 Wei Tu1,2,3†
Wei Tu1,2,3† Xiaojun Xiao2†Jiahua Lu2†Xiaoyu Liu2
Xiaojun Xiao2†Jiahua Lu2†Xiaoyu Liu2 Eryi Wang1,2Ruyi Yuan2
Eryi Wang1,2Ruyi Yuan2 Rongjun Wan3,4Yingchun Shen3Damo Xu1,2
Rongjun Wan3,4Yingchun Shen3Damo Xu1,2 Pingchang Yang1,2Miao Gong1,2
Pingchang Yang1,2Miao Gong1,2 Peisong Gao3*
Peisong Gao3* Shau-Ku Huang1,5*
Shau-Ku Huang1,5*- 1Department of Respiratory & Allergy, Third Affiliated Hospital of Shenzhen University, Shenzhen, China
- 2The State Key Laboratory of Respiratory Disease for Allergy, Shenzhen Key Laboratory of Allergy & Immunology, Shenzhen University School of Medicine, Shenzhen, China
- 3Johns Hopkins Asthma and Allergy Center, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 4Department of Respiratory Medicine, Xiangya Hospital, Central South University, Changsha, China
- 5National Institute of Environmental Health Sciences, National Health Research Institutes, Miaoli, Taiwan
Background: Metal components of environmental PM2.5 are associated with the exacerbation of allergic diseases like asthma. In our recent hospital-based population study, exposure to vanadium is shown to pose a significant risk for current asthma, but the causal relationship and its underlying molecular mechanisms remain unclear.
Objective: We sought to determine whether vanadium co-exposure can aggravate house dust mite (HDM)-induced allergic airway inflammation and remodeling, as well as investigate its related mechanisms.
Methods: Asthma mouse model was generated by using either vanadium pentoxide (V2O5) or HDM alone or in combination, in which the airway inflammation and remodeling was investigated. The effect of V2O5 co-exposure on HDM-induced epithelial-derived cytokine release and oxidative stress (ROS) generation was also examined by in vitro analyses. The role of ROS in V2O5 co-exposure-induced cytokine release and airway inflammation and remodeling was examined by using inhibitors or antioxidant.
Results: Compared to HDM alone, V2O5 co-exposure exacerbated HDM-induced airway inflammation with increased infiltration of inflammatory cells and elevated levels of Th1/Th2/Th17 and epithelial-derived (IL-25, TSLP) cytokines in the bronchoalveolar lavage fluids (BALFs). Intriguingly, V2O5 co-exposure also potentiated HDM-induced airway remodeling. Increased cytokine release was further supported by in vitro analysis in human bronchial epithelial cells (HBECs). Mechanistically, ROS, particularly mitochondrial-derived ROS, was significantly enhanced in HBECs after V2O5 co-exposure as compared to HDM challenge alone. Inhibition of ROS with its inhibitor N-acetyl-L-cysteine (NAC) and mitochondrial-targeted antioxidant MitoTEMPO blocked the increased epithelial release caused by V2O5 co-exposure. Furthermore, vitamin D3 as an antioxidant was found to inhibit V2O5 co-exposure-induced increased airway epithelial cytokine release and airway remodeling.
Conclusions: Our findings suggest that vanadium co-exposure exacerbates epithelial ROS generation that contribute to increased allergic airway inflammation and remodeling.
Introduction
The incidence of allergic asthma is increasing worldwide in recent decades (1), and has been considered to be a major global public health concern (2). Environmental pollutants have been suggested to be one of the major risk factors contributing to the increased incidence of asthma (3, 4). Particularly, the metal components of environmental PM2.5, such as vanadium, mercury, lead, copper, cadmium and arsenic, have been reported to affect the severity of allergic respiratory diseases (5, 6). Of these, vanadium exposure has been reported to cause occupational bronchial asthma and bronchitis (7, 8). Vanadium levels in air PM2.5 have been associated with the increased respiratory symptoms of children (9) and elevated levels of exhaled FeNO (10, 11). We have recently demonstrated that in a hospital-based case-control study, vanadium exposure posed a significant risk for current asthma, particularly for severe asthma, and that its urinary level correlated with the levels of Nε-(hexanoyl)-lysine (HEL; an oxidative stress marker) and circulating IL-8, as well as with the 3- and 7-day accumulated levels of ambient PM2.5 prior to the clinical visit (12). Furthermore, we and others have suggested interactions between environmental pollutants and allergens in the context of respiratory diseases including asthma (13–15). This suggests that vanadium exposure may contribute to the exacerbation of allergic asthma.
As a transition metal, vanadium is widely distributed not only in nature but also in various heavy industries such as steel and petroleum. Incomplete combustion of fuels such as petroleum is the main source of vanadium pollution in nature. It is well-recognized that exposure to an excessive amount of vanadium can cause damage and metabolic changes in multiple systems including respiratory system (16, 17). Recent epidemiological studies have indicated that vanadium exposure may be associated with impaired pulmonary function parameters and reduced asthma control among children with preexisting asthma (18). Studies with mouse models have suggested that exposure to vanadium pentoxide (V2O5) gives rise to airway smooth muscle thickening, airway mucous cell metaplasia, and an increased proliferation of peribronchiolar myofibroblasts, thereby leading to the development of airway fibrosis and remodeling (19). Mechanistically, exposure to vanadium has been associated with altered DNA methylation of allergic and proinflammatory asthma genes implicated in air pollution related asthma (20). Most interestingly, oxidative stress (ROS) has been suggested as a possible mechanism underlying the vanadium exposure-induced pathological effects (21, 22).
ROS as a central inflammatory mediator has been associated with the aggravation of the occurrence and development of lung diseases (23) and the severity of asthma (24). Of interest, exposure to vanadium has been shown to cause cell apoptosis and mouse lung inflammation through ROS generation (25). Vanadium exposure can also induce oxidative stress and cellular senescence in human lung fibrosis by In vitro analysis (26). These findings were supported by similar studies that silica nanoparticles can induce lung inflammation through ROS signaling and its mediated lysosome impairment and autophagy dysfunction (27), and ozone induces lung inflammation through mitochondrial ROS (28, 29). Our recent studies indicated that benzo(a)pyrene (BaP), a ubiquitous environmental pollutant (30), co-exposure significantly induced HDM-induced lung inflammation via ROS-mediated release of proinflammatory cytokines (13). These findings raise the possibility that vanadium co-exposure with HDM may exacerbate HDM-induced airway inflammation and remodeling through triggering epithelial ROS generation.
In the present study, we generated asthma mouse model by using either vanadium or HDM alone or in combination and investigated their effects on airway inflammation and remodeling. Especially, we investigated the effect of vanadium co-exposure on HDM-induced epithelial-derived cytokine release and ROS generation by both in vivo and in vitro analyses. Most importantly, we explored the role of ROS inhibitors and antioxidant vitamin D3 (VD3) in attenuating the vanadium co-exposure-induced cytokine release and airway inflammation and remodeling.
Materials and methods
Mice
Balb/c mice were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). All mice were maintained at Shenzhen University under specific pathogen-free conditions. All experiments were conducted using age- and sex-matched 6- to 8-week-old male and female mice. The experimental protocols were reviewed and approved by the animal Care and Use Committee in Shenzhen University and were in accordance with the guidelines and regulations of the institution.
Generation of asthma mouse model
Mouse asthma model was constructed as previously described and also illustrated in Figure 1A (31). Briefly, mice were sensitized by intraperitoneal injection with 50 µg of house dust mite extract (HDM, Greer Laboratories, Lenoir, NC) supplemented with 2 mg of adjuvant aluminum hydroxide (Al(OH)3) (Thermo Fisher Scientific, Cleveland, OH, USA) in 200 µl PBS at day 1, 3 and 7. During the sensitization, mice were also administered intranasally every day with or without 10 µg of vanadium pentoxide (V2O5, the most common form of vanadium; Sigma-Aldrich, Inc., St. Louis, Mo, USA) per mouse in 20 µl of PBS. The mice were challenged intranasally with 50 µg of HDM per mouse in 20 µl of PBS from day 15 to 21 with or without 10 µg of V2O5 per mouse. All mice were sacrificed 24 h after the last challenge. In some cases, the mice were pre-treated with 100 µg/kg cholecalciferol (vitamin D3, Sigma-Aldrich, St. Louis, MO) dissolved in coconut oil (Sigma-Aldrich, St. Louis, MO) via oral administration 1 h before every single challenge. PBS-treated age- and gender-matched mice were taken as control.
