 Marco M. Rodari
Marco M. Rodari Nadine Cerf-Bensussan
Nadine Cerf-Bensussan Marianna Parlato
Marianna Parlato- Université Paris-Cité, Institute Imagine, Laboratory of Intestinal Immunity, INSERM U1163, Paris, France
The transforming growth factor-β (TGF-β) family of cytokines exerts pleiotropic functions during embryonic development, tissue homeostasis and repair as well as within the immune system. Single gene defects in individual component of this signaling machinery cause defined Mendelian diseases associated with aberrant activation of TGF-β signaling, ultimately leading to impaired development, immune responses or both. Gene defects that affect members of the TGF-β cytokine family result in more restricted phenotypes, while those affecting downstream components of the signaling machinery induce broader defects. These rare disorders, also known as TGF-β signalopathies, provide the unique opportunity to improve our understanding of the role and the relevance of the TGF-β signaling in the human immune system. Here, we summarize this elaborate signaling pathway, review the diverse clinical presentations and immunological phenotypes observed in these patients and discuss the phenotypic overlap between humans and mice genetically deficient for individual components of the TGF-β signaling cascade.
Introduction
The TGF-β family comprises a group of pleiotropic cytokines exerting a broad range of responses on both immune and non-immune cells. The key role of TGF-β in immunity has been established in genetically-engineered mice. This cytokine orchestrates T lymphocyte regulation by limiting effector T cell functions and promoting regulatory T cell development and functions. In humans, genome wide association studies (GWAS) have linked the SMAD3 locus - a key component of the TGF-β pathway - to predisposition to inflammatory bowel disease (IBD) and asthma (1). In addition, observations in patients carrying single gene defects in individual components of the TGF-β family (such as TGF-β1) (2) and their downstream signaling components (such as TGFβ-R1/2 or SMAD2/3) have further underscored the role of TGF-β in the pathogenesis of intestinal inflammation and allergic diseases. In addition to gene defects that directly affect the TGF-β cytokine family signaling, others affect TGF-β responses indirectly by impacting the nuclear transport of signaling components (such as IPO8) (3), or by interfering with cross-regulatory non-canonical pathways (such as ERBIN or JNK1) (4, 5). Here, we provide an overview of the TGF-β signaling pathway (Figure 1) and review Mendelian disorders affecting components of the TGF-β signaling machinery, also known as TGF-β signalopathies (6). These defects commonly result in a complex syndrome combining craniofacial, cardiovascular and skeletal defects, features that have been extensively reviewed elsewhere (6, 7). Here, we intend to focus on features of immune dysregulation which are also associated with these defects and often overlooked and to discuss the possible mechanisms underlying abnormal TGF-β signaling and disease pathology.
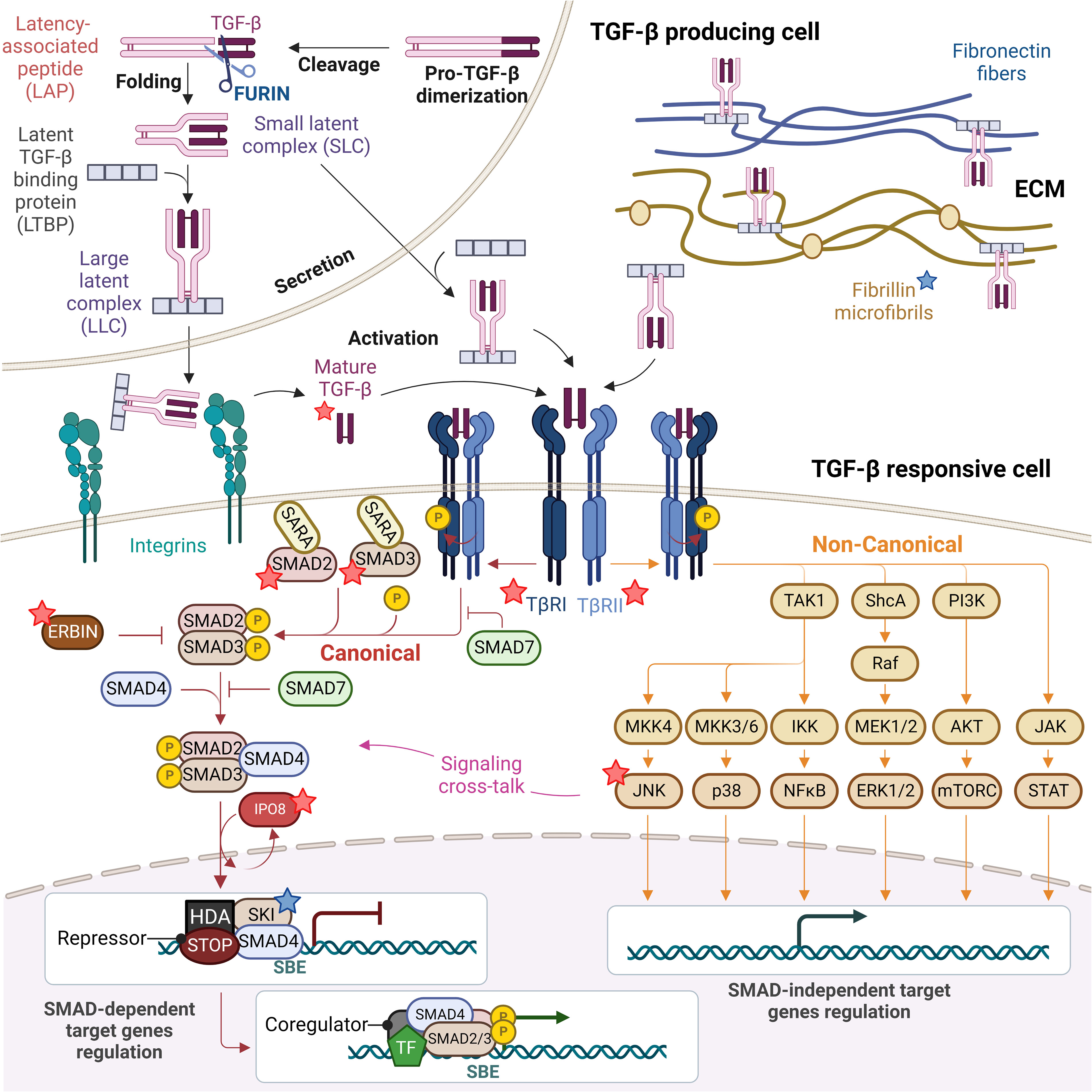
Figure 1 TGF-β signaling pathway: from TGF-β biogenesis to gene expression regulation. Mature TGF-β is secreted in form of small and large latent complexes (SLC and LLC) and associates with fibronectin fibers and fibrillin microfibrils composing the extracellular matrix (ECM), as well as with plasma membrane-associated proteins (not shown). Upon interaction with extracellular proteases and integrins on the surface of TGF-β responsive cells, mature TGF-β is released. Active TGF-β binds to type II receptors (TβRII), which recruits and activates type I receptors (TβRI). Receptor-induced phosphorylation of SMAD2 and SMAD3 induces their dissociation from SARA and formation of SMAD2/3-SMAD4 complexes which in turn translocate to the nucleus in an IPO8-mediated manner to initiate signaling. In the absence of TGF-β, SKI is bound to DNA at Smad Binding Elements (SBEs) with SMAD4, recruiting others repressors and histone deacetylases (HDA) to inhibit the expression of TGF-β target genes. SMAD6/7 attenuate ligand-induced SMAD activation, preventing SMAD2/3 recruitment to TβRI and competing with SMAD4 for binding to SMAD2/3, while ERBIN competes with SARA for binding to SMAD2/3, segregating phosphorylated SMAD2/3 in the cytoplasm. The TGF-β TβRI/TβRII complex can also activate non-canonical non-SMAD signaling pathways, including the ERK MAP kinase and the p38 MAPK and JNK pathways, the PI3K-AKT-mTOR and the JAK-STAT signaling, which overall complement SMAD-dependent signaling and strongly complicate the output of a TGF-β trigger. Proteins found mutated in TGF-β signalopathies are indicated with a star, blue if patients do not present with immune dysregulation (fibrillin and SKI), red if they present with immune dysregulation (TGF-β1, TGF-βRI and TGF-βRII, SMAD2 and SMAD3, IPO8, ERBIN and JNK).
