 Chen Lyu
Chen Lyu Yonghu Sun
Yonghu Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 November 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1055958
This article is part of the Research TopicImmunometabolism of T cells in Skin Infection, Autoimmunity and Cancer BiologyView all 7 articles
Vitiligo is a common depigmenting skin disorder characterized by the selective loss of melanocytes. Autoimmunity, genetic, environmental, and biochemical etiology have been proposed in vitiligo pathogenesis. However, the exact molecular mechanisms of vitiligo development and progression are unclear, particularly for immunometabolism. Sporadic studies have suggested mitochondrial dysfunction, enhanced oxidative stress, and specific defects in other metabolic pathways can promote dysregulation of innate and adaptive immune responses in vitiligo. These abnormalities appear to be driven by genetic and epigenetic factors modulated by stochastic events. In addition, glucose and lipid abnormalities in metabolism have been associated with vitiligo. Specific skin cell populations are also involved in the critical role of dysregulation of metabolic pathways, including melanocytes, keratinocytes, and tissue-resident memory T cells in vitiligo pathogenesis. Novel therapeutic treatments are also raised based on the abnormalities of immunometabolism. This review summarizes the current knowledge on immunometabolism reprogramming in the pathogenesis of vitiligo and novel treatment options.
Vitiligo is an autoimmune skin disease characterized by skin pigmentation loss due to functional melanocyte dysregulation. Many studies suggest that vitiligo is associated with insulin resistance, lipid abnormalities, and other metabolic disorders (1–3). Vitiligo affects approximately 0.5% to 2% of the world’s population (4). Various mechanisms have been proposed to explain the destruction of melanocytes in vitiligo, including genetics, autoimmune response, oxidative stress, and the production of inflammatory mediators (5). However, the exact pathogenesis of immunometabolism abnormalities in vitiligo is not fully understood. In 2016, vitiligo was further classified into three categories: nonsegmental vitiligo (NSV), segmental vitiligo (SV), and undetermined/unclassified (6). NSV is the most common type and has been reclassified as a systemic rather than a depigmented skin disease (7). Regulation of the innate immune response and B-cell differentiation and their activation was demonstrated in NSV. Especially, NSV was reported to be more pronounced than SV (8, 9). Several systemic metabolic disorders suggest a close association with NSV, such as type I diabetes, atherosclerotic cardiovascular disease, and metabolic syndrome (10–12).
Metabolic reprogramming is the modification of different metabolic processes to regulate the function and phenotype of immune cells to meet the regulation and execution of immune functions (13). The focus is on the effects of different metabolic pathways on immune cell activation, differentiation, and function, including glucose metabolism and lipid metabolism (14). Metabolic reprogramming of innate and adaptive immune elements might contribute to the disturbed immune homeostasis in vitiligo. The translational implications of these findings are enormous and may lead to novel therapeutic targets.
Reactive oxygen species (ROS) induced oxidative stress triggers a wide range of abnormal organelle functions, disrupts metabolic pathways, and impairs defense mechanisms against oxidant shocks. It plays a crucial role in cellular events such as inflammatory responses, altered cell growth, and apoptosis (Figure 1) (15–20).
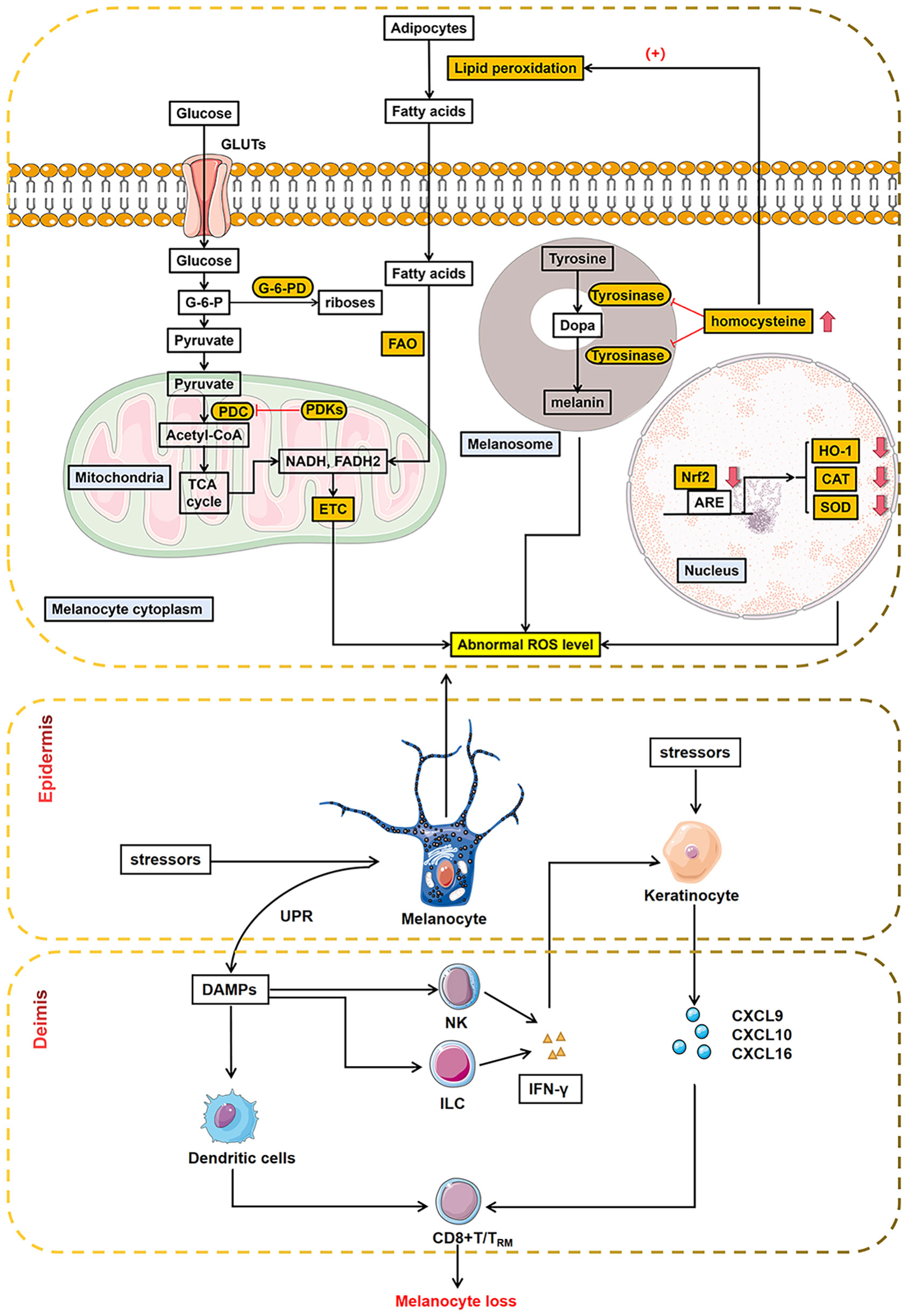
Figure 1 Abnormal metabolic processes in melanocytes during oxidative stress. Oxidative stress triggered abnormal glycolipid metabolism within melanocytes, disrupting metabolic pathways. Melanocytes with mitochondrial disorders, abnormal melanin biosynthesis, and dysregulated metabolic pathway produce abnormal ROS, exacerbating oxidative stress. The orange-boxed portion of the figure shows outlier segments, as reported in recent literature. In addition, melanocytes interact with different immune cells in innate and adaptive immunity, leading to melanocyte loss ultimately. ARE, antioxidant response element; CAT, catalase; HO-1, heme oxygenase-1; Nrf2, nuclear factor E2-related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase; GLUTs, glucose transporters; PDC, pyruvate dehydrogenase complex; PDKs, pyruvate dehydrogenase kinase; ETC, electron transport chain; FAO, fatty acid oxidation; G-6-PD, glucose-6-phosphate dehydrogenase; UPR, unfolded protein response; DAMPs, damage-associated molecular patterns; NK, natural killer cells; ILC, innate lymphoid cells; IFN-γ, inteferon-γ; CXCL9, C-X-C motif chemokine ligand 9; CXCL10, C-X-C motif chemokine ligand 10; CXCL16, C-X-C motif chemokine ligand 16.
