 Yusi Chen
Yusi Chen Li Tang
Li Tang- State Key Laboratory of Proteomics, National Center for Protein Sciences, Beijing Proteome Research Center, Beijing Institute of Lifeomics, Beijing, China
Non-parenchymal cells (NPCs) and parenchymal cells (PCs) collectively perform tissue-specific functions. PCs play significant roles and continuously adjust the intrinsic functions and metabolism of organs. Tissue-resident macrophages (TRMs) are crucial members of native NPCs in tissues and are essential for immune defense, tissue repair and development, and homeostasis maintenance. As a plastic-phenotypic and prevalent cluster of NPCs, TRMs dynamically assist PCs in functioning by producing cytokines, inflammatory and anti-inflammatory signals, growth factors, and proteolytic enzymes. Furthermore, the PCs of tissues modulate the functional activity and polarization of TRMs. Dysregulation of the PC‐TRM crosstalk axis profoundly impacts many essential physiological functions, including synaptogenesis, gastrointestinal motility and secretion, cardiac pulsation, gas exchange, blood filtration, and metabolic homeostasis. This review focuses on the PC‐TRM crosstalk in mammalian vital tissues, along with their interactions with tissue homeostasis maintenance and disorders. Thus, this review highlights the fundamental biological significance of the regulatory network of PC‐TRM in tissue homeostasis.
Introduction
Tissues can be considered a collection of cell clusters and intercellular substances. The communication between non-parenchymal cells (NPCs) and parenchymal cells (PCs) creates different organ functions. As basic cellular units, PCs play significant roles and continuously regulate the intrinsic functions and metabolism of tissues (1–4). Hepatocytes account for approximately 60% of total liver cells and 80% of liver tissue volume and perform a series of metabolic functions in the liver (2). Cardiomyocytes account for about 30% of whole cardiac cells in the heart, which drive cardiac contraction and relaxation (3). Alveoli are the functional units of the lungs performing gas exchange (4). The alveolar wall, the main structure of the alveoli, is composed of specialized alveolar type I cells that provide an extensive surface area for gas exchange with the surrounding capillaries and specialized alveolar type II cells that secrete surfactants and other proteins (4). These specialized PCs support the liver in orchestrating systemic metabolism, the heart in regulating blood circulation, and the lung in exchanging carbon dioxide and oxygen. Therefore, they are the primary cells responsible for the organ’s primary function and are essential to tissue homeostasis and systemic physiological processes.
In addition to PCs, NPCs are another large group of cells that make up tissues, such as hepatic stellate cells and Kupffer cells in the liver, macrophages and lymphocytes in the spleen, alveolar macrophages and monocytes in the lung, and glial cells in the CNS. NPCs act as mechanical scaffolds to guide parenchymal repair and regeneration, maintain substance metabolism and nutrition balance, regulate transmitter function, and participate in the immune response (2, 5–7). For example, hepatocytes perform the primary metabolic functions in the liver, whereas NPCs serve regulatory functions, such as pathogen clearance, apoptotic cell phagocytosis, and cytokine secretion (8).
These tissue elements arrange and interconnect to form a particular tissue (9). In tissue, the cell‐cell crosstalk develops a “mutualistic” relationship and produces a specific function output (9). Regarding the organism as an ecosystem, the circulation of matter and energy flow is relatively stable under steady-state conditions. In the short term, the out-of-balance fluctuations can be self-corrected to maintain relative stability. Nevertheless, the regulatory balance of homeostasis can be exhausted in the long term (9, 10). Moreover, certain specific cells will be recruited to generate proper signals and bring the fluctuations back to equilibrium (9–11).
Homeostatic regulation operates based on negative feedback mechanisms that correct deviations of the system state variables from the desired range or setpoint values. When variations are over-large, homeostatic mechanisms are insufficient to maintain system stability. In such cases, inflammatory signals complement homeostatic regulation and enforce the return to homeostasis (9, 10). As a plastic-phenotypic cluster of NPCs, macrophages dynamically participate in signal communication with surrounding cells by producing cytokines, inflammatory and anti-inflammatory signals, growth factors, and proteolytic enzymes (1). Tissue-resident macrophage (TRM) populations stem from yolk sac-derived erythromyeloid progenitors (YS-EMPs) or fetal liver monocytes, which self-renew and proliferate in the steady state (10–12), whereas the niche of TRMs can be replaced with the macrophages generated from bone marrow-derived monocytes (BM-monocytes) in a non-steady state. Some TRMs, such as intestinal macrophages, can be gradually supplemented by Ly6Chi monocyte-derived macrophages during development (10–12). Over the years, the regulation of macrophages and PCs has gradually attracted increasing attention. Growing research has demonstrated that disrupting the balance of macrophage pools triggers tissue homeostasis and development (13–18).
This review briefly summarizes the phenotypes and functions of TRMs in seven organs, focusing on communication with PCs in steady and non-steady states, and discusses how their crosstalk maintains organ homeostasis. Exploring the relationship between PCs and TRMs in homeostasis maintenance may increase our understanding of the formation of non-homeostatic conditions.
Microglia and neurons coordinate CNS homeostasis
In the CNS, embryonic yolk-sac progenitors generate erythro-myeloid progenitors (EMPs, c-Kit+ CD45+ CX3CR1– CSF1R+ F4/80–) and subsequently differentiate into embryonic microglia (11, 19–22). Microglia (CD45low/int F4/80low/int CX3CR1+ CD11b+) are the first line of defense against infections in the CNS (23). In addition, microglia also contribute to CNS development and homeostasis, such as apoptotic neuron phagocytosis, neuron development, vasculature development, and neuronal circuit formation (24–26). Under physiological conditions, microglia are in a resting state and on standby (25). However, “resting” microglia exist in a process-bearing and ramified phenotype, progressing toward and actively engulfing synapses (“synaptic pruning”) to control their number and maintain proper neuronal functions (27, 28). Additionally, microglia regulate programmed cell death, axon fasciculation, neurite formation, and synaptogenesis (29, 30).
The signal communication between microglia and neurons greatly depends on microglial signaling molecules (31–34). Neurons talk to microglia through “off” and “on” signals, respectively (Figure 1) (35). The “off” signals include neurotransmitters, neurotrophins, and transforming growth factor β (TGFβ), which can keep microglia quiescent. The “on” signals include glutamate, chemokines, purines, and triggering receptors expressed on myeloid cells 2 (TREM2) that may be induced by inflammation (35). These signals activate microglia toward a beneficial or detrimental phenotype to regulate neurons under pathological conditions (35–38).
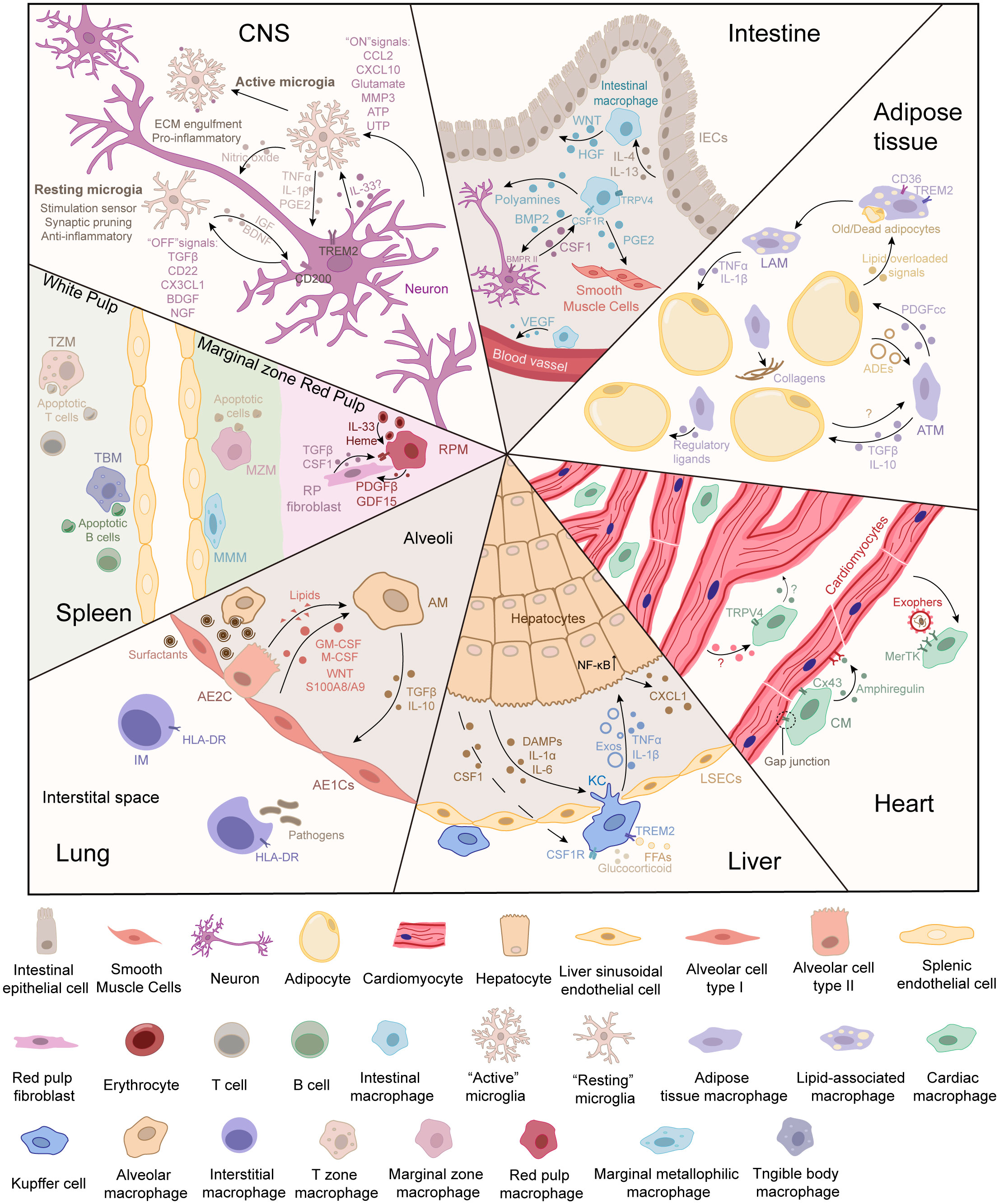
Figure 1 Cell communication between tissue-resident macrophages and parenchymal cells in different tissues. The parenchymal cells can be deemed the “primary cells,” which are responsible for performing the primary function of the tissue. During functions, parenchymal cells can release signals to inform the demand for or the accumulation of metabolites. Tissue-resident macrophages can be deemed “supportive cells,” which sense signals from the environment and parenchymal cells. In turn, tissue-resident macrophages respond to cell demand or modify the microenvironment to maintain the normal physiological functions of primary cells. The crosstalk between tissue-resident macrophages and parenchymal cells maintains tissue homeostasis.
