 Renata de Freitas Saito
Renata de Freitas Saito Luciana Nogueira de Sousa Andrade
Luciana Nogueira de Sousa Andrade Silvina Odete Bustos
Silvina Odete Bustos Roger Chammas
Roger Chammas- Centro de Investigação Translacional em Oncologia (LIM24), Departamento de Radiologia e Oncologia, Faculdade de Medicina da Universidade de São Paulo and Instituto do Câncer do Estado de São Paulo, São Paulo, Brazil
To become resistant, cancer cells need to activate and maintain molecular defense mechanisms that depend on an energy trade-off between resistance and essential functions. Metabolic reprogramming has been shown to fuel cell growth and contribute to cancer drug resistance. Recently, changes in lipid metabolism have emerged as an important driver of resistance to anticancer agents. In this review, we highlight the role of choline metabolism with a focus on the phosphatidylcholine cycle in the regulation of resistance to therapy. We analyze the contribution of phosphatidylcholine and its metabolites to intracellular processes of cancer cells, both as the major cell membrane constituents and source of energy. We further extended our discussion about the role of phosphatidylcholine-derived lipid mediators in cellular communication between cancer and immune cells within the tumor microenvironment, as well as their pivotal role in the immune regulation of therapeutic failure. Changes in phosphatidylcholine metabolism are part of an adaptive program activated in response to stress conditions that contribute to cancer therapy resistance and open therapeutic opportunities for treating drug-resistant cancers.
Introduction
Cancer cells are characterized by their eximious ability to adapt and survive within harsh microenvironments (poor oxygenation and nutrient deprivation). Cancer metabolic plasticity is among the adaptive responses that allow tumor development in these conditions and also contribute to therapy resistance. The first tumor metabolic adaptation was identified by Otto Warburg in the 1920s, who described that cancer cells have an exacerbated glucose uptake and glycolysis accompanied by increased lactate production even under aerobic conditions (1). Since this pioneering work, known as the “Warburg effect”, much effort has been made to exploit the unique features of tumor metabolic phenotypes and metabolic reprogramming that is currently well-recognized as one of the hallmarks of cancer (2, 3). In recent years, lipid metabolism reprogramming has received renewed interest in the cancer field, and compelling evidence reveals the contribution of lipid remodeling in regulating the hallmarks of cancer (4).
Uncontrolled cell division exhibited by cancer cells introduces a cellular metabolic challenge, since it is necessary to double the total biomass (nucleic acid, proteins, and lipids) to support the mitotic cell division of a single cell into two equal-sized daughter cells. Cancer cells reprogram their metabolism from catabolism to anabolism to attend to this energetic and biomass demand to fuel cell proliferation (5). Among the biomolecules that compose total cell biomass, lipids have received fewer research efforts mainly due to their extremely diverse structure that turns their detection and quantification an analytical challenge. However, this scenario has changed due to technological progress in analytical approaches for lipid investigation that helped to gain a comprehensive look at the complexity and singularity of tumor lipid metabolism (6). Advances in two main analytical techniques, magnetic resonance spectroscopy (MRS) and mass spectrometry (MS) often coupled to liquid chromatography (LC) systems, contributed to the identification of abnormal choline (Cho) metabolism in tumors. Over the past four decades, accumulating evidence of MRS studies evaluating total choline (tCho) metabolite levels in cancer cells, notably free choline (Cho), phosphocholine (PCho), and glycerophosphocholine (GPC), revealed the importance of choline metabolism in tumor biology. Almost every tumor cell type investigated showed increased levels of tCho metabolites compared to non-malignant counterparts (7–15).
Cho-containing phospholipids are the most abundant phospholipids in eukaryotic cell membranes, and phosphatidylcholine (PtdCho) is the predominant phospholipid (<50%) in most mammalian membranes. Notably, cancer cells accumulate Cho-containing metabolites that are precursors or breakdown products of PtdCho to fuel their anabolic phenotype with building blocks and to promote intracellular processes that contribute to drug resistance. Additionally, hydrolysis of PtdCho generates lipid mediators that exert an intercellular crosstalk favoring cancer cell survival, proliferation, and immune modulation that culminate in resistance to therapy (Figure 1). Here, we highlight the role of PtdCho as a molecular link between altered choline metabolism and cancer therapy resistance. We start defining PtdCho-mediated protumoral signaling in a cancer cell perspective and further extend our discussion on the immune modulation of PtdCho-derived lipid mediators. In addition, we list some studied therapeutic strategies to intervene in the PtdCho metabolism and emphasize the importance to increase the knowledge of this lipid metabolism due to the complexity of the intracellular and intercellular signaling of PtdCho-mediated resistance to therapy.
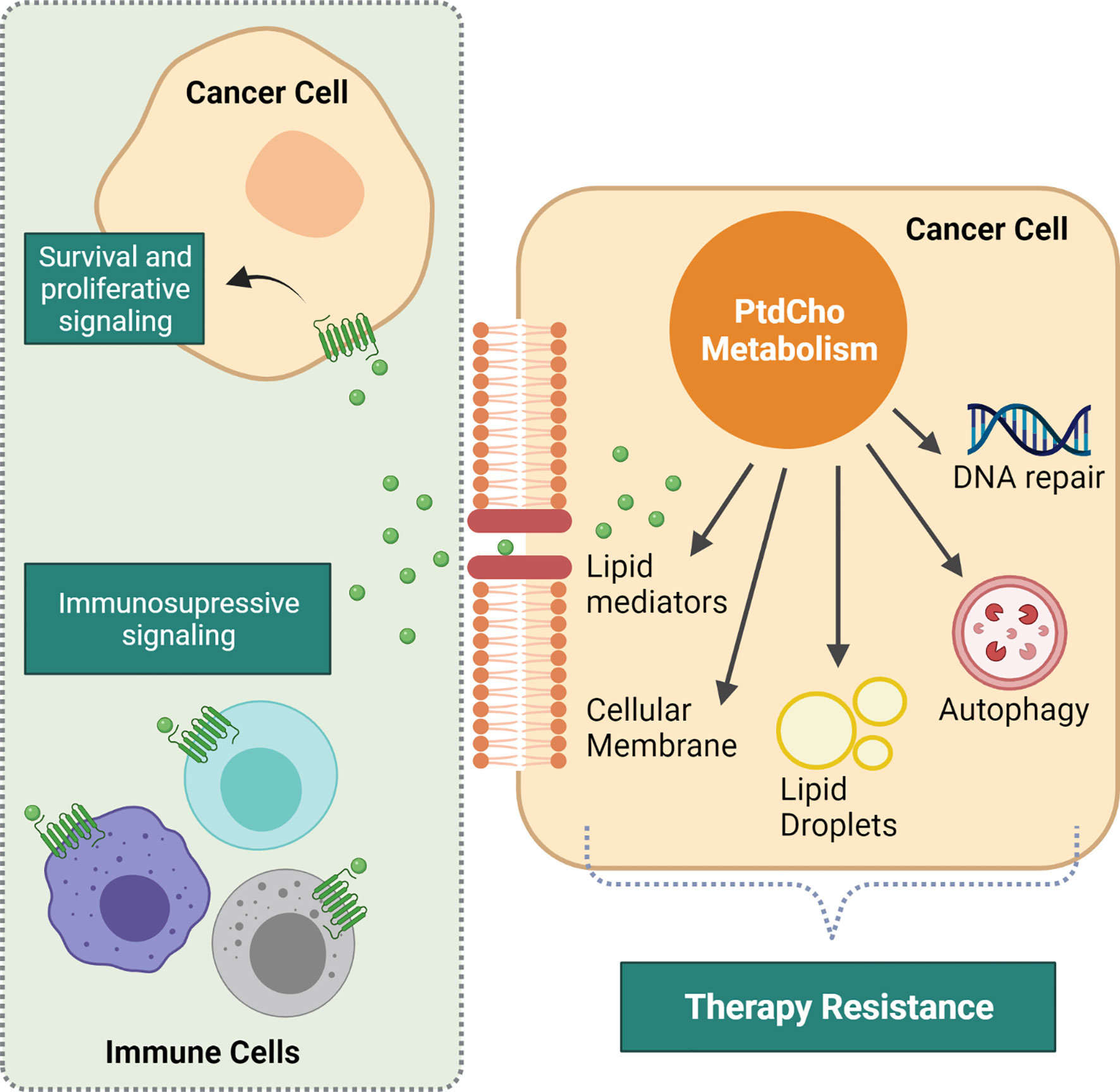
Figure 1 Intracellular and intercellular consequences of altered phosphatidylcholine (PtdCho) metabolism that impacts response to therapy. Increased PtdCho metabolism supports cancer cell accelerated growth by providing the major cellular membrane component. Additionally, PtdCho promotes intracellular events that mediate resistance to therapy, such as DNA repair, lipid droplet synthesis, and autophagy process. PtdCho-derived lipid mediators are prominent drivers of resistance. They are recognized by their cognate receptors present both in cancer cells and immune microenvironment cells, driving cancer cell survival and proliferation and promoting immunosuppression. Created with BioRender.com.
Phosphatidylcholine Metabolism and Cancer
PtdCho is a glycerophospholipid consisting of a choline headgroup and a phosphate group substituent linked to two fatty acid chains (Figure 1). Of note, choline is an essential nutrient obtained from dietary sources or by degradation of choline-containing lipids, and once inside the cell, the main fate of choline is PtdCho synthesis. Considering that cancer cells exhibit elevated levels of choline-containing lipids, it is appropriate to assume that cancer cells have efficient lipidic feedback to sustain an elevated choline metabolism.
To understand how and why cancer cells accumulate choline metabolites, either PtdCho precursors or products, we start summarizing the biosynthetic pathway of this lipid. Over 65 years ago, Eugene Kennedy elucidated the de novo biosynthetic pathway of PtdCho, known as Kennedy pathway or CDP-choline pathway (Figure 2) (16). PtdCho is predominantly synthesized through the CDP-choline pathway in all mammalian cells with choline as the first substrate of a sequential cascade of enzymatic alterations that result in PtdCho formation. In this pathway, choline obtained from an external medium or available in the cytosol by the breakdown of choline-containing compounds is phosphorylated by choline kinase (ChoK). In the rate-limiting second step, phosphocholine (PCho) is converted into the high‐energy intermediate CDP-choline by the enzyme CTP: phosphocholine cytidylyltransferase (CCT). Subsequently, the enzyme CDP-choline cholinetransferase (CPT) catalyzes the final reaction using CDP-choline and diacylglycerol (DAG) to form PtdCho (17).
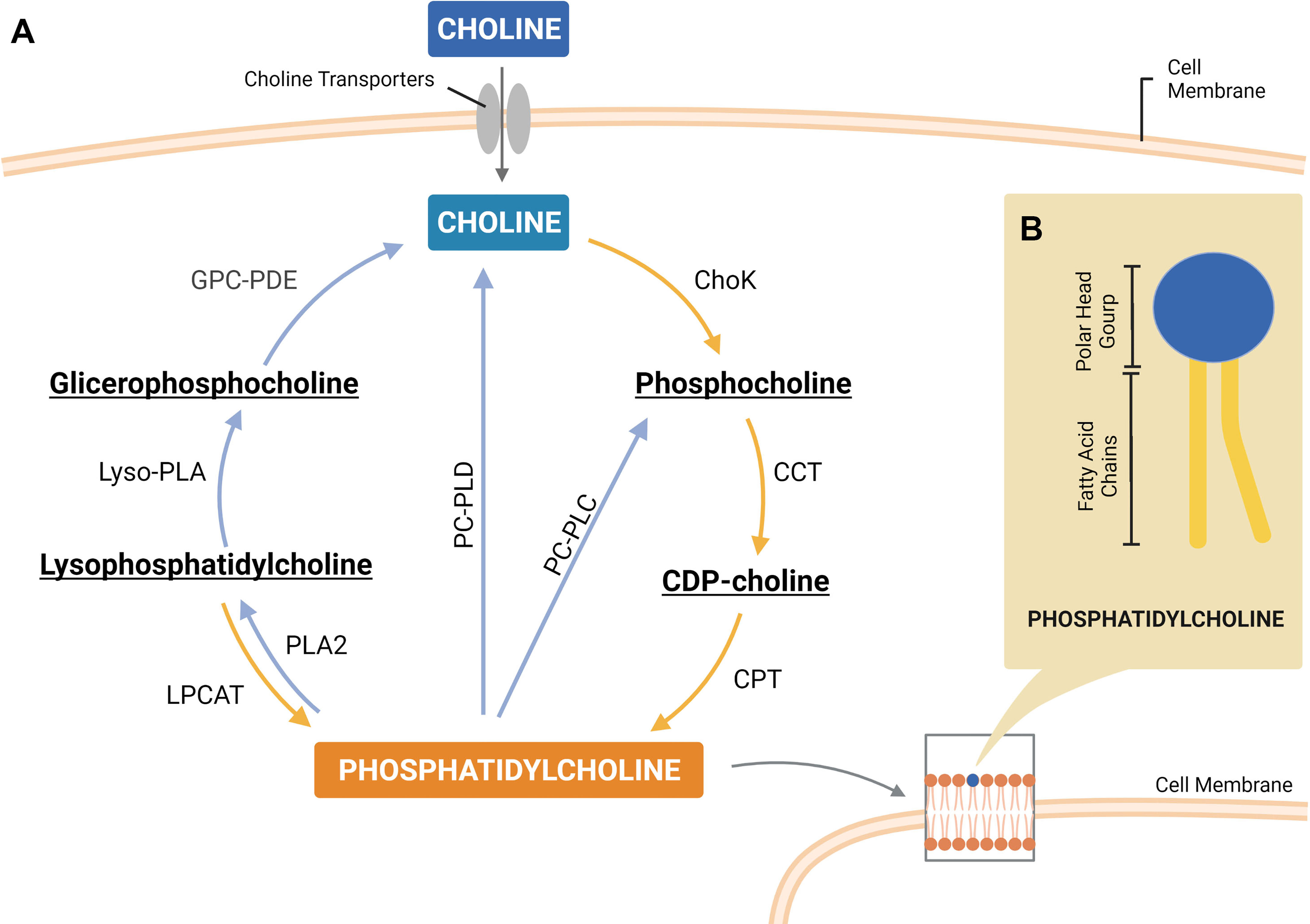
Figure 2 A simplified scheme of the phosphatidylcholine (PtdCho) cycle, highlighting the enzymes involved in PtdCho synthesis (Kennedy pathway) and catabolism (A). Representative structure of PtdCho consisted of a choline head group and a phosphate group (polar head group) linked to two fatty acid chains by a glycerol moiety (B). ChoK, choline kinase; CCT, CTP:phosphocholine cytidylyltransferase; CPT, CDP-choline cholinetransferase; LPCAT, lysophosphatidylcholine acyltransferase; PLA2, phospholipase A2; PC-PLC, phosphatidylcholine-specific phospholipase C; PC-PLD, phosphatidylcholine-specific phospholipase D; lyso-PLA, lysophospholipase; GPC-PDE, glycerophosphocholine phosphodiesterase. Created with BioRender.com.
After the final step of PtdCho synthesis, which occurs in the endoplasmic reticulum (ER) membrane, this phospholipid is transported and delivered to other organelles, such as cell membrane, by different inter-organelle mechanisms of transport (18). PtdCho is asymmetrically distributed across the lipid bilayer membrane and is enriched in the outer leaflet, comprising 40%–50% of total phospholipids. PtdCho also serves as a precursor of two other major membrane phospholipids, sphingomyelin (SM) and phosphatidylethanolamine (PtdEth). Thus, PtdCho has a crucial role as a direct or indirect source of structural building blocks for cellular membranes. However, PtdCho is more than a structural component of mammalian membranes, it is also an important source of lipid second messengers. PtdCho catabolism generates signaling molecules such as phosphatidic acid (PA), DAG, lyso-PC, and arachidonic acid (AA) that have protumoral effects. Additionally, degradation of PtdCho releases choline for replenishment of intermediates in the CDP-choline pathway. It is reasonable to postulate that the PtdCho cycle of synthesis and catabolism (Figure 2) supports the maintenance of the proliferative phenotype of cancer cells and contributes to protumoral characteristics that allow tumor progression and culminate in resistance to therapy.
The Molecular Origins of Deregulated Choline Metabolism in Cancer
The role of increased levels of Cho metabolites was initially interpreted as a means to meet the demands of fast-growing cancer cells. Indeed, increased consumption of choline and secretion of PtdCho by cancer cells are positively correlated with cell proliferation rates (19). However, Daly et al. (20) demonstrated in vitro that proliferative non-malignant cells maintain lower PCho and tCho levels compared to cancer cells (20), revealing that altered choline metabolism is not only supportive to cell proliferation but is also linked to malignant transformation and cancer progression (11). This assertion is supported by in vitro studies showing that both tCho and PCho levels increase in the malignant transformation of human mammary (21) and prostate (7) epithelial cells. Additionally, PCho accumulation is also associated with a more aggressive cancer phenotype (7, 21–23). The knowledge about the molecular mechanisms that regulate choline metabolism in cancer is growing, and we underline some of these mechanistic insights.
Increased Choline Uptake by Cancer Cells
Considering that fatty acids are substrates to PtdCho synthesis, the increased ratio of PtdCho biosynthesis in cancer can be in part a response to the enhanced fatty acid synthesis frequently observed in cancer cells (24). Additionally, it is intuitive thinking that one of the causes of cholinic phenotype is an enhanced ability of importing free extracellular choline by tumor cells. Of note, choline does not cross cell membranes by passive diffusion, being dependent on a choline transport system composed of four transporter families categorized according to their affinity for choline, high-affinity choline transporter 1, choline transporter-like proteins, polyspecific organic cation transporters, and organic cation/carnitine transporters. Several studies have underlined increased expression of each subtype of choline transporter in different human cancer cell lines in comparison with normal counterparts (11, 25). Given the fact that choline uptake is a rate‐limiting step in phospholipid metabolism and a prerequisite for cancer cell proliferation, the inhibition of choline transporters in cancer cells results in lower levels of intracellular choline accompanied by cell death induction (26, 27). As a consequence of decreased intracellular choline levels, there is also a reduction of PtdCho and PCho levels. In this context, cancer cells hydrolyze sphingomyelin as a compensatory response to maintain the generation of PtdCho and PCho. However, sphingomyelinase‐catalyzed hydrolysis of sphingomyelin also generates apoptosis-inducing factor ceramide, which activates caspase-3 and results in apoptotic cell death induction (28). Notably, the increased capacity of the cancer cell to import extracellular choline is a major contributor to the cholinic phenotype. However, further molecular characterization is needed to define what orchestrates the different combinations of choline transporters and how they lead to enhanced choline transport in cancer cells to drive the discovery of potential cancer targets.
Enhanced Activity of Choline Metabolic Enzymes Mediated by Oncogenic Regulation
The PtdCho cycle is composed of a network of enzymes whose expression and activity can be modulated by genetic alterations present in cancer cells. Increased intracellular levels of PCho in cancer cells are mainly attributed to upregulation of ChoK enzyme and also partially derived from phosphatidylcholine-specific phospholipase C (PC-PLC) and phosphatidylcholine-specific phospholipase D (PC-PLD) enhanced activity.
Accordingly, the ChoKα isoform, which catalyzes the phosphorylation of Cho to PCho, is upregulated in epithelial ovarian (13), breast (25), bladder (29), and colon (22) cancer cells. In addition to this metabolic function, in vitro and in vivo pieces of evidence show that ChoKα overexpression contributes to tumor progression, metastasis, and aggressiveness (29). Moreover, ChoKα overexpression has a prognostic significance and predicts poor prognosis of colorectal cancer (30), early-stage non-small cell lung cancer (31), and hepatocellular carcinoma patients (32). Importantly, PCho can also be generated by the breakdown of PtdCho through PC-PLC activity. PC-PLC is upregulated in ovarian and breast cancer cells of different subtypes and accounts for 20%–50% of intracellular PCho production (15, 33). Moreover, in vitro inhibition of PC-PLC activity resulted in cell proliferation arrest in both cancer models (13, 34). In particular, a decrease of migration and invasion potential, together with a loss of mesenchymal traits, was observed after treatment of breast cancer cells with a PC-PLC inhibitor (15). Additionally, PC-PLD hydrolyzes PtdCho in PA and free choline, which can reenter the CDP pathway and generate choline intermediates. PC-PLD expression is elevated in diverse cancer types, such as gastric (35), breast (36, 37), epithelial ovarian (13), and melanoma (38). Evidence shows that PC-PLD regulates multiple tumor cell events, such as cell transformation, proliferation, survival, and migration (39).
