 Christopher Schorr
Christopher Schorr Fabiana Perna
Fabiana Perna
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 December 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1085978
This article is part of the Research TopicNew biomarkers in hematological disease and allogeneic cell transplant complicationsView all 14 articles
Acute Myeloid Leukemia (AML) is an aggressive myeloid malignancy associated with high mortality rates (less than 30% 5-year survival). Despite advances in our understanding of the molecular mechanisms underpinning leukemogenesis, standard-of-care therapeutic approaches have not changed over the last couple of decades. Chimeric Antigen Receptor (CAR) T-cell therapy targeting CD19 has shown remarkable clinical outcomes for patients with acute lymphoblastic leukemia (ALL) and is now an FDA-approved therapy. Targeting of myeloid malignancies that are CD19-negative with this promising technology remains challenging largely due to lack of alternate target antigens, complex clonal heterogeneity, and the increased recognition of an immunosuppressive bone marrow. We carefully reviewed a comprehensive list of AML targets currently being used in both proof-of-concept pre-clinical and experimental clinical settings. We analyzed the expression profile of these molecules in leukemic as well normal tissues using reliable protein databases and data reported in the literature and we provide an updated overview of the current clinical trials with CAR T-cells in AML. Our study represents a state-of-art review of the field and serves as a potential guide for selecting known AML-associated targets for adoptive cellular therapies.
Acute myeloid leukemia (AML) is an aggressive, rapidly progressing hematological malignancy. The 5-year overall survival rate of AML is only 27%, accounting for approximately 2% of all cancer deaths in the United States (1). While Chimeric Antigen Receptor (CAR) T-cell therapies have shown remarkable success in the treatment of acute lymphoblastic leukemia (ALL), demonstrating an impressive 82% response rate in relapsed & refractory B-cell ALL, developing CAR T-cells for AML has been challenging largely due to the lack of specific and suitable target antigens, complex clonal heterogeneity, and an increased recognition of immunosuppressive regulators in the bone marrow microenvironment. As a result, there is no formal FDA approval for CAR T-cell therapy in AML thus far.
CARs are synthetic receptors that retarget and reprogram T cells (2). Unlike the physiological T-cell receptor, which engages HLA-peptide complexes, CARs bind to native cell surface molecules without requiring antigen processing or HLA expression for tumor recognition (3), two mechanisms that promote immune cell evasion (4). An ideal CAR T-cell target is a cell surface protein that is highly expressed in all patients and by all malignant cells within the patients, including cancer stem cells. Surface targets that play a critical role in the pathogenesis of AML are more likely to block and challenge cancer cell mechanisms of survival and progression. As importantly, an ideal CAR T-cell target should not be expressed or very minimally expressed in normal tissues of the body including normal hematopoietic stems cells (HSCs) (5, 6).
In this manuscript, we carefully and systematically reviewed the characteristics of all molecules that have been proposed thus far as potential target antigens for CAR T-cell therapy of AML, both in pre-clinical and clinical settings. We describe the expression profile of these molecules in leukemic and normal cells beyond normal counterparts (i.e. HSPCs) when rigorous data were available. Furthermore, we provide an updated overview of the current AML CAR T-cell therapies worldwide, offering a state-of-art review of an unmet and urgent need in this emerging field.
No ideal CAR T-cell therapy target (i.e. a cell surface molecule exclusively expressed on AML cells including leukemic stem cells and absent in any normal tissues) for AML patients has been identified, yet. In current clinical practice, CD33 and CD123 represent the most commonly utilized molecules for CAR T-cell engineering. This type of targets includes normal proteins overexpressed in AML and thus, they are classified as tumor-associated antigens (TAA) and are generally expressed in normal tissues as well, raising concerns of on-target off-tumor toxicity. Targeting myeloid antigens is more challenging than CD19 for lymphoid malignancies as myeloid targets are generally expressed on normal myeloid progenitor cells as well as HSCs and thus, targeting this type of tumor-associated antigens may lead to significant myelotoxicity. Over-expressed antigens can often be further classified into myeloid (CD123, CLL-1, CD33, CD64, CD116, CD13, CD93) or re-purposed lymphoid (CD19, CD7, CD70) markers. Neoantigens in AML are few and mainly intracellularly located. This is believed to be due to the fact that AML bears fewer albeit frequently recurring genetic mutations compared to solid tumors (7). Potentially cancer-specific targets are those derived from alternative mRNA splicing such as CD44v6 or molecules that are up-regulated or subcellularly re-localized upon cellular stress (PR1 peptide, GRP78) or chemical induction (CD38, FRβ).
We reviewed all these types of AML targets that have been proposed as CAR T-cell targets in AML either in proof-of-principle strategies that we call pre-clinical or more rigorous strategies actively being used in clinical trials. We specifically looked at the expression profile of these molecules in both leukemic and normal cells based on extensive review of current literature and mining large datasets (Table 1). We carefully reviewed all available clinical trial data published on clinicaltrials.gov for CAR T-cell therapy used in refectory/relapsed (r/r) AML patients. We provide an overview of all clinical trials showing enrollment status, type of adoptive cellular therapy (T-cell, γδT-cell, or NK cell), phase classification, and recruiting sample size for each AML CAR target (Table 2) as of October 7th 2022.
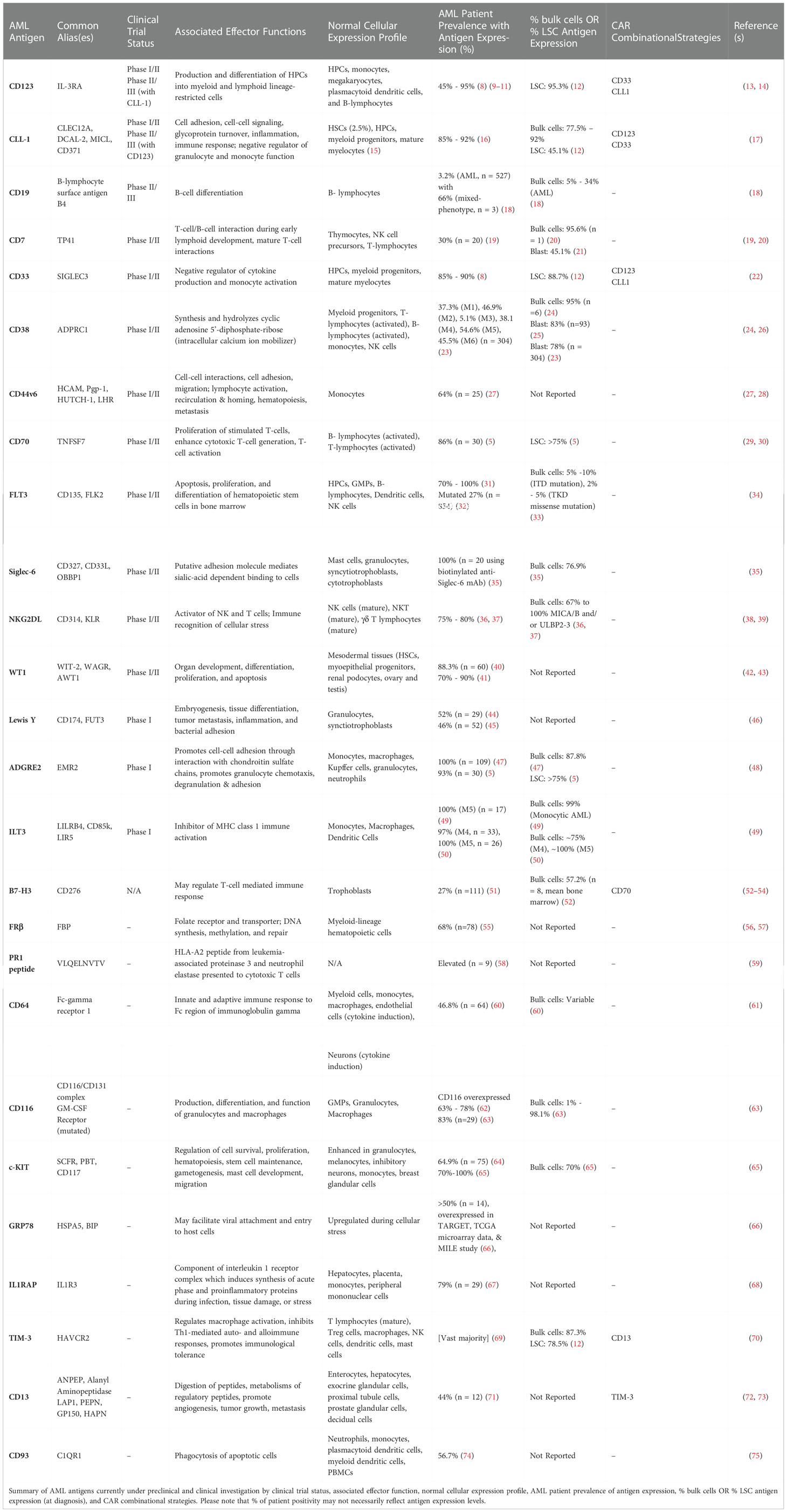
Table 1 Characteristics of AML target antigens proposed for CAR T-cell therapy: .
