
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 27 December 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1070068
This article is part of the Research TopicAdvances in primary Immunodeficiencies (Inborn Errors of Immunity) in Central-Eastern Europe: Volume IIView all 20 articles
 Adriana Margarit-Soler1*
Adriana Margarit-Soler1* Àngela Deyà-Martínez2,3,4
Àngela Deyà-Martínez2,3,4 Juan Torres Canizales5
Juan Torres Canizales5 Alexandru Vlagea5Ana García-García2,3,4Júlia Marsal1Maria Trabazo Del Castillo1
Alexandru Vlagea5Ana García-García2,3,4Júlia Marsal1Maria Trabazo Del Castillo1 Sílvia Planas6Sílvia Simó7,8
Sílvia Planas6Sílvia Simó7,8 Ana Esteve-Sole2,3,4María Suárez-Lledó Grande9,10,11Isabel Badell1,12
Ana Esteve-Sole2,3,4María Suárez-Lledó Grande9,10,11Isabel Badell1,12 Montserrat Rovira Tarrats1,9,10Francesc Fernández-Avilés9,10
Montserrat Rovira Tarrats1,9,10Francesc Fernández-Avilés9,10 Laia Alsina2,3,4,11*
Laia Alsina2,3,4,11*Cytotoxic T-lymphocyte antigen-4 (CTLA-4) haploinsufficiency is a T-cell hyperactivation disorder that can manifest with both immunodeficiency and immune dysregulation. Approximately one-third of patients may present mild symptoms and remain stable under supportive care. The remaining patients may develop severe multiorgan autoimmunity requiring lifelong immunosuppressive treatment. Hematopoietic stem cell transplantation (HSCT) is potentially curable for patients with treatment-resistant immune dysregulation. Nevertheless, little experience is reported regarding the management of complications post-HSCT. We present case 1 (CTLA-4 haploinsufficiency) and case 2 (CTLA-4 insufficiency-like phenotype) manifesting with severe autoimmunity including cytopenia and involvement of the central nervous system (CNS), lung, and gut and variable impairment of humoral responses. Both patients underwent HSCT for which the main complications were persistent mixed chimerism, infections, and immune-mediated complications [graft-versus-host disease (GVHD) and nodular lung disease]. Detailed management and outcomes of therapeutic interventions post-HSCT are discussed. Concretely, post-HSCT abatacept and human leukocyte antigen (HLA)-matched sibling donor lymphocyte infusions may be used to increase T-cell donor chimerism with the aim of correcting the immune phenotype of CTLA-4 haploinsufficiency.
Regulatory T cell (Treg) defects are conditions included within the category of primary immune regulatory disorders (PIRDs) (1). They are defined by quantitative or qualitative impairment of the Treg compartment, predisposing to severe multiorgan autoimmunity with or without susceptibility to infections (2–4). Cytotoxic T-lymphocyte antigen-4 (CTLA-4) haploinsufficiency is included in this group, since the reduced surface availability of CTLA-4 ultimately results in Treg cell dysfunction (5). The three entities that negatively affect CTLA-4 function in human disease are CTLA-4, lipopolysaccharide-responsive and beige like anchor protein (LRBA), and DEF6 deficiencies (5). These three have been grouped under the term immune checkpoint defects (6). They share common features of dual symptoms of immune dysregulation and immune deficiency, with variable disease expressivity even in individuals with the same mutation (5, 7). Shared phenotypic manifestations of immune dysregulation include autoimmune cytopenia, enteropathy, and lymphoproliferation (5, 8, 9). This triad combination is not commonly seen in other PIRDs (5).
The long-term therapeutic approach for affected patients is challenging (5, 10). Approximately one-third of patients may present mild symptoms and remain stable under supportive care (8). The remaining patients may develop severe multiorgan autoimmunity requiring lifelong immunosuppressive treatment with Treg-sparing immunosuppression [mammalian target of rapamycin (mTOR) inhibitors] or targeted soluble CTLA-4-Ig (abatacept, belatacept) (10, 11). Currently, hematopoietic stem cell transplantation (HSCT) is offered to these patients with treatment-resistant immune dysregulation (1, 12). Nevertheless, little experience in HSCT in these conditions is reported (10), mostly in LRBA deficiency (13). Doubts regarding bridge or remission induction therapy (11, 13, 14), conditioning for the transplant (15), and post-HSCT chimerism goals remain.
Detailed clinical observations can provide insight into the challenges of HSCT management in this subgroup of PIRD patients and more so when they are diagnosed worldwide, including Eastern Europe (5), and the HSCT approach can be variable. Thus, we present case 1 (CTLA-4 haploinsufficiency) and case 2 (CTLA-4 insufficiency-like phenotype), both manifested with autoimmune cytopenia, enteropathy, and lymphoproliferation, with typical lung and central nervous system (CNS) involvement, and variable impairment of humoral responses. Both patients underwent HSCT, and the main complications were mixed chimerism, infections, and immune-mediated complications.
