 Qingqing Jia
Qingqing Jia Shihua Li
Shihua Li Xiao-Jiang Li
Xiao-Jiang Li Peng Yin
Peng Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 December 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1088124
This article is part of the Research Topic The Molecular Basis of Programmed Cell Death and Neuroinflammation in Neurodegenerative Diseases View all 10 articles
Huntington’s disease (HD) is a progressive neurodegenerative disease characterized by preferential loss of neurons in the striatum in patients, which leads to motor and cognitive impairments and death that often occurs 10-15 years after the onset of symptoms. The expansion of a glutamine repeat (>36 glutamines) in the N-terminal region of huntingtin (HTT) has been defined as the cause of HD, but the mechanism underlying neuronal death remains unclear. Multiple mechanisms, including inflammation, may jointly contribute to HD pathogenesis. Altered inflammation response is evident even before the onset of classical symptoms of HD. In this review, we summarize the current evidence on immune and inflammatory changes, from HD animal models to clinical phenomenon of patients with HD. The understanding of the impact of inflammation on HD would help develop novel strategies to treat HD.
Huntington’s disease (HD) is a devastated neurodegenerative disorder caused by a trinucleotide CAG repeat expansion in exon 1 of HD gene encoding for huntingtin (HTT) protein (1–3). The normal HTT gene contains less than 36 CAG repeats, whereas mutant HTT carriers 40 or more CAG repeats with complete penetrance. Expansions varying from 36 to 39 CAG repeats may also result in HD, but with incomplete penetrance (1). The CAG repeat expansion encodes an expanded polyglutamine (polyQ) tract in mutant HTT that elicits neuronal loss in HD (4). One of the important mechanisms for neurodegeneration in HD is the inflammation caused by mutant HTT, which is the focus of this review for discussion. Generally, mutant HTT causes the inflammatory response in both of the central nervous system (CNS) and peripheral tissues directly. Immunomodulatory messengers can cross the blood-brain barrier (BBB) in both directions, with inflammation spreading from the periphery to the CNS or vice versa (5, 6). The degree of microgliosis and astrogliosis correlates with disease severity directly, and result in an increased production of inflammatory mediators. The increase in microglial activation seemed to be a very early event that occurs before the neuropathological changes. Pathological processes underlying HD, such as excitotoxicity or oxidative stress, induce the morphological change of microglia by a constant expression of inflammatory mediators (7). The secreted pro-inflammatory cytokines can damage nerve cells, leading to their death (8). Astrocytes are also susceptible to inflammatory processes in the brain, becoming activated locally by dead neurons or pathogens. For example, the mutant HTT expressed in glial cells, primarily astrocytes in HD mouse models, could reduce their neuroprotective function (9, 10). However, in some conditions, the neuroinflammation can benefit neuronal tissue by promoting cell debris clearance (11). This review will summarize the growing evidence on inflammatory changes, including the pros and cons of pathogenic mutant HTT, from HD animal models to patients’ clinical phenotypes. The better understanding of the impact of neuroinflammation cytokines on HD will open new venues for the development of novel therapeutic targets.
Drosophila melanogaster has been used as an early HD animal model to identify metabolic biomarkers at pre-symptomatic and symptomatic stages of the disease. In vivo experiments have revealed that the ectopic overexpression of mutant human HTT (exon 1 with expanded CAG repeats) in the neural tissue of transgenic flies causes neurodegeneration (12, 13). However, there was limited work on neuroinflammation in Drosophila model of HD. Lin et al. observed that the expression of mutant HTT (Q93) in hemocytes did not directly affect the animal survival, but the numbers of circulating hemocytes were significantly decreased, leading to an impaired immune response against pathogenic invasion. Mutant HTT caused the altered production of cytokines, such as upd3, dome, tep1, totA, totB, and totC, and consequently the immune dysregulation in Drosophila (14).
Identification of the genetic mutation for HD has accelerated the establishment of various transgenic or knock-in HD mouse models, in which human mutant HD gene is inserted randomly into the mouse genome or precisely into the endogenous mouse HTT gene. In HD transgenic mice, the animals expressed a full-length or a fragment of the mutant HTT gene in addition to the two normal copies of the endogenous mouse HTT gene (15). Common transgenic models of HD are divided as follow. Firstly, N-terminal HTT fragment models with expanded CAG-repeats, such as R6/2, R6/1 or N171-82Q, present an earlier onset of motor and cognitive abnormalities (16). The R6/1 and R6/2 transgenic mice were the first transgenic model developed to study HD, which express exon 1 of the human HD gene with around 115 and 150 CAG repeats respectively (16). Secondly, full-length transgenic mouse models carry mutant HTT in a yeast artificial chromosome (YAC) or bacterial artificial chromosome (BAC), which present a slow progression of the disease with comparable neuropathology and motor-related behavior changes (17, 18). Despite the wealth of different transgenic HD mice available currently, the R6/2 and YAC128 mouse strains are widely used to study many pathological aspects of HD (19). Symptoms of motor impairment in R6/2 mice appear around 3-6 weeks of age. A continuous loss of weight results in death between 11-14 weeks of age (20–22). In YAC128 mice with its full length mutant HTT spanning about 120 CAG repeats (18), hypoactivity is first seen until the age of 8 months. Additionally, progressive gait abnormalities, ataxia, hind limb clasping, and a progressive decline in the forced motor function occur over time (23). In HD knock-in mice, the pathological CAG-repeat is integrated into the mouse HTT gene in a homozygous or heterozygous manner. Because knock-in mice carry the mutation in its appropriate genetic and protein context, they are the most faithful genetic model of human disease. The widely used HdhQ150 and zQ175 mouse models exemplarily belong to this group (24–26). Knock-in mice exhibit more slow progression and age-dependent development of behavioral, pathological, cellular, and molecular abnormalities, which makes these animals valuable for studying age-dependent neuropathology and symptoms.
Based on the various transgenic or knock-in HD mouse models, many researchers reported the important role of the immune system in HD. Träger et al. investigated the myeloid cells from different HD mouse models of R6/2, HdhQ150 and YAC128, to assess whether they are similarly hyperresponsive as HD patient cells. They found that blood CD11b+ cells isolated from 12-week-old R6/2 and 22-month-old HdhQ150 are hyper-reactive upon LPS stimulation in vitro, replicating the phenotype observed in human cells. Analyzing the cytokine profile in 3-month-old YAC128 mice, peritoneal macrophages were found to produce high levels of IL-6 upon the standard endotoxin stimulation. These mouse models recapitulate altered cytokine profiles identified in HD patients, and therefore they could be used to model the immune phenotype (27). Pido-Lopez et al. revealed that the increased TNF-α and IL-12 levels in the striatum of 14-week-old R6/2 mice while IL-12 and IL-1β decreased in the cortex (28). In contrast, Godavarthi et al. found no differences in cytokine expression, including MCP-1, IL-6, IL-10, TNF-α, interferon-γ and IL-12, in the brain samples of R6/2 mice compared with the age-matched control. However, the Iba-1 immunostaining did not reveal any significant increase in the numbers of microglia in the cortex and striatum of R6/2 mice at 12 weeks, an age when these mice show severe symptoms (29). Recently, the fractalkine signaling axis has been explored in the 8-20 weeks old R6/1 HD mouse, and the expression levels of cx3cl1 have been significantly decreased (30), which may lead to an increase in synaptic pruning mediated by microglia, as the total density of PSD-95 puncta decreased and the density of PSD-95 puncta in Iba1 stained microglia increased significantly (30). Furthermore, administering CX3CL1 protein was able to rescue R6/1 mice from long-term depression. It appears that striatal synaptic plasticity dysfunction in pre-symptomatic R6/1 mice is caused by a reduction in CX3CL1 (30), which is consistent with the previous research that CX3CL1 regulates microglia engulfment of synapse (31). Additionally, quinolinic acid (QUIN) has been associated with neuroinflammation in various neurological diseases (32). It is possible that the activation of microglia and astroglia may result in a production of the excitotoxic metabolites, such as the kynurenine pathway, before morphological markers of glial activation are apparent (33–35). Thus, abnormalities in the kynurenic acid pathway are an interesting aspect of HD pathology. QUIN and 3-hydroxykynurenine (3-HK) are neurotoxic metabolites that are elevated in the cortex of HD patients at the early stages of disease progression (36). Especially the 3-HK levels were elevated in the striatum, cortex and cerebellum in the R6/2 starting at 4 weeks of age significantly and selectively, and both 3-HK and QUIN levels were increased in the striatum and cortex in HdhQ92 and HdhQ111 at 15 months and YAC128 mouse at 8 months (35).
