 Chuyu Wu1,2
Chuyu Wu1,2 Tao Pang
Tao Pang Cun-Jin Zhang
Cun-Jin Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 January 2023
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1077335
Fungal infection or proliferation in our body is capable of initiation of strong inflammation and immune responses that result in different consequences, including infection-trigged organ injury and inflammation-related remote organ dysfunction. Fungi associated infectious diseases have been well recognized in the clinic. However, whether fungi play an important role in non-infectious central nervous system disease is still to be elucidated. Recently, a growing amount of evidence point to a non-negligible role of peripheral fungus in triggering unique inflammation, immune response, and exacerbation of a range of non-infectious CNS disorders, including Multiple sclerosis, Neuromyelitis optica, Parkinson’s disease, Alzheimer’s disease, and Amyotrophic lateral sclerosis et al. In this review, we summarized the recent advances in recognizing patterns and inflammatory signaling of fungi in different subsets of immune cells, with a specific focus on its function in CNS autoimmune and neurodegeneration diseases. In conclusion, the fungus is capable of triggering unique inflammation by multiple mechanisms in the progression of a body of CNS non-infectious diseases, suggesting it serves as a key factor and critical novel target for the development of potential therapeutic strategies.
Fungal infection or proliferation in our body is capable of initiation of strong inflammation and immune responses that result in different consequences, including infection trigged organ injury and inflammation related remote organ dysfunction. Normally, only a very small percentage of fungi can cause human infections. It has been known that the incidence of fungal infections of the central nervous system (CNS) increased significantly in the past few years (1, 2). This is possibly due to the widely use of immunosuppressive therapies (e.g., chemotherapy or corticosteroid treatment), the stem cells and organ transplants, and the spread of HIV/AIDS (3). There are various clinical manifestations, mainly meningitis, encephalitis, hydrocephalus, brain abscess, and stroke syndrome. The pathogens of CNS fungal infections include yeasts, dimorphic fungi, and filamentous fungi (1). Cryptococcus neoformans, Aspergillus, and Candida albicans are the most common pathogens. Fungi usually enter the human body through the respiratory tract and spread to the CNS via the bloodstream. Immunocompetent individuals can also become infected as a result of surgery, trauma, intravenous drug use, or contaminated medical supplies. Fungi are usually disseminated to the CNS from adjacent tissues (sinusoid structures, mastoid processes, or orbital orbits) (3).
The blood-brain barrier (BBB) prevents chemicals and germs from passing through, thereby safeguarding the CNS. C. albicans crosses the BBB through both the in-paracellular pathway and the transcellular pathway (4). More evidence implicates that C. albicans enter the brain through the transcellular pathway. C. albicans can cross the BBB by binding to the heat shock protein gp96 on microvascular endothelial cells (5). and this process requires the interaction of the invasin Als3 with gp96 to induce fungal endocytosis.
Cryptococci also cross the BBB in multiple ways. After initially adhering to the BBB, Cryptococcus neoformans forms biofilm-like cell clusters on the brain unit. Such aggregated cells are able to penetrate the BBB without severely affecting endothelial cells tight junctions or forming pores in the layer of endothelial cells (6). The “Trojan horse” approach by Cryptococcus to infiltrate the brain has been demonstrated by in vitro experiments that phagocytes holding engulfed viable C. neoformans to pass the endothelial cell layer of the brain (7, 8). Additionally, Aspergillus fumigatus mycotoxin gliotoxin penetrates and damages the human blood-brain barrier in vitro (9).
In addition to fungal-associated infectious diseases, recent advances have also suggested a remarkable role of fungi in several non-infectious autoimmune diseases and neurodegeneration diseases, such as Multiple sclerosis (MS), Neuromyelitis optica spectrum disorder (NMOSD), Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS). While the classic pathogenic feature of MS is the presenting of the death of oligodendrocyte and CNS demyelination, an outcome of neuron death has believed to be the cause of AD, PD, and ASL. Of note, the common characteristics of these non-infectious diseases are sterile inflammation that takes place in CNS. However, whether fungi play a critical role in these disorders has been unclear. Here, we outline recent studies on the role and mechanisms of fungi in these non-infectious CNS autoimmune and neurodegeneration disorders.
Although Fungi infection or proliferation is a common event, the population of fungi is usually efficiently cleared or suppressed under an intact immune system and homeostasis condition. The fungi-triggered immunity includes both the innate and adaptive immune systems. In fungal infections, the innate immune response plays an important role in preventing the onset of fungal infection, while the adaptive immune response is closely associated with recovery from fungal disease. Innate immunity constitutes the first line of host defense, which includes physical barriers such as skin and mucosa, the complement system, antimicrobial peptides (AMPs), and cell-mediated protection, recognizing and initiating an inflammatory response to invade or inhibit the fungi populations.
Activation of innate immune response relies on a lineage-encoded receptor, also known as a pattern recognition receptor (PRR), which recognizes a variety of pathogen-associated molecules (pathogen-associated molecular patterns, PAMPs) expressed by invading microorganisms. PRRs are primarily expressed by antigen-presenting cells (APCs), including dendritic cells (DC) and macrophages. PAMPs are known to play a crucial function in pathogens-cell interaction and are often shared by whole classes of microorganisms. In addition, PAMPs are either structural determinants or necessary for pathogenicity. Both Candida and Aspergillus undergo a constant morphological switch between yeasts and hyphae, resulting in variable fungal PAMPs exposure. The cell wall of C. albicans contains carbohydrate polymers and glycoproteins, including chitin, β-1,3- and β-1,6-glucans, and N- and O-linked mannan (10). In contrast, the Aspergillus cell wall is mainly composed of chitin, α-glucans, β-1,3-1,4-glucans, and galactomannans. An important feature of the Aspergillus conidial cell wall is the hydrophobic rodlets in outer layer. During swelling of the conidial cell wall and subsequent germination, the hydrophobic layer changes into a hydrophilic structure exposing polysaccharides. Different from C. albicans and Aspergillus, the cell wall of Cryptococcus is composed of glucan, chitin, chitosan, mannoprotein and pigment melanin. Of note, its cell wall is wrapped with an exopolysaccharide capsule, which masks the available PAMPs in the cell wall, resulting in limited recognition of Cryptococcus by many PRRs (11). During fungal infection and invasion, the first critical step for an effective immune response is the recognition of invaders. Various PRRs expressed on different cells are competent in recognizing fungal invaders and trigger effective signaling pathways and cellular responses (Figure 1).
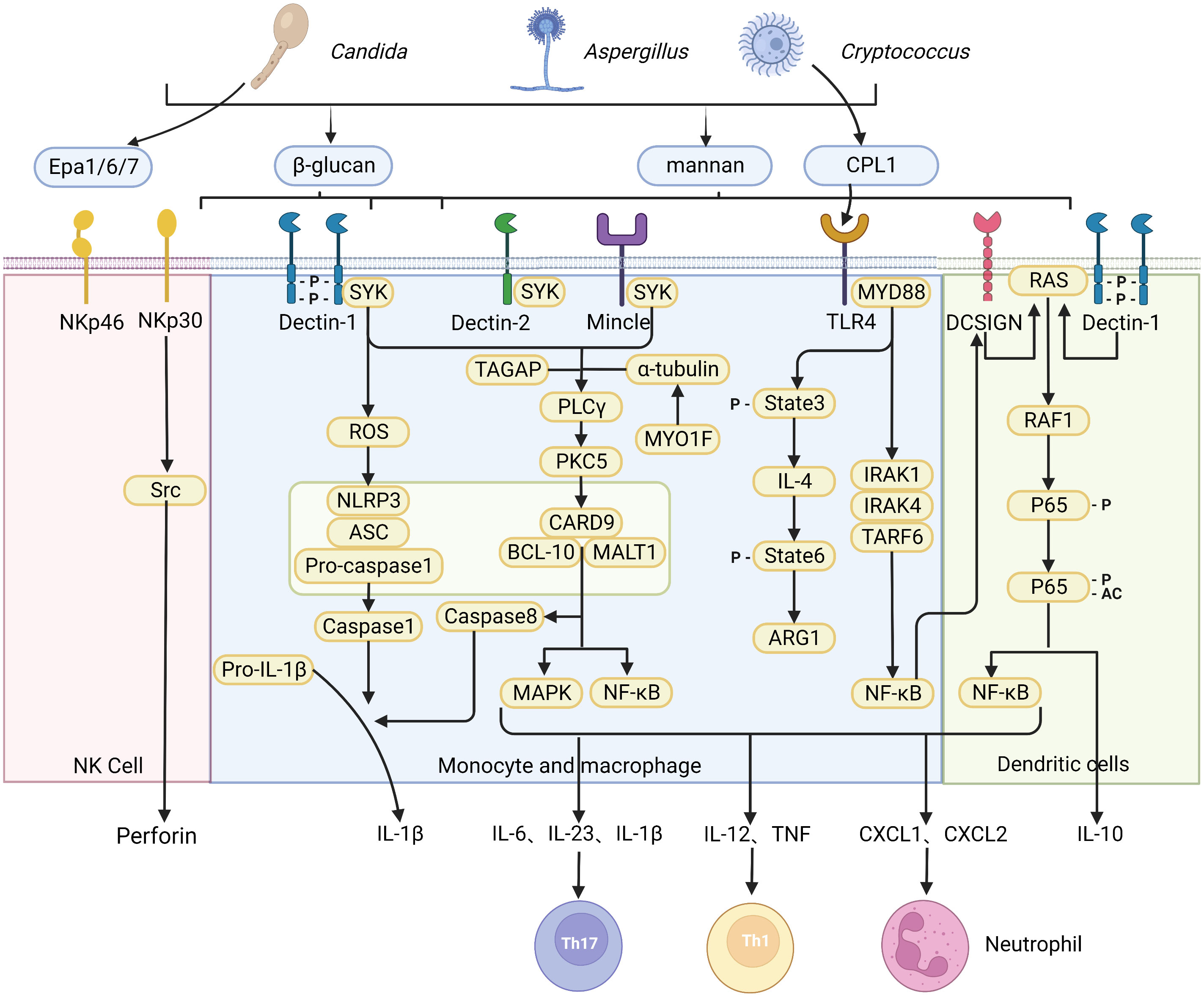
Figure 1 Recognition of fungi by pattern recognition receptors. A diagrammatic representation of pattern recognition receptors involved in the recognition of various fungal species, and the cellular responses triggered by ligand-receptor binding. The CLR family of receptors recognize fungal carbohydrates and result in activation of the SYK–CARD9 axis, formation of the CARD9–BCL10–MALT1 complex and activating nuclear NF-κB and MAPK signaling pathways, which in turn activate synthesis and release of pro-inflammatory cytokines and chemokines including IL-6, IL-1β, IL-23, IL-12, CXCL1 and CXCL2. The release of chemokines and cytokines that promote CD4+ T cell differentiation into specific T helper cell. The NOD-like receptor NLRP3 forms inflammasome complex with ASC and caspase-1 that results in production of IL-1β. In addition, dectin-1 signaling engages the CARD9–BCL10–MALT1 complex to promote non-canonical inflammasome activation that results in IL-1β production via caspase-8. In contrast, NK cells equipment with receptor NKp30 and NKp46 as a microbial pattern-recognition receptor to recognize fungi. C-type lectin DC-SIGN modulates TLR signaling via Raf-1 kinase-dependent acetylation of NF-κB in DCs. (Created with Biorender.com).
