 Thierry Gauthier
Thierry Gauthier Wanjun Chen
Wanjun Chen- Mucosal Immunology Section, National Institute of Dental and Craniofacial Research (NIDCR), National Institutes of Health (NIH), Bethesda, MD, United States
Macrophages are essential innate immune cells that contribute to host defense during infection. An important feature of macrophages is their ability to respond to extracellular cues and to adopt different phenotypes and functions in response to these stimuli. The evidence accumulated in the last decade has highlighted the crucial role of metabolic reprogramming during macrophage activation in infectious context. Thus, understanding and manipulation of macrophage immunometabolism during infection could be of interest to develop therapeutic strategies. In this review, we focus on 5 major metabolic pathways including glycolysis, pentose phosphate pathway, fatty acid oxidation and synthesis, tricarboxylic acid cycle and amino acid metabolism and discuss how they sustain and regulate macrophage immune function in response to parasitic, bacterial and viral infections as well as trained immunity. At the end, we assess whether some drugs including those used in clinic and in development can target macrophage immunometabolism for potential therapy during infection with an emphasis on SARS-CoV2 infection.
Introduction
Macrophages are professional phagocytes patrolling most of the tissues, helping to maintain homeostasis and contributing to the first line of defense against pathogens (1). They are notably characterized by a high plasticity and ability to change their phenotype in response to different environmental stimuli (2). Macrophages derive either from an embryonic origin (deriving from the yolk-sac or the liver and maintained throughout the life by self-renewal) or originate from monocyte precursors (differentiating in the tissue after their infiltration) (3). Macrophages notably counter invading pathogens by recognizing defined pathogen associated molecular patterns (also known as PAMP) by a system of pathogen recognition receptors (PRR). Different classes of PRR have been so far described: the ALR (for Absent in melanoma 2 (AIM2)-like receptors) the CLR (for C-type lectin receptors), the NLR (for NOD-like receptors), the RLR (for RIG-I-like receptors), the TLR (for Toll-like receptors) and the cGAS (cyclic GMP–AMP Synthase)-STING (Stimulator of Interferon Genes) signaling (4, 5). ALR are composed of AIM2 and IFI16 (Interferon (IFN)-Inducible protein 16) which can sense cytosolic and nuclear DNA by assembling inflammasomes (6, 7). RLR are composed of RIG-I (Retinoic acid-inducible gene I), MDA-5 (Melanoma differentiation factor-5) and LGP-2 (Laboratory of genetics and physiology-2) which detect viral RNA and DNA. NOD1 (Nucleotide-binding oligomerization domain-containing protein 1) and 2 belong to the NLR and recognize gram positive and negative bacteria. TLR are the best described family of PRR and 10 members belong to this family in human (TLR1 to 10) and 12 in mice (TLR1 to 13 except TLR 10). They recognize a wide variety of PAMP including bacterial, parasitic and viral ligands (4, 8–10). Finally, the cGAS-STING pathway can recognize microbial and cytosolic DNA (11). While all these PRR trigger different molecular signaling, they will lead to the generation of an innate immune response via the production of pro-inflammatory molecules (cytokines, chemokines and DAMP (Damage Associated Molecular Patterns). The PRR recognizing viral PAMP will also trigger the secretion of type I interferon (IFN) which are crucial molecules in the antiviral response (4, 10, 12). These signaling cascades lead to the activation of different components of innate and adaptive immune responses, host cell metabolism and phagocytosis (13, 14). An important hallmark of macrophages is their plasticity in response to environmental cues. During bacterial and viral infections, the PRR stimulation and the pro-inflammatory micro-environment will enable macrophages to be activated toward a pro-inflammatory phenotype (also called classically activated macrophages or M1). On the other hand, a parasitic infection will result in the differentiation of alternatively activated macrophages, notably through the effects of IL-4 and IL-13 (also called anti-inflammatory phenotype, or M2). These different polarized states will help the macrophages to sustain their functions during homeostasis and in diseases, including infections (15–17). Notably, a growing body of evidence show that macrophages change their activation state through reprogramming of the metabolism. These metabolic changes, not only provide energy but also sustain changes in function and phenotype (18). In this review, we will highlight the main metabolic pathways and discuss how they regulate macrophages functions in response to different types of infections. We will also assess how the innate immune memory of macrophages during infections (called trained immunity) can be supported by changes in metabolism. Finally, we will envision the possibility of targeting macrophage immunometabolism as a possible therapeutic target to infections.
Main Metabolic Pathways Used by Macrophages
Cell intrinsic metabolic changes are required in all cells to metabolize nutrients to help their survival, proliferation and differentiation. Five major pathways are used by macrophages to generate energy: i.e. the glycolysis, the tricarboxylic acid (TCA) cycle, the pentose-phosphate pathway (PPP), the fatty acid metabolism [including the fatty acid oxidation (FAO) and the fatty acid synthesis (FAS)] and the amino acid metabolism. In addition to generating energy, macrophages also produce intermediates metabolites that support their phenotype reprogramming in response to external stimuli. Interestingly, these diverse metabolic pathways are closely linked to each other and interconnected as described below. In most cells, glucose is the primary source of energy. Once entering the cells through its transporters, glucose is broken down by glycolysis. Along all these different steps, glycolysis can be diverted to provide metabolites for the PPP pathway or the generation of amino acids, but its primary fate will be to enter the TCA cycle and finally feed the OXPHOS to generate energy in the form of ATP. While it has been thought for decades that the purpose of metabolic pathways is to generate energy, it now appears that producing intermediates metabolites is also important for cellular and molecular signaling (Figure 1). We will discuss in more detail the different steps of these five major pathways in the next sections.
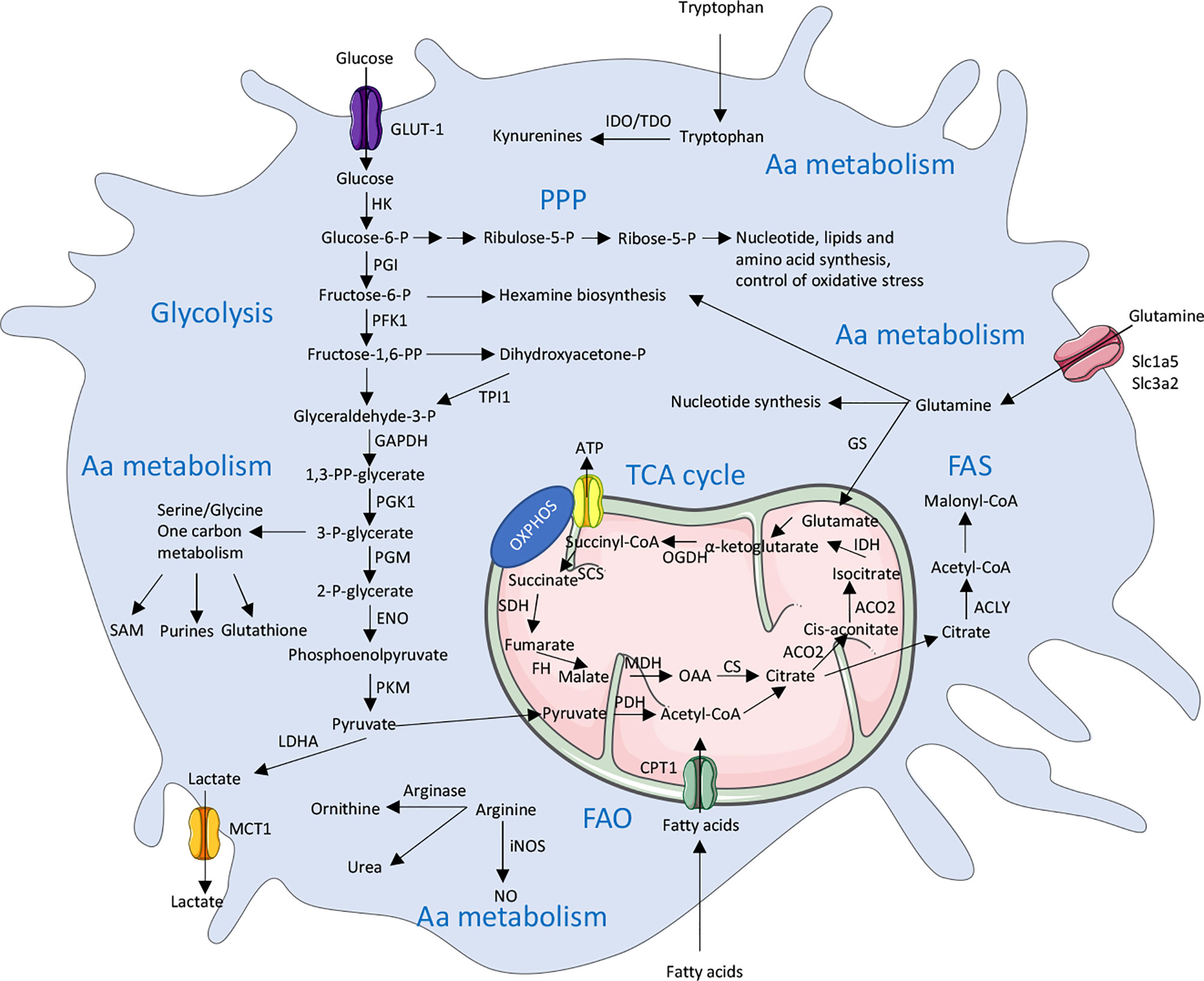
Figure 1 Overview of the main metabolic pathways used by macrophages. There are 5 major pathways used by macrophages to provide energy in cells including glycolysis, TCA (Tricarboxylic acid) cycle, PPP (Pentose phosphate pathway), FAS (Fatty acid synthesis) and FAO (Fatty acid oxidation) and amino acid (Aa) metabolism. These pathways are highly interconnected and are tightly regulated in immune cells, including macrophages. ACLY, ATP citrate lyase; ACO2, Aconitase 2; ATP, Adenosine triphosphate; CPT1, Carnitine palmitoyltransferase 1; CS, Citrate synthase; ENO, Enolase; FH, Fumarase; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; GLUT1, Glucose transporter 1; HK, Hexokinase; GS, Glutamine synthetase; IDH, Isocitrate dehydrogenase; IDO, Indoleamine 2,3-dioxygenase; LDHA, Lactate dehydrogenase; MCT1, Monocarboxylate transporter 1; MDH, Malate dehydrogenase; NO, Nitric oxide; iNOS, inducible NO synthase; OAA, Oxaloacetate; OGDH, α-ketoglutarate dehydrogenase; OXPHOS, Oxidative phosphorylation; P, Phosphate; PDH, Pyruvate dehydrogenase; PFK1,Phosphofructokinase 1; PGK1, Phosphoglycerate kinase 1; PGI, Phosphoglucoisomerase; PGM, Phosphoglycerate mutase; PKM, Pyruvate kinase muscle isotype; PP, bisphosphate; SAM, S-Adenosyl methionine; SCS, Succinyl coenzyme A synthetase; SDH, Succinate dehydrogenase; SLC, Solute carrier; TDO, Tryptophan 2,3-dioxygenase; TPI1, Triosephosphate isomerase 1.
The Glycolytic Pathway
In macrophages, glucose from the extracellular environment typically enters the cell through the glucose transporter GLUT1 (Glucose transporter 1) [encoded by the gene Slc2a1 (Solute Carrier 2a1)] to fulfill glycolysis (19, 20) (Figure 1). Glucose is further catalyzed by the Hexokinases (Hk1-4) which phosphorylates glucose into glucose-6-phosphate. The glucose-6-phosphate then enters the glycolysis (through the form of fructose-6-phosphate) or the PPP (which will be discussed below). Fructose-6-phosphate can also be used by the phosphofructokinases (Pfkl, m, p) into glycolysis or diverted toward the hexosamine biosynthesis pathway. This pathway will lead to the generation of UDP-GlcNAc that is the substrate used for the glycosylation reactions (O- and N-GlcNAcylation). A downstream metabolite of glycolysis, the glyceraldehyde-3-phosphate can also lead to the generation of glycerol-3-phosphate and the biosynthesis of diverse lipids. Another possible break into the glycolysis is to enter the serine and glycine pathway from 3-phosphoglycerate. Serine can further be converted into folate to generate one-carbon units. The final glycolytic enzyme is the pyruvate kinase (PKM1 and 2 are the main isoforms in most tissues) which catalyzes the conversion of phosphoenolpyruvate into pyruvate. Pyruvate is then converted into 2 major metabolites. The first one is lactate which is generated by the lactate dehydrogenase (LDHA and B), and finally exported to outside the cell, in a process that is called aerobic glycolysis. While this process was originally described to occur in cancer cells due to a defect in mitochondria, this aerobic glycolysis clearly occurs during normal cellular processes in immune cells including macrophages. It appears that, despite generating less adenosine triphosphate (ATP) per molecule of glucose used, this mechanism can sustain a rapid activation of immune cells and preserve the redox balance through a tight control of the NADH levels. The second pathway is for the pyruvate to be oxidized in the mitochondria by the pyruvate dehydrogenase (PDH) which convert it into acetyl-coA to enter the TCA cycle (21–23).
The TCA Cycle
The TCA cycle (also called Krebs cycle or citrate cycle) occurs into the mitochondria (Figure 1). It is initiated with the generation of acetyl-coA coming from three possible sources: the pyruvate from glycolysis, the fatty acyl-coA from fatty acids and the acetate (either coming from acetate metabolism or extracellular uptake). The acetyl-coA will, in combination with oxaloacetate, generate citrate which will be further oxidized into the TCA cycle. Citrate can also be exported to the cytosol to generate itaconate or to be hydrolyzed by ATP-citrate lyase (ACLY) in cytosolic acetyl-coA which will fuel the fatty acid and cholesterol synthesis (for the generation of new membranes) or will contribute to protein acetylation (notably histone acetylation) (24). Interestingly, cytosolic citrate can also exert a negative feedback on glycolysis by inhibiting, directly or indirectly, PFK and HK enzymes (25). The major products generated by the TCA cycle are NADH and FADH2 which can be transferred into the electron transport chain to support the oxidative phosphorylation (OXPHOS) and the efficient generation of ATP (26).
The Pentose Phosphate Pathway
The glucose-6-phosphate (G6P) generated by hexokinases can be metabolized to enter the glycolysis or be directed into the PPP (which occurs in the cytosol) (Figure 1). The PPP is divided into 2 phases; an oxidative phase which will give raise to the reduction of NADP+ (Nicotinamide adenine dinucleotide phosphate) into NADPH linked to the conversion of G6P into ribulose-5-phosphate (R5P); a non-oxidative phase will generate ribose-5-phosphate. NADPH is an essential cofactor for the generation of antioxidants, ROS and NO, but also to generate lipids and nucleotides. The R5P is a precursor of nucleotides and amino acids synthesis (27). NADPH can further be used by the Fatty acid synthase to promote the generation of fatty acids or by the enzyme NADPH oxidase 2 (NOX2) to generate reactive oxygen species (ROS) ultimately leading to an oxidative burst (28, 29).
The Fatty Acid Metabolism
Fatty acid Oxidation (FAO) is the most efficient producer of energy for the cell since a single molecule of fatty acid can generate as much as 100 molecules of ATP. The short chain fatty acids can passively enter the mitochondria, while the medium and long chain fatty acids need to be imported by the ligation to coA, which is then exchanged by carnitine palmitoyltransferase 1A (CPT-1A) upon mitochondrial transfer (Figure 1). The carnitine conjugated to the fatty acid is then shuttled into the mitochondria and the carnitine is removed by the CPT2 to give a molecule of fatty acid acyl-coA. The oxidation of this fatty acid will lead to produce large amounts of acetyl-coA, NADH and FADH2 which are used to augment the TCA cycle and the OXPHOS to generate ATP.
Fatty acid synthesis (FAS), on the other hand, uses precursors from the other metabolic pathways (glycolysis, TCA cycle and PPP) to generate lipids. Notably, the acetyl-coA is transformed into malonyl-coA by the acetyl-coA carboxylases. Seven molecules of malonyl-coA are then condensated to generate palmitate (the initial product of fatty acid synthesis) by the enzyme Fatty Acid Synthase. Palmitate, a 16 carbons saturated molecule, is then elongated and desaturated to generate fatty acid of diverse size and degrees of saturation (30, 31).