Mechanistic investigation revealed that PtdCho metabolic enzyme activation is dependent on oncogenic signaling pathways, mainly the oncogene ras that affects the activities of ChoK, PC-PLC, and PC-PLD enzymes. Glunde et al. (11) describe the oncogenic signaling pathways involved in the regulation of choline metabolism enzymes (11). In this sense, ras-transformed cells exhibit increased ChoK activity accompanied by increased levels of its product, PtdCho (40–42). Further investigation revealed that in mammalian cells, the mechanism of ChoK regulation by ras implies the involvement of two ras effectors, RAL GTPase guanine nucleotide dissociation stimulator (RALGDS) and Phosphoinositide 3-kinase (PI3K) signaling (41). Several studies underline that the oncogene ras also regulates the activity of PC-PLD enzyme (39, 43, 44). Moreover, the enzyme PC-PLC is a downstream target of Ras, and its activation plays an important role in inducing Ras-mediated mitogenic signaling (45, 46). Thus, oncogene-driven activation of ChoK, PC-PLD, and PC-PLC enzymes increases PtdCho synthesis and degradation, leading to the accumulation of energy-rich molecules and providing sources for cancer cell proliferation.
The Consequences of Deregulated Choline Metabolism in Cancer
Phosphatidylcholine Accumulation Confers Metabolic Flexibility to Cancer Cells’ Survival Under Stress Conditions
Cancer choline metabolism is also modulated by harsh tumor microenvironment (TME) conditions, mainly hypoxic and nutritional stress. A large number of studies indicate increased levels of tCho-containing compounds in cancer cells (47). It is interesting to stress out that these choline metabolites were observed to be heterogeneously distributed in tumor sections (48–50). This is of particular interest, as Glunde et al. (51) demonstrated in vivo that hypoxic areas of human prostate tumor xenografts contain increased tCho levels (51). Additionally, they reported that in vitro exposure of prostate cancer cells to hypoxia generates increased levels of PCho and tCho as well as increased expression of ChoK. They also provided mechanistic insights into how hypoxia induces choline metabolism by demonstrating that Hypoxia Inducible Factor-1, HIF-1 directly binds to ChoKα promoter region. Similarly, intermittent hypoxia also upregulates ChoK in rat pheochromocytoma PC-12 cells (52). In contrast, opposing evidence shows a decrease in choline levels in cancer cells exposed to hypoxic conditions without loss of cell viability (53). A hypoxia-mediated inhibition of choline phosphorylation has also been demonstrated in cancer cells (53, 54) as a result of a decrease in ChoK expression and activity mediated by HIF-1α (55).
Importantly, Glunde et al. (51) observed that not all tumor areas with high tCho levels were colocalized with hypoxic regions, indicating that other tumor environment conditions can also modulate choline metabolism. Acidosis can elicit opposite effects in PtdCho synthesis once in vitro evidence shows that it inhibits ChoK but enhances CCT (CTP:phosphocholine cytidylyltransferase) enzyme activity with a net increase of PtdCho pool in low pH conditions (56). However, another in vitro evidence shows that acidosis decreases PCho levels, reinforcing that ChoK is inhibited by low pH, but also increases GPC levels, indicating enhanced PtdCho degradation (57). Of note, GPC production from PtdCho catabolism is mediated by lysophospholipase, and phospholipase A2 catalyzed reactions with the generation of free fatty acids. It is interesting to note that acidosis can inhibit glycolysis in human cancers (58) and, as a consequence, result not only in a reduction of ATP generation but also in decreased amounts of acetyl-CoA, which feeds the tricarboxylic acid (TCA) cycle for aerobic respiration. Thus, these pieces of evidence drive us to suggest that during acidosis, decreased PtdCho synthesis and increased PtdCho breakdown allow cancer cells to fuel beta-oxidation of fatty acids as a source of acetyl-CoA (Figure 3).
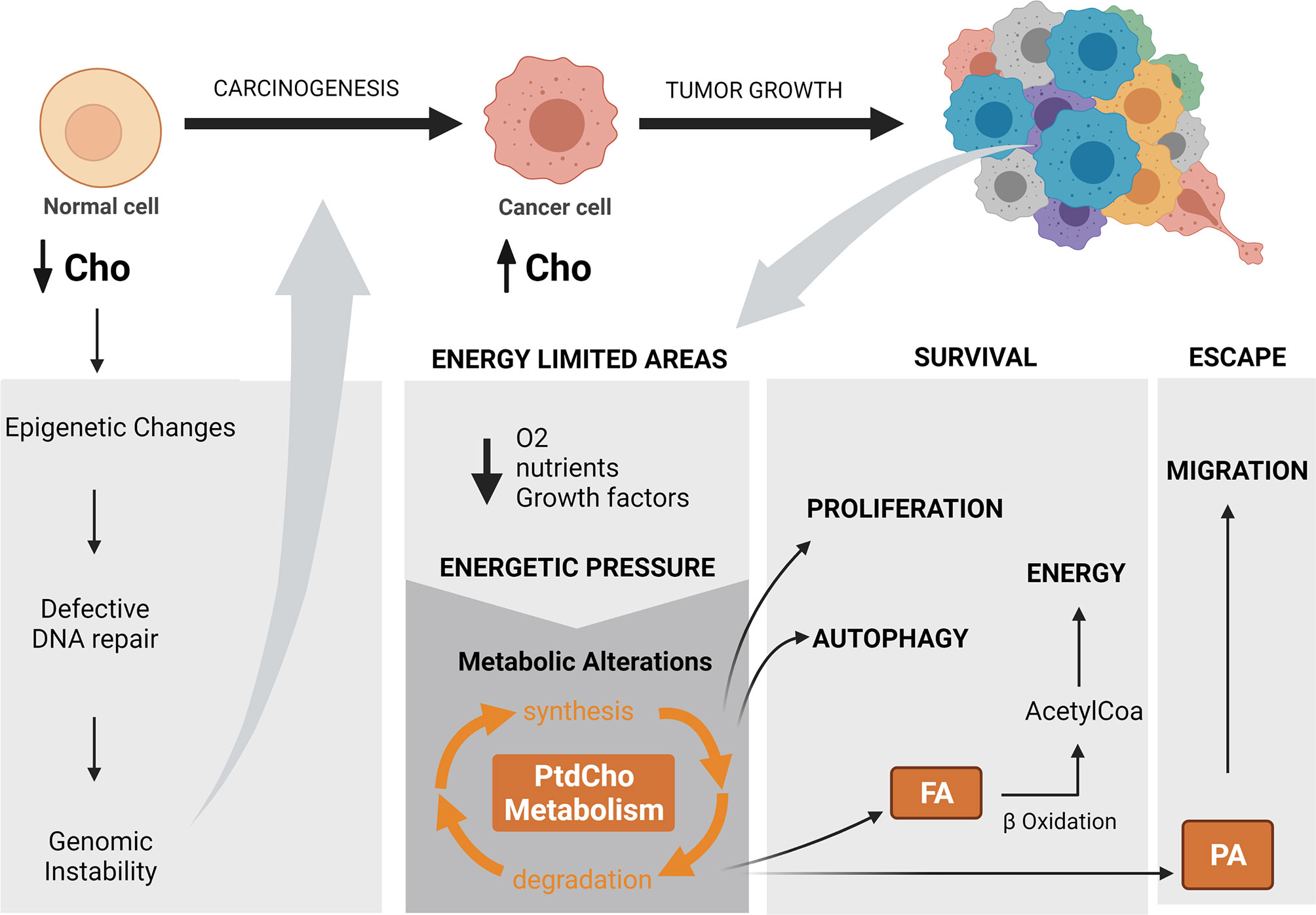
Figure 3 The impact of altered choline metabolism in carcinogenesis and tumor progression. Altered choline metabolism promotes carcinogenesis through modulating DNA repair gene expression by methylation and generating genomic instability. Energy-limited areas arise with tumor growth and exert an energetic pressure that leads to metabolic reprogramming and results in diverse metabolic phenotypes. Altered choline metabolism is among these phenotypes. In particular, phosphatidylcholine metabolism regulates adaptative cellular processes, such as proliferation and autophagy that allow cancer cell survival with limited energetic sources and in parallel induce migration as an escape route toward an energy-privileged area. Cho, choline; PtdCho, phosphatidylcholine; FA, fatty acid; PA, phosphatidic acid; O2, oxygen.Created with BioRender.com.
In this sense, ample evidence supports the notion that in aerobic conditions, cancer cells exhibit increased choline uptake to activate anabolic metabolic pathways to sustain their high proliferative rates. On the contrary, hypoxic conditions have been reported to diminish choline uptake while enhancing glucose and acetate in cancer cells (53). Kamphorst et al. (59) demonstrated that during hypoxia, acetate is the major additional source of carbon donor for acetyl-CoA, and the generation of this precursor for fatty acid biosynthesis allows cancer cells to maintain lipogenesis and proliferation under hypoxic conditions (59). Interestingly, the in vitro study by Yoshimoto et al. (60) revealed that the rate of acetate incorporation in tumor cells under hypoxia is superior to the rate observed in normal cells, and the metabolic fate of acetate in hypoxic cells was preferentially incorporated into PtdCho (60). Importantly, they also showed that this increased acetate uptake and lipid incorporation were positively correlated with growth activity. Thus, cancer cells might have these metabolic adaptations to maintain the anabolism of fatty acids, which require acetyl-CoA units to compensate for inhibition of glycolytic ATP and acetyl-CoA production by activation of fatty acids as a source of energy.
Phosphatidylcholine Metabolism Promotes a Tumor Escape Route From Energetic Stress
There is a correlation between survival and cancer cell migration under stress conditions, suggesting that cancer cells in addition to surviving and suppressing cell death also trigger a migration phenotype to escape from stressful regions (61). This raises the question of how migration is triggered by stressful conditions and if enhanced choline metabolism could be a linker of these phenomena.
Oxygen- and nutrient-deprived areas arise as a consequence of inadequate blood supplies during solid tumor growth and impose an energetic pressure on cancer cells. To survive, cancer cells need to first suppress cell death induced by these stress conditions and ultimately provide means for obtaining energy. One possibility is that cancer cells with increased amounts of choline compounds have the advantage of obtaining energy and building blocks from the degradation of these lipids. Alternatively, cancer cells in parallel induce migration to sites where nutrition could be found, and choline compound storage can contribute to this process by their breakdown products. In this sense, Zheng et al. (62) revealed that under stress caused by serum withdrawal, MDA-MB-231 human breast cancer cells exhibited increased PC-PLD enzyme activity concomitant to an enhanced migration and invasion potential (62). Compelling evidence gained from PLD2 overexpression (63), an isoform of PC-PLD gene, in low-invasive breast cancer cells resulted in the conversion of these cells into a highly aggressive phenotype with increased capacity of lung metastasis formation, which was inhibited by two different small-molecule inhibitors of PC-PLD activity (63). Animals deficient in another PC-PLD gene isoform, PLD1, or treated with a small-molecule inhibitor of PLD1 activity, exhibited reduced tumor growth, angiogenesis, and metastasis (63). Aberrant expression of both PC-PLD isoforms has been detected in different cancers and linked to cancer cell survival and a pro-metastatic phenotype through activation of different signaling pathways revised in Yao et al. (64). Thus, a stressful tumor environment can drive PtdCho degradation, in particular through PC-PLD hydrolysis, which contributes to a cancer cell migration program. These data indicate PC-PLD as a potential target for cancer therapy and point toward a small-molecule dual inhibitor of PLD1 and PLD2 as a promising strategy.
Phosphatidylcholine and Phosphatidylcholine-Derived Lipid Mediators Regulate Cancer Cell Growth and Survival
PtdCho metabolism has been linked to opposing cellular events, cell proliferation, and cell death. Noticeably, the cell cycle controls PtdCho homeostasis to avoid an excess or deficit of membranes. Essentially, PtdCho metabolism is modulated during the cell cycle and is characterized by a high rate of PtdCho degradation and resynthesis in the G1 phase, a reduced PtdCho turnover that leads to doubling PtdCho amounts in the S phase, and a cessation of PtdCho metabolism in G2/M phases (65). In the opposite direction, the first evidence of a direct link between cell death and PtdCho synthesis was a report showing that Chinese hamster ovary (CHO) cells with a mutation in the CCT enzyme resulted in PtdCho depletion and concomitant apoptosis induction (66). The molecular mechanism by which PtdCho depletion is sensed and transduced to cell death has not yet been fully elucidated; however, evidence shows that inhibition of PtdCho synthesis triggers apoptosis through a mechanism that involves the activation of an endoplasmic reticulum, ER stress response (67, 68). Moreover, PtdCho is a substrate for sphingomyelin (SM) synthesis, and the final step of this biosynthetic route involves the exchange of the phosphocholine head group from PtdCho to ceramide. Yen et al. (69) demonstrated that in parallel to the intracellular decrease of PtdCho, SM levels also decrease and the apoptosis inducer ceramide accumulates (69).
More recently, studies have implicated lipid metabolism in the non-apoptotic cell death process of ferroptosis. This process is characterized by the accumulation of iron-dependent lethal lipid peroxides (LPOs) that can be generated from the oxidation of phospholipids, such as arachidonoyl and adrenoyl, by the catalysis of acyl-CoA synthetase long-chain family member 4 (ACSL4), LPCAT3, and 15-lipoxygenase (15-LOX/ALOX15) (70, 71). A detailed underlying mechanism of ferroptosis in cancer biology was reviewed by Li and Li (72). Chemotherapy and mainly ionizing radiation (IR) therapy generate reactive oxygen species (ROS) that can target lipid peroxidation and cause ferroptosis induction. Of interest, IR was reported to induce ferroptosis, and inhibition of ACSL4 enzyme activity reverted IR-induced ferroptosis and promoted radioresistance (73, 74). While oxidative metabolites from arachidonoyl and adrenoyl can generate "find me signals" and elicits an antitumor response, ferroptotic cancer cells have increased PTGS2 gene expression, which encodes cyclooxygenase 2 (COX-2) to produce prostaglandin E2 (PGE2), a major pro-inflammatory factor (75). These data suggest that ferroptosis and lipid metabolism may be involved in resistance to therapy. Further research to expand the understanding of the unique features of ferroptosis will unveil the therapeutic windows to precisely target this process.
Importantly, PtdCho depletion can indirectly interfere in cell viability once it is an important source of lipid mediators that are known to regulate cell growth, such as PA and DAG. Accordingly, a balance between mitogenic and antimitogenic lipid mediators derived from PtdCho can dictate the fate of cells toward cell proliferation, arrest, or death (76). This is of particular interest in oncology once several antitumoral drugs, including chemotherapy and radiotherapy, induce an increase in proapoptotic ceramide levels and parallel mitogenic DAG levels (77).
The Impact of Cancer Therapy Response on Phosphatidylcholine Metabolism
Since the cholinic phenotype, characterized by elevated PCho and high tCho-containing metabolites, is considered a metabolic hallmark of cancer (11, 21), some groups started to explore the use of choline metabolite biomarkers to monitor tumor response (78). Several reports have shown that choline-containing metabolites are modulated by antitumoral therapy (79–83). Nishio et al. (81) showed that PtdCho levels were decreased by 50% in human lung adenocarcinoma cells treated with cisplatin in vitro (81). Additionally, a decrease in PCho levels and an increase in GPC levels were observed in breast cancer cells in vitro in response to doxorubicin (79) and in vivo after docetaxel treatment (84). In contrast, chemotherapy was also reported to increase choline metabolites. Notably, PtdCho levels were increased in breast cancer cells by doxorubicin treatment (79) and in human colon cancer cells and tumor xenografts by histone deacetylase (HDAC) inhibitors (85). Additionally, increased levels of CDP-choline were observed in human promyelocytic leukemia (HL-60) and Chinese hamster ovary (CHO-K1) after treatment with several cytotoxic drugs (86). PCho levels were also increased in human neutrophils undergoing apoptosis (87). Overall, these divergent data indicate that changes in choline metabolism can be treatment-specific and cellular context-dependent.
Concerning enzymatic inhibitors, in pediatric glioblastoma, PCho, tCho, and choline kinase alpha (ChoKα) protein levels decreased upon PI3K pathway inhibition, whereas an increase in PCho, glycerophosphocholine (GPC), and tCho was observed in response to temozolomide (TMZ). Since these metabolic changes can be monitored by non-invasive techniques like NMR, the authors suggested that monitoring Cho metabolism might represent a potential biomarker for monitoring response in pediatric gliomas (88). Furthermore, choline and PCho metabolism can also be altered in response to certain treatments such as histone HDAC, phospholipase Cγ1, Mitogen Activated Protein Kinase, MAPK, PI3K, and Heat Shock Protein 90, HSP90 inhibitors (89–95). Regarding the use of HDAC, which is approved for cutaneous T-cell lymphoma treatment, Beloueche-Babari et al. (85) showed that HDAC inhibition led to an increase in de novo phosphocholine synthesis that was accompanied by ChoKα expression in colon and prostate carcinoma cells in vitro and in vivo (85). This modification in choline metabolism is also observed in response to radiotherapy. In xenograft pancreatic tumors, an increase in choline and a decrease in glycerophosphocholine + phosphocholine in comparison to the normal pancreas was reported by a study in 2013 (96). Moreover, the authors observed that, in response to different doses of radiotherapy, choline levels were diminished and glycerophosphocholine + phosphocholine increased.
Although there are apparent discrepancies about the increase or decrease in some choline-containing metabolites, 1H-MRS imaging of tCho levels in many cancers has been used to improve treatment monitoring and therapy strategy, as also proposed by Katz-Brull et al. (97), Mignion et al. (98), and Al-Saffar et al. (99) (97–99). In a retrospective study, patients with locally advanced breast cancer that responded or did not to neoadjuvant chemotherapy were differentiated by a reduction in tCho levels (100). In line with this finding, Meisamy et al. (78) reported a reduction in PCho levels as early as 24 h after the first treatment in locally advanced breast cancer patients who responded to doxorubicin chemotherapy, while it remained the same or increased in non-responders (78). An early response to therapy associated with a reduction of tCho was also observed in prostate cancer (101, 102). In contrast, a transient increase in choline compounds was observed in Breast Cancer gene 1, BRCA-1 mouse mammary tumors sensitive to docetaxel treatment (103). These results imply that quantitative changes in tCho levels could be a parameter to predict early tumor response, which would be valuable to guide the clinician in determining an alteration in the dosage of the drug and administration of alternative drugs or offer surgery options to non-responders.
Considering that PCho concentration correlates strongly with cell proliferation (104), one hypothesis is that a decrease in choline metabolites after therapy may reflect cell cycle arrest. However, the molecular basis of how chemotherapy interferes in choline metabolism has been investigated to clarify the molecular mechanisms behind this effect. Accordingly, a cisplatin-induced decrease in PtdCho levels was attributed to an increase in PC-PLC activity (81) and doxorubicin-induced decrease in PCho levels to downregulation of PLD1, ChoKα, and glycerophosphodiester phosphodiesterase domain containing 6 (GDPD6) enzymes (79). Thus, therapy-induced PCho and PtdCho increased levels could reflect an increase in de novo synthesis through ChoK activity and/or a decrease in the degradative pathways mediated by PC-PLC or PC-PLD activity. The decrease in PCho levels observed posttreatment is frequently coupled with an increase in GPC levels. Considering that PCho is an anabolite and GPC a catabolite of PtdCho, a decrease in PCho/GPC ratio after treatment implies a net increase in PtdCho turnover. In line with this, evidence shows that HDAC inhibitors result in a net augmentation of PtdCho by positively modulating the expression of CTP-PC cytidylyltransferase, the rate-limiting enzyme in PtdCho biosynthesis together with the observation that PtdCho breakdown product GPC is decreased after HDAC inhibitor treatment (85, 105). Interestingly, the augmentation of PtdCho was not translated to increases in cell volume, suggesting that it was not used to synthesize new outer membrane. In line with this, the accumulation of PtdCho can be an important reservoir of PtdCho-derived lipid mediators that can drive cancer survival and resistance to therapy.