Among different pattern recognition receptors (PRRs), C-Type lectin receptors (CLRs) are the most common receptors that recognize fungi, while Toll-like receptor (TLR) and Nod-like receptor (NLR) play a secondary role. The Members of the CLR superfamily include Dectin-1, Dectin-2, macrophage C-type lectin, macrophage-induced C-type lectin, mannose receptor, melanin-sensing C-type lectin, and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (12). CLRs recognize fungal carbohydrates such as β-glucans, α-mannan, hyphal mannose, glycolipids, and chitin that are present within the cell wall (13), but can also recognize other components such as melanin (14). Recognition of fungal cell wall components by various CLRs leads to activation of splenic tyrosine kinase (Syk) in DC and macrophages (15), triggering assembly of the CARD9-BCL10-MALT1 (CBM) complex, which further leads to activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways, which in turn activates synthesis and release of pro-inflammatory cytokines and chemokines including IL-6, IL-1β, IL-23, IL-12, CXCL1, CXCL2 and GM-CSF, among others. CLR intracellular signaling pathways promote maturation and migration of DC to the draining lymph nodes to activate naïve T cells. This process is dependent on the release of DC-derived chemokines and cytokines that promote CD4+ T cell differentiation into various T helper cell subsets, including Th1, Th2, Th9, Th17, and regulatory T cells (Tregs). Besides, the antifungal adaptive T cell immunity is also activated at the same time.
Others and our groups have shown that CARD9 is crucial for maintaining gut homeostasis with commensal organisms and preventing opportunistic fungi from invading tissue. One of the crucial pieces of evidence is that the abnormalities of the CARD9 pathway often result in the increase of the fungi population, even severe fungi infections. Moreover, autosomal recessive CARD9 deficiency in human leads to profound defects in the production of pro-inflammatory cytokines in response to stimuli specific to fungi, including IL-6, IL-1β, GM-CSF, and TNFα (16). Similar results are also confirmed in animal model. It has been shown that mice with CARD9 genetic deletion produce less pro-inflammatory cytokines, which impairs myeloid cell survival after exposure to C. albicans, a significant inducer of antifungal immunoglobulin G(IgG) (17). Furthermore, CARD9 and CX3CR1 double-positive macrophages regulate innate immunity and are necessary for the generation of antifungal IgG (Figure 2) (18). In contrast to the critical role of CARD9 in peripheral, CARD9 also engages the protective antifungal immunity within the CNS. It was reported that human CARD9 deficiency causes fungal-specific CNS-targeted infection susceptibility (Figure 2). In all, PRRs and CARD9 signaling play indispensable roles in fungi-mediated immunity.
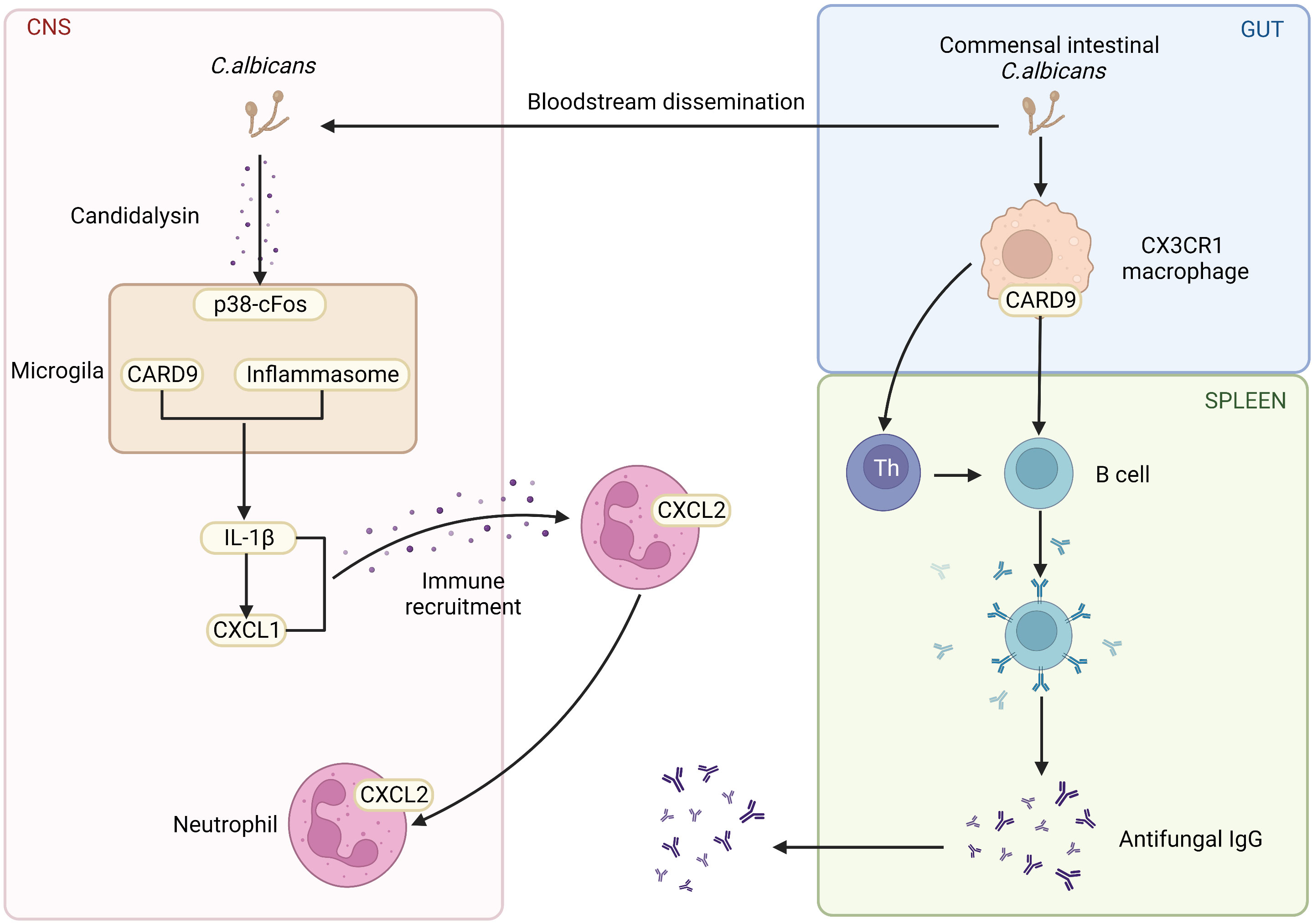
Figure 2 The role of CARD9 pathway in the brain and gut. Commensal Intestinal C. albicans induce the production of antifungal IgG. CARD9 and CX3CR1 double-positive macrophages promote the generation of antifungal IgG. CARD9-deficiency increases the susceptibility of fungal-specific CNS-targeted infection. In the fungal-infected CNS, microglia rely on CARD9 for producing IL-1β and CXCL1, which recruits neutrophils to clear the fungi. (Created with Biorender.com).
