 Hossein Halimi
Hossein Halimi Shirin Farjadian
Shirin Farjadian- Department of Immunology, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran
Based on the structural and signaling roles of cholesterol, which are necessary for immune cell activity, high concentrations of cholesterol and its metabolites not only trigger malignant cell activities but also impede immune responses against cancer cells. To proliferate and evade immune responses, tumor cells overcome environmental restrictions by changing their metabolic and signaling pathways. Overexpression of mevalonate pathway enzymes and low-density lipoprotein receptor cause elevated cholesterol synthesis and uptake, respectively. Accordingly, cholesterol can be considered as both a cause and an effect of cancer. Variations in the effects of blood cholesterol levels on the outcome of different types of cancer may depend on the stage of cancer. However, positive effects of cholesterol-lowering drugs have been reported in the treatment of patients with some malignancies.
Introduction
In addition to the physical role of fatty acids, they play multifaceted roles in cancer. Depending on their composition, lipids may have inflammatory or anti-inflammatory impacts on the tumor microenvironment (1, 2).
The functions of cholesterol in cancer were first noted in the 1900s. Webb reported that cholesterol crystallization in normal cells led to malignancy (3), and other researchers subsequently found alterations in blood cholesterol levels in patients with cancer (4, 5). Recently, cholesterol was considered not only a risk factor, but also a prognostic factor in cancer (6), as high-density lipoprotein (HDL) cholesterol was reported to have impact on patient survival and cancer relapse (7). Indeed, cholesterol accumulation has been observed in different tumor cells (8, 9), through the upregulation of cholesterol biosynthesis or via increased uptake. Many of cancer cells overexpress low-density lipoprotein receptor (LDLR) compared to normal cells, and evade negative feedback mechanisms for enhancing cholesterol uptake – a phenomenon which leads to their rapid proliferation (10–13).
Cholesterol is crucial to triggering immune responses as one of the components of the lipid raft, which can recruit receptors and signaling molecules (14). Proteins are also localized in the lipid raft via the palmitoylation or glycosylphosphatidylinositol (GPI) anchor (15). As a result of cholesterol accumulation, tumor cells have higher membrane levels of cholesterol, and this structure plays a role in tumor cell growth, adhesion, migration, invasion and apoptosis (16).
Indeed, Cholesterol metabolism is associated with cancer stemness. Cancer stem cells are a population within cancer cells related to progression and metastasis (17). Ehmsen et al. demonstrated that raised de novo cholesterol synthesis is a feature of breast cancer stem cells and inhibiting cholesterol synthesis impedes the growth of cancer stem cells (18).
Cholesterol is a signaling molecule that in turn affects other signaling pathways such as PI3K and Hedgehog (19). The metabolism of cholesterol impacts the cancer-associated immune system through altering responses such as the induction of exhausted T cells, changing macrophage phenotype, and affecting antigen presentation by dendritic cells (DCs) (20).
Targeting cholesterol biosynthesis or uptake is a new area in cancer therapy. In efforts to regulate blood cholesterol levels as well as research in other treatment modalities such as immunotherapy, further insights into cholesterol biosynthesis and uptake may be applied in strategies to prevent tumor relapse or metastasis, and may extend patient survival (21).
Cholesterol synthesis
Although most human cells are able to synthesize cholesterol, the liver is the leading site of de novo cholesterol synthesis. This pathway comprises of more than 20 enzymes (22), and starts with the conversion of two acetyl-CoA by thiolase and then further conversion of acetyl-CoA into 3-hydroxy-3-methylglutaryl (HMG)-CoA. This is followed by the production of other intermediates such as isopentyl pyrophosphate, farnesyl pyrophosphate, squalene, lanosterol, and finally cholesterol (20). Elucidating the cholesterol synthesis pathway is important because mediators and intermediate enzymes affect the cancer process. For example, C-terminal farnesylation of proteins leads to the formation of a hydrophobic tail, thus enabling lipid–protein and protein–protein interaction (23–25). Indeed, farnesylation is a necessary step for RAS oncoprotein activation and cell membrane binding. Moreover, inhibition of this process was shown to have anti-tumor effects (26, 27). Thus, current evidence suggests that deeper knowledge of the cholesterol synthesis pathway can point the way to novel cancer treatments and the potential to extend patients’ lifespans.
Cancer-associated cholesterol metabolism
Cholesterol concentrations within the cell are precisely controlled through the regulation of absorption and synthesis (Figure 1). Cholesterol absorption is firmly controlled by regulating the amount of its receptor. High levels of cholesterol prevent the activation of SREBPs (sterol regulatory-element binding proteins), i.e. membrane-bound transcription factors that activate genes involved in cholesterol synthesis, thereby reducing LDLR expression (28, 29). In addition, PCSK9 (proprotein convertase subtilisin/kexin type 9) binds to LDLR and thus triggers its breakdown (30). Cholesterol synthesis is also tightly controlled, as high cholesterol levels cause the degradation of squalene monooxygenase and HMG-CoA reductase, which catalyzes the conversion of squalene to 2,3 oxidosqualene and HMG-CoA to mevalonate as the precursors of cholesterol (31, 32).
