
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 12 January 2023
Sec. Cytokines and Soluble Mediators in Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1067885
 Paola Antonello1,2
Paola Antonello1,2 Diego U. Pizzagalli1,3
Diego U. Pizzagalli1,3 Mathilde Foglierini1,4
Mathilde Foglierini1,4 Serena Melgrati1,2
Serena Melgrati1,2 Egle Radice1Sylvia Thelen1
Egle Radice1Sylvia Thelen1 Marcus Thelen1*
Marcus Thelen1*Chemotaxis is an essential physiological process, often harnessed by tumors for metastasis. CXCR4, its ligand CXCL12 and the atypical receptor ACKR3 are overexpressed in many human cancers. Interfering with this axis by ACKR3 deletion impairs lymphoma cell migration towards CXCL12. Here, we propose a model of how ACKR3 controls the migration of the diffused large B-cell lymphoma VAL cells in vitro and in vivo in response to CXCL12. VAL cells expressing full-length ACKR3, but not a truncated version missing the C-terminus, can support the migration of VAL cells lacking ACKR3 (VAL-ko) when allowed to migrate together. This migration of VAL-ko cells is pertussis toxin-sensitive suggesting the involvement of a Gi-protein coupled receptor. RNAseq analysis indicate the expression of chemotaxis-mediating LTB4 receptors in VAL cells. We found that LTB4 acts synergistically with CXCL12 in stimulating the migration of VAL cells. Pharmacologic or genetic inhibition of BLT1R markedly reduces chemotaxis towards CXCL12 suggesting that LTB4 enhances in a contact-independent manner the migration of lymphoma cells. The results unveil a novel mechanism of cell-to-cell-induced migration of lymphoma.
Cell migration is a widely conserved biological process, but the involved intracellular pathways are poorly understood. Directional migration or chemotaxis is orchestrated by distinct cell surface receptors, which guide cells along gradients of increasing concentrations of attractants (1). Chemokines, which constitute the largest family of chemoattractants, share a basic isoelectric point that allows them to be presented on cell surfaces and extracellular matrices (2, 3). They induce chemotaxis by binding to cognate G-protein-coupled receptors (GPCRs), which upon ligand binding activate via pertussis toxin-sensitive G proteins downstream pathways leading to cell migration (4, 5). In addition to conventional G-protein coupled chemokine receptors, chemokines bind to seven-transmembrane domain receptors, which do not activate G-proteins, called atypical chemokine receptors (ACKR) (3–5). ACKR3 acts mainly as a decoy with high affinity for the chemokine CXCL12 and with lower affinity for CXCL11, which are also ligands for the conventional receptors CXCR4 and CXCR3, respectively (6, 7). The physiological importance of the CXCL12/ACKR3/CXCR4 axis is underlined by the fact that absence of either component causes perinatal death in mice severely disrupting central nervous system and heart development (8–11).
Chemotaxis is governed by a chemokine-producing source, often constituted by stromal cells, and a juxtaposed sink that removes chemokines, either cells that express ACKRs or other mechanisms of degradation. Removal of chemoattractants is essential to maintain gradients and to avoid blurring of the source. Chemokine gradients are established over a relatively short distance of about 100 to 150 µm (12). Therefore, chemotaxis is not involved in long distance cell displacements as observed in passive blood-flow mediated cell movement (13).
In neoplasms, the chemokine system plays a promiscuous role depending on the type of cancer, the cellular and environmental context, and the involved chemokine axis. The chemokine system can influence disease progression in a beneficial or detrimental way (14–21). In many solid cancers altered expression levels of ACKR3 correlate with poor prognosis and tumor metastasis (22–29). In addition, the CXCL12/ACKR3/CXCR4 axis was proposed to be involved in lymphoma (30–32). Diffuse large B cell lymphoma (DLBCL) arise in the germinal center when B cells undergo somatic hypermutation and class-switch recombination. During these events, genetic alterations can cause malignant tumor transformation leading to GC-derived B cell lymphoma (33). We have previously shown that GC derived DLBCL VAL cells, require the functional expression of ACKR3 to disseminate into distant organs in NOD/SCID mice (32).
Tumor spreading is a multistage process where cancer cells undergo transformation, leave the primary tumor and migrate into adjacent and distant tissues, where they proliferate (34–38). It was previously assumed that cancers metastasize through single tumor cells escaping from the primary site arriving at distant tissues and propagating there. However, in the last decade, emerging evidence suggests that metastases can disseminate with a more complex mechanism called collective cell migration (39). This process describes the presence of leading cells that trace the way into adjacent tissues for the migration of follower cells (40) or induce the movement of cohesive clusters (39). Collective migration has been extensively studied to describe the metastasis of solid tumors but is seldom used to describe the movement of hematological cancers in a contact independent manner. Through chemokine sequestration expression of scavenger receptors can contribute to the formation of self-generated gradients (40, 41).
The mechanisms by which ACKR3, CXCR4 and CXCL12 induce the dissemination of VAL cells in a synergetic manner have not been addressed. Here, we demonstrate that in the presence of CXCL12 wild type VAL cells (VAL-wt), which can express ACKR3 on the cell surface, create cues that in vitro can be sensed by CXCR4+ follower VAL-wt. Moreover, VAL-wt support in vitro chemotaxis of VAL cells in which ACKR3 was genetically deleted (VAL-ko). The latter by themselves are not able to migrate in response to CXCL12. In vivo VAL-wt can induce the migration of CXCR4+ VAL-ko, contributing to the infiltration of draining lymph nodes. Using a novel computational analysis of 3D migration, we show that in the presence of CXCL12 VAL-wt stimulate cell-to-cell-induced migration presumably through the release of leukotriene B4 (LTB4). Moreover, the release of LTB4 from VAL cells induces cell polarization and chemotaxis in an autocrine and paracrine fashion. We propose a novel mechanism by which ACKR3 sustains the movement of otherwise migration incompetent cells and partially reveal the molecular components of this pathway.
The atypical chemokine receptor ACKR3 is known to be upregulated on both solid cancers and blood neoplasms (22, 24, 30, 31). Here we investigated its expression and functionality on two DLBCL lines, VAL and DOHH2. Flow cytometry was performed on wild type DLBCL cells and VAL cells in which ACKR3 was genetically ablated (VAL-ko). In the cells with deleted ACKR3 cyan fluorescent protein (CFP) was introduced under constitutively active spleen focus-forming virus (SFFV) promoter, which allows to easily identify cells in tissues and mixed cultures (32). We measured surface expression on these cells of ACKR3, CXCR4 and CCR7. All cells expressed similar levels of CXCR4 on the plasma membrane (Figure 1A). Conversely, ACKR3 was expressed only on a fraction of VAL-wt (10-18%), despite their clonal nature and continuous mRNA levels of the receptor (32). When cells were sorted for ACKR3 surface expression and cultured, the mixed phenotype reestablished rapidly within 2-3 weeks. However, cells negative for ACKR3 at sorting were more refractory for acquiring ACKR3 surface expression during this time (Figure S1A). However, during prolonged culture (6 weeks) the cell established the initial phenotype. Similarly, only a small fraction of DOHH2 expressed ACKR3 on the plasma membrane (Figure 1A). Interestingly, CCR7 was predominantly expressed on the surface of wild type DLBCL (VAL-wt and DOHH2), which were negative for ACKR3 plasma membrane expression. Genetic deletion of ACKR3 on VAL cells had no effect on CXCR4 expression. Surprisingly, CCR7 transcripts were less frequent in VAL-ko (117 ± 23 vs. 898 ± 32, (n=5)) and surface expression was markedly reduced on VAL-ko (Figure 1A).
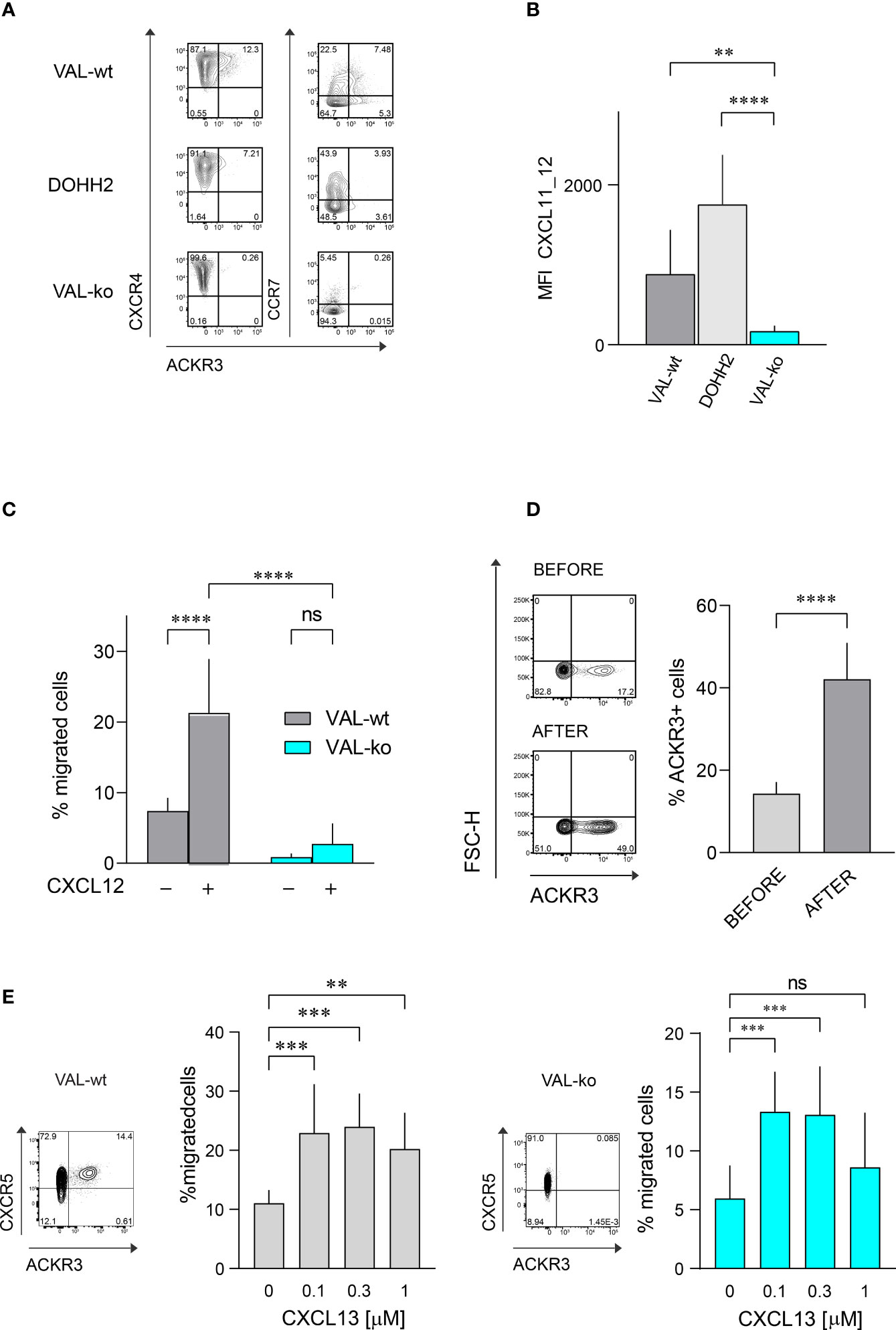
Figure 1 Chemokine receptor expression and function on DLBCL cells. (A) ACKR3 (mAb 11G8), CXCR4 (mAb 12G5) and CCR7 (mAb G043H7) surface expression on VAL-wt, DOHH2, and VAL-ko was measured by flow cytometry. Representative plots of three independent experiments. (B) Uptake of the chimeric chemokine CXCL11_12 AF647 by DLBCL. Mean fluorescence intensity (MFI) of DLBCL incubated with 50 nM of CXCL11_12 for 45 min at 37°C. MFI (by flow cytometry) reports the mean ± SD (Dunnett’s multiple comparisons, ONE-WAY ANOVA **p ≤ 0.01, ****p ≤ 0.0001) of four independent experiments performed in triplicates. (C) Transendothelial migration of lymphoma cells. VAL cell migration through mLEC towards 10 nM CXCL12 was measured after 8h incubation. Cell migration was calculated as percentage of the input (% migrated cells). Cumulative data from three independent experiments measured in triplicates (means ± SD, Turkey’s multiple comparison ONE-WAY ANOVA ns (not significant), p≥0.05, ****p ≤ 0.0001). (D) (Left) ACKR3 (mAb 11G8) surface expression on VAL-wt cells before (upper panel, before) and after transendothelial migration (lower panel, after) towards 10 nM CXCL12 as in (C). Representative plots from ten independent experiments performed in duplicates. (Right) Percentage of cells expressing ACKR3 on the plasma membrane before and after migration. Cumulative data from five different experiments measured in triplicates (means ± SD, unpaired t-test, ****p ≤ 0.0001). (E) Left side: flow cytometry plot showing ACKR3 (mAb 11G8) and CXCR5 (mAb J252D4) surface expression on VAL-wt (left panel). Transendothelial migration of VAL-wt towards increasing concentrations of CXCL13 (0.1, 0.3 and 1 µM) (bar chart). Right side: flow cytometry plot showing ACKR3 (mAb 11G8) and CXCR5 (mAb J252D4) surface expression on VAL-ko (left panel). Transendothelial migration of VAL-ko through mLEC towards increasing concentrations of CXCL13 (0.1, 0.3 and 1 µM) (bar chart). Cell migration was calculated as described above. Bar charts show cumulative data of three independent experiments performed in triplicates (means ± SD, Dunnett’s multiple comparisons, ONE WAY ANOVA, **p ≤ 0.01, ***p ≤ 0.001).
