 Faraz Ahmad1†‡
Faraz Ahmad1†‡ Anshu Rani2‡
Anshu Rani2‡ Anwar Alam1‡
Anwar Alam1‡ Sheeba Zarin1
Sheeba Zarin1 Saurabh Pandey3
Saurabh Pandey3 Hina Singh4
Hina Singh4 Seyed Ehtesham Hasnain4,5*
Seyed Ehtesham Hasnain4,5* Nasreen Zafar Ehtesham1*
Nasreen Zafar Ehtesham1*- 1Laboratory of Infection Biology and Cell Signaling, Indian Council of Medical Research (ICMR)-National Institute of Pathology, New Delhi, India
- 2Kusuma School of Biological Sciences, Indian Institute of Technology, Delhi (IIT-D), New Delhi, India
- 3Department of Biochemistry, Jamia Hamdard, New Delhi, India
- 4Department of Biochemical Engineering and Biotechnology, Indian Institute of Technology, Delhi (IIT-D), New Delhi, India
- 5Department of Life Science, School of Basic Sciences and Research, Sharda University, Greater Noida, India
Mycobacterium tuberculosis (Mtb) is the causative agent of human tuberculosis (TB) which primarily infects the macrophages. Nearly a quarter of the world’s population is infected latently by Mtb. Only around 5%–10% of those infected develop active TB disease, particularly during suppressed host immune conditions or comorbidity such as HIV, hinting toward the heterogeneity of Mtb infection. The aerosolized Mtb first reaches the lungs, and the resident alveolar macrophages (AMs) are among the first cells to encounter the Mtb infection. Evidence suggests that early clearance of Mtb infection is associated with robust innate immune responses in resident macrophages. In addition to lung-resident macrophage subsets, the recruited monocytes and monocyte-derived macrophages (MDMs) have been suggested to have a protective role during Mtb infection. Mtb, by virtue of its unique cell surface lipids and secreted protein effectors, can evade killing by the innate immune cells and preferentially establish a niche within the AMs. Continuous efforts to delineate the determinants of host defense mechanisms have brought to the center stage the crucial role of macrophage phenotypical variations for functional adaptations in TB. The morphological and functional heterogeneity and plasticity of the macrophages aid in confining the dissemination of Mtb. However, during a suppressed or hyperactivated immune state, the Mtb virulence factors can affect macrophage homeostasis which may skew to favor pathogen growth, causing active TB. This mini-review is aimed at summarizing the interplay of Mtb pathomechanisms in the macrophages and the implications of macrophage heterogeneity and plasticity during Mtb infection.
Introduction
Tuberculosis (TB), the oldest global pandemic since prehistoric times, is caused by Mycobacterium tuberculosis (Mtb) which has co-evolved with humans for around 70,000 years (1, 2). While the exact evolutionary age of Mtb is contentious, it has been a cause of significant concern at least since Neolithic human expansion (3–5). As per the current estimates, a quarter of the world’s population has a latent form of TB (6) and around 10% of them may develop active TB during their lifetime (7). Annually, nearly 10 million people are affected with TB which causes nearly 1.2 million deaths (8). The situation is further complicated due to increasing numbers (21%, 0.46 million) of drug-resistant TB (9). Moreover, the control and management of TB has currently been affected due to the unprecedented COVID-19 pandemic ravaging the world (10). As a result, there have been a decrease in notifications and treatment trends and an enhanced mortality for TB. The WHO has therefore flagged concerns about the retardation in the milestones envisaged for the END-TB program (11). With decreased notification and treatment trends and enhanced mortality reported, the COVID-19 pandemic has been suggested to have reversed the years of progress made in TB control efforts (11).
The reductive evolution of the genome has enabled Mtb to evolve into a more virulent and successful pathogen (12, 13). The reductive evolution of Mtb from its evolutionarily close and mildly virulent species including M. kansasii and M. marinum has been reported (14–16). The actual size of the genome in common ancestors of mycobacteria is ambiguous; hence, it remains an open question whether a large number of open reading frames (ORFs) were lost during the reductive evolution.
Macrophages are the frontline cells of innate defense and are present in every major tissue. Macrophages play crucial roles in maintaining tissue integrity, homeostasis, and wound repair and regulating inflammatory processes (17, 18). Mtb also makes efficient use of macrophage heterogeneity and plasticity for productive infection and dissemination. Following infection through the aerosol route, mostly alveolar macrophages (AMs) in the lungs harbor Mtb. To survive immune or drug pressure, Mtb can acquire and maintain a “metabolically slowed” latent infection phase within the macrophages, which are the primary innate immune responders for eliminating the intracellular pathogens (19–24). However, it is largely unclear which macrophage subtype(s) Mtb prefers for its latent residency program. Mtb, thriving within the macrophages, has evolved a number of mechanisms to evade or counter the host immune response (25). As a result, macrophages serve as a suitable niche for the survival of Mtb, making it one of the most successful pathogens (26).
Macrophages are characterized based on their functional and spatial heterogeneity. For example, Kupffer cells that populate the liver and the glial cells or microglia present in the brain are both subtypes of macrophages. Ontologically, the resident macrophages that arise from the embryonic yolk sac remain within their designated tissue spaces for a lifetime or differentiate from the bone marrow (fetal liver at the prenatal stage)-derived myeloid mononuclear cells, giving rise to mature macrophages in virtually all the tissues (27, 28).
The primary organ for TB infection is the lungs which are predominantly populated by fetal liver monocytes, bone-marrow-derived resident AMs, and self-renewing macrophages that originate from the yolk sac (27–30). In response to inflammation, the recruited blood monocytes can differentiate into AMs in the tissue microenvironment and are termed as recruited AMs (31, 32). Interestingly, in a mouse model of mixed AMs, embryonic host-derived and donor-derived postnatal macrophages displayed minor (0.1% of all the genes) yet conserved differences in transcriptomic signature and exhibited overlapping functional attributes (29). Further studies have revealed significant differences in the metabolic, proliferative, and inflammatory states of the resident and recruited AMs. Macrophages derived from the circulatory monocytes are more proliferative and pro-inflammatory, are short-lived, and derive energy primarily from glycolysis (32).
The interstitial macrophages (IMs) are relatively stable and short-lived as compared with the AMs (33) and are localized in either the alveolar interstitial or peribronchial regions (31). Recent studies have described two distinct lineages of IMs consisting of 1) Lyve1lowMHC-IIhigh IMs with a role in antigen presentation and 2) Lyve1highMHC-IIlow perivascular IMs involved in wound healing and tissue repair (31). These subsets of IMs are conserved in mice and humans (33–35).
Depending on the activation status, macrophages were initially categorized into M1 type with pro-inflammatory attributes and M2 type with anti-inflammatory features (36, 37). However, the dichotomy of M1 and M2 types is now considered an oversimplification of the complex functional heterogeneity of the macrophages (38). Recent studies have shown diversity in macrophage populations which do not exhibit typical characteristics of either the M1 or M2 sublineages (39, 40). Therefore, a dynamic classification is needed to incorporate different subsets of macrophages, which may characterize beyond the dimorphic M1/M2 paradigm. The present classification of macrophages does not account for the microenvironmental conditioning and immunological stimulus which direct M1/M2 diversification. A clear-cut demarcation of the M1/M2 subset becomes obscure due to the coexistence of diverse stimuli in the inflamed tissues (41).
Hence, it was proposed to categorize macrophages based on the effector molecules they produce (Figure 1), for example, M(IFN-γ), M(IL-4), or M(IL-10) (42). M1-activated macrophage markers include inducible nitric oxide synthase (iNOS)/eNOS, IFN-γ, STAT-4, T-bet, SOCS3, CCR7, and CCL19/21. M1-activated macrophages have the absence (or low expression) of arginase-1/2, CD206, CD163, MerTK, STAT-3/6, Ym1/2, Fizz1, and MRC1 markers, which are predominantly expressed in M2-biased macrophages (43–48). M1 macrophages were also subcategorized as classical and innate activated macrophages, M1a and M1b, respectively (49).
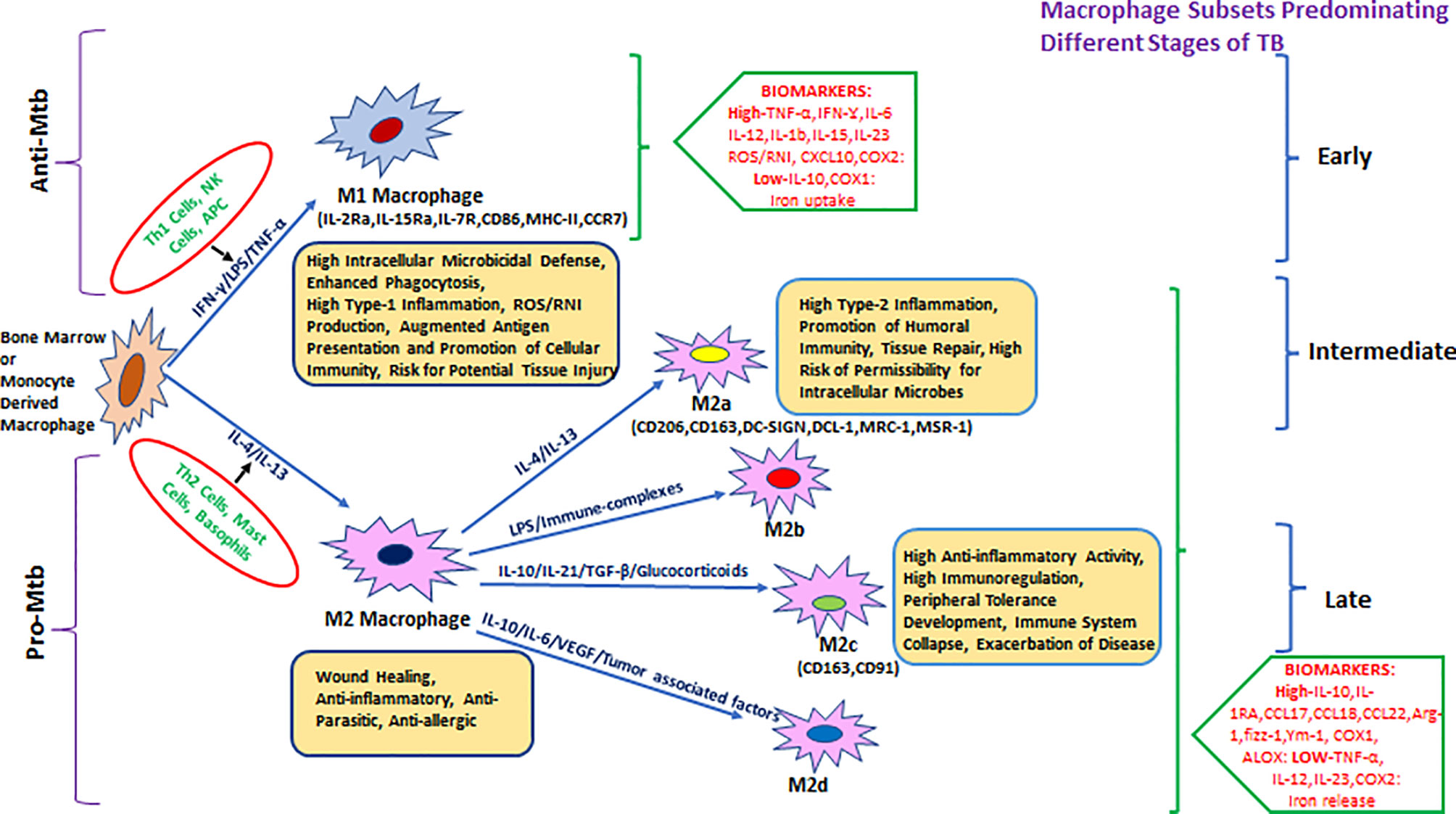
Figure 1 Heterogeneity of bone-marrow- or monocyte-derived macrophages and their physiological roles with reference to TB. The fate and function of recruited macrophages are generally shaped with influence from the local environment, stimulatory signals, and type of infection. Effector cells, including Th1/NK cells and APCs, displaying antigenic peptides from intracellular pathogens (or due to stimulation with LPS/IFN-γ), give rise to the M1 type of pro-inflammatory macrophages that either clear or restrain (through granulomatous response) intracellular infections including Mycobacterium tuberculosis (Mtb). M1 macrophages are generally characterized by a high level of pro-inflammatory mediators such as TNF-α, IFN-γ, IL-6, IL-12, IL-1β, IL-15, IL-23, CXCL-10, COX-2, and ROS/RNI and a low level of immune-regulatory molecules including IL-10, IL-4, TGF-β, and COX-1. In parallel, during invasion by an extracellular pathogen, Th2 cells/mast cells/basophils (or stimulation with IL-4, IL-10, IL-13, and immune complexes) act to differentiate macrophages toward the M2 state that generally ensure clearance of extracellular parasites. M2 macrophages are generally characterized by the production of a high level of IL-10, IL-1Ra, CCL17, CCL18, CCL22, Arg-1, fizz-1, Ym-1, COX-1, ALOX, etc. and a low level of TNF-α, IL-12, IL-23, and COX2 among others and further divided into various subtypes such as M2a, M2b, M2c, and the most recently described M2d. They are known for their role in promoting intracellular infections, for example, AMs in Mtb infection.
M2-activated macrophages showed greater diversity as compared with the M1 macrophages. Several subtypes of M2 macrophages such as the M2a/b/c and M2d are categorized based on different activation states and associated cytokine/chemokine signaling. M2a macrophages express CD206 (mannose receptor), differentiate in response to IL-4 and IL-13 (mainly produced by Th2 cells, mast cells, and basophils), and can downregulate pro-inflammatory responses (36, 47).
M2b macrophages, activated by immune complexes and TLR agonists, produce both pro- and anti-inflammatory cytokines (49). M2b differentiation is induced by IL-1R ligand or exposure to LPS. This phenotype is marked by a low expression of IL-12 and a high expression of IL-10 which favors Th2 type immune response (50).
M2c macrophages express anti-inflammatory cytokines such as IL-10, IL-21, TGF-β, or glucocorticoids. M2c macrophages are upregulated during the scavenging activity of the cellular debris and are associated with tissue remodeling (36, 47). Mtb-permissive M2c macrophages that get differentiated via activation of IL-10/STAT-3 signaling display angiogenic characteristics and are implicated in TB pathogenesis (43, 51).
