 Robert C. Sterner
Robert C. Sterner Rosalie M. Sterner
Rosalie M. Sterner- 1School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 2Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
Traumatic spinal cord injury (SCI) is a devastating condition that is often associated with significant loss of function and/or permanent disability. The pathophysiology of SCI is complex and occurs in two phases. First, the mechanical damage from the trauma causes immediate acute cell dysfunction and cell death. Then, secondary mechanisms of injury further propagate the cell dysfunction and cell death over the course of days, weeks, or even months. Among the secondary injury mechanisms, inflammation has been shown to be a key determinant of the secondary injury severity and significantly worsens cell death and functional outcomes. Thus, in addition to surgical management of SCI, selectively targeting the immune response following SCI could substantially decrease the progression of secondary injury and improve patient outcomes. In order to develop such therapies, a detailed molecular understanding of the timing of the immune response following SCI is necessary. Recently, several studies have mapped the cytokine/chemokine and cell proliferation patterns following SCI. In this review, we examine the immune response underlying the pathophysiology of SCI and assess both current and future therapies including pharmaceutical therapies, stem cell therapy, and the exciting potential of extracellular vesicle therapy.
1. Introduction
Traumatic spinal cord injury (SCI) is severely debilitating and is associated with substantial financial and emotional costs (1–3). In the United States, >12,000 patients annually suffer from SCI, with approximately 270,000 SCI patients across North America (1, 3, 4). The pathophysiology of SCI is complex with the initial trauma (primary injury) causing acute cell dysfunction/death that is further propagated by secondary injury cascades (5, 6). Among the secondary injury mechanisms, overactivation of the systemic immune response and neuroinflammation are key determinants of the extent of injury (6, 7). More specifically, the inflammatory cascade following SCI is complex and involves both the adaptive and innate immune responses, several cell types, and many inflammatory cytokines including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNFα) (8, 9). Although the immune response following SCI also has many beneficial effects, it is thought that the large-scale inflammatory response is a key factor in causing neural degeneration (8, 9). Management of traumatic spinal cord injury (tSCI) patients has mainly focused on the timing of surgical decompression in which recent studies have demonstrated that early surgical decompression of SCI patients within 8-12 hours following SCI is associated with improved neurological outcomes based on American Spinal Injury Impairment Scale (AIS) improvements (10, 11). Given the pivotal role the immune response plays in dictating the extent of secondary injury and neural degeneration, selectively targeting the immune response to SCI to promote neural regeneration instead of degeneration is a premier therapeutic target to improve functional outcomes (12). In order to develop neuro-immunological therapies for SCI, however, a detailed characterization of the immune response following SCI is necessary. Over the last two decades several studies have characterized the complex cytokine/chemokine cascades and cell infiltration patterns (7). In this review, we examine the immune response including the cytokine/chemokine cascades as well as the cell proliferation patterns underlying SCI and evaluate current and future therapies including pharmaceutical therapies, stem cell therapy, and extracellular vesicle therapy.
2. Pathophysiology of SCI
2.1. Primary versus secondary phases of injury
The pathophysiology underlying SCI occurs in two phases, the primary and secondary phases of injury. The primary injury phase is caused by the initial mechanical forces delivered to the spinal cord at the time of injury (5, 6). These mechanical forces cause direct structural damage to the surrounding neuronal tissue and vasculature tissue resulting in acute cell dysfunction and cell death (5, 6, 13). Broadly, four types of primary injury mechanisms have been described: (a) impact plus persistent compression; (b) impact alone; (c) distraction; and (d) laceration/transection (5, 6). Among the four mechanisms of primary injury, impact plus persistent compression is most common and frequently occurs via burst fractures (5, 6). In contrast to the primary phase of injury which occurs within a short window of time, studies have shown that the secondary phase of injury continues for days to weeks or even months after SCI (12–14). The secondary phase of injury propagates the acute cell dysfunction and cell death initially caused by the primary injury. Several secondary injury mechanisms (Figure 1) have been shown to contribute to propagation of acute cell dysfunction and cell death including neuroinflammation, ischemia, free radical formation, lipid peroxidation, blood CNS barrier break down, edema, release of proteases, and excitotoxicity (12–14). Importantly, the immune response plays a profound role in the propagation of secondary injury after SCI.
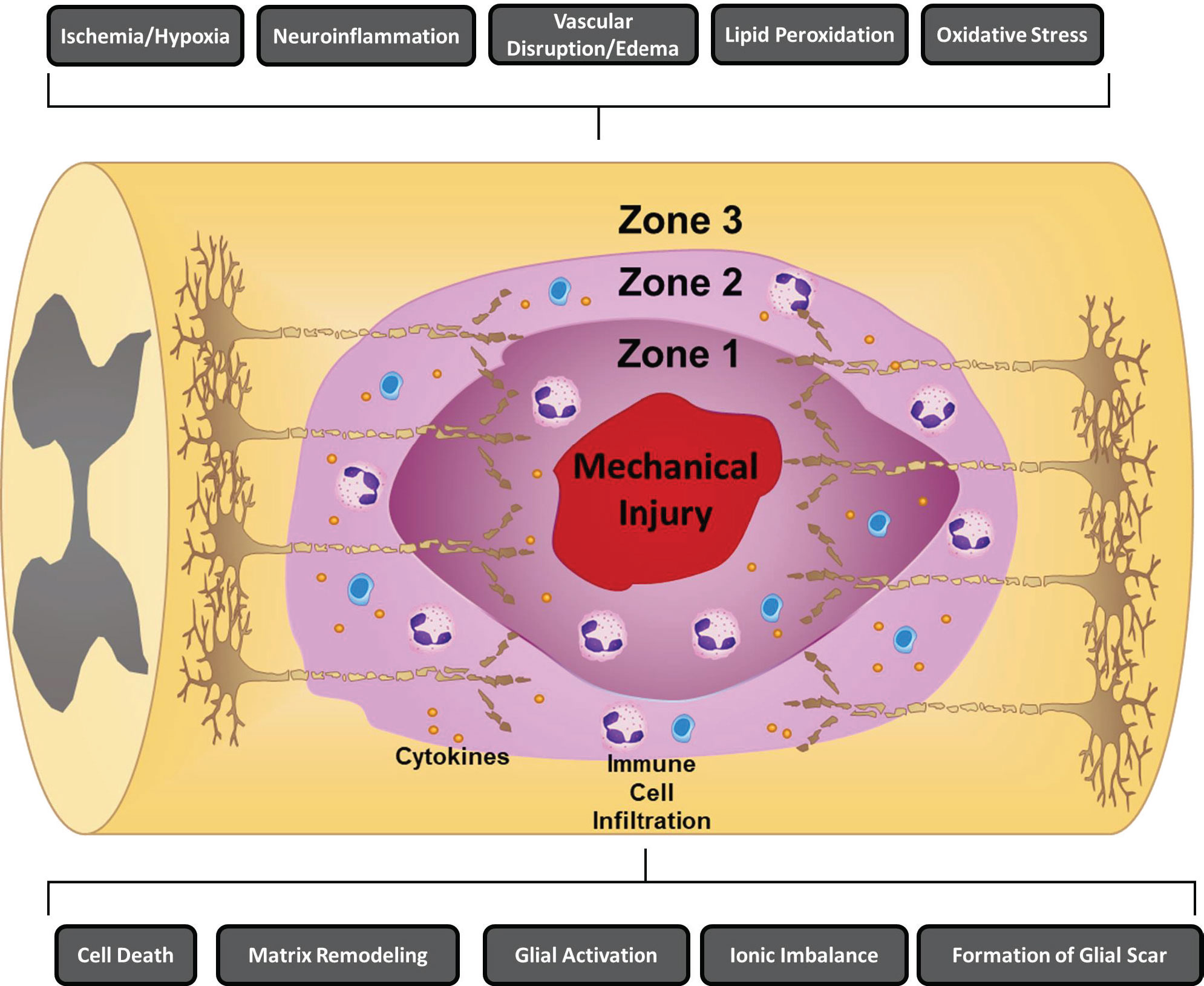
Figure 1 Mechanisms of secondary injury following traumatic spinal cord injury. Several key secondary mechanisms of injury that have been shown to contribute to the propagation of acute cell dysfunction and cell death including ischemia/hypoxia, neuroinflammation, vascular disruption/edema, lipid peroxidation, oxidative stress, cell death, ionic imbalance, formation of a glial scar, glial activation, and matrix remodeling are depicted. Following SCI, three zones that differ in tissue quality form including: (1) Zone 1, which is mainly a product of the initial trauma and is defined by regions of necrosis, inflammation, and cysts; (2) Zone 2, is characterized by regions of incomplete injury that are still accompanied by immune cell infiltration, axonal swelling, and Wallerian degeneration; and (3) Zone 3, which are histologically intact areas (15, 16).
Following SCI, three zones that differ in tissue quality form including: (1) Zone 1, which is mainly a product of the initial trauma and is defined by regions of necrosis, inflammation, and cysts; (2) Zone 2, is characterized by regions of incomplete injury that are still accompanied by immune cell infiltration, axonal swelling, and Wallerian degeneration; and (3) Zone 3, which are histologically intact areas (15, 16). Among the zones, Zone 1 is the region that has the highest level of damage with the lowest chance of neuron survival/recovery (12, 15, 16). Neuroinflammation has been shown to be a critical secondary injury mechanism that directly alters the progression of lesions and cell function across all three zones (12). Although surgical decompression is the first line therapy for spinal cord compression following SCI, studies have shown that the severity of injury is dependent on the extent of secondary damage (10, 12, 17). Therefore, therapeutic strategies have focused on strategies to modulate the immune response after SCI in order to promote neuro-regeneration and neuroprotection. This review will focus on examining the immune response underlying the pathophysiology of SCI and assesses both current and future neuro-immunological therapies.
2.2. SCI induced immune depression versus autoimmunity
After SCI, both immune depression and autoimmunity commonly co-occur (18, 19). The CNS exerts control over the immune system through several pathways including the hardwired fibers of the autonomic nervous system (19, 20). Throughout the body, both central and peripheral autonomic nervous system sensors function to relay information about the status of the body’s immune system (19, 20). Due to SCI, the CNS interaction with the immune system is disrupted resulting in a significant systemic decrease in immune function with reports of several different functional alterations in macrophages, T and B cells, and natural killer (NK) cells (20, 21). This systemic decrease in immune function after SCI is termed spinal cord injury-induced immune deficiency syndrome (SCI-IDS) (12, 21). Although the precise mechanism(s) causing SCI-IDS are not completely clear, patients with SCI-IDS are at a significantly higher risk of developing complications such as infections, pneumonia, and urinary tract infections (12, 21).
One recent study suggested that the mechanism underlying post-SCI immune suppression may be due to the fact that after SCI, significant plasticity develops below the injury site within the autonomic spinal circuitry resulting in a sympathetic anti-inflammatory reflex (22). Furthermore, the authors showed that chemogenetic silencing of this reflex prevents post-SCI immune suppression and could be a promising therapeutic approach in the future (22). Although there are several negative effects of SCI-IDS, immunosuppression following SCI may actually be advantageous and serve a protective function (12, 23). SCI damages the blood-spinal cord barrier and spine parenchyma, which exposes cells of the adaptive immune system including B and T lymphocytes to CNS antigens (12, 23). Exposure of the adaptive immune system to CNS antigens can then trigger autoimmune responses and the production of autoantibodies that have been shown to worsen pathology within the spinal cord (12, 23). Notably, studies in mice suggest that SCI especially impairs the production of new antibody responses but preserves existing immunity (18, 24). Thus, autoantibodies identified after SCI may have existed prior to the injury (18). This hypothesis was supported by a study that demonstrated a set of naturally occurring auto-antibodies expanded following SCI (18). Although it is clear that trauma induced autoimmunity has the potential to contribute pathologically, the triggering mechanisms and molecular signatures will require further investigation in the future.
2.3. Inflammatory cascade after SCI
Following CNS injury, neuroinflammation is a result of innate immune system activation that is mediated by cytokines and chemokines released by astrocytes, resident microglia, endothelial cells, and peripherally derived immune cells (14). Several studies have clearly demonstrated that within hours of CNS injury, the inflammatory cytokines IL-1, IL-6, and TNF are all upregulated (7, 25). Upregulation of these potent inflammatory cytokines subsequently triggers substantial infiltration of macrophages, microglia, and neutrophils (7). Furthermore, these infiltrating immune cells continue to produce and secrete additional chemokines and cytokines that modulate the immune response. The size of the primary insult is a critical factor in determining the magnitude of neuroinflammation (26). In the case of SCI, additional cell death is often caused by overactivation of the immune response. Therefore, in order to develop therapies that can selectively dampen the overactive immune response, the immune response following SCI must be thoroughly characterized. In this review, we briefly review the literature on the systemically upregulated cytokines derived from the blood/cerebrospinal fluid (CSF), which may have clinical utility as biomarkers and focus on the timeline of the local cytokine/chemokine cascade and cell infiltration patterns (Figure 2) (32).
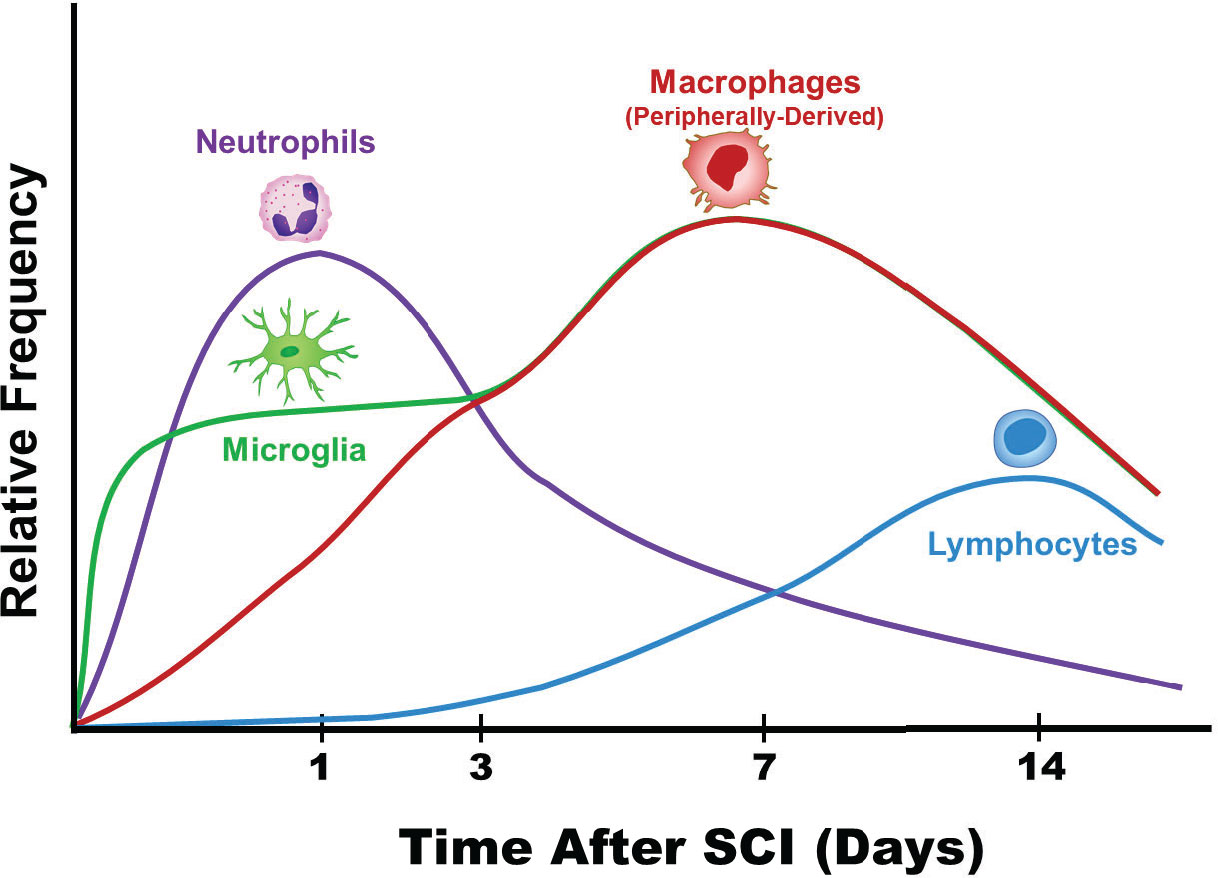
Figure 2 Patterns of immune cell infiltration following spinal cord injury. The order and timing (7, 27–31) of various immune cell infiltrates is depicted (32). The level of neutrophils peak at approximately 24 hours after SCI and decrease over the course of the next 7-10 days. Comparatively, lymphocytes peak much later and at lower levels compared to neutrophils. The resident macrophages, microglia, are early responders after SCI. Eventually, microglia become indistinguishable from the peripherally derived macrophages from a morphological standpoint.
2.4. Cytokine patterns from serum and CSF as biomarkers
Overall, the patterns of cytokine and chemokine upregulation following SCI are generally similar in the serum and CSF compared to the patterns within the injured tissue itself. Understanding the tempo and the major players of the immune response in SCI could allow for the identification of biomarkers from patient’s serum and CSF that could guide management of SCI. Within 6 hours after SCI, a rat model showed that CSF levels of IL-1, IL-10, IL-17a, IFN-γ, and TNFα were all significantly elevated (7, 33). Furthermore, the serum levels of IL-1β, IL-6, and TNFα, remained elevated throughout the first week after SCI in rats (7, 34, 35). A clinical study examining changes in serum biomarkers found that within 24 hours after SCI, there were significant elevations in structural proteins including tau, S100β, and GFAP as well as elevations in the cytokines IL-6, IL-8, and MCP-1 (7, 36). One study reported decreased serum levels of IL-1β and IL-10 after 14 days following SCI while another study found persistently elevated IL-1β and no change in IL-10 28 days after SCI (7, 36). Notably, this study (36) also reported persistent serum elevations in TNFα 28 days after SCI and downregulation of IL-4 at 28 days. Importantly, clinical studies have revealed that lower levels of serum and CSF IL-6, IL-8, tau, S100b, GFAP, and MCP-1, are associated with significantly higher AIS grade improvement and neurological recovery compared to patients that did not improve (37, 38). In these studies, the most severely injured patients typically had the highest levels of NSE tau, S100b, and GFAP (37, 38). In the future, additional clinical studies will be needed to further validate these proteins as true biomarkers that could enhance clinical decision-making.
Below, we discuss the timeline of the inflammatory cascade following SCI including cytokine/chemokine regulation and cell infiltration patterns (Figure 2).
3. Timeline of the cytokine/chemokine cascade and cell infiltration
Within the first 24 hours following tSCI, proinflammatory cytokines including TNFα, IL-1β, and IL-6 appear to be upregulated and are likely the key mediators of injury (25, 33, 39–45). Studies have also suggested that other cytokines including IL-7, growth-related oncogene (GRO), macrophage inflammatory protein 1-alpha (MIP-1α), and monocyte chemoattractant protein 1 (MCP-1) are upregulated while IL-4, IL-10, IL-13, and TGF- β1 remain at baseline levels within the first 24 hours after tSCI (33, 43, 46). Here, we provide an in-depth review of the patterns of cytokine/chemokine regulation and cell infiltration patterns within the first 24 hours and examine the inflammatory cascade over the course of the first two weeks following tSCI.
3.1. 1 Hour after SCI
Within minutes after the primary SCI, secondary injury mechanisms begin to cause significant damage (7, 47, 48). More specifically, edema, oxidative damage, cell permeabilization, excitotoxicity (glutamate), ischemic injury due to microvascular supply destruction, and proapoptotic signaling among other factors all contribute to substantial cell dysfunction and cell death (7, 47, 48). Dysregulation in intracellular calcium also causes injury and further cell death as dysregulated intracellular calcium results in calpain activation causing mitochondrial dysfunction (7, 48–50).
Although in the early phases of injury the resident immune cells of the CNS, microglia, respond in a protective manner, quickly microglia morph into proinflammatory cells that secrete cytokines that trigger peripheral immune cell infiltration (27, 51–53). For example, one study showed that microglia and astrocytes start expressing TNFα mRNA and IL-1β within a half hour following SCI (25). Notably, the number of TNFα positive cells peaked 1 hour following SCI (25).
3.2. 1-3 Hours after SCI
Within 1-3 hours following tSCI, TNFα, IL-1β, and IL-6 are believed to be the key mediators. mRNA colocalization studies have demonstrated that microglia, neurons, and astrocytes synthesize TNFα, IL-1β, and IL-6 during the early hours after SCI in a mouse model (7, 25). In five other studies, the levels of TNFα in the early hours after SCI were also substantially increased (39–41, 54, 55). Three of these studies showed a significantly elevated level of IL-1β during the early hours of SCI; however, two studies using enzyme-linked immunosorbent assays (ELISAs) reported that there was not an increase in IL-1β during the early hours of injury (34, 39–41, 54). Hellebrand et al. (7) suggested that this inconsistency may be due to the time necessary to synthesize the full protein product as well as the time required for caspase 1 to proteolytically process it to its active form. Studies assessing IL-6 upregulation observed similar results to the IL-1β pattern (39–41, 54).