CD4+ T cells are thought to play an indispensable role in fungi-triggered immune responses. While this response contributes to the recovery of both superficial and invasive fungal infections, an overactive immune response could also result in or aggregate the development of several diseases. Unique T cell responses are aggressively induced in response to lysates and peptides of C. albicans or A. fumigatus, delivered by autologous monocytes. The differentiation and activation of various T cell subsets depend on the activities of APCs which recognize fungi through CLRs. Among the T cell subsets, Th1 cells secrete IFN-γ, GM-CSF, and TNF, which affect the maturation and killing capacity of phagocytes and the function of APC. TNF synergizes with IFN- to cause the generation of ROS of macrophage in vitro, which is hypothesized to be a factor in the growth arrest of intracellular fungal infections such as Coccidioides immitis and Histoplasma capsulatum in vivo (19, 20). As another T cell subset in addition to Th1, Th17 cells release specific signature cytokines including IL-17A, IL-17F, and IL-22 that aid in fungicidal activity, neutrophil trafficking, and the activation of AMPs from epithelial and keratinocyte cells protect against fungal overgrowth (21). In mice, Th17 cells have also been demonstrated to promote the production of Immunoglobulin A (IgA), an immunoglobulin that is crucial for protection and homeostasis at mucosal surfaces (22, 23). Moreover, Th17 cells support host immunological homeostasis against microbiota, at least in part, by inducing epithelial pIgR expression through IL-17, which boosts IgA secretion into the lumen (23). Of note, Th17 cells are capable of converting into T follicular helper cells (TFH), which may be essential for inducing the development of IgA-producing B cells (22). Thus, Candida-specific Th17 cells may play an essential role in fungal intestinal homeostasis through their ability to convert into TFH cells. In addition, studies have also shown that the gut mycobiome may be involved in providing protection against other pathogens at distal sites. For instance, Th17 cell responses in humans are specifically induced by C. albicans. Cross-reactivity to C. albicans causes the production of Th17 cells that attack other fungi. Both C. albicans and cross-reactive Th17 cells are increased by intestinal inflammation (24). Numerous novel molecules involved in antifungal signaling pathways have been recently identified by others and our group, such as MYO1F (25), STIM1 and TRIM31 (Table 1). Several antifungal drugs have also been developed based on newly discovered antifungal molecules (Table 2).
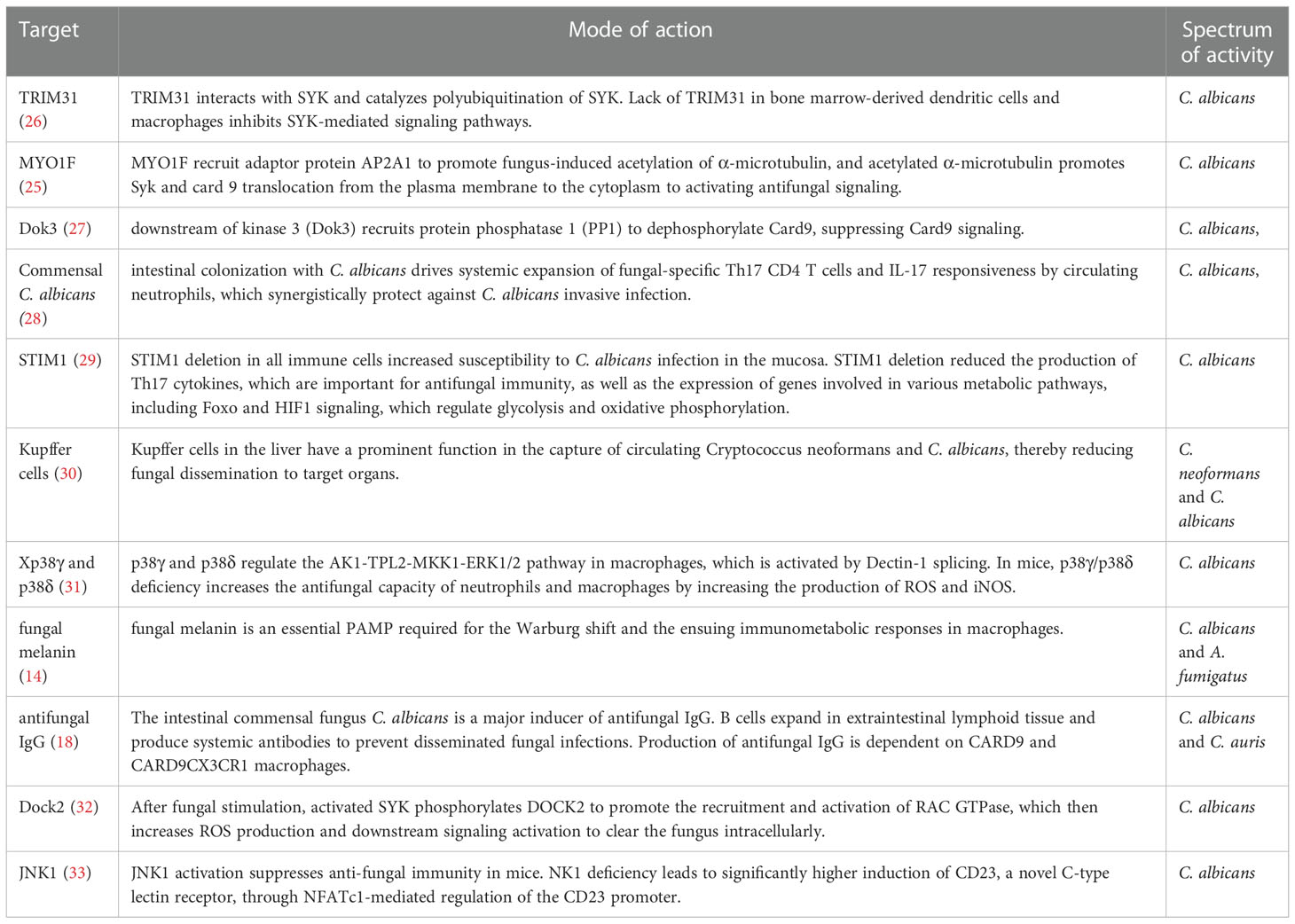
Table 1 Antifungal signaling pathways discovered in the last 5 years.
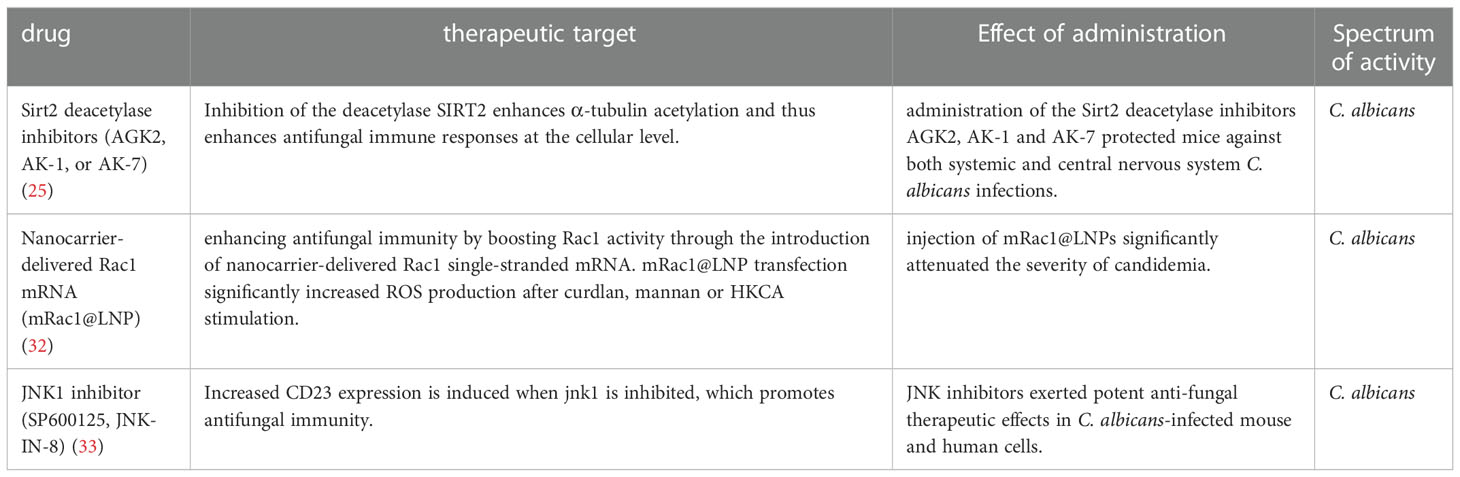
Table 2 Antifungal drugs discovered in the last 5 years.
In addition to T cell subpopulations, microglia and natural killer (NK) cells also play important roles in fungi immunity partially by collaboration with CD4+ T cell. Microglia exhibit strong responses to various species of fungi, including C. neoformans and C. albicans (34, 35), utilizing the receptor GPR43 (36), PI3 kinase signaling (37), and inducible nitric oxide synthase (iNOS) (38) for C. neoformans phagocytosis and killing. Moreover, there are other APCs in the brain, such as astrocytes and recruited DCs (38), that may also be involved in the induction of specific T-cell immunity and CD4+ T migration to the brain through an increase in TNF, CCL2, and CCL5 in experimental cryptococcosis (39). In the fungal-infected CNS, microglia is capable of producing IL-1β and CXCL1 by the fungal-secreted toxin Candidalysin, which recruits neutrophils to clear the fungi (40, 41). Besides, NK cells are capable of using the activating receptor NKp30 as a microbial PRR to recognize fungi and activate cytolytic pathways. NKp30 recognizes β-1,3-glucan, which induces Src family kinase signal transduction, cytotoxic granule trafficking, and synapse formation (42). NKp46 is another kind of NK activating receptor, which recognizes the Epa adhesins (43). Although fungi-associated immunity is complex, fine-turning of the overactive immune response is important to control the infection and reduce inflammatory injury.
