 Xiaojie Song
Xiaojie Song Jiannan Ma
Jiannan Ma
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 01 December 2022
Sec. Multiple Sclerosis and Neuroimmunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1050688
Purpose: To facilitate the identification of myelin-oligodendrocyte glycoprotein (MOG) antibody-associated diseases in pediatric autoimmune encephalitis without demyelination, we explored the clinical characteristics of patients having MOG antibody-positive pediatric autoimmune encephalitis without demyelination in Children’s Hospital of Chongqing Medical University, China.
Methods: We reviewed patients’ medical records from January 2019 to June 2022 and retrospectively analyzed clinical manifestations, brain magnetic resonance imaging (MRI) findings, laboratory findings, treatments and outcomes of children with autoimmune encephalitis who tested positive for MOG antibodies in serum but for whom demyelination was not detected on MRI.
Results: Eighteen patients (6 boys, 12 girls; median age: 103.2 (range: 36–160) months) were included: 15 tested positive for MOG antibodies in both serum and cerebrospinal fluid (CSF); three tested positive only in serum. The most common clinical symptoms were altered mental status (18/18), fever (16/18), headache (14/18), seizures (6/18) and focal neurologic deficits (5/18). All patients had CSF pleocytosis (median count: 74/µL, range: 14–380/µL); five patients had elevated CSF protein levels (median: 0.85 g/L, range: 0.53–1.48 g/L) simultaneously. CSF glucose levels were normal in all patients. Abnormal electroencephalogram (EEG) results were found in 12 patients: generalized or focal slowing (9/12), focal epileptic discharges (2/12), and generalized slowing and focal epileptic discharges (1/12). Twelve of the 18 patients showed hyperintense T2-weighted lesions on brain MRI in the cortex (6), basal ganglia (5), thalamus (3), cerebellum (4), and brainstem (2). All patients received immunotherapy and had favorable outcomes at discharge (modified Rankin scale score: <2). Three children relapsed once; however, all children had good outcomes at the last follow-up.
Conclusion: MOG antibody-positive pediatric autoimmune encephalitis without demyelination is mainly characterized by prolonged fever, altered mental status, headache, mild-to-moderate increase in cell count in the CSF, and normal or abnormal brain MRI, which may involve any part outside the white matter without specificity. All patients with MOG antibody-positive pediatric autoimmune encephalitis without demyelination had favorable outcomes after immunotherapy, while a few patients relapsed once.
Myelin-oligodendrocyte glycoprotein (MOG) is an important component of myelin in the central nervous system (CNS) and is expressed at the external surface of myelin and the plasma membranes of oligodendrocytes (1). The antibody to MOG is involved in various demyelinating disorders of CNS, including acute disseminated encephalomyelitis (ADEM), optic neuritis (ON), myelitis, and neuromyelitis optic spectrum disorder (NMOSD) (2, 3). Multinational studies have identified serum anti-MOG antibody positivity in approximately 40% of children with acute demyelinating syndrome (1, 4).
In recent years, the anti-MOG antibody has been reported to be associated with various types of autoimmune encephalitis (AE) without demyelination, such as cortical encephalitis, brainstem encephalitis, and even AE with normal findings on brain magnetic resonance imaging (MRI) (5–10). As more and more MOG antibody-associated phenotypes emerged, the concept of MOG antibody-associated diseases (MOGAD) was proposed (1). However, relevant reports about MOG antibody-associated AE without demyelination were usually case reports, and there were few pertinent studies with large samples, especially including Asian children. Therefore, in this study, we assessed the cases of 18 children having MOG antibody-positive AE without demyelination in a single center in southwest China.
Medical records of children who were hospitalized at Children’s Hospital of Chongqing Medical University between January 2019 and June 2022 were reviewed. Those who met the following criteria were included in the study: (i) patients who tested positive for MOG antibodies in the serum, (ii) patients who fulfilled the criteria for possible AE (11), (iii) patients whose brain MRI revealed no demyelination (i.e. no involvement of the cerebral white matter), and (iv) patients for whom alternative causes (e.g., intracranial infections or autoimmune diseases) could be reasonably excluded.