The role of alterations in the antioxidant system in the pathogenesis of vitiligo has been extensively studied (21). Low levels of catalase (CAT), glutathione peroxidase (GPx), glucose-6-phosphate dehydrogenase (G6PD), and superoxide dismutase (SOD) have been demonstrated in the epidermis and blood of vitiligo patients (22–29). In addition, polymorphisms in the CAT gene and reduced catalase activity have also been associated with several metabolic diseases, such as diabetes, hypertension, and peroxisomal/peroxisomal diseases (30–39). All of the above etiologies can lead to chronic oxidative stress (OS) state that causes melanocytes to be programmed for cellular senescence. Senescent melanocytes exhibit a specific “senescence-associated secretory phenotype (SASP),” which recruits immune cells to remove “senescent melanocytes” by expressing various cytokines (40).
On the other hand, the chronic OS will produce an excessive accumulation of ROS, which in turn induces the production of damage-associated molecular patterns (DAMPs) and the release of melanosomal antigens that activate innate immunity (41). Natural killer (NK) cells and innate lymphoid cells (ILC) secret interferon-γ (IFN-γ) to induce expression of C-X-C motif chemokine receptor (CXCR)3B on melanocyte surface and release of C-X-C motif chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10), and C-X-C motif chemokine ligand 11 (CXCL11) from keratinocyte and melanocyte. CXCL10 then activates CXCR3B and triggers the apoptosis of melanocytes (42). What’s more, melanocyte specific CD8+ T cells are activated through HSP70i activation and antigen presentation by mature dendritic cells (43, 44). In response to cytokine and chemokine stimuli, CD8+ T cells are recruited to induce melanocyte apoptosis (45, 46). Furthermore, dysfunctional regulatory T cells (Tregs) in patients with active vitiligo contribute to pathogenesis by impairing the suppressive activity of CD8+ T cells (47, 48).
These findings suggest that oxidative stress plays a vital role in the pathogenesis of vitiligo. Abnormalities in ROS serve as a bridge between oxidative stress and immune response.
Mitochondrial dysfunction is considered essential in losing redox homeostasis (high spontaneous ROS production and lack of antioxidant network) in vitiligo (49–51). A study by Bhattiet et al. mentioned that oxidative stress leads to ROS deposition in cells and tissues by interacting with mitochondrial and cellular components such as DNA, proteins, and lipids leading to mitochondrial dysfunction (52). Dysfunctional mitochondria in melanocytes and CD8+ T cells are significant for the induction of melanocyte apoptosis.
Studies of melanocytes and peripheral blood mononuclear cells (PBMCs) from vitiligo patients have shown a defect in complex I formation (51, 53). Due to low concentrations and abnormal distribution of cardiolipin of the mitochondrial electron transport chain (mETC), the stability of the mitochondrial supercomplex was impaired, and the level of glycolytic phosphorylation was decreased (54). Consequently, the production of ATP is reduced, and mitochondrial ROS are highly emitted. Interestingly, cardiolipin replacement rescues normal mitochondrial function in vitiligo patients (54). However, more studies are needed to determine the reason for the low concentration of mitochondrial cardiolipin in melanocytes and CD8+ T cells and how to restore average concentrations of this lipid in vivo.
In vitiligo patients, NADPH oxidase isoform 4 (NOX4) in the mitochondria produces a large amount of H2O2 (55). However, higher NOX4 activity depletes NADPH molecules used as cofactors for CAT, GPX, and thioredxin reductase (TrxR) in H2O2 buffer activity (56). In addition, excess H2O2 impairs CAT, GPX, and TrxR buffering action. Meanwhile, it causes mitochondrial swelling and changes in mitochondrial morphology upon loss of calcium metabolism. Besides inhibiting the mitochondrial autophagy process, morphological altered mitochondrial hyperactivated P53 and enhanced SOD enzyme activity (28, 57). Thus, the highly damaged mitochondria continue to produce large amounts of ROS, which induce melanocyte apoptosis via mitochondrial cytochrome c release or massive recruitment of CD8+ T cells.
In addition to serving as essential sites for the synthesis, folding, modification, and trafficking of proteins, the endoplasmic reticulum (ER) senses cellular stress and regulates cell survival or death (58). Under unstressed conditions, three ER stress sensors (inositol-requiring enzyme 1α (IRE1α), PKR like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) (59–62)) mainly bind to GRP78(78 kDa glucose-regulated protein), helping to keep it inactive. In the presence of differentiation stimuli, abnormal protein folding in the ER activates the unfolded protein response (UPR) signaling, which can be alleviated by global translational attenuation, induction of molecular chaperones, misfolded protein degradation by endoplasmic reticulum-associated degradation (ERAD), and apoptosis (63). Accumulation of misfolded proteins increases the production of BiP/GRP78, which initiates XBP1 splicing and induces ATF6 signaling (64, 65).
As the primary immune cells in autoimmunity in vitiligo, the differentiation, proliferation, and homeostasis of CD8+ T cells are also influenced by UPR signaling. CD4−/CD8 − progenitor T cells do not display the UPR but greatly increase it during maturation as CD4+/CD8+ T cells. After differentiation into CD4+ T cells, the UPR is suppressed again (64). Infection of mice with lymphocytic choriomeningitis virus (LCMV) leads to upregulation of spliced and unspliced XBP1, further enhancing CD8+ T cell differentiation (65). In addition, the UPR of ER stress signaling was activated during epidermal keratinocyte differentiation (66–68). C-X-C motif chemokine ligand 16 (CXCL16) levels are enhanced by IRE1α/XBP1s signaling in stressed keratinocytes, which are involved in recruiting CD8+ T cells to vitiligo lesions (69).
ER stress has also been implicated in the regulation of Tregs. Human Tregs clones produced increased interleukin-10 (IL-10) when treated with thapsigargin, an activator of ER stress and UPR, in an eIF2α phosphorylation-dependent manner (70). Loss of ATF4 leads to an increase in Forkhead box P3 (Foxp3) mRNA expression in mouse CD4+ T cells differentiated under regulatory conditions in a high oxidative environment (71). Recently, nuclear factor of activated T cells (NFAT) and Foxp3 levels have been reported to be reduced in Tregs from patients with generalized vitiligo, which may impair Tregs function and decrease IL-10 and cytotoxic T-lymphocyte-associated antigen-4 (CTLA4) levels (72–74). However, the exact mechanism between Tregs and ER stress in the pathogenesis of vitiligo remains to be investigated.
What’s more, prolonged ER stress and a defective UPR may lead to the activation of inflammatory transcriptional programs and the release of proinflammatory cytokines, generating further ER stress and oxidative stress. Notably, UPR-induced autophagy is speculated to stimulate the production of exosomes and soluble inflammatory signals that are proinflammatory and promote autoimmunity (46). However, autophagy is essentially a protective response in the face of cellular stress, and defective autophagy increases melanocyte sensitivity to oxidative stress (75). The effects of ROS also extend to other macromolecules in the ER, such as calreticulin (CRT). Calreticulin (CRT), a ubiquitous ER protein regulating intracellular Ca2+ homeostasis, was positively correlated with the lesion size and duration of vitiligo in patients (44). Zhang et al. reported that oxidative stress drove the redistribution of CRT from the ER lumen to the cell membrane, thereby improving the immunogenicity of stressed melanocytes and inducing apoptosis (76). In addition to the release of proinflammatory cytokines, melanocyte apoptosis may lead to the release of misfolded/unfolded proteins that have the potential to act as autoantigens and may be recognized by immune cells as DAMPs. Antigen-presenting cells (APCs) may process and present altered proteins/peptides, generating novel epitopes and activating target B and T cells, leading to an autoimmune response against melanocytes (44, 76).
The UPR activated by ER stress plays a crucial role in regulating and maintaining innate and adaptive immunity in vitiligo. Further studies on tissue/cell type-specific UPR are warranted to provide more comprehensive evidence-based support for charting the panorama of vitiligo pathogenesis.
Various glucose pathways defects have been implicated in vitiligo. Clinical investigations can also reveal a significantly increased risk of diabetes in patients with vitiligo. In this regard, Studies by Mubki, Gopal et al. described a higher prevalence of elevated fasting glucose levels in patients with vitiligo. They concluded that the prevalence of diabetes in patients with vitiligo was statistically significant (77, 78). Several studies have also found an increased prevalence of vitiligo in patients with type II diabetes (79, 80).
