 Wei Jiang
Wei Jiang Li-Xi Zhang2
Li-Xi Zhang2 Ming Dong
Ming Dong- 1Department of Neurology and Neuroscience Center, The First Hospital of Jilin University, Changchun, China
- 2Department of Thyroid Surgery, The First Hospital of Jilin University, Changchun, China
- 3Department of Neurosurgery, The First Hospital of Jilin University, Changchun, China
- 4Department of Ophthalmology, The Second Hospital of Jilin University, Changchun, China
Increasing evidence suggests that epigenetic mechanisms have great potential in the field of pain. The changes and roles of epigenetics of the spinal cord and dorsal root ganglia in the chronic pain process may provide broad insights for future pain management. Pro-inflammatory cytokines and chemokines released by microglia and astrocytes, as well as blood-derived macrophages, play critical roles in inducing and maintaining chronic pain, while histone modifications may play an important role in inflammatory metabolism. This review provides an overview of neuroinflammation and chronic pain, and we systematically discuss the regulation of neuroinflammation and histone modifications in the context of chronic pain. Specifically, we analyzed the role of epigenetics in alleviating or exacerbating chronic pain by modulating microglia, astrocytes, and the proinflammatory mediators they release. This review aimed to contribute to the discovery of new therapeutic targets for chronic pain.
1. Introduction
Chronic pain includes inflammatory pain and neuropathic pain (NP). Inflammatory pain refers to peripheral tissue damage and persistent inflammation and neuropathic pain is a pathological adaptation of the peripheral nervous system (PNS) or central nervous system (CNS) (1). They are characterized by persistent nociceptive hypersensitivity (2). Chronic pain symptoms include an excessive increase in pain caused by painful stimuli (hyperalgesia) and pain caused by stimuli that do not usually cause pain (allodynia) (3). Current analgesics, such as opioids and nonsteroidal anti-inflammatory drugs (NSAIDS), relieve pain symptoms by inhibiting neuronal activity. However, their effects are unsatisfactory, and these drugs have serious toxicity and dose-limiting side effects (4–6). Due to the complexity of the mechanisms associated with chronic pain, researchers’ incomplete understanding of the mechanism reduces the therapeutic effect. Previous studies on the pathological mechanisms of chronic pain mainly focused on the expression of neuronal channels, receptors, and neurotransmitters (7). More research is needed on the activation of non-neurons and the expression of pro-inflammatory neuromodulators.
Neuroinflammation can occur both in the PNS and CNS. Activation of glial cells and the release and interaction of inflammatory mediators are emerging as key mechanisms for chronic pain. Local inflammatory response, including the infiltration of hematogenous immune cells and induction of various cytokines, can participate in the pathological response of chronic peripheral nerve injury. Pain can be alleviated by inhibiting the activation of inflammatory cells and the synthesis or accumulation of inflammatory factors, or by blocking the interaction between inflammatory factors and pain receptors in nociceptors. Inflammation and nerve damage can lead to the hyperexcitability of neurons. Then, a positive feedback loop between peripheral and central inflammatory cells, chemokines, and cytokines maintains neuroinflammation and chronic pain.
Accumulating evidence has shown that molecular changes in chronic pain are controlled by epigenetic mechanisms. Epigenetic modifications induce or suppresses gene expression without altering the DNA sequence, including DNA methylation, post-translational histone modifications, and expression of microRNAs (miRNAs). Histone tails are modified by acetylation and methylation. They regulate chromatin structure and function and gene expression through post-transcriptional modifications (PTMs) of the N-terminal histone tails of nucleosomes (8, 9). The histone acetyltransferase (HAT) family acts on lysine residues, leading to transcriptional activation. Conversely, the histone deacetylase family (HDACs) deacetylates histones, leading to transcriptional repression (10). Different from histone acetylation, histone methylation can lead to both gene repression and activation of gene transcription because it can act on different sites of amino acid residues. For example, methylation of Lys9 or Lys27 that occurs in histone H3 typically represses gene expression, whereas methylation of H3 Lys4, Lys36, or Lys79 typically inhibits gene activation (11). Chromatin modification is not limited to simple DNA methylation or histone modification; the activation or suppression of a gene usually involves a large number of histone co-modifications and DNA methylation.
Many studies have demonstrated that histone modifications can affect the generation and maintenance of chronic pain (12–15). The link between histone modification-induced inflammatory cells and cytokine production inhibition and pain has been reported in various disease models (16, 17). How does the inheritance of histone modifications alleviate pain by suppressing inflammation? In this review, we describe the changes in inflammatory cells and cytokines observed after inflammatory and neurological injury, which are modulated by histone modifications. Despite the increasing importance of epigenetics in the field of pain, discoveries in this area are still limited, and more research is needed to elucidate the molecular pathways behind the pain. We believe that histone modification during inflammation may provide a new therapeutic approach for treatment-resistant chronic pain.