M2d macrophage phenotypes were characterized from Fra-1-mediated differentiation of RAW264.7 macrophages upon induction with tumor cells. M2d macrophages have a characteristic low expression of IL-12 and high expression of IL-6 and IL-10 and exhibit immunosuppressive features of tumor-associated macrophages (TAMs) (52). M2d macrophages are also derived by co-stimulation with TLR and adenosine receptor agonists and are characterized by high levels of IL-10, VEGF, and iNOS expression, which are independent of IL-4Rα expression (53, 54).
Despite all these findings, characterization of the exact surface markers for distinguishing monocyte/macrophage subsets is challenging due to overlapping markers and due to the existence of hybrid subpopulations co-expressing both M1/M2 markers (55, 56). Therefore, renewed attempts are required to precisely describe cell surface markers which can define and delineate different macrophage subsets.
In Mtb infection, the enormous macrophage heterogeneity makes it difficult to delineate the macrophage subtypes that are protective or pathogenic in nature. In Mtb-infected lungs, distinct macrophage subtypes such as the AMs, monocyte-derived macrophages (MDMs), and IMs have been identified (30). In the initial stage of infection, AMs act as a niche for Mtb (57); later on, AMs move to the pulmonary interstitium to disseminate infection to other cell types including the recruited macrophages (58). The macrophages in murine lungs display additional heterogeneity with the presence of three distinct subsets of IMs: (IM)-1, 2, and 3 (34). Although IMs have a protective role in TB (59, 60), it is possible that some IM subsets can be potentially pathogenic which needs to be investigated. Additionally, a separate subset of lipid-rich foamy macrophages (FMs) has also been characterized in the Mtb-infected lungs (discussed later) (23, 61). Also, a recent study has identified distinct subsets of macrophages in the lungs of tumor-bearing mice, which do not fall under the purview of the common M1/M2 paradigm (40). The exact roles of these novel subsets of macrophage in Mtb infection are yet to be established and may be of significant interest in future studies.
Sequel of Infection After Inhalation of Aerosolized Mtb
The AMs are the first responder cells that encounter the aerosolized Mtb. AMs are poor at processing and presenting antigen to the T cells and produce minimal amounts of anti-mycobacterial effector molecules including the reactive oxygen and nitrogen species (62). Depletion of AMs in mice resulted in reduced Mtb burden and enhanced survival of animals (63). Upon encounter with Mtb, AMs engulf the bacilli in a phagosome which may fuse with the acidic lysosome. This process, called phagolysosome maturation, is subverted by Mtb to escape lysosomal sequestration and killing (64–66). Recent reports suggest that Mtb has evolved to not only survive but also replicate within phagosomes (67). Mtb perforates the phagosomal membrane and leaks out to the cytosol of the macrophage (68–70) where it can replicate or cause necrosis of the infected cells to disseminate, thereby infecting bystander cells (71, 72). Analogous to the lytic and lysogenic phases of the virus life cycle, Mtb probably represents a two-stage intracellular growth model. In the first stage of intracellular growth, Mtb resides in the phagosome where it replicates. In the subsequent stage, it reaches the cytosol where it can replicate and disseminate. However, the mechanistic details on how Mtb manages to replicate in the cytosol are largely unclear and need to be explored.
Mtb disrupts membrane-compartment integrity through the ESX-1 (73–75) and phthiocerol dimycocerosates (PDIM) (76) dependent mechanisms, both of which are generally absent in avirulent mycobacteria including the vaccine strain M. bovis BCG (77). ESX-1 is a component of the Mtb-specific type VII secretion system (T7SS) consisting of subclusters, ESX-1–5 (75, 78). The main effector of the Mtb ESX-1 system, ESX-A (ESAT-6), along with its substrate ESX-B (CFP-10) can cause membrane perforation (72, 79–81). The other components of the ESX system, ESX-3, ESX-H, and ESX-G, can also block phagolysosome maturation by inhibiting the ESCRT (endosomal sorting complex required for transport) assembly (82, 83). Mtb can also modulate apoptotic pathways (84–86) and autophagy (64, 86–89). The ESX-1 system effectors, including ESAT-6 and espB, suppress autophagy to favor mycobacterial survival inside the host cells (90, 91). ESAT-6 is known to induce apoptosis in Mtb-infected macrophages by inducing ROS production (92). The role of ESAT-6 is equally established in causing membranolytic activities and necrotic death of the infected host cells (72, 79–81). AcpM (Rv2244), an acyl career protein of Mtb, inhibits the ROS/JNK signaling pathway to arrest macrophage apoptosis, which can have potential implications in virulence and pathogenesis of mycobacteria (93). Moreover, Mtb-infected macrophages attain the M2 phenotype that produces IL-10 and lowers the ER stress to block apoptosis, thereby favoring intramacrophage bacillary survival (94).
Thus, Mtb has evolved multiple strategies to breach the innate immune defenses and can modulate macrophages into a permissive niche for its quiescent growth. The inhibition of phagolysosome maturation, dissemination via translocation to the cytosol, and modulation of programmed cell death mechanisms are all part of its defense strategies.
Macrophage Heterogeneity in TB
Samuel Behar and coworkers had demonstrated the role of innate immune cells including macrophages for the clearance of Mtb in an aerogenic infection model (95). The study showed a positive correlation between the augmented of protection against TB and early lymphatic dissemination of Mtb in resistant B6 mice, as compared with the susceptible strains. The study also showed that clearance of Mtb was associated with its rapid systemic spread, which causes potent immune priming and anti-mycobacterial response. Interestingly, T and B lymphocytes had no role in protection, which emphasized the crucial role of macrophages and other innate immune cells as primary defenders against TB infections (95).
Early clearance of Mtb has been associated with heightened innate immune responses and trained immunity (96, 97), notwithstanding the host genetic variability (98). Trained immunity, caused by epigenetic and metabolic reprogramming of innate immunity, confers cross-protection to the host against various pathogens (99). Trained immunity is non-specific and maintains a short-term memory that is independent of a somatic gene rearrangement scheme of adaptive immune cells (100). Monocytes/macrophages and NK cells are the major cells involved in trained immunity-mediated defense in TB and other infections (96, 97, 101–104).
In a study on human PBMC-derived macrophages, distinct DNA methylation patterns were observed in BCG-vaccinated responders compared with non-responders. Promoter sequences of genes responsible for immune responses showed loss of methylation, which corroborated with increased ex-vivo anti-mycobacterial activity (105). In mice, intravenous BCG administration led to epigenetic and metabolic reprogramming of hematopoietic stem cells, resulting in enhanced myelopoiesis at the expense of lymphopoiesis (106). Preferential myelopoiesis in BCG-immunized mice gave rise to monocytes and macrophages with “trained immunity” features that were associated with protection against Mtb infection in vitro and in vivo (106). In another report, priming of human monocytes (and mice) with fungal cell wall PAMP (β-glucan) resulted in enhanced protection against unrelated TB infection via IL-1 signaling-dependent trained immunity (107).
In mice, monocytes and macrophages display at least two distinct phenotypes based on the level of expression of Ly6c, a surface marker present on the cells of myeloid origin (108–110). Ly6chigh monocytes are recruited to the site of inflammation, while the Ly6clow subset patrols the blood vessels for vascular integrity (110, 111). Monocytes that infiltrate the site of inflammation or injured tissues can differentiate into cells that are either pro- or anti-inflammatory, depending on the microenvironmental stimulus. Ly6chigh pro-inflammatory monocytes convert into anti-inflammatory M2 macrophages and affected the significant regression of the atherosclerotic plaque (112) or inflammation-induced damage of liver tissue during chronic infection caused by Schistosoma mansoni (113). Inflammatory Ly6chigh monocytes/macrophages have a protective role against Brucella abortus infection (114), in contrast to their detrimental role in controlling visceral leishmaniasis caused by Leishmania donovani (115). Although Ly6c+(high/low) monocytes and monocyte-derived macrophages were significantly mobilized in TB-infected mice (111) and contributed to rBCG30 vaccine-induced protection (116), their exact role in the protection or pathogenesis of TB is yet to be fully elucidated. Infection with Mtb can modulate the macrophage from a pro-inflammatory to an anti-inflammatory cell type (25, 117), and the recruited monocyte-derived permissive M2 macrophages may also contribute to this pool; however, it is yet to be established. Two excellent reviews have been published on the functional and phenotypical heterogeneity of the cells of the mononuclear phagocyte system in the context of TB, which can be perused for a greater understanding of the topic (68, 118).
Interactions of Lung Alveolar Macrophages With Mtb
The upper regions of the lungs are constantly exposed to particulate stimulants like dust, pollen, and organic and inorganic particles as well as microbes (29, 119, 120). Macrophages can sense the pathogen/damage-associated molecular patterns (PAMPs/DAMPs) using the cell surface pathogen recognition receptors (PRRs) and initiate appropriate immune responses for clearance or containment of an underlying stimulant. Recurrent exposure causes persistent innate immune activation in the lungs. A subset of lung macrophages plays a regulatory/suppressive role in limiting the collateral immunopathological consequences (62, 119). AMs, given their predominant immunoregulatory role, are involved in taming excessive inflammation during Mtb infection (30, 62, 121). Despite their host-protective roles, AMs serve as a niche for Mtb and help subdue immune surveillance for Mtb clearance (58–60, 122). Mtb can subvert continuous innate cell resistance by masking its crucial PAMPs beneath the stealth coat of specialized PDIM lipids on its surface (120). It was suggested that PDIMs protect the Mtb from recognition and killing by highly phagocytic iNOS+ M1 macrophages and facilitate its smooth passage to the distal regions of the lungs (120). Mtb traverses through the pathogen-eliminating environment in the upper respiratory tract and preferentially resides in the distal ends of the lungs, and the mechanism by which Mtb reaches its preferred niche is largely unclear.
A two-pronged explanation of this phenomenon has been proposed based on the zebrafish model of TB. First, Mtb evades scrutiny by the mycobactericidal iNOS+ macrophages in the lungs by using cell surface PDIM lipids in a TLR2/MyD88-dependent manner (120). Secondly, virulent mycobacteria (such as M. marinum) exploit a unique lipid effector, phenolic glycolipid (PGL), to secrete CCL2, a chemokine ligand for CCR2, which recruits permissive macrophages to the infection site in a STING–CCL2–CCR2-dependent manner. This same mechanism may also facilitate the transfer of Mtb from the lung-resident AMs to the recruited permissive monocytes/macrophages for survival and dissemination (123).
Interestingly, a transcriptional repressor coded by Mtb Rv3167c negatively regulates PDIM expression, and a loss-of-function mutant (Mtb ΔRv3167c) was demonstrated to have an enhanced ability to escape the phagosome to the cytosol with augmented autophagic and necrotic cell death (124). The observed effects were attributed to the enhanced PDIM levels and were reversed in the double deletion mutant (Mtb ΔRv3167c Δmmpl7) with impaired PDIM production, confirming the central role of PDIM behind the enhanced virulence (124). More recently, PDIMs have been implicated in Mtb infection of the epithelial/endothelial cells (125, 126). Apart from other effectors that partner with PDIM in the Mtb virulence program, the ESX-1 operon effectors are also determined to be essential for PDIM conferred virulence to Mtb (127). BCG can produce PDIM but cannot escape phagosome due to a lack of RD components including ESX-1. Exploiting this fact, it was shown that transforming BCG with ESX-1 enables it to escape the phagosome, confirming that both PDIMs and ESX-1 are required for mycobacteria to escape the phagosomes (76). Concomitantly, multiple mutant strains impaired in producing PDIMs (ΔppsD, Δmas, ΔdrrC, Δhrp1, and Δrv0712) were inefficient in secreting ESX-1 effectors, highlighting the co-dependability of PDIMs and ESX-1 system proteins for mycobacterial virulence (128).
In addition to the recruited permissive macrophages, the lungs (murine) are home to the highly heterogeneous macrophage population (34, 40). The latest insight into the heterogeneity of lung macrophages in TB has been provided in a study by Cohen et al. (58) The study revealed an unexpected role of AMs and demonstrated that AMs translocate Mtb away from the alveolar space to the interstitium before the arrival of recruited myeloid cells in mice lungs. This is orchestrated jointly by Mtb ESX-1 components and host MyD88/IL-1R/ASC-mediated inflammasome signaling. The unexpected role of Mtb-infected AMs in traversing the epithelial boundary to deliver bacilli to the recruited monocytes/macrophages in interstitial space has renewed the interest in redefining the macrophage realm in TB-infected lungs. Plausibly, the newly identified monocyte-derived recruited AMs (31, 32) may overlap with the population identified by Cohen et al. (58) which are responsible for Mtb dissemination.
The Role of Recruited Interstitial Macrophages in TB
Recently, Russell and coworkers defined the dynamics, phenotype, and role of different macrophage subsets in the lungs of Mtb-infected mice using fluorescent Mtb reporter strains and macrophage transcriptomics data (59, 60). They demonstrated that Mtb predominantly inhabits the lung’s resident AMs for their unchecked growth, while monocyte-derived macrophage subsets (IMs) restrict Mtb survival in the lungs. It is apparent that Mtb faces less stress in AMs than in the IMs, making AMs a permissive niche for bacilli. These two macrophage subsets have shown distinct inflammatory states as a result of adopting different metabolic programs. The immunometabolic circuit of TB-loaded macrophages is described in Figure 2. It was demonstrated that AMs predominantly sustained fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS), while pro-inflammatory IMs were committed to glycolysis for their sustenance. It has been shown that recruitment of macrophages is CCL2 dependent, and in CCL2-deficient mice, the migration and transformation of circulatory monocytes into the lung’s IMs is abolished with concomitant loss of Mtb infection control (59). The results were divergent from the earlier reports in the zebrafish infection model of M. marinum, where the Mtb lipid PGL exploits the host CCL2–CCR2 axis to recruit permissive macrophages which serve as a niche for Mtb growth (120, 123). These divergent findings need to be interpreted with caution as both studies were done in different model systems with different species of virulent mycobacteria (59, 120). This example also insinuates caution while translating findings from one model system to another and in humans ultimately. Additionally, it should also be noted that majority of the studies were carried out on cell lines in vitro which are not the exact representation of the complex macrophage landscape in tissues (129).