The Amino Acid Metabolism
Amino acids availability is crucial for multiple aspects of cell biological functions. Since there is a large number of different amino acids, there are different pathways leading to the utilization and generation of amino acids. They can be divided into two categories: the essential amino acids which cannot be synthesized by the human body (and therefore need to be taken from nutrition) and the non-essential amin acids which can be synthesized by the body (32).
An important amino acid for the macrophage behavior is glutamine. Glutamine enters the cell through a diverse range of Slc transporter including Slc1a5 (Solute carrier 1a5) and Slc3a2, which are highly expressed in macrophages (33) (Figure 1). Glutamine can then contribute to the generation of nucleotides or UDP-GlcNac or enter the mitochondria to generate glutamate (34). The glutamate can generate glutathione (which can help to control the redox balance) or be converted into α-ketoglutarate to enter the TCA cycle. The glutamate is also a donor for the generation of many different amino acids (35).
Serine is a central hub for cell metabolism. As described previously, it can be converted from the glycolytic metabolite 3-PG (3-phosphoglycerate). The conversion of Serine to Glycine is an outcome of Serine generation which can later lead to production of glutathione. Serine is also a major source for the one-carbon metabolism pathway which will serve as a building block for S-adenosylmethionine (and the regulation of protein methylation), nucleotides, NAD(P)H, and ATP. Finally, this pathway can also fuel the folate metabolism leading to the production of purines (36).
Another important amino acid in term of immunometabolism is Arginine. Arginine can be produced by many different pathways (including extracellular uptake and intracellular production) to support cell growth and proliferation (37). An important feature, in macrophages, is the ability of arginine to be catalyzed either by NOS (Nitric Oxide Synthase) to generate NO (Nitric oxide) and citrulline or to be catalyzed by Arginase 1 (Arg1) to ornithine and urea (38). Of note, Arg1 has been long described to be expressed by anti-inflammatory M2 macrophages while the expression of iNOS has been demonstrated to be a marker of pro-inflammatory M1 macrophages (38).
L-tryptophan is also an essential amino acid coming from dietary intake. A small fraction of tryptophan is used to the production of proteins and neurotransmitters; however, the major part is used to fuel the kynurenine pathway which give raise to several metabolites. The first step of this reaction is the conversion of tryptophan into N-formylkynurenine, which is catalyzed by the rate-limiting enzymes IDO1,2 (Indoleamine-2,3-dioxygenase 1 and 2) and TDO (Tryptophan-2,3-dioxygenase) (39, 40). Interestingly, it appears that the tryptophan metabolism in macrophages promotes immune tolerance by increasing the generation of M2 macrophages and by depleting extracellular tryptophan, thus modulating T cell functions (41, 42).
While all these metabolic pathways are described as distinct entities, they are highly interdependent and inter-regulated demonstrating a tight and complex regulation of cellular metabolism. A good example of this complex regulation is the kinase serine/threonine kinase mTOR (mammalian Target of Rapamycin). mTOR is composed of 2 different complexes mTORC1 and 2 (mTOR Complex). mTOR activation has been widely demonstrated to be regulated by the level of amino acid in the cell but mTOR is also regulated by the levels of glucose, oxygen and DNA damage (43, 44). Downstream, mTOR can regulate lipid synthesis or the PPP through SREBP (sterol responsive element binding protein) and the glycolysis through the transcription factor Hif-1α (Hypoxia induced factor 1 alpha), both of which can transcriptionally activate genes that encode enzymes belonging to these pathways (45, 46).
Importantly, these different pathways are used by all mammalian cells to generate energy and intermediate metabolites. However, different cells can modulate the use of these pathways to adapt their function, development, or proliferation. In the context of macrophage immunology, it appears that, despite the fact they are long-lived non proliferative cells, pro- and anti-inflammatory macrophages use different pathways to meet their needs, differentiate into M1 and M2 macrophages and perform their function. More specifically, it has now been demonstrated that the pro-inflammatory M1 macrophages rely on an increased dependency on glycolysis and PPP, while they remodel largely their TCA cycle and depend less on OXPHOS to generate energy. This will allow them to sustain an inflammatory phenotype (increased phagocytosis, production of pro-inflammatory cytokines and chemokines, NO and ROS production, and enhanced bacterial killing). On the other hand, the anti-inflammatory M2 macrophages will use the TCA cycle, the FAO and the OXPHOS to generate energy, while relying less on glycolysis. They will also promote the glutamine metabolism and arginase activity. This will promote the expression of M2 markers, the production of anti-inflammatory cytokines and their pro-repair functions. Interestingly, it appears that other pro- or anti-inflammatory immune cells (for example Th1/TH17 versus Treg) use similar pathways than macrophages to sustain their pro- (glycolysis, PPP) and anti-inflammatory (TCA cycle, OXHPOS, FAO) phenotypes. This highlights the crucial role of the microenvironmental cues to modulate immune cell metabolism and functions (18, 47). We will now describe in details how macrophages adapt their metabolic responses during different types of infections.
Macrophage Immunometabolism in Infections
Role of Macrophage Immunometabolism During Parasitic Infection
Parasitic infections such as helminths and protozoans represent a major health concern in developing countries. According to the US Center for Disease Control and Prevention (CDC), malaria (caused by a protozoan named Plasmodium) is responsible for the death of 600 000 people each year, mainly in sub-Saharan Africa. Helminths, in the other side, could infect up to 1.5 billion people worldwide and lead to diverse manifestations like diarrhea, respiratory symptoms, asthma-like symptoms, as well as neurologic and motor disorders (16, 48, 49).
Macrophage Metabolism in Helminth Infection and IL-4 Dependent Polarization
Helminths generate a type 2 immune response in the infected organs, inducing the release of high levels of IL-4 and IL-13, which will instruct the macrophages to adopt an alternatively activated macrophages (50, 51). IL4 and IL13 can be produced by T cells, innate lymphoid cells (ILC), basophils and eosinophils. AAM express typical markers like RELMα (Resistin-like molecule-alpha), VEGF (Vascular Endothelial Growth Factor), Arg1, YM1, IGF-1 (Insulin-like Growth Factor 1) or TGF-β (Transforming Growth Factor Beta) which will help them to control parasite confinement in granulomas and their clearance, as well as tissue repair and control of the immune response. In addition, the transcription factors associated with this AAM phenotype include STAT6 (Signal Transducer and Activator of Transcription 6), GATA3 (GATA binding protein 3) or PPARγ (Peroxisome Proliferator-Activated Receptor gamma) (50, 52) (Figure 2).
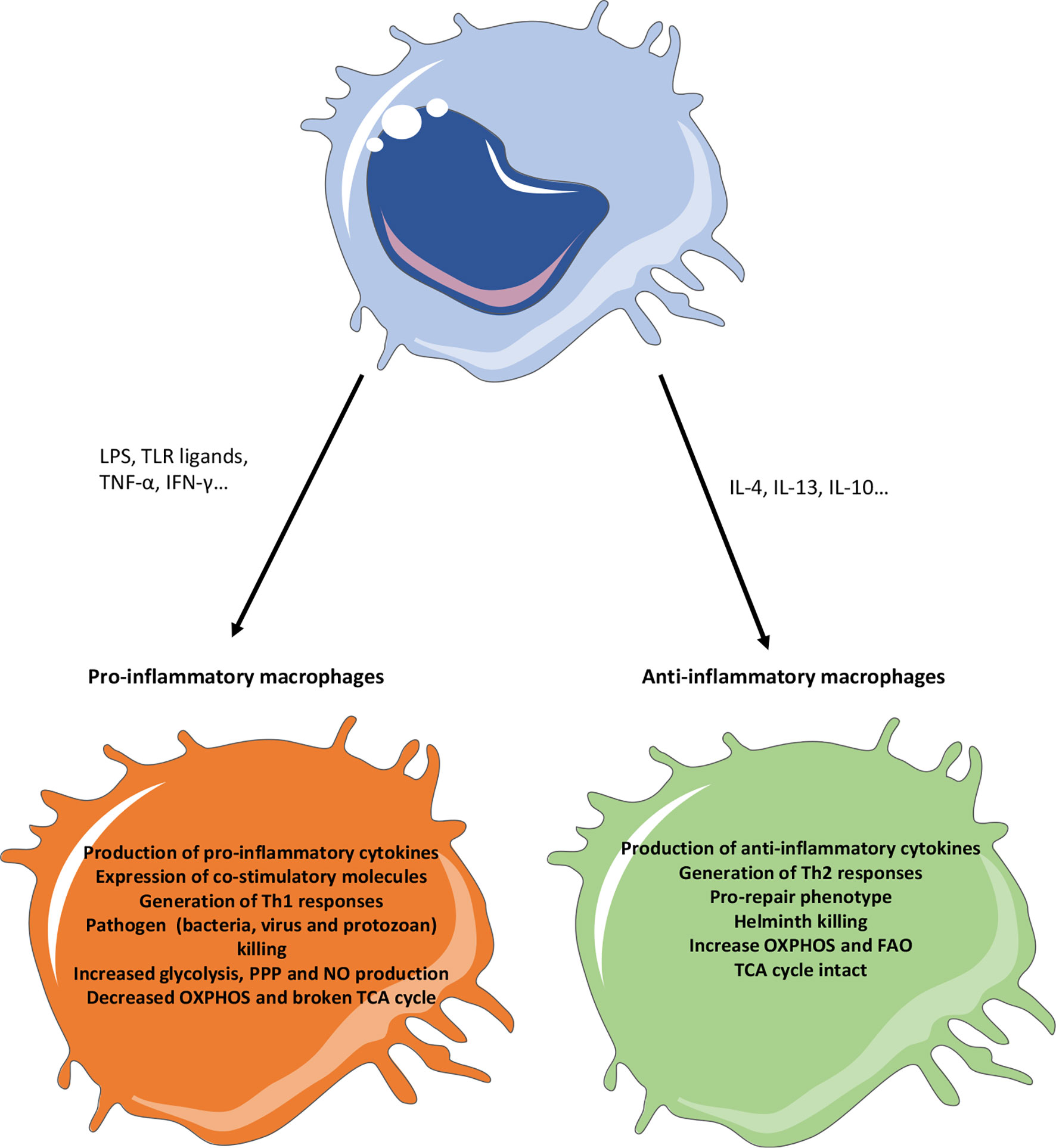
Figure 2 Phenotypic characteristics of pro- versus anti-inflammatory macrophages. Pro-inflammatory stimuli (like TLR ligands or pro-inflammatory cytokines) will generate a pro-inflammatory response in macrophages, notably characterized by the production of pro-inflammatory cytokines, the expression of co-stimulatory molecules and a Th1 response. On the other hand, anti-inflammatory stimuli (like IL4, IL13 or IL10) will promote a pro-repair phenotype in macrophages notably caracterized by the production of anti-inflammatory and pro-resolutive factors and the generation of a Th2 response. In the context of infection, the generation of pro-inflammatory macrophages will promote their killing activity but microbes will try to promote the generation of anti-inflammatory phenotype to escape these responses. Anti-inflammatory macrophages, while promoting infections in general, will have a strong anti-helminth effect. Metabolically, the pro-inflammatory macrophages use glycolysis and PPP to produce energy and have a broken TCA cycle. Instead, anti-inflammatory macrophages use the FAO and OXPHOS to provide cellular energy. FAO, Fatty acid oxidation; IFN, Interferon; IL, Interleukin; LPS, Lipopolysaccharide; OXPHOS, Oxidative phosphorylation; NO, Nitric oxide; PPP, Pentose phosphate pathway; TCA, Tricarboxylic acid; Th, T helper; TLR, Toll like receptor; TNFα, Tumor necrosis factor alpha.
The up-regulation of FAO is a crucial feature of AAM (53, 54). This is orchestrated by a STAT6-PPARγ/PGC-1β signaling pathway, finally leading to the expression of specific markers of AAM and their survival (53, 55, 56). The main source of fatty acids for IL4 treated macrophages is through uptake of fatty acids via the scavenger receptor CD36 or through the lysosomal lipolysis via lysosomal acid lipase, which both sustain the expression of alternative markers (57). Interestingly, during H. polygyrus infection, the inhibition of lipolysis block AAM differentiation and the elimination of the parasite in an IL4 setting (57). However, two recent publications challenged the previous findings demonstrating that FAO is indispensable for AAM polarization. These publications demonstrated that the genetic depletion of Cpt1a and Cpt2 does not inhibit the IL4 induced polarization (58, 59). They also observed that the widely used FAO inhibitor etomoxir, at the doses commonly used to inhibit FAO, inhibits IL4 polarization by targeting the CoA metabolism instead of the FAO (59). In fact, despite its effect observed at low-dose (FAO inhibitor), a high dose of etomoxir can disrupt intracellular CoA homeostasis therefore leading to block the IL4-induced polarization. The example of etomoxir is of great interest for the field of immunometabolism. In fact, many of the findings in this field rely on the use of inhibitor, some of which could have several off-targets, and tightly controlling the dose used when performing experiments appears to be of crucial importance. This also highlights the necessity to confirm the findings observed by using inhibitors with other techniques like gene knock-down and to proceed carefully in the interpretation of the results. The alternatively activated macrophages also increase glycolysis in response to IL4 in a manner dependent on an AKT-mTORC2-IRF4 (Interferon regulatory factor 4) signaling pathway (60, 61). Interestingly, the loss of mTORC2 in macrophages during helminth infection by H. polygyrus (Heligmosomoides polygyrus) lead to lose the AAM polarization and their ability to clear the infection (61). A possible outcome for this increased glycolysis is to feed, together with glutamine, the hexosamine biosynthetic pathway to promote protein glycosylation. In fact, the inhibition of this pathway with tunicamycin (an inhibitor of N-glycosylation) prevent the expression of some AAM markers (54). Another outcome of this increased glycolysis is possibly to fuel lipid synthesis, acetyl-coA production and TCA cycle (54, 62). The pool of acetyl-coA (notably coming from the cleavage of cytosolic citrate from the enzyme ACLY which is activated in an AKT-mTORC1 pathway) is used by macrophage in this context to promote the histone acetylation of IL-4 inducible genes (60). However, some recent publications suggest that the link between glycolysis and AAM might be more complex. This question has been raised because some publications demonstrated that some inhibitors used to study the role of glycolysis (ACLY inhibitor and 2-DG) have a broader effect than just inhibit their primary target (63). In fact, glucose depletion or galactose treatment, while affecting glycolysis, does not affect the AAM polarization. In the meantime, 2-DG, which can suppress both glycolysis and AAM polarization, likely affect the AAM polarization by modulating the ATP levels and the JAK-STAT6 signaling (64). Thus, several questions about the role of glycolysis during IL-4 polarization and helminth infection remain unanswered. Firstly, how the glycolysis pathway is up-regulated during AAM; secondly, what is the exact role of glycolysis during AAM polarization and how it affects helminth infection; and finally, which metabolic pathway the different glycolytic metabolites are preferentially fueling.
In the meantime, IL4 activation limits the use of the PPP in macrophages by increasing the expression of the sedoheptulose kinase CARKL (Carbohydrate kinase-like) that limits the production of sedoheptulose-7-phosphate, thus promoting an alternative activation (65). However, the mechanisms by which the PPP interacts with the IL-4 polarization is not fully understood and needs to be further investigated.
IL4 treated macrophages generate ATP through an oxidative TCA cycle coupled to OXPHOS (54) (Table 1). To fuel the TCA cycle, AAM will use the glutaminolysis or the FAO. The degradation of glutamine through glutaminolysis in IL-4 treated macrophages will generate α-ketoglutarate which will promote the AAM polarization through 3 different mechanisms: 1) it is used to fuel the FAO; 2) it induces an epigenetic reprogramming demethylation of H3K27 on the promoters of AAM-specific genes; 3) it favors PHD activity leading to the inhibition of NFkB pathway (66).
