
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 November 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1049301
This article is part of the Research Topic The Immune Escape Mechanism and Novel Immunotherapeutic Strategies of Leukemia View all 14 articles
 Carsten Riether1,2*†
Carsten Riether1,2*†Adult bone marrow (BM) hematopoietic stem cells (HSCs) are maintained in a quiescent state and sustain the continuous production of all types of blood cells. HSCs reside in a specialized microenvironment the so-called HSC niche, which equally promotes HSC self-renewal and differentiation to ensure the integrity of the HSC pool throughout life and to replenish hematopoietic cells after acute injury, infection or anemia. The processes of HSC self-renewal and differentiation are tightly controlled and are in great part regulated through cellular interactions with classical (e.g. mesenchymal stromal cells) and non-classical niche cells (e.g. immune cells). In myeloid leukemia, some of these regulatory mechanisms that evolved to maintain HSCs, to protect them from exhaustion and immune destruction and to minimize the risk of malignant transformation are hijacked/disrupted by leukemia stem cells (LSCs), the malignant counterpart of HSCs, to promote disease progression as well as resistance to therapy and immune control. CD4+ regulatory T cells (Tregs) are substantially enriched in the BM compared to other secondary lymphoid organs and are crucially involved in the establishment of an immune privileged niche to maintain HSC quiescence and to protect HSC integrity. In leukemia, Tregs frequencies in the BM even increase. Studies in mice and humans identified the accumulation of Tregs as a major immune-regulatory mechanism. As cure of leukemia implies the elimination of LSCs, the understanding of these immune-regulatory processes may be of particular importance for the development of future treatments of leukemia as targeting major immune escape mechanisms which revolutionized the treatment of solid tumors such as the blockade of the inhibitory checkpoint receptor programmed cell death protein 1 (PD-1) seems less efficacious in the treatment of leukemia. This review will summarize recent findings on the mechanisms by which Tregs regulate stem cells and adaptive immune cells in the BM during homeostasis and in leukemia.
During the last decade a series of studies have been performed to decipher the role and contribution of non-hematopoietic (osteoblasts, stromal and endothelial cells, pericytes, Schwann cells and sympathetic neurons) as well as mature hematopoietic cells to the hematopoietic stem cell (HSC) niche in the bone marrow (BM). All these different cell types were allocated as components of the HSC niche and even as critical regulators of HSC function. Especially intensive work on cells of non-hematopoietic origin by many different groups unraveled the composition of the HSC niche and the cellular and molecular interactions exchanged between its different stromal and endothelial components and their contribution to HSC function (1). Recent work has highlighted the importance of mature immune cells as constituents of the HSC niche (1). Regulatory T cells (Tregs) have been initially identified as a specialized T cell subpopulation which suppresses immune responses to guarantee immune homeostasis and self-tolerance. Recently, however, Tregs have been shown to also control non-lymphoid processes, insulin resistance in visceral fat, muscle repair and to prevent lung damage (2). In the BM microenvironment Treg frequencies are higher than in other secondary lymphoid organs suggesting a role for Tregs in shaping an immunosuppressive BM microenvironment which may protect HSCs from elimination and damage. However, the current understanding of Tregs in the regulation of HSCs and/or the HSC niche under steady state conditions is still limited. During leukemia development, the immune microenvironment in the BM undergoes massive changes with especially immunosuppressive Tregs infiltrating or accumulating. These Tregs may be able to directly promote leukemogenesis or to protect LSCs from elimination by activated immune cells. However, like for HSCs, the actual interaction of Tregs with LSCs remains in part elusive. Therefore, in this review we discuss recent insights into the communication between Tregs, HSCs and LSCs during health and hematological disease.
Under homeostatic conditions, the majority of HSCs in the BM are in a quiescent state to prevent HSC exhaustion and to maintain and protect their self-renewal capacity (1). Fundamental HSC characteristics such as self-renewal, differentiation and proliferation are crucially regulated by hematopoietic and stromal components of the HSC niche. Perivascular regions in the BM form distinct niches and have been identified to regulate HSC quiescence and the supply of lineage-committed progenitors (1). Especially NG2-expressing pericytes maintain HSCs in a quiescent state as depletion of NG2-expressing cells resulted in cycling of HSCs, loss of long-term repopulating HSCs and altered HSC localization in the BM. In contrast, cycling HSCs are found near sinusoids and more specifically to sinusoids associated with stem cell factor (SCF) - producing leptin receptor-positive (LEPR+) cells. Depletion of SCF in LEPR+ cells reduced BM HSC numbers (3). Consequently, it is postulated that once entering cell cycle quiescent HSCs translocate from a NG2+ periarteriolar to a LEPR+ perisinusoidal region of the HSC niche. In addition, the low oxygen-permeability of arteriolar blood vessels in the BM promotes HSC quiescence by reducing the accumulation of reactive oxygen species (ROS) in HSCs in contrast to highly oxygen-permeable sinusoids which confer proliferation and differentiation signals to the HSCs (4–6). Furthermore, endothelial cells have been identified together with LEPR+ cells as the main sources of SCF and CXC-chemokine ligand 12 (CXCL-12) required for HSC maintenance in normal young and adult BM (1). Thus, HSCs reside in a hypoxic perivascular niche in which endothelial cells and stromal cells express distinct factors that promote HSC maintenance (1, 3).