To assess the functionality of ACKR3 we took advantage of a previously developed specific chemokine uptake assay (42). CXCL11_12 AF647 is a fluorescent chimeric chemokine side-specific labeled at the C-terminus, composed of the N-terminus of CXCL11 and the main body of CXCL12. The chimera specifically binds ACKR3, but not CXCR3 or CXCR4, the respective cognate receptors for CXCL11 and CXCL12. VAL-wt and DOHH2 readily internalized the chimera (Figure 1B). Hence, despite the limited surface expression of ACKR3 (Figure 1A), the receptor appears functional on both DLBCL lines. The observation is in agreement with the notion that the receptor cycles between the plasma membrane and endocytic compartments (6). As expected, VAL-ko did not scavenge the chimera and displayed low unspecific binding (Figure 1B and Figure S1B). Chemokine scavenging and receptor internalization requires the intact C-terminus of ACKR3 (6). We reintroduced into VAL-ko ACKR3 with a truncated C-terminus by stable transfection resulting in VAL-ACKR3-ΔC cells (VAL-ACKR3-ΔC). VAL-ACKR3-ΔC poorly scavenged CXCL11_12 AF647 indicating that the C-terminus of ACKR3 is required for scavenging and receptor internalization (Figures S1B, C), consistent with previous findings (6, 43).
Next, we assessed the chemotactic response towards CXCL12. Chemotaxis assays were performed as previously described with transwell plates coated with a monolayer of endothelial cells grown on a collagen-based extracellular matrix (32). The presence of endothelial cells optimizes the migration of lymphoma cells. VAL cells were allowed to migrate through a layer of mouse lymphatic endothelial cells (mLEC) for 8h in the absence and presence of an optimal concentration of CXCL12 (10 nM) in the lower compartment. In agreement with our previous findings (32), under these conditions about 20% of VAL-wt migrated into the lower compartment in the presence of chemokine, which accounts for a 3-fold increase compared to unstimulated chemokinesis. By contrast, VAL-ko, which express equal surface levels of CXCR4 showed no response to CXCL12, but also no spontaneous migration (Figure 1C). The observation was confirmed with genetically different clones to exclude off-target effects on cell migration through editing of ACKR3 by CRISPR/Cas9. Chemokinesis of VAL cells through mLEC was not inhibited in the presence of the CXCR4 antagonist AMD3100, suggesting a basal migratory activity that was not mediated by CXCR4 (Figure S1D). Similarly, VAL-ACKR3-ΔC, which express comparable CXCR4 levels on the surface (Figure S1Ei) did not perform CXCL12-dependent migration (Figure S1Eii). The observation suggests that the C-terminus is necessary for ACKR3 activity during VAL cell migration.
We next compared the surface expression of ACKR3 on VAL-wt before and after migration towards CXCL12. The fraction of cells, which expressed ACKR3 at the plasma membrane increased from about 17% to 40-50% after chemotaxis (Figure 1D). The observation could indicate that cells, which express ACKR3 at the plasma membrane, migrate more efficiently. However, it cannot be excluded that cells upregulate surface expression of the receptor during migration. To get a better insight we sorted ACKR3+ and ACKR3- VAL cells and analyzed their migratory properties in chemotaxis assays. The efficiency of cells expressing ACKR3 at the surface was notably higher compared to cells that did not present ACKR3 at the plasma membrane. Moreover, VAL-wt lacking ACKR3 surface expression did not upregulate the receptor during migration (Figure S1F). Nevertheless, in ACKR3 negative VAL-wt the mRNA of the receptor is not absent compared to the genetically modified VAL-ko (32). Hence, it cannot be excluded that ACKR3 expressed on endosomal structures somehow contributes to the migration. All together, these findings suggest that functional ACKR3 plasma membrane expression is necessary to support efficiently CXCR4-dependent cell migration.
In order to exclude a general inhibitory effect on VAL cell migration by ACKR3 deletion we examined the chemotaxis towards CXCL13, the ligand for CXCR5, which is expressed on both, VAL-wt and VAL-ko (Figure 1E). With increasing concentrations of CXCL13 we observed a typical bell-shaped migration response by both cell types (Figure 1E), indicating that VAL-ko retain the capability to sense a chemokine gradient, polarize and migrate. In addition, we explored the ability of VAL-wt and VAL-ko to migrate in response to stimulation of CCR7 with CCL19. VAL-wt, which heterogeneously express CCR7 (Figure 1A), were sorted for CCR7 surface expression and tested separately. CCR7+ VAL-wt, which do not express ACKR3 on the surface (Figure 1A), migrated more efficiently than the CCR7- ACKR3+ sorted cells (Figure S1G). The data indicate that the moderate chemotaxis towards CCL19 correlates with CCR7 surface expression. In line with this, VAL-ko cells, which do not express CCR7 (Figure 1A) did not migrate towards CCL19 (Figure S1G).
The observation that VAL-wt, which do not express ACKR3 on the surface, can migrate towards CXCL12 in the presence of cells exposing the receptor on the plasma membrane was intriguing (Figure 1D). Previous data showed that in zebrafish ACKR3 expressed in trans on stromal cells can support the CXCL12-mediated migration of primordial germ cells (44). It is plausible that ACKR3 positive VAL-wt, when locally scavenging CXCL12, shape micro-gradients that are sensed by CXCR4+ VAL cells that do not express ACKR3 on the surface. The hypothesis implies that ACKR3 scavenging activity is necessary for VAL cell migration. To address the assumption, we used mLEC transwell assays to measure CXCL12-induced chemotaxis of VAL-ko in the presence of VAL-wt. We mixed VAL-wt and CFP+ VAL-ko in a 1:1 ratio, which enables the identification of the two cell types by flow cytometry. The mixture was allowed to migrate towards different concentrations of CXCL12 (Figure 2A). CFP+ VAL-ko in the 1:1 mixture with VAL-wt showed a typical bell-shaped migratory response towards CXCL12, whereas VAL-ko cells alone poorly responded (Figure 2A). Moreover, in the mixture, the CFP+ VAL-ko cells migrated with a similar efficiency as VAL-wt cells (Figures 2A, B). Next, we addressed the role of the C-terminus of ACKR3 in promoting the migration of VAL-ko. VAL-ACKR3-ΔC, barely supported VAL-ko migration towards CXCL12 in a 1:1 mixture, highlighting the importance the C-terminus for scavenging (Figure S1C), migration (Figure S1Eii) and stimulation in trans (Figure 2B). It is conceivable that the scavenging activity is essential for chemotaxis of ACKR3 expressing cells and to support migration of VAL-ko, however it cannot be excluded that the C-terminus mediates additional intracellular signaling unrelated to receptor trafficking. The later assumption is in line with previous findings (9) where it was shown that phosphorylation-deficient (C-terminus) ACKR3 does not scavenge chemokines but rescues the lethal phenotype of receptor-deficient mice.
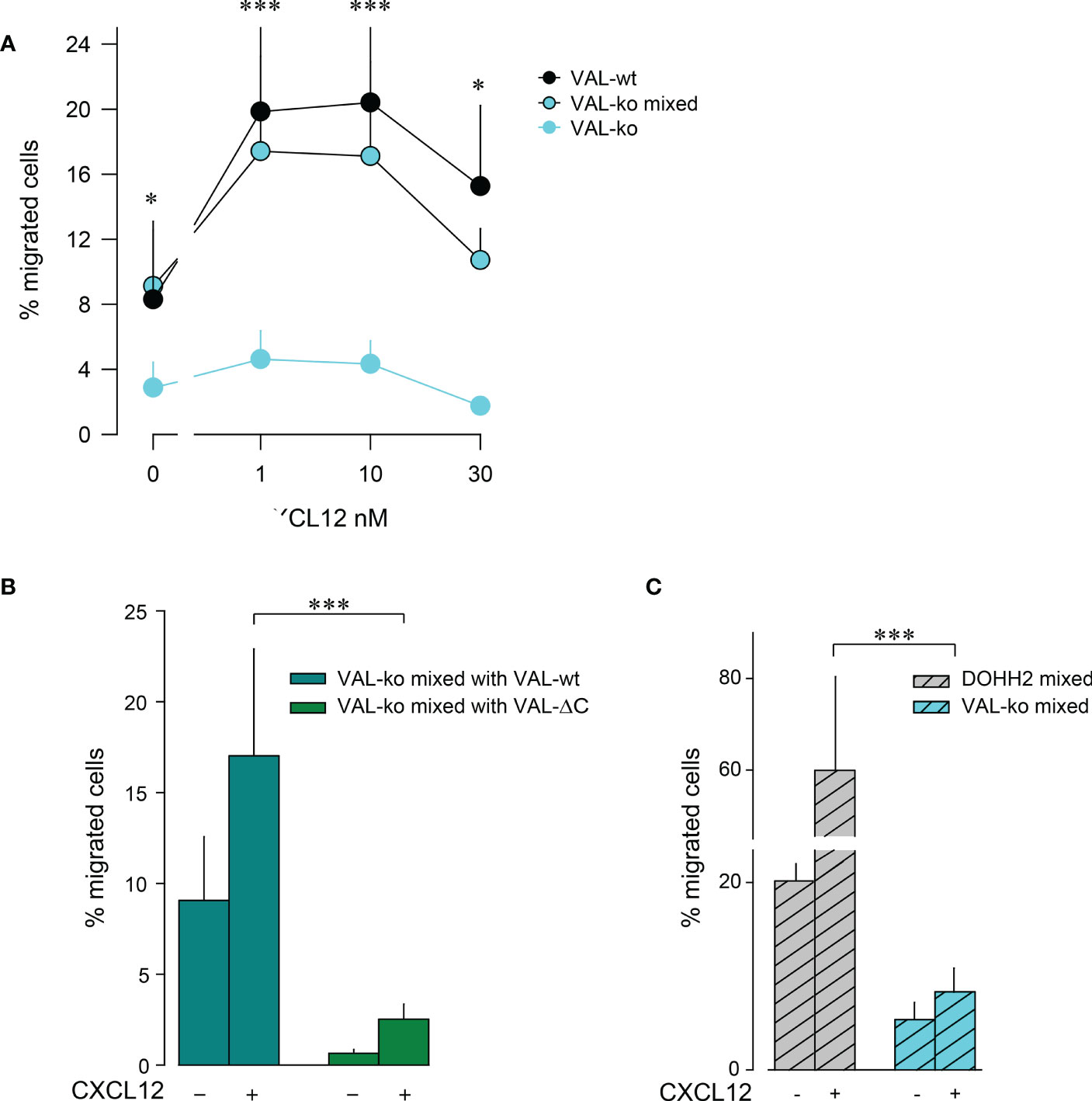
Figure 2 VAL-wt cells induce the migration of VAL-ko cells. (A) Transendothelial migration of VAL cells (as in Figure 1) towards different concentrations of CXCL12 (1, 10, 30 nM). Cyan symbols and cyan lines (VAL-ko) displays % migrated VAL-ko in the absence of VAL-wt. The % migrated VAL-ko (gated on CFP) in a 1:1 mixture with VAL-wt was calculated from the input of VAL-ko in the mixture (VAL-ko mixed, cyan filled symbols and black lines). Black symbols: % migrated VAL-wt calculated from the respective input. Calculation by flow cytometry as in Figure 1. Cumulative data of three independent experiments measured in triplicates (means ± SD, Dunn’s multiple comparisons ONE WAY ANOVA, *p ≤ 0.05, ***p ≤ 0.001) (B) Transendothelial migration of VAL cells towards 10 nM CXCL12 or medium. VAL-ko were mixed 1:1 with VAL-wt (dark cyan) or VAL-ΔC (green) and the % migrated VAL-ko was calculated from the input of VAL-ko in the mixture. Quantification was performed as described in Figure 1. Cumulative data of three independent experiments measured in triplicates (means ± SD, Turkey’s multiple comparison, ONE WAY ANOVA, ***p ≤ 0.001). (C) Migration of DOHH2 cells. DOHH2 cells were mixed 1:1 with VAL-ko and the mixture allowed to migrate for 8 h towards 10 nM CXCL12 or medium. The % migrated DOHH2 (grey bars) and of VAL-ko (cyan bars) were calculated from the respective inputs in the mixture. Experiments performed in triplicates, representative example of three independent experiments (means ± SD, Turkey’s multiple comparison, ONE WAY ANOVA ***p ≤ 0.001).