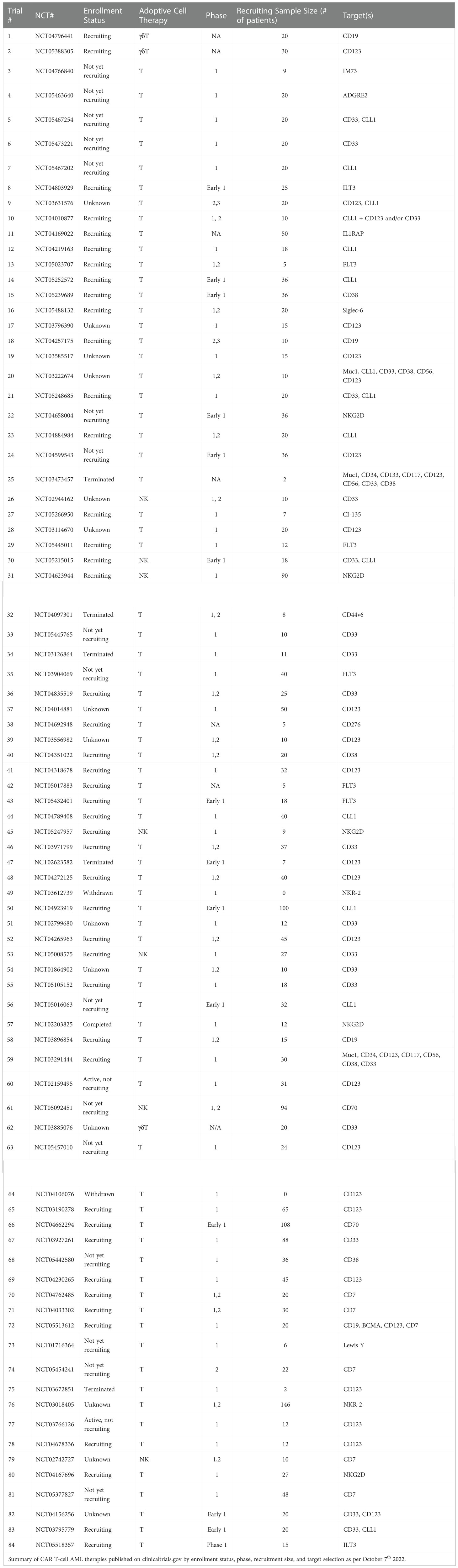
Table 2 Clinical Trials with CAR T-cells in AML patients as of October 7th 2022.
CD123, also known as IL-3Rα, is a membrane protein that is highly expressed in ~80% of AML patients (although varied levels of expression across patients) and is closely related to AML patient prognosis. CD123 overexpression has been identified on leukemic stem cells (LSCs) and AML blast cells with minimal expression on normal HSCs. CD123 expression has been reported on 95% of LSCs (12). In addition to AML, CD123 has been implicated in other myeloid malignancies such as Chronic Myelomonocytic Leukemia (CMML), Myelodysplastic Syndrome (MDS), Myeloproliferative Neoplasms (MPN), and Chronic Myeloid Leukemia (CML), and blastic plasmacytoid dendritic cell neoplasm (BPDCN) (76). Stimulation of CD123 with IL-3 functions as an important promoter of proliferation and differentiation of hematopoietic progenitor cells (HPCs) into both myeloid-derived and lymphoid-restricted lineages. CD123 is expressed on IL-3-responsive cells such as HPCs, monocytes, megakaryocytes, plasmacytoid dendritic cells, and B-lymphocytes. Several studies have targeted CD123 in AML including recombinant fusion proteins, anti-CD123 neutralizing monoclonal antibodies, and CAR T-cell therapies. In the field of CAR T-cell therapy for AML, CD123 is currently the most utilized target with over 20 clinical trials either completed or ongoing. In one study by Khawanky et al., third-generation anti-CD123 CAR T-cells showed significant anti-AML activity when combined with DNA methyltransferase inhibitor, AZA (13). Currently, several anti-CD123 CAR T-cell therapies are in phase I and phase I/II clinical trials including combinatorial targeting with anti-CLL-1 CAR T-cell therapy in active phase III. As of this writing, there are two terminated anti-CD123 CAR T-cell trials. Seven r/r AML patients were enrolled in phase I clinical trial at the University of Pennsylvania, United States by Gill et al. in which patients were given “biodegradable” T-cells transiently expressing anti-CD123 CAR (77). These T-cells were ex vivo electroporated with anti-CD123 mRNA to express anti-CD123 CAR. Despite no treatment-related death nor any clinical hematologic suppression, there was no reduction in CD123 expressing cells in the bone marrow, and all patients who were treated had disease progression before day 28.
CLL-1 or CLEC12A is a C-type lectin-like receptor that has a critical role in cell adhesion, cell-to-cell signaling, inflammation, and serves as a negative regulator of granulocyte and monocyte function. CLL-1 is a commonly studied marker of AML due to its relatively high expression on both leukemic stem cells (approximately 45%) and leukemic blasts (between 77.5% to 92%) with a high proportion of AML patients (> 85%) expressing CLL-1. Clinical measurement of CLL-1 has been considered a reliable marker for minimal residual disease and risk stratification/prognosis of AML patients (78). In a phase I clinical trial at Nankai University, China by Jin et al., ten r/r AML patients treated with anti-CLL-1 CAR T-cells demonstrated a 70% complete remission or complete remission with incomplete hematological recovery (CR/CRi) (17). In addition to CD123, anti-CLL-1 CAR T-cells has been combined with anti-CD33 CAR T-cells, however clinical results have yet to be published.
CD19, also known as B-lymphocyte surface antigen B4, is a marker of B-cell differentiation and has restricted expression on B-lymphocytes. CD19 is critical for regulating intrinsic B-cell activation thresholds via B-cell receptor-dependent and independent signaling pathways (79). CAR T-cells targeting CD-19 have been well studied in the context of ALL, but less so for AML. Aberrant expression of CD19 has been identified in AML subtypes with the t(8:21) translocation and can often present in cases without this genetic aberration (80).
In a study of 527 AML patients, the overall CD19 expression was 3.2%, with 17 out of 527 AML patients expressing CD19. CD19 was expressed in 2 out of 3 mixed-phenotype acute leukemia patients (66%) (18). Bulk cells have been shown to express anywhere between 5% to 34% of CD19 in these patients. While strategies of CAR-T cell targeting of CD19 for AML have been limited, one study for CD19+ AML patients has made it to phase 2 & 3 clinical trial and is actively recruiting at the Sheba Medical Center, Israel.
CD7 is a glycoprotein that is functionally essential for T-cell and T-cell/B-cell interactions during early lymphoid development. Expression of CD7 is common on thymocytes, NK cell precursor cells, and T-lymphocytes. In one study CD7 expression was found in approximately 30% of AML patients (n = 30) with high expression linked with more aggressive disease status and resistance to traditional therapy (19). Elhadi et al. showed that CD7 positivity was detected on 45% of AML Sudanese patients (n = 51) with relatively minimal expression compared to other markers; the mean positivity among all cases reported was 27.4% (21). While positive for CD7, minimal expression of CD7 on AML blast cells on patients limits their potential enrollment in clinical trials. In a 2019 study, Gomes-Silva et al. demonstrated that a CD7-directed CAR in CD7 gene-knockout T cells could effectively eliminate CD7+ leukemic cells in mice while protecting against significant systemic myeloablation (19). Efforts to genetically remove endogenous CD7 for an “off-the-shelf” anti-CD7 CAR-T cell therapy have been made to limit effector T cell fratricide (81). In a phase I clinical trial at Soochow University, China, one patient with a complex karyotype, TP53 deletion, FLT3-ITD mutation, and SKAP2-RUNKX1 fusion gene, expressed blasts with 95.6% CD7 positivity (20). After seventeen days post anti-CD7 CAR T-cell infusion, the patient achieved morphologic leukemia-free state. Despite a consequent grade 3 cytokine release syndrome (CRS), the patient did not experience organ failure or immune effector cell-associated neurotoxicity syndrome (ICANS) and received allo-HSC transplantation with CR without MRD.