Moreover, microglia sense and catabolize neuron-derived extracellular ATP during neuronal activation (39, 40). This activates microglia in a region-specific manner, leading to the suppression of neuronal activity (39, 40). Interleukin (IL)-33 is a member of the IL-1 cytokine family that is generally secreted into the nucleus. It can activate nuclear factor κB (NF-κB) signaling in target cells after being released into the extracellular space (41). During the early stages of postnatal synaptic maturation, the expression of IL-33 is increased only in the spinal and thalamic astrocytes of the gray matter (42). In the adult brain, IL-33 is widely expressed in the corpus callosum, hippocampus, thalamus, granular layer, and cerebellum white matter (43). Recent research suggests that microglia engulf the extracellular matrix (ECM) under the regulation of neuronal IL-33 in the adult hippocampus (43).
Microglia maintain a dynamic relationship with neurons. The “quiescent” microglia are more like “nannies” that take care of the growth and development of neurons. When neurons encounter stressful conditions, microglia become “fighters” against the hostile environment. Sometimes the weapons of these warriors may accidentally injure innocent victims. To confront pathogen invasion, microglia are activated and release several bioactive molecules to strike down pathogenic bacteria (44–46). These active molecules from amoeboid-like microglia may mis-strike the healthy neurons. The extracellular ATP released by neurons during neuronal activation is sensed and catabolized by microglia (39). This activates microglia in a highly region-specific manner, leading to the suppression of neuronal activity (39). In brief, ATP promotes the recruitment of microglial protrusions whereas the microglial ectoenzyme CD39 hydrolyzes ATP into AMP (40). AMP is converted into adenosine by CD73 and subsequently suppresses neuronal responses (40). Additionally, the “resting” microglia prevent sympathetic overactivation by maintaining Kv4.3 (a potassium channel) on presympathetic neurons (47).
Acting as a “double-edged sword,” microglia play a pivotal role in maintaining tissue homeostasis while partially promoting neurological disease development when exposed to external and internal insults (36, 48, 49). A release of diverse nucleotides accompanies nerve injury, and some of these nucleotides act as “find/eat-me” signals in mediating neuron-glial interplay (46). As mentioned above, the nucleotides ATP and ADP are predominant signal transmitters in mechanical stimulation-induced intercellular Ca2+ wave (ICW) communication by acting on P2Y12/13 receptors in BV-2 microglia (46). Once microglia are activated, they participate in developing, spreading, and potentiating low-grade neuroinflammation (50). The inflammatory-activated glial cells exhibit cellular changes that alter their communication with each other and neurons and render neurons more excitable (50). Thus, pain transmission is enhanced and prolonged (50). In neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD), a unique population of microglia is termed disease-associated microglia (DAM) (21). Similar to DAM, aged microglia exhibit an elevated expression of transcripts upregulated in neurodegenerative diseases, including Cxcr4, Clec7a, Axl, Lgals3, and MHC-II, which are linked to neuronal loss and exacerbation of the disease (51, 52).
Overall, strategic communication exists in the microglia–neuron axis in physiological and pathological states. However, how dysfunctional microglia–neuronal communication affects disease progression or onset at various stages is still unclear. Using the high-dimensional techniques, a map of microglial diversity has been described on a temporal and spatial axis (24, 53–55). Thus, it is better to use various research methods to comprehensively understand the multi-omics states of condition-specific microglia, which may be essential for understanding the physiological heterogeneity of microglia–neuron interactions and controlling CNS diseases.
Crosstalk between macrophages and epithelial cells maintains gastrointestinal motility and secretion
The gastrointestinal (GI) tract, consisting of the small intestine (SI) and colon, is the center of nutrient digestion. Intestinal macrophages are an abundant immune member in the gut and play a crucial role as function keepers and adjusters (56, 57). Different from Kupffer cells and microglia, intestinal resident macrophages (CD45+ F4/80+ CD64+ CX3CR1hi/int CD11b+) are derived from fetal liver monocytes and gradually become supplemented by Ly6Chi monocytes during development (57–59). They maintain self-renewal and reside mainly in the lamina propria (LP macrophages, CD11c+ CD14+) and muscularis (muscularis macrophages, CD11clo CD4+ TIMD4+ RELMα+) (Figure 1) (57–59).
Gastrointestinal secretion is essential for the movement and absorption of nutrients and ions across intestinal epithelial cells (IECs) (60). Intestinal macrophages significantly influence epithelial integrity and mucosal permeability by secreting cytokines to IECs and submucosal neurons (60). A recent study demonstrated that monocyte-derived PTGER4+ intestinal macrophages promote the healing and repair of the intestinal mucosa by CXCL1 secretion (61). Moreover, macrophages found in the submucosa can maintain the integrity of the submucosal vasculature (62, 63). The intestinal macrophages at the distal colon protrude into the epithelium via balloon-like protrusions that prevent the absorption of fungal toxins to preserve mucosal integrity (63).
Gut motility is potentially modulated by crosstalk among enteric neurons, intestinal macrophages, and smooth muscle cells (64). For example, the interaction between muscularis macrophages and intestinal smooth muscle cells is mediated by TRPV4 channels (65). After activating TRPV4 signaling, muscularis macrophages release prostaglandin E2 (PGE2), which directly activates intestinal smooth muscle cells to trigger muscle contraction in a paracrine manner (65). In addition, a subset of intestinal macrophages that reside in the lamina propria (LP) is responsible for the clearance of apoptotic and senescent epithelial cells (66). They promote epithelial integrity by expressing metalloproteinases and cytokines that stimulate tissue remodeling and the renewal of epithelial stem cells, such as hepatocyte growth factor (HGF), PGE2, and WNT ligands (66). Moreover, intestinal macrophages and enteric neurons interact through bone morphogenetic protein (BMP) and colony-stimulating factor 1 (CSF1). Previous research has shown that macrophage-derived BMP2 promotes neuronal activity (67). Reciprocally, enteric neurons can maintain the self-renewal capability of intestinal macrophages through CSF1 (64). Then, local macrophages can adjust intestinal muscle contraction by inducing the production of neurotransmitters, thereby controlling peristalsis (67).
The unique environment of the GI tract is likely to shape the heterogeneity of intestinal macrophages (both resident and recruited macrophages) (56). However, except for immune functions, how the spatial molecular communication of intestinal macrophages senses signals and regulates IECs or other cells to coordinate gut activity remains unclear. How intestinal macrophage subsets play heterogeneous roles in various gastrointestinal diseases also remains unknown. Several chronic inflammatory conditions affect the GI tract and are referred to as inflammatory bowel disease (IBD) (68, 69). IBD is characterized by recurrent bouts of inflammation in the GI tract (68, 69). Endogenous damage-associated molecular patterns (DAMPs) released from injured intestinal epithelial cells activate intestinal macrophages to release abnormal inflammatory factors, recruit monocytes, and promote their proinflammatory transformation, consequently aggravating inflammation and tissue damage (68, 69). In addition, at the onset of the disease, the intestinal epithelium is damaged and the glial cells and neurons in the enteric ganglia are injured or overactivated, resulting in gastrointestinal motility disorders (69). Nevertheless, depleting mature intestinal macrophages alone can cause the death of intestinal epithelial cells and inflammation (63). How macrophage function is affected by gastrointestinal inflammation remains to be studied in the future.
Adipose tissue macrophages and adipocytes regulate energy metabolism
Adipose tissues are the primary reservoir for storing energy substrates and have adapted to respond rapidly to caloric fluctuations. According to physiological functions, morphology, characteristics, and localizations, adipose tissues are divided into three types: brown adipose tissue (BAT), beige adipose tissue, and white adipose tissue (WAT) (70–72). Adipocytes are the main site of energy metabolism in adipose tissues, such as energy intake and fatty acid release, which have been intensively studied. In WAT, macrophages comprise 30%–50% of the immune cells (70, 73). Through scRNA-seq analysis, five subpopulations of adipose tissue macrophages (ATMs, CD45+ F4/80+ CD11b+ CD64+) have been found, including vascular-associated macrophages (VAMs, CD9– MHC-IIlo LYVE1hi), lipid-associated macrophages (LAMs, CD9+ MHC-IIhi LYVE1lo), infiltrated monocyte-derived macrophages (CD11b+ Ly6C+), and two additional minor subpopulations of ATMs (73–75). ATMs are derived from YS-EMPs and BM monocytes and are mainly distributed around adipocytes (76).