The contribution of the BM microenvironment in the leukemogenesis of leukemias and other hematopoietic neoplasms has been demonstrated in recent years (7). In hematological malignancies and with age, not only hematopoietic cells accumulate genetic alterations but also the HSC niche undergoes an extensive re-organization and thereby contributes to the generation of hematopoietic disorders such as leukemia (8). Nevertheless, despite these findings generated in experimental models, direct evidence that initial lesions in cells of the HSC niche trigger the development of the disease is still lacking for human leukemia.
In contrast, evidence that leukemic cells actively create an aberrant leukemic niche has been extensively generated in parallel to the description of the normal HSC niche. Normal HSCs and LSCs compete for the same region of the endosteal HSC niche (9–12). Through their localization in the normal HSC niche, LSCs co-opt evolutionary conserved signaling cascades of the BM microenvironment to further support the growth of the leukemia (7). In addition, malignant hematopoietic cells interact specifically with stromal and endothelial elements of the HSC niche to induce remodeling. As a consequence, the re-modelled BM microenvironment becomes supportive for AML LSCs and negatively affects healthy hematopoiesis and, specifically, HSCs (8–14). For example, genes in AML MSCs are in general hypomethylated which is accompanied by an altered expression of genes related to inflammation and CXCL-12 signaling (9). Similar results on the provision of pro-inflammatory signals by MSCs to promote leukemogenesis have been described in murine AML and CML models (10). Furthermore, pro-inflammatory cytokines such as interleukin (IL)-6 and TNF produced by leukemia cells have a central role in the promotion of disease development by promoting differentiation of leukemic progenitors and by simultaneous disruption and alteration of normal HSC function (8, 13, 14). Single cell RNA-sequencing further revealed that MSCs in AML have an impaired potential to differentiate into osteoblasts and to secrete key molecules required for HSCs maintenance such as CXCL-12 and SCF (9). The accumulation of these altered osteoprogenitors/osteoblasts, also characterized by increased production of inflammatory cytokines, modulates the endosteal HSC niche in a way that leukemogenesis is supported while the capacity to support normal hematopoiesis and HSCs is lost (11, 12).
Two recently published studies also highlighted the impact of alterations in the BM vasculature in the endosteal region for the maintenance of LSCs and the loss of HSCs. Increased permeability and reduced perfusion in the endosteal region resulted in hypoxic changes, the overproduction of ROS and nitric oxid, loss of barrier function and cell death. Inhibition of nitric oxid production reduced vascular permeability and preserved normal HSC function (15). Furthermore, enhanced inflammatory signaling in leukemia cells in the endosteal niche was associated with a reduction in vessels and HSCs (13). Importantly, a recent transcriptomic analysis revealed that pathologic crosstalk exists not only between AML and different niche cells but also within niche fractions (15).
The sympathetic nervous system (SNS) also promotes AML development. However, in contrast to normal hematopoiesis (16), nerves fibers get disrupted during AML development resulting in a reduction of HSC-promoting NG2+ niche cells and instruction of Nestin+ stromal cells to generate osteoblasts (17). Furthermore, recent preclinical work indicated a role for the pro-inflammatory cytokine IL-1β in the loss of sympathetic nerve fibers in the BM during MPN development. During early stages of the disease, JAK2V617F-expressing HSCs secrete IL-1β resulting in apoptosis of sympathetic fibers which was further associated with reduced numbers of Nestin-expressing MSCs and concomitant expansion of mutant HSCs (18). Overall, these studies indicate that LSCs, like HSCs, are dependent on signals derived from the HSC niche. Additionally, LSCs actively contribute to the re-organization of the niche from a microenvironment supportive for HSCs into an HSC unfriendly, aberrant leukemic microenvironment.
Self-renewal and maintenance of HSCs are not exclusively regulated by endothelial and stromal elements of the BM niche. As a primary and secondary lymphoid organ, the BM harbors a variety of different mature and immature cells of hematopoietic origin such as T cells, B cells monocytes/macrophages and neutrophils (7). Apart from developing and mature B cell subsets which occupy specific niches in the BM (19), most other lymphocyte subsets are widely distributed and can in general not be allocated to a specific location in the BM stroma or parenchyma (7). In addition, billions of leukocytes circulate every day through the BM. Recent studies highlighted the substantial contribution of multiple of these immune cell subsets in the regulation of the HSC niche (Table 1). Especially monocytes and macrophages but also different T cell subset have been shown to mediate the mobilization of HSCs indirectly by modulating MSCs (34–36, 39). Monocytes and macrophages comprise around 0.4% of total BM cells and are widely distributed throughout the BM.