To extend the observation of DLBCL migration we measured mLEC transwell chemotaxis of DOHH2 cells in response to CXCL12. DOHH2 cells migrated more efficiently than VAL-wt, but also displayed enhanced chemokinesis. When DOHH2 cells were mixed with VAL-ko cells they induced chemokinesis of VAL-ko cells and barely supported chemotaxis towards CXCL12 (Figure 2C). Taken together, the data suggest that expression of full-length ACKR3 on leader cells is needed to induce the CXCL12-dependent cell-to-cell-induced migration of CXCR4+ VAL-ko.
We used a previously reported localized tumor xenograft model in NOD/SCID mice (32) to investigate lymphoma cell migration in vivo. We injected 107 lymphoma cells subcutaneously in the flanks of NOD/SCID mice. All animals showed a comparable increase in body weight and tumor size (Figure 2SA). After 20-21 days, animals were sacrificed and the mass of the primary tumor was measured. No significant differences in mass were observed between tumors grown from wild type DLBCL, VAL-wt cells and DOHH2, VAL-ko and the respective mixtures of wild type and VAL-ko (Figure 3A). NOD/SCID mice barely developed sizable lymph nodes due to the lack of endogenous lymphocytes. Injection of wild type DLBCL cells led to measurable mass of the draining lymph node. By contrast, the draining lymph nodes of tumors from VAL-ko remained hardly detectable (Figure 3A). However, an increase in lymph node mass was observed, when 1:1 mixtures of wild type DLBCL and VAL-ko were injected (Figure 3A). To test whether under these conditions VAL-ko contributed to the increase in mass of the draining lymph node, we used flow cytometry to measure the presence of CFP+ VAL-ko. While VAL-ko alone did not migrate to the axillary lymph node, in the presence of VAL-wt or DOHH2 the CFP+ VAL-ko could readily be detected (Figure 3B). Hence, like in vitro, also in vivo wild type lymphoma cells appeared to promote the migration of ACKR3 deficient VAL-ko. In the mixture preferentially ACKR3 positive VAL-wt migrated to the draining lymph node along with VAL-wt, which do not express ACKR3 at the surface and CFP+ VAL-ko (Figure 3C). Consistent with our in vitro observations (Figure 1D) the proportion of VAL-wt expressing ACKR3 on the surface was markedly increased from 18% to over 50% in the draining lymph node. By contrast, the expression of CCR7 was unaltered on VAL-wt and remained essentially absent on CFP+ VAL-ko, suggesting that the receptor may not contribute to draining lymph node infiltration (Figure S2B).
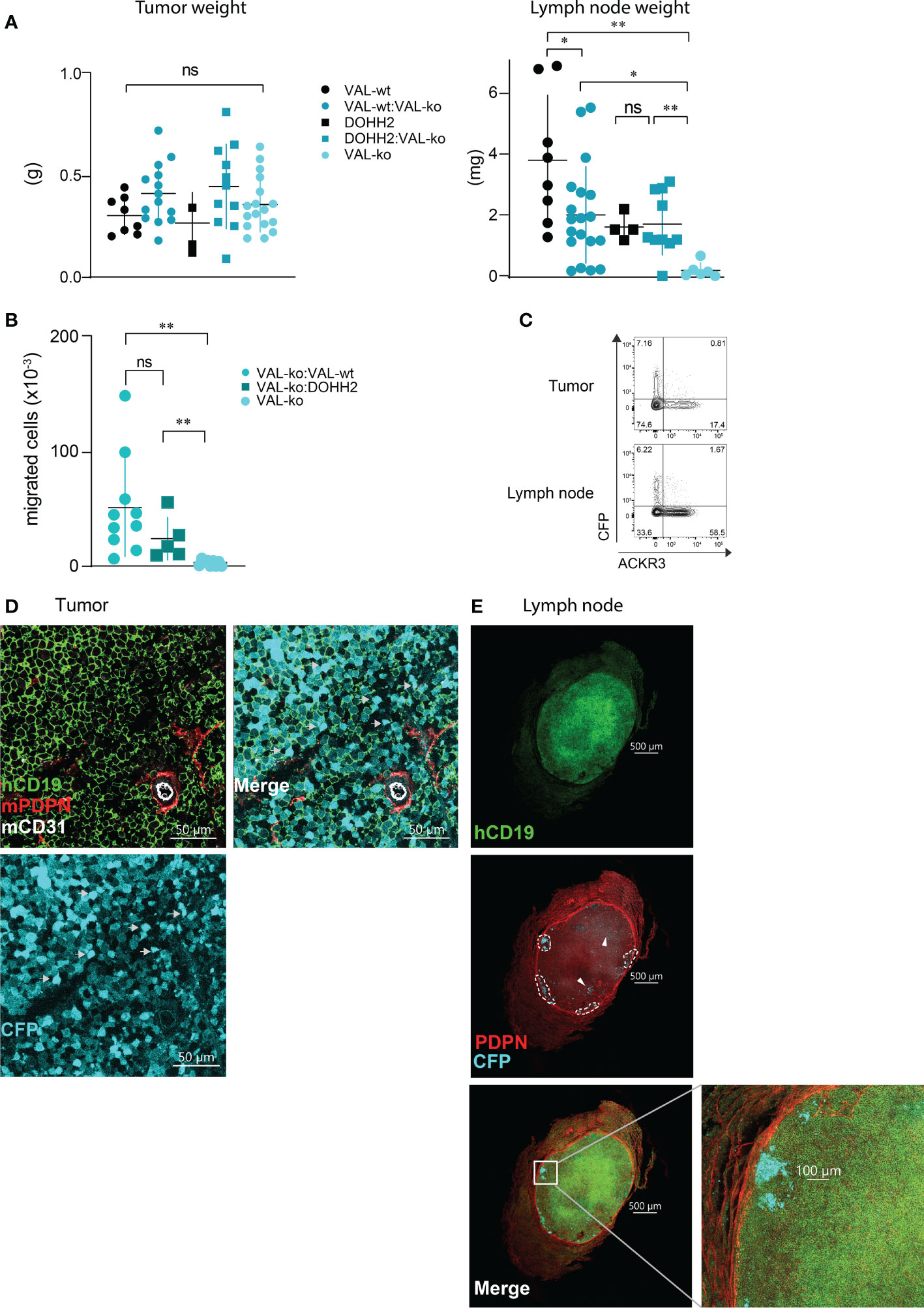
Figure 3 Migration of VAL-ko from the primary tumor to the draining lymph node is enhanced in the presence of ACKR3 expressing cells. (A) Tumor (left) and lymph node (right) weights from NOD/SCID common γ-chainko mice injected with 107 VAL-wt, VAL-ko and DOHH2 cells alone and as 1:1 mixture VAL-wt:VAL-ko and DOHH2:VAL-ko. Weights are reported as means ± SD from three independent xenograft experiments with 4-5 animals per group. Tumor and lymph node t-test single comparison: ns, p≥0.05, *p≤ 0.05, **p ≤ 0.01. (B) Total number of CFP+ VAL-ko present in the draining axillary lymph node from the same experiments. Lymph node cells were analyzed by flow cytometry and gated for hCD19+ (HIB19 or SJ25C1), mCD45- (30-F11) and CFP+ expression (means ± SD, t-test single comparison, ns, p≥0.05 **P ≤ 0.01). (C) Surface expression of ACKR3 on VAL-wt cells and CFP expression in VAL-ko extracted from a representative localized tumor (upper panel) and from the corresponding lymph node (lower panel). Representative data from one experiment shown in A (n= 4-5). (D) Confocal images of sections of a tumor generated by injecting a 1:1 mixture of VAL-wt and VAL-ko (CFP+). Tumors were fixed for 48h in a solution of 4% paraformaldehyde and Vibratome sections stained overnight with anti-hCD19 (mAb HIB19) anti-mPDPN (mAb 8.1.1) and anti-mCD31 (mAb MEC13.3). (E) Confocal images of sections of a draining lymph node from a tumor generated by injecting a 1:1 mixture of VAL-wt and VAL-ko (CFP+). Lymph nodes were fixed for 5h in a solution 4% paraformaldehyde and Vibratome sections were stained overnight with anti-hCD19 Ab (mAb HIB19), anti-mPDPN (mAb 8.1.1).
Immunofluorescence (IF) analysis of sections from tumors of VAL-wt:VAL-ko mixtures by confocal microscopy revealed that VAL-wt and VAL-ko are randomly distributed in the tumor mass. Using podoplanin (PDPN) and CD31 staining for mouse lymphatic and endothelial vessels we found a similar localization for both cell populations (Figure 3D). By contrast, IF analysis of axillary lymph node sections from mice bearing VAL-wt:Val-ko tumors revealed striking differences in the distribution of the two cell populations. Human CD19+ VAL-wt almost completely populated the entire draining lymph node. By contrast, CFP+ VAL-ko were found at the edge of the lymph node, close to the sub-capsular sinus in clusters surrounded by VAL-wt cells (Figure 3E). Taken together, the data indicate that like in vitro lymphoma cells can also promote the migration of VAL-ko in vivo.