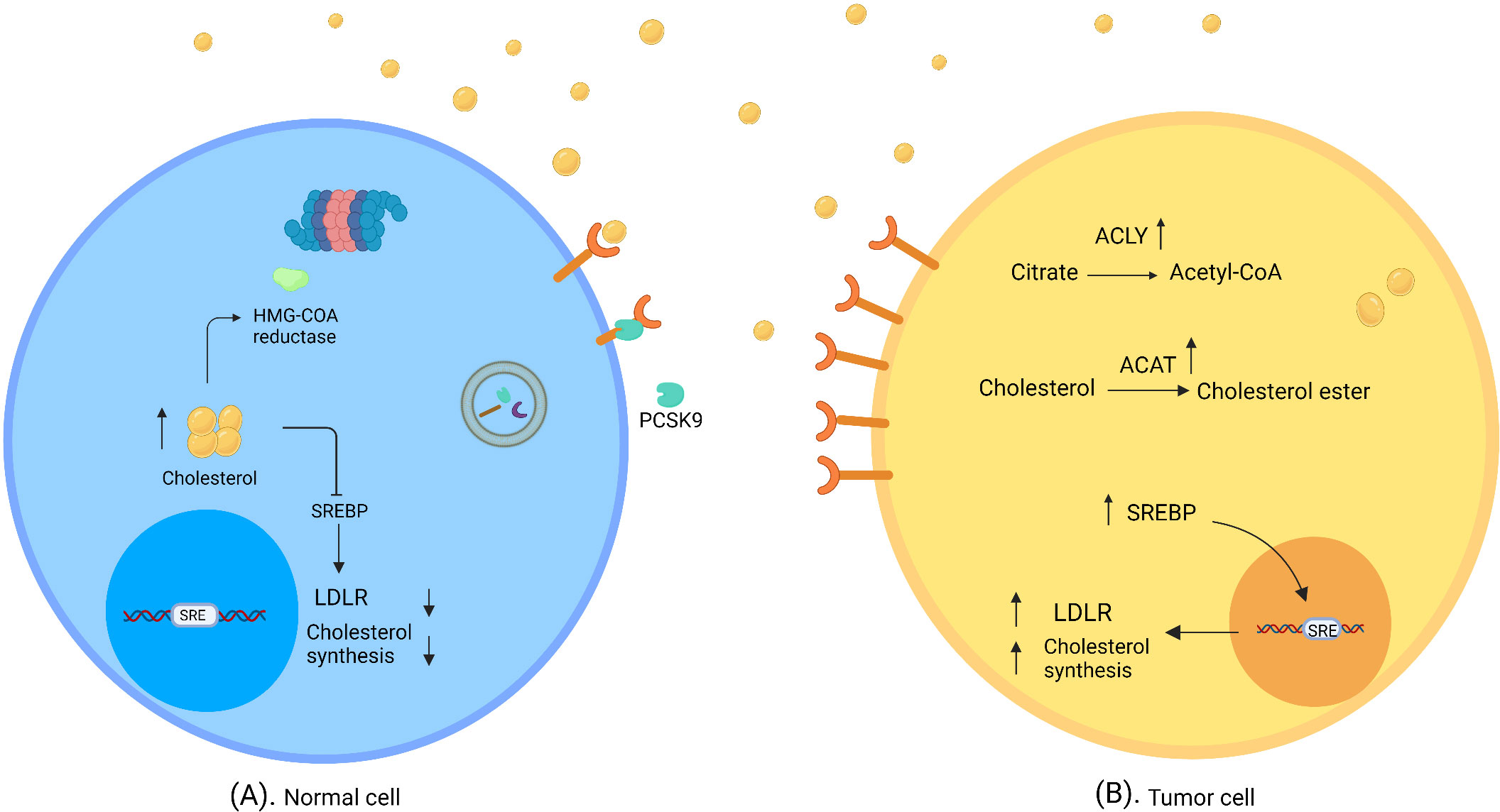
Figure 1 Normal and tumor cells in cholesterol-rich environment. (A) In normal cells, a high concentration of cholesterol triggers regulatory mechanisms of cholesterol synthesis and uptake, which downregulate SREBP and degrade HMG- CoA reductase. (B) Tumor cells evade feedback mechanisms in a cholesterol-rich environment. SREBP shows high activity; thus LDLR expression and/or cholesterol synthesis are elevated. ACAT catalyzes the synthesis of cholesterol ester; therefore, cholesterol accumulates in tumor cells. ATP citrate lyase converts citrate to acetyl-CoA and induces fatty acid synthesis (created with biorender.com).
To utilize more cholesterol, cancer cells evade the feedback mechanisms of cholesterol inhibition. In this connection it was found that prostate cancer cells overexpress LDLR and SREBP in cholesterol-rich conditions in the absence of cholesterol regulatory mechanisms (10).
Cholesterol absorption is more energy-saving in cancer cells than de novo synthesis. Accordingly, in large-cell lymphoma, malignant cells rely more on cholesterol uptake through the overexpression of LDLR rather than de novo synthesis (33). This might be explained by the higher LDLR expression in inflammatory conditions, which enables tumor cells to accumulate more cholesterol (34). In addition, prostate cancer cells were reported to accumulate excess cholesterol from the environment through the overexpression of HDLR, SR-B1 (35); however, advanced-stage prostate cancer cells were reported to be dependent on cholesterol synthesis (36).
Another factor that supports cancer progression through cholesterol synthesis is ATP citrate lyase (ACLY) enzyme, which converts citrate to acetyl-CoA. Overexpression of this enzyme has been reported in different kinds of tumor (37).
Because of insufficient vascularization and a high rate of growth, some cancers such as glioblastoma exist in a hypoxic and lipid-limited microenvironment. In this condition, SREBP2 overexpression induces cholesterol synthesis (38, 39).
Cholesterol ester is a storage form of cholesterol catalyzed by acyl-CoA: cholesterol acyltransferase (ACAT), which plays an important role in cancer biology. Cholesterol acyltransferase overexpression has been detected in cancer tissues, and it appears that cancer cells use this storage source for their proliferation (40). In addition, lysosomal acid lipase breaks down cholesterol ester to generate fatty acids; the overexpression of this enzyme and its relation to cancer progression have been reported in renal cell carcinoma (41).
Oxysterols, i.e. oxygenated sterol derivatives such as different forms of hydroxycholesterols, are bioactive molecules which affect cancer cell behavior (42). A role for 27-hydroxycholesterol has been shown in breast cancer progression through VEGF overexpression and STAT3 activation (43).
Cholesterol and cancer cell signaling
Hedgehog pathway
The Hedgehog signaling pathway is an essential process which enables appropriate cell differentiation during vertebrate embryonic development. It is also necessarily activated in circumstances such as wound healing and tissue repair. It has been shown that this pathway is involved in cancer growth, and that cholesterol plays a crucial role in the reactivation of this pathway through binding to smoothened transmembrane protein (44–46).
Wnt pathway
The Wnt pathway is responsible for stem cell renewal and organogenesis. This pathway is also involved in the development of malignancies such as leukemia and gastrointestinal cancer. The Wnt protein interacts with a heterodimeric receptor including Frizzled and LRP5/6 proteins. Dishevelled and other signaling molecules are then recruited to the receptor and transduce the signal through activated β-catenin (47, 48). It has been indicated that cholesterol can bind to the PDZ domain of scaffold proteins including NHERF1/EBP50, and then activate Wnt signaling pathways (49, 50). An additional way in which cholesterol plays a role in cancer biology is by triggering the Wnt pathway through its interaction with Dishevelled scaffold protein (51).
mTORC1 pathway
The mTORC1 pathway is a key factor in cell growth and survival. It is also suggested that mTORC1 is involved in cancer cell proliferation. This signaling pathway is responsible for lipid synthesis in malignant cells via SREBP2 upregulation (52, 53).
Cholesterol can increase tumor cell proliferation through activation of the mTORC1 pathway at the lysosomal surface by SLC38A9 (solute carrier family 38 member 9), a lysosomal transmembrane protein that senses cholesterol in addition to arginine, with independent motifs (54).
Relevance of cholesterol and cancer-associated immune cell
Although cholesterol is essential for the formation of the immunological synapse through receptor clustering and recruitment of signaling molecules, high amounts cholesterol have a negative effect on immune responses (55) (Figure 2).