Patients’ clinical features, MRI findings, laboratory findings, treatment, and outcomes were analyzed retrospectively. After discharge, all patients were followed up by outpatient visits. Severity at onset and outcome at the last clinical evaluation were evaluated with the modified Rankin scale (mRS) (12). This study was approved by the ethics committee of Children’s Hospital of Chongqing Medical University. Written informed consent was obtained from the children’s parents or legal guardians.
MOG antibodies in serum and CSF were tested by using a fixed cell-based assay involving HEK293 cells co-transfected with human MOG and pcDNA3.1-EGFP.
Eighteen patients who met the inclusion criteria were included: fifteen and three patients tested positive for MOG antibodies in both serum and CSF and in serum only, respectively. The demographic and clinical characteristics of the 18 patients are shown in Table 1. The median patient age was 103.2 months (range: 36–160 months), and 6 patients were male (33.3%). All patients had no previous history of disease in the central nervous system.
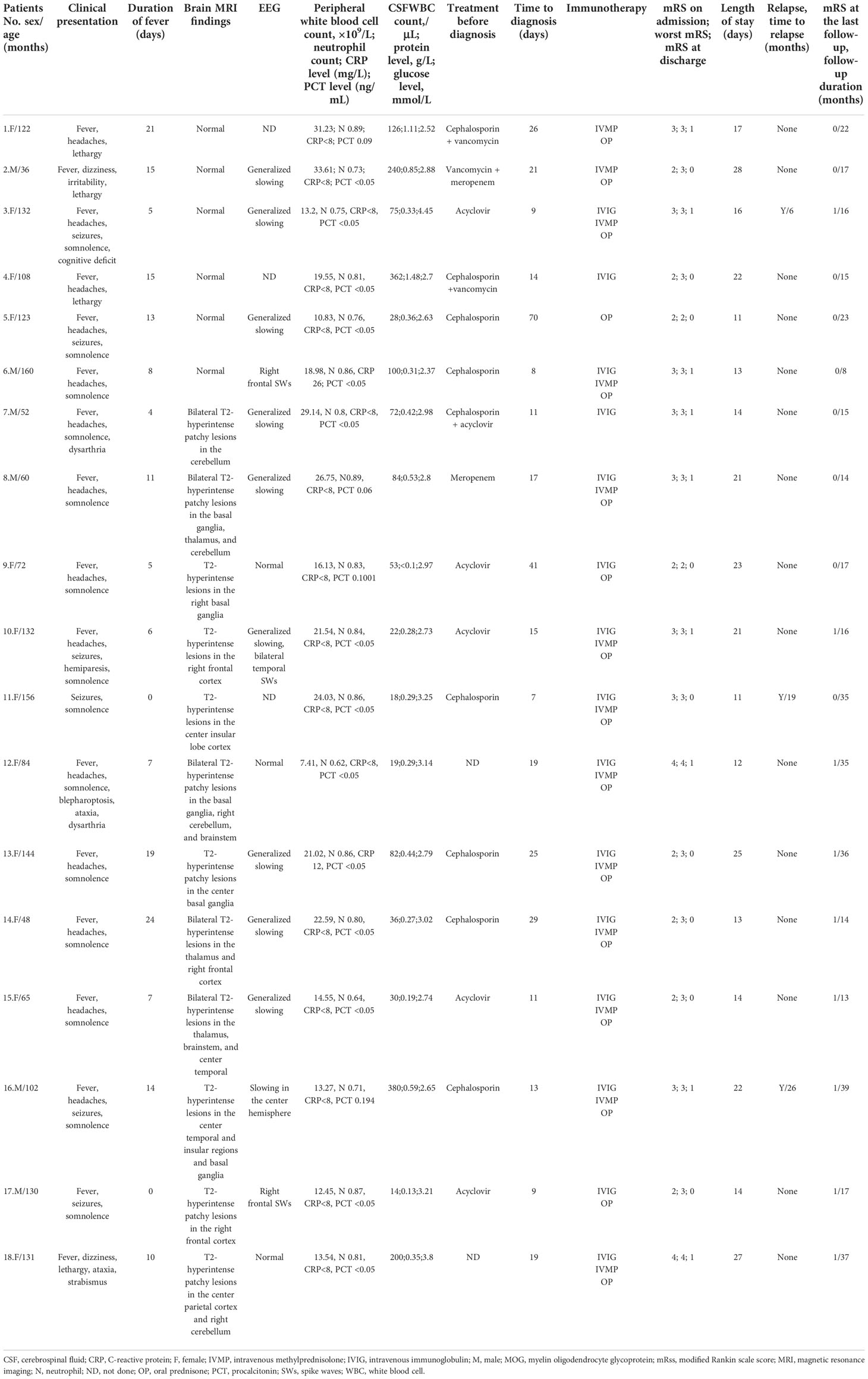
Table 1 Detailed demographic and clinical characteristics of 18 patients having MOG antibody-positive pediatric autoimmune encephalitis without demyelination.