Changes in Phosphatidylcholine Metabolism Contribute to Drug Resistance
Treatment failure in cancer patients is closely related to the development of drug resistance. Thus, it is crucial to elucidate the molecular processes that lead to drug resistance to intervene in these events and improve patient response to therapy. Few hints about lipid remodeling involvement in tumor resistance were reported several years ago. Back in the 1970s, Schlager and Ohanian (106), using guinea pig tumoral cells, observed that the metabolic inhibitors actinomycin D and Adriamycin were able to increase cell sensitivity to antibody-complement killing (106). Interestingly, this effect was accompanied by a reduction in PtdCho incorporation, among other lipids, into cellular organelles such as ER, nuclear membrane, mitochondria, and microsomes, suggesting that lipid synthesis might be involved in tumor resistance. The cellular mechanisms involved in acquired and intrinsic resistance are diverse and complex, and the understanding of how lipid metabolism modulates these pathways is still largely unknown. Overexpression of multidrug resistance (MDR) proteins is found in several tumor types and is associated with increased resistance to drug compounds due to the active efflux of a broad range of chemical molecules. In 1997, Bosch et al. (107) showed that PtdCho is a substrate for MDR1 P-glycoprotein (PgP) in T-cell leukemia resistant cells that might be responsible for the altered lipid composition between sensitive and resistant tumor cells as well as the inefficacy of treatments based on liposome delivery (107). In breast cancer resistant cells, tamoxifen, a broadly used agent in hormone therapy for estrogen-positive breast tumors, inhibited the uptake of choline probably by its action as an antagonist of PgP. Although the impact of this blockade had not been evaluated under these circumstances, the authors speculated that tamoxifen can interfere in choline metabolism (108). A study by Ramu et al. (109) revealed that the incorporation of choline in phosphocholine is decreased in drug-resistant leukemia cells in comparison to the parental cells (109). The authors also found that PtdCho synthesis could be restored through the inhibition of MDR inhibitors such as verapamil, indicating that sensitive and resistant tumor cells present different membrane lipid compositions that correlate to their sensitivity to a range of drugs used in cancer treatment impacting on the outcome. In 1992, Dubois and Tapiero (110) demonstrated an alteration in phospholipid metabolism characterized by an increase in PtdCho synthesis from PtdEth exclusively in leukemia resistant cells (110).
Some years later, the correlation between Cho/PtdCho, plasma membrane lipid composition, and drug sensitivity was demonstrated by others using different experimental approaches. Riedel et al. showed that proliferating pre-malignant Chang cells were more resistant to the FB1(fumonisin B1)-induced cytotoxicity compared to primary hepatocytes. Differences in lipid content, including lower PtdCho levels in Chang cells, which imply a more rigid plasma membrane, were partially responsible for this differential cell response to FB1. This finding reinforces the notion that lipid composition changes along with cell transformation and tumor progression, interfering in tumor response to cytotoxic therapy (111). In 2009, it was demonstrated that the upregulation of Cho transporter CHT1 and ChoK was involved in acquired resistance to chemotherapy in glioblastoma (GBM) (112). Concerning radiotherapy, Desoubzdanne et al. (113) compared choline metabolism between glioma radiosensitive and radioresistant cells (113). As reported by Vanpouille et al. (112), the authors found higher Cho and PCho levels and a global PtdCho metabolism more active in radioresistant cells. In this sense, NMR spectroscopy has also been used to investigate if changes in choline metabolism are associated with the MDR phenomenon. It has been demonstrated that choline metabolite spectra detected by 31P NMR are indeed different in resistant (drug-selected) cancer cells compared to drug-sensitive cells. In a model of MCF-7 human breast cancer cell induction of MDR with Adriamycin, a combined analysis of both 1H and 31P NMR spectra revealed that sensitive cells showed higher PCho concentrations than resistant cells, but choline levels were similar (114). In agreement, in an in vivo study with murine mammary adenocarcinoma, NMR revealed that Adriamycin-sensitive tumors have increased PCho and GPC levels compared to Adriamycin-resistant tumors (115). Additionally, in the same study, treatment of tumors with Adriamycin decreased PCho and GPC levels only in Adriamycin-sensitive tumors. On the contrary, another piece of evidence shows that docetaxel-sensitive tumors exhibited a lower level of choline compounds compared to their resistant counterparts (103). This inconsistency in choline metabolism change in MDR reinforces that these changes may depend strongly on the drug used for MDR induction and/or cancer cell type.
Albeit not universal, an increase in choline metabolites would likely be a predictive marker of drug resistance, and PtdCho metabolic enzymes are a linker of these phenomena. Evidence shows that breast cancer cells treated with doxorubicin increased PCho/GPC ratio caused by a downregulation of the enzymes PLD1, GDPD6, and ChoKα (Figure 2). Importantly, silencing of the metabolic enzymes PLD1 and ChoKα sensitized breast cancer cells to doxorubicin and specific GDPD6 silencing counteracted doxorubicin migration induction (79). Considering the role of ChoKα enzyme in the generation of PCho, high levels were consistently observed in cancer cells, and overexpression of this enzyme mediated an increase in MCF-7 breast cancer cell resistance to 5-fluorouracil together with a substantial increase in PCho level (116). Moreover, silencing of ChoKα enhanced the sensitivity of epithelial ovarian cancer to chemotherapeutic agents, such as platinum, doxorubicin, and paclitaxel (117). Importantly, the same sensitization effect of ChoKα silencing was observed in a drug-resistant context with platinum-resistant SKOV3 cell line (117). Following these findings, a study identified a group of super-enhancers (SEs) that are abnormally activated in castration-resistant prostate cancer resistant to enzalutamide antiandrogen drug. Among these SEs was the choline phosphotransferase 1 (CHPT1) gene, which encodes cholinephosphotransferase 1 (CPT) protein that catalyzes the last step of PtdCho synthesis (118). Indeed, CHPT1 has been shown overexpressed in cancer and associated with tumor growth (119). Taken together, all these reports demonstrate that cancer therapy modulates the expression of PtdCho metabolic enzymes, which alter choline metabolite levels and render cancer cells resistant to treatment.
Phosphatidylcholine as a Precursor of Lipid Mediators Involved in Therapy Resistance
PtdCho turnover (catabolism) is mediated by phospholipases (A2, C, and D), generating both choline-containing phospholipids (e.g., PhoC, GPC, and choline), that can be reutilized for PtdCho biosynthesis and lipid mediators that regulate multiple protumoral signaling pathways. The list of dysregulated bioactive lipids that have been shown to contribute to tumor biology includes AA, eicosanoids, DAG, PA, lysophosphatidic acid (LPA), platelet-activating factor (PAF), ceramide, sphingosine, and other lysosphingolipids (120). This list continues to grow, and here we highlight PtdCho-derived lipid mediators emerging as lipid second messengers involved in resistance to therapy (Figure 4).
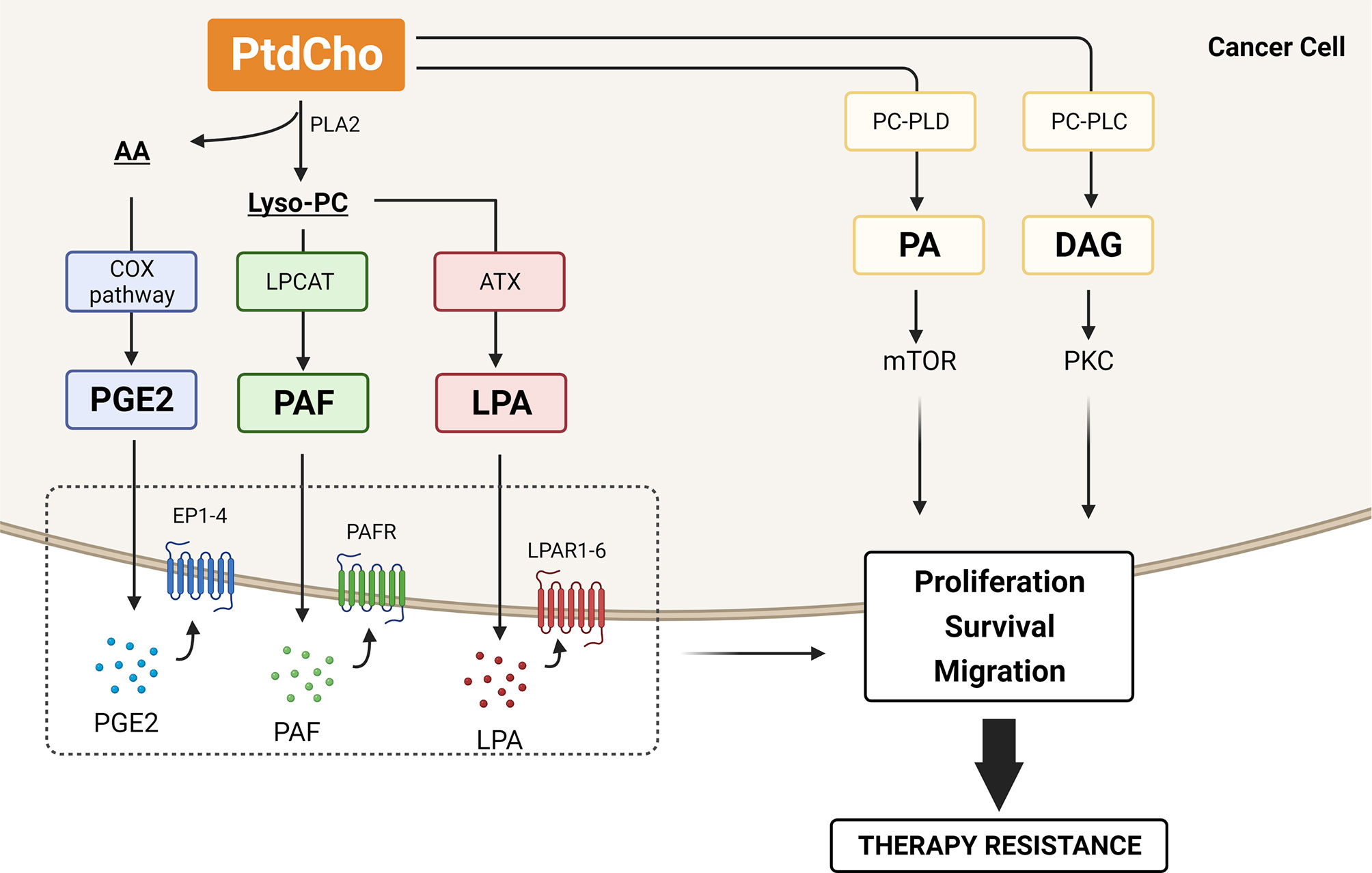
Figure 4 Lipid mediators generated by phosphatidylcholine (PtdCho) turnover that contribute to therapy resistance. PtdCho is hydrolyzed by phospholipase A2 (PLA2), resulting in the production of lysophosphatidylcholine (lyso-PC) and arachidonic acid (AA). The COX2 enzymes catalyze the conversion of AA to prostaglandin E2 (PGE2), and lyso-PC acetyltransferases (LPCATs) convert lyso-PC into platelet-activating factor (PAF). Alternatively, lyso-PC can be hydrolyzed by autotaxin (ATX) and generate lysophosphatidic acid (LPA). These three lipid mediators, PGE2, PAF, and LPA, are secreted and bind to their cognate receptors EP1-4, PAFR, and LPAR1-6, respectively, promoting cancer cell proliferation, survival, and migration. PtdCho is also hydrolyzed by phosphatidylcholine phospholipase C (PC-PLC) and D (PC-PLD), generating diacylglycerol (DAG) and phosphatidic acid (PA). DAG activates the protein kinase C (PKC) pathway, and PA is crucial for mTOR activity, promoting cancer cell proliferation and survival. All these catabolic products of PtdCho have been involved in therapy resistance. Created with BioRender.com.
More than 50% of PtdCho synthesized in the Kennedy pathway is remodeled through the Lands cycle (121). In this remodeling pathway, PtdCho is hydrolyzed by phospholipase A2 (PLA2), resulting in the production of lysophosphatidylcholine (lyso-PC) and AA. Once released, AA participates in the biosynthesis of eicosanoids such as prostaglandins and leukotrienes. Notably, AA is metabolized through the enzyme COX-2 into the terminal product PGE2. Elevated levels of COX-2 and PGE2 are frequently observed in many cancers and are associated with cancer initiation, progression, and resistance to therapy. Mechanistically, the activated COX-2/PGE2 pathway leads to therapy resistance mainly through affecting the TME by inducing epithelial–mesenchymal transition (EMT), suppressing anticancer immunity, and regulating cancer stem cell (CSC) homeostasis (122). Several reports with diverse cancer cell lines have shown that EMT is promoted by COX-2-induced PGE2 production, of which inhibition reverts this phenomenon (123–125). Moreover, EMT promoted by COX-2/PGE2 axis confers resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor erlotinib (126).
Under physiological conditions, another PtdCho-derived lipid mediator generated from PLA2 activity, lyso-PC, is rapidly metabolized or reacylated to avoid the cytolytic induction caused by high intracellular concentrations due to its amphipathic property. The reacylation of lyso-PC is performed by lysophosphatidylcholine acyltransferases (LPCATs) by adding fatty acids at the sn-2 position to yield PtdCho, which rapidly gets recycled by the Lands cycle. These cycles of deacylation and reacylation of PtdCho modify the fatty acid composition of the phospholipids de novo generated in the Kennedy pathway and produce membrane asymmetry and diversity. Intracellular Lyso-PC concentration is also regulated by its hydrolysis through the enzymatic activity of autotaxin (ATX), an enzyme with lysophospholipase D activity, that generates LPA. Several cancers exhibit the activity of ATX enhanced and consequent increased levels of LPA (127) that is associated with cancer development and poor prognosis (128). Several reports have exploited the ATX–LPA signaling on cancer cell protection against chemotherapy and radiotherapy. Minami et al. (129) showed that LPA signaling via LPA receptor (LPAR5) regulates the resistance to cisplatin and dacarbazine in a melanoma cell line (129). Additionally, ATX–LPA signaling was reported to protect colon cancer cells from cisplatin and 5-fluorouracil-induced apoptosis (130) and to decrease cisplatin cytotoxic effect in human ovarian cancer cells (131). Inhibition of ATX activity reverts the protective effect of LPA on Taxol-induced apoptosis in breast cancer cells (132). The mechanism involved in ATX–LPA axis attenuation of chemotherapy-induced cell death includes the activation of PI3K–Akt survival pathway (132) and stabilization of nuclear factor E2-related factor 2 (Nrf2) transcription factor. Nrf2 increases the transcription of multidrug-resistant transporters and antioxidant genes, counteracting the chemotherapy-induced oxidative damage (133). Similarly, ATX inhibition enhances the radiotherapy-induced apoptosis in breast cancer cells (134) and attenuates radiation-induced survival, invasion, and angiogenesis in glioblastoma cells (135). LPA-mediated therapy resistance could also be attributed to its role in regulating tumor-associated macrophage (TAM) formation and tumor immunity (136–138).
In addition, to convert lyso-PC into PtdCho, LPCAT enzymes reacetylate lyso-PC and generate another lipid that is a potent cellular mediator, platelet-activating factor (PAF). Four enzymes (LPCAT1–LPCAT4) constitute the LPCAT family and, despite all LPCAT members being involved in lyso-PC conversion into PtdCho, only LPCAT1 and LPCAT2 are known to play an important role in PAF production (139, 140). We have demonstrated in vitro that the simultaneous silencing of all four LPCAT transcripts by modular nucleic acid nanoparticles resulted in lyso-PC (lyso-PAF 16:0 and 18:0) accumulation and enhanced the radiation cytotoxic effect in melanoma cells. We suggest that interfering in LPCAT-mediated signaling disturbs the generation of lyso-PAF, and PAF and contributes to cancer cell sensitization (141). PAF generally refers to alkyl-PAF, the most active form of PAF; however, abundant amounts of an acyl analog of PAF (acyl-PAF) is concomitantly generated with the alkyl PAF species. For a long time, acyl-PAF was considered an inactive PAF analog (142). Intriguingly, Chaithra et al. (143) have demonstrated in vitro and in vivo that acyl-PAF dampens PAF-R signaling and suppresses the action of alkyl-PAF (143). Accordingly, besides exerting their actions through a single PAF receptor (PAF-R), this pair of lipid mediators has opposite effects as inflammatory set-point modulators. The acyl-PAF has been neglected in PAF biology studies in the oncology field, and it is crucial to address the complex interplay between PAFR, alkyl-PAF, acyl-PAF, and their common catabolic enzyme PAF acetylhydrolase (PAF-AH) to unravel the role of PAF/PAFR signaling pathway. PAF is implicated in cancer progression by triggering inflammation and promoting proliferation, survival, metastasis, angiogenesis, and immune-suppressive responses (144). Elevated levels of PAF or increased PAF-R expression was observed in response to various stimuli, including therapeutic agents (145). As outlined, cisplatin increases PAF-R expression, and its inhibition by a PAF-R antagonist resulted in the chemosensitization of melanoma (146) and ovarian (147) cancer cells in vitro and in vivo. Additionally, PAF is generated following the treatment of B16F10 melanoma cells with chemotherapic agents such as etoposide, cisplatin, and melphalan. Importantly, elevated levels of PAF and oxidized lipids with PAF-R agonist activity were detected after the treatment with these drugs as a result of their common ability to induce ROS (148). Similar results were obtained after exposure of melanoma and cervical cancer cells to irradiation (149, 150). Interesting, in a murine melanoma model with a dual injection of B16F10 cells, treatment of one tumor with irradiation or chemotherapy augmented the growth of the untreated tumor in a PAF-R-dependent manner (150) (148). This evidence offers important insight into the systemic role of PAF and PAF-R agonists on negative regulation of therapy efficacy. In this sense, increased activation of PAF–PAFR axis impacts chemo/radioresistance through inducing immunosuppression by modulating regulatory T cells (Tregs) in a COX-2–dependent process (148). These results have driven the investigation of PAF/PAF-R axis in the tumor repopulation phenomenon.
The most prominent consequence of anticancer therapy is a massive induction of cell death frequently associated with a residual number of surviving tumor cells with the capacity to repopulate the tumor. The molecular mechanism involved in tumor repopulation has been investigated, and in 2011, Huang et al. (151) showed that in radiotherapy-induced apoptotic cancer cells, activated caspase-3 activates cPLA-2 and results in increased levels of PGE2, which as mentioned above can trigger protumoral signaling pathways and stimulate the growth of surviving tumor cells culminating in tumor repopulation (151). Interestingly, compelling evidence has indicated that PAF is at least partially responsible for this mitogenic effect of dying cells. Bachi et al. (152) reported that co-injection of apoptotic cells and a subtumorigenic dose of melanoma cells promote the tumor growth, and this phenomenon was inhibited by PAF-R antagonists (152). The following study showed that irradiated TC-1 cells promote the in vitro proliferation of TC-1 viable cells that was diminished by PAF-R antagonist treatment. In the same study, in an in vivo repopulation assay with a model of a human carcinoma cell line expressing (KBP) or not (KBM) PAF-R, the co-injection of live KBP cells and irradiated-induced dying KBM cells resulted in faster tumor growth compared with co-injection of a mixture of live and irradiated KBM cells (153).
Another catabolic route of PtdCho is mediated by PC-PLC enzyme that hydrolyzes PtdCho into PCho and DAG. The latter is probably the best-studied second messenger in cancer biology. It has been shown that the transformation of cells with oncogenes, such as ras, results in a prolonged and persistent elevation in DAG levels. Moreover, DAG activates the protein kinase C (PKC) pathway that is involved in several protumoral pathways, including cell cycle progression, tumorigenesis, and metastatic dissemination (154). Another lipid mediator, PA, is generated from the PtdCho hydrolysis mediated by PC-PLD. The mammalian target of rapamycin (mTOR) was reported as the main target of PA in cancer cells (155). The stability and activity of mTOR complexes depend on interaction with PA and result in signals for cancer cell survival (156). PA interacts with mTOR in a manner that is competitive with the mTOR inhibitor rapamycin, and as a consequence, elevated PC-PLD activity, frequently observed in tumors, confers rapamycin resistance (157). Upregulation of PLD2 was observed in multidrug-resistant colon and breast cancer cells, suggesting that PC-PLD could provide a survival signal involved in therapy resistance (158).
In line with the experimental observations mentioned above, it is not unreasonable to assume that cancer cells exhibit a prominent PtdCho degradation. The fact that PtdCho degradative enzyme activity (46, 159, 160) and PtdCho-derived mediator levels are frequently found elevated in tumors (161), cancer cells with increased PtdCho degradation hijack PtdCho-derived lipid mediators to favor tumor progression and enhance therapy resistance.