To investigate further the migration and the cooperation between cells, we used a 3D migration and quantitative computational (45) analysis system. Cells were embedded in a collagen matrix and chemotaxis towards CXCL12 was followed by time-lapse imaging (Figure 4A). Tracks of migrating cells were generated automatically by computational analysis and used to extrapolate data of statistical significance to calculate displacement, track length and velocity of single cells. Single cell data were pooled into experimental groups for comparison (Figure 4B). The analysis confirmed that VAL-ko are unable to migrate towards CXCL12 (Figure S3A) while the mixture of migrating VAL-ko:VAL-wt is mobile and migration competent (Figure S3B). Displacement and track length of VAL-ko in the mixture is comparable to that of VAL-wt (Figure 4B). However, cells in the mixture appeared to migrate with an intermediate velocity being faster than VAL-ko cells alone, but slower than VAL-wt cells (Figure 4B). Similarly, plotting the tracks on common origin revealed that (i) VAL-wt in the mixture are highly motile, (ii) VAL-ko essentially do not move, while (iii) the migratory capacity of VAL-ko is partially restored in the presence of VAL-wt (Figure 4C). The results are consistent with the transwell assays and the in vivo data, demonstrating that the presence of ACKR3 is essential to promote migration in cis and in trans.
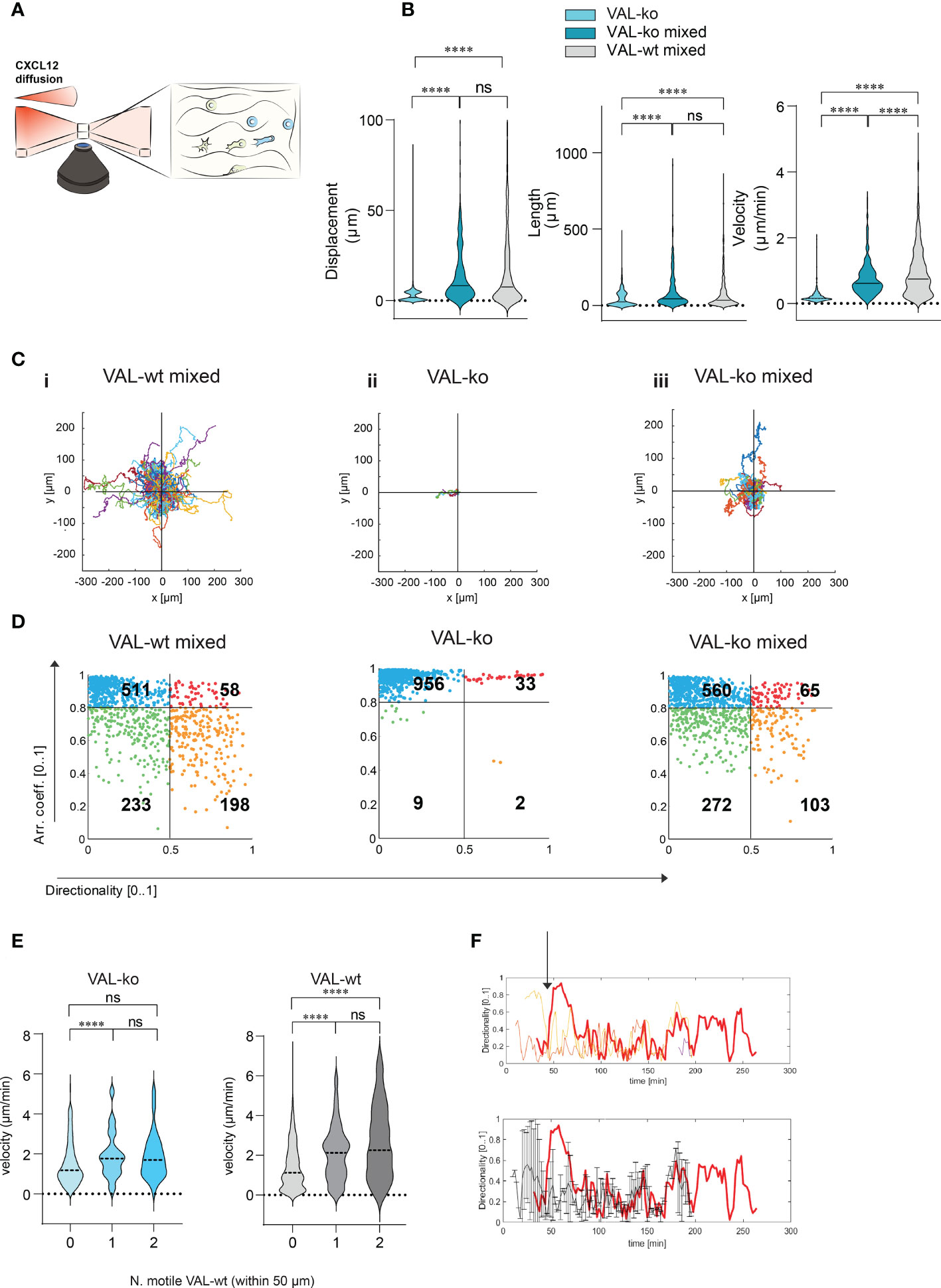
Figure 4 CXCL12 stimulated VAL-wt increase the 3D motility and directional migration of VAL-ko. (A) Schematic representation of 3D migration chamber showing a chemotactic gradient (CXCL12 diffusion) from left to right. (B) Violin plots showing displacement, length and velocity of VAL-ko alone (light cyan), VAL-ko in a 1:1 mixture with VAL-wt (VAL-ko mixed, dark cyan) and VAL-wt in the same mixture (VAL-wt mixed, grey). Tracking analyses were performed using Imaris software (means, Turkey’s multiple comparisons test, ONE-WAY ANOVA ns, p≥0.05, ****p ≤ 0.0001). (C) Plots of tracks with a common origin. (i) VAL-wt mixed, (ii) VAL-ko alone, (iii) VAL-ko mixed. (D) Scatter plots of directionality vs arrest coefficients of 1000 VAL-wt mixed, VAL-ko alone, VAL-ko mixed. Gates with differently colored dots indicate four different motility patterns. (E) Violin plots of changes in velocity of VAL-ko and VAL-wt, relative to the number of motile (0, 1, 2) VAL-wt in a 50 µm radius (means, Mann-Whitney t-test ns, p≥0.05, ****p ≤ 0.0001). (F) Directionality changes over time of a VAL-ko (red thick line) with respect to directionality changes of two VAL-wt in a 50 µm radius (orange and red thin lines, upper plot). The lower plot shows the same directionality changes of a single VAL-ko (red thick line), compared to the average of directionality value of VAL cells in the 50 µm radius.
To better characterize differences in motility, we analyzed the behavior of individual cells over time. We applied an algorithm using mobility values to generate plots that distinguish four populations according to their migratory behavior. We quantified instantaneous motility measures over time, by dividing each track into fragments (tracklets), and performed a tracklet-based action analysis as previously described (46). From the motility measures, we computed the continuous arrest coefficient (46), that is the proportion of time during which a cell does not move. Cells with low motility are associated with high values of arrest coefficient. Moreover, we computed the instantaneous directionality (46), which was associated with high values for polarized cells migrating along straight trajectories. We assigned these coefficients to 1000 tracklets of each group: VAL-wt and VAL-ko in the mixture (separately analyzing CFP- and CFP+ cells) and VAL-ko alone (Figure 4D). For VAL-wt, in a 1:1 mixture with VAL-ko, 233 tracklets displayed low arrest coefficients and low directionality, which indicates that these cells perform chemokinesis (green gate), whereas 511 tracklets were associated with immobile cells, characterized by tracklets with a high arrest coefficient and low directionality (blue gate). Tracklets of cells, which protruded lamellipods, hence visibly polarized, but showed slow or no migration (red gate 58 tracklets), were defined by a high arrest coefficient and high directionality (deduced from the polarity). Finally, 198 tracklets (orange gate) displayed low arrest coefficients and high directionality and were associated with cells that performed a pronounced directional migration. VAL-ko when placed alone in the chamber were found to be mostly immobile (956 tracklets in the blue gate). However, VAL-ko in the mixture displayed similar motility as VAL-wt with slightly less directional migration ability (103 vs. 198 tracklets). Our algorithm applied to a large number of tracklets confirmed the motility of VAL-wt and their ability to promote the migration of VAL-ko.
We further analyzed whether the velocity of VAL-ko and VAL-wt migration depends on a motile VAL-wt in the direct neighborhood. Figure 4E shows that a VAL-ko in the presence of at least one motile VAL-wt within a radius of 50 µm migrated with significantly higher velocity compared to VAL-ko where no motile VAL-wt were in close proximity. We observed that the increase in velocity of migration of VAL-ko was similar whether one or two motile VAL-wt were in the neighborhood, suggesting no additive effect. The same analysis performed for the velocity of VAL-wt revealed that the motility of the cells was also influenced by the presence of a motile VAL-wt within a radius of 50 µm. The finding suggested that VAL-ko and VAL-wt might perform a secondary migration, which could be triggered either by microgradients of CXCL12 created by ACKR3+ VAL-wt or by another stimulus that is released during the migration of VAL-wt along a CXCL12 gradient.
By analyzing in depth the tracks of VAL-ko we saw that the cells often migrate to a spot where a VAL-wt had briefly paused and left (Movies S1-3). In addition, we measured the changes in directionality of VAL-ko and VAL-wt in close proximity and plotted the data over time (Figure 4F). We found that the changes in directionality of VAL-ko were in most cases preceded by a change in VAL-wt directionality (Figure 4F upper panel). Moreover, the migration of a VAL-ko was similar to the overall movement of motile cells in a radius of 50 µm (Figure 4F, lower panel). The observations support the assumption that the migration of VAL-ko depends on the movement of VAL-wt, in line with the hypothesis that DLBCL invade tissues through cell-to-cell-induced migration as a heterogeneous population of cells.
Pertussis toxin (PTX) treatment inhibits the transmigration of VAL-wt towards CXCL12 but has no effect on the VAL cell chemokinesis (Figure S4A). We next mixed PTX-pretreated VAL-wt in a 1:1 ratio with untreated VAL-ko and subjected them to transmigration assays towards CXCL12. As expected, inhibition of GiPCRs in VAL-wt abolished their migration towards CXCL12 in the mixture with VAL-ko (Figure 5Ai). Moreover, pretreatment of VAL-wt cells with PTX significantly reduced their ability to promote the migration of untreated VAL-ko cells (Figure 5Aii). To investigate the possible involvement of a Gi-protein coupled chemotactic receptor in the migration of VAL-ko cells, we preincubated VAL-ko with pertussis toxin and mixed them with untreated VAL-wt. Preincubation of VAL-ko with PTX inhibited their migration (Figure 5Aiii), suggesting that the migration of VAL-ko in the presence of VAL-wt depends on the activation of a chemotactic Gi/o-protein coupled receptor.