The most common clinical symptoms were altered mental status (18/18, lethargy in 4, somnolence in 14); fever (16/18) lasting for 4–24 days (median: 10.5 days); headaches (14/18); seizures (6/18); and focal neurologic deficits (5/18), including dysarthria (2/18), ataxia (2/18), extraocular muscle disorder (2/18), and hemiparesis (1/18). All the patients had no movement disorder, psychiatric/behavioural symptoms, or dysautonomia.
Seventeen in 18 patients had leukocytosis in peripheral blood (median: 19.55 × 109 cells/L, range: 10.83–33.61 × 109 cells/L, normal value: 4–10 × 109 cells/L). C-reaction protein (CRP) and procalcitonin (PCT) were mildly elevated in two patients (range: 12–26 mg/L, normal value: <8 mg/L) and 4 patients (range: 0.06–0.194 ng/mL, normal value: <0.05 ng/mL), respectively. All patients had CSF pleocytosis (median: 74/µL, range: 14–380/µL, normal value: ≤5/µL); five patients had elevated CSF protein levels (median: 0.85 g/L, range: 0.53–1.48 g/L, normal value: ≤0.45 g/L). All patients had normal CSF glucose levels. Bacterial culture and polymerase chain reaction for herpes simplex virus (HSV) and Epstein-Barr virus (EBV) in the CSF were negative for all patients.
Electroencephalogram (EEG) was performed in 15 patients, revealing generalized or focal slowing in nine children, focal epileptic discharges in two children, and generalized slowing combined with focal epileptic discharges in one child, and normal EEG in three patients.
Brain MRI findings were showed in Table 1. Brain MRIs were performed for all 18 patients. Hyperintense lesions on T2-weighted images were noted in 12 patients (Figures 1 A–F), while the findings were negative in six patients. The distribution of lesions was unilateral in six patients and bilateral in six patients. The abnormal T2-hyperintense lesions were located in the cortex (n = 6, including parietal [2/6], temporal [3/6], frontal [1/6], and insular [2/6] regions), basal ganglia (n = 5), thalamus (n = 3), cerebellum (n = 4), and brainstem (n = 2). Brain MRI showed isolated lesions in six patients (isolated cortex [n = 3], isolated cerebellum [n = 1], and isolated basal ganglia [n = 2]), combined lesions in six other patients (cortex combined with basal ganglia or thalamus [n = 3, one involved the brainstem], cortex combined with the cerebellum [n = 1], basal ganglia combined with the thalamus and cerebellum [n = 1], and basal ganglia combined with the brainstem and cerebellum [n = 1]). Gadolinium -enhanced brain MRI was performed in 17 patients (except patient 5), 16 of whom had no enhancement, and only one patient (patient 8) had slightly enhancement in the right cerebellum. Diffusion Weighted Imaging (DWI) was performed in all patients, but diffusion restriction was observed in none of them.