3.3. 3-6 hours after SCI
Although there appear to be a few differences in the adaptive immune system among rodents, a majority of literature has shown a significant increase in proinflammatory cytokines (7, 25, 40, 56–59). More specifically, within the window of 3-6 hours after SCI, several studies have shown that TNFα, IL-1β, and IL-6 are the primary cytokines significantly elevated (7, 25, 40, 56–59). In addition, most studies agree that within the 3-6 hours window there is a slight lag in upregulation of IL-1β and IL-6 relative to TNFα and IL-1a (7, 25, 40, 41, 57–59). The increase in cytokine production in microglia and astrocytes also results in increased peripheral immune cell invasion. For instance, neutrophils start to arrive within 4-6 hours after SCI and function to prepare the region for repair through the production of proteolytic and oxidative enzymes (28, 29). The overwhelming number of neutrophils, however, can cause significant tissue damage (28, 29).
At 3 hours following SCI, the number of cells expressing TNFα mRNA within the center of the injury is significantly increased relative to control cells; however, the number of cells expressing TNFα mRNA was 66% lower compared to the 1 hour time point (25). Additional evidence suggests that TNFα levels return closer to baseline following rapid onset as two studies reported no significant increase at 4 hours following SCI (40, 54).
Even though anti-inflammatory cytokines could decrease the levels of proinflammatory cytokines, studies suggest that following SCI, the levels of anti-inflammatory cytokines are either low or absent (7). Theoretically, IL-10 is an anti-inflammatory cytokine that could reduce pro-inflammatory cytokine levels and IL-4/IL-13 are anti-inflammatory cytokines that can trigger alternative macrophage activation (7, 60, 61). Studies using rodent models have shown that IL-13 was increased at 4 hours following SCI in rats but not in mice, and IL-10 was increased in mice but not in rats (40, 57, 58, 60–64). Finally, Xiong et al (65) reported increases in 4-hydroxynonenal (4-HNE), which is formed during lipid peroxidation, starting at 3 hours following SCI (7, 65).
3.4. 6-12 after SCI
Within the 6-12 hour time period, the proinflammatory environment continues facilitating increased microglia proliferation/recruitment and increased peripheral immune cell invasion (7). Studies agree that from 6-12 hours after SCI, TNFα, IL-1β, and IL-6 continue to be upregulated while IL-4, IL-10, and IL-13 remain at baseline levels (25, 33, 39–45). In addition, other immune mediators that are significantly increased during the 6-12 hour window include macrophage inflammatory protein 1-alpha (MIP-1a), monocyte chemoattractant protein 1 (MCP-1), C–X–C motif chemokine ligand 1 (CXCL1), growth-related oncogene (GRO), and RANTES (Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted) (33, 43, 46).
Initially, neutrophils are the most common infiltrating cell type (30). Overactivation of neutrophils can be quite destructive to tissue as these cells release substantial amounts of neurotoxic substances including chemokines, enzymes, reactive oxygen species (ROS), and reactive nitrogen species (RNS) (9, 29, 66, 67). At approximately 8 hours after SCI, apoptosis from the inflammatory response peaks in neuronal cells (68, 69).
3.5. 12-24 hours after SCI
During the 12-24 hour window a majority of studies show: (a) upregulated levels of the proinflammatory cytokines TNFα, IL-1β, and IL-6; (b) elevated levels of IL-7, GRO, MIP-1α, and MCP-1; and (c) baseline levels of TGF-β1, IL-4, IL-10, and IL-13 (39, 45, 46, 56, 70–72). At 24 hours following SCI, the level of neutrophils peak (Figure 2) (31). Neutrophils infiltrating the site of injury function to clear debris, proteases release reactive oxygen species, and neutrophils secrete myeloperoxidase, elastase, and proteases (31). In glial cells, apoptosis peaks at 24 hours following SCI (68, 69).
One study in rats showed that lymphocytes start to collect near blood vessels within gray matter as early as 6 hours after SCI (52). Lymphocytes that infiltrate the site of injury produce IL-1β, IL-6, TNFα, and LIF at 12 hours post injury (25). The levels of the neurotoxic 4-HNE peak at 24 hours and for two weeks remained elevated (65).
3.6. 24 hours-7 days after SCI
In a rat model of SCI, flow cytometry showed that the initial phase of cellular inflammation at 1 day after SCI consisted of an early neutrophil peak; at 7 days after SCI a peak of macrophages/microglia; and at 9 days after SCI a peak of T-cells (73). In mice, although neutrophils enter the site of injury at 6 hours after SCI, levels do not peak until 14 days after injury and remain for up to two weeks (27). At day 5 after SCI, phagocytic macrophages are most commonly localized in areas of necrosis while microglia are mainly at the margins (7, 74). Microglia are highly dynamic and during the first week after SCI have been shown to proliferate extensively (7, 74). Furthermore, at the border of the site of injury between infiltrating peripherally-derived macrophages and astrocytes, microglia form a dense cellular interface (7, 74). Due to the initial injury, axons start to retract at day 2 and macrophages also trigger a late phase of axon retraction (75–77).
After 2 days following SCI, co-localization studies showed that TNFα mRNA levels had returned to a level that was not significantly different from the level of mice that had received control laminectomies (7, 25). The average number of cells expressing IL-6 mRNA decreased from 24 hours-4 days after SCI to an almost zero cells 7 days after injury (7, 25). Similarly, the number of IL-1β positive cells decreased at 2, 4, and 7 days following SCI (7, 25). Notably, although the number of cells expressing the above transcripts decreased, several studies in both rats and mice have reported increased levels of TNFα, IL-1β, and IL-6 at 1, 3, and 7 days following SCI (7). In addition, GRO has been reported to be significantly increased at 1, 3, and 7 days following SCI (46, 71, 78).
During the first week of injury, studies have shown that there is a significant increase in production of chemokines that recruit T-cells, monocytes, and dendritic cells to the lesion (7). Previous studies have shown upregulation of MIP-1α at 1 and 3 days following SCI, CXCL1 expression is increased at 3 and 7 days following SCI, and RANTES is significantly upregulated at 3 days following SCI (43, 71, 78–81). While there have been some conflicting results, a majority of studies show that MCP-1 mRNA is increased in mice 1 day after SCI with return to baseline 7 days following SCI (82, 83). Among rats and mice, it is important to note that there have been some contrasting behaviors (7). Cytokines that have been reported to have at least somewhat contrasting behaviors among rats versus mice include IL-2, IL-4, IL-5, IL-13, and IFN-γ (7, 70, 80, 81, 84). A majority of studies have suggested that IL-10 is not significantly increased until 3 days following SCI although some reports have not observed changes in IL-10 levels after SCI in both mice and rats (40, 42, 79). Finally, TGF-β1 was reported to be upregulated at 3 and 7 days following SCI (7, 39).
3.7. 7-14 days after SCI and beyond
Macrophages and peripherally derived macrophages peak at 7 days following SCI and persist for months after the injury in mice, rats, and humans (15, 60, 73, 74, 85). At 14 days following SCI injury, Mukhumedshina et al. (78) found that IL-1a, IL-2, MIP-1a, and GRO were significantly increased in rats while IL-2, IL-5, IL-13, IL-17a, IL-18, and GM-CSF were significantly decreased (78). In mice on the other hand, IL-1a, IL-4, IL-7, IL-12, IL-15, MIP-1a, MCP-1, and RANTES were significantly upregulated (79, 84, 86). Mice also were found to have a second surge of cells expressing IL-1β and TNFα at 14 days after SCI (25). Rats, however, did not undergo a second surge at 14 days after SCI (78, 87). Future studies examining the cytokines profiles after 14 days in SCI are necessary as there is less data compared to other time frames.
3.8. Cellular reactions and cytokine signaling at 14 days after SCI and beyond
Astrocytes are a subtype of CNS glial cells that function to maintain neurons and the blood spinal cord barrier (7, 88). Initially, reactive astrocytes flock to the injury site to aid in tissue repair but eventually become scar forming astrocytes (7, 88). These scar forming astrocytes form a glial scar surrounding the injury site. The layer of astrocytes around the lesion are defined by increased expression of glial fibrillary acidic protein (GFAP)/intermediate filaments, cellular hypertrophy, and process extension (7, 88). Overall, the glial scar has immune cells in the center with macrophages interacting with pericytes, which are the main scar connective tissue source around the edges and astrocytes that surround the periphery (74, 89).
The question of whether T cells mediate wound healing or result in secondary degeneration is controversial (7, 15, 90–92). T cells mediate adaptive immunity and appear to infiltrate into the spinal cord at somewhat different times depending on the species of animal (7). Studies have shown that chemokines CXCL10 and RANTES are critical for the proliferation and cytokine production of T cells (7, 93). Recent evidence suggests that T cells appear to play more of a destructive role after SCI as Gonzalez et al. (93) demonstrated that tissue preservation and functional recovery were improved when T cell infiltration was decreased by neutralizing the chemoattractant CXCL10. In a rat model, cytotoxic T-cells were predominant compared to regulatory T cells (>90% versus 10%, respectively) further suggesting a negative role of T cells (94). In addition, secondary injury was aggravated after SCI by T cell perforin as perforin destroyed the blood spinal cord barrier triggering infiltration of inflammatory cytokines (95).
4. Current and future therapies for SCI
4.1. Pharmacological therapies
To date, we do not fully understand the clinical utility and efficacy of several immunotherapies and pharmacological agents following tSCI. These immunotherapies have the potential to improve outcomes in tSCI patients by modulating the immune response to injury and promoting regeneration, however, further preclinical and clinical studies are needed. Although our current understanding of the precise immunological mechanisms by which some of these therapies may potentially improve functional outcomes is limited, here we provide the state of the field and future directions of pharmacological therapies in tSCI.
4.2. Corticosteroids
The clinical utility of methylprednisolone in the treatment of traumatic SCI is controversial (48, 96–98). Theoretically, corticosteroids could prevent secondary damage after SCI and be beneficial by preserving the ultrastructure of the spinal cord by decreasing the injury induced, decreasing free radical catalyzed lipid peroxidation, enhancing impulse conduction and neuronal excitability, improving blood flow, reducing oxidative stress, and modulating the immune response (99, 100). Three landmark multicenter, double-blinded, randomized controlled trials, National Spinal Cord Injury Study I (NASCIS-I), NASCIS-II, and NASCIS-III were performed in 1984, 1990, and 1997 (96, 101, 102). NASCIS-I sought to assess the methylprednisolone dosage following SCI, but there was no placebo group in this trial due to the belief that methylprednisolone was beneficial, and it would be unethical to withhold methylprednisolone therapy (101). Significant debate has been centered around the results of the NASCIS-II which randomized 487 patients with acute traumatic SCI to receive an initial bolus of 30 mg/kg of methylprednisolone followed by an infusion of 5.4 mg/kg per hour for 23 hours or treatment with placebo or naloxone (48, 96–98). Although there was no significant benefit in the neurological outcomes among the 162 patients receiving methylprednisolone within 12 hours, analysis of a group of 65 of these patients that received methylprednisolone within 8 hours after SCI significantly showed improved neurological function at 6 months (96, 103). Therefore, the authors concluded that treatment with methylprednisolone within 8 hours improved recovery of neurological function after SCI (96). Advocates of this study tend to highlight the lack of high-quality evidence currently available within the literature while critics point to the potential arbitrary 8-hour time point, the magnitude of treatment effects, and the potent issues with losses to follow-up (98, 103–108). A prospective randomized clinical trial demonstrated no neurological benefits (109). Several class III medical evidence studies have demonstrated the beneficial effects of methylprednisolone therapy in SCI (96, 101, 102, 110, 111). Critics of these studies, however, suggest that these studies are limited by small sample sizes derived retrospectively from larger study populations and/or incomplete data sets (112–115). Another potential concern of using methylprednisolone in the treatment of acute SCI is the potential for harmful side effects (96, 101, 102, 110, 111, 116). More specifically, three Class I studies reported wound infection, hyperglycemia requiring insulin administration, and GI hemorrhage as statistically significant side effects (96, 101, 102, 110, 111, 117, 118).
Although the clinical utility of these trials remains controversial, the methylprednisolone treatment protocols outlined in NASCIS II and III are still used today (96, 102). Early critics of the NASCIS advocated that methylprednisolone therapy be considered “a treatment option” rather than “standard of care” (96, 97, 101, 102). In 2013, the routine administration of methylprednisolone in the management of acute traumatic SCI was not recommended by “Guidelines for the Management of Acute Cervical Spine and Spinal Cord Injuries” (97, 119–121). The use of methylprednisolone among surgeons has decreased based on studies surveying the practice habits of surgeons, a 2018 study suggested that a majority of surgeons worldwide (52.9%) still use methylprednisolone in the treatment of acute SCI (122–127). In the future, further studies assessing the effect of steroid therapy on the neurological outcomes of SCI patients will be necessary in order to determine its clinical utility.
4.3. Cyclooxygenase inhibitors
The use of non-steroidal anti-inflammatory drugs (NSAIDs) following acute SCI injury appears promising as NSAIDs have been suggested to decrease spinal cord inflammation and edema, improve motor function with minimal side effects, and increase axonal sprouting (128). The effectiveness of NSAIDs in acute SCI is likely due to their ability to inhibit Rho-A (128). In animal models after traumatic SCI, two non-steroidal anti-inflammatory drugs that can be utilized in order to maintain spinal cord blood flow are ibuprofen and meclofenamate (129, 130). Similar results have been reported with the use of both a prostacyclin analogue and a thromboxane inhibitor (131, 132). Other studies have suggested that COX-2 expression is significantly increased within the damaged rat spinal cord tissue after contusion injury, and inhibition of COX-2 enhances functional outcomes (129, 133). Small sample size and heterogeneity creates problems with studies, and the therapeutic effects seen in animals may not be the same in patients (128, 131). Given the potential for improved outcomes and the low risk for adverse side effects, well-designed prospective studies evaluating ibuprofen and indomethacin are needed in the future.
4.4. Minocycline
Minocycline is a chemically modified second generation tetracycline antibiotic that has the ability to penetrate the blood brain barrier (134, 135). Minocycline has been shown to provide neuroprotective effects and has been shown to have anti-inflammatory, antioxidant, and anti-apoptotic properties (131, 134, 136). In animal models of SCI, TBI, and cerebral ischemia, minocycline has been shown to protect white matter structures, motor neurons, and oligodendrocytes (137, 138). Studies suggest that minocycline following SCI had significant anti-inflammatory and anti-apoptotic effects by preventing the activation of TNFα, IL-1β, COX-2, and MMPs decreasing caspase-1 and caspase-3 levels (131, 139, 140). Minocycline has also been reported to alleviate excitotoxicity and has been suggested to inhibit the p38 mitogen activated protein kinase pathway in microglial cells (137, 141, 142).
Tissue sparing, reduction of the lesion, inhibiting oligodendroglia and neuronal apoptosis, reducing inflammation, and enhancing neurological and histological outcomes could possibly be achieved with minocycline treatment (131, 143). Studies in mice have suggested that minocycline therapy is associated with substantial improvements in long term functional outcomes in which administration of minocycline over a 4 week recovery time course increased Beattie, Basso, and Bresnahan scores at 20 days following injury (144). A 2008 study in rats is not in agreement with other preclinical and clinical studies such as Pinzon et al. (143, 145) which found that minocycline therapy did not generate tissue sparing or improved behavioral outcomes (131, 143). In clinical studies, minocycline treatment improves outcomes in acute incomplete cervical SCI patients (131, 135). A randomized, double bind, placebo-controlled, single center phase II clinical trial examined treatment with IV minocycline (n=27) for 7 days versus placebo (n=25) treatment in patients with SCI. This study showed that minocycline therapy was safe and feasible. In addition, there was a trend toward improvements in motor scores especially in tetraplegic patients. Currently, a multicenter phase III randomized controlled trial (NCT01828203) is assessing the impact minocycline therapy versus placebo therapy on acute (<12 hours) SCI patient recovery at 3 months and one year after injury. Minocycline therapy, therefore, appears to be promising for SCI, as it works via several mechanisms; however, more well-designed trials are necessary to definitively conclude the role of minocycline in SCI management.
4.5. Chondroitinase ABC enzyme
The axons of patients with SCI do not regenerate appropriately (74, 89, 131, 146). Glial reaction occurs at the site of injury, resulting in formation of a glial scar (74, 89, 131, 146). Furthermore, microglia, oligodendrocytes, and their precursors (which include astrocytes, meningeal cells, and myelin fragments) are recruited due to the glial reaction (14, 69, 89, 131, 146). Many of these cells release factors that halt axonal regeneration (74, 89, 131, 146). Chondroitin sulfate proteoglycans (CSPG) are one major class of axonal growth inhibitors that may play a key role in regeneration failure (131, 146–148). Importantly, chondroitinase ABC (ChABC) is an enzyme that could be critical in overcoming CSPG mediated inbition as ChABC digests glycosaminoglycans on CSPGs (146–149).
Animal studies have demonstrated that treatment with ChABC after SCI can dramatically improve functional recovery and regeneration of both sensory axons and the corticospinal cord (131, 150, 151). Mechanistically, ChABC most likely results in growth promoting effects through causing increased germination of spare axons, formation of new synaptic connections beneath the sites of injury, and removing perineuronal nets (152). In addition to these mechanisms, ChABC also has been shown to have immunoregulatory activity (153). More specifically, Didangelos et al. showed that ChABC increased expression of the anti-inflammatory cytokine IL-10 and reduced expression of the pro-inflammatory cytokine IL-12B (153).
In rat models, administration of ChABC promoted both improved recovery of proprioceptive and locomotor behaviors (151). In addition, below the lesion treatment with ChABC resulted in restoration of postsynaptic activity following electrical stimulation of corticospinal neurons (151). The benefits of ChABC for SCI have been clearly demonstrated in rodent models of: (a) SCI, (b) stroke, and (c) nigrostriatal injury as well as in cats with SCI (131, 154–156). A recent study in dogs with severe chronic SCI strongly supported a beneficial effect of intraspinal injection of chABC (157). Importantly, there was no evidence of long-term adverse effects with this therapy (157). Given the impressive improvements of functional outcomes with ChABC treatment and the lack of reported harmful effects, human SCI ChABC trials should be conducted in the future.
4.6. Monosialotetrahexosylganglioside (GM-1)
Glycosphingolipids exert several critical functions in cellular differentiation, interaction and immune response (158). Theoretically, the benefit of glycosphingolipid GM-1 stems from the ability of GM-1 to stimulate tyrosine kinase receptors, resulting in improved regeneration and enhanced neuronal plasticity (100, 131). A randomized, double blind, prospective phase II trial demonstrated significant improvement of 1-year ASIA motor scores in patients that were treated with daily GM-1 ganglioside for 18-32 days following SCI (135, 159). A randomized, double-blind, sequential multicenter phase III clinical trial examined the effect of two doses of Sygen (monosialotetrahexosylganglioside GM1 sodium salt) compared to placebo in 797 patients with acute SCI (160). Although there appeared to be accelerated bladder, bowel, and motor recovery following Sygen treatment within the first three months, ultimately there was no significant effects after the study ended (135, 160). The time of administration of GM-1 ganglioside has been postulated to be a key factor in the observed variation among the two trials (135). Furthermore, a Cochrane Database Systematic Review described significant weaknesses in data collection and presentation methodologies in these trials (135, 161). This review concluded that there is no available evidence to support treatment with gangliosides to reduce the death rate or improve the quality of life in SCI and to date no follow-up studies have been performed (161).
4.7. Neuroimmunophilin ligands
Neuroimmunophilin ligands are a class of compounds that have great potential for the treatment for SCI. To date, however, the results of neuroimmunophilin ligand therapy for SCI have been conflicting. Cyclosporin A and FK-506, which act as neuroimmunophilin ligands, have shown protective properties in ischemia, neurodegenerative disorders, and in trauma (131, 162, 163). In neuronal tissues, NIL-A, which is currently in Phase II clinical trials for SCI, binds with FK506 binding protein (FKBP)-12 (131, 164). Neurophilin ligand V10367 binds to FK-506 binding protein and was investigated in a preclinical model (131, 165). V10367 resulted in increased neuroregeneration in the CNS and peripheral nervous system (165). Tacrolimus in addition to its immunosuppressive effects also has neurotrophic activity and has been shown to enhance nerve regeneration in peripheral nerve injury (166, 167). Due to the potential of neuroimmunophilin ligands in the treatment of nerve injury, further preclinical studies are needed to clarify their potential role in the treatment of tSCI.
4.8. Anti-Nogo-A antibodies (ATI-355)
Nogo-A is neuritite growth inhibitory myelin protein which plays a key role in inhibition of neurite outgrowth and limits neurological recovery (146, 168–170). Treatment with anti-Nogo-A antibody after SCI in rat models has been shown to significantly improve recovery of locomotor training and results in superior spinal cord reorganization and regeneration (171, 172). A macaque model showed similar results (131, 173). Another study examined the therapeutic outcomes of anti-Nogo-A antibody therapy on the corticospinal tract after acute, 1 week, or 2 week delayed intrathecal anti-Nogo-A antibody infusions (131, 174). In this study, Gonzenbach et al. (174) find that treatment of SCI with anti-Nogo antibodies in rodents is limited to less than 2 weeks. A multicenter Phase I open-label cohort study assessed intrathecal administration of the human anti-Nogo-A antibody ATI355 in acute, complete traumatic paraplegia and tetraplegia (175). Administration of anti-Nogo antibodies was well tolerated and in 7 of 19 patients with tetraplegia conversion from complete to incomplete SCI injury occurred (175).