Table 1 Immune cell subsets present in the BM stem cell niche and their reported effect on the HSCs and HSC niche function.
The BM microenvironment does not only provide HSCs with key signals for survival, quiescence and self-renewal (1) but it also ensures protection against harmful signals that may drive HSCs differentiation, exhaustion or destruction (7). Tregs is one cellular subset, ideally suited to protect against the negative impact for example of inflammation by shaping an immune-suppressive environment. The BM is considered as a reservoir for Tregs in healthy individuals (40). Under homeostatic conditions, Tregs locate close to sinusoids and close to HSCs located at the endosteal surface in the BM (25) and comprise around 0.5% of all BM mononuclear cells and around one third of all CD4+ T cells in the BM (41). Thereby, Tregs are significantly enriched in frequency in the BM compared to other secondary lymphoid organs such as the spleen or the lymph nodes (40, 42). In contrast to blood and skin, BM Tregs are phenotypically characterized by the expression of CD45RA, C-X-C chemokine receptor type 4 (CXCR4) and the high expression of FOXP3 and CD25 (40, 43). Functionally, BM Tregs have a superior immunosuppressive activity compared to their blood counterparts (40). The BM retains these CXCR4-expressing Tregs through CXCL-12. After adoptive transfer into NSG mice, human Tregs preferentially homed into the BM which harbored elevated levels of CXCL-12. Blockade of the CXCR4/CXCL-12 interaction via monoclonal antibody (mAb) abolished the retention of Tregs in the BM but not the spleen (40). Therefore, it is speculated that the BM may represent a niche where naive Tregs accumulate to mature, survive, and/or undergo homeostatic proliferation while maintaining their naive phenotype. Furthermore, BM-resident Tregs may contribute to the formation of the immune-privileged perivascular HSC niche and shield the stem cell compartment from elimination by immune cells, excessive inflammation as well as cell-death.
Total CD4+ T cells (T helper cells and Tregs) secret hematopoietic-related cytokines and are essential for hematopoiesis stimulation during infection and hematologic recovery after BM transplantation [reviewed in (44)]. In addition, early observations that 1) STAT-4–/– mice, which harbor predominately Th2 cells, have decreased numbers of cycling progenitors (21) and 2) that adoptive transfer of total CD4+ T cells into common gamma chain-deficient mice restored impaired myeloid differentiation indicated that CD4+ T cells regulate HSPC activity and basal hematopoiesis (20). These findings have been recently extended by Hernández-Malmierca and co-workers who demonstrated that the presentation of immunogenic antigens on MHC class II by HSPCs triggers an interaction with CD4+ T cells resulting in stem cell differentiation and depletion of immunogenic HSPCs (22).
Most of the effects observed upon transfer or depletion of total CD4+ T cells may most likely be mediated directly or indirectly by immunomodulatory Tregs. Tregs regulate IL-3 mediated differentiation of myeloid progenitors under homeostatic conditions. Depletion of Tregs in situ and co-transplantation of CD4+ FOXP3+ Tregs demonstrated that BM Tregs promote myeloid colony formation in vitro and myeloid differentiation of HSCs in vivo (28). Importantly, the cognate interaction between Tregs and HSPCs was dependent on MHC class II expression suggesting a role for antigen-specific activation of Tregs in the regulation of HSCs in steady-state. In line with these initial findings, Kim et al. demonstrated that Foxp3 mutant scurfy mice have an enhanced granulopoiesis at the expense of B cell lymphopoiesis. However, this effect of myeloid differentiation was not mediated directly via Tregs but indirectly via the production of myeloid differentiation-related cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF, and IL-6 by activated effector T cells (45). In addition, it was demonstrated that Tregs regulate the production of IL-7, a fundamental growth factor for lymphopoiesis, by intracellular adhesion molecule 1 (ICAM-1)-expressing perivascular stromal cells in the BM (27). In line with these findings, Camacho et al. recently demonstrated that BM Tregs regulate hematopoiesis indirectly through modulation of stromal cell function via secretion of IL-10 (29) (Figure 1 and Tables 1, 2). The role of Tregs in the regulation of stem cells, however, is not a unique feature for the BM and can be further extrapolated to multiple non-lymphoid tissues such as for example the skin, muscle, adipose tissue and brain (46–49). Consequently, accumulating scientific evidence supports the concept that Tregs control stem cells, their proliferation and self-renewal.