We present an 18-year-old man with CTLA-4 haploinsufficiency [CTLA4 frameshift mutation c.342_342delC, reported in Schwab et al. (8), subject 42] with low CTLA-4 expression in FoxP3+ CD4 T cell (Supplementary Figure S1) (16). Patient presented autoimmune and/or inflammatory disorders such as autoimmune hemolytic anemia (AIHA), idiopathic thrombocytopenic purpura (ITP), granulomatous-lymphocytic interstitial lung disease (GLILD), and autoimmune encephalitis, requiring multiple immunosuppressants as shown in Figure 1. HSCT was indicated due to partial response of these immune dysregulatory manifestations despite targeted treatment. His immune deficiency and dysregulation activity (IDDA) score (11) prior to HSCT was 17.6. Main characteristics of his baseline disease and HSCT are presented in Table 1. The patient received HSCT from his identical human leukocyte antigen (HLA) sister, both sharing blood type and positivity for cytomegalovirus (CMV). The European Group for Blood and Marrow Transplantation (EBMT) guidelines were followed for HSCT using treosulfan, fludarabine, and thiotepa as conditioning treatment and cyclosporin and mycophenolate for graft-versus-host disease (GVHD) prophylaxis (17). The clinical course was complicated with low donor lymphoid chimerism and an episode of lung lesions that posed the differential diagnosis with GLILD. Both resolved with the combination of donor lymphocyte infusions (DLIs) from the identical HLA sister and expert management of immune modulation including abatacept introduction post-HSCT, as described below. The timeline is depicted in Figure 1.
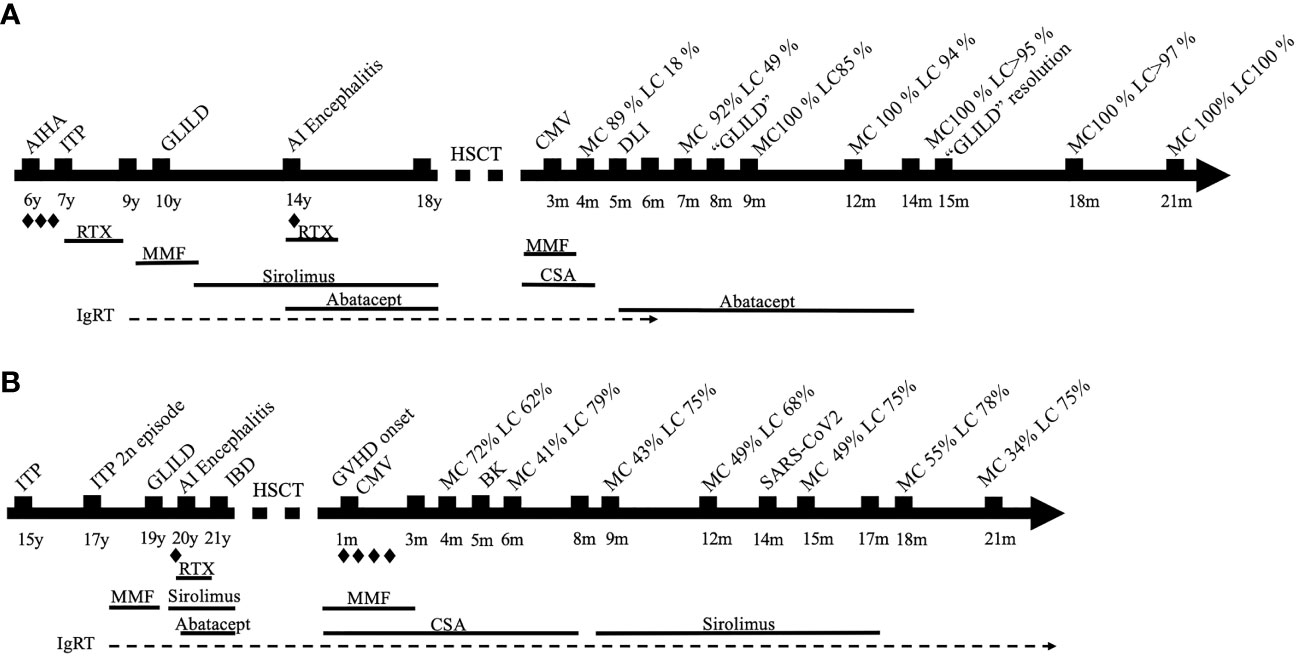
Figure 1 (A) Timeline of case 1. (B) Timeline of case 2. Steroids; AIHA, autoimmune hemolytic anemia; ITP, immune thrombocytopenic purpura; AI, autoimmune; HSCT, hematopoietic stem cell transplantation; CMV, cytomegalovirus; MC, myeloid chimerism; LC, lymphoid chimerism; CSA, cyclosporin; DLI, donor lymphocyte infusion; GLILD, granulomatous-lymphocytic interstitial lung disease; RTX, rituximab; IgRT, immunoglobulin replacement therapy; IBD, inflammatory bowel disease; GVHD, graft-versus-host disease.