2. Microglia and astrocytes: “Relay station” for epigenetic treatment of chronic pain
The role of neuroinflammation in chronic pain state is widely recognized. Microglia and astrocytes in the CNS, and satellite glia in the dorsal root and trigeminal ganglia are involved in chronic pain (18). Microglia and astrocytes, particularly microglia, are discussed in this review. Activation of microglia and astrocytes in the spinal cord and dorsal root ganglion (DRG)leads to the production of proinflammatory cytokines/chemokines, resulting in an inflammatory cascade (19). Activated microglia have both pro-inflammatory (M1) and anti-inflammatory (M2) functions. M1 microglia express pro-inflammatory molecules, whereas M2 microglia expressed anti-inflammation molecules (20). Neuroinflammation is a defense response, and anti-inflammatory cytokines and proinflammatory factors regulate these pain states. Epigenetic modifications of pro-inflammatory and anti-inflammatory factors have increased our understanding of the mechanisms underlying pain. Microglia and astrocytes have close connections to neurons and communicate with them in a bidirectional manner. Upon activation, microglia and astrocytes regulate the interaction between the nervous system and the immune system by secreting soluble mediators, including chemokines (21). This inflammatory response eventually lowers the triggering thresholds of A-σ- and C-fiber nociceptors, leading to chronic pain (22). Numerous studies have shown that microglia and astrocytes play key roles in the development of neuropathic and acute inflammatory pain (23–26). As promoters of chronic pain, non-neuronal immune cells may provide therapeutic ideas for future treatment (27).
Inflammatory mediators are involved in the production and maintenance of many forms of pain, and their importance is well-established. Chemokines are expressed in astrocytes and microglia in the spinal cord in the NP model (28). Moreover, studies have shown that the CX3CL1/CX3CR1 signaling pathway can regulate neuron-microglia interactions in the spinal cord, thus mediating the development of neuropathic pain (28, 29). Histone acetylation, deacetylation, and methylation have been shown to modulate chronic pain through inflammatory mediators (14, 30–32). Histone acetylation regulates chemokine expression in the CNS and peripheral tissues (29, 33, 34). Histone modifications, including phosphorylation, acetylation, and methylation, can occur in specific regions of the promoters of the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and IL-1β and the anti-inflammatory cytokine IL-10 (35–37). Histone modifications reduce inflammation by decreasing the expression of some pro-inflammatory mediators, including IL-1β and TNF-α, and increasing the production of anti-inflammatory factors, including IL-10 (38, 39). IL-10 overexpression can reduce the expression of pro-inflammatory factor TNF-α in microglia, thereby alleviating inflammatory pain (40). On the contrary, IL-1β, IL-6, and TNF-α are involved in the activation of microglia and astrocytes (41, 42), leading to peripheral nerve injury or chronic pain caused by inflammatory responses (43–45). HDAC inhibitors have been shown to improve symptoms in several animal models of inflammatory diseases (46, 47). Data show that HDAC can be continuously expressed in mouse microglia and astrocytes, and its inhibition inhibits the neuroinflammatory response (48). Monocyte chemotactic protein-3 (MCP-3), also known as CCL7, increases after sciatic nerve ligation. MCP-3 is regulated by IL-6. Intrathecal administration of IL-6 decreased Lys27 H3 trimethylation at the McP-3 gene promoter, resulting in increased MCP-3 expression (1). Furthermore, MCP-3 expression is increased in astrocytes but not in microglia or neurons. Hence, we discuss the histone modification regulation of cytokines or chemokines through glial cells that regulate the production and maintenance of chronic pain. In recent years, numerous studies have focused on this issue. Epigenetic regulation of the activation of inflammatory cells and the expression of inflammatory mediators may be a future target for the prevention and treatment of pain.
3. Histone acetylation and methylation regulate chronic pain through neuro-inflammation in spinal systems
Histone acetylation primarily relies on two key enzymes, HATs and HDACs. A dynamic balance exists between HATs and HDACs to maintain stable gene expression. Nerve injury or neuroinflammatory infiltration can lead to the upregulation of histone acetylase or histone deacetylase, leading to pain. Here, we discuss the dual role and underlying mechanism of histone acetylation in the development of chronic pain. HATs reduce the electrostatic interactions between histones and DNA, thus making chromosome transcription easier. The role of HDACs is to suppress gene transcription. The molecular mechanism, in summary, involves the removal of acetyl groups, which increases the positive charge of histone tails, thereby increasing the affinity of DNA binding, leading to chromosome densification and inhibiting transcription (49, 50). Histone acetylation and deacetylation are processes of dynamic equilibrium, and when these enzymes are activated or inhibited, they lead to many diseases, including neurological dysfunction. We briefly discuss the role and possible mechanisms of histone acetylation, histone deacetylation, and histone methylation in chronic pain through neuroinflammation.