4.9. VX-210 (Cethrin)
The Rho signaling pathway plays a key role in neuronal growth inhibition as well as regulating the cytoskeleton and motility (176). C3 transferase, an enzyme from Clostridium botulinum, inhibits Rho signaling by locking RhoA in an inactive state (176). In animal models of SCI, local application of C3 transferase induced long distance cortico-spinal regeneration and improved functional outcomes (176, 177). C3 transferase therapy with wild-type C3 transferse was limited, however, by its very low levels of cell penetration (177, 178). Thus, a recombinant version that readily crosses the dura was developed BA-210 (Cethrin) (177). In addition to promoting axonal regeneration and neuroprotection, Cethrin also appears to modify the adverse immune reaction following SCI (177–179). A multicenter phase I/IIa clinical trial involving cervical and thoracic SCI patients taking 0.3 mg to 9 mg BA-210 (later VX-210) showed that BA-210 treatment significantly improved motor recovery (178, 179). More specifically, the largest changes in motor recovery were observed in cervical injury patients taking 3 mg BA-210 in which 31% of cervical injury patients, 66% of patients taking 3mg BA-210,and 6% of thoracic injury patients converted from ASIA A to AISA C or D (178, 179). A phase Iib/III study assessing whether application of VX-210 to the overlying dura would improve motor outcomes following cervical SCI was stopped prematurely as the primary efficacy end-point was not met (180).
4.10. Anti-CD11d Antibodies
The CD11/CD18 family of integrins are localized on the surface of leukocytes and have been shown to bind both human intercellular adhesion molecule-3 (ICAM-3) and human/rat vascular cell adhesion molecule-1 (VCAM-1) (181–184). Following injury, damaged endothelial cells upregulate VCAM-1 and ICAM-3 which facilitates leukocyte binding and migration to the site of injury (184, 185). Preclinical studies have demonstrated the promising potential of a monoclonal antibody, 217L, which binds to the CD11d subunit of the CD11/CD18 integrins (184). Initial preclinical studies in rats demonstrated that anti-CD11d therapy significantly decreased number of both neutrophils and macrophages within the cord following SCI (186, 187). Furthermore, anti-CD11d therapy substantially decreased secondary damage and resulted in superior autonomic and locomotor recovery as well as decreased neuropathic pain (188–191). Similarly, other studies have also suggested that decreasing the number of neutrophils is neuroprotective (29, 46, 85, 192–194). A CD11d monoclonal antibody study in mice, however, reported that depletion of neutrophils after SCI through the use of Ly-6G (RB6-8C5) mAb impeded rather than enhanced functional recovery (195). A follow-up study examining the therapeutic effectiveness of anti-CD11d treatment in mice demonstrated that neutrophil infiltration was reduced by 61% at 72 hours after SCI and significantly improved functional outcomes and decreased secondary damage (184).
In addition to the role of anti-CD11d therapy in decreasing the infiltration of neutrophils, studies have suggested that anti-CD11d therapy improves neurological outcomes, provides neuroprotective effects decreases pain, significantly reduces histopathological damage, and improves intraspinal serotonergic innervation patterns (190, 196). A recent study also suggested that anti-CD11d therapy decreases secondary damage by significantly reducing free radical formation (197). Given the promising results from preclinical studies, future clinical trials assessing the efficacy of anti-CD11d in humans are needed. Importantly, the mechanism of injury and injury severity may be key determinants of the efficacy of anti-CD11d therapy following SCI (198). More specifically, using a rat model Geremia et al. (198) suggested that anti-CD11d therapy is most efficacious in cases of modest injury severity with minimal non-penetrating injury and frank hemorrhage into the spinal cord (198). Thus, clinical studies assessing efficacy of anti-CD11d therapy with careful consideration of the injury severity and mechanism of injury are necessary in the future.
4.11. B-Cell depletion therapies
Following SCI, a robust B cell response results in the production of pathogenic antibodies that impedes neurological recovery (199, 200). The role of B cells in the pathogenesis of SCI is further supported by the fact that functional outcomes are improved in B cell deficient RAG knockout mice following SCI (201). Based on this principle, selective B-cell depletion may be neuroprotective following SCI (201, 202). Casili et al. (202) assessed the effects of antibody mediated B cell depletion in mice using a glycoengineered anti-muCD20 antibody (18B12). Casili et al. (202) demonstrated that antibody mediated B cell depletion in mice improved functional recovery, slowed neuronal death and hindlimb motor dysfunction, and substantially inhibited the NFkB-dependent production of pro-inflammatory molecules. In the future, clinical studies are necessary in order to assess efficacy of anti-CD20 antibodies including rituximab or obinutuzumab after SCI (202, 203).
4.12. Targeting T-cells: trafficking, infiltration, and depletion
After SCI, T cells can directly or indirectly effect neurons, glia, or other CNS cells through the production of tumor necrosis factor (TNF)-α, interleukins (ILs), or other proinflammatory cytokines that play key roles in cytotoxic cell damage (92, 202, 204). Studies have demonstrated that therapeutic strategies targeting T cell infiltration and trafficking are neuroprotective (205). More specifically, antagonism or neutralizing antibodies against CXCL10, a known T cell recruiter, has been shown to improve functional outcomes, decrease T cell infiltration, decrease neuronal death, and significantly increase regeneration of axons (8, 206, 207). Another promising immunosuppressive therapy following SCI, involves the use of fingolimod, which targets sphingosine 1-phosphate receptor (S1P1), induces internalization of S1P1, decreases the number of circulating lymphocytes, and prevents trafficking of lymphocytes into tissues by sequestering lymphocytes within lymph nodes (208, 209). Locally administered fingolimod has been shown to result in superior functional recovery, decreases reactive gliosis, and prevents neuronal cell death (203, 210). Systemic treatment of fingolimod after SCI has been shown to prevent neuroinflammation and significantly improve both bladder and motor function (211, 212). Given the promising results of T cell targeting therapies, further clinical studies are necessary in order to assess the efficacy of CXCL10 antagonisms and fingolimod after SCI.
4.13. Macrophage transplantation
Previously, it has been shown that the macrophage immune response to injury was both delayed and blunted in the CNS of mature rodents compared to the more regenerative peripheral nervous system (213–215). Rapalino et al. (216) showed that local injection of homologous macrophages activated ex vivo through incubation with homologous peripheral nerves triggered partial motor recovery in rats with complete spinal cord transection (216, 217). Incubation of macrophages with autologous skin has been shown to result in equivalent effects and is thought to produce an “alternatively activated wound-healing phenotype” (213, 218). More specifically, in rats with a contused spinal cord, injection of macrophages activated with skin resulted in decreased spinal cyst formation and enhanced motor recovery (213, 218). This study also demonstrated that the skin activated macrophages had increased levels of cell surface markers that are characteristic of antigen presenting cells and macrophage secretion of brain-derived neurotrophic factor and interleukin-1b (213, 218). Autologous macrophage therapy has exciting potential due to the key functions macrophages play including clearing tissue debris from the site of injury, ability to modulate the immune system and influence spinal cord neurons, glial cells, and immune cells (217, 218). Safety studies in animals reported no short-or long-term adverse effects or toxicity in animals treated with macrophages to the Food and Drug Administration under Investigational New drug application No. 8427 (217).
Based on preclinical studies, an open-label phase I clinical trial assessing the safety and tolerability of treatment with incubated autologous macrophages in acute complete SCI patients was conducted (217). In this trial 3 of 8 subjects demonstrated conversion from AIS A to AIS C and overall the therapy was well tolerated (217). A single-blinded primary outcome randomized controlled trial involving six treatment centers in the United States and Israel, however, did not demonstrate a significant difference in the primary outcome among the autologous incubated macrophage treatment group (n=26) and the control group (n=16) (213). In the future, additional studies and further protocol optimization will be necessary to assess the efficacy of treatment of SCI with autologous incubated macrophages.
4.14. Hepatocyte growth factor
Hepatocyte growth factor (HGF) binds to c-Met and plays key roles in protection, regeneration, homeostasis, and exerts anti-inflammatory effects in several organs (219). Studies in rodent models have suggested that treatment with HGF results in the neuroprotective effects, enhanced angiogenesis, reduction of the lesion size, improved neuronal survival, and a decrease in production of oligodendrocytes (131, 220, 221). Furthermore, injection of HGF expression vector into rat spinal cords resulted in higher rates of survival, regeneration of axons and oligodendrocytes, and increased angiogenesis (131, 222). In a primate model of cervical SCI, significant improvements in hand dexterity were seen after HGF treatment (221, 223). Another study showed that exogenous treatment of HGF decreased astrocyte activation, decreased glial scar formation, decreased leukocyte infiltration, and produced anti-inflammatory effects (222). A recent double blind, randomized phase I/II clinical trial demonstrated the safety of treatment with recombinant human HGF and a larger phase III trial is necessary in the future in order to assess efficacy (224).
4.15. Neurotrophic factors
Neurotrophic factors are molecules that control the growth, survival, proliferation, and differentiation of neurons (225). In addition, neurotrophic factors play key roles in modulating immune responses and autoimmunity (226). Neurotrophic factors are often delivered with the use of collagenases (152). Delivering neurotrophic factors via collagenases has been reported to be more effective compared to delivery via direct injection, growth factor-saturated gel, or continuous injection (131, 152, 227). Ex vivo therapy can also be used when a patient’s cells are removed, genetically modified to synthesize specific neurotrophic factors, expanded in culture, and retransplanted into the patient (131, 152). The benefit of this route is that it provides localized, high-dose growth factors on delivery; however, initial studies suggest that ex vivo therapy is not ideal at stimulating distal axonal growth following initial axon growth (227).
4.16. Fibroblast growth factors
Fibroblast growth factors are a family of growth factors that are present in both the central and peripheral nervous system that play key roles in determining cell fate, differentiation, migration, and display neuroprotective effects (228–230). In addition to their neuroprotective effects, FGFs also may have some immunomodulatory effects as one approach utilizing acidic FGF (aFGF) combined with peripheral nerve grafts in rats following transection SCI increased levels of IL-4, IL-10, and IL-13 (9, 231). Other studies have shown FGFs protect against excitotoxicity and prevent the production of free radicals (131, 232). Treatment with FGF1 and FGF2 has been shown to significantly increase the growth and survival of various types of neurons including dopaminergic, cerebellar, hippocampal, and isolated sensory neuronal cells (143, 228, 233–235). Furthermore, both keratinocyte growth factor and basic FGF treatments appears to provide neuroprotection following SCI (131, 236). A prospective, open label uncontrolled clinical study demonstrated the safety, feasibility, modest improvements in functional outcomes in chronic SCI patients at 48 months that were treated with survival nerve grafts with fibrin glue containing acidic FGF (aFGF) (131, 237). Another clinical trial involving nine patients with cervical SCI treated with direct implantation of fibrin glue with aFGF for over six months showed significantly improved ASIA motor and sensory scale scores between the preoperative and follow-up (6-months postoperative) (131, 238). In addition, a prospective, open-label, uncontrolled clinical trial with 60 recruited patients with SCI demonstrated substantial improvement in AISA motor and sensory scale scores after 24 months following FGF therapy (239).
4.17. Granulocyte colony-stimulating factor
Granulocyte colony-stimulating factor (G-CSF) is a cytokine that in addition to its role in inducing proliferation, survival, and development of granulocyte lineage cells has been shown to play key roles in functional recovery following neuronal injury (240, 241). More specifically, studies have shown that G-CSF decreases both neuron and oligodendrocyte apoptosis, suppresses inflammatory cytokines (reduces TNFα and IL-1β in the spinal cord lesion), inhibits glutamate excitotoxicity, enhances angiogenesis, and promotes proliferation/mobilization of neuronal cells (131, 242–245). Based on these studies, phase I/IIa clinical trials were conducted and confirmed both the feasibility and safety of G-CSF therapy following SCI (246–248). The sentence should now read: "A phase II multicenter, prospective, non-blinded, nonrandomized clinical trial assessing patients with acute cervical SCI (within 48 hours after injury) receiving G-CSF therapy versus control therapy showed significant improvement (P<0.01) of motor paralysis and ASIA scores even at 1 year after injury in patients receiving G-CSF therapy compared to patients receiving control therapy (249). A recent phase 3 prospective, randomized, double-blind, and placebo controlled comparative trial was conducted and failed to show significant changes in ASIA motor scores from baseline to three months after G-CSF therapy (245). Additional randomized controlled trials will be needed in the future in order to determine the therapeutic benefits of SCI. Immunological pharmaceutical clinical studies are summarized in Table 1.
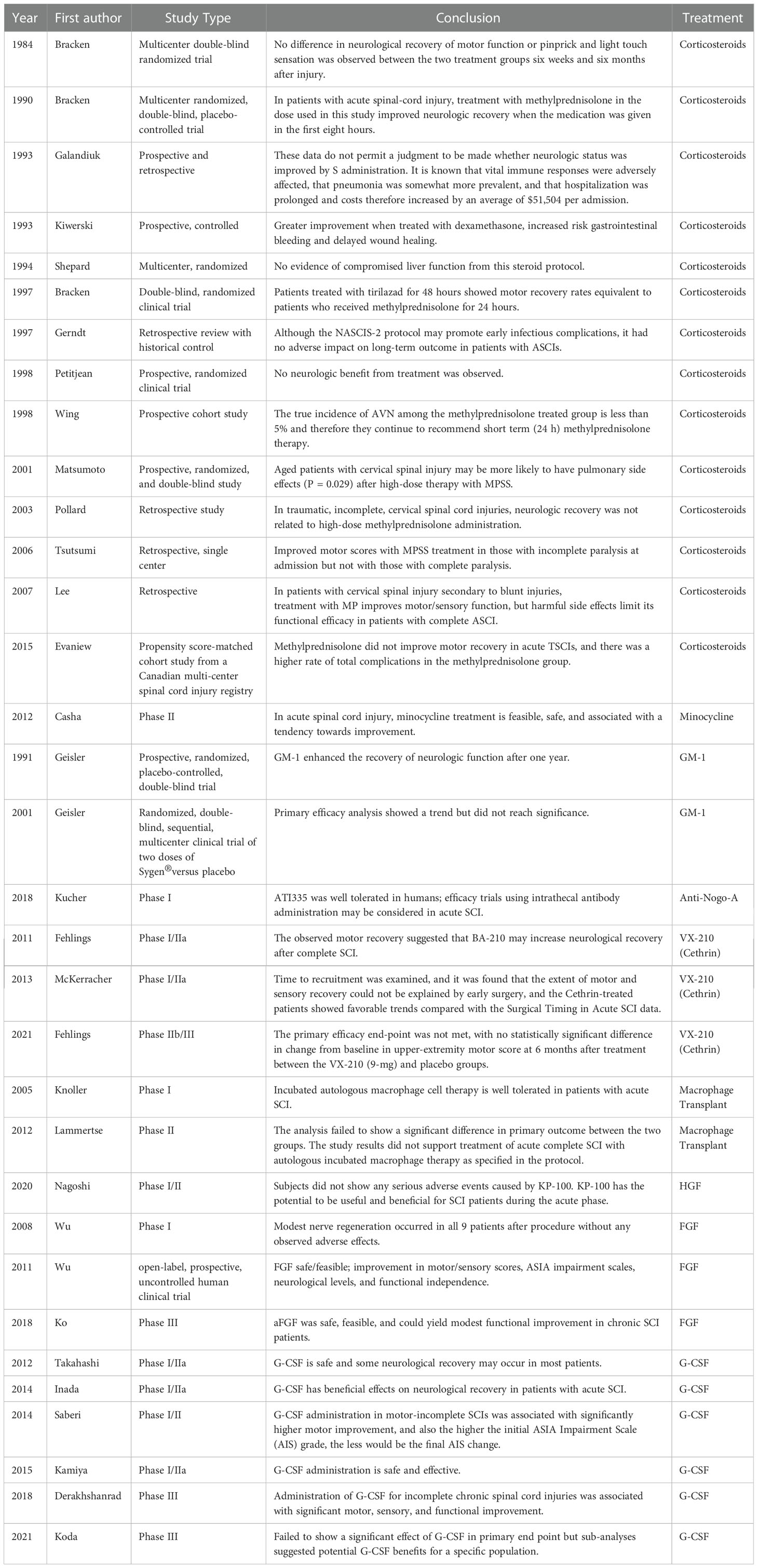
Table 1 Immunological pharmaceutical clinical studies summary table.
5. Stem cell therapy for SCI: Recent developments, limitations, and future directions
Stem cell therapy has potential to be revolutionary in the management of SCI as stem cell therapy can possibly regenerate neurological networks, modulate neuroinflammation, and ameliorate damage. Although many transplanted stem cell clinical trials for traumatic SCI patients are in the early stages (phase 1/2), there recently have been some promising results (250). Several studies have demonstrated safety and early evidence of potential efficacy of transplanted mesenchymal stem cells (MSCs), human neural stem/progenitor cells (hNSPC) autologous human Schwann cell (ahSCs), human umbilical cord-derived Wharton jelly mesenchymal stromal cells (WJ-MSCs), and autologous bone marrow derived mononuclear cells administered through multiple different routes (251–273). Transplanted stems cells exert their effects via three distinct mechanisms including: (1) cell replacement (274–276), characterized by the differentiation of stem cells into neuronal or vascular cells to compensate for the decreased function due to injury; (2) stem cell regeneration (31, 277, 278), in which transplantation of stem cells triggers the regeneration of the patient’s neuronal stem cells; and (3) functional multipotency (279), in which transplanted stem cells secrete a multitude of trophic factors which ameliorate damage to the nervous system or assists in the regeneration of new neuronal circuits. Interestingly, Kishk et al. suggested a potential contraindication of autologous MSCs in patients with a past medical history of myelitis (280).
Currently, the majority of clinical trials have focused on stem cell transplant in severely injured SCI patients (ASIA A) that are >6 months after SCI (chronic stage) (250). To date, there is minimal hope for spontaneous recovery, and there are no effective treatments for chronic stage (>6 months following injury) ASIA A patients, which makes the potential benefits of stem cell transplantation an attractive option (250). The results of trials of stem cell transplantation in chronic stage SCI patients have varied with some trials demonstrating recovery rates as high as 100% while other trials have shown no improvement based on ASIA impairment scale (256, 281, 282). Within the studies that suggested no improvement based on AISA impairment scale, patients did show some improvement on somatosensory evoked potential (SEP) and motor-evoked potential (MEP) testing suggesting some benefit in these patients (250, 281, 282). Other studies have suggested higher rates of SCI recovery after receiving stem cell therapy in matched-control patients (283, 284). In a group of 70 patients randomly divided, 34% of patients that were treated with intrathecal bone-marrow-derived mesenchymal stromal cell therapy demonstrated improvement in AISA grade compared to 0% of patients in the control group (250, 285). Similarly, in a group of 34 ASIA A SCI that were randomly divided into a control group, rehabilitation group, and a cell transplantation group, only patients that received cell transplantation showed significant motor, sensory, and urinary recovery compared to their status prior to treatment (250, 286).
Although a majority of preclinical animal studies have focused on delivering stem cell therapy within the acute phase after SCI (within the first 24 hours), there are relatively few clinical trials that examine stem cell therapy within the first 24 hours after SCI (287). Due to the unforeseen requirement for stem cells such as human umbilical cord mesenchymal stromal cells (MSCs) for stem cell therapy following traumatic SCI, it is unfortunately not feasible to use autologous material as the cells would first need to be expanded ex vivo (288). Thus, stem cell therapy during the acute phase of traumatic SCI requires the use of allogenic material. Two complete injury ASIA A patients that were treated with allogenic human umbilical cord mesenchymal stromal cells that were transplanted using a collagen scaffold showed functional recovery to an incomplete injury (ASIA C) (289). More trials in the future will be necessary to assess the potential benefits of acute phase stem cell transplant in traumatic SCI patients. One potential confounder to consider in the acute phase when assessing the efficacy of stem cell therapy is that within the acute phase, spontaneous recover is possible (250).
Finally, some trials and studies have assessed stem cell therapy administered between 2 days and 6 months following SCI, which has been defined as the sub-acute phase (250). The results of stem cell therapy trials within the sub-acute period of SCI have produced variable results with some trials demonstrating significant improvement and another group of trials suggesting no significant recovery (264, 281, 290, 291). One study examined the effectiveness of intra-spinal bone-marrow-derived mononuclear cells in patients with complete SCI (292). This study showed that when treated within 8 weeks following SCI, 30% of patients had significant functional recovery compared to 0% when stem cell transplantation was performed at >8 weeks and 7.6% AISA improvement in matched control patients (250, 292). A different study reported that significant improvement within the stem cell therapy group compared to the control group in which 46% of AISA A patients improved to ASIA C following injection of intrathecal bone-marrow-derived mesenchymal stromal cells (BMSC) versus 15% in the control group (290).
5.1. Future of stem cell therapy in SCI
Clinical trials, preclinical studies, and meta-analyses have shown that although the efficacy of stem cell therapy is promising, many challenges remain which will require further optimization and innovative solutions (293). One of the biggest and most concerning challenges of stem cell therapy for spinal cord injury moving forward is the potential for adverse effects. Although many clinical trials have been reported as safe without mortalities or severe morbidity, tumorigenicity, immunogenicity, and cell therapy-related immunotoxicity are potentially serious consequences of stem cell therapy (294). More specifically, to date there have been at least 28 kinds of adverse effects reported (293). Some side effects reported are likely secondary to the delivery procedures rather than the cells (294). Side effects secondary to the delivery procedure itself likely include subarachnoid hemorrhage, cerebrospinal fluid leaks, and transient deterioration in sensorimotor symptoms (25, 217, 280, 294–297). Moving forward, innovative delivery strategies will need to be investigated and employed to reduce side effects and further improve efficacy. In addition, side effects not associated with the transplanted cells and/or procedures were reported and include but are not limited to vomiting, pulmonary thromboembolism, fever, body aches, urinary tract infection (217, 262, 273, 291, 292, 294, 296, 298). Unfortunately, comprehensive assessment of safety and efficacy of stem cell therapy has been challenging for SCI patients as clinical trials in the past have been limited by relatively low sample sizes and insufficient control groups (299). In the future, large preclinical studies and early clinical studies are necessary in order to assess the safety of transplantation of human stem cells. Without rigorous assessment of the safety and potential adverse effects, the widespread adaptation of stem cell therapy will continue to be hampered.