Also known as Siglec3, CD33 is a surface marker that binds and becomes stimulated by sialic acid residues such as glycoproteins or glycolipids. CD33 serves as a negative regulator of cytokine production and monocytic activation commonly expressed on HPCs, myeloid progenitors, and mature myelocytes. Present in over 85% of AML patients, CD33 has been shown to have a high expression on LSCs (88.7%) (8, 12). CD33 lack of expression on HSCs has gained importance as a potential tumor-associated antigen marker for targeting in AML. Several CAR T-cell therapies against CD33 have made it to clinical trial phases 1 and 2 with seven actively recruiting and different cell type modalities being used such as CAR T-cell, CAR γδT-cell, and CAR NK-cell. In the single terminated CD-33 CAR T-cell clinical trial at M.D. Anderson Cancer Center, United States, three adult r/r AML patients received anti-CD33 CAR T-cells at a dose level (0.3 x 10^6 cells/kg) (22). Two of the three patients developed toxicities including grade 3 tumor lysis syndrome, acute kidney injury, grade 2 mucositis, grade 1 tachycardia, and grade 2 intermittent orthostatic hypotension with increases in bilirubin and ALT/AST levels. Despite CAR T-cell detection in peripheral blood, all three patients died from AML disease progression by 31 days post CAR T-cell infusion. No dose-limiting toxicity was observed.
CD38 a surface glycoprotein involved in cell adhesion, migration, and signal transduction. Present in myeloid progenitors, B- and T-lymphocytes, monocytes, and NK cells, CD38 plays an important role in intracellular calcium ion mobilization. Pre-exposing leukemic cells with all-trans-retinoic acid (ATRA) has been shown to increase CD38 expression on AML cells and enhance the cytotoxic effects of anti-CD38 CAR T-cells in AML cell lines (26). Within the AML patient population, between 5.1% (lowest, M3) to 54.6% (highest, M5) have expression of CD38 with blast expression approximately 78% (23). Anti-CD38 CAR T-cells are currently being studied in phase I/II clinical trial. In phase I/II clinical study at Soochow University, China, by Cui et al., six patients exhibited a median CD38 blast expression of 95%. Two-thrids (66.7%) of patients in this study achieved CR/CRi within four weeks following anti-CD38 CAR T-cell infusion (24). Of the adverse events reported, 5 patients experienced CRS grade 1-2 and one patient with CRS grade 3, no patients with ICANS, and all patients with neutropenia and thrombocytopenia (cell and platelet counts less than 500/μl and 10,000/μl respectively). Interestingly, of the one of two patients who relapsed, one patient achieved a CR after second treatment with anti-CD38 CAR T-cells.
The hyaluronate receptor CD44 is a surface glycoprotein that is highly implicated in specific processes such as leukocyte activation and malignant transformation. As an adhesive receptor, CD44 mediates cell-to-cell and cell-to-extracellular matrix interactions by binding to its main ligand, hyaluronic acid, a glycosaminoglycan highly concentrated in the bone marrow niche (82). CD44 is a useful marker for prospective identification of cancer stem cells (83). While the normal isoform of CD44 is expressed ubiquitously in both normal adult and fetal tissue, certain splice-isoforms of CD44 are relatively tumor-restricted (84, 85). In particular, CD44v6 is the most well-known splice variant of CD44 and has been associated with a poorer prognosis of AML (86). Abnormal CD44 splicing and expression has been demonstrated in other hematological and non-hematological malignancies such as NHL, myeloma, gastric mucosa, colon, and pancreatic cancers (87). As an AML-targeted therapy, CD44v6 is a promising candidate due to its lack of expression on HSCs and minimal expression on normal cells (88). CD44v6 shows 64% expression in AML patients (n = 25) (27). Monoclonal antibodies against CD44 have been shown to interfere with human leukemia initiation in immunocompromised mice (89) and second-generation anti-CD44v6 CAR T-cells have some efficacy in vivo and in vitro mouse models (28). While an anti-CD44v6 CAR T-cell phase I/II clinical trial by AGS Biologics S.p.A. began in August 2019, it was terminated due to inability to close study in a clinically relevant time frame.
CD70 is a member of the TNF receptor family whose expression is present on antigen-presenting cells upon maturation. Interaction with CD27 stimulates CD70 to promote effector CD8+ T cell response, influence polarization of CD4+ T-cells, and suppress effector Th17 function (90). Within AML pathogenesis, CD70 may have some influence in activating stem cell expression programs including the Wnt signaling pathway and promoting cellular division/proliferation (91). Over 85% of AML primary patient samples have been found with CD70 expression over 75% of their LSCs being positive for CD70 (5). Sauer et al., investigated several anti-CD70 CAR constructs with different hinges (IM, LF) and costimulatory molecules (CD27, CD28, and 4-1BB) and they report significant in vitro and in-vivo tumor-killing with LF-28z and CD27z CAR T-cells in two AML xenograft models without HSC colony assay toxicity (29). Azactidine (AZA) has been shown to increase expression of CD70 through hypomethylation of the CD70 promoter (92). Enhanced CD70 expression with anti-CD70 CAR T-cells have shown promising preclinical results in vitro and in vivo models. A non-cleavable hinge in the anti-CD70 CAR construct demonstrates enhanced binding, expansion, and potent in vivo activity and synergy with AZA (30). There is currently one anti-CD70 CAR T-cell clinical trial at Zhejiang University, China recruiting in early phase I and one not yet recruiting anti-CD70 NK-cell phase I/II clinical trial at M.D. Anderson Cancer Center, United States.
FLT3 (Fms-like tyrosine kinase 3) is a well-known receptor ligand for stem cell maintenance and differentiation. FLT3 is expressed in hematopoietic progenitor cells, granulocyte-myeloid progenitor cells, B-cells, dendritic cells, and NK cells. Mutations in the FLT3 receptor can lead to abnormal myeloid development. FLT3 mutations can be classified into either internal tandem duplicates (ITD) (representing approximately 25% of AML patients) and point mutations in the tyrosine kinase domain (TKD) (approximately 5% of AML patients) (93). Between 70% of 100% of AML patients have increased expression of FLT3 with ITD mutations having increased relapse rates, reduced disease-free survival, and decreased long-term survival (31, 94). Anti-FLT3 CAR T-cells have been shown to kill FLT3+ cell lines and AML patient bone marrow mononuclear cells in vitro with more potency in FLT3-ITD cells with an effector to target (E:T) ratio of as low as 1:8 (34). There are four recruiting clinical trials for anti-FLT3 CAR T-cells, but no results have been published yet.
Siglec-6 is a putative cell adhesion molecule that binds sialic-acid on mast cells, granulocytes, syncytiotrophoblasts, and cytotrophoblasts. Siglec-6 prevents primary mast cell activation and may be relevant for mast cell activity in colorectal cancer (95). Siglec-6 has mild expression in mucosal lymphoid cells and high expression in synctiotrophoblasts of the placenta. Using flow cytometry biotinylated anti-Siglec-6 monoclonal antibodies, Jetani et al. report 100% (n = 20) AML patients expressing Siglec-6. Within this group, over 75% of bulk AML cells expressed Siglec-6. Anti-Siglec-6 CAR T-cells have shown promising in vivo and in vitro cell killing activity with E:T ratios ranging from 10:1 to 1:4 against AML cell lines including U937, MV4;11, and MOLM-13 (35). Development of CAR T-cell for Siglec-6 is currently recruiting for phase I/II clinical trial at Xuzhou Medical University, China.
Natural killer group 2, member D ligand (NKG2DL) is an activating receptor of NK cells and T lymphocytes for immune recognition of abnormal cells (i.e. tumor cells). Targeting the NKG2D/NKG2D ligand axis is a new potential avenue for both biomolecular and adoptive cellular therapies for AML. NKG2D interacts with MHC class I related molecules (upregulated during cellular stress), which in humans, consist of either MHC class I related chain (MIC) family (MICA, MICB) or UL16-binding protein (ULBP) family (ULBP1-6) (96). Stimulating NKG2D can activate immune cells to recognize the NKG2D ligands present on AML cells. Resistance to chemotherapy and immune recognition has been associated with downregulation of NKG2D. According to a 2012 study by Hilpert et al., the leukemic cells of 70% of AML patients (n = 104) were positive for at least one NKG2D ligand (36). Within 15% of total cases, AML cells expressed four or five different NKG2D ligands (36). There is only one completed clinical trial with anti-NKG2DL CAR cells. In a first in-human AML Phase 1 study at Dana-Farber Cancer Institute and National Heart, Lung, and Blood Institute (NHLBI), Baumeister et al. were to achieve appropriate safety or feasibility levels, but unfortunately unable to elicit an efficient clinical response (38). Driouk et al. demonstrate increased NKG2D CAR T-cell efficacy against AML cells in vitro upon pharmacologic NKG2D-ligand upregulation with HDAC inhibitors (39). Several other clinical trials are currently recruiting for both CAR T-cells and CAR NK-cells against NKG2D ligands.