The TGF-β signaling pathway
The human TGF-β family includes thirty-three genes that encode for homodimeric or heterodimeric secreted cytokines, including TGF-β1, TGF-β2 and TGF-β3, activins, nodals, bone morphogenetic proteins (BMPs), and growth and differentiation factors (GDFs). Identified in 1983 and best characterized (8), TGF-β1 remains the most emblematic member of the family and, as discussed below, the key actor of the family in the immune system. TGF-β1 is synthesized in the rough endoplasmic reticulum in precursor form featuring a large ~250 residue amino-signal peptide, a central pro-domain, called latency associated protein (LAP), and a short ~110 residue carboxy-terminal mature peptide. After initial biogenesis and signal peptide cleavage, two pro-peptides dimerize via intermolecular disulfide bonds and translocate into the Golgi complex, where TGF-β1 is cleaved from LAP by FURIN. The two products remain non-covalently bound and form the small latent complex (SLC) (9). In this conformation, the SLC is inactive as mature TGF-β is hidden and protected deep inside the protein structure (10). The SLC typically associates with either a family of proteins called latent TGF-β binding proteins (LTBPs), forming the large latent complex (LLC) that in turn associates with the extra-cellular matrix (ECM) upon secretion (11, 12), or with membrane proteins GARP (glycoprotein-A repetitions predominant) (13). Several mechanisms regulate the release of active TGF-β, including αV integrins expressed at the cell surface of many cell types and extracellular proteases (9, 14). Mechanistically, it has been suggested that physical traction resulting from interaction between ECM-associated latent TGF-β and cell-surface αV integrins ultimately leads to conformational changes of the latent complexes and to active TGF-β release (15). In addition, this conformational change is thought to allow higher accessibility of the latent complex to extracellular proteases (9). Overall physiological release of active TGF-β likely requires synergic actions of both integrins and proteases (16).
TGF-βs signal via a pair of transmembrane serine/threonine protein kinases known as the type I (TGF-βR1 or ALK5) and type II receptors (TGF-βR2). Binding of one of these cytokines to a TGF-βR2 dimer triggers the recruitment and subsequent transphosphorylation of TGF-βR1 which, in turn, activates the downstream canonical signaling cascade via the receptor-regulated SMADs (R-SMADs). BMP subfamily receptors phosphorylate SMAD1, SMAD5 and SMAD8, while TGF-β subfamily receptors mainly phosphorylate SMAD2 and SMAD3. Upon phosphorylation, R-SMADs dissociate from the receptor and form a heterotrimeric transcriptional complex with SMAD4. Typically, heterotrimers of SMAD2, SMAD3 and SMAD4 are formed, but complexes that contain homodimers of SMAD2 or SMAD3 may form as well and target distinct genes (17). Localization and concentration of SMADs are regulated by anchor for receptor activation (SARA) proteins which recruit non-phosphorylated SMADs to the activated receptors for phosphorylation. Upon receptor activation, phosphorylation of SMADs causes their dissociation from SARA and formation of SMAD2/3–SMAD4 complexes (18). Heteromeric SMAD complexes translocate and accumulate in the nucleus where they bind DNA directly but with low affinity and specificity or indirectly by interacting with other DNA-binding proteins to target specific genes for transcriptional regulation. Regulatory mechanisms are in place to avoid spontaneous transcriptional activation of TGF-β target genes. Thus, in absence of TGF-β stimuli, SKI inhibits the expression of TGF-β target genes by binding Smad Binding Elements (SBEs) complexed with SMAD4 and by recruiting other repressors and histone deacetylases (HDA) (19). SMADs turnover in the nucleus is tightly controlled. Nuclear SMAD levels are kept in check either by regulating protein turnover, before and after translocation into the nucleus, or by regulating the subcellular localization of SMADs. SMAD6 and SMAD7 are inhibitory molecules that suppress receptor and SMAD signaling functions. SMAD7 recruits SMURF2 ubiquitin ligases to TGF-β receptors to promote their ubiquitin-mediated degradation (20, 21). Inhibitory SMADs also associate with R-SMADs, preventing the formation of R-SMAD complexes with SMAD4. In addition, the expression of SMAD6 and SMAD7 induced by TGF-β family members also triggers negative feedback (22). SMAD interactions with distinct karyopherin proteins might also result in differential regulation of nuclear import depending on the SMAD, which may represent a complementary layer of regulation of the dynamics of SMAD-mediated transcription responses. Once in the nucleus, SMAD trimers interact with specific transcriptional factors as well as with regulatory proteins, to initiate transcription of specific target genes. Upon dephosphorylation by the nuclear phosphatase PPM1A, SMAD trimeric complexes disassemble and SMAD proteins are exported out of the nucleus by different exportins, such as RanBP3, thus facilitating termination of TGF-β signals (23, 24).