It is known that pro-inflammatory cytokines play a significant role in the development of various autoimmune diseases. Pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-1β (IL-1β), IFN-γ, and some anti-inflammatory cytokines such as interleukin-5 (IL-5) and IL-10 are increased in patients with active vitiligo (81). Inflammatory cytokines are involved in inhibiting the insulin signaling pathway by phosphorylating serine residues of the insulin receptor substrate-1, leading to insulin resistance development in vitiligo (82). Pietrzak et al. showed that impaired secretion of TNF-α, IL-6, and monocyte chemokines during glucose metabolism contributes to the induction of insulin resistance and other metabolic complications (83). We have known that CD4+ and CD8+ T cells are present at the epidermal and dermal junctions near vitiligo lesions, and CD8+ T cells have been proven to induce melanocytes loss, underscoring the role of T cell-mediated immunity in the pathogenesis of vitiligo (84). Recognizing imbalanced and dysfunctional effector T cell subsets and dysregulated cytokine production may have significant potential for targeted therapy. However, much remains to be explored about the mechanisms underlying dysregulated cytokine production in the pathogenesis of vitiligo.
In vitro studies have shown that epidermal vitiligo melanocytes have lower adenosine Triphopsphate (ATP), increased proton leakage, and altered expression of several glycolytic enzymes (hexokinase II, pyruvate dehydrogenase kinase 1, and pyruvate kinase M2) compared to healthy cells. Moreover, the defective ATP production is easily compensated by the increased activity of enzymes in glucose utilization, providing the cell with alternative substrates but not increasing energy levels but rescuing the affected activities and pathways by stabilizing the mitochondrial membrane lipid components (54). Furthermore, melanocytes in vitiligo patients are more sensitive to G6PD inhibition than normal melanocytes. A study in the Gujarat population showed a genetic and biochemical association of the G6PD 3’UTR rs1050757 polymorphism with vitiligo (85). In addition to melanocytes, keratinocytes play an essential role in oxidative stress and glycolysis processes. In a study of metabolic processes in vitiligo lesioned skin, researchers quantitatively assessed enriched metabolic pathways using gene scoring analysis. They found that Oxidative phosphorylation (OXPHOS) and glycolysis showed the most significant differences between stressed keratinocytes and the other, respectively (86). Therefore, abnormal glucose metabolism in vitiligo patients may result from an imbalance in the secretion of pro-inflammatory factors, low oxidative stress, or impaired G6PD levels.
Previous studies on a small number of patients have reported conflicting results regarding the association between vitiligo and dyslipidemia. For example, recent findings suggest that patients with vitiligo have significant metabolic disturbances in elevated cholesterol (Chol), level of triglyceride (TG), low-density lipoprotein (LDL), and leptin levels. High-density lipoprotein (HDL) levels were lower compared to healthy controls (1). However, another study of children with vitiligo found that girls with vitiligo had lower LDL and HDL levels than controls (87). Evidence of the relationship between vitiligo and abnormal lipid metabolism is rapidly growing in the literature (2, 3, 82).
Lipids are the main components of cellular membranes, which have a highly relevant role in oxidative stress. In addition, dyslipidemia and other traditional risk factors for atherosclerosis, such as diabetes, hypertension, and smoking, activate the NADPH oxidase system, leading to excess superoxide anion production and promoting oxidative stress (88). Pietrzak et al. firstly hypothesized a role for lipid peroxidation in the pathogenesis of vitiligo (89). A study by Karadag et al. showed that hyperhomocysteinemia, except a known cardiovascular risk factor, may also contribute to the development of vitiligo by inhibiting tyrosinase (90). Reported significantly higher homocysteine levels in patients with active vitiligo compared with patients with stable vitiligo, which may induce oxidative stress, ER stress, and proinflammatory cytokine expression (91). Notably, N-homocysteinylated proteins formed by N-linkage of the carboxyl group of homocysteine with the E-amino group of lysine residues of proteins may be considered neo-antigen formation, thereby inducing the destruction of melanocytes by CD8+ T cell-mediated autoimmune reactions (92).
Ferroptosis, characterized by an iron-dependent increase in oxidative stress and lipid peroxidation, has been extensively studied in various diseases (93–95). However, whether ferroptosis plays a role in melanocyte loss of vitiligo remains to be elucidated. Lipid peroxide accumulation may be fatal to the cells as an integral event in ferroptosis (96). In the analysis of epidermal tissues, it was shown that ferroptosis markers were aberrantly expressed in vitiligo patients, with glutathione peroxidase 4 (GPX4) and ferritin being downregulated at both protein and mRNA levels. In contrast, transferrin receptor protein 1 (TfR) was observed to be overexpressed in both lesional and non-lesional areas of vitiligo. In addition, the same study also showed that the treatment of erastin (an inducer of iron toxicity) decreased GPX4 and ferritin levels in human epidermal melanocytes (HEMs) while increasing TfR and Acyl-CoA synthetase long-chain family member 4 (ACSL4) expression, indicating that HEMs are sensitive to ferroptosis (97). Together, ferroptosis and melanocyte loss are characterized by mitochondrial malformations, glutathione peroxidase inactivation, p53 elevation, ROS accumulation, and lipid peroxidation (98, 99). Furthermore, ferroptosis is involved in IFN-γ- associated melanocyte destruction, whereas the role of IFN-γ in vitiligo pathogenesis has been demonstrated. However, whether ferroptosis is involved in melanocyte destruction is unknown (100, 101). As a critical cell death process, Ferroptosis may provide novel therapeutic targets and serve as a potential biomarker for vitiligo.
Abnormal lipid metabolism may also contribute to T cell dysfunction in vitiligo. Compared to effector T cells and other memory T cells, tissue-resident memory T cells (TRM) exhibit a unique metabolic panorama. It has been reported to play an essential role in the pathogenesis of host antimicrobial infections, cancer immunotherapy, and several autoimmune diseases (102–106). Currently, the focus on TRM cells may help explain the recurrence of vitiligo lesions at the same positions, as TRM cells provide rapid local defense against recurrent pathogens.
Studies in vitiligo have shown that the skin surrounding vitiligo lesions is enriched with CD8+ TRM populations expressing CD69 and CD103 during the stable and active phase (101, 107). TRM cells produce perforin, Granzyme B, and IFN-γ upon interleukin-15 (IL-15) stimulation and maintain disease in cooperation with recirculating memory T (TRCM) cells. In addition, TRM cells also secrete CXCL9 and CXCL10, which bind to CXCR3 on TRCM cells to recruit them to the skin. In addition, using a well-established model of CD8+ TRM production in the skin following cowpox virus skin immunization, researchers found that cutaneous CD8+ TRM utilized exogenous free fatty acid (FFA) internalized from the surrounding microenvironment to ensure their survival (108). This phenomenon suggested that TRM cells may establish a link between lipid metabolism and adaptive memory immunity. The above studies suggest that there may be an essential target for controlling vitiligo recurrence in modulating lipid metabolism to eliminate TRM in peripheral tissues.
Uncovering the biological mediators and the molecular mechanisms of metabolic defects in melanocyte degeneration and autoimmunity is essential for novel therapeutic targets and drugs intercepting the process of vitiligo. The experience with systemic biological therapies for psoriasis suggests that a similar approach might be successfully used in vitiligo. Promising treatments targeting the IFN-γ chemokine axis, JAK-STAT pathway, and Nrf2-ARE pathway, have recently emerged (Figure 2).