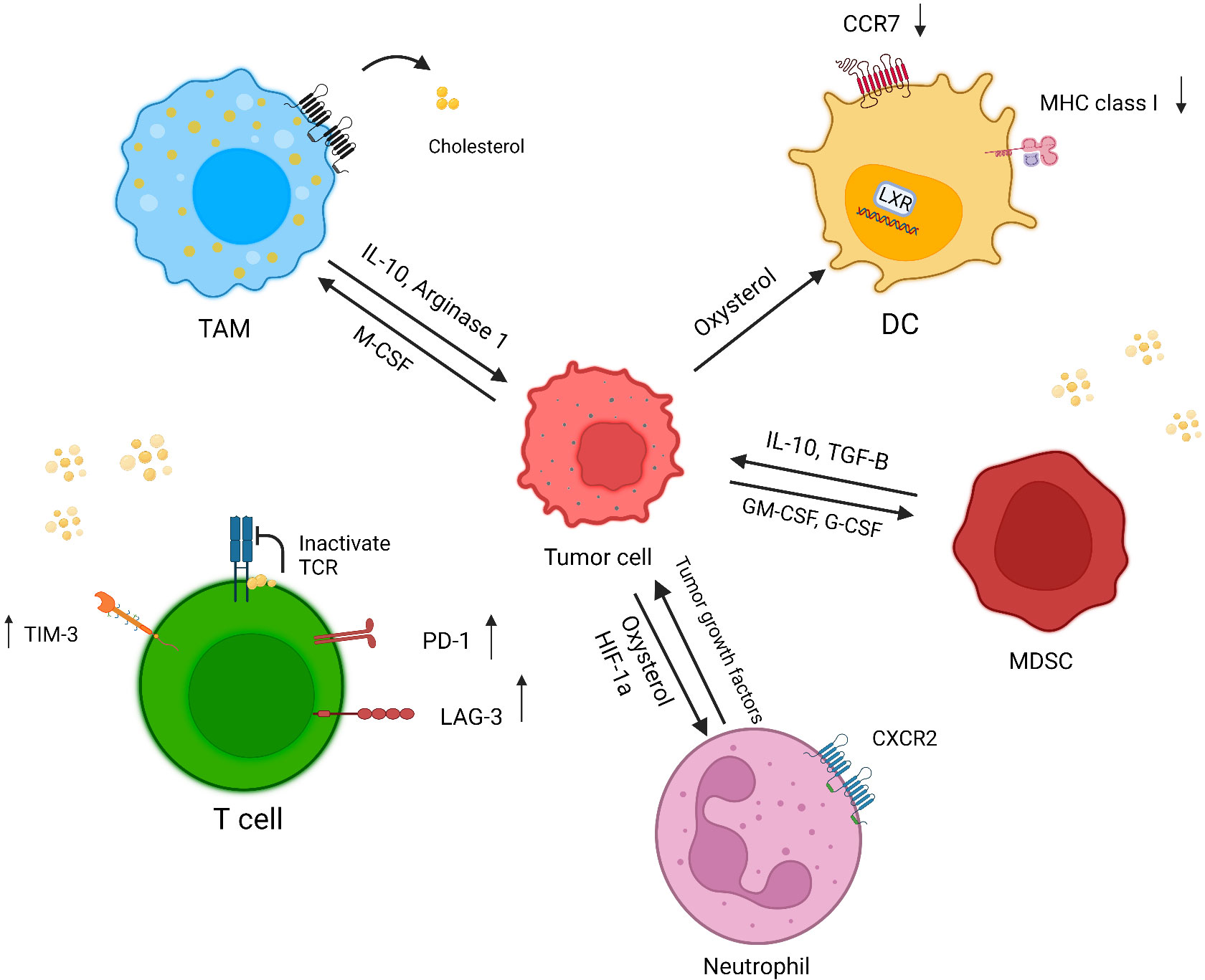
Figure 2 Effect of a cholesterol-rich tumor microenvironment on immune cells. Increased cholesterol efflux from macrophage membranes induces the M2 phenotype in tumor resident macrophages. In cholesterol-rich area, T cells overexpress exhaustion markers, and cholesterol binds to the TCR β-chain and puts T cells in a resting state. Oxysterol recruits CXCR2-positive neutrophils, which in turn induces angiogenesis and tumor growth. Tumor-released factors induce MDSC suppressive responses through cholesterol accumulation. The accumulation of cholesterol in DCs causes a reduction in MHC class I and CCR7 expression, which diminishes the immune response against tumor cells (created with biorender.com).
Macrophages
Macrophages are innate immune cells with a dual effect on cancer immunity. These cells show plasticity depending on the tumor microenvironment. At an advanced stage of cancer, macrophages convert to the M2 phenotype with tumor-promoting function (56).
Cholesterol has a pivotal effect on macrophage phenotype and function. Goossens et al. showed increased cholesterol efflux from the macrophage membrane and depletion of cholesterol from the lipid raft induced the tumor-promoting phenotype in tumor resident macrophages in a mouse model of ovarian cancer (57). Park et al. also revealed tumor-derived M-CSF activated PPARβ/δ (peroxisome proliferator-activated receptor belonging to the nuclear hormone receptor family) through increased fatty acid synthesis, thereby augmenting IL-10 and arginase 1 expression in macrophages, which resulted in cancer invasion and angiogenesis (58).
Dendritic cells
MHC molecules play a pivotal role in tumor antigen presentation to immune cells and the induction of anti-tumor responses (59, 60). Dendritic cells are innate immune cells with a central role in triggering an anti-tumor response by presenting tumor antigens to T cells. Cancer cells can downmodulate the function of DCs by increasing the concentration of cholesterol (61).
Oxysterol secretion by tumor cells affects LXRs (liver x receptors) in DCs, which leads to CCR7 reduction, hampering the migration of DCs to lymph nodes and thus reducing T cell priming (62, 63). In addition, a high amount of lipid in the tumor microenvironment increases the uptake and accumulation of lipids in DCs, which in turn reduces tumor antigen presentation by these cells (64). It is also showed that oxidized lipids reduce the expression of MHC class I on DCs (65).
T lymphocytes
Cholesterol has a dual effect on T cell function. During T cell responses, SREBPs exert an effect on cell proliferation through cholesterol synthesis (66). The results of one earlier study showed that cholesterol can bind to the transmembrane part of the TCR β-chain and keep T cells in the resting state (67). In addition, the tumor microenvironment is a cholesterol-rich milieu which leads to endoplasmic reticulum stress; as a result, T cells express more inhibitory receptors such as Tim-3, LAG3 and PD-1, and become exhausted. On the other hand cholesterol-lowering drugs such as atorvastatin were reported to decrease checkpoint inhibitors on T cells (68, 69). Although both CTLs and target cells are exposed to perforin within the synapse, high cholesterol content in the CTL cell membrane protects them against lysis by perforin (70).
In conclusion, although a sufficient concentration of cholesterol is needed for an efficient immune response, high levels of cholesterol can disrupt immune cell functioning (55).
Neutrophils
There is cross-talk between cancer cells and neutrophils, the latter of which can play an anti-tumor or tumor-promoting role. In this connection, it was shown that microenvironmental conditions such as lipid level can change neutrophil functions (71).
Tumor-secreted oxysterols recruit CXCR2+ neutrophils that can release tumor growth factors (72, 73). In addition, hypoxia-induced Hif1 can elevate the expression of CYP46A1 (cytochrome P450 family 46 subfamily A member 1). This enzyme plays a role in cholesterol metabolism in the brain, and converts cholesterol to 24s-HC oxysterol, which can pass through the blood–brain barrier. One study showed that lipid accumulation recruited proangiogenic neutrophils in a pancreatic neuroendocrine tumor model (74).
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) originate from immature or myeloid progenitor cells, and potentiate suppressive responses. Hence these cells can support cancer growth through regulatory mechanisms such as interference with T cell trafficking, release of suppressive factors (IL-10 and TGF-β), and depletion of essential T cell amino acids. Furthermore, tumor cells can induce MDSC responses through lipid-mediated mechanisms (75).