TGF-β receptors stimulation can also result in the activation of SMAD-independent pathways that are collectively referred to as non-canonical TGF-β signaling pathways (25). They include branches of ERK, JNK, p38 MAPK, NF-κB, PI3K-AKT, JAK-STAT and small GTPase pathways (10, 26, 27). Upon activation, direct phosphorylation of ShcA by TGF-βRI induces its association with Grb2/Sos and sequential Ras, MEK1/2 and ERK1/2 activation (28). Thus, initiation of the ERK pathway also depends on TGF-βRI mediated phosphorylation. In contrast, TGF-βRI kinase activity seems to be dispensable for the activation of other non-SMAD signaling pathways. Recruitment of the E3 ubiquitin ligases TRAF4 and TRAF6 onto the TGF-β receptors leads to intramolecular polyubiquitination of TRAF4 or TRAF6, and consequent recruitment and ubiquitination of the MAPKK kinase TAK1. This post-translational modification does not lead to TAK1 degradation, but instead to MAPK kinases (MKKs) phosphorylation by TAK1 and finally to activation of either JNK or p38 (29, 30). Eventually, TRAF6 can also interact with IKK proteins and trigger NF-κB signaling (31). In a similar way, TRAF6-mediated ubiquitination seems to be central for TGF-β-dependent activation of PI3K and AKT in the mTOR pathway (32). Finally, TGF-β can also regulate other non-SMAD pathways, including Rho-like GTPases. Yet, more work is needed to better elucidate the details of their TGF-β-dependent activation (10, 33, 34). When and how TGF-β stimulation can lead to SMAD-dependent or -independent signals is clearly cell- and context- dependent, but the intricate regulatory mechanisms that orchestrate these events remain to be fully deciphered. It has been shown that SMAD-dependent signaling is associated with clathrin-bound receptors, while non-SMAD signaling pathways originate from lipid-raft caveolar compartments (35), suggesting that the variability in signaling output might depend on functionally different receptor complexes (10). Several kinases, including some members of the MAP kinase family, can also phosphorylate R-SMADs, and thereby regulate their effector functions in a TGF-β-independent manner. As a consequence, R-SMADs activation can result from stimulation by cytokines distinct from TGF-β family members. Moreover, translocation of R-SMADs to the nucleus can induce a delayed activation of non-SMAD pathways via gene-expression regulation, further complicating the study of downstream molecular events (10). Overall, these multiple signaling cross-talks enable a broad diversity of responses and underlie the versatility in terms of dosage, spatiotemporal activity and cell context of this family of cytokines in regulating cellular processes. A range of inherited human diseases results from single-gene variants in several components of this tightly controlled signaling pathway. On one end of the range, these gene defects lead to exclusively developmental defects, while on the other end, they are restricted almost purely to the immune system. Therefore, comparing the phenotypic consequences of pathogenic variants in genes encoding TGF-β receptors or downstream components of the signaling machinery, and in genes encoding members of the family (TGF-β1, TGF-β2, and TGF-β3) enable to differentiate and appreciate the role of those individual cytokines in immune and non-immune cells in humans.
Mendelian disorders affecting TGF-β signaling and presenting with immune dysregulation
The term “TGF-β signalopathies” defines a number of Mendelian disorders associated with inappropriate activation of TGF-β signaling and ultimately impacting developmental processes, immune responses, or both. A large group of these gene defects result in complex syndromes of variable expressivity encompassing a broad spectrum of cardiovascular, craniofacial, and skeletal features associated or not with impaired immune regulation (6) (Table 1). Those, include, among others, Loeys–Dietz syndrome (LDS) which is caused by heterozygous loss-of-function (LOF) variants in TGFBR1 (MIM: 190181), TGFBR2 (MIM: 190182), TGFB2 (MIM: 190220) or TGFB3 (MIM: 190230), SMAD2 (MIM: 601366) or SMAD3 (MIM: 603109) and Marfan syndrome (MFS) (MIM: 154700) caused by variants in FBN1, encoding fibrillin 1, the main component of extracellular matrix microfibrils that scaffolds latent TGF-β (Figure 1). Clinically, MFS and LDS show overlapping cardiovascular and skeletal manifestations. Yet, in contrast to LDS patients, immune regulation is not affected in Marfan patients, likely because fibrillin 1 is mainly expressed in connective and soft tissues, and muscles, while it is almost absent in sites that are exposed to environmental immune triggers such as lungs and intestine, or in sites of adaptive immune priming such as lymph nodes and spleen. Similarly, Shprintzen-Goldberg syndrome (MIM: 182212), which is caused by heterozygous variants in SKI features all the craniofacial, skeletal and cardiovascular manifestations of LDS but no immune dysregulation, likely due to the expression pattern of SKI. Indeed, expression of this negative regulator of the pathway, at least in vessels, seems to be regionally confined and temporally limited at early stages of development in mice (41). Contrasting with this group of diseases, the more recently identified TGF-β1 deficiency results in a prominent immune phenotype underscoring the central immunoregulatory role of this cytokine.
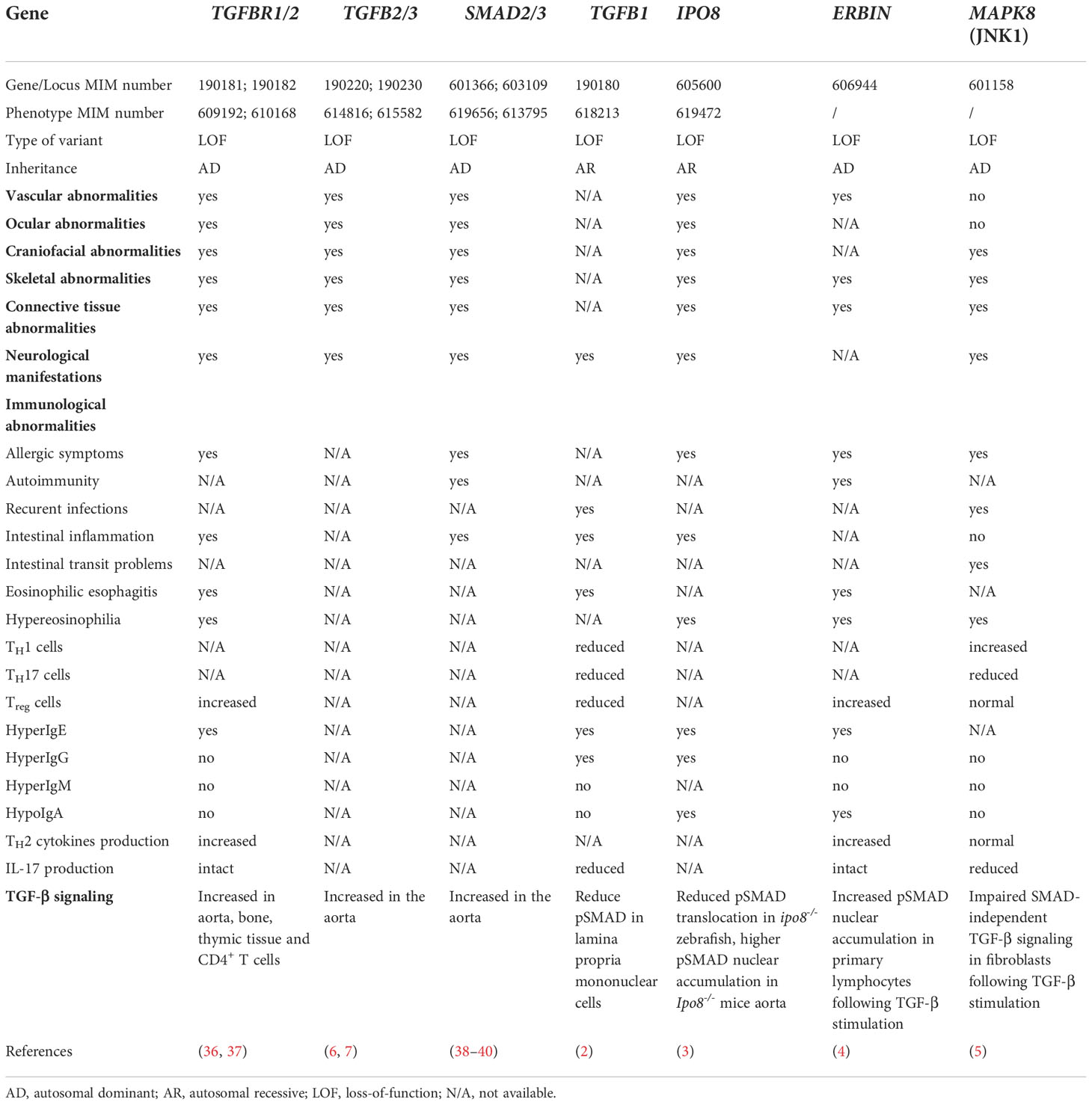
Table 1 Clinical features presenting in patients with TGF-β signalopathies.