ATMs account for 5%–10% of stromal cells in lean adipose tissue (76). The crosstalk between macrophages and adipocytes coordinates the functions of adipose tissues (Figure 1) (70). When sensing excessive free fatty acids (FFAs), ATMs facilitate the secretion of PDGFcc to increase lipid storage in white adipocytes (77). PDGFcc blockade redirects unstored lipids in BAT and increases thermogenesis (77). Old adipocytes send out “find-me” and “eat-me” signals, which trigger phagocytosis and IL-6 secretion by macrophages (70, 78, 79). The phenotype of ATMs changes under different conditions. Alternatively activated (M2) ATMs may be predominant in physiological homeostasis, and classically activated (M1) ATMs are increased in conditions of obesity (70). M2 macrophages rebuild the microenvironment and regulate systemic glucose homeostasis via TGFβ (80). In addition, M2 ATMs can affect adipocyte thermogenesis, contributing to the regulation of energy storage and ready response to energy demands in WAT (71). M2-derived slit guidance ligand 3 (Slit3) stimulates the release of norepinephrine by binding to the specific receptor in sympathetic neurons, thereby improving adipocyte thermogenesis for cold adaptation (81). In addition to maintaining metabolic homeostasis, ATMs also orchestrate the source of some bona fide adipocytes by promoting the hematopoietic-to-mesenchymal transition (82). CD206+ ATMs are predominantly M2 macrophages, and ablation of these ATMs improves systemic insulin sensitivity through TGFβ signaling (81).
However, under conditions of obesity, chemokines secreted by hypertrophic adipocytes recruit large numbers of monocytes that differentiate into ATMs, which account for 40%–50% of the stromal cell population (76). Recruited monocyte-derived ATMs often surround damaged adipocytes and form a crown-like structure (CLS) (83–85). These ATMs clear dead cell debris and lipid droplets and contribute to maintaining the integrity of adipose tissue (76). During CLS formation, adipocyte death locally induces ATM metabolic activation and increased lipid metabolism, which may be involved in meta-inflammation development (86, 87). In hypertrophic adipocytes, monocyte-derived macrophages act as early sensors of metabolic changes and produce tumor necrosis factor α (TNFα) and IL-1β, which mediate hepatosteatosis and insulin resistance (77, 88). Moreover, the signals released from adipose tissues, such as exosomes (adipocyte-derived exosomes, ADEs), adipokines, cytokines, and lipids, can affect peripheral tissues and macrophages in an endocrine manner (77, 89). Interestingly, ADEs carrying Sonic Hedgehog (Shh) promote the development of insulin resistance by stimulating macrophage activation to secrete inflammatory cytokines (89, 90). ADEs also act as carriers of miR-34a, exacerbating obesity-induced systemic inflammation and metabolic dysregulation (89, 90). However, during high-fat-diet (HFD)-induced epididymal white adipose tissue (eWAT) remodeling, ATMs are most closely associated with blood vessels, preventing the dysregulation of ECM composition and the formation of tissue fibrosis (91).
As mentioned above, the regulation of physiological and metabolic homeostasis and the inflammatory response in adipose tissue have been described. However, more detailed molecular mechanisms of the macrophage‐adipocyte crosstalk and their roles in obesity-related diseases still need to be investigated. In addition, how the macrophage populations surrounding the distinct parts of adipose tissue accumulate and function differently is still unclear.
Kupffer cells collaborate with hepatocytes to contribute to liver homeostasis
The liver is a multitasking organ that assumes diversified functions, such as protein synthesis, lipid metabolism, detoxication, and amino acid metabolism (2). In mice, there are two types of liver macrophages: yolk sac (YS)-derived macrophages and monocyte-derived macrophages (92–95). Specifically, KCs (CX3CR1– TIMD4+ CLEC4F+) are the only YS-derived macrophages in the liver (93). Self-renewing KCs are distributed along the hepatic sinusoids (Figure 1). Hepatic stellate cells (HSCs), hepatocytes (HCs), and endothelial cells (ECs) compose the KC niche and imprint identity (96). It has been reported that stimulatory signals in the tissue environment contribute to hepatic macrophage differentiation (97). During liver development, EMPs occupy most liver niches and are generated in KCs, whose identity and self-renewal are maintained through BMP9/BMP10/ALK1 signaling and Smad4-dependent pathways (98–102).
Macrophages play a pivotal role in maintaining immune defense and liver homeostasis. An increasing number of studies have suggested that the crosstalk axis of KC‐HC modulates metabolic homeostasis. Lipid metabolism is a critical functional feature of the liver. During fasting and feeding, the liver regulates lipid fluxes through lipogenic and oxidative pathways to adjust to the altered energy state. In the physiological state, excess lipids are mainly stored by adipose tissue and not the liver. KC-derived IL-1β contributes to suppressing the expression of hepatokines in hepatocytes and lipolysis in adipose tissue (103). This suggests that macrophages can promote the proper storage of excess lipids and play an essential role in liver–adipose tissue communication. TNFα, another proinflammatory cytokine from macrophages, suppresses the nuclear translocation of GR and the ketogenesis pathway in HCs (104). Thus, hepatic ketogenesis is inhibited when the body has enough energy sources during feeding (104). During fasting, macrophage GR suppresses the expression of TNFα (104). The limited production of TNFα promotes the mutual intercellular crosstalk between liver macrophages and HCs, directly influencing glucocorticoid signaling and ketogenesis by reshaping the hepatic transcriptional response to coordinate fasting homeostasis (104). In contrast, HCs generate acetoacetate (AcAc) from fatty acid-derived acetyl-CoA via a series of enzymatic reactions (105). AcAc acts as a shuttle between HCs and M2 macrophages (105). These studies suggest that crosstalk between HCs and liver macrophages is related not only to cytokines but also to cellular metabolites. Liver macrophages can produce exosomes containing insulin-sensitizing miR-690 that directly inhibits de novo lipogenesis and insulin resistance in HCs through the miR-690–Nadk axis (106, 107). However, the accumulation of anti-inflammatory macrophages in the liver may drive insulin resistance by increasing cytokine secretion (108, 109). Additionally, KCs were found to act as central regulators in cholesterol homeostasis. Under iron overload, KCs transfer LDL-derived cholesterol to HCs in an Abca1-dependent manner (110). Moreover, macrophages can synthesize anti-inflammatory fatty acids by activating the LXR signaling pathway and SREBP1 signaling pathway, regulating the functions of surrounding HCs in a paracrine manner (111, 112).
However, KCs may exert dual actions on lipid metabolism in hepatocytes. The FFAs released from adipose tissues promote hepatic triglyceride storage, and fatty acid oxidation is inhibited by KC-derived IL-1β in a PPARα-dependent manner (113). The increased secretion of KC-derived IL-1β promotes hepatocyte damage and the progression of ethanol-induced liver diseases (114). Additionally, pyroptotic hepatocytes release IL-1β to stimulate KCs; in turn, KC-derived proinflammatory signals amplify liver inflammation (115). Moreover, DAMPs are sensed by KCs, leading to the release of KC-secreted tumor necrosis factor α (TNFα) to promote chemokine expression in HCs (116). Under HFD conditions, CD11b+ F4/80+ macrophage-derived TNFα triggers Sarm1-dependent sympathetic neuropathy and insulin insensitivity in HCs (117). Lipid overload in HCs induces lipotoxicity and oxidative stress, resulting in damage to HCs with the concomitant release of DAMPs (118). HC-derived FFAs induce the production of IL-1β mtDNA in KCs (119). Reciprocally, this may aggravate the accumulation of hepatic lipids and fatty degeneration (119, 120).
KCs also play a dual role in the immunocompetent mouse model of acute hepatitis B viral (HBV) infection. The stimulation of KCs with IL-6 or TNFα suppresses the expression of LSECtin and accelerates the clearance of liver adenovirus. In contrast, the activation of IL-4, IL-10, or IFN-g in KCs upregulates LSECtin expression and delays viral clearance (121). Additionally, macrophages engulf apoptotic cells and produce anti-inflammatory/tissue repair factors in an LSECtin-dependent manner in IBD (122). However, LSECtin is upregulated in the liver after HBV infection, implying that KCs are hijacked by HBV and have to protect the liver from inflammation by delaying viral clearance (121).
Briefly, we introduce the signal communications between KCs and HCs in metabolic homeostasis and inflammation. Under non-homeostatic conditions, the hepatic niche occupied by KCs is gradually supplemented by Ly6Chi monocyte-derived macrophages (10–12). The differential functions of KCs and monocyte-derived macrophages in diseases, such as NAFLD, remain unclear. In addition, studies on the crosstalk between KCs and HCs in homeostasis have focused more on the unsteady state than the steady state. There is still plenty to discover about the KC‐HC interaction axis.
Cardiac macrophages and cardiomyocytes maintain cardiac pulsation and energetics
The heart is composed of four chambers and is a complex and vital organ. The cardiovascular system includes blood vessels and blood and is responsible for transporting nutrients and oxygen throughout the body and removing metabolic wastes in cells. During the steady state, cardiac macrophages (CMs, CD11b+ F4/80+ MHC-IIhigh/low CD64+ MerTK+) occupy most of the immune niche in the cardiac interstitium (Figure 1) (123, 124). Multiomics and fate-mapping studies revealed that CM subsets can be identified as TLF+ CMs, CCR2+ CMs, and MHC-IIhi CMs (123).
The heart requires the precise regulation of heterogeneous cell populations for intense metabolic and mechanical demands. The complex crosstalk between CMs and cardiomyocytes controls cardiac impulse (125). CMs are closely connected with cardiomyocytes and crucially maintain cardiac impulse conduction through gap junctions supported by Areg (coding amphiregulin) (126). During the acute phase of myocardial infarction (MI), leukocytes and monocytes are recruited to the ischemic area by CMs (127). The RNA-seq data of single AV node macrophages show that macrophages have an electrical connection with cardiomyocytes through Cx43-containing gap junctions (16, 128). These results suggest that CMs may significantly contribute to conduction abnormalities. In addition, CMs may impact morphogenesis and development in the cardiac conduction system (129, 130). For example, CM ablation in a Cd11bDTR mouse induces a progressive atrioventricular block (16). A flow cytometric analysis of Cx3cr1GFP/+ fetal hearts, combined with EGF-like module staining, revealed the active recruitment of macrophages at E12.5-16.5 and proliferation throughout the time course of cardiac development (129).