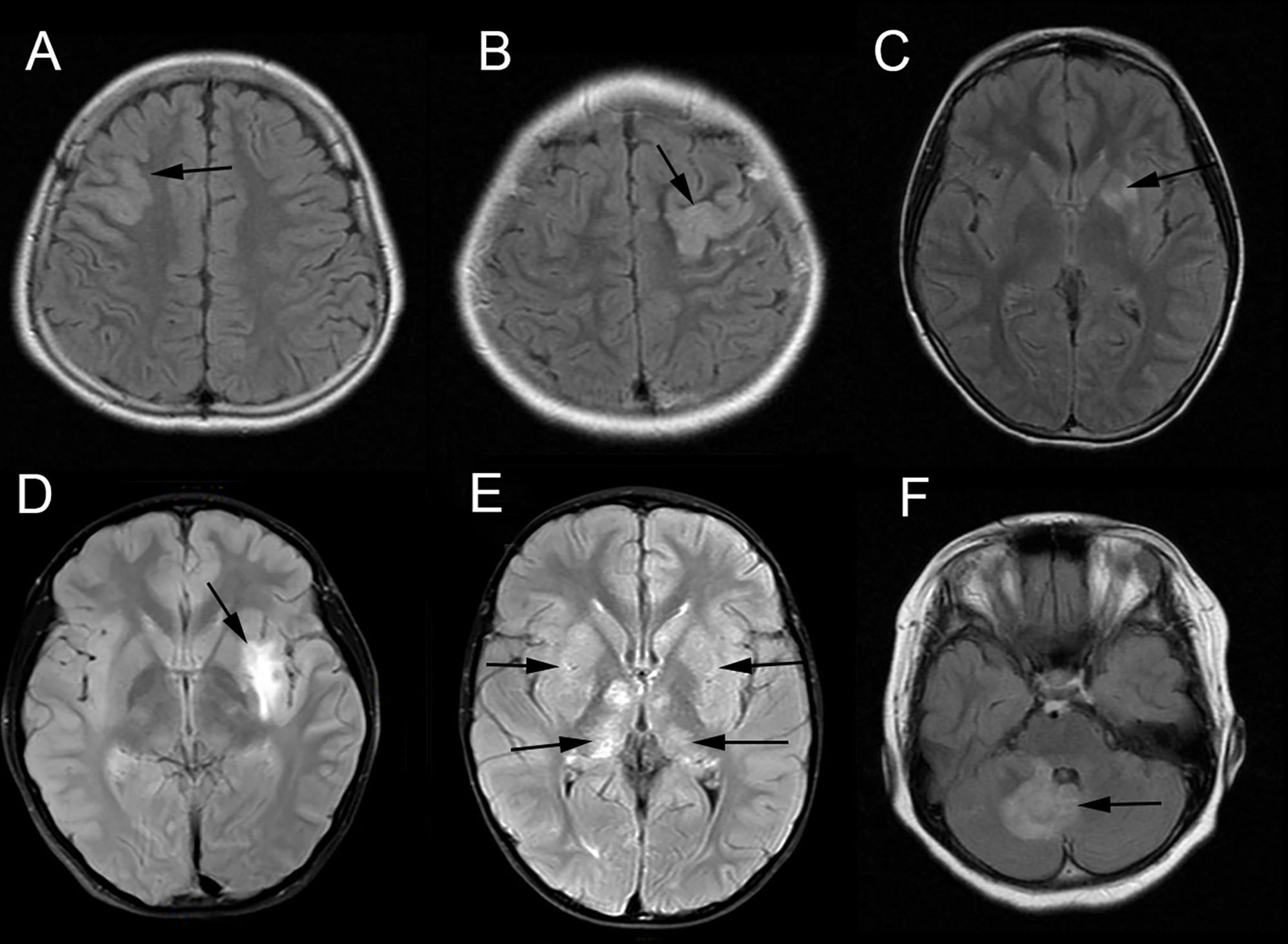
Figure 1 Brain magnetic resonance imaging (MRI) of patients having MOG antibody-positive pediatric autoimmune encephalitis without demyelination. The abnormal hyperintensity lesions on axial T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) image were observed in the right frontal cortex (A, Patient 17), left parietal cortex (B, Patient 18), left basal ganglia (C, Patient 13), left insular cortex and basal ganglia (D, Patient 16), bilateral basal ganglia and thalamus (E, Patient 8) and right cerebellum (F, Patient 18).
The treatment and outcomes at discharge in the 18 patients are shown in Table 1. Before the diagnosis of MOGAD, anti-infection therapy was administered empirically due to suspected intracranial infections. Ten patients received antibiotic therapy, five patients received acyclovir, and one patient received both antibiotics and acyclovir. However, no patient responded to anti-infection therapy. The median time to diagnosis after onset was 14.5 days (range: 7–70 days) in all patients. All patients were treated with immunotherapy after MOGAD was confirmed. In our center, the general immunotherapy program for patients with MOG antibody-positive pediatric autoimmune encephalitis without demyelination is intravenous immunoglobulin (IVIG, 400 mg/kg/day for 5 days) followed by sequential administration of intravenous methylprednisolone (IVMP, 15–20 mg/kg/day for 3–5 days) and oral prednisone (OP, 1.5–2.0 mg/kg/day). If the condition is significantly improved after the use of IVIG, the IVMP can be omitted. However, in clinic, sometimes patients’ parents would refuse to use IVIG due to economic reason or corticosteroids in consideration of their side effect. So, in our study, 15 of the 18 patients were treated with IVIG, of whom 11 were subsequently treated with IVMP followed by OP, 2 were followed by OP only, and 2 without corticosteroids. Two of the 18 patients were treated with IVMP followed by OP, and one patient was treated with OP alone. For the patients received OP, the drug was tapered off within 3-6 months. The modified Rankin scale (mRS) score decreased in all patients after immunotherapy, and all patients had favorable outcomes at discharge (mRS score < 2). The median length of stay was 16.5 days (range: 11–28 days) in all patients.