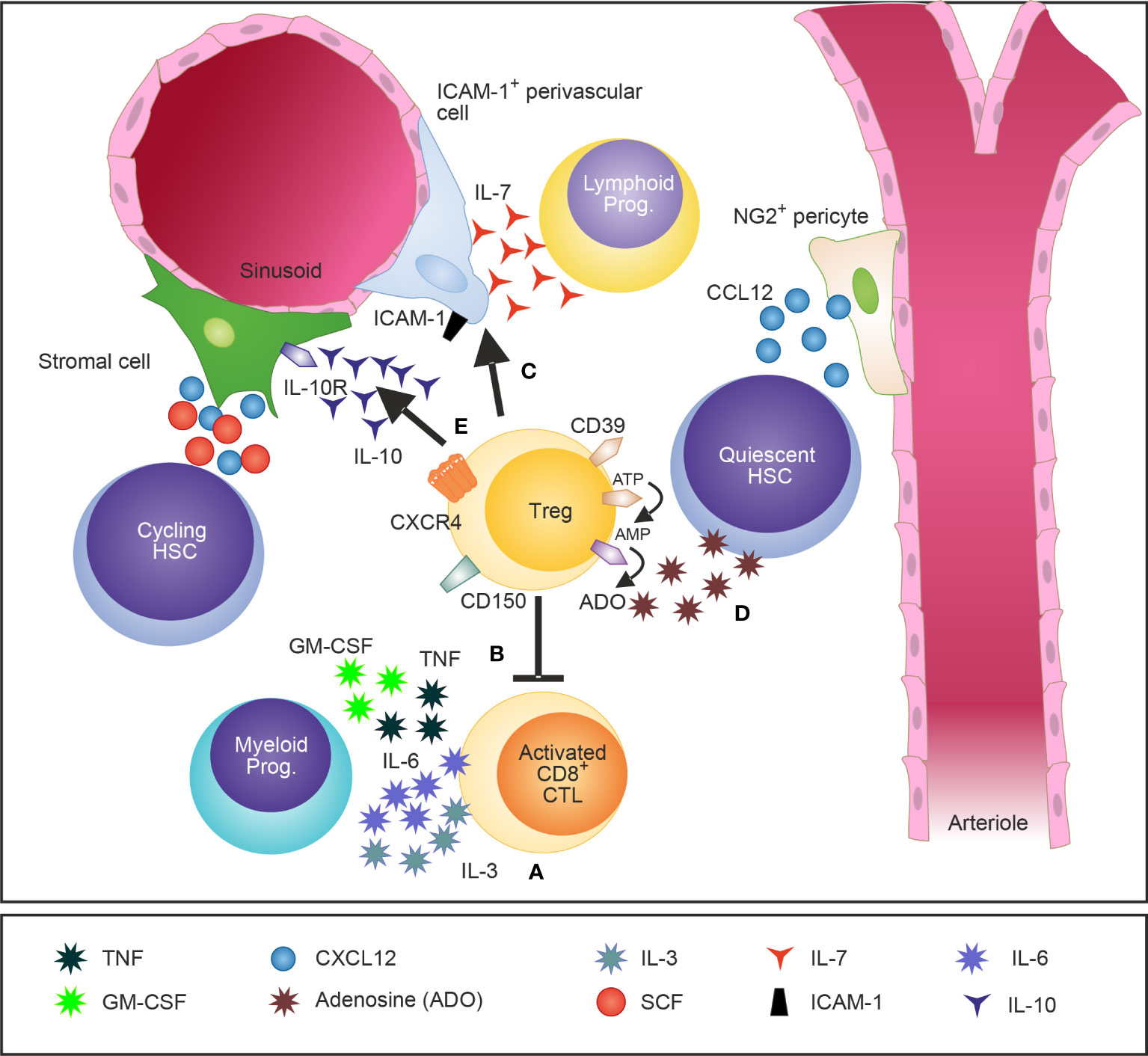
Figure 1 A schematic illustration of Tregs in the BM and their attributed effects on the HSCs niche and HSPCs. Tregs in the BM are widely distributed and localize close to the endosteum adjacent to HSCs. Tregs regulate IL-3 mediated differentiation of myeloid progenitors (A) (28) and modulate basal hematopoietic output by restricting production of myelopoiesis-promoting cytokine production by activated CD8 T cells in the BM (B) (45). Furthermore, Tregs control indirectly lymphopoiesis by mediating the release of the lymphoid cell growth and survival factor IL-7 by ICAM-1+ stromal cells of the HSC niche (C) (27). CD150+ Tregs were recently identified as crucial regulators of HSC quiescence and engraftment of HSCs into the BM after allogeneic BM transplantation (D) (26). In addition, IL-10 secreting Tregs have been shown to modulate the function of IL-10R-expressing BM stromal cells (E) (29).

Table 2 Treg-mediated immunomodulatory mechanisms in the BM niche at steady state.