TGF-β1 deficiency causes very early onset-IBD
As mentioned above, bi-allelic LOF variants in TGFB1 were recently identified in 3 patients from 2 unrelated families presenting with severe very early onset IBD, manifesting as bloody diarrhea with failure to thrive, and central nervous system (CNS) disease associated with epilepsy, brain atrophy and posterior leukoencephalopathy (2). Functionally, these variants affected TGF-β1 complex formation, secretion and/or bioavailability for signal transduction ultimately resulting in decreased downstream SMAD2/3 signaling. Recurrent infections and dermatitis were also reported in two patients altogether with reduced T-cell responses to stimulation with anti-CD3 and anti-CD28 as well as impaired T cell proliferation in response to diphtheria and tetanus toxoids. Immunophenotyping was performed only in one child and showed decreased memory Treg, TH1 and TH17 cell counts in peripheral blood, associated with reduced frequencies of Treg, TH1, TH17 and CD103+ T cells infiltrating the colon and increased serum concentration of IgG and IgE. The disease course was extremely severe as only one patient remained alive at the age of 11 years in stable condition, while in 2 others septicemia resulted in death at 25 and 39 months, respectively. Given the very severe condition of the affected children, it remains difficult to ascribe all immunological abnormalities and notably the fatal infectious complications to the lack of TGF-β1. However, the very severe IBD phenotype illustrates the key role of TGF-β1 in maintaining immune homeostasis at the intestinal barrier. Of note, Tgfb1-/- mice show no gross developmental defects but they develop immediately after birth very severe multi-organ inflammation resulting in organ failure and fatal outcome within 20-30 days (Table 2) (42–44). Overall data in humans and in mice converge to demonstrate the non-redundant role of TGF-β1 in maintaining immune homeostasis. In contrast to LDS, TGF-β1 deficiency is associated with severe CNS dysfunction. The mechanism(s) of this very severe neurological phenotype remain(s) not fully understood and the respective role of uncontrolled inflammation and of a developmental defect are not yet well-delineated.

Table 2 Mouse models for the study of immune dysregulation in TGF-β signalopathies.
Defects in TGF-β receptor 1/2 and SMAD2/3 result in Loeys-Dietz syndrome featuring atopy and intestinal inflammation
Autosomal dominant (AD) variants in genes coding for TGF-β ligands (TGFB2, TGFB3), receptors (TGFBR1, TGFBR2) or downstream effectors (SMAD2, SMAD3) (58–63) cause LDS syndrome. These gene defects share the craniofacial, cardiovascular and skeletal phenotypic core but penetrance, disease severity and immune defects are variable depending on the gene. The exact nature of the signaling defects remains difficult to appreciate. Thus, despite being inactivating when functionally tested in vitro, heterozygous variants in TGFBR1 or TGFBR2 paradoxically result in increased TGF-β signaling output, as revealed by elevated levels of phosphorylated SMAD2/3 or ERK1/2 and increased TGF-β1 levels in several tissues derived from LDS patients, including aorta, bone, and thymus (59, 61–63). Altered cell surface recycling of the mutant receptors (64), imbalance between SMAD-dependent and SMAD-independent cascades (65) or alternative ligand usage (41, 62) may explain the paradoxical increase in TGF-β signaling. Yet, more recently, data obtained in cardiovascular progenitor cell-derived smooth muscle cells (CPC-SMCs) differentiated from human induced pluripotent stem cells (hiPSC) carrying AD TGFBRs variants showed decreased, rather than enhanced, pSMAD2/3 and pAKT signaling following TGF-β1 stimulation (66), thus suggesting that LOF TGFBRs variants do indeed result in decreased pathway activation. It seems possible that epigenetic changes and compensatory mechanisms become activated overtime, thus leading to the increase in pSMAD2/3 that is observed in end-stage aortic tissues. More undifferentiated cell types might thus be better suited to interrogate the early molecular events underlying development of LDS and LDS-related disorders. Immune dysregulation with variable prevalence and severity can be observed in LDS patients. Increased risk to develop atopy including food allergies asthma, allergic rhinitis, eczema and eosinophilic gastrointestinal diseases was first reported in a cohort of TGFBR1 or TGFBR2 carriers (36) and confirmed by latter identification of additional cases (37). Consistent with the allergic manifestations, these patients had significantly increased eosinophil counts, elevated levels of both IgE and allergy-associated type 2 cytokines IL-5 and IL-13 which contrasted with normal non-IgE antibody isotypes. As previously reported in aortic tissues, a paradoxical increase in TGF-β signaling was observed in CD4+ T lymphocytes (36). Accordingly, and consistent with the role of TGF-β in driving Treg cells generation (67), but at odd with the loss of tolerance, LDS patients had increased frequency of peripheral FOXP3+ Treg cells with normal ability to suppress effector T cell proliferation (36). In addition, increased prevalence of IBD has also been reported in 2 unrelated cohorts of LDS patients (58, 68). Overall, the paradoxical enhancement of TGF-β signaling observed in LDS patients with monoallelic LOF variants in TGFBR1 or TGFBR2 makes it difficult to compare observations in humans with data available in mice, where complete ablation of Tgfbr1 or Tgfbr2 in T cells causes a rapidly fatal autoimmune disease characterized by lymphoproliferation, increased T cell activation and cytotoxic differentiation, and drastic Treg cells decrease in the periphery (Table 2) (46–49).