Multiple sclerosis (MS) is a prototypical autoimmune neuroinflammatory disease of the CNS characterized by multifocal demyelination and neurodegeneration. The autoimmune response of MS is pathologically characterized by pro-inflammatory cell infiltration in the CNS, initiated by Th1/Th17 cells and promoting the proliferation of innate immune cells and B cells. The onset of MS is thought to be caused by the activation of peripheral CNS reactive T cells (44). The two main hypotheses are the identification of immune cross-reactivity of T cells with foreign antigens and the leakage of CNS-derived antigens into deep cervical lymph nodes. The etiology of this incurable disease is unknown. Many microbes have been investigated as potential causes of MS, such as the Epstein Barr virus and some bacteria (45). In recent years, Genome-wide association studies have identified several antifungal immune genes associated with autoimmune disease (46, 47). Significant enrichment of MS susceptibility loci is evident in many different immune cell types and tissues, but lack of enrichment in the CNS profiles at the tissue level. Although the adaptive immune system has been proposed to play a significant role in MS pathogenesis (48), analysis of the genetic map of MS patients demonstrates that many elements of innate immunity, such as NK cells and DCs, also show strong enrichment of MS susceptibility genes (48). These findings provide a genetic link between innate immune dysregulation and susceptibility to autoimmune disease. However, the mechanisms by which dysregulation of innate immune signaling pathways leads to autoimmune disease are unclear.
Recent studies and our group have also suggested that fungi may play an important role in the pathogenesis of MS (25, 49, 50). It has been reported that fungi can be detected in the brain specimens of MS patients by using nested PCR assays and next-generation sequencing. Among fungal DNA of different species, Trichosporon mucoid was found in the majority of MS patients (51), and Candida is the most closely related to multiple sclerosis as reported (52–54). C. albicans isolated from MS patients had higher specific enzymatic activity and were positively correlated with the severity of MS (52). In addition, MS patients had a higher rate of candida infection than the healthy controls (51). For example, the HLA-DRB1*15 allele group is the most important genetic risk factor for multiple sclerosis (55) and is also a risk factor linked to fungal infections (56). The evidence supported that Candida species infection may be associated with increased odds of MS. It reported that C. albicans infection before the induction of experimental autoimmune encephalomyelitis (EAE), the classic animal model of MS, promotes the EAE onset and aggravates the disease severity (57). In addition to C. albicans, EAE was also aggravated in mice infected with non-C. albicans strains C. glabrata and C. krusei (58), suggesting a critical role of fungi in promoting EAE disease progression.
Although the mechanism by which immune cells are directed into the CNS is unknown, it is believed that activation of peripheral T cells triggers the expression of adhesion molecules and chemokines, thereby facilitating the migration of immune cells. MS lesions show increased expression of endothelial adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and dual immunoglobulin domain-containing cell adhesion molecule (DICAM) (59, 60). These adhesion molecules and their ligands promote T cell migration in the BBB. In VLA-4-deficient mice, auto-reactive T cells fail to migrate to the CNS (61). adhesion molecule DICAM expression is increased on circulating CD4 T cells in patients with active RRMS and PMS disease course, while expression of ligands of DICAM is increased in BBB endothelium and MS lesions. DICAM promotes Th17 crossing during autoimmune neuroinflammation in the BBB (60).
It has been proposed that fungal toxins may play a role in the destruction of astrocytes and oligodendrocytes, resulting in characteristic myelin degradation (62). Gliotoxin is the main and the most potent mycotoxin that is secreted by A. fumigatus. Gliotoxin may contribute to the cerebral invasion processes of Aspergillus fumigatus by altering the BBB integrity (9). Once the toxin reaches the BBB, astrocytes, and oligodendrocytes surrounding the BBB will become targets for attack. Astrocytes play an integral role in maintaining barrier integrity, while oligodendrocytes provide nutritional maintenance for myelin. The result includes disruption of the BBB, enhanced CNS inflammation, degradation of myelin, and exacerbation of MS (Figure 3). This hypothesis is also supported by the fact that Gliotoxin can increase neuroinflammation of EAE in non-neuronal sites (63). In addition, chitinase can hydrolyze chitin, a component of fungal cell wall but not present in bacterial and mammalian cells, while elevated chitinase in cerebrospinal fluid is an important biomarker of MS (64, 65). Besides, calprotectin is elevated in the cerebrospinal fluid of MS patients, and amyloid deposits are found in the brains of MS patients (66) (67), both of which are associated with antifungal immunity, suggesting fungal infection in the CNS of MS patients is a critical event for disease.
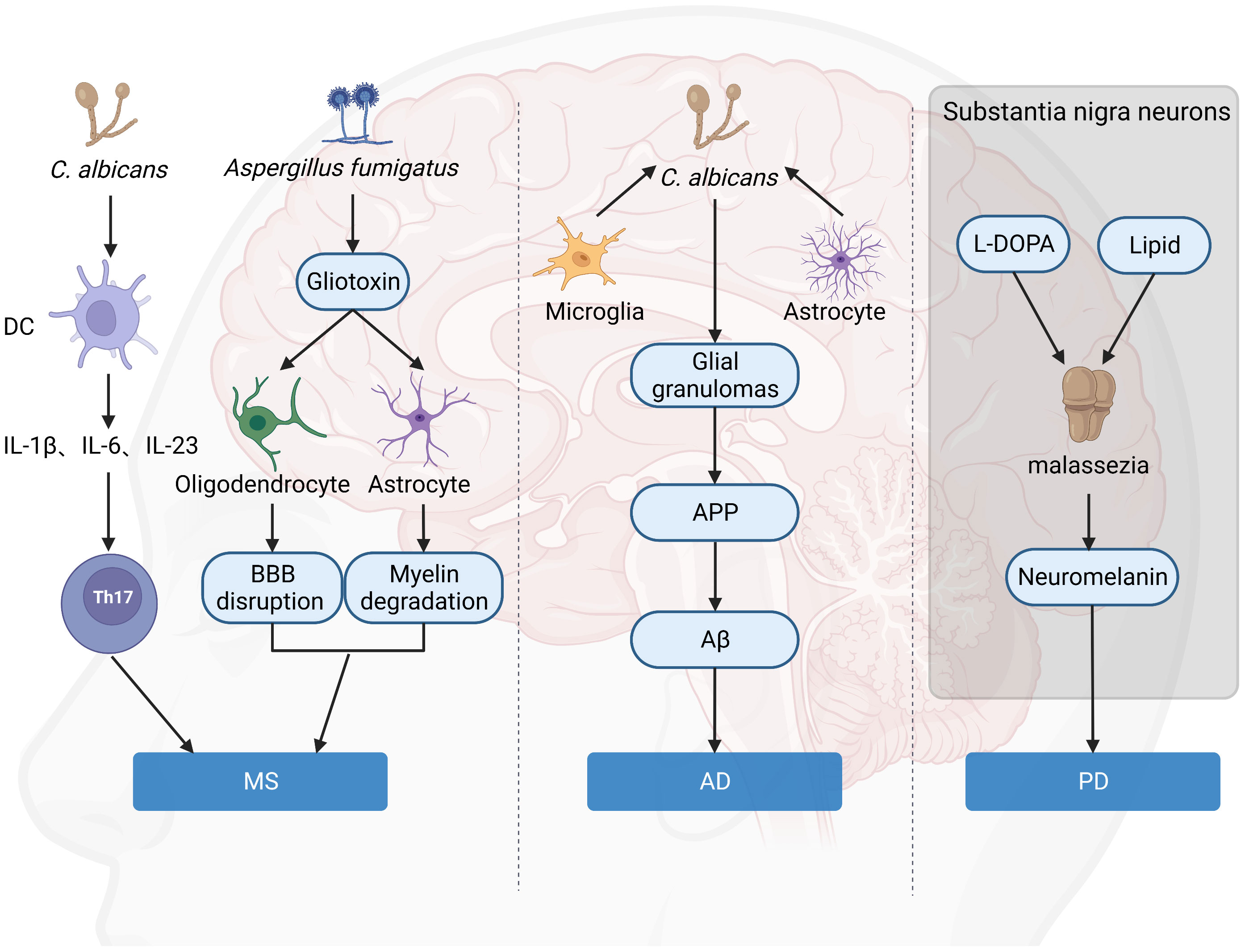
Figure 3 Hypothesis of the role of fungi in brain disorders. Gliotoxin attacks on astrocytes and oligodendrocytes around the BBB lead to BBB destruction and myelin degradation, which aggravates MS. C. albicans is a significant inducer of human Th17 cells, and TH17 exacerbates MS. C. albicans infection of the brain causes activated microglia and astrocytes to accumulate around yeast aggregates, forming fungal-induced glial granulomas. APP aggregates at the periphery of these granulomas, and APP cleavage leads to the production of Aβ. Malassezia consumes intracellular lipid droplets of neurons in L-DOPA-rich substantia nigra and produces neuromelanin, leading to exacerbation of PD. (Created with Biorender.com).
Although fungi have been detected in the CNS of MS patients, attacks on the immune system by fungi in the periphery may also exacerbate MS. ITS sequencing of MS patients and healthy controls showed that Aspergillus and Saccharomyces were significantly increased in the gut microbiome of MS patients (68). Alterations in the gut mycobiome in MS patients lead to changes in various types of immune cells. Aspergillus are positively correlated with B cells and activated CD16 DCs in MS patients, while Saccharomyces was positively correlated with circulating basophils and negatively correlated with regulatory B cells (68). Immune cells infiltrating in the CNS of MS are thought to be initiated in the periphery and reactivated in the CNS by local APCs (69). Many drugs for MS work through this. For example, alemtuzumab and ocrelizumab work by depleting peripheral immune cells, and natalizumab works thought blocking cell migration (44). Foreign antigens in tissues such as intestine, lung, and skin are captured by APCs, which migrate to draining lymph nodes and may activate pathogen-reactive T cells that cross-react with CNS self-antigens (70). After they are activated and polarized into pro-inflammatory cells, these self-reactive T cells migrate to the CNS, causing tissue damage.
