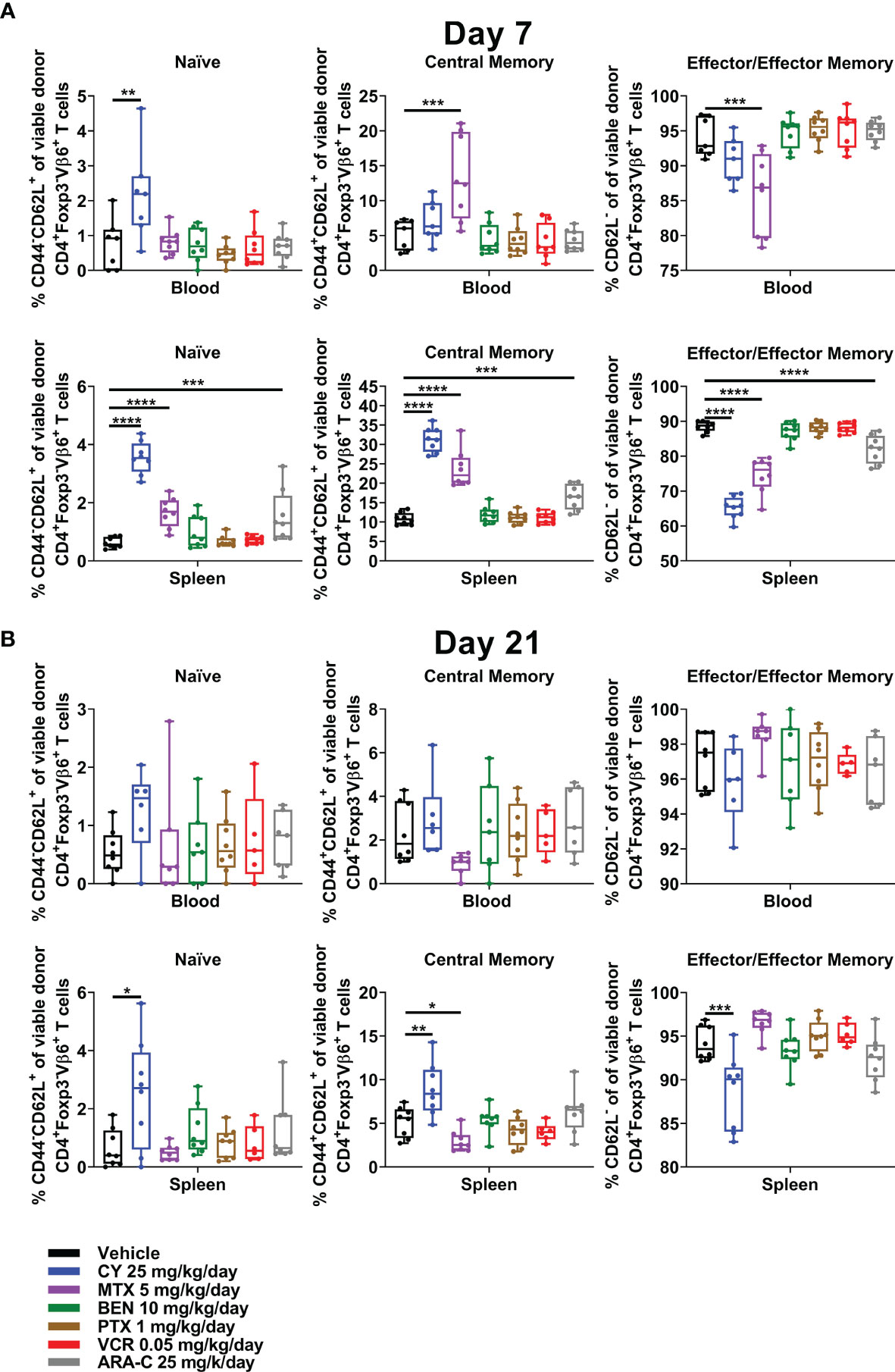
Figure 7 CY uniquely restrains T-cell differentiation at both early and later timepoints. Mice were transplanted, treated with PBS or a chemotherapeutic on days +3 and +4, and euthanized for flow cytometric assessment at day +7 or +21 as in Figure 3. (A) At day +7, CY decreased percentages of CD4+Foxp3-Vβ6+ T cells that were phenotypically effector/effector memory (CD62L-). Consequently, percentages of naïve (CD44-CD62L+) and central memory (CD44+CD62L+) CD4+Foxp3-Vβ6+ T cells were increased by CY at day +7. This same effect was achieved to a lesser extent after both MTX and ARA-C. (B) This restrained differentiation was persistent after CY at day +21 but was completely lost after MTX, wherein differentiation seemed to be overall accelerated. Combined results from two independent experiments are shown with n = 4/group/experiment except for VCR (n = 6 total) in (B) due to excess early deaths. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 on one-way ANOVA followed by the Holm-Sidak post hoc test using the vehicle-treated group as the control. Only significant results are shown; all other comparisons between treatment groups and the vehicle group are non-significant.