3.1. Histone acetylation promotes pain
Histone hypoacetylation decreases the response to nerve injury. Correspondingly, histone hyperacetylation increases the response to nerve injury, and blocking histone acetylation is effective in relieving pain (34, 49, 51). A total of 11 HDAC proteins are divided into three categories: Category I includes HDACs 1, 2, 3, and 8; Category IIa includes HDACs 4, 5, 7, and 9; Category IIb includes HDACs 6 and 10; Category IV includes HDAC 11 (52). CX3CL1 is involved in paclitaxel-induced neuropathic pain (PINP), and blocking CX3CL1 significantly inhibits microglial activation and mechanical allodynia. Inhibition of the nuclear factor-κB (NF-κB) pathway significantly inhibits H4 acetylation in the CX3CL1 promoter region and inhibits CX3CL1 upregulation, thereby alleviating pain. NF-κB can also be co-expressed with specific microglial markers; therefore, microglia may play an important role in neuropathy of pain (53). Zhang et al. demonstrated that inflammatory pain and NP resulted in increased histone acetylation levels in the brainstem nucleus raphe magnus (NRM) in a time-dependent manner (54). JNJ-26481585, a new pan-HDAC inhibitor, induces mechanical hypersensitivity in mice. In addition, IL-1 β expression in mouse macrophage RAW 264.7 was increased after JNJ-26481585 treatment in vitro (55). Histone acetylation can not only upregulate the pain state through inflammatory factors but also through signaling pathways in glial cells. In one study, paclitaxel-induced neuropathic pain (PINP) increased the activity of the κB subunit P65 and its binding to the IRF8 promoter region. These effects lead to the hyperacetylation of histone H3 in the IRF8 promoter region, which promotes paclitaxel-induced allodynia pain and IRF8 transcription and expression in the rat dorsal horn (19). Previous studies have shown that overexpression of IRF8 can enhance cytokines in microglia, and knockdown of IRF8 can attenuate mechanical allodynia caused by nerve injury (56).
Microglia are macrophage-like cells that reside in the central nervous system (57). Histone modification modulates pain through inflammatory intervention by microglia, similarly, acetylation regulation interferes with pain states by affecting the expression of several molecules in the chemokine system of peripheral tissue, and these chemokines can act on macrophages in peripheral tissues to affect pain (34). The C-X-C chemokine ligand type 2 [macrophage inflammatory protein 2 (MIP-2)]/C-X-C chemokine receptor type 2 (CXCR2) axis targets neutrophils and macrophages in the injured sciatic nerve (SCN) and plays an important role in neuropathic pain. High expression of these proteins is controlled by the hyperacetylation of histone H3 in neutrophils and macrophage nuclei in the SCN. HAT inhibitors can inhibit the upregulation of MIP-2 and CXCR2, thereby preventing NP (34). CCL2 and CCL3 are upregulated in macrophage immune cells in the injured SCN through lysine 9-acetylated histone H3 (H3K9Ac) and lysine 4-trimethylated H3 [H3K4me (3)]. Similarly, histone acetyltransferase inhibitors can inhibit the upregulation of CCLs and CCRs. These chemokine cascades are amplified and can be involved in chronic inflammation, causing pain after nerve injury (58).
3.2. Histone deacetylation promotes pain
Microglia may be involved in neuropathy of chronic pain. Treadmill running has been suggested to activate the increase in acetylated histone H3K9 in microglia, thereby promoting the production of the anti-inflammatory factor IL-10, downregulating pro-inflammatory cytokines, and ultimately improving pain (39).
D-hydroxybutyric acid (DBHB) is an HDAC inhibitor that enhances histone acetylation after spinal cord injury (SCI) in mice. DBHB can inhibit the activation of microglia and astrocyte, reduce the expression of proinflammatory factors, improve inflammatory response, and play a protective role after SCI injury (59). Similarly, HDAC inhibitors have been demonstrated to increase the acetylation of the GAD65 promoter, and global histone hyperacetylation counterbalances the inflammatory pain effect. GAD65 is used for GABA synthesis in synaptic vesicles, The downregulation of GAD65 activity leads to impaired synaptic inhibition of GABA and increased neuronal excitability, thereby promoting the occurrence of pain (60). Chronic constriction injury (CCI) can cause mechanical allodynia, hyperalgesia, and increased TNF-α levels. Oral administration of sodium butyrate, an HDAC inhibitor, significantly attenuates the pro-inflammatory cytokine TNF-α and reduces injury-related pain behaviors (61). The regulation of inflammatory factors through histone modifications is a future direction of research. Activation of JNK-c-Jun signaling was also accompanied by an increase in HDAC1 expression. A novel selective HDACI, LG325, blocked the stimulation of c-Jun phosphorylation. Double immunostaining showed that p-JNK co-localized with p-C-Jun and HDAC1 in astrocytes in the spinal dorsal horn (62). The JNK signaling pathway is important for the induction and maintenance of chronic pain (63). Similarly, HDAC2, another subtype of HDAC, is expressed in astrocytes and elevated in neuropathic pain (64), and HDAC2 mRNA is found in cortical microglia (65). In contrast, HDAC2 was shown to be unexpressed in both microglia and astrocytes in the adult mouse brain without neuropathic pain (66). These results suggest that HDAC may inhibit the expression of pain related genes in astrocytes. Cell-specific expression in the CNS may be responsible for this phenomenon. Attenuating pain by reducing the production of pro-inflammatory factors may be a good target in the future. Whether HDAC can be used as a target for the treatment of chronic pain through glial cells still needs to be studied.