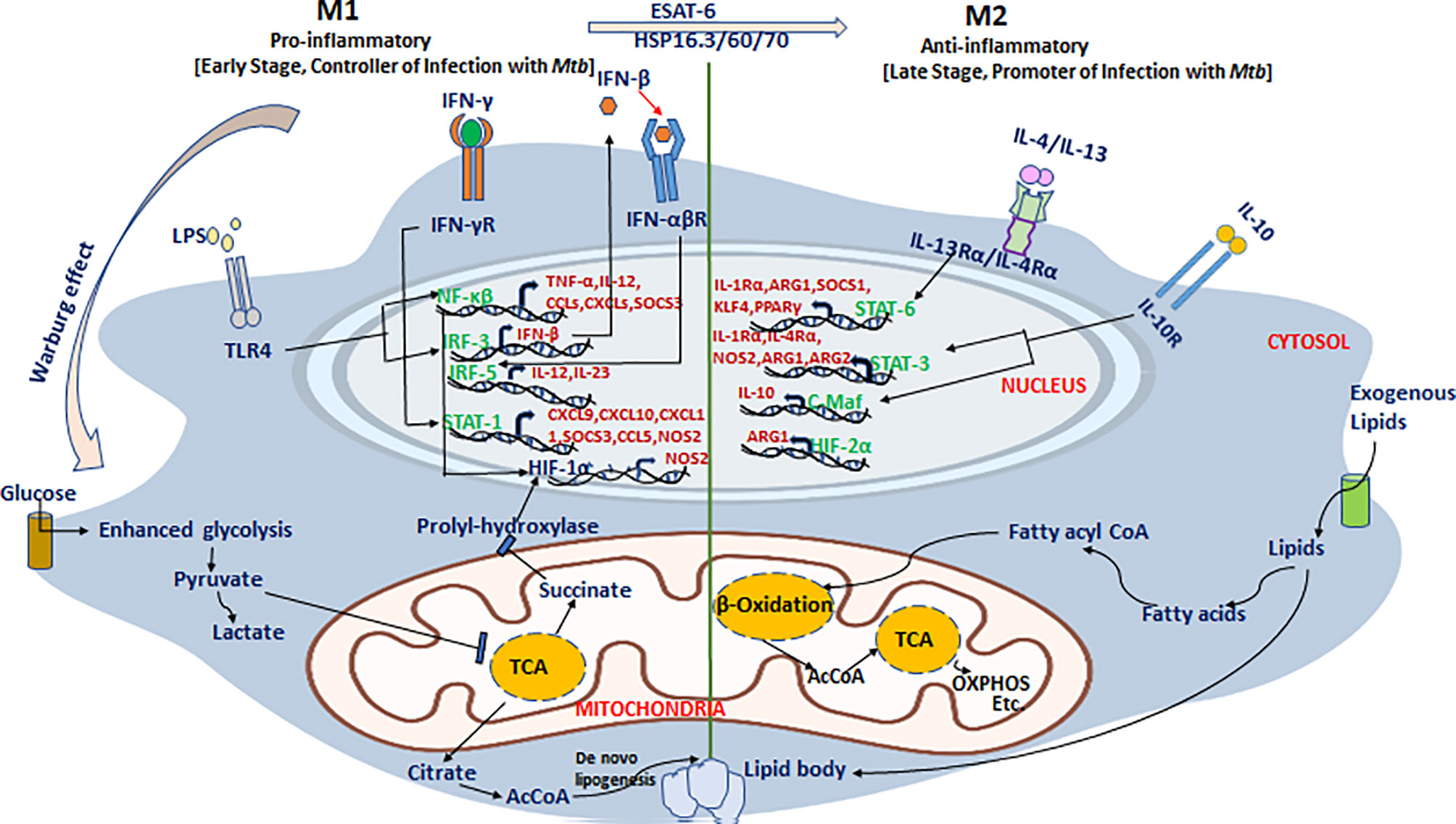
Figure 2 An immunometabolic circuit that dictates macrophage fate and function in tuberculosis. M1 macrophages that restrict Mtb proliferation are generally glycolytically active and utilize more glucose to meet increased energy demand as a result of enhanced proliferation. M1 macrophages skip the tricarboxylic acid (TCA) cycle and prefer energetically favored lactate production from pyruvate even in the absence of oxygen (Warburg effect) to which support rapid cellular turnover and generation of anti-mycobacterial oxidative burst. Instead of the TCA cycle, they utilize the pentose phosphate pathway and citric acid cycle for extra-mitochondrial utilization of available fatty acids to meet increased energy demands to support activation of pro-inflammatory genes including NF-κβ, IRF-3/5, STAT-1, and HIF-1α and their downstream effectors such as TNF-α, IL-12, IFN-β/γ, CCL-5, and CXCL-4/9/10/11 to restrict Mtb growth. In stark contrast, both AMs and M2 macrophages that support Mtb persistence/proliferation elect cost-inefficient mitochondrial oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) pathways to meet cellular demands. Mtb exploit these pathways to hijack host cells and utilizes the host’s own lipids to thrive in hibernation for longer periods. In M2 macrophages, it induces the cellular expression of anti-inflammatory mediators including STAT-3/6, C-Maf, and HIF-2α that stimulate the production of downstream anti-inflammatory effectors including IL-10, IL-1Rα, IL-4Rα, Arg-1/2, and PPAR-γ among others and thus make a permissive environment for Mtb growth as well persistence.
Nonetheless, the exact deciding factor(s) for the fate of mycobacteria-loaded macrophage is unknown. It could be dependent on the ontogeny of macrophages (embryonic or bone-marrow-derived) or the tissue microenvironment. The bone-marrow-egressed monocyte-derived macrophages are important innate responder cells during Mtb infection. The monocyte mobilization to Mtb-infected lungs is the result of emergency monopoiesis in bone marrow, rather than recruitment from blood (111). The classical Ly6chigh monocytes can replenish the local macrophage reservoir in Mtb-infected lungs, differentiate into protective IMs, and support local innate resistance against Mtb (59). Recruited inflammatory monocytes (Ly6chigh) enter the lung parenchyma and give rise to multiple subsets of macrophages and DCs. Among them, CD11chighCD11b+Ly6chigh DCs are involved in the transport of Mtb to the lungs draining mediastinal lymph nodes (111). Interestingly, Mtb hampers antigen processing and presentation by delaying the migration of local DCs to the draining lymph nodes during early infection. It uses this strategy as a means to delay the arrival of adaptive immune cells in infected lungs by ~10–12 days in mice (130) and up to 6 weeks in humans (7) to establish a chronic infection. Plausibly, the recruited monocyte-derived DCs in Mtb-infected lungs may represent a host strategy to pose a counter to Mtb-mediated suppression of adaptive immune priming of naive DCs and T cells in the local lymph nodes.
Mtb Promotes Biased M2 Shift in Macrophages to Dampen Host Immune Defense
Usually, bacterial pathogens induce a pro-inflammatory immune milieu that modulates macrophage polarization toward the M1 state, which can clear acute infections (47). Intracellular bacterial pathogens including Mtb (61), Listeria monocytogenes (131), and Brucella abortus (114) modulate macrophages toward an anti-inflammatory M2 state. This M1/M2 bias synchronizes intracellular bacterial fitness to favor either persistence or proliferation. Macrophages may kill Mtb or serve as a reservoir for intracellular Mtb persistence and proliferation and may be involved in immune regulation as well (87). M1–M2 switch in macrophages skews the transition from acute to chronic infection. Mtb has evolved strategies to escape M1-activated macrophage killings and drive phenotypic switch to the M2 polarization state to promote chronic infection (43, 117).
In an acute Mtb infection, the macrophage switches to the M1-polarized state and activates downstream PRR signaling to produce multiple host-protective effectors including reactive oxygen (ROS) and nitrogen (NO) species (47) and host defensins such as cathelicidin-related anti-mycobacterial peptide (Cramp) (132), all of which help kill Mtb. M1 macrophages initiate granuloma formation as a means to contain the infection. Overt M1 activation induces an exaggerated pro-inflammatory response that hampers tissue homeostasis and granuloma intactness. As a result, M1 macrophages skew toward M2 polarization to regulate inflammation and to promote tissue repair during Mtb infection progression in the chronic phase (121, 133). This plastic behavior during macrophage polarization depends on the distinct immune microenvironment, a result of extensive metabolic reshuffling. Macrophages undergo a metabolic shift from aerobic glycolysis to mitochondrial OXPHOS and glutamine metabolism (134) cater to its energy requirements during Mtb infection. In the lung lesions of TB patients, M1 macrophages are mostly present in non-granulomatous sites, whereas M2 macrophages predominate necrotic as well as non-necrotic granulomatous zones, which point to the principal role of M2 macrophages in granulomatous reactions as well as possible M1–M2 transition during infection (135).
ESAT-6 plays a significant role to skew M2 polarization for Mtb survival during an infection (94). Serine proteases (thrombin and trypsin) (136) and heat shock proteins Hsp70 (DnaK), Hsp60, and Hsp16.3 of Mtb are also involved in M2 polarization to shield the bacteria from the host immune pressure (137–139). ER stress-mediated apoptosis is typical in M1 macrophages to control infection (94). Apoptosis induces a drastic T-cell response which is less favorable for Mtb growth and dissemination (140). In contrast, cell death via necrosis, together with M2 shift in early infected macrophages, provides a permissive environment for Mtb proliferation (47). Thus, macrophage polarization is a crucial mechanism of virulence and pathogenesis of Mtb with contribution of multiple host and pathogen effectors, which needs to be explored in greater detail.
Differential Metabolism in M1/M2-Polarized Macrophages
Macrophage metabolic plasticity plays a significant role in disease pathology (141). Macrophage polarization defines the distinct metabolic profile of macrophages that drives differential macrophage activation (142). M1 cells actively use aerobic glycolysis (known as the Warburg effect in cancer cells) for bioenergetics and biosynthetic intermediates and also induce pentose phosphate pathway for ROS production. In contrast to M1, M2 cells prefer mitochondrial OXPHOS and glutamine metabolism as carbon and nitrogen sources, similar to the resting macrophages (36, 134, 143, 144). Arginine metabolism is involved in the differential regulation of macrophage polarization. L-arginine is a common substrate for iNOS as well as arginase-1. M1 macrophages produce iNOS that catabolizes arginine into L-citrulline and NO, which mediate cytotoxicity to control bacterial infection. Arginase-1 produces polyamine, L-ornithine, and urea linked to the wound-healing activity of M2 macrophages (145, 146). During TB, both iNOS (M1) and arginase (M2) compete for arginine, and this competition eventually shapes the dominant macrophage phenotype and functionality (M1 or M2), which is crucial for mycobacterial control (134). Lipid metabolism is also found to be differentially regulated in macrophage polarization. The COX-2 gene is upregulated in M1 macrophages, whereas COX-1 is upregulated in M2 macrophages (147). Membrane phospholipid-derived downstream metabolites of arachidonic acid (AA), prostaglandins, leukotrienes, and lipoxins play a diverse and essential role in shaping the immune response in TB, primarily by modulating cell death type (148–152). The delicate balance between the production and bioavailability of lipoxin A-4 (LXA-4) and prostaglandin E-2 (PGE-2) largely dictates the programmed death in Mtb-infected macrophage (148, 149). Mtb causes macrophage necrosis by inducing LXA-4 production and inhibiting PGE-2 (148). PGE-2 production is critical for apoptotic death of macrophage (148) and regulates anti-TB immune response primarily through its engagement with E prostanoid-2 (EP-2) receptor (153). While EP-2 promotes type 2 immune response (154), mice deficient in EP-2 develop pathogenic Th17 and Treg responses associated with poor clearance of Mtb.
Iron utilization is a classic hallmark of Mtb infection with the predominant role of PE/PPE proteins (155–161). M1 macrophages can sequester iron by high ferritin and low ferroportin ratio and also via heme uptake to maintain the bacteriostatic effect. This scenario is reversed in M2 macrophages, which reduces iron storage and releases iron to favor tissue repair and cell remodeling (162, 163). Heme is a cellular reserve of iron and Mtb is known to utilize heme for intracellular subsistence (159, 161, 164, 165). Heme oxygenase-1 (HO-1) has been reported to be upregulated in Mtb-infected mice, rabbits, and macaques (166). The induction of HO-1 is dependent on NADPH oxidase-dependent ROS production and nuclear translocation of the transcription factor NRF-2, which is mediated by ESAT-6 (166). Inhibition of HO-1 controls Mtb infection in macrophages in vitro and in mice via upregulation of NOS-2/IFN-γ and enhanced expansion of T lymphocytes (167, 168), which might be the result of macrophage reprogramming toward the pro-inflammatory M1 state. Furthermore, the essential Mtb gene ripA rewires the macrophage metabolism toward glycolysis by inhibiting mitochondrial oxidative phosphorylation (169). The glycolysis induced in RipA-treated RAW264.7 macrophages indicates RipA-mediated modulation of the macrophage phenotype which, however, was associated with poor intramacrophage growth control of recombinant Mycobacterium smegmatis expressing Mtb RipA. This observation, while underlining an important role of Mtb RipA, also motivates fresh investigations into the macrophage modulatory roles of unexplored Mtb effectors beyond common ESX system proteins.
Macrophage M1/M2 Paradigm: Conflicting Evidence From Human TB
Much that has been said and known about the heterogeneity of macrophages in TB is learned from animal models, primarily mice. Very few studies are available or being done that aim to characterize monocyte/macrophage dynamics in human lungs, a primary site of Mtb infection. This might be, in part, due to the ethical and anatomical challenges associated with collecting tissue/BAL samples from human lungs. While the M1/M2 dichotomy is largely established in the lungs and spleen of mice, the macrophage identity is not as demarcated in humans, and considerably divergent phenotypical heterogeneity has been reported in macrophages from various anatomical origins (55, 56, 122).
Emerging reports have indicated the presence of mixed macrophage phenotype (co-expressing both M1/M2 markers) in a range of conditions (55, 56, 170). AMs from human lungs, obtained from two geographically distinct populations of the UK and Malawi, were immunophenotyped. These AMs displayed mixed expression of M1 (CD80/86) and M2 (CD163/206) markers, which challenges the established dichotomy of the macrophage phenotypical identity (56). Another study has reported circulatory macrophage (CD204+CD163+CD206+TLR4+CD80+CD86+) and monocyte (CD14+CD206+CD163+CD204+TLR4+CD80+CD86+) populations that expressed both M1/M2 markers in systemic sclerosis patients and in interstitial lung disease (ILD) (55). In the context of TB, Lavelette and colleagues used microarray and qRT-PCR-based sequential approach to demonstrate differential immune response trajectories in BAL macrophages, obtained from human TB patients infected with clinical Mtb strains (170). Interestingly, differential response to two clinical Mtb strains belonging to LAM (Latin American and Mediterranean) lineage in pulmonary (the lung’s AMs) and extrapulmonary (splenic macrophages or SMs) sites was observed, with AMs showing a predominantly pro-inflammatory phenotype and SMs displaying a largely attenuated response. Moreover, when compared with uninfected macrophages from healthy controls, the TB-infected AMs displayed an attenuated transcriptomic response and regulation of critical gene sets related to anti-TB responses including ISGs, IFITs, and GBPs (IFN pathway), AIM2 (inflammasome) FCGR1A (fc receptor pathway), and TREM (myeloid receptor pathway), which suggest modulation of macrophage-specific immune responses. In addition to the tissue-specific macrophage responses, the two clinical isolates (UT127 and UT205) induced contrasting macrophage transcriptomic responses in human macrophages (22 and 5 genes induced, respectively). Differential responses were attributed to the altered virulence profile of the two isolates, despite belonging to the same lineage (LAM) (171), which suggest altered host immune trajectory as a plausible function of intrahost microevolution of the Mtb bacilli. However, the drug resistance profile of these isolates (which is unknown) might have provided additional correlates to the observed differential responses. Nonetheless, of the two isolates, one induced apoptotic cell death (UT127), while the other triggered necrosis (UT205), which itself attests to their altered virulence profile. Interestingly, the one that induced necrosis (UT205) displayed attenuated transcriptomic responses in macrophages as well, suggesting its higher virulence and immunosuppressive abilities.