Phosphatidylcholine-Coated Lipid Droplets Confer Resistance to Therapy
Lipid droplets (LDs) are predominantly formed by triacylglycerol (TAG) and PtdCho. In the last years, the role of these organelles in cancer has been well recognized, and more recently, some groups have made an effort to understand their role in tumor resistance. LD formation and accumulation were found in some drug-resistant cell lines, raising the possibility that these organelles might confer resistance to therapy (162–165). The association of choline metabolism and LD was demonstrated by the presence of active LPCAT1 and LPCAT2 in LDs by Moessinger et al. in 2011 (166). Recently, Cotte et al. (167) demonstrated that 5-fluorouracil and oxaliplatin-induced lipid droplet formation in colorectal cancer cell lines was supported by the enzyme LPCAT2 (167). Moreover, it was observed that LPCAT2-dependent lipid droplets conferred resistance to chemotherapy in these cells, and this effect could be reversed by inhibition of LD biogenesis, indicating the potential of LPCAT2 as a target to increase chemotherapy efficacy.
Choline Metabolites Can Modulate DNA Methylation and DNA Repair
Some studies conducted in normal cells showed the relationship between choline and DNA. Due to the presence of three methyl groups in the nitrogen atom, choline can donate these methyl groups to the formation of S-adenosylmethionine (SAM) that is the main methyl donor for the epigenetic alteration in DNA and histones (168, 169). In 2004, Niculescu et al. (170) observed an increase in cyclin-dependent kinase inhibitor 3 (CDKN3) levels in choline-deficient neuroblastoma cells due to its hypomethylation leading to a reduction in proliferation, revealing that choline can interfere with tumorigenesis as a modulator of DNA methylation (170). A few years later, this finding was corroborated by studies using rodent models that showed that diets low in choline led to an increase in spontaneous hepatocarcinoma (revised in 171). Additionally, Kovacheva et al. demonstrated that choline deficiency was responsible for DNA methyltransferase 1 (DNMT1) overexpression due to its hypomethylation, which led to a global DNA hypermethylation in rats (172). These studies suggest that choline might contribute to methyl metabolism and DNA methylation and gene regulation in carcinogenesis and tumor progression.
Furthermore, it is known that the effect of cytotoxic therapy is mainly dependent on nuclear DNA damage extension and the DNA repair capacity of tumor cells to remove these lesions, and choline metabolism can interfere in this process. In 2007, Mori et al. (173) observed that under ChoK knockdown, the death ratio of 5-fluorouracil-treated breast cancer cells increased and, at the molecular levels, this effect was accompanied by a decrease in the expression of some DNA repair-related genes such as RAD23 that is known to participate in the nucleotide excision repair pathway (173). Using a rodent model to study carcinogenesis, choline deficiency was found to be correlated with the silencing of some tumor suppressor genes including the DNA repair genes BRCA1 and hMLH1, indicating that this metabolite also modulates DNA stability (174).
Phosphatidylcholine Metabolic Enzymes and Receptor Tyrosine Kinase Activation
In the last years, genome sequencing from tumor cells led to the identification of oncogenic mutations that are responsible for tumor cell survival and growth. Some of these mutations were found to be druggable, and blockade of the signaling pathways governed by them had improved cancer treatment in these cases. A noticeable example is the receptor tyrosine kinases (RTKs) that are often constitutively activated in different tumor types. Interestingly, some choline metabolites seem to participate in some of these oncogenic pathways controlled by RTKs. A possible correlation between RTK and choline metabolism was demonstrated by Pisanu et al. (175). The authors verified an increase in PC-PLC activity and PCho content in Human epidermal growth factor receptor 2, HER2--overexpressing ovarian cancer cells. Previously, Paris et al. (176) showed PC-PLC accumulation in the plasma membrane of HER2-overexpressing breast tumor cells (176). PC-PLC inhibition caused a downregulation in HER2 levels due to HER2 internalization that impaired its return to the cell membrane and the activation of HER2 signaling pathways. Moreover, the authors demonstrated that PC-PLC is physically associated with both HER2 and EGFR, and blockade of PC-PLC was able to reduce cell proliferation even in trastuzumab-resistant cells. These results provide evidence that PC-PLC is a promising target to counteract the oncogenic effect of HER2 amplification mainly in breast and ovary malignancies. Concerning the other molecules from choline metabolism, in 2012, it was shown that EGFR interacts with choline kinase α2 (ChoKα2) (177). The authors observed that c-Src-dependent phosphorylation sites of CHKA2 are necessary for EGF-dependent cell growth, suggesting that ChoKα may be an effective target for the treatment of tumors that overexpress EGFR and c-Src. In prostate cancer, the enzyme ChoKα was proposed to be a chaperone for androgen receptor, since its transcriptional activity was dependent on ChoKα. The inhibition of these choline kinases caused a decrease in cell proliferation in vitro, tumor growth, and metastasis in vivo, demonstrating its potential as a target for prostate cancer treatment (178). A recent work by Lin et al. (179) also described the association between ChoKα and EGFR in hepatocarcinoma (179). The authors observed that the pro-metastatic effect of ChoKα is mediated by its binding to EGFR, promoting its dimerization and AKT activation. Additionally, ChoKα overexpression promoted resistance to EGFR-targeted drugs both in vitro and in vivo, and the dual inhibition of ChoKα/mammalian target of rapamycin complex 2, mTORC2 might overcome the resistance to EGFR-targeted therapy in these tumors.
Still, under this context, PAF metabolite also interacts with EGFR in cancer cells. In ovarian tumor cells, PAF increased EGFR phosphorylation via PLCβ, intracellular Ca2+, Src, and the ADAM-mediated release of EGFR ligand HB-EGF, showing the interaction between PAFR and EGFR signaling pathways (147). More recently, one of the enzymes responsible for PAF production, LPCAT1, was also shown to be required for EGFR signaling. In GBM cells, EGFRvIII altered cell lipid composition through LPCAT1 that is, in turn, upregulated by EGFR. Knockdown of LPCAT1 was able to reduce tumor growth in vivo, indicating that targeting LPCAT1 can be a promising strategy to treat or reduce tumor recurrence in amplified EGFRvIII GBMs (180). These studies demonstrate the potentially actionable role of the choline-related enzyme in cancer treatment.
Phosphatidylcholine Contributes to Autophagy-Induced Drug Resistance
Autophagy is a catabolic mechanism that plays an important role in the lysosomal degradation of protein aggregates, macromolecules, and damages organelles to recycle cellular components and sustain cell metabolism. This dynamic cellular self-digestion has a dual role in cancer cells, acting as a tumor suppressor or tumor promoter, depending on cancer type and stage. Cancer cells display activation of diverse processes to overcome stress, among them, autophagy. This program provides metabolic needs and helps cancer cells to sustain tumor viability and promote drug resistance (181, 182). While autophagy has been plenty studied, the role of lipids in this process is in its early stages, in part, due to the technical challenge of working with lipids. Phospholipids derived from the Kennedy pathway play an important role in the first phases of autophagy. PtdEth acts as an anchor of the microtubule-associated light protein light chain 3 (LC3), essential to cargo selection and autophagosome biogenesis (183). Recently, choline phospholipids (ChoPL), composed of phosphatidylcholines, sphingomyelin, and lysophosphatidylcholines, were reported in the autophagosome assembly. In this study, Andrejeva et al. (184) demonstrated that autophagy induced by anticancer drugs, followed by the incorporation of 13C-labeled choline, resulted in a high de novo synthesis of ChoPL in cancer cells. Moreover, to investigate the mechanism responsible for this process, they used MT58 cells that hold a temperature-sensitive mutation in the rate-limiting enzyme of PtdCho synthesis, CTP: phosphocholine cytidylyltransferase α1 (CCTα1). They showed that the loss of CCTα1 activity revokes autophagy and impairs cells to sustain autophagosome formation for extended periods of autophagy (184). By this, novel studies have also shown the importance of a second human CTP: phosphocholine cytidylyltransferase, CCTβ3 enzyme. In short-term starved cells, CCTβ3 is recruited to the autophagosome membrane to activate PtdCho synthesis and induce omegasome expansion. Despite that, CCTβ3 did not cause a meaningful upregulation of autophagy. However, opposite effects were observed in cells submitted to long periods of starvation, indicating that CCTβ3 is critical in the PtdCho synthesis to sustain prolonged autophagy (185). Additionally, the induction of autophagy in CCTβ-null cancer cells was significantly suppressed, and such effect was reversed by rescued expression of CCTβ3. Interestingly, the re-expression of CCTβ3 increased cell survival after starvation, indicating the relevance of PtdCho metabolism to autophagy activation and the subsequent impact in survival and resistance to therapy of cancer cells. In this sense, it was demonstrated that treatment with ChoK inhibitors, as B-3D and EB-3P, in liver cancer cells caused the reduction of autophagy components and induced apoptosis (186). Thus, it is likely that elevated levels of choline phospholipids observed in cancer cholinic phenotype may sustain drug-induced cytoprotective autophagy, which favors therapy resistance.
Modulation of the Immune Microenvironment by Phosphatidylcholine-Derived Lipid Mediators
The studies that characterized the aberrant choline metabolism in cancer cells were based on two-dimensional (2D) tissue culture models. These reductionist models fail to reflect the complexities of TMEs that can influence cancer metabolic pathways. In line with this, Mori et al. (187) identified differences in Cho metabolites loads, especially PC and tCho, between cancer cells maintained in 2D monolayer culture and the corresponding tumor xenografts (187). This study reveals the importance of the TME in modulating choline metabolism. As mentioned above, TME conditions such as hypoxic, acidic, and areas of cell death can modulate Cho metabolic pathways. Additionally, altered tumoral PtdCho metabolism can mediate the interaction of cancer cells with TME components, such as immune cells, regulating the immune responses. Nishiyama-Naruke and Curi (188) found that PtdCho is incorporated by macrophages at higher rates than lymphocytes; afterward, it is secreted and transferred to these latest cells, promoting an antiproliferative effect (188). Lyso-PC, a class of lipids derived from the cleavage of PtdCho, can be recognized in the context of CD1d by a subpopulation of human T lymphocytes, called natural killer T (NKT) cells (189). Fox et al. (189) identified LPC as a self-antigen responsible for the activation of human NKT cells, specifically the subgroup known as invariant NKT (iNKT). In a murine context, PtdCho was determined to be in complex with murine CD1d by the crystal structure study; however, this last study did not address the activation of murine NKT by CD1d-mediated presentation of PtdCho (190). There is little information about the role of NKT cells in the TME compared to NK cells. However, differences in the distributions and phenotypic and metabolic profiles of NK vs. NKT cells have been observed in breast cancer and melanoma progression (191). Liu et al. (191) have demonstrated that NKT cells are exhausted in advanced cancers, contributing to the suppressive TME. A major contributor to tumor progression and the main obstacle for successful tumor immunotherapy is the suppression or dysfunction of immune cells, and PtdCho-derived lipid mediators play a role as an intercellular signal during tumor immune responses, promoting regulatory functions of diverse immune cells (Figure 5). It is worth noting that lipid mediators exert their biological effects by binding to cognate receptors (192), which can be expressed in cancer cells and stromal cells. Considering that several PtdCho-derived lipid mediators can be generated by both cells, mainly immune cells, we will further extend our discussion on the impact of PtdCho-derived mediators on therapy resistance by exploiting the role of these lipid mediators in the crosstalk between cancer and immune cells.
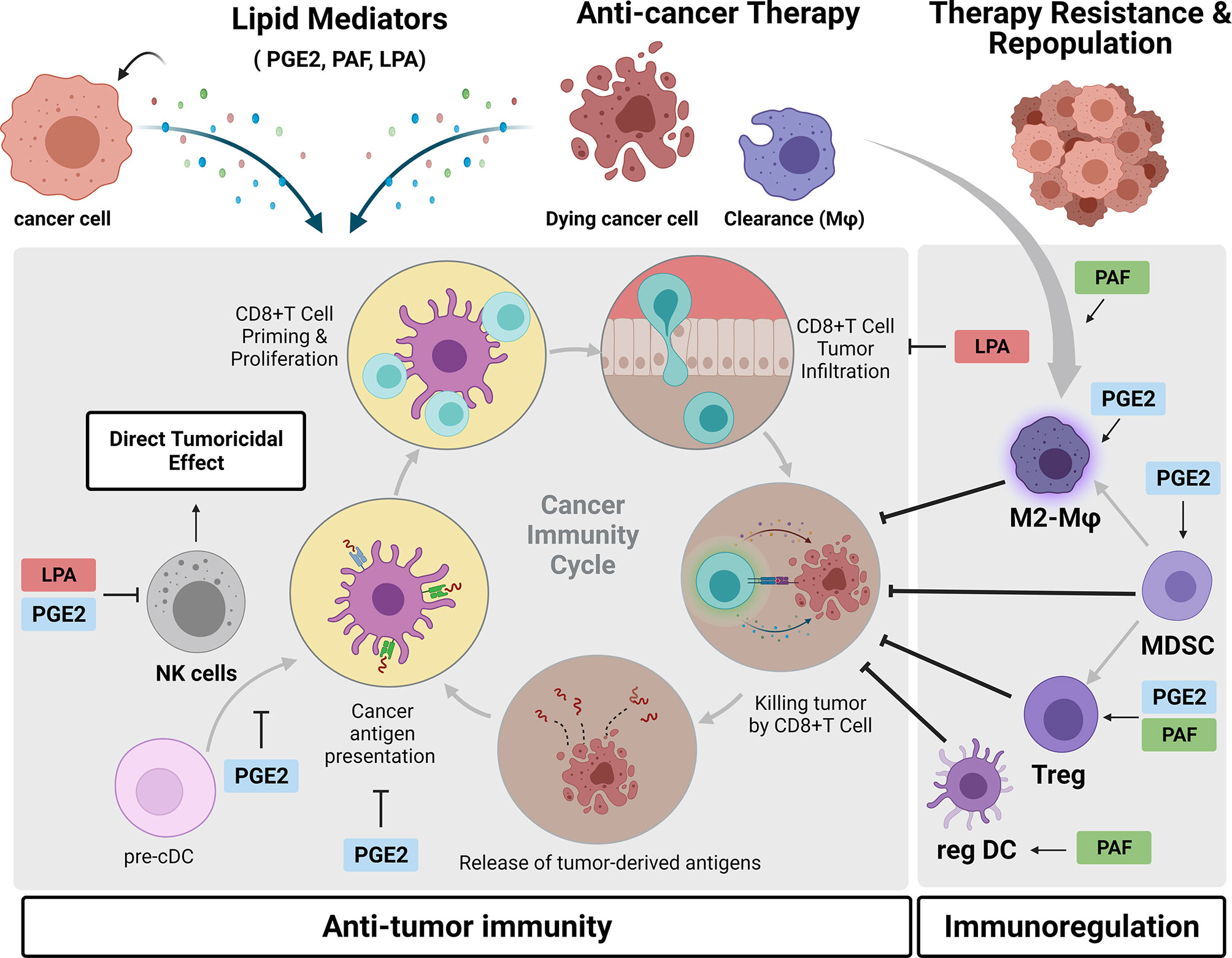
Figure 5 Crosstalk between cancer cells and the immune microenvironment mediated by phosphatidylcholine (PtdCho)-derived lipid mediators. During tumor progression and in response to anticancer treatments, cancer cells generate PtdCho-derived lipid mediators, such as prostaglandin E2 (PGE2), platelet-activating factor (PAF), and LPA. Once released in the tumor microenvironment, they bind to their cognate receptors present in diverse immune cells, inhibiting the antitumor immunity and promoting immunoregulation. These lipid mediators exert a complex interplay between tumor and immune cells that contributes to therapy resistance and tumor repopulation. Created with BioRender.com.
There is extensive literature describing that COX-2/PGE2 axis triggers tumor immune evasion in multiple ways leading to disease progression and therapy resistance (193). Tumor-derived PGE2 promotes the activity of the main immunosuppressive cells in the TME, such as myeloid-derived suppressor cells (MDSCs) (194, 195), M2-like macrophages (196, 197), and Tregs (198, 199). PGE2 is also reported to promote inactivation of antitumor immune response by directly impairing NK activity (200) and also inhibition of NK–dendritic cell (DC) crosstalk, which is crucial for DC recruitment into the tumor (201). Modulatory effects of PGE2 on DC have also been described and show that PGE2-primed DC has increased production of the anti-inflammatory cytokine interleukin 10 (IL-10) and decreased antigen-presenting cell, APC activity, inducing the development of a tolerogenic subset of DCs (202) (203). Recently, evidence shows that tumor-derived PGE2 promoted programmed cell death protein ligand 1 (PD-L1) expression on tumor-infiltrating myeloid cells and, therefore, plays an important role in tumor escape from anti-PD-L1 immunotherapies (204). Altogether, these effects of PGE2 drives tumor to a non-T cell-inflamed status, a crucial refractory condition to cancer immunotherapies. Indeed, COX-2 inhibition and consequently diminished levels of PGE2 reduce the infiltration of MDSC in the TME along with a lymphocyte-mediated antitumor response (194, 195). A study with viral vectors engineered to target PGE2 demonstrated that this viral therapy was able to reduce intratumoral MDSC and sensitize tumors to anti-PD-1 treatment (205). Several in vivo studies have demonstrated an antitumoral effect of selective inhibitors of the prostaglandin E receptor 4 (EP4), one of four PGE2 receptors. These EP4 antagonists suppressed tumor growth by NK cell function reactivation and DC repopulation together with an increase in CD8+ T-cell frequency while decreasing M2-like macrophage polarization (206). Similarly, the effect of switching from an immunosupressive response to antitumor response was observed with inhibition of EP2 receptor (207).
PAF is another PtdCho-derived lipid mediator with immunoregulatory activity. The main idea is that therapy-induced PAF/PAFR axis activation could result in systemic immunosuppression that reduces therapy efficacy. In line with this, evidence shows that PAFR is essential in the clearance of apoptotic cells and induces a regulatory phenotype of macrophages (208, 209). Another piece of evidence also shows that implanted tumors in mice that do not express PAFR (PAFR KO) exhibited higher infiltration of M1-like (CD11c+) and lower M2-like (CD206+) macrophages (153). Similarly, PAFR activation in DC has been shown to induce a regulatory phenotype of these cells characterized by an increase in IL-10 and PGE2 production, which was blocked by PAFR antagonists (210). These data suggest that either the phagocytosis of therapy-induced apoptotic cells or the binding of therapy-generated PAF to macrophages or DCs results in an M2-like phenotype (TAMs) and regulatory DCs, respectively, in the TME. Moreover, Tregs and MDSCs participate in the PAF-mediated increased growth of B16F10 melanoma tumors. Sahu et al. (211) reported that UVB-generated PAFR agonists potentiate the tumor growth of B16F10 cells, a phenomenon that was reversed by depletion of Tregs via anti-CD25 neutralizing antibodies (211).
Recently, emerging evidence of LPA has addressed the role of this lipid mediator in the crosstalk between cancer cells and TME cells (137). It has been demonstrated that LPA negatively modulates antitumor immunity via suppressing NK activity (212), inhibiting CD8+ T-cell infiltration and activity (138, 213). Interestingly, it was reported that a predominant source of LPA production in the TME is derived from a consecutive action by platelet-activating factor acetylhydrolase (PAF-AH) and ATX in TAM (214). In parallel, LPA has been reported to mediate TAM formation by activating the PI3K/AKT/mTOR signaling pathway through LPAR receptor activation, describing an LPA vicious cycle that contributes to malignant features of ovarian cancer (136). Although emerging data on immunomodulatory actions of LPA in the context of cancer immunity have been reported, there are open questions of how LPA regulates other TME cells, including Tregs, MDSCs, TAMs, and CD4+ T cells.