After discharge, all patients were followed up by outpatient visits. The follow-up duration was between 8 and 39 months, with the average being 21.67 months. During follow-up, three patients relapsed once (patient 3, patient 11, patient 16). The patient 3 presented with seizure once 6 months after discharge. Her repeated brain MRI showed T2-hyperintense lesions in the right thalamus after relapsed, EEG had no positive finding, and MOG antibodies in both serum and CSF were positive. The patient 11 suffered seizure once 9 months after discharge with positive MOG antibodies in both serum and CSF, but she had normal EEG and brain MRI findings. The patient 16 presented with optic neuritis in the right eye with MOG antibodies positive in serum 26 months after discharge. His checkerboard visual evoked potential showed increased latency time and decreased amplitude of p100 wave for the right eye, but his brain and optic nerve MRI finding were normal. All the three patients experienced symptom relief after receiving IVMP followed by OP. Further, those three patients were also treated with immunosuppressive treatments (such as mycophenolate mofetil (MMF) or azathioprine) to prevent relapses. All patients had favorable outcomes at the last follow-up (mRS score < 2).
In this study, we evaluated the clinical characteristics of 18 pediatric patients having autoimmune encephalitis with positive MOG antibodies in the serum but without demyelination on brain MRI. To our knowledge, this is the first study on Asian children to specifically report the clinical characteristics of the spectrum of MOG antibody-positive pediatric autoimmune encephalitis without demyelination.
In a prospective study, Armangue et al. (13) showed that among 296 children with definite or possible encephalitis other than ADEM, MOG antibodies were more common than NMDAR antibodies (22 [7.4%] VS 14 [4.7%]), and surpassed all neuronal antibodies combined. During the same period (January 2019 to June 2022), 88 patients were diagnosed with anti-NMDAR encephalitis in our center. Not all patients with suspected autoimmune encephalitis were tested for anti-MOG antibodies in our retrospective study. Therefore, whether the anti-MOG antibody is the most common antibody in Asian patients having autoimmune encephalitis without demyelination needs to be confirmed in more prospective studies.
In this study, the main clinical manifestations in patients having MOG antibody-positive autoimmune encephalitis without demyelination were altered mental status, prolonged fever, and headaches. However, these clinical manifestations are non-specific. In contrast, anti-NMDAR encephalitis, which was considered the most common type of autoimmune encephalitis in children, is characterized by behavioral problems and/or movement disorders (11, 14).
CSF tests revealed abnormal findings in all patients in our study. All patients had CSF pleocytosis, and a few (5/18) had elevated CSF protein levels. No patient had an abnormal glucose level. In the study by Armangue et al. (13), all encephalitis patients with anti-MOG antibody positivity other than ADEM had varying degrees of CSF leukocytosis. However, none of them showed elevated protein levels.
Interestingly, almost all patients (17/18) had leukocytosis in peripheral blood, with half of them having a cell count higher than 20 × 109/L. Based on our experience, leukocytosis in peripheral blood is rare in other types of autoimmune encephalitis (e.g., anti-NMDAR encephalitis). Therefore, many of our patients were suspected of having bacterial meningitis and administered antibiotics for a long time before the diagnosis of MOGAD, and the longest time to diagnosis was approximately 70 days. However, the following findings are especially important. (1) No patient showed significantly increased levels of the inflammatory indicators CRP and PCT. In fact, most patients had normal levels of these inflammatory indicators. (2) CSF glucose levels were normal in all patients. (3) Finally, no patient responded to antibiotic therapy.