In murine BM transplantation models, experimental data demonstrated that Tregs of the host not only suppresses graft-versus host disease but also favors hematopoietic reconstitution and HSC engraftment (50, 51). Similarly, co-transplantation of recipients alloantigen-specific Tregs together with HSC facilitated long-term hematopoietic reconstitution (52). In addition, Tregs activated ex vivo with allogeneic APCs also induced long-term tolerance and reconstitution of BM grafts in an antigen-specific manner in vivo (53). A potential role for Tregs in hematopoiesis was further demonstrated in murine allogeneic BM transplantation experiments where depletion of host Tregs resulted in loss of engraftment of stem and progenitor cells to the BM suggesting that Tregs promote hematopoiesis and protect HSCs from elimination (25). In addition, a specific BM Treg subset characterized by the expression the surface markers CD150 and CD39 was recently identified as regulator of HSC quiescence and engraftment of HSCs into the BM after allogeneic BM transplantation. These BM Tregs reside near HSCs in the BM niche and protect HSCs from oxidative stress by secretion of adenosine. In addition, co-transfer of BM Tregs but not splenic Tregs together with allogeneic HSCs prevented the rejection of the graft (26). Mechanistically, long-term hematopoietic reconstitution has been shown to crucially depend on MHC-II signaling in a canine BM transplantation model as delivery of an anti-MHC-II mAb directly after BM transplantation prevented engraftment and hematopoietic reconstitution (54). In contrast, Huss and co-worker demonstrated in various murine allogeneic BM transplantation models that immunological but not hematopoietic recovery is impaired in recipient mice lacking MHC class II expression or treated with an anti-MHC II mAb (55) (Figure 1 and Table 1).
In humans, the role of Tregs in HSC engraftment and long-term reconstitution after BM transplantation is still unclear. While depletion of total T cells from allogeneic BM resulted in rejection of grafts in a majority of the patients, the clinical benefit of Tregs in the graft still has to be demonstrated [reviewed in (50)]. Several clinical studies correlating the number of Tregs in the graft retrospectively with transplantation outcome delivered varying results and could not attribute a fundamental role of Tregs to long-term hematopoietic reconstitution. Similarly, conflicting data have been generated on the number of Tregs detected in vivo post-transplantation and transplantation outcome. Overall, these data imply a role for Tregs in the regulation of HSC function in mice while the role of Tregs on human hematopoiesis must still be validated.
Experimental and clinical evidence indicates that myeloid leukemias are controlled by cells of the adaptive immune system to a certain extent (56, 57). Leukemic cells in myeloid malignancies are immunogenic, express HLA class I restricted leukemia epitopes and may be recognized by activated cytotoxic CD8+ T cells (CTLs) (7, 56, 57). LSCs and bulk leukemia cells express major histocompatibility and co-stimulatory molecules suggesting that LSCs can be recognized and interact directly with T cells (7, 56).
In addition, immune responses against myeloid leukemias as well as leukemia-antigen specific CD8+ CTLs have been observed in clinical and experimental studies (7, 56). However, activated CD8+ CTLs in myeloid leukemias are dysfunctional and fail to eliminate LSCs in vivo (23, 24, 58–60). In CML, T cell exhaustion at diagnosis has been associated with an increased expression of immune inhibitory receptors and a reduced capacity to produce Th1-cytokines compared to patients in remission (59–63). Similar to CML, gene expression profiling of CD8+ CTLs from AML patients at diagnosis indicate that CTLs in intermediate and high risk AML are less functional than in favorable risk AML and exhibit an exhaustion immune signature (24). CD8+ CTL dysfunction was attributed to epigenetic silencing of activating immune checkpoint receptors rather than due to signaling by immune inhibitory immune checkpoint receptors (64). The phenomenon could in part explain the limited efficacy of antibodies that block inhibitory immune check-point inhibitors in AML. Furthermore, instead of promoting elimination, CD8+ CTL-related mechanism may also contribute to the expansion of LSCs and leukemia (23, 24). For example, secretion of the hematopoietic cytokine IL-3 has been shown to expand and maintain leukemia stem and progenitor cells in favorable risk AML patients, while AML cells in more aggressive forms of AML seem to develop largely independent of CD8+ T cell help (24). A systemic impairment/exhaustion of T cells and especially programmed cell death protein 1 (PD-1) and Eomes-expressing memory stem T cells has also been associated with relapse after allogeneic HSCT (65). Furthermore, response to chemotherapy correlated with the immune signatures indicative for the restoration of T cell function (58).