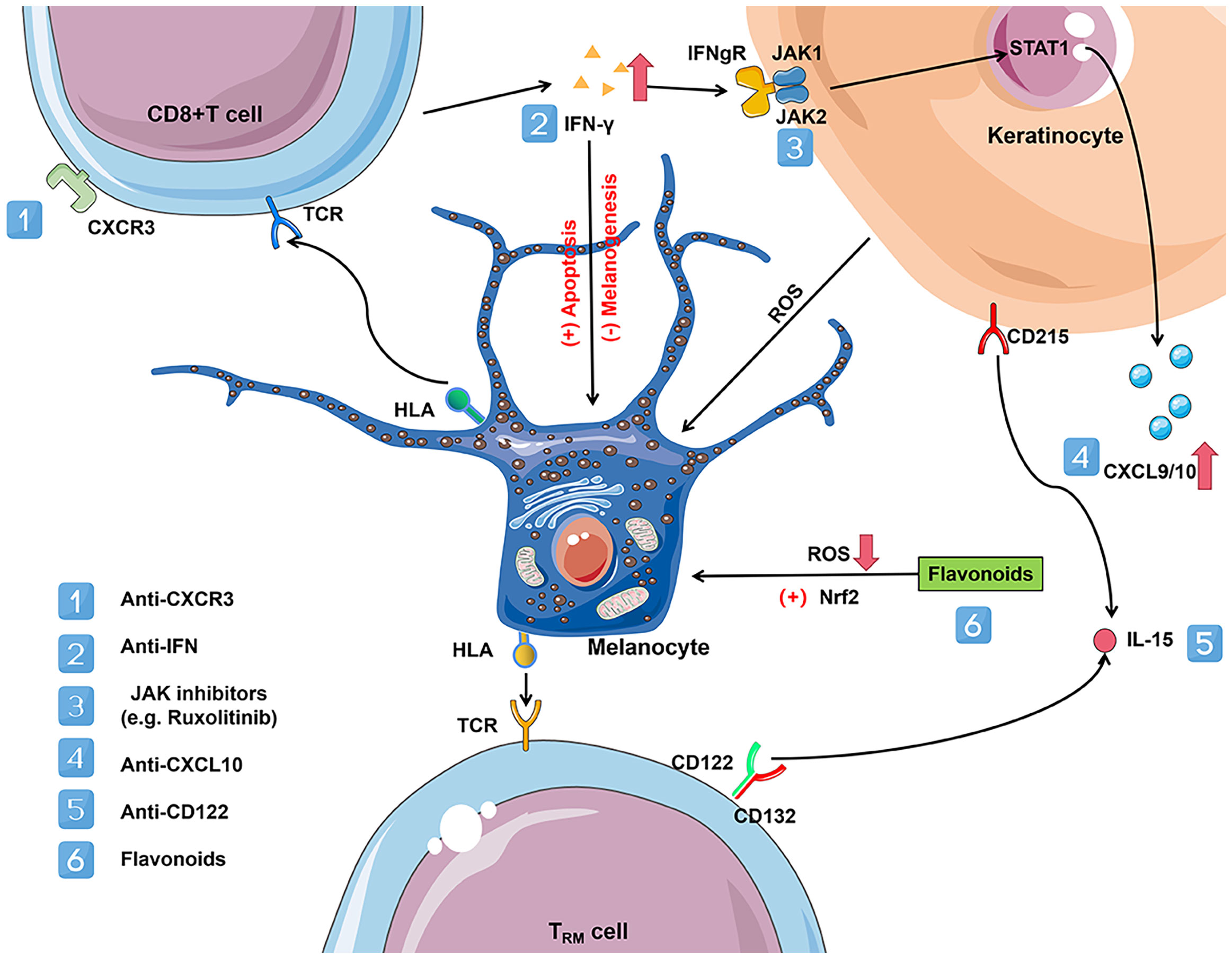
Figure 2 Vitiligo immunometabolism promising targets. CD8+ T cells in vitiligo lesions can produce a variety of cytokines, including IFN-γ. Elevated levels of IFN-γ disrupt glucose metabolism and promote melanocyte apoptosis. IFN-γ binds with IFNgR, activates the JAK-STAT pathway, and leads to the secretion of CXCL9 and CXCL10 in the skin. JAK-STAT signaling pathway activation plays a vital role in various metabolic disorders and can be treated with JAK inhibitors (e.g. ruxolitinib) acting on JAK1 and JAK2. CXCL9 promotes massive recruitment of melanocyte-specific CD8+ T cells to the skin via the cognate receptor CXCR3, while CXCL10 promotes their localization within the epidermis and their effector functions, which increases inflammation through a positive feedback loop. Activation of CXCR3B by CXCL10 induces apoptosis of melanocytes. However, depleting antibodies acting on CXCR3 could reduce the number of C8+ T cells, thereby reversing the disease. In addition, flavonoids act by activating the Nrf2/ARE signaling pathway and inhibiting NFκB activation by reducing the extent of oxidative stress in melanocytes. Established vitiligo lesions are maintained by melanocyte-reactive TRM cells that maintain longevity in the skin via IL-15-dependent survival signals. TRM is linked to lipid metabolism due to their reliance on exogenous free fatty acid (FFA) uptake to maintain their residence in the skin. The use of antibodies against CD122, the β subunit of the IL-15 receptor, in a mouse model of vitiligo effectively inhibits TRM production. IL-15 and its receptor may be a target for vitiligo treatment. CXCR3, C-X-C motif chemokine receptor 3; CXCL9/10, C-X-C motif chemokine ligand 9/10; IFN-γ, inteferon-γ; IL-15, interleukin-15; IFNgR, inteferon-gamma receptor; TRM, resident memory T cells; TCR, T cell receptor; HLA, human leukocyte antigen; STAT1, signal transducer and activator of transcription protein 1; JAK, janus kinases.
The reduction in the level of IFN-γ, a key cytokine of the immune system in glucose metabolism, can improve glucose metabolism, as demonstrated in a mouse model with low IFN-γ levels (109). Furthermore, according to functional studies in a mouse model, IFN-γ, inteferon-gamma receptor (IFNgR), signal transducer and activator of transcription protein 1 (STAT1), C-X-C motif chemokine ligand 10 (CXCL10), and C-X-C motif chemokine receptor 3 (CXCR3) are also critical for developing hypopigmentation in vitiligo (46, 107, 110). Targeting CXCR3 with depleting antibodies in a mouse model reduced the number of self-reactive T cells and reversed vitiligo manifestations (111). Functional studies using conditional STAT 1 knockout mice have shown that keratinocyte-derived chemokines and IFN-γ signaling drive vitiligo and proper homing of auto-reactive T cells to the epidermis. In contrast, epidermal immune cells, such as endogenous T cells, Langerhans cells, and γδ T cells, are not required (112). IFN-γ inhibits melanogenesis and directly induces melanocyte apoptosis (113). A recent single-cell sequencing study of vitiligo showed that fibroblast pairs from different sites IFN-γ responsiveness determine its ability to recruit CD8+ T cells. The study also demonstrates that the degree of upregulation of CXCL9 and CXCL10 was higher in the high-incidence area. By the Cre-loxP system, they constructed an IFN-γ receptor knockout mice which no longer have vitiligo (114). Thus, the IFN- γ axis or potential targets for treating vitiligo.
In recent years, evidence has been increasing that abnormalities in the JAK-STAT pathway led to metabolic disorders. Janus kinase 2 (JAK2) is associated with central obesity and increased waist circumference (115). Mice lacking janus kinase 3 (JAK3) exhibit metabolic disorders such as insulin resistance, weight gain, increased fasting insulin and glucose levels, decreased glucose tolerance, and hepatic steatosis (116). Recent reports suggest that JAK inhibitors have good therapeutic promise in vitiligo. A recent case report found that a JAK inhibitor successfully improved glucose levels in a 19-year-old patient with type I diabetes (117). Clinical Trials of topical JAK inhibitor Ruxolitinib cream met the primary endpoint in both pivotal phases 3 clinical trials with critical secondary endpoints, with a significantly higher proportion of the trial group achieving at least 75% improvement in the F-VASI score (a quantitative measure to assess skin symptoms in patients with vitiligo) relative to baseline than the placebo group at week 24 of treatment (118). Ruxolitinib cream is currently the first Food and Drug Administration (FDA) approved vitiligo therapy for the topical treatment of non-segmental vitiligo in adults and pediatric patients over 12 years of age (119). In addition, Shiu et al. implies that combining therapies that reverse defects in keratinocytes’ metabolism with JAK inhibitors could be a novel approach to treating vitiligo (86).
In vitiligo, inactive Nrf2 signaling is ineffective in protecting HEM from H2O2-induced OS damage and, therefore, rarely prevents melanocyte apoptosis by mediating its downstream antioxidant gene heme oxygenase-1 (HO-1) (120). Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activation has emerged as an important target for protection against OS-related skin diseases such as skin photodamage, and vitiligo. Nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) is a significant regulator of pro-inflammatory responses and is considered a redox-regulated transcription factor that can be activated by H2O2 (121). By catalyzing the phosphorylation and degradation of IkB at specific amino acid residues, NFkB can translocate from the cytoplasm to the nucleus and further activate immune mediators such as IL-6 and IL-8 (122). These NFκB-regulated inflammatory factors accelerate islet β-cell apoptosis, cause insulin resistance in peripheral cells, and play an essential role in the pathogenesis of diabetes (123). However, the initiation of the Nrf2/ARE pathway can effectively inhibit NFκB activation. In addition, Nrf2 can inhibit the expression of TNFα, inducible nitric oxidesynthase (iNOS), and cyclooxygenase-2 (COX2) by suppressing pro-inflammatory genes. Among them, HO-1 can significantly inhibit TNFα activity and suppress the phosphorylation of the NFκB pathway promoter nuclear factor kappa-B inhibitory protein kinase (IKK)β, thereby suppressing inflammation (21). NFκB can also counteract the transcriptional activity of Nrf2 and suppress the expression of HO-1 (124). Flavonoids such as Ginkgo biloba extract EGb761 and afzelin can protect melanocytes from OS-induced apoptosis by enhancing the Nrf2 signaling pathway and its downstream antioxidant (125, 126).