HATs and HDACs seem to have different effects on chronic pain. There are many factors responsible for this difference, such as different doses and cell types, which lead to different effects of HDAC inhibitors. TSA, as a broad-spectrum HDAC inhibitor, has different pro-inflammatory and anti-inflammatory effects on macrophages within a certain concentration range (67). Furthermore, with respect to intrathecal injection, oral administration is not as effective as local injection, and there are more unknown influencing factors (68–70). Histone modification is a complex process. Most importantly, histone acetylases or deacetylases do not increase or decrease NP alone but act as acetyl agents to affect chronic pain by increasing or decreasing the expression of specific genes. Acetylation can bind to promoters of different genes, leading to acetylation or deacetylation of genes, and the product of gene expression determines whether chronic pain is aggravated or alleviated. Therefore, understanding the acetylation or deacetylation of corresponding gene promoters caused by histone acetylases or deacetylases can provide new guidance for the treatment of chronic pain. We should focus on specific HDACs and HATs in the future, hoping to treat pain by targeting different cells or even different genetic targets using highly specific inhibitors.
Although histone acetylation and deacetylation are effective in alleviating chronic pain symptoms, they have major limitations. First, most inhibitors act both centrally and peripherally, raising concerns regarding drug toxicity. Second, many of these compounds require long-term, high-dose treatment that increases side effects and reduces patient compliance. These factors should be considered when using these compounds in the context of pain. Histone acetylation is not only involved in HATs and HDACs but may also involve other epigenetic forms. Its complex and highly cell-specific nature suggests that we still have a long way to go (71). Therefore, we cannot simply assume that histone acetylation and deacetylation affect gene transcription alone. This is also what we discussed above: HDAC can have two interactive ways on chronic pain.
3.3. Histone methylation regulates pain
Histone methyltransferases and demethylases are also altered in inflammation-mediated neuropathic pain. G9a, a histone H3K9 dimethyl transferase, catalyzes H3K9 dimethylation (H3K9me2), which is associated with gene silencing (72). Inhibition of G9a can reverse Mu opioid receptor (MOR, encoded by Oprm1) downregulation in the DRG induced by spinal nerve ligation (SNL) and enhances the effect of morphine on pain hypersensitivity induced by nerve injury (73). Enhancer of zeste homolog 2 (EZH2) leads to gene silencing by catalyzing di-methylation and tri-methylation of histone H3 on lysine 27 (H3K27Me2/3) and has been demonstrated to be expressed in microglia (74). As an EZH2 inhibitor, EPZ-6438 controls important inflammatory gene targets by regulating the promoter levels of interferon regulators and transcriptional activators. Therefore, EPZ-6438 may be an effective treatment for neuroinflammatory diseases associated with microglial activation (75). After partial sciatic nerve ligation (PSL), the number of microglia expressing EZH2 increased significantly in the spinal cord of rats (37). Activation of microglia leads to increased overall levels of EZH2 and tri-methylated H3K27 in the L4 and L5 spinal dorsal horn, and excessive production of TNF-α and IL-β in the spinal dorsal horn contributes to NP. Intrathecal injection of an EZH2 inhibitor can relieve chronic pain (37). MCP-3 (CCL7) expression increased in IL-6 sensitive astrocytes after CCI in mice. This was due to the reduced H3K27 trimethylation of the MCP-3 promoter. Increased MCP-3 expression activates spinal microglia expressing CCR2, thereby inducing central sensitization. The CCR2 receptor expressed in spinal cord neurons and microglia may be induced by MCP3, thereby enhancing neuronal sensitivity (76).
Granulocyte macrophage colony-stimulating factor (GM-CSF) is a key pro-inflammatory cytokine. In mouse models of arthritis and inflammatory pain, GM-CSF up-regulated IRF4 expression by enhancing JMJD3 (histone H3K27 demethylase) activity (77, 78). IRF4 regulates the formation of CCL17 and then mediates the proinflammatory and analgesic effects of GM-CSF. After SCI, JMJD3 expression is upregulated and MMP-3 and MMP-9 gene expressions are regulated. In addition, inhibition of JMJD3 inhibited the expression of matrix metalloprotease-3 (MMP-3) and MMP-9 genes and significantly reduced the permeability of the blood-spinal cord barrier (BSCB) (79). Subsequently, increased JMJD3 expression in the cauda equina of rats with lumbar spinal stenosis-induced chronic NP was demonstrated. JMJD3 infiltrates cauda equina macrophages after injury, mediates neuroinflammation, and increases the permeability of the blood-nerve barrier (80). We summarize the key studies focusing on the links between histone modification with chronic pain in Table 1. These results suggest a potential therapeutic approach for the treatment of inflammatory pain and NP.