In contrast to CD8+ T cells, the role of CD4+ T cells in the control of leukemia has been studied less intensively (7). Nevertheless, even though less studied, several independent reports demonstrated that myeloid leukemia cells are competent to process and present endogenous immunogenic leukemia peptides in the context of HLA class II resulting in leukemia antigen-specific CD4+ T cell response [reviewed in (7)]. Recently, the HLA class I and class II ligandome was characterized using immunoprecipitation and mass spectrometry in CD34+ stem/progenitor cells from AML and CML patients and CD34+CD38- healthy controls (66, 67). For AML, the analysis revealed 36 AML-specific leukemia antigens. Importantly, the top-ranked HLA class II leukemia antigens were successfully validated in T cell activation assays in vitro (67). A similar analysis for CML identified 44 CML-associated HLA class II antigens (66). A clinical vaccination study which assessed the immunogenicity and anti-tumor activity of a vaccine cocktail comprising 4 b3a2-petides restricted to HLA I and one b3a2-petide restricted to HLA II in b3a2-espressing CML patients with stable measurable residual disease, demonstrated immunological activity of the vaccine as illustrated by CML-peptide specific CD4+ T cells in vitro in 13 of 14 evaluable patients (68). Furthermore, vaccination of a 63-year old woman which displayed residual CML cells after cessation of IFN-α therapy with a immunogenic b2a2 breakpoint derived peptide able to bind several HLA-DR molecules induced a complete molecular response in blood and BM (69). Overall, these data suggest that leukemia-antigen specific CD4+ T cells may potentially also contribute to anti-tumoral immunity in leukemia.
In AML, T cells appear to infiltrate the BM microenvironment, but their efficacy to control leukemia and eliminate LSCs is limited. In general, the BM microenvironment in AML may be considered as immune cold (70, 71). Analysis of an 18-gene tumor inflammation signature in solid tumor and AML samples using data from the Cancer Genome Atlas revealed that AML patients have a low expression of this signature which is associated with an immune cold microenvironment (70). Similarly, transcriptomic, and proteomic analysis of AML BM cells demonstrated that 70% of AML patients display an immune-depleted signature (71). Because LSCs reside primarily in the BM, the generation of an immunosuppressive/immune cold environment may be considered as a central mechanism how LSCs escape immune control. However, several different mechanisms may contribute to generation of an immune cold environment and the subsequent immune escape in myeloid leukemia.
One mechanism how LSCs can escape immunosurveillance is through the activation of immune checkpoint pathways. Immune checkpoint pathways play a fundamental role in the regulation of immune homeostasis (72). The balance between stimulatory and inhibitory signals determines the amplitude and quality of the antigen-specific T cell responses (72). Immune checkpoint inhibitors targeting the co-inhibitory molecules cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1 have been approved for the treatment of various solid tumors and lymphoma (72). Increased expression of the inhibitory ligands for T-cell–regulating checkpoints such as CTLA-4, PD-1, B7-H3 and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) was reported in AML and was associated with T cell exhaustion and poor prognosis (62, 63, 73). This CD8+ T cell exhaustion was in part reversible by blocking the immune checkpoint molecules PD-1, CTLA-4, and TIM-3 (63) or by OX-40 co-stimulation (62). However, in patients, immune checkpoint inhibitors against CTLA-4 and PD-1 seem to be less effective in AML compared to solid tumors (74). One possible explanation for this lack of potency in AML is that BM-infiltrating T cells in AML expressing various inhibitory receptors, such as TIM-3 and lymphocyte-activation gene 3 (LAG-3). Another explanation could be the limited or restricted surface expression of PD-L1 in de novo AML (73, 75) and variable kinetic of PD-L1 expression during the progression of the disease. For example, PD-L1 expression increases on AML cells during relapse and could represent an adaptive immune escape mechanism (74). However, it also unclear whether the increased expression of immune inhibitory receptors reflects T cell exhaustion or whether it is indicative for differentiated effector T population. Currently several clinical trials with different antibodies targeting the PD-1/PD-L1 interaction are ongoing in combination with CTLA-4 inhibition, in combination with chemotherapy, or in combination with hypomethylating agents that upregulate the expression of PD-L1 on leukemia cells (76).
Because the efficacy of immune checkpoint inhibitor treatments in myeloid malignancies is limited, other immune-regulatory mechanisms in the BM may be in place. Studies in mice and humans identified the accumulation of Tregs as a potential major immune-regulatory mechanism in myeloid leukemia. Several clinical studies reported increased frequencies of Tregs in blood and/or BM at diagnosis, after allogeneic hematopoietic stem cell transplantation (HSCT) or during induction chemotherapy in myeloid leukemias (77, 78).