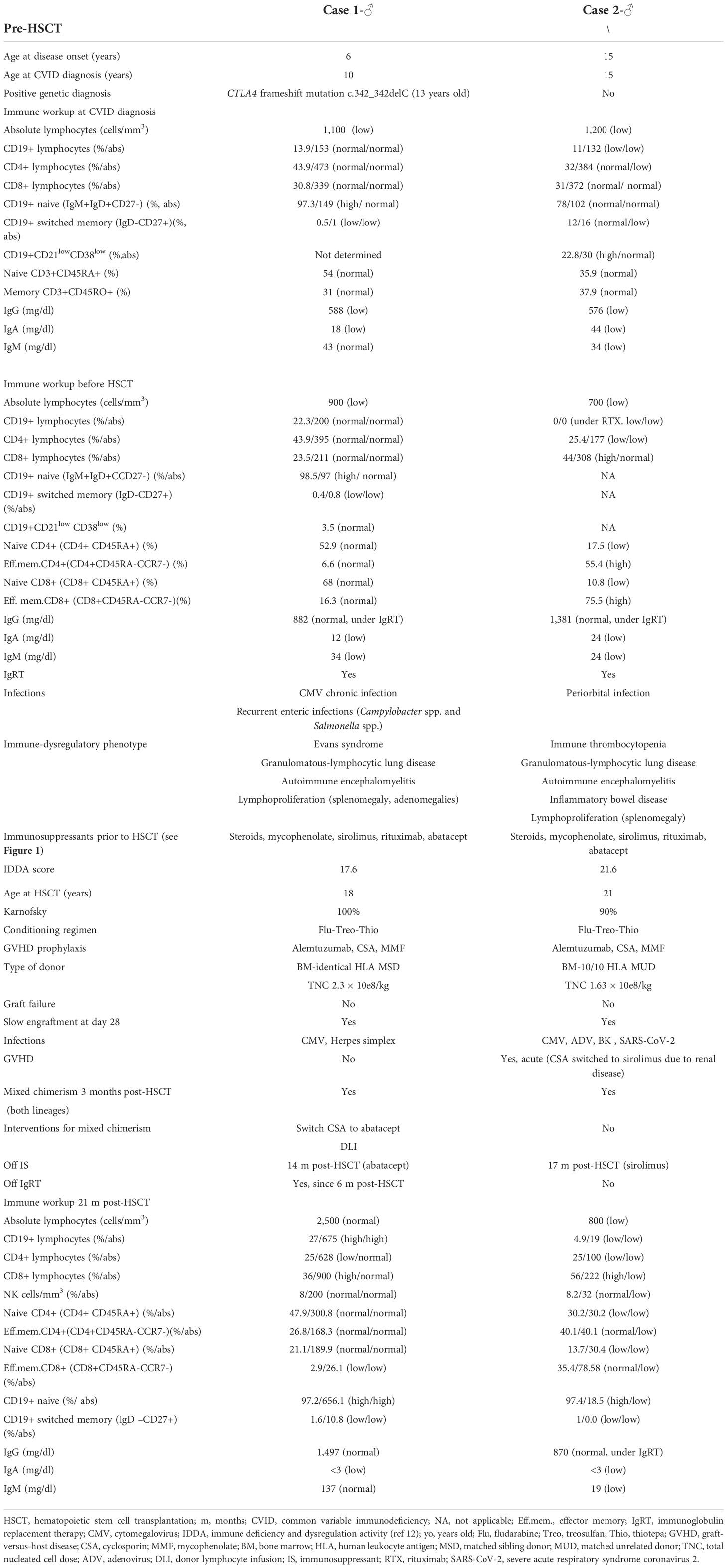
Table 1 Table 1 Summary of the two cases.
Three months post-procedure, split chimerism showed myeloid lineage of 89% and lymphoid lineage of 18% from the donor. In order to improve the chimerism, cyclosporin was quickly withdrawn over 4 weeks and abatacept as a target therapy was started to control the dysregulated lymphocytes that could be left from the recipient. Three weeks later, chimerism remained very low in the lymphoid lineage (18%); therefore, the patient received three DLIs (total dose CD3 1.3 × 108) from his sister with no signs of GVHD after that. Seven months after transplant, 3 months after stopping cyclosporin and switching to abatacept, and 1.5 months after DLI, chimerism improved, showing 92% of myeloid and 49% of lymphoid from the donor.
Eight months after transplantation, the patient presented fever and diarrhea. Infectious screening was positive for Campylobacter coli in a stool culture. After 5 days of fever and on antibiotics, computed tomography (CT) scan showed multiple lung nodules suggesting an infection or lymphoproliferative disorder (Figure 2). The study was completed with brain magnetic resonance imaging, positron emission tomography (PET) scan, bronchoalveolar lavage (BAL), and bone marrow aspirate. Multiple adenopathies and splenomegaly were seen. Bone marrow aspirate was normal. Epstein–Barr virus (EBV) was positive in blood and in the BAL, although at a very low number of copies (<250 copies/ml), and the patient did not present other signs of posttransplant lymphoproliferative disorder (PTLD).