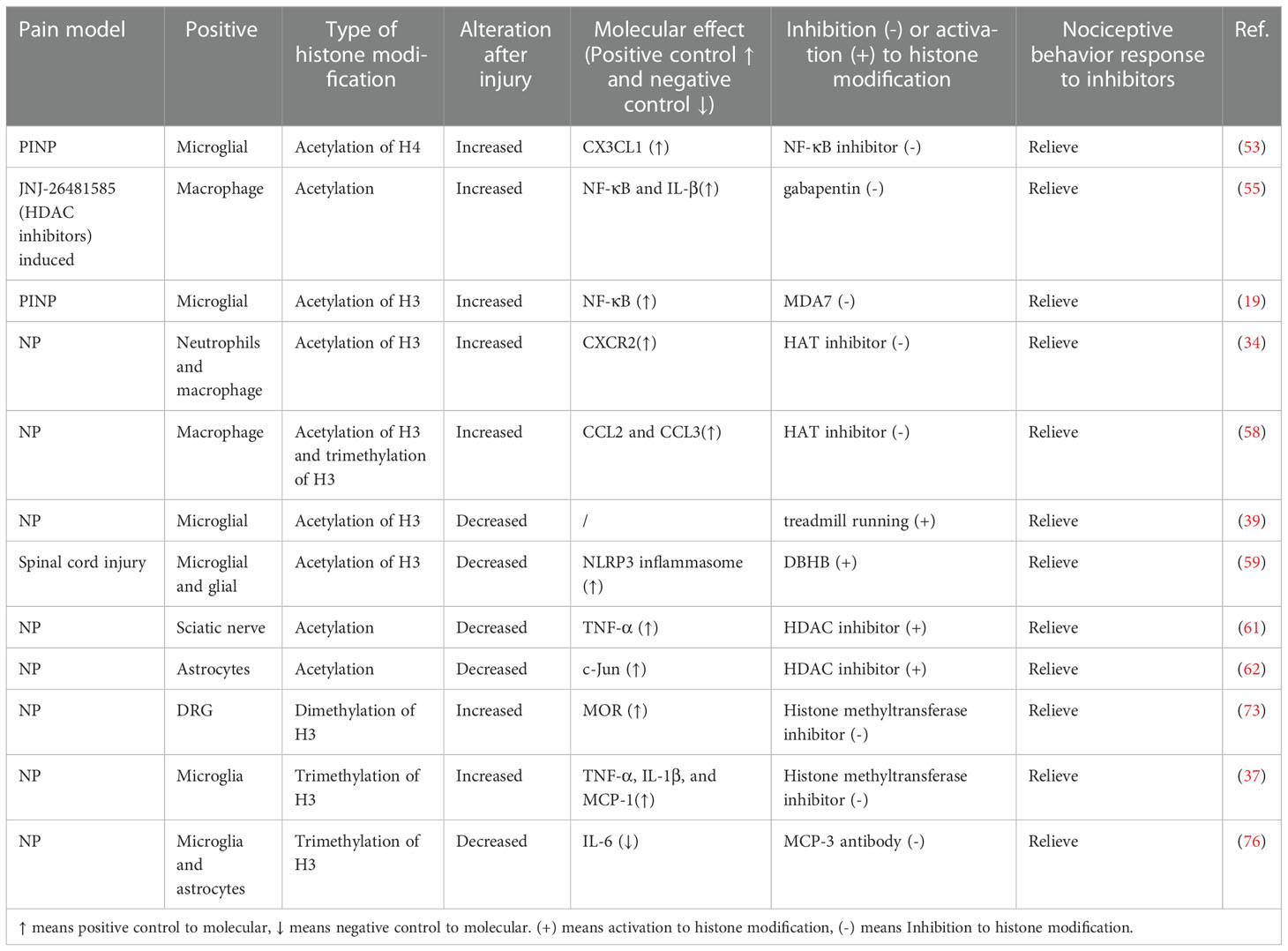
Table 1 Roles of histone modification in chronic in rodent models.
4. Conclusions and future studies
It is clear that we are at this initial stage, where there is no consensus in the literature and some contradictory findings have been described. In the field of pain, the contributions of genetic and epigenetic mechanisms have been widely recognized. We believe that the epigenetic mechanism of chronic pain will be further explored in the future and there is great potential for the epigenetic intervention of inflammatory system genes to improve pain. Currently, our discussion of rodent models, which poses a challenge for clinical transformation, is still in the preclinical stage. Further studies on epigenetics and neuroinflammation are needed to find preclinical transformational management of neuropathic pain. Epigenetic mechanisms controlling chronic pain may offer a wide range of potential therapeutic targets. As a potential targeted intervention, epigenetic inheritance may improve the symptoms of chronic pain by suppressing inflammation, preventing injury sensitization.