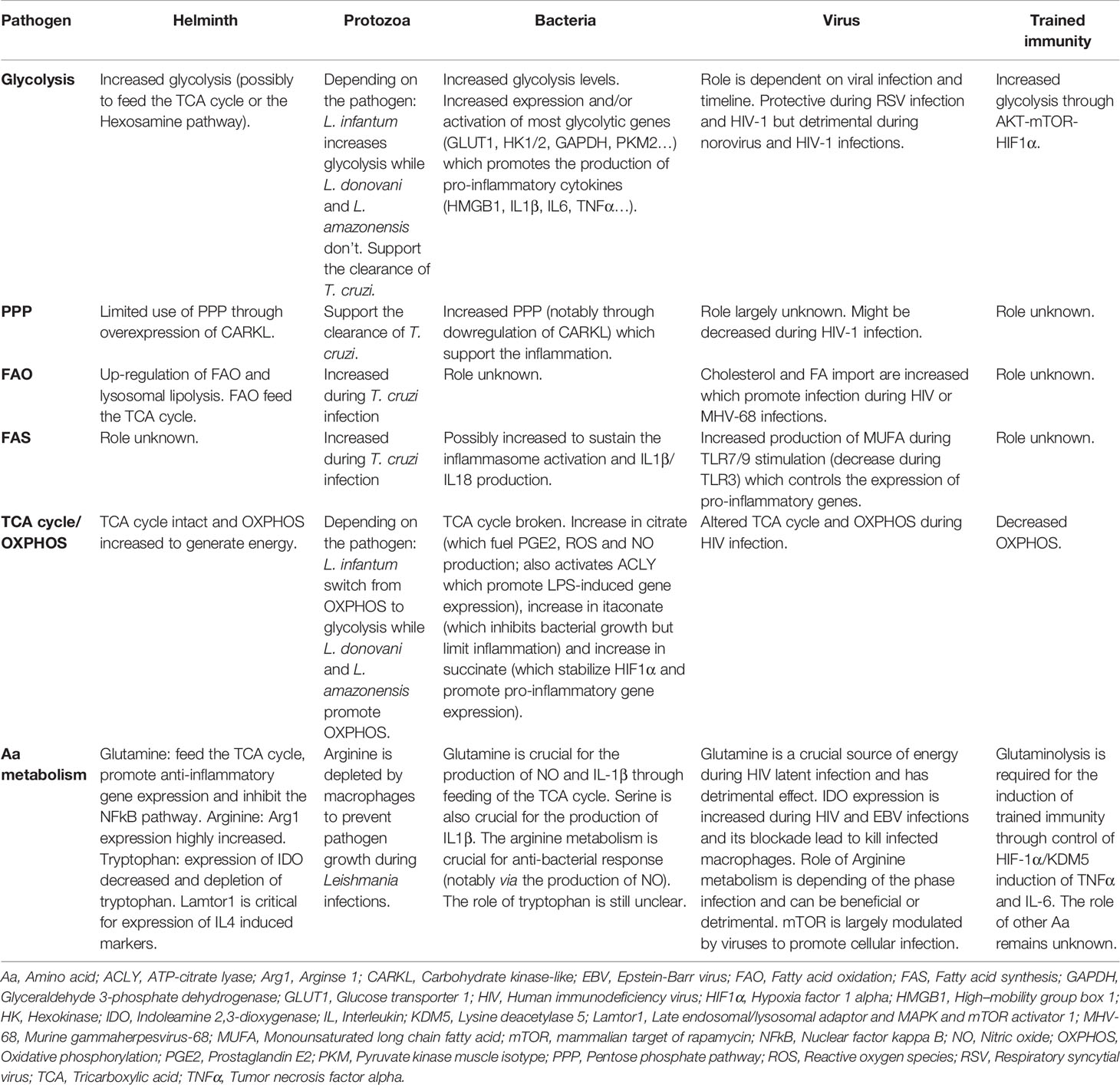
Table 1 Metabolic changes induced during pathogen infections.
A crucial regulator of AAM polarization is the protein LAMTOR1 (Late endosomal/lysosomal adaptor and MAPK and mTOR activator 1). LAMTOR1 is a component of the mTORC1 complex which is necessary for the recruitment of mTORC1 to the lysosome in response to amino acid stimulation (67). Importantly, macrophages deficient for LAMTOR1 or depleted in amino acids in the media are completely unable to express the main IL-4 induced markers (e.g., Arginase I, Mannose receptor, IL10 and RELMα) demonstrating a decisive role of amino acids for the polarization (68). IL4 treated macrophages metabolize arginine to urea and ornithine via an increased expression of the Arg1 (69). Arg1 expression is induced by a STAT6-Cebp/β (CCAAT-enhancer-binding proteins beta) (70, 71). While Arg1 is one of the most used markers to define AAM, its exact role in macrophage polarization remains largely unexplored. Some studies suggest that, notably through the synthesis of polyamines, Arg1 might promote tissue repair during infections (72, 73). Downstream of arginine, the polyamine-eIF5-hypusine pathway regulates IL4 mediated polarization of macrophages and the blockade of this pathway inhibits the protective effect of IL4 during H. polygyrus infection (74). On the other hand, the resistance to infection by the helminth Trichuris muris is unaffected by the deletion of Arg1 in macrophages suggesting more complex roles of arginine metabolism during helminth infection. In this context, macrophages treated by IL4 downregulate the expression of IDO and promote the expression of the immunoregulatory phenylalanine oxidase IL4L1 (IL4-induced gene 1) leading to the depletion of tryptophan (75, 76). However, the consequence of this depletion is largely unexplored. One suggestion is that the depletion of tryptophan in the micro-environment remove a source of energy used by helminths.
Macrophage Metabolism During Protozoan Infection
Macrophages also play a crucial role in the immune responses to protozoan infections. During protozoan infections, macrophage polarization toward a pro-inflammatory phenotype will play a role in the clearance of the pathogen. Macrophages will use the respiratory burst and production of ROS (Reactive oxygen species), NO (Nitric oxide) and pro-inflammatory cytokines (TNFα, IL6, IFNγ) as ways to kill the pathogen and initiate an adaptive immune response if needed (77). Macrophages are responsible for the destruction of the parasites, yet paradoxically also provide a way for parasite to replicate. In fact, protozoan could polarize macrophages toward an anti-inflammatory phenotype (similar to the one induced by IL4 during helminth infection) to escape the killing by macrophages and favor their replication (78). Notably, the expression of Arg1, Mannose receptor (also called CD206) or PPARγ in macrophages are detrimental to the host response during Leishmania infections (79–81).
At basal state, macrophage infection with L. donovani and L. amazonensis increases the OXPHOS levels, without affecting the glycolysis, which is linked to an increase in the production of pro-inflammatory cytokines and chemokines (82) (Table 1). During L. infantum infection, macrophages transiently increase aerobic glycolysis which is followed by a later sustained increased in OXPHOS through a SIRT1 (Sirtuin 1)-LKB1 (Liver kinase B1)-AMPK (AMP-activated protein kinase) pathway. In this context, the deletion of SIRT1 or AMPK in mice led to promote parasite clearance (83). Interestingly, arginine, which is a crucial metabolite for the growth of Leishmania parasite, is depleted by macrophages (either through NO or polyamines). To counterbalance the depletion of arginine in infected macrophages, Leishmania induces the overexpression of many arginine transporter. These findings demonstrate that the interaction between host and pathogen metabolism is crucial to control the infection (84). During Trypanosoma cruzi infection, the glycolysis and OXPHOS do not appear to be modulated in macrophages (82). However, FAO and lipids production are increased, and they promote the pathogen replication. This appears to be dependent on the ability of T. cruzi to promote the expression of LDLR (Low Density Lipoprotein Receptor) therefore leading to the accumulation of LDL and cholesterol into the cells. However, the exact downstream mechanisms remain to be elucidated (85, 86). Interestingly, IFN-γ treatment of macrophages infected with T. cruzi support the up-regulation of a glycolysis-PPP axis important for the production of ROS and NO and the clearance of the pathogen (87). T. brucei produces large amounts of indolepyruvate (a transamination product of tryptophan). This metabolite reduces the host level of HIF1α and the production of IL1β as well as the glycolysis during LPS-induced inflammation (88). While the links between helminth infection and macrophages start to be understood (mostly due to the study of IL4 treated macrophages), further mechanistic studies will be necessary to decipher how macrophages modulate their metabolism to fight protozoa infection compared to how the pathogen modulate their metabolism to favor its survival.
Role of Macrophage Immunometabolism During Bacterial Infection
During encounter of bacteria, macrophages are able to sense the pathogen through the system of PAMP-PRR. A well described PAMP is LPS that signals through TLR4. These stimuli will generate a pro-inflammatory phenotype of macrophages typically characterized by their ability to kill pathogens and elicit an adaptive immune response via antigen presentation. The macrophages express high levels of co-stimulatory molecules like CD40 (Cluster of differentiation 40), CD80, CD86, as well as MHC-II (Major Histocompatibility Complex II) to perform antigen presentation. They also produce pro-inflammatory cytokines such as TNFα, IL6, IL1β, IL12 and IL23 which will promote a TH1 (T helper 1) response leading to the production of IFNγ. The production of these cytokines will also further polarize the macrophages toward a more pro-inflammatory profile through a positive feedback loop. Th1 cells, through production of IFNγ, will reinforce this pro-inflammatory polarization of macrophages notably by enhancing their ability to clear the bacteria (via increased phagocytosis, autophagy, phagolysosomal maturation and promoting cytokines production). In the meantime, the enhanced production of NO and ROS will also provide mechanisms to enhance bacterial clearance. The expression of these factors is controlled by a network of transcription factors including NFkB (Nuclear factor kappa B), STAT1 and 3, HIF1α or the IRFs (15, 23, 89). While the principle of immunometabolism has been documented in many different contexts, the most well studied one is during the bacterial infections associated with LPS stimulation (coupled or not to IFNγ stimuli) (Table 1).
It has been shown that macrophages treated by LPS+IFNγ largely up-regulate their aerobic glycolysis to provide energy to the cell in a more efficient way (54). This increase is mediated by the glucose transporter GLUT1 which is up-regulated after bacterial stimuli. Interestingly, the deletion or up-regulation of Slc2a1 result in changes in the expression of many inflammatory genes (for example nos2, serpine1, mcp-1) (19, 20). The first step of glycolysis is the generation of glucose-6-phosphate by the Hexokinases (and mostly in immune cells by HK1 and HK2). HK1 is regulated by mTORC1 and HK1-induced glycolysis is necessary for the activation of the inflammasome (90). HK2 exerts a similar effect through its localization in the mitochondrial membrane. The release of HK2 from the outer membrane of the mitochondria is a sufficient event to trigger the activation of the NLRP3 inflammasome and the IL1β/IL18 production (91). The rate-limiting enzyme PFKL has been identified as a negative regulator of the oxidative burst in the context of Staphylococcus aureus infection. When PFKL is deleted, glucose is diverted in the PPP rather than entering glycolysis and sustains the production of NADPH finally leading to enhanced bactericidal activity through an unregulated respiratory burst (92). Another glycolytic enzyme, aldolase can also play a role in macrophage immunometabolism. In fact, treatment of macrophages with itaconate, a well-known anti-bacterial product which is a by-product of citrate and TCA activity, induces the inhibition of aldolase during LPS stimulation and prevents the production of IL1β demonstrating that aldolase promotes the production of IL1β (93). Downstream of aldolase, LPS can also regulate GAPDH through the malonylation of its lysine 213 which will regulate its activity and its binding to the TNFα mRNA, leading to an enhance translation and cytokine production (94). Moreover, macrophage treated with 4-Octyl itaconate modulates GAPDH activity and glycolysis leading to a decrease in LPS-induced inflammation in vitro and in a sepsis model (95). Another article linked GAPDH to an anti-inflammatory response in mice. In fact, treatment of LPS-induced sepsis mice with GAPDH lead to decrease inflammation and improve survival in a not fully understood mechanism suggesting that the regulation of GAPDH might be a tight point of control for the inflammatory response. However, in this study, the authors injected high levels (10 mg/kg) of GAPDH originated from rabbit muscle in a systemic manner suggesting that this effect might not be relevant to decipher the physiological role of GAPDH in inflammation (96). α-enolase, which catalyzes the conversion of 2-phosphoglycerate into phosphoenolpyruvate has also been described to be expressed at the surface of monocytes and macrophages from rheumatoid arthritis patients and a mouse model of arthritis. The activation of surface α-enolase trigger the production of inflammatory factors (TNFα, IL1β, IFNγ and PGE2) and could be detrimental for the pathology but could be beneficial during bacterial infection (97). Finally, PKM2, the major isoform of pyruvate kinases expressed in macrophages, is also up-regulated after LPS treatment. Interestingly, the activity of PKM2 is also regulated by its ability to dimerize or tetramerize (while other PK isoforms only exists as tetramers). PKM2 tetramer acts as a pyruvate kinase enzyme and therefore it regulates glycolysis, while the PKM2 dimer can exist in different localizations and has a moonlighting function (it can notably regulates the mitochondria and ER functions or regulates gene expression in the nucleus) (98). LPS induces the tetramerization of PKM2 leading to decrease its nuclear localization and the expression of glycolytic and HIF-1α-induced genes. However, its dimerization promotes the expression of pro-inflammatory genes. These findings are also applicable in vivo since the activation of PKM2 decreases the inflammation and the bacterial load in a model of LPS-sepsis and a model of Salmonella typhimurium infection (99). The knockdown or inhibition of PKM2 confirmed that PKM2 is crucial for the inflammatory effect of LPS since it also inhibits the NLRP3 and AIM2 inflammasomes activation, the HMGB1 (High–mobility group box 1) release and improves the survival of the mice in a model of LPS-induced septic shock (100, 101). Moreover, the inhibition of LDHA (the final step enzyme of glycolysis) might also protect cells against LPS induction of pro-inflammatory genes (102). Finally, most of the glycolytic enzymes are overexpressed after LPS treatment in a HIF-1α-dependent manner, suggesting the regulation of immune responses through glycolytic enzymes might be much wider than previously thought and might also be tightly regulated by the network of transcription factors expressed (47, 103). However, many of the findings obtained to prove the role of glycolysis in supporting inflammatory functions of LPS-treated macrophages rely on the use of inhibitors that may possibly be non-specific (e.g., 2-DG as it has been described before) or on the deletion of transcription factors playing a broad role in the generation of proper immune response (e.g., HIF1α). Thus, it needs more studies and development of specific tools to target glycolysis to fully understand the role of glycolysis in the development of anti-bacterial responses.
The increase in glycolysis in these cells is substantially rerouted toward the PPP by downregulating the inhibitory sedoheptulose kinase CARKL. This increase in PPP in turn supports pro-inflammatory macrophage phenotype. It might promote NADPH production necessary for the NADPH oxidase and iNOS activity thus supporting an antibacterial function (65, 104).
While IL4 treated macrophages are not link to changes in the TCA cycle, LPS-treated macrophages largely remodel their TCA cycle through breaks at several key points of the cycle leading to accumulation of citrate, succinate and itaconate (54, 104, 105). Accumulation of citrate is due to a decreased expression of IDH (Isocitrate dehydrogenase) and increased expression of CIC (Citrate carrier) (leading to its removal from the mitochondria) (54, 106). Citrate can then be utilized to fuel the production of PGE2 (Prostaglandin E2), NO and ROS (through increased FAS and NADPH production) (106). Another crucial role of citrate is to promote histone acetylation via ACLY and the expression of LPS responsive genes (105–108). The next break occurs at the level of itaconate. Itaconate production is enhanced because of the increased expression of IRG1 (Immune-Responsive Gene 1) (54, 109). Itaconate inhibits directly the growth of bacteria like Salmonella enterica and Mycobacterium tuberculosis (Mtb) by targeting the isocitrate lyase demonstrating a strong anti-bacterial effect (109). Despite this effect, itaconate has an anti-inflammatory effect on macrophage activation triggered by LPS+IFNγ treatment by limiting the production of pro-inflammatory factors including IL1β, IL6, IL12, NO or HIF1α. This occurs through different mechanisms that include the regulation of succinate oxidation (and the level of OXPHOS), the activation of a KEAP1 (Kelch-like ECH-associated protein 1)-NRF2 (Nuclear factor erythroid 2-related factor 2) pathway and the control of the ATF3 (Activating transcription factor 3)-IkBζ (Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor zeta) (110, 111). The last break of TCA cycle in response to LPS occurs at the level of succinate. Succinate production is highly induced after LPS treatment in a glutamine dependent manner, which stabilizes HIF-1α and the expression of IL1β (104). Succinate can also promote this pathway through the succinylation of PKM2 by SIRT5. Once PKM2 is succinylated it can form a heterodimer with HIF1α and promote the expression of IL1β (112). Another mechanism by which succinate can activate HIF1α is through its oxidation, coupled with an increased mitochondrial potential membrane, which increases ROS production and thus the stability of HIF1α (113). Interestingly, succinate can also promote inflammation through an autocrine and a paracrine manner via its release into the extracellular milieu and its sensing by the receptor GPR91 (114).