The antifungal immunity and the immune response to exacerbate MS largely overlap, leading to speculation that fungi be responsible for the onset or exacerbation of MS (71). While Th17 cells are involved in the pathogenesis of several autoimmune diseases, including MS (72), TH17 cells also play an important role in antifungal immunity (73). Our recent studies suggested that fungi activate the peripheral macrophage in an EPHB2 pathway and lead to the secretion of robust inflammatory cytokines, such as IL-23, IL-12, and IL-1β, which directly promote the activation of peripheral Th1 and Th17 (50). As a critical receptor, Dectin-1 plays an important role during the above process. Dectin-1 is capable of inducing the production of IL-1β, which directly activates Th17 cellular response during fungal infections via CARD9- and NF-κB signaling. According to recent research by M. Elizabeth Deerhake, Dectin-1 and its downstream signaling molecule CARD9 not only played a role in fungal infection but were also involved in EAE pathogenesis. Of note, Dectin-1 inhibits EAE progression, whereas CARD9 play the opposite role. Dectin-1 promotes myeloid cell-astrocyte crosstalk in EAE by upregulating Oncostatin M through a CARD9-independent mechanism, suggesting an important function of Dectin-1 in EAE disease (74). Human carrying TAGAP mutation has increased susceptibility to fungi infection. Similarly, mice lacking TAGAP mount inefficient innate and adaptive immune responses featured with impaired cytokine release by macrophage and Th17 cell differentiation during C. albicans infection. TAGAP-defective mice exhibit significantly attenuated onset and severity in the EAE model. TAGAP is phosphorylated by EPHB2 at tyrosine 310 in macrophage after stimulation with Dectin ligands, which bridges proximal Dectin-induced EPHB2 activity to downstream CARD9-mediated signaling pathways (50). The current pieces of evidences suggest that fungi may promote MS via direct invasion to CNS or regulation of the peripheral T cells, which could migrate to the brain and promote the disease progression, suggesting fungi signaling may be the possible target for MS treatment.
Neuromyelitis optica spectrum disorders (NMOSD) are a group of CNS autoimmune diseases characterized by recurrent episodes of optic neuritis and long-segment transverse myelitis (75). In 2004, the discovery of IgG1 against aquaporin 4 (AQP4) made NMOSD a separate disease entity from MS (76). AQP4 antibodies were clinically detectable in the serum and cerebrospinal fluid of approximately 70%-80% of NMOSD patients. The exact cause of NMO is not yet known. However, most experts believe that NMO is caused by dysfunction of immune tolerance, which refers to the ability of the immune system to distinguish its own cells or proteins from potentially foreign substances. It is believed that self-reactive T and B cells, autoantibodies, complement and leukocytes play a role in NMO (77).
The pathogenesis of NMO is influenced by a combination of genetic and environmental factors, including infectious agents. Several infectious diseases can trigger or exacerbate autoimmunity in NMO. Although there is no direct evidence linking NMO to fungal infections, some studies have shown that C. albicans can slightly promote the degree of lymphocyte proliferation in PBMC of NMO patients, suggesting that fungal infections may also exacerbate NMO (78).
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the presence of amyloid beta (Aβ), accumulating extracellularly as amyloid plaques and phosphorylation of tau proteins, and neurofibrillary tangles (NFTs) in the brain, affecting neuronal connectivity and functioning, which results in a progressive loss of brain function (79). Studies have shown that intestinal colonization of mucosal fungi induces the systemic release of IL-17A, which increases mouse responses to social stimuli by mediating the IL-17R pathway in neurons (80). AD-related chronic neuroinflammation is manifested by the activation of microglia to produce pro-inflammatory cytokines and chemokines, including IL-1β, IL-6, TNF-α, CC motif chemokine ligand-5 (CCL5), and macrophage inflammatory protein-1α (MIP-1α) by TLR2, TLR4 and inflammasome NALP3 signals (81, 82).
Although the cause of Alzheimer’s disease is still uncertain, multiple studies have shown a relationship between microbial infections and AD. Pathogens such as HSV-1, EBV, Chlamydia pneumoniae, and Helicobacter pylori have been detected in serum, cerebrospinal fluid (CSF) and brain tissues of AD patients (83). Extensive studies have detected the presence of different fungal species or fungal proteins in the brains of AD patients (84–86). Chitin is a N-acetylglucosamine polymer that is found in the fungal cell wall. Chitin-like structures have been identified in Alzheimer’s disease brains by fluorochrome dye calcofluor (87). Others created and tested specific antibodies that detect the fungal proteins enolase and -tubulin and show the presence of two fungal proteins, enolase and α-tubulin, as well as the polysaccharide chitin, in brain tissue of AD patients (86). Control subject brain slices were typically negative for staining with the three antibodies. Furthermore, levels of chitinase, a human enzyme generated by macrophages, are pretty high in the serum and CSF of AD patients. These data support the concept that AD may be caused by a disseminated fungal infection.
With the discovery of the antimicrobial properties of Aβ against fungal and bacterial infections (88), a new AD hypothesis has emerged, called the “antimicrobial protection hypothesis” (89) (Figure 3). This hypothesis suggests that the deposition of Aβ plaques in the brain can initiate an early innate immune response. Under normal conditions, Aβ can entrap and neutralize invading pathogens. However, this pathway is overactivated in AD, leading to persistent inflammation, neurodegeneration, and Aβ plaque deposition. Intravenous injection of small numbers of C. albicans cells results in highly localized encephalitis characterized by the accumulation of activated microglia and astrocytes around the aggregated fungus, forming fungal-induced glial granulomas. Amyloid precursor protein (APP) accumulates at the periphery of these granulomas, and the cleavage of APP leads to the production of Aβ. C. albicans in the CNS activates the transcription factor NF-κB and induces the production of IL-1β, IL-6, and tumor necrosis factor, further upregulating the excessive synthesis of Aβ. Mice infected with C. albicans exhibit mild memory impairment, which resolves after fungal clearance (90). All of this evidence suggested a direct connection between fungi and AD onset and progression.
Amyotrophic lateral sclerosis (ALS) is one of the most common forms of motor neuron disease (MND) in adults. ALS is a fatal illness that causes selective and progressive degeneration of motor neurons in the brain, brainstem, and spinal cord. Motor neuron degeneration causes muscular weakness and, in most cases, death due to respiratory failure (91). Approximately 90% of ALS cases are spontaneous, with 5-10% caused by genetic abnormalities (familial) (92).
In a recent study, the researchers discovered a variety of fungi in ALS patients’ cerebrospinal fluid and brain tissue. A number of fungus species were detected using PCR analysis and next-generation sequencing of DNA isolated from frozen brain tissue, including Candida, Malassezia, Fusarium, Botrytis, Trichoderma, and Cryptococcus (93). In eleven individuals with ALS, immunohistochemistry utilizing a battery of antifungal antibodies identified fungal formations such as yeast and hyphae in the motor cortex, medulla, and spinal cord (94). Recently, it has been hypothesized that the pathogenesis of ALS may be related to neurotoxins produced by the fungus (95). In response to some fungal neurotoxins, neurons increase glutamate production in vivo. Excessive glutamate release from neurons is one of the hallmarks of ALS, and it has been related to the death of motor neurons as a result of glutamate receptor activation (95–97). Researchers collected data from 923 U.S. counties and found a significant correlation between MND mortality and well water prevalence in those counties (98). Therefore, a hypothesis is proposed that the pathogenesis of ALS may be an opportunistic fungal infection caused by a fungus in the well water (97).
Parkinson’s Disease (PD) is a progressive and debilitating disease marked by motor dysfunction and the loss of dopaminergic neurons in the substantia nigra pars compacta. This progressive loss of neurons is also associated with the release of the misfolded and aggregated protein α-synuclein (αSyn), which is expressed primarily in neurons throughout the brain (99). It has been shown that gut microbes promote the release of αSyn in mouse models and that gut microbes from PD patients impair motor function significantly more than the microbiota of healthy controls (100). One of the most noticeable aspects of PD is neuroinflammation. However, the cause is yet unknown. Neuroinflammation is mainly characterized by persistent and excessive microgliosis and astrogliosis in the substantia nigra (101). αSyn inhibits microglia autophagy extracellularly and promotes neurodegeneration in a PD mouse model (102). Knockdown of autophagy-related gene 5 (Atg5) in microglia promotes neuroinflammation and dopaminergic neurodegeneration, which is associated with the regulation of NLRP3 inflammasome activation (102, 103).
By using nested PCR analysis and next-generation sequencing to identify specific fungal species in various CNS regions from people with PD, strong evidence for polymicrobial infections in the central nervous system of PD patients is presented. Most of the identified fungal species belonged to the genera Candida, Fusarium, Botrytis, and Malassezia (104). Recently an association between Malassezia and PD has been discovered (105). By searching the published literature in PubMed and EMBASE databases and analyzing the sources of heterogeneity using sensitivity analysis and meta-regression, Malassezia was found to be positively associated with PD risk with a significant P<0.01 (106). While Malassezia is the most common fungal genus in the human microbiome, over proliferation of Malassezia causes seborrheic dermatitis (SD), a common benign inflammatory disease of the skin. SD is prevalent in 50% of Parkinson’s disease patients but just 3% of controls (107, 108). The local skin of PD patients is characterized by an increased ratio of sebum excretion, which plays a role in SD by stimulating yeast reproduction and enzyme synthesis (108). PD risk-associated genes frequently affect lipid metabolism. Polymorphisms in GBA, PINK1, and LRRK2 (109) are three important genetic risk factors for PD, in the meanwhile increasing the concentration of intracellular lipids. Malassezia is a lipid-dependent fungus (110), and if Malassezia reaches the CNS, adequate access to lipids may allow them to colonize or over-proliferate.