Author contributions
WJ and X-YT designed the study, reviewed the literature, and drafted the manuscript. L-XZ helped in writing and revising the manuscript. PY and MD contributed significantly to the revision of the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31872772) and the Natural Science Foundation of Jilin Province of China (Grant No. 20200201606JC) to MD.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends Neurosci (2015) 38(4):237–46. doi: 10.1016/j.tins.2015.02.001
2. Zhuo M. Cortical excitation and chronic pain. Trends Neurosci (2008) 31(4):199–207. doi: 10.1016/j.tins.2008.01.003
3. Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol (2014) 13(9):924–35. doi: 10.1016/s1474-4422(14)70102-4
4. O'Brien JB, Roman DL. Novel treatments for chronic pain: moving beyond opioids. Transl Res (2021) 234:1–19. doi: 10.1016/j.trsl.2021.03.008
5. Koller G, Schwarzer A, Halfter K, Soyka M. Pain management in opioid maintenance treatment. Expert Opin Pharmacother (2019) 20(16):1993–2005. doi: 10.1080/14656566.2019.1652270
6. Ribeiro H, Rodrigues I, Napoleão L, Lira L, Marques D, Veríssimo M, et al. Non-steroidal anti-inflammatory drugs (NSAIDs), pain and aging: Adjusting prescription to patient features. BioMed Pharmacother (2022) 150:112958. doi: 10.1016/j.biopha.2022.112958
7. Krames ES. The dorsal root ganglion in chronic pain and as a target for neuromodulation: a review. Neuromodulation (2015) 18(1):24–32. doi: 10.1111/ner.12247
8. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med (2017) 49(4):e324. doi: 10.1038/emm.2017.11
9. Li W, Nagaraja S, Delcuve GP, Hendzel MJ, Davie JR. Effects of histone acetylation, ubiquitination and variants on nucleosome stability. Biochem J (1993) 296(Pt 3):737–44. doi: 10.1042/bj2960737
10. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
11. Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, et al. Epigenetics in the nervous system. J Neurosci (2008) 28(46):11753–9. doi: 10.1523/jneurosci.3797-08.2008
12. Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain (2010) 6:51. doi: 10.1186/1744-8069-6-51
13. Chiechio S, Copani A, Zammataro M, Battaglia G, RWt G, Nicoletti F. Transcriptional regulation of type-2 metabotropic glutamate receptors: an epigenetic path to novel treatments for chronic pain. Trends Pharmacol Sci (2010) 31(4):153–60. doi: 10.1016/j.tips.2009.12.003
14. Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, RWt G, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol (2009) 75(5):1014–20. doi: 10.1124/mol.108.054346
15. Cherng CH, Lee KC, Chien CC, Chou KY, Cheng YC, Hsin ST, et al. Baicalin ameliorates neuropathic pain by suppressing HDAC1 expression in the spinal cord of spinal nerve ligation rats. J Formos Med Assoc (2014) 113(8):513–20. doi: 10.1016/j.jfma.2013.04.007
16. Montagna P. The primary headaches: genetics, epigenetics and a behavioural genetic model. J Headache Pain (2008) 9(2):57–69. doi: 10.1007/s10194-008-0026-x
17. Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem (2011) 22(5):450–8. doi: 10.1016/j.jnutbio.2010.03.014
18. Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain (2013) 154 Suppl 1(0 1):S10–s28. doi: 10.1016/j.pain.2013.06.022
19. Wu J, Hocevar M, Bie B, Foss JF, Naguib M. Cannabinoid type 2 receptor system modulates paclitaxel-induced microglial dysregulation and central sensitization in rats. J Pain (2019) 20(5):501–14. doi: 10.1016/j.jpain.2018.10.007
20. Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis (2013) 4(3):e525. doi: 10.1038/cddis.2013.54
21. Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci (2001) 24(8):450–5. doi: 10.1016/s0166-2236(00)01854-3
22. Waxman SG. The molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neurons. Pain (1999) Suppl 6:S133–s40. doi: 10.1016/s0304-3959(99)00147-5
23. Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature (2005) 438(7070):1017–21. doi: 10.1038/nature04223
24. Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature (2003) 424(6950):778–83. doi: 10.1038/nature01786
25. Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem (2003) 86(6):1534–44. doi: 10.1046/j.1471-4159.2003.01969.x
26. Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain (2011) 134(Pt 4):1127–39. doi: 10.1093/brain/awr025
27. Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci (2006) 26(16):4308–17. doi: 10.1523/jneurosci.0003-06.2006
28. Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain (2005) 6(7):434–8. doi: 10.1016/j.jpain.2005.02.001
29. Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci (2004) 20(5):1150–60. doi: 10.1111/j.1460-9568.2004.03593.x
30. Kim CS, Hwang CK, Choi HS, Song KY, Law PY, Wei LN, et al. Neuron-restrictive silencer factor (NRSF) functions as a repressor in neuronal cells to regulate the mu opioid receptor gene. J Biol Chem (2004) 279(45):46464–73. doi: 10.1074/jbc.M403633200
31. Uchida H, Ma L, Ueda H. Epigenetic gene silencing underlies c-fiber dysfunctions in neuropathic pain. J Neurosci (2010) 30(13):4806–14. doi: 10.1523/jneurosci.5541-09.2010