TRM is inextricably linked to lipid metabolism due to its dependence on exogenous FFA uptake to maintain its residency in the skin. Functional CD8+ TRM has been found in both stable and active vitiligo, suggesting that those cells maintained in stable disease could explain disease reactivation (127). Thus, intercepting critical metabolic pathways that deplete TRM may be promising, such as trimetazidine, which blocks mitochondrial β-oxidation of FFA and reduces TRM lifespan in the skin (108, 128). In preclinical mice models of vitiligo, the use of antibodies against CD122 (the β-subunit of the IL-15 receptor, expressed on TRM cells) effectively inhibits TRM production. IL-15 and its receptor might be a promising target for vitiligo treatment (129). However, the precise molecular mechanisms involved in this phenomenon and the putative clinical implications of this T-cell regulation remain to be determined, as some discrepancies exist between in vivo and in vitro findings.
In addition to their beneficial effects on the primary and secondary prevention of cardiovascular events, lipid-lowering agents statins may also attenuate vitiligo disease activity. As an inhibitor of the pro-inflammatory cytokine, they protect against H2O2-induced apoptosis and ROS accumulation. In addition, they inhibit CD8+ T cell action in melanocytes through IFN-γ and even block the T cells involved in the T generation of the PI3K/AKT signaling pathway (130–132).
Due to significant metabolic alterations in tumor cells, oncologists have long expected to treat tumors by inhibiting upregulated metabolic pathways, but the actual effects have been suboptimal. Compared to the tumor cell that needs to be cleared entirely, the normal number and proportion of immune cells still need to be maintained to keep immune homeostasis in autoimmune diseases. However, multiple energy metabolism is altered in autoimmune disease pathogenesis, increasing treatment difficulty. Since immunometabolism research in vitiligo is still unclear, there is still much to be discovered about related treatments, and their therapeutic effects need to be confirmed by more studies.
Immunometabolism has emerged as a major and popular area of research in immunological disease, focusing on cellular metabolism and metabolic programs of specific cells, have advanced our understanding of pathogenesis and novel therapeutic targets. Although substantial progress has been made in the pathogenesis of vitiligo, there are still many questions remain to be answered. Immunometabolism in the pathogenesis of vitiligo is one of the most attractive research areas, which raised by the melanocytes from vitiligo patients are more susceptible to oxidative stress and finally lead to activation of the innate immune response and subsequently to adaptive immune response through activation of autoreactive cytotoxic CD8+ T cells. However, the mechanisms of how immunometabolism change skin melanocyte and immune cell infiltrations and function are not sufficiently explored and reviewed.
Here in this study, we reviewed the oxidative stress related physiological progress and specific defects in the metabolic pathways, which promote dysregulation of innate and adaptive immune responses in vitiligo. These abnormalities appear to be driven by genetic and epigenetic factors modulated by stochastic events. In addition to extensive descriptions of abnormalities in immunometabolism of vitiligo, recent studies support the critical role of dysregulation of metabolic pathways in various skin immune cells, including keratinocytes, tissue-resident memory T cells, and innate lymphoid cells of vitiligo pathogenesis.
Identifying therapeutic targets is a key goal for the research field. Currently, treatments targeting the IFN-γ chemokine axis, JAK-STAT pathway, and Nrf2-ARE pathway, have been recently emerged and reviewed. This review summarizes the current knowledge on immunometabolism reprogramming in the pathogenesis of vitiligo and gives an overview of future vitiligo treatment. Future studies may incorporate the effect of the microenvironment on immune cell metabolism and finding new approaches to understand the diversity of metabolic programs in specific cell population and ultimately for discovery of immune modulation and novel therapeutic targets.
CL drafted and edited the manuscript; YS edited and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Taishan Scholars Program of Shandong Province (tsqn201909141), Shandong Provincial Youth Science and Technology Talents Support Plan (ZR2020YQ56), the National Natural Science Foundation of China (81502736,82073441, 81874244).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Aryanian Z, Shirzadian A, Farzaneh S, Goodarzi A, Azizpour A, Hatami P. Metabolic derangement in patients with vitiligo: a cross-sectional study. J Investig Med (2022) 70(4):963–6. doi: 10.1136/jim-2021-002062
2. Kang P, Zhang WG, Ji ZH, Shao ZJ, Li CY. Association between vitiligo and relevant components of metabolic syndrome: A systematic review and meta-analysis. J Dtsch Dermatol Ges (2022) 20(5):629–41. doi: 10.1111/ddg.14717
3. Chuang KW, Chang HC. Association between vitiligo and metabolic syndrome: A systematic review and meta-analysis. J Dtsch Dermatol Ges (2022) 20(2):218–21. doi: 10.1111/ddg.14652
4. Picardo M, Dell'Anna ML, Ezzedine K, Hamzavi I, Harris JE, Parsad D, et al. Vitiligo. Nat Rev Dis Primers (2015) 1:15011. doi: 10.1038/nrdp.2015.11
5. Bergqvist C, Ezzedine K. Vitiligo: A review. Dermatology (2020) 236(6):571–92. doi: 10.1159/000506103
6. Geel NSR. Acquired pigmentary disorders. In: Griffiths C BJ, Bleiker T, Chalmers R, Creamer D, editors. Rook’s textbook of dermatology. Chichester, West Sussex: John Wiley & Sons Inc (2016).
7. Roberts GHL, Santorico SA, Spritz RA. The genetic architecture of vitiligo. Pigment Cell Melanoma Res (2020) 33(1):8–15. doi: 10.1111/pcmr.12848
8. Wang P, Li Y, Nie H, Zhang X, Shao Q, Hou X, et al. The changes of gene expression profiling between segmental vitiligo, generalized vitiligo and healthy individual. J Dermatol Sci (2016) 84(1):40–9. doi: 10.1016/j.jdermsci.2016.07.006
9. Speeckaert R, Lambert J, Bulat V, Belpaire A, Speeckaert M, van Geel N. Autoimmunity in segmental vitiligo. Front Immunol (2020) 11:568447. doi: 10.3389/fimmu.2020.568447
10. Chang HC, Lin MH, Huang YC, Hou TY. The association between vitiligo and diabetes mellitus: A systematic review and meta-analysis. J Am Acad Dermatol (2019) 81(6):1442–5. doi: 10.1016/j.jaad.2019.06.022
11. Ataş H, Gönül M. Increased risk of metabolic syndrome in patients with vitiligo. Balkan Med J (2017) 34(3):219–25. doi: 10.4274/balkanmedj.2016.1005
12. Tanacan E, Atakan N. Higher incidence of metabolic syndrome components in vitiligo patients: a prospective cross-sectional study. Bras Dermatol (2020) 95(2):165–72. doi: 10.1016/j.abd.2019.07.006
13. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
14. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
15. Azzazi Y, Mostafa WZ, Sayed KS, Alhelf M, Safwat M, Mahrous A, et al. Support for increased cardiovascular risk in non-segmental vitiligo among egyptians: A hospital-based, case-control study. Pigment Cell Melanoma Res (2021) 34(3):598–604. doi: 10.1111/pcmr.12941