Neuromelanin is present in large quantities in neurons of the substantia nigra (111). Although the exact role of neuromelanin in PD pathogenicity is unknown, neurons containing this pigment appear to die as the disease progresses (112). Unlike peripheral melanin, neuromelanin is frequently associated with lipids (113). In vitro, Malassezia was found to produce melanin from exogenous L-DOPA (the precursor of dopamine) (114), and in addition, L-DOPA triggered mycelial growth, allowing it to penetrate host cells and tissues. It is possible that Malassezia consumes intracellular lipid droplets from these neurons in naturally L-DOPA-rich substantia nigra to produce neuromelanin leading to exacerbation of the disease (105) (Figure 3). All of the above results suggested fungi may be a critical factor in neuron death and PD development. However, how to target fungi-mediated effects in PD is still to be explored.
While fungi infection has been studied for a long time, the mechanism and signaling regarding the anti-fungi are still partially understood. In addition, the previous research on fungi infection mainly focuses on infectious diseases, with very limited studies on other brain disorders. In the current review, we summarized the advanced results and supported a critical role of fungi roles in CNS autoimmune and neurodegeneration disorders, such as Multiple sclerosis, Neuromyelitis optica spectrum disorder, Alzheimer’s disease, Parkinson’s disease, and Amyotrophic lateral sclerosis et al. Of note, the gut microbiota has been suggested with above CNS disorders. However, fungi have received much less attention than bacteria since they make up a smaller fraction of the human microbiome. Despite the fact that the significance of fungal infections has been highlighted in recent years, the detailed mechanism and target cells still need further exploration. In addition, the over-accumulation of fungi is also associated with specific gene mutations, such as TAGAP. The isolation and identification of fungi in the brain have revealed fresh information about the involvement of fungi in neurological illnesses. With these findings, antifungal therapy may become a potential treatment for MS and other neurological disorders.
Currently, our knowledge about the effect of fungi on CNS autoimmune and neurodegeneration-associated brain disorders is still largely unclear. Further demonstrating the role of fungi in these diseases requires in-depth studies, such as antifungal treatment of patients with neurological diseases et al.
C-JZ, TP and M-LJ conceived the study. C-JZ and CW wrote the manuscript. RJ provides important suggestions for the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (82022019, 82101414), Nanjing special fund for Medical Science and Technology Development Projects for Distinguished Young Scholars (JQX19005 to C-JZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nathan CL, Emmert BE, Nelson E, Berger JR. CNS fungal infections: A review. J Neurol Sci (2021) 422:117325. doi: 10.1016/j.jns.2021.117325
2. Reyes EY, Shinohara ML. Host immune responses in the central nervous system during fungal infections. Immunol Rev (2022) 311(1):50–74. doi: 10.1111/imr.13101
3. Góralska K, Blaszkowska J, Dzikowiec M. Neuroinfections caused by fungi. Infection. (2018) 46(4):443–59. doi: 10.1007/s15010-018-1152-2
4. Sheppard DC, Filler SG. Host cell invasion by medically important fungi. Cold Spring Harbor Perspect Med (2015) 5(1):a019687. doi: 10.1101/cshperspect.a019687
5. Liu Y, Mittal R, Solis NV, Prasadarao NV, Filler SG. Mechanisms of candida albicans trafficking to the brain. PloS pathogens. (2011) 7(10):e1002305. doi: 10.1371/journal.ppat.1002305
6. Kim J, Lee K-T, Lee JS, Shin J, Cui B, Yang K, et al. Fungal brain infection modelled in a human-neurovascular-unit-on-a-chip with a functional blood-brain barrier. Nat BioMed Eng. (2021) 5(8):830–46. doi: 10.1038/s41551-021-00743-8
7. Sorrell TC, Juillard P-G, Djordjevic JT, Kaufman-Francis K, Dietmann A, Milonig A, et al. Cryptococcal transmigration across a model brain blood-barrier: Evidence of the Trojan horse mechanism and differences between cryptococcus neoformans var. grubii strain H99 and cryptococcus gattii strain R265. Microbes infection. (2016) 18(1):57–67. doi: 10.1016/j.micinf.2015.08.017
8. Santiago-Tirado FH, Onken MD, Cooper JA, Klein RS, Doering TL. Trojan Horse transit contributes to blood-brain barrier crossing of a eukaryotic pathogen. MBio. (2017) 8(1):e02183–16. doi: 10.1128/mBio.02183-16
9. Patel R, Hossain MA, German N, Al-Ahmad AJ. Gliotoxin penetrates and impairs the integrity of the human blood-brain barrier in vitro. Mycotoxin Res (2018) 34(4):257–68. doi: 10.1007/s12550-018-0320-7
10. Speakman EA, Dambuza IM, Salazar F, Brown GD. T Cell antifungal immunity and the role of c-type lectin receptors. Trends Immunol (2020) 41(1):61–76. doi: 10.1016/j.it.2019.11.007
11. Drummond RA. Neuro-immune mechanisms of anti-cryptococcal protection. J Fungi (Basel) (2017) 4(1):4. doi: 10.3390/jof4010004
12. Salazar F, Brown GD. Antifungal innate immunity: A perspective from the last 10 years. J Innate Immun (2018) 10(5-6):373–97. doi: 10.1159/000488539
13. Sancho D, Reis e Sousa C. Signaling by myeloid c-type lectin receptors in immunity and homeostasis. Annu Rev Immunol (2012) 30:491–529. doi: 10.1146/annurev-immunol-031210-101352
14. Gonçalves SM, Duarte-Oliveira C, Campos CF, Aimanianda V, Ter Horst R, Leite L, et al. Phagosomal removal of fungal melanin reprograms macrophage metabolism to promote antifungal immunity. Nat Commun (2020) 11(1):2282. doi: 10.1038/s41467-020-16120-z
15. Mócsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat Rev Immunol (2010) 10(6):387–402. doi: 10.1038/nri2765
16. Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood J Am Soc Hematology. (2013) 121(13):2385–92. doi: 10.1182/blood-2012-08-450551
17. Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. (2006) 442(7103):651–6. doi: 10.1038/nature04926
18. Doron I, Leonardi I, Li XV, Fiers WD, Semon A, Bialt-DeCelie M, et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell (2021) 184(4):1017–31.e14. doi: 10.1016/j.cell.2021.01.016
19. Kroetz DN, Deepe GS. The role of cytokines and chemokines in histoplasma capsulatum infection. Cytokine. (2012) 58(1):112–7. doi: 10.1016/j.cyto.2011.07.430
20. Castro-Lopez N, Hung C-Y. Immune response to coccidioidomycosis and the development of a vaccine. Microorganisms. (2017) 5(1):13. doi: 10.3390/microorganisms5010013
21. De Luca A, Zelante T, D'Angelo C, Zagarella S, Fallarino F, Spreca A, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol (2010) 3(4):361–73. doi: 10.1038/mi.2010.22
22. Hirota K, Turner J-E, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of Th17 cells in peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol (2013) 14(4):372–9. doi: 10.1038/ni.2552
23. Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol (2012) 189(9):4666–73. doi: 10.4049/jimmunol.1200955
24. Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against candida albicans. Cell. (2019) 176(6):1340–55.e15. doi: 10.1016/j.cell.2019.01.041
25. Sun W, Ma X, Wang H, Du Y, Chen J, Hu H, et al. MYO1F regulates antifungal immunity by regulating acetylation of microtubules. Proc Natl Acad Sci U.S.A. (2021) 118(30):e2100230118. doi: 10.1073/pnas.2100230118
26. Wang X, Zhang H, Shao Z, Zhuang W, Sui C, Liu F, et al. TRIM31 facilitates K27-linked polyubiquitination of SYK to regulate antifungal immunity. Signal Transduct Target Ther (2021) 6(1):298. doi: 10.1038/s41392-021-00711-3
27. Loh JT, Xu S, Huo JX, Kim SS-Y, Wang Y, Lam K-P. Dok3-protein phosphatase 1 interaction attenuates Card9 signaling and neutrophil-dependent antifungal immunity. J Clin Invest. (2019) 129(7):2717–29. doi: 10.1172/JCI126341
28. Shao T-Y, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, et al. Commensal candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe (2019) 25(3):404–17.e6. doi: 10.1016/j.chom.2019.02.004
29. Kahlfuss S, Kaufmann U, Concepcion AR, Noyer L, Raphael D, Vaeth M, et al. STIM1-mediated calcium influx controls antifungal immunity and the metabolic function of non-pathogenic Th17 cells. EMBO Mol Med (2020) 12(8):e11592. doi: 10.15252/emmm.201911592
30. Coelho C, Drummond RA. Kupffer cells mediate systemic antifungal immunity. Trends Immunol (2019) 40(12):1071–3. doi: 10.1016/j.it.2019.11.001
31. Alsina-Beauchamp D, Escós A, Fajardo P, González-Romero D, Díaz-Mora E, Risco A, et al. Myeloid cell deficiency of p38γ/p38δ protects against candidiasis and regulates antifungal immunity. EMBO Mol Med (2018) 10(5):e8485. doi: 10.15252/emmm.201708485