Taken together, all these reports demonstrate that a variety of PtdCho-derived lipid mediators support an immunosuppressive TME that compromises the therapeutic efficacy of anticancer treatments. As outlined, chemo and radiotherapy-induced tumor cell debris generates lipid mediators, in particular PGE2 (151, 215) and PAF (153), that create a protumorigenic TME, favoring the growth of residual surviving cancer cells. This has unveiled an insight into the mechanism behind the tumoral repopulation process and has driven the investigation of whether stimulating the clearance of therapy-generated debris could mitigate this phenomenon. Emerging evidence indicates that the chronic inflammation associated with tumor growth is promoted by a failure in the resolution of inflammation (216, 217), a process coordinated by specialized pro-resolving mediators (SPMs), such as resolvins, a family of endogenous lipid mediators that counteract pro-inflammatory cytokines and increase the macrophage-mediated clearance of cell debris. Sulciner et al. (218) reported that resolvins (RvD1, RvD2, or RvE1) inhibit therapy-generated debris stimulation of tumor growth (219). Interestingly, increased levels of resolvin (RevE1) were detected in the plasma of healthy individuals after administration of aspirin (219). Moreover, low-dose aspirin inhibited experimental tumor growth and metastasis by triggering SPM generation, identifying a resolving receptor-dependent mechanism of aspirin chemopreventive activity (220). These findings have unveiled an exciting pro-resolving strategy to enhance the effectiveness of current cancer therapies and prevent a recurrence.
Extracellular Vesicles as a Communication Route of Phosphatidylcholine Metabolites Between Cancer Cells and Tumor Microenvironment Cells
The modification in lipid cellular composition observed in cancer cells has also been noticed in the extracellular vesicles (EVs) secreted by them. EVs are nanostructures delimited by a lipid bilayer that carry a range of biologically active macromolecules like RNAs, DNA, protein, lipids, and cytokines. These spherical structures can bind to the plasma membrane or be engulfed by recipient cells, leading to a reprogramming that affects their functionality in the TME. Exosomes, a type of EVs that originated from multivesicular bodies, with bioactive lipids, such as PGE2α, PGE1, and PGE2, are secreted by macrophages and tumor cells into the TME (221).
sPLA2, cPLA2, iPLA2, COX-1, COX-2, AA, and PGE2 were already identified in tumor-derived EVs (204, 222–224). Concerning PGE2, EVs carrying this prostaglandin were shown to be associated with immune escape (204) and release of pro-inflammatory cytokines responsible for MDSC recruitment in breast cancer microenvironment (225, 226). In addition, the blockade of PGE2/EP4 signaling reduced the secretion of EVs by basal mammary epithelial stem cells while promoting the release of EVs and CSC-associated proteins from transformed mesenchymal breast cancer cells, modulating tumor progression. Although the relationship between choline metabolism and EVs is still an unexplored field, one might propose that lipid metabolism indeed affects the production and secretion of EVs, as well as interactions with the recipient cells, and more evidence showing the consequences of altered choline metabolism in tumor-derived EVs and their effects in the TME is a matter of time (227).
Choline Metabolism and Cancer Diagnosis
The abnormal choline metabolism frequently described in cancer stimulated the development of strategies to evaluate this differential metabolic alteration in cancer diagnosing. One technique that has been used to quantify the metabolic profile of tumor tissues is a high-resolution magic angle spinning (HR-MAS) proton magnetic resonance spectroscopy (1H MRS). 1H MRS helps in the detection of increased choline expression and CHKa activity in cancer cells compared to those in non-tumoral cells, making it a potential biomarker to diagnose cancer and a strategy to follow treatment response (228). Although 1H MRS exhibits high sensitivity, the adoption of reliable tCho quantification in the clinics is challenging due to spatial localization errors and overlapping signals from PC, GPC, and Cho. On the contrary, the use of 31P-MRS spectra allows the individual detection of PC, GPC, and GPE metabolites but has lower sensitivity compared with that of 1H MRS (229, 230). Thus, the improvement and the combination of both techniques can be used to do cross-calibration and obtain more accurate results. While several studies use 1H MRS and 31P-MRS to aid in the diagnosis of different types of cancers (228, 231, 232), their use in the clinics is not yet broadly applied.
Another technique to detect increased choline metabolism is positron emission tomography/computed tomography (PET/CT) imaging with tracers. Along with the development of radiolabeled choline analogs, PET imaging, combining metabolic activity and anatomical structure (CT), has gained importance to visualize choline metabolism, providing more definitive diagnostic information (227). The main tracers available in the clinics and approved by the U.S. Food & Drug Administration (FDA) to use in PET imaging are [11C]-choline, [18F]-fluoroethylcholine, and [18F]-fluoromethylcholine. Still, there are no guidelines yet for image acquisition, and they are not widely available due to the high cost and the need for further development (233, 234). Currently, the combination of PET/magnetic resonance imaging (MRI) is being evaluated, since it could have complementary functions that provide more robust data (227).
Alternatives to Specifically Target Phosphatidylcholine Metabolism to Treat Cancer
Based on the protumoral effects associated with aberrant choline activity in tumors, investigations have been conducted to target several components of choline metabolism. A well-explored drug target is the inhibition of ChoK activity. ChoK inhibitors or ChoKα gene silencing by RNA interference has been developed to target ChoK, the enzyme responsible for sustaining PCho biosynthesis (235). Interestingly, studies have shown that downregulation of ChoKα decreased epithelial ovarian cancer cell aggressiveness and increased drug sensitivity (117, 236). Moreover, in ovarian cancer cells, ChoKα impairment overcomes Tumor Necrosis Factor (TNF)-Related Apoptosis-Inducing Ligand (TRAIL) resistance (237). Similar results were also obtained with different pharmacological ChoK inhibitors as hemicholinium-3 (HC-3), a choline transport blocker that presents high toxicity in vivo (238) and chemically modified HC-3 structures, MN58b and RSM932A (also TCD-717). Further modification in MN58b and RSM932A produced novel inhibitors, such as EB-3D and EB-3P, respectively (235). These inhibitors exhibited anticancer activity and decreased cell proliferation in preclinical models (186, 239, 240). RSM932A inhibitor resulted in the most prominent in vivo antitumoral effect, retarding tumor growth in mouse xenograft without associated toxicity (240). RSM932A was the first inhibitor to enter a phase I clinical trial in patients with advanced solid tumors, and although this study has been ended, no data are available yet (186).
There are also other inhibitors targeting several components of the choline metabolism, such as PC-PLD1, PC-PLC, and choline transporters (Table 1). Recently, a novel strategy was designed to modify cancer cell membranes to prevent tumor proliferation. The investigators synthesized PtdCho-reversed choline phosphate lipid-modified with a PD-L1 antibody. Then, this structure was loaded in nanoparticles along with drugs to interact with melanoma cell membranes interfering in its functionality and rigidity, therefore reducing tumor growth and migration (247). Curiously, although inhibitors, drugs, and strategies to target PtdCho pathway have been generated, there is still no established molecule for use in the clinic, and resistance to ChoK inhibitor-induced antitumor effects has also been reported (248). This notion reinforces that it is necessary to investigate more selective and efficient inhibitors of the PtdCho pathway. To that end, it is crucial to clarify the association between local and systemic measurements of PtdCho and their metabolites. Systemic changes can be assessed by lipid quantification in cancer patient serum; however, in vivo measurements of these lipids in the TME is still a challenge. Considering that these lipids are susceptible to degradation or acetylation reactions, serum measurements do not necessarily correspond to TME levels. Thus, one of the most interesting remaining questions is how serum levels of PtdCho and their derivatives correlate with the actual concentration of these molecules within the local TME and their effects. Methodology improvement in the in vivo lipid measurement and strategies to specifically target lipid enzymatic synthesis in cancer cells will allow the study of PtdCho tumoral local effects and will be critical to determine the precise therapeutic window to effectively target this lipid pathway.
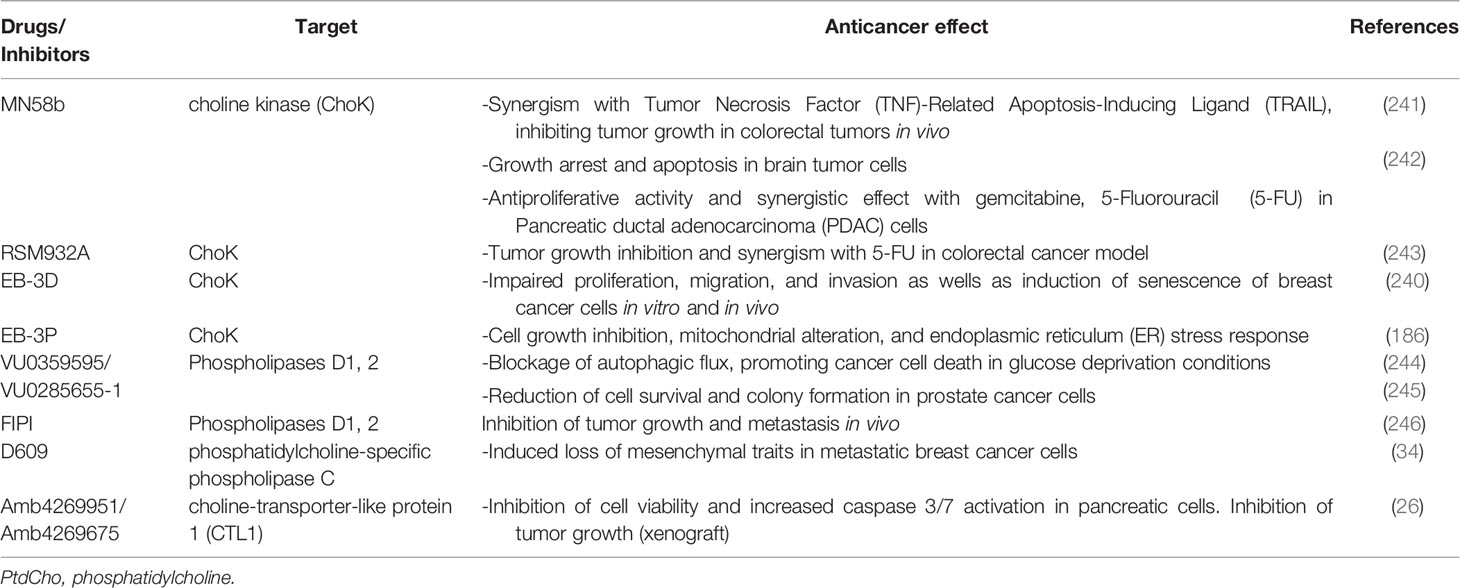
Table 1 Targeting PtdCho metabolism—current strategies for experimental cancer control and treatment.
Conclusion
Abnormal choline metabolism drives cancer cell growth, survival, proliferation, and resistance to therapies in part due to the metabolism of PtdCho, which generates lipid mediators that in turn interfere with immune cell functions. These specific lipid mediators are also produced by immune cells and mediate complex crosstalk that results in immunoregulation and the development of therapy resistance. Controlling lipid metabolism represents a promising strategy for both the inhibition of therapy-induced tumor repopulation and the generation of a sustained antitumor immune response. The development of strategies toward cancer control and treatment through interference with PtdCho metabolism, however, relies on finding the right window of opportunity (when and for how long) for effective treatment.
Author Contributions
All authors participated in the review conceptual design. RS, LA, and SB wrote, reviewed, and edited the article. RC revised and edited the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants CNPq 426714/2016-0 and 305700/2017-0 and FAPESP/SPRINT 17/50029-6.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Warburg O. On the Origin of Cancer Cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell (2012) 21(3):297–308. doi: 10.1016/j.ccr.2012.02.014
4. Molendijk J, Robinson H, Djuric Z, Hill MM. Lipid Mechanisms in Hallmarks of Cancer. Mol Omics (2020) 16(1):6–18. doi: 10.1039/C9MO00128J
5. Faubert B, Solmonson A, Deberardinis RJ. Metabolic Reprogramming and Cancer Progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
6. Tumanov S, Kamphorst JJ. Recent Advances in Expanding the Coverage of the Lipidome. Curr Opin Biotechnol (2017) 43:127–33. doi: 10.1016/j.copbio.2016.11.008
7. Ackerstaff E, Pflug BR, Nelson JB, Bhujwalla ZM. Detection of Increased Choline Compounds With Proton Nuclear Magnetic Resonance Spectroscopy Subsequent to Malignant Transformation of Human Prostatic Epithelial Cells. Cancer Res (2001) 61(9):3599–603.
8. Beloueche-Babari M, Workman P, Leach MO. Exploiting Tumor Metabolism for non-Invasive Imaging of the Therapeutic Activity of Molecularly Targeted Anticancer Agents. Cell Cycle (2011) 10(17):2883–93. doi: 10.4161/cc.10.17.17192
9. Esseridou A, di Leo G, Sconfienza LM, Caldiera V, Raspagliesi F, Grijuela B, et al. In Vivo Detection of Choline in Ovarian Tumors Using 3d Magnetic Resonance Spectroscopy. Invest Radiol (2011) 46(6):377–82. doi: 10.1097/RLI.0b013e31821690ef
10. Glunde K, Bhujwalla ZM. Metabolic Tumor Imaging Using Magnetic Resonance Spectroscopy. Semin Oncol (2011) 38(1):26–41. doi: 10.1053/j.seminoncol.2010.11.001
11. Glunde K, Bhujwalla ZM, Ronen SM. Choline Metabolism in Malignant Transformation. Nat Rev Cancer (2011) 11(12):835–48. doi: 10.1038/nrc3162
12. Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D'Ascenzo S, et al. Alterations of Choline Phospholipid Metabolism in Ovarian Tumor Progression. Cancer Res (2005) 65(20):9369–76. doi: 10.1158/0008-5472.CAN-05-1146
13. Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Di Vito M, et al. Activation of Phosphatidylcholine Cycle Enzymes in Human Epithelial Ovarian Cancer Cells. Cancer Res (2010) 70(5):2126–35. doi: 10.1158/0008-5472.CAN-09-3833
14. Podo F, Canevari S, Canese R, Pisanu ME, Ricci A, Iorio E. MR Evaluation of Response to Targeted Treatment in Cancer Cells. NMR BioMed (2011) 24(6):648–72. doi: 10.1002/nbm.1658
15. Podo F, Paris L, Cecchetti S, Spadaro F, Abalsamo L, Ramoni C, et al. Activation of Phosphatidylcholine-Specific Phospholipase C in Breast and Ovarian Cancer: Impact on MRS-Detected Choline Metabolic Profile and Perspectives for Targeted Therapy. Front Oncol (2016) 6:171. doi: 10.3389/fonc.2016.00171
16. Kennedy EP, Weiss SB. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J Biol Chem (1956) 222(1):193–214. doi: 10.1016/S0021-9258(19)50785-2
17. Gibellini F, Smith TK. The Kennedy Pathway–De Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life (2010) 62(6):414–28. doi: 10.1002/iub.337
18. Vance JE. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic (2015) 16(1):1–18. doi: 10.1111/tra.12230
19. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science (2012) 336(6084):1040–4. doi: 10.1126/science.1218595
20. Daly PF, Lyon RC, Faustino PJ, Cohen JS. Phospholipid Metabolism in Cancer Cells Monitored by 31P NMR Spectroscopy. J Biol Chem (1987) 262(31):14875–8. doi: 10.1016/S0021-9258(18)48107-0
21. Aboagye EO, Bhujwalla ZM. Malignant Transformation Alters Membrane Choline Phospholipid Metabolism of Human Mammary Epithelial Cells. Cancer Res (1999) 59(1):80–4.
22. Nakagami K, Uchida T, Ohwada S, Koibuchi Y, Suda Y, Sekine T, et al. Increased Choline Kinase Activity and Elevated Phosphocholine Levels in Human Colon Cancer. Jpn J Cancer Res (1999) 90(4):419–24. doi: 10.1111/j.1349-7006.1999.tb00764.x
23. Smith TA, Bush C, Jameson C, Titley JC, Leach MO, Wilman DE, et al. Phospholipid Metabolites, Prognosis and Proliferation in Human Breast Carcinoma. NMR BioMed (1993) 6(5):318–23. doi: 10.1002/nbm.1940060506
24. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
25. Eliyahu G, Kreizman T, Degani H. Phosphocholine as a Biomarker of Breast Cancer: Molecular and Biochemical Studies. Int J Cancer (2007) 120(8):1721–30. doi: 10.1002/ijc.22293
26. Hirai K, Watanabe S, Nishijima N, Shibata K, Hase A, Yamanaka T, et al. Molecular and Functional Analysis of Choline Transporters and Antitumor Effects of Choline Transporter-Like Protein 1 Inhibitors in Human Pancreatic Cancer Cells. Int J Mol Sci (2020) 21(15):5190. doi: 10.3390/ijms21155190
27. Watanabe S, Nishijima N, Hirai K, Shibata K, Hase A, Yamanaka T, et al. Anticancer Activity of Amb4269951, a Choline Transporter-Like Protein 1 Inhibitor, in Human Glioma Cells. Pharmaceuticals (Basel) (2020) 13(5):104. doi: 10.3390/ph13050104
28. Inazu M. Choline Transporter-Like Proteins CTLs/SLC44 Family as a Novel Molecular Target for Cancer Therapy. Biopharm Drug Dispos (2014) 35(8):431–49. doi: 10.1002/bdd.1892
29. Hernando E, Sarmentero-Estrada J, Koppie T, Belda-Iniesta C, Ramírez de Molina V, Cejas P, et al. A Critical Role for Choline Kinase-Alpha in the Aggressiveness of Bladder Carcinomas. Oncogene (2009) 28(26):2425–35. doi: 10.1038/onc.2009.91
30. Hu L, Wang RY, Cai J, Feng D, Yang GZ, Xu QG, et al. Overexpression of CHKA Contributes to Tumor Progression and Metastasis and Predicts Poor Prognosis in Colorectal Carcinoma. Oncotarget (2016) 7(41):66660–78. doi: 10.18632/oncotarget.11433
31. Ramírez De Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Tarón M, Ramírez de Molina V, Cejas P, et al. Expression of Choline Kinase Alpha to Predict Outcome in Patients With Early-Stage non-Small-Cell Lung Cancer: A Retrospective Study. Lancet Oncol (2007) 8(10):889–97. doi: 10.1016/S1470-2045(07)70279-6
32. Kwee SA, Hernandez B, Chan O, Wong L. Choline Kinase Alpha and Hexokinase-2 Protein Expression in Hepatocellular Carcinoma: Association With Survival. PloS One (2012) 7(10):e46591. doi: 10.1371/journal.pone.0046591
33. Spadaro F, Ramoni C, Mezzanzanica D, Miotti S, Alberti P, Cecchetti S, et al. Phosphatidylcholine-Specific Phospholipase C Activation in Epithelial Ovarian Cancer Cells. Cancer Res (2008) 68(16):6541–9. doi: 10.1158/0008-5472.CAN-07-6763
34. Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, et al. Inhibition of Phosphatidylcholine-Specific Phospholipase C Results in Loss of Mesenchymal Traits in Metastatic Breast Cancer Cells. Breast Cancer Res (2012) 14(2):R50. doi: 10.1186/bcr3151
35. Uchida N, Okamura S, Kuwano H. Phospholipase D Activity in Human Gastric Carcinoma. Anticancer Res (1999) 19(1B):671–5.
36. Noh DY, Ahn SJ, Lee RA, Park IA, Kim JH, Suh PG, et al. Overexpression of Phospholipase D1 in Human Breast Cancer Tissues. Cancer Lett (2000) 161(2):207–14. doi: 10.1016/S0304-3835(00)00612-1
37. Uchida N, Okamura S, Nagamachi Y, Yamashita S. Increased Phospholipase D Activity in Human Breast Cancer. J Cancer Res Clin Oncol (1997) 123(5):280–5. doi: 10.1007/BF01208639
38. Oka M, Kageshita T, Ono T, Goto A, Kuroki T, Ichihashi M, et al. Protein Kinase C Alpha Associates With Phospholipase D1 and Enhances Basal Phospholipase D Activity in a Protein Phosphorylation-Independent Manner in Human Melanoma Cells. J Invest Dermatol (2003) 121(1):69–76. doi: 10.1046/j.1523-1747.2003.12300.x
39. Xu L, Frankel P, Jackson D, Rotunda T, Boshans RL, D'Souza-Schorey C, et al. Elevated Phospholipase D Activity in H-Ras- But Not K-Ras-Transformed Cells by the Synergistic Action of RalA and ARF6. Mol Cell Biol (2003) 23(2):645–54. doi: 10.1128/MCB.23.2.645-654.2003
40. Janardhan S, Srivani P, Sastry GN. Choline Kinase: An Important Target for Cancer. Curr Med Chem (2006) 13(10):1169–86. doi: 10.2174/092986706776360923
41. Ramírez de Molina A, Penalva V, Lucas L, Lacal JC. Regulation of Choline Kinase Activity by Ras Proteins Involves Ral-GDS and PI3K. Oncogene (2002) 21(6):937–46. doi: 10.1038/sj.onc.1205144
42. Ratnam S, Kent C. Early Increase in Choline Kinase Activity Upon Induction of the H-Ras Oncogene in Mouse Fibroblast Cell Lines. Arch Biochem Biophys (1995) 323(2):313–22. doi: 10.1006/abbi.1995.9959
43. Carnero A, Cuadrado A, Del Peso L, Lacal JC. Activation of Type D Phospholipase by Serum Stimulation and Ras-Induced Transformation in NIH3T3 Cells. Oncogene (1994) 9(5):1387–95.