16. Zhao Y, Zhang CF, Rossiter H, Eckhart L, Konig U, Karner S, et al. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J Invest Dermatol (2013) 133(6):1629–37. doi: 10.1038/jid.2013.26
17. Speeckaert R, Dugardin J, Lambert J, Lapeere H, Verhaeghe E, Speeckaert MM, et al. Critical appraisal of the oxidative stress pathway in vitiligo: A systematic review and meta-analysis. J Eur Acad Dermatol Venereol (2018) 32(7):1089–98. doi: 10.1111/jdv.14792
18. Liu Z, Pouli D, Alonzo CA, Varone A, Karaliota S, Quinn KP, et al. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Sci Adv (2018) 4(3):eaap9302. doi: 10.1126/sciadv.aap9302
19. Abdel-Malek ZA, Jordan C, Ho T, Upadhyay PR, Fleischer A, Hamzavi I, et al. The enigma and challenges of vitiligo pathophysiology and treatment. Pigment Cell Melanoma Res (2020) . 33(6):778–87. doi: 10.1111/pcmr.12878
20. Jimbow K, Chen H, Park JS, Thomas PD. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. Br J Dermatol (2001) 144(1):55–65. doi: 10.1046/j.1365-2133.2001.03952.x
21. Mansuri MS, Jadeja SD, Singh M, Laddha NC, Dwivedi M, Begum R. The catalase gene promoter and 5'-untranslated region variants lead to altered gene expression and enzyme activity in vitiligo. Br J Dermatol (2017) 177(6):1590–600. doi: 10.1111/bjd.15681
22. Em S, Laddha NC, Chatterjee S, Gani AR, Malek RA, Shah BJ, et al. Association of catalase T/C exon 9 and glutathione peroxidase codon 200 polymorphisms in relation to their activities and oxidative stress with vitiligo susceptibility in Gujarat population. Pigment Cell Res (2007) 20(5):405–7. doi: 10.1111/j.1600-0749.2007.00406.x
23. Mansuri MS, Laddha NC, Dwivedi M, Patel D, Alex T, Singh M, et al. Genetic variations (Arg5Pro and Leu6Pro) modulate the structure and activity of GPX1 and genetic risk for vitiligo. Exp Dermatol (2016) 25(8):654–7. doi: 10.1111/exd.13007
24. Yildirim M, Baysal V, Inaloz HS, Kesici D, Delibas N. The role of oxidants and antioxidants in generalized vitiligo. J Dermatol (2003) 30(2):104–8. doi: 10.1111/j.1346-8138.2003.tb00356.x
25. Agrawal D, Shajil EM, Marfatia YS, Begum R. Study on the antioxidant status of vitiligo patients of different age groups in Baroda. Pigment Cell Res (2004) 17(3):289–94. doi: 10.1111/j.1600-0749.2004.00149.x
26. Hazneci E, Karabulut AB, Ozturk C, Batcioglu K, Dogan G, Karaca S, et al. A comparative study of superoxide dismutase, catalase, and glutathione peroxidase activities and nitrate levels in vitiligo patients. Int J Dermatol (2005) 44(8):636–40. doi: 10.1111/j.1365-4632.2004.02027.x
27. Shajil EM, Begum R. Antioxidant status of segmental and non-segmental vitiligo. Pigment Cell Res (2006) 19(2):179–80. doi: 10.1111/j.1600-0749.2006.00299.x
28. Laddha NC, Dwivedi M, Gani AR, Shajil EM, Begum R. Involvement of superoxide dismutase isoenzymes and their genetic variants in progression of and higher susceptibility to vitiligo. Free Radic Biol Med (2013) 65:1110–25. doi: 10.1016/j.freeradbiomed.2013.08.189
29. Agrawal S, Kumar A, Dhali TK, Majhi SK. Comparison of oxidant-antioxidant status in patients with vitiligo and healthy population. Kathmandu Univ Med J (KUMJ) (2014) 12(46):132–6. doi: 10.3126/kumj.v12i2.13660
30. Röhrdanz E KR. Alterations of antioxidant enzyme expression in response to hydrogen peroxide. Free Radic Biol Med (1998) 24:27–38. doi: 10.1016/s0891-5849(97)00159-7
31. Hu Y, Huang J, Li Y, Jiang L, Ouyang Y, Li Y, et al. Cistanche deserticola polysaccharide induces melanogenesis in melanocytes and reduces oxidative stress via activating NRF2/HO-1 pathway. J Cell Mol Med (2020) 24(7):4023–35. doi: 10.1111/jcmm.15038
32. Goth L, Rass P, Pay A. Catalase enzyme mutations and their association with diseases. Mol Diagn (2004) 8(3):141–9. doi: 10.1007/BF03260057
33. Góth L EJ. Hereditary catalase deficiencies and increased risk of diabetes. Lancet (2000) 356 (9244):1820–1. doi: 10.1016/S0140-6736(00)03238-4
34. Jiang Z, Akey JM, Shi J, Xiong M, Wang Y, Shen Y, et al. A polymorphism in the promoter region of catalase is associated with blood pressure levels. Hum Genet (2001) 109(1):95–8. doi: 10.1007/s004390100553
35. Goulas A, Fidani L, Kotsis A, Mirtsou V, Petersen RC, Tangalos E, et al. An association study of a functional catalase gene polymorphism, -262C–>T, and patients with alzheimer's disease. Neurosci Lett (2002) 330(2):210–3. doi: 10.1016/S0304-3940(02)00780-2
36. Casp CB, She JX, McCormack WT. Genetic association of the catalase gene (CAT) with vitiligo susceptibility. Pigment Cell Res (2002) 15(1):62–6. doi: 10.1034/j.1600-0749.2002.00057.x
37. Liu L, Li C, Gao J, Li K, Zhang R, Wang G, et al. Promoter variant in the catalase gene is associated with vitiligo in Chinese people. J Invest Dermatol (2010) 130(11):2647–53. doi: 10.1038/jid.2010.192
38. Gavalas NG, Akhtar S, Gawkrodger DJ, Watson PF, Weetman AP, Kemp EH. Analysis of allelic variants in the catalase gene in patients with the skin depigmenting disorder vitiligo. Biochem Biophys Res Commun (2006) 345(4):1586–91. doi: 10.1016/j.bbrc.2006.05.063
39. Rashighi M, Harris JE. Vitiligo pathogenesis and emerging treatments. Dermatol Clin (2017) 35(2):257–65. doi: 10.1016/j.det.2016.11.014
40. Ohtani N. Deciphering the mechanism for induction of senescence-associated secretory phenotype (SASP) and its role in aging and cancer development. J Biochem (2019) 166(4):289–95. doi: 10.1093/jb/mvz055
41. Richmond JM, Frisoli ML, Harris JE. Innate immune mechanisms in vitiligo: danger from within. Curr Opin Immunol (2013) 25(6):676–82. doi: 10.1016/j.coi.2013.10.010
42. Tulic MK, Cavazza E, Cheli Y, Jacquel A, Luci C, Cardot-Leccia N, et al. Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo. Nat Commun (2019) 10(1):2178. doi: 10.1038/s41467-019-09963-8
43. Jacquemin C, Rambert J, Guillet S, Thiolat D, Boukhedouni N, Doutre MS, et al. Heat shock protein 70 potentiates interferon alpha production by plasmacytoid dendritic cells: relevance for cutaneous lupus and vitiligo pathogenesis. Br J Dermatol (2017) 177(5):1367–75. doi: 10.1111/bjd.15550
44. Xie H, Zhou F, Liu L, Zhu G, Li Q, Li C, et al. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci (2016) 81(1):3–9. doi: 10.1016/j.jdermsci.2015.09.003
45. Rashighi M, Harris JE. Interfering with the IFN-γ/CXCL10 pathway to develop new targeted treatments for vitiligo. Ann Transl Med (2015) 3(21):343. doi: 10.3978/j.issn.2305-5839.2015.11.36
46. Harris JE. Cellular stress and innate inflammation in organ-specific autoimmunity: lessons learned from vitiligo. Immunol Rev (2016) 269(1):11–25. doi: 10.1111/imr.12369
47. Ben Ahmed M, Zaraa I, Rekik R, Elbeldi-Ferchiou A, Kourda N, Belhadj Hmida N, et al. Functional defects of peripheral regulatory T lymphocytes in patients with progressive vitiligo. Pigment Cell Melanoma Res (2012) 25(1):99–109. doi: 10.1111/j.1755-148X.2011.00920.x
48. Lin M, Zhang BX, Shen N, Dong XJ, Zhang C, Qi XY, et al. Regulatory T cells from active non-segmental vitiligo exhibit lower suppressive ability on CD8+CLA+ T cells. Eur J Dermatol (2014) 24(6):676–82. doi: 10.1684/ejd.2014.2436
49. Kodydková J, Vávrová L, Kocík M, Žák A. Human catalase, its polymorphisms, regulation and changes of its activity in different diseases. Folia Biol (Praha) (2014) 60(4):153–67.