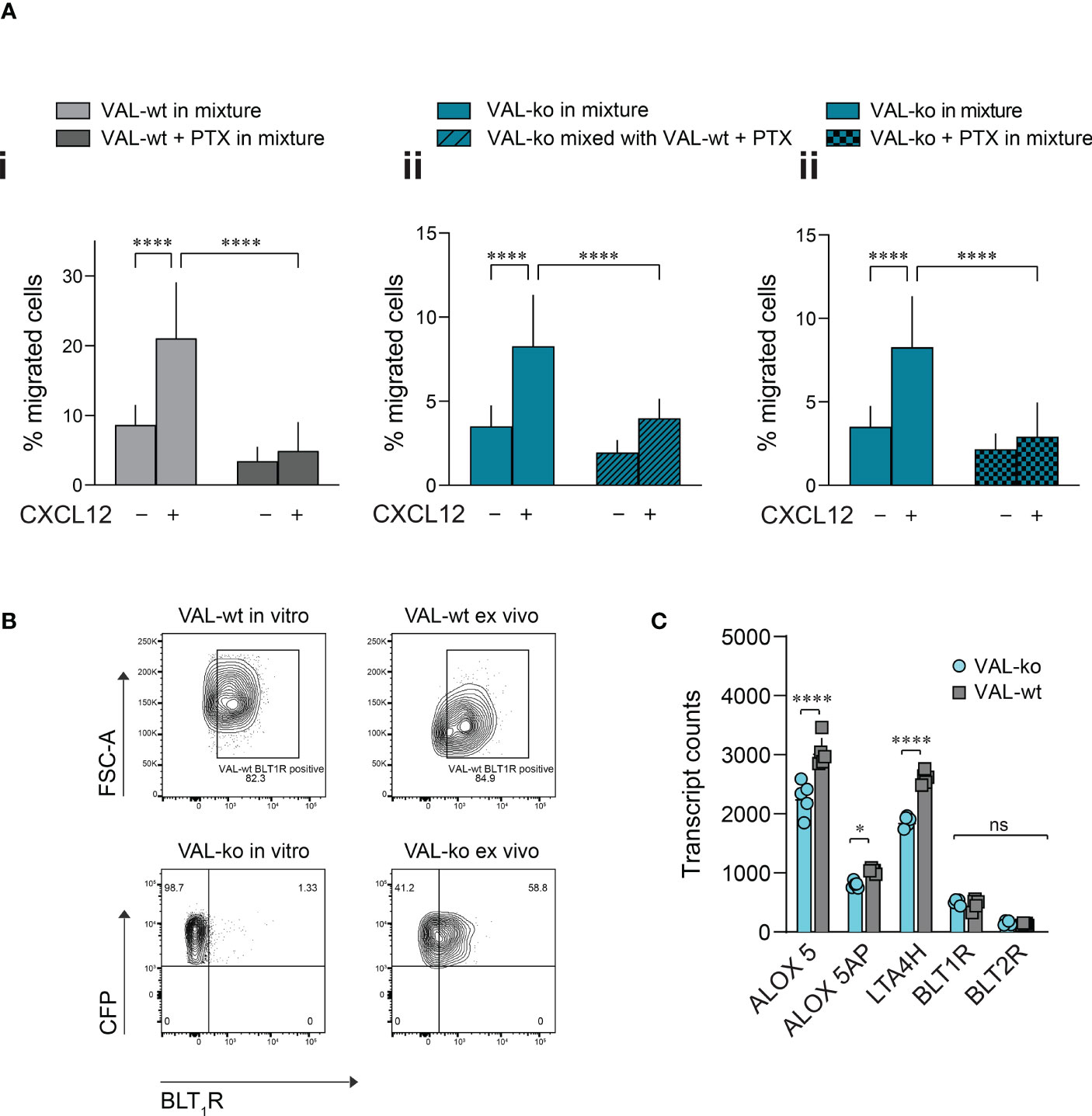
Figure 5 VAL-ko migration in the presence of VAL-wt is PTX sensitive. (A) Transmigration towards 10 nM CXCL12 was performed and calculated as described in Figure 1. (i) VAL-wt untreated (light grey) or treated with PTX (dark grey) in the mixture with untreated VAL-ko. (ii) VAL-ko mixed with untreated (left, plain bars) or PTX-treated VAL-wt (right, hatched bars). (iii) VAL-ko untreated (left, plain bars) or PTX-treated (right, hatched bars) mixed with untreated VAL-wt. Cumulative data of three independent experiments performed in triplicates (means ± SD, Turkey’s multiple comparisons test, ONE WAY ANOVA, ****p ≤ 0.0001). (B) BLT1R (mAb 203/14F11) surface expression on VAL-wt (upper panels) and VAL-ko (lower panel) cultured in vitro (left panels) or ex vivo (right panels). Representative plots of three different flow cytometry analyses. (C) Transcript levels of the lipoxygenases ALOX5 and ALOX5AP, the hydrolase LTA4H and the LTB4 receptors BLT1R and BLT2R of VAL-wt and VAL-ko (means ± SD, Šídák multiple comparison, 2way ANOVA ns p≥0.05, *p ≤ 0.05, ****p ≤ 0.0001) ns (not significant).
To identify such chemotactic receptor, we performed RNA-seq analyses of the transcriptomes of VAL-wt and VAL-ko. Comparing the data revealed that in general the expression levels of GPCRs transcripts showed no gross differences between the two cell types (Figure S4B). Of the known chemotactic receptors, the transcripts of CCR7 were higher expressed in VAL-wt (log2 fold 2.9) in agreement with our FACS data (Figure 1A, Figure S4B). GPR183, also known as EBI2, a chemotactic receptor for dihydroxysterols (47–49) was markedly higher expressed in VAL-wt (log2 fold 4.1) (Figure S4B). However, we could not observe a chemotactic response of VAL cells towards dihydroxysterols (not shown). Both cell types expressed similar transcript levels of CXCR5 and CXCR4 (Figure S4B) in agreement with the flow cytometry data and the chemotaxis experiments. In addition, we found similar levels of transcripts for two Gi-protein coupled receptors for leukotriene B4 (LTB4), namely BLT1R (BLT1R) and BLT2R (BLT2R) (Figure S4B), although the transcripts of both receptors were less frequent than those of chemokine receptors. The transcript levels of both receptors were similar in VAL-wt and VAL-ko, however BLT2R transcripts were slightly higher in both cell types. BLT1R is well known to mediate chemotaxis of leukocytes in response to LTB4 (50), while the ubiquitously expressed BLT2R is more involved in regulating inflammatory responses, rather than cell migration (51, 52). We therefore measured the surface expression of BLT1R on VAL-wt and VAL-ko by flow cytometry. Despite similar transcript levels, VAL-wt expressed higher levels of BLT1R than VAL-ko in in vitro culture (Figure 5B). Interestingly, VAL-ko extracted from local tumors of NOD/SCID mice, showed a marked increase of surface BLT1R surface expression (up to 60% positive cells) (Figure 5B), suggesting that the tumor microenvironment promoted the upregulation of surface BLT1R, either from intracellular stores or via de novo translation. VAL-wt ex vivo did not display changes at surface expression of BLT1R compared to VAL-wt cultured in vitro (Figure 5B). The transcriptome analysis indicated further that several critical enzymes of the LTB4 synthesis pathway, such as the hydrolase LTA4H, the lipoxygenases ALOX5 and ALOX5AP were significantly more frequent in VAL-wt compared to VAL-ko (Figure 5C). We also sorted DOHH2 cells for ACKR3 surface expression and measured the transcriptomes. Both populations (ACKR+ vs ACKR-) expressed comparable levels of the transcripts for BLT1R (424.6 ± 66 vs 332 ± 56.5), LTA4H (3012 ± 102.7 vs 2894 ± 208.5), ALOX5 (1870 ± 154 vs 1819 ± 237) and ALOX5AP (688.2 ± 154 vs 708.1 ± 236) like VAL-wt.
The expression of enzymes involved in LTB4 production suggested that CXCL12 and LTB4 could act synergistically in stimulating VAL-wt migration through a positive feedback loop. Moreover, it could explain why cells, which are in the proximity of a LTB4 secreting cell, respond more efficiently to CXCL12 (Figure 4E). To test the possible synergistic involvement of the BLT1R in cell-to-cell-induced migration of VAL cells, we measured transwell chemotaxis of VAL-wt and VAL-ko towards CXCL12 in the presence of the BLT1R inhibitor, BIIL315. BIIL315 markedly attenuated CXCL12-stimulated migration of VAL-wt. Moreover, the inhibitor abolished VAL-wt-mediated migration of VAL-ko in response to CXCL12 (Figure 6A). The observation suggested the involvement of the LTB4/BLT1R axis in the cell-to-cell-induced migration of VAL cells. However, the reduced migration of VAL-ko could also result from the lower number of VAL-wt migrating in the mixture. To measure LTB4-stimulated chemotaxis, we embedded VAL cells in a collagen matrix coating the transwell to exclude metabolic interference of an endothelial cell layer. In general, under these conditions chemotaxis of VAL cells was less efficient. We observed that LTB4 alone did not induce migration of VAL cells (Figure 6B). However, CXCL12- stimulated chemotaxis was markedly enhanced in the presence of LTB4. Similarly, CXCL12-induced migration of DOHH2 cells was markedly enhanced in the presence of LTB4 (Figure S4C). The synergistic effect of LTB4 was impaired in the presence of the BIIL315 confirming the involvement of BLT1R (Figure 6B).
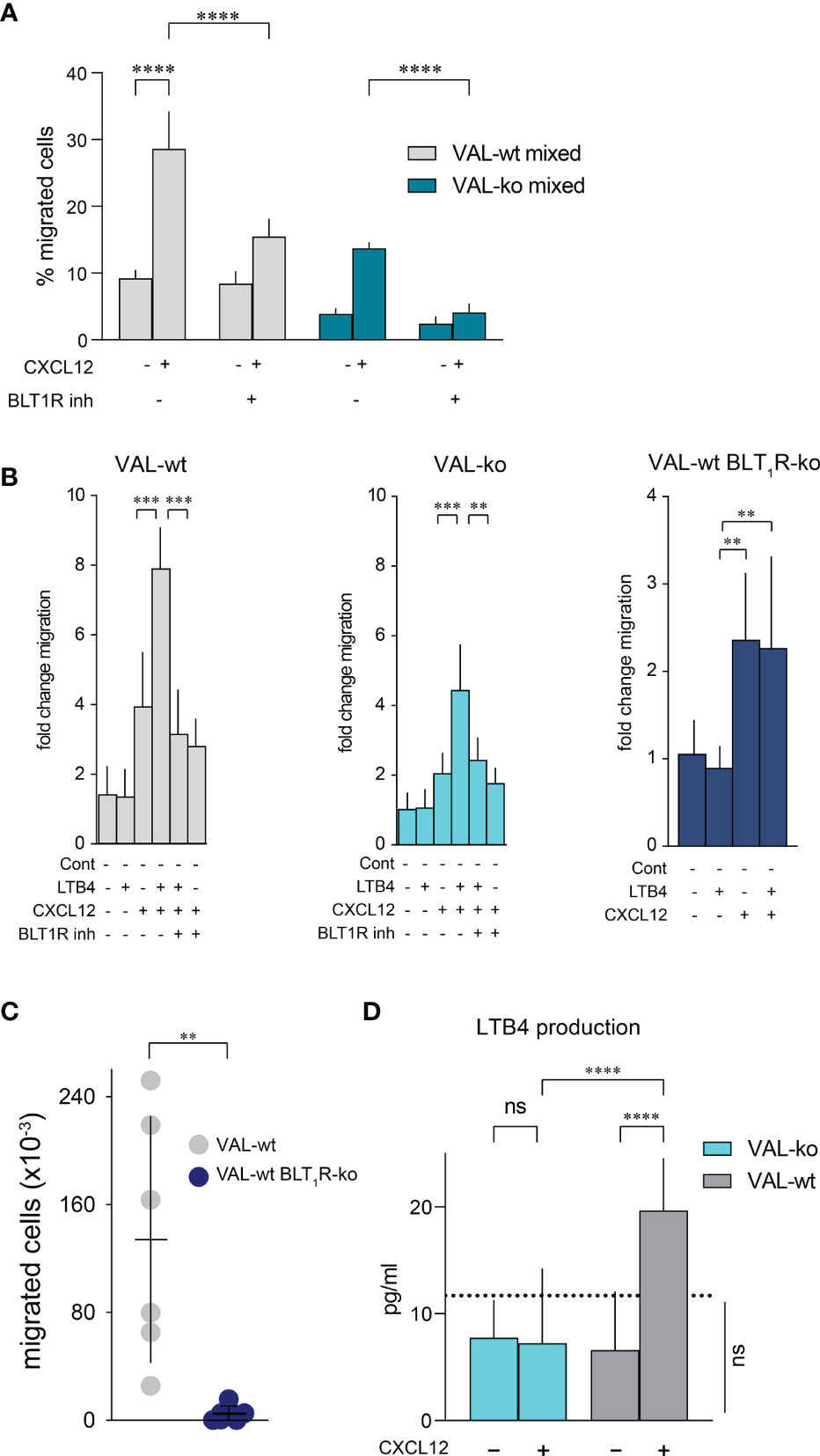
Figure 6 CXCL12 and LTB4 synergistically induce the migration of DLBCL. (A) Transmigration through mLEC was performed and calculated as described in Figure 1. Migration towards 10 nM CXCL12 of VAL-wt in a 1:1 mixture with VAL-ko (VAL-wt mixed, grey bars) and VAL-ko in a 1:1 mixture with VAL-wt (VAL-ko mixed, dark cyan bars) was measured in the absence and presence of 1 µM of the BLT1R inhibitor, BIIL315. Cumulative data of three independent experiments performed in triplicates (means ± SD, ONE WAY ANOVA, Turkey’s multiple comparisons test, ****p ≤ 0.0001). (B) VAL cells were embedded in a collagen matrix on the transwell insert. Fold change of VAL-wt (left, grey bars), VAL-ko (middle, cyan bars) and VAL-wt with deleted blt1r gene (right, blue bars) migrating in response to 10nM LTB4 and 10 nM CXCL12 in the absence and presence of 1 µM BLT1R inhibitor (BIIL315) as indicated. Cumulative data from three experiments performed in quadruplicates (means ± SD, Student’s t-test **p ≤ 0.01 ***p ≤ 0.001). (C) Total number of VAL-wt (gray dots) and VAL-wt lacking BLT1R (dark blue dots) present in the draining axillary lymph node. Lymph node cells were analyzed by flow cytometry and gated for hCD19+ (HIB19 or SJ25C1) and mCD45- (30-F11) expression (means ± SD, t-test single comparison, **p ≤ 0.01). (D) LTB4 production (pg/mL) by VAL-wt and VAL-ko in absence or presence of 10 nM CXCL12. Values below the dashed line are under the sensitivity level. Cumulative data of three independent experiments performed in duplicates or quadruplicates (mean ± SD, ONE WAY ANOVA, Turkey’s multiple comparison test, ****p ≤ 0.0001).