In a latest attempt to characterize the spatial heterogeneity of macrophage identity and function at the primary site of TB infection, Pisu et al. recently defined at least four different subsets, each of the interstitial macrophages (IM 1–4) and alveolar macrophages (AM 1–4), in human bronchoalveolar lavage (BAL) and in mice lungs (122). The study, employing single-cell transcriptomic analysis of macrophages infected with fluorescence reporter strains for intramacrophage Mtb fitness, clearly defined substantial plasticity among lung macrophages. Both Mtb-permissive and Mtb-restrictive macrophage subsets were spotted among the lung’s resident AMs (AM2 being the most restrictive) and recruited IMs (IM2 being the most permissive). In addition, the study also identified CD11clow IMs/AMs in the lungs as predominantly permissive to Mtb growth which allows the development of drug tolerance as well (122). Of note, IMs and AMs were labeled restrictive and permissive, respectively, to Mtb growth in a previous study by the same group (60). Nevertheless, these lines of evidence provide important insights into the macrophage heterogeneity and plasticity in TB-infected lungs from mice and humans. These data clearly suggest that both the IM and AM sublineages of Mtb-infected macrophages contain permissive and controller subpopulations. It is therefore time to reconstruct the simplified dimorphic view of macrophage heterogeneity and to assess the enormous phenotypical and functional polymorphism present in Mtb-infected macrophage which may have far-ranging implications.
In addition to macrophage ontogeny, significant heterogeneity in Mtb-infected macrophages has been suggested to be due to the genetic differences among the infecting strains and lineage (171–174). Mtb isolates belonging to the hypervirulent modern strains have been associated with a lower inflammatory response in vitro by human or murine macrophages due to the predominance of anti-inflammatory M2-like monocytes/macrophages (175, 176).
Macrophage M2 Polarization and Drug Resistance in Mtb
Classically activated M1 polarization displays pro-inflammatory and antimicrobicidal activities (129). In contrast, alternatively activated M2 polarization plays a pivotal role in circumventing host immune defense during Mtb infection and may result in MDR/XDR-TB treatment failure to favor persistent infection (135). M1/M2 polarization of macrophages has a crucial role in the progression or regression of TB infection as a result of pro- or anti-inflammatory responses they exert, respectively. It was recently demonstrated that the M2 polarization rate and the M2 to M1 polarization ratio were significantly higher in the MDR/XDR-TB group as compared with the drug-susceptible TB group (177), reflecting the crucial role of macrophage polarization in drug resistance development. Thus, it may be an attractive host-directed avenue to modulate macrophage phenotype in drug-resistant (or even drug-sensitive) TB infection, with or without chemotherapy.
Macrophage Plasticity in Mtb Infection
The majority of macrophages are not terminally differentiated and are poised to reprogram either homeostatically or in response to infection and other stimuli such as cytokines, growth factors, hormones, small molecules, and metabolites including prostaglandins and leukotrienes. In TB-infected mice, recruited macrophages have a dynamic expansion and differentiation program, even during the chronic stage of infection (111). Moreover, substantial diversity and plasticity of macrophages have been observed in tuberculous granuloma (46, 178) which is spatially organized (133). The imbalance of this spatially organized granulomatous structure, required for containing Mtb, is reminiscent of progression to active TB and is largely dictated by macrophage polarization metrics in the granuloma microenvironment (179). Mtb transforms macrophages into multiple subtypes through a variety of mechanisms and exploits them for its survival.
Macrophage Reprogramming Into Epithelioid Cell, Multinucleated Giant Cell, and Foamy Cell
The macrophage at the core of granuloma undergoes a series of morphological changes including epithelioid cell differentiation to form “epithelioid” cells like macrophages. These epithelioid cells can further develop into multinucleated giant cells (MGCs), probably due to cell–cell fusion or cytokinesis failure (180–182). One of the well-characterized virulence schemes of Mtb is the modulation and differentiation of macrophages into lipid-rich foam cells (23, 51, 61, 183) that serve as a niche for Mtb persistence. In Mtb infection progression, macrophages convert into foam cells by importing and accumulating host lipids, mainly low-density lipoproteins (LDLs) and cholesterol (23, 51, 61, 183–185). In the following subsections, we will be discussing the origin and functionality of these macrophage subtypes which have been extensively studied and characterized for their role in TB progression.
Epithelioid Cells or Histiocytes
Mtb reprograms macrophages for granuloma formation via E-cadherin-dependent mesenchymal–epithelial transition. Epithelioid cells are characterized as hypertrophic, flattened in appearance, containing diffused cytoplasm and elongated nuclei with the interdigitating cell membrane, which enable the cell to stick and form an epithelioid barrier to persistently capture available antigen (186). These inflammation- or infection-induced histopathological transformations in macrophage microanatomy resemble an epithelial cell, thus acquiring the name epithelioid cells or histiocytes (187). Macrophage-derived epithelioid cells form the central scaffold of organized granulomas meant to be less accessible to immune cells and, thus, provide a favorable niche for mycobacterial persistence (186, 188–190). E-cadherin expression in motile mesenchymal cells induces transformation into the epithelial structure via the process of mesenchymal–epithelial transition (or vice versa) in tissue development (191) and cancer (192). Cronan et al. demonstrated that epithelial reprogramming is analogous to the mesenchymal–epithelial transition and is conserved within the tuberculous granuloma of mice and humans (186). They confirmed that the macrophage forms adherence junctions, desmosomes, and tight junctions as a prestructure for stable granuloma formation and speeds up infection trajectory. Host angiogenic signaling has also been implicated in macrophage epithelioid transition and formation of tuberculous granulomas with an important role of vascular endothelial growth factor-receptor (VEGF-R) (193). Therapeutic inhibition of granuloma vascularization and angiogenesis using VEGF-R antagonists not only reduces M. marinum infection in zebrafish but also synergizes with the anti-TB drugs rifampicin and metronidazole to improve bacillary clearance (193). Recently, signal transducer and activator of transcription (STAT)-6 signaling is found to be absolutely necessary for macrophage epithelialization and granuloma formation (194). By analyzing single-cell RNA-sequencing data from M. marinum and Mtb granulomas from zebrafish and macaques, respectively, the study concluded that the strong type-2 signaling mediated via the IL-4R/STAT-6 derives macrophage epithelialization and granuloma formation and is largely unaffected by the presence of robust type-1 immune signals.
Multinucleated Giant Cells
MGCs are polykaryons of monocytic origin, where multiple monocytes (or macrophages) fuse and differentiate into specialized MGCs (180, 195). Macrophage fusion occurs in the granulomatous region to form MGCs during mycobacterial infection. Mtb cell wall lipids, trehalose dimycolate (TDM), and lipomannan (LM) have been described to derive the formation of MGCs (183). In addition, macrophage exposure to cytokines IL-4 and IL-13 induces MGC formation (180). MGCs also possess a specialized ability to uptake large and opsonized complement particles mediated by CR3 signaling (196). However, in the context of mycobacterial infection, it was reported that MGCs contain very few bacilli and may be unable to phagocytose invading bacilli (190, 197). Classical and alternatively activated MGCs have been reported in acute and chronic TB infections, respectively (195). It is thus possible that MGCs typical of M2-type macrophages may enable rapid progression of chronic infection in TB. However, due to the paucity of credible evidence regarding the defined role and function of MGCs, they may continue to be considered a pathological hallmark of mycobacterial and other granulomatous infections.
Interestingly, the differentiation of MGCs has been shown to occur only in virulent Mtb complex organisms and not with other avirulent mycobacteria (197). MGCs are integral to granuloma formation and have a role in maintaining latency or conferring tolerance to Mtb. Recently, MGCs have been suggested to have originated from the common monocyte progenitor (CMoP) or inducible monocyte progenitor (iMoP) population in circulation (198). Their poor phagocytic activity and absence in disseminated TB during immunosuppression is intriguing and suggestive of their role as a niche for latent Mtb (198). MGCs contain plenty of cholesterol and other fatty acids, a preferred energy source for intracellular Mtb persistence (184). Moreover, TLR2-mediated Mtb modulation of macrophages induces excess NO production, leading to DNA damage and impaired p53 function and consequential establishment and differentiation of permissive MGCs (199). While these reports highlight that MGCs serve as a niche for mycobacterial survival, some earlier reports have demonstrated the protective role of MGCs in TB. Specifically, cytokines IFN-γ and IL-3, in combination with some other factors, have induced MGCs that limit mycobacterial infection in vitro (200, 201). In the face of these contrasting reports, more studies are required to assess the exact role of MGCs in mycobacterial pathogenesis, particularly in the context of humans. This gap in knowledge about the functional role of MGCs is an active area for future research.
Foamy Macrophages
FMs are lipid-enriched macrophages formed due to uptake of LDL or oxidized LDL via LDL receptor or scavenger receptor-A (SR-A) and CD36, respectively, in response to TLR2 activation by mycobacterial components, pro-inflammatory chemokines, and cytokines (23, 61, 183, 202). The lipid-rich environment of FMs allows Mtb transition into the latent phase with ample access to nutrients in the form of intracellular lipids (203).
FMs can also arise through phagocytosis of platelets by monocytes. Platelets, when co-cultured with monocytes in the presence of mycobacteria, induce the formation of multinucleated giant foam cells (185). Interestingly, despite their increased phagocytic activity and BCG uptake, FMs display predominantly the M2 phenotype and produce IL-10 abundantly (185).
LDL and lipid-loaded platelets break down into triacylglycerol, phospholipids, and cholesterol (183). In mycobacterial infection, cholesterol gets accumulated within the macrophages in the form of lipid droplets or effluxes via ATP-binding cassette (ABC) transporters (183, 204). The ABC transporters, ABCA1 and G1, are key mediators of cholesterol efflux and their absence exacerbates FM formation (183). Accumulated cholesterol also modulates inflammatory responses by producing leukotrienes and prostaglandins (183, 205, 206). Mtb also utilizes the host cholesterol using the mce4 locus (analogous to mammalian ABC transporters) for its survival and persistence during the chronic phase of infection, which supports its preference to induce differentiation of host lipid-rich macrophages (184).
In addition, the triacylglycerol synthase 1 (Tgs1) of Mtb helps in the accumulation of triacylglycerol (TAG)-derived fatty acids/triglycerides in macrophages (207). Mtb’s cell wall long-chain fatty acid and oxygenated mycolic acid induce human macrophage differentiation into FMs, which serve as a nutrient reservoir for the persistence of dormant Mtb for longer periods (22).
The Heterogeneity and Plasticity of Macrophages in Tuberculous Granuloma
Small aerosol droplets containing Mtb from infected carriers enter the lungs through inhalation where it is phagocytosed by AMs (26). Mtb remains unhindered within AMs and recruits permissive macrophages through coordinated usage of surface-expressed lipid PGL/PDIM (120). PDIM is a cell wall-derived component present exclusively in the pathogenic strains of mycobacteria (208, 209) and all clinical isolates of Mtb (26). The conical shape of PDIM augments membrane fusion to allow efficient Mtb uptake via endophagocytosis (210). PDIM is transferred from the Mtb cell wall to the macrophage’s lumen through the formation of PDIM aggregates in transient membrane stalks and enhances the non-bilayer phase followed by an endocytic uptake and phagosome formation (210). The phagosomal encasement consists of both the host and Mtb lipids, where Mtb survives for an extended period and disseminates to distant locations (210, 211). Mtb, once able to invade AMs in the upper airways, finally settles down to the extreme ends of the lungs (26, 120). Here, it replicates and triggers necrotic death of the infected cells, thereby infecting bystander cells (30, 71) and causing a cascade of immune reactions (212). These local inflammatory reactions and release of myriad chemokines/cytokines recruit neutrophils (30), MDMs (111), and DCs (213) that engulf and transport Mtb to the draining lymph nodes to activate T-cell-mediated adaptive immune response (130). By the time the adaptive immune cells get activated and reach the infected lungs, the cells of the innate immune system and local non-immune cells cordon the Mtb in a granulomatous structure, where adaptive T and B cells further add to make the outermost layer (190). Thus, granuloma forms during the critical time window when innate immunity fails to contain the growing Mtb burden, following initial exposure, and adaptive immunity cannot respond within time. Traditionally, granuloma has been seen as a host-protective measure to contain Mtb infection (133, 214); however, growing evidence also suggests its pro-mycobacterial role (186, 188–190, 194).
Macrophages, especially AMs, act as a perfect ecosystem for intracellular adaptation of Mtb and to achieve persistent infection. Mtb-encapsulated granulomas are a highly organized structure containing a pro-inflammatory core and surrounding periphery with a strong anti-inflammatory signature (133, 214), thus holding a pro- and anti-inflammatory balance to keep itself intact and non-disseminative. The formation of caseous necrotic granuloma is the hallmark of uncontrolled Mtb infection and TB disease progression (190, 215). The classic necrotic granuloma has a necrotic core containing extracellular bacteria surrounded by epithelioid histiocytes and macrophages and an outer cuff of macrophages intermixed with lymphocytes (214). As granuloma matures, a number of cell-like granulocytes, monocytes, DCs, B cells, T cells, NK cells, and fibroblasts migrate to the structure and surround the macrophage core (141, 216, 217). This may cause exaggerated and uncontrolled inflammation which induces pathophysiological changes at the final stage of granuloma formation and is associated with tissue damage and morbidity (141). In a C3HeB/FeJ mouse model of tuberculous granuloma, the central caseous necrotic regions are reminiscent of immune defense and restrain the TB bacilli. However, dysregulated inflammation-induced neutrophil and lymphocyte infiltration causes liquefaction of packed granulomas that led to mycobacterial dissemination and development of active TB (218, 219).