MDSCs are heterogeneous cells which are divided into two major groups: polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). Lectin-type oxidized LDL receptor 1 (Lox-a1) has been identified as one of the newest PMN-MDSC markers, which is evidence of a probable suppressive effect of cholesterol on these cells (76). Moreover, tumor-secreted G-CSF and GM-CSF induce the expression of lipid transporters on MDSCs through STAT3 and STAT5 signaling; consequently, elevated lipid absorption and accumulation trigger a suppressive response of MDSCs (77), Table 1.
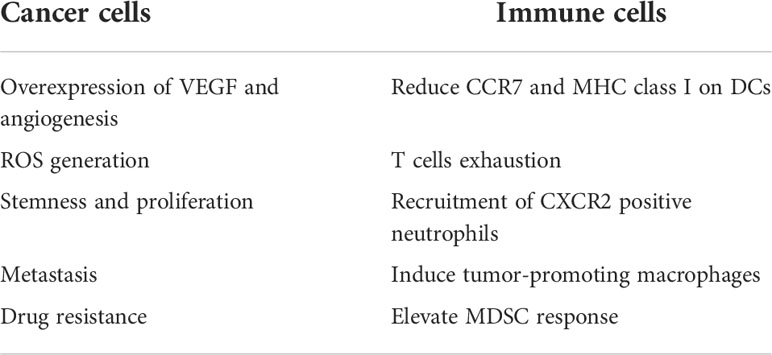
Table 1 Effects of high cholesterol levels on cancer cells and immune cells.
The effect of cholesterol on different types of cancer
Hypercholesterolemia is considered a risk factor for the induction of different types of cancer. Moreover, high cholesterol levels have also been shown to play a role in cancer progression and metastasis.
Colon cancer
Blood concentrations of cholesterol in patients with colon cancer were reported to be lower than in healthy individuals (78, 79). The higher expression of LDLR by colon cancer cells results in decreased blood cholesterol levels, and colon cancer surgery can elevate cholesterol levels after 1 year (11).
LDL increases intestinal inflammation and the proliferation of malignant colon cells through the generation of reactive oxygen species (ROS) and activation of the MAPK signaling pathway. In addition, LDL promotes malignant colon cell migration and induces stemness genes such as Sox2 and Oct4 in these cells (13).
Breast cancer
There is controversy regarding the relevance of cholesterol levels in breast cancer, although cholesterol is generally considered a risk factor. A recent report noted increased body mass index (BMI) as an important factor associated with breast cancer (80). Moreover, a cholesterol-rich diet reportedly elevates the risk of breast cancer. In this study, they found that high levels of cholesterol in patients with breast cancer are related to a poor outcome and a higher malignant cell proliferation rate (81).
In this connection, abundant expression of LDLR was seen in breast cancer cells (82), and LDL was reported to have an impact on breast cancer cell proliferation in association with overexpression of Akt and ERK pathway intermediates (83). Moreover, overexpression of CYP27A1 (sterol 27-hydroxylase), which produces 27-hydroxycholesterol, was reported in patients with breast cancer. This oxysterol downregulates the expression of P53,which in turn results in epithelial–mesenchymal transition and initiates the metastatic cascade (84, 85).
Prostate cancer
There is controversy regarding the correlation between blood cholesterol levels and prostate cancer (86–88). High cholesterol synthesis and elevated LDLR expression may play a role in the outcome of this cancer (36); in this connection, high LDLR expression has been demonstrated in prostate cancer. Also, elevated amounts of cholesterol and cyclin E overexpression were detected inside of nucleus; hence cholesterol may play a role in the cell cycle of prostate cancer cells (89). In addition, androgen is necessary for prostate cancer cell proliferation, and prostate cancer cells were reported to gradually gain androgen synthesis potential by themselves in cholesterol-rich conditions (90, 91).
Hepatocellular cancer
The liver is the primary source of cholesterol synthesis in the body (92). Many studies have documented high cholesterol concentrations in patients with hepatocellular cancer. It has been suggested that malignant cells are responsible for elevated cholesterol levels in these patients (93, 94). In this connection, the impairment of cholesterol feedback mechanisms was shown to have a substantial impact on lipid production in malignant liver cells (95).
Lung cancer
It is currently hard to delineate specific correlations between lung cancer and blood cholesterol levels. The results of some studies showed that patients with low cholesterol levels have lower survival (96, 97). In contrast, one report suggested that elevated cholesterol is a risk factor for lung cancer (98). In this connection, a recent study suggested that moderate cholesterol levels can prevent lung cancer, whereas high and low cholesterol concentrations are both risk factors for this malignancy (99). The results of an in vitro study showed that 25-hydroxycholesterol promoted lung adenocarcinoma cell migration and invasion (100). It remains to be seen whether or not elevated cholesterol is a hallmark of lung cancer.
Pancreatic cancer
The association between blood cholesterol concentration and pancreatic cancer remains unclear. Although one study found high cholesterol intake to correlated with pancreatic cancer (101), other research, in contrast, reported that low cholesterol levels result in an increased risk (102).
Overexpression of ACAT1 is related to cholesterol ester accumulation in tumor cells and unwanted cell survival (103); furthermore, the higher absorption of cholesterol by cancer cells than by normal cells is associated with LDLR overexpression (12).
Ovarian cancer
Contradictory results have appeared regarding blood cholesterol levels and ovarian cancer (104, 105). In one ovarian cancer cell line, high ACAT1 expression was shown, leading to cholesterol ester accumulation. Furthermore, ACAT1 inhibition was reported to suppress cancer cell proliferation (106). Other research documented the importance of HMG-CoA reductase in ovarian cancer, finding that genetically proxied inhibition of HMG-CoA reductase was related to a lower rate of ovarian cancer (107).
Hematologic malignancies
Studies of the relationship between blood cholesterol levels and leukemia have yielded conflicting results. Lower cholesterol concentrations were reported in patients with chronic lymphocytic leukemia (CLL) and acute lymphocytic leukemia (ALL) than in healthy controls (108, 109), but a different report documented high cholesterol levels in patients with CLL (110). The results of another study demonstrated that in patients with CLL, elevated SREBP2 expression resulted in increased LDLR, thus cholesterol accumulation in the tumor cell cytoplasm was noted as a possible cause of this cancer (111).
The role of anticholesteremic agents in cancer treatment and patient survival
Cholesterol serves as the component involved in drug resistance in cancer (6). Drug-resistance cells elevate cholesterol uptake and biosynthesis. Increased cholesterol content in cancer cell membrane changes the signaling pathway because of the interaction of cholesterol and different receptors on the surface of these cells. Furthermore, high cholesterol levels convert the entrance drug pathway in malignant cells (112), for example gefitinib-resistant non-small cell lung cancer (NSCLC) cell lines possess higher cholesterol in their lipid raft than gefitinib-sensitive cell lines, as gefitinib exerts a better function on cholesterol-depleted cells (113).
Taken together, surveying a drug that adjusts cholesterol levels for achieving practical and effective treatment.