The altered immune system function, not only in the CNS but also peripherally, has been implicated in HD pathogenesis (6, 37). To elucidate peripheral inflammatory activation in HD, Björkqvist et al. measured the levels of multiple cytokines in the serum of R6/2, Hdh150Q/150Q and YAC128 mice. In 12-week-old R6/2 mice, IL-6, IL-10, IL-1β, and IL-12p70 were significantly increased as compared to control animals. However, the IL-6, IL-10, and IL-12p70 were significantly elevated in 22-month-old Hdh150Q/150Q mice. In the YAC128 mouse model of HD at 12 months of age, the similar elevations in serum IL-6 and IL-8 were observed (6). Similary, Chang et al. detected the levels of inflammatory markers in the plasma of R6/2 mice. IL-6 levels in R6/2 mice at different disease stages (9, 11 and 13 weeks) were higher than those in the age-matched wild-type (WT) littermates, with higher levels of MMP-9 and TGF-β1 compared with their WT littermates from 11 weeks. In contrast, the plasma level of IL-18 (11-13 weeks old) was lower than control mice (38). Furthermore, Disatnik et al. also measured the levels of inflammatory cytokines such as TNF and IL-6, the levels of both these cytokines were two times higher in the plasma of 13 weeks old R6/2 mice than WT mice (39). Interestingly, treatment with P110 (a selective peptide inhibitor, of excessive mitochondrial fission) for 8 weeks was efficient in reducing the levels of these inflammatory levels to the level in WT mice (39, 40). A study including 9-12 months old YAC128 mice serum samples revealed the significant increases in IL-8 and IL-10 levels. However, the elevation of IL-6 in serum of YAC128 mice occurred at 12 months of age whereas the level of IFN-γ increased in serum samples from YAC128 mice were at 6-9 months old (41). More recently, Podlacha et al. also reported the significant elevation of levels of inflammatory markers, such as IL-6, TNF-α, IL-1β and IL-12, in R6/1 mouse peripheral blood. This allows for a more objective assessment of particular biomarkers in course of a slower progression of symptoms in HD mice. At early stages of disease, the anti-inflammatory defense mechanism was significantly impaired, with a marked decrease in IL-10 levels (42). Valadão et al. also investigated the immune changes in many peripheral organs of the 12-month-old BAC HD model. They found significant changes in cytokine levels in all organs analyzed, including heart, liver, spleen and kidney. Levels of IL-6 and IL-12p70 were increased in the heart of BAC HD mice as compared with WT animals. In the liver samples of BAC HD mice, enhanced IL-12p70 and TNF-α levels were observed. In the spleen, there was an increase in the levels of the cytokine IL-4, but a decrease in the levels of IL-5 and IL-6 in BAC HD mice. However the increased level of IL-6 was exhibited in kidney (43).
Considering that pro-inflammatory cytokine IL-6 may actively influence the disease course in HD, an excessive IL-6 release was also detected in YAC128 mice at 12 months and R6/2 mice at 9, 11 and 13 weeks of age (6, 38). Bouchard et al., administered an IL-6 neutralizing antibody to R6/2 mice to test whether IL-6 influences the disease course. In their work, treatment with the specific antibody in R6/2 mice reduced weight loss at late stages and partially rescued motor deficits on the rotarod performance as compared with the control IgG treatment (44). Following this line of reasoning, Wertz et al. tested the hypothesis that IL-6 deficiency would be protective against the effects of mutant huntingtin and therefore generated the R6/2 mice model lacking IL-6. Contrary to previous studies, the lack of IL-6 exacerbated R6/2 associated behavioral phenotypes. Based on a single nuclear RNA sequencing of striatal cell types, it was evident that IL-6 deficiency affected normal regulation of various genes associated with synaptic function, as well as the BDNF receptor Ntrk2 (45).
Although relatively shorter experimental period and lower housing cost of using rodents are the advantages of studying mouse models of HD, the data on activation of the inflammatory system in HD mouse models are sparse and contradictory, possibly due to the considerable pathological differences between different transgenic strains and at different disease stages. The immunocytochemical staining of GFAP or Iba-1 has been widely used to identify gliosis, an early CNS damage that is associated with neuroinflammation in HD (46–49). Different levels of gliosis activation were observed in mouse models with full-length or a fragment HTT at different ages. Yu et al. systematically compared that the N171-82Q mouse striatum and found a GFAP increase, at 3 months of age and became prominent at 4-5 months of age, indicated by intense labeling throughout astroglial cell bodies and their fibrous processes. Although the striatum of R6/2 mice at 12 weeks of age displayed some increased GFAP staining, the overall number of GFAP-labeled glial cells was not increased in R6/2 mice compared with that of their littermates or other HD mice. In R6/1 mice even at 9 months of age, there was still little GFAP staining in the striatum (50). Reiner et al. also compared the cortex (22 and 30 weeks) and striatum (30 weeks) of R6/2 chimeras, which displayed some glial cells with increasingly upregulated GFAP staining, much later than pure R6/2 mice that died at 12-15 weeks of age (51). Gatto et al. found that the neuroinflammatory processes with activated astrocyte GFAP occurred in the 11- and 30-week-old R6/1 mice (52). In 17-week-old R6/2 mouse model, reactive astrocytes with processes enveloping degenerating neurons was seen, though no accompanying inflammatory response or increased macrophages and microglia was observed (53). Simmons et al. also reported that the Iba-1 positive cells did not obviously differ in both R6/2 and WT mice at the age of <7 weeks, but increased in R6/2 mice older than 8 weeks (54). While Hdh150Q knock-in mice at 27-30 weeks of age also did not show intense GFAP immunoreactivity, a significant increase in GFAP immunoreactivity was found in the striatum of Hdh150Q mice at 14 months of age, as reported previously (26, 50). In HD KI mice expressing shorter CAG repeats (HdhQ92 or HTTQ111), no gliosis was found up to 17-18 months of age (55, 56). However, in 18-20 months old Hdh175/175 mouse, the expression of full-length mHTT in microglia was able to promote pro-inflammatory transcription (57). Different extents of gliosis seen in HD mice are likely associated with genetic background, CAG repeats in the HD gene, housing and experimental conditions. However, the data in HD patients show that the inflammatory response is activated at the very early disease stage (6). It is also necessary to point out that most of rodent models fail to mimic the overt and typic neurodegeneration seen in HD patients (58–60). The lack of overt neuronal loss and robust gliosis in HD mouse models prevents the rigorous evaluation of the therapeutic effects on neurodegeneration. The summarized evidence on gliosis activation, including the pros and cons observations, was listed in Table 1.

Table 1 Changes in glial cells in different HD mouse models at multiple ages.