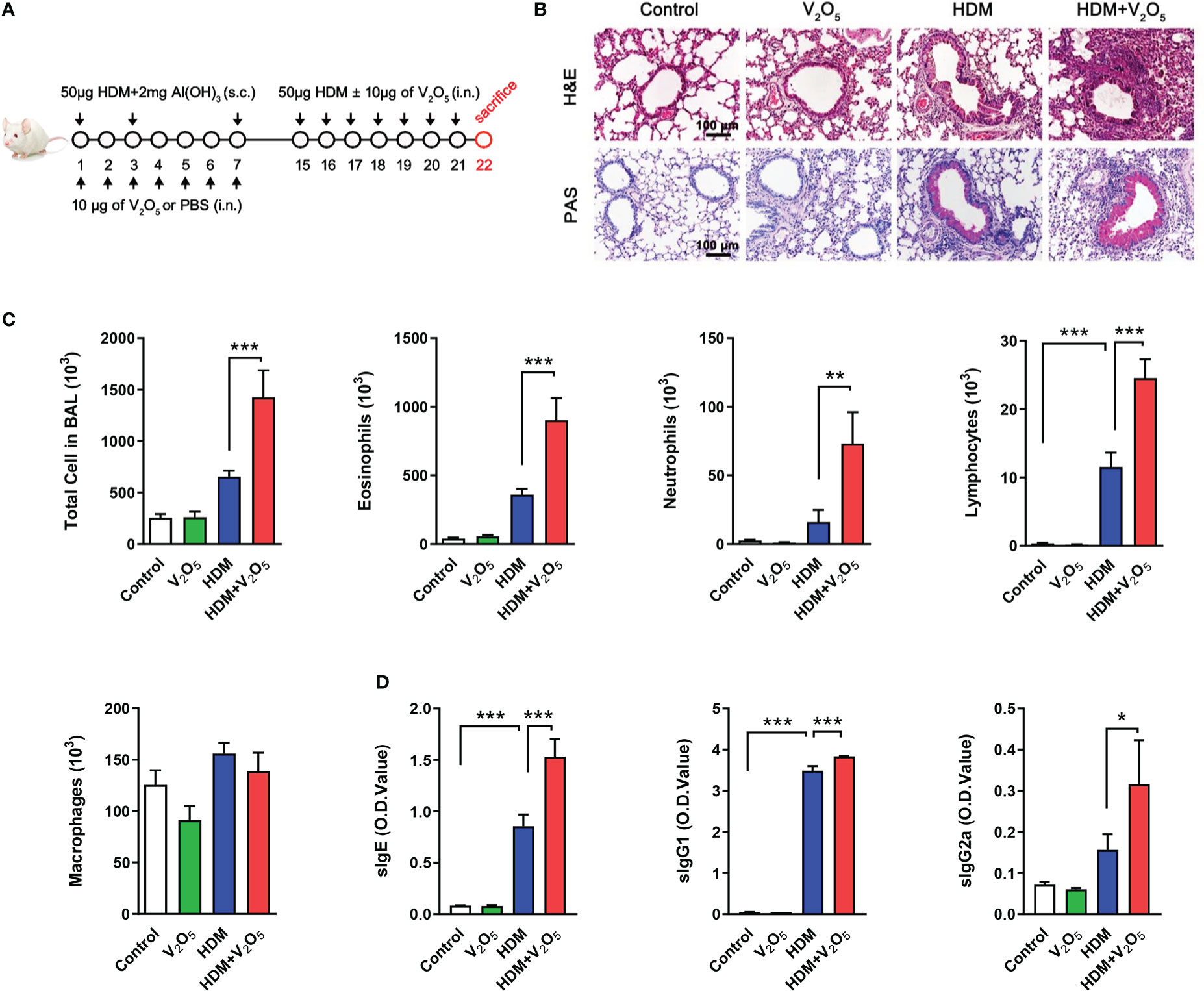
Figure 1 V2O5 co-exposure exacerbates HDM-induced allergic lung inflammation. (A), Protocol for the generation of mouse model of asthma. Mice were sensitized by intraperitoneal injection with 50 µg of house dust mite extract (HDM) supplemented with 2 mg of Al(OH)3 in 200 µl PBS at day 1, 3 and 7. During the sensitization, mice were also administered intranasally every day with or without 10 µg of V2O5 per mouse in 20 µl of PBS. The mice were challenged intranasally with 50 µg of HDM per mouse in 20 µl of PBS from day 15 to 21 with or without 10 µg of V2O5 per mouse. All mice were sacrificed 24 h after the last challenge. (B), H&E and periodic acid-Schiff (PAS) staining of lung tissue. (C), Cell differential analysis of Bronchoalveolar lavage (BAL) fluid from mice. (D), ELISAs for levels of HDM-specific IgE, IgG1 and IgG2a in serum. n=7-10/group. Data represent mean ± SEM of two independent experiments. *P < 0.05, ** P < 0.01, *** P < 0.001.
Broncheoalveolar lavage fluids
Broncheoalveolar lavage fluids (BALFs) was collected by instillation of 0.8 ml of PBS through a tracheal cannula. BALF samples were centrifuged at 1500 rpm for 5 min at 4°C. Supernatants were collected and stored for cytokine measurements. The number of total cells, eosinophils, neutrophils, lymphocytes and macrophages was determined by flow cytometry as previously described (32). Briefly, cells were blocked with CD16/32 antibody for 15 min, then incubated for 30 min with antibodies as following: anti-Siglet F-PE (S17007L, BioLegend), anti-Mac-3- FITC (M3/84, BioLegend), anti-Gr-1- APC (RB6-8C5, BioLegend), anti-CD3ϵ PerCP-Cyanine5.5 (145-2C11, BioLegend). Cells were analyzed on a FACSCalibur cytometer (BD Biosystems). SSChighSiglec-F+Mac-3- and SSChighSiglec-F+Mac-3+ cells were identified as eosinophils and alveolar macrophages, respectively. FSChighSSChigh Gr-1+cells were recognized as neutrophils. FSClowSSClowCD3+ cells were identified as lymphocytes.
Histological analysis
Lung tissues were fixed with 4% of formalin for 24 h. After dehydration, lung tissues were embedded in paraffin, and then cut into 4 µm sections. To evaluate lung inflammation, sections were deparaffinized and dehydrated, and then subjected to H&E and periodic acid–Schiff (PAS) staining. Masson trichrome staining was performed with a kit (Electron Microscopy Sciences, Halfield, PA, USA) under the manufacturer’s instructions.
ELISA
Concentrations of cytokines (IL-4, IL-5, IL-13, IL-17A, IFN-ƴ, TSLP, IL-25 and IL-33) in cell-free BAL were measured by enzyme-linked immunosorbent assay (ELISA) under the manual instructions of ELISA kits (4A Biotech Co., Ltd., Beijing, China) and (Thermo Fisher Scientific, Cleveland, OH, USA). Serum HDM-specific IgE, IgG1 and IgG2a (sIgE, sIgG1 and sIgG2a) were detected by ELISA as previously described (13). Serum was diluted at 1:4 for IgE, 1:800 for IgG1, and 1:40 for IgG2a.
Cell culture
Human bronchial epithelial cells (HBECs) were cultured in medium which contains a 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F-12 (DMEM/F-12; Gibco, Invitrogen corporation, Beijing, China) supplemented with 10% fetal bovine serum (FBS) (Gibco, Invitrogen corporation, Beijing, China) and 1% penicillin-streptomycin (Gibco, Invitrogen corporation, Beijing, China). The cells were maintained at 37°C with a humidified atmosphere containing 5% CO2.
CCK-8 assay
HBE cells were seeded into 96-well plate (5x103/well) and cultured overnight. Then cell viability was measured using Cell Counting Kit-8 (CCK-8; Kumamoto, Japan) according to the protocol. Briefly, cells were treated with different doses of V2O5 with/without HDM (50 µg/ml) for 24 h. Discard the medium and wash the cells with PBS twice, and then add CCK-8 work solution (diluted with medium, 100 µl/well) to each well of the plate. After incubation at 37°C for 2 h, absorbance at 450 nm was measured with a microplate reader (Thermo Fisher Scientific, Cleveland, OH, USA).
ROS measurement
ROS in the lung tissues and HBECs were measured by using flow cytometry and immunostaining, respectively. For ROS in lung tissues, lung tissues were minced and then ground slightly. Single-cell suspensions were obtained after filtration with 40 µm of cell strainers (Millipore). Single cells were stained with 5 µM of CM-H2DCFDA (Thermo Fisher Scientific) at 37°C for 30 min, and then the ROS generation was measured by flow cytometry. For detection of intracellular superoxide in lung sections, frozen sections were incubated with 5 µM of dihydroethidium (DHE, Thermo Fisher Scientific) 37°C for 30 min, and then fluorescent signal was observed under microscope (Nikon, Chiyoda, Japan). For ROS in HBECs, these treated HBECs were incubated with 5 µM of CM-H2DCFDA or 5 µM of mitoSOX (Thermo Fisher Scientific) at 37°C for 20 min. Total and mitochondrial ROS production were analyzed under microscope.
Immunofluorescence staining
After de-paraffin, dehydration and antigen retrieval, the slides were blocked with blocking buffer at room temperature for 1h. Then tissues were incubated with primary antibodies against α-SMA (Sigma-Aldrich, St. Louis, MO) or vimentin (Cell Signaling Technology, Beverly, MA, USA) at 4°C overnight. After three washes with TBST, secondary antibody incubation was followed at room temperature for 1h. The nuclei were counterstained with 4′,6-Diamidino-2-phenylindole dihydrochloride at room temperature for 10 minutes. The sections were mounted with fluoromount aqueous mounting medium (Sigma-Aldrich, St. Louis, MO) and imaged under microscope (Nikon, Japan). To determine the fluorescent signal, four different high-power fields from each slide were quantified with Fiji (National Institutes of Health) and presented as mean fluorescence intensity per square micrometer.
Statistical analysis
Statistical significance among experimental groups were analyzed by One-way ANOVA with Student-Newman-Keuls test using GraphPad Prism version 5.1 software (GraphPad Software, San Diego, CA, USA). Data were represented as mean ± SEM. A p-value <0.05 were considered statistically significant for all analyses.
Results
V2O5 co-exposure aggravates HDM-induced airway inflammation
To explore whether vanadium co-exposure can aggravate allergic lung inflammation, we constructed a mouse model of asthma with either HDM or V2O5 or co-exposure following the protocol as illustrated in Figure 1A. Compared to HDM alone, histological analysis demonstrated that mice treated with both HDM and V2O5 co-exposure showed increased airway inflammation as characterized by dense peri-bronchial inflammatory cell infiltrates (H&E, upper panel) and goblet cell hyperplasia (PAS, lower panel) (Figure 1B). The increased airway inflammation caused by V2O5 co-exposure, but not V2O5 alone, was further supported by the increased inflammatory cells in BAL samples, including the number of total cells, eosinophils, neutrophils, lymphocytes, and macrophages (Figure 1C). Furthermore, serum levels of house dust mite specific IgE (sIgE), sIgG1, and sIgG2a were also increased in V2O5 co-exposure treated mice as compared to those treated with HDM alone (Figure 1D). The results suggest that V2O5 co-exposure can aggravate HDM-induced airway inflammation.
V2O5 co-exposure aggravates HDM-induced epithelial cytokine release
Next, we investigated whether vanadium co-exposure can aggravate HDM-induced cytokine release. As compared to HDM alone, V2O5 co-exposure showed significantly increased levels of IL-4, IL-13, and IFNγ in BALFs. No significant increase was noted for IL-5 and IL-17 (Figure 2A). Furthermore, we specifically investigated whether V2O5 co-exposure can aggravate HDM-induced the release of epithelial-derived cytokines. As expected, higher levels of IL-25 and TSLP were observed in BAL samples of V2O5 co-exposure-treated mice as compared to those treated with HDM alone (Figure 2B). While there is no difference in the level of IL-33 (Figure 2B). To further confirm whether V2O5 co-exposure can aggravate HDM-induced epithelial-derived cytokine release, we performed in vitro analyses by treating HBECs with either V2O5 or HDM or in combination. Cell viability was firstly examined by CCK-8 assay in HBECs treated with HDM (50 μg/ml) or HDM with different doses of V2O5 (0.125, 0.25, 0.5 and 1 µg/ml) for 24 h. As shown in Figure 2C, no significant effect on cell viability was noted for HDM-treated HBECs and in HBECs treated with HDM (50 μg/ml) and V2O5 at a dose from 0.125 to 0.5 µg/ml. A modest reduction in cell viability was observed for V2O5 co-exposure at a dose of 1 µg/ml. HBECs were then treated with either HDM (50 μg/ml) or V2O5 (0.5 µg/ml) for 24 h and pro-inflammatory cytokines and chemokines were measured in supernatants by ELISA. Compared to HDM, V2O5 co-exposure led to increased levels of IL-25, TSLP, IL-8, CCL2 and CCL20 in supernatants of HBECs (Figure 2D). Taken together, both in vivo and in vitro analyses demonstrate that vanadium co-exposure can aggravate cytokine release, particularly those epithelial-derived cytokines and chemokines.