44. Jiang H, Lu Z, Luo JQ, Wolfman A, Foster DA. Ras Mediates the Activation of Phospholipase D by V-Src. J Biol Chem (1995) 270(11):6006–9. doi: 10.1074/jbc.270.11.6006
45. Cai H, Erhardt P, Troppmair J, Diaz-Meco MT, Sithanandam G, Rapp UR, et al. Hydrolysis of Phosphatidylcholine Couples Ras to Activation of Raf Protein Kinase During Mitogenic Signal Transduction. Mol Cell Biol (1993) 13(12):7645–51. doi: 10.1128/mcb.13.12.7645-7651.1993
46. Bjørkøy G, Overvatn A, Diaz-Meco MT, Moscat J, Johansen T. Evidence for a Bifurcation of the Mitogenic Signaling Pathway Activated by Ras and Phosphatidylcholine-Hydrolyzing Phospholipase C. J Biol Chem (1995) 270(36):21299–306. doi: 10.1074/jbc.270.36.21299
47. Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM. Choline Phospholipid Metabolism in Cancer: Consequences for Molecular Pharmaceutical Interventions. Mol Pharm (2006) 3(5):496–506. doi: 10.1021/mp060067e
48. Jacobs MA, Barker PB, Bottomley PA, Bhujwalla Z, Bhujwalla Z, Bluemke DA. Proton Magnetic Resonance Spectroscopic Imaging of Human Breast Cancer: A Preliminary Study. J Magn Reson Imaging (2004) 19(1):68–75. doi: 10.1002/jmri.10427
49. Stadlbauer A, Gruber S, Nimsky C, Fahlbusch R, Hammen T, Buslei R, et al. Preoperative Grading of Gliomas by Using Metabolite Quantification With High-Spatial-Resolution Proton MR Spectroscopic Imaging. Radiology (2006) 238(3):958–69. doi: 10.1148/radiol.2382041896
50. Zakian KL, Sircar K, Hricak H, Chen HN, Shukla-Dave A, Eberhardt S, et al. Correlation of Proton MR Spectroscopic Imaging With Gleason Score Based on Step-Section Pathologic Analysis After Radical Prostatectomy. Radiology (2005) 234(3):804–14. doi: 10.1148/radiol.2343040363
51. Glunde K, Shah T, Winnard PT, Raman V, Takagi T, Vesuna F, et al. Hypoxia Regulates Choline Kinase Expression Through Hypoxia-Inducible Factor-1 Alpha Signaling in a Human Prostate Cancer Model. Cancer Res (2008) 68(1):172–80. doi: 10.1158/0008-5472.CAN-07-2678
52. Prabhakar NR. Oxygen Sensing During Intermittent Hypoxia: Cellular and Molecular Mechanisms. J Appl Physiol (1985) (2001) 90(5):1986–94. doi: 10.1152/jappl.2001.90.5.1986
53. Hara T, Bansal A, Degrado TR. Effect of Hypoxia on the Uptake of [Methyl-3h]Choline, [1-14c] Acetate and [18F]FDG in Cultured Prostate Cancer Cells. Nucl Med Biol (2006) 33(8):977–84. doi: 10.1016/j.nucmedbio.2006.08.002
54. Bansal A, Shuyan W, Hara T, Harris RA, Degrado TR. Biodisposition and Metabolism of [(18)F]fluorocholine in 9L Glioma Cells and 9L Glioma-Bearing Fisher Rats. Eur J Nucl Med Mol Imaging (2008) 35(6):1192–203. doi: 10.1007/s00259-008-0736-y
55. Bansal A, Harris RA, Degrado TR. Choline Phosphorylation and Regulation of Transcription of Choline Kinase α in Hypoxia. J Lipid Res (2012) 53(1):149–57. doi: 10.1194/jlr.M021030
56. Sarri E, Garcia-Dorado D, Abellan A, Soler-Soler J. Effects of Hypoxia, Glucose Deprivation and Acidosis on Phosphatidylcholine Synthesis in HL-1 Cardiomyocytes. CTP:phosphocholine Cytidylyltransferase Activity Correlates With Sarcolemmal Disruption. Biochem J (2006) 394(Pt 1):325–34. doi: 10.1042/BJ20050834
57. Galons JP, Job C, Gillies RJ. Increase of GPC Levels in Cultured Mammalian Cells During Acidosis. A 31p MR Spectroscopy Study Using a Continuous Bioreactor System. Magn Reson Med (1995) 33(3):422–6. doi: 10.1002/mrm.1910330317
58. Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, et al. The Genomic Analysis of Lactic Acidosis and Acidosis Response in Human Cancers. PloS Genet (2008) 4(12):e1000293. doi: 10.1371/journal.pgen.1000293
59. Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative Analysis of Acetyl-CoA Production in Hypoxic Cancer Cells Reveals Substantial Contribution From Acetate. Cancer Metab (2014) 2:23. doi: 10.1186/2049-3002-2-23
60. Yoshimoto M, Waki A, Yonekura Y, Sadato N, Murata T, Omata N, et al. Characterization of Acetate Metabolism in Tumor Cells in Relation to Cell Proliferation: Acetate Metabolism in Tumor Cells. Nucl Med Biol (2001) 28(2):117–22. doi: 10.1016/S0969-8051(00)00195-5
61. Nikolaou S, Machesky LM. The Stressful Tumour Environment Drives Plasticity of Cell Migration Programmes, Contributing to Metastasis. J Pathol (2020) 250(5):612–23. doi: 10.1002/path.5395
62. Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, et al. Phospholipase D Couples Survival and Migration Signals in Stress Response of Human Cancer Cells. J Biol Chem (2006) 281(23):15862–8. doi: 10.1074/jbc.M600660200
63. Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Phospholipase D. (PLD) Drives Cell Invasion, Tumor Growth and Metastasis in a Human Breast Cancer Xenograph Model. Oncogene (2013) 32(49):5551–62. doi: 10.1038/onc.2013.207
64. Yao Y, Wang X, Li H, Fan J, Qian X, Xu Y. Phospholipase D as a Key Modulator of Cancer Progression. Biol Rev Camb Philos Soc (2020) 95(4):911–35. doi: 10.1111/brv.12592
65. Jackowski S. Cell Cycle Regulation of Membrane Phospholipid Metabolism. J Biol Chem (1996) 271(34):20219–22. doi: 10.1074/jbc.271.34.20219
66. Cui Z, Houweling M, Chen MH, Record M, Chap H, Vance DE, et al. A Genetic Defect in Phosphatidylcholine Biosynthesis Triggers Apoptosis in Chinese Hamster Ovary Cells. J Biol Chem (1996) 271(25):14668–71. doi: 10.1074/jbc.271.25.14668
67. Sanchez-Lopez E, Zimmerman T, GOMEZ DEL Pulgar T, Moyer MP, Lacal Sanjuan JC, Cebrian A. Choline Kinase Inhibition Induces Exacerbated Endoplasmic Reticulum Stress and Triggers Apoptosis via CHOP in Cancer Cells. Cell Death Dis (2013) 4:e933. doi: 10.1038/cddis.2013.453
68. van der Sanden MH, Houweling M, van Golde LM, Vaandrager AB. Inhibition of Phosphatidylcholine Synthesis Induces Expression of the Endoplasmic Reticulum Stress and Apoptosis-Related Protein CCAAT/enhancer-Binding Protein-Homologous Protein (CHOP/GADD153). Biochem J (2003) 369(Pt 3):643–50. doi: 10.1042/bj20020285
69. Yen CL, Mar MH, Zeisel SH. Choline Deficiency-Induced Apoptosis in PC12 Cells is Associated With Diminished Membrane Phosphatidylcholine and Sphingomyelin, Accumulation of Ceramide and Diacylglycerol, and Activation of a Caspase. FASEB J (1999) 13(1):135–42. doi: 10.1096/fasebj.13.1.135
70. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol (2015) 10(7):1604–9. doi: 10.1021/acschembio.5b00245
71. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
72. Li D, Li Y. The Interaction Between Ferroptosis and Lipid Metabolism in Cancer. Signal Transduct Target Ther (2020) 5(1):108. doi: 10.1038/s41392-020-00216-5
73. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res (2020) 30(2):146–62. doi: 10.1038/s41422-019-0263-3
74. Lövey J, Nie D, Tóvári J, Kenessey I, Tímár J, Kandouz M, et al. Radiosensitivity of Human Prostate Cancer Cells can be Modulated by Inhibition of 12-Lipoxygenase. Cancer Lett (2013) 335(2):495–501. doi: 10.1016/j.canlet.2013.03.012
75. Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
76. Flores I, Jones DR, Mérida I. Changes in the Balance Between Mitogenic and Antimitogenic Lipid Second Messengers During Proliferation, Cell Arrest, and Apoptosis in T-Lymphocytes. FASEB J (2000) 14(13):1873–5. doi: 10.1096/fj.99-1066fje
77. Bettaïeb A, Plo I, Mansat-de Mas V, Quillet-Mary A, Levade T, Laurent G, et al. Daunorubicin- and Mitoxantrone-Triggered Phosphatidylcholine Hydrolysis: Implication in Drug-Induced Ceramide Generation and Apoptosis. Mol Pharmacol (1999) 55(1):118–25. doi: 10.1124/mol.55.1.118
78. Meisamy S, Bolan PJ, Baker EH, Bliss RL, Gulbahce E, Everson LI, et al. Neoadjuvant Chemotherapy of Locally Advanced Breast Cancer: Predicting Response With In Vivo (1)H MR Spectroscopy–a Pilot Study at 4 T. Radiology (2004) 233(2):424–31. doi: 10.1148/radiol.2332031285
79. Cheng M, Rizwan A, Jiang L, Bhujwalla ZM, Glunde K. Molecular Effects of Doxorubicin on Choline Metabolism in Breast Cancer. Neoplasia (2017) 19(8):617–27. doi: 10.1016/j.neo.2017.05.004
80. Jagannathan NR, Kumar M, Seenu V, Coshic O, Dwivedi SN, Julka PK, et al. Evaluation of Total Choline From in-Vivo Volume Localized Proton MR Spectroscopy and its Response to Neoadjuvant Chemotherapy in Locally Advanced Breast Cancer. Br J Cancer (2001) 84(8):1016–22. doi: 10.1054/bjoc.2000.1711
81. Nishio K, Sugimoto Y, Fujiwara Y, Ohmori T, Morikage T, Takeda Y, et al. Phospholipase C-Mediated Hydrolysis of Phosphatidylcholine is Activated by Cis-Diamminedichloroplatinum(Ii). J Clin Invest (1992) 89(5):1622–8. doi: 10.1172/JCI115758
82. Ruiz-Cabello J, Cohen JS. Phospholipid Metabolites as Indicators of Cancer Cell Function. NMR BioMed (1992) 5(5):226–33. doi: 10.1002/nbm.1940050506
83. Sharma U, Baek HM, Su MY, Jagannathan NR. In Vivo 1h MRS in the Assessment of the Therapeutic Response of Breast Cancer Patients. NMR BioMed (2011) 24(6):700–11. doi: 10.1002/nbm.1654
84. Morse DL, Raghunand N, Sadarangani P, Murthi S, Job C, Day S, et al. Response of Choline Metabolites to Docetaxel Therapy is Quantified In Vivo by Localized (31)P MRS of Human Breast Cancer Xenografts and In Vitro by High-Resolution (31)P NMR Spectroscopy of Cell Extracts. Magn Reson Med (2007) 58(2):270–80. doi: 10.1002/mrm.21333
85. Beloueche-Babari M, Arunan V, Troy H, Te Poele RH, te Fong AC, Jackson LE, et al. Histone Deacetylase Inhibition Increases Levels of Choline Kinase α and Phosphocholine Facilitating Noninvasive Imaging in Human Cancers. Cancer Res (2012) 72(4):990–1000. doi: 10.1158/0008-5472.CAN-11-2688
86. Williams SN, Anthony ML, Brindle KM. Induction of Apoptosis in Two Mammalian Cell Lines Results in Increased Levels of Fructose-1,6-Bisphosphate and CDP-Choline as Determined by 31P MRS. Magn Reson Med (1998) 40(3):411–20. doi: 10.1002/mrm.1910400311
87. Nunn AV, Barnard ML, Bhakoo K, Murray J, Chilvers EJ, Bell JD. Characterisation of Secondary Metabolites Associated With Neutrophil Apoptosis. FEBS Lett (1996) 392(3):295–8. doi: 10.1016/0014-5793(96)00839-3
88. Al-Saffar NM, Marshall LV, Jackson LE, Balarajah G, Eykyn TR, Agliano A, et al. Lactate and Choline Metabolites Detected In Vitro by Nuclear Magnetic Resonance Spectroscopy are Potential Metabolic Biomarkers for PI3K Inhibition in Pediatric Glioblastoma. PloS One (2014) 9(8):e103835. doi: 10.1371/journal.pone.0103835
89. Al-Saffar NM, Jackson LE, Raynaud FI, Clarke PA, Ramírez de Molina A, Lacal JC, et al. The Phosphoinositide 3-Kinase Inhibitor PI-103 Downregulates Choline Kinase Alpha Leading to Phosphocholine and Total Choline Decrease Detected by Magnetic Resonance Spectroscopy. Cancer Res (2010) 70(13):5507–17. doi: 10.1158/0008-5472.CAN-09-4476
90. Beloueche-Babari M, Arunan V, Jackson LE, Perusinghe N, Sharp SY, Workman P, et al. Modulation of Melanoma Cell Phospholipid Metabolism in Response to Heat Shock Protein 90 Inhibition. Oncotarget (2010) 1(3):185–97. doi: 10.18632/oncotarget.125
91. Beloueche-Babari M, Jackson LE, Al-Saffar NM, Eccles SA, Raynaud FI, Workman P, et al. Identification of Magnetic Resonance Detectable Metabolic Changes Associated With Inhibition of Phosphoinositide 3-Kinase Signaling in Human Breast Cancer Cells. Mol Cancer Ther (2006) 5(1):187–96. doi: 10.1158/1535-7163.MCT-03-0220
92. Beloueche-Babari M, Jackson LE, Al-Saffar NM, Workman P, Leach MO, Ronen SM. Magnetic Resonance Spectroscopy Monitoring of Mitogen-Activated Protein Kinase Signaling Inhibition. Cancer Res (2005) 65(8):3356–63. doi: 10.1158/10.1158/0008-5472.CAN-03-2981
93. Beloueche-Babari M, Peak JC, Jackson LE, Tiet MY, Leach MO, Eccles SA. Changes in Choline Metabolism as Potential Biomarkers of Phospholipase C{gamma}1 Inhibition in Human Prostate Cancer Cells. Mol Cancer Ther (2009) 8(5):1305–11. doi: 10.1158/1535-7163.MCT-09-0039
94. Chung YL, Troy H, Banerji U, Jackson LE, Walton MI, Stubbs M, et al. Magnetic Resonance Spectroscopic Pharmacodynamic Markers of the Heat Shock Protein 90 Inhibitor 17-Allylamino,17-Demethoxygeldanamycin (17AAG) in Human Colon Cancer Models. J Natl Cancer Inst (2003) 95:1624–33. doi: 10.1093/jnci/djg084
95. Chung YL, Troy H, Kristeleit R, Aherne W, Jackson LE, Atadja P, et al. Noninvasive Magnetic Resonance Spectroscopic Pharmacodynamic Markers of a Novel Histone Deacetylase Inhibitor, LAQ824, in Human Colon Carcinoma Cells and Xenografts. Neoplasia (2008) 10(4):303–13. doi: 10.1593/neo.07834
96. He XH, Li WT, Gu YJ, Yang BF, Deng HW, Yu YH, et al. Metabonomic Studies of Pancreatic Cancer Response to Radiotherapy in a Mouse Xenograft Model Using Magnetic Resonance Spectroscopy and Principal Components Analysis. World J Gastroenterol (2013) 19(26):4200–8. doi: 10.3748/wjg.v19.i26.4200
97. Katz-Brull R, Lavin PT, Lenkinski RE. Clinical Utility of Proton Magnetic Resonance Spectroscopy in Characterizing Breast Lesions. J Natl Cancer Inst (2002) 94(16):1197–203. doi: 10.1093/jnci/94.16.1197
98. Mignion L, Danhier P, Magat J, Porporato PE, Masquelier J, Gregoire V, et al. Non-Invasive In Vivo Imaging of Early Metabolic Tumor Response to Therapies Targeting Choline Metabolism. Int J Cancer (2016) 138(8):2043–9. doi: 10.1002/ijc.29932
99. Al-Saffar NMS, Troy H, Wong Te Fong AC, Paravati R, Jackson LE, Gowan S, et al. Metabolic Biomarkers of Response to the AKT Inhibitor MK-2206 in Pre-Clinical Models of Human Colorectal and Prostate Carcinoma. Br J Cancer (2018) 119(9):1118–28. doi: 10.1038/s41416-018-0242-3
100. Danishad KK, Sharma U, Sah RG, Seenu V, Parshad R, Jagannathan NR. Assessment of Therapeutic Response of Locally Advanced Breast Cancer (LABC) Patients Undergoing Neoadjuvant Chemotherapy (NACT) Monitored Using Sequential Magnetic Resonance Spectroscopic Imaging (MRSI). NMR BioMed (2010) 23(3):233–41. doi: 10.1002/nbm.1436
101. Fei B, Wang H, Wu C, Chiu SM. Choline PET for Monitoring Early Tumor Response to Photodynamic Therapy. J Nucl Med (2010) 51(1):130–8. doi: 10.2967/jnumed.109.067579
102. Kurhanewicz J, Vigneron DB, Hricak H, Parivar F, Nelson SJ, Shinohara K, et al. Prostate Cancer: Metabolic Response to Cryosurgery as Detected With 3d H-1 MR Spectroscopic Imaging. Radiology (1996) 200(2):489–96. doi: 10.1148/radiology.200.2.8685346
103. van Asten JJ, Vettukattil R, Buckle T, Rottenberg S, van Leeuwen F, Bathen TF, et al. Increased Levels of Choline Metabolites are an Early Marker of Docetaxel Treatment Response in BRCA1-Mutated Mouse Mammary Tumors: An Assessment by Ex Vivo Proton Magnetic Resonance Spectroscopy. J Transl Med (2015) 13:114. doi: 10.1186/s12967-015-0458-4
104. Smith TA, Eccles S, Ormerod MG, Tombs AJ, Titley JC, Leach MO. The Phosphocholine and Glycerophosphocholine Content of an Oestrogen-Sensitive Rat Mammary Tumour Correlates Strongly With Growth Rate. Br J Cancer (1991) 64(5):821–6. doi: 10.1038/bjc.1991.407
105. Banchio C, Lingrell S, Vance DE. Role of Histone Deacetylase in the Expression of CTP:phosphocholine Cytidylyltransferase Alpha. J Biol Chem (2006) 281(15):10010–5. doi: 10.1074/jbc.M513503200
106. Schlager SI, Ohanian SH. Plasma Membrane and Intracellular Lipid Synthesis in Tumor Cells Rendered Sensitive to Humoral Immune Killing After Treatment With Metabolic Inhibitors. J Natl Cancer Inst (1979) 63(6):1475–83.