The brain MRI of six patients was normal. Armangue et al. (13) have reported two cases of encephalitis with normal brain MRI findings and anti-MOG antibody positivity. However, they did not describe the specific clinical characteristics of these two patients. In our study, the main clinical manifestations of the aforementioned six patients were fever, a slightly altered mental status, and headaches (5/6); pleocytosis in the CSF; and normal CSF glucose levels. Given these clinical manifestations, the condition needs to be differentiated from viral meningitis and systemic diseases [e.g., systemic lupus erythematosus (SLE), Kawasaki disease] with meningeal involvement. However, five of these patients had a fever that lasted for more than 1 week (up to 21 days) before immunotherapy, which is not consistent with viral meningitis because viral meningitis is self-limited (duration is usually less than 1 week). None of these 6 patients had rashes, lymphadenopathy, other common manifestations of systemic diseases, and elevated levels of the inflammatory indicator CRP. Therefore, systemic disease was not considered. The brain MRI of the other 12 patients was abnormal. However, the lesions had no specificity and involved the cerebral cortex, basal ganglia, thalamus, cerebellum, or brainstem. Such lesions may be isolated (isolated cortex, cerebellum, or basal ganglia) or combined (e.g., involving the cortex combined with basal ganglia or thalamus or basal ganglia combined with thalamus). Therefore, differentiating them from the lesions associated with viral encephalitis based on brain imaging alone may be difficult. Hence, we tested CSF for HSV and EBV to exclude HSV and EBV encephalitis). Most patients were empirically treated with anti-infection therapy before the diagnosis of MOGAD. Therefore, the detection of MOG antibodies was crucial for confirming the diagnosis in these patients.
All of our patients had a good response to intravenous immunotherapy and had a good prognosis at discharge (mRS score < 2). This finding was consistent with those reported for most patients with MOG antibody-positive demyelinating syndromes. It’s worth mentioning that the immunotherapy strategy for our patients differs slightly from available consensus recommendations of pediatric MOGAD due to the previous suspicions of intracranial infections in these patients (15). However, in our study, three patients treated with the corticosteroids alone also experienced remission, which suggested the IVIG treatment might not be necessary for some of our patients. During follow-up, three patients relapsed; the rate was comparable to that of MOG antibody-positive demyelinating syndromes (16.7% VS 33%) (13). These three patients improved rapidly after re-administration of intravenous immunotherapy and had a good prognosis at the final follow-up.
The pathological mechanism underlying the generation of anti-MOG antibodies in pediatric autoimmune encephalitis without demyelination is unknown. Theoretically, the anti-MOG antibody, as a kind of extracellular antigen located on the surface of myelin, should only be associated with inflammatory demyelinating diseases of the white matter in the central nervous system (such as ADEM and ON) (16). Some previous reports have described cortical encephalitis and meningoencephalitis without demyelination and positive results for anti-MOG antibodies (6–8, 11). Therefore, for encephalitis without demyelination, the anti-MOG antibody itself may not be directly associated with the encephalitis and another unknown autoimmune disorder coexisting with anti-MOG antibody positivity may be responsible for the encephalitis. In addition, MOG is not only an important component of the myelin sheath but also a type of myelin inhibitor of axonal regrowth (17). Whether it is another possible pathological mechanism that even though anti-MOG antibodies do not lead to demyelination, may damage the structures and functions of neurons through destroying the function of MOG as a kind of inhibitor of axonal regrowth. Nevertheless, this is only a hypothesis, and more animal and clinical research is needed to prove it.
Our study had some limitations. First, we did not detect neuronal surface antibodies (such as anti-NMDAR antibodies) in all patients. However, we did not think our patients had coexisting neuronal surface antibodies since pediatric autoimmune encephalitis with neuronal surface antibodies mainly manifests as behavioral problems and/or movement disorders. Second, the follow-up period was relatively short for some patients. Hence, determination of the true recurrence rate of MOG antibody-positive pediatric autoimmune encephalitis without demyelination will require a longer follow-up. Finally, our study was a single-center retrospective study with a small number of cases. Hence, more prospective multicenter studies with larger samples are needed in the future.