In CML, numbers and frequencies of effector Tregs in peripheral blood and BM are also increased in CML patients at diagnosis and in patients refractory to tyrosine kinase inhibitor (TKI) treatment (59–61). These effector Tregs are characterized by the expression of the immune checkpoints PD-1, TIM-3 and CTLA-4, all markers for an increased suppressive activity (61). Anatomically, Tregs in CML are widely distributed throughout the BM parenchyma in mice and humans. In human CML, the majority of Tregs were localized close to CD8+ CTLs and not close to CD34+ CML stem/progenitor cells (30). Treatment with the TKIs Imatinib, Dasatinib and Nilotinib demonstrated an inhibitory effect of these TKIs on the quantity of Tregs in CML patients. Tregs were especially reduced in patients who achieved a complete cytogenetic response (59, 79). Similarly, Imatinib-treated CML patients in complete molecular remission (CMR) displayed a selective depletion of effector Tregs which was accompanied by an increase in effector/memory CD8+ CTLs in contrast to CMR patients who did not reach a complete molecular remission (80). Furthermore, a successful maintenance of treatment-free-remission (TFR) is associated with reduced numbers of Tregs (81–83). In the frame of the JALSG-STIM213 trial, the frequency of effector Tregs in the peripheral blood has been identified as potential biomarker for TFR after Imatinib stop (84). Importantly, tyrosine kinase inhibitor (TKI) treatment responses were more potent in patients with less exhausted T cells (60). Because the state of T cell dysfunction at diagnosis is reverted in patients that reach a deep molecular response (60), one may hypothesize that off-target immune-modulatory effects of TKIs trigger already pre-existing anti-tumoral immune responses or interfere with immunosuppressive cells as Tregs. In solid tumors, tyrosine kinase inhibitor (TKI) treatment with Imatinib and Dasatinib has been shown to reduce immunosuppressive immunoindolamine-2, 3-dioxygenase (IDO) expression, a key factor in the generation of Tregs, and to increase the numbers of intratumoral CD8+ CTLs (85, 86). Future therapeutic approaches may therefore aim at an expansion and effector function of CTLs through targeting of Tregs. For example, Tregs expressing the tumor necrosis factor 4 (TNFRSF4, alias OX-40) have recently been identified as the key regulator of immune escape of CML LSCs in mice (30). Stimulation of Tnfrsf4-signaling did not deplete Tregs but reduced the capacity of Tregs to protect LSCs from granzyme-mediated CD8+ CTL- killing in a murine CML model.
Like in CML (30), Tregs in the BM of AML patients are widely distributed throughout the BM parenchyma and are occasionally located close to clusters of CD8+ T cells (Figure 2). Tregs frequencies in blood and BM are significantly elevated in adult and pediatric AML patients compared to Tregs from healthy donors and further increase at relapse (87–89). In general, increased levels of Tregs at diagnosis correlate with poor response to induction chemotherapy and relapse after allogeneic HSCT (78, 90). In contrast to baseline AML biopsies, higher numbers of Tregs early after induction therapy are associated with higher complete remission rates and better overall survival. This discrepancy may be attributed to the different cellular composition of the BM microenvironment early after chemotherapy and at an advanced stage of the