To further confirm the synergism between BLT1R and CXCR4 signaling we deleted the blt1r gene on all alleles in VAL-wt using CRISPR/Cas9 technology (Figure S4D) and measured transwell migration in response to CXCL12 and LTB4. Deletion of the blt1r gene completely abrogated the LTB4-mediated enhancement of CXCL12-stimulated migration (Figure 6B). By contrast, CXCR13-stimulated migration of VAL-wt, VAL-ko and VAL-wt with deletion of blt1r was not enhanced in the altered of LTB4 (Figure S4E). Together the observations indicate a synergistic effect of LTB4 on CXCR4/CXCL12- mediated VAL cell migration in vitro (Figure 6B). To confirm the relevance of the finding in vivo we injected VAL-wt and VAL-wt BLT1R ko (cells lacking BLT1R) into the flanks of mice. Tumors from both cell lines grew equally and had similar weights on the day of sacrifice (Figure S4F). However, while the draining axillary lymph nodes were readily detected in animals injected with VAL-wt, these were essentially absent when mice were inoculated with VAL-wt BLT1R ko cells (Figure 6C). The results are in line with the in vitro observations indicating that LTB4/BLT1R signaling is required for cell-to-cell-induced migration.
We next measured the release of LTB4 by VAL-wt and VAL-ko cells. Stimulation of CXCR4 with CXCL12 leads to the PTX-sensitive activation of phosphoinositide lipase C and via phospholipase A2 to the formation of arachidonic acid, a critical precursor of LTB4. Inhibiting 5-lipoxigenase with Demethylnobiletin blocks LTB4 formation (53). In line with the induction of cell-to-cell-induced migration, CXCL12 stimulated in VAL-wt a significant over background (~ 10 pg/mL) release of LTB4 (Figure 6D). By contrast, LTB4 production was not observed in VAL-ko and was fully inhibited by treatments with PTX or Demethylnobiletin (data not shown).
We demonstrate here that CXCR4-mediated migration of lymphoma cells towards CXCL12 requires the expression of functional ACKR3 at the cell surface. The conclusion is further supported by the finding that VAL-wt, which express ACKR3 at the surface, enhance the migration of cells that do not express ACKR3 at the plasma membrane, but contain the transcripts for the receptor, a finding that is reproducible both in vitro and in vivo. Moreover, VAL-wt can induce CXCL12-dependent chemotaxis of otherwise migration-deficient VAL-ko in vitro. The same stimulatory effect of ACKR3+ VAL cells is also observed in vivo, where VAL-wt promote lymph node infiltration of VAL-ko. These observations indicate that ACKR3 can act in trans, not necessarily being expressed on the same cell as the chemotaxis mediating CXCR4. Computer-assisted quantitative analysis of 3D migration hinted the presence of a secondary chemotactic cue created by VAL-wt, which induces the migration of VAL-ko. RNAseq data revealed the presence of transcripts for the expression of the chemotactic receptor BLT1R. In fact, we demonstrate that the BLT1R ligand LTB4 supports CXCR4-mediated chemotaxis acting synergistically with CXCL12 in triggering VAL cell chemotaxis. Moreover, upon stimulation with CXCL12, VAL-wt secrete LTB4, hence creating a positive feedback loop for contact-independent cell-to-cell-induced migration.
ACKR3 surface expression is heterogeneous on VAL cells. In culture the majority of cells do not express the receptor on the plasma membrane, despite the presence of similar levels of transcripts in receptor positive and negative cells. It is plausible that in ACKR3- cells the receptor may reside in endosomes. In order to support CXCR4-mediated migration, however, ACKR3 has to be expressed on the cell surface. Removing the C-terminus of ACKR3 causes its plasma membrane localization on all cells, but the truncation renders the receptor incapable to enhance CXCR4-dependent chemotaxis. The finding suggests that the C-terminus is not only involved in receptor localization, trafficking and chemokine scavenging, but is also part of a signaling cascade leading to LTB4 production. Genetic ablation of ACKR3 in VAL cells almost completely abrogates their chemotactic response to CXCL12, albeit continuous expression of CXCR4. The inhibition of CXCR4/CXCL12 signaling in VAL-ko was not observed when chemotaxis was stimulated via the CXCL13/CXCR5 axis, indicating a selective interference of ACKR3 in the CXCR4 signaling pathway.
Heterodimerization of chemokine receptors has been suggested to alter their respective activities (54–57). However, functional interaction of chemokine receptors may depend on the cellular context. It was reported that CCR7 inactivates CXCR4 (56) while in another report CXCR4 expression was shown to be required for stable CCR7 expression (58). Our observation that CCR7 is downregulated in VAL-ko despite unaltered CXCR4 expression may indicates that ACKR3 is involved in CCR7 expression. On the other side it has been proposed that ACKR3 can form heterodimers on the plasma membrane with CXCR4 altering its activity (8, 59). We demonstrate here that ACKR3 expression in trans is sufficient to support CXCR4-mediated chemotaxis of VAL-ko in vitro and in vivo. Furthermore, our 3D time-lapse migration analysis strongly supports the view that the CXCR4-stimulated migration of ACKR3-deficient cells is contact independent. Interestingly, in many instances, regardless of ACKR3 expression, cells arrest at spots where a leader cell stopped before and typically followed the direction. The data further support a mechanism of cell-to-cell-induced migration that is contact-independent. In line with these observations is the finding that cells in a radius of 50 µm are sufficient to enhance the migration of slow-moving cells. Our data suggest that LTB4 could be the mediator of the contact-independent enhancement of CXCL12-stimulated migration of ACKR3-deficent cells. In fact, LTB4 synergistically enhances CXCL12-mediated migration of VAL cells and pharmacologic inhibition of BLT1R or genetic abrogation of the receptor reverts the effect. The assumption is in agreement with reports showing that B-cells can secrete LTB4 (60, 61) and with studies showing that neutrophils secrete LTB4 to induce swarming. Nevertheless, the LTB4-mediated migration reported here differs from neutrophil swarming over long distances since the effect of LTB4 appears to be confined to less than 50 µm (62, 63). Moreover, it was reported that LTB4 upregulates chemotactic receptors in leukocytes promoting chemotaxis toward their respective ligands (64), confirming the presence of a positive feed-back between LTB4 release and chemotaxis. The involvement of LTB4 in the recruitment of neutrophils in combination with other chemoattractants to inflamed joints was also shown (65, 66). Our data suggest that the synergistic effect of LTB4 and CXCL12 is specific for CXCR4-induced chemotaxis since LTB4 has no effect on CXCR5-mediated migration of VAL-wt. Nevertheless, we cannot fully exclude that CXCL12 scavenging by ACKR3 creates microgradients, which somehow modify CXCR4/CXCL12 signaling on ACKR3 negative cells. Further studies are required to describe the molecular mechanism that supports the release of LTB4 upon ACKR3/CXCR4 stimulation.
Using a diffuse xenograft model we showed that spreading of VAL cells into tissues, such as brain, bone marrow and spleen, requires ACKR3 expression (32). Given the poor prognosis of DLBCL patients with brain infiltrations (67, 68), understanding the underlying mechanism is of great clinical relevance. Here we report that ACKR3 is required for DLBCL to escape from local tumors to move to draining lymph nodes resembling the spread of malignant cells from lumps or extra nodal sites. Hence, targeting ACKR3-mediated tumor dissemination is appealing. Our data showing the involvement of the LTB4/BLT1R axis in VAL cell migration, could open a way for the development of novel therapeutic approaches in the treatment of DLBCL, potentially in combination with inhibitors of ACKR3 (69–72).
VAL and DOHH2 cells were cultured in RPMI-1640 supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS), 1% Penicillin/Streptomycin (P/S), 1% GlutaMAX. 1% NEAA, 1% Sodium-Pyruvate and 50 µM β-mercaptoethanol all from Thermo Fisher. Primary murine lymphatic endothelial cells (mLEC) were isolated from lymph nodes of C57BL/6 mice and cultured in αMEM (Thermo Fisher) supplemented with 10% heat-inactivated FBS, 1% P/S. Cells were cultured in humidified air at 37° C with 5% CO2.
Freshly harvested lymph nodes were harvested from C57BL/6 mice, and incubated in digestion mixture containing RPMI-1640, 0.25 mg/mL Liberase TL (Roche, Cat: 5401020001), 200 Kunits/mL DNAse I (Sigma-Aldrich, Cat: 10104159001) at 37° C for 1h with occasional shaking. Single cell suspensions were strained using a 70 µm cell strainer (Corning), centrifuged and resuspended in αMEM. Cells were plated on 6-well plates coated with 10 µg/mL PureCol (Collagen, Sigma-Aldrich) and human 10 µg/mL plasma Fibronectin (Sigma-Aldrich). After 3 hours non-adhering cells were removed. 5-7 days later, when >80% confluence was achieved, cells were detached using Accutase (Biological Industries). Under these conditions blood endothelial cells die and the remaining cells are composed of fibroblastic reticular cells (FRCs) (~40%) and LECs. LECs were purified by staining with APC-conjugated anti-mouse CD31 (Biolegend, clone 390) and positively selected using anti-APC microbeads according to manufacturer’s instructions (Miltenyi Biotec).
ACKR3 and the C-terminus deficient variant ACKR3-ΔC (6) were cloned into CDH-IF1-MCS-COP-GFP vector and nucleofected (Lonza) into CFP+ VAL-ko (32) giving rise to VAL-wt rescue and VAL-ACKR3-ΔC. The transfected constructs expressed GFP after a T2A cleavage sequence, which was used for clonal selection by flow cytometry and single cell sorting (FACS-ARIA).