32. Ma X, Tan X, Yu B, Sun W, Wang H, Hu H, et al. DOCK2 regulates antifungal immunity by regulating RAC GTPase activity. Cell Mol Immunol (2022) 19(5):602–18. doi: 10.1038/s41423-021-00835-0
33. Zhao X, Guo Y, Jiang C, Chang Q, Zhang S, Luo T, et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat Med (2017) 23(3):337–46. doi: 10.1038/nm.4260
34. Barluzzi R, Brozzetti A, Delfino D, Bistoni F, Blasi E. Role of the capsule in microglial cell–cryptococcus neoformans interaction: impairment of antifungal activity but not of secretory functions. Med mycology. (1998) 36(4):189–97. doi: 10.1111/j.1365-280X.1998.00126.x
35. Blasi E, Mazzolla R, Barluzzi R, Bistoni F. Microglial cell-mediated anti-candida activity: temperature, ions, protein kinase c as crucial elements. J Neuroimmunology. (1991) 34(1):53–60. doi: 10.1016/0165-5728(91)90098-R
36. Preissler J, Grosche A, Lede V, Le Duc D, Krügel K, Matyash V, et al. Altered microglial phagocytosis in GPR34-deficient mice. Glia. (2015) 63(2):206–15. doi: 10.1002/glia.22744
37. Song X, Tanaka S, Cox D, Lee SC. Fcγ receptor signaling in primary human microglia: Differential roles of PI-3K and Ras/ERK MAPK pathways in phagocytosis and chemokine induction. J leukocyte Biol (2004) 75(6):1147–55. doi: 10.1189/jlb.0403128
38. Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R. S100B expression in and effects on microglia. Glia. (2001) 33(2):131–42. doi: 10.1002/1098-1136(200102)33:2<131::AID-GLIA1012>3.0.CO;2-D
39. Neal LM, Xing E, Xu J, Kolbe JL, Osterholzer JJ, Segal BM, et al. CD4+ T cells orchestrate lethal immune pathology despite fungal clearance during cryptococcus neoformans meningoencephalitis. MBio. (2017) 8(6):e01415–17. doi: 10.1128/mBio.01415-17
40. Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9 microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat Immunol (2019) 20(5):559–70. doi: 10.1038/s41590-019-0377-2
41. Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PloS Pathogens. (2015) 11(12):e1005293. doi: 10.1371/journal.ppat.1005293
42. Li SS, Ogbomo H, Mansour MK, Xiang RF, Szabo L, Munro F, et al. Identification of the fungal ligand triggering cytotoxic PRR-mediated NK cell killing of cryptococcus and candida. Nat Commun (2018) 9(1):751. doi: 10.1038/s41467-018-03014-4
43. Vitenshtein A, Charpak-Amikam Y, Yamin R, Bauman Y, Isaacson B, Stein N, et al. NK cell recognition of candida glabrata through binding of NKp46 and NCR1 to fungal ligands Epa1, Epa6, and Epa7. Cell Host Microbe (2016) 20(4):527–34. doi: 10.1016/j.chom.2016.09.008
44. Ostkamp P, Deffner M, Schulte-Mecklenbeck A, Wünsch C, Lu IN, Wu GF, et al. A single-cell analysis framework allows for characterization of CSF leukocytes and their tissue of origin in multiple sclerosis. Sci Trans Med (2022) 14(673):eadc9778. doi: 10.1126/scitranslmed.adc9778
45. McKay KA, Jahanfar S, Duggan T, Tkachuk S, Tremlett H. Factors associated with onset, relapses or progression in multiple sclerosis: A systematic review. Neurotoxicology. (2017) 61:189–212. doi: 10.1016/j.neuro.2016.03.020
46. Shi G, Wang T, Li S, Cheng Y, Sheng P, Fan Y, et al. TLR2 and TLR4 polymorphisms in southern Chinese psoriasis vulgaris patients. J Dermatol Sci (2016) 83(2):145–7. doi: 10.1016/j.jdermsci.2016.04.014
47. Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. (2018) 67(1):108–19. doi: 10.1136/gutjnl-2016-312135
48. International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science (2019) 365(6460):eaav7188. doi: 10.1126/science.aav7188.60
49. Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, Waubant E. The gut microbiome in human neurological disease: A review. Ann Neurol (2017) 81(3):369–82. doi: 10.1002/ana.24901
50. Chen J, He R, Sun W, Gao R, Peng Q, Zhu L, et al. TAGAP instructs Th17 differentiation by bridging dectin activation to EPHB2 signaling in innate antifungal response. Nat Commun (2020) 11(1):1913. doi: 10.1038/s41467-020-15564-7
51. Alonso R, Fernández-Fernández AM, Pisa D, Carrasco L. Multiple sclerosis and mixed microbial infections. direct identification of fungi and bacteria in nervous tissue. Neurobiol Dis (2018) 117:42–61. doi: 10.1016/j.nbd.2018.05.022
52. Saroukolaei SA, Ghabaee M, Shokri H, Badiei A, Ghourchian S. The role of candida albicans in the severity of multiple sclerosis. Mycoses. (2016) 59(11):697–704. doi: 10.1111/myc.12489
53. Benito-León J, Pisa D, Alonso R, Calleja P, Díaz-Sánchez M, Carrasco L. Association between multiple sclerosis and candida species: evidence from a case-control study. Eur J Clin Microbiol Infect Dis (2010) 29(9):1139–45. doi: 10.1007/s10096-010-0979-y
54. Pisa D, Alonso R, Jiménez-Jiménez FJ, Carrasco L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur J Clin Microbiol Infect diseases. (2013) 32(6):795–801. doi: 10.1007/s10096-012-1810-8
55. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J autoimmunity. (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010
56. Chauhan B, Santiago L, Hutcheson PS, Schwartz HJ, Spitznagel E, Castro M, et al. Evidence for the involvement of two different MHC class II regions in susceptibility or protection in allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol (2000) 106(4):723–9. doi: 10.1067/mai.2000.109913
57. Fraga-Silva TFC, Mimura LAN, Marchetti CM, Chiuso-Minicucci F, França TGD, Zorzella-Pezavento SFG, et al. Experimental autoimmune encephalomyelitis development is aggravated by candida albicans infection. J Immunol Res (2015) 2015:635052. doi: 10.1155/2015/635052
58. Fraga-Silva T, Munhoz-Alves N, Mimura LAN, Oliveira L, Figueiredo-Godoi LMA, Garcia MT, et al. Systemic infection by non- species affects the development of a murine model of multiple sclerosis. J Fungi (Basel) (2022) 8(4):386. doi: 10.3390/jof8040386
59. Ransohoff RM. Mechanisms of inflammation in MS tissue: adhesion molecules and chemokines. J Neuroimmunology. (1999) 98(1):57–68. doi: 10.1016/S0165-5728(99)00082-X
60. Charabati M, Grasmuck C, Ghannam S, Bourbonnière L, Fournier AP, Lécuyer M-A, et al. DICAM promotes T17 lymphocyte trafficking across the blood-brain barrier during autoimmune neuroinflammation. Sci Trans Med (2022) 14(626):eabj0473.
61. Parker Harp CR, Archambault AS, Cheung M, Williams JW, Czepielewski RS, Duncker PC, et al. Neutrophils promote VLA-4-dependent b cell antigen presentation and accumulation within the meninges during neuroinflammation. Proc Natl Acad Sci U S A. (2019) 116(48):24221–30. doi: 10.1073/pnas.1909098116
62. Purzycki CB, Shain DH. Fungal toxins and multiple sclerosis: A compelling connection. Brain Res Bull (2010) 82(1-2):4–6. doi: 10.1016/j.brainresbull.2010.02.012
63. Fraga-Silva TFC, Mimura LAN, Leite LCT, Borim PA, Ishikawa LLW, Venturini J, et al. Gliotoxin aggravates experimental autoimmune encephalomyelitis by triggering neuroinflammation. Toxins (Basel) (2019) 11(8):443. doi: 10.3390/toxins11080443
64. Vega K, Kalkum M. Chitin, chitinase responses, and invasive fungal infections. Int J Microbiol (2012) 2012:920459. doi: 10.1155/2012/920459
65. Verbeek M, Notting E, Faas B, Claessens-Linskens R, Jongen P. Increased cerebrospinal fluid chitotriosidase index in patients with multiple sclerosis. Acta neurologica scandinavica. (2010) 121(5):309–14. doi: 10.1111/j.1600-0404.2009.01242.x
66. Berg-Hansen P, Vandvik B, Fagerhol M, Holmøy T. Calprotectin levels in the cerebrospinal fluid reflect disease activity in multiple sclerosis. J neuroimmunology (2009) 216(1-2):98–102. doi: 10.1016/j.jneuroim.2009.09.006
67. Gentile A, Mori F, Bernardini S, Centonze D. Role of amyloid-β CSF levels in cognitive deficit in MS. Clin Chim Acta (2015) 449:23–30. doi: 10.1016/j.cca.2015.01.035
68. Shah S, Locca A, Dorsett Y, Cantoni C, Ghezzi L, Lin Q, et al. Alterations of the gut mycobiome in patients with MS. EBioMedicine. (2021) 71:103557. doi: 10.1016/j.ebiom.2021.103557
69. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol (2021) 20(6):470–83. doi: 10.1016/S1474-4422(21)00063-6
70. Gran B, Hemmer B, Vergelli M, McFarland HF, Martin R. Molecular mimicry and multiple sclerosis: degenerate T-cell recognition and the induction of autoimmunity. Ann Neurology. (1999) 45(5):559–67. doi: 10.1002/1531-8249(199905)45:5<559::AID-ANA3>3.0.CO;2-Q
71. Benito-León J, Laurence M. The role of fungi in the etiology of multiple sclerosis. Front In Neurology. (2017) 8:535. doi: 10.3389/fneur.2017.00535
72. Moser T, Akgün K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: Therapeutic implications. Autoimmun Rev (2020) 19(10):102647. doi: 10.1016/j.autrev.2020.102647