Discussion
Using our previously described MHC-haploidentical murine HCT model (5), we tested the relative efficacy of five other chemotherapeutics (MTX, BEN, PTX, VCR, and ARA-C) in comparison with cyclophosphamide when given as GVHD prophylaxis early post-transplant. We demonstrated that PTCy not only was superior to all other tested chemotherapeutics in ameliorating severe GVHD clinically and histopathologically, but also showed that some of the effects of PTCy on T-cell subsets appear unique. Similar to PTCy, the other partially effective chemotherapeutics, MTX and ARA-C, did constrain conventional T-cell numerical recovery at day +21 and facilitated preferential recovery of Tregs at day +21. But, unlike PTCy, MTX and ARA-C did not reduce percentages of alloreactive CD4+ conventional T cells that were activated (as measured by CD25 expression) or proliferating at day +7 and only restrained alloreactive CD4+ conventional T-cell differentiation at day +7 (not day +21); indeed, MTX was associated with a more differentiated phenotype at day +21 compared with even vehicle-treated mice. These findings provide further support for reduced alloreactive CD4+ conventional T-cell proliferation at day +7 and preferential Treg recovery at day +21 as potential biomarkers for effective GVHD control by PTCy as identified in our previous publications on optimal dosing and timing of PTCy (5, 6). Additionally, we propose restrained alloreactive CD4+ conventional T-cell activation and differentiation as possible additional elements that may further explain PTCy’s superior efficacy in preventing GVHD. The discrepancy between activation markers of CD25 and phospho-STAT5 positivity within CD4+ conventional T cells in PTCy-treated mice may suggest that activation is incomplete or impaired after PTCy, and we are working to better understand this phenomenon in the laboratory.
It is difficult to determine if PTCy causes preferential deletion of differentiated effector/effector memory alloreactive CD4+ conventional T cells or has a direct effect on alloreactive CD4+ conventional T cells that prevents their subsequent differentiation. Although this distinction is impossible to definitively tease out with this model system, the continued increased percentages of naïve/central memory cells and decreased percentages of effector/effector memory cells at day +21 seen in PTCy-treated mice would support the latter possibility. We are continuing to study in the laboratory the nature of the alloreactive T-cell dysfunction induced by PTCy that may contribute to GVHD prevention and restrained T-cell proliferation and differentiation.
PTCy previously was thought to work via the selective elimination of alloreactive T cells, as these cells would be rapidly proliferating in the early post-transplant time period. However, this hypothesis was extrapolated from data in MHC-matched murine skin-allografting models that were extremely contextual and had questionable relevance to HCT (5, 57–60). Our recently published work disproved this hypothesis in HCT, showing that alloreactive T cells persisted after PTCy administration even at high, intolerable PTCy doses (5). Both in our current and prior studies (5, 6), we did find a small transient reduction in the percentages of alloreactive CD4+ conventional T cells after PTCy in some organs, and here we also found that CD25-expressing activated alloreactive CD4+ conventional T cells were reduced. We cannot exclude that PTCy in some organs may preferentially diminish alloreactive T cells, particularly activated and proliferating alloreactive T cells, but any such effect is minor, incomplete, and short-lived. Alternatively, this effect could be explained by relative restraint of activation, proliferation, and expansion of surviving alloreactive T cells rather than selective killing, consistent with our findings of similar pSTAT5 signaling but reduced CD25 expression in PTCy-treated mice. Even so, all our prior and current data clearly show that alloreactive T cells are not selectively eliminated by PTCy (5, 61).
In retrospect, the lack of selective alloreactive T-cell elimination should not be surprising since cyclophosphamide, as an alkylator, is a non-cell-cycle-specific chemotherapeutic. Thus, it appears that PTCy may be killing a substantial percentage of T cells in a dose-dependent manner, but this effect is broad and not selective for alloreactive T cells (5, 61). Interestingly, none of the chemotherapeutics tested in these experiments selectively eliminated alloreactive T cells, despite the use of some cell-cycle-specific chemotherapeutic agents; by contrast, the percentages of alloreactive T cells were even higher after MTX at day +7. Even though none of these other chemotherapeutics greatly affected either the relative percentages of alloreactive T cells or global T-cell proliferation, all drugs except MTX did slightly reduce the proliferation of activated (CD25+) alloreactive CD4+ conventional T cells at day +7. Overall though, the decline was modest and not associated with substantial clinical or histopathologic reduction in GVHD severity for most drugs.