In line with the role of SMAD3 downstream all 3 TGF-βs, autoimmune features and allergic manifestations have been also reported in a cohort of heterozygous SMAD3 carriers (38). In marked contrast, the regulation of the immune response does not seem to be affected in LDS patients carrying variants in TGFB2/3. The lack of immune phenotype in the latter patients is in keeping with data obtained in Tgfb2-/- and Tgfb3-/- mice (69–71). Thus, these mice present perinatal mortality with multiple but little overlapping developmental defects affecting a broad range of organs, but no immune defects. Overall, these data indicate that, in humans as in mice, the three TGF-β isoforms cannot compensate each other. While TGF-β1 is key to maintain homeostasis in the immune system, TGF-β2 and TGF-β3 play complementary roles in connective tissue development.
Aberrant regulation of TGF-β signaling due to defects in STAT3-ERBIN-SMAD2/3 complex leads to atopic predisposition
ERBB2-interacting protein (ERBIN), a member of the leucine-rich repeat and PDZ domain (LAP) family, is a SMAD-binding (72) and SARA-interacting protein (73) that has been shown to negatively regulate TGF-β signaling activation. ERBIN binds and segregates phosphorylated SMAD2/3 in the cytoplasm, preventing their translocation in the nucleus thereby dampening SMAD2/3-dependent transcription. SARA recruits non-phosphorylated SMAD2/3 to activated receptors to promote their phosphorylation and competes with SMAD2/3 for binding to ERBIN (73) (Figure 1). Therefore, the concentrations of these three components of the pathway and their relative binding affinities modulate the output of TGF-β signaling. Accordingly, deregulation of TGF-β signaling is observed in patients carrying heterozygous LOF variants in ERBIN (4). These patients present with a connective tissue syndrome including vascular abnormalities, as well as with immunological manifestations, including eczema, atopy, eosinophilic esophagitis (EoE) with hypereosinophilia and elevated IgE (4, 74). Hypertrophic cardiomyopathy observed in one patient could be recapitulated in Erbin deficient mice (75). Interestingly, these mice are more susceptible to experimental colitis and spontaneously develop low grade intestinal inflammation and epithelial injury over time but the role of TGF-β signaling in these manifestations was not explored (Table 2) (55). In primary CD4+ T lymphocytes from ERBIN patients, reduced ERBIN expression did not affect total pSMAD2/3 levels in response to TGF-β stimulation, but rather resulted in increased nuclear accumulation of pSMAD2/3, consistent with the role of ERBIN in segregating activated pSMAD2/3 in the cytoplasm. In keeping with the increased sensitivity to TGF-β conferred by ERBIN deficiency and with the key role of TGF-β in Treg cells development, patients displayed increased counts of peripheral Treg cells. Moreover, in vitro differentiation of iTreg cells from naïve CD4+ T cells was increased compared to controls. Dysregulation of TGF-β signaling was associated with increased IL-4Rα expression and increased activation of the IL-4-GATA3 axis upon weak TCR stimulation of naïve ERBIN deficient CD4+ T lymphocytes. Increased ex vivo production of TH2 cytokines (IL-4, IL-5 and IL-13) by ERBIN deficient memory CD45RO+ CD4+ lymphocytes further suggest skewed type 2 immune responses and might account for the allergic manifestations observed in the patients. Accordingly, amelioration of the refractory allergic eosinophilic inflammation could be achieved by IL-4Rα blockade in a 16 yo ERBIN patient (74). Interestingly, IL-6/STAT3 and IL-11/STAT3 activation suppresses TGF-β signaling via formation of the STAT3-ERBIN-SMAD2/3 complex with subsequent SMAD2/3 retention in the nucleus and inhibition of the TGF-β signaling, thus highlighting the molecular intersection between JAK-STAT pathway activation and repression of TGF-β signaling (4, 76). Accordingly, enhanced SMAD signaling is reported in patients carrying dominant-negative STAT3 variants, who show, alike ERBIN patients, elevated IgE and EoE. Overall these observations establish a link between increased TGF-β activity, impaired STAT3 activation and immune dysregulation.
Impaired SMADs translocation due to IPO8 deficiency causes Loeys-Dietz like syndrome
Nuclear translocation to the nucleus of activated R-SMADs is a regulated process which is key for transduction of TGF-β signals. Some data have shown that the shuttling of SMADs downstream TGF-β signal depends on a member of the karyopherin family of nuclear transport receptors, importin 8 (IPO8) (77, 78). Karyopherins use RANGTP to translocate cargos through the nuclear pore complexes and once in the nucleus, they release the cargo and finally return to the cytoplasm (79). Three groups have concurrently reported biallelic LOF variants as cause of a complex syndrome overlapping with LDS, including early-onset thoracic aortic aneurysm and structural cardiac defects, joint hypermobility, skin laxity, and hernias, dysmorphic features, skeletal abnormalities, and developmental delay (3, 56, 80). As in LDS, immune dysregulation has been observed in some patients, including predisposition to allergic or intestinal inflammatory diseases, hyperIgE, hyperIgG, hypoIgA, and hypereosinophilia (3). Evidence in zebrafish and in mouse (Table 2), indicate that Ipo8 deficiency can disrupt TGF-β and or BMP signaling and thereby explain the vascular, skeletal and immunological anomalies observed in the patients. Yet, whether IPO8 loss results in increased or decreased pSMADs shuttling with consequent increased or decreased TGF-β pathway activation is still unclear. Immunohistochemistry analysis of ascending aortic sections in Ipo8-/- mice revealed increased nuclear pSMAD2/3 as compared to WT animals. Along with the observation of decreased expression of TGF-β negative regulator Smad7, this might suggest increased pathway activation (56). In contrast, decreased pSMAD1/5/9 nuclear accumulation was found in ipo8-/- zebrafish embryos. Accordingly, expression of transcripts encoding smad7 and other TGF-β dependent genes were reduced in 13 hpf and 24 hpf zebrafish embryos, pointing to decreased - rather than increased - pathway activation in this model (3). Taken together, defective SMAD translocation might be considered as an early event underlying disease development, as it can be observed in undifferentiated cell types, while compensatory mechanisms acting overtime might lead to increased SMADs phosphorylation in end-stage tissues. Of note, the development of thoracic aortic aneurysms development is recapitulated in Ipo8 deficient mice but their immune system has not been studied (56). Further investigation will be necessary to define whether and how defective IPO8-dependent nuclear translocation may underlie immune dysregulation in IPO8 deficient patients.