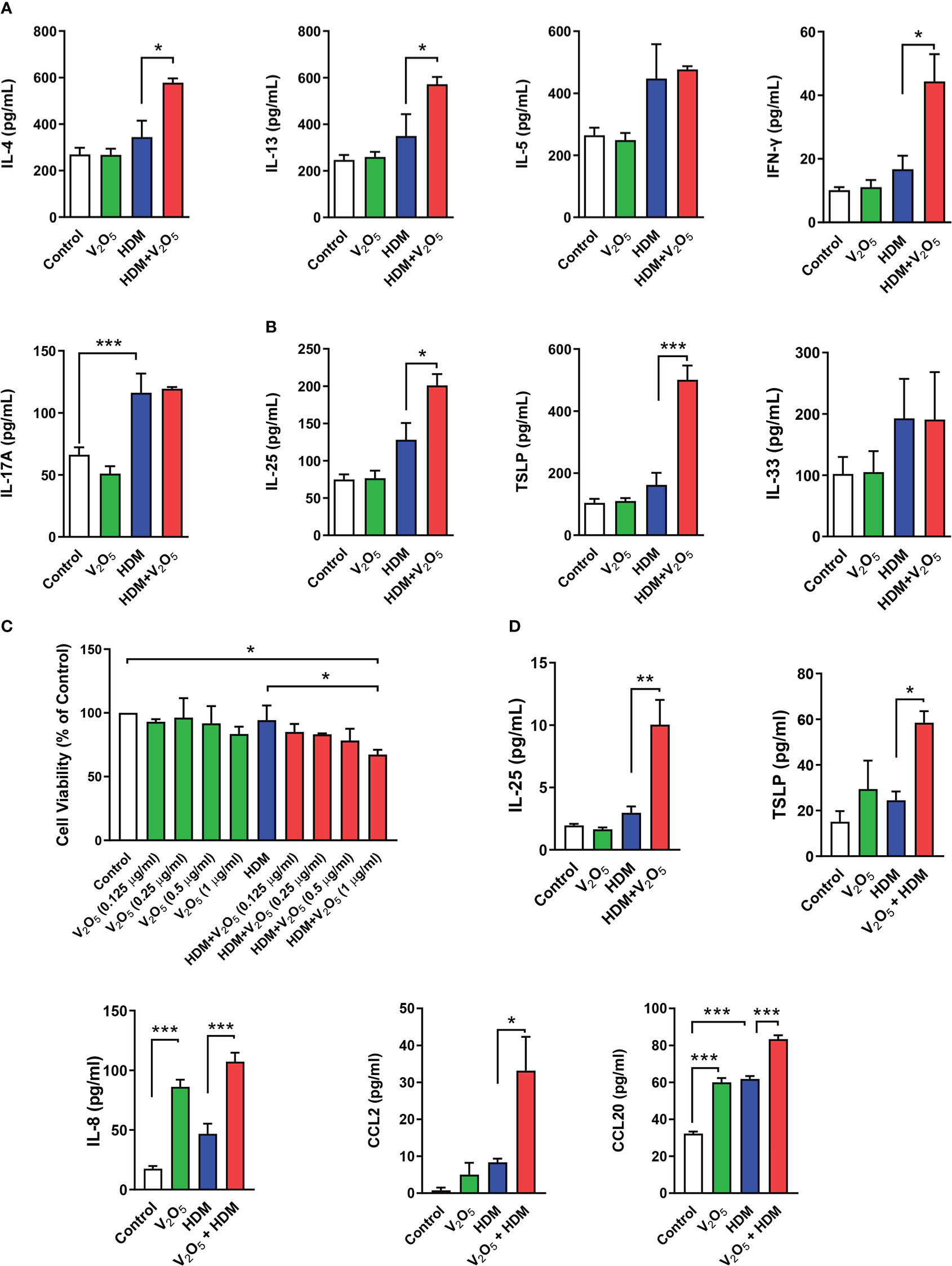
Figure 2 V2O5 co-exposure aggravates HDM-induced epithelial cytokine release. (A), ELISAs for levels of Th1, Th2, and Th17 cytokines in BAL samples. (B), ELISAs for levels of epithelial-derived cytokines in BAL samples. (C), CCK-8 assay in HBECs treated with HDM (50 μg/ml) or HDM with different doses of V2O5 (0.125, 0.25, 0.5 and 1 µg/ml) for 24 h. (D), ELISAs for levels of epithelial-derived cytokines and chemokines in supernatants of HBECs treated with either V2O5 (0.5 µg/ml) or HDM (50 μg/ml) alone or in combination for 24h. A-B: 7-10/group, C-D: three independent experiments. Data represent mean ± SEM. *P < 0.05, ** P < 0.01, *** P < 0.001.
V2O5 co-exposure aggravates HDM-induced airway remodeling
Studies have suggested that V2O5 exposure at 200 µg/animal can lead to the development of airway fibrosis and remodeling in a rat model (19). Thus, we examined whether V2O5 exposure can aggravate the HDM-induced airway fibrosis and remodeling. Masson trichrome staining was first performed in the lung tissues. Mice with V2O5 (10µg/mouse) co-exposure showed dramatic increase in airway thickening and collagen deposition/fibrosis as compared to those treated with either HDM or V2O5 alone (Figures 3A, B). The increased airway fibrosis was further confirmed by means of immunofluorescence staining with α-SMA and vimentin. Mice with V2O5 co-exposure showed a significant increase in the expression of both α-SMA (Figures 3C, D) and vimentin (Figures 3E, F) in the airways. Taken together, these data suggest that V2O5 co-exposure can aggravate HDM-induced airway fibrosis/remodeling.
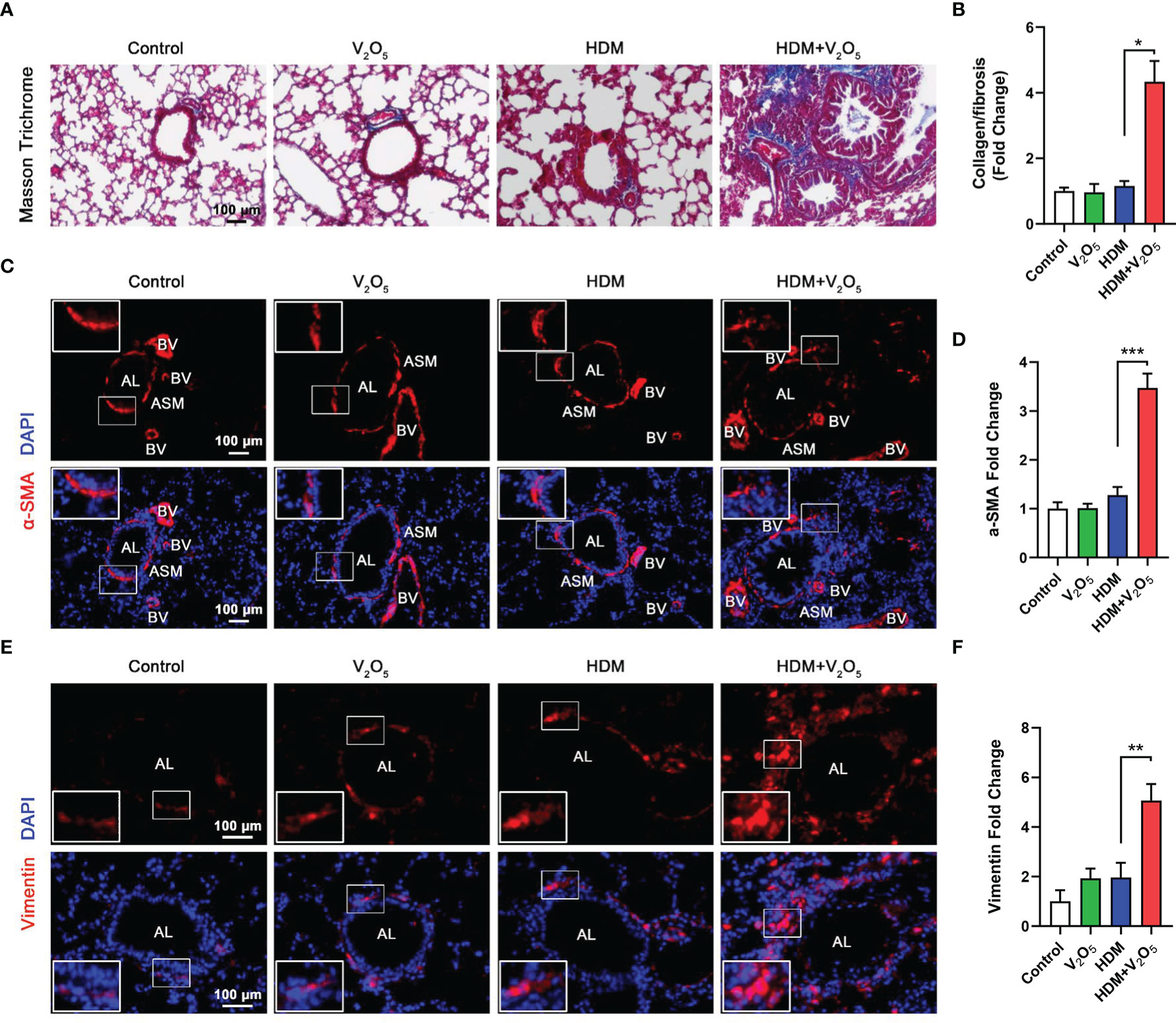
Figure 3 V2O5 co-exposure aggravates HDM-induced airway remodeling. (A), Masson trichrome staining in the lung tissues. (B), quantification for Masson trichrome staining in (A). (C), Immunofluorescence staining with α-SMA in the lung tissues of mouse model. (D), Quantitative analysis of florescent signals in (C), (E), Immunofluorescence staining with vimentin in the lung tissues of mouse model. (F), Quantitative analysis of florescent signals in (E). AL, airway lumen; ASM, airway smooth muscle; BV, bronchial vein. n=6-8/group. Data represent mean ± SEM of three independent experiments. *P < 0.05, ** P < 0.01, *** P < 0.001.