CRISPR/Cas-9 ribonucleoproteins (crRNPs) targeting blt1r gene were delivered to VAL-wt cells by transfection with NEON nucleofector (Invitrogen, MA, USA) at 1’600 V, 10 ms, 3 pulses, using the provided buffer R. 106 cells were used with the 10-μl tip transfection. The crRNAs (/AltR1/rCrArU rGrArG rUrCrU rArGrA rCrCrG rCrUrC rArCrG rUrUrU rUrArG rArGrC rUrArU rGrCrU/AltR2/”), tracrRNAs (IDT, USA) and HiFi Cas9 Nuclease V3 (IDT, USA) transfection was performed as indicated in the manufacturer’s instruction (IDT, USA). Deletion of blt1r was confirmed by genomic sequencing of the locus.
For flow cytometry cells were suspended in MACS buffer (phosphate buffered saline (PBS), 2% FBS, 2mM EDTA) and stained for 15 minutes on ice with the indicated antibodies. For chemokine uptake, 50 nM of fluorescently labelled chemokine (42, 73) was added to cells for 45 min at 37°C. Mean fluorescence intensity (MFI) of the fluorophore were determined by flow cytometry (Fortessa or FACSCanto (BD) and FlowJo software.
Lymphoma cell migration was measured in transmigration assays using 5 µm transwell inserts. mLECs were seeded on polycarbonate inserts coated with 10 µg/mL fibronectin and 10 µg/mL collagen. Monolayers of endothelial cells were grown by adding medium to the upper compartment, leaving the lower compartment empty.
For LTB4-induced migration lymphoma cells were embedded in a collagen matrix formed by 1.6 mg/mL PureCol, 0.36% PBS supplemented with 0.36% FBS, 0.036% P/S, 1.5 µg/mL recombinant human ICAM-1/CD54 Fc chimera (R&D systems) at 4° C. To induce collagen polymerization the temperature was slowly raised over 45 min to 37°C. Then medium was added on the upper compartment of the transwell inserts for 24 h before the experiment. When indicated, embedded cells were pretreated with 1 µM BIIL315 (OpnMe-Boehringer Ingelheim) for one hour at 37°C. The medium of the upper compartment was then replaced with fresh medium containing 1 µM BIIL315, which was also present in the lower compartment during the experiment.
Lymphoma cells were pretreated with 10 µM AMD3100 (Sigma-Aldrich) for one hour at 37° C and transwell migration was performed in the presence of the same concentration of the inhibitor in the upper and lower compartment. Cells were treated with 2 µg/mL pertussis toxin (PTX) (Sigma-Aldrich) for two hours at 37°C., washed and added to inserts coated with endothelial cells without toxin.
Migration was stimulated with chemoattractants at the indicated concentrations in the lower compartment. Cells were allowed to migrate to the lower chamber for 8 hours. After a brief centrifugation (1 min, 250 x g) of the inserts, cells were collected by centrifugation and resuspended in 100 µL of MACS buffer. For cell number determination cells were counted for 50 sec by flow cytometry.
Cells were resuspended at 106 cells mL-1 in PBS and incubated for 30 minutes on ice. Cells were centrifuged at 350g for 3 minutes, re-suspended at 107 cells mL-1 in plain RPMI and incubated for 30 minutes at 37°C. Secretion of LTB4 was stopped by centrifugation at 4° C (350g for 3 minutes) and supernatants collected. LTB4 ELISA was performed as indicated in the manufacturer’s instruction (Enzo Life Sciences).
Lymphoma cell migration in 3D chemotaxis were performed using µ-Slide Chemotaxis (Ibidi). Cells were embedded in a collagen matrix as described above. To optimize and standardize the architecture of the collagen fibers we slowly induced polymerization by precooling and extending the warming rate from 4°C (liquid) to 37°C (solid) of the collagen matrix (74) (75). The IBD chambers were loaded at 4°C and slowly warmed up to room temperature (20 min) before placing into a 37°C cell incubator. Total warming time to 37°C was about 40 min. The condition allows the formation of thin fibers with homogenous diameter, length and density and prevents the formation of fibers in hierarchical structures (74) (75). After polymerization complete medium (65 µL) was added on both side-reservoirs. Cells embedded in the collagen matrix in the µ-Slide chamber were incubated in a humidified environment at 37°C and 5% CO2 for one day. After 24 hours 15 µL of 400 nM CXCL12 were added to one of the reservoirs and migration observed for 6 hours by time-lapse video microscopy (ImageXpress® Molecular Devices) at 2 minutes or 20 seconds time intervals between frames.
The localized mouse xenograft model was described previously (32). 8–10-week-old mice were injected subcutaneously with 107 VAL or DOHH2 cells in PBS, or only PBS for control groups. All in vivo experiments were performed following the rules of the Swiss Federal Veterinary Office guidelines and authorized by the Animal Studies Committee of Cantonal Veterinary.
A day before culling 50 µL of Evans Blue (2 µg/mL) were injected subcutaneously on the flank close to the xenograft do visualize lymphatic vessels. Localized tumors and draining axillary lymph node were surgically removed and disintegrated with 150 µm Sefar Nitex filters and cells processed for flow cytometry analysis as described (76). Cells of the disintegrated lymph node were resuspended in 150 µL of MACS buffer and analyzed by flow cytometry.
Tumors and lymph nodes were collected and fixed with a solution of 4% paraformaldehyde and washed with PBS, 1% FBS and 0.05% NaN3. The organs were embedded in low melting agarose (Sigma-Aldrich) and 50-100 µm sections were prepared with a Leica VT1200S Vibratome. Images were acquired with a confocal microscope (Leica SP5) and analyzed with Imaris software (Bitplane).
For each condition, five biological replicates of cells were separately cultured, treated and sorted. DLBCL VAL cells were stained with 5 µg/mL anti-human ACKR3 (mAb 11G8). at 4°C for 15 min. Cells were washed twice with MACS Buffer and sorted (5x105 cells) for ACKR3 positivity or negativity in 500 µL of RNA-later (Sigma-Aldrich). RNA-later lysates were centrifuged at 2500g x 5 minutes. The Zymo column RNA Isolation kit was used for RNA extraction (Zymo). Transcripts were processed for RNAseq with E7765L NEBNext Ultra II Directional RNA Library Prep, Sample Purification Beads and E7490L NEBNext® Poly(A) mRNA Magnetic Isolation Module. Poly-A RNA sequencing was performed with Illumina NextSeq500. RNA quantification was performed using Salmon v1.4.0 with hg38 transcriptome reference (77). The expression levels of the transcripts were then compared in the R package DESeq2 v1.28.0 (78).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169144.
The animal study was reviewed and approved by Swiss Federal Veterinary Office guidelines and authorized by the Animal Studies Committee of Cantonal Veterinary.
Conceptualization: PA, MT. Methodology: PA, DP, MT. Investigation: PA, SM, ER, ST. Formal Analysis: MF. Visualization: PA, MT. Supervision: MT. Writing - original draft: PA, DP. Writing - review & editing: PA, MT. All authors contributed to the article and approved the submitted version.
The study was supported by the Swiss Cancer league (KFS-4223-08-2017-R (MT)), the Novartis Foundation for medical-biological Research and The Helmut Horten Foundation.
The authors thank Diego Morone (IRB, Bellinzona), David Jarrossay (IRB, Bellinzona) and Andrea Rinaldi (IOR, Bellinzona) for help with imaging, flow cytometry and transcriptome analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1067885/full#supplementary-material
1. Mayor R, Etienne-Manneville S. The front and rear of collective cell migration. Nat Rev Mol Cell Biol (2016) 17:97–109. doi: 10.1038/nrm.2015.14
3. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International union of pharmacology. LXXXIX. update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2014) 66:1–79. doi: 10.1124/pr.113.007724
5. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol (2008) 9:953–9. doi: 10.1038/ni.f.207
6. Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG, et al. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLos. One (2010) 5:e9175. doi: 10.1371/journal.pone.0009175
7. Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem (2005) 280:35760–6. doi: 10.1074/jbc.M508234200
8. Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U. S. A (2007) 104:14759–64. doi: 10.1073/pnas.0702229104
9. Saaber F, Schutz D, Miess E, Abe P, Desikan S, Ashok KP, et al. ACKR3 regulation of neuronal migration requires ACKR3 phosphorylation, but not beta-arrestin. Cell Rep (2019) 26:1473–88. doi: 10.1016/j.celrep.2019.01.049
10. Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, et al. Impaired b-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA (1998) 95:9448–53. doi: 10.1073/pnas.95.16.9448
11. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature (1998) 393:595–9. doi: 10.1038/31269
12. Tweedy L, Thomason PA, Paschke PI, Martin K, Machesky LM, Zagnoni M, et al. Seeing around corners: Cells solve mazes and respond at a distance using attractant breakdown. Science (2020) 369:eaay9792. doi: 10.1126/science.aay9792
13. Tweedy L, Insall RH. Self-generated gradients yield exceptionally robust steering cues. Front Cell Dev Biol (2020) 8:133. doi: 10.3389/fcell.2020.00133
14. Massara M, Bonavita O, Mantovani A, Locati M, Bonecchi R. Atypical chemokine receptors in cancer: friends or foes? J Leukoc. Biol (2016) 99:927–33. doi: 10.1189/jlb.3MR0915-431RR
15. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
16. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410:50–6. doi: 10.1038/35065016
17. Mollica Poeta V, Massara M, Capucetti A, Bonecchi R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front Immunol (2019) 10:379. doi: 10.3389/fimmu.2019.00379
18. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity (2021) 54:859–74. doi: 10.1016/j.immuni.2021.01.012
19. Balkwill FR. The chemokine system and cancer. J Pathol (2012) 226:148–57. doi: 10.1002/path.3029
20. Hawkins OE, Richmond A. The dynamic yin-yang interaction of CXCR4 and CXCR7 in breast cancer metastasis. Breast Cancer Res (2012) 14:103. doi: 10.1186/bcr3092
21. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4:540–50. doi: 10.1038/nrc1388
22. Freitas C, Desnoyer A, Meuris F, Bachelerie F, Balabanian K, Machelon V. The relevance of the chemokine receptor ACKR3/CXCR7 on CXCL12-mediated effects in cancers with a focus on virus-related cancers. Cytokine Growth Factor Rev (2014) 25:307–16. doi: 10.1016/j.cytogfr.2014.04.006
23. Sanchez-Martin L, Sanchez-Mateos P, Cabanas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med (2013) 19:12–22. doi: 10.1016/j.molmed.2012.10.004
24. Neves M, Fumagalli A, van den Bor J, Marin P, Smit MJ, Mayor F. The role of ACKR3 in breast, lung, and brain cancer. Mol Pharmacol (2019) 96:819–25. doi: 10.1124/mol.118.115279
25. Azab AK, Sahin I, Azab F, Moschetta M, Mishima Y, Burwick N, et al. CXCR7-dependent angiogenic mononuclear cells trafficking regulates tumor progression in multiple myeloma. Blood (2014) 124:1905–14. doi: 10.1182/blood-2014-02-558742
26. Brennecke P, Arlt MJ, Muff R, Campanile C, Gvozdenovic A, Husmann K, et al. Expression of the chemokine receptor CXCR7 in CXCR4-expressing human 143B osteosarcoma cells enhances lung metastasis of intratibial xenografts in SCID mice. PLos. One (2013) 8:e74045. doi: 10.1371/journal.pone.0074045
27. Goguet-Surmenian E, Richard-Fiardo P, Guillemot E, Benchetrit M, Gomez-Brouchet A, Buzzo P, et al. CXCR7-mediated progression of osteosarcoma in the lungs. Br J Cancer (2013) 109:1579–85. doi: 10.1038/bjc.2013.482
28. Laskin DL, Kimura T, Sakakibara S, Riley DJ, Berg RA. Chemotactic activity of collagen-like polypeptides for human peripheral blood neutrophils. J Leukoc. Biol (1986) 39:255–66. doi: 10.1002/jlb.39.3.255
29. Stacer AC, Fenner J, Cavnar SP, Xiao A, Zhao S, Chang SL, et al. Endothelial CXCR7 regulates breast cancer metastasis. Oncogene (2015) 35:1716–24. doi: 10.1038/onc.2015.236
30. Melo RCC, Longhini AL, Bigarella CL, Baratti MO, Traina F, Favaro P, et al. CXCR7 is highly expressed in acute lymphoblastic leukemia and potentiates CXCR4 response to CXCL12. PLos. One (2014) 9:e85926. doi: 10.1371/journal.pone.0085926
31. Kim HY, Lee SY, Kim DY, Moon JY, Choi YS, Song IC, et al. Expression and functional roles of the chemokine receptor CXCR7 in acute myeloid leukemia cells. Blood Res (2015) 50:218–26. doi: 10.5045/br.2015.50.4.218
32. Puddinu V, Casella S, Radice E, Thelen S, Dirnhofer S, Bertoni F, et al. ACKR3 expression on diffuse large b cell lymphoma is required for tumor spreading and tissue infiltration. Oncotarget (2017) 8:85068–84. doi: 10.18632/oncotarget.18844
33. Basso K, Dalla-Favera R. Germinal centres and b cell lymphomagenesis. Nat Rev Immunol (2015) 15:172–84. doi: 10.1038/nri3814
34. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158:1110–22. doi: 10.1016/j.cell.2014.07.013
35. Nagai T, Ishikawa T, Minami Y, Nishita M. Tactics of cancer invasion: solitary and collective invasion. J Biochem (2020) 167:347–55. doi: 10.1093/jb/mvaa003
36. Vilchez Mercedes SA, Bocci F, Levine H, Onuchic JN, Jolly MK, Wong PK. Decoding leader cells in collective cancer invasion. Nat Rev Cancer (2021) 21:592–604. doi: 10.1038/s41568-021-00376-8
37. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res (2010) 70:5649–69. doi: 10.1158/0008-5472.CAN-10-1040
38. Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science (2016) 352:167–9. doi: 10.1126/science.aaf6546
39. Yang Y, Zheng H, Zhan Y, Fan S. An emerging tumor invasion mechanism about the collective cell migration. Am J Transl Res (2019) 11:5301–12.