Next, the properties and characteristics of the stem cells themselves must be rigorously assessed in order to continue to improve efficacy. Arguably the most critical factor in response to stem cell therapy is the origin of the stem cells used in the therapy (269, 300, 301). In addition to the studies examining MSCs, studies have also suggested potential feasibility and safety of human neural stem cell transplantation, but the clinical efficacy of such therapies requires further study. Similarly, studies examining autologous human Schwann cell transplantation have demonstrated feasibility, although currently they show unclear benefit (296, 302). Multiple studies have also demonstrated safety and feasibility of olfactory mucosa autografts (297, 303). To date, factors such as cell survival and integration are hurdles that potentially explain some of the heterogenous results among clinical trials must be improved through more carefully standardizing experimental designs.
The dose of stem cells also appears to be an especially critical factor and has varied significantly among trials from 106 to 1010 cells (250, 304). The route of administration of stem cells also appears to have a significant impact on therapy as intraspinal approaches result in the highest level of cell engraftment but is highly invasive compared to intravenous approaches which have been suggested to result in the lowest number of cells within the damaged lesion (250, 264). Furthermore, the optimal timing of stem cell transplantation in SCI remains to be determined. One pitfall to stem cell therapy moving forward is that a majority of clinical trials are single-centered investigator-oriented trials and therefore, further standardization including standardization between agencies will be necessary moving forward (250). Stem cell clinical studies are summarized in Table 2. In the future, clear and standardized transplantation strategies, cell numbers, treatment times, storage protocols, and consistent generation protocols which produce genetically stable and consistent therapeutic cells will be critical to unlock the potential efficacy of stem cell therapy for spinal cord injury
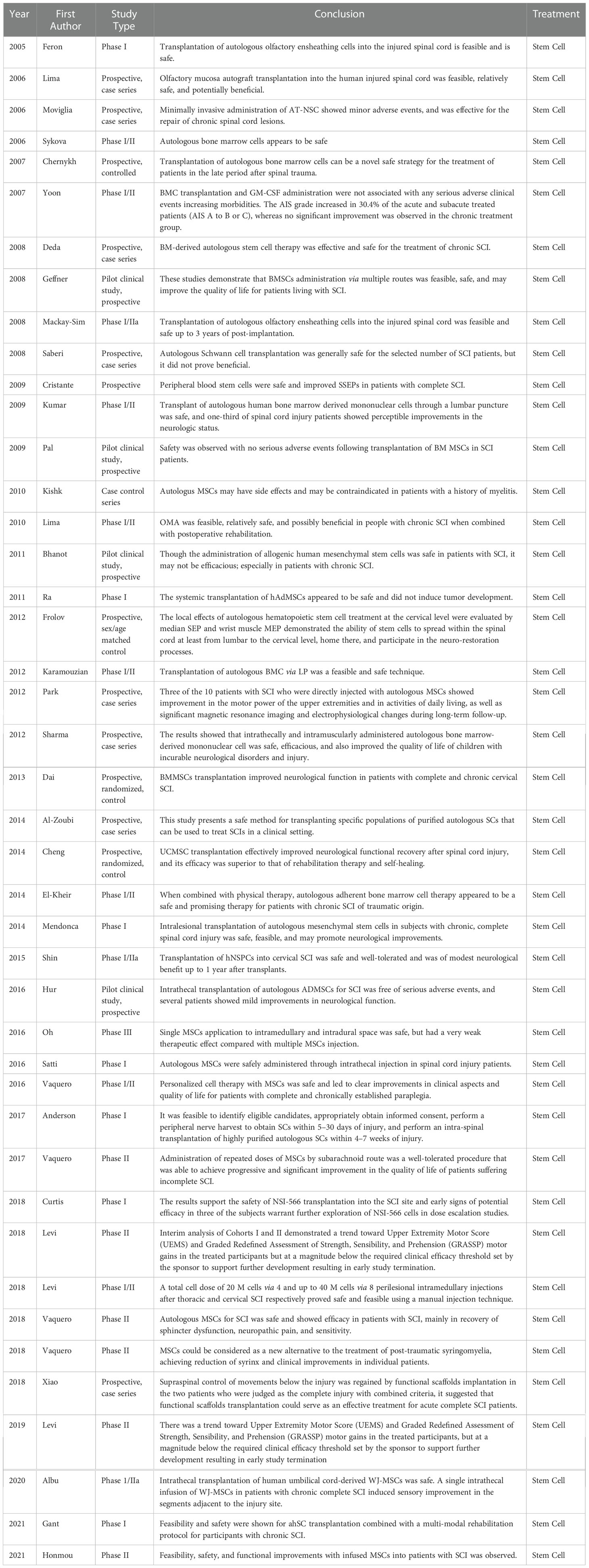
Table 2 Stem cells clinical studies summary table.
Importantly, recent evidence suggests that the biological and therapeutic effects of stem cell therapy including MSCs are contained within extracellular vesicles and secreted factors (305–307). Next, we review the exciting potential of EV therapy.
6. Extracellular vesicle therapy for SCI: An exciting new direction
Extracellular vesicles (EVs) are cell derived, nanosized sacs that are encapsulated with a lipid bilayer and enriched for nucleic acids, lipids, and proteins (308, 309). EVs are most commonly classified based on their route of biogenesis and size in which exosomes (40-150 nm) are produced via the endolysosomal pathway, microvesicles (150-1000 nm) are blebbed from the cell membrane, and apoptotic bodies (1,000-5,000 nm) are released as a part of programmed cell death from the plasma membrane (14). EVs have been shown to be released from almost all cells and have been isolated from almost all body fluids (310–313). Importantly, exosomes play a crucial role in intercellular communication in diverse cellular processes including immune response (314, 315). The initiation signal for the peripheral immune response following traumatic SCI is unclear (14, 316). Both neuronal and humoral hypotheses have been investigated; however, to date no consistent molecular candidates have been identified that fully account for initiation of the peripheral immune response (14, 316–318). Recent evidence suggests that EVs may be the missing link as they mediate communicate between distant organs (14, 319). Furthermore, EVs are attractive for therapeutic purposes because accumulating evidence suggests that the potential therapeutic benefit of MSC stem cell therapy is primarily due to the secreted factors including EVs and is not due to the direct cell-cell interactions within the injured tissues (305–307). EVs also have exciting therapeutic potential due to their ability to target specific cell types and their inherently biocompatibility (319, 320). Below, we review the recent developments and outline the future directions necessary in order to apply this potentially revolutionary therapy to SCI patients.
Reports have suggested that traumatic SCI induces a significant increase in plasma-derived EVs during the acute phase of injury (14). To date, however, studies characterizing EVs after SCI are lacking compared to the number of studies investigating EVs following traumatic brain injury (TBI) (14, 319). It is important to emphasize that although TBI data may provide some insight into the role of EV signaling following SCI injury, the impact of EVs on lesion progression and the overall impact on the acute phase response likely differ in traumatic SCI compared to TBI due to factors such as: (a) anatomical differences in the distribution of gray and white matter; (b) the phenotype and distribution of microglia in the spine compared to the brain; (c) increased local CXC chemokine expression and neutrophil recruitment to the spinal cord compared to the brain; and (d) greater breakdown of the blood spinal cord barrier compared to the blood brain barrier (14, 321). One study showed that EVs isolated from deceased SCI patients had a proinflammatory phenotype as EVs were enriched for inflammasome associated proteins including caspase-1, NLRP1, and ASC (322). In 2020, two studies reported EV-associated RNA-sequencing data from the serum of rats at 1 and 7 days postinjury (323, 324). Another study showed dynamic alterations in EVs in a mouse model following a thoracic contusion SCI (325). In this study, Khan et al. (325) found that there was a robust increase in plasma tetraspanin CD81+ extracellular vesicles after SCI and an overall decrease in total plasma EV. A significant decrease in CD81 surface expression on astrocytes was also observed by Khan et al. (325) at the site of injury suggesting that these cells may release CD81 positive EVs into the blood.
Astrocytes had previously been suggested to regulate the acute phase response (APR) (326). Based on this evidence, plasma EVs from mice with SCI or from uninjured control mice were intracerebroventricularly injected into healthy mice to assess the role of circulating EVs in promoting inflammation in recipient target organs (319, 325). Injection of EVs from SCI mice into healthy mice resulted in increased expression of several key inflammatory genes including astrocyte reactivity markers within 24 hours after injection of SCI EVs compared to injected EVs from uninjured mice (319, 325, 327). In addition, flow cytometry showed increased intracellular IL-1β and IL-1α levels in brain astrocytes from injected SCI Evs compared to EVs from uninjured mice (319, 325, 327). Therefore, this recent data suggests that astrocytes may play a key role in the production of EVs and regulating the APR following SCI (319, 325). In order to design and optimize EV therapies for traumatic SCI, it will be imperative for future studies to focus on characterizing the circulating population of EVs in SCI and to characterize the pathophysiological functions of EVs in SCI as there may be key differences in the role of EVs in SCI versus TBI.
To date, most studies have employed EVs from mesenchymal stem cells and have consistently shown improved recovery of function and behavior deficits in SCI and TBI models (306, 328, 329). For example, Jia et al. reported that injecting sonic hedgehog (Shh)-overexpressing bone mesenchymal stem cell derived exosomes was an effective therapy in a rat SCI model as there was an improvement in hind limb motor function based on Basso-Beattie-Bresnahan scores relative to both untreated and control bone mesenchymal stem cell derived exosome treatment groups (330). In addition, Li et al. (331) reported significant nerve recovery through attenuation of inflammation and oxidation in a rat transection SCI model receiving human mesenchymal stem cell derived exosomes immobilized in an adhesive hydrogel. Interestingly, Guo et al. (332) reported that intranasal administration of mesenchymal stem cell derived exosomes could cross the blood brain barrier, travel to area of the SCI, and when loaded with phosphatase and tensin homolog small interfering RNA, these exosomes decreased astrogliosis, decreased microgliosis, improved axonal growth, and increased neovascularization.
In addition to the consistently improved recovery of function in SCI models administered EVs, EVs are attractive compared to cell-based therapies as they mitigate concerns of uncontrolled proliferation or differentiation of cellular transplants and immunogenicity (319, 320). Excitingly, EV therapy compared to administration of their parental cells appears to result in superior outcomes as a recent study demonstrated that intravenous administration of human umbilical cord mesenchymal stromal cell (hUC-MSC) derived extracellular vesicles resulted in more effective modulation of the systemic immune response and more efficiently reduced scarring and inflammation compared to administration of parental MSCs (333). Other studies have also reported markedly reduced inflammation and improved functional outcomes with MSC-derived EV administration following traumatic SCI (334–336). In addition to the role of EVs modulating the systemic immune response, the potential of a higher local concentration and more rapid contact with the tissue lesion may also facilitate improved outcomes compared to cell therapies although more studies are needed (288, 333). Interestingly, reduction of circulating neutrophils and retention of monocytes within the spleen have been suggested to result in improvement in locomotor function in a SCI model administered MSC-EVs (306). Previous studies have shown that MSC-EVs localize to the spleen, and splenectomies have been associated with improved neurological outcomes in SCI models (14, 306, 337, 338). Future studies to assess whether the benefit of MSC EVs would remain in SCI models that had undergone splenectomy are needed (14).
6.1. Principles of design, limitations, and future directions of EV therapy
Arguably the cell source of the EVs is the most critical factor in manufacturing EVs for therapeutic purposes as the phenotype of EVs including the cell-surface proteins, cytosolic proteins, and composition of the nucleic acids that are available to be packaged into EVs are at least partly dictated by the cell source (339–342). For instance, the EV content of one critical class of small ncRNAs and miRNAs is significantly different among cell sources suggesting the importance of cell source as an EV design criterion (343, 344).
The cell culture conditions and microenvironment are also critical in order to tune the therapeutic bioactivity of EVs (319). Parameters including cell density seeding, cell age/passage, and the collection frequency of EVs have been reported to result in significant changes in production rates and the therapeutic activities in several different cell types (319, 345). For instance, EVs isolated from stressed astrocytes that were oxygen and glucose deprived resulted in neuroprotection in vitro most likely due to an increase in EV associated miR-92b-3p content compared to astrocytes that were not oxygen and glucose deprived (346, 347). The microenvironment cells are grown in also appears to be a critical factor as neural progenitor cells grown in proinflammatory conditions caused significant changes in RNA and protein levels with one study demonstrating enrichment of membrane bound IFN-γ and IFN-γ receptor 1 (IFNGR1) complexes that can induce key regulators of neuroinflammation and cell proliferation such as IFNGR1 and STAT1 (348). The employment of 3D printing could also be critical as 3D printing biomaterials will facilitate construction of complex precisely controlled microenvironments that could optimize EV production (319, 349, 350).
EVs are advantageous for therapeutic purposes as they protect nucleic acids and other bioactive components from degradation over long distances while also ensuring delivery to the cytosol of the target cell without triggering an auto-immune response (319). Although EVs hold great promise for transporting small interfering RNAs (siRNAs), miRNAs, mRNAs, and antisense oligonucleotides, in the case of some critical ncRNAs, the loading can be relatively low with as few as one bioactive copy of a given miRNA per EV (319, 351–353). Thus, it may be necessary to use high or repeated EV doses or employ cargo loading control strategies in situations where the dose of bioactive cargo is insufficient to adequately modulate gene expression (319, 351–353). Multiple studies have reported methods to load CNS-specific miRNA cargo into non-resident CNS cell types using genetic modification and subsequent over expression (14, 319). Other studies have used sonication, coincubation of EVs with desired cargo, and lipid or cholesterol functionalization (354–356).
One of the most important limitations of EVs to address for therapeutic purposes is the isolation protocols and scalability. Currently, several EV isolation strategies, each with their own advantages, have been utilized including differential centrifugation, ultrafiltration, precipitation, size exclusion chromatography, immunoisolation, and density gradient separation (357, 358). Ideally, an EV isolation protocol would be readily scalable to produce EVs with high purity for therapeutic purposes. The issue is that methods that result in a high purity can often have a relatively low yield and protocols with relatively high yields often co-isolate various protein aggregates, lipoproteins, and other impurities (357, 358). Therefore, the isolation protocol must always be optimized for yield and purity when manufacturing EVs for therapeutic purposes. Currently in the field, most groups are isolating EVs by differential centrifugation followed by ultracentrifugation (359). In situations where high purity is needed, further density-based purification using a sucrose or iodixanol centrifugation gradient can be used (360).
Finally, the route of administration appears to be a critical factor in EV therapy. More specifically, a 2021 study demonstrated that intralesional injection of EVs in a SCI rat model resulted in a more robust improvement of BBB score and sub-score compared to EVs delivered intravenously (288). Although intravenous injection is less invasive, delivering EVs intravenously will require higher doses of EVs as EVs can accumulate at off-target sites including the liver, spleen, and lungs, and systemic delivery of EVs intravenously results in delayed delivery of therapeutic EVs to the site of injury (288, 319, 333). The half-life of EVs is also an important to consider as a study observed a half-life of approximately 30 minutes in vivo in EVs loaded with dye after intravenous delivery (358). Therefore, intralesional injection could facilitate improved outcomes by more rapidly delivering a higher dose of therapeutic EVs to the lesion while minimizing off target accumulation and systemic degradation (7, 47, 48).
7. Discussion
SCI is a devasting event that has a complex pathophysiology. Following the primary mechanical injury, several secondary mechanisms of injury propagate cell dysfunction/cell death resulting in worse SCI patient outcomes. Among the many mechanisms of secondary injury, the systemic inflammatory response and neuroinflammation play pivotal roles in determining the severity of injury and patient outcomes. Following tSCI, the immune response likely exerts both beneficial and harmful roles and as previously discussed both the innate and adaptive immune systems have been implicated in secondary injury progression in tSCI. Thus, selectively targeting the immune response is a premier therapeutic approach as developing immunotherapeutics for SCI that shift the inflammatory cascade towards wound healing and away from secondary injury progression could drastically improve functional outcomes.
Recently, as discussed in this review, several studies have characterized the cytokine/chemokine cascades and the patterns of immune cell infiltration after SCI. Although there are some differences among humans, rats, and mice and further characterization is necessary, this work will facilitate therapeutic design and identification of potential biomarkers. Overall, TNFα, IL-1β, and IL-6 have been shown to be key mitigators of the immune response in the early hours/days following SCI. Although progress over the past two decades has significantly improved our understanding of the cytokine/chemokine cascades following tSCI, further characterization which identifies the timing of inflammatory cascades and the transit process of both innate and adaptive immune responses is necessary. More specifically, preclinical and clinical studies are needed that further characterize these inflammatory cascades and transit processes in the context of “beneficial” versus “harmful” responses to injury. A detailed molecular understanding of the inflammatory cascades underlying maladaptive injury responses versus wound healing responses would then allow for the design of novel immunotherapeutics or the repurposing of existing agents to shift maladaptive immune responses towards wound healing and thus potentially drastically improve outcomes. Over the next few decades, it is likely that immunomodulatory pharmaceuticals will be involved in managing SCI patients given the potential benefits. Due to the complex pathophysiology of SCI, however, multiple and synergistic immunotherapies may be required to promote recovery and halt maladaptive immune responses that promote secondary injury. In addition, clinical research and methods to improve targeted delivery to specific organs or cell types in a safe and stable manner will need to be developed and investigated.
Currently, there are multiple promising potential pharmaceutical agents and preclinical/clinical studies have reported no harmful effects and have suggested impressive benefits with NSAID/cyclooxygenase inhibitor, ChABC, and G-CSF therapy after SCI. Given these impressive improvements in functional results with no apparent harmful effects, we suggest a pressing need for clinical trials examining the role of these therapies in SCI. Two other pharmaceuticals, methylprednisolone and GM-1, appear to have minimal or questionable benefit in multiple studies and based off these studies should likely not routinely be used in SCI.
Furthermore, development of reliable SCI biomarkers that could predict outcomes and aid both medical management as well the surgical management of SCI patients is crucial. Due to the complicated pathophysiology underling SCI with likely both beneficial and detrimental immune responses following SCI, development of reliable predictive biomarkers has been challenging. Although recent studies have identified several proteins that appear to correlate with disease severity and patient outcomes including CSF IL-6, IL-8, tau, S100b, GFAP, and MCP-1, further studies are necessary in order to validate these proteins as true biomarkers that will guide clinical decision making.
Finally, given the complexity of SCI pathophysiology it is likely that these patients could benefit from not only immunotherapeutic agents but also combining other synergistic therapies including stem cell therapies/extracellular vesicle therapies and neuromodulation, spinal stimulation, and prosthetic devices which are beyond the scope of this review. In order to best apply combinations of these potentially synergistic therapies, further clinical and preclinical research investigating the efficacy, adverse effects, and biomarkers or parameters for reliable patient selection will be critical moving forward. In the case of stem cell transplantation, clinical trials have shown some promising results; however, overall, the results appear heterogenous. Further standardization including standardization between agencies will be necessary in the future. In addition, recent accumulating evidence suggests that the biological and therapeutic effects of stem cell therapy maybe due to secreted factors including EVs. Recent studies also suggest that EV therapy results in superior outcomes and is advantageous in that EV therapy mitigates concerns of immunogenicity and uncontrolled proliferation/differentiation that are associated with cellular transplants. Although the EV therapy has the potential to be revolutionary, several challenges as described in this review need to be addressed before it becomes a mainstream therapy. While challenges in developing pharmaceutical, stem cell, and EV therapies for SCI remain, new strategies and potential solutions continue to evolve which may provide a path forward to better the outcomes of SCI patients.
Author contributions
RCS and RMS designed, wrote, edited, and approved the final version of the manuscript. RMS supervised the study. All authors contributed to the article and approved the submitted version.
Funding
RCS was supported by 1 F30 MH124284-01.