73. Hernández-Santos N, Gaffen SL. Th17 cells in immunity to candida albicans. Cell Host Microbe (2012) 11(5):425–35. doi: 10.1016/j.chom.2012.04.008
74. Deerhake ME, Danzaki K, Inoue M, Cardakli ED, Nonaka T, Aggarwal N, et al. Dectin-1 limits autoimmune neuroinflammation and promotes myeloid cell-astrocyte crosstalk via Card9-independent expression of oncostatin m. Immunity (2021) 54(3):484–98.e8. doi: 10.1016/j.immuni.2021.01.004
75. Pittock SJ, Zekeridou A, Weinshenker BG. Hope for patients with neuromyelitis optica spectrum disorders - from mechanisms to trials. Nat Rev Neurol (2021) 17(12):759–73. doi: 10.1038/s41582-021-00568-8
76. Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol (2012) 11(6):535–44. doi: 10.1016/S1474-4422(12)70133-3
77. Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, et al. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm (2020) 7(5):e841. doi: 10.1212/NXI.0000000000000841
78. Barros PO, Linhares UC, Teixeira B, Kasahara TM, Ferreira TB, Alvarenga R, et al. High in vitro immune reactivity to escherichia coli in neuromyelitis optica patients is correlated with both neurological disabilities and elevated plasma lipopolysaccharide levels. Hum Immunol (2013) 74(9):1080–7. doi: 10.1016/j.humimm.2013.06.016
79. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer Disease. Nat Rev Dis Primers. (2021) 7(1):33. doi: 10.1038/s41572-021-00269-y
80. Leonardi I, Gao IH, Lin WY, Allen M, Li XV, Fiers WD, et al. Mucosal fungi promote gut barrier function and social behavior via type 17 immunity. Cell. (2022) 185(5):831–46.e14. doi: 10.1016/j.cell.2022.01.017
81. Hemonnot A-L, Hua J, Ulmann L, Hirbec H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front In Aging Neurosci (2019) 11:233. doi: 10.3389/fnagi.2019.00233
82. Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem Int (2016) 99:110–32. doi: 10.1016/j.neuint.2016.06.011
83. Nemergut M, Batkova T, Vigasova D, Bartos M, Hlozankova M, Schenkmayerova A, et al. Increased occurrence of treponema spp. and double-species infections in patients with alzheimer's disease. Sci Total Environ (2022) 844:157114. doi: 10.1016/j.scitotenv.2022.157114
84. Alonso R, Pisa D, Aguado B, Carrasco L. Identification of fungal species in brain tissue from alzheimer’s disease by next-generation sequencing. J Alzheimer's Disease. (2017) 58(1):55–67. doi: 10.3233/JAD-170058
85. Alonso R, Pisa D, Fernández-Fernández AM, Carrasco L. Infection of fungi and bacteria in brain tissue from elderly persons and patients with alzheimer’s disease. Front Aging Neurosci (2018) 10:159. doi: 10.3389/fnagi.2018.00159
86. Pisa D, Alonso R, Rábano A, Horst MN, Carrasco L. Fungal enolase, β-tubulin, and chitin are detected in brain tissue from alzheimer’s disease patients. Front Microbiol (2016) 7:1772. doi: 10.3389/fmicb.2016.01772
87. Castellani RJ, Siedlak SL, Fortino AE, Perry G, Ghetti B, Smith MA. Chitin-like polysaccharides in alzheimer's disease brains. Curr Alzheimer Res (2005) 2(4):419–23. doi: 10.2174/156720505774330555
88. Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of alzheimer’s disease. Sci Trans Med (2016) 8(340):340ra72–ra72. doi: 10.1126/scitranslmed.aaf1059
89. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of alzheimer's disease. Alzheimer's Dementia. (2018) 14(12):1602–14. doi: 10.1016/j.jalz.2018.06.3040
90. Wu Y, Du S, Johnson JL, Tung H-Y, Landers CT, Liu Y, et al. Microglia and amyloid precursor protein coordinate control of transient candida cerebritis with memory deficits. Nat Commun (2019) 10(1):58. doi: 10.1038/s41467-018-07991-4
91. Guo W, Vandoorne T, Steyaert J, Staats KA, Van Den Bosch L. The multifaceted role of kinases in amyotrophic lateral sclerosis: genetic, pathological and therapeutic implications. Brain. (2020) 143(6):1651–73. doi: 10.1093/brain/awaa022
92. Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. Lancet. (2022) 400(10360):1363–80. doi: 10.1016/S0140-6736(22)01272-7
93. Alonso R, Pisa D, Fernández-Fernández AM, Rábano A, Carrasco L. Fungal infection in neural tissue of patients with amyotrophic lateral sclerosis. Neurobiol Disease. (2017) 108:249–60. doi: 10.1016/j.nbd.2017.09.001
94. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, et al. Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int J Biol Sci (2015) 11(5):546–58. doi: 10.7150/ijbs.11084
95. French PW, Ludowyke R, Guillemin GJ. Fungal neurotoxins and sporadic amyotrophic lateral sclerosis. Neurotox Res (2019) 35(4):969–80. doi: 10.1007/s12640-018-9980-5
96. Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal (2009) 11(7):1587–602. doi: 10.1089/ars.2009.2444
97. French PW, Ludowyke RI, Guillemin GJ. Fungal-contaminated grass and well water and sporadic amyotrophic lateral sclerosis. Neural Regener Res (2019) 14(9):1490–3. doi: 10.4103/1673-5374.255959
98. Schwartz GG, Klug MG. Motor neuron disease mortality rates in U.S. states are associated with well water use. Amyotroph Lateral Scler Frontotemporal Degener (2016) 17(7-8):528–34. doi: 10.1080/21678421.2016.1195409
99. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. (2016) 353(6301):777–83. doi: 10.1126/science.aag2590
100. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinson's disease. Cell (2016) 167(6):1469–80.e12. doi: 10.1016/j.cell.2016.11.018
101. Sarkar S, Nguyen HM, Malovic E, Luo J, Langley M, Palanisamy BN, et al. Kv1.3 modulates neuroinflammation and neurodegeneration in parkinson's disease. J Clin Invest (2020) 130(8):4195–212. doi: 10.1172/JCI136174
102. Tu H-Y, Yuan B-S, Hou X-O, Zhang X-J, Pei C-S, Ma Y-T, et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of parkinson's disease. Aging Cell (2021) 20(12):e13522. doi: 10.1111/acel.13522
103. Qin Y, Qiu J, Wang P, Liu J, Zhao Y, Jiang F, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of parkinson's disease. Brain Behav Immun (2021) 91:324–38. doi: 10.1016/j.bbi.2020.10.010
104. Pisa D, Alonso R, Carrasco L. Parkinson's disease: A comprehensive analysis of fungi and bacteria in brain tissue. Int J Biol Sci (2020) 16(7):1135–52. doi: 10.7150/ijbs.42257
105. Laurence M, Benito-León J, Calon F. Parkinson's disease. Front In Neurology. (2019) 10:758. doi: 10.3389/fneur.2019.00758
106. Wang H, Liu X, Tan C, Zhou W, Jiang J, Peng W, et al. Bacterial, viral, and fungal infection-related risk of parkinson's disease: Meta-analysis of cohort and case-control studies. Brain Behav (2020) 10(3):e01549. doi: 10.1002/brb3.1549
107. Ravn A-H, Thyssen JP, Egeberg A. Skin disorders in parkinson's disease: potential biomarkers and risk factors. Clin Cosmet Investig Dermatol (2017) 10:87–92. doi: 10.2147/CCID.S130319
108. Arsic Arsenijevic VS, Milobratovic D, Barac AM, Vekic B, Marinkovic J. Kostic VS. a laboratory-based study on patients with parkinson's disease and seborrheic dermatitis: the presence and density of malassezia yeasts, their different species and enzymes production. BMC Dermatol (2014) 14:5. doi: 10.1186/1471-5945-14-5
109. Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol (2013) 9(8):445–54. doi: 10.1038/nrneurol.2013.132
110. Theelen B, Cafarchia C, Gaitanis G, Bassukas ID, Boekhout T, Dawson TL. Malassezia ecology, pathophysiology, and treatment. Med Mycology (2018) 56(suppl_1):S10–25. doi: 10.1093/mmy/myx134
111. Fedorow H, Tribl F, Halliday G, Gerlach M, Riederer P, Double KL. Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to parkinson's disease. Prog Neurobiol (2005) 75(2):109–24. doi: 10.1016/j.pneurobio.2005.02.001
112. Haining RL, Achat-Mendes C. Neuromelanin, one of the most overlooked molecules in modern medicine, is not a spectator. Neural Regener Res (2017) 12(3):372–5. doi: 10.4103/1673-5374.202928
113. Engelen M, Vanna R, Bellei C, Zucca FA, Wakamatsu K, Monzani E, et al. Neuromelanins of human brain have soluble and insoluble components with dolichols attached to the melanic structure. PloS One (2012) 7(11):e48490. doi: 10.1371/journal.pone.0048490
Keywords: fungus, brain disorders, multiple sclerosis, signaling, disease
Citation: Wu C, Jiang M-L, Jiang R, Pang T and Zhang C-J (2023) The roles of fungus in CNS autoimmune and neurodegeneration disorders. Front. Immunol. 13:1077335. doi: 10.3389/fimmu.2022.1077335
Received: 22 October 2022; Accepted: 30 December 2022;
Published: 26 January 2023.
Edited by:
Yaping Yan, Shaanxi Normal University, ChinaReviewed by:
Guillaume Tabouret, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceCopyright © 2023 Wu, Jiang, Jiang, Pang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cun-Jin Zhang, emhhbmdjdW5qaW41MTZAMTYzLmNvbQ==; Mei-Ling Jiang, amlhbmdtZWlsaW5nMTIxNUAxNjMuY29t; Tao Pang, dHBhbmdAY3B1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.