V2O5 co-exposure potentiates HDM-induced ROS generation
ROS have been considered as important mediators contributing to oxidative damage and chronic inflammation in allergic diseases (32, 33). To explore the underlying mechanism by which V2O5 co-exposure aggravates HDM-induced airway inflammation and remodeling, we investigated whether V2O5 co-exposure could enhance the HDM-induced ROS generation. Firstly, single cells were prepared from lung tissues of asthma mouse model at the end of the treatment/challenge protocol and the relative level of ROS production was evaluated by flow cytometry with CM-H2DCFDA. Compared to control, significantly higher level of ROS was found in the HDM group, which was further augmented by V2O5 co-exposure (Figure 4A). The enhanced ROS generation by V2O5 co-exposure was further supported by the in vivo analyses of ROS production in the lung tissues by dihydrothidium (DHE) immunostaining (Figure 4B). Lung tissues from V2O5 co-exposure treated mice had a significant increase in ROS levels as compared to those from HDM-treated mice. These findings were further confirmed by in vitro analysis in HBECs. Compared to untreated HBECs, treatment with either HDM or V2O5 at different doses induced ROS generation as determined by immunostaining with CM-H2DCFDA (Figure 4C). Notably, the increased ROS were further potentiated by V2O5 co-exposure with HDM. The same pattern was also observed for mitochondrial ROS, one of the major sources of ROS (33), in those treated and un-treated HBECs (Figure 4D). Compared to HDM or V2O5 treatment, V2O5 co-exposure induced increased production of mitochondrial ROS as assessed by immunostaining with MitoSOX, a fluorescent mitochondrial ROS reporter dye. Collectively, our findings support the notion that V2O5 co-exposure potentiates HDM-induced ROS generation, particularly mitochondrial ROS production.
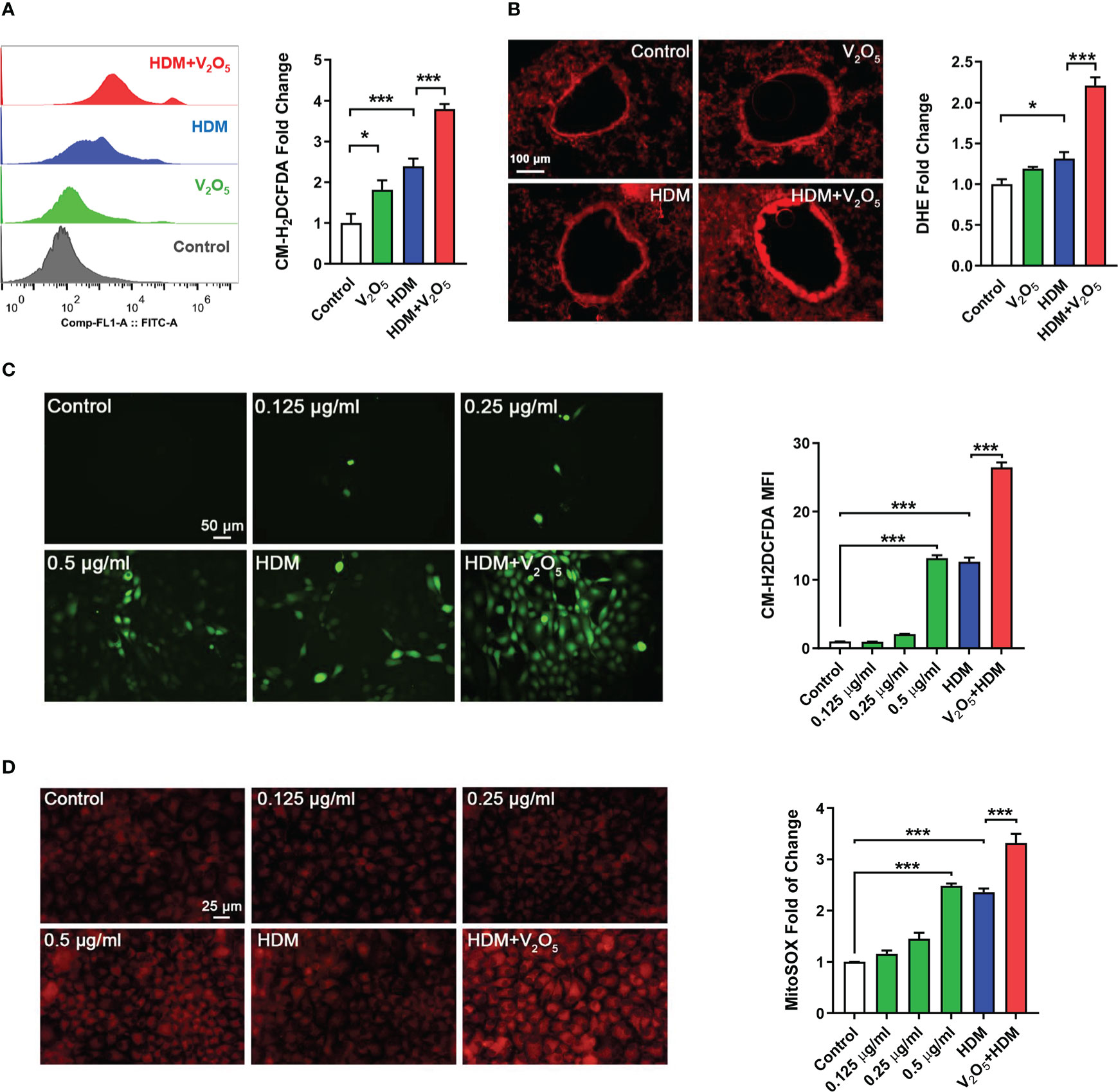
Figure 4 V2O5 co-exposure potentiates HDM-induced ROS generation. (A), ROS production in single cell from lung tissues of asthma mouse model as detected by flow cytometry analysis with CM-H2DCFDA and quantified as fold changes. (B), Representative immunofluorescence images of ROS expression in the lung tissues with dihydrothidium (DHE) immunostaining. (C, D), Representative immunofluorescence images of ROS expression with CM-H2DCFDA (C) or MitoSOX (D) in HBECs exposed to either V2O5 (0.5 µg/ml) or HDM (50 μg/ml) alone or in combination for 24h. A-B: 5-8/group, C-E: Three independent experiments. Data represent mean ± SEM. *P < 0.05, *** P < 0.001.
ROS inhibition alleviates the V2O5 co-exposure induced cytokine release from epithelial cells
To examine the role of ROS in V2O5 co-exposure-induced airway inflammation, we specifically investigated whether ROS play a role in V2O5 co-exposure induced cytokine release from epithelial cells. HBECs were pre-treated with NAC, a widely used ROS inhibitor, and then treated with either V2O5 or HDM or co-exposure. Supernatants were harvested after 24 h and cytokines and chemokines in supernatants were measured. Similar to the previous findings that V2O5 co-exposure significantly upregulated HDM-induced IL-25, TSLP, IL-8, CCL2, and CCL20 production (Figure 5). In contrast, pre-treatment with NAC almost completely blocked the V2O5 co-exposure-induced significant increase in those cytokines and chemokines. The same pattern was also seen when HBECs were pre-treated with mitoTEMPO, a mitochondria-targeted antioxidant (34). These results implicate that vanadium co-exposure induces epithelial cytokine and chemokine release through ROS generation.
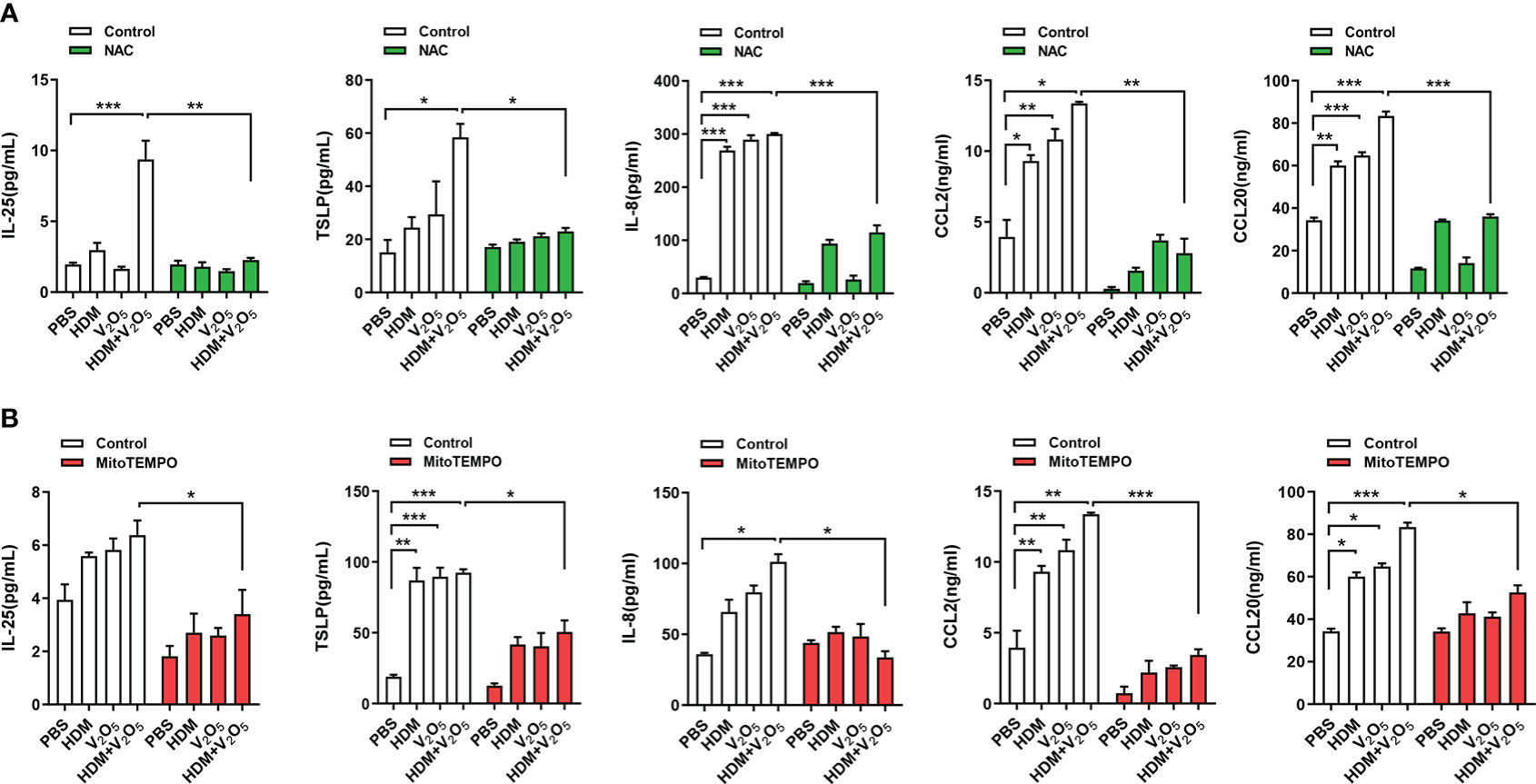
Figure 5 ROS inhibition alleviates the V2O5 co-exposure induced epithelial cytokine and chemokine release. (A), ELISAs for levels of cytokine and chemokines in supernatants of ROS inhibitor NAC pre-treated HBECs treated with V2O5 (0.5 µg/ml) or HDM (50 μg/ml) alone or in combination for 24h. (B), ELISAs for levels of cytokine and chemokines in supernatants of mitochondria-targeted antioxidant mitoTEMPO pre-treated HBECs treated with V2O5 (0.5 µg/ml) or HDM (50 μg/ml) alone or in combination for 24h. Data represent mean ± SEM of three independent experiments. *P < 0.05, ** P < 0.01, *** P < 0.001.