Moreover, no chemotherapeutic induced pan T-cell depletion at day +7 compared with vehicle-treated mice, which may in part be attributable to intermediate rather than maximally tolerated doses being tested as intermediate doses were the most optimal for survival for each chemotherapeutic. Even so, we have previously shown that very high doses of PTCy, which did greatly reduce T-cell counts at day +7, resulted in preferential survival of alloreactive T cells at day +7 and actually led at day +21 to a tremendous rebound in alloreactive T-cell counts, consequent blunting of regulatory T-cell recovery, and worse GVHD in our model compared with intermediate dosing (5). Indeed, early results from a PTCy dose de-escalation clinical study at our institution suggest that intermediate-dose PTCy maintains excellent protection against acute GVHD (62). Whether such results also are true for other chemotherapeutics is unknown, but is important to understand, particularly given the recent attempts to implement post-transplantation bendamustine clinically (17, 18).
A limitation of this study is that all the alternative chemotherapeutics tested were administered on days +3/+4 to best compare against PTCy each chemotherapeutic’s effects on GVHD and immune subsets; PTCy is administered in this manner in clinical practice and administration on days +3/+4 is among the most effective PTCy dosing schedules in our MHC-haploidentical murine HCT model (6). We have hypothesized that PTCy administration on days +3/+4 is particularly effective because during this critical window specific T-cell subsets are metabolically primed, based on differential dynamic expression of ALDH and ABC transporter activity, for differential sensitivity to cyclophosphamide (3–7, 9). In previous experiments using our MHC-haploidentical murine HCT model, earlier administration of PTCy on days +1/+2 was associated with a less robust decrease in alloreactive CD4+ conventional T-cell proliferation at day +7 and later dosing on days +5/+6 was associated with a blunted relative recovery of Tregs by day +21, likely contributing in either case to the lower efficacy seen when compared with PTCy on days +3/+4 (6). Clinical success of PTCy was not achieved until the dose and timing were better optimized; in fact, early clinical studies using serial administration for 100 days of low dose PTCy suggested partial efficacy that was inferior to cyclosporine when each was combined with methylprednisolone (63). Moreover, it is possible that other chemotherapeutics may be more effective when given in different dosing schedules, and such optimal dosing schedules might vary depending on the relative metabolism of each drug. For example, MTX’s current dosing schedule for acute GVHD prophylaxis, which was derived from animal studies (45, 64, 65) and then tested clinically (46, 47, 66), is distinct from PTCy with administration on days +1, +3, +6, +/- +11. Nevertheless, additional studies on optimal timing for administration of other chemotherapeutics would be necessary to determine if the day +3/+4 timing is universally optimal or whether another chemotherapeutic administered in a different schedule or in combination with PTCy may produce similar or superior results to PTCy alone.
Our data showed no clear association between drug metabolism or resistance pathways and effective GVHD control. We had specifically chosen chemotherapeutics with varying involvement of ABC transporters and ALDH in resistance pathways (Table 1) to explore their relative efficacy. Interestingly, resistance patterns of the partially effective drugs (CY, MTX, and ARA-C) diverge (8, 12–14, 20–23), but may explain why each had differing effects on T-cell subset activation, proliferation, differentiation, and recovery kinetics. Yet, resistance patterns are not very dissimilar for the ineffective drugs compared with the effective drugs (Table 1) (8, 12–14, 19–36), suggesting either a small therapeutic index to effectively prevent GVHD or that the cellular impact of these drugs and/or their metabolism may be more complicated than we currently appreciate. Furthermore, MTX still facilitated the preferential recovery of Tregs by day +21 and even expanded Tregs at day +7. However, when considering ALDH versus ABC transporter activity, ABC transporters, which Tregs lack (67), likely play a comparatively larger role in resistance to MTX (Table 1) (12–14, 21–23, 30, 31, 36). By contrast, MTX has not been reported to be metabolized via ALDH which appears important for mediating Treg resistance to cyclophosphamide (3, 4). Even so, the recovery kinetics of Tregs in MTX-treated mice were distinct from all other chemotherapeutics as was the balance of CD4+ versus CD8+ T cells. MTX uniquely facilitated increased percentages of Tregs at day +7, which may have mitigated effects of the increased percentages of activated (CD25+) alloreactive CD4+ conventional T cells at that timepoint. The divergence in some immunologic effects of MTX and PTCy suggests that GVHD control by MTX may occur via a different mechanism than PTCy and may explain why MTX is only effective for acute GVHD clinically, while PTCy can prevent both acute and chronic GVHD.