Statins (HMG-CoA reductase inhibitors) are a group of drugs that can help to lower LDL cholesterol. The advantages of statins for cancer monotherapy or combination therapy have been widely reported. It is suggested that statins reduce cancer cell growth through AKT suppression (114), promote cancer cell apoptosis through Bax overexpression and Bcl2 inhibition (115), and also exert an effect on cell cycle regulatory elements and thus reduce cancer cell proliferation (116).
Numerous studies have documented the beneficial effects of statins on cancer recurrence and mortality (117); for example, lipophilic statins were found to decrease breast cancer recurrence (118).
Other research showed that statin together with palliative care can reduce the mortality rate in patients with hepatocellular carcinoma (119). In addition, statins were found to reduce the mortality rate of colorectal cancer (120, 121). Another study reported beneficial effects of statins on cancer mortality both before and after diagnosis (122). However, statins may have no benefits for patients with advanced cancer (123).
Regarding the interaction of statins with other treatments for cancer, cholesterol-lowering drugs were found to be safe for use along with immunotherapy (124), chemotherapy (125), and radiotherapy (126).
Conclusion
Cholesterol has a dual effect on the immune response depending on its concentration. High serum levels of cholesterol are related to a higher risk of cancer progression because of their effect on signaling pathways. Tumor cells have complex interactions with their surrounding environment; in generally, tumor cell metabolic pathways are modified by increased cholesterol synthesis and/or uptake. Cholesterol-rich environments change immune cell phenotypes and functions, such that these cells may support cancer cell survival.
Reports of the correlations between blood cholesterol levels and different types of cancer are conflicting. Elevated cholesterol uptake by malignant cells may be associated with low blood cholesterol levels, whereas high blood cholesterol levels in patients with cancer may be viewed as a result of increased cholesterol synthesis in tumor cells. Therefore, cholesterol can potentially act as a cause or an effect of cancer, although the type and stage of the tumor should not be overlooked.
In light of the effects of high cholesterol levels in cancer biology, prescribing cholesterol-lowering drugs may be valuable in cancer therapy. To reduce the side effects of these drugs and achieve better outcomes, targeting the pathways of cancer cell cholesterol metabolism is a potentially helpful avenue for further research.
Author contributions
Both authors contributed equally to this article and approved the submitted version.
Acknowledgments
We thank Dr. P. Mokaram, Professor of Biochemistry, for her critical review of this manuscript and K. Shashok (AuthorAID in the Eastern Mediterranean) for improving the use of English.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fritsche KL. The science of fatty acids and inflammation. Adv Nutr (2015) 6(3):293S–301S. doi: 10.3945/an.114.006940
2. Amirkhani Z, Alavi M, Kalani M, Alavianmehr A, Farjadian S. Immunomodulatory effects of omega-3 fatty acids in patients with differentiated thyroid cancer before or after radioiodine ablation. Iran J Immunol (2022) 19(1):71–90. doi: 10.22034/IJI.2022.92648.2163
3. Webb J. Cancer, its nature and its treatment. Lancet (1901) 158(4076):976–8. doi: 10.1016/S0140-6736(01)73258-8
4. Mattick WL, Buchwald K. Blood cholesterol studies in cancer, II. with investigations as to possible diagnostic relations. J Cancer Res (1928) 12(3):236–45. doi: 10.1158/jcr.1928.236
5. Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS. Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years' follow up. BMC Cancer (2012) 12:25. doi: 10.1186/1471-2407-12-25
6. Yan A, Jia Z, Qiao C, Wang M, Ding X. Cholesterol metabolism in drug−resistant cancer. Int J Oncol (2020) 57(5):1103–15. doi: 10.3892/ijo.2020.5124
7. Zhou P, Li B, Liu B, Chen T, Xiao J. Prognostic role of serum total cholesterol and high-density lipoprotein cholesterol in cancer survivors: A systematic review and meta-analysis. Clin Chim Acta (2018) 477:94–104. doi: 10.1016/j.cca.2017.11.039
8. Sharma B, Gupta V, Dahiya D, Kumar H, Vaiphei K, Agnihotri N. Clinical relevance of cholesterol homeostasis genes in colorectal cancer. Biochim Biophys Acta Mole Cell Biol Lipids (2019) 1864(10):1314–27. doi: 10.1016/j.bbalip.2019.06.008
9. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab (2014) 19(3):393–406. doi: 10.1016/j.cmet.2014.01.019
10. Chen Y, Hughes-Fulford M. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int J Cancer (2001) 91(1):41–5. doi: 10.1002/1097-0215(20010101)91:1<41:AID-IJC1009>3.0.CO;2-2
11. Niendorf A, Nägele H, Gerding D, Meyer-Pannwitt U, Gebhardt A. Increased LDL receptor mRNA expression in colon cancer is correlated with a rise in plasma cholesterol levels after curative surgery. Int J Cancer (1995) 61(4):461–4. doi: 10.1002/ijc.2910610405
12. Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A (2015) 112(8):2473–8. doi: 10.1073/pnas.1421601112
13. Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, et al. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK signaling pathway activation. Cell Physiol Biochem (2017) 42(2):729–42. doi: 10.1159/000477890
14. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol (2000) 1(1):31–9. doi: 10.1038/35036052
15. Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, et al. Cholesterol and its metabolites in tumor growth: Therapeutic potential of statins in cancer treatment. Front Endocrinol (2019) 9:807. doi: 10.3389/fendo.2018.00807
16. Vona R, Iessi E, Matarrese P. Role of cholesterol and lipid rafts in cancer signaling: a promising therapeutic opportunity? Front Cell Dev Biol (2021) 9:622908. doi: 10.3389/fcell.2021.622908
17. Kim HY, Bae SJ, Choi JW, Han S, Bae SH, Cheong JH, et al. Cholesterol synthesis is important for breast cancer cell tumor sphere formation and invasion. Biomedicines (2022) 10(8):1908. doi: 10.3390/biomedicines10081908
18. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL, et al. Increased cholesterol biosynthesis is a key characteristic of breast cancer stem cells influencing patient outcome. Cell Rep (2019) 27(13):3927–3938.e6. doi: 10.1016/j.celrep.2019.05.104
19. Deng CF, Zhu N, Zhao TJ, Li HF, Gu J, Liao DF, et al. Involvement of LDL and ox-LDL in cancer development and its therapeutical potential. Front Oncol (2022) 12:803473. doi: 10.3389/fonc.2022.803473.