Impaired JNK1-dependent MAPK signaling pathway cause chronic mucocutaneous candidiasis and connective tissue disorder
A heterozygous LOF variant in MAPK8, the gene encoding the c-Jun N-terminal kinase 1 (JNK1) was recently reported in a three-generation kindred with a complex syndrome combining chronic mucocutaneous candidiasis and a connective tissue disorder but no evidence of aortic aneurysm or dissection (5). JNK1 is one of the three kinases of the JNK family and a member of the mitogen-activated protein kinase (MAPK) pathway (81). As shown in Figure 1, TGF-β can induce the activation of MAPK kinases (specifically MKK4 and MKK7) resulting in JNK1 phosphorylation. In turn, JNK1 phosphorylates activator protein-1 (AP-1) transcription factors, including c-Jun and ATF-2, and ultimately triggers SMAD2/3-independent transcription (82). Accordingly, fibroblasts derived from patients carrying heterozygous MAPK8 LOF variants showed impaired c-Jun/ATF-2 signaling in response to TGFβ but normal ERK1/2, p38, and SMAD2/3 activation. Dysregulation of TGF-β-dependent signals was suggested to account for the connective tissue disorders. Of note, patients also displayed higher proportions of TH1 cells and lower proportions of TH17 cells than controls, while frequencies of TH2 and Treg cells were normal. In addition, naïve CD4+ T cells from patients carrying the heterozygous MAPK8 variant produced less IL-17A and IL-17F in TH17-polarizing conditions in vitro. Overall these results are consistent with the role played by JNK1 in TH17-cell activation and differentiation in mice (Table 2) (57, 83) and with TGF-β requirement for TH17 differentiation in both mice (84) and humans (85–87) and suggest that impaired TH17 development underlies the predisposition to chronic mucocutaneous candidiasis.
Conclusions
Patients carrying single gene defects in individual components of the TGF-β signaling machinery are invaluable resources to dissect the relative contribution, signaling cross-talks, specificity and redundancy of each of those components in the orchestration of the immune response. Human TGF-β1 deficiency results in very early onset severe IBD, demonstrating the non-redundant role of TGF-β1 in suppressing intestinal inflammation and supporting the hypothesis that the aberrant TGF-β1/SMAD signaling observed in active lesions of IBD patients participate in disease pathogenesis (88). Increased Smad7 associated with downregulation of the TGF-β signaling, shown to amplify gut inflammatory responses in those patients (89), could be targeted by specific antisense oligonucleotides (mongersen), which however provided conflicting results in phase II/III studies (89–91). Pathogenic monoallelic variants in TGFBR1/2 as well as in SMAD3 predispose to IBD, strengthening the key role of the SMAD3 canonical pathway for maintaining intestinal homeostasis, a role previously supported by GWAS studies in common multifactorial forms of IBD (1) as well as by Smad3 inactivation in mice (53, 54). Strikingly, these gene defects also predispose to allergy, overall suggesting that TGF-β is instrumental in humans to control immune responses to environmental triggers. While TGF-β1 deficiency may benefit from supplementation by this cytokine, the complexity of TGF-β signaling remains a major challenge to define appropriate therapeutic strategies in other defects impairing either chains of the receptor or downstream elements of the signaling cascade.
Author contributions
MR, NC-B and MP wrote the manuscript. MR prepared the tables and the figure with BioRender. All authors contributed to the article and approved the submitted version.
Funding
This work has received funding from Agence Nationale de la Recherche (ANR-22-CE17-0040). MR is supported by ED562 fellowship from University of Paris Cité.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
2. Kotlarz D, Marquardt B, Barøy T, Lee WS, Konnikova L, Hollizeck S, et al. Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat Genet (2018) 50:344–8. doi: 10.1038/S41588-018-0063-6
3. Ziegler A, Duclaux-Loras R, Revenu C, Charbit-Henrion F, Begue B, Duroure K, et al. Bi-allelic variants in IPO8 cause a connective tissue disorder associated with cardiovascular defects, skeletal abnormalities, and immune dysregulation. Am J Hum Genet (2021) 108:1126–37. doi: 10.1016/j.ajhg.2021.04.020
4. Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med (2017) 214:669–80. doi: 10.1084/jem.20161435
5. Li J, Ritelli M, Ma CS, Rao G, Habib T, Corvilain E, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-β. Sci Immunol (2019) 4 4(41):eaax7965. doi: 10.1126/sciimmunol.aax7965
6. Cannaerts E, van de Beek G, Verstraeten A, Van Laer L, Loeys B. TGF-β signalopathies as a paradigm for translational medicine. Eur J Med Genet (2015) 58:695–703. doi: 10.1016/J.EJMG.2015.10.010
7. Rodrigues Bento J, Meester J, Luyckx I, Peeters S, Verstraeten A, Loeys B. The genetics and typical traits of thoracic aortic aneurysm and dissection. Annu Rev Genomics Hum Genet. (2022) 23:223–253. doi: 10.1146/annurev-genom-111521
8. Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: A historical perspective. Cold Spring Harb Perspect Biol (2016) 8(7):a021865. doi: 10.1101/cshperspect.a021865
9. Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-Related proteins. Cold Spring Harb Perspect Biol (2016) 8:a021907. doi: 10.1101/cshperspect.a021907
10. Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal (2019) 12(570):eaav5183. doi: 10.1126/scisignal.aav5183
11. Kanzaki T, Olofsson A, Morén A, Wernstedt C, Hellman U, Miyazono K, et al. TGF-beta 1 binding protein: A component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell (1990) 61:1051–61. doi: 10.1016/0092-8674(90)90069-q
12. Koli K, Hyytiäinen M, Ryynänen MJ, Keski-Oja J. Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp Cell Res (2005) 310:370–82. doi: 10.1016/j.yexcr.2005.08.008
13. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PloS One (2008) 3:e2705. doi: 10.1371/journal.pone.0002705
14. Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi: 10.1146/ANNUREV-IMMUNOL-032713-120257
15. Wipff P-J, Rifkin DB, Meister J-J, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol (2007) 179:1311–23. doi: 10.1083/jcb.200704042
16. Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol (2002) 157:493–507. doi: 10.1083/jcb.200109100
17. Lucarelli P, Schilling M, Kreutz C, Vlasov A, Boehm ME, Iwamoto N, et al. Resolving the combinatorial complexity of smad protein complex formation and its link to gene expression. Cell Syst (2018) 6:75–89.e11. doi: 10.1016/j.cels.2017.11.010
18. Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell (1998) 95:779–91. doi: 10.1016/s0092-8674(00)81701-8
19. Tecalco-Cruz AC, Ríos-López DG, Vázquez-Victorio G, Rosales-Alvarez RE, Macías-Silva M. Transcriptional cofactors ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct Target Ther (2018) 3:15. doi: 10.1038/s41392-018-0015-8
20. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell (2000) 6:1365–75. doi: 10.1016/s1097-2765(00)00134-9
21. Zhang J, Zhang X, Xie F, Zhang Z, van Dam H, Zhang L, et al. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell (2014) 5:503–17. doi: 10.1007/s13238-014-0058-8
22. Miyazawa K, Miyazono K. Regulation of TGF-β family signaling by inhibitory smads. Cold Spring Harb Perspect Biol (2017) 9(3):a022095. doi: 10.1101/cshperspect.a022095
23. Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, et al. PPM1A functions as a smad phosphatase to terminate TGFβ signaling. Cell (2006) 125:915–28. doi: 10.1016/J.CELL.2006.03.044
24. Dai F, Lin X, Chang C, Feng X-H. Nuclear export of Smad2 and Smad3 by RanBP3 facilitates termination of TGF-β signaling. Dev Cell (2009) 16(3):345–57. doi: 10.1016/j.devcel.2009.01.022
25. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol (2012) 13:616–30. doi: 10.1038/nrm3434
26. Luo K. Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harb Perspect Biol (2017) 9(1):a022137. doi: 10.1101/cshperspect.a022137
27. Zhang YE. Non-smad signaling pathways of the TGF-β family. Cold Spring Harb Perspect Biol (2017) 9(2):a022129. doi: 10.1101/cshperspect.a022129
28. Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, et al. TGF-beta activates erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J (2007) 26:3957–67. doi: 10.1038/sj.emboj.7601818
29. Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates smad-independent activation of JNK and p38 by TGF-beta. Mol Cell (2008) 31:918–24. doi: 10.1016/j.molcel.2008.09.002
30. Zhang L, Zhou F, García de Vinuesa A, de Kruijf EM, Mesker WE, Hui L, et al. TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol Cell (2013) 51:559–72. doi: 10.1016/j.molcel.2013.07.014
31. Hamidi A, von Bulow V, Hamidi R, Winssinger N, Barluenga S, Heldin C-H, et al. Polyubiquitination of transforming growth factor β (TGFβ)-associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J Biol Chem (2012) 287:123–33. doi: 10.1074/jbc.M111.285122
32. Hamidi A, Song J, Thakur N, Itoh S, Marcusson A, Bergh A, et al. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci Signal (2017) 10(486):eaal4186. doi: 10.1126/scisignal.aal4186
33. Zhang YE. Non-smad pathways in TGF-beta signaling. Cell Res (2009) 19:128–39. doi: 10.1038/CR.2008.328
34. Ozdamar B, Bose R, Barrios-Rodiles M, Wang H-R, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFß receptors controls epithelial cell plasticity. Science (2005) 307:1603–9. doi: 10.1126/science.1105718
35. Meyer C, Godoy P, Bachmann A, Liu Y, Barzan D, Ilkavets I, et al. Distinct role of endocytosis for smad and non-smad TGF-β signaling regulation in hepatocytes. J Hepatol (2011) 55(2):369–78. doi: 10.1016/j.jhep.2010.11.027
36. Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, et al. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med (2013) 5(195):195ra94. doi: 10.1126/SCITRANSLMED.3006448
37. Felgentreff K, Siepe M, Kotthoff S, von Kodolitsch Y, Schachtrup K, Notarangelo LD, et al. Severe eczema and hyper-IgE in loeys-dietz-syndrome - contribution to new findings of immune dysregulation in connective tissue disorders. Clin Immunol (2014) 150:43–50. doi: 10.1016/j.clim.2013.11.008
38. Aubart M, Gobert D, Aubart-Cohen F, Detaint D, Hanna N, D’Indya H, et al. Early-onset osteoarthritis, charcot-Marie-Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: An unrecognized and life threatening condition. PloS One (2014) 9:e96387. doi: 10.1371/journal.pone.0096387
39. Laterza D, Ritelli M, Zini A, Colombi M, Dell’acqua ML, Vandelli L, et al. Novel pathogenic TGFBR1 and SMAD3 variants identified after cerebrovascular events in adult patients with loeys-dietz syndrome. Eur J Med Genet. (2019) 62(10):103727. doi: 10.1016/j.ejmg.2019.103727
40. Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan syndrome and related disorders: 25 years of gene discovery. Hum Mutat (2016) 37:524–31. doi: 10.1002/HUMU.22977
41. Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D, et al. Mutations in the TGF-β repressor SKI cause shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet (2012) 44:1249–54. doi: 10.1038/ng.2421
42. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature (1992) 359:693–9. doi: 10.1038/359693a0
43. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci (1993) 90:770–4. doi: 10.1073/PNAS.90.2.770
44. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-β1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med (2005) 201:1061–7. doi: 10.1084/jem.20042276
45. Li MO, Wan YY, Flavell RA. T Cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity (2007) 26:579–91. doi: 10.1016/j.immuni.2007.03.014
46. Konkel JE, Maruyama T, Carpenter AC, Xiong Y, Zamarron BF, Hall BE, et al. Control of the development of CD8αα+ intestinal intraepithelial lymphocytes by TGF-β. Nat Immunol (2011) 12:312–9. doi: 10.1038/ni.1997
47. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen WJ. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol (2008) 9:632–40. doi: 10.1038/ni.1607
48. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity (2006) 25:455–71. doi: 10.1016/j.immuni.2006.07.011
49. Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity (2006) 25:441–54. doi: 10.1016/j.immuni.2006.07.012
50. Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell (1998) 92:797–808. doi: 10.1016/s0092-8674(00)81407-5
51. Nomura M, Li E. Smad2 role in mesoderm formation, left–right patterning and craniofacial development. Nature (1998) 393:786–90. doi: 10.1038/31693
52. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-β–mediated regulation of regulatory T plasticity and Th1 development. J Immunol (2010) 185:842–55. doi: 10.4049/jimmunol.0904100
53. Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol (1999) 19:2495–504. doi: 10.1128/MCB.19.4.2495
54. Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J (1999) 18:1280–91. doi: 10.1093/emboj/18.5.1280
55. Shen T, Li S, Cai L-D, Liu J-L, Wang C-Y, Gan W-J, et al. Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget (2018) 9:12035–49. doi: 10.18632/ONCOTARGET.23925