Vitamin D3 as an antioxidant alleviates vanadium co-exposure-induced airway inflammation
Vitamin D3 (VD3) supplementation has been shown to protect against oxidative stress processes in the OVA-induced airway inflammation (35). Thus, we hypothesized that VD3 could inhibit the V2O5 co-exposure-induced ROS generation and subsequent airway inflammation. Vitamin D3 (VD3, 100 μg/kg) was used to treat mice before every single challenge followed the protocol as illustrated in Figure 6A. To confirm whether pre-treatment with VD3 as an antioxidant can inhibit V2O5 co-exposure-induced ROS generation, we detected ROS generation in single cell suspension from lung tissues of asthma mouse model by flow cytometry analysis (Figure 6B) and in lung tissues by immunofluorescence staining (Figure 6C). As expected, both flow cytometry analysis and immunofluorescence staining consistently showed increased ROS generation in V2O5 co-exposured mice but its level was significantly reduced when those mice were pre-treated with VD3. H&E and PAS staining demonstrated that VD3 treatment reduced V2O5 co-exposure induced lung inflammation and mucus production (Figure 6D). Significant reductions were noted for the numbers of total inflammatory cells and eosinophils in BAL samples (Figure 6E). The same pattern was also noted for serum levels of HDM sIgE and sIgG2a (Figure 6F). Similarly, mice pretreated with VD3 showed clear reduction for IL-4 in BAL samples as compared to V2O5 co-exposure-treated mice (Figure 6G). In contrast, significant reductions were observed for TSLP, and IL-25 in BAL samples of VD3-pre-treated mice compared with V2O5 co-exposure-treated mice (Figure 6H). These preliminary findings suggest that VD3 can, at least partially, alleviate vanadium co-exposure-induced cytokine release and airway inflammation.
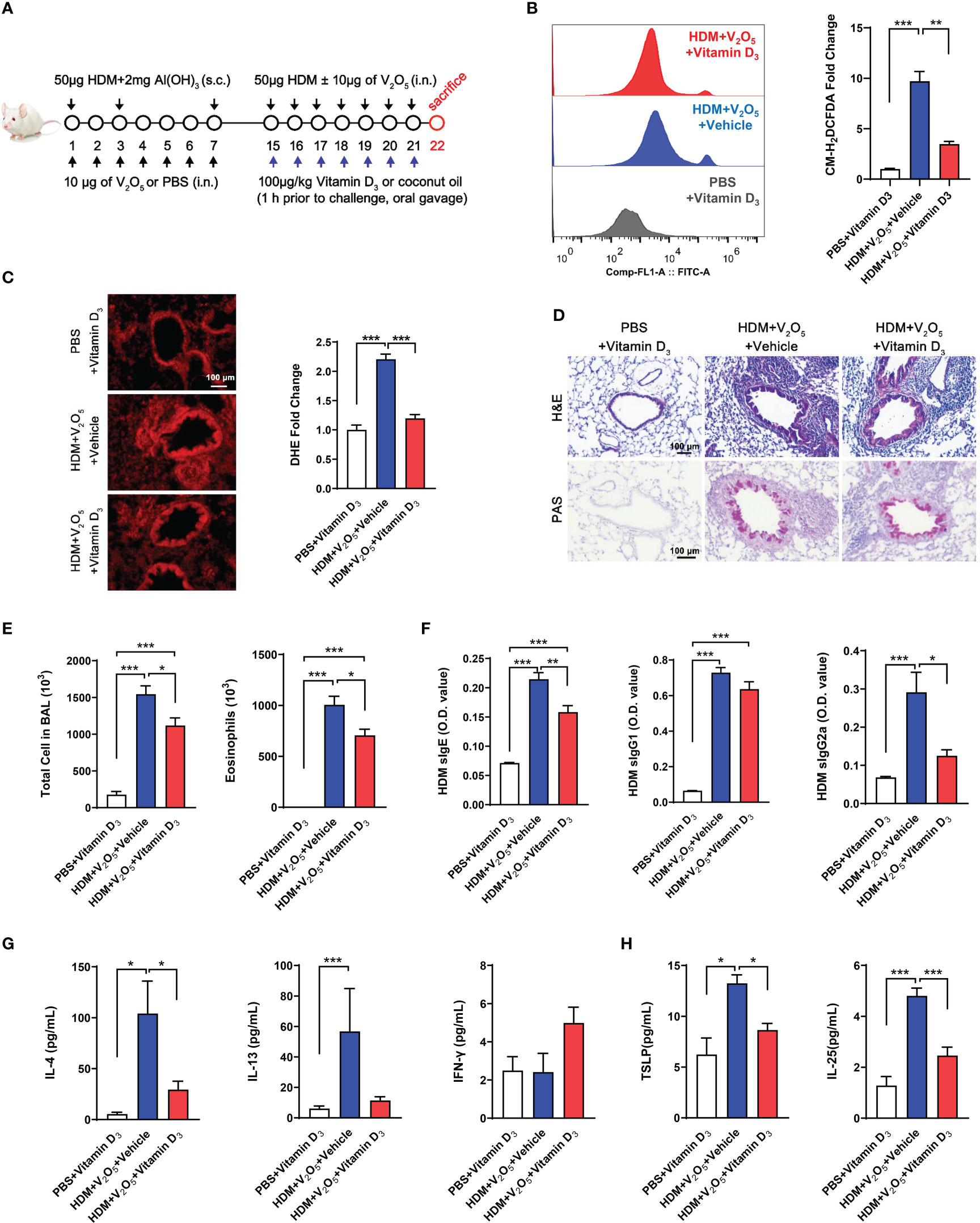
Figure 6 Vitamin D3 alleviates vanadium co-exposure-induced airway inflammation. (A), Protocol for the generation of mouse model of asthma. (B), ROS production in single cell from lung tissues of asthma mouse model as detected by flow cytometry analysis with CM-H2DCFDA and quantified as fold changes. (C), Representative immunofluorescence images of ROS expression in the lung tissues with dihydrothidium (DHE) immunostaining. (D), H&E and PAS staining of lung tissue. (E), Cell differential analysis of Bronchoalveolar lavage (BAL) fluid from mice. (F), ELISAs for levels of HDM-specific IgE, IgG1 and IgG2a in serum. (G), ELISAs for levels of Th1 and Th2 cytokines in BAL samples. (H), ELISAs for levels of epithelial-derived cytokines in BAL samples. n=6-11/group. Data represent mean ± SEM of two independent experiments. *P < 0.05, ** P < 0.01, *** P < 0.001.
Antioxidant vitamin D3 inhibits vanadium co-exposure-induced airway remodeling
Next, we investigated whether pre-treatment with VD3 could inhibit V2O5 co-exposure-induced airway fibrosis/remodeling. Masson trichrome staining was performed in the lung tissues. Mice pretreated with VD3 showed a significant inhibition of airway thickening and collagen deposition/fibrosis caused by V2O5 co-exposure (Figures 7A, B). The reduced airway fibrosis was further confirmed by immunofluorescence staining with α-SMA and vimentin. Expression of both α-SMA (Figures 7C, D) and vimentin (Figures 7E, F) was reduced in the airways of VD3 pre-treated mice as compared to those treated with V2O5 co-exposure. Collectively, these data suggest that VD can inhibit vanadium co-exposure-induced airway fibrosis/remodeling.
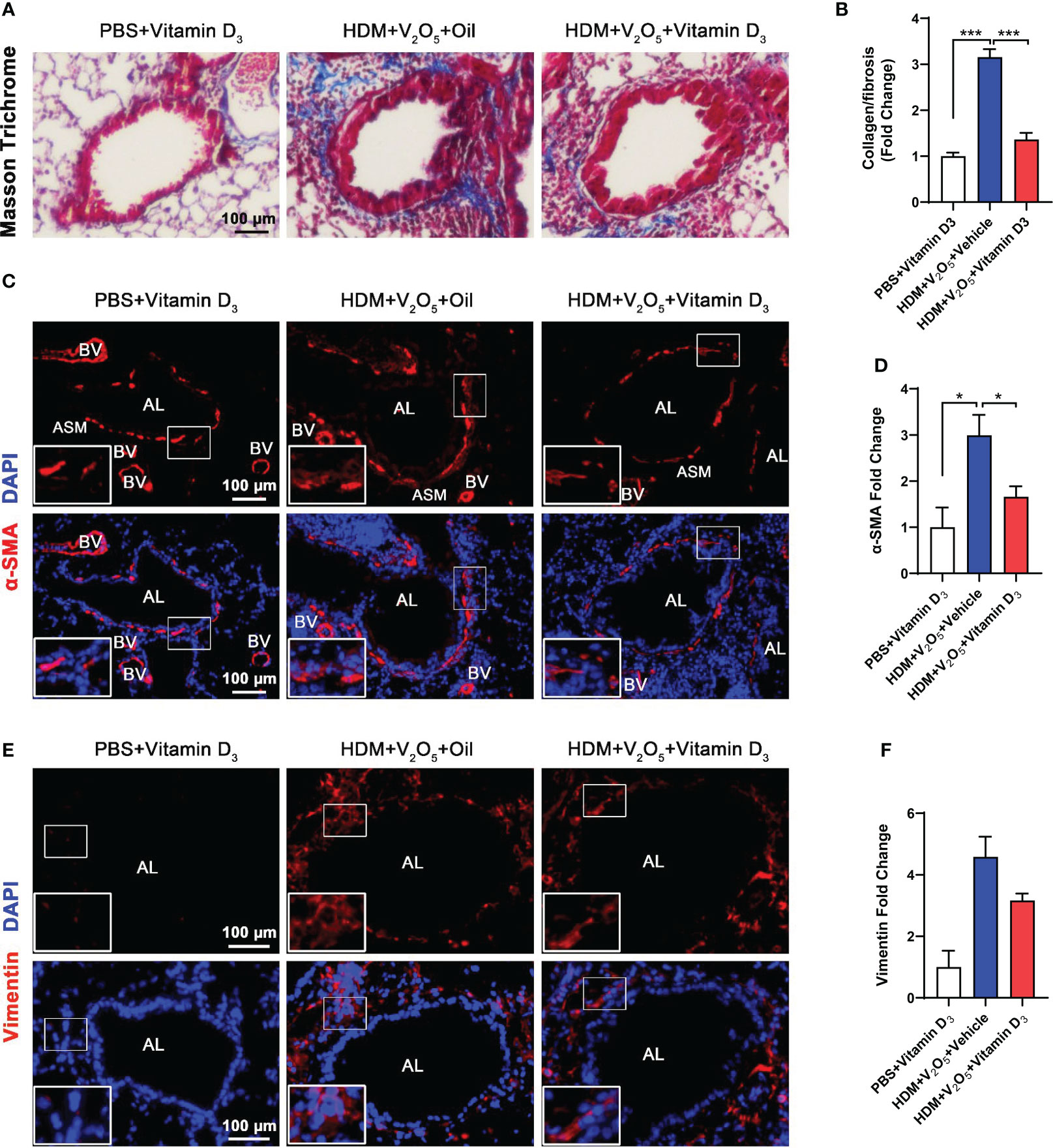
Figure 7 Antioxidant vitamin D3 inhibits vanadium co-exposure-induced airway remodeling. (A), Representative immunofluorescence images of airway fibrosis with Masson trichrome staining. (B), Quantification for Masson trichrome staining in (A). (C), Representative immunofluorescence images of α-SMA expression in the lung tissues of mouse model. (D), Quantitative analysis of florescent signals in (C), (E), Representative immunofluorescence images of vimentin expression in the lung tissues of mouse model. (F), Quantitative analysis of florescent signals in (E). n=6-8/group. Data represent mean ± SEM of two independent experiments. *P < 0.05, < 0.01, *** P < 0.001.