The biological differences between humans and mice may account for the failure of some mouse models to replicate pathology seen in humans. Thus, it is possible that larger transgenic animal models may be able to mimic important neurodegenerative features. However, an HD transgenic sheep model (OVT73) was created, which does not exhibit many of the overt phenotypes observed in HD patients and was thought to be a model of prodromal or early-stage HD (65). By using CRISPR/Cas9, Yan et al. established the 140Q KI pig that endogenously expresses full-length mutant HTT (66). Importantly, the increased immunohistochemical staining of GFAP was firstly observed in the dorsal caudate nucleus and putamen of the 4-5-month-old F0 KI and F1 KI pigs. Also, immunostaining with the antibody to Iba-1 revealed a marked increase of the microglial cells in KI pig brain, which is more abundant in the striatum than in the cortex. Quantification of the number of different types of cells revealed that the KI striatum had the most severe loss of NeuN-positive cells and the highest increase in glial cell numbers, which was not observed in the age-matched HD KI mouse (66, 67). Additionally, Valekova et al. used transgenic HD minipig, which was generated by injecting lentiviral vectors carrying truncated mutant huntingtin genes that encode 124 glutamine repeats integrated into chromosome 1q24-q25 and transmitted through successive three generations (68), for investigating multiple cytokines (69). At the same time, various cytokines were analyzed in the secretomes of microglia and blood monocytes, as well as in the cerebrospinal fluid (CSF) and serum collected from pre-symptomatic HD minipigs. A decline in IFN-α was observed in CSF collected from an early time at 9 months of age and lasted at least up to 36 months. The transgenic minipigs at 36 months of age had lower levels of IL-10 in the CSF. IFN-α and IL-10 levels were also decreased in secretome of microglia, whilst elevated IL-8 and IL-1β levels were secreted by primary microglia that were isolated from the HD minipigs at 36 months of age. In serum samples collected from 36-month-old HD minipigs had significantly higher levels of IL-8 than WT ones (69).
Besides, transgenic non-human primate models expressing the disease genes were established, transgenic HD rhesus monkeys, which express exon 1 mutant HTT with 84Q under the control of the human ubiquitin promoter, were generated by injecting lentiviruses into fertilized oocytes to express mutant HTT (70). HD transgenic monkeys with 84Q die postnatally, and this early death was associated with the overexpression of N-terminal mutant HTT. Despite their early death, some transgenic monkeys developed key clinical HD features including dystonia, chorea, and seizure (70). Interestingly, the 5-year-old transgenic HD monkeys expressing N-terminal HTT 509 amino acids with approximately 67-72Q under the human HTT promoter (71) exhibited increased pro-inflammatory cytokines and higher induction of immune pathway genes, such as inflammatory response factor IL-6, TNF-α, and C-Reactive Protein (CRP) (72) (Figure 1).
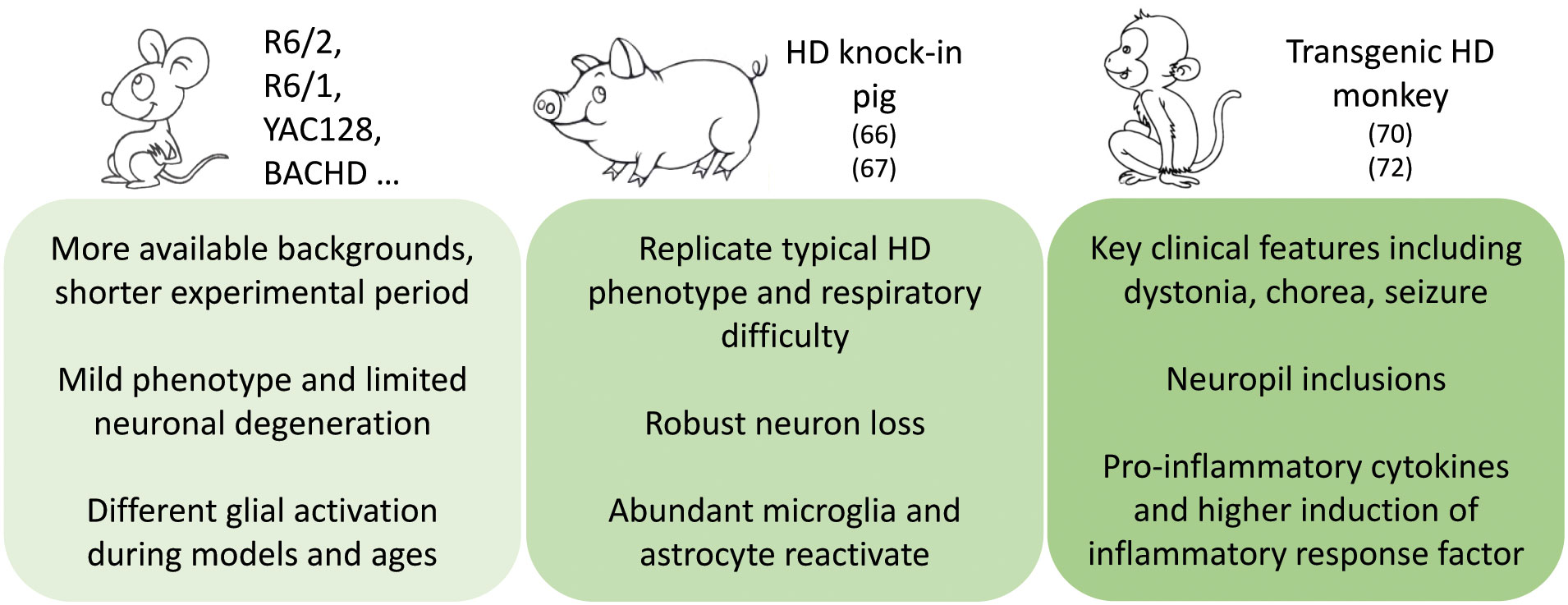
Figure 1 Pathogenic insights from and phenotype differences between animal models expressing mutant HTT.
Robust evidence regarding neuroinflammation in HD comes from postmortem studies of brain tissues from patients with HD. Evidence for involvement of inflammation in the pathogenesis of HD includes upregulation of inflammatory cytokines or chemokines and activation of the complement system. Björkqvist et al. have reported the increase of multiple cytokines in the plasma (IL-1β, IL-4, IL-6, IL-8, TNF-α, IL-10) and striatum (IL-6, IL-8 and TNF-α) of HD patients (6). Interestingly, a significant increase in IL-6 plasma levels was found in pre-symptomatic HD mutation carriers 16 years prior to the predicted onset of the disease, suggesting that inflammatory changes occurs very early in the disease process (6). According to Dalrymple et al., the level of IL-6 in HD patient plasma was increased significantly (37). Likewise, HD patients also exhibit increased expression of IL-6, IL-8, and matrix metalloproteinase-9 (MMP-9) in their cortex and cerebellum (73). Remarkably, MCP1/CCL2 and IL-10 mRNA levels were significantly higher in the striatum of HD patients than controls (73). Gang et al. also quantified the plasma concentrations of IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNF-α from HD patients. Patients with HD have significantly lower plasma concentrations of IL-4, a marker of responses from T-helper-2 cells, than healthy controls. In contrast, no significant differences was observed in the plasma concentrations of IFN-γ, IL-1β, IL-2, IL-6, IL-8, IL-10, IL-12p70, IL-13 or TNF-α in HD patients (74). Recently, von Essen MR et al. reported the immune abnormalities before motor onset of disease (75). Proinflammatory cytokines, including IL-17, were detected in the CSF of HTT mutation carriers, as well as the increased IL-7 consumption before motor onset of HD. Moreover, they reported an increased prevalence of IL-17 producing Th17.1 cells in the CSF of HTT mutation carriers, predominantly in pre-motor manifest individuals. There was a negative correlation between intrathecal Th17.1 cell frequency and the progression of HD, suggesting that Th17.1 cells play an important role at the early stages of the disease. Moreover, the author found that the balance of pro-inflammatory and regulatory T cells was skewed. This skewing further favors a pro-inflammatory environment in the CSF of HTT mutation carriers (75).