Conflict of interest
RMS is an inventor on patents related to CAR-T cell therapy licensed to Humanigen through Mayo Clinic. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen Y, Tang Y, Vogel LC, DeVivo MJ. Causes of spinal cord injury. Top Spinal Cord Inj Rehabil (2013) 19:1–8. doi: 10.1310/sci1901-1
2. French DD, Campbell RR, Sabharwal S, Nelson AL, Palacios PA, Gavin-Dreschnack D. Health care costs for patients with chronic spinal cord injury in the veterans health administration. J Spinal Cord Med (2007) 30:477–81. doi: 10.1080/10790268.2007.11754581
3. Singh A, Tetreault L, Kalsi-Ryan S, Nouri A, Fehlings MG. Global prevalence and incidence of traumatic spinal cord injury. Clin Epidemiol (2014) 6:309–31. doi: 10.2147/CLEP.S68889
4. Merritt CH, Taylor MA, Yelton CJ, Ray SK. Economic impact of traumatic spinal cord injuries in the united states. Neuroimmunology Neuroinflamm (2019) 6:9. doi: 10.20517/2347-8659.2019.15
5. Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front Neurol (2019) 10:282. doi: 10.3389/fneur.2019.00282
6. Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, et al. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol (2001) 24:254–64. doi: 10.1097/00002826-200109000-00002
7. Hellenbrand DJ, Quinn CM, Piper ZJ, Morehouse CN, Fixel JA, Hanna AS. Inflammation after spinal cord injury: a review of the critical timeline of signaling cues and cellular infiltration. J Neuroinflamm (2021) 18:284. doi: 10.1186/s12974-021-02337-2
8. Glaser J, Gonzalez R, Sadr E, Keirstead HS. Neutralization of the chemokine CXCL10 reduces apoptosis and increases axon sprouting after spinal cord injury. J Neurosci Res (2006) 84:724–34. doi: 10.1002/jnr.20982
9. Garcia E, Aguilar-Cevallos J, Silva-Garcia R, Ibarra A. Cytokine and growth factor activation In vivo and In vitro after spinal cord injury. Mediators Inflammation (2016) 2016:9476020. doi: 10.1155/2016/9476020
10. Sterner RC, Brooks NP. Early decompression and short transport time after traumatic spinal cord injury are associated with higher American spinal injury association impairment scale conversion. Spine (2022) 47:59–66. doi: 10.1097/BRS.0000000000004121
11. Burke JF, Yue JK, Ngwenya LB, Winkler EA, Talbott JF, Pan JZ, et al. Ultra-early (<12 hours) surgery correlates with higher rate of American spinal injury association impairment scale conversion after cervical spinal cord injury. Neurosurgery (2019) 85:199–203. doi: 10.1093/neuros/nyy537
12. Chio JCT, Xu KJ, Popovich P, David S, Fehlings MG. Neuroimmunological therapies for treating spinal cord injury: Evidence and future perspectives. Exp Neurol (2021) 341:113704. doi: 10.1016/j.expneurol.2021.113704
13. Lambert C, Cisternas P, Inestrosa NC. Role of wnt signaling in central nervous system injury. Mol Neurobiol (2016) 53:2297–311. doi: 10.1007/s12035-015-9138-x
14. Yates AG, Anthony DC, Ruitenberg MJ, Couch Y. Systemic immune response to traumatic CNS injuries–are extracellular vesicles the missing link? Front Immunol (2019) 10:2723. doi: 10.3389/fimmu.2019.02723
15. Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, et al. The cellular inflammatory response in human spinal cords after injury. Brain (2006) 129:3249–69. doi: 10.1093/brain/awl296
16. Ahuja CS, Wilson JR, Nori S, Kotter MRN, Druschel C, Curt A, et al. Traumatic spinal cord injury. Nat Rev Dis Primers (2017) 3:1–21. doi: 10.1038/nrdp.2017.18
17. Huang S, Liu X, Zhang J, Bao G, Xu G, Sun Y, et al. Expression of peroxiredoxin 1 after traumatic spinal cord injury in rats. Cell Mol Neurobiol (2015) 35:1217–26. doi: 10.1007/s10571-015-0214-6
18. Arevalo-Martin A, Grassner L, Garcia-Ovejero D, Paniagua-Torija B, Barroso-Garcia G, Arandilla AG, et al. Elevated autoantibodies in subacute human spinal cord injury are naturally occurring antibodies. Front Immunol (2018) 9:2365. doi: 10.3389/fimmu.2018.02365
19. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci (2005) 6:775–86. doi: 10.1038/nrn1765
20. Brommer B, Engel O, Kopp MA, Watzlawick R, Müller S, Prüss H, et al. Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain (2016) 139:692–707. doi: 10.1093/brain/awv375
21. Riegger T, Conrad S, Liu K, Schluesener HJ, Adibzahdeh M, Schwab JM. Spinal cord injury-induced immune depression syndrome (SCI-IDS). Eur J Neurosci (2007) 25:1743–7. doi: 10.1111/j.1460-9568.2007.05447.x
22. Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y. Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci (2016) 19:784–7. doi: 10.1038/nn.4289
23. Ankeny DP, Popovich PG. Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience (2009) 158:1112–21. doi: 10.1016/j.neuroscience.2008.07.001
24. Oropallo MA, Held KS, Goenka R, Ahmad SA, O’Neill PJ, Steward O, et al. Chronic spinal cord injury impairs primary antibody responses but spares existing humoral immunity in mice. J Immunol (2012) 188:5257–66. doi: 10.4049/jimmunol.1101934
25. Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol (2007) 500:267–85. doi: 10.1002/cne.21149
26. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem (2016) 139 Suppl 2:136–53. doi: 10.1111/jnc.13607
27. Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol (2006) 494:578–94. doi: 10.1002/cne.20827
28. Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol (1998) 151:77–88. doi: 10.1006/exnr.1998.6785
29. Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, et al. Role of neutrophils in spinal cord injury in the rat. Neuroscience (1997) 79:1177–82. doi: 10.1016/s0306-4522(97)00011-0
30. Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res (2008) 86:1944–58. doi: 10.1002/jnr.21659
31. Neirinckx V, Cantinieaux D, Coste C, Rogister B, Franzen R, Wislet-Gendebien S. Concise review: Spinal cord injuries: How could adult mesenchymal and neural crest stem cells take up the challenge? Stem Cells (2014) 32:829–43. doi: 10.1002/stem.1579
32. Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol (2008) 209:378–88. doi: 10.1016/j.expneurol.2007.06.009
33. Cox A, Varma A, Barry J, Vertegel A, Banik N. Nanoparticle estrogen in rat spinal cord injury elicits rapid anti-inflammatory effects in plasma, cerebrospinal fluid, and tissue. J Neurotrauma (2015) 32:1413–21. doi: 10.1089/neu.2014.3730
34. Hasturk AE, Yilmaz ER, Turkoglu E, Arikan M, Togral G, Hayirli N, et al. Potential neuroprotective effect of anakinra in spinal cord injury in an in vivo experimental animal model. Neurosci (Riyadh) (2015) 20:124–30. doi: 10.17712/nsj.2015.2.20140483
35. Celik H, Karatay M, Erdem Y, Yildirim AE, Sertbas I, Karatay E, et al. The biochemical, histopathological and clinical comparison of the neuroprotective effects of subcutaneous adalimumab and intravenous methylprednisolone in an experimental compressive spinal cord trauma model. Turk Neurosurg (2016) 26:622–31. doi: 10.5137/1019-5149.JTN.13210-14.1
36. Kwon BK, Stammers AMT, Belanger LM, Bernardo A, Chan D, Bishop CM, et al. Cerebrospinal fluid inflammatory cytokines and biomarkers of injury severity in acute human spinal cord injury. J Neurotrauma (2010) 27:669–82. doi: 10.1089/neu.2009.1080
37. Ahadi R, Khodagholi F, Daneshi A, Vafaei A, Mafi AA, Jorjani M. Diagnostic value of serum levels of GFAP, pNF-h, and NSE compared with clinical findings in severity assessment of human traumatic spinal cord injury. Spine (Phila Pa 1976) (2015) 40:E823–830. doi: 10.1097/BRS.0000000000000654
38. Leister I, Haider T, Mattiassich G, Kramer JLK, Linde LD, Pajalic A, et al. Biomarkers in traumatic spinal cord injury–technical and clinical considerations: A systematic review. Neurorehabil Neural Repair (2020) 34:95–110. doi: 10.1177/1545968319899920
39. Streit WJ, Semple-Rowland SL, Hurley SD, Miller RC, Popovich PG, Stokes BT. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp Neurol (1998) 152:74–87. doi: 10.1006/exnr.1998.6835
40. Stammers AT, Liu J, Kwon BK. Expression of inflammatory cytokines following acute spinal cord injury in a rodent model. J Neurosci Res (2012) 90:782–90. doi: 10.1002/jnr.22820
41. Wang J, Chen J, Jin H, Lin D, Chen Y, Chen X, et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J Cell Mol Med (2019) 23:3214–23. doi: 10.1111/jcmm.14196
42. Sandrow-Feinberg HR, Zhukareva V, Santi L, Miller K, Shumsky JS, Baker DP, et al. PEGylated interferon-beta modulates the acute inflammatory response and recovery when combined with forced exercise following cervical spinal contusion injury. Exp Neurol (2010) 223:439–51. doi: 10.1016/j.expneurol.2010.01.009
43. Marsh DR, Flemming JMP. Inhibition of CXCR1 and CXCR2 chemokine receptors attenuates acute inflammation, preserves gray matter and diminishes autonomic dysreflexia after spinal cord injury. Spinal Cord (2011) 49:337–44. doi: 10.1038/sc.2010.127
44. van Neerven S, Mey J, Joosten EA, Steinbusch HW, van Kleef M, Marcus M a. E, et al. Systemic but not local administration of retinoic acid reduces early transcript levels of pro-inflammatory cytokines after experimental spinal cord injury. Neurosci Lett (2010) 485:21–5. doi: 10.1016/j.neulet.2010.08.051
45. Francos-Quijorna I, Amo-Aparicio J, Martinez-Muriana A, López-Vales R. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia (2016) 64:2079–92. doi: 10.1002/glia.23041
46. Tonai T, Shiba K, Taketani Y, Ohmoto Y, Murata K, Muraguchi M, et al. A neutrophil elastase inhibitor (ONO-5046) reduces neurologic damage after spinal cord injury in rats. J Neurochem (2001) 78:1064–72. doi: 10.1046/j.1471-4159.2001.00488.x
47. Blight AR. Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma (1985) 2:299–315. doi: 10.1089/cns.1985.2.299
48. Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J (2004) 4:451–64. doi: 10.1016/j.spinee.2003.07.007
49. Schanne FA, Kane AB, Young EE, Farber JL. Calcium dependence of toxic cell death: a final common pathway. Science (1979) 206:700–2. doi: 10.1126/science.386513
50. Oyinbo CA. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars) (2011) 71:281–99.
51. Hines DJ, Hines RM, Mulligan SJ, Macvicar BA. Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia (2009) 57:1610–8. doi: 10.1002/glia.20874
52. Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol (2002) 61:623–33. doi: 10.1093/jnen/61.7.623
53. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci (2005) 8:752–8. doi: 10.1038/nn1472
54. Pan JZ, Ni L, Sodhi A, Aguanno A, Young W, Hart RP. Cytokine activity contributes to induction of inflammatory cytokine mRNAs in spinal cord following contusion. J Neurosci Res (2002) 68:315–22. doi: 10.1002/jnr.10215
55. Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, et al. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma (1999) 16:851–63. doi: 10.1089/neu.1999.16.851
56. Bao W, Lin Y, Chen Z. The peripheral immune system and traumatic brain injury: Insight into the role of T-helper cells. Int J Med Sci (2021) 18:3644–51. doi: 10.7150/ijms.46834
57. Sánchez-Ventura J, Amo-Aparicio J, Navarro X, Penas C. BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J Neuroinflamm (2019) 16:124. doi: 10.1186/s12974-019-1511-7
58. Tai P-A, Chang C-K, Niu K-C, Lin M-T, Chiu W-T, Lin C-M. Attenuating experimental spinal cord injury by hyperbaric oxygen: stimulating production of vasculoendothelial and glial cell line-derived neurotrophic growth factors and interleukin-10. J Neurotrauma (2010) 27:1121–7. doi: 10.1089/neu.2009.1162
59. Hall JCE, Priestley JV, Perry VH, Michael-Titus AT. Docosahexaenoic acid, but not eicosapentaenoic acid, reduces the early inflammatory response following compression spinal cord injury in the rat. J Neurochem (2012) 121:738–50. doi: 10.1111/j.1471-4159.2012.07726.x
60. Hellenbrand DJ, Reichl KA, Travis BJ, Filipp ME, Khalil AS, Pulito DJ, et al. Sustained interleukin-10 delivery reduces inflammation and improves motor function after spinal cord injury. J Neuroinflamm (2019) 16:93. doi: 10.1186/s12974-019-1479-3
61. Junttila IS. Tuning the cytokine responses: An update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol (2018) 9:888. doi: 10.3389/fimmu.2018.00888
62. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
63. McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine (2015) 75:38–50. doi: 10.1016/j.cyto.2015.05.023
64. Thompson CD, Zurko JC, Hanna BF, Hellenbrand DJ, Hanna A. The therapeutic role of interleukin-10 after spinal cord injury. J Neurotrauma (2013) 30:1311–24. doi: 10.1089/neu.2012.2651
65. Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem (2007) 100:639–49. doi: 10.1111/j.1471-4159.2006.04312.x
66. Popovich PG, van Rooijen N, Hickey WF, Preidis G, McGaughy V. Hematogenous macrophages express CD8 and distribute to regions of lesion cavitation after spinal cord injury. Exp Neurol (2003) 182:275–87. doi: 10.1016/s0014-4886(03)00120-1
67. Bethea JR, Dietrich WD. Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol (2002) 15:355–60. doi: 10.1097/00019052-200206000-00021
68. Lu J, Ashwell KW, Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine (Phila Pa 1976) (2000) 25:1859–66. doi: 10.1097/00007632-200007150-00022
69. Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci (1997) 17:5395–406. doi: 10.1523/JNEUROSCI.17-14-05395.1997
70. Cheng P, Kuang F, Zhang H, Ju G, Wang J. Beneficial effects of thymosin β4 on spinal cord injury in the rat. Neuropharmacology (2014) 85:408–16. doi: 10.1016/j.neuropharm.2014.06.004
71. Austin JW, Afshar M, Fehlings MG. The relationship between localized subarachnoid inflammation and parenchymal pathophysiology after spinal cord injury. J Neurotrauma (2012) 29:1838–49. doi: 10.1089/neu.2012.2354
72. Lee SI, Jeong SR, Kang YM, Han DH, Jin BK, Namgung U, et al. Endogenous expression of interleukin-4 regulates macrophage activation and confines cavity formation after traumatic spinal cord injury. J Neurosci Res (2010) 88:2409–19. doi: 10.1002/jnr.22411
73. Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain (2010) 133:433–47. doi: 10.1093/brain/awp322
74. Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle M-E, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun (2019) 10:518. doi: 10.1038/s41467-019-08446-0
75. Gensel JC, Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res (2015) 1619:1–11. doi: 10.1016/j.brainres.2014.12.045
76. Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, et al. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol (2014) 254:109–20. doi: 10.1016/j.expneurol.2014.01.013
77. Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci (2008) 28:9330–41. doi: 10.1523/JNEUROSCI.2488-08.2008
78. Mukhamedshina YO, Akhmetzyanova ER, Martynova EV, Khaiboullina SF, Galieva LR, Rizvanov AA. Systemic and local cytokine profile following spinal cord injury in rats: A multiplex analysis. Front Neurol (2017) 8:581. doi: 10.3389/fneur.2017.00581
79. Yu WR, Fehlings MG. Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: implications for translational, clinical application. Acta Neuropathol (2011) 122:747–61. doi: 10.1007/s00401-011-0882-3
80. Ellman DG, Degn M, Lund MC, Clausen BH, Novrup HG, Flæng SB, et al. Genetic ablation of soluble TNF does not affect lesion size and functional recovery after moderate spinal cord injury in mice. Mediators Inflammation (2016) 2016:2684098. doi: 10.1155/2016/2684098
81. Ellman DG, Lund MC, Nissen M, Nielsen PS, Sørensen C, Lester EB, et al. Conditional ablation of myeloid TNF improves functional outcome and decreases lesion size after spinal cord injury in mice. Cells (2020) 9:E2407. doi: 10.3390/cells9112407
82. Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res (2002) 68:691–702. doi: 10.1002/jnr.10269
83. Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci (1997) 9:1422–38. doi: 10.1111/j.1460-9568.1997.tb01497.x
84. Bao C, Wang B, Yang F, Chen L. Blockade of interleukin-7 receptor shapes macrophage alternative activation and promotes functional recovery after spinal cord injury. Neuroscience (2018) 371:518–27. doi: 10.1016/j.neuroscience.2017.10.022
85. Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol (2003) 462:223–40. doi: 10.1002/cne.10736
86. Kerr BJ, Girolami EI, Ghasemlou N, Jeong SY, David S. The protective effects of 15-deoxy-delta-(12,14)-prostaglandin J2 in spinal cord injury. Glia (2008) 56:436–48. doi: 10.1002/glia.20630
87. Liu G, Fan G, Guo G, Kang W, Wang D, Xu B, et al. FK506 attenuates the inflammation in rat spinal cord injury by inhibiting the activation of NF-κB in microglia cells. Cell Mol Neurobiol (2017) 37:843–55. doi: 10.1007/s10571-016-0422-8
88. Okada S, Hara M, Kobayakawa K, Matsumoto Y, Nakashima Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci Res (2018) 126:39–43. doi: 10.1016/j.neures.2017.10.004
89. Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science (2011) 333:238–42. doi: 10.1126/science.1203165
90. Crutcher KA, Gendelman HE, Kipnis J, Perez-Polo JR, Perry VH, Popovich PG, et al. Debate: “is increasing neuroinflammation beneficial for neural repair?” J Neuroimmune Pharmacol (2006) 1:195–211. doi: 10.1007/s11481-006-9021-7
91. Hauben E, Gothilf A, Cohen A, Butovsky O, Nevo U, Smirnov I, et al. Vaccination with dendritic cells pulsed with peptides of myelin basic protein promotes functional recovery from spinal cord injury. J Neurosci (2003) 23:8808–19. doi: 10.1523/JNEUROSCI.23-25-08808.2003
92. Jones TB, Hart RP, Popovich PG. Molecular control of physiological and pathological T-cell recruitment after mouse spinal cord injury. J Neurosci (2005) 25:6576–83. doi: 10.1523/JNEUROSCI.0305-05.2005
93. Gonzalez R, Glaser J, Liu MT, Lane TE, Keirstead HS. Reducing inflammation decreases secondary degeneration and functional deficit after spinal cord injury. Exp Neurol (2003) 184:456–63. doi: 10.1016/s0014-4886(03)00257-7
94. Wu Y, Lin Y-H, Shi L-L, Yao Z-F, Xie X-M, Jiang Z-S, et al. Temporal kinetics of CD8+ CD28+ and CD8+ CD28- T lymphocytes in the injured rat spinal cord. J Neurosci Res (2017) 95:1666–76. doi: 10.1002/jnr.23993
95. Liu Z, Zhang H, Xia H, Wang B, Zhang R, Zeng Q, et al. CD8 T cell-derived perforin aggravates secondary spinal cord injury through destroying the blood-spinal cord barrier. Biochem Biophys Res Commun (2019) 512:367–72. doi: 10.1016/j.bbrc.2019.03.002
96. Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. results of the second national acute spinal cord injury study. N Engl J Med (1990) 322:1405–11. doi: 10.1056/NEJM199005173222001
97. Evaniew N, Noonan VK, Fallah N, Kwon BK, Rivers CS, Ahn H, et al. Methylprednisolone for the treatment of patients with acute spinal cord injuries: A propensity score-matched cohort study from a Canadian multi-center spinal cord injury registry. J Neurotrauma (2015) 32(21):1674–1683. doi: 10.1089/neu.2015.3963.