An additional interesting finding of this study is that BEN was ineffective in ameliorating severe and fatal GVHD in our MHC-haploidentical HCT model. Recently, post-transplantation BEN has shown promise in pre-clinical murine HCT studies (16) and mixed results in early phase clinical trials (17, 18). It is possible that our conflicting findings may be explained in part due to differences in models or relative doses of CY or BEN, which were higher in the CB6F1→CAF1/J model (16) than we have determined to be optimal at preventing GVHD in our B6C3F1→B6D2F1 model (5). Additionally, a recent clinical study examining post-transplantation BEN showed that patients developed severe cytokine release syndrome, often manifesting with liver dysfunction, at unacceptably high rates (18). This may parallel the ascites secondary to liver failure that developed in our BEN-treated mice. Ultimately, our results and the recent clinical study (18) suggest the need for caution in considering BEN as a suitable, safe, and effective alternative to PTCy until more mature clinical data are available.
In conclusion, our results provide further insight into the mechanisms of PTCy and the biology of GVHD prevention. The clinical and immunologic effects of cyclophosphamide given in the early post-transplant period appear unique and not fully reproducible by another alkylating agent or four other chemotherapeutics of multiple classes given over the same dosing schedule. Our data show that effective GVHD prophylaxis is associated with distinctive effects on constraining alloreactive conventional T-cell numerical reconstitution and facilitating preferential Treg recovery at day +21, but also uncover that PTCy uniquely restrains alloreactive CD4+ conventional T-cell proliferation and differentiation. To what extent these findings hold true in patients, particularly those receiving adjunct immunosuppression beyond PTCy or to patients undergoing combined HCT/solid organ transplantation (68, 69), requires further exploration.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by National Cancer Institute Animal Care and Use Committee.
Author Contributions
CK designed the study. AH contributed to the study design. AH, NN, SK, RF, and AP performed the experiments. AH and CK analyzed the data. ME performed blinded assessments of histopathology. DV designed and performed the statistical analyses. All authors interpreted the data. AH and CK designed and created the tables and figures. AH and CK wrote the manuscript, and all authors revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (NIH), and the Lasker Foundation. This work was made possible through the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Foundation, Genentech, the American Association for Dental Research, and the Colgate-Palmolive Company. None of these supporting institutions or companies were involved in the study design, data collection, data analysis, data interpretation, manuscript preparation, or the decision to submit the manuscript for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Devorah Gallardo for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.796349/full#supplementary-material
References
1. Szydlo R, Goldman JM, Klein JP, Gale RP, Ash RC, Bach FH, et al. Results of Allogeneic Bone Marrow Transplants for Leukemia Using Donors Other Than HLA-Identical Siblings. J Clin Oncol (1997) 15(5):1767–77. doi: 10.1200/jco.1997.15.5.1767
2. Kanakry CG, Fuchs EJ, Luznik L. Modern Approaches to HLA-Haploidentical Blood or Marrow Transplantation. Nat Rev Clin Oncol (2016) 13(2):132. doi: 10.1038/nrclinonc.2015.234
3. Kanakry CG, Ganguly S, Zahurak M, Bolanos-Meade J, Thoburn C, Perkins B, et al. Aldehyde Dehydrogenase Expression Drives Human Regulatory T Cell Resistance to Posttransplantation Cyclophosphamide. Sci Transl Med (2013) 5(211):211ra157. doi: 10.1126/scitranslmed.3006960
4. Ganguly S, Ross DB, Panoskaltsis-Mortari A, Kanakry CG, Blazar BR, Levy RB, et al. Donor CD4+ Foxp3+ Regulatory T Cells are Necessary for Posttransplantation Cyclophosphamide-Mediated Protection Against GVHD in Mice. Blood (2014) 124(13):2131–41. doi: 10.1182/blood-2013-10-525873
5. Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Gress RE, Kanakry CG. Post-Transplantation Cyclophosphamide Prevents Graft-Versus-Host Disease by Inducing Alloreactive T Cell Dysfunction and Suppression. J Clin Invest (2019) 129(6):2357–73. doi: 10.1172/JCI124218
6. Wachsmuth LP, Patterson MT, Eckhaus MA, Venzon DJ, Kanakry CG. Optimized Timing of Post-Transplantation Cyclophosphamide in MHC-Haploidentical Murine Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2020) 26(2):230–41. doi: 10.1016/j.bbmt.2019.09.030
7. Patterson MT, Wachsmuth LP, Panjabi A, Kanakry JA, Luznik L, Kanakry CG. Mechanisms of CD8+ T-Cell Resistance to Post-Transplantation Cyclophosphamide (PTCy). Biol Blood Marrow Transplant (2019) 25(3, Supplement):S331–S2. doi: 10.1016/j.bbmt.2018.12.535
8. Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and Cancer: Golden Anniversary. Nat Rev Clin Oncol (2009) 6(11):638–47. doi: 10.1038/nrclinonc.2009.146
9. Kanakry CG, Ganguly S, Luznik L. Situational Aldehyde Dehydrogenase Expression by Regulatory T Cells may Explain the Contextual Duality of Cyclophosphamide as Both a Pro-Inflammatory and Tolerogenic Agent. Oncoimmunology (2015) 4(3):e974393. doi: 10.4161/2162402X.2014.974393
10. Köck K, Grube M, Jedlitschky G, Oevermann L, Siegmund W, Ritter CA, et al. Expression of Adenosine Triphosphate-Binding Cassette (ABC) Drug Transporters in Peripheral Blood Cells: Relevance for Physiology and Pharmacotherapy. Clin Pharmacokinetics (2007) 46(6):449–70. doi: 10.2165/00003088-200746060-00001
11. Muzio G, Maggiora M, Paiuzzi E, Oraldi M, Canuto RA. Aldehyde Dehydrogenases and Cell Proliferation. Free Radical Biol Med (2012) 52(4):735–46. doi: 10.1016/j.freeradbiomed.2011.11.033
12. Gottesman MM, Fojo T, Bates SE. Multidrug Resistance in Cancer: Role of ATP–Dependent Transporters. Nat Rev Cancer (2002) 2(1):48–58. doi: 10.1038/nrc706
13. Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC Transporters as Mediators of Drug Resistance and Contributors to Cancer Cell Biology. Drug Resist Update (2016) 26:1–9. doi: 10.1016/j.drup.2016.03.001
14. Januchowski R, Wojtowicz K, Zabel M. The Role of Aldehyde Dehydrogenase (ALDH) in Cancer Drug Resistance. BioMed Pharmacother (2013) 67(7):669–80. doi: 10.1016/j.biopha.2013.04.005
15. Owens AH Jr., Santos GW. The Effect of Cytotoxic Drugs on Graft-Versus-Host Disease in Mice. Transplantation (1971) 11(4):378–82. doi: 10.1097/00007890-197104000-00004
16. Stokes J, Hoffman EA, Zeng Y, Larmonier N, Katsanis E. Post-Transplant Bendamustine Reduces Gvhd While Preserving Gvl in Experimental Haploidentical Bone Marrow Transplantation. Br J Haematol (2016) 174(1):102–16. doi: 10.1111/bjh.14034
17. Katsanis E, Maher K, Roe DJ, Simpson RJ. Progressive Substitution of Posttransplant Cyclophosphamide With Bendamustine: A Phase I Study in Haploidentical Bone Marrow Transplantation. eJHaem (2020) 1(1):286–92. doi: 10.1002/jha2.20
18. Moiseev I, Bondarenko S, Morozova E, Vlasova Y, Dotsenko A, Epifanovskaya O, et al. Graft-Versus-Host Disease Prophylaxis With Post-Transplantation Bendamustine in Patients With Refractory Acute Leukemia: A Dose-Ranging Study. Transplant Cell Ther (2021) 27:601.e1-.e7. doi: 10.1016/j.jtct.2021.03.032
19. van Tellingen O, Buckle T, Jonker JW, van der Valk MA. Beijnen JH. P-Glycoprotein and Mrp1 Collectively Protect the Bone Marrow From Vincristine-Induced Toxicity. vivo Br J Cancer (2003) 89(9):1776–82. doi: 10.1038/sj.bjc.6601363
20. Yang W, Xie J, Hou R, Chen X, Xu Z, Tan Y, et al. Disulfiram/Cytarabine Eradicates a Subset of Acute Myeloid Leukemia Stem Cells With High Aldehyde Dehydrogenase Expression. Leukemia Res (2020) 92:106351. doi: 10.1016/j.leukres.2020.106351
21. Breedveld P, Zelcer N, Pluim D, Sönmezer O, Tibben MM, Beijnen JH, et al. Mechanism of the Pharmacokinetic Interaction Between Methotrexate and Benzimidazoles: Potential Role for Breast Cancer Resistance Protein in Clinical Drug-Drug Interactions. Cancer Res (2004) 64(16):5804–11. doi: 10.1158/0008-5472.Can-03-4062
22. Inoue K, Yuasa H. Molecular Basis for Pharmacokinetics and Pharmacodynamics of Methotrexate in Rheumatoid Arthritis Therapy. Drug Metab Pharmacokinet (2014) 29(1):12–9. doi: 10.2133/dmpk.dmpk-13-rv-119
23. Norris MD, De Graaf D, Haber M, Kavallaris M, Madafiglio J, Gilbert J, et al. Involvement of MDR1 P-Glycoprotein in Multifactorial Resistance to Methotrexate. Int J Cancer (1996) 65(5):613–9. doi: 10.1002/(sici)1097-0215(19960301)65:5<613::Aid-ijc10>3.0.Co;2-8
24. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting Multidrug Resistance in Cancer. Nat Rev Drug Discovery (2006) 5(3):219–34. doi: 10.1038/nrd1984
25. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat Rev Cancer (2018) 18(7):452–64. doi: 10.1038/s41568-018-0005-8
26. Vassalli G. Aldehyde Dehydrogenases: Not Just Markers, But Functional Regulators of Stem Cells. Stem Cells Int (2019) 2019:3904645. doi: 10.1155/2019/3904645
27. Croker AK, Allan AL. Inhibition of Aldehyde Dehydrogenase (ALDH) Activity Reduces Chemotherapy and Radiation Resistance of Stem-Like Aldhhicd44⁺ Human Breast Cancer Cells. Breast Cancer Res Treat (2012) 133(1):75–87. doi: 10.1007/s10549-011-1692-y
28. Nakahata K, Uehara S, Nishikawa S, Kawatsu M, Zenitani M, Oue T, et al. Aldehyde Dehydrogenase 1 (ALDH1) is a Potential Marker for Cancer Stem Cells in Embryonal Rhabdomyosarcoma. PloS One (2015) 10(4):e0125454. doi: 10.1371/journal.pone.0125454
29. Huang RS, Murry DJ, Foster DR. Role of Xenobiotic Efflux Transporters in Resistance to Vincristine. Biomedicine Pharmacother (2008) 62(2):59–64. doi: 10.1016/j.biopha.2007.04.011
30. Aslibekyan S, Brown EE, Reynolds RJ, Redden DT, Morgan S, Baggott JE, et al. Genetic Variants Associated With Methotrexate Efficacy and Toxicity in Early Rheumatoid Arthritis: Results From the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Pharmacogenomics J (2014) 14(1):48–53. doi: 10.1038/tpj.2013.11
31. Karasik A, Váradi A, Szeri F. In Vitro Transport of Methotrexate by Drosophila Multidrug Resistance-Associated Protein. PloS One (2018) 13(10):e0205657. doi: 10.1371/journal.pone.0205657
32. Strumberg D, Harstrick A, Doll K, Hoffmann B, Seeber S. Bendamustine Hydrochloride Activity Against Doxorubicin-Resistant Human Breast Carcinoma Cell Lines. Anti-Cancer Drugs (1996) 7(4):415–21. doi: 10.1097/00001813-199606000-00007
33. Fukuda Y, Schuetz JD. and Their Role in Nucleoside and Nucleotide Drug Resistance. Biochem Pharmacol (2012) 83(8):1073–83. doi: 10.1016/j.bcp.2011.12.042
34. Gandhi V. Metabolism and Mechanisms of Action of Bendamustine: Rationales for Combination Therapies. Semin Oncol (2002) 29(4 Suppl 13):4–11. doi: 10.1053/sonc.2002.34872
35. Kawasoe M, Yamamoto Y, Okawa K, Funato T, Takeda M, Hara T, et al. Acquired Resistance of Leukemic Cells to Arac is Associated With the Upregulation of Aldehyde Dehydrogenase 1 Family Member A2. Exp Hematol (2013) 41(7):597–603.e2. doi: 10.1016/j.exphem.2013.03.004
36. Wang Z, Zhou Q, Kruh GD, Gallo JM. Dose-Dependent Disposition of Methotrexate in Abcc2 and Abcc3 Gene Knockout Murine Models. Drug Metab Dispos (2011) 39(11):2155–61. doi: 10.1124/dmd.111.041228
37. Fotoohi AK, Albertioni F. Mechanisms of Antifolate Resistance and Methotrexate Efficacy in Leukemia Cells. Leukemia Lymphoma (2008) 49(3):410–26. doi: 10.1080/10428190701824569
38. Guo W, Healey JH, Meyers PA, Ladanyi M, Huvos AG, Bertino JR, et al. Mechanisms of Methotrexate Resistance in Osteosarcoma. Clin Cancer Res (1999) 5(3):621–7.
39. Cros E, Jordheim L, Dumontet C, Galmarini CM. Problems Related to Resistance to Cytarabine in Acute Myeloid Leukemia. Leukemia Lymphoma (2004) 45(6):1123–32. doi: 10.1080/1042819032000159861
40. Cai J, Damaraju VL, Groulx N, Mowles D, Peng Y, Robins MJ, et al. Two Distinct Molecular Mechanisms Underlying Cytarabine Resistance in Human Leukemic Cells. Cancer Res (2008) 68(7):2349–57. doi: 10.1158/0008-5472.Can-07-5528
41. Galletti E, Magnani M, Renzulli ML, Botta M. Paclitaxel and Docetaxel Resistance: Molecular Mechanisms and Development of New Generation Taxanes. ChemMedChem (2007) 2(7):920–42. doi: 10.1002/cmdc.200600308
42. Zhang Y, Yang S-H, Guo X-L. New Insights Into Vinca Alkaloids Resistance Mechanism and Circumvention in Lung Cancer. Biomedicine Pharmacother (2017) 96:659–66. doi: 10.1016/j.biopha.2017.10.041
43. Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol Resistance Related to Microtubules. Oncogene (2003) 22(47):7280–95. doi: 10.1038/sj.onc.1206934
44. Friedman HS, Colvin OM, Kaufmann SH, Ludeman SM, Bullock N, Bigner DD, et al. Cyclophosphamide Resistance in Medulloblastoma. Cancer Res (1992) 52(19):5373–8.