Discussion
Evidence is provided here in support of the hypothesis that environmental pollutants coexisted with allergens has the potential to exacerbate airway inflammation and remodeling. In particular, we found that V2O5 co-exposure enhanced HDM-induced allergic lung inflammation and airway remodeling by in a well-established mouse model of asthma (13, 14). Both in vivo and in vitro analyses demonstrated that V2O5 co-exposure amplified HDM-induced epithelial-derived cytokine release. To explore the underlying molecular mechanisms, we found that the V2O5 co-exposure-aggravated oxidative stress is essential in the V2O5 co-exposure-induced epithelial cytokine release. Importantly, inhibition of ROS with ROS inhibitors and antioxidant VD3 attenuated the V2O5 co-exposure-induced airway inflammation and remodeling. This study suggests, therefore that ROS play a central role in vanadium co-exposure evoked HDM-induced epithelial cell cytokine and chemokine release contributing to the increased airway inflammation and remodeling.
Recent studies suggest that environmental pollutants co-exposure with allergens may contribute to the increased incidence of asthma (3, 4). Interestingly, DEP alone had no clear effects on the major phenotypes of allergic asthma (36). However, increased allergic responses were found when DEPs were co-exposed with HDM, including airway hyperresponsiveness, upregulated levels of HDM-specific IgE, and recruitment of eosinophils and Th2/Th17 cells (36–38). These findings were supported by our previous studies that benzo(a)pyrene (BaP), a common indoor air pollutant, co-exposure significantly exacerbated Der f 1-induced airway resistance, airway inflammation, and secretion of pro-inflammatory cytokines (13, 14). Vanadium, one of the major components of environmental PM2.5, can be found in fresh water, ground water and potable water (<10 nM) (39). The average concentrations of vanadium in the air ranges from 0.1 ng/m3 (eastern Pacific) to 0.72 ng/m3 (rural northwest Canada) (40). However, industrial production and burning of fuel oil in rural and urban air led to an increased concentration of vanadium in rural and urban air up to 75 ng/m3 and 1000 ng/m3, respectively (39, 40). Vanadium exposure has been shown to cause occupational bronchial asthma and bronchitis (7, 8), and has been significantly associated with severe respiratory symptoms of children (9) and higher levels of exhaled FeNO (10, 11). Moreover, our recent hospital-based case-control study also revealed that increased vanadium exposure posed a significant risk for current asthma, correlating with the levels of oxidative stress marker, circulating IL-8, and the accumulated levels of ambient PM2.5, providing further environmental and epidemiological evidence (12).
In this study, we found that relatively short-term vanadium exposure alone, similar to DEP (36), had no effect on the major phenotypes of airway inflammation. However, significant exacerbation of allergic airway inflammation was found for vanadium co-exposure with HDM as assessed by inflammatory infiltrates, goblet cell hyperplasia, inflammatory cells in BAL samples, and serum levels of house dust mite specific IgE (sIgE), sIgG1, and sIgG2a. These findings suggest that vanadium co-exposure could aggravate HDM-induced airway inflammation.
Production and release of epithelial-derived cytokines have been considered as one of the early trigger signals to link epithelial injury to downstream inflammatory responses (41). Thus, in addition to Th1, Th2, and Th17 cytokines, we specifically examined epithelial-derived cytokines IL-25, TSLP, and IL-33. As expected, V2O5 co-exposure induced higher levels of IL-25 and TSLP in BAL samples when compared with those treated with HDM alone. Notably, no difference was observed for IL-33, which may suggest distinct mechanisms for the vanadium co-exposure-promoted epithelial cytokine release. This finding was further supported by in vitro analysis. V2O5 co-exposure significantly enhanced the release of epithelial IL-25, TSLP, IL-8, CCL2 and CCL20 as compared to those treated with HDM alone. These results were also consistent with our previous report that BaP co-exposure can potentiate HDM-induced cytokine release from airway epithelial cells (13). These findings raise the possibility that the increased epithelial cytokine release caused by vanadium co-exposure may contribute to the enhanced Th2-associated airway inflammation in asthma.
Further, we explored the possible mechanisms regarding the vanadium co-exposure-induced cytokine release. ROS have been considered as central inflammatory mediators that contribute to oxidative damage, chronic inflammation in allergic diseases (32, 33, 42–44), and the severity of asthma (24). Our previous studies have also suggested that BaP co-exposure significantly induced HDM-induced lung inflammation via ROS-mediated release of proinflammatory cytokines (13). Thus, it is possible that vanadium co-exposure with HDM may exacerbate HDM-induced ROS generation, leading to epithelial cytokine release and subsequent airway inflammation. Indeed, exposure to vanadium has been shown to induce ROS generation that contribute to cell apoptosis, barrier dysfunction and subsequent airway inflammation (25, 45–47). Furthermore, in vitro analysis with also provide evidence that vanadium treatment can evoke oxidative stress and cellular senescence in human lung fibroblasts (26). In this study, we provided evidence that vanadium co-exposure potentiated HDM-induced ROS generation, particularly mitochondrial ROS production by both in vivo and in vitro analyses. To further determine the role of ROS in vanadium co-exposure-induced airway inflammation, we specifically investigated its role in vanadium co-exposure induced epithelial cytokine and chemokine release. Notably, pre-treatment with both NAC as a ROS inhibitor and mitoTEMPO as a mitochondria-targeted antioxidant (34) almost completely blocked the vanadium co-exposure-induced cytokine and chemokine release from human primary airway epithelial cells. These findings further support the notion that ROS play a critical role in oxidative damage and epithelial cytokine and chemokine release. Mechanistically, we have provided evidence that ROS regulate allergic airway inflammation and asthma through controlling either autophagy/mitophagy (48) or NLRP3 inflammasome activation (49). Thus, it is likely that the ROS-mediated autophagy/mitophagy or NLRP3 inflammasome plays an important role in vanadium co-exposure-induced allergic airway inflammation and asthma, which warrant a further investigation in the future.
Vitamin D3 has been reported to have a broad range of biological activities, including nutrition, anti-infective effect, and antioxidant stress function (35, 50, 51). VD3 supplementation is a relatively cheap, easy and non-invasive intervention in asthma management. VD3 supplementation has been associated with reduced incidence of asthma attacks (52) and severe exacerbation (53). Increased serum levels of VD3 have been shown to correlate with better pulmonary function and improved quality of life in pediatric asthmatics (54). Recent study has proved experimental evidence that VD3 supplementation protects against oxidative stress in the OVA-induced mouse model of asthma (35). In this study, we provided new evidence that pretreatment with vitamin D3 could, at least partially, inhibit V2O5 co-exposure-induced ROS production and airway inflammation. Particularly, both TSLP and IL-25 showed statistically significant reduction in vitamin D3 treated mice with V2O5 co-exposure. These findings implicate that VD3 supplementation could be used routinely to prevent lung tissue oxidative damage and inflammation caused by environmental pollutants co-exposure with allergens.
In addition to airway inflammation, we have also examined the effects of vanadium co-exposure on HDM-induced airway fibrosis/remodeling in these mouse model of asthma. Recent studies have changed our understanding of asthma as purely an inflammation and suggested that alterations within the structural cells of the airway (airway remodeling), together with abnormal immune responses, lead to the full manifestation of asthma (55–58). Airway remodeling is a gradual progression in symptoms and irreversible decline in lung function due to the structural changes in the airway walls (59–61). Studies have demonstrated that vanadium exposure can lead to the development of airway fibrosis and remodeling (19). Our study provided further supporting evidence that vanadium co-exposure could significantly potentiate HDM-induced airway remodeling, and importantly, pre-treatment with VD3 could reverse such effects. This offers a great promise to prevent/inhibit environmental pollutant-evoked allergen-induced fibrotic remodeling and development of asthma.
Taken together, we provide evidence that vanadium co-exposure can potentially potentiate and/or amplify HDM-induced allergic lung inflammation and airway remodeling. Mechanistically, we found that ROS function as a molecular link for vanadium co-exposure and epithelial cytokine release that contribute to the Th2-associated airway inflammation and airway remodeling (Figure 8). Of great relevance for the clinic, our finding reveals that ROS inhibitors and antioxidant VD3 can be used to prevent or treat environmental pollutant co-exposure promoted allergen-induced airway inflammation and remodeling. Our observations provide the basis for future study to investigate how ROS regulate airway inflammation and remodeling, and to further determine whether targeting ROS is an efficient strategy to prevent or treat environmental pollutant-mediated exacerbation of allergic airway inflammation and remodeling.