107. Bosch I, Dunussi-Joannopoulos K, Wu RL, Furlong ST, Croop J. Phosphatidylcholine and Phosphatidylethanolamine Behave as Substrates of the Human MDR1 P-Glycoprotein. Biochemistry (1997) 36(19):5685–94. doi: 10.1021/bi962728r
108. Kiss Z. Tamoxifen Inhibits Uptake and Metabolism of Ethanolamine and Choline in Multidrug-Resistant, But Not in Drug-Sensitive, MCF-7 Human Breast Carcinoma Cells. Ks, C. FEBS Lett (1995) 360:165–8. doi: 10.1016/0014-5793(95)00094-P
109. Ramu A, Ramu N, Rosario LM. Circumvention of Multidrug-Resistance in P388 Cells is Associated With a Rise in the Cellular Content of Phosphatidylcholine. Biochem Pharmacol (1991) 41(10):1455–61. doi: 10.1016/0006-2952(91)90561-I
110. Dubois G, Tapiero H. Variation of Choline-Substituted Lipid Metabolism in Doxorubicin-Resistant Leukemia Cells. BioMed Pharmacother (1992) 46(10):485–9. doi: 10.1016/0753-3322(92)90006-S
111. Riedel S, Abel S, Burger HM, van der Westhuizen L, Swanevelder S, Gelderblom WC. Differential Modulation of the Lipid Metabolism as a Model for Cellular Resistance to Fumonisin B1-Induced Cytotoxic Effects In Vitro. Prostaglandins Leukot Essent Fatty Acids (2016) 109:39–51. doi: 10.1016/j.plefa.2016.04.006
112. Vanpouille C, Jeune LE, Kryza D, Clotagatide A, Janier M, Dubois F, et al. Influence of Multidrug Resistance on (18)F-FCH Cellular Uptake in a Glioblastoma Model. Eur J Nucl Med Mol Imaging (2009) 36(8):1256–64. doi: 10.1007/s00259-009-1101-5
113. Desoubzdanne D, Claparols C, Martins-Froment N, Zedde C, Balayssac S, Gilard V, et al. Analysis of Hydrophilic and Lipophilic Choline Compounds in Radioresistant and Radiosensitive Glioblastoma Cell Lines by HILIC-ESI-MS/MS. Anal Bioanal Chem (2010) 398(6):2723–30. doi: 10.1007/s00216-010-4196-4
114. Kaplan O, van Zijl PC, Cohen JS. Information From Combined 1H and 31P NMR Studies of Cell Extracts: Differences in Metabolism Between Drug-Sensitive and Drug-Resistant MCF-7 Human Breast Cancer Cells. Biochem Biophys Res Commun (1990) 169(2):383–90. doi: 10.1016/0006-291X(90)90343-L
115. Evelhoch JL, Keller NA, Corbett TH. Response-Specific Adriamycin Sensitivity Markers Provided by In Vivo 31p Nuclear Magnetic Resonance Spectroscopy in Murine Mammary Adenocarcinomas. Cancer Res (1987) 47(13):3396–401.
116. Shah T, Wildes F, Penet MF, Winnard PT, Glunde K, Artemov D, et al. Choline Kinase Overexpression Increases Invasiveness and Drug Resistance of Human Breast Cancer Cells. NMR BioMed (2010) 23(6):633–42. doi: 10.1002/nbm.1510
117. Granata A, Nicoletti R, Tinaglia V, de Cecco L, Pisanu ME, Ricci A. Choline Kinase-Alpha by Regulating Cell Aggressiveness and Drug Sensitivity is a Potential Druggable Target for Ovarian Cancer. Br J Cancer (2014) 110(2):330–40. doi: 10.1038/bjc.2013.729
118. Wen S, He Y, Wang L, Zhang J, Quan C, Niu Y, et al. Aberrant Activation of Super Enhancer and Choline Metabolism Drive Antiandrogen Therapy Resistance in Prostate Cancer. Oncogene (2020) 39(42):6556–71. doi: 10.1038/s41388-020-01456-z
119. Ting YL, Sherr D, Degani H. Variations in Energy and Phospholipid Metabolism in Normal and Cancer Human Mammary Epithelial Cells. Anticancer Res (1996) 16(3B):1381–8.
120. Sulciner ML, Gartung A, Gilligan MM, Serhan CN, Panigrahy D. Targeting Lipid Mediators in Cancer Biology. Cancer Metastasis Rev (2018) 37(2-3):557–72. doi: 10.1007/s10555-018-9754-9
121. Shindou H, Hishikawa D, Harayama T, Yuki K, Shimizu T. Recent Progress on Acyl CoA: Lysophospholipid Acyltransferase Research. J Lipid Res (2009) 50(Suppl):S46–51. doi: 10.1194/jlr.R800035-JLR200
122. Tong D, Liu Q, Wang LA, Xie Q, Pang J, Huang Y, et al. The Roles of the COX2/PGE2/EP Axis in Therapeutic Resistance. Cancer Metastasis Rev (2018) 37(2-3):355–68. doi: 10.1007/s10555-018-9752-y
123. Bozzo F, Bassignana A, Lazzarato L, Boschi D, Gasco A, Bocca C, et al. Novel Nitro-Oxy Derivatives of Celecoxib for the Regulation of Colon Cancer Cell Growth. Chem Biol Interact (2009) 182(2-3):183–90. doi: 10.1016/j.cbi.2009.08.006
124. Fujii R, Imanishi Y, Shibata K, Sakai N, Sakamoto K, Shigetomi S, et al. Restoration of E-Cadherin Expression by Selective Cox-2 Inhibition and the Clinical Relevance of the Epithelial-To-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. J Exp Clin Cancer Res (2014) 33:40. doi: 10.1186/1756-9966-33-40
125. Liu XJ, Chen ZF, Li HL, Hu ZN, Liu M, Tian AP, et al. Interaction Between Cyclooxygenase-2, Snail, and E-Cadherin in Gastric Cancer Cells. World J Gastroenterol (2013) 19(37):6265–71. doi: 10.3748/wjg.v19.i37.6265
126. St John MA. Inflammatory Mediators Drive Metastasis and Drug Resistance in Head and Neck Squamous Cell Carcinoma. Laryngoscope (2015) 125(Suppl 3):S1–11. doi: 10.1002/lary.24998
127. Tang X, Benesch MGK, Brindley DN. Role of the Autotaxin-Lysophosphatidate Axis in the Development of Resistance to Cancer Therapy. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(8):158716. doi: 10.1016/j.bbalip.2020.158716
128. Zeng R, Li B, Huang J, Zhong M, Li L, Duan C, et al. Lysophosphatidic Acid is a Biomarker for Peritoneal Carcinomatosis of Gastric Cancer and Correlates With Poor Prognosis. Genet Test Mol Biomarkers (2017) 21(11):641–8. doi: 10.1089/gtmb.2017.0060
129. Minami K, Ueda N, Maeda H, Ishimoto K, Otagaki S, Tsujiuchi T. Modulation of Chemoresistance by Lysophosphatidic Acid (LPA) Signaling Through LPA. Biochem Biophys Res Commun (2019) 517(2):359–63. doi: 10.1016/j.bbrc.2019.07.092
130. Sun H, Ren J, Zhu Q, Kong FZ, Wu L, Pan BR. Effects of Lysophosphatidic Acid on Human Colon Cancer Cells and its Mechanisms of Action. World J Gastroenterol (2009) 15(36):4547–55. doi: 10.3748/wjg.15.4547
131. Frankel A, Mills GB. Peptide and Lipid Growth Factors Decrease Cis-Diamminedichloroplatinum-Induced Cell Death in Human Ovarian Cancer Cells. Clin Cancer Res (1996) 2(8):1307–13.
132. Samadi N, Gaetano C, Goping IS, Brindley DN. Autotaxin Protects MCF-7 Breast Cancer and MDA-MB-435 Melanoma Cells Against Taxol-Induced Apoptosis. Oncogene (2009) 28(7):1028–39. doi: 10.1038/onc.2008.442
133. Venkatraman G, Benesch MG, Tang X, Dewald J, McMullen TP, Brindley DN. Lysophosphatidate Signaling Stabilizes Nrf2 and Increases the Expression of Genes Involved in Drug Resistance and Oxidative Stress Responses: Implications for Cancer Treatment. FASEB J (2015) 29(3):772–85. doi: 10.1096/fj.14-262659
134. Tang X, Wuest M, Benesch MGK, Dufour J, Zhao Y, Curtis JM, et al. Inhibition of Autotaxin With GLPG1690 Increases the Efficacy of Radiotherapy and Chemotherapy in a Mouse Model of Breast Cancer. Mol Cancer Ther (2020) 19(1):63–74. doi: 10.1158/1535-7163.MCT-19-0386
135. Bhave SR, Dadey DY, Karvas RM, Ferraro DJ, Kotipatruni RP, Jaboin JJ, et al. Autotaxin Inhibition With PF-8380 Enhances the Radiosensitivity of Human and Murine Glioblastoma Cell Lines. Front Oncol (2013) 3:236. doi: 10.3389/fonc.2013.00236
136. Feng Y, Xiao M, Zhang Z, Cui R, Jiang X, Wang S, et al. Potential Interaction Between Lysophosphatidic Acid and Tumor-Associated Macrophages in Ovarian Carcinoma. J Inflammation (Lond) (2020) 17:23. doi: 10.1186/s12950-020-00254-4
137. Lee SC, Dacheux MA, Norman DD, Balázs L, Torres RM, Augelli-Szafran CE, et al. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers (Basel) (2020) 12(5):1202. doi: 10.3390/cancers12051202
138. Mathew D, Kremer KN, Strauch P, Tigyi G, Pelanda R, Torres RM. LPA. Front Immunol (2019) 10:1159. doi: 10.3389/fimmu.2019.01159
139. Harayama T, Shindou H, Shimizu T. Biosynthesis of Phosphatidylcholine by Human Lysophosphatidylcholine Acyltransferase 1. J Lipid Res (2009) 50(9):1824–31. doi: 10.1194/jlr.M800500-JLR200
140. Shindou H, Hishikawa D, Nakanishi H, Harayama T, Ishii S, Taguchi R, et al. A Single Enzyme Catalyzes Both Platelet-Activating Factor Production and Membrane Biogenesis of Inflammatory Cells. Cloning and Characterization of Acetyl-CoA : LYSO-PAF Acetyltransferase. J Biol Chem (2007) 282(9):6532–9. doi: 10.1074/jbc.M609641200
141. Saito RF, Rangel MC, Halman JR, Chandler M, de Sousa Andrade LN, Odete-Bustos S, et al. Simultaneous Silencing of Lysophosphatidylcholine Acyltransferases 1-4 by Nucleic Acid Nanoparticles (NANPs) Improves Radiation Response of Melanoma Cells. Nanomedicine (2021) 36:102418. doi: 10.1016/j.nano.2021.102418
142. Marathe GK, Chaithra VH, Ke LY, Chen CH. Effect of Acyl and Alkyl Analogs of Platelet-Activating Factor on Inflammatory Signaling. Prostaglandins Other Lipid Mediat (2020) 151:106478. doi: 10.1016/j.prostaglandins.2020.106478
143. Chaithra VH, Jacob SP, Lakshmikanth CL, Sumanth MS, Abhilasha KV, Chen CH, et al. Modulation of Inflammatory Platelet-Activating Factor (PAF) Receptor by the Acyl Analogue of PAF. J Lipid Res (2018) 59(11):2063–74. doi: 10.1194/jlr.M085704
144. Travers JB, Rohan JG, Sahu RP. New Insights Into the Pathologic Roles of the Platelet-Activating Factor System. Front Endocrinol (Lausanne) (2021) 12:624132. doi: 10.3389/fendo.2021.624132
145. Chammas R, de Sousa Andrade LN, Jancar S. Oncogenic Effects of PAFR Ligands Produced in Tumours Upon Chemotherapy and Radiotherapy. Nat Rev Cancer (2017) 17(4):253. doi: 10.1038/nrc.2017.15
146. Onuchic AC, Machado CM, Saito RF, Rios FJ, Jancar S, Chammas R. Expression of PAFR as Part of a Prosurvival Response to Chemotherapy: A Novel Target for Combination Therapy in Melanoma. Mediators Inflammation (2012) 2012:175408. doi: 10.1155/2012/175408
147. Yu Y, Zhang X, Hong S, Zhang M, Cai Q, Jiang W, et al. The Expression of Platelet-Activating Factor Receptor Modulates the Cisplatin Sensitivity of Ovarian Cancer Cells: A Novel Target for Combination Therapy. Br J Cancer (2014) 111(3):515–24. doi: 10.1038/bjc.2014.323
148. Sahu RP, Ocana JA, Harrison KA, Ferracini M, Touloukian CE, Al-Hassani M, et al. Chemotherapeutic Agents Subvert Tumor Immunity by Generating Agonists of Platelet-Activating Factor. Cancer Res (2014) 74(23):7069–78. doi: 10.1158/0008-5472.CAN-14-2043
149. da Silva-Junior IA, Dalmaso B, Herbster S, Lepique AP, Jancar S. Platelet-Activating Factor Receptor Ligands Protect Tumor Cells From Radiation-Induced Cell Death. Front Oncol (2018) 8:10. doi: 10.3389/fonc.2018.00010
150. Sahu RP, Harrison KA, Weyerbacher J, Murphy RC, Konger RL, Garrett JE, et al. Radiation Therapy Generates Platelet-Activating Factor Agonists. Oncotarget (2016) 7(15):20788–800. doi: 10.18632/oncotarget.7878
151. Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, et al. Caspase 3-Mediated Stimulation of Tumor Cell Repopulation During Cancer Radiotherapy. Nat Med (2011) 17(7):860–6. doi: 10.1038/nm.2385
152. Bachi AL, Dos Santos LC, Nonogaki S, Jancar S, Jasiulionis MG. Apoptotic Cells Contribute to Melanoma Progression and This Effect is Partially Mediated by the Platelet-Activating Factor Receptor. Mediators Inflammation (2012) 2012:610371. doi: 10.1155/2012/610371
153. da Silva Junior IA, Stone SC, Rossetti RM, Jancar S, Lepique AP. Modulation of Tumor-Associated Macrophages (TAM) Phenotype by Platelet-Activating Factor (PAF) Receptor. J Immunol Res (2017) 2017:5482768. doi: 10.1155/2017/5482768
154. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein Kinase C and Cancer: What We Know and What We do Not. Oncogene (2014) 33(45):5225–37. doi: 10.1038/onc.2013.524
155. Foster DA. Phosphatidic Acid and Lipid-Sensing by mTOR. Trends Endocrinol Metab (2013) 24(6):272–8. doi: 10.1016/j.tem.2013.02.003
156. Foster DA, Xu L. Phospholipase D in Cell Proliferation and Cancer. Mol Cancer Res (2003) 1(11):789–800.
157. Chen Y, Zheng Y, Foster DA. Phospholipase D Confers Rapamycin Resistance in Human Breast Cancer Cells. Oncogene (2003) 22(25):3937–42. doi: 10.1038/sj.onc.1206565
158. Fiucci G, Czarny M, Lavie Y, Zhao D, Berse B, Blusztajn JK, et al. Changes in Phospholipase D Isoform Activity and Expression in Multidrug-Resistant Human Cancer Cells. Int J Cancer (2000) 85(6):882–8. doi: 10.1002/(SICI)1097-0215(20000315)85:6<882::AID-IJC24>3.0.CO;2-E
159. Larrodera P, Cornet ME, Diaz-Meco MT, Lopez-Barahona M, Diaz-Laviada I, Guddal PH, et al. Phospholipase C-Mediated Hydrolysis of Phosphatidylcholine is an Important Step in PDGF-Stimulated DNA Synthesis. Cell (1990) 61(6):1113–20. doi: 10.1016/0092-8674(90)90074-O
160. Martin A, Duffy PA, Liossis C, Gomez-Muñoz A, O'Brien L, Stone JC, et al. Increased Concentrations of Phosphatidate, Diacylglycerol and Ceramide in Ras- and Tyrosine Kinase (Fps)-Transformed Fibroblasts. Oncogene (1997) 14(13):1571–80. doi: 10.1038/sj.onc.1200987
161. Xu Y. Lysophospholipid Signaling in the Epithelial Ovarian Cancer Tumor Microenvironment. Cancers (Basel) (2018) 10(7):727. doi: 10.3390/cancers10070227
162. Montopoli M, Bellanda M, Lonardoni F, Ragazzi E, Dorigo P, Froldi G, et al. “Metabolic Reprogramming” in Ovarian Cancer Cells Resistant to Cisplatin. Curr Cancer Drug Targets (2011) 11(2):226–35. doi: 10.2174/156800911794328501
163. Penrose H, Heller S, Cable C, Makboul R, Chadalawada G, Chen Y, et al. Epidermal Growth Factor Receptor Mediated Proliferation Depends on Increased Lipid Droplet Density Regulated via a Negative Regulatory Loop With FOXO3/Sirtuin6. Biochem Biophys Res Commun (2016) 469(3):370–6. doi: 10.1016/j.bbrc.2015.11.119
164. Schlaepfer IR, Hitz CA, Gijón MA, Bergman BC, Eckel RH, Jacobsen BM. Progestin Modulates the Lipid Profile and Sensitivity of Breast Cancer Cells to Docetaxel. Mol Cell Endocrinol (2012) 363(1-2):111–21. doi: 10.1016/j.mce.2012.08.005
165. Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, et al. Blocking Lipid Synthesis Overcomes Tumor Regrowth and Metastasis After Antiangiogenic Therapy Withdrawal. Cell Metab (2014) 20(2):280–94. doi: 10.1016/j.cmet.2014.05.022
166. Moessinger C, Kuerschner L, Spandl J, Shevchenko A, Thiele C. Human Lysophosphatidylcholine Acyltransferases 1 and 2 are Located in Lipid Droplets Where They Catalyze the Formation of Phosphatidylcholine. J Biol Chem (2011) 286(24):21330–9. doi: 10.1074/jbc.M110.202424
167. Cotte AK, Aires V, Fredon M, Limagne E, Derangère V, Thibaudin M, et al. Lysophosphatidylcholine Acyltransferase 2-Mediated Lipid Droplet Production Supports Colorectal Cancer Chemoresistance. Nat Commun (2018) 9(1):322. doi: 10.1038/s41467-017-02732-5
168. Bekdash RA. Neuroprotective Effects of Choline and Other Methyl Donors. Nutrients (2019) 11(12):2995. doi: 10.3390/nu11122995
169. Zeisel S. Choline, Other Methyl-Donors and Epigenetics. Nutrients (2017) 9(5);445. doi: 10.3390/nu9050445
170. Niculescu MD, Yamamuro Y, Zeisel SH. Choline Availability Modulates Human Neuroblastoma Cell Proliferation and Alters the Methylation of the Promoter Region of the Cyclin-Dependent Kinase Inhibitor 3 Gene. J Neurochem (2004) 89(5):1252–9. doi: 10.1111/j.1471-4159.2004.02414.x
171. Zeisel SH. Dietary Choline Deficiency Causes DNA Strand Breaks and Alters Epigenetic Marks on DNA and Histones. Mutat Res (2012) 733(1-2):34–8. doi: 10.1016/j.mrfmmm.2011.10.008
172. Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, et al. Gestational Choline Deficiency Causes Global and Igf2 Gene DNA Hypermethylation by Up-Regulation of Dnmt1 Expression. J Biol Chem (2007) 282(43):31777–88. doi: 10.1074/jbc.M705539200
173. Mori N, Glunde K, Takagi T, Raman V, Bhujwalla ZM. Choline Kinase Down-Regulation Increases the Effect of 5-Fluorouracil in Breast Cancer Cells. Cancer Res (2007) 67(23):11284–90. doi: 10.1158/0008-5472.CAN-07-2728
174. Pogribny IP, Shpyleva SI, Muskhelishvili L, Bagnyukova TV, James SJ, Beland FA. Role of DNA Damage and Alterations in Cytosine DNA Methylation in Rat Liver Carcinogenesis Induced by a Methyl-Deficient Diet. Mutat Res (2009) 669(1-2):56–62. doi: 10.1016/j.mrfmmm.2009.05.003
175. Pisanu ME, Ricci A, Paris L, Surrentino E, Liliac L, Bagnoli M, et al. Monitoring Response to Cytostatic Cisplatin in a HER2(+) Ovary Cancer Model by MRI and In Vitro and In Vivo MR Spectroscopy. Br J Cancer (2014) 110(3):625–35. doi: 10.1038/bjc.2013.758
176. Paris L, Cecchetti S, Spadaro F, Abalsamo L, Lugini L, Pisanu ME, et al. Inhibition of Phosphatidylcholine-Specific Phospholipase C Downregulates HER2 Overexpression on Plasma Membrane of Breast Cancer Cells. Breast Cancer Res (2010) 12(3):R27. doi: 10.1186/bcr2575
177. Miyake T, Parsons SJ. Functional Interactions Between Choline Kinase α, Epidermal Growth Factor Receptor and C-Src in Breast Cancer Cell Proliferation. Oncogene (2012) 31(11):1431–41. doi: 10.1038/onc.2011.332
178. Asim M, Massie CE, Orafidiya F, Pértega-Gomes N, Warren AY, Esmaeili M, et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J Natl Cancer Inst (2016) 108(5):djv371. doi: 10.1093/jnci/djv371