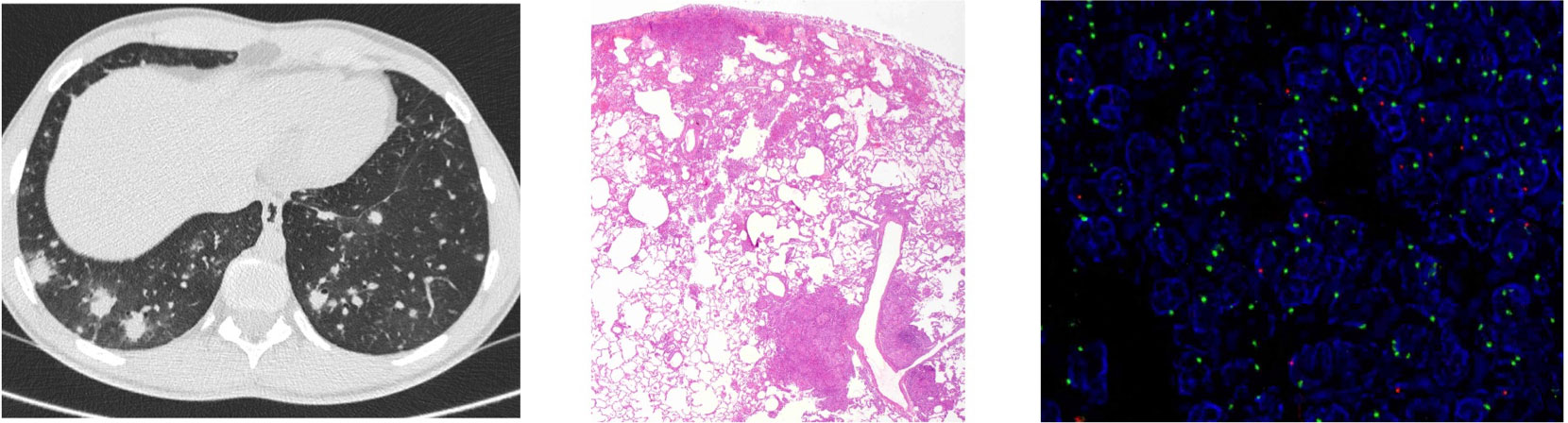
Figure 2 Study of lung lesions of case 1. First image computed tomography (CT) scan showing multiple lung nodules. Second image wedge luyng biopsy demonstrated a non-necrotizing granulomatous inflammation concentrated bronchovasculocentrically and paraseptally [hematoxylin and eosin (H&E), 2x]. Third image fluorescencein-situ hybridization (FISH) using locus specific indentifier (LSI) SRY. In the lymphocytic infiltrates, up to 27% of the nuclei showed a green dot (CEP X) and a red dot (SRY), and 73% of the nuclei showed two green dots (CEP X), thus indicating that the majority of the lymphocytic infiltrate corresponded to the female donor.
A lung wedge biopsy was performed, demonstrating non-necrotizing granulomatous inflammation concentrated bronchovasculocentrically and paraseptally, made up of histiocytes admixed with a lymphocytic component, predominantly T-cell CD4+. Microbiological tests ruled out infection. These findings were suggestive of a GLILD flare (18). To confirm which T cells (female donor vs. male patient) were driving this inflammation, fluorescence in situ hybridization (FISH) X/Y was performed in the lung tissue to assess the chimerism in situ. The same pattern as in the blood (Figure 2) was observed, ruling out enrichment of the patient’s T cells in the granulomas. Lung lesions presented remission within 2 weeks without further intervention. The patient remained on abatacept as the sole immunosuppression.
Fifteen months post-HSCT, the patient presented good clinical evolution with an improvement in the CT scan and respiratory test. Chimerism presented sustained improvement, maintaining 100% and >95% in myeloid and lymphoid lineages, respectively. In this context, abatacept was discontinued.
Currently, 21 months posttransplantation, CT scan and pulmonary function tests are within normal range and the patient’s chimerism is 100% in both lineages. He is off immunosuppression with good immune reconstitution and also off immunoglobulin replacement therapy (IgRT) (Table 1).
We present a 21-year-old man with common variable immunodeficiency (CVID) and a phenotype of immune dysregulation similar to CTLA-4 haploinsufficiency characterized by autoimmune cytopenia, inflammatory bowel disease (IBD), lymphoproliferation (GLILD), autoimmune encephalitis, and humoral deficiency requiring multiple immunosuppressants (Figure 1). No genetic defect was identified in a gene panel including inborn errors of immunity (IEIs) and PIRD-related genes (SureSelect Custom Constitutional Panel 17 Mb, Agilent), and an array comparative genomic hybridization (aCGH) was normal. The patient fulfilled the European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis for Common Variable Immunodeficiency (19) (Table 1). However, CTLA-4 expression in FoxP3+ CD4+ T cells resembled that of CTLA-4-deficient patients, with a marked decrease in the CTLA-4hiFoxP3+CD4+ T cells (Supplementary Figure S1). HSCT was indicated due to a partial control of immune dysregulation despite sirolimus and abatacept. The IDDA score prior to HSCT was 21.6. The patient received HSCT from an HLA-identical (10/10) unrelated donor with the same blood type. The patient was CMV-positive, while the donor was negative. The clinical course was complicated by GVHD, mixed chimerism, and infectious episodes, as described below. The timeline is depicted in Figure 1.
One month post-HSCT, he developed grade 2 GVHD requiring steroids and poor engraftment requiring granulocyte colony-stimulating factor and eltrombopag. He also presented CMV infection requiring preemptive treatment with foscarnet, cidofovir, and specific T-lymphocyte infusion. Thereafter, CMV infection was controlled, but he developed BK hemorrhagic cystitis, community-acquired pneumonia, and a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) upper respiratory tract infection.
Mixed chimerism persisted around 60%–70% in the lymphocytic lineage (Figure 1). Cyclosporin was switched to sirolimus for the treatment of GVHD at 8 months post-HSCT due to renal dysfunction. Lymphoid chimerism remained in similar range, with a slight increase to 75%. At 17 months post-HSCT, with controlled GVHD, immunosuppression was slowly discontinued with no complications. As the patient maintained a stable lymphoid chimerism above 70%–75% and persistent grade 2 GVHD, no further interventions were performed.
Currently, 21 months post-HSCT, the patient is off immunosuppression. He still presents mixed donor chimerism of 34% in granulocytes and 75% in T lymphocytes without autoimmune episodes. His immune cellular and humoral reconstitution is still incomplete, requiring IgRT and antimicrobial prophylaxis (Table 1).