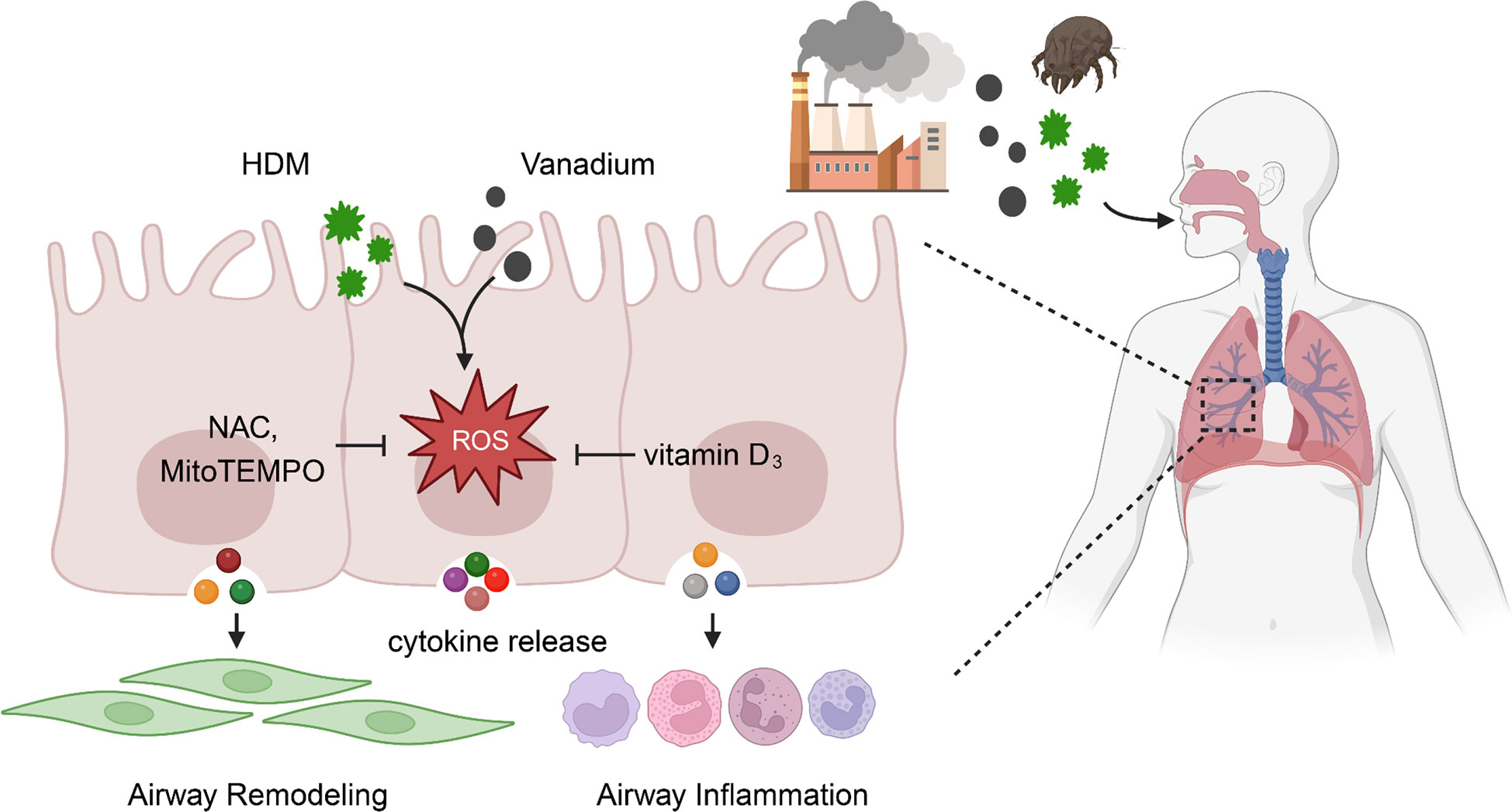
Figure 8 ROS functions as a molecular link for vanadium co-exposure with house dust mite (HDM) and epithelial cytokine release that contribute to airway inflammation and remodeling.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The experimental protocols were reviewed and approved by the animal Care and Use Committee in Shenzhen University and were in accordance with the guidelines and regulations of the institution.
Author contributions
WT, XX, JL, XL, RY, RW, YS, and MG performed experiments, analyzed data, and review the manuscript. WT and PG wrote the manuscript. S-KH and PG designed and supervised the study, and wrote the manuscript. EW, DX, and PY provided intellectual input and aided in the experimental design. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work was supported by grants from The National Natural Science Foundation (NO.31900667, 81929001, 82073950), Science and Technology Planning Project of Guangdong Province (2020A1515010029), International Cooperation Program of Shenzhen (NO.GJHZ20190822095605512, SGDX20201103095609027), Science and Technology Program of Shenzhen (No. JCYJ2019081217 1617278, JCYJ20200109143435556, JCYJ20220531102217038, JCYJ 20180306171550045), Science and Technology Program of Nanshan (LHTD20180007), State Key Laboratory of Respiratory Disease (SKLRD-Z-202111, SKLRD-OP-201910, SKLRD-Z-202216). This work was also supported by grants from the US National Institutes of Health (NIH) (1R01AI153331 and R01AI141642 to PG) and National Health Research Institutes (EO-111-PP-10), Academia Sinica (AS-SS-111-02), Taiwan, and Shenzhen Science and Technology Peacock Team Project (KQTD20170331145453160). These funding agencies had no role in study design, the collection and analysis of data, the decision to publish or the preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
HDM, House dust mite; ROS, Reactive oxygen species; HBEC, Human bronchial epithelial cell; BALF, Bronchoalveolar lavage fluid; NAC, N-acetyl-L-cysteine; V2O5, Vanadium pentoxide; VD3, Vitamin D3; BaP, Benzo(a)pyrene; PM2.5, Particulate matter 2.5; HEL, Nε-(hexanoyl)-lysine; Al(OH)3, Adjuvant aluminum hydroxide; DMEM/F-12, Dulbecco’s modified Eagle’s medium and Ham’s F-12; FBS, Fetal bovine serum; CCK-8, Cell Counting Kit-8; DEP, Diesel exhaust particles.
References
1. Woollen L, Paudyal P. Practitioner-based complementary and alternative therapies in the management of asthma in children and adolescents: A systematic review and meta-analysis. Lancet (2015) 386:S80. doi: 10.1016/S0140-6736(15)00918-6
2. To T, Stanojevic S, Moores G, Gershon AS, Bateman ED, Cruz AA, et al. Global asthma prevalence in adults: findings from the cross-sectional world health survey. BMC Public Health (2012) 12:204. doi: 10.1186/1471-2458-12-204
3. Nurmatov U, Tagieva N, Semple S, Devereux G, Sheikh A. Volatile organic compounds and risk of asthma and allergy: A systematic review and meta-analysis of observational and interventional studies. Prim Care Respir J (2013) 22(1):PS9–15. doi: 10.4104/pcrj.2013.00010
4. Lee JH, Lee HS, Park MR, Lee SW, Kim EH, Cho JB, et al. Relationship between indoor air pollutant levels and residential environment in children with atopic dermatitis. Allergy Asthma Immunol Res (2014) 6(6):517–24. doi: 10.4168/aair.2014.6.6.517
5. Gavett SH, Haykal-Coates N, Copeland LB, Heinrich J, Gilmour MI. Metal composition of ambient PM2.5 influences severity of allergic airways disease in mice. Environ Health Perspect (2003) 111(12):1471–7. doi: 10.1289/ehp.6300
6. Jin L, Xie J, Wong CKC, Chan SKY, Abbaszade G, Schnelle-Kreis J, et al. Contributions of city-specific fine particulate matter (PM2.5) to differential in vitro oxidative stress and toxicity implications between Beijing and guangzhou of China. Environ Sci Technol (2019) 53(5):2881–91. doi: 10.1021/acs.est.9b00449
7. Musk AW, Tees JG. Asthma caused by occupational exposure to vanadium compounds. Med J Aust (1982) 1(4):183–4. doi: 10.5694/j.1326-5377.1982.tb132251.x
8. Levy BS, Hoffman L, Gottsegen S. Boilermakers' bronchitis. respiratory tract irritation associated with vanadium pentoxide exposure during oil-to-coal conversion of a power plant. J Occup Med (1984) 26(8):567–70. doi: 10.1097/00043764-198408000-00009
9. Patel MM, Hoepner L, Garfinkel R, Chillrud S, Reyes A, Quinn JW, et al. Ambient metals, elemental carbon, and wheeze and cough in new York city children through 24 months of age. Am J Respir Crit Care Med (2009) 180(11):1107–13. doi: 10.1164/rccm.200901-0122OC
10. Rosa MJ, Perzanowski MS, Divjan A, Chillrud SN, Hoepner L, Zhang H, et al. Association of recent exposure to ambient metals on fractional exhaled nitric oxide in 9-11 year old inner-city children. Nitric Oxide (2014) 40:60–6. doi: 10.1016/j.niox.2014.05.008
11. Godri Pollitt KJ, Maikawa CL, Wheeler AJ, Weichenthal S, Dobbin NA, Liu L, et al. Trace metal exposure is associated with increased exhaled nitric oxide in asthmatic children. Environ Health (2016) 15(1):94. doi: 10.1186/s12940-016-0173-5
12. Wu C-C, Wang C-C, Chung W-Y, Sheu C-C, Yang Y-H, Cheng M-Y, et al. Environmental risks and sphingolipid signatures in adult asthma and its phenotypic clusters: A multicentre study. Thorax (2022) 2022:thoraxjnl–2021-218396. doi: 10.1136/thoraxjnl-2021-218396
13. Wang E, Liu X, Tu W, Do DC, Yu H, Yang L, et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy (2019) 74(9):1675–90. doi: 10.1111/all.13784
14. Wang E, Tu W, Do DC, Xiao X, Bhatti SB, Yang L, et al. Benzo(a)pyrene enhanced dermatophagoides group 1 (Der f 1)-induced TGFbeta1 signaling activation through the aryl hydrocarbon receptor-RhoA axis in asthma. Front Immunol (2021) 12:643260. doi: 10.3389/fimmu.2021.643260
15. Lam HCY, Jarvis D, Fuertes E. Interactive effects of allergens and air pollution on respiratory health: A systematic review. Sci total Environ (2021) 757:143924. doi: 10.1016/j.scitotenv.2020.143924
16. Fortoul TI, Rodriguez-Lara V, Gonzalez-Villalva A, Rojas-Lemus M, Cano-Gutierrez G, Ustarroz-Cano M, et al. Vanadium inhalation in a mouse model for the understanding of air-suspended particle systemic repercussion. J BioMed Biotechnol (2011) 2011:951043. doi: 10.1155/2011/951043
17. Fortoul TI, Rodriguez-Lara V, González-Villalva A, Rojas-Lemus M, Cano-Gutiérrez G, Ustarroz-Cano M, et al. Inhalation of vanadium pentoxide and its toxic effects in a mouse model. Inorganica Chimica Acta (2014) 420:8–15. doi: 10.1016/j.ica.2014.03.027
18. Madrigal JM, Persky V, Jackson BP, Bain A, Siemer M, Pappalardo AA, et al. Assessment of metal concentrations and associations with pulmonary function among children with asthma in Chicago, Illinois. Int J Environ Res Public Health (2021) 18(14):1–12. doi: 10.3390/ijerph18147279
19. Bonner JC, Rice AB, Moomaw CR, Morgan DL. Airway fibrosis in rats induced by vanadium pentoxide. Am J Physiol Lung Cell Mol Physiol (2000) 278(1):L209–16. doi: 10.1152/ajplung.2000.278.1.L209
20. Jung KH, Torrone D, Lovinsky-Desir S, Perzanowski M, Bautista J, Jezioro JR, et al. Short-term exposure to PM2.5 and vanadium and changes in asthma gene DNA methylation and lung function decrements among urban children. Respir Res (2017) 18(1):63. doi: 10.1186/s12931-017-0550-9
21. Espinosa-Zurutuza M, Gonzalez-Villalva A, Albarran-Alonso JC, Colin-Barenque L, Bizarro-Nevares P, Rojas-Lemus M, et al. Oxidative stress as a mechanism involved in kidney damage after subchronic exposure to vanadium inhalation and oral sweetened beverages in a mouse model. Int J Toxicol (2018) 37(1):45–52. doi: 10.1177/1091581817745504
22. Yeh CC, Wu JY, Lee GL, Wen HT, Lin P, Kuo CC. Vanadium derivative exposure promotes functional alterations of VSMCs and consequent atherosclerosis via ROS/p38/NF-kappaB-mediated IL-6 production. Int J Mol Sci (2019) 20(24):1–19. doi: 10.3390/ijms20246115