Cytokine signaling mediates the conversion of macrophages into heterogeneous phenotypes within the granuloma encasing TB bacilli. A computational model defines the metrics of macrophage polarization as a function of cytokine signaling (179). It examined the ratio of the temporal expression of STAT-1 and NF-κβ (pro-inflammatory) to STAT-3 (anti-inflammatory) in the macrophage and concluded that the expression level of NF-κβ can dictate macrophage or granuloma polarization and outcome in TB, whether protective or disseminative.
In an in-vitro differentiated model of tuberculous granuloma from human MDMs, M1 polarization of macrophages predominated the early stage of granuloma formation and decreased over time, while M2 polarization gradually increased during the late stage of granuloma formation (135). The study also utilized lung tissues from TB patients to show predominant M2 polarization of macrophages in necrotic and non-necrotic granulomatous lesions, whereas non-granulomatous sections were mixed, populated with both M1 and M2 macrophages (135). Thus, tuberculous granulomas are highly plastic with spatial and temporal heterogeneity which dictates the eventual outcome of Mtb infection.
Although the plasticity of macrophages has long been appreciated, this dogma is getting challenged based on recent epigenetics and single-cell transcriptomics studies that hypothesize that the plasticity of macrophages is lost due to an extended residency in a particular tissue type or restricted to favor tissue homeostasis (31) or reprogram epigenetically depending on the tissue microenvironment and local inflammatory and metabolic signals during steady state or inflammation (220, 221).
Concluding Remarks
The co-evolution of Mtb and its human host is in progress from prehistoric times (2). Despite numerous efforts, Mtb eradication is still a far-reaching target due to multiple factors. As macrophages are considered to be the primary niche for replication and persistence of Mtb, macrophage heterogeneity and plasticity allow them to be in multiple phenotypic and metabolic states which are skewed in favor of Mtb in the granuloma. However, recent reports emanating from epigenetics and single-cell transcriptomics studies have contradicted the established dogma about the plasticity of macrophages. Fresh evidence suggests that the plasticity of macrophages is lost due to an extended residency in a particular tissue type or is restricted to favor tissue homeostasis (31). Studies aimed at defining the actual dynamics of complex transitional states of macrophage populations based on tissue microenvironment and epigenetic landscape will be key to pinpoint the macrophage subsets as protective or pathogenic in TB. Nonetheless, strategic manipulation of macrophage activity and its phenotypic states can be effective to counter Mtb. Also, this may open new frontiers to clear off the persisting reservoir of Mtb, which is a source of continued supply of new infection cycles.
Author Contributions
FA, NE and SH conceived the idea of this review. FA, AR, AA, SZ, SP, HS, SH and NE have contributed to the writing of this manuscript. All the authors have reviewed and approved the final manuscript. SH and NE are co-corresponding authors.
Funding
SH and NE are supported by the North-East Grants BT/PR23099/NER/95/632/2017 and BT/PR23155/NER/95/634/2017 of the Department of Biotechnology, MoS&T, GoI. Intramural funds of the ICMR-National Institute of Pathology are gratefully acknowledged.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
FA acknowledges the fellowship support from the Department of Biotechnology. AR and SZ acknowledge the Council of Scientific and Industrial Research, Government of India, for fellowship support. SH is the National Science Chair of the Science & Engineering Research Board, Department of Science & Technology, Ministry of Science and Technology (MoS&T), Government of India (GoI). SH is a JC Bose National Fellow, Department of Science and Technology, Government of India, and is a Robert Koch Fellow, Robert Koch Institute, Berlin, Germany.
References
1. Brites D, Gagneux S. Co-Evolution of Mycobacterium Tuberculosis and Homo Sapiens. Immunol Rev (2015) 264:6–24. doi: 10.1111/imr.12264
2. Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-Of-Africa Migration and Neolithic Coexpansion of Mycobacterium Tuberculosis With Modern Humans. Nat Genet (2013) 45:1176–82. doi: 10.1038/ng.2744
3. Bos KI, Harkins KM, Herbig A, Coscolla M, Weber N, Comas I, et al. Pre-Columbian Mycobacterial Genomes Reveal Seals as a Source of New World Human Tuberculosis Provided Samples for Analysis HHS Public Access. Nature (2014) 514:494–7. doi: 19.1038/nature13591
4. Sabin S, Herbig A, Vågene ÅJ, Ahlström T, Bozovic G, Arcini C, et al. A Seventeenth-Century Mycobacterium Tuberculosis Genome Supports a Neolithic Emergence of the Mycobacterium Tuberculosis Complex. Genome Biol (2020) 21:201. doi: 10.1186/s13059-020-02112-1
5. Hershkovitz I, Donoghue HD, Minnikin DE, May H, Lee OY, Feldman M, et al. Tuberculosis Origin: The Neolithic Scenario. Tuberculosis (2015) 95:S122–6. doi: 10.1016/j.tube.2015.02.021
6. Houben RMGJ, Dodd PJ. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PloS Med (2016) 13:e1002152. doi: 10.1371/journal.pmed.1002152
7. Behr MA, Edelstein PH, Ramakrishnan L. Revisiting the Timetable of Tuberculosis. BMJ (2018) 362:k2738. doi: 10.1136/bmj.k2738
9. Chakaya JM, Marais B, du Cros P, Ntoumi F, Mfinanga S, Kapata N, et al. Programmatic Versus Personalised Approaches to Managing the Global Epidemic of Multidrug-Resistant Tuberculosis. Lancet Respir Med (2020) 8:334–5. doi: 10.1016/S2213-2600(20)30104-1
10. Singh J, Ehtesham NZ, Hasnain SE. Two Parallel Pandemics: The Challenges Faced by Countries With COVID-19 and TB. Int J Tuberc Lung Dis (2020) 24:1319–20. doi: 10.5588/ijtld.20.0592
12. Rahman SA, Singh Y, Kohli S, Ahmad J, Ehtesham NZ, Tyagi AK, et al. Comparative Analyses of Nonpathogenic, Opportunistic, and Totally Pathogenic Mycobacteria Reveal Genomic and Biochemical Variabilities and Highlight the Survival Attributes of Mycobacterium Tuberculosis. MBio (2014) 5:1–11. doi: 10.1128/mBio.02020-14
13. Ahmed N, Dobrindt U, Hacker J, Hasnain SE. Genomic Fluidity and Pathogenic Bacteria: Applications in Diagnostics, Epidemiology and Intervention. Nat Rev Microbiol (2008) 6:387–94. doi: 10.1038/nrmicro1889
14. Stinear TP, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, et al. Reductive Evolution and Niche Adaptation Inferred From the Genome of Mycobacterium Ulcerans, the Causative Agent of Buruli Ulcer. Genome Res (2007) 17:192–200. doi: 10.1101/gr.5942807
15. Sapriel G, Brosch R, Bapteste E. Shared Pathogenomic Patterns Characterize a New Phylotype, Revealing Transition Toward Host-Adaptation Long Before Speciation of Mycobacterium Tuberculosis. Genome Biol Evol (2019) 11:2420–38. doi: 10.1093/gbe/evz162
16. Wang J, McIntosh F, Radomski N, Dewar K, Simeone R, Enninga J, et al. Insights on the Emergence of Mycobacterium Tuberculosis From the Analysis of Mycobacterium Kansasii. Genome Biol Evol (2015) 7:856–70. doi: 10.1093/gbe/evv035
17. Wynn T, Chawla A, Pollard JW. Macrophage Biology in Development, Homeostasis and Disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
18. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage Heterogeneity in Tissues: Phenotypic Diversity and Functions. Immunol Rev (2014) 262:36–55. doi: 10.1111/imr.12223
19. Dahl JL, Krau CN, Boshoff HM, Doan B, Foley K, Avarbock D, et al. The Role of RelMtb-Mediated Adaptation to Stationary Phase in Long-Term Persistence of Mycobacterium Tuberculosis in Mice. Proc Natl Acad Sci U S A (2003) 100:10026–31. doi: 10.1073/pnas.1631248100
20. Hingley-Wilson SM, Sambandamurthy VK, Jacobs WR. Survival Perspectives From the World’s Most Successful Pathogen, Mycobacterium Tuberculosis. Nat Immunol vol (2003) 4:949–55. doi: 10.1038/ni981
21. Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic Carbon Flow of Tricarboxylic Acid Cycle Intermediates Is Critical for Mycobacterium Tuberculosis to Establish and Maintain Infection. Proc Natl Acad Sci U S A (2010) 107:9819–24. doi: 10.1073/pnas.1000715107
22. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy Macrophages From Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. Tuberculosis Persistence. PloS Pathog (2008) 4:1–14. doi: 10.1371/journal.ppat.1000204
23. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium Tuberculosis-Driven Targeted Recalibration of Macrophage Lipid Homeostasis Promotes the Foamy Phenotype. Cell Host Microbe (2012) 12:669–81. doi: 10.1016/j.chom.2012.09.012
24. McKinney JD, Höner zuBentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium Tuberculosis in Macrophages and Mice Requires the Glyoxylate Shunt Enzyme Isocitrate Lyase. Nature (2000) 406:735. doi: 10.1038/35021074
25. Queval CJ, Brosch R, Simeone R. The Macrophage: A Disputed Fortress in the Battle Against Mycobacterium Tuberculosis. Front Microbiol (2017) 8:1–11. doi: 10.3389/fmicb.2017.02284
26. Cambier CJ, Falkow S, Ramakrishnan L. Host Evasion and Exploitation Schemes of Mycobacterium Tuberculosis. Cell (2014) 159:1497–509. doi: 10.1016/j.cell.2014.11.024
27. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
28. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
29. Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, et al. Transcriptome Analysis Highlights the Conserved Difference Between Embryonic and Postnatal-Derived Alveolar Macrophages. Blood (2015) 126:1357–66. doi: 10.1182/blood-2015-01-624809
30. Srivastava S, Ernst JD, Desvignes L. Beyond Macrophages: The Diversity of Mononuclear Cells in Tuberculosis. Immunol Rev (2014) 262:179–92. doi: 10.1111/imr.12217
31. Guilliams M, Svedberg FR. Does Tissue Imprinting Restrict Macrophage Plasticity? Nat Immunol (2021) 22:118–27. doi: 10.1038/s41590-020-00849-2
32. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell Origin Dictates Programming of Resident Versus Recruited Macrophages During Acute Lung Injury. Am J Respir Cell Mol Biol (2017) 57:294–306. doi: 10.1165/rcmb.2017-0061OC
33. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two Distinct Interstitial Macrophage Populations Coexist Across Tissues in Specific Subtissular Niches. Sci (80- ) (2019) 363:eaau0964. doi: 10.1126/science.aau0964
34. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am J Respir Cell Mol Biol (2017) 57:66–76. doi: 10.1165/rcmb.2016-0361OC
35. Evren E, Ringqvist E, Willinger T. Origin and Ontogeny of Lung Macrophages: From Mice to Humans. Immunology (2020) 160:126–38. doi: 10.1111/imm.13154
36. Brundu S FA. Polarization and Repolarization of Macrophages. J Clin Cell Immunol (2015) 06:1–10. doi: 10.4172/2155-9899.1000319
37. Wynn T, Chawla A, Pollard JW. Origins and Hallmarks of Macrophages: Development, Homeostasis, and Disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
38. Nahrendorf M, Swirski FK. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res (2016) 119:414–7. doi: 10.1161/CIRCRESAHA.116.309194
39. Chávez-Galán L, Olleros ML, Vesin D, Garcia I. Much More Than M1 and M2 Macrophages, There are Also CD169+ and TCR+ Macrophages. Front Immunol (2015) 6:1–15. doi: 10.3389/fimmu.2015.00263
40. Poczobutt JM, De S, Yadav VK, Nguyen TT, Li H, Sippel TR, et al. Expression Profiling of Macrophages Reveals Multiple Populations With Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J Immunol (2016) 196:2847–59. doi: 10.4049/jimmunol.1502364
41. Cassetta L, Cassol E, Poli G. Macrophage Polarization in Health and Disease. Sci World J (2011) 11:2391–402. doi: 10.1100/2011/213962
42. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
43. Lastrucci C, Bénard A, Balboa L, Pingris K, Souriant S, Poincloux R, et al. Tuberculosis is Associated With Expansion of a Motile, Permissive and Immunomodulatory CD16+ Monocyte Population via the IL-10/STAT3 axis. Cell Res (2015) 25:1333–51. doi: 10.1038/cr.2015.123
44. McClean CM, Tobin DM. Macrophage Form, Function, and Phenotype in Mycobacterial Infection: Lessons From Tuberculosis and Other Diseases. Pathog Dis (2016) 74:ftw068. doi: 10.1093/femspd/ftw068
45. Arnold CE. A Critical Role for Suppressor of Cytokine Signalling 3 in Promoting M1 Macrophage Activation and Function in vitro and in vivo. Immunology (2014) 141:96–110. doi: 10.1111/imm.12173
46. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in Tuberculous Granulomas Are Delineated by Distinct Populations of Macrophage Subsets and Expression of Nitric Oxide Synthase and Arginase Isoforms. J Immunol (2013) 191:773–84. doi: 10.4049/jimmunol.1300113
47. Thiriot JD, Martinez-Martinez YB, Endsley JJ, Torres AG. Hacking the Host: Exploitation of Macrophage Polarization by Intracellular Bacterial Pathogens. Pathog Dis (2020) 78:1–14. doi: 10.1093/femspd/ftaa009
48. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
49. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage Activation and Polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
50. Anderson CF, Mosser DM. A Novel Phenotype for an Activated Macrophage: The Type 2 Activated Macrophage. J Leukoc Biol (2002) 72:101–6. doi: 10.1189/jlb.72.1.101
51. Genoula M, Marín Franco JL, Dupont M, Kviatcovsky D, Milillo A, Schierloh P, et al. Formation of Foamy Macrophages by Tuberculous Pleural Effusions Is Triggered by the Interleukin-10/Signal Transducer and Activator of Transcription 3 Axis Through ACAT Upregulation. Front Immunol (2018) 9:1–17. doi: 10.3389/fimmu.2018.00459
52. Wang Q. Fra-1 Protooncogene Regulates IL-6 Expression in Macrophages and Promotes the Generation of M2d Macrophages. Cell Res (2010) 20:701–12. doi: 10.1038/cr.2010.52
53. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-Like Phenotype Is Independent of Interleukin-4 Receptor Alpha (IL-4rα) Signaling. Inflammation (2013) 36:921–31. doi: 10.1007/s10753-013-9621-3
54. Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of Plcβ2 by Endotoxin Plays a Role in the Adenosine A 2A Receptor-Mediated Switch of Macrophages From an Inflammatory to an Angiogenic Phenotype. Am J Pathol (2009) 175:2439–53. doi: 10.2353/ajpath.2009.090290
55. Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A Circulating Cell Population Showing Both M1 and M2 Monocyte/Macrophage Surface Markers Characterizes Systemic Sclerosis Patients With Lung Involvement. Respir Res (2018) 19:1–12. doi: 10.1186/s12931-018-0891-z
56. Mitsi E, Kamng'ona R, Rylance J, Solórzano C, Jesus Reiné J, Mwandumba HC, et al. Human Alveolar Macrophages Predominately Express Combined Classical M1 and M2 Surface Markers in Steady State. Respir Res (2018) 19:1–4. doi: 10.1186/s12931-018-0777-0
57. Srivastava S, Ernst JD, D L. Beyond Macrophages: The Diversity of Mononuclear Cells in Tuberculosis. Immunol Rev (2014) 262:179–92. doi: 10.1111/imr.12217
58. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar Macrophages Provide an Early Mycobacterium Tuberculosis Niche and Initiate Dissemination. Cell Host Microbe (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
59. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium TuberculosisIn Vivo Segregates With Host Macrophage Metabolism and Ontogeny. J Exp Med (2018) 215:1135–52. doi: 10.1084/jem.20172020
60. Pisu D, Huang L, Grenier JK, Russell DG. Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep (2020) 30:335–50.e4. doi: 10.1016/j.celrep.2019.12.033
61. Mahajan S, Dkhar HK, Chandra V, Dave S, Nanduri R, Janmeja AK, et al. Mycobacterium Tuberculosis Modulates Macrophage Lipid-Sensing Nuclear Receptors Pparγ and TR4 for Survival. J Immunol (2012) 188:5593–603. doi: 10.4049/jimmunol.1103038
62. Rajaram MVS, Ni B, Dodd CE, Schlesinger LS. Macrophage Immunoregulatory Pathways in Tuberculosis. Semin Immunol (2014) 26:471–85. doi: 10.1016/j.smim.2014.09.010
63. Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, et al. Depletion of Alveolar Macrophages Exerts Protective Effects in Pulmonary Tuberculosis in Mice. J Immunol (2001) 166:4604–11. doi: 10.4049/jimmunol.166.7.4604
64. Upadhyay S, Mittal E, Philips JA. Tuberculosis and the Art of Macrophage Manipulation. Pathog Dis (2018) 76:1–12. doi: 10.1093/femspd/fty037
65. Sun J. Mycobacterial Nucleoside Diphosphate Kinase Blocks Phagosome Maturation in Murine Raw 264.7 Macrophages. PloS One (2010) 5:1–12. doi: 10.1371/journal.pone.0008769
66. Armstrong JA, Hart D. Response of Cultured Macrophages to Mycobacterium Tuberculosis, With Observations on Fusion of Lysosomes With Phagosomes. J Exp Med (1971) 134:713–40. doi: 10.1084/jem.134.3.713
67. Bussi C, Gutierrez MG. Mycobacterium Tuberculosis Infection of Host Cells in Space and Time. FEMS Microbiol Rev (2019) 43:341–61. doi: 10.1093/femsre/fuz006
68. Lugo-Villarino G, Neyrolles O. Manipulation of the Mononuclear Phagocyte System by Mycobacterium Tuberculosis. Cold Spring Harb Perspect Med (2014) 4:a018549–a018549. doi: 10.1101/cshperspect.a018549
69. Russell DG. Mycobacterium Tuberculosis : Here Today, and Here Tomorrow. Nat Rev Mol Cell Biol (2001) 2:569–78. doi: 10.1038/35085034
70. Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, et al. The Primary Mechanism of Attenuation of Bacillus Calmette-Guérin is a Loss of Secreted Lytic Function Required for Invasion of Lung Interstitial Tissue. Proc Natl Acad Sci USA (2003) 100:12420–5. doi: 10.1073/pnas.1635213100
71. Sun J, Siroy A, Lokareddy RK, Speer A, Doornbos KS, Cingolani G, et al. The Tuberculosis Necrotizing Toxin Kills Macrophages by Hydrolyzing NAD. Nat Struct Mol Biol (2015) 22:672–8. doi: 10.1038/nsmb.3064
72. Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, et al. Phagosomal Rupture by Mycobacterium Tuberculosis Results in Toxicity and Host Cell Death. PloS Pathog (2012) 8:e1002507. doi: 10.1371/journal.ppat.1002507
73. Ates LS, Houben ENG, Bitter W. Type VII Secretion: A Highly Versatile Secretion System. Microbiol Spectr (2016) 4:1–21. doi: 10.1128/microbiolspec.VMBF-0011-2015
74. Målen H, Berven FS, Fladmark KE, Wiker HG. Comprehensive Analysis of Exported Proteins Frommycobacterium Tuberculosis H37Rv. Proteomics (2007) 7:1702–18. doi: 10.1002/pmic.200600853
75. Tiwari S, Casey R, Goulding CW, Hingley-Wilson S, Jacobs WR Jr. Infect and Inject: How Mycobacterium Tuberculosis Exploits Its Major Virulence-Associated Type VII Secretion System, ESX-1. Microbiol Spectr (2019) 7:113–26. doi: 10.1128/9781683670261.ch8
76. Augenstreich J, Arbues A, Simeone R, Haanappel E, Wegener A, Sayes F, et al. ESX-1 and Phthiocerol Dimycocerosates of Mycobacterium Tuberculosis Act in Concert to Cause Phagosomal Rupture and Host Cell Apoptosis. Cell Microbiol (2017) 19:1–19. doi: 10.1111/cmi.12726
77. Gordon SV. Identification of Variable Regions in the Genomes of Tubercle Bacilli Using Bacterial Artificial Chromosome Arrays. Mol Microbiol (1999) 32:643–55. doi: 10.1046/j.1365-2958.1999.01383.x
78. Tran HKR, Grebenc DW, Klein TA, Whitney JC. Bacterial Type VII Secretion: An Important Player in Host-Microbe and Microbe-Microbe Interactions. Mol Microbiol (2021) 115:478–89. doi: 10.1111/mmi.14680
79. De Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, et al. ESAT-6 From Mycobacterium Tuberculosis Dissociates From its Putative Chaperone CFP-10 Under Acidic Conditions and Exhibits Membrane-Lysing Activity. J Bacteriol (2007) 189:6028–34. doi: 10.1128/JB.00469-07
80. Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, et al. Mycobacterial ESX-1 Secretion System Mediates Host Cell Lysis Through Bacterium Contact-Dependent Gross Membrane Disruptions. Proc Natl Acad Sci USA (2017) 114:1371–6. doi: 10.1073/pnas.1620133114
81. Gao LY. A Mycobacterial Virulence Gene Cluster Extending RD1 is Required for Cytolysis, Bacterial Spreading and ESAT-6 Secretion. Mol Microbiol (2004) 53:1677–93. doi: 10.1111/j.1365-2958.2004.04261.x
82. Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, et al. Mycobacterium Tuberculosis Type VII Secreted Effector EsxH Targets Host ESCRT to Impair Trafficking. PloS Pathog (2013) 9:e1003734. doi: 10.1371/journal.ppat.1003734
83. Philips JA. Mycobacterial Manipulation of Vacuolar Sorting. Cell Microbiol (2008) 10:2408–15. doi: 10.1111/j.1462-5822.2008.01239.x
84. Divangahi M, Behar SM, Remold H. Dying to Live: How the Death Modality of the Infected Macrophage Affects Immunity to Tuberculosis Vol. vol. 783. Divangahi M, editor. New York: Springer (2013) p. 103–20.
85. Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium Tuberculosis Strains Evade Apoptosis of Infected Alveolar Macrophages. J Immunol (2000) 164:2016–20. doi: 10.4049/jimmunol.164.4.2016
86. Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, et al. Mycobacterium Tuberculosis nuoG Is a Virulence Gene That Inhibits Apoptosis of Infected Host Cells. PloS Pathog (2007) 3:0972–80. doi: 10.1371/journal.ppat.0030110
87. Dey B, Bishai WR. Crosstalk Between Mycobacterium Tuberculosis and the Host Cell. Semin Immunol (2014) 26:486–96. doi: 10.1016/j.smim.2014.09.002
88. Zulauf KE, Sullivan JT, Braunstein M. The SecA2 Pathway of Mycobacterium Tuberculosis Exports Effectors That Work in Concert to Arrest Phagosome and Autophagosome Maturation. PloS Pathog (2018) 14:1–29. doi: 10.1371/journal.ppat.1007011
89. Shariq M, Quadir N, Sheikh JA, Singh AK, Bishai WR, Ehtesham NZ, et al. Post Translational Modifications in Tuberculosis: Ubiquitination Paradox. Autophagy (2021) 17:814–7. doi: 10.1080/15548627.2020.1850009
90. Wong KW. The Role of ESX-1 in Mycobacterium Tuberculosis Pathogenesis. Tuberc Tuber Bacillus Second Ed (2017) 5:627–34. doi: 10.1128/9781555819569.ch29
91. Behura A, Mishra A, Chugh S, Mawatwal S, Kumar A, Manna D, et al. ESAT-6 Modulates Calcimycin-Induced Autophagy Through microRNA-30a in Mycobacteria Infected Macrophages. J Infect (2019) 79:139–52. doi: 10.1016/j.jinf.2019.06.001
92. Lin J. Early Secreted Antigenic Target of 6-kDa of Mycobacterium Tuberculosis Promotes Caspase-9/Caspase-3-Mediated Apoptosis in Macrophages. Mol Cell Biochem (2019) 457:179–89. doi: 10.1007/s11010-019-03522-x
93. Paik S, Choi S, Lee KI, Back YW, Son YJ, Jo EK, et al. Mycobacterium Tuberculosis Acyl Carrier Protein Inhibits Macrophage Apoptotic Death by Modulating the Reactive Oxygen Species/C-Jun N-Terminal Kinase Pathway. Microbes Infect (2019) 21:40–9. doi: 10.1016/j.micinf.2018.06.005
94. Lim YJ, Yi MH, Choi JA, Lee J, Han JY, Jo SH, et al. Roles of Endoplasmic Reticulum Stress-Mediated Apoptosis in M1-Polarized Macrophages During Mycobacterial Infections. Sci Rep (2016) 6:1–11. doi: 10.1038/srep37211
95. Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium Tuberculosis Is Influenced by Host Factors and Precedes the Initiation of T-Cell Immunity. Infect Immun (2002) 70:4501–9. doi: 10.1128/IAI.70.8.4501-4509.2002
96. Verrall AJ, Schneider M, Alisjahbana B, Apriani L, van Laarhoven A, Koeken VACM, et al. Early Clearance of Mycobacterium Tuberculosis Is Associated With Increased Innate Immune Responses. J Infect Dis (2020) 221:1342–50. doi: 10.1093/infdis/jiz147
97. Joosten SA, van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE, et al. Mycobacterial Growth Inhibition is Associated With Trained Innate Immunity. J Clin Invest (2018) 128:1837–51. doi: 10.1172/JCI97508
98. Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological Mechanisms of Human Resistance to Persistent Mycobacterium Tuberculosis Infection. Nat Rev Immunol (2018) 18:575–89. doi: 10.1038/s41577-018-0025-3
99. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and Its Role in Health and Disease. Nat Rev Immunol vol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
100. Netea MG, van der Meer JWM. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe (2017) 21:297–300. doi: 10.1016/j.chom.2017.02.003
101. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-Lasting Effects of BCG Vaccination on Both Heterologous Th1/Th17 Responses and Innate Trained Immunity. J Innate Immun (2014) 6:152–8. doi: 10.1159/000355628
102. Koeken VACM, Verrall AJ, Netea MG, Hill PC, van Crevel R. Trained Innate Immunity and Resistance to Mycobacterium Tuberculosis Infection. Clin Microbiol Infect vol (2019) 25:1468–72. doi: 10.1016/j.cmi.2019.02.015
103. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects Against Experimental Viral Infection in Humans Through the Induction of Cytokines Associated With Trained Immunity. Cell Host Microbe (2018) 23:89–100.e5. doi: 10.1016/j.chom.2017.12.010
104. Ciarlo E, Heinonen T, Théroude C, Asgari F, Le Roy D, Netea MG, et al. Trained Immunity Confers Broad-Spectrum Protection Against Bacterial Infections. J Infect Dis (2020) 222:1869–81. doi: 10.1093/infdis/jiz692
105. Verma D, Parasa VR, Raffetseder J, Martis M, Mehta RB, Netea M, et al. Anti-Mycobacterial Activity Correlates With Altered DNA Methylation Pattern in Immune Cells From BCG-Vaccinated Subjects. Sci Rep (2017) 7:1–10. doi: 10.1038/s41598-017-12110-2
106. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity Against Tuberculosis. Cell (2018) 172:176–90.e19. doi: 10.1016/j.cell.2017.12.031
107. Moorlag S. J. C. F. M., Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, et al. β-Glucan Induces Protective Trained Immunity Against Mycobacterium Tuberculosis Infection: A Key Role for IL-1. Cell Rep (2020) 31:107634. doi: 10.1016/j.celrep.2020.107634
108. Rose S, Misharin A, Perlman H. A Novel Ly6C/Ly6G-Based Strategy to Analyze the Mouse Splenic Myeloid Compartment. Cytom Part A (2012) 81A:343–50. doi: 10.1002/cyto.a.22012
109. Reynolds G, Haniffa M. Human and Mouse Mononuclear Phagocyte Networks: A Tale of Two Species? Front Immunol (2015) 6:1–15. doi: 10.3389/fimmu.2015.00330
110. Geissmann F, Jung S, Littman DR. Blood Monocytes Consist of Two Principal Subsets With Distinct Migratory Properties. Immunity (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
111. Norris BA, Ernst JD. Mononuclear Cell Dynamics in M. Tuberculosis Infection Provide Opportunities for Therapeutic Intervention. PloS Pathog (2018) 14:e1007154. doi: 10.1371/journal.ppat.1007154
112. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, et al. Inflammatory Ly6Chi Monocytes and Their Conversion to M2 Macrophages Drive Atherosclerosis Regression. J Clin Invest (2017) 127:2904–15. doi: 10.1172/JCI75005
113. Girgis NM. Ly6Chigh Monocytes Become Alternatively Activated Macrophages in Schistosome Granulomas With Help From CD4+ Cells. PloS Pathog (2014) 10:e1004080. doi: 10.1371/journal.ppat.1004080
114. Xavier MN, Winter MG, Spees AM, den Hartigh AB, Nguyen K, Roux CM, et al. Pparγ-Mediated Increase in Glucose Availability Sustains Chronic Brucella Abortus Infection in Alternatively Activated Macrophages. Cell Host Microbe (2013) 14:159–70. doi: 10.1016/j.chom.2013.07.009
115. Terrazas C, Varikuti S, Oghumu S, Steinkamp HM, Ardic N, Kimble J, et al. Ly6Chi Inflammatory Monocytes Promote Susceptibility to Leishmania Donovani Infection. Sci Rep (2017) 7:1–10. doi: 10.1038/s41598-017-14935-3
116. Ahmad F, Umar MS, Khan N, Jamal F, Gupta P, Zubair S, et al. Immunotherapy With 5, 15-DPP Mediates Macrophage M1 Polarization and Modulates Subsequent Mycobacterium Tuberculosis Infectivity in rBCG30 Immunized Mice. Front Immunol (2021) 12:706727. doi: 10.3389/fimmu.2021.706727
117. Queval CJ, Song OR, Deboosère N, Delorme V, Debrie AS, Iantomasi R, et al. STAT3 Represses Nitric Oxide Synthesis in Human Macrophages Upon Mycobacterium Tuberculosis Infection. Sci Rep (2016) 6:29297. doi: 10.1038/srep29297
118. Pahari S, Kaur G, Negi S, Aqdas M, Das DK, Bashir H, et al. Reinforcing the Functionality of Mononuclear Phagocyte System to Control Tuberculosis. Front Immunol (2018) 9:1–17. doi: 10.3389/fimmu.2018.00193
119. MacLean JA, Xia W, Pinto CE, Zhao L, Liu HW, Kradin RL. Sequestration of Inhaled Particulate Antigens by Lung Phagocytes: A Mechanism for the Effective Inhibition of Pulmonary Cell-Mediated Immunity. Am J Pathol (1996) 148:657–66.
120. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria Manipulate Macrophage Recruitment Through Coordinated Use of Membrane Lipids. Nature (2014) 505:218–22. doi: 10.1038/nature12799
121. Sheedy FJ, Divangahi M. Targeting Immunometabolism in Host Defence Against Mycobacterium Tuberculosis. Immunology (2021) 162:145–59. doi: 10.1111/imm.13276
122. Pisu D, Huang L, Narang V, Theriault M, Lê-Bury G, Lee B, et al. Single Cell Analysis of M. Tuberculosis Phenotype and Macrophage Lineages in the Infected Lung. J Exp Med (2021) 218:e20210615. doi: 10.1084/jem.20210615
123. Cambier CJ, O’Leary SM, O’Sullivan MP, Keane J, Ramakrishnan L. Phenolic Glycolipid Facilitates Mycobacterial Escape From Microbicidal Tissue-Resident Macrophages. Immunity (2017) 47:552–565.e4. doi: 10.1016/j.immuni.2017.08.003
124. Quigley J, Hughitt VK, Velikovsky CA, Mariuzza RA, El-Sayed NM, Briken V, et al. The Cell Wall Lipid PDIM Contributes to Phagosomal Escape and Host Cell Exit of Mycobacterium Tuberculosis. MBio (2017) 8:e00148-17. doi: 10.1128/mBio.00148-17
125. Cambier CJ, Banik SM, Buonomo JA, Bertozzi CR. Spreading of a Mycobacterial Cell Surface Lipid Into Host Epithelial Membranes Promotes Infectivity. Elife (2020) 9:1–68. doi: 10.7554/eLife.60648
126. Lerner TR. Phthiocerol Dimycocerosates Promote Access to the Cytosol and Intracellular Burden of Mycobacterium Tuberculosis in Lymphatic Endothelial Cells. BMC Biol (2018) 16:1–13. doi: 10.1186/s12915-017-0471-6
127. Rens C, Chao JD, Sexton DL, Tocheva EI, Av-Gay Y. Roles for Phthiocerol Dimycocerosate Lipids in Mycobacterium Tuberculosis Pathogenesis. Microbiol (United Kingdom) (2021) 167:1–14. doi: 10.1099/mic.0.001042
128. Barczak AK, Avraham R, Singh S, Luo SS, Zhang WR, Bray MA, et al. Systematic, Multiparametric Analysis of Mycobacterium Tuberculosis Intracellular Infection Offers Insight Into Coordinated Virulence. PloS Pathog (2017) 13:1–27. doi: 10.1371/journal.ppat.1006363
129. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage Polarization in Inflammatory Diseases. Int J Biol Sci (2014) 10:520–9. doi: 10.7150/ijbs.8879
130. Redford PS, Murray PJ, O’Garra A. The Role of IL-10 in Immune Regulation During M. Tuberculosis Infection. Mucosal Immunol (2011) 4:261–70. doi: 10.1038/mi.2011.7
131. Abdullah Z, Geiger S, Nino-Castro A, Böttcher JP, Muraliv E, Gaidt M, et al. Lack of Pparγ in Myeloid Cells Confers Resistance to Listeria Monocytogenes Infection. PloS One (2012) 7:1–15. doi: 10.1371/journal.pone.0037349
132. Gupta S, Winglee K, Gallo R, Bishai WR. Bacterial Subversion of cAMP Signalling Inhibits Cathelicidin Expression, Which is Required for Innate Resistance to Mycobacterium Tuberculosis. J Pathol (2017) 242:52–61. doi: 10.1002/path.4878
133. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory Signaling in Human Tuberculosis Granulomas is Spatially Organized. Nat Med (2016) 22:531–8. doi: 10.1038/nm.4073
134. Shi L, Jiang Q, Bushkin Y, Subbian S, Tyagi S. Biphasic Dynamics of Macrophage Immunometabolism During Mycobacterium Tuberculosis Infection. MBio (2019) 10:1–19. doi: 10.1128/mBio.02550-18
135. Huang Z, Luo Q, Guo Y, Chen J, Xiong G, Peng Y, et al. Mycobacterium Tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas. In Vitro PloS One (2015) 10:e0129744. doi: 10.1371/journal.pone.0129744
136. García-González G, Sánchez-González A, Hernández-Bello R, González GM, Franco-Molina MA, Coronado-Cerda EE, et al. Triggering of Protease-Activated Receptors (PARs) Induces Alternative M2 Macrophage Polarization With Impaired Plasticity. Mol Immunol (2019) 114:278–88. doi: 10.1016/j.molimm.2019.08.004
137. Lopes RL, Borges TJ, Araújo JF, Pinho NG, Bergamin LS, Battastini AM, et al. Extracellular Mycobacterial DnaK Polarizes Macrophages to the M2-Like Phenotype. PloS One (2014) 9:1–16. doi: 10.1371/journal.pone.0113441
138. Zhang Y, Li S, Liu Q, Long R, Feng J, Qin H, et al. Mycobacterium Tuberculosis Heat-Shock Protein 16.3 Induces Macrophage M2 Polarization Through Ccrl2/Cx3cr1. Inflammation (2020) 43:487–506. doi: 10.1007/s10753-019-01132-9
139. Parveen N. Endocytosis of Mycobacterium Tuberculosis Heat Shock Protein 60 Is Required to Induce Interleukin-10 Production in Macrophages. J Biol Chem (2013) 288:24956–71. doi: 10.1074/jbc.M113.461004
140. Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid Pathways Regulate Adaptive Immunity to Mycobacterium Tuberculosis. Nat Immunol (2010) 11:751–8. doi: 10.1038/ni.1904
141. Wilson JL, Mayr HK, Weichhart T. Metabolic Programming of Macrophages: Implications in the Pathogenesis of Granulomatous Disease. Front Immunol (2019) 10:1–22. doi: 10.3389/fimmu.2019.02265
142. O’Neill LAJ, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
143. Galvan-Pena S, O’Neill LAJ. Metabolic Reprograming in Macrophage Polarization. Front Immunol (2014) 5:1–6. doi: 10.3389/fimmu.2014.00420
144. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate Fate in Activated Macrophages: A Comparison Between Innate, Classic, and Alternative Activation. J Immunol (2010) 185:605–14. doi: 10.4049/jimmunol.0901698
145. El-Gayar S, Thüring-Nahler H, Pfeilschifter J, Röllinghoff M, Bogdan C. Translational Control of Inducible Nitric Oxide Synthase by IL-13 and Arginine Availability in Inflammatory Macrophages. J Immunol (2003) 171:4561–8. doi: 10.4049/jimmunol.171.9.4561
146. Modolell M, Choi BS, Ryan RO, Hancock M, Titus RG, Abebe T, et al. Local Suppression of T Cell Responses by Arginase-Induced L-Arginine Depletion in Nonhealing Leishmaniasis. PloS Negl Trop Dis (2009) 3:e480. doi: 10.1371/journal.pntd.0000480
147. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional Profiling of the Human Monocyte-To-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J Immunol (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
148. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al, et al. Mycobacterium Tuberculosis Evades Macrophage Defenses by Inhibiting Plasma Membrane Repair. Nat Immunol (2009) 10:899–906. doi: 10.1038/ni.1758
149. Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid Mediators in Innate Immunity Against Tuberculosis: Opposing Roles of PGE2 and LXA4 in the Induction of Macrophage Death. J Exp Med (2008) 205:2791–801. doi: 10.1084/jem.20080767
150. Peres CM, de Paula L, Medeiros AI, Sorgi CA, Soares EG, Carlos D, et al. Inhibition of Leukotriene Biosynthesis Abrogates the Host Control of Mycobacterium Tuberculosis. Microbes Infect (2007) 9:483–9. doi: 10.1016/j.micinf.2007.01.006
151. Peres-Buzalaf C, de Paula L, Frantz FG, Soares EM, Medeiros AI, Peters-Golden M, et al. Control of Experimental Pulmonary Tuberculosis Depends More on Immunostimulatory Leukotrienes Than on the Absence of Immunosuppressive Prostaglandins. Prostaglandins Leukot Essent Fat Acids (2011) 85:75–81. doi: 10.1016/j.plefa.2011.04.024
152. Dietzold J, Gopalakrishnan A, Salgame P. Duality of Lipid Mediators in Host Response Against Mycobacterium Tuberculosis: Good Cop, Bad Cop. F1000Prime Rep (2015) 7:1–8. doi: 10.12703/P7-29
153. Kaul V, Bhattacharya D, Singh Y, Van Kaer L, Peters-Golden M, Bishai WR, et al. An Important Role of Prostanoid Receptor EP2 in Host Resistance to Mycobacterium Tuberculosis Infection in Mice. J Infect Dis (2012) 206:1816–25. doi: 10.1093/infdis/jis609
154. Kaul V, Van Kaer L, Das G, Das J. Prostanoid Receptor 2 Signaling Protects T Helper 2 Cells From BALB/c Mice Against Activation-Induced Cell Death. J Biol Chem (2012) 287:25434–9. doi: 10.1074/jbc.C111.324707
155. Tullius MV, Nava S, Horwitz MA. PPE37 Is Essential for Mycobacterium Tuberculosis Heme-Iron Acquisition (HIA), and a Defective PPE37 in Mycobacterium Bovis BCG Prevents HIA. Infect Immun (2019) 87:1–26. doi: 10.1128/IAI.00540-18
156. Ahmad J, Farhana A, Pancsa R, Arora SK, Srinivasan A, Tyagi AK, et al. Contrasting Function of Structured N-Terminal and Unstructured C-Terminal Segments of Mycobacterium Tuberculosis PPE37 Protein. MBio (2018) 9:1–18. doi: 10.1128/mBio.01712-17
157. Serafini A, Pisu D, Palù G, Rodriguez GM, Manganelli R. The ESX-3 Secretion System Is Necessary for Iron and Zinc Homeostasis in Mycobacterium Tuberculosis. PloS One (2013) 8:1–15. doi: 10.1371/annotation/56401af3-74aa-4c4f-882c-06481b29aa94
158. Mitra A, Speer A, Lin K, Ehrt S, Niederweis M. PPE Surface Proteins Are Required for Heme Utilization by Mycobacterium Tuberculosis. MBio (2017) 8:1–14. doi: 10.1128/mBio.01720-16
159. Mitra A, Ko Y-H, Cingolani G, Niederweis M. Heme and Hemoglobin Utilization by Mycobacterium Tuberculosis. Nat Commun (2019) 10:4260. doi: 10.1038/s41467-019-12109-5
160. Ehtram A, Shariq M, Ali S, Quadir N, Sheikh JA, Ahmad F, et al. Teleological Cooption of Mycobacterium Tuberculosis PE/PPE Proteins as Porins: Role in Molecular Immigration and Emigration. Int J Med Microbiol (2021) 311:151495. doi: 10.1016/j.ijmm.2021.151495
161. Banerjee S, Farhana A, Ehtesham NZ, Hasnain SE. Iron Acquisition, Assimilation and Regulation in Mycobacteria. Infect Genet Evol (2011) 11:825–38. doi: 10.1016/j.meegid.2011.02.016
162. Cairo G, Recalcati S, Mantovani A, Locati M. Iron Trafficking and Metabolism in Macrophages: Contribution to the Polarized Phenotype. Trends Immunol (2011) 32:241–7. doi: 10.1016/j.it.2011.03.007
163. Muraille E, Leo O, Moser M. Th1/Th2 Paradigm Extended: Macrophage Polarization as an Unappreciated Pathogen-Driven Escape Mechanism? Front Immunol (2014) 5:1–12. doi: 10.3389/fimmu.2014.00603
164. Banerjee S, Nandyala AK, Raviprasad P, Ahmed N, Hasnain SE. Iron-Dependent RNA-Binding Activity of Mycobacterium Tuberculosis Aconitase. J Bacteriol (2007) 189:4046–52. doi: 10.1128/JB.00026-07
165. Farhana A, Kumar S, Rathore SS, Ghosh PC, Ehtesham NZ, Tyagi AK, et al. Mechanistic Insights Into a Novel Exporter-Importer System of Mycobacterium Tuberculosis Unravel Its Role in Trafficking of Iron. PloS One (2008) 3:e2087. doi: 10.1371/journal.pone.0002087
166. Rockwood N, Costa DL, Amaral EP, Du Bruyn E, Kubler A, Gil-Santana L, et al. Mycobacterium Tuberculosis Induction of Heme Oxygenase-1 Expression Is Dependent on Oxidative Stress and Reflects Treatment Outcomes. Front Immunol (2017) 8:1–15. doi: 10.3389/fimmu.2017.00542
167. Costa DL, Namasivayam S, Amaral EP, Arora K, Chao A, Mittereder LR, et al. Pharmacological Inhibition of Host Heme Oxygenase-1 Suppresses Mycobacterium Tuberculosis InfectionIn Vivo by a Mechanism Dependent on T Lymphocytes. MBio (2016) 7:1–6. doi: 10.1128/mBio.01675-16
168. Costa DL, Amaral EP, Namasivayam S, Mittereder LR, Fisher L, Bonfim CC, et al. Heme Oxygenase-1 Inhibition Promotes Ifnγ- and NOS2-Mediated Control of Mycobacterium Tuberculosis Infection. Mucosal Immunol (2021) 14:253–66. doi: 10.1038/s41385-020-00342-x
169. Shariq M, Quadir N, Sharma N, Singh J, Sheikh JA, Khubaib M, et al. Mycobacterium Tuberculosis RipA Dampens TLR4-Mediated Host Protective Response Using a Multi-Pronged Approach Involving Autophagy, Apoptosis, Metabolic Repurposing, and Immune Modulation. Front Immunol (2021) 12:1–19. doi: 10.3389/fimmu.2021.636644
170. Lavalett L, Ortega H, Barrera LF. Human Alveolar and Splenic Macrophage Populations Display a Distinct Transcriptomic Response to Infection With Mycobacterium Tuberculosis. Front Immunol (2020) 11:1–16. doi: 10.3389/fimmu.2020.00630
171. Baena A. Differential Determinants of Virulence in Two Mycobacterium Tuberculosis Colombian Clinical Isolates of the LAM09 Family. Virulence (2019) 10:695–710. doi: 10.1080/21505594.2019.1642045
172. Manca C, Tsenova L, Barry 3rd CE, Bergtold A, Freeman S, Haslett PA, et al. Mycobacterium Tuberculosis CDC1551 Induces a More Vigorous Host ResponseIn Vivo and In Vitro, But Is Not More Virulent Than Other Clinical Isolates. J Immunol (1999) 162:6740–6.