40. Sengupta S, Parent CA, Bear JE. The principles of directed cell migration. Nat Rev Mol Cell Biol (2021) 22:529–47. doi: 10.1038/s41580-021-00366-6
41. Torphy RJ, Sun Y, Lin R, Caffrey-Carr A, Fujiwara Y, Ho F, et al. GPR182 limits antitumor immunity via chemokine scavenging in mouse melanoma models. Nat Commun (2022) 13(1):97. doi: 10.1038/s41467-021-27658-x
42. Ameti R, Melgrati S, Radice E, Cameroni E, Hub E, Thelen S, et al. Characterization of a chimeric chemokine as a specific ligand for ACKR3. J Leukoc. Biol (2018) 104:391–400. doi: 10.1002/JLB.2MA1217-509R
43. Hoffmann F, Mueller W, Schuetz D, Penfold ME, Wong YH, Schulz S, et al. Rapid uptake and degradation of CXCL12 depend on CXCR7 carboxyl-terminal Serine/Threonine residues. J Biol Chem (2012) 287:28362–77. doi: 10.1074/jbc.M111.335679
44. Boldajipour B, Mahabaleshwar H, Kardash E, Reichman-Fried M, Blaser H, Minina S, et al. Control of chemokine-guided cell migration by ligand sequestration. Cell (2008) 132:463–73. doi: 10.1016/j.cell.2007.12.034
45. Antonello P, Thelen M, Krause R, Pizzagalli DU. A pipeline to track unlabeled cells in wide migration chambers using pseudofluorescence. bioRxiv (2022). doi: 10.1101/2022.01.26.476896
46. Pizzagalli DU, Latino I, Pulfer A, Palomino-Segura M, Virgilio T, Farsakoglu Y, et al. Characterization of the dynamic behavior of neutrophils following influenza vaccination. Front Immunol (2019) 10:2621. doi: 10.3389/fimmu.2019.02621
47. Benned-Jensen T, Norn C, Laurent S, Madsen CM, Larsen HM, Arfelt KN, et al. Molecular characterization of oxysterol binding to the Epstein-Barr virus-induced gene 2 (GPR183). J Biol Chem (2012) 287:35470–83. doi: 10.1074/jbc.M112.387894
48. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475:524–7. doi: 10.1038/nature10280
49. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct b-cell migration through EBI2. Nature (2011) 475:519–23. doi: 10.1038/nature10226
50. Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature (1997) 387:620–4. doi: 10.1038/42506
51. Yokomizo T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J Biochem (2015) 157:65–71. doi: 10.1093/jb/mvu078
52. Saeki K, Yokomizo T. Identification, signaling, and functions of LTB4 receptors. Semin Immunol (2017) 33:30–6. doi: 10.1016/j.smim.2017.07.010
53. Bas E, Recio MC, Giner RM, Manez S, Cerda-Nicolas M, Rios JL. Anti-inflammatory activity of 5-o-demethylnobiletin, a polymethoxyflavone isolated from sideritis tragoriganum. Planta Med (2006) 72:136–42. doi: 10.1055/s-2005-873191
54. Gahbauer S, Pluhackova K, Bockmann RA. Closely related, yet unique: Distinct homo- and heterodimerization patterns of G protein coupled chemokine receptors and their fine-tuning by cholesterol. PLos. Comput Biol (2018) 14:e1006062. doi: 10.1371/journal.pcbi.1006062
55. Poltavets V, Faulkner JW, Dhatrak D, Whitfield RJ, McColl SR, Kochetkova M. CXCR4-CCR7 heterodimerization is a driver of breast cancer progression. Life (Basel) (2021) 11(10):1049. doi: 10.3390/life11101049
56. Mcheik S, Van EN, De PC, Gales C, Parmentier M, Springael JY. Coexpression of CCR7 and CXCR4 during b cell development controls CXCR4 responsiveness and bone marrow homing. Front Immunol (2019) 10:2970. doi: 10.3389/fimmu.2019.02970
57. Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, et al. Hetero-oligomerization of CCR2, CCR5 and CXCR4 and the protean effects of "selective"-antagonists. J Biol Chem (2009) 284:31270–9. doi: 10.1074/jbc.M109.054809
58. Hayasaka H, Kobayashi D, Yoshimura H, Nakayama EE, Shioda T, Miyasaka M. The HIV-1 Gp120/CXCR4 axis promotes CCR7 ligand-dependent CD4 T cell migration: CCR7 homo- and CCR7/CXCR4 hetero-oligomer formation as a possible mechanism for up-regulation of functional CCR7. PLos. One (2015) 10:e0117454. doi: 10.1371/journal.pone.0117454
59. Decaillot FM, Kazmi MA, Lin Y, Ray-Saha S, Sakmar TP, Sachdev P. CXCR7/CXCR4 heterodimer constitutively recruits {beta}-arrestin to enhance cell migration. J Biol Chem (2011) 286:32188–97. doi: 10.1074/jbc.M111.277038
60. Odlander B, Claesson HE. Formation of leukotriene B4 and monohydroxy acids in slices of porcine thyroid gland. Acta Endocrinol (Copenh) (1988) 117:463–9. doi: 10.1530/acta.0.1170463
61. Odlander B, Jakobsson PJ, Rosen A, Claesson HE. Human b and T lymphocytes convert leukotriene A4 into leukotriene B4. biochem. Biophys Res Commun (1988) 153:203–8. doi: 10.1016/S0006-291X(88)81209-9
62. Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, et al. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev Cell (2012) 22:1079–91. doi: 10.1016/j.devcel.2012.02.003
63. Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature (2013) 498:371–5. doi: 10.1038/nature12175
64. Del Prete A, Shao WH, Mitola S, Santoro G, Sozzani S, Haribabu B. Regulation of dendritic cell migration and adaptive immune response by leukotriene B4 receptors: A role for LTB4 in up-regulation of CCR7 expression and function. Blood (2007) 109:626–31. doi: 10.1182/blood-2006-02-003665
65. Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med (2006) 203:829–35. doi: 10.1084/jem.20052349
66. Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med (2006) 203:837–42. doi: 10.1084/jem.20052371
67. Muringampurath-John D, Jaye DL, Flowers CR, Saxe D, Chen Z, Lechowicz MJ, et al. Characteristics and outcomes of diffuse large b-cell lymphoma presenting in leukaemic phase. Br J Haematol. (2012) 158:608–14. doi: 10.1111/j.1365-2141.2012.09209.x
68. Zahid MF, Khan N, Hashmi SK, Kizilbash SH, Barta SK. Central nervous system prophylaxis in diffuse large b-cell lymphoma. Eur J Haematol. (2016) 97:108–20. doi: 10.1111/ejh.12763
69. Aspnes GE, Menhaji-Klotz E, Boehm M, Londregan AT, Lee ECY, Limberakis C, et al. Discovery and evaluation of non-basic small molecule modulators of the atypical chemokine receptor CXCR7. Bioorg. Med Chem Lett (2021) 50:128320. doi: 10.1016/j.bmcl.2021.128320
70. Pouzol L, Baumlin N, Sassi A, Tunis M, Marrie J, Vezzali E, et al. ACT-1004-1239, a first-in-class CXCR7 antagonist with both immunomodulatory and promyelinating effects for the treatment of inflammatory demyelinating diseases. FASEB J (2021) 35:e21431. doi: 10.1096/fj.202002465R
71. Richard-Bildstein S, Aissaoui H, Pothier J, Schafer G, Gnerre C, Lindenberg E, et al. Discovery of the potent, selective, orally available CXCR7 antagonist ACT-1004-1239. J Med Chem (2020) 63:15864–82. doi: 10.1021/acs.jmedchem.0c01588
72. Zabel BA, Lewen S, Berahovich RD, Jaen JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol Cancer (2011) 10:73. doi: 10.1186/1476-4598-10-73
73. Moepps B, Thelen M. Monitoring scavenging activity of chemokine receptors. Methods Enzymol (2016) 570:87–118. doi: 10.1016/bs.mie.2015.11.003
74. Sung KE, Su G, Pehlke C, Trier SM, Eliceiri KW, Keely PJ, et al. Control of 3-dimensional collagen matrix polymerization for reproducible human mammary fibroblast cell culture in microfluidic devices. Biomaterials (2009) 30:4833–41. doi: 10.1016/j.biomaterials.2009.05.043
75. Kreger ST, Bell BJ, Bailey J, Stites E, Kuske J, Waisner B, et al. Polymerization and matrix physical properties as important design considerations for soluble collagen formulations. Biopolymers (2010) 93:690–707. doi: 10.1002/bip.21431
76. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
77. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods (2017) 14:417–9. doi: 10.1038/nmeth.4197
Keywords: ACKR3, CXCR4, CXCR5, chemokine, atypical chemokine receptor, leukotriene B4, lymphoma characters
Citation: Antonello P, Pizzagalli DU, Foglierini M, Melgrati S, Radice E, Thelen S and Thelen M (2023) ACKR3 promotes CXCL12/CXCR4-mediated cell-to-cell-induced lymphoma migration through LTB4 production. Front. Immunol. 13:1067885. doi: 10.3389/fimmu.2022.1067885
Received: 12 October 2022; Accepted: 22 December 2022;
Published: 12 January 2023.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Ronen Alon, Weizmann Institute of Science, IsraelCopyright © 2023 Antonello, Pizzagalli, Foglierini, Melgrati, Radice, Thelen and Thelen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcus Thelen, bWFyY3VzLnRoZWxlbkBpcmIudXNpLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.