Currently, the management of CTLA-4 haploinsufficiency and similar diseases characterized by marked T-cell activation is still in discussion. Bridge or remission induction therapy (11, 13, 14), transplant conditioning (15), and post-HSCT chimerism goals need to be refined. The two cases reported here are representative of this group of patients who fail to respond to conservative immunosuppressant treatment and move to HSCT with high levels of immune dysregulation. Their post-HSCT outcome and management highlight the importance of an individualized approach to achieve maximum, if not full, lymphoid chimerism to ensure disease remission and complete immune reconstitution.
Over the last decade, allo-HSCT outcomes in IEI have improved significantly. The survival rate for conventional IEI transplants is now approaching 90% (17, 20, 21). HSCT is also being offered to young adults with high rates of success (22). This is due to improved donor selection, better management of HSCT complications, and optimized supportive care (20, 23, 24). Still, high levels of hyperinflammation prior to transplant may promote a greater incidence of alloreactivity disorders post-HSCT (25, 26), and has already been shown to worsen HSCT outcomes, with a greater risk of GVHD (as observed in case 2) and toxicity, and impaired immune reconstitution (25). Also, although the same conditioning and GVHD prophylaxis were given in both cases, they received different types of graft: case 1 received marrow from his HLA-identical sister and case 2 received marrow from a 10/10 HLA unrelated donor. Still, some immune disparities could be present and interfere in the immune reconstitution phase, as described before (25, 27). In addition, patient 2 presented a higher IDDA score and higher number of effector memory CD4+ cells prior to HSCT. All these factors may have contributed to the different post-HSCT outcome in both patients.
In our cases, HLA-matched donors (one related, one unrelated) and bone marrow source were used. In this sense, it is important to bear in mind that because of the genetic nature of most IEIs, genetic screening of family donors is warranted regardless of symptoms, since PIRDs can display a late or variable clinical onset, as typically described for CTLA-4 haploinsufficiency (8). In order to minimize the higher incidence of alloreactivity, the role of biologic modifiers or targeted therapies as a bridge to HSCT in PIRDs is an important field to explore (14). In our center, both patients received targeted immunosuppression including abatacept until 2 weeks prior to transplantation. Currently, one of the main questions that arise is the dichotomy between lifelong immune modulation vs. HSCT for IEI and PIRD (28).
Current recommendations do not identify patients with CTLA-4 haploinsufficiency who might benefit from long-term targeted immunomodulation vs. HSCT nor the optimal timing for HSCT (10). HSCT outcomes must be balanced with the risks of disease. Reports of HSCT for CTLA-4 haploinsufficiency are scarce. They illustrate that HSCT can be effective (8, 29). Schwab et al. (8) reported 12 transplanted patients among 90 symptomatic CTLA4 mutation carriers undergoing allo-HSCT between 10 and 50 years of age. Main indications were uncontrollable cytopenia, enteropathy, and lymphoma with additional autoimmune disorders involving lymphoproliferative and infectious complications (8). Nine of the 12 patients (75%) are alive, three of them more than 5 years after HSCT and currently well without medication. Slatter et al. (29) described eight pediatric patients with CTLA-4 haploinsufficiency who underwent HSCT. All received transplants from 10/10 HLA-matched unrelated donors following a reduced-intensity conditioning regimen; the outcome was 50% GVHD, 25% autoimmune disorders, and an overall survival rate of 75% (29). In the report by Chan et al. (24), 13 of the 226 transplanted patients were CTLA-4 haploinsufficiencies, but no specific subgroup data analysis could be performed. The informed nature of decision-making for clinicians, patients, and families in these ill-defined situations is improved if clinical outcome data of defined patients with defined treatments are reported. For this purpose, a systematic description of patients with CTLA-4 haploinsufficiency undergoing HSCT is needed. This description should include IDDA score prior to HSCT. The IDDA score (11) can be used to compare semiquantitative values in one individual over time (clinical course, longitudinally) or between individuals or cohorts at a specific time point (cross-sectionally). The new IDDA version includes broader manifestations of immune dysregulation, factors that indicate the quality of life and need for supportive care, and the occurrence of malignancies (30). This is critical because the absence of ongoing medication and quality of life are important features that are rarely quantified, and these may be better in patients who have undergone transplantation (31–33). Also, a detailed description of immunosuppressants used prior to and post-HSCT and the dynamics of T-cell chimerism are necessary to enable comparisons of therapeutic approaches. In our patients, disease evolution prior to HSCT was 12 and 6 years, respectively; the patients had a high disease burden with IDDA scores >15 and had received multiple courses of immunosuppressants with only partial or transient responses. Despite the non-compelling abovementioned HSCT data, the transplant choice was made by the patients, mainly motivated by chronic disease fatigue and the low quality of life of two active young adults.
More and more adolescents and young adults (AYAs) with IEI are referred for HSCT (22, 34, 35). For these patients, aging leads to early end organ damage, reduced quality of life, and early death (36). Transplant decision is challenging, as they have survived childhood with conservative management, but HSCT needs to be considered before further deterioration. Both of our reported patients have received adult-stage HSCT with reduced toxicity regimen, and the decision to transplant was based on the lack of disease control despite long-term use of two targeted immunomodulators. For newly identified diseases with alternative targeted therapies (14, 37–39) such as CTLA-4 fusion proteins, careful follow-up of different treatment cohorts is necessary to determine the best treatment modalities in the long-term. The safety and efficacy of abatacept for adult patients with CTLA-4 insufficiency or LRBA deficiency are currently being evaluated in a phase 2 clinical trial (ABACHAI) (40).