Wilms Tumor 1 (WT1) has been extensively studied over several decades in its deterministic role of AML pathophysiology. Either overexpression or mutation of WT1 leading to cancer has led to its interesting dual-role as both tumor suppressor and oncogene, depending on the context (97). WT1 signaling is essential to organ development of mesodermal tissue including HSCs, myoepithelial progenitor cells, and kidney podocytes. Between 70% to 90% of AML patients have elevated expression of WT1 on AML cells (41). One phase I/II clinical trial at University College, London, has been completed with anti-WT1 CAR T-cells demonstrating a Relapse-Free Survival (RFS) of 100% after 44 months following infusion in 12 patients compared to 54% RFS in 88 patients without CAR T-cell therapy after allogenic HSC transplantation (42).
Lewis Y is a surface glycoprotein present on granulocytes and synctiotrophblasts during embryogenesis. Lewis Y antigen is a difucosylated oligosaccharide and its presence is highly correlated with the pathological staging and prognosis of epithelial ovarian cancer (98). Lewis Y is suggested to promote tumor survival, invasion, and metastasis (99). Within AML patients, reports indicate that up to approximately 50% of patients have expression of Lewis Y on leukemic cells. In the first AML CAR T-cell therapy trial a phase 1 clinical study at Peter MacCallum Cancer Center, Australia, with anti-Lewis Y CAR T-cells for r/r AML patients, Ritchie et al. demonstrate mild feasibility and safety for targeting Lewis Y and CAR T cell persistence up to 10 months in proven sites of disease (leukemia cutis and bone marrow) (46). Although limited efficacy was observed overall, this was an important study to assess the potential of CAR-T therapy in AML patients and its use with limited hematological suppression. Neeson et al. engineered anti-Lewis Y CAR T-cells using autologous T-cells based on the Lewis-Y ex-vivo transduction and expansion protocol, showing a cytolytic response against Lewis Y positive AML cells via production of interferon-γ (100).
ADGRE2 or EMR2 is a cell adhesion protein that interacts with chondroitin sulfate chains and promotes granulocyte chemotaxis, and degranulation. ADGRE2 expression is present on granulocytes, monocytes, macrophages, Kupffer cells, and neutrophils. Expression of ADGRE2 in AML was first identified in a set of four candidate targeting proteins by Perna et al. with 93% of patients (n = 30) having greater than 75% protein expression on LSCs (5). Haubner et al. recently created an anti-ADGRE2 CAR T-cell model with a chimeric-costimulatory receptor against CLEC12A (48). In this, both ADGRE2 and CLEC12A must be present in the tumor for the CAR T-cell to be activated. This gating strategy is proposed to maintain T-cell cytotoxicity and tumor-killing efficacy within the tumor microenvironment and lead to long-term potentiation of the infiltrating CAR T-cells. Using NSG in-vivo xenografts of engineered low-expressing ADGRE2 MOLM13 AML cells to model antigen escape, Haubner et al. showed ADGRE2 and CLEC12A had increased performance than alternative dual (OR-gated) ADGRE2 CLEC12A CAR T-cells with functional persistence >70 days. There is one clinical trial that has not yet begun recruiting at Zhejiang University, China.
ILT3 or LILRB4 is an inhibitor of MHC class-I immune activation and present on monocytes, macrophages, dendritic cells, endothelial cells, and osteoclasts. ILT proteins are structurally and functionally similar to the killer cell immunoglobulin-like receptors (KIR) expressed on NK cell and T-lymphocytes. Expression of ILT3 from antigen-presenting cells can induce anergy of CD4+ helper T-lymphocytes and differentiation of CD8+ T suppressor cells. ILT3 expression has been identified in both M4 and M5 AML subtypes (FAB classification) with M4 and M5 patients showing approximately 60% and 100% positivity (49, 50). While prevalence of ILT3 positivity is high in both M4 and M5, its expression intensity may change during leukemic disease progression (101, 102). Preclinical evaluation of an anti-ILT3 CAR T-cell model has demonstrated efficient effector function in vitro and in vivo against ILT3+ AML cells (MV4-11, PriAML-1, THP-1, PriAML-2, MOLM13, PriMonoctye) with an E:T ratio of 5:1 resulting in between approximately 50% to 90% specific cell lysis compared to only approximately 20% specific cell lysis in non-transduced control T-cells after 4 hours. Toxicity to CD34+ umbilical cord blood cells was also not present (49). An early phase 1 clinical trial at Zheijang Provincial People’s Hospital, China is currently recruiting patients with M4 and M5 AML, however results have yet to be published. Concurrently, an anti-LILBR4 chimeric synthetic T cell receptor and antigen receptor (STAR), which incorporates an antigen recognition domain and constant regions of a TCR, is actively recruiting in phase I clinical investigation at Hebei Yanda Ludopei Hospital, China and results have yet to be published.
B7-H3, also known as CD273, is a type I transmembrane protein that was initially first characterized as a T-lymphocyte stimulating protein. However, recent studies have suggested that B7-H3 serves as an inhibitor to T-cell cytotoxicity and promoter of immune evasion within cancer cells (103). B7-H3 is expressed on multiple cancer types including AML blasts (53). Several groups have begun to investigate B7-H3 as a potentially new immune checkpoint therapy with analogous function to the PD-1/PD-L1 immune tolerance axis (104–106). Within a cohort of AML patients, 27% had positivity for B7-H3 with highest expression in the M3 and M5 subtypes (51). Anti-B7-H3 CAR T-cells have shown promising results in preclinical in vitro and in vivo models (52, 53, 106). Lichtman et al. demonstrate an anti-B7-H3 CAR T-cell model that shows significant interferon-γ and IL-2 release with cytotoxicity against OCl-AML2, OCI-AML3, THP1, and U937 along with primary AML patient samples with E:T ratio of 1:5 after 48 hours (52). Yang et al. have shown efficacy with tandem anti-B7-H3 and anti-CD70 CAR T-cells in vitro and in vivo (54). At an E:T ratio of 8:1, the tandem CAR T-cells has approximately 50% and 60% specific cell killing compared to single CD70 CAR T-cell 35% and 40% specific cell killing for NHI-460 and A375 AML cell lines respectively.
Folate Receptor β (FRβ) is a receptor and transporter of folate which is a necessary molecule for DNA synthesis, methylation, and repair. FRβ represents a potentially useful marker for AML due to its relatively limited expression in myeloid-lineage hematopoietic cells. Approximately 70% of AML patients (n = 20) have shown to be positive for FRβ (55). While there is some concern for off-tumor toxicity against normal myeloid-lineage cells, anti-FRβ CAR T-cells have been reported to have mild cytotoxic efficacy in vitro against FRβ-expressing AML cell lines with minimal toxicity against normal CD34+ HSCs (56). To address the limited capacity of anti-FRβ CAR T-cells to kill low-expressing FRβ AML, addition of ATRA increasing FRβ expression enhances CAR T-cell tumor-killing. High affinity anti-FRβ AML CAR T-cells demonstrate >50% cell killing in C30-FRβ expressing, THP1, and MV411 AML cell lines with minimal killing in HL60 at E:T ratios 5:1 and 1:1 (57). FRβ CAR-T cells show limited toxicity against CD34- bone marrow cells in CFU assays.
PR1 peptide is a 9 amino-acid long peptide derived from the myeloid proteinase 3 (PR3) and neutrophil elastase (NE). Stress-induced presentation of PR1 peptide in AML patients allows for cytotoxic T lymphocytes to bind via the MHC-HLA/TCR interaction. Targeting PR1 in AML is a promising choice due to both PR3 and NE high expression in myeloid malignancies including AML, MDS, and CML and very minimal expression in other tissues. Therapies such as a PR1 -peptide vaccine, anti-PR1/HLA-A2 antibody, and anti-PR1 CAR T-cells have been under development (58, 59, 107). Compared to a control CMV pp65 peptide HLA-expressing T2 cells, anti-PR1-peptide CAR T-cells (derived from the TCR-like 8F4 antibody) show increased % cell killing against PR1 peptide HLA-expressing T2 cells at E:T ratios between 4:1 to 0.5:1 (59). Additionally, anti-PR1 peptide CAR T-cells show rapid and effacing killing of AML in vitro.
CD64 or Fc-gamma receptor 1 is a high-affinity receptor present in monocytes, macrophages, and neutrophils that mediates many immunological responses including phagocytosis, cytokine release, antigen presentation, and cellular cytotoxicity (108). CD64 is widely expressed in patient AML samples and serves as a potential target for CAR T-cell therapy. Thus far, reporting of CD64 expression in AML samples has remained variable with approximately 46.8% AML patients (n = 64) positive for CD64 (60). Blast % positive with CD64 is highest in the M5 subtype, but also present in M3, M2, and M4 with 80%, 58%, and 20% respectively. In both in vitro and in vivo models, anti-CD64 CAR T-cells demonstrate limited myeloid cell suppression and lack of toxicity to CD34+ HSCs (61). T-cells transduced with anti-CD64 CAR show approximately 80% specific cell lysis against U937 and THP-1 AML cell lines at E:T ratio 1:2 with control T-cells showing minimal killing effect (61).