20. Mayengbam SS, Singh A, Pillai AD, Bhat MK. Influence of cholesterol on cancer progression and therapy. Trans Oncol (2021) 14(6):101043. doi: 10.1016/j.tranon.2021.101043
21. Zhang H, Zhao W, Li X, He Y. Cholesterol metabolism as a potential therapeutic target and a prognostic biomarker for cancer immunotherapy. OncoTargets Ther (2021) 14:3803–12. doi: 10.2147/OTT.S315998
22. Giacomini I, Gianfanti F, Desbats MA, Orso G, Berretta M, Prayer-Galetti T, et al. Cholesterol metabolic reprogramming in cancer and its pharmacological modulation as therapeutic strategy. Front Oncol (2021) 11:682911. doi: 10.3389/fonc.2021.682911
23. Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, et al. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic ras signaling by inducing cytoplasmic accumulation of inactive ras-raf complexes. J Biol Chem (1995) 270(45):26802–6. doi: 10.1074/jbc.270.45.26802
24. Novelli G, D’Apice MR. Protein farnesylation and disease. J inherited Metab Dis (2012) 35(5):917–26. doi: 10.1007/s10545-011-9445-y
25. Kobayashi E, Kondo S, Dochi H, Moriyama-Kita M, Hirai N, Komori T, et al. Protein farnesylation on nasopharyngeal carcinoma, molecular background and its potential as a therapeutic target. Cancers (Basel) (2022) 14(12):2826. doi: 10.3390/cancers14122826
26. Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci U S A (1989) 86(21):8323–7. doi: 10.1073/pnas.86.21.8323
27. Nagasu T, Yoshimatsu K, Rowell C, Lewis MD, Garcia AM. Inhibition of human tumor xenograft growth by treatment with the farnesyl transferase inhibitor B956. Cancer Res (1995) 55(22):5310–4.
28. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem (2018) 87:783. doi: 10.1146/annurev-biochem-062917-011852
29. Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, Lottenberg AM. Molecular pathways underlying cholesterol homeostasis. Nutrients (2018) 10(6):760. doi: 10.3390/nu10060760
30. Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem (2009) 284(42):28856–64. doi: 10.1074/jbc.M109.037085
31. Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem (2003) 278(52):52479–90. doi: 10.1074/jbc.M310053200
32. Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab (2011) 13(3):260–73. doi: 10.1016/j.cmet.2011.01.015
33. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature (2019) 567(7746):118–22. doi: 10.1038/s41586-019-0945-5
34. He M, Zhang W, Dong Y, Wang L, Fang T, Tang W, et al. Pro-inflammation NF-κB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J Exp Clin Cancer Res (2017) 36(1):15. doi: 10.1186/s13046-017-0490-8
35. Schörghofer D, Kinslechner K, Preitschopf A, Schütz B, Röhrl C, Hengstschläger M, et al. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod Biol Endocrinol (2015) 13(1):88. doi: 10.1186/s12958-015-0087-z
36. Stopsack KH, Gerke TA, Andrén O, Andersson SO, Giovannucci EL, Mucci LA, et al. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis (2017) 38(8):806–11. doi: 10.1093/carcin/bgx058
37. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J medicinal Chem (2018) 157:1276–91. doi: 10.1016/j.ejmech.2018.09.001
38. Lewis C, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid-and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene (2015) 34(40):5128–40. doi: 10.1038/onc.2014.439
39. Kondo A, Yamamoto S, Nakaki R, Shimamura T, Hamakubo T, Sakai J, et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep (2017) 18(9):2228–42. doi: 10.1016/j.celrep.2017.02.006
40. Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat (2010) 122(3):661–70. doi: 10.1007/s10549-009-0594-8
41. Wang J, Tan M, Ge J, Zhang P, Zhong J, Tao L, et al. Lysosomal acid lipase promotes cholesterol ester metabolism and drives clear cell renal cell carcinoma progression. Cell Prolif (2018) 51(4):e12452. doi: 10.1111/cpr.12452
42. Huang B, Song B-l, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab (2020) 2(2):132–41. doi: 10.1038/s42255-020-0174-0
43. Zhu D, et al. The ROS-mediated activation of STAT-3/VEGF signaling is involved in the 27-hydroxycholesterol-induced angiogenesis in human breast cancer cells. Toxicol Lett (2016) 264:79–86. doi: 10.1016/j.toxlet.2016.11.006
44. Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, et al. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell (2016) 166(5):1176–1187.e14. doi: 10.1016/j.cell.2016.08.003
45. Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, et al. Cholesterol activates the G-protein coupled receptor smoothened to promote hedgehog signaling. Elife (2016) 5:e20304. doi: 10.7554/eLife.20304
46. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the hedgehog signaling pathway in cancer: A comprehensive review. Bosnian J basic Med Sci (2018) 18(1):8–20. doi: 10.17305/bjbms.2018.2756
47. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene (2017) 36(11):1461–73. doi: 10.1038/onc.2016.304
48. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis (2008) 4(2):68–75. doi: 10.4161/org.4.2.5851
49. Sheng R, Chen Y, Yung Gee H, Stec E, Melowic HR, Blatner NR, et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat Commun (2012) 3(1):1249. doi: 10.1038/ncomms2221
50. Vaquero J, Nguyen Ho-Bouldoires TH, Clapéron A, Fouassier L. Role of the PDZ-scaffold protein NHERF1/EBP50 in cancer biology: from signaling regulation to clinical relevance. Oncogene (2017) 36(22):3067–79. doi: 10.1038/onc.2016.462
51. Sheng R, Kim H, Lee H, Xin Y, Chen Y, Tian W, et al. Cholesterol selectively activates canonical wnt signalling over non-canonical wnt signalling. Nat Commun (2014) 5:4393. doi: 10.1038/ncomms5393
52. Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Bioscience (2020) 10:31. doi: 10.1186/s13578-020-00396-1
53. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene (2016) 35(10):1250–60. doi: 10.1038/onc.2015.179
54. Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science (2017) 355(6331):1306–11. doi: 10.1126/science.aag1417
55. King RJ, Singh PK, Mehla K. The cholesterol pathway: Impact on immunity and cancer. Trends Immunol (2022) 43(1):78–92. doi: 10.1016/j.it.2021.11.007
56. Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The role of macrophages in cancer development and therapy. Cancers (Basel) (2021) 13(08):1946. doi: 10.3390/cancers13081946
57. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab (2019) 29(6):1376–1389.e4. doi: 10.1016/j.cmet.2019.02.016
58. Park J, Lee SE, Hur J, Hong EB, Choi JI, Yang JM, et al. M-CSF from cancer cells induces fatty acid synthase and PPARβ/δ activation in tumor myeloid cells, leading to tumor progression. Cell Rep (2015) 10(9):1614–25. doi: 10.1016/j.celrep.2015.02.024
59. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer (2021) 21(5):298–312. doi: 10.1038/s41568-021-00339-z