For some IEIs, a certain level of mixed chimerism is sufficient to improve the patients’ well-being. But is there a minimum level of donor T-cell chimerism required to correct the immune phenotype of PIRD whose immune pathology is T-cell activation? Chimerism was designed to monitor the percentage of the donor cells after the infusion and not to monitor the disease baseline or malignant relapse (41). Full donor T-cell chimerism in LRBA deficiency has been shown to be positively linked to the probability of remission, although data on the relevance of donor chimerism for cure are still limited (11, 13, 42, 43). In many IEIs, stable mixed donor chimerism does not lead to graft rejection, and it may suffice to correct the underlying immunodeficiency (44). However, it has been shown that donor myeloid chimerism is important for long-term immune recovery of T and B lymphocytes and adequate immune function after HSCT (45). In case 2, low mixed myeloid chimerism was a concern, although no DLIs were considered due to the high risk of GVHD and to donor availability. Some data suggest that mixed chimerism can cause persistent autoimmunity or autoinflammation in these patients (46–48). From the report by Slatter et al. (29), six of the eight patients are alive and well with donor chimerism ranging 85%–100%. So, for patients with CTLA-4 haploinsufficiency who present mixed chimerism post-HSCT, it seems reasonable to aim at high, or even full, T-cell chimerism. In cases of no active GVHD, like in case 1, weaning from the immunosuppressant drugs and performing DLI, especially in matched family donors, could be considered. In case 1, abatacept was also started to immunomodulate the patient’s CTLA-4 haploinsufficient lymphocytes. The use of abatacept prior to HSCT is to modulate autologous activated T cells to control the underlying disease. Its use in the posttransplant phase could be beneficial both in controlling the remaining autologous T cells to reduce the risk of disease flare, but also to modulate allo-reactive donor T cells to reduce the risk of GVHD development (49, 50). On the other hand, if the patient presents GVHD, such as case 2, the approach to slowly withdraw the immunosuppressant drug, or even switch to a Treg-sparing regimen such as sirolimus, can be considered. It is difficult to ascertain what the main reasons are for such a different outcome. Our hypothesis is that GVHD development in case 2 [linked to a matched unrelated donor (MUD) and higher levels of inflammation at the time of HSCT] was one of the main determinants in the different management of mixed chimerism and final outcome, since it obliged to a certain level of immunosuppression and interfered with immune reconstitution.
Beyond chimerism, during the early phase of PIRD patient transplant, the recipient’s dysregulated lymphocytes are still a concern, especially in mixed chimerism. Therefore, close monitoring of inflammation and autoimmune complications is crucial for early flare recognition until chimerism and immune reconstitution are complete. In case 1, it is difficult to determine whether the lung nodular lesions were a flare of his GLILD in the context of persistent low chimerism and his being off abatacept (no infection or PTLD was demonstrated, and he was respiratory asymptomatic so suspicion of an immune reconstitution phenomenon was reduced). Also, no CT had been performed in the first 8 months post-HSCT to enable comparisons. The predominance of donor T cells in the nodules along with the improved chimerism thereafter might explain the quick resolution of the lung nodules in a GLILD. In patient 2, currently with 75% of lymphoid chimerism, no disease flare has been observed while off immunosuppression. Immunological biomarkers are still to be defined in the monitoring of PIRD patients’ immune reconstitution and detection of immune dysregulation post-HSCT. Functional studies on the underlying genetic defect (i.e., transendocytosis test for checkpoint deficiencies) might be of interest in cases of persistent mixed lymphoid chimerism (6, 8, 51).
CTLA-4 haploinsufficiency encompasses a heterogeneous and often complex group of patients requiring an individualized therapeutic approach. Detailed descriptions of case reports and HSCT outcomes are crucial to identify strategies to improve allo-HSCT outcomes and help delimit the target T-cell chimerism and thereby avoid disease flares. These strategies may include the use of targeted immunomodulators not only prior to but also post-HSCT. Furthermore, we need to define post-HSCT-specific monitoring and evaluate improvements of disease burden with specific scores. All of the foregoing is necessary to enrich the informed nature of the decision-making in lifelong management of children and adults with these diseases.
Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AM-S and ÀD-M: These authors share first authorship; AM-S, ÀD-M, and JT: These authors contributed equally to this work; FF-A and LA: These authors share last authorship. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the projects PI18/00223, FI19/00208 and PI21/00211 of LA, integrated in the Plan Nacional de I+D+I and cofinanced by the ISCIII – Subdirección General de Evaluación y Fomento de la Investigación Sanitaria – and the Fondo Europeo de Desarrollo Regional (FEDER), by Pla Estratègic de Recerca i Innovació en Salut (PERIS), Departament de Salut, Generalitat de Catalunya (SLT006/17/00199 to LA), by a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (IN[17]_BBM_CLI_0357) to LA, by a 2017 Beca de Investigación de la Sociedad Española De Inmunología Clínica Alergología y Asma Pediátrica to LA, and by a 2022 Convocatòria de Beques de Recerca IRSJD – Carmen de Torres 2022 to LA (2022AR-IRSJD-CdTorres) and CERCA Programme/Generalitat de Catalunya.