98. Coleman WP, Benzel D, Cahill DW, Ducker T, Geisler F, Green B, et al. A critical appraisal of the reporting of the national acute spinal cord injury studies (II and III) of methylprednisolone in acute spinal cord injury. J Spinal Disord (2000) 13:185–99. doi: 10.1097/00002517-200006000-00001
99. Hall ED, Braughler JM. Glucocorticoid mechanisms in acute spinal cord injury: a review and therapeutic rationale. Surg Neurol (1982) 18:320–7. doi: 10.1016/0090-3019(82)90140-9
100. Hurlbert RJ, Hadley MN, Walters BC, Aarabi B, Dhall SS, Gelb DE, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery (2013) 72 Suppl 2:93–105. doi: 10.1227/NEU.0b013e31827765c6
101. Bracken MB, Collins WF, Freeman DF, Shepard MJ, Wagner FW, Silten RM, et al. Efficacy of methylprednisolone in acute spinal cord injury. JAMA (1984) 251:45–52. doi: 10.1001/jama.1984.03340250025015
102. Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. results of the third national acute spinal cord injury randomized controlled trial. national acute spinal cord injury study. JAMA (1997) 277:1597–604. doi: 10.1001/jama.1997.03540440031029
103. Bracken MB. Steroids for acute spinal cord injury. Cochrane Database Syst Rev (2012) 1:CD001046. doi: 10.1002/14651858.CD001046.pub2
104. Hurlbert RJ. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg (2000) 93:1–7. doi: 10.3171/spi.2000.93.1.0001
105. Fehlings MG, Wilson JR, Cho N. Methylprednisolone for the treatment of acute spinal cord injury: counterpoint. Neurosurgery (2014) 61 Suppl 1:36–42. doi: 10.1227/NEU.0000000000000412
106. Sun X, Briel M, Busse JW, You JJ, Akl EA, Mejza F, et al. Credibility of claims of subgroup effects in randomised controlled trials: systematic review. BMJ (2012) 344:e1553. doi: 10.1136/bmj.e1553
107. Akl EA, Briel M, You JJ, Sun X, Johnston BC, Busse JW, et al. Potential impact on estimated treatment effects of information lost to follow-up in randomised controlled trials (LOST-IT): systematic review. BMJ (2012) 344:e2809. doi: 10.1136/bmj.e2809
108. Gerndt SJ, Rodriguez JL, Pawlik JW, Taheri PA, Wahl WL, Micheals AJ, et al. Consequences of high-dose steroid therapy for acute spinal cord injury. J Trauma (1997) 42:279–84. doi: 10.1097/00005373-199702000-00017
109. Petitjean ME, Pointillart V, Dixmerias F, Wiart L, Sztark F, Lassié P, et al. [Medical treatment of spinal cord injury in the acute stage]. Ann Fr Anesth Reanim (1998) 17:114–22. doi: 10.1016/s0750-7658(98)80058-0
110. Pointillart V, Petitjean ME, Wiart L, Vital JM, Lassié P, Thicoipé M, et al. Pharmacological therapy of spinal cord injury during the acute phase. Spinal Cord (2000) 38:71–6. doi: 10.1038/sj.sc.3100962
111. Matsumoto T, Tamaki T, Kawakami M, Yoshida M, Ando M, Yamada H. Early complications of high-dose methylprednisolone sodium succinate treatment in the follow-up of acute cervical spinal cord injury. Spine (Phila Pa 1976) (2001) 26:426–30. doi: 10.1097/00007632-200102150-00020
112. Kiwerski JE. Application of dexamethasone in the treatment of acute spinal cord injury. Injury (1993) 24:457–60. doi: 10.1016/0020-1383(93)90149-z
113. Pollard ME, Apple DF. Factors associated with improved neurologic outcomes in patients with incomplete tetraplegia. Spine (Phila Pa 1976) (2003) 28:33–9. doi: 10.1097/00007632-200301010-00009
114. Tsutsumi S, Ueta T, Shiba K, Yamamoto S, Takagishi K. Effects of the second national acute spinal cord injury study of high-dose methylprednisolone therapy on acute cervical spinal cord injury-results in spinal injuries center. Spine (Phila Pa 1976) (2006) 31:2992–2996; discussion 2997. doi: 10.1097/01.brs.0000250273.28483.5c
115. Lee H-C, Cho D-Y, Lee W-Y, Chuang H-C. Pitfalls in treatment of acute cervical spinal cord injury using high-dose methylprednisolone: a retrospect audit of 111 patients. Surg Neurol (2007) 68 Suppl 1:S37–41; discussion S41-42. doi: 10.1016/j.surneu.2007.06.085
116. Wing PC, Nance P, Connell DG, Gagnon F. Risk of avascular necrosis following short term megadose methylprednisolone treatment. Spinal Cord (1998) 36:633–6. doi: 10.1038/sj.sc.3100647
117. Galandiuk S, Raque G, Appel S, Polk HC. The two-edged sword of large-dose steroids for spinal cord trauma. Ann Surg (1993) 218:419–425; discussion 425-427. doi: 10.1097/00000658-199310000-00003
118. Shepard MJ, Bracken MB. The effect of methylprednisolone, naloxone, and spinal cord trauma on four liver enzymes: observations from NASCIS 2. national acute spinal cord injury study. Paraplegia (1994) 32:236–45. doi: 10.1038/sc.1994.43
119. Chappell ET. Pharmacological therapy after acute cervical spinal cord injury. Neurosurgery (2002) 51:855–856; author reply 856.
120. Walters BC, Hadley MN, Hurlbert RJ, Aarabi B, Dhall SS, Gelb DE, et al. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery (2013) 60:82–91. doi: 10.1227/01.neu.0000430319.32247.7f
121. Hugenholtz H, Cass DE, Dvorak MF, Fewer DH, Fox RJ, Izukawa DMS, et al. High-dose methylprednisolone for acute closed spinal cord injury–only a treatment option. Can J Neurol Sci (2002) 29:227–35. doi: 10.1017/s0317167100001992
122. Falavigna A, Quadros FW, Teles AR, Wong CC, Barbagallo G, Brodke D, et al. Worldwide steroid prescription for acute spinal cord injury. Global Spine J (2018) 8:303–10. doi: 10.1177/2192568217735804
123. Hurlbert RJ, Moulton R. Why do you prescribe methylprednisolone for acute spinal cord injury? a Canadian perspective and a position statement. Can J Neurol Sci (2002) 29:236–9. doi: 10.1017/s0317167100002006
124. Schroeder GD, Kwon BK, Eck JC, Savage JW, Hsu WK, Patel AA. Survey of cervical spine research society members on the use of high-dose steroids for acute spinal cord injuries. Spine (Phila Pa 1976) (2014) 39:971–7. doi: 10.1097/BRS.0000000000000297
125. Hurlbert RJ, Hamilton MG. Methylprednisolone for acute spinal cord injury: 5-year practice reversal. Can J Neurol Sci (2008) 35:41–5. doi: 10.1017/s031716710000754x
126. Miekisiak G, Kloc W, Janusz W, Kaczmarczyk J, Latka D, Zarzycki D. Current use of methylprednisolone for acute spinal cord injury in Poland: survey study. Eur J Orthop Surg Traumatol (2014) 24 Suppl 1:S269–273. doi: 10.1007/s00590-014-1422-3
127. Druschel C, Schaser K-D, Schwab JM. Current practice of methylprednisolone administration for acute spinal cord injury in Germany: a national survey. Spine (Phila Pa 1976) (2013) 38:E669–677. doi: 10.1097/BRS.0b013e31828e4dce
128. Lambrechts MJ, Cook JL. Nonsteroidal anti-inflammatory drugs and their neuroprotective role after an acute spinal cord injury: A systematic review of animal models. Global Spine J (2021) 11:365–77. doi: 10.1177/2192568220901689
129. Domon Y, Kitano Y, Makino M. Analgesic effects of the novel α₂δ ligand mirogabalin in a rat model of spinal cord injury. Pharmazie (2018) 73:659–61. doi: 10.1691/ph.2018.8550
130. Chaves RH de F, de Souza CC, Furlaneto IP, Teixeira RKC, de Oliveira CP, Rodrigues E de M, et al. Influence of tramadol on functional recovery of acute spinal cord injury in rats. Acta Cir Bras (2018) 33:1087–94. doi: 10.1590/s0102-865020180120000006
131. Zhang Y, Al Mamun A, Yuan Y, Lu Q, Xiong J, Yang S, et al. Acute spinal cord injury: Pathophysiology and pharmacological intervention (Review). Mol Med Rep (2021) 23:1–18. doi: 10.3892/mmr.2021.12056
132. Harada N, Taoka Y, Okajima K. Role of prostacyclin in the development of compression trauma-induced spinal cord injury in rats. J Neurotrauma (2006) 23:1739–49. doi: 10.1089/neu.2006.23.1739
133. Ko W-K, Kim SJ, Jo M-J, Choi H, Lee D, Kwon IK, et al. Ursodeoxycholic acid inhibits inflammatory responses and promotes functional recovery after spinal cord injury in rats. Mol Neurobiol (2019) 56:267–77. doi: 10.1007/s12035-018-0994-z
134. Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain (2012) 135:1224–36. doi: 10.1093/brain/aws072
135. Donovan J, Kirshblum S. Clinical trials in traumatic spinal cord injury. Neurotherapeutics (2018) 15:654–68. doi: 10.1007/s13311-018-0632-5
136. Shultz RB, Zhong Y. Minocycline targets multiple secondary injury mechanisms in traumatic spinal cord injury. Neural Regener Res (2017) 12:702–13. doi: 10.4103/1673-5374.206633
137. Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH, Kim YC, et al. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci (2007) 27:7751–61. doi: 10.1523/JNEUROSCI.1661-07.2007
138. Guimarães JS, Freire MAM, Lima RR, Picanço-Diniz CW, Pereira A, Gomes-Leal W. Minocycline treatment reduces white matter damage after excitotoxic striatal injury. Brain Res (2010) 1329:182–93. doi: 10.1016/j.brainres.2010.03.007
139. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35:495–516. doi: 10.1080/01926230701320337
140. Zhang H, Xiang Z, Duan X, Jiang JL, Xing YM, Zhu C, et al. Antitumor and anti-inflammatory effects of oligosaccharides from cistanche deserticola extract on spinal cord injury. Int J Biol Macromol (2019) 124:360–7. doi: 10.1016/j.ijbiomac.2018.11.132
141. Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem (2006) 96:314–23. doi: 10.1111/j.1471-4159.2005.03520.x
142. Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci (2001) 21:2580–8. doi: 10.1523/JNEUROSCI.21-08-02580.2001
143. Rabchevsky AG, Patel SP, Springer JE. Pharmacological interventions for spinal cord injury: where do we stand? how might we step forward? Pharmacol Ther (2011) 132:15–29. doi: 10.1016/j.pharmthera.2011.05.001
144. Squair JW, Ruiz I, Phillips AA, Zheng MMZ, Sarafis ZK, Sachdeva R, et al. Minocycline reduces the severity of autonomic dysreflexia after experimental spinal cord injury. J Neurotrauma (2018) 35:2861–71. doi: 10.1089/neu.2018.5703
145. Pinzon A, Marcillo A, Pabon D, Bramlett HM, Bunge MB, Dietrich WD. A re-assessment of erythropoietin as a neuroprotective agent following rat spinal cord compression or contusion injury. Exp Neurol (2008) 213:129–36. doi: 10.1016/j.expneurol.2008.05.018
146. Anjum A, Yazid MD, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, et al. Spinal cord injury: Pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci (2020) 21:7533. doi: 10.3390/ijms21207533
147. Fry EJ, Chagnon MJ, López-Vales R, Tremblay ML, David S. Corticospinal tract regeneration after spinal cord injury in receptor protein tyrosine phosphatase sigma deficient mice. Glia (2010) 58:423–33. doi: 10.1002/glia.20934
148. Kim J-W, Ha K-Y, Molon JN, Kim Y-H. Bone marrow-derived mesenchymal stem cell transplantation for chronic spinal cord injury in rats: comparative study between intralesional and intravenous transplantation. Spine (Phila Pa 1976) (2013) 38:E1065–1074. doi: 10.1097/BRS.0b013e31829839fa
149. Lee H, McKeon RJ, Bellamkonda RV. Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci (2010) 107:3340–5. doi: 10.1073/pnas.0905437106
150. Nout YS, Ferguson AR, Strand SC, Moseanko R, Hawbecker S, Zdunowski S, et al. Methods for functional assessment after C7 spinal cord hemisection in the rhesus monkey. Neurorehabil Neural Repair (2012) 26:556–69. doi: 10.1177/1545968311421934
151. Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature (2002) 416:636–40. doi: 10.1038/416636a
152. Weissmiller AM, Wu C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl Neurodegener (2012) 1:14. doi: 10.1186/2047-9158-1-14
153. Didangelos A, Iberl M, Vinsland E, Bartus K, Bradbury EJ. Regulation of IL-10 by chondroitinase ABC promotes a distinct immune response following spinal cord injury. J Neurosci (2014) 34:16424–32. doi: 10.1523/JNEUROSCI.2927-14.2014
154. Kwok JCF, Afshari F, García-Alías G, Fawcett JW. Proteoglycans in the central nervous system: plasticity, regeneration and their stimulation with chondroitinase ABC. Restor Neurol Neurosci (2008) 26:131–45.
155. McRae PA, Porter BE. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem Int (2012) 61:963–72. doi: 10.1016/j.neuint.2012.08.007
156. Grimpe B, Silver J. A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci (2004) 24:1393–7. doi: 10.1523/JNEUROSCI.4986-03.2004
157. Hu HZ, Granger N, Pai SB, Bellamkonda RV, Jeffery ND. Therapeutic efficacy of microtube-embedded chondroitinase ABC in a canine clinical model of spinal cord injury. Brain (2018) 141:1017–27. doi: 10.1093/brain/awy007
158. Zhang T, de Waard AA, Wuhrer M, Spaapen RM. The role of glycosphingolipids in immune cell functions. Front Immunol (2019) 10:90. doi: 10.3389/fimmu.2019.00090
159. Geisler FH, Dorsey FC, Coleman WP. Correction: recovery of motor function after spinal-cord injury–a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med (1991) 325:1659–60. doi: 10.1056/NEJM199106273242601
160. Geisler FH, Coleman WP, Grieco G, Poonian D, Sygen Study Group. The sygen multicenter acute spinal cord injury study. Spine (Phila Pa 1976) (2001) 26:S87–98. doi: 10.1097/00007632-200112151-00015
161. Chinnock P, Roberts I. Gangliosides for acute spinal cord injury. Cochrane Database Syst Rev (2005) 2, CD004444. doi: 10.1002/14651858.CD004444.pub2
162. Kawakami M. Molecular dissection of cyclosporin a’s neuroprotective effect reveals potential therapeutics for ischemic brain injury. Brain Sci (2013) 3:1325–56. doi: 10.3390/brainsci3031325
163. Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin a and FKBP-FK506 complexes. Cell (1991) 66:807–15. doi: 10.1016/0092-8674(91)90124-h
164. Gold BG, Densmore V, Shou W, Matzuk MM, Gordon HS. Immunophilin FK506-binding protein 52 (not FK506-binding protein 12) mediates the neurotrophic action of FK506. J Pharmacol Exp Ther (1999) 289:1202–10.
165. Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals (2008) 16:318–25. doi: 10.1159/000123041
166. Mekaj A, Mekaj Y. The role of pharmacological agents in nerve regeneration after peripheral nerve repair. IntechOpen (2017) 166:147–74. doi: 10.5772/intechopen.68378
167. Madsen JR, MacDonald P, Irwin N, Goldberg DE, Yao GL, Meiri KF, et al. Tacrolimus (FK506) increases neuronal expression of GAP-43 and improves functional recovery after spinal cord injury in rats. Exp Neurol (1998) 154:673–83. doi: 10.1006/exnr.1998.6974
168. Zörner B, Schwab ME. Anti-nogo on the go: from animal models to a clinical trial. Ann N Y Acad Sci (2010) 1198 Suppl 1:E22–34. doi: 10.1111/j.1749-6632.2010.05566.x
169. Schwab ME. Functions of nogo proteins and their receptors in the nervous system. Nat Rev Neurosci (2010) 11:799–811. doi: 10.1038/nrn2936
170. Buchli AD, Schwab ME. Inhibition of nogo: a key strategy to increase regeneration, plasticity and functional recovery of the lesioned central nervous system. Ann Med (2005) 37:556–67. doi: 10.1080/07853890500407520
171. Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, et al. Nogo-a antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol (2005) 58:706–19. doi: 10.1002/ana.20627
172. Chen K, Marsh BC, Cowan M, Al’Joboori YD, Gigout S, Smith CC, et al. Sequential therapy of anti-Nogo-A antibody treatment and treadmill training leads to cumulative improvements after spinal cord injury in rats. Exp Neurol (2017) 292:135–44. doi: 10.1016/j.expneurol.2017.03.012
173. Freund P, Wannier T, Schmidlin E, Bloch J, Mir A, Schwab ME, et al. Anti-Nogo-A antibody treatment enhances sprouting of corticospinal axons rostral to a unilateral cervical spinal cord lesion in adult macaque monkey. J Comp Neurol (2007) 502:644–59. doi: 10.1002/cne.21321
174. Gonzenbach RR, Zoerner B, Schnell L, Weinmann O, Mir AK, Schwab ME. Delayed anti-nogo-a antibody application after spinal cord injury shows progressive loss of responsiveness. J Neurotrauma (2012) 29:567–78. doi: 10.1089/neu.2011.1752
175. Kucher K, Johns D, Maier D, Abel R, Badke A, Baron H, et al. First-in-Man intrathecal application of neurite growth-promoting anti-Nogo-A antibodies in acute spinal cord injury. Neurorehabil Neural Repair (2018) 32:578–89. doi: 10.1177/1545968318776371
176. Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci (2002) 22:6570–7. doi: 10.1523/JNEUROSCI.22-15-06570.2002
177. Lord-Fontaine S, Yang F, Diep Q, Dergham P, Munzer S, Tremblay P, et al. Local inhibition of rho signaling by cell-permeable recombinant protein BA-210 prevents secondary damage and promotes functional recovery following acute spinal cord injury. J Neurotrauma (2008) 25:1309–22. doi: 10.1089/neu.2008.0613
178. Fehlings MG, Theodore N, Harrop J, Maurais G, Kuntz C, Shaffrey CI, et al. A phase I/IIa clinical trial of a recombinant rho protein antagonist in acute spinal cord injury. J Neurotrauma (2011) 28:787–96. doi: 10.1089/neu.2011.1765
179. McKerracher L, Anderson KD. Analysis of recruitment and outcomes in the phase I/IIa cethrin clinical trial for acute spinal cord injury. J Neurotrauma (2013) 30:1795–804. doi: 10.1089/neu.2013.2909
180. Fehlings MG, Chen Y, Aarabi B, Ahmad F, Anderson KD, Dumont T, et al. A randomized controlled trial of local delivery of a rho inhibitor (VX-210) in patients with acute traumatic cervical spinal cord injury. J Neurotrauma (2021) 38:2065–72. doi: 10.1089/neu.2020.7096
181. Grayson MH, van der Vieren M, Sterbinsky SA, Gallantin WM, Hoffman P, Staunton D, et al. alphadbeta2 integrin is a ligand for vascular cell adhesion molecule-1. Int Arch Allergy Immunol (1999) 118:263–4. doi: 10.1159/000024094
182. Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, Grayson MH, et al. The leukocyte integrin alpha d beta 2 binds VCAM-1: evidence for a binding interface between I domain and VCAM-1. J Immunol (1999) 163:1984–90. doi: 10.4049/jimmunol.163.4.1984
183. Van der Vieren M, Le Trong H, Wood CL, Moore PF, St John T, Staunton DE, et al. A novel leukointegrin, alpha d beta 2, binds preferentially to ICAM-3. Immunity (1995) 3:683–90. doi: 10.1016/1074-7613(95)90058-6
184. Geremia NM, Bao F, Rosenzweig TE, Hryciw T, Weaver L, Dekaban GA, et al. CD11d antibody treatment improves recovery in spinal cord-injured mice. J Neurotrauma (2012) 29:539–50. doi: 10.1089/neu.2011.1976
185. Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci (1999) 11:3648–58. doi: 10.1046/j.1460-9568.1999.00792.x
186. Mabon PJ, Weaver LC, Dekaban GA. Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol (2000) 166:52–64. doi: 10.1006/exnr.2000.7488
187. Saville LR, Pospisil CH, Mawhinney LA, Bao F, Simedrea FC, Peters AA, et al. A monoclonal antibody to CD11d reduces the inflammatory infiltrate into the injured spinal cord: a potential neuroprotective treatment. J Neuroimmunol (2004) 156:42–57. doi: 10.1016/j.jneuroim.2004.07.002
188. Bao F, Chen Y, Dekaban GA, Weaver LC. An anti-CD11d integrin antibody reduces cyclooxygenase-2 expression and protein and DNA oxidation after spinal cord injury in rats. J Neurochem (2004) 90:1194–204. doi: 10.1111/j.1471-4159.2004.02580.x
189. Bao F, Chen Y, Dekaban GA, Weaver LC. Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J Neurochem (2004) 88:1335–44. doi: 10.1046/j.1471-4159.2003.02240.x
190. Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, et al. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci (2004) 24:4043–51. doi: 10.1523/JNEUROSCI.5343-03.2004
191. Oatway MA, Chen Y, Bruce JC, Dekaban GA, Weaver LC. Anti-CD11d integrin antibody treatment restores normal serotonergic projections to the dorsal, intermediate, and ventral horns of the injured spinal cord. J Neurosci (2005) 25:637–47. doi: 10.1523/JNEUROSCI.3960-04.2005
192. Gorio A, Madaschi L, Zadra G, Marfia G, Cavalieri B, Bertini R, et al. Reparixin, an inhibitor of CXCR2 function, attenuates inflammatory responses and promotes recovery of function after traumatic lesion to the spinal cord. J Pharmacol Exp Ther (2007) 322:973–81. doi: 10.1124/jpet.107.123679
193. Hamada Y, Ikata T, Katoh S, Nakauchi K, Niwa M, Kawai Y, et al. Involvement of an intercellular adhesion molecule 1-dependent pathway in the pathogenesis of secondary changes after spinal cord injury in rats. J Neurochem (1996) 66:1525–31. doi: 10.1046/j.1471-4159.1996.66041525.x
194. Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-alpha. J Neurochem (2007) 102:900–12. doi: 10.1111/j.1471-4159.2007.04643.x
195. Stirling DP, Liu S, Kubes P, Yong VW. Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci (2009) 29:753–64. doi: 10.1523/JNEUROSCI.4918-08.2009
196. Hurtado A, Marcillo A, Frydel B, Bunge MB, Bramlett HM, Dietrich WD. Anti-CD11d monoclonal antibody treatment for rat spinal cord compression injury. Exp Neurol (2012) 233:606–11. doi: 10.1016/j.expneurol.2010.11.015
197. Bao F, Dekaban GA, Weaver LC. Anti-CD11d antibody treatment reduces free radical formation and cell death in the injured spinal cord of rats. J Neurochem (2005) 94:1361–73. doi: 10.1111/j.1471-4159.2005.03280.x
198. Geremia NM, Hryciw T, Bao F, Streijger F, Okon E, Lee JHT, et al. The effectiveness of the anti-CD11d treatment is reduced in rat models of spinal cord injury that produce significant levels of intraspinal hemorrhage. Exp Neurol (2017) 295:125–34. doi: 10.1016/j.expneurol.2017.06.002
199. Ankeny DP, Guan Z, Popovich PG. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest (2009) 119:2990–9. doi: 10.1172/JCI39780
200. Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic b lymphocyte activation and lupus-like autoantibody synthesis. J Neurochem (2006) 99:1073–87. doi: 10.1111/j.1471-4159.2006.04147.x
201. Wu B, Matic D, Djogo N, Szpotowicz E, Schachner M, Jakovcevski I. Improved regeneration after spinal cord injury in mice lacking functional T- and b-lymphocytes. Exp Neurol (2012) 237:274–85. doi: 10.1016/j.expneurol.2012.07.016
202. Casili G, Impellizzeri D, Cordaro M, Esposito E, Cuzzocrea S. B-cell depletion with CD20 antibodies as new approach in the treatment of inflammatory and immunological events associated with spinal cord injury. Neurotherapeutics (2016) 13:880–94. doi: 10.1007/s13311-016-0446-2
203. Putatunda R, Bethea JR, Hu W-H. Potential immunotherapies for traumatic brain and spinal cord injury. Chin J Traumatology (2018) 21:125–36. doi: 10.1016/j.cjtee.2018.02.002
204. Nitsch R, Pohl EE, Smorodchenko A, Infante-Duarte C, Aktas O, Zipp F. Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J Neurosci (2004) 24:2458–64. doi: 10.1523/JNEUROSCI.4703-03.2004
205. Clausen F, Lorant T, Lewén A, Hillered L. T Lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma (2007) 24:1295–307. doi: 10.1089/neu.2006.0258
206. Ghirnikar RS, Lee YL, Eng LF. Chemokine antagonist infusion promotes axonal sparing after spinal cord contusion injury in rat. J Neurosci Res (2001) 64:582–9. doi: 10.1002/jnr.1110
207. Gonzalez R, Hickey MJ, Espinosa JM, Nistor G, Lane TE, Keirstead HS. Therapeutic neutralization of CXCL10 decreases secondary degeneration and functional deficit after spinal cord injury in mice. Regener Med (2007) 2:771–83. doi: 10.2217/17460751.2.5.771
208. Healy LM, Antel JP. Sphingosine-1-Phosphate receptors in the central nervous and immune systems. Curr Drug Targets (2016) 17:1841–50. doi: 10.2174/1389450116666151001112710
209. Chiba K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther (2005) 108:308–19. doi: 10.1016/j.pharmthera.2005.05.002
210. Wang J, Wang J, Lu P, Cai Y, Wang Y, Hong L, et al. Local delivery of FTY720 in PCL membrane improves SCI functional recovery by reducing reactive astrogliosis. Biomaterials (2015) 62:76–87. doi: 10.1016/j.biomaterials.2015.04.060
211. Norimatsu Y, Ohmori T, Kimura A, Madoiwa S, Mimuro J, Seichi A, et al. FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am J Pathol (2012) 180:1625–35. doi: 10.1016/j.ajpath.2011.12.012
212. Lee KD, Chow WN, Sato-Bigbee C, Graf MR, Graham RS, Colello RJ, et al. FTY720 reduces inflammation and promotes functional recovery after spinal cord injury. J Neurotrauma (2009) 26:2335–44. doi: 10.1089/neu.2008.0840
213. Lammertse DP, Jones LAT, Charlifue SB, Kirshblum SC, Apple DF, Ragnarsson KT, et al. Autologous incubated macrophage therapy in acute, complete spinal cord injury: results of the phase 2 randomized controlled multicenter trial. Spinal Cord (2012) 50:661–71. doi: 10.1038/sc.2012.39
214. Perry VH, Brown MC, Gordon S. The macrophage response to central and peripheral nerve injury. a possible role for macrophages in regeneration. J Exp Med (1987) 165:1218–23. doi: 10.1084/jem.165.4.1218
215. Zeev-Brann AB, Lazarov-Spiegler O, Brenner T, Schwartz M. Differential effects of central and peripheral nerves on macrophages and microglia. Glia (1998) 23:181–90. doi: 10.1002/(sici)1098-1136(199807)23:3<181::aid-glia1>3.0.co;2-8
216. Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med (1998) 4:814–21. doi: 10.1038/nm0798-814
217. Knoller N, Auerbach G, Fulga V, Zelig G, Attias J, Bakimer R, et al. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine (2005) 3:173–81. doi: 10.3171/spi.2005.3.3.0173
218. Bomstein Y, Marder JB, Vitner K, Smirnov I, Lisaey G, Butovsky O, et al. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J Neuroimmunol (2003) 142:10–6. doi: 10.1016/s0165-5728(03)00260-1
219. Matsumoto K, Funakoshi H, Takahashi H, Sakai K. HGF–met pathway in regeneration and drug discovery. Biomedicines (2014) 2:275–300. doi: 10.3390/biomedicines2040275
220. Kitamura K, Iwanami A, Nakamura M, Yamane J, Watanabe K, Suzuki Y, et al. Hepatocyte growth factor promotes endogenous repair and functional recovery after spinal cord injury. J Neurosci Res (2007) 85:2332–42. doi: 10.1002/jnr.21372
221. Kitamura K, Fujiyoshi K, Yamane J-I, Toyota F, Hikishima K, Nomura T, et al. Human hepatocyte growth factor promotes functional recovery in primates after spinal cord injury. PloS One (2011) 6:e27706. doi: 10.1371/journal.pone.0027706
222. Kitamura K, Nagoshi N, Tsuji O, Matsumoto M, Okano H, Nakamura M. Application of hepatocyte growth factor for acute spinal cord injury: The road from basic studies to human treatment. Int J Mol Sci (2019) 20:E1054. doi: 10.3390/ijms20051054
223. Ohta Y, Takenaga M, Hamaguchi A, Ootaki M, Takeba Y, Kobayashi T, et al. Isolation of adipose-derived Stem/Stromal cells from cryopreserved fat tissue and transplantation into rats with spinal cord injury. Int J Mol Sci (2018) 19:E1963. doi: 10.3390/ijms19071963
224. Nagoshi N, Tsuji O, Kitamura K, Suda K, Maeda T, Yato Y, et al. Phase I/II study of intrathecal administration of recombinant human hepatocyte growth factor in patients with acute spinal cord injury: A double-blind, randomized clinical trial of safety and efficacy. J Neurotrauma (2020) 37:1752–8. doi: 10.1089/neu.2019.6854
225. Skaper SD. Neurotrophic factors: An overview. Methods Mol Biol (2018) 1727:1–17. doi: 10.1007/978-1-4939-7571-6_1
226. Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol (2009) 29:43–68. doi: 10.1615/critrevimmunol.v29.i1.20
227. Hodgetts SI, Harvey AR. Neurotrophic factors used to treat spinal cord injury. Vitam Horm (2017) 104:405–57. doi: 10.1016/bs.vh.2016.11.007
228. Zhou Y, Wang Z, Li J, Li X, Xiao J. Fibroblast growth factors in the management of spinal cord injury. J Cell Mol Med (2018) 22:25–37. doi: 10.1111/jcmm.13353
229. Yun Y-R, Won JE, Jeon E, Lee S, Kang W, Jo H, et al. Fibroblast growth factors: biology, function, and application for tissue regeneration. J Tissue Eng (2010) 2010:218142. doi: 10.4061/2010/218142
230. Awad BI, Carmody MA, Steinmetz MP. Potential role of growth factors in the management of spinal cord injury. World Neurosurg (2015) 83:120–31. doi: 10.1016/j.wneu.2013.01.042
231. Kuo H-S, Tsai M-J, Huang M-C, Chiu C-W, Tsai C-Y, Lee M-J, et al. Acid fibroblast growth factor and peripheral nerve grafts regulate Th2 cytokine expression, macrophage activation, polyamine synthesis, and neurotrophin expression in transected rat spinal cords. J Neurosci (2011) 31:4137–47. doi: 10.1523/JNEUROSCI.2592-10.2011
232. Lee Y, Kim C-Y, Lee HJ, Kim JG, Han DW, Ko K, et al. Two-step generation of oligodendrocyte progenitor cells from mouse fibroblasts for spinal cord injury. Front Cell Neurosci (2018) 12:198. doi: 10.3389/fncel.2018.00198
233. Clarke WE, Berry M, Smith C, Kent A, Logan A. Coordination of fibroblast growth factor receptor 1 (FGFR1) and fibroblast growth factor-2 (FGF-2) trafficking to nuclei of reactive astrocytes around cerebral lesions in adult rats. Mol Cell Neurosci (2001) 17:17–30. doi: 10.1006/mcne.2000.0920
234. Teng YD, Mocchetti I, Taveira-DaSilva AM, Gillis RA, Wrathall JR. Basic fibroblast growth factor increases long-term survival of spinal motor neurons and improves respiratory function after experimental spinal cord injury. J Neurosci (1999) 19:7037–47. doi: 10.1523/JNEUROSCI.19-16-07037.1999
235. Koshinaga M, Sanon HR, Whittemore SR. Altered acidic and basic fibroblast growth factor expression following spinal cord injury. Exp Neurol (1993) 120:32–48. doi: 10.1006/exnr.1993.1038
236. Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci (2000) 20:5012–23. doi: 10.1523/JNEUROSCI.20-13-05012.2000
237. Ko C-C, Tu T-H, Wu J-C, Huang W-C, Tsai Y-A, Huang S-F, et al. Functional improvement in chronic human spinal cord injury: Four years after acidic fibroblast growth factor. Sci Rep (2018) 8:12691. doi: 10.1038/s41598-018-31083-4
238. Wu J-C, Huang W-C, Tsai Y-A, Chen Y-C, Cheng H. Nerve repair using acidic fibroblast growth factor in human cervical spinal cord injury: a preliminary phase I clinical study. J Neurosurg Spine (2008) 8:208–14. doi: 10.3171/SPI/2008/8/3/208
239. Wu J-C, Huang W-C, Chen Y-C, Tu T-H, Tsai Y-A, Huang S-F, et al. Acidic fibroblast growth factor for repair of human spinal cord injury: a clinical trial. J Neurosurg Spine (2011) 15:216–27. doi: 10.3171/2011.4.SPINE10404
240. Nicola NA, Metcalf D, Matsumoto M, Johnson GR. Purification of a factor inducing differentiation in murine myelomonocytic leukemia cells. identification as granulocyte colony-stimulating factor. J Biol Chem (1983) 258:9017–23. doi: 10.1016S0021-9258(18)32158-6
241. Roberts AW. G-CSF: a key regulator of neutrophil production, but that’s not all! Growth Factors (2005) 23:33–41. doi: 10.1080/08977190500055836
242. Gupta V, Mesa RA, Deininger MWN, Rivera CE, Sirhan S, Brachmann CB, et al. A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis. Haematologica (2017) 102:94–102. doi: 10.3324/haematol.2016.148924
243. Derakhshanrad N, Saberi H, Yekaninejad MS, Joghataei MT, Sheikhrezaei A. Granulocyte-colony stimulating factor administration for neurological improvement in patients with postrehabilitation chronic incomplete traumatic spinal cord injuries: a double-blind randomized controlled clinical trial. J Neurosurg Spine (2018) 29:97–107. doi: 10.3171/2017.11.SPINE17769
244. Nishio Y, Koda M, Kamada T, Someya Y, Kadota R, Mannoji C, et al. Granulocyte colony-stimulating factor attenuates neuronal death and promotes functional recovery after spinal cord injury in mice. J Neuropathol Exp Neurol (2007) 66:724–31. doi: 10.1097/nen.0b013e3181257176
245. Koda M, Hanaoka H, Fujii Y, Hanawa M, Kawasaki Y, Ozawa Y, et al. Randomized trial of granulocyte colony-stimulating factor for spinal cord injury. Brain (2021) 144:789–99. doi: 10.1093/brain/awaa466
246. Takahashi H, Yamazaki M, Okawa A, Sakuma T, Kato K, Hashimoto M, et al. Neuroprotective therapy using granulocyte colony-stimulating factor for acute spinal cord injury: a phase I/IIa clinical trial. Eur Spine J (2012) 21:2580–7. doi: 10.1007/s00586-012-2213-3
247. Kamiya K, Koda M, Furuya T, Kato K, Takahashi H, Sakuma T, et al. Neuroprotective therapy with granulocyte colony-stimulating factor in acute spinal cord injury: a comparison with high-dose methylprednisolone as a historical control. Eur Spine J (2015) 24:963–7. doi: 10.1007/s00586-014-3373-0
248. Saberi H, Derakhshanrad N, Yekaninejad MS. Comparison of neurological and functional outcomes after administration of granulocyte-colony-stimulating factor in motor-complete versus motor-incomplete postrehabilitated, chronic spinal cord injuries: a phase I/II study. Cell Transplant (2014) 23 Suppl 1:S19–23. doi: 10.3727/096368914X684943
249. Inada T, Takahashi H, Yamazaki M, Okawa A, Sakuma T, Kato K, et al. Multicenter prospective nonrandomized controlled clinical trial to prove neurotherapeutic effects of granulocyte colony-stimulating factor for acute spinal cord injury: analyses of follow-up cases after at least 1 year. Spine (Phila Pa 1976) (2014) 39:213–9. doi: 10.1097/BRS.0000000000000121
250. Yamazaki K, Kawabori M, Seki T, Houkin K. Clinical trials of stem cell treatment for spinal cord injury. Int J Mol Sci (2020) 21:E3994. doi: 10.3390/ijms21113994
251. Sharma A, Gokulchandran N, Chopra G, Kulkarni P, Lohia M, Badhe P, et al. Administration of autologous bone marrow-derived mononuclear cells in children with incurable neurological disorders and injury is safe and improves their quality of life. Cell Transplant (2012) 21:79–90. doi: 10.3727/096368912X633798
252. Kumar AA, Kumar SR, Narayanan R, Arul K, Baskaran M. Autologous bone marrow derived mononuclear cell therapy for spinal cord injury: A phase I/II clinical safety and primary efficacy data. Exp Clin Transplant (2009) 7:241–8.
253. Vaquero J, Zurita M, Rico MA, Aguayo C, Bonilla C, Marin E, et al. Intrathecal administration of autologous mesenchymal stromal cells for spinal cord injury: Safety and efficacy of the 100/3 guideline. Cytotherapy (2018) 20:806–19. doi: 10.1016/j.jcyt.2018.03.032
254. Oh SK, Choi KH, Yoo JY, Kim DY, Kim SJ, Jeon SR. A phase III clinical trial showing limited efficacy of autologous mesenchymal stem cell therapy for spinal cord injury. Neurosurgery (2016) 78:436–447; discussion 447. doi: 10.1227/NEU.0000000000001056
255. Mendonça MVP, Larocca TF, de Freitas Souza BS, Villarreal CF, Silva LFM, Matos AC, et al. Safety and neurological assessments after autologous transplantation of bone marrow mesenchymal stem cells in subjects with chronic spinal cord injury. Stem Cell Res Ther (2014) 5:126. doi: 10.1186/scrt516
256. Deda H, Inci MC, Kürekçi AE, Kayihan K, Ozgün E, Ustünsoy GE, et al. Treatment of chronic spinal cord injured patients with autologous bone marrow-derived hematopoietic stem cell transplantation: 1-year follow-up. Cytotherapy (2008) 10:565–74. doi: 10.1080/14653240802241797
257. Vaquero J, Zurita M, Rico MA, Bonilla C, Aguayo C, Fernández C, et al. Repeated subarachnoid administrations of autologous mesenchymal stromal cells supported in autologous plasma improve quality of life in patients suffering incomplete spinal cord injury. Cytotherapy (2017) 19:349–59. doi: 10.1016/j.jcyt.2016.12.002
258. Ra JC, Shin IS, Kim SH, Kang SK, Kang BC, Lee HY, et al. Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells Dev (2011) 20:1297–308. doi: 10.1089/scd.2010.0466
259. Cristante AF, Barros-Filho TEP, Tatsui N, Mendrone A, Caldas JG, Camargo A, et al. Stem cells in the treatment of chronic spinal cord injury: evaluation of somatosensitive evoked potentials in 39 patients. Spinal Cord (2009) 47:733–8. doi: 10.1038/sc.2009.24
260. Geffner LF, Santacruz P, Izurieta M, Flor L, Maldonado B, Auad AH, et al. Administration of autologous bone marrow stem cells into spinal cord injury patients via multiple routes is safe and improves their quality of life: comprehensive case studies. Cell Transplant (2008) 17:1277–93. doi: 10.3727/096368908787648074
261. Shin JC, Kim KN, Yoo J, Kim I-S, Yun S, Lee H, et al. Clinical trial of human fetal brain-derived neural Stem/Progenitor cell transplantation in patients with traumatic cervical spinal cord injury. Neural Plast (2015) 2015:630932. doi: 10.1155/2015/630932
262. Hur JW, Cho T-H, Park D-H, Lee J-B, Park J-Y, Chung Y-G. Intrathecal transplantation of autologous adipose-derived mesenchymal stem cells for treating spinal cord injury: A human trial. J Spinal Cord Med (2016) 39:655–64. doi: 10.1179/2045772315Y.0000000048
263. Satti HS, Waheed A, Ahmed P, Ahmed K, Akram Z, Aziz T, et al. Autologous mesenchymal stromal cell transplantation for spinal cord injury: A phase I pilot study. Cytotherapy (2016) 18:518–22. doi: 10.1016/j.jcyt.2016.01.004
264. Syková E, Homola A, Mazanec R, Lachmann H, Konrádová SL, Kobylka P, et al. Autologous bone marrow transplantation in patients with subacute and chronic spinal cord injury. Cell Transplant (2006) 15:675–87. doi: 10.3727/000000006783464381
265. Albu S, Kumru H, Coll R, Vives J, Vallés M, Benito-Penalva J, et al. Clinical effects of intrathecal administration of expanded Wharton jelly mesenchymal stromal cells in patients with chronic complete spinal cord injury: a randomized controlled study. Cytotherapy (2021) 23:146–56. doi: 10.1016/j.jcyt.2020.08.008
266. Gant KL, Guest JD, Palermo AE, Vedantam A, Jimsheleishvili G, Bunge MB, et al. Phase 1 safety trial of autologous human schwann cell transplantation in chronic spinal cord injury. J Neurotrauma (2022) 39:285–99. doi: 10.1089/neu.2020.7590
267. Honmou O, Yamashita T, Morita T, Oshigiri T, Hirota R, Iyama S, et al. Intravenous infusion of auto serum-expanded autologous mesenchymal stem cells in spinal cord injury patients: 13 case series. Clin Neurol Neurosurg (2021) 203:106565. doi: 10.1016/j.clineuro.2021.106565
268. Park JH, Kim DY, Sung IY, Choi GH, Jeon MH, Kim KK, et al. Long-term results of spinal cord injury therapy using mesenchymal stem cells derived from bone marrow in humans. Neurosurgery (2012) 70:1238–1247; discussion 1247. doi: 10.1227/NEU.0b013e31824387f9
269. Dai G, Liu X, Zhang Z, Yang Z, Dai Y, Xu R. Transplantation of autologous bone marrow mesenchymal stem cells in the treatment of complete and chronic cervical spinal cord injury. Brain Res (2013) 1533:73–9. doi: 10.1016/j.brainres.2013.08.016
270. Moviglia GA, Fernandez Viña R, Brizuela JA, Saslavsky J, Vrsalovic F, Varela G, et al. Combined protocol of cell therapy for chronic spinal cord injury. report on the electrical and functional recovery of two patients. Cytotherapy (2006) 8:202–9. doi: 10.1080/14653240600736048
271. Vaquero J, Zurita M, Rico MA, Aguayo C, Fernandez C, Rodriguez-Boto G, et al. Cell therapy with autologous mesenchymal stromal cells in post-traumatic syringomyelia. Cytotherapy (2018) 20:796–805. doi: 10.1016/j.jcyt.2018.04.006
272. Frolov AA, Bryukhovetskiy AS. Effects of hematopoietic autologous stem cell transplantation to the chronically injured human spinal cord evaluated by motor and somatosensory evoked potentials methods. Cell Transplant (2012) 21:49–55. doi: 10.3727/096368912X633761
273. Vaquero J, Zurita M, Rico MA, Bonilla C, Aguayo C, Montilla J, et al. An approach to personalized cell therapy in chronic complete paraplegia: The puerta de hierro phase I/II clinical trial. Cytotherapy (2016) 18:1025–36. doi: 10.1016/j.jcyt.2016.05.003
274. Lee J, Kuroda S, Shichinohe H, Ikeda J, Seki T, Hida K, et al. Migration and differentiation of nuclear fluorescence-labeled bone marrow stromal cells after transplantation into cerebral infarct and spinal cord injury in mice. Neuropathology (2003) 23:169–80. doi: 10.1046/j.1440-1789.2003.00496.x
275. Novikova LN, Brohlin M, Kingham PJ, Novikov LN, Wiberg M. Neuroprotective and growth-promoting effects of bone marrow stromal cells after cervical spinal cord injury in adult rats. Cytotherapy (2011) 13:873–87. doi: 10.3109/14653249.2011.574116
276. Gao S, Guo X, Zhao S, Jin Y, Zhou F, Yuan P, et al. Differentiation of human adipose-derived stem cells into neuron/motoneuron-like cells for cell replacement therapy of spinal cord injury. Cell Death Dis (2019) 10:597. doi: 10.1038/s41419-019-1772-1
278. Marrow stromal cells form guiding strands in the injured spinal cord and promote recovery (Accessed February 1, 2022).