The role of fatty acid in macrophages treated by LPS is poorly understood. While the FAO seems to not be important for macrophage polarization, the fatty acid synthesis might play a role. In fact, LPS-treated macrophages increased their production of triglycerides which is associated with an increase in CD36 expression (115). The production of FA is regulated through a UCP2 (Uncoupling Protein 2)-FASN (Fatty acid synthase) axis which can trigger the activation of the NLRP3 inflammasome and the production of IL1β and IL18. The regulation of this pathway improves the survival in a model of polymicrobial sepsis (116). Salmonella infection promotes the expression of PPARδ in macrophages. This will induce a switch from a glycolytic metabolism toward FA metabolism in host cells and allow Salmonella to use the available glucose to promote its replication (117). Mycobacterium tuberculosis, through an IFNγ-HIF1α axis, promotes the formation of lipid droplets. Lipid droplets are not used by the bacteria for replication, but are rather used by macrophages to promote the production of PGE2 and LXB4 (Lipoxin B4) to support host defense (118). Live Mycobacterium tuberculosis (in contrast the dead or attenuated bacteria) also shifted the mitochondrial and glycolytic metabolism toward quiescence and induces a higher dependency of mitochondria to use exogenous fatty acid as a source of energy (119).
Amino acids also have a role in the regulation of macrophages during bacterial infection. Glutamine is a crucial metabolite for the production of NO as well as for IL1β (120, 121). Of note, a similar phenomenon also occurs in macrophages activated by BCG (122). Glutamine can feed the TCA cycle and is notably responsible for the increase of succinate observed after LPS treatment by inducing a GABA shunt. Inhibiting this glutamine-induced GABA shunt protects mice against LPS-induced sepsis and S. Typhymurium infection in mice (104). While the role of this metabolite is less described, serine is required for the optimal expression of IL1β gene and the blockade of de novo serine synthesis improve survival in a model of LPS-induced sepsis (123). The role of tryptophan metabolism is more controversial. Studies have demonstrated that LPS or IFNγ can induce the expression of IDO and the degradation of tryptophan could have an anti-bacterial effect (47). However, another group reported that IL4L1 can block the LPS effect in macrophages through induction of tryptophan catabolism, suggesting that its exact role needs to be further studied (76). Finally, the arginine metabolism is another crucial pathway to modulate the anti-bacterial response by regulating the balance between citrulline/NO (notably via iNOS) and the levels of ornithine and urea (notably via ARG1) (38, 124). It also appears that Mycobacterium tuberculosis regulates several amino acid transporters and metabolic enzymes but the exact mechanisms by which it affects the host response versus the bacterial survival remains to be elucidated (125).
Despite the well described effect of LPS, other bacterial infections have been described to modulate the host metabolism. LPS has been widely used to study macrophage immunometabolism because it is a simple way to mimic bacterial infections and it shares functional similarities with other TLR ligands. However, it is a single product of gram-negative bacteria (and therefore does not mimic the possible effect of gram-positive bacteria) and it does not mimic the complex in vivo settings in which several TLR ligands and inflammatory mediators, as well as several types of bacteria, can modulate immunometabolism. Besides LPS, infection of human macrophages with Legionella Pneumophila induces aerobic glycolysis and the inhibition of glycolysis by 2-DG reduces bacterial replication. The OXPHOS is however largely suppressed due to mitochondrial fragmentation through accumulation of DNM1L (Dynamin 1 like) (126, 127). L. Pneumophila also induces the production of itaconate by IRG1 which promotes the bacterial clearance as a host protective mechanism (128). Besides these effects, L. Pneumophila modulates the expression of genes involved in lipid and amino acid metabolism but more studies are needed to precisely define the roles of these genes (129). During Mycobacterium tuberculosis infection, interstitial and alveolar macrophages both have different roles and metabolism. In mice, interstitial macrophages use the glycolysis to differentiate toward a pro-inflammatory phenotype and control bacterial growth as well as the mice survival. On the other hand, alveolar macrophages use the fatty acid oxidation and are not able to control bacterial infection. Blocking glycolysis with 2-DG enhances bacterial replication while blocking FAO with etomoxir decreases bacterial replication (130–132). Interestingly, the control of glycolysis in alveolar macrophages appear to be dependent on miR-21 which controls the expression of PFKM and IL1β (133). Finally, and similarly to other pathogens, Staphylococcus aureus has to ability to modulate the host metabolism which has been recently reviewed recently (134–137). Another important metabolic regulation of bacterial infection through metabolism is that bacteria can reroute the macrophage metabolism to use nutrients for their own use which is nicely described elsewhere and might be a key in the macrophage response to infections (138–141).
Role of Macrophage Immunometabolism During Viral Infection
During viral infections, macrophages will elicit a pro-inflammatory response similar to what have been described during bacterial infection. Coupled to this, macrophages will also start producing type I interferons (interferon alpha and beta). The sensing of ssRNA, dsRNA and unmethylated DNA with CpG motifs via TLR3, 7 and 9, respectively, will trigger type I interferon production by macrophages. The RLR family members will also recognize viral motifs and mediate the production of type I interferons (142). Type I interferons will therefore signal through their receptors (Interferon-α/β receptor 1 and 2) which will lead to the activation of the PI3K, MAPKs, STATs and IRF9 ultimately leading to the induction of the ISGs (Interferon Stimulated Genes). Theses ISGs include genes implicated in the mount of antiviral responses, inflammation, pro- and anti-apoptotic molecules as well as regulation of translation and RNA turnover. Type I interferons can be produced by a broad range of cells including macrophages, dendritic cells, epithelial cells, fibroblasts, as well as plasmacytoid dendritic cells (which is the primary source of interferons during viral infection). Type I interferon will therefore signal in the abovementioned cells as well as in T and B cells (143, 144). Interestingly, while macrophages were not supposed to be the primarily source of type I interferon producers, it clearly appears that they are able to control viral infection through production of IFNα/β and are among the first responders during viral infections (145, 146).
The exact role of glycolysis during viral infection remains unclear. First of all, the expression of GLUT1 is increased in monocytes from HIV infected patients and is linked to an increase in glucose uptake and the generation of pro-inflammatory monocytes (147) (Table 1). HIV-I infection in monocytes/macrophages also promotes the expression of HK1 and its localization to the mitochondria thus protecting the cells from apoptosis (148). However, a previous study reported that HIV-I infection of macrophages decreased the glucose uptake and the levels of several glycolytic intermediate (149). These discrepancies suggest that the timeline of infection as well as other parameters like cell differentiation status (monocytes vs macrophages) and the phenotype (pro- vs anti-inflammatory) might play a part for the regulation of glycolysis into HIV-infected macrophages and need to be further studied (150). During Dengue virus infection, the expression of GLUT1 and HK2 are increased as well as the level of early (from glucose to glyceraldehyde 3 phosphate) glycolytic metabolites (G6P, F6P), while the levels of late (from glyceraldehyde 3 phosphate to pyruvate) glycolytic metabolites is increased at shorter time (10 h) and decreased later (48 h). Interestingly, glycolysis supports the viral replication and inhibition of glycolysis using oxamate (a competitive inhibitor of LDH) and 2-DG blocks the viral replication (151). A similar phenomenon occurs during murine norovirus infection (152). While the authors of these two papers did not study further mechanisms, they hypothesized that an increase in glycolysis could promote the generation of biomolecules needed for their replication such as lipids, ATP or NADH. The role of glycolysis during viral infection might be virus dependent. In fact, during VSV (Vesicular stomatitis virus) infection, glycolysis is increased through a type I IFN dependent pathway. The expression of several glycolytic enzymes is increased in this context, and more particularly the expression of PFKFB3 which supports the viral phagocytosis and protects the mice during RSV (Respiratory syncytial virus) infection in vivo (153). Finally, during SARS-CoV2 infection, macrophages largely increase their glycolytic levels which promotes the viral replication and the production of pro-inflammatory cytokines. Mechanistically, the infection induces the production of mROS leading to the stabilization of HIF1α, thus promoting glycolysis. Interestingly, these changes in metabolism inhibit T cell responses and reduce epithelial cell survival (154).
Little is known regarding the role of PPP during viral infections in macrophages. A pioneer study underlined that 6PG and S7P are decreased during HIV-1 infection and the ratio NADP/NADPH is largely decreased (149). However, the functional role of these changes in PPP remains unknown and will have to be further studied. Importantly, it has to be noted that virus, similarly to what has been described above for bacteria, can hijack the host glycolysis (and metabolism in general) in an attempt to use these nutrients to sustain their replication and survival in the host (155, 156).
Similarly, the function of TCA cycle is poorly defined. During HIV infection, the levels of the TCA cycle metabolites are unchanged (except for malate which is increased) (149). However, macrophages surviving HIV infection present an altered TCA cycle and OXPHOS (157).
On the other hand, the activation of macrophages by viruses and their products largely remodels their lipid pool. The stimulation of macrophages with TLR3, 7 and 9 agonists to mimic viral infection modulates the lipid composition of nearly all lipid classes. These changes largely occur through a MyD88 and TRIF signaling pathway and the interferon signaling is also a requirement to these changes. Interestingly, the TLR3 and TLR7/9 signaling differentially modulate the fatty acid synthesis respectively because of their use of the TRIF (TIR-domain-containing adapter-inducing interferon-β) and MyD88 (Myeloid differentiation primary response 88) signaling pathways. While TLR3 stimulation decreased the generation of saturated long chain fatty acid (SFA) and monounsaturated long chain fatty acid (MUFA), the TLR7/9 stimulations increased these levels. Mechanistically, the MyD88-NRF2 (Nuclear factor erythroid 2-related factor 2)/SREBP (Sterol regulatory element-binding protein) axis induces the expression of stearoyl-CoA desaturases 1 and 2 which negatively controls the inflammation (Il1b, Il6 and Cxcl1 expression notably) through an increased production of these MUFAs (158). While this study was performed using only TLR agonists, other publications in different cell types suggest that targeting fatty acid during different types of viral infections might be a strategy to control viral replication (159–161). Type I interferons can also promote the import of cholesterol and long chain fatty acid during murine gammaherpesvirus-68 (MHV-68) infection (but also with HIV). In this setting, blocking the lipid import protects from viral infection through production of type I interferon in a STING-dependent manner (162). Moreover, the infection of macrophages by HIV impairs the cholesterol efflux through a Nef (Negative regulatory factor)-ABCA1 (ATP-binding cassette A1) pathway inducing the formation of foam cells. In the meantime, the activation of TLR8 in macrophages by ssRNA from HIV reinforces this foam cell phenotype through the production of TNFα (163, 164). Besides these effects, the cholesterol has also largely been demonstrated to be crucial for the virus entry in the cell and the anti-viral response (165–168). A growing number of evidence showed that SARS-CoV2 infection is linked to a reprogramming in lipid metabolism with cholesterol playing a crucial role. Indeed, membranes rich in cholesterol are a point of entry of SARS-CoV2 in the cells (169). The enzyme cholesterol 25-hydroxylase (CH25H, belonging to the ISGs) is highly induced during SARS-CoV2 infection and restricts viral infection by depleting cholesterol on the plasma membrane (170, 171). Moreover, SARS-CoV2 promotes the expression of several lipid synthesis modulators (including SREBP1/2, CD36, PPARγ or DGAT-1) leading to the production of cholesterol and lipid droplets. Blockade of this pathway can decrease both the viral replication and the inflammatory response induced by SARS-CoV2 (172, 173).
The modulation of amino acid uptake and production is also a crucial regulator during viral infections. The amino acid glutamine is the main source of energy during HIV latent infection along with glutamate and α-ketoglutarate and blocking the use of these metabolites induces the death of latency infected macrophages (157). EBV (Epstein-Barr virus) and HIV infections both induce the expression of IDO in macrophages in an IL6 and TNFα dependent manner thus inhibiting the activation of T cell activation. Inhibition of IDO eventually lead to the elimination of the macrophages infected by viruses (174, 175). The arginine metabolism is regulated in a complex manner during viral infection. The activation of innate immune responses by viruses induces the production of NO by macrophages and other immune cells (176). But some viruses like the Sendai virus will try to limit the production of NO as a mechanism to escape host responses (177). In fact, while beneficial at first, a sustained production of NO over the time will lead to damage the host tissues and inhibits the Th1 responses (176); (178). Arginine, in the other hand, is a critical metabolite for the replication of viruses and the inhibition of Arg1 reduces the viral replication and ability to infect the host cells (179). However, Arg1 might also promote the tissue repair after viral infection [for an extensive review about the role of arginine metabolism see (180)]. Finally, viruses can target mTOR to modulate the innate immune responses. For example, Vaccinia virus encode the protein F17 which as the ability to disrupt the mTOR complex in the Golgi which will block the activation of STING (Stimulator of interferon genes) and the generation of an interferon-mediated immune response (181, 182). Additionally, viruses can modulate mTORC1 to inhibit host protein translation or promote the translation of their own mRNAs (183–185).
Role of Macrophage Immunometabolism During Trained Immunity
The immune system is classically divided into two arms: the innate immune system and the adaptive immune system. Scientists assumed for a long time that only the adaptive immune system has an immunological memory, and that the innate immune system was only able to sense pathogens in a partially unspecific manner (through the PRR) which does not last over the time. However, this concept has recently been largely challenged. In fact, it appears that innate immune cells do have an ability to develop a broad immunological memory that lasts over the time (and could even be antigen-specific in some cases) (186, 187). These memory-like responses are now well known as trained immunity (188–190). Overall, trained immunity will induce an enhanced inflammatory response in response to secondary stimuli marked by the increased ability of monocytes to produce inflammatory cytokines (notably TNFα and IL6) trough sustained changes in metabolism. In human monocytes, the stimulation with β-glucan followed by 7 days of resting period leads to a decreased level of OXPHOS, increased glucose consumption and lactate production (Table 1). An AKT-mTOR-HIF1α pathway is responsible for this increase in glycolysis and its blocking (either by pharmacological or genetic inhibition) abrogates this trained immunity and is protective in lethal models of C. albicans and S. aureus infections (191). Glutaminolysis is also required for the induction of trained immunity through its ability to sustain the production of fumarate which will modulate the stability of HIF1α and KDM5 (Lysine deacetylase 5) activity, thus promoting the epigenetic reprogramming at the promoter of IL6 and TNFα (192). The cholesterol pathway is also linked to the induction of trained immunity through the induction of mevalonate production (192, 193). Mechanistically, mevalonate promotes the function of IGF1R (Insulin-like growth factor 1 receptor) and mTOR leading to subsequent epigenetic changes (193). A similar glycolysis-AKT-mTOR-epigenetic pathway is also involved during BCG (Bacille Calmette-Guerin)-induced trained immunity demonstrating that this pathway might be a general process during different type of stimuli inducing trained immunity (194). Interestingly, a similar phenomenon is observed in hematopoietic myeloid progenitors and increases the myelopoiesis (195). At the opposite of the concept of trained immunity is the concept of immune tolerance (or immunoparalysis) which induced a persistent tolerance in macrophages over the time, notably in response to LPS (186, 189). Tolerant monocytes from sepsis patients show an impair levels of OXPHOS and glycolysis and IFNγ can restore the metabolic defects through activation of mTOR (196). The state of tolerance is induced by itaconate and β-glucan can revert this state of tolerance by blocking the expression of Irg1 and increasing the expression of Sdh eventually reversing the immunoparalysis (197, 198).