In particular, a significant elevation of chemokines (C-C motif) ligand (CCL)-2, CCL4, CCL11, CCL13, CCL26 has been detected in the plasma from HD patients (76). Wild et al. have also reported that CCL11 and related chemokines may directly contribute to the neurodegenerative processes in CNS. As a result of analyzing plasma levels of cytokines in two separate cohorts of HD patients, the elevated levels of CCL11 and CCL26 were found in HD group from their first cohort of 65 HD patients (76). There were also significant differences in CCL11 and CCL26 levels across all HD clinical stages. In their second cohort of 68 HD patients and 26 healthy controls, plasma CCL11 and CCL26 levels were significantly increased with more advanced HD cases. In addition, CCL11 levels were positively correlated with standardized assessments of motor impairment while negatively correlated with functional scores (76, 77). Thus, it is worth performing future studies of both CCL11 and CCL26 as potential biomarkers in HD. It has been shown that the complement factors C1QC, C2, and C3 are also increased in CSF samples taken from living HD patients in comparison with controls (78). Singhrao et al. also found the complement activation, and increased mRNA levels of complement proteins in HD brains (79). Increased protein expression of complement components C7 and C9, complement inhibitor cluster proteins, and acute phase protein α-2-macroglobulin were shown in HD patient plasma and CSF (37).
Neuroinflammation can be modulated by neuron-glial signaling through various soluble factors, such as cluster of differentiation CD22 (80), CD47 (81), CD200 (82, 83), the family of CD300 receptors (84) and CX3CL1 (85). CX3CL1 produced in neurons is the sole member of the CX3C family of chemokines (86), and it is known that CX3CL1 is the only ligand binding to CX3CR1, a 7 transmembrane domain class A G-protein coupled receptor that is expressed in microglia, monocytes, natural killer cells (NK), T cells and smooth muscle cells (87). Interestingly, CX3CL1 was also found to be an important novel factor in HD pathogenesis and survival following a network analysis of microarray data from human post-mortem tissue (88). An investigation of the fractalkine signaling axis in HD patients revealed that cx3cl1 gene expression was significantly reduced in their putamen (30) (Figure 2).

Figure 2 Schematic summary of brain and peripheral inflammatory changes in HD mice and patients.
The accumulating evidences supports that inflammation plays a key role in neurodegenerative diseases and has stimulated the use of immunotherapeutic strategies to modulate neuroinflammatory diseases. Several preclinical and clinical trials of potential immunomodulatory drugs have been investigated in HD, such as minocycline and cannabinoids, laquinimod, TNF-α inhibitors, anti-SEMA4D monoclonal antibody, gangliosides and so on.
It has also been demonstrated that minocycline and cannabinoids have anti-inflammatory properties, even though they are not a member of the classical definition of anti-inflammatory drugs. R6/2 mice were administered with the anti-inflammatory tetracycline minocycline showed improvements in behavioral and neuropathological deficits (89, 90), which is also supported by an excitotoxic rat model of HD (91). A clinical study revealed that minocycline at 100 and 200 mg/day was well tolerated for 8 weeks. The study involved 60 patients who were randomly assigned to receive placebo (n = 23), minocycline 100 mg/day (n = 18), or minocycline 200 mg/day (n = 19). However, there was no effect on the UHDRS (Unified HD Rating Scale) scores, with no efficacy observed (92). Cannabidiol was considered safe and well tolerated during a 6 weeks clinical trial conducted on 15 patients affected by HD. However, clinical outcomes did not show the significant improvement (93).
The immunomodulator laquinimod (LAQ) could downregulate both the production of pro-inflammatory cytokines in peripheral blood mononuclear cells and neuroglial activation in the brain, which was initially known as an immunomodulatory agent and was used to treat multiple sclerosis. For example, the neuroprotective effect of LAQ is well supported by its therapeutic effects on animal models of neuroinflammatory diseases, such as the experimental autoimmune encephalomyelitis (EAE) (94, 95), and by alleviation of demyelination in different diseases (96–99). LAQ has also been shown to improve behavioral phenotypes and white matter integrity in many HD mouse models (100–104). Especially, LAQ rescues evidence of cortico-striatal neurodegeneration, demyelination of white matter, and behavioral deficits in YAC128 HD mice (105). A mild ameliorative effect of LAQ was also observed in R6/2 mice with motor function deficits and striatal neuropathology (106). Thus, the therapeutic effect of LAQ is therefore thought to be due to an anti-inflammation effect (98, 103). Although the precise mechanism of action of LAQ is unclear, Dobson et al. demonstrated that LAQ significantly dampened the release of hyper-reactive cytokines from stimulated premanifest and manifest HD patient monocytes, and may exert its neuroprotective effects by promoting BDNF production (94). However, LAQ had no efficacy observed in the Phase 2 clinical trials of HD patients unfortunately. (https://clinicaltrials.gov/ct2/show/NCT02215616). Thus, whether LAQ can be used to treat HD patients remains to be verified.
TNF-α is a multifunctional cytokine associated with cellular proliferation, differentiation, inflammation, immune responses and apoptosis (107). Based on the evidence of increased levels of TNF-α in HD, one study investigated the therapeutic potential of DN-TNF-α (XPro1595), which demonstrated that intracerebroventricular (ICV) injection of DN-TNF-α modulates neuroinflammation, caspase activation, mHTT aggregate burden, and motor function deficit in R6/2 HD transgenic mice (108). However, the clinical efficacy of this molecule in human HD warrants further investigation based on the fact that DN-TNF-α’s systemic injection rather than an ICV injection shows lesser efficacy on motor function in R6/2 mice (109). However, in a study of R6/2 mice carried out by Pido-Lopez et al., the systemic injection of etanercept, a drug that inhibits TNF-α, dampened the levels of TNF-α in plasma and other peripheral proinflammatory molecules such as IL-1β and IL-6. However, the level of TNF-α and IL-6 expression in the striatum is not affected by etanercept treatment. According to the follow-up study, etanercept partially reduced brain atrophy, but failed to improve HD related functional and cognitive deficits in R6/2 mice (110).
Semaphorin 4D (SEMA4D), also called CD100, is chemorepulsive axonal guidance and immunoregulatory transmembrane signaling molecule. It signals via three receptor subtypes of Plexin-B1 (PLXNB1), Plexin-B2 (PLXNB2) and CD72. In the CNS, SEMA4D interacts with PLXNB1 in neuronal cells via Rho-GTPases-RhoA and R-Ras GTPase-activating protein activities, inducing axonal growth cone collapse (111, 112). In previous studies, the treatment on YAC128 HD transgenic mice with neutralizing SEMA4D antibody has been shown to ameliorate the neuropathological deficits and behavioral symptoms (111). However, it is still unclear whether SEMA4D inhibition will be beneficial in human HD.
Besides, therapeutic administration of the brain gangliosides, GM1, has also been used to provide neuroprotection in models of neuronal injury and neurodegeneration of Alzheimer’s Disease (AD) (113), Parkinson’s Disease (PD) (114, 115) and HD (116). Furthermore, the intraventricular administration of GM1 showed profound disease-modifying effects across HD mouse models exhibiting varying genetic backgrounds, and there is a reduction in mutant HTT levels after GM1 administration (116). The treatment of R6/2 mice with GM1 slows down the white matter atrophy and body weight loss, while the motor functions have been significantly improved. The administration of GM1 also ameliorated psychiatric-like and cognitive dysfunctions and gait abnormalities. It was also shown that GM1 administration improves psychiatric-like and cognitive dysfunctions in YAC128 mice (116). Prados et al. reported an efficacy of the compound betulinic acid hydroxamate (BAH), a hypoximimetic derivative of betulinic acid, against the striatal HD neurodegeneration. In striatal STHdhQ111/Q111 cells, BAH stabilized HIF-1α protein and protected against mitochondrial toxin-induced cytotoxicity. Pharmacokinetic analyses showed that BAH had a good brain penetrability in 3-nitropropionic acid-treated mouse model with striatal neurodegeneration, improved the clinical symptoms, prevented neuronal loss, decreased reactive astrogliosis and microgliosis, and inhibited the upregulation of proinfammatory markers in the brain (117). Purushothaman et al. also reported the neuropharmacological protective effect of Baicalein (BC) against the Quinolinic Acid (QA)-induced HD-like rat models that displayed the psychological and behavioural changes. This study proved that BC is efficient to revive the level of enzymatic and non-enzymatic antioxidants and mitochondrial complexes by decreasing a number of inflammatory mediators such as Malondialdehyde (MDA), protein carbonyls and Nitric Oxide. It also restores the amount of BDNF and GDNF, thereby preventing the neurophysiological changes (118). Taking together, these findings show that targeting cytokines might assist in resolving neuroinflammation, but evidence suggests that merely suppressing the neuroinflammatory processes would be insufficient to restore functional capacity in HD.