CD116 is the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor that complexes with CD131 to respond to GM-CSF which is a potent stimulator of myeloblast development and lymphoid production. GM-CSF is primarily located on neutrophils, eosinophils, monocytes, and macrophages. Mutated GM-CSF has been reported in juvenile myelomonocytic leukemia. CD116 is overexpressed in 63% to 78% of AML cases and Hasegawa et al. report a wide range of % blast positivity from 1% to 87.7% in AML patients (n = 29) with various subtypes (62). Hasegawa et al. use a mutated GM-CSF CAR at residue 21 to target CD116+ AML cells in vitro. These anti-CD116 CAR T-cells demonstrate durable cytotoxicity against AML cells (Kasumi-1, HL-60, ShinAML, THP-1, and MV4-11) preclinically with E:T ratios of 1:10 and 1:5 having reliable cell killing effects after 5 day co-culture with AML M4 and THP-1 M5 AML cell lines (63).
Stem cell factor (SCF) and its cognate receptor tyrosine kinase (c-kit), also known as CD117, is an important regulator of many hematological cellular processes including cell survival, proliferation, hematopoiesis, and stem cell maintenance. C-kit is highly expressed in HPCs and upregulated in most primary AML cells. Enhancement of c-kit can occur in granulocyte, melanocytes, inhibitory neurons, and monocytes. C-kit expression has been reported between 64% to 100% of AML patients with median bulk AML expression of 70% (64, 65). The generation and validation of anti-c-kit CAR T-cells in vitro and in vivo models show both CAR T-cell directed killing against AML cells and primary HPCs (65). Anti-c-kit CAR T-cells eliminated c-kit-mixed negative/low, c-kit intermediate, and c-kit high target cells at an E:T ratio of 1:10. While myeloablation is inevitable, Myburgh et al. propose this platform to be a potential bridging therapy for pre-transplant AML patients.
Glucose-regulated-protein 78 (GRP78) is a regulator of the unfolded protein response and may facilitate viral attachment and entry into host cells. GRP78 acts as a promising target for AML CAR T-cell therapy due to its normal residence in the endoplasmic reticulum in healthy cells and stress-induced transportation to the cell surface when a cell undergoes tumorigenesis. Over 50% of AML patients (n =14) have been reported to express GRP78 with gene overexpression also present in large databases such as TARGET, TCGA, and MILE (66). CAR T-cell against GRP78 show ability to recognize and kill GRP78 positive AML cells without toxicity to HPCs (66). Increased surface expression of GRP78 with dasatinib improves effector function of anti-GRP78 CAR T-cells. For single anti-GRP78 CAR expression, approximately 70% specific cell lysis occurred against MOLM13 AML cells compared to approximately 10% control T-cells at an E:T ratio of 0.5:1 (66).
Interleukin-1 receptor accessory protein (IL1RAP) is an indispensable type 1 receptor for IL-1 signaling. IL1RAP is widely expressed by multiple subtypes of AML and serves as a putative marker for leukemic stem cells (109). IL1RAP induces synthesis of the acute phase and proinflammatory proteins during infection, tissue damage, or stress. IL1RAP interacts with both FLT3 and c-KIT, two receptor tyrosine kinases with roles in AML pathogenesis. Approximately 80% of AML patients (n = 29) have expression of IL1RAP on blasts (67). Third-generation anti-IL1RAP CAR T-cells show significant cytotoxicity against primary AML at both time of diagnosis and relapse (68). Anti-IL1RAP CAR T-cells demonstrate significant tumor cell killing HL-60, MOLM-13, Mono-Mac-6, and KU812 AML cell lines.
T-cell immunoglobulin mucin-3 (TIM-3) is a surface molecule expressed in interferon gamma (IFNγ)-producing T lymphocytes. TIM-3 has been identified as a marker of LSCs with lack of expression on normal CD34+CD38- HSCs but not CD34+CD38- LSCs in most types of AML except for acute promyelocytic leukemia (M3) (110). Kikushige et al. report that a vast majority of AML patients have increased expression of TIM-3 on bulk AML cells (41). Haubner et al. report bulk cell expression of TIM-3 to be 87.3% and LSC expression 78.5%. Anti-TIM-3 CAR T-cells exhibit effective AML cell killing and eradication of LSCs in vitro and in vivo models (70).
CD13 or Alanyl Aminopeptidase is a plasma membrane peptidase enzyme that digests peptides on the surface of the small intestine. CD13 also has less clear function in proximal tubular cells and is thought to be involved in the metabolism of peptides that serve regulatory functions in a diverse array of cell types including macrophages, granulocytes, and neurons. Defects in CD13 have been an implicated cause of various leukemia and lymphoma. Khurram et al. report 44% of AML patients (n = 12) having aberrant expression of CD13 on blast cells along with patients with B-ALL and T-ALL (71). Due to the off-target expression of CD13 on gastrointestinal cells, He et al. deploy the use of a switchable CAR-compatible nanobody, Nb157, anti-CD13 CAR T-cell in vitro against THP-1 cells and CD13+ K562 cells (72). In vivo modeling demonstrated significant tumor reduction after day 21 switch injection and by day 52, all tumors were undetectable. Over 35% of HSCs and progenitors were detected in the BM of post-conditioned and HSC-transplanted mice following treatment of low dose (0.1 mg/kg) CAR T-cells.
CD93 or C1QR1 is a cell-surface glycoprotein and type 1 membrane protein identified as a myeloid cell-specific marker. CD93 is thought to be involved in immunological clearance of apoptotic cells. Approximately 55% of AML patients have differential expression of CD93 and CD93 is associated with genomic rearrangements of the MLL (mixed lineage leukemia) gene (111). Richards et al. recently demonstrate a NOT-gated anti-CD93 CAR T-cells that can recognize CD123 and not engage thus limiting the cytotoxic effects of endothelial cell CAR T-cell recognition and killing (75).
To provide a comprehensive analysis of these AML targets we also annotated the expression of each antigen in a large panel of normal tissues based on the integration of three publicly available protein databases such as the Human Protein Map, Human Protein Atlas and Proteomic Database (Figure 1). The methodology to merge these datasets and obtain an approximate cut-off for low, medium and high expression levels was previously described in Perna F. et al., Cancer Cell 2017 (5). We believe that this annotation of antigen protein expression represents a valuable resource to predict potential on-target off-tumor effects and help the selection of a suitable antigen based on the current available list of targets.
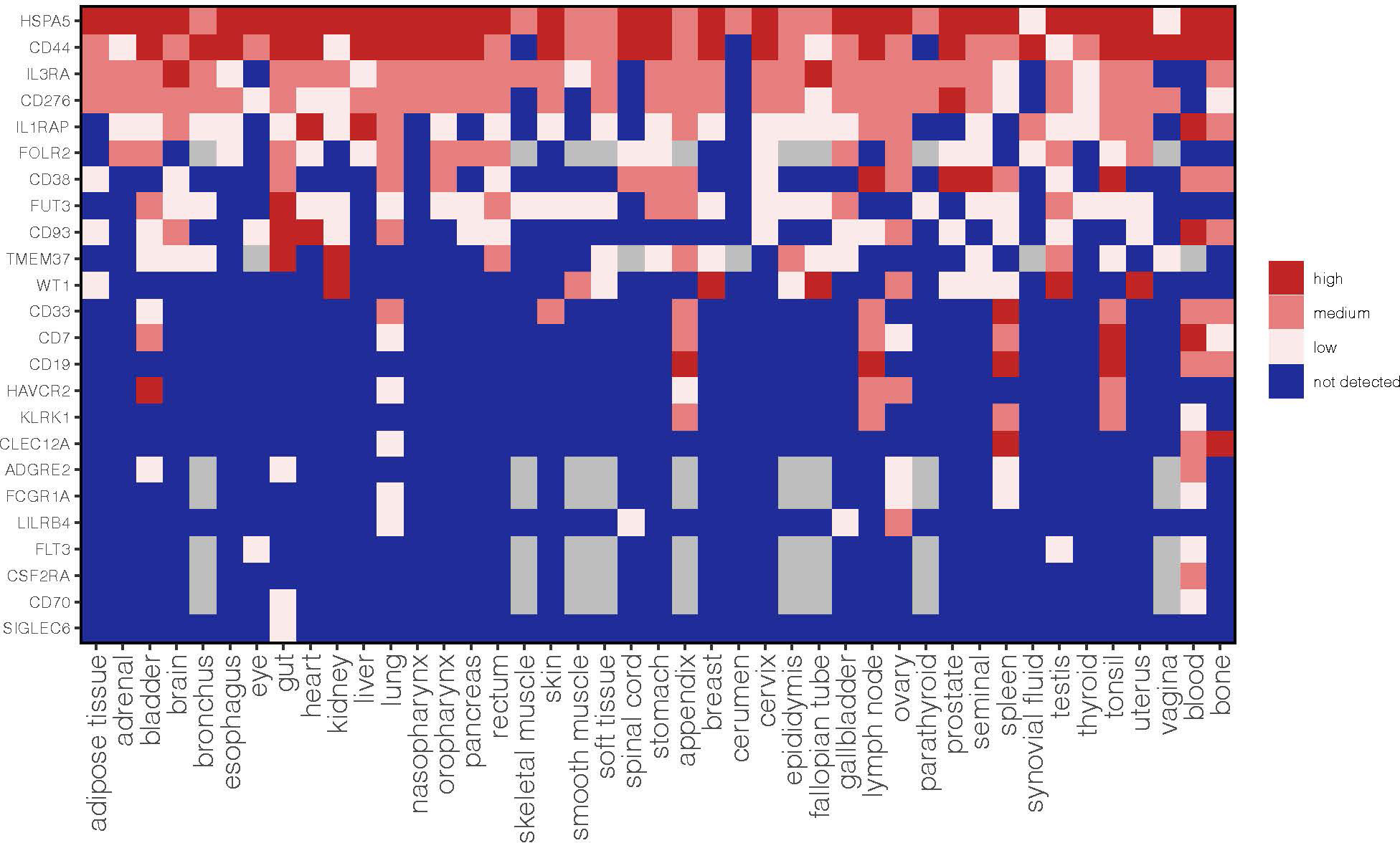
Figure 1 Expression profile of AML target antigens previously reported as AML CAR targets in normal tissues. CD44 refers to aggregate of CD44 isoforms rather than the CD44v6 CAR target. This is an updated figure from Perna F et al., Cancer Cell 2017 (5).