50. Dell'Anna ML, Maresca V, Briganti S, Camera E, Falchi M, Picardo M. Mitochondrial impairment in peripheral blood mononuclear cells during the active phase of vitiligo. J Invest Dermatol (2001) 117(4):908–13. doi: 10.1046/j.0022-202x.2001.01459.x
51. Dell'Anna ML, Ottaviani M, Albanesi V, Vidolin AP, Leone G, Ferraro C, et al. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J Invest Dermatol (2007) 127(5):1226–33. doi: 10.1038/sj.jid.5700700
52. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis (2017) 1863(5):1066–77. doi: 10.1016/j.bbadis.2016.11.010
53. Dell'Anna ML, Urbanelli S, Mastrofrancesco A, Camera E, Iacovelli P, Leone G, et al. Alterations of mitochondria in peripheral blood mononuclear cells of vitiligo patients. Pigment Cell Res (2003) 16(5):553–9. doi: 10.1034/j.1600-0749.2003.00087.x
54. Dell'Anna ML, Ottaviani M, Kovacs D, Mirabilii S, Brown DA, Cota C, et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci Rep (2017) 7(1):13663. doi: 10.1038/s41598-017-13961-5
55. Barygina V, Becatti M, Lotti T, Moretti S, Taddei N, Fiorillo C. Treatment with low-dose cytokines reduces oxidative-mediated injury in perilesional keratinocytes from vitiligo skin. J Dermatol Sci (2015) 79(2):163–70. doi: 10.1016/j.jdermsci.2015.05.003
56. Dell'Anna ML, Ottaviani M, Bellei B, Albanesi V, Cossarizza A, Rossi L, et al. Membrane lipid defects are responsible for the generation of reactive oxygen species in peripheral blood mononuclear cells from vitiligo patients. J Cell Physiol (2010) 223(1):187–93. doi: 10.1002/jcp.22027
57. Gasparro FP. The role of PUVA in the treatment of psoriasis. photobiology issues related to skin cancer incidence. Am J Clin Dermatol (2000) 1(6):337–48. doi: 10.2165/00128071-200001060-00002
58. Ramirez MU, Hernandez SR, Soto-Pantoja DR, Cook KL. Endoplasmic reticulum stress pathway, the unfolded protein response, modulates immune function in the tumor microenvironment to impact tumor progression and therapeutic response. Int J Mol Sci (2019) 21(1):169. doi: 10.3390/ijms21010169
59. Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J (1998) 17(19):5708–17. doi: 10.1093/emboj/17.19.5708
60. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell (1999) 10(11):3787–99. doi: 10.1091/mbc.10.11.3787
61. Huang G, Yao J, Zeng W, Mizuno Y, Kamm KE, Stull JT, et al. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J Cell Sci (2006) 119(Pt 1):153–61. doi: 10.1242/jcs.02731
62. Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell (2008) 134(5):769–81. doi: 10.1016/j.cell.2008.06.037
63. Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol (2009) 187(4):525–36. doi: 10.1083/jcb.200907074
64. Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, et al. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem (2008) 283(26):17954–61. doi: 10.1074/jbc.M801395200
65. Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol (2008) 181(8):5433–41. doi: 10.4049/jimmunol.181.8.5433
66. Saheki Y, De Camilli P. Endoplasmic reticulum-plasma membrane contact sites. Annu Rev Biochem (2017) 86:659–84. doi: 10.1146/annurev-biochem-061516-044932
67. Celli A, Sanchez S, Behne M, Hazlett T, Gratton E, Mauro T. The epidermal Ca(2+) gradient: Measurement using the phasor representation of fluorescent lifetime imaging. Biophys J (2010) 98(5):911–21. doi: 10.1016/j.bpj.2009.10.055
68. Celli A, Mackenzie DS, Crumrine DS, Tu CL, Hupe M, Bikle DD, et al. Endoplasmic reticulum Ca2+ depletion activates XBP1 and controls terminal differentiation in keratinocytes and epidermis. Br J Dermatol (2011) 164(1):16–25. doi: 10.1111/j.1365-2133.2010.10046.x
69. Riedhammer C, Weissert R. Antigen presentation, autoantigens, and immune regulation in multiple sclerosis and other autoimmune diseases. Front Immunol (2015) 6:322. doi: 10.3389/fimmu.2015.00322
70. Franco A, Almanza G, Burns JC, Wheeler M, Zanetti M. Endoplasmic reticulum stress drives a regulatory phenotype in human T-cell clones. Cell Immunol (2010) 266(1):1–6. doi: 10.1016/j.cellimm.2010.09.006
71. Yang X, Xia R, Yue C, Zhai W, Du W, Yang Q, et al. ATF4 regulates CD4(+) T cell immune responses through metabolic reprogramming. Cell Rep (2018) 23(6):1754–66. doi: 10.1016/j.celrep.2018.04.032
72. Giri PS, Dwivedi M, Laddha NC, Begum R, Bharti AH. Decreased suppression of CD8(+) and CD4(+) T cells by peripheral regulatory T cells in generalized vitiligo due to reduced NFATC1 and FOXP3 proteins. Exp Dermatol (2020) 29(8):759–75. doi: 10.1111/exd.14157
73. Giri PS, Dwivedi M, Laddha NC, Begum R, Bharti AH. Altered expression of nuclear factor of activated T cells, forkhead box P3, and immune-suppressive genes in regulatory T cells of generalized vitiligo patients. Pigment Cell Melanoma Res (2020) 33(4):566–78. doi: 10.1111/pcmr.12862
74. Giri PS, Patel S, Begum R, Dwivedi M. Association of FOXP3 and GAGE10 promoter polymorphisms and decreased FOXP3 expression in regulatory T cells with susceptibility to generalized vitiligo in Gujarat population. Gene (2021) 768:145295. doi: 10.1016/j.gene.2020.145295
75. He Y, Li S, Zhang W, Dai W, Cui T, Wang G, et al. Dysregulated autophagy increased melanocyte sensitivity to H(2)O(2)-induced oxidative stress in vitiligo. Sci Rep (2017) 7:42394. doi: 10.1038/srep42394
76. Zhang Y, Liu L, Jin L, Yi X, Dang E, Yang Y, et al. Oxidative stress-induced calreticulin expression and translocation: new insights into the destruction of melanocytes. J Invest Dermatol (2014) 134(1):183–91. doi: 10.1038/jid.2013.268
77. Thamer M, Ahmed A, Sanjeev M, Salma A, Mohammed Y, Mohammed A. Association of vitiligo with anemia, vitamin B12 deficiency, diabetes mellitus, and thyroid dysfunction in Saudi Arab patients: A case control study. J Dermatol Dermatologic Surg (2017) 21(2):72–6. doi: 10.1016/j.jdds.2017.06.001
78. Gopal KV, Rao GR, Kumar YH. Increased prevalence of thyroid dysfunction and diabetes mellitus in Indian vitiligo patients: A case-control study. Indian Dermatol Online J (2014) 5(4):456–60. doi: 10.4103/2229-5178.142493
79. Afkhami-Ardekani M, Ghadiri-Anari A, Ebrahimzadeh-Ardakani M, Zaji N. Prevalence of vitiligo among type 2 diabetic patients in an Iranian population. Int J Dermatol (2014) 53(8):956–8. doi: 10.1111/ijd.12148
80. Raveendra L, Hemavathi RN, Rajgopal S. A study of vitiligo in type 2 diabetic patients. Indian J Dermatol (2017) 62(2):168–70. doi: 10.4103/ijd.IJD_360_16
81. Mitra S, De Sarkar S, Pradhan A, Pati AK, Pradhan R, Mondal D, et al. Levels of oxidative damage and proinflammatory cytokines are enhanced in patients with active vitiligo. Free Radic Res (2017) 51(11-12):986–94. doi: 10.1080/10715762.2017.1402303
82. Karadag AS, Tutal E, Ertugrul DT. Insulin resistance is increased in patients with vitiligo. Acta Derm Venereol (2011) 91(5):541–4. doi: 10.2340/00015555-1141
83. Pietrzak A, Bartosińska J, Dybiec E, Chodorowska G, Krasowska D, Hercogova J, et al. Hepato-splenic and lipid profile abnormalities–do they exist in children affected with vitiligo? Acta Dermatovenerol Croat (2014) 22(1):19–25.
84. van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol (2009) 129(9):2220–32. doi: 10.1038/jid.2009.32
85. Mansuri MS, Singh M, Jadeja SD, Begum R. Association of glucose 6-phosphate dehydrogenase (G6PD) 3'UTR polymorphism with vitiligo and in vitro studies on G6PD inhibition in melanocytes. J Dermatol Sci (2019) 93(2):133–5. doi: 10.1016/j.jdermsci.2018.12.001
86. Shiu J, Zhang L, Lentsch G, Flesher JL, Jin S, Polleys C, et al. Multimodal analyses of vitiligo skin identify tissue characteristics of stable disease. JCI Insight (2022) 7(13):e154585. doi: 10.1172/jci.insight.154585
87. Pietrzak A, Lecewicz-Toruń B, Urban J. Comparison of serum lipid in girls affected with vitiligo and control group. Ann Univ Mariae Curie Sklodowska Med (2000) 55:269–74.