173. Homolka S, Niemann S, Russell DG, Rohde KH. Functional Genetic Diversity Among Mycobacterium Tuberculosis Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes During Intracellular Survival. PloS Pathog (2010) 6:1–17. doi: 10.1371/journal.ppat.1000988
174. Gao Q. Gene Expression Diversity Among Mycobacterium Tuberculosis Clinical Isolates. Microbiology (2005) 151:5–14. doi: 10.1099/mic.0.27539-0
175. Portevin D, Gagneux S, Comas I, Young D. Human Macrophage Responses to Clinical Isolates From the Mycobacterium Tuberculosis Complex Discriminate Between Ancient and Modern Lineages. PloS Pathog (2011) 7:e1001307. doi: 10.1371/journal.ppat.1001307
176. Tram TTB, Nhung HN, Vijay S, Hai HT, Thu DDA, Ha VTN, et al. Virulence of Mycobacterium Tuberculosis Clinical Isolates Is Associated With Sputum Pre-Treatment Bacterial Load, Lineage, Survival in Macrophages, and Cytokine Response. Front Cell Infect Microbiol (2018) 8:417. doi: 10.3389/fcimb.2018.00417
177. Cho HJ. Different Macrophage Polarization Between Drug-Susceptible and Multidrug-Resistant Pulmonary Tuberculosis. BMC Infect Dis (2020) 20:1–10. doi: 10.1186/s12879-020-4802-9
178. Marakalala MJ, Martinez FO, Plüddemann A, Gordon S. Macrophage Heterogeneity in the Immunopathogenesis of Tuberculosis. Front Microbiol (2018) 9:1–15. doi: 10.3389/fmicb.2018.01028
179. Marino S. Macrophage Polarization Drives Granuloma Outcome During Mycobacterium Tuberculosis Infection. Infect Immun (2015) 83:324–38. doi: 10.1128/IAI.02494-14
180. Helming L, Gordon S. The Molecular Basis of Macrophage Fusion. Immunobiology (2008) 212:785–93. doi: 10.1016/j.imbio.2007.09.012
181. Helming L, Gordon S. Molecular Mediators of Macrophage Fusion. Trends Cell Biol (2009) 19:514–22. doi: 10.1016/j.tcb.2009.07.005
182. Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar , et al. DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell (2016) 167:1264–80.e18. doi: 10.1016/j.cell.2016.09.054
183. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy Macrophages and the Progression of the Human Tuberculosis Granuloma. Nat Immunol (2009) 10:943–8. doi: 10.1038/ni.1781
184. Pandey AK, Sassetti CM. Mycobacterial Persistence Requires the Utilization of Host Cholesterol. Proc Natl Acad Sci USA (2008) 105:4376–80. doi: 10.1073/pnas.0711159105
185. Feng Y, Dorhoi A, Mollenkopf HJ, Yin H, Dong Z, Mao L, et al. Platelets Direct Monocyte Differentiation Into Epithelioid-Like Multinucleated Giant Foam Cells With Suppressive Capacity Upon Mycobacterial Stimulation. J Infect Dis (2014) 210:1700–10. doi: 10.1093/infdis/jiu355
186. Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity (2016) 45:861–76. doi: 10.1016/j.immuni.2016.09.014
187. Adams DO. The Structure of Mononuclear Phagocytes DifferentiatingIn Vivo. I. Sequential Fine and Histologic Studies of the Effect of Bacillus Calmette-Guerin (BCG). Am J Pathol (1974) 76:17–48.
188. Dorhoi A, Kaufmann SHE. Perspectives on Host Adaptation in Response to Mycobacterium Tuberculosis: Modulation of Inflammation. Semin Immunol (2014) 26:533–42. doi: 10.1016/j.smim.2014.10.002
189. Philips JA, Ernst JD. Tuberculosis Pathogenesis and Immunity. Annu Rev Pathol Mech Dis (2012) 7:353–84. doi: 10.1146/annurev-pathol-011811-132458
190. Ramakrishnan L. Revisiting the Role of the Granuloma in Tuberculosis. Nat Rev Immunol (2012) 12:352–66. doi: 10.1038/nri3211
191. Bryant DM, Mostov KE. From Cells to Organs: Building Polarized Tissue. Nat Rev Mol Cell Biol (2008) 9:887–901. doi: 10.1038/nrm2523
192. Craene B. De & Berx, G. Regulatory Networks Defining EMT During Cancer Initiation and Progression. Nat Rev Cancer (2013) 13:97–110. doi: 10.1038/nrc3447
193. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of Host Angiogenic Signalling Limits Mycobacterial Growth. Nature (2015) 517:612–5. doi: 10.1038/nature13967
194. Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, et al. A non-Canonical Type 2 Immune Response Coordinates Tuberculous Granuloma Formation and Epithelialization. Cell (2021) 184:1757–74.e14. doi: 10.1016/j.cell.2021.02.046
195. Brooks PJ, Glogauer M, McCulloch CA. An Overview of the Derivation and Function of Multinucleated Giant Cells and Their Role in Pathologic Processes. Am J Pathol (2019) 189:1145–58. doi: 10.1016/j.ajpath.2019.02.006
196. Milde R, Ritter J, Tennent GA, Loesch A, Martinez FO, Gordon S, et al. Multinucleated Giant Cells Are Specialized for Complement-Mediated Phagocytosis and Large Target Destruction. Cell Rep (2015) 13:1937–48. doi: 10.1016/j.celrep.2015.10.065
197. Lay G, Poquet Y, Salek-Peyron P, Puissegur MP, Botanch C, Bon H, et al. Langhans Giant Cells From M. Tuberculosis-Induced Human Granulomas Cannot Mediate Mycobacterial Uptake. J Pathol (2007) 211:76–85. doi: 10.1002/path.2092
198. Lösslein AK, Lohrmann F, Scheuermann L, Gharun K, Neuber J, Kolter J, et al. Monocyte Progenitors Give Rise to Multinucleated Giant Cells. Nat Commun (2021) 12:2027. doi: 10.1038/s41467-021-22103-5
199. Gharun K, Senges J, Seidl M, Lösslein A, Kolter J, Lohrmann F, et al. Mycobacteria Exploit Nitric Oxide-Induced Transformation of Macrophages Into Permissive Giant Cells. EMBO Rep (2017) 18:2144–59. doi: 10.15252/embr.201744121
200. Byrd TF. Multinucleated Giant Cell Formation Induced by IFN-γ/IL-3 Is Associated With Restriction of VirulentMycobacterium Tuberculosiscell to Cell Invasion in Human Monocyte Monolayers. Cell Immunol (1998) 188:89–96. doi: 10.1006/cimm.1998.1352
201. Gasser A, Möst J. Generation of Multinucleated Giant CellsIn Vitro by Culture of Human Monocytes With Mycobacterium Bovis BCG in Combination With Cytokine- Containing Supernatants. Infect Immun (1999) 67:395–402. doi: 10.1128/IAI.67.1.395-402.1999
202. D’Avila H, Maya-Monteiro CM, Bozza PT. Lipid Bodies in Innate Immune Response to Bacterial and Parasite Infections. Int Immunopharmacol (2008) 8:1308–15. doi: 10.1016/j.intimp.2008.01.035
203. Neyrolles O, et al. Is Adipose Tissue a Place for Mycobacterium Tuberculosis Persistence? PloS One (2006) 1:e43. doi: 10.1371/journal.pone.0000043
204. Kondo E, Kanai K. A Suggested Role of a Host-Parasite Lipid Complex in Mycobacterial Infection. Jpn J Med Sci Biol (1976) 29:199–210. doi: 10.7883/yoken1952.29.199
205. D’Avila H. Mycobacterium Bovis Bacillus Calmette-Guérin Induces TLR2-Mediated Formation of Lipid Bodies: Intracellular Domains for Eicosanoid SynthesisIn Vivo. J Immunol (2006) 176:3087–97. doi: 10.4049/jimmunol.176.5.3087
206. Silva AR, Pacheco P, Vieira-de-Abreu A, Maya-Monteiro CM, D'Alegria B, Magalhães KG, et al. Lipid Bodies in Oxidized LDL-Induced Foam Cells are Leukotriene-Synthesizing Organelles: A MCP-1/CCL2 Regulated Phenomenon. Biochim Biophys Acta - Mol Cell Biol Lipids (2009) 1791:1066–75. doi: 10.1016/j.bbalip.2009.06.004
207. Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium Tuberculosis Uses Host Triacylglycerol to Accumulate Lipid Droplets and Acquires a Dormancy-Like Phenotype in Lipid-Loaded Macrophages. PloS Pathog (2011) 7:e1002093. doi: 10.1371/journal.ppat.1002093
208. Cox JS, Chen B, McNeil M, Jacobs WRJ. Complex Lipid Determines Tissue-Specific Replication of Mycobacterium Tuberculosis In Mice. Nature (1999) 402:79–83. doi: 10.1038/47042
209. Yu J, Tran V, Li M, Huang X, Niu C, Wang D, et al. Both Phthiocerol Dimycocerosates and Phenolic Glycolipids are Required for Virulence of Mycobacterium Marinum. Infect Immun (2012) 80:1381–9. doi: 10.1128/IAI.06370-11
210. Augenstreich J, Haanappel E, Ferré G, Czaplicki G, Jolibois F, Destainville N, et al. The Conical Shape of DIM Lipids Promotes Mycobacterium Tuberculosis Infection of Macrophages. Proc Natl Acad Sci USA (2019) 116:25649–58. doi: 10.1073/pnas.1910368116
211. Gawrisch K. Mycobacterium Tuberculosis Enters Macrophages With Aid From a Bacterial Lipid. Proc Natl Acad Sci USA (2019) 116:25372–3. doi: 10.1073/pnas.1918900116
212. O’Garra A. The Immune Response in Tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
213. Marino S. Dendritic Cell Trafficking and Antigen Presentation in the Human Immune Response to Mycobacterium Tuberculosis. J Immunol (2004) 173:494–506. doi: 10.4049/jimmunol.173.1.494
214. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in Tuberculous Granulomas Are Delineated by Distinct Populations of Macrophage Subsets and Expression of Nitric Oxide Synthase and Arginase Isoforms. J Immunol (2013) 191:773–84. doi: 10.4049/jimmunol.1300113
215. Wang L, Shang X, Qi X, Ba D, Lv J, Zhou X, et al. Clinical Significance of M1/M2 Macrophages and Related Cytokines in Patients With Spinal Tuberculosis. Dis Markers (2020) 2020:1–8. doi: 10.1155/2020/2509454
216. Petersen HJ, Smith AM. The Role of the Innate Immune System in Granulomatous Disorders. Front Immunol (2013) 4:1–11. doi: 10.3389/fimmu.2013.00120
217. Pagán AJ, Ramakrishnan L. The Formation and Function of Granulomas. Annu Rev Immunol (2018) 36:639–65. doi: 10.1146/annurev-immunol-032712-100022
218. Marzo E. Damaging Role of Neutrophilic Infiltration in a Mouse Model of Progressive Tuberculosis. Tuberculosis (2014) 94:55–64. doi: 10.1016/j.tube.2013.09.004
219. Cardona P-J. What We Have Learned and What We Have Missed in Tuberculosis Pathophysiology for a New Vaccine Design: Searching for the “Pink Swan”. Front Immunol (2017) 8:1–11. doi: 10.3389/fimmu.2017.00556
220. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes are Shaped by the Local Microenvironment. Cell (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
Keywords: Mycobacterium tuberculosis, innate immunity, macrophage heterogeneity, phenotype switching, metabolic reprogramming, trained immunity
Citation: Ahmad F, Rani A, Alam A, Zarin S, Pandey S, Singh H, Hasnain SE and Ehtesham NZ (2022) Macrophage: A Cell With Many Faces and Functions in Tuberculosis. Front. Immunol. 13:747799. doi: 10.3389/fimmu.2022.747799
Received: 26 July 2021; Accepted: 30 March 2022;
Published: 06 May 2022.
Edited by:
Suzie Hingley-Wilson, University of Surrey, United KingdomReviewed by:
Shahram Salek-Ardakani, Pfizer, United StatesLuis F. Barrera, University of Antioquia, Colombia
Fernando O. Martinez, University of Surrey, United Kingdom
Tom Mendum, University of Surrey, United Kingdom
Copyright © 2022 Ahmad, Rani, Alam, Zarin, Pandey, Singh, Hasnain and Ehtesham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Ehtesham Hasnain, c2V5ZWRoYXNuYWluQGdtYWlsLmNvbQ==; c2V5ZWQuaGFzbmFpbkBzaGFyZGEuYWMuaW4=; c2VoQGRiZWIuaWl0ZC5hYy5pbg==; Nasreen Zafar Ehtesham, bnplaHRlc2hhbUBnbWFpbC5jb20=; bmFzcmVlbnplaHRlc2hhbS5uaXBAZ292Lmlu
†Present address: Faraz Ahmad, Department of Ophthalmology, School of Medicine, University of Missouri, Columbia, MO, United States
‡These authors have contributed equally to this work