60. Ashouri E, Rajalingam K, Barani S, Farjadian S, Ghaderi A, Rajalingam R, et al. Coexistence of inhibitory and activating killer-cell immunoglobulin-like receptors to the same cognate HLA-C2 and Bw4 ligands confer breast cancer risk. Sci Rep (2021) 11(1):7932. doi: 10.1038/s41598-021-86964-y
61. Wylie B, Macri C, Mintern JD, Waithman J. Dendritic cells and cancer: from biology to therapeutic intervention. Cancers (Basel) (2019) 11(4):521. doi: 10.3390/cancers11040521
62. Lin CY, Gustafsson JÅ. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer (2015) 15(4):216–24. doi: 10.1038/nrc3912
63. Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-mediated liver X receptor-α activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med (2010) 16(1):98–105. doi: 10.1038/nm.2074
64. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
65. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol (2014) 192(6):2920–31. doi: 10.4049/jimmunol.1302801
66. Kidani Y, lsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element–binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14(5):489–99. doi: 10.1038/ni.2570
67. Swamy M, Beck-Garcia K, Beck-Garcia E, Hartl FA, Morath A, Yousefi OS, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity (2016) 44(5):1091–101. doi: 10.1016/j.immuni.2016.04.011
68. Okoye I, Namdar A, Xu L, Crux N, Elahi S. Atorvastatin downregulates co-inhibitory receptor expression by targeting ras-activated mTOR signalling. Oncotarget (2017) 8(58):98215–32. doi: 10.18632/oncotarget.21003
69. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–156.e5. doi: 10.1016/j.cmet.2019.04.002
70. Rudd-Schmidt JA, Hodel AW, Noori T, Lopez JA, Cho HJ, Verschoor S, et al. Lipid order and charge protect killer T cells from accidental death. Nat Commun (2019) 10(1):5396. doi: 10.1038/s41467-019-13385-x
71. Hedrick CC, Malanchi I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat Rev Immunol (2022) 22(3):173–87. doi: 10.1038/s41577-021-00571-6
72. Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, Paniccia A, et al. The oxysterol–CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med (2013) 210(9):1711–28. doi: 10.1084/jem.20130440
73. Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun (2017) 8(1):864. doi: 10.1038/s41467-017-00910-z
74. Soncini M, Corna G, Moresco M, Coltella N, Restuccia U, Maggioni D, et al. 24-hydroxycholesterol participates in pancreatic neuroendocrine tumor development. Proc Natl Acad Sci U S A (2016) 113(41):E6219–27. doi: 10.1073/pnas.1613332113
75. Yang Y, Li C, Liu T, Dai X, Bazhin AV. Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front Immunol (2020) 11:1371. doi: 10.3389/fimmu.2020.01371
76. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
77. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology (2017) 6(10):e1344804. doi: 10.1080/2162402X.2017
78. Neugut AI, Johnsen CM, Fink DJ. Serum cholesterol levels in adenomatous polyps and cancer of the colon: a case-control study. JAMA (1986) 255(3):365–7. doi: 10.1001/jama.1986.03370030085033
79. Zhang X, Zhao XW, Liu DB, Han CZ, Du LL, Jing JX, et al. Lipid levels in serum and cancerous tissues of colorectal cancer patients. World J Gastroenterol (2014) 20(26):8646–52. doi: 10.3748/wjg.v20.i26.8646
80. Liu K, Zhang W, Dai Z, Wang M, Tian T, Liu X, et al. Association between body mass index and breast cancer risk: Evidence based on a dose–response meta-analysis. Cancer Manage Res (2018) 10:143–51. doi: 10.2147/CMAR.S144619
81. Li C, Yang L, Zhang D, Jiang W. Systematic review and meta-analysis suggest that dietary cholesterol intake increases risk of breast cancer. Nutr Res (2016) 36(7):627–35. doi: 10.1016/j.nutres.2016.04.009
82. Liu J, Xu A, Lam KS, Wong NS, Chen J, Shepherd PR, et al. Cholesterol-induced mammary tumorigenesis is enhanced by adiponectin deficiency: role of LDL receptor upregulation. Oncotarget (2013) 4(10):1804–18. doi: 10.18632/oncotarget.1364
83. Rodrigues dos Santos C, et al. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis (2014) 13:16. doi: 10.1186/1476-511X-13-16
84. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science (2013) 342(6162):1094–8. doi: 10.1126/science.1241908
85. Raza S, Ohm JE, Dhasarathy A, Schommer J, Roche C, Hammer KD, et al. The cholesterol metabolite 27-hydroxycholesterol regulates p53 activity and increases cell proliferation via MDM2 in breast cancer cells. Mol Cell Biochem (2015) 410(1–2):187–95. doi: 10.1007/s11010-015-2551-7
86. YuPeng L, YuXue Z, PengFei L, Cheng C, YaShuang Z, DaPeng L, et al. Cholesterol levels in blood and the risk of prostate cancer: A meta-analysis of 14 prospective StudiesBlood cholesterol and prostate cancer: A meta-analysis. Cancer Epidemiol Biomarkers Prev (2015) 24(7):1086–93. doi: 10.1158/1055-9965.EPI-14-1329
87. Jamnagerwalla J, Howard LE, Allott EH, Vidal AC, Moreira DM, Castro-Santamaria R, et al. Serum cholesterol and risk of high-grade prostate cancer: results from the REDUCE study. Prostate Cancer prostatic Dis (2018) 21(2):252–9. doi: 10.1038/s41391-017-0030-9
88. Kitahara CM, Berrington de González A, Freedman ND, Huxley R, Mok Y, Jee SH, et al. Total cholesterol and cancer risk in a large prospective study in Korea. J Clin Oncol (2011) 29(12):1592–8. doi: 10.1200/JCO.2010.31.5200
89. Singh G, Sankanagoudar S, Dogra P, Chandra NC. Interlink between cholesterol & cell cycle in prostate carcinoma. Indian J Med Res (2017) 146(Suppl 2):S38–44. doi: 10.4103/ijmr.IJMR_1639_15
90. Mostaghel EA, Solomon KR, Pelton K, Freeman MR, Montgomery RB. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PloS One (2012) 7(1):e30062. doi: 10.1371/journal.pone.0030062
91. Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol (2008) 295(1-2):115–20. doi: 10.1016/j.mce.2008.08.013
92. Malhotra P, Gill RK, Saksena S, Alrefai WA. Disturbances in cholesterol homeostasis and non-alcoholic fatty liver diseases. Front Med (Lausanne) (2020) 7:467. doi: 10.3389/fmed.2020.00467
93. Venturini I, Amedei R, Modonesi G, Cosenza R, Miglioli L, Cioni G, et al. May plasma cholesterol level be considered a neoplastic marker in liver disease from cirrhosis to hepatocellular carcinoma? Ital J Gastroenterol Hepatol (1999) 31(1):61–5.
94. Hirayama C, Yamanishi Y, Irisa T. Serum cholesterol and squalene in hepatocellular carcinoma. Clinica Chimica Acta (1979) 91(1):53–7. doi: 10.1016/0009-8981(79)90470-4
95. Bricker LA, Morris HP, Siperstein MD. Loss of the cholesterol feedback system in the intact hepatoma-bearing rat. J Clin Invest (1972) 51(2):206–15. doi: 10.1172/JCI106805
96. Li JR, Zhang Y, Zheng JL. Decreased pretreatment serum cholesterol level is related with poor prognosis in resectable non-small cell lung cancer. Int J Clin Exp Pathol (2015) 8(9):11877–83.