The contribution of neuroinflammation to neurodegeneration has previously been defined. However, the role of CNS and peripheral inflammatory changes in HD remains poorly understood. This is because neuroinflammatory and neuroimmune reactions can be beneficial or detrimental, and there are various interactions between diverse brain cell types and the signaling cascades triggered in HD. Various families of cytokines and cytokine receptors, growth factors, and chemokines influence the apoptotic or survival pathways of neurons and the degree of inflammatory processes in the CNS. It is unclear whether inflammatory changes are caused by neurodegeneration or represent an independent pathological mechanism. Thus, it is important to refine our understanding of these more specific immune and inflammatory mechanisms involved in HD.
Although HD mouse models have been widely used to investigate HD pathogenesis and neuropathology, there are various differences in neurodegenerative pathophysiology between rodent models and clinical patients. In particular, neuronal inflammatory responses were found in HD patients at the early disease stage but were not consistently seen in many mouse models of HD at young ages. Given the lack of obvious neurodegeneration phenotypes in most genetic mouse models, the demand for establishing large animal models to study neurodegenerative diseases is well-appreciated. Investigation of large animal models would be highly valuable for understanding the novel pathogenic mechanisms and identifying neuroinflammation alterations, which may not be uncovered in small animals, though there are challenges and limitations that are largely stemmed from the costly and time-consuming investigation.
In addition, it remains to be investigated whether peripheral immune response and inflammatory alterations mirror the changes and the putative pathways in the CNS in HD. Yet, whether the inflammatory response is an active or a reactive (or both) mechanism in HD pathophysiology remains controversial. Further mechanistic studies are need and would require use of multiple animal models including those large animal models that can more closely mimic the pathological changes in HD patients. Advancing our understanding of the involvement of the immune system in HD pathophysiology would help identify a valid target for new therapeutic interventions to halt the progression of HD.
QJ and PY wrote the manuscript. X-JL and SL edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by The National Natural Science Foundation of China (31872779, 81830032, 32270564); Department of Science and Technology of Guangdong Province (2021ZT09Y007; 2018B030337001); Guangzhou Key Research Program on Brain Science (202007030008); Guangdong Basic and Applied Basic Research (2022A1515011205); Fundamental Research Funds for The Central Universities (21622113).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington's disease chromosomes. Cell (1993) 72(6):971–83. doi: 10.1016/0092-8674(93)90585-E
2. Li S-H, Li X-J. Huntingtin–protein interactions and the pathogenesis of huntington's disease. Trends Genet (2004) 20(3):146–54. doi: 10.1016/j.tig.2004.01.008
3. van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: Widespread pathology in huntington's disease. Lancet Neurol (2009) 8(8):765–74. doi: 10.1016/s1474-4422(09)70178-4
4. Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci (2000) 23:217. doi: 10.1146/annurev.neuro.23.1.217
5. Ellrichmann G, Reick C, Saft C, Linker RA. The role of the immune system in huntington’s disease. Clin Dev Immunol (2013) 2013:541259. doi: 10.1155/2013/541259
6. Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in huntington's disease. J Exp Med (2008) 205(8):1869–77. doi: 10.1084/jem.20080178
7. Sapp E, Kegel K, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, et al. Early and progressive accumulation of reactive microglia in the huntington disease brain. J Neuropathology Exp Neurol (2001) 60(2):161–72. doi: 10.1093/jnen/60.2.161
8. Pavese N, Gerhard A, Tai Y, Ho A, Turkheimer F, Barker R, et al. Microglial activation correlates with severity in huntington disease: A clinical and pet study. Neurology (2006) 66(11):1638–43. doi: 10.1212/01.wnl.0000222734.56412.17
9. Shin J-Y, Fang Z-H, Yu Z-X, Wang C-E, Li S-H, Li X-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol (2005) 171(6):1001–12. doi: 10.1083/jcb.200508072
10. Bradford J, Shin J-Y, Roberts M, Wang C-E, Sheng G, Li S, et al. Mutant huntingtin in glial cells exacerbates neurological symptoms of huntington disease mice. J Biol Chem (2010) 285(14):10653–61. doi: 10.1074/jbc.M109.083287
11. Kaushik DK, Basu A. A friend in need may not be a friend indeed: Role of microglia in neurodegenerative diseases. CNS Neurol Disord Drug Targets (2013) 12(6):726–40. doi: 10.2174/18715273113126660170
12. Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in drosophila. Nature (2001) 413(6857):739–43. doi: 10.1038/35099568
13. Song W, Smith MR, Syed A, Lukacsovich T, Barbaro BA, Purcell J, et al. Morphometric analysis of huntington’s disease neurodegeneration in drosophila. Tandem Repeats Genes Proteins Disease. Springer (2013) 1017:41–57. doi: 10.1007/978-1-62703-438-8_3
14. Lin Y-H, Maaroufi HO, Ibrahim E, Kucerova L, Zurovec M. Expression of human mutant huntingtin protein in drosophila hemocytes impairs immune responses. Front Immunol (2019) 10:2405. doi: 10.3389/fimmu.2019.02405
15. Menalled LB. Knock-in mouse models of huntington’s disease. NeuroRx (2005) 2(3):465–70. doi: 10.1602/neurorx.2.3.465
16. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the hd gene with an expanded cag repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell (1996) 87(3):493–506. doi: 10.1016/S0092-8674(00)81369-0
17. Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu X-H, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in bachd mice. J Neurosci (2008) 28(24):6182–95. doi: 10.1523/JNEUROSCI.0857-08.2008
18. Slow EJ, Van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, et al. Selective striatal neuronal loss in a Yac128 mouse model of huntington disease. Hum Mol Genet (2003) 12(13):1555–67. doi: 10.1093/hmg/ddg169
19. Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, et al. Chronology of behavioral symptoms and neuropathological sequela in R6/2 huntington's disease transgenic mice. J Comp Neurol (2005) 490(4):354–70. doi: 10.1002/cne.20680
20. Lüesse H-G, Schiefer J, Spruenken A, Puls C, Block F, Kosinski CM. Evaluation of R6/2 hd transgenic mice for therapeutic studies in huntington's disease: Behavioral testing and impact of diabetes mellitus. Behav Brain Res (2001) 126(1-2):185–95. doi: 10.1016/S0166-4328(01)00261-3
21. Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, et al. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human huntington's disease mutation. J Neurosci (2000) 20(13):5115–23. doi: 10.1523/JNEUROSCI.20-13-05115.2000
22. Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, et al. Characterization of progressive motor deficits in mice transgenic for the human huntington’s disease mutation. J Neurosci (1999) 19(8):3248–57. doi: 10.1523/JNEUROSCI.19-08-03248.1999
23. Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the Yac128 mouse model of huntington disease. Hum Mol Genet (2005) 14(24):3823–35. doi: 10.1093/hmg/ddi407
24. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of huntington's disease with 140 cag repeats. J Comp Neurol (2003) 465(1):11–26. doi: 10.1002/cne.10776
25. Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of huntington’s disease: Zq175. PloS One (2012) 7(12):e49838. doi: 10.1371/journal.pone.0049838
26. Lin C-H, Tallaksen-Greene S, Chien W-M, Cearley JA, Jackson WS, Crouse AB, et al. Neurological abnormalities in a knock-in mouse model of huntington’s disease. Hum Mol Genet (2001) 10(2):137–44. doi: 10.1093/hmg/10.2.137
27. Träger U, Andre R, Magnusson-Lind A, Miller JR, Connolly C, Weiss A, et al. Characterisation of immune cell function in fragment and full-length huntington's disease mouse models. Neurobiol Dis (2015) 73:388–98. doi: 10.1016/j.nbd.2014.10.012
28. Pido-Lopez J, Andre R, Benjamin AC, Ali N, Farag S, Tabrizi SJ, et al. In vivo neutralization of the protagonist role of macrophages during the chronic inflammatory stage of huntington’s disease. Sci Rep (2018) 8(1):1–14. doi: 10.1038/s41598-018-29792-x
29. Godavarthi SK, Narender D, Mishra A, Goswami A, Rao SN, Nukina N, et al. Induction of chemokines, mcp-1, and kc in the mutant huntingtin expressing neuronal cells because of proteasomal dysfunction. J neurochemistry (2009) 108(3):787–95. doi: 10.1111/j.1471-4159.2008.05823.x
30. Kim A, García-García E, Straccia M, Comella-Bolla A, Miguez A, Masana M, et al. Reduced fractalkine levels lead to striatal synaptic plasticity deficits in huntington’s disease. Front Cell Neurosci (2020) 14:163. doi: 10.3389/fncel.2020.00163
31. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination Via Adam10 and fractalkine signaling. Nat Neurosci (2019) 22(7):1075–88. doi: 10.1038/s41593-019-0419-y
32. Maddison DC, Giorgini F eds. The kynurenine pathway and neurodegenerative disease. Seminars in cell & developmental biology. (2015) 40:134–41. doi: 10.1016/j.semcdb.2015.03.002
33. Stoy N, Mackay G, Forrest C, Christofides J, Egerton M, Stone T, et al. Tryptophan metabolism and oxidative stress in patients with huntington's disease. J neurochemistry (2005) 93(3):611–23. doi: 10.1111/j.1471-4159.2005.03070.x
34. Guidetti P, Reddy PH, Tagle DA, Schwarcz R. Early kynurenergic impairment in huntington's disease and in a transgenic animal model. Neurosci Lett (2000) 283(3):233–5. doi: 10.1016/S0304-3940(00)00956-3
35. Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, et al. Elevated brain 3-hydroxykynurenine and quinolinate levels in huntington disease mice. Neurobiol Dis (2006) 23(1):190–7. doi: 10.1016/j.nbd.2006.02.011
36. Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade huntington's disease. Neurobiol Dis (2004) 17(3):455–61. doi: 10.1016/j.nbd.2004.07.006
37. Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Björkqvist M, Petersén Å, et al. Proteomic profiling of plasma in huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res (2007) 6(7):2833–40. doi: 10.1021/pr0700753
38. Chang K-H, Wu Y-R, Chen Y-C, Chen C-M. Plasma inflammatory biomarkers for huntington’s disease patients and mouse model. Brain behavior Immun (2015) 44:121–7. doi: 10.1016/j.bbi.2014.09.011
39. Disatnik M-H, Joshi AU, Saw NL, Shamloo M, Leavitt BR, Qi X, et al. Potential biomarkers to follow the progression and treatment response of huntington’s disease. J Exp Med (2016) 213(12):2655–69. doi: 10.1084/jem.20160776
40. Qi X, Qvit N, Su Y-C, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci (2013) 126(3):789–802. doi: 10.1242/jcs.114439
41. Kwan W, Magnusson A, Chou A, Adame A, Carson MJ, Kohsaka S, et al. Bone marrow transplantation confers modest benefits in mouse models of huntington's disease. J Neurosci (2012) 32(1):133–42. doi: 10.1523/JNEUROSCI.4846-11.2012
42. Podlacha M, Pierzynowska K, Gaffke L, Jerzemowska G, Piotrowska E, Węgrzyn G. Behavioral-and blood-based biomarkers for huntington's disease: Studies on the R6/1 mouse model with prospects for early diagnosis and monitoring of the disease. Brain behavior immunity-health (2022) 23:100482. doi: 10.1016/j.bbih.2022.100482
43. Valadão PAC, da Silva Oliveira B, Joviano-Santos JV, Vieira ÉLM, Rocha NP, Teixeira AL, et al. Inflammatory changes in peripheral organs in the bachd murine model of huntington's disease. Life Sci (2019) 232:116653. doi: 10.1016/j.lfs.2019.116653
44. Bouchard J, Truong J, Bouchard K, Dunkelberger D, Desrayaud S, Moussaoui S, et al. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of huntington's disease. J Neurosci (2012) 32(50):18259–68. doi: 10.1523/JNEUROSCI.4008-12.2012
45. Wertz MH, Pineda SS, Lee H, Kulicke R, Kellis M, Heiman M. Interleukin-6 deficiency exacerbates huntington’s disease model phenotypes. Mol neurodegeneration (2020) 15(1):1–8. doi: 10.1186/s13024-020-00379-3
46. Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in huntington's disease. J Neuropathology Exp Neurol (1995) 54(1):105–20. doi: 10.1097/00005072-199501000-00013
47. Sharp AH, Ross CA. Neurobiology of huntington's disease. Neurobiol Dis (1996) 3(1):3–15. doi: 10.1006/nbdi.1996.0002
48. Etxeberria-Rekalde E, Alzola-Aldamizetxebarria S, Flunkert S, Hable I, Daurer M, Neddens J, et al. Quantification of huntington’s disease related markers in the R6/2 mouse model. Front Mol Neurosci (2021) 13:617229. doi: 10.3389/fnmol.2020.617229
49. Palpagama TH, Waldvogel HJ, Faull RL, Kwakowsky A. The role of microglia and astrocytes in huntington’s disease. Front Mol Neurosci (2019) 12:258. doi: 10.3389/fnmol.2019.00258
50. Yu Z-X, Li S-H, Evans J, Pillarisetti A, Li H, Li X-J. Mutant huntingtin causes context-dependent neurodegeneration in mice with huntington's disease. J Neurosci (2003) 23(6):2193–202. doi: 10.1523/JNEUROSCI.23-06-02193.2003
51. Reiner A, Del Mar N, Deng YP, Meade CA, Sun Z, Goldowitz D. R6/2 neurons with intranuclear inclusions survive for prolonged periods in the brains of chimeric mice. J Comp Neurol (2007) 505(6):603–29. doi: 10.1002/cne.21515
52. Gatto RG, Weissmann C, Amin M, Angeles-López QD, García-Lara L, Castellanos LCS, et al. Evaluation of early microstructural changes in the R6/1 mouse model of huntington's disease by ultra-high field diffusion Mr imaging. Neurobiol Aging (2021) 102:32–49. doi: 10.1016/j.neurobiolaging.2021.02.006
53. Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of huntington's disease. Proc Natl Acad Sci (2000) 97(14):8093–7. doi: 10.1073/pnas.110078997
54. Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of huntington's disease. Glia (2007) 55(10):1074–84. doi: 10.1002/glia.20526
55. Wheeler VC, White JK, Gutekunst C-A, Vrbanac V, Weaver M, Li X-J, et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in hdh Q92 and hdh Q111 knock-in mice. Hum Mol Genet (2000) 9(4):503–13. doi: 10.1093/hmg/9.4.503