Data regarding protein target associated effector function, % AML patient expression, and AML cell (blast/LSC) expression were gathered through a systematic review of CAR T-cell AML publications available to our knowledge. Additionally, the summarized list of AML CAR products was made through searching of keywords “AML” with “CAR” or “STAR” in the clinicaltrials.gov public database available on October 7th, 2022. All clinical trials available were included. Surface protein expression of each AML target was extracted from the Human Protein Atlas Database, Human Protein Map, and Human Protein Atlas.
To date, twenty-six AML-associated target antigens have been proposed as targets for CAR T-cell therapy of AML, with many more likely on their way. Through an extensive review of the current literature and the clinicaltrials.gov website, we report the fifteen targets that are currently or have been investigated within a clinical trial with CAR T-cells in AML patients. While the excitement surrounding the advancement of novel AML CAR T-cell antigens is warranted, the validation of many preclinical designs has yet to be completed, and the results of many clinical studies have yet to be published. Thus far, nearly all investigational CAR T-cell products published have shown variable, yet moderate clinical efficacy or significant toxicity. There are many considerations related to this outcome such as the clonal complexity of AML and an evolving toxicity profile as novel CAR T-cell targets enter the clinical landscape. Understanding the AML-specific mechanisms that drive leukemogenesis remains a top priority for the identification of biologically and therapeutically relevant cell surface targets that could be used for curative CAR T-cell therapies tackling the critical mechanisms of leukemia initiation and progression. Leukemia-specific mechanisms such as epigenetic alterations and alternative mRNA splicing that are driven by frequently recurring genetic mutations may expand the CAR T-cell target space that is currently limited in AML. This attractive type of targets that may derive from AML genetic mutations may include post-translational modifications or splice variants of cell surface proteins. Meanwhile, to reduce toxicity and enhance the safety of CAR T-cell therapy targeting leukemia-associated target antigens such as CD33 or CD123 and preventing/predicting on-target/off-tumor effects an appropriate target antigen selection and validation remain critical steps in the development of AML CAR T-cell therapy and that can be integrated within the control of CAR activity by suicide genes or switch-off designs, optimization of CAR construct designs and/or combination approaches. Further, technologies such as deep-scale proteomic analyses with Mass-Spectrometry continue to advance and investigations of protein dysregulation in AML pathogenesis may soon uncover a new list of AML CAR antigens.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
FP received research funding from the Leukemia Research Foundation, IUSM, Lonza, and NGM Biopharmaceuticals. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. CS is a William J. Wright Scholar.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rai KR, Holland JF, Glidewell OJ, Weinberg V, Brunner K, Obrecht JP, et al. Treatment of acute myelocytic leukemia: A study by cancer and leukemia group B. Blood (1981) 58(6):1203–12.
2. Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol (2015) 33:9–15. doi: 10.1016/j.coi.2015.01.002
3. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature (2017) 545(7655):423–31. doi: 10.1038/nature22395
4. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MTL, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med (2009) 361(5):478–88. doi: 10.1056/NEJMoa0811036
5. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell (2017) 32(4):506–519.e5. doi: 10.1016/j.ccell.2017.09.004
6. Perna F. Safety starts with selecting the targets. Mol Ther (2021) 29(2):424–5. doi: 10.1016/j.ymthe.2021.01.011
7. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
8. Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J (2014) 4:e218. doi: 10.1038/bcj.2014.39
9. Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, et al. Elevated expression of IL-3Rα in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood (2002) 100(8):2980–8. doi: 10.1182/blood-2002-03-0852
10. Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. biomark Res (2014) 2(1):4. doi: 10.1186/2050-7771-2-4
11. Bras AE, de Haas V, van Stigt A, Jongen-Lavrencic M, Beverloo HB, Te Marvelde JG, et al. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytometry B Clin Cytom. (2019) 96(2):134–42. doi: 10.1002/cyto.b.21745
12. Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia (2019) 33(1):64–74. doi: 10.1038/s41375-018-0180-3
13. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun (2021) 12(1):6436. doi: 10.1038/s41467-021-26683-0
14. Sugita M, Galetto R, Zong H, Ewing-Crystal N, Trujillo-Alonso V, Mencia-Trinchant N, et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat Commun (2022) 13(1):2227. doi: 10.1038/s41467-022-29668-9
15. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol OncolJ Hematol Oncol (2019) 12(1):41. doi: 10.1186/s13045-019-0726-5
16. Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to c-type lectin-like molecule 1. Mol Ther (2017) 25(9):2202–13. doi: 10.1016/j.ymthe.2017.05.024
17. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol OncolJ Hematol Oncol (2022) 15(1):88. doi: 10.1186/s13045-022-01308-1
18. Ma G, Wang Y, Ahmed T, Zaslav AL, Hogan L, Avila C, et al. Anti-CD19 chimeric antigen receptor targeting of CD19+ acute myeloid leukemia. Leuk Res Rep (2018) 9:42–4. doi: 10.1016/j.lrr.2018.03.002
19. Gomes-Silva D, Atilla E, Atilla PA, Mo F, Tashiro H, Srinivasan M, et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol Ther (2019) 27(1):272–80. doi: 10.1016/j.ymthe.2018.10.001
20. Cao X, Dai H, Cui Q, Li Z, Shen W, Pan J, et al. CD7-directed CAR T-cell therapy: a potential immunotherapy strategy for relapsed/refractory acute myeloid leukemia. Exp Hematol Oncol (2022) 11(1):67. doi: 10.1186/s40164-022-00318-6
21. Osman IM, Humeida AAK, Eltayeb O, Abdelrhman I, Elhadi TA. Flowcytometric immunophenotypic characterization of acute myeloid leukemia (AML) in Sudan. Int J Hematol Disord (2015) 2(1):10–7. doi: 10.1309/2FYJ-00BE-R8N0-HMPQ
22. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia (2021) 35(11):3282–6. doi: 10.1038/s41375-021-01232-2
23. Keyhani A, Huh YO, Jendiroba D, Pagliaro L, Cortez J, Pierce S, et al. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk Res (2000) 24(2):153–9. doi: 10.1016/S0145-2126(99)00147-2
24. Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, et al. CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol OncolJ Hematol Oncol (2021) 14(1):82. doi: 10.1186/s13045-021-01092-4
25. Terstappen LW, Safford M, Unterhalt M, Könemann S, Zurlutter K, Piechotka K, et al. Flow cytometric characterization of acute myeloid leukemia: IV. comparison to the differentiation pathway of normal hematopoietic progenitor cells. Leukemia (1992) 6(10):993–1000.