45. Lochte HL Jr., Levy AS, Guenther DM, Thomas ED, Ferrebee JW. Prevention of Delayed Foreign Marrow Reaction in Lethally Irradiated Mice by Early Administration of Methotrexate. Nature (1962) 196:1110–1. doi: 10.1038/1961110a0
46. Storb R, Deeg HJ, Thomas ED, Appelbaum FR, Buckner CD, Cheever MA, et al. Marrow Transplantation for Chronic Myelocytic Leukemia: A Controlled Trial of Cyclosporine Versus Methotrexate for Prophylaxis of Graft-Versus-Host Disease. Blood (1985) 66(3):698–702. doi: 10.1182/blood.V66.3.698.698
47. Storb R, Deeg HJ, Whitehead J, Appelbaum F, Beatty P, Bensinger W, et al. Methotrexate and Cyclosporine Compared With Cyclosporine Alone for Prophylaxis of Acute Graft Versus Host Disease After Marrow Transplantation for Leukemia. New Engl J Med (1986) 314(12):729–35. doi: 10.1056/nejm198603203141201
48. Chao NJ, Schmidt GM, Niland JC, Amylon MD, Dagis AC, Long GD, et al. Cyclosporine, Methotrexate, and Prednisone Compared With Cyclosporine and Prednisone for Prophylaxis of Acute Graft-Versus-Host Disease. New Engl J Med (1993) 329(17):1225–30. doi: 10.1056/nejm199310213291703
49. Chao NJ, Snyder DS, Jain M, Wong RM, Niland JC, Negrin RS, et al. Equivalence of 2 Effective Graft-Versus-Host Disease Prophylaxis Regimens: Results of a Prospective Double-Blind Randomized Trial. Biol Blood Marrow Transplant (2000) 6(3):254–61. doi: 10.1016/s1083-8791(00)70007-3
50. Lobo ED, Balthasar JP. Pharmacokinetic-Pharmacodynamic Modeling of Methotrexate-Induced Toxicity in Mice. J Pharm Sci (2003) 92(8):1654–64. doi: 10.1002/jps.10431
51. Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W, et al. Treatment of Acute Leukemia With Unmanipulated HLA-Mismatched/Haploidentical Blood and Bone Marrow Transplantation. Biol Blood Marrow Transplant (2009) 15(2):257–65. doi: 10.1016/j.bbmt.2008.11.025
52. Johnson DR, Finch RA, Lin ZP, Zeiss CJ, Sartorelli AC. The Pharmacological Phenotype of Combined Multidrug-Resistance Mdr1a/1b- and Mrp1-Deficient Mice. Cancer Res (2001) 61(4):1469–76.
53. Le Garrec D, Gori S, Luo L, Lessard D, Smith DC, Yessine MA, et al. Poly(N-Vinylpyrrolidone)-Block-Poly(D,L-Lactide) as a New Polymeric Solubilizer for Hydrophobic Anticancer Drugs: In Vitro and In Vivo Evaluation. J Controlled release: Off J Controlled Release Soc (2004) 99(1):83–101. doi: 10.1016/j.jconrel.2004.06.018
54. Vassileva V, Grant J, De Souza R, Allen C, Piquette-Miller M. Novel Biocompatible Intraperitoneal Drug Delivery System Increases Tolerability and Therapeutic Efficacy of Paclitaxel in a Human Ovarian Cancer Xenograft Model. Cancer Chemother Pharmacol (2007) 60(6):907–14. doi: 10.1007/s00280-007-0449-0
55. Leach WB, Laster WR Jr, Mayo JG, Griswold DP Jr, Schabel FM Jr. Toxicity Studies in Mice Treated With 1-Beta-D-Arabinofuranosylcytosine (Ara-C). Cancer Res (1969) 29(3):529–35.