97. Chang AK, Barrett-Connor E, Edelstein S. Low plasma cholesterol predicts an increased risk of lung-cancer in elderly women. Prev Med (1995) 24(6):557–62. doi: 10.1006/pmed.1995.1089
98. Shekelle RB, Rossof AH, Stamler J. Dietary cholesterol and incidence of lung cancer: the Western electric study. Am J Epidemiol (1991) 134(5):480–4. doi: 10.1093/oxfordjournals.aje.a116119
99. Lyu Z, Li N, Wang G, Su K, Li F, Guo LW, et al. Association between total cholesterol and risk of lung cancer incidence in men: a prospective cohort study. Zhonghua Liu Xing Bing Xue Za Zhi (2018) 39(5):604–8. doi: 10.3760/cma.j.issn.0254-6450.2018.05.012
100. Chen L, Zhang L, Xian G, Lv Y, Lin Y, Wang Y. 25-hydroxycholesterol promotes migration and invasion of lung adenocarcinoma cells. Biochem Biophys Res Commun (2017) 484(4):857–63. doi: 10.1016/j.bbrc.2017.02.003
101. Chen H, Qin S, Wang M, Zhang T, Zhang S. Association between cholesterol intake and pancreatic cancer risk: evidence from a meta-analysis. Sci Rep (2015) 5:8243. doi: 10.1038/srep08243
102. Chen WC, Boursi B, Mamtani R, Yang YX. Total serum cholesterol and pancreatic cancer: A nested case–control StudyTotal serum cholesterol and pancreatic cancer. Cancer Epidemiol Biomarkers Prev (2019) 28(2):363–9. doi: 10.1158/1055-9965.EPI-18-0421
103. Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene (2016) 35(50):6378–88. doi: 10.1038/onc.2016.168
104. Helzlsouer KJ, Alberg AJ, Norkus EP, Morris JS, Hoffman SC, Comstock GW. Prospective study of serum micronutrients and ovarian cancer. J Natl Cancer Institute (1996) 88(1):32–7. doi: 10.1093/jnci/88.1.32
105. Onwuka JU, Okekunle AP, Olutola OM, Akpa OM, Feng R. Lipid profile and risk of ovarian tumours: a meta-analysis. BMC Cancer (2020) 20(1):200. doi: 10.1186/s12885-020-6679-9
106. Ayyagari VN, Wang X, Diaz-Sylvester PL, Groesch K, Brard L. Assessment of acyl-CoA cholesterol acyltransferase (ACAT-1) role in ovarian cancer progression–an in vitro study. PloS One (2020) 15(1):e0228024. doi: 10.1371/journal.pone.0228024
107. Yarmolinsky J, Bull CJ, Vincent EE, Robinson J, Walther A, Smith GD, et al. Association between genetically proxied inhibition of HMG-CoA reductase and epithelial ovarian cancer. JAMA (2020) 323(7):646–55. doi: 10.1001/jama.2020.0150
108. Yavasoglu I, Sargin G, Yilmaz F, Altındag S, Akgun G, Tombak A, et al. Cholesterol levels in patients with chronic lymphocytic leukemia. J Natl Med Assoc (2017) 109(1):23–7. doi: 10.1016/j.jnma.2016.11.006
109. Baroni S, Scribano D, Zuppi C, Pagano L, Leone G, Giardina B. Prognostic relevance of lipoprotein cholesterol levels in acute lymphocytic and nonlymphocytic leukemia. Acta haematologica (1996) 96(1):24–8. doi: 10.1159/000203710
110. Chow S, Buckstein R, Spaner DE. A link between hypercholesterolemia and chronic lymphocytic leukemia. Leukemia lymphoma (2016) 57(4):797–802. doi: 10.3109/10428194.2015.1088651
111. Sankanagoudar S, Singh G, Mahapatra M, Kumar L, Chandra NCH. Cholesterol homeostasis in isolated lymphocytes: A differential correlation between male control and chronic lymphocytic leukemia subjects. Asian Pacific J Cancer prevention: APJCP (2017) 18(1):23–30. doi: 10.22034/APJCP.2017.18.1.23
112. Abdulla N, Vincent CT, Kaur M. Mechanistic insights delineating the role of cholesterol in epithelial mesenchymal transition and drug resistance in cancer. Front Cell Dev Biol (2021) 9:728325. doi: 10.3389/fcell.2021.728325
113. Chen Q, Pan Z, Zhao M, Wang Q, Qiao C, Miao L, et al. High cholesterol in lipid rafts reduces the sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J Cell Physiol (2018) 233(9):6722–32. doi: 10.1002/jcp.26351
114. Beckwitt CH, Shiraha K, Wells A. Lipophilic statins limit cancer cell growth and survival, via involvement of akt signaling. PloS One (2018) 13(5):e0197422. doi: 10.1371/journal.pone.0197422
115. Spampanato C, De Maria S, Sarnataro M, Giordano E, Zanfardino M, Baiano S, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of bax and down-regulation of BCL-2 gene expression. Int J Oncol (2012) 40(4):935–41. doi: 10.3892/ijo.2011.1273
116. Relja B, et al. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int J Mol Med (2010) 26(5):735–41. doi: 10.3892/ijmm_00000520
117. Beckwitt CH, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res (2018) 20(1):1–11.
118. Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, et al. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J Natl Cancer Inst (2011) 103(19):1461–8. doi: 10.1093/jnci/djr291
119. Shao JY-H, Lee FP, Chang CL, Wu SY. Statin-based palliative therapy for hepatocellular carcinoma. Medicine (2015) 94(42):e1801. doi: 10.1097/MD.0000000000001801
120. Lash TL, Riis AH, Ostenfeld EB, Erichsen R, Vyberg M, Ahern TP, et al. Associations of statin use with colorectal cancer recurrence and mortality in a Danish cohort. Am J Epidemiol (2017) 186(6):679–87. doi: 10.1093/aje/kww245
121. Gray RT, Coleman HG, Hughes C, Murray LJ, Cardwell CR. Statin use and survival in colorectal cancer: Results from a population-based cohort study and an updated systematic review and meta-analysis. Cancer Epidemiol (2016) 45:71–81. doi: 10.1016/j.canep.2016.10.004
122. Zhong S, Zhang X, Chen L, Ma T, Tang J, Zhao J. Statin use and mortality in cancer patients: Systematic review and meta-analysis of observational studies. Cancer Treat Rev (2015) 41(6):554–67. doi: 10.1016/j.ctrv.2015.04.005
123. Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, et al. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PloS One (2018) 13(12):e0209486. doi: 10.1371/journal.pone.0209486
124. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr, et al. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol (2020) 27(Suppl 2):S87–97. doi: 10.3747/co.27.5223
125. Behranvand N, Nasri F, Zolfaghari Emameh R, Khani P, Hosseini A, Garssen J, et al. Chemotherapy: a double-edged sword in cancer treatment. Cancer Immunol Immunother (2022) 71(3):507–26. doi: 10.1007/s00262-021-03013-3
Keywords: cholesterol, immune cells, cancer, anticholesteremic agents, immunometabolism
Citation: Halimi H and Farjadian S (2022) Cholesterol: An important actor on the cancer immune scene. Front. Immunol. 13:1057546. doi: 10.3389/fimmu.2022.1057546
Received: 29 September 2022; Accepted: 04 November 2022;
Published: 21 November 2022.
Edited by:
Huashan Shi, Sichuan University, ChinaReviewed by:
Xiaocan Guo, Harvard Medical School, United StatesCopyright © 2022 Halimi and Farjadian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shirin Farjadian, c2hpcmluZW5hdG9yQGdtYWlsLmNvbQ==