26. Yoshida T, Mihara K, Takei Y, Yanagihara K, Kubo T, Bhattacharyya J, et al. All-trans retinoic acid enhances cytotoxic effect of T cells with an anti-CD38 chimeric antigen receptor in acute myeloid leukemia. Clin Transl Immunol (2016) 5(12):e116. doi: 10.1038/cti.2016.73
27. Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood (2013) 122(20):3461–72. doi: 10.1182/blood-2013-04-493361
28. Stornaiuolo A, Valentinis B, Sirini C, Scavullo C, Asperti C, Zhou D, et al. Characterization and functional analysis of CD44v6.CAR T cells endowed with a new low-affinity nerve growth factor receptor-based spacer. Hum Gene Ther (2021) 32(13–14):744–60. doi: 10.1089/hum.2020.216
29. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood (2021) 138(4):318–30. doi: 10.1182/blood.2020008221
30. Leick MB, Silva H, Scarfò I, Larson R, Choi BD, Bouffard AA, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell (2022) 40(5):494–508.e5. doi: 10.1016/j.ccell.2022.04.001
31. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood (2002) 100(5):1532–42. doi: 10.1182/blood-2002-02-0492
32. Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the united kingdom medical research council AML 10 and 12 trials. Blood (2001) 98(6):1752–9. doi: 10.1182/blood.V98.6.1752
33. Cheng J, Qu L, Wang J, Cheng L, Wang Y. High expression of FLT3 is a risk factor in leukemia. Mol Med Rep (2018) 17(2):2885–92. doi: 10.3892/mmr.2017.8232
34. Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol OncolJ Hematol Oncol (2018) 11(1):60. doi: 10.1186/s13045-018-0603-7
35. Jetani H, Navarro-Bailón A, Maucher M, Frenz S, Verbruggen C, Yeguas A, et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood (2021) 138(19):1830–42. doi: 10.1182/blood.2020009192
36. Hilpert J, Grosse-Hovest L, Grünebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol Baltim Md (1950) 189(3):1360–71. doi: 10.4049/jimmunol.1200796
37. Nowbakht P, Ionescu MCS, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood (2005) 105(9):3615–22. doi: 10.1182/blood-2004-07-2585
38. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res (2019) 7(1):100–12. doi: 10.1158/2326-6066.CIR-18-0307
39. Driouk L, Gicobi JK, Kamihara Y, Rutherford K, Dranoff G, Ritz J, et al. Chimeric antigen receptor T cells targeting NKG2D-ligands show robust efficacy against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.580328
40. Ujj Z, Buglyó G, Udvardy M, Beyer D, Vargha G, Biró S, et al. WT1 expression in adult acute myeloid leukemia: Assessing its presence, magnitude and temporal changes as prognostic factors. Pathol Oncol Res POR. (2016) 22(1):217–21. doi: 10.1007/s12253-015-0002-0
41. Lambert J, Lambert J, Thomas X, Marceau-Renaut A, Micol JB, Renneville A, et al. Early detection of WT1 measurable residual disease identifies high-risk patients, independent of transplantation in AML. Blood Adv (2021) 5(23):5258–68. doi: 10.1182/bloodadvances.2021004322
42. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T Cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med (2019) 25(7):1064–72. doi: 10.1038/s41591-019-0472-9
43. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular wilms tumor 1 antigen. Leukemia (2017) 31(8):1788–97. doi: 10.1038/leu.2016.373
44. Peinert S, Ritchie DS, Hoenemann D, Harrison SJ, Guru P, Kershaw MH, et al. Targeting Lewis y-positive multiple myeloma and acute myeloid leukemia with gene-modified T cells demonstrating memory phenotype. Blood (2008) 112(11):3900. doi: 10.1182/blood.V112.11.3900.3900
45. Peinert S, Prince HM, Guru PM, Kershaw MH, Smyth MJ, Trapani JA, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis y antigen. Gene Ther (2010) 17(5):678–86. doi: 10.1038/gt.2010.21
46. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther J Am Soc Gene Ther (2013) 21(11):2122–9. doi: 10.1038/mt.2013.154
47. Shahswar R, Kloos A, Gabdoulline R, Mohanty S, Koenecke C, Wichmann M, et al. Frequency and clinical characteristics associated with putative CAR targets ADGRE2, CCR1, CD70, and LILRB2 in acute myeloid leukemia. Blood (2018) 132(Supplement 1):5259. doi: 10.1182/blood-2018-99-113037
48. Haubner S, Mansilla-Soto J, Nataraj S, He X, Park J, Wang X, et al. ADCLEC.SYN1, a combinatorial car platform for AML (2021). Available at: https://library.ehaweb.org/eha/2021/eha2021-virtualcongress/324658/sascha.haubner.adclec.syn1.a.combinatorial.car.platform.for.aml.html?f=listing%3D3%2Abrowseby%3D8%2Asortby%3D1%2Amedia%3D1.
49. John S, Chen H, Deng M, Gui X, Wu G, Chen W, et al. A novel anti-LILRB4 CAR-T cell for the treatment of monocytic AML. Mol Ther J Am Soc Gene Ther (2018) 26(10):2487–95. doi: 10.1016/j.ymthe.2018.08.001
50. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature (2018) 562(7728):605–9. doi: 10.1038/s41586-018-0615-z
51. Guery T, Roumier C, Berthon C, Renneville A, Preudhomme C, Quesnel B. B7-H3 protein expression in acute myeloid leukemia. Cancer Med (2015) 4(12):1879–83. doi: 10.1002/cam4.522
52. Lichtman EI, Du H, Shou P, Song F, Suzuki K, Ahn S, et al. Preclinical evaluation of B7-H3–specific chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Clin Cancer Res (2021) 27(11):3141–53. doi: 10.1158/1078-0432.CCR-20-2540
53. Zhang Z, Jiang C, Liu Z, Yang M, Tang X, Wang Y, et al. B7-H3-Targeted CAR-T cells exhibit potent antitumor effects on hematologic and solid tumors. Mol Ther Oncolytics. (2020) 17:180–9. doi: 10.1016/j.omto.2020.03.019
54. Yang M, Tang X, Zhang Z, Gu L, Wei H, Zhao S, et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics (2020) 10(17):7622–34. doi: 10.7150/thno.43991
55. Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood (2002) 100(2):594–602. doi: 10.1182/blood.V100.2.594
56. Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood (2015) 125(22):3466–76. doi: 10.1182/blood-2014-11-612721
57. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia (2016) 30(6):1355–64. doi: 10.1038/leu.2016.35
58. Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti–PR1/HLA-A2 T-cell receptor–like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood (2011) 117(16):4262–72. doi: 10.1182/blood-2010-07-299248
59. Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy (2016) 18(8):985–94. doi: 10.1016/j.jcyt.2016.05.001
60. Dunphy CH, Tang W. The value of CD64 expression in distinguishing acute myeloid leukemia with monocytic differentiation from other subtypes of acute myeloid leukemia: a flow cytometric analysis of 64 cases. Arch Pathol Lab Med (2007) 131(5):748–54. doi: 10.5858/2007-131-748-TVOCEI
61. Sun X, Wang G, Zuo S, Niu Q, Chen X, Feng X. Preclinical evaluation of CD64 as a potential target for CAR-t-cell therapy for acute myeloid leukemia. J Immunother. (2022) 45(2):67–77. doi: 10.1097/CJI.0000000000000406
62. Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K, Schmetzer H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol (2004) 72(2):89–106. doi: 10.1046/j.0902-4441.2003.00184.x
63. Hasegawa A, Saito S, Narimatsu S, Nakano S, Nagai M, Ohnota H, et al. Mutated GM-CSF-based CAR-T cells targeting CD116/CD131 complexes exhibit enhanced anti-tumor effects against acute myeloid leukaemia. Clin Transl Immunol (2021) 10(5):e1282. doi: 10.1002/cti2.1282
64. El-Meligui YM, Elrhman HEA, Salahuddin A, Hamouda MA, Kassem AB. Correlation study on HLA-DR and CD117 (c-kit) expressions: Its prognosis and treatment response in acute myeloid leukemia patients. Pharmacogenomics Pers Med (2021) 14:381–93. doi: 10.2147/PGPM.S268986
65. Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia (2020) 34(10):2688–703. doi: 10.1038/s41375-020-0818-9
66. Hebbar N, Epperly R, Vaidya A, Thanekar U, Moore SE, Umeda M, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun (2022) 13(1):587. doi: 10.1038/s41467-022-28243-6
67. Askmyr M, Ågerstam H, Hansen N, Gordon S, Arvanitakis A, Rissler M, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood (2013) 121(18):3709–13. doi: 10.1182/blood-2012-09-458935
68. Trad R, Warda W, Alcazer V, Neto da Rocha N, Berceanu A, Nicod C, et al. Chimeric antigen receptor T-cells targeting IL-1RAP: a promising new cellular immunotherapy to treat acute myeloid leukemia. J Immunother Cancer. (2022) 10(7):e004222. doi: 10.1136/jitc-2021-004222
69. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell (2010) 7(6):708–17. doi: 10.1016/j.stem.2010.11.014
70. Lee WHS, Ye Z, Cheung AMS, Goh YPS, Oh HLJ, Rajarethinam R, et al. Effective killing of acute myeloid leukemia by TIM-3 targeted chimeric antigen receptor T cells. Mol Cancer Ther (2021) 20(9):1702–12. doi: 10.1158/1535-7163.MCT-20-0155
71. Khurram MM, Jafri SA, Mannan A, Nadeem A, Jamal A. FREQUENCY OF ABERRANT EXPRESSION OF CD MARKERS IN CASES OF ACUTE LEUKEMIA. Med J Islam World Acad Sci (2010) 6:55–60.