For patients with suspected encephalitis manifesting as altered mental status, prolonged fever, headaches, and leukocytosis in the CSF, anti-MOG antibody-associated diseases should be concerned and the anti-MOG antibody test should be performed early to avoid a delayed diagnosis, even for those whose brain MRI is normal or shows no demyelination lesions. Our study shows that patients having MOG antibody-positive pediatric autoimmune encephalitis without demyelination usually have favorable outcomes after immunotherapy, while a few may relapse.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Ethics Committee of Children’s Hospital of Chongqing Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
XS collected clinical records, analyzed and interpreted the data, drafted and revised the manuscript. JM designed and conceptualized the study and revised the manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol (2021) 20(9):762–72. doi: 10.1016/S1474-4422(21)00218-0.2
2. Ramanathan S, Dale RC, Brilot F. Anti-MOG antibody: The history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev (2016) 15(4):307–24. doi: 10.1016/j.autrev.2015.12.004
3. Di Pauli F, Berger T. Myelin oligodendrocyte glycoprotein antibody-associated disorders: Toward a new spectrum of inflammatory demyelinating CNS disorders? Front Immunol (2018) 9:2753. doi: 10.3389/fimmu.2018.02753
4. López-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-Associated disorders. JAMA Neurol (2018) 75(11):1355–63. doi: 10.1001/jamaneurol.2018.1814
5. Wegener-Panzer A, Cleaveland R, Wendel EM, Baumann M, Bertolini A, Häusler M, et al. Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies. Neurol Neuroimmunol Neuroinflamm (2020) 7(4):e731. doi: 10.1212/NXI.0000000000000731
6. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm (2017) 4(2):e322. doi: 10.1212/NXI.0000000000000322
7. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Sharma M, Nicole MW. Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES): characterization of a distinct clinico-radiographic syndrome. J Neurol (2019) 266(10):2481–7. doi: 10.1007/s00415-019-09440-8
8. Doig D, McNamara C, Mewasingh L, Beri S, Jones B, Kachramanoglou C, et al. Autoimmune cortical encephalitis in two children with anti-myelin oligodendrocyte glycoprotein (MOG) antibody. J Neurol (2021) 268(3):1096–101. doi: 10.1007/s00415-020-10260-4
9. Bruijstens AL, Lechner C, Flet-Berliac L, Deiva K, Neuteboom RF, Hemingway C, et al. Paediatric MOG consortium consensus: Part 1 - classification of clinical phenotypes of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol (2020) 29:2–13. doi: 10.1016/j.ejpn.2020.10.006
10. Song X, Ma J, Li X, Jiang L. Children suspected of having intracranial infection with normal brain magnetic resonance imaging may be associated with the myelin oligodendrocyte glycoprotein antibody. Brain Dev (2022) 44(4):281–6. doi: 10.1016/j.braindev.2021.12.009
11. Cellucci T, Van Mater H, Graus F, Muscal E, Gallentine W, Klein-Gitelman MS, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflamm (2020) 7(2):e663. doi: 10.1212/NXI.0000000000000663
12. Patel N, Rao VA, Heilman-Espinoza ER, Lai R, Quesada RA, Flint AC. Simple and reliable determination of the modified rankin scale score in neurosurgical and neurological patients: the mRS-9Q. Neurosurgery. (2012) 71(5):971–5. doi: 10.1227/NEU.0b013e31826a8a56
13. Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol (2020) 19(3):234–46. doi: 10.1016/S1474-4422(19)30488-0
14. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Celluci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol (2016) 15(4):391–404. doi: 10.1016/S1474-4422(15)00401-9
15. Bruijstens AL, Wendel EM, Lechner C, Bartels F, Finke C, Breu M, et al. Paediatric MOG consortium consensus: Part 5 - treatment of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol (2020) 29:41–53. doi: 10.1016/j.ejpn.2020.10.005
16. Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol (1987) 139:4016–21.
Keywords: autoimmune encephalitis, myelin-oligodendrocyte glycoprotein antibody, MOG antibody-associated diseases, brain magnetic resonance imaging, children
Citation: Song X and Ma J (2022) Clinical characteristics of myelin-oligodendrocyte glycoprotein antibody-positive pediatric autoimmune encephalitis without demyelination: A case series. Front. Immunol. 13:1050688. doi: 10.3389/fimmu.2022.1050688
Received: 22 September 2022; Accepted: 18 November 2022;
Published: 01 December 2022.
Edited by:
Margherita Nosadini, University of Padua, ItalyCopyright © 2022 Song and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiannan Ma, bWpuMDIzQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.