Similar to TRMs in other tissues, cardiac macrophages have a potent phagocytic capacity to remove necrotic debris that prevents myocardial infarct (MI)-induced arrhythmias (131). Treatment with a CSF1R inhibitor or the depletion of recruited CMs increases post-MI ventricular tachycardia, ventricular fibrillation burden, and myocyte death (131). Nevertheless, as highly specialized cells in the heart, cardiomyocytes contain large numbers of mitochondria and eject redundant mitochondria and other materials in subcellular vesicles to partly solve the intense energetic demand (132). Taken together, resident CMs crucially contribute to cardiac homeostasis maintenance.
However, in cardiac diseases, the TRM niche is occupied by CCR2+ monocyte-derived macrophages (131). The recruited CMs may initiate the inflammatory cascade that promotes tissue injury and suppresses tissue repair. Thus, different CM subsets serve different roles that require more precise approaches to understanding their character and functions during pre- and postnatal developmental stages (131). Regarding spatial distribution, the intratissue heterogeneity of CMs has not been clarified (123, 128). The disturbance of CM–cardiomyocyte communication may involve a series of heart diseases, including hypertension, ischemia, arrhythmias, and myocarditis (123). Whether CMs distribute homogeneously and how macrophage phenotypes change during disease progression have not been elucidated.
The macrophage‐alveolar epithelial cell axis regulates pulmonary functions
The lungs mainly carry out gas exchange in alveoli, which are rich in connective tissues such as capillaries, elastic fibers, mesh fibers, and collagen fibers. There are two kinds of epithelial cells on the alveolar surface: type I alveolar epithelial cells (AE1Cs) and type II alveolar epithelial cells (AE2Cs) (4). To combat foreign pathogens, many immune defenders, primarily macrophages, are located in the lungs (Figure 1) (4, 10–12). Pulmonary macrophages phagocytize surfactants, inhaled stimuli/invaders, and apoptotic and fragmented cells to maintain lung homeostasis (133, 134). Different microenvironments shape resident macrophages as distinct populations, such as alveolar macrophages (AMs, CD64+ MerTK+ F4/80+ SiglecFhi/– CD169+/–) and interstitial macrophages (IMs, LYVE1lo/hi CD64+ MerTK+ F4/80+ SiglecF–) (135). AMs and IMs reside in two anatomical compartments and perform slightly different functions (135, 136). IMs are located near many non-hematopoietic cells (136). In addition, IMs are not as abundant as AMs and have lower phagocytic potential (137, 138). Thus, IMs act as a second line of defense against invaders (137, 138).
AMs are located in the lumen of the alveoli and are surrounded by AE1Cs, AE2Cs, and stromal cells (135, 136). In 1-week-old mice, alveolar epithelial cell-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) provides the instructive cytokine signal for AMs to thrive (139). Mature AMs adhere tightly to the luminal side of alveolar epithelial cells, continuously capturing and phagocytosing large amounts of inhaled pathogens and particles without triggering an influx of neutrophils and excessive inflammation (140). The accumulation of surfactants on the alveolar space increases surface tension and leads to alveolar collapse and respiratory failure (141, 142). AMs are responsible for avoiding unnecessary inflammation and capturing and metabolizing surfactants to maintain the biomechanical homeostasis of the lungs (134, 135, 141). Additionally, AMs contribute to inflammatory-associated or non-inflammatory responses through the macrophage–epithelial cell axis to regulate lung homeostasis (133–136). In rats, AMs transport miR-21-5p to tracheal epithelial cells by exosomes that can promote the epithelial–mesenchymal transition (EMT) (143). In mice, cytokines released by macrophages regulate the transcription factor CEBPB in pulmonary epithelial cells (144).
Conversely, pulmonary epithelial cells also influence macrophages. The epithelium-derived WNT and S100A8/A9 regulate the phenotypes and functions of macrophages (145). In vitro, AMs lost the expression of genes involved in adhesion molecules, lipid metabolism, TGFβ signaling, and oxygen response (146). However, when the cultured cells are transferred back into the lungs, the ex vivo expanded AMs can reacquire their in vivo expression profile and identity (146). These findings suggest a potential role for epithelial cells in the maintenance of the AM phenotype (146).
In the airways of chronic obstructive pulmonary disease (COPD) patients, the accumulated oxidized lipids in pulmonary epithelial cells may reduce the phagocytotic ability of AMs (147). With overloaded phagocytosis, AMs trigger inflammation by producing chemokines and proinflammatory cytokines that recruit and activate neutrophils, further contributing to lung damage and systemic “cytokine storms” (136). Although these macrophages have been extensively studied, there are still numerous questions. The mechanisms of the macrophage‐epithelial cell axis in different diseases and the use of macrophage transplantation as an immunotherapeutic approach still require further investigation.
Red pulp macrophages and fibroblasts orchestrate splenic homeostasis
As the largest secondary lymphoid organ in the body, the spleen also functions as a blood reservoir and filter and participates in immune defense, iron homeostasis, and cell reservoirs (for red blood cells, monocytes, plasmablasts, thrombocytes, and long-lived memory B cells) (11, 13, 148, 149). The splenic resident macrophages contain several subsets, such as red pulp macrophages (RPMs, VCAM-1hi F4/80+ CD68hi), marginal zone macrophages (MZMs, SIGN-R1+, or SIGN-R1-), marginal metallophilic macrophages (MMMs, CD169+ MHC-II+ MOMA-1+), tingible body macrophages (TBMs, CD68+ MFG-E+ MerTK+ Tim4+ CD36+), and T zone macrophages (TZMs, MerTK+ CX3CR1+) (11, 149). They reside in the different locations of the spleen, with distinct developmental origins, phenotypes, and functions (11, 149) (Table 1).
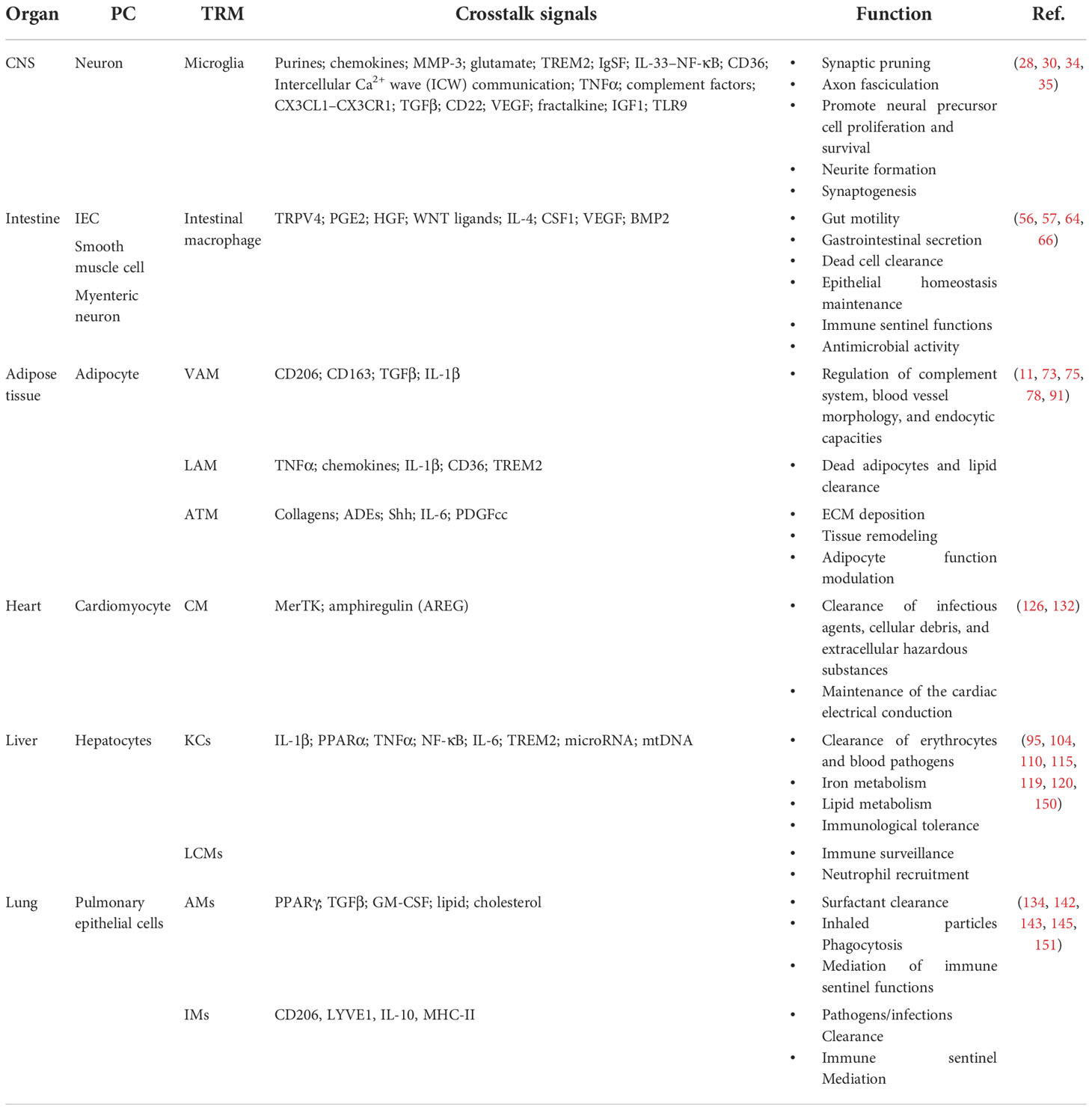
Table 1 The classification and function of the PC–TRM crosstalk in tissue homeostasis.