Targeting Macrophage Immunometabolism as a Potential Therapeutic Target for Infections: an Emphasis on COVID-19
Macrophages have, for a long time, be considered potential targets to control immune responses in a wide variety of diseases (199, 200). As new research shed light on the role of immunometabolism in macrophages, it becomes clear that targeting the immunometabolism in macrophages can be a therapeutic target during the development of many diseases including infections. A current focus of research in the past few months has been the development of drugs and vaccines for the SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) infection that leads to the development of COVID-19 (Coronavirus disease 2019) and firstly appeared in December 2019 in Wuhan, China. In fact, the development of potential therapeutics is critically needed since it already affected more than two hundred fifty million people worldwide and led to 5,284,432 deaths (according to the daily WHO report on December 7th). As other viral infections, COVID-19 induces the development of an immune response in which the innate immune cells (and notably macrophages) are the first line of defense (201). Several studies have been reported that the progression to severe forms of infection by COVID-19 (but this also true for many viral and bacterial infections like the development of sepsis) is associated with an overt and dysregulated production of inflammatory factors like IL1β, IL6, TNFα, IFNγ, GMCSF, CCL2, CCL3, CCL4, CXCL10 and many others (202, 203). This cytokine release syndrome (or cytokine storm) is responsible for damages during infections and more particularly into the lungs of patients of sepsis and COVID-19 [also called Acute respiratory distress syndrome (ARDS)] and plays a major role in the related deaths observed in patients with severe conditions (204–207). Importantly, the monocyte/macrophage system is largely remodeled during acute SARS-CoV2 infection with an increased proportion of inflammatory monocyte infiltration in patients with severe condition and macrophages harboring a highly pro-inflammatory phenotype (208, 209). Interestingly, a similar phenomenon occurs during SARS-CoV infection (210, 211). As discussed before, many of these parameters are regulated by metabolism suggesting that targeting metabolism might be a therapeutic strategy to protect against this overt inflammation during severe infections and more particularly during COVID-19. Five drugs targeting metabolism are currently in use clinically to treat different diseases and could be used to prevent the cytokine storm: dimethylfumarate (DMF), metformin, methotrexate, rapamycin and dexamethasone (Table 2).
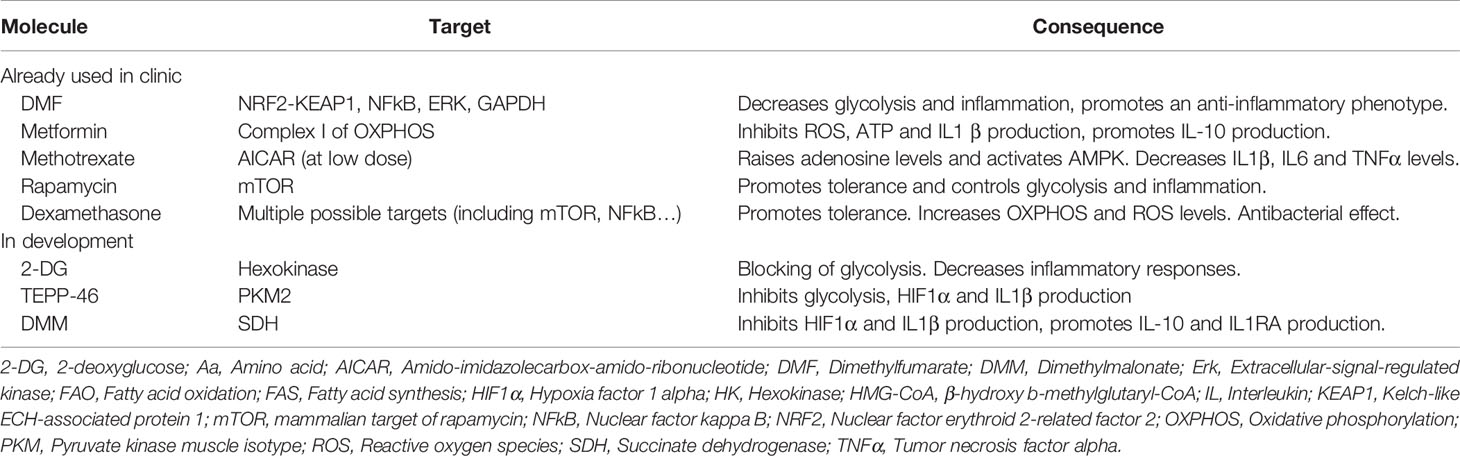
Table 2 Potential therapeutic molecules for the treatment of infections.
Firstly, DMF (a fumarate analog and NRF2 activator currently used for the treatment of multiple sclerosis) notably inhibits NFkB, ERK (Extracellular-signal-regulated kinase) and other signaling pathway. In macrophages, DMF will notably activates NRF2 to protect the cells from oxidative stress and promote an anti-inflammatory phenotype (212). DMF also acts on glycolysis since it decreases the activity of GAPDH suggesting therefore that DMF could be repurposed to modulate immunometabolism during infectious diseases (213). Similarly to DMF, 4-OI (an itaconate analog not used in clinic for the moment) target the NRF2-KEAP1 pathway and might prevent the cytokine storm during acute infections (214). These 2 analogs work by mimicking their respective metabolites suggesting that fumarate and itaconate modulation could be interesting targets to dampen inflammation in infectious settings. Interestingly, a recent report suggested that these 2 drugs might have a potent antiviral and anti-inflammatory activity during COVID-19 (DOI:10.21203/rs.3.rs-31855/v1, under review in Virology).
Metformin is a first-line treatment for the treatment of type 2 diabetes and metabolic disorders notably through a glucose lowering effect. Interestingly, metformin has also an immunomodulatory function. Both these functions depends on the ability of metformin to activate AMPK. This occurs through the ability of metformin to inhibits the complex I of ETC which controls the production of ATP and ROS. The blockade of ATP generation will lead to an increase in AMP or ADP/ATP ratio which will consequently activate AMPK (215). Metformin is known to suppress the production of IL1β and promotes the production of IL10 in response to LPS (216). Interestingly, metformin has been used in the 1940’s as an antimalarial drug as well as to treat influenza and might show interesting properties in the treatment of M. tuberculosis and COVID-19 (217–220). Importantly, three recent studies have suggested that metformin could be used as a therapeutic during HIV, SARS-CoV2 and Mtb infections (219, 221–223). In all pathologies, metformin use has been linked to an improved survival in diabetes and obese patients. Mechanistically, metformin reprograms the immunometabolism of CD4 and CD8 T cells which lead to a modulation of viral replication and enhanced the immune responses. However, whether macrophages can be targeted by metformin remains to be studied (219, 223). Metformin has therefore be proposed to possibly be a treatment for several bacterial, protozoal and viral infections (https://doi.org/10.1002/dmrr.2975), and several clinical trials to assess the use of metformin during HIV (NCT04500678, NCT02383563, NCT02659306, NCT04930744) Mtb (NCT04930744) or SARS-CoV2 (NCT04510194) infections are now ongoing. However, despite a relative good safety profile, metformin is associated with several side effects (notably at the cutaneous and gastro-intestinal tract levels) and approximately 5% of the patients have to discontinue the treatment (224).
At high dose, the methotrexate is an inhibitor of the DHFR (Dihydrofolase reductase) which will block the downstream inhibitors of the folate pathway eventually leading to the inhibition of nucleotide synthesis. At lower dose, methotrexate is inhibiting AICAR (Amido-imidazolecarbox-amido-ribonucleotide) leading to the increased production of the anti-inflammatory factor adenosine. Methotrexate can induce the activation of AMPK and further inhibit the production of IL1β, IL6 and TNFα in macrophages in response to LPS (and can also inhibit the activation of pro-inflammatory B and T cells and promote the generation of Treg) suggesting a potential therapeutic efficacy to control the overt inflammation during SARS-CoV2 infection (225–227). The effect of methotrexate on viral-induced inflammation has been or is currently being tested in two cohorts of SARS-CoV2 and HIV patients and will require further investigations (NCT01949116, NCT04352465). Methotrexate is currently used as an anti-tumoral, anti-psoriatic and anti-arthritic drug. However, due to its immunomodulatory effects, methotrexate is associated to an increased level of infection in rheumatoid arthritis patients. Moreover, its use is also linked to hepatotoxicity, pulmonary toxicity, nephrotoxicity, hematologic toxicity as well as gastrointestinal side effects and carcinogenicity and 20-30% of patients have to stop the usage of this drug due to these side effects (which could remain for up to 5 years) therefore emphasizing the need of more research to determine its potential use to treat infections (228).
Another interesting target is to modulate mTOR by using the inhibitor rapamycin (or similar mTOR inhibitors like everolimus, vistusertib or AZD8055). It has been demonstrated that rapamycin can protect mice against inflammation and death (through control of macrophages) in a model of CLP-induced sepsis (229). Moreover, derivatives of rapamycin were shown to reduce the rate of infection to influenza in elderly in without side effects (230). These findings have led to the hypothesis that rapamycin might be a potential target to treat infections and might be of great interest in severe forms of COVID-19 (231, 232). mTOR has also been hypothesized to be a therapeutic target during Mtb, T cruzi or HIV infections (233–235) and the safety and efficacy of sirolimus is currently being tested in a clinical trial as a Covid-19 treatment (NCT04461340). As an immunosuppressant, blocking mTOR (notably with the use of sirolimus) is linked to development of cancer (especially lymphoma and skin cancer), infections and other adverse events including hyperglycemia and dyslipidemia (236).
Finally, Dexamethasone, a synthetic glucocorticoid with anti-inflammatory and immunosuppressive properties is widely used to treat inflammatory conditions. Dexamethasone acts largely through macrophages by decreasing their production of pro-inflammatory factors (like CCL2, TNFα, COX-2…) (237–239). Dexamethasone is able to promote bacterial phagocytosis and killing by human macrophages in vitro and is protective in a model of LPS-sepsis (240, 241). A possible mechanism of action is also to increase the expression of OXPHOS genes and to promote the production of ROS by macrophages finally leading to suppress the T cell responses (242, 243). Interestingly, dexamethasone has recently been determined to be the first drug to save lives in the SARS-CoV2 infection. The RECOVERY trial enrolled 2100 patients treated with low to intermediate doses of dexamethasone (6 mg per day for 10 days) compared to patients receiving standard care. The survival rate was improved by 30% in patients receiving invasive ventilation and by 20% in patients receiving oxygen support (without invasive mechanical ventilation) (244). However, as a corticosteroid with an immunosuppressive action, dexamethasone has been described to have several side effects and the dose used appears to be critical (245).
Besides the drugs already approved in clinic, the development of drugs inhibiting the metabolites implied in the mounting of an immune response can be targeted as well (Table 2). For example, the development of 2-DG (HK2 inhibitor, currently tested in 219 clinical trials currently), TEPP-46 (PKM2 activator) or dimethylmalonate (DMM, SDH inhibitor, tested in phase 2 clinical trials currently) might be an important advance in the development of immunometabolic inhibitors (226). These finding provide a crucial understanding on how using drugs targeting macrophage immunometabolism (notably to prevent the cytokine storm) might be used as therapeutic targets during infections and more specifically during SARS-CoV2 Infection.
Concluding Remarks
The past decade has seen a great development in our understanding on how the metabolism can regulate immune responses. Macrophages have been demonstrated to be a key player in how immunometabolism regulates the mount of a proper immune response during different types of infections. Although the modulation of metabolism has been largely described in vitro in response to LPS and IL4, its role in different complex microenvironment and more importantly in vivo remains largely poorly understood. Moreover, much of the data has been published in mice and the potential to target metabolism in humans must be further studied. While the different major metabolic pathways are seen as a unique block, they all intersect each other and a break in a unique metabolite might largely affect the cell behavior rendering the things more complex. Despite these limitations, the possibility to target metabolism in macrophages to control infectious disease has shown a great potential and might play an important role in the finding for a cure of different infections, including COVID-19. Indeed, the modulation of macrophages phenotype is a promising target since, contrary to many other immune cell types, it can be targeted in a specific manner through the use of liposomes or other cellular “backpack”, thus limiting specificity and side effects (246, 247). Based on these facts, it is likely that an increase in macrophage immunometabolism understanding will provide new insights to cure infections.
Author Contributions
TG and WC drafted and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Intramural Research Program of NIDCR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pollard JW. Trophic Macrophages in Development and Disease. Nat Rev Immunol (2009) 9(4):259–70. doi: 10.1038/nri2528
2. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
3. Epelman S, Lavine KJ, Randolph GJ. Origin and Functions of Tissue Macrophages. Immunity (2014) 41(1):21–35. doi: 10.1016/j.immuni.2014.06.013
4. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
5. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
6. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 Recognizes Cytosolic Dsdna and Forms a Caspase-1-Activating Inflammasome With ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
7. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host Microbe (2011) 9(5):363–75. doi: 10.1016/j.chom.2011.04.008
8. Takeda K, Akira S. Toll-Like Receptors in Innate Immunity. Int Immunol (2005) 17(1):1–14. doi: 10.1093/intimm/dxh186
9. Ashour DS. Toll-Like Receptor Signaling in Parasitic Infections. Expert Rev Clin Immunol (2015) 11(6):771–80. doi: 10.1586/1744666X.2015.1037286
10. Jang JH, Shin HW, Lee JM, Lee HW, Kim EC, Park SH. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediators Inflammation (2015) 2015:794143. doi: 10.1155/2015/794143
11. Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of Cgas-STING Signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
12. Kumar H, Kawai T, Akira S. Toll-Like Receptors and Innate Immunity. Biochem Biophys Res Commun (2009) 388(4):621–5. doi: 10.1016/j.bbrc.2009.08.062
13. Weiss G, Schaible UE. Macrophage Defense Mechanisms Against Intracellular Bacteria. Immunol Rev (2015) 264(1):182–203. doi: 10.1111/imr.12266
14. Ren Y, Khan FA, Pandupuspitasari NS, Zhang S. Immune Evasion Strategies of Pathogens in Macrophages: The Potential for Limiting Pathogen Transmission. Curr Issues Mol Biol (2017) 21:21–40.
15. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11(11):723–37. doi: 10.1038/nri3073
16. Sica A, Erreni M, Allavena P, Porta C. Macrophage Polarization in Pathology. Cell Mol Life Sci (2015) 72(21):4111–26. doi: 10.1007/s00018-015-1995-y
17. Gordon S, Pluddemann A. Tissue Macrophages: Heterogeneity and Functions. BMC Biol (2017) 15(1):53. doi: 10.1186/s12915-017-0392-4
18. Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: Where are We (Going)? Trends Immunol (2017) 38(6):395–406. doi: 10.1016/j.it.2017.03.001
19. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
20. Freemerman AJ, Zhao L, Pingili AK, Teng B, Cozzo AJ, Fuller AM, et al. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype are Fueled by GLUT1. J Immunol (2019) 202(4):1265–86. doi: 10.4049/jimmunol.1800002
21. Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
22. Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity (2013) 38(4):633–43. doi: 10.1016/j.immuni.2013.04.005
23. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
24. Zaidi N, Swinnen JV, Smans K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res (2012) 72(15):3709–14. doi: 10.1158/0008-5472.CAN-11-4112
25. Yalcin A, Telang S, Clem B, Chesney J. Regulation of Glucose Metabolism by 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatases in Cancer. Exp Mol Pathol (2009) 86(3):174–9. doi: 10.1016/j.yexmp.2009.01.003
26. Anderson NM, Mucka P, Kern JG, Feng H. The Emerging Role and Targetability of the TCA Cycle in Cancer Metabolism. Protein Cell (2018) 9(2):216–37. doi: 10.1007/s13238-017-0451-1
27. Patra KC, Hay N. The Pentose Phosphate Pathway and Cancer. Trends Biochem Sci (2014) 39(8):347–54. doi: 10.1016/j.tibs.2014.06.005
28. Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
29. Panday A, Sahoo MK, Osorio D, Batra S. NADPH Oxidases: An Overview From Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol (2015) 12(1):5–23. doi: 10.1038/cmi.2014.89
30. Jakobsson A, Westerberg R, Jacobsson A. Fatty Acid Elongases in Mammals: Their Regulation and Roles in Metabolism. Prog Lipid Res (2006) 45(3):237–49. doi: 10.1016/j.plipres.2006.01.004
31. Rohrig F, Schulze A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat Rev Cancer (2016) 16(11):732–49. doi: 10.1038/nrc.2016.89
32. Lopez MJ, Mohiuddin SS. Biochemistry, Essential Amino Acids. Treasure Island (FL: StatPearls (2021).
33. Tavakoli S, Downs K, Short JD, Nguyen HN, Lai Y, Jerabek PA, et al. Characterization of Macrophage Polarization States Using Combined Measurement of 2-Deoxyglucose and Glutamine Accumulation: Implications for Imaging of Atherosclerosis. Arterioscler Thromb Vasc Biol (2017) 37(10):1840–8. doi: 10.1161/ATVBAHA.117.308848
34. Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, et al. The Hexosamine Biosynthetic Pathway Couples Growth Factor-Induced Glutamine Uptake to Glucose Metabolism. Genes Dev (2010) 24(24):2784–99. doi: 10.1101/gad.1985910
35. Altman BJ, Stine ZE, Dang CV. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat Rev Cancer (2016) 16(11):749. doi: 10.1038/nrc.2016.114
36. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and Glycine Metabolism in Cancer. Trends Biochem Sci (2014) 39(4):191–8. doi: 10.1016/j.tibs.2014.02.004
37. Wu G, Morris SM Jr Arginine Metabolism: Nitric Oxide and Beyond. Biochem J (1998) 336(Pt 1):1–17. doi: 10.1042/bj3360001
38. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front Immunol (2014) 5:532. doi: 10.3389/fimmu.2014.00532
39. Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: Tryptophan’s Metabolites in Exercise, Inflammation, and Mental Health. Science (2017) 357(6349). doi: 10.1126/science.aaf9794
40. Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat Rev Drug Discov (2019) 18(5):379–401. doi: 10.1038/s41573-019-0016-5
41. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J Exp Med (1999) 189(9):1363–72. doi: 10.1084/jem.189.9.1363
42. Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The Role of Indoleamine 2,3-Dioxygenase (IDO) in Immune Tolerance: Focus on Macrophage Polarization of THP-1 Cells. Cell Immunol (2014) 289(1-2):42–8. doi: 10.1016/j.cellimm.2014.02.005
43. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 Tumor Suppressor Negatively Regulates Mtor Signaling. Cancer Cell (2004) 6(1):91–9. doi: 10.1016/j.ccr.2004.06.007
44. Saxton RA, Sabatini DM. Mtor Signaling in Growth, Metabolism, and Disease. Cell (2017) 169(2):361–71. doi: 10.1016/j.cell.2017.03.035
45. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP Activity is Regulated by Mtorc1 and Contributes to Akt-Dependent Cell Growth. Cell Metab (2008) 8(3):224–36. doi: 10.1016/j.cmet.2008.07.007
46. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a Metabolic Gene Regulatory Network Downstream of Mtor Complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
47. O’Neill LA, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
48. Faz-Lopez B, Morales-Montor J, Terrazas LI. Role of Macrophages in the Repair Process During the Tissue Migrating and Resident Helminth Infections. BioMed Res Int (2016) 2016:8634603. doi: 10.1155/2016/8634603
49. Rolot M, Dewals BG. Macrophage Activation and Functions During Helminth Infection: Recent Advances From the Laboratory Mouse. J Immunol Res (2018) 2018:2790627. doi: 10.1155/2018/2790627
50. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
51. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 Directly Signals Tissue-Resident Macrophages to Proliferate Beyond Homeostatic Levels Controlled by CSF-1. J Exp Med (2013) 210(11):2477–91. doi: 10.1084/jem.20121999
52. Gause WC, Wynn TA, Allen JE. Type 2 Immunity and Wound Healing: Evolutionary Refinement of Adaptive Immunity by Helminths. Nat Rev Immunol (2013) 13(8):607–14. doi: 10.1038/nri3476
53. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
54. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules That Regulate Macrophage Polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
55. Coste A, Dubourdeau M, Linas MD, Cassaing S, Lepert JC, Balard P, et al. Ppargamma Promotes Mannose Receptor Gene Expression in Murine Macrophages and Contributes to the Induction of This Receptor by IL-13. Immunity (2003) 19(3):329–39. doi: 10.1016/S1074-7613(03)00229-2
56. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-Specific Ppargamma Controls Alternative Activation and Improves Insulin Resistance. Nature (2007) 447(7148):1116–20. doi: 10.1038/nature05894
57. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-Intrinsic Lysosomal Lipolysis is Essential for Alternative Activation of Macrophages. Nat Immunol (2014) 15(9):846–55. doi: 10.1038/ni.2956
58. Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty Acid Oxidation in Macrophage Polarization. Nat Immunol (2016) 17(3):216–7. doi: 10.1038/ni.3366
59. Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting Coa Homeostasis. Cell Metab (2018) 28(3):490–503 e497. doi: 10.1016/j.cmet.2018.06.001
60. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-Mtorc1 Signaling Regulates Acly to Integrate Metabolic Input to Control of Macrophage Activation. Elife (2016) 5. doi: 10.7554/eLife.11612
61. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic Reprogramming Mediated by the Mtorc2-IRF4 Signaling Axis is Essential for Macrophage Alternative Activation. Immunity (2016) 45(4):817–30. doi: 10.1016/j.immuni.2016.09.016
62. Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a Central Regulator of Immunometabolic Function. Nat Immunol (2016) 17(9):1046–56. doi: 10.1038/ni.3532
63. Namgaladze D, Zukunft S, Schnutgen F, Kurrle N, Fleming I, Fuhrmann D, et al. Polarization of Human Macrophages by Interleukin-4 Does Not Require ATP-Citrate Lyase. Front Immunol (2018) 9:2858. doi: 10.3389/fimmu.2018.02858
64. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic Stimulation is Not a Requirement for M2 Macrophage Differentiation. Cell Metab (2018) 28(3):463–475 e464. doi: 10.1016/j.cmet.2018.08.012
65. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization Through Control of Glucose Metabolism. Cell Metab (2012) 15(6):813–26. doi: 10.1016/j.cmet.2012.04.023
66. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. Alpha-Ketoglutarate Orchestrates Macrophage Activation Through Metabolic and Epigenetic Reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
67. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag Complex Targets Mtorc1 to the Lysosomal Surface and is Necessary for its Activation by Amino Acids. Cell (2010) 141(2):290–303. doi: 10.1016/j.cell.2010.02.024
68. Kimura T, Nada S, Takegahara N, Okuno T, Nojima S, Kang S, et al. Polarization of M2 Macrophages Requires Lamtor1 That Integrates Cytokine and Amino-Acid Signals. Nat Commun (2016) 7:13130. doi: 10.1038/ncomms13130
69. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase Induction by Suppressors of Nitric Oxide Synthesis (IL-4, IL-10 and PGE2) in Murine Bone-Marrow-Derived Macrophages. Biochem Biophys Res Commun (1995) 206(2):667–73. doi: 10.1006/bbrc.1995.1094
70. Serrat N, Pereira-Lopes S, Comalada M, Lloberas J, Celada A. Deacetylation of C/Ebpbeta is Required for IL-4-Induced Arginase-1 Expression in Murine Macrophages. Eur J Immunol (2012) 42(11):3028–37. doi: 10.1002/eji.201242413
71. Osorio EY, Travi BL, da Cruz AM, Saldarriaga OA, Medina AA, Melby PC. Growth Factor and Th2 Cytokine Signaling Pathways Converge at STAT6 to Promote Arginase Expression in Progressive Experimental Visceral Leishmaniasis. PloS Pathog (2014) 10(6):e1004165. doi: 10.1371/journal.ppat.1004165
72. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential Regulation of Nitric Oxide Synthase-2 and Arginase-1 by Type 1/Type 2 Cytokines In Vivo: Granulomatous Pathology is Shaped by the Pattern of L-Arginine Metabolism. J Immunol (2001) 167(11):6533–44. doi: 10.4049/jimmunol.167.11.6533
73. Yang Z, Ming XF. Functions of Arginase Isoforms in Macrophage Inflammatory Responses: Impact on Cardiovascular Diseases and Metabolic Disorders. Front Immunol (2014) 5:533. doi: 10.3389/fimmu.2014.00533
74. Puleston DJ, Buck MD, Klein Geltink RI, Kyle RL, Caputa G, O’Sullivan D, et al. Polyamines and Eif5a Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab (2019) 30(2):352–363 e358. doi: 10.1016/j.cmet.2019.05.003
75. Musso T, Gusella GL, Brooks A, Longo DL, Varesio L. Interleukin-4 Inhibits Indoleamine 2,3-Dioxygenase Expression in Human Monocytes. Blood (1994) 83(5):1408–11. doi: 10.1182/blood.V83.5.1408.bloodjournal8351408
76. Yue Y, Huang W, Liang J, Guo J, Ji J, Yao Y, et al. IL4I1 is a Novel Regulator of M2 Macrophage Polarization That can Inhibit T Cell Activation via L-Tryptophan and Arginine Depletion and IL-10 Production. PloS One (2015) 10(11):e0142979. doi: 10.1371/journal.pone.0142979
77. Stafford JL, Neumann NF, Belosevic M. Macrophage-Mediated Innate Host Defense Against Protozoan Parasites. Crit Rev Microbiol (2002) 28(3):187–248. doi: 10.1080/1040-840291046731
78. Tomiotto-Pellissier F, Bortoleti B, Assolini JP, Goncalves MD, Carloto ACM, Miranda-Sapla MM, et al. Macrophage Polarization in Leishmaniasis: Broadening Horizons. Front Immunol (2018) 9:2529. doi: 10.3389/fimmu.2018.02529
79. Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, et al. Arginase I Induction by Modified Lipoproteins in Macrophages: A Peroxisome Proliferator-Activated Receptor-Gamma/Delta-Mediated Effect That Links Lipid Metabolism and Immunity. Mol Endocrinol (2008) 22(6):1394–402. doi: 10.1210/me.2007-0525
80. Abebe T, Hailu A, Woldeyes M, Mekonen W, Bilcha K, Cloke T, et al. Local Increase of Arginase Activity in Lesions of Patients With Cutaneous Leishmaniasis in Ethiopia. PloS Negl Trop Dis (2012) 6(6):e1684. doi: 10.1371/journal.pntd.0001684
81. Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, et al. Mannose Receptor High, M2 Dermal Macrophages Mediate Nonhealing Leishmania Major Infection in a Th1 Immune Environment. J Exp Med (2018) 215(1):357–75. doi: 10.1084/jem.20171389
82. Ty MC, Loke P, Alberola J, Rodriguez A, Rodriguez-Cortes A. Immuno-Metabolic Profile of Human Macrophages After Leishmania and Trypanosoma Cruzi Infection. PloS One (2019) 14(12):e0225588. doi: 10.1371/journal.pone.0225588
83. Moreira D, Rodrigues V, Abengozar M, Rivas L, Rial E, Laforge M, et al. Leishmania Infantum Modulates Host Macrophage Mitochondrial Metabolism by Hijacking the SIRT1-AMPK Axis. PloS Pathog (2015) 11(3):e1004684. doi: 10.1371/journal.ppat.1004684
84. Goldman-Pinkovich A, Balno C, Strasser R, Zeituni-Molad M, Bendelak K, Rentsch D, et al. An Arginine Deprivation Response Pathway is Induced in Leishmania During Macrophage Invasion. PloS Pathog (2016) 12(4):e1005494. doi: 10.1371/journal.ppat.1005494
85. Johndrow C, Nelson R, Tanowitz H, Weiss LM, Nagajyothi F. Trypanosoma Cruzi Infection Results in an Increase in Intracellular Cholesterol. Microbes Infect (2014) 16(4):337–44. doi: 10.1016/j.micinf.2014.01.001
86. Miao Q, Ndao M. Trypanosoma Cruzi Infection and Host Lipid Metabolism. Mediators Inflammation (2014) 2014:902038. doi: 10.1155/2014/902038
87. Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma Cruzi in Macrophages. Front Immunol (2018) 9:202. doi: 10.3389/fimmu.2018.00202
88. McGettrick AF, Corcoran SE, Barry PJ, McFarland J, Cres C, Curtis AM, et al. Trypanosoma Brucei Metabolite Indolepyruvate Decreases HIF-1alpha and Glycolysis in Macrophages as a Mechanism of Innate Immune Evasion. Proc Natl Acad Sci USA (2016) 113(48):E7778–87. doi: 10.1073/pnas.1608221113
89. Labonte AC, Tosello-Trampont AC, Hahn YS. The Role of Macrophage Polarization in Infectious and Inflammatory Diseases. Mol Cells (2014) 37(4):275–85. doi: 10.14348/molcells.2014.2374
90. Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, et al. Mtorc1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep (2015) 12(1):102–15. doi: 10.1016/j.celrep.2015.05.046
91. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell (2016) 166(3):624–36. doi: 10.1016/j.cell.2016.05.076
92. Graham DB, Becker CE, Doan A, Goel G, Villablanca EJ, Knights D, et al. Functional Genomics Identifies Negative Regulatory Nodes Controlling Phagocyte Oxidative Burst. Nat Commun (2015) 6:7838. doi: 10.1038/ncomms8838
93. Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, et al. S-Glycosylation-Based Cysteine Profiling Reveals Regulation of Glycolysis by Itaconate. Nat Chem Biol (2019) 15(10):983–91. doi: 10.1038/s41589-019-0323-5
94. Galvan-Pena S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, et al. Malonylation of GAPDH is an Inflammatory Signal in Macrophages. Nat Commun (2019) 10(1):338. doi: 10.1038/s41467-018-08187-6
95. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH to Exert Anti-Inflammatory Effects. Nat Commun (2019) 10(1):5091. doi: 10.1038/s41467-019-13078-5
96. Takaoka Y, Goto S, Nakano T, Tseng HP, Yang SM, Kawamoto S, et al. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Prevents Lipopolysaccharide (LPS)-Induced, Sepsis-Related Severe Acute Lung Injury in Mice. Sci Rep (2014) 4:5204. doi: 10.1038/srep05204