The authors gratefully acknowledge all team members, especially the nursery team, who dedicate their best efforts to taking care of transplant patients. In addition, we would like to thank Eva Rodriguez Nunez, Biomedical Scientist from the Anatomical Pathology Department. We also would like to thank both patients for allowing the publication of their case details.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1070068/full#supplementary-material
1. Chan AY, Torgerson TR. Primary immune regulatory disorders: A growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol (2020) 20(6):582–90. doi: 10.1097/ACI.0000000000000689
2. Cepika AM, Sato Y, Liu JMH, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol (2018) 142(6):1679–95. doi: 10.1016/j.jaci.2018.10.026
3. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol (2016) 36(1):33–45. doi: 10.1007/s10875-015-0224-7
4. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for malignancies in 131 affected CTLA4 mutation carriers. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02012
5. Gámez-Díaz L, Seidel MG. Different apples, same tree: Visualizing current biological and clinical insights into CTLA-4 insufficiency and LRBA and DEF6 deficiencies. Front Pediatr (2021) 9. doi: 10.3389/fped.2021.662645
6. Gámez-Díaz L, Grimbacher B. Immune checkpoint deficiencies and autoimmune lymphoproliferative syndromes. BioMed J (2021) 44(4):400–11. doi: 10.1016/j.bj.2021.04.005
7. Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: New genetic diseases of CTLA-4 checkpoint insufficiency. Blood (2016) 128(8):1037–42. doi: 10.1182/blood-2016-04-712612
8. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4–insufficient subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
9. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Sci (1979) (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
10. Egg D, Rump IC, Mitsuiki N, Rojas-Restrepo J, Maccari ME, Schwab C, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol (2022) 149(2):736–46. doi: 10.1016/j.jaci.2021.04.039
11. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol (2020) 145(5):1452–63. doi: 10.1016/j.jaci.2019.12.896
12. Flinn AM, Gennery AR. Primary immune regulatory disorders: Undiagnosed needles in the haystack? Orphanet J Rare Dis (2022) 17(1):99. doi: 10.1186/s13023-022-02249-1
13. Seidel MG, Böhm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol (2018) 141(2):770–775.e1. doi: 10.1016/j.jaci.2017.04.023
14. Arnold DE, Chellapandian D, Leiding JW. The use of biologic modifiers as a bridge to hematopoietic cell transplantation in primary immune regulatory disorders. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.692219
15. Shaw P, Shizuru J, Hoenig M, Veys P. Conditioning perspectives for primary immunodeficiency stem cell transplants. Front Pediatr (2019) 7. doi: 10.3389/fped.2019.00434
16. Deyà-Martínez A, Esteve-Solé A, Vélez-Tirado N, Celis V, Costa J, Cols M, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol (2018) 29(4):425–32. doi: 10.1111/pai.12890
17. Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transplant (2021) 56(9):2052–62. doi: 10.1038/s41409-021-01378-8
18. Dhalla F, Lochlainn DJm, Chapel H, Patel SY. Histology of interstitial lung disease in common variable immune deficiency. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.605187
19. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European society for immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract (2019) 7(6):1763–70. doi: 10.1016/j.jaip.2019.02.004
20. Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: Entering a new century, do we do better? J Allergy Clin Immunol (2010) 126(3):602–610.e11. doi: 10.1016/j.jaci.2010.06.015
21. Neven B, Ferrua F. Hematopoietic stem cell transplantation for combined immunodeficiencies, on behalf of IEWP-EBMT. Front Pediatr (2020) 7. doi: 10.3389/fped.2019.00552
22. Marçais A, Mahlaoui N, Neven B, Lanternier F, Catherinot É, Salvator H, et al. Curative allogeneic hematopoietic stem cell transplantation following reduced toxicity conditioning in adults with primary immunodeficiency. Bone Marrow Transplant (2022) 57(10):1520–30. doi: 10.1038/s41409-022-01739-x
23. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. New Engl J Med (2014) 371(5):434–46. doi: 10.1056/nejmoa1401177
24. Chan AY, Leiding JW, Liu X, Logan BR, Burroughs LM, Allenspach EJ, et al. Hematopoietic cell transplantation in patients with primary immune regulatory disorders (PIRD): A primary immune deficiency treatment consortium (PIDTC) survey. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00239
25. Toubai T, Mathewson ND, Magenau J, Reddy P. Danger signals and graft-versus-host disease: Current understanding and future perspectives. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00539
26. Ghimire S, Weber D, Mavin E, Wang Xn, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00079
27. Fürst D, Neuchel C, Tsamadou C, Schrezenmeier H, Mytilineos J. HLA matching in unrelated stem cell transplantation up to date. Transfusion Med Hemotherapy (2019) 46(5):326–36. doi: 10.1159/000502263
28. Cooper MA, Zimmerman O, Nataraj R, Wynn RF. Lifelong immune modulation versus hematopoietic cell therapy for inborn errors of immunity. J Allergy Clin Immunol Pract (2021) 9(2):628–39. doi: 10.1016/j.jaip.2020.11.055
29. Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol (2016) 138(2):615–619.e1. doi: 10.1016/j.jaci.2016.01.045