56. Van Gucht I, Meester JAN, Bento JR, Bastiaansen M, Bastianen J, Luyckx I, et al. A human importin-β-related disorder: Syndromic thoracic aortic aneurysm caused by bi-allelic loss-of-function variants in IPO8. Am J Hum Genet (2021) 108:1115–25. doi: 10.1016/j.ajhg.2021.04.019
57. Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science (1998) 282:2092–5. doi: 10.1126/science.282.5396.2092
58. Guerrerio AL, Frischmeyer-Guerrerio PA, Huang C, Wu Y, Haritunians T, McGovern DPB, et al. Increased prevalence of inflammatory bowel disease in patients with mutations in genes encoding the receptor subunits for TGFβ. Inflammation Bowel Dis (2016) 22:2058–62. doi: 10.1097/MIB.0000000000000872
59. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet (2005) 37:275–81. doi: 10.1038/NG1511
60. van de Laar IMBH, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JMA, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet (2011) 43:121–6. doi: 10.1038/ng.744
61. Boileau C, Guo D-C, Hanna N, Regalado ES, Detaint D, Gong L, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of marfan syndrome. Nat Genet (2012) 44:916–21. doi: 10.1038/ng.2348
62. Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet (2012) 44:922–7. doi: 10.1038/ng.2349
63. Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol (2015) 65:1324–36. doi: 10.1016/j.jacc.2015.01.040
64. Lin J, Vora M, Kane NS, Gleason RJ, Padgett RW. Human marfan and marfan-like syndrome associated mutations lead to altered trafficking of the type II TGFβ receptor in caenorhabditis elegans. PloS One (2019) 14:e0216628. doi: 10.1371/journal.pone.0216628
65. Bolar N, Van Laer L, Loeys BL. Marfan syndrome: From gene to therapy. Curr Opin Pediatr (2012) 24:498–504. doi: 10.1097/MOP.0B013E3283557D4C
66. Zhou D, Feng H, Yang Y, Huang T, Qiu P, Zhang C, et al. hiPSC modeling of lineage-specific smooth muscle cell defects caused by TGFBR1 A230T variant, and its therapeutic implications for loeys-Dietz syndrome. Circulation (2021) 144:1145–59. doi: 10.1161/CIRCULATIONAHA.121.054744
67. Konkel JE, Jin W, Abbatiello B, Grainger JR, Chen W. Thymocyte apoptosis drives the intrathymic generation of regulatory T cells. Proc Natl Acad Sci U.S.A. (2014) 111:E465–73. doi: 10.1073/pnas.1320319111
68. Naviglio S, Arrigo S, Martelossi S, Villanacci V, Tommasini A, Loganes C, et al. Severe inflammatory bowel disease associated with congenital alteration of transforming growth factor beta signaling. J Crohn’s Colitis (2014) 8:770–4. doi: 10.1016/J.CROHNS.2014.01.013/2/8-8-FIG022.JPEG
69. Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet (1995) 11:409–14. doi: 10.1038/ng1295-409
70. Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet (1995) 11:415–21. doi: 10.1038/ng1295-415
71. Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development (1997) 124:2659–70. doi: 10.1242/dev.124.13.2659
72. Dai F, Chang C, Lin X, Dai P, Mei L, Feng X-H. Erbin inhibits transforming growth factor beta signaling through a novel smad-interacting domain. Mol Cell Biol (2007) 27:6183–94. doi: 10.1128/MCB.00132-07
73. Sflomos G, Kostaras E, Panopoulou E, Pappas N, Kyrkou A, Politou AS, et al. ERBIN is a new SARA-interacting protein: Competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J Cell Sci (2011) 124:3209–22. doi: 10.1242/jcs.062307
74. Droghini HR, Abonia JP, Collins MH, Milner JD, Lyons JJ, Freeman AF, et al. Targeted IL-4Rα blockade ameliorates refractory allergic eosinophilic inflammation in a patient with dysregulated TGF-β signaling due to ERBIN deficiency. J Allergy Clin Immunol Pract (2022) 10:1903–6. doi: 10.1016/j.jaip.2022.01.012
75. Rachmin I, Tshori S, Smith Y, Oppenheim A, Marchetto S, Kay G, et al. Erbin is a negative modulator of cardiac hypertrophy. Proc Natl Acad Sci (2014) 111:5902–7. doi: 10.1073/pnas.1320350111
76. Hu Y, Chen H, Duan C, Liu D, Qian L, Yang Z, et al. Deficiency of erbin induces resistance of cervical cancer cells to anoikis in a STAT3-dependent manner. Oncogenesis (2013) 2:e52–2. doi: 10.1038/oncsis.2013.18
77. Xu L, Yao X, Chen X, Lu P, Zhang B, Ip YT. Msk is required for nuclear import of TGF-β/BMP-activated smads. J Cell Biol (2007) 178:981–94. doi: 10.1083/JCB.200703106
78. Yao X, Chen X, Cottonham C, Xu L. Preferential utilization of Imp7/8 in nuclear import of smads. J Biol Chem (2008) 283:22867–74. doi: 10.1074/JBC.M801320200
79. Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol (2007) 8:195–208. doi: 10.1038/nrm2114
80. Bertoli-Avella AM, Kandaswamy KK, Khan S, Ordonez-Herrera N, Tripolszki K, Beetz C, et al. Combining exome/genome sequencing with data repository analysis reveals novel gene–disease associations for a wide range of genetic disorders. Genet Med (2021) 23:1551–68. doi: 10.1038/S41436-021-01159-0
81. Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta (2007) 1773:1341–8. doi: 10.1016/j.bbamcr.2006.12.009
82. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol (2007) 19:142–9. doi: 10.1016/j.ceb.2007.02.001
83. Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, et al. Transcription factor smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity (2011) 34:741–54. doi: 10.1016/j.immuni.2011.02.021
84. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
85. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9:641–9. doi: 10.1038/ni.1610
86. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol (2008) 9:650–7. doi: 10.1038/ni.1613
87. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature (2008) 454:350–2. doi: 10.1038/nature07021
88. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest (2001) 108:601–9. doi: 10.1172/JCI12821
89. Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and crohn’s disease. N Engl J Med (2015) 372:1104–13. doi: 10.1056/NEJMoa1407250
90. Bewtra M, Lichtenstein GR. Mongersen and SMAD-7 inhibition, not a lucky 7 for patients with IBD: When trial design is as important as disease therapy. Am J Gastroenterol (2020) 115:687–8. doi: 10.14309/ajg.0000000000000564
Keywords: TGF-β, immune dysregulation, Loeys-Dietz syndrome, TGF-β signalopathies, inflammatory bowel disease
Citation: Rodari MM, Cerf-Bensussan N and Parlato M (2022) Dysregulation of the immune response in TGF-β signalopathies. Front. Immunol. 13:1066375. doi: 10.3389/fimmu.2022.1066375
Received: 10 October 2022; Accepted: 11 November 2022;
Published: 09 December 2022.
Edited by:
Elena Wen-Yuan Hsieh, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Peter Hsu, Children’s Hospital at Westmead, AustraliaRong Tang, Xiangya Hospital, Central South University, China
Copyright © 2022 Rodari, Cerf-Bensussan and Parlato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianna Parlato, bWFyaWFubmEucGFybGF0b0BpbnNlcm0uZnI=