The crosstalk between RPMs and fibroblasts is shown in Figure 1. RPMs reside in the splenic cord and are closely associated with RP fibroblasts (152). PCs are a group of cells responsible for the primary functions of tissues. The spleen mainly acts as a blood filter (selectively removing circulating pathogens, dysfunctional red blood cells, and immune complexes), blood storage site, and blood volume regulator (13, 148). Unlike other tissues (those of the CNS, liver, lung, etc.), the distinction between PCs and supportive cells may still be unclear in the spleen.
The red pulp is composed of fibroblasts and reticular fibers that form a complex framework of open blood circulation, allowing for the selective removal of senescent and dysfunctional red blood cells (13). Therefore, from blood storage and blood volume regulation functions, fibroblasts are similar to PCs, and RPMs act as supportive cells. RPMs can support the survival, proliferation, and ECM secretion of RP fibroblasts via trophic factors (13, 148, 149). RPMs communicate with RP fibroblasts by expressing TGFβ and progranulin, and RP fibroblasts express TGFβ-RIII (a coreceptor for active TGFβ) and TNFRSF1A/B for survival (152, 153). RPMs also regulate the survival and proliferation of PDGFRα/β+ RP fibroblasts by producing PDGFβ (154, 155). In addition, RPMs can also regulate the reticular structure of RP through the production of proteases and the modulation of fibroblastic activity (11). Thus, RPMs are involved in controlling the quality of blood filtration indirectly.
Both immune defense and the maintenance of iron homeostasis are essential functions of the spleen, in which RPMs play a crucial role (152, 156). RPMs are PCs that function in blood filtration, whereas fibroblasts act as supportive cells and are critical regulators of macrophage homeostasis in RP. WT1+ reticular fibroblasts regulate the proliferation and location of RPMs through the production of CSF1 (152). Activation of the transcription factor Spi-C and heme oxygenase (HO)-1 is required for intracellular heme breakdown and free iron release from RPMs (156, 157). This molecular mechanism can neutralize the toxic effects of heme and metabolize iron (156, 157). Thus, RPMs can degrade the toxic cargo when senescent red blood cells are captured (156, 157).
Additionally, extramedullary hematopoiesis is supported by RPMs, and their absence impairs the recovery of normal red blood cell counts (152). Stress erythropoiesis causes the rapid production of mature erythrocytes. Previous research has indicated that RPMs can release a critical regulator (called GDF15) to expand the stress erythropoietic niches (158, 159). RPMs induce RP fibroblast-secreted BMP4 to maintain a suitable microenvironment and produce GDF15 to promote stress erythropoiesis in the spleen (158, 159).
However, since the spleen is a vital lymphoid organ for clearing blood pathogens, researchers mainly focus on the function of spleen macrophages in removing bacteria and regulating their interaction with other immune cells as well as the interaction between macrophages and fibroblasts in the spleen. The intercommunication between splenic macrophages and fibroblasts in splenic homeostasis and diseases is still unclear.
Conclusions
With the development and escalation of sequencing techniques, the genetic landscapes of different tissues have been mapped and are continuously improved through single-cell transcriptome and spatial metabolomics (2, 99, 160–163). Information on PC‐NPC interactions is constantly being mined (99, 150, 151, 160–167). Research on the communication between them has found that the PC‐TRM crosstalk is instrumental in maintaining overall tissue homeostasis through cell membrane receptors, inflammatory or anti-inflammatory cytokines, metabolites, and extracellular vesicles (Table 1). Meizlish and colleagues put forward the following definition of tissue homeostasis: a collection of circuits regulating specific variables within the tissue microenvironment (166). The values of regulated variables are monitored by a controller (166). TRMs are homeostatic controllers that can monitor fluctuating environmental signals directly or indirectly and react in certain ways, such as pathogen clearance, apoptotic cell phagocytosis, ECM modification, and cytokine secretion (1, 166).
The functional demand can be deemed the deviation of a homeostatic variable, and signals are the proxies of homeostatic variations that report on practical demands (1). From this, we posit that PCs sense and reshape the functional demand during environmental fluctuations. While TRMs act as environmental “sensors” and “gatekeepers,” they have strong plasticity and motility for phenotype reshaping to respond to the variational signals of the environment and PCs. Macrophages can express certain substances through negative feedback signals and responsive PCs to bring the off-balance value back to its equilibrium point. For instance, neurons perform impulse conduction continuously and discharge “excess ATP” (40). However, the surrounding microglia sense the “excess ATP” and generate negative feedback signals to prevent neuronal overactivation (40). Furthermore, CMs sense and ingest cardiomyocyte-derived vesicles to avoid the accumulation of harmful extracellular substances (132). In the lung, the capture and metabolism of surfactants via AMs are critical for maintaining lung biomechanics (141, 142). In turn, pulmonary epithelial cells regulate macrophage phenotypes and functions through the WNT/β-catenin pathway and epithelial-derived S100A8/A9 (145). Splenic RP fibroblasts regulate the proliferation and location of RPMs through CSF1 to maintain iron homeostasis in the spleen, whereas RPMs can support the survival and proliferation of fibroblasts and regulate the functions of RP fibroblasts (152, 156–159).
The role of TRMs cannot be unilaterally defined as “good” or “bad” but depends on the signals from the microenvironment and peripheral cells. In KCs, FFA-induced NLRP3 inflammatory body activation promotes the production of proinflammatory IL-1β (119, 120). In contrast, KCs induced by IL-4/IL-13 produce M2-type exosomes and regulate insulin resistance of HCs through the miR-690–Nadk axis (106). Additionally, KC-derived TNFα has been confirmed as one of the inducers of HC steatosis (117). However, a recent study showed that KCs can regulate ketone generation in HCs during fasting and maintain hepatic and systemic metabolism (104). Similarly, the effect of macrophages on adipocyte metabolism is not one-fold. M2-like macrophages affect adipogenesis, and the heat production of adipocytes helps regulate energy storage in WAT to respond to energy needs (71, 80). Under pathological conditions, adipocytokines stimulate M1 macrophages, which aggravate insulin resistance, obesity-induced inflammation, and metabolic disorders (89). Among these, the adjustment mechanism of the threshold points of the transformation from “favorable” to “unfavorable” is still a “mystery.” Therefore, further investigations are necessary to clarify the “mutual benefit” or “mutual restraint” relationship between PCs and TRMs.
In recent years, bioinformatics techniques combined with transcriptome, proteomics, and spatial data have been widely used to predict intercellular communications and map cell space (2, 99, 160–163). Thus, we can obtain evidence for the heterogeneity of PCs and TRMs in various tissues and the differences in cell‐cell communication in different regions. Additionally, we should further understand the transformation or regulation mechanisms involved in communication under physiological and pathological conditions. With the gradual deepening of our understanding of “zonation,” it is worth exploring what regulatory mechanisms may exist for cell interactions between different anatomical regions in tissue in the future.
Author contributions
YC and LT contributed to the conception of this work. YC drafted the manuscript and prepared figures using Adobe Illustrator 2021. LT supervised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31900632, 31900666, 82225009).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol (2016) 17(1):9–17. doi: 10.1038/ni.3320
2. Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol (2019) 16(7):395–410. doi: 10.1038/s41575-019-0134-x
3. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther (2010) 128(1):191–227. doi: 10.1016/j.pharmthera.2010.04.005
4. Barkauskas CE, Chung MI, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development (2017) 144(6):986–97. doi: 10.1242/dev.140103
5. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci (2020) 21(3):139–52. doi: 10.1038/s41583-020-0263-9
6. Marrone J, Danielli M, Gaspari CI, Capiglioni AM, Marinelli RA. Aquaporin gene transfer for hepatocellular cholestasis. Biochimie (2021) 188:12–5. doi: 10.1016/j.biochi.2021.03.016
7. Norris GT, Kipnis J. Immune cells and CNS physiology: Microglia and beyond. J Exp Med (2019) 216(1):60–70. doi: 10.1084/jem.20180199
8. Ding C, Li Y, Guo F, Jiang Y, Ying W, Li D, et al. A cell-type-resolved liver proteome. Mol Cell Proteomics (2016) 15(10):3190–202. doi: 10.1074/mcp.M116.060145
9. Medzhitov R. The spectrum of inflammatory responses. Science (2021) 374(6571):1070–5. doi: 10.1126/science.abi5200