56. Kovalenko M, Milnerwood A, Giordano J, St Claire J, Guide JR, Stromberg M, et al. HTT Q111/+ huntington’s disease knock-in mice exhibit brain region-specific morphological changes and synaptic dysfunction. J Huntington's Dis (2018) 7(1):17–33. doi: 10.3233/JHD-170282
57. Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, et al. Mutant huntingtin promotes autonomous microglia activation Via myeloid lineage-determining factors. Nat Neurosci (2014) 17(4):513–21. doi: 10.1038/nn.3668
58. Deng H-X, Siddique T. Transgenic mouse models and human neurodegenerative disorders. Arch Neurol (2000) 57(12):1695–702. doi: 10.1001/archneur.57.12.1695
59. Crook ZR, Housman D. Huntington's disease: Can mice lead the way to treatment? Neuron (2011) 69(3):423–35. doi: 10.1016/j.neuron.2010.12.035
60. Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci (2018) 21(10):1370–9. doi: 10.1038/s41593-018-0236-8
61. Paldino E, D’Angelo V, Sancesario G, Fusco FR. Pyroptotic cell death in the R6/2 mouse model of huntington’s disease: New insight on the inflammasome. Cell Death Discovery (2020) 6(1):1–12. doi: 10.1038/s41420-020-00293-z
62. Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, et al. Microglial Cb2 cannabinoid receptors are neuroprotective in huntington's disease excitotoxicity. Brain (2009) 132(11):3152–64. doi: 10.1093/brain/awp239
63. Franciosi S, Ryu JK, Shim Y, Hill A, Connolly C, Hayden MR, et al. Age-dependent neurovascular abnormalities and altered microglial morphology in the Yac128 mouse model of huntington disease. Neurobiol Dis (2012) 45(1):438–49. doi: 10.1016/j.nbd.2011.09.003
64. Hickey MA, Kosmalska A, Enayati J, Cohen R, Zeitlin S, Levine MS, et al. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in huntington's disease mice. Neuroscience (2008) 157(1):280–95. doi: 10.1016/j.neuroscience.2008.08.041
65. Jacobsen JC, Bawden CS, Rudiger SR, McLaughlan CJ, Reid SJ, Waldvogel HJ, et al. An ovine transgenic huntington's disease model. Hum Mol Genet (2010) 19(10):1873–82. doi: 10.1093/hmg/ddq063
66. Yan S, Tu Z, Liu Z, Fan N, Yang H, Yang S, et al. A huntingtin knockin pig model recapitulates features of selective neurodegeneration in huntington's disease. Cell (2018) 173(4):989–1002.e13. doi: 10.1016/j.cell.2018.03.005
67. Bai D, Yin P, Zhang Y, Sun F, Chen L, Lin L, et al. Lack of association of somatic cag repeat expansion with striatal neurodegeneration in hd knock-in animal models. Hum Mol Genet (2021) 30(16):1497–508. doi: 10.1093/hmg/ddab129
68. Baxa M, Hruska-Plochan M, Juhas S, Vodicka P, Pavlok A, Juhasova J, et al. A transgenic minipig model of huntington's disease. J Huntingtons Dis (2013) 2(1):47–68. doi: 10.3233/jhd-130001
69. Valekova I, Jarkovska K, Kotrcova E, Bucci J, Ellederova Z, Juhas S, et al. Revelation of the ifnα, il-10, il-8 and il-1β as promising biomarkers reflecting immuno-pathological mechanisms in porcine huntington's disease model. J Neuroimmunol (2016) 293:71–81. doi: 10.1016/j.jneuroim.2016.02.012
70. Yang S-H, Cheng P-H, Banta H, Piotrowska-Nitsche K, Yang J-J, Cheng EC, et al. Towards a transgenic model of huntington’s disease in a non-human primate. Nature (2008) 453(7197):921–4. doi: 10.1038/nature06975
71. Chan AW, Jiang J, Chen Y, Li C, Prucha MS, Hu Y, et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic huntington disease monkey model from infancy to adulthood. PloS One (2015) 10(5):e0122335. doi: 10.1371/journal.pone.0122335
72. Raper J, Bosinger S, Johnson Z, Tharp G, Moran SP, Chan AWS. Increased irritability, anxiety, and immune reactivity in transgenic huntington's disease monkeys. Brain Behav Immun (2016) 58:181–90. doi: 10.1016/j.bbi.2016.07.004
73. Silvestroni A, Faull RL, Strand AD, Möller T. Distinct neuroinflammatory profile in post-mortem human huntington's disease. Neuroreport (2009) 20(12):1098–103. doi: 10.1097/WNR.0b013e32832e34ee
74. Du G, Dong W, Yang Q, Yu X, Ma J, Gu W, et al. Altered gut microbiota related to inflammatory responses in patients with huntington’s disease. Front Immunol (2021) 11:603594. doi: 10.3389/fimmu.2020.603594
75. von Essen MR, Hellem MN, Vinther-Jensen T, Ammitzbøll C, Hansen RH, Hjermind LE, et al. Early intrathecal T helper 17.1 cell activity in huntington disease. Ann Neurol (2020) 87(2):246–55. doi: 10.1002/ana.25647
76. Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi SJ, et al. Abnormal peripheral chemokine profile in huntington’s disease. PloS currents (2011) 3:1–9. doi: 10.1371/currents.RRN1231
77. Huber AK, Giles DA, Segal BM, Irani DN. An emerging role for eotaxins in neurodegenerative disease. Clin Immunol (2018) 189:29–33. doi: 10.1016/j.clim.2016.09.010
78. Fang Q, Strand A, Law W, Faca VM, Fitzgibbon MP, Hamel N, et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with huntington disease. Mol Cell Proteomics (2009) 8(3):451–66. doi: 10.1074/mcp.M800231-MCP200
79. Singhrao S, Neal J, Morgan B, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in huntington's disease. Exp Neurol (1999) 159(2):362–76. doi: 10.1006/exnr.1999.7170
80. Mott RT, Ait-Ghezala G, Town T, Mori T, Vendrame M, Zeng J, et al. Neuronal expression of Cd22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia (2004) 46(4):369–79. doi: 10.1002/glia.20009
81. Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, et al. Microrna profiling of multiple sclerosis lesions identifies modulators of the regulatory protein Cd47. Brain (2009) 132(12):3342–52. doi: 10.1093/brain/awp300
82. Lago N, Pannunzio B, Amo-Aparicio J, López-Vales R, Peluffo H. Cd200 modulates spinal cord injury neuroinflammation and outcome through Cd200r1. Brain Behavior Immun (2018) 73:416–26. doi: 10.1016/j.bbi.2018.06.002
83. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with Ox2 (Cd200). Science (2000) 290(5497):1768–71. doi: 10.1126/science.290.5497.1768
84. Borrego F. The Cd300 molecules: An emerging family of regulators of the immune system. Blood J Am Soc Hematol (2013) 121(11):1951–60. doi: 10.1182/blood-2012-09-435057
85. Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and Cx3cr1-expressing microglia. Proc Natl Acad Sci (1998) 95(18):10896–901. doi: 10.1073/pnas.95.18.10896
86. Nomiyama H, Osada N, Yoshie O. Systematic classification of vertebrate chemokines based on conserved synteny and evolutionary history. Genes to Cells (2013) 18(1):1–16. doi: 10.1111/gtc.12013
87. Sheridan GK, Murphy KJ. Neuron–glia crosstalk in health and disease: Fractalkine and Cx3cr1 take centre stage. Open Biol (2013) 3(12):130181. doi: 10.1098/rsob.130181
88. Chandrasekaran S, Bonchev D. Network analysis of human post-mortem microarrays reveals novel genes, micrornas, and mechanistic scenarios of potential importance in fighting huntington's disease. Comput Struct Biotechnol J (2016) 14:117–30. doi: 10.1016/j.csbj.2016.02.001
89. Stack EC, Smith KM, Ryu H, Cormier K, Chen M, Hagerty SW, et al. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic huntington's disease mice. Biochim Biophys Acta (BBA)-Molecular Basis Dis (2006) 1762(3):373–80. doi: 10.1016/j.bbadis.2005.11.002
90. Wang X, Zhu S, Drozda M, Zhang W, Stavrovskaya IG, Cattaneo E, et al. Minocycline inhibits caspase-independent and-dependent mitochondrial cell death pathways in models of huntington's disease. Proc Natl Acad Sci (2003) 100(18):10483–7. doi: 10.1073/pnas.1832501100