72. He X, Feng Z, Ma J, Zhang X, Ling S, Cao Y, et al. CAR T cells targeting CD13 controllably induce eradication of acute myeloid leukemia with a single domain antibody switch. Leukemia (2021) 35(11):3309–13. doi: 10.1038/s41375-021-01208-2
73. He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood (2020) 135(10):713–23. doi: 10.1182/blood.2019002779
74. Coustan-Smith E, Song G, Shurtleff S, Yeoh AEJ, Chng WJ, Chen SP, et al. Universal monitoring of minimal residual disease in acute myeloid leukemia. JCI Insight (2017) 3(9):e98561. doi: 10.1172/jci.insight.98561
75. Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discovery (2021) 2(6):648–65. doi: 10.1158/2643-3230.BCD-20-0208
76. Lane AA. Targeting CD123 in AML. Clin Lymphoma Myeloma Leuk. (2020) (Suppl 1):20:S67–8. doi: 10.1016/S2152-2650(20)30466-3
77. Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, et al. Treating relapsed / refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood (2017) 130:1359. doi: 10.1182/blood.V130.Suppl_1.1359.1359
78. Morsink LM, Walter RB, Ossenkoppele GJ. Prognostic and therapeutic role of CLEC12A in acute myeloid leukemia. Blood Rev (2019) 34:26–33. doi: 10.1016/j.blre.2018.10.003
79. Wang K, Wei G, Liu D. CD19: a biomarker for b cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol (2012) 1(1):36. doi: 10.1186/2162-3619-1-36
80. Francis J, Dharmadhikari AV, Sait SNJ, Deeb G, Wallace PK, Thompson JE, et al. CD19 expression in acute leukemia is not restricted to the cytogenetically aberrant populations. Leuk Lymphoma. (2013) 54(7):1517–20. doi: 10.3109/10428194.2012.754096
81. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An ‘off-the-shelf’ fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia (2018) 32(9):1970–83. doi: 10.1038/s41375-018-0065-5
82. Avigdor A, Goichberg P, Shivtiel S, Dar A, Peled A, Samira S, et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood (2004) 103(8):2981–9. doi: 10.1182/blood-2003-10-3611
83. Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. (2011) 11(4):254–67. doi: 10.1038/nrc3023
84. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol (2003) 4(1):33–45. doi: 10.1038/nrm1004
85. Bendall LJ, Bradstock KF, Gottlieb DJ. Expression of CD44 variant exons in acute myeloid leukemia is more common and more complex than that observed in normal blood, bone marrow or CD34+ cells. Leukemia (2000) 14(7):1239–46. doi: 10.1038/sj.leu.2401830
86. Legras S, Günthert U, Stauder R, Curt F, Oliferenko S, Kluin-Nelemans HC, et al. A strong expression of CD44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood (1998) 91(9):3401–13. doi: 10.1182/blood.V91.9.3401
87. Günthert U, Hofmann M, Rudy W, Reber S, Zöller M, Haussmann I, et al. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell (1991) 65(1):13–24. doi: 10.1016/0092-8674(91)90403-L
88. Neu S, Geiselhart A, Sproll M, Hahn D, Kuçi S, Niethammer D, et al. Expression of CD44 isoforms by highly enriched CD34-positive cells in cord blood, bone marrow and leukaphereses. Bone Marrow Transplant. (1997) 20(7):593–8. doi: 10.1038/sj.bmt.1700940
89. Nicolis di Robilant B, Casucci M, Falcone L, Camisa B, Gentner B, Naldini L, et al. CD44v6 is required for In vivo tumorigenesis of human AML and MM cells: Role of microenvironmental signals and therapeutic implications. Blood (2013) 122(21):605. doi: 10.1182/blood.V122.21.605.605
90. Corgnac S, Perret R, Zhang L, Mach JP, Romero P, Donda A. iNKT/CD1d-antitumor immunotherapy significantly increases the efficacy of therapeutic CpG/peptide-based cancer vaccine. J Immunother Cancer. (2014) 2(1):39. doi: 10.1186/s40425-014-0039-8
91. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin AL, Hoepner S, et al. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J Exp Med (2017) 214(2):359–80. doi: 10.1084/jem.20152008
92. Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med (2020) 26(9):1459–67. doi: 10.1038/s41591-020-0910-8
93. Kennedy VE, Smith CC. FLT3 mutations in acute myeloid leukemia: Key concepts and emerging controversies. Front Oncol (2020). doi: 10.3389/fonc.2020.612880
94. Castaño-Bonilla T, Alonso-Dominguez JM, Barragán E, Rodríguez-Veiga R, Sargas C, Gil C, et al. Prognostic significance of FLT3-ITD length in AML patients treated with intensive regimens. Sci Rep (2021) 11(1):20745. doi: 10.1038/s41598-021-00050-x
95. Yu Y, Blokhuis BRJ, Diks MAP, Keshavarzian A, Garssen J, Redegeld FA. Functional inhibitory siglec-6 is upregulated in human colorectal cancer-associated mast cells. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02138
96. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one.” Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00476
97. Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica (2016) 101(6):672–9. doi: 10.3324/haematol.2015.141796
98. Liu J, Lin B, Hao Y, Qi Y, Zhu L, Li F, et al. Lewis Y antigen promotes the proliferation of ovarian carcinoma-derived RMG-I cells through the PI3K/Akt signaling pathway. J Exp Clin Cancer Res (2009) 28(1):154. doi: 10.1186/1756-9966-28-154
99. Zhuang H, Hu Z, Tan M, Zhu L, Liu J, Liu D, et al. Overexpression of Lewis y antigen promotes human epididymis protein 4-mediated invasion and metastasis of ovarian cancer cells. Biochimie (2014) 105:91–8. doi: 10.1016/j.biochi.2014.06.022
100. Neeson P, Shin A, Tainton KM, Guru P, Prince HM, Harrison SJ, et al. Ex vivo culture of chimeric antigen receptor T cells generates functional CD8+ T cells with effector and central memory-like phenotype. Gene Ther (2010) 17(9):1105–16. doi: 10.1038/gt.2010.59
101. Chien KS, Class CA, Montalban-Bravo G, Wei Y, Sasaki K, Naqvi K, et al. LILRB4 expression in chronic myelomonocytic leukemia and myelodysplastic syndrome based on response to hypomethylating agents. Leuk Lymphoma. (2020) 61(6):1493–9. doi: 10.1080/10428194.2020.1723014
102. Zhao L, Cheng B, Xiong J, Ma D, Liu X, Wang L, et al. Protein arginine methyltransferase 5 promotes the migration of AML cells by regulating the expression of leukocyte immunoglobulin-like receptor B4. BioMed Res Int (2021) 2021:7329072. doi: 10.1155/2021/7329072
103. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. B7H3 as a promoter of metastasis and promising therapeutic target. Front Oncol (2018) 8. doi: 10.3389/fonc.2018.00264
104. Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci (2020) 16(11):1767–73. doi: 10.7150/ijbs.41105
105. Zhou WT, Jin WL. B7-H3/CD276: An emerging cancer immunotherapy. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.701006
106. Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol OncolJ Hematol Oncol (2021) 14(1):21. doi: 10.1186/s13045-020-01024-8
107. Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia (2017) 31(3):697–704. doi: 10.1038/leu.2016.254
108. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol (2008) 8(1):34–47. doi: 10.1038/nri2206
109. Mitchell K, Barreyro L, Todorova TI, Taylor SJ, Antony-Debré I, Narayanagari SR, et al. IL1RAP potentiates multiple oncogenic signaling pathways in AML. J Exp Med (2018) 215(6):1709–27. doi: 10.1084/jem.20180147
110. Akashi K. TIM-3 is a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. In: Nakao K, Minato N, Uemoto S, editors. Innovative medicine: Basic research and development. Tokyo: Springer (2015). Available at: http://www.ncbi.nlm.nih.gov/books/NBK500347/.
Keywords: CAR (chimeric antigen receptor) T cells, AML – acute myeloid leukaemia, target antigen, clinical trial, efficacy, safety
Citation: Schorr C and Perna F (2022) Targets for chimeric antigen receptor T-cell therapy of acute myeloid leukemia. Front. Immunol. 13:1085978. doi: 10.3389/fimmu.2022.1085978
Received: 31 October 2022; Accepted: 30 November 2022;
Published: 20 December 2022.
Edited by:
Sophie Paczesny, Medical University of South Carolina, United StatesReviewed by:
Peihua Lu, Lu Daopei Hospital, ChinaCopyright © 2022 Schorr and Perna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiana Perna, ZmFicGVybmFAaXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.