23. Tlili M, Rouatbi S, Sriha B, Ben Rhouma K, Sakly M, Vaudry D, et al. Pituitary adenylate cyclase-activating polypeptide reverses ammonium metavanadate-induced airway hyperresponsiveness in rats. Oxid Med Cell Longev (2015) 2015:787561. doi: 10.1155/2015/787561
24. Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy (2012) 8(4):694–5. doi: 10.4161/auto.19315
25. Wang L, Medan D, Mercer R, Overmiller D, Leornard S, Castranova V, et al. Vanadium-induced apoptosis and pulmonary inflammation in mice: Role of reactive oxygen species. J Cell Physiol (2003) 195(1):99–107. doi: 10.1002/jcp.10232
26. He X, Jarrell ZR, Liang Y, Ryan Smith M, Orr ML, Marts L, et al. Vanadium pentoxide induced oxidative stress and cellular senescence in human lung fibroblasts. Redox Biol (2022) 55:102409. doi: 10.1016/j.redox.2022.102409
27. Wang M, Li J, Dong S, Cai X, Simaiti A, Yang X, et al. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part Fibre Toxicol (2020) 17(1):23. doi: 10.1186/s12989-020-00353-3
28. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res (2018) 19(1):230. doi: 10.1186/s12931-018-0931-8
29. Wiegman CH, Li F, Ryffel B, Togbe D, Chung KF. Oxidative stress in ozone-induced chronic lung inflammation and emphysema: a facet of chronic obstructive pulmonary disease. Front Immunol (2020) 11:1957. doi: 10.3389/fimmu.2020.01957
30. Infante PF. The IARC october 2009 evaluation of benzene carcinogenicity was incomplete and needs to be reconsidered. Am J Ind Med (2011) 54(2):157–64. doi: 10.1002/ajim.20887
31. Xue JM, Zhao MZ, Ma F, Li SS, Mo LH, Zeng XH, et al. Survivin facilitates T-helper 2-biased inflammation in the airway. Int Forum Allergy Rhinol (2019) 9(6):656–64. doi: 10.1002/alr.22301
32. Qu J, Do DC, Zhou Y, Luczak E, Mitzner W, Anderson ME, et al. Oxidized CaMKII promotes asthma through the activation of mast cells. JCI Insight (2017) 2(1):e90139. doi: 10.1172/jci.insight.90139
33. Qu J, Li Y, Zhong W, Gao P, Hu C. Recent developments in the role of reactive oxygen species in allergic asthma. J Thorac Dis (2017) 9(1):E32–43. doi: 10.21037/jtd.2017.01.05
34. Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol (2015) 52(1):106–15. doi: 10.1165/rcmb.2013-0519OC
35. Adam-Bonci TI, Bonci EA, Parvu AE, Herdean AI, Mot A, Taulescu M, et al. Vitamin d supplementation: Oxidative stress modulation in a mouse model of ovalbumin-induced acute asthmatic airway inflammation. Int J Mol Sci (2021) 22(13):1–20. doi: 10.3390/ijms22137089
36. Maes T, Provoost S, Lanckacker EA, Cataldo DD, Vanoirbeek JAJ, Nemery B, et al. Mouse models to unravel the role of inhaled pollutants on allergic sensitization and airway inflammation. Respir Res (2010) 11(1):7. doi: 10.1186/1465-9921-11-7
37. Acciani TH, Brandt EB, Khurana Hershey GK, Le Cras TD. Diesel exhaust particle exposure increases severity of allergic asthma in young mice. Clin Exp Allergy (2013) 43(12):1406–18. doi: 10.1111/cea.12200
38. Brandt EB, Biagini Myers JM, Acciani TH, Ryan PH, Sivaprasad U, Ruff B, et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J Allergy Clin Immunol (2015) 136(2):295–303.e7. doi: 10.1016/j.jaci.2014.11.043
39. Rehder D. The role of vanadium in biology. Metallomics (2015) 7(5):730–42. doi: 10.1039/c4mt00304g
40. Sabbioni E, Kuèera J, Pietra R, Vesterberg O. A critical review on normal concentrations of vanadium in human blood, serum, and urine. Sci Total Environ (1996) 188(1):49–58. doi: 10.1016/0048-9697(96)05164-9
41. Patel NN, Kohanski MA, Maina IW, Workman AD, Herbert DR, Cohen NA. Sentinels at the wall: epithelial-derived cytokines serve as triggers of upper airway type 2 inflammation. Int Forum Allergy Rhinol (2019) 9(1):93–9. doi: 10.1002/alr.22206
42. Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. J Immunol (2009) 183(8):5379–87. doi: 10.4049/jimmunol.0900228
43. Iyer D, Mishra N, Agrawal A. Mitochondrial function in allergic disease. Curr Allergy Asthma Rep (2017) 17(5):29. doi: 10.1007/s11882-017-0695-0
44. Pattnaik B, Bodas M, Bhatraju NK, Ahmad T, Pant R, Guleria R, et al. IL-4 promotes asymmetric dimethylarginine accumulation, oxo-nitrative stress, and hypoxic response-induced mitochondrial loss in airway epithelial cells. J Allergy Clin Immunol (2016) 138(1):130–41. doi: 10.1016/j.jaci.2015.11.036
45. Wang A, Keita AV, Phan V, McKay CM, Schoultz I, Lee J, et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am J Pathol (2014) 184(9):2516–27. doi: 10.1016/j.ajpath.2014.05.019
46. Li R, Kou X, Geng H, Xie J, Yang Z, Zhang Y, et al. Effect of ambient PM(2.5) on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol (2015) 28(3):408–18. doi: 10.1021/tx5003723
47. Qiu YN, Wang GH, Zhou F, Hao JJ, Tian L, Guan LF, et al. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol Environ Saf (2019) 167:178–87. doi: 10.1016/j.ecoenv.2018.08.050
48. Zhang Y, Do DC, Hu X, Wang J, Zhao Y, Mishra S, et al. CaMKII oxidation regulates cockroach allergen-induced mitophagy in asthma. J Allergy Clin Immunol (2021) 147(4):1464–77.e11. doi: 10.1016/j.jaci.2020.08.033
49. Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis (2014) 5:e1498. doi: 10.1038/cddis.2014.460
50. Zhang H, Liu Y, Fang X, Gu L, Luo C, Chen L, et al. Vitamin D3 protects mice from diquat-induced oxidative stress through the NF-kappaB/Nrf2/HO-1 signaling pathway. Oxid Med Cell Longev (2021) 2021:6776956. doi: 10.1155/2021/6776956
51. Almeida Moreira Leal LK, Lima LA, Alexandre de Aquino PE, Vitamin D. (VD3) antioxidative and anti-inflammatory activities: Peripheral and central effects. Eur J Pharmacol (2020) 879:1–12. doi: 10.1016/j.ejphar.2020.173099
52. Wang M, Liu M, Wang C, Xiao Y, An T, Zou M, et al. Association between vitamin d status and asthma control: A meta-analysis of randomized trials. Respir Med (2019) 150:85–94. doi: 10.1016/j.rmed.2019.02.016
53. Solidoro P, Bellocchia M, Aredano I, Mattei A, Pivetta E, Patrucco F, et al. Asthmatic patients with vitamin d deficiency have decreased exacerbations after vitamin replacement. Nutrients (2017) 9(11):1–14. doi: 10.3390/nu9111234
54. Bai YJ, Dai RJ. Serum levels of vitamin a and 25-hydroxyvitamin D3 (25OHD3) as reflectors of pulmonary function and quality of life (QOL) in children with stable asthma: A case-control study. Med (Baltimore) (2018) 97(7):e9830. doi: 10.1097/MD.0000000000009830
55. Holgate ST, Roberts G, Arshad HS, Howarth PH, Davies DE. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc Am Thorac Soc (2009) 6(8):655–9. doi: 10.1513/pats.200907-072DP
56. Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev (2011) 242(1):205–19. doi: 10.1111/j.1600-065X.2011.01030.x
57. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med (2012) 18(5):684–92. doi: 10.1038/nm.2737
58. Bacharier LB, Mori A, Kita H. Advances in asthma, asthma-COPD overlap, and related biologics in 2018. J Allergy Clin Immunol (2019) 144(4):906–19. doi: 10.1016/j.jaci.2019.08.021
59. Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol (2011) 128(3):451–62. doi: 10.1016/j.jaci.2011.04.047
60. Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest (1999) 104(8):1001–6. doi: 10.1172/JCI8124
Keywords: vanadium, house dust mite, airway inflammation, airway remodeling, ROS, vitamin D3
Citation: Tu W, Xiao X, Lu J, Liu X, Wang E, Yuan R, Wan R, Shen Y, Xu D, Yang P, Gong M, Gao P and Huang S-K (2023) Vanadium exposure exacerbates allergic airway inflammation and remodeling through triggering reactive oxidative stress. Front. Immunol. 13:1099509. doi: 10.3389/fimmu.2022.1099509
Received: 15 November 2022; Accepted: 22 December 2022;
Published: 11 January 2023.
Edited by:
Xin Chen, University of Macau, Macau SAR, ChinaCopyright © 2023 Tu, Xiao, Lu, Liu, Wang, Yuan, Wan, Shen, Xu, Yang, Gong, Gao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shau-Ku Huang, c2todWFuZzFAZ21haWwuY29t; Peisong Gao, cGdhbzFAamhtaS5lZHU=
†These authors have contributed equally to this work