179. Lin XM, Hu L, Gu J, Wang RY, Li L, Tang J, et al. Choline Kinase α Mediates Interactions Between the Epidermal Growth Factor Receptor and Mechanistic Target of Rapamycin Complex 2 in Hepatocellular Carcinoma Cells to Promote Drug Resistance and Xenograft Tumor Progression. Gastroenterology (2017) 152(5):1187–202. doi: 10.1053/j.gastro.2016.12.033
180. Bi J, Ichu TA, Zanca C, Yang H, Zhang W, Gu Y, et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab (2019) 30(3):525–538.e528. doi: 10.1016/j.cmet.2019.06.014
181. Chang H, Zou Z. Targeting Autophagy to Overcome Drug Resistance: Further Developments. J Hematol Oncol (2020) 13(1):159. doi: 10.1186/s13045-020-01000-2
182. Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, et al. Neutral Lipid Stores and Lipase PNPLA5 Contribute to Autophagosome Biogenesis. Curr Biol (2014) 24(6):609–20. doi: 10.1016/j.cub.2014.02.008
183. Thukral L, Sengupta D, Ramkumar A, Murthy D, Agrawal N, Gokhale RS. The Molecular Mechanism Underlying Recruitment and Insertion of Lipid-Anchored LC3 Protein Into Membranes. Biophys J (2015) 109(10):2067–78. doi: 10.1016/j.bpj.2015.09.022
184. Andrejeva G, Gowan S, Lin G, Wong Te Fong AL, Shamsaei E, Parkes HG, et al. Phosphatidylcholine Synthesis is Required for Autophagosome Membrane Formation and Maintenance During Autophagy. Autophagy (2020) 16(6):1044–60. doi: 10.1080/15548627.2019.1659608
185. Ogasawara Y, Cheng J, Tatematsu T, Uchida M, Murase O, Yoshikawa S, et al. Long-Term Autophagy is Sustained by Activation of Cctβ3 on Lipid Droplets. Nat Commun (2020) 11(1):4480. doi: 10.1038/s41467-020-18153-w
186. Sola-Leyva A, López-Cara LC, Ríos-Marco P, Ríos A, Marco C, Carrasco-Jiménez MP, et al. Choline Kinase Inhibitors EB-3D and EB-3p Interferes With Lipid Homeostasis in HepG2 Cells. Sci Rep (2019) 9(1):5109. doi: 10.1038/s41598-019-40885-z
187. Mori N, Wildes F, Takagi T, Glunde K, Bhujwalla ZM. The Tumor Microenvironment Modulates Choline and Lipid Metabolism. Front Oncol (2016) 6:262. doi: 10.3389/fonc.2016.00262
188. Nishiyama-Naruke A, Curi R. Phosphatidylcholine Participates in the Interaction Between Macrophages and Lymphocytes. Am J Physiol Cell Physiol (2000) 278(3):C554–560. doi: 10.1152/ajpcell.2000.278.3.C554
189. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of Lyso-Phospholipids by Human Natural Killer T Lymphocytes. PloS Biol (2009) 7(10):e1000228. doi: 10.1371/journal.pbio.1000228
190. Giabbai B, Sidobre S, Crispin MD, Sanchez-Ruìz Y, Bachi A, Kronenberg M, et al. Crystal Structure of Mouse CD1d Bound to the Self Ligand Phosphatidylcholine: A Molecular Basis for NKT Cell Activation. J Immunol (2005) 175:977–84. doi: 10.4049/jimmunol.175.2.977
191. Liu X, Li L, Si F, Huang L, Zhao Y, Zhang C, et al. NK and NKT Cells Have Distinct Properties and Functions in Cancer. Oncogene (2021) 40(27):4521–37. doi: 10.1038/s41388-021-01880-9
192. Shimizu T. Lipid Mediators in Health and Disease: Enzymes and Receptors as Therapeutic Targets for the Regulation of Immunity and Inflammation. Annu Rev Pharmacol Toxicol (2009) 49:123–50. doi: 10.1146/annurev.pharmtox.011008.145616
193. Johnson AM, Kleczko EK, Nemenoff RA. Eicosanoids in Cancer: New Roles in Immunoregulation. Front Pharmacol (2020) 11:595498. doi: 10.3389/fphar.2020.595498
194. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 Blockade Suppresses Gliomagenesis by Inhibiting Myeloid-Derived Suppressor Cells. Cancer Res (2011) 71(7):2664–74. doi: 10.1158/0008-5472.CAN-10-3055
195. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in Myeloid Suppressor Cells is Induced by COX-2 in Lung Carcinoma. J Exp Med (2005) 202(7):931–9. doi: 10.1084/jem.20050715
196. Heusinkveld M, de vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, et al. M2 Macrophages Induced by Prostaglandin E2 and IL-6 From Cervical Carcinoma are Switched to Activated M1 Macrophages by CD4+ Th1 Cells. J Immunol (2011) 187(3):1157–65. doi: 10.4049/jimmunol.1100889
197. Ylöstalo JH, Bartosh TJ, Coble K, Prockop DJ. Human Mesenchymal Stem/Stromal Cells Cultured as Spheroids are Self-Activated to Produce Prostaglandin E2 That Directs Stimulated Macrophages Into an Anti-Inflammatory Phenotype. Stem Cells (2012) 30(10):2283–96. doi: 10.1002/stem.1191
198. Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor Cyclooxygenase-2/Prostaglandin E2-Dependent Promotion of FOXP3 Expression and CD4+ CD25+ T Regulatory Cell Activities in Lung Cancer. Cancer Res (2005) 65(12):5211–20. doi: 10.1158/0008-5472.CAN-05-0141
199. Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+CD4+CD25+ Adaptive Regulatory T Cells Express Cyclooxygenase-2 and Suppress Effector T Cells by a Prostaglandin E2-Dependent Mechanism. J Immunol (2006) 177(1):246–54. doi: 10.4049/jimmunol.177.1.246
200. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE (2)) Suppresses Natural Killer Cell Function Primarily Through the PGE(2) Receptor Ep4. Cancer Immunol Immunother (2011) 60(11):1577–86. doi: 10.1007/s00262-011-1064-9
201. Harizi H. Reciprocal Crosstalk Between Dendritic Cells and Natural Killer Cells Under the Effects of PGE2 in Immunity and Immunopathology. Cell Mol Immunol (2013) 10(3):213–21. doi: 10.1038/cmi.2013.1
202. Harizi H, Juzan M, Pitard V, Moreau JF, Gualde N. Cyclooxygenase-2-Issued Prostaglandin E(2) Enhances the Production of Endogenous IL-10, Which Down-Regulates Dendritic Cell Functions. J Immunol (2002) 168(5):2255–63. doi: 10.4049/jimmunol.168.5.2255
203. Gualde N, Harizi H. Prostanoids and Their Receptors That Modulate Dendritic Cell-Mediated Immunity. Immunol Cell Biol (2004) 82(4):353–60. doi: 10.1111/j.0818-9641.2004.01251.x
204. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 Pathway Regulates PD-L1 Expression in Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells. Proc Natl Acad Sci U.S.A. (2017) 114(5):1117–22. doi: 10.1073/pnas.1612920114
205. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell (2016) 30(1):108–19. doi: 10.1016/j.ccell.2016.05.012
206. Take Y, Koizumi S, Nagahisa A. Prostaglandin E Receptor 4 Antagonist in Cancer Immunotherapy: Mechanisms of Action. Front Immunol (2020) 11:324. doi: 10.3389/fimmu.2020.00324
207. Sun X, Li Q. Prostaglandin EP2 Receptor: Novel Therapeutic Target for Human Cancers (Review). Int J Mol Med (2018) 42(3):1203–14. doi: 10.3892/ijmm.2018.3744
208. de Oliveira SI, Fernandes PD, Amarante Mendes JG, Jancar S. Phagocytosis of Apoptotic and Necrotic Thymocytes is Inhibited by PAF-Receptor Antagonists and Affects LPS-Induced COX-2 Expression in Murine Macrophages. Prostaglandins Other Lipid Mediat (2006) 80(1-2):62–73. doi: 10.1016/j.prostaglandins.2006.04.002
209. Ferracini M, Rios FJ, Pecenin M, Jancar S. Clearance of Apoptotic Cells by Macrophages Induces Regulatory Phenotype and Involves Stimulation of CD36 and Platelet-Activating Factor Receptor. Mediators Inflammation (2013) 2013:950273. doi: 10.1155/2013/950273
210. Koga MM, Bizzarro B, Sá-Nunes A, Rios FJ, Jancar S. Activation of PAF-Receptor Induces Regulatory Dendritic Cells Through PGE2 and IL-10. Prostaglandins Leukot Essent Fatty Acids (2013) 89(5):319–26. doi: 10.1016/j.plefa.2013.09.003
211. Sahu RP, Turner MJ, Dasilva SC, Rashid BM, Ocana JA, Perkins SM, et al. The Environmental Stressor Ultraviolet B Radiation Inhibits Murine Antitumor Immunity Through its Ability to Generate Platelet-Activating Factor Agonists. Carcinogenesis (2012) 33(7):1360–7. doi: 10.1093/carcin/bgs152
212. Lagadari M, Truta-Feles K, Lehmann K, Berod L, Ziemer M, Idzko M, et al. Lysophosphatidic Acid Inhibits the Cytotoxic Activity of NK Cells: Involvement of Gs Protein-Mediated Signaling. Int Immunol (2009) 21(6):667–77. doi: 10.1093/intimm/dxp035
213. Matas-Rico E, Frijlink E, van der Haar Àvila I, Menegakis A, van Zon M, Morris AJ, et al. Autotaxin Impedes Anti-Tumor Immunity by Suppressing Chemotaxis and Tumor Infiltration of CD8+ T Cells. bioRxiv (2021) 37(7):110013. doi: 10.1101/2020.02.26.966291
214. Reinartz S, Lieber S, Pesek J, Brandt DT, Asafova A, Finkernagel F, et al. Cell Type-Selective Pathways and Clinical Associations of Lysophosphatidic Acid Biosynthesis and Signaling in the Ovarian Cancer Microenvironment. Mol Oncol (2019) 13(2):185–201. doi: 10.1002/1878-0261.12396
215. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-Induced Tumour Repopulation Abrogates Bladder Cancer Chemoresistance. Nature (2015) 517(7533):209–13. doi: 10.1038/nature14034
216. Lawrence T. Inflammation and Cancer: A Failure of Resolution? Trends Pharmacol Sci (2007) 28(4):162–5. doi: 10.1016/j.tips.2007.02.003
217. Zhang Q, Zhu B, Li Y. Resolution of Cancer-Promoting Inflammation: A New Approach for Anticancer Therapy. Front Immunol (2017) 8:71. doi: 10.3389/fimmu.2017.00071
218. Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, et al. Resolvins Suppress Tumor Growth and Enhance Cancer Therapy. J Exp Med (2018) 215(1):115–40. doi: 10.1084/jem.20170681
219. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical Assignment, Antiinflammatory Properties, and Receptor for the Omega-3 Lipid Mediator Resolvin E1. J Exp Med (2005) 201(5):713–22. doi: 10.1084/jem.20042031
220. Gilligan MM, Gartung A, Sulciner ML, Norris PC, Sukhatme VP, Bielenberg DR, et al. Aspirin-Triggered Proresolving Mediators Stimulate Resolution in Cancer. Proc Natl Acad Sci U.S.A. (2019) 116(13):6292–7. doi: 10.1073/pnas.1804000116
221. Kim J, Hong SW, Kim S, Kim D, Hur DY, Jin DH, et al. Cyclooxygenase-2 Expression is Induced by Celecoxib Treatment in Lung Cancer Cells and is Transferred to Neighbor Cells via Exosomes. Int J Oncol (2018) 52(2):613–20. doi: 10.3892/ijo.2017.4227
222. Boilard E. Extracellular Vesicles and Their Content in Bioactive Lipid Mediators: More Than a Sack of microRNA. J Lipid Res (2018) 59(11):2037–46. doi: 10.1194/jlr.R084640
223. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science (2010) 327(5965):580–3. doi: 10.1126/science.1181928
224. Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes Account for Vesicle-Mediated Transcellular Transport of Activatable Phospholipases and Prostaglandins. J Lipid Res (2010) 51(8):2105–20. doi: 10.1194/jlr.M003657
225. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
226. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of Myeloid-Derived Suppressor Cells by Tumor Exosomes. Int J Cancer (2009) 124(11):2621–33. doi: 10.1002/ijc.24249
227. Glunde K, Penet MF, Jiang L, Jacobs MA, Bhujwalla ZM. Choline Metabolism-Based Molecular Diagnosis of Cancer: An Update. Expert Rev Mol Diagn (2015) 15(6):735–47. doi: 10.1586/14737159.2015.1039515
228. Fuss TL, Cheng LL. Evaluation of Cancer Metabolomics Using Ex Vivo High Resolution Magic Angle Spinning (HRMAS) Magnetic Resonance Spectroscopy (MRS). Metabolites (2016) 6(1):11. doi: 10.3390/metabo6010011
229. Gogiashvili M, Nowacki J, Hergenröder R, Hengstler JG, Lambert J, Edlund K. HR-MAS NMR Based Quantitative Metabolomics in Breast Cancer. Metabolites (2019) 9(2):229. doi: 10.3390/metabo9020019
230. Iorio E, Podo F, Leach MO, Koutcher J, Blankenberg FG, Norfray JF. A Novel Roadmap Connecting the. Eur Radiol Exp (2021) 5(1):5. doi: 10.1186/s41747-020-00192-z
231. Bae S, Ulrich CM, Neuhouser ML, Malysheva O, Bailey LB, Xiao L, et al. Plasma Choline Metabolites and Colorectal Cancer Risk in the Women’s Health Initiative Observational Study. Cancer Res (2014) 74(24):7442–52. doi: 10.1158/0008-5472.CAN-14-1835
232. Choi JS, Baek HM, Kim S, Kim MJ, Youk JH, Moon HJ, et al. Magnetic Resonance Metabolic Profiling of Breast Cancer Tissue Obtained With Core Needle Biopsy for Predicting Pathologic Response to Neoadjuvant Chemotherapy. PloS One (2013) 8(12):e83866. doi: 10.1371/journal.pone.0083866
233. Wallitt KL, Khan SR, Dubash S, Tam HH, Khan S, Barwick TD. Clinical PET Imaging in Prostate Cancer. Radiographics (2017) 37(5):1512–36. doi: 10.1148/rg.2017170035
234. Witney TH, Alam IS, Turton DR, Smith G, Carroll L, Brickute D, et al. Evaluation of Deuterated 18F- and 11C-Labeled Choline Analogs for Cancer Detection by Positron Emission Tomography. Clin Cancer Res (2012) 18(4):1063–72. doi: 10.1158/1078-0432.CCR-11-2462
235. Gokhale S, Xie P. ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies. Pharmaceutics (2021) 13(6):911. doi: 10.3390/pharmaceutics13060911
236. Bagnoli M, Granata A, Nicoletti R, Krishnamachary B, Bhujwalla ZM, Canese R, et al. Choline Metabolism Alteration: A Focus on Ovarian Cancer. Front Oncol (2016) 6:153. doi: 10.3389/fonc.2016.00153
237. Rizzo A, Satta A, Garrone G, Cavalleri A, Napoli A, Raspagliesi F, et al. Choline Kinase Alpha Impairment Overcomes TRAIL Resistance in Ovarian Cancer Cells. J Exp Clin Cancer Res (2021) 40(1):5. doi: 10.1186/s13046-020-01794-6
238. Inazu M, Yamada T, Kubota N, Yamanaka T. Functional Expression of Choline Transporter-Like Protein 1 (CTL1) in Small Cell Lung Carcinoma Cells: A Target Molecule for Lung Cancer Therapy. Pharmacol Res (2013) 76:119–31. doi: 10.1016/j.phrs.2013.07.011
239. Lacal JC, Campos JM. Preclinical Characterization of RSM-932A, a Novel Anticancer Drug Targeting the Human Choline Kinase Alpha, an Enzyme Involved in Increased Lipid Metabolism of Cancer Cells. Mol Cancer Ther (2015) 14(1):31–9. doi: 10.1158/1535-7163.MCT-14-0531
240. Mariotto E, Viola G, Ronca R, Persano L, Aveic S, Bhujwalla ZM, et al. Choline Kinase Alpha Inhibition by EB-3d Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers (Basel) (2018) 10(10):391. doi: 10.3390/cancers10100391
241. Kumar M, Arlauckas SP, Saksena S, Verma G, Ittyerah R, Pickup S, et al. Magnetic Resonance Spectroscopy for Detection Of Choline Kinase Inhibition in The Treatment of Brain Tumors. Mol Cancer Ther (2015) 14(4):899–908. doi: 10.1158/1535-7163.MCT-14-0775
242. Mazarico JM, Sánchez-Arévalo Lobo VJ, Favicchio R, Greenhalf W, Costello E, Carrillo-de Santa Pau E, et al. Choline Kinase Alpha (CHKα) as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma: Expression, Predictive Value, and Sensitivity to Inhibitors. Mol Cancer Ther (2016) 15(2):323–33. doi: 10.1158/1535-7163.MCT-15-0214
243. de la Cueva A, Ramírez de Molina A, Alvarez-Ayerza N, Ramos MA, Cebrian A, Del Pulgar TG, et al. Combined 5-FU and ChoKα Inhibitors as a New Alternative therapy of Colorectal Cancer: Evidence in Human Tumor-Derived Cell Lines and Mouse Xenografts. PloS One (2013) 8(6):e64961. doi: 10.1371/journal.pone.0064961
244. Cai M, He J, Xiong J, Tay LWR, Wang Z, Rog C, et al. Phospholipase D1-Regulated Autophagy Supplies Free Fatty Acids to Counter Nutrient Stress in Cancer Cells. Cell Death Dis (2016) 7(11):e2448. doi: 10.1038/cddis.2016.355
245. Noble AR, Maitland NJ, Berney DM, Rumsby MG. Phospholipase D Inhibitors Reduce Human Prostate Cancer Cell Proliferation and Colony Formation. Br J Cancer (2018) 118(2):189–99. doi: 10.1038/bjc.2017.391
246. Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo JA, et al. Key Roles for the Lipid Signaling Enzyme Phospholipase d1 in the Tumor Microenvironment During Tumor Angiogenesis and Metastasis. Sci Signal (2012) 5(249):ra79. doi: 10.1126/scisignal.2003257
247. Li S, Mei W, Wang X, Jiang S, Yan X, Liu S, et al. Choline Phosphate Lipid Insertion and Rigidification of Cell Membranes for Targeted Cancer Chemo-Immunotherapy. Chem Commun (Camb) (2021) 57(11):1372–5. doi: 10.1039/D0CC08011J
248. Ramírez de Molina A, de la Cueva A, Machado-Pinilla R, Rodriguez-Fanjul V, Gomez del Pulgar T, Cebrian A, et al. Acid Ceramidase as a Chemotherapeutic Target to Overcome Resistance to the Antitumoral Effect of Choline Kinase α Inhibition. Curr Cancer Drug Targets (2012) 12(6):617–24. doi: 10.2174/156800912801784811
Keywords: lipid metabolism, phosphatidylcholine, lipid mediators, immunoregulation, immune microenvironment, cancer drug resistance
Citation: Saito RF, Andrade LNS, Bustos SO and Chammas R (2022) Phosphatidylcholine-Derived Lipid Mediators: The Crosstalk Between Cancer Cells and Immune Cells. Front. Immunol. 13:768606. doi: 10.3389/fimmu.2022.768606
Received: 31 August 2021; Accepted: 13 January 2022;
Published: 15 February 2022.
Edited by:
Gabriela Brumatti, Walter and Eliza Hall Institute of Medical Research, AustraliaReviewed by:
Luisa Magalhaes, Universidade Federal de Minas Gerais, BrazilMenglin Cheng, Johns Hopkins University, United States
Jeffrey B. Travers, Wright State University, United States
Copyright © 2022 Saito, Andrade, Bustos and Chammas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Chammas, cmNoYW1tYXNAdXNwLmJy