32. Imai S, Saeki M, Yanase M, Horiuchi H, Abe M, Narita M, et al. Change in microRNAs associated with neuronal adaptive responses in the nucleus accumbens under neuropathic pain. J Neurosci (2011) 31(43):15294–9. doi: 10.1523/jneurosci.0921-11.2011
33. Xu T, Zhang XL, Ou-Yang HD, Li ZY, Liu CC, Huang ZZ, et al. Epigenetic upregulation of CXCL12 expression mediates antitubulin chemotherapeutics-induced neuropathic pain. Pain (2017) 158(4):637–48. doi: 10.1097/j.pain.0000000000000805
34. Kiguchi N, Kobayashi Y, Maeda T, Fukazawa Y, Tohya K, Kimura M, et al. Epigenetic augmentation of the macrophage inflammatory protein 2/C-X-C chemokine receptor type 2 axis through histone H3 acetylation in injured peripheral nerves elicits neuropathic pain. J Pharmacol Exp Ther (2012) 340(3):577–87. doi: 10.1124/jpet.111.187724
35. Hill EV, Ng TH, Burton BR, Oakley CM, Malik K, Wraith DC. Glycogen synthase kinase-3 controls IL-10 expression in CD4(+) effector T-cell subsets through epigenetic modification of the IL-10 promoter. Eur J Immunol (2015) 45(4):1103–15. doi: 10.1002/eji.201444661
36. Lee CG, Kwon HK, Sahoo A, Hwang W, So JS, Hwang JS, et al. Interaction of ets-1 with HDAC1 represses IL-10 expression in Th1 cells. J Immunol (2012) 188(5):2244–53. doi: 10.4049/jimmunol.1101614
37. Yadav R, Weng HR. EZH2 regulates spinal neuroinflammation in rats with neuropathic pain. Neuroscience (2017) 349:106–17. doi: 10.1016/j.neuroscience.2017.02.041
38. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U.S.A. (2002) 99(5):2995–3000. doi: 10.1073/pnas.052702999
39. Kami K, Taguchi S, Tajima F, Senba E. Histone acetylation in microglia contributes to exercise-induced hypoalgesia in neuropathic pain model mice. J Pain (2016) 17(5):588–99. doi: 10.1016/j.jpain.2016.01.471
40. Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther (2008) 15(3):183–90. doi: 10.1038/sj.gt.3303054
41. Klein MA, Möller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia (1997) 19(3):227–33. doi: 10.1002/(sici)1098-1136(199703)19
42. Yan HQ, Banos MA, Herregodts P, Hooghe R, Hooghe-Peters EL. Expression of interleukin (IL)-1 beta, IL-6 and their respective receptors in the normal rat brain and after injury. Eur J Immunol (1992) 22(11):2963–71. doi: 10.1002/eji.1830221131
43. Ramer MS, Murphy PG, Richardson PM, Bisby MA. Spinal nerve lesion-induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain (1998) 78(2):115–21. doi: 10.1016/s0304-3959(98)00121-3
44. Mailhot B, Christin M, Tessandier N, Sotoudeh C, Bretheau F, Turmel R, et al. Neuronal interleukin-1 receptors mediate pain in chronic inflammatory diseases. J Exp Med (2020) 217(9). doi: 10.1084/jem.20191430
45. Gao Y, Bai L, Zhou W, Yang Y, Zhang J, Li L, et al. PARP-1-regulated TNF-α expression in the dorsal root ganglia and spinal dorsal horn contributes to the pathogenesis of neuropathic pain in rats. Brain Behav Immun (2020) 88:482–96. doi: 10.1016/j.bbi.2020.04.019
46. Chung YL, Lee MY, Wang AJ, Yao LF. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther (2003) 8(5):707–17. doi: 10.1016/s1525-0016(03)00235-1
47. Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med (2005) 11(1-12):1–15. doi: 10.2119/2006-00005.Dinarello
48. Faraco G, Pittelli M, Cavone L, Fossati S, Porcu M, Mascagni P, et al. Histone deacetylase (HDAC) inhibitors reduce the glial inflammatory response in vitro and in vivo. Neurobiol Dis (2009) 36(2):269–79. doi: 10.1016/j.nbd.2009.07.019
49. Khangura RK, Bali A, Jaggi AS, Singh N. Histone acetylation and histone deacetylation in neuropathic pain: An unresolved puzzle? Eur J Pharmacol (2017) 795:36–42. doi: 10.1016/j.ejphar.2016.12.001
50. Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem (2007) 76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114
51. Zhu XY, Huang CS, Li Q, Chang RM, Song ZB, Zou WY, et al. p300 exerts an epigenetic role in chronic neuropathic pain through its acetyltransferase activity in rats following chronic constriction injury (CCI). Mol Pain (2012) 8:84. doi: 10.1186/1744-8069-8-84
52. Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol (2004) 338(1):17–31. doi: 10.1016/j.jmb.2004.02.006
53. Li D, Huang ZZ, Ling YZ, Wei JY, Cui Y, Zhang XZ, et al. Up-regulation of CX3CL1 via nuclear factor-κB-dependent histone acetylation is involved in paclitaxel-induced peripheral neuropathy. Anesthesiology (2015) 122(5):1142–51. doi: 10.1097/aln.0000000000000560
54. Zhang Z, Cai YQ, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med (2011) 17(11):1448–55. doi: 10.1038/nm.2442
55. Capasso KE, Manners MT, Quershi RA, Tian Y, Gao R, Hu H, et al. Effect of histone deacetylase inhibitor JNJ-26481585 in pain. J Mol Neurosci (2015) 55(3):570–8. doi: 10.1007/s12031-014-0391-7
56. Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep (2012) 1(4):334–40. doi: 10.1016/j.celrep.2012.02.014
57. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