88. Steffen Y, Vuillaume G, Stolle K, Roewer K, Lietz M, Schueller J, et al. Cigarette smoke and LDL cooperate in reducing nitric oxide bioavailability in endothelial cells via effects on both eNOS and NADPH oxidase. Nitric Oxide (2012) 27(3):176–84. doi: 10.1016/j.niox.2012.06.006
89. Pietrzak A, Bartosinska J, Hercogova J, Lotti TM, Chodorowska G. Metabolic syndrome in vitiligo. Dermatol Ther (2012) 25 Suppl 1:S41–3. doi: 10.1111/dth.12012
90. Karadag AS, Tutal E, Ertugrul DT, Akin KO, Bilgili SG. Serum holotranscobalamine, vitamin B12, folic acid and homocysteine levels in patients with vitiligo. Clin Exp Dermatol (2012) 37(1):62–4. doi: 10.1111/j.1365-2230.2011.04142.x
91. Jadeja SD, Mansuri MS, Singh M, Patel H, Marfatia YS, Begum R. Association of elevated homocysteine levels and methylenetetrahydrofolate reductase (MTHFR) 1298 A > C polymorphism with vitiligo susceptibility in Gujarat. J Dermatol Sci (2018) 90(2):112–22. doi: 10.1016/j.jdermsci.2018.01.003
92. Jakubowski H. Molecular basis of homocysteine toxicity in humans. Cell Mol Life Sci (2004) 61(4):470–87. doi: 10.1007/s00018-003-3204-7
93. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun (2020) 11(1):433. doi: 10.1038/s41467-020-14324-x
94. Herbette S, Roeckel-Drevet P, Drevet JR. Seleno-independent glutathione peroxidases. more than simple antioxidant scavengers. FEBS J (2007) 274(9):2163–80. doi: 10.1111/j.1742-4658.2007.05774.x
95. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10(1):1617. doi: 10.1038/s41467-019-09277-9
96. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
97. Wu X, Jin S, Yang Y, Lu X, Dai X, Xu Z, et al. Altered expression of ferroptosis markers and iron metabolism reveals a potential role of ferroptosis in vitiligo. Pigment Cell Melanoma Res (2022) 35(3):328–41. doi: 10.1111/pcmr.13032
98. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
99. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
100. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
101. Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med (2014) 6(223):223ra23. doi: 10.1126/scitranslmed.3007811
102. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HSc, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest (2017) 127(11):4031–41. doi: 10.1172/JCI93396
103. Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol (2017) 18(8):940–50. doi: 10.1038/ni.3775
104. Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol (2017) 2(10):eaam6346. doi: 10.1126/sciimmunol.aam6346
105. Boniface K, Jacquemin C, Darrigade AS, Dessarthe B, Martins C, Boukhedouni N, et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol (2018) 138(2):355–64. doi: 10.1016/j.jid.2017.08.038
106. Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun (2017) 8:15221. doi: 10.1038/ncomms15221
107. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol (2012) 132(7):1869–76. doi: 10.1038/jid.2011.463
108. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543(7644):252–6. doi: 10.1038/nature21379
109. Greer RL, Dong X, Moraes AC, Zielke RA, Fernandes GR, Peremyslova E, et al. Akkermansia muciniphila mediates negative effects of IFNgamma on glucose metabolism. Nat Commun (2016) 7:13329. doi: 10.1038/ncomms13329
110. Agarwal P, Rashighi M, Essien KI, Richmond JM, Randall L, Pazoki-Toroudi H, et al. Simvastatin prevents and reverses depigmentation in a mouse model of vitiligo. J Invest Dermatol (2015) 135(4):1080–8. doi: 10.1038/jid.2014.529
111. Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE. CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol (2017) 137(4):982–5. doi: 10.1016/j.jid.2016.10.048
112. Richmond JM, Bangari DS, Essien KI, Currimbhoy SD, Groom JR, Pandya AG, et al. Keratinocyte-derived chemokines orchestrate T-cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol (2017) 137(2):350–8. doi: 10.1016/j.jid.2016.09.016
113. Ng CY, Chiu YC, Chan YP, Lin YJ, Chung PH, Chung WH, et al. Skin interstitial fluid and plasma multiplex cytokine analysis reveals IFN-gamma signatures and granzyme b as useful biomarker for activity, severity and prognosis assessment in vitiligo. Front Immunol (2022) 13:872458. doi: 10.3389/fimmu.2022.872458
114. Xu Z, Chen D, Hu Y, Jiang K, Huang H, Du Y, et al. Anatomically distinct fibroblast subsets determine skin autoimmune patterns. Nature (2022) 601(7891):118–24. doi: 10.1038/s41586-021-04221-8
115. Ge D, Gooljar SB, Kyriakou T, Collins LJ, Swaminathan R, Snieder H, et al. Association of common JAK2 variants with body fat, insulin sensitivity and lipid profile. Obes (Silver Spring) (2008) 16(2):492–6. doi: 10.1038/oby.2007.79
116. Mishra J, Verma RK, Alpini G, Meng F, Kumar N. Role of janus kinase 3 in predisposition to obesity-associated metabolic syndrome. J Biol Chem (2015) 290(49):29301–12. doi: 10.1074/jbc.M115.670331
117. Chaimowitz NS, Ebenezer SJ, Hanson IC, Anderson M, Forbes LR. STAT1 gain of function, type 1 diabetes, and reversal with JAK inhibition. N Engl J Med (2020) 383(15):1494–6. doi: 10.1056/NEJMc2022226
118. Incyte announces positive results from phase 3 TRuE-V program evaluating ruxolitinib cream in patients with vitiligo (2021). Available at: https://www.businesswire.com/news/home/20210517005235/en.
119. Sheikh A, Rafique W, Owais R, Malik F, Ali E. FDA Approves ruxolitinib (Opzelura) for vitiligo therapy: A breakthrough in the field of dermatology. Ann Med Surg (Lond) (2022) 81:104499. doi: 10.1016/j.amsu.2022.104499
120. Jian Z, Li K, Song P, Zhu G, Zhu L, Cui T, et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo. J Invest Dermatol (2014) 134(8):2221–30. doi: 10.1038/jid.2014.152
121. de Oliveira-Marques V, Cyrne L, Marinho HS, Antunes F. A quantitative study of NF-kappaB activation by H2O2: relevance in inflammation and synergy with TNF-alpha. J Immunol (2007) 178(6):3893–902. doi: 10.4049/jimmunol.178.6.3893
122. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-kappaB: A blossoming of relevance to human pathobiology. Cell (2017) 168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012
123. Blackwell TS, Christman JW. The role of nuclear factor-kappa b in cytokine gene regulation. Am J Respir Cell Mol Biol (1997) 17(1):3–9. doi: 10.1165/ajrcmb.17.1.f132
124. Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant (2014) 29 Suppl 1:i19–24. doi: 10.1093/ndt/gft224
125. Zhang S, Yi X, Su X, Jian Z, Cui T, Guo S, et al. Ginkgo biloba extract protects human melanocytes from H2 O2 -induced oxidative stress by activating Nrf2. J Cell Mol Med (2019) 23(8):5193–9. doi: 10.1111/jcmm.14393
126. Jung E, Kim JH, Kim MO, Lee SY, Lee J. Melanocyte-protective effect of afzelin is mediated by the Nrf2-ARE signalling pathway via GSK-3beta inactivation. Exp Dermatol (2017) 26(9):764–70. doi: 10.1111/exd.13277
127. Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol (2019) 139(4):769–78. doi: 10.1016/j.jid.2018.10.032
128. Pan Y, Kupper TS. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol (2018) 9:1347. doi: 10.3389/fimmu.2018.01347
129. Richmond JM, Strassner JP, Zapata L. Jr, Garg M, Riding RL, Refat MA, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med (2018) 10(450):eaam7710. doi: 10.1126/scitranslmed.aam7710
130. Noel M, Gagne C, Bergeron J, Jobin J, Poirier P. Positive pleiotropic effects of HMG-CoA reductase inhibitor on vitiligo. Lipids Health Dis (2004) 3:7. doi: 10.1186/1476-511X-3-7
131. Namazi MR. Statins: novel additions to the dermatologic arsenal? Exp Dermatol (2004) 13(6):337–9. doi: 10.1111/j.0906-6705.2004.00208.x
Keywords: vitiligo, immunometabolism, oxidative stress, glucose metabolism, lipid metabolism, immunotherapy
Citation: Lyu C and Sun Y (2022) Immunometabolism in the pathogenesis of vitiligo. Front. Immunol. 13:1055958. doi: 10.3389/fimmu.2022.1055958
Received: 28 September 2022; Accepted: 24 October 2022;
Published: 10 November 2022.
Edited by:
Zhu Shen, Guangdong Academy of Medical Sciences, ChinaReviewed by:
Shahnawaz Jadeja, University College Dublin, IrelandCopyright © 2022 Lyu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonghu Sun, c3VvaGFuZG9uZ0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.