56. Palo AK, Sahoo D, Choudhury RC. Cytosine Arabinoside-Induced Cytogenotoxicity in Bone Marrow and Spermatogonial Cells of Mice and its Potential Transmission Through the Male Germline. Mutat Res (2009) 673(1):29–36. doi: 10.1016/j.mrgentox.2008.11.007
57. Mayumi H, Good RA. Dependency of Cyclophosphamide-Induced Skin Allograft Tolerance on Age of Adult Recipient Mice. Transplantation (1988) 46(3):451–2. doi: 10.1097/00007890-198809000-00024
58. Mayumi H, Good RA. Long-Lasting Skin Allograft Tolerance in Adult Mice Induced Across Fully Allogeneic (Multimajor H-2 Plus Multiminor Histocompatibility) Antigen Barriers by a Tolerance-Inducing Method Using Cyclophosphamide. J Exp Med (1989) 169(1):213–38. doi: 10.1084/jem.169.1.213%JJournalofExperimentalMedicine
59. Mayumi H, Good RA. The Necessity of Both Allogeneic Antigens and Stem Cells for Cyclophosphamide-Induced Skin Allograft Tolerance in Mice. Immunobiology (1989) 178(4):287–304. doi: 10.1016/S0171-2985(89)80053-1
60. Mayumi H, Umesue M, Nomoto K. Cyclophosphamide-Induced Immunological Tolerance: An Overview. Immunobiology (1996) 195(2):129–39. doi: 10.1016/S0171-2985(96)80033-7
61. Nunes NS, Kanakry CG. Mechanisms of Graft-Versus-Host Disease Prevention by Post-Transplantation Cyclophosphamide: An Evolving Understanding. Front Immunol (2019) 10:2668. doi: 10.3389/fimmu.2019.02668
62. McAdams MJ, Hyder M, Dimitrova D, Sadler JL, McKeown C, Steinberg SM, et al. Phase I/II Study of Reduced Dosing of Post-Transplantation Cyclophosphamide (PTCy) After HLA-Haploidentical Bone Marrow Transplantation. In: Ash Annual Meeting & Exposition. Atlanta, Georgia: American Society of Hematology.
63. Santos G, Tutschka P, Brookmeyer R, Saral R, Beschorner W, Bias W. Cyclosporine Plus Methylprednisolone as Prophylaxis for Graft-Versus-Host Disease: A Randomized Double-Blind Study in Patients Undergoing Allogeneic Marrow Transplantation. Clin Transplant (1987) 1:21.
64. Storb R, Epstein RB, Graham TC, Thomas ED. Methotrexate Regimens for Control of Graft-Versus-Host Disease in Dogs With Allogeneic Marrow Grafts. Transplantation (1970) 9(3):240–6. doi: 10.1097/00007890-197003000-00007
65. Storb R, Rudolph RH, Kolb HJ, Graham TC, Mickelson E, Erickson V, et al. Marrow Grafts Between Dl-a-Matched Canine Littermates. Transplantation (1973) 15(1):92–100. doi: 10.1097/00007890-197301000-00014
66. Storb R, Deeg HJ, Pepe M, Appelbaum F, Anasetti C, Beatty P, et al. Methotrexate and Cyclosporine Versus Cyclosporine Alone for Prophylaxis of Graft-Versus-Host Disease in Patients Given HLA-Identical Marrow Grafts for Leukemia: Long-Term Follow-Up of a Controlled Trial. Blood (1989) 73(6):1729–34. doi: 10.1182/blood.V73.6.1729.1729
67. Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, et al. Human Regulatory T Cells Lack the Cyclophosphamide-Extruding Transporter ABCB1 and are More Susceptible to Cyclophosphamide-Induced Apoptosis. Eur J Immunol (2014) 44(12):3614–20. doi: 10.1002/eji.201444879
68. Chen YB, Elias N, Heher E, McCune JS, Collier K, Li S, et al. Haploidentical Hematopoietic Cell and Kidney Transplantation for Hematological Malignancies and End-Stage Renal Failure. Blood (2019) 134(2):211–5. doi: 10.1182/blood.2019000775
Keywords: graft-versus-host disease (GVHD), haploidentical, alloreactive, bendamustine, methotrexate, regulatory (Treg) cell, post-transplantation cyclophosphamide (PTCy), allogeneic hematopoietic cell transplantation (HCT)
Citation: Hadjis AD, Nunes NS, Khan SM, Fletcher RE, Pohl AdP, Venzon DJ, Eckhaus MA and Kanakry CG (2022) Post-Transplantation Cyclophosphamide Uniquely Restrains Alloreactive CD4+ T-Cell Proliferation and Differentiation After Murine MHC-Haploidentical Hematopoietic Cell Transplantation. Front. Immunol. 13:796349. doi: 10.3389/fimmu.2022.796349
Received: 16 October 2021; Accepted: 06 January 2022;
Published: 15 February 2022.
Edited by:
Dietlinde Wolf, University of Miami, United StatesReviewed by:
John Koreth, Dana–Farber Cancer Institute, United StatesRoberto Crocchiolo, Niguarda Ca’ Granda Hospital, Italy
Copyright © 2022 Hadjis, Nunes, Khan, Fletcher, Pohl, Venzon, Eckhaus and Kanakry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher G. Kanakry, Y2hyaXN0b3BoZXIua2FuYWtyeUBuaWguZ292