58. Kiguchi N, Kobayashi Y, Saika F, Kishioka S. Epigenetic upregulation of CCL2 and CCL3 via histone modifications in infiltrating macrophages after peripheral nerve injury. Cytokine (2013) 64(3):666–72. doi: 10.1016/j.cyto.2013.09.019
59. Qian J, Zhu W, Lu M, Ni B, Yang J. D-β-hydroxybutyrate promotes functional recovery and relieves pain hypersensitivity in mice with spinal cord injury. Br J Pharmacol (2017) 174(13):1961–71. doi: 10.1111/bph.13788
60. Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci (2002) 22(15):6724–31. doi: 10.1523/jneurosci.22-15-06724.2002
61. Kukkar A, Singh N, Jaggi AS. Attenuation of neuropathic pain by sodium butyrate in an experimental model of chronic constriction injury in rats. J Formos Med Assoc (2014) 113(12):921–8. doi: 10.1016/j.jfma.2013.05.013
62. Sanna MD, Galeotti N. The HDAC1/c-JUN complex is essential in the promotion of nerve injury-induced neuropathic pain through JNK signaling. Eur J Pharmacol (2018) 825:99–106. doi: 10.1016/j.ejphar.2018.02.034
63. Sanna MD, Stark H, Lucarini L, Ghelardini C, Masini E, Galeotti N. Histamine H4 receptor activation alleviates neuropathic pain through differential regulation of ERK, JNK, and P38 MAPK phosphorylation. Pain (2015) 156(12):2492–504. doi: 10.1097/j.pain.0000000000000319
64. Maiarù M, Morgan OB, Tochiki KK, Hobbiger EJ, Rajani K, Overington DW, et al. Complex regulation of the regulator of synaptic plasticity histone deacetylase 2 in the rodent dorsal horn after peripheral injury. J Neurochem (2016) 138(2):222–32. doi: 10.1111/jnc.13621
65. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
66. Yao ZG, Zhang L, Huang L, Zhu H, Liu Y, Ma CM, et al. Regional and cell-type specific distribution of HDAC2 in the adult mouse brain. Brain Struct Funct (2013) 218(2):563–73. doi: 10.1007/s00429-012-0416-3
67. Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the toll-like receptor 4 agonist LPS. J Leukoc Biol (2010) 87(6):1103–14. doi: 10.1189/jlb.0509363
68. Jain S, Malinowski M, Chopra P, Varshney V, Deer TR. Intrathecal drug delivery for pain management: recent advances and future developments. Expert Opin Drug Delivery (2019) 16(8):815–22. doi: 10.1080/17425247.2019.1642870
69. Ver Donck A, Vranken JH, Puylaert M, Hayek S, Mekhail N, Van Zundert J. Intrathecal drug administration in chronic pain syndromes. Pain Pract (2014) 14(5):461–76. doi: 10.1111/papr.12111
70. Leppert W, Krajnik M, Wordliczek J. Delivery systems of opioid analgesics for pain relief: a review. Curr Pharm Des (2013) 19(41):7271–93. doi: 10.2174/138161281941131219130127
71. Selvi BR, Mohankrishna DV, Ostwal YB, Kundu TK. Small molecule modulators of histone acetylation and methylation: a disease perspective. Biochim Biophys Acta (2010) 1799(10-12):810–28. doi: 10.1016/j.bbagrm.2010.09.005
72. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129(4):823–37. doi: 10.1016/j.cell.2007.05.009
73. Zhang Y, Chen SR, Laumet G, Chen H, Pan HL. Nerve injury diminishes opioid analgesia through lysine methyltransferase-mediated transcriptional repression of μ-opioid receptors in primary sensory neurons. J Biol Chem (2016) 291(16):8475–85. doi: 10.1074/jbc.M115.711812
74. Penas C, Navarro X. Epigenetic modifications associated to neuroinflammation and neuropathic pain after neural trauma. Front Cell Neurosci (2018) 12:158. doi: 10.3389/fncel.2018.00158
75. Arifuzzaman S, Das A, Kim SH, Yoon T, Lee YS, Jung KH, et al. Selective inhibition of EZH2 by a small molecule inhibitor regulates microglial gene expression essential for inflammation. Biochem Pharmacol (2017) 137:61–80. doi: 10.1016/j.bcp.2017.04.016
76. Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain (2013) 136(Pt 3):828–43. doi: 10.1093/brain/aws330
77. Lee MC, Saleh R, Achuthan A, Fleetwood AJ, Förster I, Hamilton JA, et al. CCL17 blockade as a therapy for osteoarthritis pain and disease. Arthritis Res Ther (2018) 20(1):62. doi: 10.1186/s13075-018-1560-9
78. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest (2016) 126(9):3453–66. doi: 10.1172/jci87828
79. Lee JY, Na WH, Choi HY, Lee KH, Ju BG, Yune TY. Jmjd3 mediates blood-spinal cord barrier disruption after spinal cord injury by regulating MMP-3 and MMP-9 expressions. Neurobiol Dis (2016) 95:66–81. doi: 10.1016/j.nbd.2016.07.015
Keywords: chronic pain, inflammation, histone modification, gene expression, epigenetic, treatment
Citation: Jiang W, Zhang L-X, Tan X-Y, Yu P and Dong M (2023) Inflammation and histone modification in chronic pain. Front. Immunol. 13:1087648. doi: 10.3389/fimmu.2022.1087648
Received: 02 November 2022; Accepted: 29 December 2022;
Published: 13 January 2023.
Edited by:
Carla Marie Cuda, Northwestern University, United StatesReviewed by:
Yuki Saito, Sapporo Medical University, JapanCopyright © 2023 Jiang, Zhang, Tan, Yu and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Yu, eXBlbmdAamx1LmVkdS5jbg==; Ming Dong, ZG9uZ2dlQGpsdS5lZHUuY24=