91. Ryu J, Choi H, McLarnon J. Combined minocycline plus pyruvate treatment enhances effects of each agent to inhibit inflammation, oxidative damage, and neuronal loss in an excitotoxic animal model of huntington’s disease. Neuroscience (2006) 141(4):1835–48. doi: 10.1016/j.neuroscience.2006.05.043
92. Huntington Study Group. Minocycline safety and tolerability in huntington disease. Neurology (2004) 63(3):547–9. doi: 10.1212/01.wnl.0000133403.30559.ff
93. Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, et al. Controlled clinical trial of cannabidiol in huntington's disease. Pharmacol Biochem Behav (1991) 40(3):701–8. doi: 10.1016/0091-3057(91)90386-g
94. Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the cns of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol (2012) 251(1-2):14–24. doi: 10.1016/j.jneuroim.2012.06.005
95. Kaye J, Piryatinsky V, Birnberg T, Hingaly T, Raymond E, Kashi R, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci U.S.A. (2016) 113(41):E6145–e52. doi: 10.1073/pnas.1607843113
96. Coskun M, Vermeire S, Nielsen OH. Novel targeted therapies for inflammatory bowel disease. Trends Pharmacol Sci (2017) 38(2):127–42. doi: 10.1016/j.tips.2016.10.014
97. Gentile A, Musella A, De Vito F, Fresegna D, Bullitta S, Rizzo FR, et al. Laquinimod ameliorates excitotoxic damage by regulating glutamate re-uptake. J Neuroinflamm (2018) 15(1):5. doi: 10.1186/s12974-017-1048-6
98. Katsumoto A, Miranda AS, Butovsky O, Teixeira AL, Ransohoff RM, Lamb BT. Laquinimod attenuates inflammation by modulating macrophage functions in traumatic brain injury mouse model. J Neuroinflamm (2018) 15(1):26. doi: 10.1186/s12974-018-1075-y
99. Wilmes AT, Reinehr S, Kühn S, Pedreiturria X, Petrikowski L, Faissner S, et al. Laquinimod protects the optic nerve and retina in an experimental autoimmune encephalomyelitis model. J Neuroinflamm (2018) 15(1):183. doi: 10.1186/s12974-018-1208-3
100. Ellrichmann G, Blusch A, Fatoba O, Brunner J, Reick C, Hayardeny L, et al. Laquinimod treatment in the R6/2 mouse model. Sci Rep (2017) 7(1):4947. doi: 10.1038/s41598-017-04990-1
101. Garcia-Miralles M, Hong X, Tan LJ, Caron NS, Huang Y, To XV, et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the Yac128 model of huntington disease. Sci Rep (2016) 6:31652. doi: 10.1038/srep31652
102. Garcia-Miralles M, Yusof N, Tan JY, Radulescu CI, Sidik H, Tan LJ, et al. Laquinimod treatment improves myelination deficits at the transcriptional and ultrastructural levels in the Yac128 mouse model of huntington disease. Mol Neurobiol (2019) 56(6):4464–78. doi: 10.1007/s12035-018-1393-1
103. Varrin-Doyer M, Zamvil SS, Schulze-Topphoff U. Laquinimod, an up-and-Coming immunomodulatory agent for treatment of multiple sclerosis. Exp Neurol (2014) 262 Pt A:66–71. doi: 10.1016/j.expneurol.2014.04.002
104. Yin P, Liu Q, Pan Y, Yang W, Yang S, Wei W, et al. Phosphorylation of myelin regulatory factor by Prkg2 mediates demyelination in huntington's disease. EMBO Rep (2020) 21(6):e49783. doi: 10.15252/embr.201949783
105. Ehrnhoefer DE, Caron NS, Deng Y, Qiu X, Tsang M, Hayden MR. Laquinimod decreases bax expression and reduces caspase-6 activation in neurons. Exp Neurol (2016) 283(Pt A):121–8. doi: 10.1016/j.expneurol.2016.06.008
106. Ellrichmann G, Blusch A, Fatoba O, Brunner J, Reick C, Hayardeny L, et al. Author correction: Laquinimod treatment in the R6/2 mouse model. Sci Rep (2019) 9(1):4960. doi: 10.1038/s41598-018-37926-4
107. Locksley RM, Killeen N, Lenardo MJ. The tnf and tnf receptor superfamilies: Integrating mammalian biology. Cell (2001) 104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9
108. Hsiao HY, Chiu FL, Chen CM, Wu YR, Chen HM, Chen YC, et al. Inhibition of soluble tumor necrosis factor is therapeutic in huntington's disease. Hum Mol Genet (2014) 23(16):4328–44. doi: 10.1093/hmg/ddu151
109. Fatoba O, Ohtake Y, Itokazu T, Yamashita T. Immunotherapies in huntington's disease and A-synucleinopathies. Front Immunol (2020) 11:337. doi: 10.3389/fimmu.2020.00337
110. Pido-Lopez J, Tanudjojo B, Farag S, Bondulich MK, Andre R, Tabrizi SJ, et al. Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of huntington's disease by etanercept treatment. Sci Rep (2019) 9(1):7202. doi: 10.1038/s41598-019-43627-3
111. Southwell AL, Franciosi S, Villanueva EB, Xie Y, Winter LA, Veeraraghavan J, et al. Anti-semaphorin 4d immunotherapy ameliorates neuropathology and some cognitive impairment in the Yac128 mouse model of huntington disease. Neurobiol Dis (2015) 76:46–56. doi: 10.1016/j.nbd.2015.01.002
112. Ito Y, Oinuma I, Katoh H, Kaibuchi K, Negishi M. Sema4d/Plexin-B1 activates gsk-3beta through r-ras gap activity, inducing growth cone collapse. EMBO Rep (2006) 7(7):704–9. doi: 10.1038/sj.embor.7400737
113. Blennow K, Davidsson P, Wallin A, Frcdman P, Gottfries C, Månsson J, et al. Differences in cerebrospinal fluid gangliosides between “Probable alzheimer’s disease” and normal aging. Aging Clin Exp Res (1992) 4(4):301–6. doi: 10.1007/BF03324111
114. Schneider J. Gm1 ganglioside in the treatment of parkinson's disease a. Ann New York Acad Sci (1998) 845(1):363–73. doi: 10.1111/j.1749-6632.1998.tb09688.x
115. Schneider JS, Aras R, Williams CK, Koprich JB, Brotchie JM, Singh V. Gm1 ganglioside modifies A-synuclein toxicity and is neuroprotective in a rat A-synuclein model of parkinson’s disease. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-42847-x
116. Alpaugh M, Galleguillos D, Forero J, Morales LC, Lackey SW, Kar P, et al. Disease-modifying effects of ganglioside Gm1 in huntington's disease models. EMBO Mol Med (2017) 9(11):1537–57. doi: 10.15252/emmm.201707763
117. Prados ME, Correa-Sáez A, Unciti-Broceta JD, Garrido-Rodríguez M, Jimenez-Jimenez C, Mazzone M, et al. Betulinic acid hydroxamate is neuroprotective and induces protein phosphatase 2a-dependent hif-1α stabilization and post-transcriptional dephosphorylation of prolyl hydrolase 2. Neurotherapeutics (2021) 18(3):1849–61. doi: 10.1007/s13311-021-01089-4
118. Purushothaman B, Sumathi T. 5, 6, 7 trihydroxy flavone armoured neurodegeneration caused by quinolinic acid induced huntington’s like disease in rat striatum-reinstating the level of brain neurotrophins with special reference to cognitive-socio behaviour, biochemical and histopathological aspects. Neurosci Res (2022) 174:25–35. doi: 10.1016/j.neures.2021.08.003

Keywords: Huntington’s disease, immune response, inflammation, animal models, immune therapies
Citation: Jia Q, Li S, Li X-J and Yin P (2022) Neuroinflammation in Huntington’s disease: From animal models to clinical therapeutics. Front. Immunol. 13:1088124. doi: 10.3389/fimmu.2022.1088124
Received: 03 November 2022; Accepted: 29 November 2022;
Published: 22 December 2022.
Edited by:
Ramkumar Mathur, University of North Dakota, United StatesCopyright © 2022 Jia, Li, Li and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Jiang Li, eGpsaTMzQGpudS5lZHUuY24=; Peng Yin, eWlucGVuZzE3N0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.