10. Nobs SP, Kopf M. Tissue-resident macrophages: guardians of organ homeostasis. Trends Immunol (2021) 42(6):495–507. doi: 10.1016/j.it.2021.04.007
11. Cox N, Pokrovskii M, Vicario R, Geissmann F. Origins, biology, and diseases of tissue macrophages. Annu Rev Immunol (2021) 39:313–44. doi: 10.1146/annurev-immunol-093019-111748
12. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity (2016) 44(3):439–49. doi: 10.1016/j.immuni.2016.02.024
13. Bellomo A, Gentek R, Golub R, Bajénoff M. Macrophage-fibroblast circuits in the spleen. Immunol Rev (2021) 302(1):104–25. doi: 10.1111/imr.12979
14. Cipriani G, Terhaar ML, Eisenman ST, Ji S, Linden DR, Wright AM, et al. Muscularis propria macrophages alter the proportion of nitrergic but not cholinergic gastric myenteric neurons. Cell Mol Gastroenterol Hepatol (2019) 7(3):689–691.e4. doi: 10.1016/j.jcmgh.2019.01.005
15. Mandal P, McCormick AL, Mocarski ES. TNF signaling dictates myeloid and non-myeloid cell crosstalk to execute MCMV-induced extrinsic apoptosis. Viruses (2020) 12(11):1221. doi: 10.3390/v12111221
16. Munshi NV. Resident macrophages: Near and dear to your heart. Cell (2017) 169(3):376–7. doi: 10.1016/j.cell.2017.04.002
17. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity (2019) 51(4):655–670.e8. doi: 10.1016/j.immuni.2019.09.002
18. Terada M, Horisawa K, Miura S, Takashima Y, Ohkawa Y, Sekiya S, et al. Kupffer cells induce notch-mediated hepatocyte conversion in a common mouse model of intrahepatic cholangiocarcinoma. Sci Rep (2016) 6:34691. doi: 10.1038/srep34691
19. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci (2014) 15(5):300–12. doi: 10.1038/nrn3722
20. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol (2017) 18(4):385–92. doi: 10.1038/ni.3703
21. Wright-Jin EC, Gutmann DH. Microglia as dynamic cellular mediators of brain function. Trends Mol Med (2019) 25(11):967–79. doi: 10.1016/j.molmed.2019.08.013
22. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol (2018) 9:697. doi: 10.3389/fimmu.2018.00697
23. Streit WJ. Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci (2006) 29(9):506–10. doi: 10.1016/j.tins.2006.07.001
24. De Biase LM, Bonci A. Region-specific phenotypes of microglia: The role of local regulatory cues. Neuroscientist (2019) 25(4):314–33. doi: 10.1177/1073858418800996
25. Kierdorf K, Prinz M. Microglia in steady state. J Clin Invest (2017) 127(9):3201–9. doi: 10.1172/JCI90602
26. Sominsky L, De Luca S, Spencer SJ. Microglia: Key players in neurodevelopment and neuronal plasticity. Int J Biochem Cell Biol (2018) 94:56–60. doi: 10.1016/j.biocel.2017.11.012
27. Xie D, He M, Hu X. Microglia/macrophage diversities in central nervous system physiology and pathology. CNS Neurosci Ther (2019) 25(12):1287–9. doi: 10.1111/cns.13257
28. Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol (2017) 79:619–43. doi: 10.1146/annurev-physiol-022516-034406
29. O'Farrell K, Fagan E, Connor TJ, Harkin A. Inhibition of the kynurenine pathway protects against reactive microglial-associated reductions in the complexity of primary cortical neurons. Eur J Pharmacol (2017) 810:163–73. doi: 10.1016/j.ejphar.2017.07.008
30. Raffo-Romero A, Arab T, Al-Amri IS, Le Marrec-Croq F, Van Camp C, Lemaire Q, et al. Medicinal leech CNS as a model for exosome studies in the crosstalk between microglia and neurons. Int J Mol Sci (2018) 19(12):4124. doi: 10.3390/ijms19124124
31. Lee JW, Nam H, Kim LE, Jeon Y, Min H, Ha S, et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy (2019) 15(5):753–70. doi: 10.1080/15548627.2018.1556946
32. Raghuraman R, Karthikeyan A, Wei WL, Dheen ST, Sajikumar S. Activation of microglia in acute hippocampal slices affects activity-dependent long-term potentiation and synaptic tagging and capture in area CA1. Neurobiol Learn Mem (2019) 163:107039. doi: 10.1016/j.nlm.2019.107039
33. Shen X, Venero JL, Joseph B, Burguillos MA. Caspases orchestrate microglia instrumental functions. Prog Neurobiol (2018) 171:50–71. doi: 10.1016/j.pneurobio.2018.09.007
34. Yeh H, Ikezu T. Transcriptional and epigenetic regulation of microglia in health and disease. Trends Mol Med (2019) 25(2):96–111. doi: 10.1016/j.molmed.2018.11.004
35. Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci (2007) 30(11):596–602. doi: 10.1016/j.tins.2007.08.007
36. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
37. Marinelli S, Basilico B, Marrone MC, Ragozzino D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol (2019) 94:138–51. doi: 10.1016/j.semcdb.2019.05.017
38. Miao W, Zhao Y, Huang Y, Chen D, Luo C, Su W, et al. IL-13 ameliorates neuroinflammation and promotes functional recovery after traumatic brain injury. J Immunol (2020) 204(6):1486–98. doi: 10.4049/jimmunol.1900909
39. Hackett TA. Adenosine A1 receptor mRNA expression by neurons and glia in the auditory forebrain. Anat Rec (2018) 301(11):1882–905. doi: 10.1002/ar.23907
40. Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, et al. Negative feedback control of neuronal activity by microglia. Nature (2020) 586(7829):417–23. doi: 10.1038/s41586-020-2777-8
41. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity (2015) 42(6):1005–19. doi: 10.1016/j.immuni.2015.06.006
42. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science (2018) 359(6381):1269–73. doi: 10.1126/science.aal3589
43. Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell (2020) 182(2):388–403.e15. doi: 10.1016/j.cell.2020.05.050
44. Illes P, Rubini P, Ulrich H, Zhao Y, Tang Y. Regulation of microglial functions by purinergic mechanisms in the healthy and diseased CNS. Cells (2020) 9(5):1108. doi: 10.3390/cells9051108
45. Lyu Q, Pang X, Zhang Z, Wei Y, Hong J, Chen H. Microglial V-set and immunoglobulin domain-containing 4 protects against ischemic stroke in mice by suppressing TLR4-regulated inflammatory response. Biochem Biophys Res Commun (2020) 522(3):560–7. doi: 10.1016/j.bbrc.2019.11.077
46. Jiang P, Xing F, Guo B, Yang J, Li Z, Wei W, et al. Nucleotide transmitters ATP and ADP mediate intercellular calcium wave communication via P2Y12/13 receptors among BV-2 microglia. PloS One (2017) 12(8):e0183114. doi: 10.1371/journal.pone.0183114
47. Bi Q, Wang C, Cheng G, Chen N, Wei B, Liu X, et al. Microglia-derived PDGFB promotes neuronal potassium currents to suppress basal sympathetic tonicity and limit hypertension. Immunity (2022) 55(8):1466–1482.e9. doi: 10.1016/j.immuni.2022.06.018
48. Rawji KS, Young AMH, Ghosh T, Michaels NJ, Mirzaei R, Kappen J, et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol (2020) 139(5):893–909. doi: 10.1007/s00401-020-02129-7
49. Bottai D, Adami R, Paroni R, Ghidoni R. Brain cancer-activated microglia: A potential role for sphingolipids. Curr Med Chem (2020) 27(24):4039–61. doi: 10.2174/0929867326666190506120213
50. Block L. Glial dysfunction and persistent neuropathic postsurgical pain. Scand J Pain (2016) 10:74–81. doi: 10.1016/j.sjpain.2015.10.002
51. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun (2015) 3:31. doi: 10.1186/s40478-015-0203-5
52. Mangalmurti A, Lukens JR. How neurons die in alzheimer's disease: Implications for neuroinflammation. Curr Opin Neurobiol (2022) 75:102575. doi: 10.1016/j.conb.2022.102575
53. Young AMH, Kumasaka N, Calvert F, Hammond TR, Knights A, Panousis N, et al. A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat Genet (2021) 53(6):861–8. doi: 10.1038/s41588-021-00875-2
54. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature (2019) 566(7744):388–92. doi: 10.1038/s41586-019-0924-x
55. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron (2019) 101(2):207–223.e10. doi: 10.1016/j.neuron.2018.12.006
56. Chiaranunt P, Tai SL, Ngai L, Mortha A. Beyond immunity: Underappreciated functions of intestinal macrophages. Front Immunol (2021) 12:749708. doi: 10.3389/fimmu.2021.749708
57. Yip JLK, Balasuriya GK, Spencer SJ, Hill-Yardin EL. The role of intestinal macrophages in gastrointestinal homeostasis: Heterogeneity and implications in disease. Cell Mol Gastroenterol Hepatol (2021) 12(5):1701–18. doi: 10.1016/j.jcmgh.2021.08.021
58. Guilliams M, Thierry GR, Bonnardel J, Bajenoff M. Establishment and maintenance of the macrophage niche. Immunity (2020) 52(3):434–51. doi: 10.1016/j.immuni.2020.02.015
59. Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med (2018) 215(6):1507–18. doi: 10.1084/jem.20180019
60. Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med (2017) 49(5):e338. doi: 10.1038/emm.2017.20
61. Na YR, Jung D, Stakenborg M, Jang H, Gu GJ, Jeong MR, et al. Prostaglandin e receptor PTGER4-expressing macrophages promote intestinal epithelial barrier regeneration upon inflammation. Gut (2021) 70(12):2249–60. doi: 10.1136/gutjnl-2020-322146
62. De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell (2018) 175(2):400–415.e13. doi: 10.1016/j.cell.2018.07.048
63. Chikina AS, Nadalin F, Maurin M, San-Roman M, Thomas-Bonafos T, Li XV, et al. Macrophages maintain epithelium integrity by limiting fungal product absorption. Cell (2020) 183(2):411–428.e16. doi: 10.1016/j.cell.2020.08.048
64. Muller PA, Koscsó B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell (2014) 158(2):300–13. doi: 10.1016/j.cell.2014.04.050