97. Bae S, Kim H, Lee N, Won C, Kim HR, Hwang YI, et al. Alpha-Enolase Expressed on the Surfaces of Monocytes and Macrophages Induces Robust Synovial Inflammation in Rheumatoid Arthritis. J Immunol (2012) 189(1):365–72. doi: 10.4049/jimmunol.1102073
98. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, Function and Expression and Regulation. Cell Biosci (2019) 9:52. doi: 10.1186/s13578-019-0317-8
99. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1alpha Activity and IL-1beta Induction and is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
100. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat Commun (2014) 5:4436. doi: 10.1038/ncomms5436
101. Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, et al. PKM2-Dependent Glycolysis Promotes NLRP3 and AIM2 Inflammasome Activation. Nat Commun (2016) 7:13280. doi: 10.1038/ncomms13280
102. Song YJ, Kim A, Kim GT, Yu HY, Lee ES, Park MJ, et al. Inhibition of Lactate Dehydrogenase a Suppresses Inflammatory Response in RAW 264.7 Macrophages. Mol Med Rep (2019) 19(1):629–37. doi: 10.3892/mmr.2018.9678
103. Liu L, Lu Y, Martinez J, Bi Y, Lian G, Wang T, et al. Proinflammatory Signal Suppresses Proliferation and Shifts Macrophage Metabolism From Myc-Dependent to HIF1alpha-Dependent. Proc Natl Acad Sci USA (2016) 113(6):1564–9. doi: 10.1073/pnas.1518000113
104. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
105. Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-Like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity (2019) 51(6):997–1011 e1017. doi: 10.1016/j.immuni.2019.11.009
106. Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A Key Role of the Mitochondrial Citrate Carrier (SLC25A1) in Tnfalpha- and Ifngamma-Triggered Inflammation. Biochim Biophys Acta (2014) 1839(11):1217–25. doi: 10.1016/j.bbagrm.2014.07.013
107. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-Citrate Lyase is Essential for Macrophage Inflammatory Response. Biochem Biophys Res Commun (2013) 440(1):105–11. doi: 10.1016/j.bbrc.2013.09.037
108. Palmieri EM, Spera I, Menga A, Infantino V, Porcelli V, Iacobazzi V, et al. Acetylation of Human Mitochondrial Citrate Carrier Modulates Mitochondrial Citrate/Malate Exchange Activity to Sustain NADPH Production During Macrophage Activation. Biochim Biophys Acta (2015) 1847(8):729–38. doi: 10.1016/j.bbabio.2015.04.009
109. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Natl Acad Sci USA (2013) 110(19):7820–5. doi: 10.1073/pnas.1218599110
110. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
111. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic Properties of Itaconate and Derivatives Regulate the Ikappabzeta-ATF3 Inflammatory Axis. Nature (2018) 556(7702):501–4. doi: 10.1038/s41586-018-0052-z
112. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep (2017) 19(11):2331–44. doi: 10.1016/j.celrep.2017.05.065
113. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–470 e413. doi: 10.1016/j.cell.2016.08.064
114. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 Senses Extracellular Succinate Released From Inflammatory Macrophages and Exacerbates Rheumatoid Arthritis. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
115. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of Triglyceride Accumulation in Activated Macrophages. J Leukoc Biol (2012) 92(4):829–39. doi: 10.1189/jlb.1111537
116. Moon JS, Lee S, Park MA, Siempos II, M. Haslip PJ, Yun M, et al. UCP2-Induced Fatty Acid Synthase Promotes NLRP3 Inflammasome Activation During Sepsis. J Clin Invest (2015) 125(2):665–80. doi: 10.1172/JCI78253
117. Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, et al. Salmonella Require the Fatty Acid Regulator Ppardelta for the Establishment of a Metabolic Environment Essential for Long-Term Persistence. Cell Host Microbe (2013) 14(2):171–82. doi: 10.1016/j.chom.2013.07.010
118. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid Droplet Formation in Mycobacterium Tuberculosis Infected Macrophages Requires IFN-Gamma/HIF-1alpha Signaling and Supports Host Defense. PloS Pathog (2018) 14(1):e1006874. doi: 10.1371/journal.ppat.1006874
119. Cumming BM, Addicott KW, Adamson JH, Steyn AJ. Mycobacterium Tuberculosis Induces Decelerated Bioenergetic Metabolism in Human Macrophages. Elife (2018) 7. doi: 10.7554/eLife.39169
120. Wallace C, Keast D. Glutamine and Macrophage Function. Metabolism (1992) 41(9):1016–20. doi: 10.1016/0026-0495(92)90130-3
121. Bellows CF, Jaffe BM. Glutamine is Essential for Nitric Oxide Synthesis by Murine Macrophages. J Surg Res (1999) 86(2):213–9. doi: 10.1006/jsre.1999.5713
122. Murphy C, Newsholme P. Importance of Glutamine Metabolism in Murine Macrophages and Human Monocytes to L-Arginine Biosynthesis and Rates of Nitrite or Urea Production. Clin Sci (Lond) (1998) 95(4):397–407. doi: 10.1042/cs0950397
123. Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, et al. Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab (2019) 29(4):1003–1011 e1004. doi: 10.1016/j.cmet.2019.01.014
124. Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, et al. Sustained Generation of Nitric Oxide and Control of Mycobacterial Infection Requires Argininosuccinate Synthase 1. Cell Host Microbe (2012) 12(3):313–23. doi: 10.1016/j.chom.2012.07.012
125. Jiang Q, Shi L. Coordination of the Uptake and Metabolism of Amino Acids in Mycobacterium Tuberculosis-Infected Macrophages. Front Immunol (2021) 12:711462. doi: 10.3389/fimmu.2021.711462
126. Escoll P, Song OR, Viana F, Steiner B, Lagache T, Olivo-Marin JC, et al. Legionella Pneumophila Modulates Mitochondrial Dynamics to Trigger Metabolic Repurposing of Infected Macrophages. Cell Host Microbe (2017) 22(3):302–316 e307. doi: 10.1016/j.chom.2017.07.020
127. Price JV, Jiang K, Galantowicz A, Freifeld A, Vance RE. Legionella Pneumophila is Directly Sensitive to 2-Deoxyglucose-Phosphate via its Uhpc Transporter But is Indifferent to Shifts in Host Cell Glycolytic Metabolism. J Bacteriol (2018) 200(16). doi: 10.1128/JB.00176-18
128. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. Ifns Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection via IRG1-Derived Itaconic Acid. PloS Pathog (2016) 12(2):e1005408. doi: 10.1371/journal.ppat.1005408
129. Price CT, Abu Kwaik Y. The Transcriptome of Legionella Pneumophila-Infected Human Monocyte-Derived Macrophages. PloS One (2014) 9(12):e114914. doi: 10.1371/journal.pone.0114914
130. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1alpha is an Essential Mediator of IFN-Gamma-Dependent Immunity to Mycobacterium Tuberculosis. J Immunol (2016) 197(4):1287–97. doi: 10.4049/jimmunol.1600266
131. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium Tuberculosis In Vivo Segregates With Host Macrophage Metabolism and Ontogeny. J Exp Med (2018) 215(4):1135–52. doi: 10.1084/jem.20172020
132. Vrieling F, Kostidis S, Spaink HP, Haks MC, Mayboroda OA, Ottenhoff THM, et al. Analyzing the Impact of Mycobacterium Tuberculosis Infection on Primary Human Macrophages by Combined Exploratory and Targeted Metabolomics. Sci Rep (2020) 10(1):7085. doi: 10.1038/s41598-020-62911-1
133. Hackett EE, Charles-Messance H, O’Leary SM, Gleeson LE, Munoz-Wolf N, Case S, et al. Mycobacterium Tuberculosis Limits Host Glycolysis and IL-1beta by Restriction of PFK-M via Microrna-21. Cell Rep (2020) 30(1):124–136 e124. doi: 10.1016/j.celrep.2019.12.015
134. Yamada KJ, Heim CE, Aldrich AL, Gries CM, Staudacher AG, Kielian T. Arginase-1 Expression in Myeloid Cells Regulates Staphylococcus Aureus Planktonic But Not Biofilm Infection. Infect Immun (2018) 86(7). doi: 10.1128/IAI.00206-18
135. Horn CM, Kielian T. Crosstalk Between Staphylococcus Aureus and Innate Immunity: Focus on Immunometabolism. Front Immunol (2020) 11:621750. doi: 10.3389/fimmu.2020.621750
136. Wong Fok Lung T, Monk IR, Acker KP, Mu A, Wang N, Riquelme SA, et al. Staphylococcus Aureus Small Colony Variants Impair Host Immunity by Activating Host Cell Glycolysis and Inducing Necroptosis. Nat Microbiol (2020) 5(1):141–53. doi: 10.1038/s41564-019-0597-0
137. Yamada KJ, Heim CE, Xi X, Attri KS, Wang D, Zhang W, et al. Monocyte Metabolic Reprogramming Promotes Pro-Inflammatory Activity and Staphylococcus Aureus Biofilm Clearance. PloS Pathog (2020) 16(3). doi: 10.1371/journal.ppat.1008354
138. Kentner D, Martano G, Callon M, Chiquet P, Brodmann M, Burton O, et al. Shigella Reroutes Host Cell Central Metabolism to Obtain High-Flux Nutrient Supply for Vigorous Intracellular Growth. Proc Natl Acad Sci USA (2014) 111(27):9929–34. doi: 10.1073/pnas.1406694111
139. Czyz DM, Willett JW, Crosson S. Brucella Abortus Induces a Warburg Shift in Host Metabolism That is Linked to Enhanced Intracellular Survival of the Pathogen. J Bacteriol (2017) 199(15). doi: 10.1128/JB.00227-17
140. Sprenger M, Kasper L, Hensel M, Hube B. Metabolic Adaptation of Intracellular Bacteria and Fungi to Macrophages. Int J Med Microbiol (2018) 308(1):215–27. doi: 10.1016/j.ijmm.2017.11.001
141. Russell DG, Huang L, VanderVen BC. Immunometabolism at the Interface Between Macrophages and Pathogens. Nat Rev Immunol (2019) 19(5):291–304. doi: 10.1038/s41577-019-0124-9
142. Kawai T, Akira S. Innate Immune Recognition of Viral Infection. Nat Immunol (2006) 7(2):131–7. doi: 10.1038/ni1303
143. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
144. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
145. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, et al. Alveolar Macrophages are the Primary Interferon-Alpha Producer in Pulmonary Infection With RNA Viruses. Immunity (2007) 27(2):240–52. doi: 10.1016/j.immuni.2007.07.013
146. Hogner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, et al. Macrophage-Expressed IFN-Beta Contributes to Apoptotic Alveolar Epithelial Cell Injury in Severe Influenza Virus Pneumonia. PloS Pathog (2013) 9(2):e1003188. doi: 10.1371/journal.ppat.1003188
147. Palmer CS, Anzinger JJ, Zhou J, Gouillou M, Landay A, Jaworowski A, et al. Glucose Transporter 1-Expressing Proinflammatory Monocytes are Elevated in Combination Antiretroviral Therapy-Treated and Untreated HIV+ Subjects. J Immunol (2014) 193(11):5595–603. doi: 10.4049/jimmunol.1303092
148. Sen S, Kaminiski R, Deshmane S, Langford D, Khalili K, Amini S, et al. Role of Hexokinase-1 in the Survival of HIV-1-Infected Macrophages. Cell Cycle (2015) 14(7):980–9. doi: 10.1080/15384101.2015.1006971
149. Hollenbaugh JA, Munger J, Kim B. Metabolite Profiles of Human Immunodeficiency Virus Infected CD4+ T Cells and Macrophages Using LC-MS/MS Analysis. Virology (2011) 415(2):153–9. doi: 10.1016/j.virol.2011.04.007
150. Palmer CS, Henstridge DC, Yu D, Singh A, Balderson B, Duette G, et al. Emerging Role and Characterization of Immunometabolism: Relevance to HIV Pathogenesis, Serious Non-AIDS Events, and a Cure. J Immunol (2016) 196(11):4437–44. doi: 10.4049/jimmunol.1600120
151. Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J Virol (2015) 89(4):2358–66. doi: 10.1128/JVI.02309-14
152. Passalacqua KD, Lu J, Goodfellow I, Kolawole AO, Arche JR, Maddox RJ, et al. Glycolysis is an Intrinsic Factor for Optimal Replication of a Norovirus. mBio (2019) 10(2). doi: 10.1128/mBio.02175-18
153. Jiang H, Shi H, Sun M, Wang Y, Meng Q, Guo P, et al. PFKFB3-Driven Macrophage Glycolytic Metabolism is a Crucial Component of Innate Antiviral Defense. J Immunol (2016) 197(7):2880–90. doi: 10.4049/jimmunol.1600474
154. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-Cov-2 Infection and Monocyte Response Through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab (2020) 32(3):437–46.e5. doi: 10.2139/ssrn.3606770
155. Mayer KA, Stockl J, Zlabinger GJ, Gualdoni GA. Hijacking the Supplies: Metabolism as a Novel Facet of Virus-Host Interaction. Front Immunol (2019) 10:1533. doi: 10.3389/fimmu.2019.01533
156. Thaker SK, Ch’ng J, Christofk HR. Viral Hijacking of Cellular Metabolism. BMC Biol (2019) 17(1):59. doi: 10.1186/s12915-019-0678-9
157. Castellano P, Prevedel L, Valdebenito S, Eugenin EA. HIV Infection and Latency Induce a Unique Metabolic Signature in Human Macrophages. Sci Rep (2019) 9(1):3941. doi: 10.1038/s41598-019-39898-5
158. Hsieh WY, Zhou QD, York AG, Williams KJ, Scumpia PO, Kronenberger EB, et al. Toll-Like Receptors Induce Signal-Specific Reprogramming of the Macrophage Lipidome. Cell Metab (2020) 32(1):128–43.e5. doi: 10.1016/j.cmet.2020.05.003
159. Kapadia SB, Chisari FV. Hepatitis C Virus RNA Replication is Regulated by Host Geranylgeranylation and Fatty Acids. Proc Natl Acad Sci USA (2005) 102(7):2561–6. doi: 10.1073/pnas.0409834102
160. Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, et al. Systems-Level Metabolic Flux Profiling Identifies Fatty Acid Synthesis as a Target for Antiviral Therapy. Nat Biotechnol (2008) 26(10):1179–86. doi: 10.1038/nbt.1500
161. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue Virus Nonstructural Protein 3 Redistributes Fatty Acid Synthase to Sites of Viral Replication and Increases Cellular Fatty Acid Synthesis. Proc Natl Acad Sci USA (2010) 107(40):17345–50. doi: 10.1073/pnas.1010811107
162. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I Ifn Signaling. Cell (2015) 163(7):1716–29. doi: 10.1016/j.cell.2015.11.045
163. Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, et al. Human Immunodeficiency Virus Impairs Reverse Cholesterol Transport From Macrophages. PloS Biol (2006) 4(11):e365. doi: 10.1371/journal.pbio.0040365
164. Bernard MA, Han X, Inderbitzin S, Agbim I, Zhao H, Koziel H, et al. HIV-Derived Ssrna Binds to TLR8 to Induce Inflammation-Driven Macrophage Foam Cell Formation. PloS One (2014) 9(8):e104039. doi: 10.1371/journal.pone.0104039
165. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The Transcription Factor STAT-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity (2013) 38(1):106–18. doi: 10.1016/j.immuni.2012.11.004
166. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity (2013) 38(1):92–105. doi: 10.1016/j.immuni.2012.11.005
167. Schoggins JW, Randall G. Lipids in Innate Antiviral Defense. Cell Host Microbe (2013) 14(4):379–85. doi: 10.1016/j.chom.2013.09.010
168. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol Suppresses Interleukin-1-Driven Inflammation Downstream of Type I Interferon. Science (2014) 345(6197):679–84. doi: 10.1126/science.1254790
169. Li X, Zhu W, Fan M, Zhang J, Peng Y, Huang F, et al. Dependence of SARS-Cov-2 Infection on Cholesterol-Rich Lipid Raft and Endosomal Acidification. Comput Struct Biotechnol J (2021) 19:1933–43. doi: 10.1016/j.csbj.2021.04.001
170. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, et al. Cholesterol 25-Hydroxylase Inhibits SARS-Cov-2 and Other Coronaviruses by Depleting Membrane Cholesterol. EMBO J (2020) 39(21):e106057. doi: 10.15252/embj.2020106057
171. Zang R, Case JB, Yutuc E, Ma X, Shen S, Gomez Castro MF, et al. Cholesterol 25-Hydroxylase Suppresses SARS-Cov-2 Replication by Blocking Membrane Fusion. Proc Natl Acad Sci USA (2020) 117(50):32105–13. doi: 10.1073/pnas.2012197117