30. Seidel MG, Tesch VK, Yang L, Hauck F, Horn AL, Smolle MA, et al. The immune deficiency and dysregulation activity (IDDA2.1 ‘Kaleidoscope’) score and other clinical measures in inborn errors of immunity. J Clin Immunol (2022) 42(3):484–98. doi: 10.1007/s10875-021-01177-2
31. Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, et al. Long-term outcome and lineage-specific chimerism in 194 patients with wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood (2011) 118(6):1675–84. doi: 10.1182/blood-2010-11-319376
32. Booth C, Gaspar HB, Thrasher AJ. Gene therapy for primary immunodeficiency. Curr Opin Pediatr (2011) 23(6):659–66. doi: 10.1097/MOP.0b013e32834cd67a
33. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol (2013) 132(5):1150–5. doi: 10.1016/j.jaci.2013.05.031
34. Burns SO, Morris EC. How I use allogeneic HSCT for adults with inborn errors of immunity. Blood (2021) 138(18):1666–76. doi: 10.1182/blood.2020008187
35. Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: A prospective multicentre study. Lancet (2014) 383(9915):436–48. doi: 10.1016/s0140-6736(13)62069-3
36. Albert MH, Hauck F, Wiebking V, Aydin S, Notheis G, Koletzko S, et al. Allogeneic stem cell transplantation in adolescents and young adults with primary immunodeficiencies. J Allergy Clin Immunol Pract (2018) 6(1):298–301.e2. doi: 10.1016/j.jaip.2017.07.045
37. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190(1):1–7. doi: 10.1111/cei.12997
38. Yang L, Xue X, Chen X, Wu J, Yang X, Xu L, et al. Abatacept is effective in Chinese patients with LRBA and CTLA4 deficiency. Genes Dis (2021) 8(5):662–8. doi: 10.1016/j.gendis.2020.03.001
39. Lee S, Moon JS, Lee CR, Kim HE, Baek SM, Hwang S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol (2016) 137(1):327–30. doi: 10.1016/j.jaci.2015.08.036
40. Krausz M, Uhlmann A, Rump IC, Ihorst G, Goldacker S, Sogkas G, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: A non controlled phase 2 clinical trial. Contemp Clin Trials Commun (2022) 30:101008. doi: 10.1016/j.conctc.2022.101008
41. Liesveld JL, Rothberg PG. Mixed chimerism in SCT: conflict or peaceful coexistence? Bone marrow transplant. (2008) 42(5):297–310. doi: 10.1038/bmt.2008.212
42. Bakhtiar S, Fekadu J, Seidel MG, Gambineri E. Allogeneic hematopoietic stem cell transplantation for congenital immune dysregulatory disorders. Front Pediatr (2019) 7. doi: 10.3389/fped.2019.00461
43. Laberko A, Gennery AR. Clinical considerations in the hematopoietic stem cell transplant management of primary immunodeficiencies. Expert Rev Clin Immunol (2018) 14(4):297–306. doi: 10.1080/1744666x.2018.1459189
44. Gennery AR, Lankester A. Long term outcome and immune function after hematopoietic stem cell transplantation for primary immunodeficiency. Front Pediatrics Front Media S.A (2019) 7. doi: 10.3389/fped.2019.00381
45. Cavazzana-Calvo M, Carlier F, le Deist F, Morillon E, Taupin P, Gautier D, et al. Long-term T-cell reconstitution after hematopoietic stem-cell transplantation in primary t-cell–immunodeficient patients is associated with myeloid chimerism and possibly the primary disease phenotype. Blood (2007) 109(10):4575–81. doi: 10.1182/blood-2006-07-029090
46. Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol (2017) 139(3):1046–9. doi: 10.1016/j.jaci.2016.09.040
47. Seidel MG, Böhm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol (2018) 141(2):770–775.e1. doi: 10.1016/j.jaci.2017.04.023
48. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol (2018) 141(2):704–717.e5. doi: 10.1016/j.jaci.2017.03.049
49. Hossain MS, Kunter GM, El-Najjar VF, Jaye DL, Al-Kadhimi Z, Taofeek OK, et al. PD-1 and CTLA-4 up regulation on donor T cells is insufficient to prevent GvHD in allo-HSCT recipients. PloS One (2017) 12(9):e0184254. doi: 10.1371/journal.pone.0184254
50. Zhu F, Zhong XM, Qiao J, Liu Q, Sun HY, Chen W, et al. Cytotoxic T lymphocyte antigen-4 down-regulates T helper 1 cells by increasing expression of signal transducer and activator of transcription 3 in acute graft-versus-Host disease. Biol Blood Marrow Transplantation (2016) 22(2):212–9. doi: 10.1016/j.bbmt.2015.11.003
Keywords: CTLA-4, primary immunodeficiency, hematopoietic stem cell transplantation, abatacept, immune reconstitution, chimerism
Citation: Margarit-Soler A, Deyà-Martínez À, Canizales JT, Vlagea A, García-García A, Marsal J, Del Castillo MT, Planas S, Simó S, Esteve-Sole A, Grande MS-L, Badell I, Tarrats MR, Fernández-Avilés F and Alsina L (2022) Case report: Challenges in immune reconstitution following hematopoietic stem cell transplantation for CTLA-4 insufficiency-like primary immune regulatory disorders. Front. Immunol. 13:1070068. doi: 10.3389/fimmu.2022.1070068
Received: 14 October 2022; Accepted: 28 November 2022;
Published: 27 December 2022.
Edited by:
Irina A. Tuzankina, Institute of Immunology and Physiology (RAS), RussiaReviewed by:
Barbara Piątosa, Children’s Memorial Health Institute (IPCZD), PolandCopyright © 2022 Margarit-Soler, Deyà-Martínez, Canizales, Vlagea, García-García, Marsal, Del Castillo, Planas, Simó, Esteve-Sole, Grande, Badell, Tarrats, Fernández-Avilés and Alsina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana Margarit-Soler, YW1hcmdhcml0c29sZXJAZ21haWwuY29t; Laia Alsina, bGFpYS5hbHNpbmFAc2pkLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.