65. Luo J, Qian A, Oetjen LK, Yu W, Yang P, Feng J, et al. TRPV4 channel signaling in macrophages promotes gastrointestinal motility via direct effects on smooth muscle cells. Immunity (2018) 49(1):107–119.e4. doi: 10.1016/j.immuni.2018.04.021
66. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
67. Yang M, Fukui H, Eda H, Kitayama Y, Hara K, Kodani M, et al. Involvement of gut microbiota in the association between gastrointestinal motility and 5‐HT expression/M2 macrophage abundance in the gastrointestinal tract. Mol Med Rep (2017) 16(3):3482–8. doi: 10.3892/mmr.2017.6955
68. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol (2019) 16:531–43. doi: 10.1038/s41575-019-0172-4
69. Delfini M, Stakenborg N, Viola MF, Boeckxstaens G. Macrophages in the gut: Masters in multitasking. Immunity (2022) 55(9):1530–48. doi: 10.1016/j.immuni.2022.08.005
70. Engin AB. Adipocyte-macrophage cross-talk in obesity. Adv Exp Med Biol (2017) 960:327–43. doi: 10.1007/978-3-319-48382-5_14
71. Frigolet ME, Gutiérrez-Aguilar R. The colors of adipose tissue. Gac Med Mex (2020) 156(2):142–9. doi: 10.24875/GMM.M20000356
72. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol (2005) 115(5):911–20. doi: 10.1016/j.jaci.2005.02.023
73. Grosjean A, Venteclef N, Dalmas E. Understanding the heterogeneity and functions of metabolic tissue macrophages. Semin Cell Dev Biol (2021) 119:130–9. doi: 10.1016/j.semcdb.2021.09.002
74. Silva HM, Báfica A, Rodrigues-Luiz GF, Chi J, Santos P, Reis BS, et al. Vasculature-associated fat macrophages readily adapt to inflammatory and metabolic challenges. J Exp Med (2019) 216(4):786–806. doi: 10.1084/jem.20181049
75. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell (2019) 178(3):686–698.e14. doi: 10.1016/j.cell.2019.05.054
76. Chakarov S, Blériot C, Ginhoux F. Role of adipose tissue macrophages in obesity-related disorders. J Exp Med (2022) 219(7):e20211948. doi: 10.1084/jem.20211948
77. Cox N, Crozet L, Holtman IR, Loyher PL, Lazarov T, White JB, et al. Diet-regulated production of PDGFcc by macrophages controls energy storage. Science (2021) 373(6550):eabe9383. doi: 10.1126/science.abe9383
78. Toita R, Kawano T, Murata M, Kang JH. Anti-obesity and anti-inflammatory effects of macrophage-targeted interleukin-10-conjugated liposomes in obese mice. Biomaterials (2016) 110:81–8. doi: 10.1016/j.biomaterials.2016.09.018
79. Sárvári AK, Doan-Xuan QM, Bacsó Z, Csomós I, Balajthy Z, Fésüs L. Interaction of differentiated human adipocytes with macrophages leads to trogocytosis and selective IL-6 secretion. Cell Death Dis (2015) 6(1):e1613. doi: 10.1038/cddis.2014.579
80. Nawaz A, Aminuddin A, Kado T, Takikawa A, Yamamoto S, Tsuneyama K, et al. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat Commun (2017) 8(1):286. doi: 10.1038/s41467-017-00231-1
81. Wang YN, Tang Y, He Z, Ma H, Wang L, Liu Y, et al. Slit3 secreted from M2-like macrophages increases sympathetic activity and thermogenesis in adipose tissue. Nat Metab (2021) 3(11):1536–51. doi: 10.1038/s42255-021-00482-9
82. Gavin KM, Majka SM, Kohrt WM, Miller HL, Sullivan TM, Klemm DJ. Hematopoietic-to-mesenchymal transition of adipose tissue macrophages is regulated by integrin β1 and fabricated fibrin matrices. Adipocyte (2017) 6(3):234–49. doi: 10.1080/21623945.2017.1314403
83. Kunz HE, Hart CR, Gries KJ, Parvizi M, Laurenti M, Dalla-Man C, et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am J Physiol Endocrinol Metab (2021) 321(1):E105–21. doi: 10.1152/ajpendo.00070.2021
84. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res (2008) 49(7):1562–8. doi: 10.1194/jlr.M800019-JLR200
85. Eguchi A, Feldstein AE. Adipocyte cell death, fatty liver disease and associated metabolic disorders. Dig Dis (2014) 32(5):579–85. doi: 10.1159/000360509
86. Hi Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U.S.A. (2018) 115(22):E5096–105. doi: 10.1073/pnas.1802611115
87. Lindhorst A, Raulien N, Wieghofer P, Eilers J, Rossi FMV, Bechmann I, et al. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis (2021) 12(6):579. doi: 10.1038/s41419-021-03872-9
88. Brykczynska U, Geigges M, Wiedemann SJ, Dror E, Böni-Schnetzler M, Hess C, et al. Distinct transcriptional responses across tissue-resident macrophages to short-term and long-term metabolic challenge. Cell Rep (2020) 30(5):1627–1643.e7. doi: 10.1016/j.celrep.2020.01.005
89. Pan Y, Hui X, Hoo RLC, Ye D, Chan C, Feng T, et al. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest (2019) 129(2):834–49. doi: 10.1172/JCI123069
90. Song M, Han L, Chen FF, Wang D, Wang F, Zhang L, et al. Adipocyte-derived exosomes carrying sonic hedgehog mediate M1 macrophage polarization-induced insulin resistance via ptch and PI3K pathways. Cell Physiol Biochem (2018) 48(4):1416–32. doi: 10.1159/000492252
91. Chen Q, Lai SM, Xu S, Tan Y, Leong K, Liu D, et al. Resident macrophages restrain pathological adipose tissue remodeling and protect vascular integrity in obese mice. EMBO Rep (2021) 22(8):e52835. doi: 10.15252/embr.202152835
92. Blériot C, Ginhoux F. Understanding the heterogeneity of resident liver macrophages. Front Immunol (2019) 10:2694. doi: 10.3389/fimmu.2019.02694
93. Hoeffel G, Ginhoux F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol (2018) 330:5–15. doi: 10.1016/j.cellimm.2018.01.001
94. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
95. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity (2017) 47(2):374–388.e6. doi: 10.1016/j.immuni.2017.07.018
96. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity (2019) 51(4):638–654.e9. doi: 10.1016/j.immuni.2019.08.017
97. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal (2014) 26(2):192–7. doi: 10.1016/j.cellsig.2013.11.004
98. Soucie EL, Weng Z, Geirsdóttir L, Molawi K, Maurizio J, Fenouil R, et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science (2016) 351(6274):aad5510. doi: 10.1126/science.aad5510
99. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell (2022) 185(2):379–396.e38. doi: 10.1016/j.cell.2021.12.018
100. Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F, et al. ALK1 signaling is required for the homeostasis of kupffer cells and prevention of bacterial infection. J Clin Invest (2022) 132(3):e150489. doi: 10.1172/JCI150489
101. Guilliams M, Scott CL. Does niche competition determine the origin of tissue-resident macrophages? Nat Rev Immunol (2017) 17(7):451–60. doi: 10.1038/nri.2017.42
102. Sawai CM, Babovic S, Upadhaya S, Knapp D, Lavin Y, Lau CM, et al. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity (2016) 45(3):597–609. doi: 10.1016/j.immuni.2016.08.007
103. Ma W, Zhao D, He F, Tang L. The role of kupffer cells as mediators of adipose tissue lipolysis. J Immunol (2019) 203(10):2689–700. doi: 10.4049/jimmunol.1900366
104. Loft A, Schmidt SF, Caratti G, Stifel U, Havelund J, Sekar R, et al. A macrophage-hepatocyte glucocorticoid receptor axis coordinates fasting ketogenesis. Cell Metab (2022) 34(3):473–486.e9. doi: 10.1016/j.cmet.2022.01.004
105. Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, et al. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab (2019) 29(2):383–398.e7. doi: 10.1016/j.cmet.2018.10.015
106. Ying W, Gao H, Dos Reis F, Bandyopadhyay G, Ofrecio JM, Luo Z, et al. MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab (2021) 33(4):781–790.e5. doi: 10.1016/j.cmet.2020.12.019
107. Gao H, Jin Z, Bandyopadhyay G, Cunha E Rocha K, Liu X, Zhao H, et al. MiR-690 treatment causes decreased fibrosis and steatosis and restores specific kupffer cell functions in NASH. Cell Metab (2022) 34(7):978–990.e4. doi: 10.1016/j.cmet.2022.05.008
108. Oh J, Riek AE, Darwech I, Funai K, Shao J, Chin K, et al. Deletion of macrophage vitamin d receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell Rep (2015) 10(11):1872–86. doi: 10.1016/j.celrep.2015.02.043
109. Qin Y, Jia L, Liu H, Ma W, Ren X, Li H, et al. Macrophage deletion of Noc4l triggers endosomal TLR4/TRIF signal and leads to insulin resistance. Nat Commun (2021) 12(1):6121. doi: 10.1038/s41467-021-26408-3
110. Demetz E, Tymoszuk P, Hilbe R, Volani C, Haschka D, Heim C, et al. The haemochromatosis gene hfe and kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur Heart J (2020) 41(40):3949–59. doi: 10.1093/eurheartj/ehaa140
111. Li P, Spann NJ, Kaikkonen MU, Lu M, Oh DY, Fox JN, et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell (2013) 155(1):200–14. doi: 10.1016/j.cell.2013.08.054
112. Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab (2017) 25(2):412–27. doi: 10.1016/j.cmet.2016.11.009
113. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology (2010) 51(2):511–22. doi: 10.1002/hep.23337
114. Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest (2017) 127(7):2829–41. doi: 10.1172/JCI90562
115. Zhang LY, Zhan DL, Chen YY, Wang WH, He CY, Lin Y, et al. Aflatoxin B1 enhances pyroptosis of hepatocytes and activation of kupffer cells to promote liver inflammatory injury via dephosphorylation of cyclooxygenase-2: an in vitro, ex vivo and in vivo study. Arch Toxicol (2019) 93(11):3305–20. doi: 10.1007/s00204-019-02572-w
116. Su L, Li N, Tang H, Lou Z, Chong X, Zhang C, et al. Kupffer cell-derived TNF-α promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis (2018) 9(3):323. doi: 10.1038/s41419-018-0377-4
117. Liu K, Yang L, Wang G, Liu J, Zhao X, Wang Y, et al. Metabolic stress drives sympathetic neuropathy within the liver. Cell Metab (2021) 33(3):666–675.e4. doi: 10.1016/j.cmet.2021.01.012
118. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci (2016) 61(5):1294–303. doi: 10.1007/s10620-016-4049-x
119. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse kupffer cells through mitochondrial DNA release. Cell Immunol (2018) 332:111–20. doi: 10.1016/j.cellimm.2018.08.006
120. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest (2021) 131(4):e135197. doi: 10.1172/JCI135197
121. Liu B, Wang M, Wang X, Zhao D, Liu D, Liu J, et al. Liver sinusoidal endothelial cell lectin inhibits CTL-dependent virus clearance in mouse models of viral hepatitis. J Immunol (2013) 190(8):4185–95. doi: 10.4049/jimmunol.1203091
122. Yang Z, Li Q, Wang X, Jiang X, Zhao D, Lin X, et al. C-type lectin receptor LSECtin-mediated apoptotic cell clearance by macrophages directs intestinal repair in experimental colitis. Proc Natl Acad Sci U.S.A. (2018) 115(43):11054–9. doi: 10.1073/pnas.1804094115
123. Zaman R, Epelman S. Resident cardiac macrophages: Heterogeneity and function in health and disease. Immunity (2022) 55(9):1549–63. doi: 10.1016/j.immuni.2022.08.009
124. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res (2019) 124(2):263–78. doi: 10.1161/CIRCRESAHA.118.314028
125. Koc A, Cagavi E. Cardiac immunology: A new era for immune cells in the heart. Adv Exp Med Biol (2021) 1312:75–95. doi: 10.1007/5584_2020_576
126. Sugita J, Fujiu K, Nakayama Y, Matsubara T, Matsuda J, Oshima T, et al. Cardiac macrophages prevent sudden death during heart stress. Nat Commun (2021) 12(1):1910. doi: 10.1038/s41467-021-22178-0
127. Petkevicius K, Bidault G, Virtue S, Jenkins B, van Dierendonck X, Dugourd A, et al. Norepinephrine promotes triglyceride storage in macrophages via beta2-adrenergic receptor activation. FASEB J (2021) 35(2):e21266. doi: 10.1096/fj.202001101R
128. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell (2017) 169(3):510–522.e20. doi: 10.1016/j.cell.2017.03.050
129. Cahill TJ, Sun X, Ravaud C, Villa Del Campo C, Klaourakis K, Lupu IE, et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development (2021) 148(3):dev194563. doi: 10.1242/dev.194563
130. Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity (2021) 54(9):2072–2088.e7. doi: 10.1016/j.immuni.2021.07.003
131. Grune J, Lewis AJM, Yamazoe M, Hulsmans M, Rohde D, Xiao L, et al. Neutrophils incite and macrophages avert electrical storm after myocardial infarction. Nat Cardiovasc Res (2022) 1(7):649–64. doi: 10.1038/s44161-022-00094-w
132. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell (2020) 183(1):94–109.e23. doi: 10.1016/j.cell.2020.08.031
133. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front Immunol (2021) 12:753940. doi: 10.3389/fimmu.2021.753940
134. Melo EM, Oliveira VLS, Boff D, Galvão I. Pulmonary macrophages and their different roles in health and disease. Int J Biochem Cell Biol (2021) 141:106095. doi: 10.1016/j.biocel.2021.106095
135. Kulikauskaite J, Wack A. Teaching old dogs new tricks? the plasticity of lung alveolar macrophage subsets. Trends Immunol (2020) 41(10):864–77. doi: 10.1016/j.it.2020.08.008
136. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity (2022) 55(9):1564–80. doi: 10.1016/j.immuni.2022.08.010j.immuni.2022.08.009
137. Hoppstädter J, Diesel B, Zarbock R, Breinig T, Monz D, Koch M, et al. Differential cell reaction upon toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respir Res (2010) 11(1):124. doi: 10.1186/1465-9921-11-124
138. Schyns J, Bureau F, Marichal T. Lung interstitial macrophages: Past, present, and future. J Immunol Res (2018) 2018:5160794. doi: 10.1155/2018/5160794
139. Gschwend J, Sherman SPM, Ridder F, Feng X, Liang HE, Locksley RM, et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J Exp Med (2021) 218(10):e20210745. doi: 10.1084/jem.20210745
140. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature (2014) 506(7489):503–6. doi: 10.1038/nature12902
141. Lopez-Rodriguez E, Gay-Jordi G, Mucci A, Lachmann N, Serrano-Mollar A. Lung surfactant metabolism: early in life, early in disease and target in cell therapy. Cell Tissue Res (2017) 367(3):721–35. doi: 10.1007/s00441-016-2520-9
142. Agudelo CW, Samaha G, Garcia-Arcos I. Alveolar lipids in pulmonary disease. a review. Lipids Health Dis (2020) 19(1):122. doi: 10.1186/s12944-020-01278-8
143. Li X, Yang N, Cheng Q, Zhang H, Liu F, Shang Y. MiR-21-5p in macrophage-derived exosomes targets Smad7 to promote epithelial mesenchymal transition of airway epithelial cells. J Asthma Allergy (2021) 14:513–24. doi: 10.2147/JAA.S307165
144. Bein K, Di Giuseppe M, Mischler SE, Ortiz LA, Leikauf GD. LPS-treated macrophage cytokines repress surfactant protein-b in lung epithelial cells. Am J Respir Cell Mol Biol (2013) 49(2):306–15. doi: 10.1165/rcmb.2012-0283OC
145. Skronska-Wasek W, Durlanik S, Le HQ, Schroeder V, Kitt K, Garnett JP, et al. The antimicrobial peptide S100A8/A9 produced by airway epithelium functions as a potent and direct regulator of macrophage phenotype and function. Eur Respir J (2022) 59(4):2002732. doi: 10.1183/13993003.02732-2020
146. Subramanian S, Busch CJ, Molawi K, Geirsdottir L, Maurizio J, Vargas Aguilar S, et al. Long-term culture-expanded alveolar macrophages restore their full epigenetic identity after transfer in vivo. Nat Immunol (2022) 23(3):458–68. doi: 10.1038/s41590-022-01146-w
147. Ween MP, White JB, Tran HB, Mukaro V, Jones C, Macowan M, et al. The role of oxidised self-lipids and alveolar macrophage CD1b expression in COPD. Sci Rep (2021) 11(1):4106. doi: 10.1038/s41598-021-82481-0
148. Golub R, Tan J, Watanabe T, Brendolan A. Origin and immunological functions of spleen stromal cells. Trends Immunol (2018) 39(6):503–14. doi: 10.1016/j.it.2018.02.007
149. A-Gonzalez N, Castrillo A. Origin and specialization of splenic macrophages. Cell Immunol (2018) 330:151–8. doi: 10.1016/j.cellimm.2018.05.005
150. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18(1):45–56. doi: 10.1038/s41423-020-00558-8
151. Cañadas O, Olmeda B, Alonso A, Pérez-Gil J. Lipid-protein and protein-protein interactions in the pulmonary surfactant system and their role in lung homeostasis. Int J Mol Sci (2020) 21(10):3708. doi: 10.3390/ijms21103708
152. Bellomo A, Mondor I, Spinelli L, Lagueyrie M, Stewart BJ, Brouilly N, et al. Reticular fibroblasts expressing the transcription factor WT1 define a stromal niche that maintains and replenishes splenic red pulp macrophages. Immunity (2020) 53(1):127–142.e7. doi: 10.1016/j.immuni.2020.06.008
153. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest (2017) 127(10):3770–83. doi: 10.1172/JCI94753
154. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
155. Kim SY, Nair MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol (2019) 97(3):258–67. doi: 10.1111/imcb.12236
156. Winn NC, Volk KM, Hasty AH. Regulation of tissue iron homeostasis: the macrophage “ferrostat”. JCI Insight (2020) 5(2):e132964. doi: 10.1172/jci.insight.132964
157. Alam Z, Devalaraja S, Li M, To T, Folkert IW, Mitchell-Velasquez E, et al. Counter regulation of spic by NF-κB and STAT signaling controls inflammation and iron metabolism in macrophages. Cell Rep (2020) 31(13):107825. doi: 10.1016/j.celrep.2020.107825
158. Bennett LF, Liao C, Quickel MD, Yeoh BS, Vijay-Kumar M, Hankey-Giblin P, et al. Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-c. Sci Signal (2019) 12(598):eaap7336. doi: 10.1126/scisignal.aap7336
159. Hao S, Xiang J, Wu DC, Fraser JW, Ruan B, Cai J, et al. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv (2019) 3(14):2205–17. doi: 10.1182/bloodadvances.2019000375
160. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572(7768):199–204. doi: 10.1038/s41586-019-1373-2
161. Bäckdahl J, Franzén L, Massier L, Li Q, Jalkanen J, Gao H, et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab (2021) 33(9):1869–1882.e6. doi: 10.1016/j.cmet.2021.07.018
162. Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, et al. Cells of the human intestinal tract mapped across space and time. Nature (2021) 597(7875):250–5. doi: 10.1038/s41586-021-03852-1
163. Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature (2017) 542(7641):352–6. doi: 10.1038/nature21065
164. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity (2019) 50(1):253–271.e6. doi: 10.1016/j.immuni.2018.11.004
165. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature (2020) 587(7835):619–25. doi: 10.1038/s41586-020-2922-4
166. Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue homeostasis and inflammation. Annu Rev Immunol (2021) 39:557–81. doi: 10.1146/annurev-immunol-061020-053734
Keywords: parenchymal cells, tissue-resident macrophages, cellular crosstalk, tissue-specific function, mammalian, tissue homeostasis
Citation: Chen Y and Tang L (2022) The crosstalk between parenchymal cells and macrophages: A keeper of tissue homeostasis. Front. Immunol. 13:1050188. doi: 10.3389/fimmu.2022.1050188
Received: 21 September 2022; Accepted: 10 November 2022;
Published: 24 November 2022.
Edited by:
Erika M. Palmieri, National Cancer Institute at Frederick (NIH), United StatesReviewed by:
Ivan Dzhagalov, National Yang Ming Chiao Tung University, TaiwanSamuel Nobs, Weizmann Institute of Science, Israel
Copyright © 2022 Chen and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Tang, dGFuZ2xpQG5jcHNiLm9yZy5jbg==