279. Ceci M, Mariano V, Romano N. Zebrafish as a translational regeneration model to study the activation of neural stem cells and role of their environment. Rev Neurosci (2018) 30:45–66. doi: 10.1515/revneuro-2018-0020
280. Kishk NA, Gabr H, Hamdy S, Afifi L, Abokresha N, Mahmoud H, et al. Case control series of intrathecal autologous bone marrow mesenchymal stem cell therapy for chronic spinal cord injury. Neurorehabil Neural Repair (2010) 24:702–8. doi: 10.1177/1545968310369801
281. Pal R, Venkataramana NK, Bansal A, Balaraju S, Jan M, Chandra R, et al. Ex vivo-expanded autologous bone marrow-derived mesenchymal stromal cells in human spinal cord injury/paraplegia: a pilot clinical study. Cytotherapy (2009) 11:897–911. doi: 10.3109/14653240903253857
282. Mackay-Sim A, Féron F, Cochrane J, Bassingthwaighte L, Bayliss C, Davies W, et al. Autologous olfactory ensheathing cell transplantation in human paraplegia: a 3-year clinical trial. Brain (2008) 131:2376–86. doi: 10.1093/brain/awn173
283. Chernykh ER, Stupak VV, Muradov GM, Sizikov MY, Shevela EY, Leplina OY, et al. Application of autologous bone marrow stem cells in the therapy of spinal cord injury patients. Bull Exp Biol Med (2007) 143:543–7. doi: 10.1007/s10517-007-0175-y
284. Féron F, Perry C, Cochrane J, Licina P, Nowitzke A, Urquhart S, et al. Autologous olfactory ensheathing cell transplantation in human spinal cord injury. Brain (2005) 128:2951–60. doi: 10.1093/brain/awh657
285. El-Kheir WA, Gabr H, Awad MR, Ghannam O, Barakat Y, Farghali HAMA, et al. Autologous bone marrow-derived cell therapy combined with physical therapy induces functional improvement in chronic spinal cord injury patients. Cell Transplant (2014) 23:729–45. doi: 10.3727/096368913X664540
286. Cheng H, Liu X, Hua R, Dai G, Wang X, Gao J, et al. Clinical observation of umbilical cord mesenchymal stem cell transplantation in treatment for sequelae of thoracolumbar spinal cord injury. J Transl Med (2014) 12:253. doi: 10.1186/s12967-014-0253-7
287. Assinck P, Duncan GJ, Hilton BJ, Plemel JR, Tetzlaff W. Cell transplantation therapy for spinal cord injury. Nat Neurosci (2017) 20:637–47. doi: 10.1038/nn.4541
288. Romanelli P, Bieler L, Heimel P, Škokić S, Jakubecova D, Kreutzer C, et al. Enhancing functional recovery through intralesional application of extracellular vesicles in a rat model of traumatic spinal cord injury. Front Cell Neurosci (2021) 15:795008. doi: 10.3389/fncel.2021.795008
289. Xiao Z, Tang F, Zhao Y, Han G, Yin N, Li X, et al. Significant improvement of acute complete spinal cord injury patients diagnosed by a combined criteria implanted with NeuroRegen scaffolds and mesenchymal stem cells. Cell Transplant (2018) 27:907–15. doi: 10.1177/0963689718766279
290. Karamouzian S, Nematollahi-Mahani SN, Nakhaee N, Eskandary H. Clinical safety and primary efficacy of bone marrow mesenchymal cell transplantation in subacute spinal cord injured patients. Clin Neurol Neurosurg (2012) 114:935–9. doi: 10.1016/j.clineuro.2012.02.003
291. Bhanot Y, Rao S, Ghosh D, Balaraju S, Radhika CR, Satish Kumar KV. Autologous mesenchymal stem cells in chronic spinal cord injury. Br J Neurosurg (2011) 25:516–22. doi: 10.3109/02688697.2010.550658
292. Yoon SH, Shim YS, Park YH, Chung JK, Nam JH, Kim MO, et al. Complete spinal cord injury treatment using autologous bone marrow cell transplantation and bone marrow stimulation with granulocyte macrophage-colony stimulating factor: Phase I/II clinical trial. Stem Cells (2007) 25:2066–73. doi: 10.1634/stemcells.2006-0807
293. Shang Z, Wang M, Zhang B, Wang X, Wanyan P. Clinical translation of stem cell therapy for spinal cord injury still premature: results from a single-arm meta-analysis based on 62 clinical trials. BMC Med (2022) 20:284. doi: 10.1186/s12916-022-02482-2
294. Huang L, Fu C, Xiong F, He C, Wei Q. Stem cell therapy for spinal cord injury. Cell Transplant (2021) 30:963689721989266. doi: 10.1177/0963689721989266
295. Zhu H, Poon W, Liu Y, Leung GK-K, Wong Y, Feng Y, et al. Phase I-II clinical trial assessing safety and efficacy of umbilical cord blood mononuclear cell transplant therapy of chronic complete spinal cord injury. Cell Transplant (2016) 25:1925–43. doi: 10.3727/096368916X691411
296. Anderson KD, Guest JD, Dietrich WD, Bartlett Bunge M, Curiel R, Dididze M, et al. Safety of autologous human schwann cell transplantation in subacute thoracic spinal cord injury. J Neurotrauma (2017) 34:2950–63. doi: 10.1089/neu.2016.4895
297. Lima C, Pratas-Vital J, Escada P, Hasse-Ferreira A, Capucho C, Peduzzi JD. Olfactory mucosa autografts in human spinal cord injury: a pilot clinical study. J Spinal Cord Med (2006) 29:191–203; discussion 204-206. doi: 10.1080/10790268.2006.11753874
298. Park HC, Shim YS, Ha Y, Yoon SH, Park SR, Choi BH, et al. Treatment of complete spinal cord injury patients by autologous bone marrow cell transplantation and administration of granulocyte-macrophage colony stimulating factor. Tissue Eng (2005) 11:913–22. doi: 10.1089/ten.2005.11.913
299. Califf RM, Zarin DA, Kramer JM, Sherman RE, Aberle LH, Tasneem A. Characteristics of clinical trials registered in ClinicalTrials.gov, 2007-2010. JAMA (2012) 307:1838–47. doi: 10.1001/jama.2012.3424
300. Gazdic M, Volarevic V, Harrell CR, Fellabaum C, Jovicic N, Arsenijevic N, et al. Stem cells therapy for spinal cord injury. Int J Mol Sci (2018) 19:1039. doi: 10.3390/ijms19041039
301. Al-Zoubi A, Jafar E, Jamous M, Al-Twal F, Al-Bakheet S, Zalloum M, et al. Transplantation of purified autologous leukapheresis-derived CD34+ and CD133+ stem cells for patients with chronic spinal cord injuries: Long-term evaluation of safety and efficacy. Cell Transplant (2014) 23:25–34. doi: 10.3727/096368914X684899
302. Saberi H, Moshayedi P, Aghayan H-R, Arjmand B, Hosseini S-K, Emami-Razavi S-H, et al. Treatment of chronic thoracic spinal cord injury patients with autologous schwann cell transplantation: An interim report on safety considerations and possible outcomes. Neurosci Lett (2008) 443:46–50. doi: 10.1016/j.neulet.2008.07.041
303. Lima C, Escada P, Pratas-Vital J, Branco C, Arcangeli CA, Lazzeri G, et al. Olfactory mucosal autografts and rehabilitation for chronic traumatic spinal cord injury. Neurorehabil Neural Repair (2010) 24:10–22. doi: 10.1177/1545968309347685
304. Levi AD, Okonkwo DO, Park P, Jenkins AL, Kurpad SN, Parr AM, et al. Emerging safety of intramedullary transplantation of human neural stem cells in chronic cervical and thoracic spinal cord injury. Neurosurgery (2018) 82:562–75. doi: 10.1093/neuros/nyx250
305. Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem (2006) 98:1076–84. doi: 10.1002/jcb.20886
306. Ruppert KA, Nguyen TT, Prabhakara KS, Toledano Furman NE, Srivastava AK, Harting MT, et al. Human mesenchymal stromal cell-derived extracellular vesicles modify microglial response and improve clinical outcomes in experimental spinal cord injury. Sci Rep (2018) 8:480. doi: 10.1038/s41598-017-18867-w
307. Zhou Y, Yamamoto Y, Xiao Z, Ochiya T. The immunomodulatory functions of mesenchymal Stromal/Stem cells mediated via paracrine activity. J Clin Med (2019) 8:E1025. doi: 10.3390/jcm8071025
308. Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest (2016) 126:1173–80. doi: 10.1172/JCI81131
309. Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci (2016) 17:160–72. doi: 10.1038/nrn.2015.29
310. Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles (2014) 3:24722. doi: 10.3402/jev.v3.24722
311. Kuharić J, Grabušić K, Tokmadžić VS, Štifter S, Tulić K, Shevchuk O, et al. Severe traumatic brain injury induces early changes in the physical properties and protein composition of intracranial extracellular vesicles. J Neurotrauma (2019) 36:190–200. doi: 10.1089/neu.2017.5515
312. Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, et al. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflamm (2017) 14:47. doi: 10.1186/s12974-017-0819-4
313. Hazelton I, Yates A, Dale A, Roodselaar J, Akbar N, Ruitenberg MJ, et al. Exacerbation of acute traumatic brain injury by circulating extracellular vesicles. J Neurotrauma (2018) 35:639–51. doi: 10.1089/neu.2017.5049
314. Zhou X, Xie F, Wang L, Zhang L, Zhang S, Fang M, et al. The function and clinical application of extracellular vesicles in innate immune regulation. Cell Mol Immunol (2020) 17:323–34. doi: 10.1038/s41423-020-0391-1
315. Bonner SE, Willms E. Intercellular communication through extracellular vesicles in cancer and evolutionary biology. Prog Biophysics Mol Biol (2021) 165:80–7. doi: 10.1016/j.pbiomolbio.2021.08.006
316. Kox M, Vaneker M, van der Hoeven JG, Scheffer G-J, Hoedemaekers CW, Pickkers P. Effects of vagus nerve stimulation and vagotomy on systemic and pulmonary inflammation in a two-hit model in rats. PloS One (2012) 7:e34431. doi: 10.1371/journal.pone.0034431
317. Konsman JP, Luheshi GN, Bluthé RM, Dantzer R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals; a functional anatomical analysis. Eur J Neurosci (2000) 12:4434–46. doi: 10.1046/j.0953-816x.2000.01319.x
318. Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci (2002) 25:154–9. doi: 10.1016/s0166-2236(00)02088-9
319. Dutta D, Khan N, Wu J, Jay SM. Extracellular vesicles as an emerging frontier in spinal cord injury pathobiology and therapy. Trends Neurosci (2021) 44:492–506. doi: 10.1016/j.tins.2021.01.003
320. Zhang B, Yeo RWY, Tan KH, Lim SK. Focus on extracellular vesicles: Therapeutic potential of stem cell-derived extracellular vesicles. Int J Mol Sci (2016) 17:174. doi: 10.3390/ijms17020174
321. Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol (2014) 258:105–11. doi: 10.1016/j.expneurol.2014.03.013
322. de Rivero Vaccari JP, Brand F, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, et al. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem (2016) 136 Suppl 1:39–48. doi: 10.1111/jnc.13036
323. Ding S-Q, Chen J, Wang S-N, Duan F-X, Chen Y-Q, Shi Y-J, et al. Identification of serum exosomal microRNAs in acute spinal cord injured rats. Exp Biol Med (Maywood) (2019) 244:1149–61. doi: 10.1177/1535370219872759
324. Ding S-Q, Chen Y-Q, Chen J, Wang S-N, Duan F-X, Shi Y-J, et al. Serum exosomal microRNA transcriptome profiling in subacute spinal cord injured rats. Genomics (2020) 112:2092–105. doi: 10.1016/j.ygeno.2019.12.003
325. Khan NZ, Cao T, He J, Ritzel RM, Li Y, Henry RJ, et al. Spinal cord injury alters microRNA and CD81+ exosome levels in plasma extracellular nanoparticles with neuroinflammatory potential. Brain Behav Immun (2021) 92:165–83. doi: 10.1016/j.bbi.2020.12.007
326. Dickens AM, Tovar-Y-Romo LB, Yoo S-W, Trout AL, Bae M, Kanmogne M, et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal (2017) 10:eaai7696. doi: 10.1126/scisignal.aai7696
327. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
328. Wang L, Pei S, Han L, Guo B, Li Y, Duan R, et al. Mesenchymal stem cell-derived exosomes reduce A1 astrocytes via downregulation of phosphorylated NFκB P65 subunit in spinal cord injury. Cell Physiol Biochem (2018) 50:1535–59. doi: 10.1159/000494652
329. Lu Y, Zhou Y, Zhang R, Wen L, Wu K, Li Y, et al. Bone mesenchymal stem cell-derived extracellular vesicles promote recovery following spinal cord injury via improvement of the integrity of the blood-spinal cord barrier. Front Neurosci (2019) 13:209. doi: 10.3389/fnins.2019.00209
330. Jia Y, Lu T, Chen Q, Pu X, Ji L, Yang J, et al. Exosomes secreted from sonic hedgehog-modified bone mesenchymal stem cells facilitate the repair of rat spinal cord injuries. Acta Neurochir (Wien) (2021) 163:2297–306. doi: 10.1007/s00701-021-04829-9
331. Li L, Zhang Y, Mu J, Chen J, Zhang C, Cao H, et al. Transplantation of human mesenchymal stem-Cell-Derived exosomes immobilized in an adhesive hydrogel for effective treatment of spinal cord injury. Nano Lett (2020) 20:4298–305. doi: 10.1021/acs.nanolett.0c00929
332. Guo S, Perets N, Betzer O, Ben-Shaul S, Sheinin A, Michaelevski I, et al. Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano (2019) 13:10015–28. doi: 10.1021/acsnano.9b01892
333. Romanelli P, Bieler L, Scharler C, Pachler K, Kreutzer C, Zaunmair P, et al. Extracellular vesicles can deliver anti-inflammatory and anti-scarring activities of mesenchymal stromal cells after spinal cord injury. Front Neurol (2019) 10:1225. doi: 10.3389/fneur.2019.01225
334. Huang J-H, Yin X-M, Xu Y, Xu C-C, Lin X, Ye F-B, et al. Systemic administration of exosomes released from mesenchymal stromal cells attenuates apoptosis, inflammation, and promotes angiogenesis after spinal cord injury in rats. J Neurotrauma (2017) 34:3388–96. doi: 10.1089/neu.2017.5063
335. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol (2020) 50:712–24. doi: 10.1002/eji.201948457
336. Noori L, Arabzadeh S, Mohamadi Y, Mojaverrostami S, Mokhtari T, Akbari M, et al. Intrathecal administration of the extracellular vesicles derived from human wharton’s jelly stem cells inhibit inflammation and attenuate the activity of inflammasome complexes after spinal cord injury in rats. Neurosci Res (2021) 170:87–98. doi: 10.1016/j.neures.2020.07.011
337. Blomster LV, Brennan FH, Lao HW, Harle DW, Harvey AR, Ruitenberg MJ. Mobilisation of the splenic monocyte reservoir and peripheral CX₃CR1 deficiency adversely affects recovery from spinal cord injury. Exp Neurol (2013) 247:226–40. doi: 10.1016/j.expneurol.2013.05.002
338. Lankford KL, Arroyo EJ, Nazimek K, Bryniarski K, Askenase PW, Kocsis JD. Intravenously delivered mesenchymal stem cell-derived exosomes target M2-type macrophages in the injured spinal cord. PloS One (2018) 13:e0190358. doi: 10.1371/journal.pone.0190358
339. Li H, Shen S, Fu H, Wang Z, Li X, Sui X, et al. Immunomodulatory functions of mesenchymal stem cells in tissue engineering. Stem Cells Int (2019) 2019:e9671206. doi: 10.1155/2019/9671206
340. Nourian Dehkordi A, Mirahmadi Babaheydari F, Chehelgerdi M, Raeisi Dehkordi S. Skin tissue engineering: wound healing based on stem-cell-based therapeutic strategies. Stem Cell Res Ther (2019) 10:111. doi: 10.1186/s13287-019-1212-2
341. Arzouni AA, Vargas-Seymour A, Nardi N, King AJF, Jones PM. Using mesenchymal stromal cells in islet transplantation. Stem Cells Transl Med (2018) 7:559–63. doi: 10.1002/sctm.18-0033
342. Shao H, Im H, Castro CM, Breakefield X, Weissleder R, Lee H. New technologies for analysis of extracellular vesicles. Chem Rev (2018) 118:1917–50. doi: 10.1021/acs.chemrev.7b00534
343. Cho KHT, Xu B, Blenkiron C, Fraser M. Emerging roles of miRNAs in brain development and perinatal brain injury. Front Physiol (2019) 10:227. doi: 10.3389/fphys.2019.00227
344. D’Anca M, Fenoglio C, Serpente M, Arosio B, Cesari M, Scarpini EA, et al. Exosome determinants of physiological aging and age-related neurodegenerative diseases. Front Aging Neurosci (2019) 11:232. doi: 10.3389/fnagi.2019.00232
345. Patel DB, Gray KM, Santharam Y, Lamichhane TN, Stroka KM, Jay SM. Impact of cell culture parameters on production and vascularization bioactivity of mesenchymal stem cell-derived extracellular vesicles. Bioeng Transl Med (2017) 2:170–9. doi: 10.1002/btm2.10065
346. Guitart K, Loers G, Buck F, Bork U, Schachner M, Kleene R. Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein. Glia (2016) 64:896–910. doi: 10.1002/glia.22963
347. Xu L, Cao H, Xie Y, Zhang Y, Du M, Xu X, et al. Exosome-shuttled miR-92b-3p from ischemic preconditioned astrocytes protects neurons against oxygen and glucose deprivation. Brain Res (2019) 1717:66–73. doi: 10.1016/j.brainres.2019.04.009
348. Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, Drago D, et al. Extracellular vesicles from neural stem cells transfer IFN-γ via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell (2014) 56:193–204. doi: 10.1016/j.molcel.2014.08.020
349. Yildirimer L, Zhang Q, Kuang S, Cheung C-WJ, Chu KA, He Y, et al. Engineering three-dimensional microenvironments towards in vitro disease models of the central nervous system. Biofabrication (2019) 11:032003. doi: 10.1088/1758-5090/ab17aa
350. Cui H, Nowicki M, Fisher JP, Zhang LG. 3D bioprinting for organ regeneration. Adv Healthc Mater (2017) 6:1601118. doi: 10.1002/adhm.201601118
351. O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA Delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol (2020) 21:585–606. doi: 10.1038/s41580-020-0251-y
352. Martellucci S, Orefice NS, Angelucci A, Luce A, Caraglia M, Zappavigna S. Extracellular vesicles: New endogenous shuttles for miRNAs in cancer diagnosis and therapy? Int J Mol Sci (2020) 21:E6486. doi: 10.3390/ijms21186486
353. Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM, et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U.S.A. (2014) 111:14888–93. doi: 10.1073/pnas.1408301111
354. Yang Y, Ye Y, Kong C, Su X, Zhang X, Bai W, et al. MiR-124 enriched exosomes promoted the M2 polarization of microglia and enhanced hippocampus neurogenesis after traumatic brain injury by inhibiting TLR4 pathway. Neurochem Res (2019) 44:811–28. doi: 10.1007/s11064-018-02714-z
355. O’Loughlin AJ, Mäger I, de Jong OG, Varela MA, Schiffelers RM, El Andaloussi S, et al. Functional delivery of lipid-conjugated siRNA by extracellular vesicles. Mol Ther (2017) 25:1580–7. doi: 10.1016/j.ymthe.2017.03.021
356. Lamichhane TN, Jeyaram A, Patel DB, Parajuli B, Livingston NK, Arumugasaamy N, et al. Oncogene knockdown via active loading of small RNAs into extracellular vesicles by sonication. Cell Mol Bioeng (2016) 9:315–24. doi: 10.1007/s12195-016-0457-4
357. Liu S, Xu X, Liang S, Chen Z, Zhang Y, Qian A, et al. The application of MSCs-derived extracellular vesicles in bone disorders: Novel cell-free therapeutic strategy. Front Cell Dev Biol (2020) 8:619. doi: 10.3389/fcell.2020.00619
358. Hercher D, Nguyen MQ, Dworak H. Extracellular vesicles and their role in peripheral nerve regeneration. Exp Neurol (2022) 350:113968. doi: 10.1016/j.expneurol.2021.113968
359. Abramowicz A, Widlak P, Pietrowska M. Proteomic analysis of exosomal cargo: the challenge of high purity vesicle isolation. Mol Biosyst (2016) 12:1407–19. doi: 10.1039/c6mb00082g
Keywords: traumatic spinal cord injury, SCI, stem cell, extracellular vesicle, vesicle, neuroimmunology, tSCI therapies
Citation: Sterner RC and Sterner RM (2023) Immune response following traumatic spinal cord injury: Pathophysiology and therapies. Front. Immunol. 13:1084101. doi: 10.3389/fimmu.2022.1084101
Received: 30 October 2022; Accepted: 19 December 2022;
Published: 06 January 2023.
Edited by:
Xiao-Yi Xiong, Chengdu University of Traditional Chinese Medicine, ChinaReviewed by:
Shengyou Li, Fourth Military Medical University, ChinaPengfei Wang, Weihai Municipal Hospital, China
Copyright © 2023 Sterner and Sterner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalie M. Sterner, U3Rlcm5lci5Sb3NhbGllQE1heW8uZWR1