172. Dias SSG, Soares VC, Ferreira AC, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, et al. Lipid Droplets Fuel SARS-Cov-2 Replication and Production of Inflammatory Mediators. PloS Pathog (2020) 16(12):e1009127. doi: 10.1371/journal.ppat.1009127
173. Lee W, Ahn JH, Park HH, Kim HN, Kim H, Yoo Y, et al. COVID-19-Activated SREBP2 Disturbs Cholesterol Biosynthesis and Leads to Cytokine Storm. Signal Transduct Target Ther (2020) 5(1):186. doi: 10.1038/s41392-020-00292-7
174. Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, et al. Inhibition of Indoleamine 2,3-Dioxygenase (IDO) Enhances Elimination of Virus-Infected Macrophages in an Animal Model of HIV-1 Encephalitis. Blood (2005) 106(7):2382–90. doi: 10.1182/blood-2005-04-1403
175. Liu WL, Lin YH, Xiao H, Xing S, Chen H, Chi PD, et al. Epstein-Barr Virus Infection Induces Indoleamine 2,3-Dioxygenase Expression in Human Monocyte-Derived Macrophages Through P38/Mitogen-Activated Protein Kinase and NF-Kappab Pathways: Impairment in T Cell Functions. J Virol (2014) 88(12):6660–71. doi: 10.1128/JVI.03678-13
176. Akaike T, Maeda H. Nitric Oxide and Virus Infection. Immunology (2000) 101(3):300–8. doi: 10.1046/j.1365-2567.2000.00142.x
177. Odkhuu E, Komatsu T, Koide N, Naiki Y, Takeuchi K, Tanaka Y, et al. Sendai Virus C Protein Limits NO Production in Infected RAW264.7 Macrophages. Innate Immun (2018) 24(7):430–8. doi: 10.1177/1753425918796619
178. Perrone LA, Belser JA, Wadford DA, Katz JM, Tumpey TM. Inducible Nitric Oxide Contributes to Viral Pathogenesis Following Highly Pathogenic Influenza Virus Infection in Mice. J Infect Dis (2013) 207(10):1576–84. doi: 10.1093/infdis/jit062
179. Sanchez MD, Ochoa AC, Foster TP. Development and Evaluation of a Host-Targeted Antiviral That Abrogates Herpes Simplex Virus Replication Through Modulation of Arginine-Associated Metabolic Pathways. Antiviral Res (2016) 132:13–25. doi: 10.1016/j.antiviral.2016.05.009
180. Burrack KS, Morrison TE. The Role of Myeloid Cell Activation and Arginine Metabolism in the Pathogenesis of Virus-Induced Diseases. Front Immunol (2014) 5:428. doi: 10.3389/fimmu.2014.00428
181. Meade N, Furey C, Li H, Verma R, Chai Q, Rollins MG, et al. Poxviruses Evade Cytosolic Sensing Through Disruption of an Mtorc1-Mtorc2 Regulatory Circuit. Cell (2018) 174(5):1143–1157 e1117. doi: 10.1016/j.cell.2018.06.053
182. Meade N, King M, Munger J, Walsh D. Mtor Dysregulation by Vaccinia Virus F17 Controls Multiple Processes With Varying Roles in Infection. J Virol (2019) 93(15). doi: 10.1128/JVI.00784-19
183. Jan E, Mohr I, Walsh D. A Cap-to-Tail Guide to Mrna Translation Strategies in Virus-Infected Cells. Annu Rev Virol (2016) 3(1):283–307. doi: 10.1146/annurev-virology-100114-055014
184. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A Diverse Range of Gene Products are Effectors of the Type I Interferon Antiviral Response. Nature (2011) 472(7344):481–5. doi: 10.1038/nature09907
185. Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, et al. Interferon-Gamma Regulates Cellular Metabolism and Mrna Translation to Potentiate Macrophage Activation. Nat Immunol (2015) 16(8):838–49. doi: 10.1038/ni.3205
186. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352(6284). doi: 10.1126/science.aaf1098
187. Dai H, Lan P, Zhao D, Abou-Daya K, Liu W, Chen W, et al. Pirs Mediate Innate Myeloid Cell Memory to Nonself MHC Molecules. Science (2020) 368(6495):1122–7. doi: 10.1126/science.aax4040
188. Netea MG, Quintin J, van der Meer JW. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe (2011) 9(5):355–61. doi: 10.1016/j.chom.2011.04.006
189. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
190. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers Nlrp3-Dependent Innate Immune Reprogramming. Cell (2018) 172(1-2):162–175 e114. doi: 10.1016/j.cell.2017.12.013
191. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. Mtor- and HIF-1alpha-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
192. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab (2016) 24(6):807–19. doi: 10.1016/j.cmet.2016.10.008
193. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic Induction of Trained Immunity Through the Mevalonate Pathway. Cell (2018) 172(1-2):135–146 e139. doi: 10.1016/j.cell.2017.11.025
194. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep (2016) 17(10):2562–71. doi: 10.1016/j.celrep.2016.11.011
195. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors is an Integral Component of Trained Immunity. Cell (2018) 172(1-2):147–161 e112. doi: 10.1016/j.cell.2017.11.034
196. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad Defects in the Energy Metabolism of Leukocytes Underlie Immunoparalysis in Sepsis. Nat Immunol (2016) 17(4):406–13. doi: 10.1038/ni.3398
197. Novakovic B, Habibi E, Wang SY, Arts RJW, Davar R, Megchelenbrink W, et al. Beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell (2016) 167(5):1354–1368 e1314. doi: 10.1016/j.cell.2016.09.034
198. Dominguez-Andres J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The Itaconate Pathway is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab (2019) 29(1):211–220 e215. doi: 10.1016/j.cmet.2018.09.003
199. Bonnefoy F, Gauthier T, Vallion R, Martin-Rodriguez O, Missey A, Daoui A, et al. Factors Produced by Macrophages Eliminating Apoptotic Cells Demonstrate Pro-Resolutive Properties and Terminate Ongoing Inflammation. Front Immunol (2018) 9:2586. doi: 10.3389/fimmu.2018.02586
200. Ponzoni M, Pastorino F, Di Paolo D, Perri P, Brignole C. Targeting Macrophages as a Potential Therapeutic Intervention: Impact on Inflammatory Diseases and Cancer. Int J Mol Sci (2018) 19(7). doi: 10.3390/ijms19071953
201. McKechnie JL, Blish CA. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe (2020) 27(6):863–9. doi: 10.1016/j.chom.2020.05.009
202. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host Response to SARS-Cov-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–1045 e1039. doi: 10.1016/j.cell.2020.04.026
203. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
204. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76(1):16–32. doi: 10.1128/MMBR.05015-11
205. Chousterman BG, Swirski FK, Weber GF. Cytokine Storm and Sepsis Disease Pathogenesis. Semin Immunopathol (2017) 39(5):517–28. doi: 10.1007/s00281-017-0639-8
206. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
207. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 Infection: The Perspectives on Immune Responses. Cell Death Differ (2020) 27(5):1451–4. doi: 10.1038/s41418-020-0530-3
208. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
209. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
210. Gralinski LE, Baric RS. Molecular Pathology of Emerging Coronavirus Infections. J Pathol (2015) 235(2):185–95. doi: 10.1002/path.4454
211. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-Cov-Infected Mice. Cell Host Microbe (2016) 19(2):181–93. doi: 10.1016/j.chom.2016.01.007
212. Han R, Xiao J, Zhai H, Hao J. Dimethyl Fumarate Attenuates Experimental Autoimmune Neuritis Through the Nuclear Factor Erythroid-Derived 2-Related Factor 2/Hemoxygenase-1 Pathway by Altering the Balance of M1/M2 Macrophages. J Neuroinflamm (2016) 13(1):97. doi: 10.1186/s12974-016-0559-x
213. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl Fumarate Targets GAPDH and Aerobic Glycolysis to Modulate Immunity. Science (2018) 360(6387):449–53. doi: 10.1126/science.aan4665
214. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature (2018) 556(7699):113–7. doi: 10.1038/nature25986
215. Ursini F, Russo E, Pellino G, D’Angelo S, Chiaravalloti A, De Sarro G, et al. Metformin and Autoimmunity: A “New Deal” of an Old Drug. Front Immunol (2018) 9:1236. doi: 10.3389/fimmu.2018.01236
216. Kelly B, Tannahill GM, Murphy MP, O’Neill LA. Metformin Inhibits the Production of Reactive Oxygen Species From NADH: Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-Activated Macrophages. J Biol Chem (2015) 290(33):20348–59. doi: 10.1074/jbc.M115.662114
217. Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. Metformin as Adjunct Antituberculosis Therapy. Sci Transl Med (2014) 6(263). doi: 10.1126/scitranslmed.3009885
218. Bailey CJ. Metformin: Historical Overview. Diabetologia (2017) 60(9):1566–76. doi: 10.1007/s00125-017-4318-z
219. Bohme J, Martinez N, Li S, Lee A, Marzuki M, Tizazu AM, et al. Metformin Enhances Anti-Mycobacterial Responses by Educating CD8+ T-Cell Immunometabolic Circuits. Nat Commun (2020) 11(1):5225. doi: 10.1038/s41467-020-19095-z
220. Sharma S, Ray A, Sadasivam B. Metformin in COVID-19: A Possible Role Beyond Diabetes. Diabetes Res Clin Pract (2020) 164:108183. doi: 10.1016/j.diabres.2020.108183
221. Lee MC, Lee CH, Lee MR, Wang JY, Chen SM. Impact of Metformin Use Among Tuberculosis Close Contacts With Diabetes Mellitus in a Nationwide Cohort Study. BMC Infect Dis (2019) 19(1):936. doi: 10.1186/s12879-019-4577-z
222. Bramante CT, Ingraham NE, Murray TA, Marmor S, Hovertsen S, Gronski J, et al. Metformin and Risk of Mortality in Patients Hospitalised With COVID-19: A Retrospective Cohort Analysis. Lancet Healthy Longev (2021) 2(1):e34–41. doi: 10.1016/S2666-7568(20)30033-7
223. Guo H, Wang Q, Ghneim K, Wang L, Rampanelli E, Holley-Guthrie E, et al. Multi-Omics Analyses Reveal That HIV-1 Alters CD4(+) T Cell Immunometabolism to Fuel Virus Replication. Nat Immunol (2021) 22(4):423–33. doi: 10.1038/s41590-021-00898-1
224. Chen X, Guo H, Qiu L, Zhang C, Deng Q, Leng Q. Immunomodulatory and Antiviral Activity of Metformin and its Potential Implications in Treating Coronavirus Disease 2019 and Lung Injury. Front Immunol (2020) 11:2056. doi: 10.3389/fimmu.2020.02056
225. Hu SK, Mitcho YL, Oronsky AL, Kerwar SS. Studies on the Effect of Methotrexate on Macrophage Function. J Rheumatol (1988) 15(2):206–9.
226. Palsson-McDermott EM, O’Neill LAJ. Targeting Immunometabolism as an Anti-Inflammatory Strategy. Cell Res (2020) 30(4):300–14. doi: 10.1038/s41422-020-0291-z
227. Safavi F, Nath A. Silencing of Immune Activation With Methotrexate in Patients With COVID-19. J Neurol Sci (2020) 215:116942. doi: 10.1016/j.jns.2020.116942
228. Wang W, Zhou H, Liu L. Side Effects of Methotrexate Therapy for Rheumatoid Arthritis: A Systematic Review. Eur J Med Chem (2018) 158:502–16. doi: 10.1016/j.ejmech.2018.09.027
229. Wang Z, Li Y, Yang X, Zhang L, Shen H, Xu W, et al. Protective Effects of Rapamycin Induced Autophagy on CLP Septic Mice. Comp Immunol Microbiol Infect Dis (2019) 64:47–52. doi: 10.1016/j.cimid.2019.01.009
230. Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, et al. TORC1 Inhibition Enhances Immune Function and Reduces Infections in the Elderly. Sci Transl Med (2018) 10(449). doi: 10.1126/scitranslmed.aaq1564
231. Omarjee L, Janin A, Perrot F, Laviolle B, Meilhac O, Mahe G. Targeting T-Cell Senescence and Cytokine Storm With Rapamycin to Prevent Severe Progression in COVID-19. Clin Immunol (2020) 216:108464. doi: 10.1016/j.clim.2020.108464
232. Zhavoronkov A. Geroprotective and Senoremediative Strategies to Reduce the Comorbidity, Infection Rates, Severity, and Lethality in Gerophilic and Gerolavic Infections. Aging (Albany NY) (2020) 12(8):6492–510. doi: 10.18632/aging.102988
233. Martin AR, Pollack RA, Capoferri A, Ambinder RF, Durand CM, Siliciano RF. Rapamycin-Mediated Mtor Inhibition Uncouples HIV-1 Latency Reversal From Cytokine-Associated Toxicity. J Clin Invest (2017) 127(2):651–6. doi: 10.1172/JCI89552
234. Rojas Marquez JD, Ana Y, Baigorri RE, Stempin CC, Cerban FM. Mammalian Target of Rapamycin Inhibition in Trypanosoma Cruzi-Infected Macrophages Leads to an Intracellular Profile That is Detrimental for Infection. Front Immunol (2018) 9:313. doi: 10.3389/fimmu.2018.00313
235. Singh P, Subbian S. Harnessing the Mtor Pathway for Tuberculosis Treatment. Front Microbiol (2018) 9:70. doi: 10.3389/fmicb.2018.00070
236. Nguyen LS, Vautier M, Allenbach Y, Zahr N, Benveniste O, Funck-Brentano C, et al. Sirolimus and Mtor Inhibitors: A Review of Side Effects and Specific Management in Solid Organ Transplantation. Drug Saf (2019) 42(7):813–25. doi: 10.1007/s40264-019-00810-9
237. Nakamura Y, Murai T, Ogawa Y. Effect of In Vitro and In Vivo Administration of Dexamethasone on Rat Macrophage Functions: Comparison Between Alveolar and Peritoneal Macrophages. Eur Respir J (1996) 9(2):301–6. doi: 10.1183/09031936.96.09020301
238. Kim BY, Son Y, Lee J, Choi J, Kim CD, Bae SS, et al. Dexamethasone Inhibits Activation of Monocytes/Macrophages in a Milieu Rich in 27-Oxygenated Cholesterol. PloS One (2017) 12(12):e0189643. doi: 10.1371/journal.pone.0189643
239. Ai F, Zhao G, Lv W, Liu B, Lin J. Dexamethasone Induces Aberrant Macrophage Immune Function and Apoptosis. Oncol Rep (2020) 43(2):427–36. doi: 10.3892/or.2019.7434
240. van der Goes A, Hoekstra K, van den Berg TK, Dijkstra CD. Dexamethasone Promotes Phagocytosis and Bacterial Killing by Human Monocytes/Macrophages In Vitro. J Leukoc Biol (2000) 67(6):801–7. doi: 10.1002/jlb.67.6.801
241. Krakauer T, Buckley M. Dexamethasone Attenuates Staphylococcal Enterotoxin B-Induced Hypothermic Response and Protects Mice From Superantigen-Induced Toxic Shock. Antimicrob Agents Chemother (2006) 50(1):391–5. doi: 10.1128/AAC.50.1.391-395.2006
242. Rachamim N, Latter H, Malinin N, Asher C, Wald H, Garty H. Dexamethasone Enhances Expression of Mitochondrial Oxidative Phosphorylation Genes in Rat Distal Colon. Am J Physiol (1995) 269(5 Pt 1):C1305–1310. doi: 10.1152/ajpcell.1995.269.5.C1305
243. Kraaij MD, van der Kooij SW, Reinders ME, Koekkoek K, Rabelink TJ, van Kooten C, et al. Dexamethasone Increases ROS Production and T Cell Suppressive Capacity by Anti-Inflammatory Macrophages. Mol Immunol (2011) 49(3):549–57. doi: 10.1016/j.molimm.2011.10.002
244. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in Hospitalized Patients With Covid-19. N Engl J Med (2021) 384(8):693–704. doi: 10.1056/NEJMoa2021436
245. Yasir M, Goyal A, Bansal P, Sonthalia S. Corticosteroid Adverse Effects. Treasure Island (FL: StatPearls (2021).
246. Kelly C, Jefferies C, Cryan SA. Targeted Liposomal Drug Delivery to Monocytes and Macrophages. J Drug Delivery (2011) 2011:727241. doi: 10.1155/2011/727241
Keywords: macrophage, immunometabolism, infections, SARS – CoV – 2, therapeutics
Citation: Gauthier T and Chen W (2022) Modulation of Macrophage Immunometabolism: A New Approach to Fight Infections. Front. Immunol. 13:780839. doi: 10.3389/fimmu.2022.780839
Received: 21 September 2021; Accepted: 07 January 2022;
Published: 26 January 2022.
Edited by:
Jan Fric, International Clinical Research Center (FNUSA-ICRC), CzechiaReviewed by:
Frederick J. Sheedy, Trinity College Dublin, IrelandMireille Laforge, U1141 Neuroprotection du cerveau en développement (INSERM), France
Copyright © 2022 Gauthier and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wanjun Chen, d2NoZW5AZGlyLm5pZGNyLm5paC5nb3Y=