disease. Furthermore, a phase IV clinical trial (NCT01347996) which assessed the efficacy of immunotherapy with histamine dihydrochloride (HDC) and low-dose IL-2 in the post-consolidation phase in AML 84 patients in first complete remission demonstrated an association of AML relapse with HDC/IL-2 treatment-induced accumulation of natural Tregs in the blood (91). Therefore, Tregs could either serve as immune-modulators and support hematopoietic recovery or induce immune escape of leukemia by limiting CTL activity. On the other hand, Tregs seem to localize close to HSCs in the niche during homeostasis and may therefore directly facilitate their homing in the BM and emergency hematopoiesis.
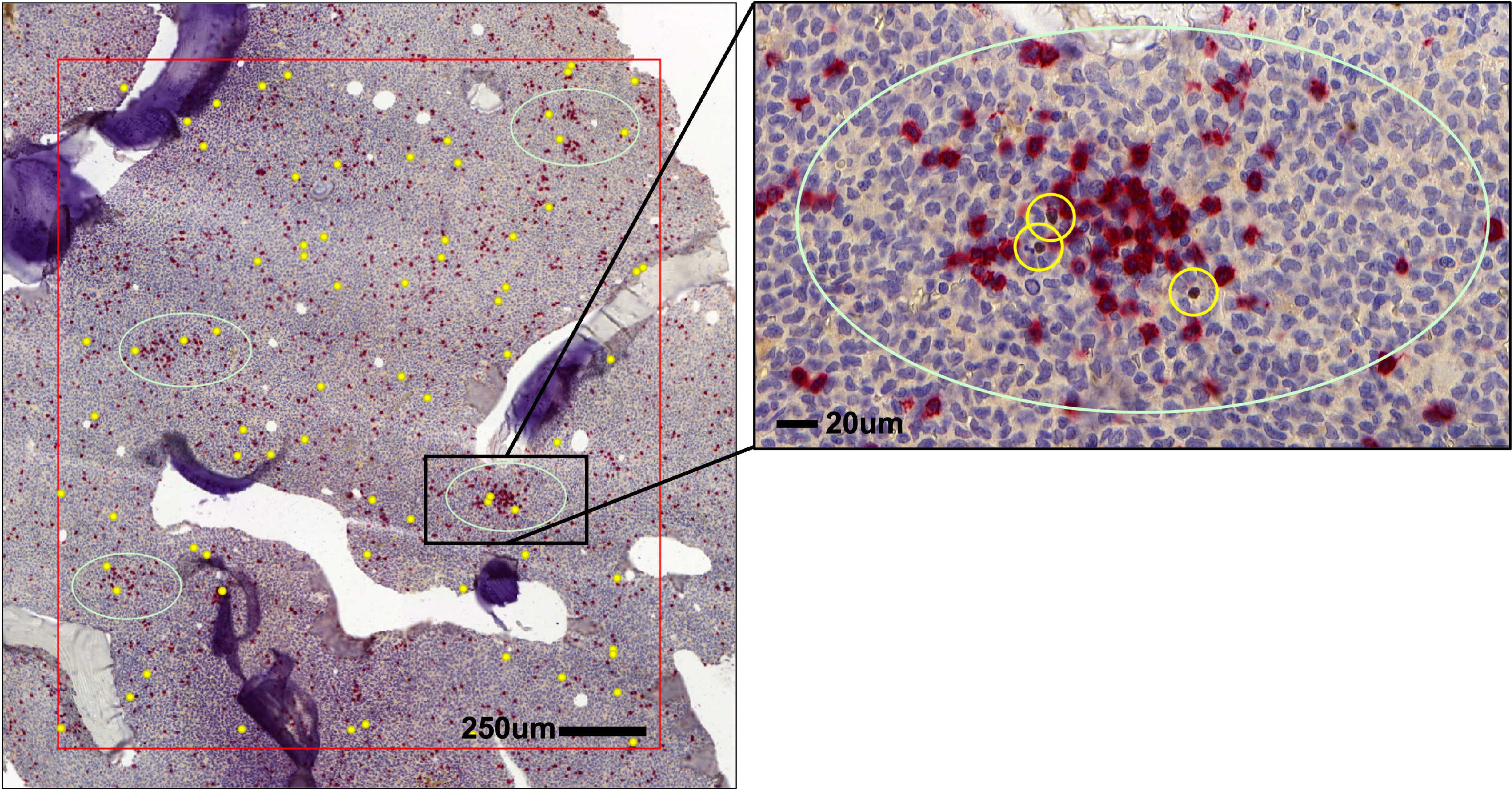
Figure 2 Distribution of Tregs and CD8+ T cells in AML bone marrow (BM). Spatial localization of FOXP3+ Tregs (brown, yellow circles) in respect to CD8+ T cells (red) in the BM of AML patients.
In contrast to CD8+ T cells, Tregs from AML patients are not exhausted but rather hyper-functional as illustrated by a higher migratory and immunosuppressive potential than Tregs from blood and BM of healthy volunteers (37). However, even though, the prevalence of Tregs in the BM and blood in AML is identical (92), BM Tregs are more suppressive than Tregs derived from blood of AML patients (77). Phenotypically, BM Tregs of AML patients express the memory marker CD45RO along with CTLA-4 and CD95 and secret low levels of the immunosuppressive cytokines IL-10 and TNF. Furthermore, BM Tregs in AML have a high turnover compared to BM Tregs from healthy donors as indicated by 8-fold higher proliferation and 4-fold higher apoptosis rate (93).
Recently, several mechanisms have been identified that promote the expansion and BM retention of Tregs in AML (37, 94–96). For example, Tregs in blood and BM of patients have been shown to express elevated levels of CXCR4 on the cell surface compared to Tregs derived from healthy donors suggesting that, like in homeostasis, the BM retains CXCR4-expressing Tregs through CXCL-12 in AML (37). Similarly, the frequency of Tregs expressing inducible T-cell co-stimulator (ICOS) has been negatively associated with outcome in AML patients (97). Co-simulation of ICOS signaling through the provision of ICOS-ligand by AML cells expanded Tregs and maintained their suppressive function in a murine C1498 AML model (97) (Figure 3). Another mechanism how Tregs may be expanded and sustained in AML is through IFN-y induced secretion of IDO by mesenchymal stromal cells (94) (MSCs, Figure 3). Blockade of IFN-y production by AML cells lowered IDO1 expression resulting in reduced Treg infiltration and leukemia engraftment. Furthermore, daunorubicin-treated AML cells induce IDO1 secretion in dendritic cells (DCs) resulting in the accumulation of PD-1-expressing Tregs (98). In the BM of newly diagnosed AML patients, high IDO expression in MSCs was associated with elevated levels of Tregs. Furthermore, an IDO1-related immune gene signature has been identified as a negative predictor for overall survival in AML (99). Similarly, MSCs derived from Fanconi anaemia patients with AML secrete high levels of prostaglandins which resulted in Treg induction and subsequent inhibition of CD8+ CTLs (100). Lastly, PD-L1 expression on AML cells have been recently demonstrated to promote the conversion and expansion of PD-1+ Tregs from conventional CD4+ T cells (95).
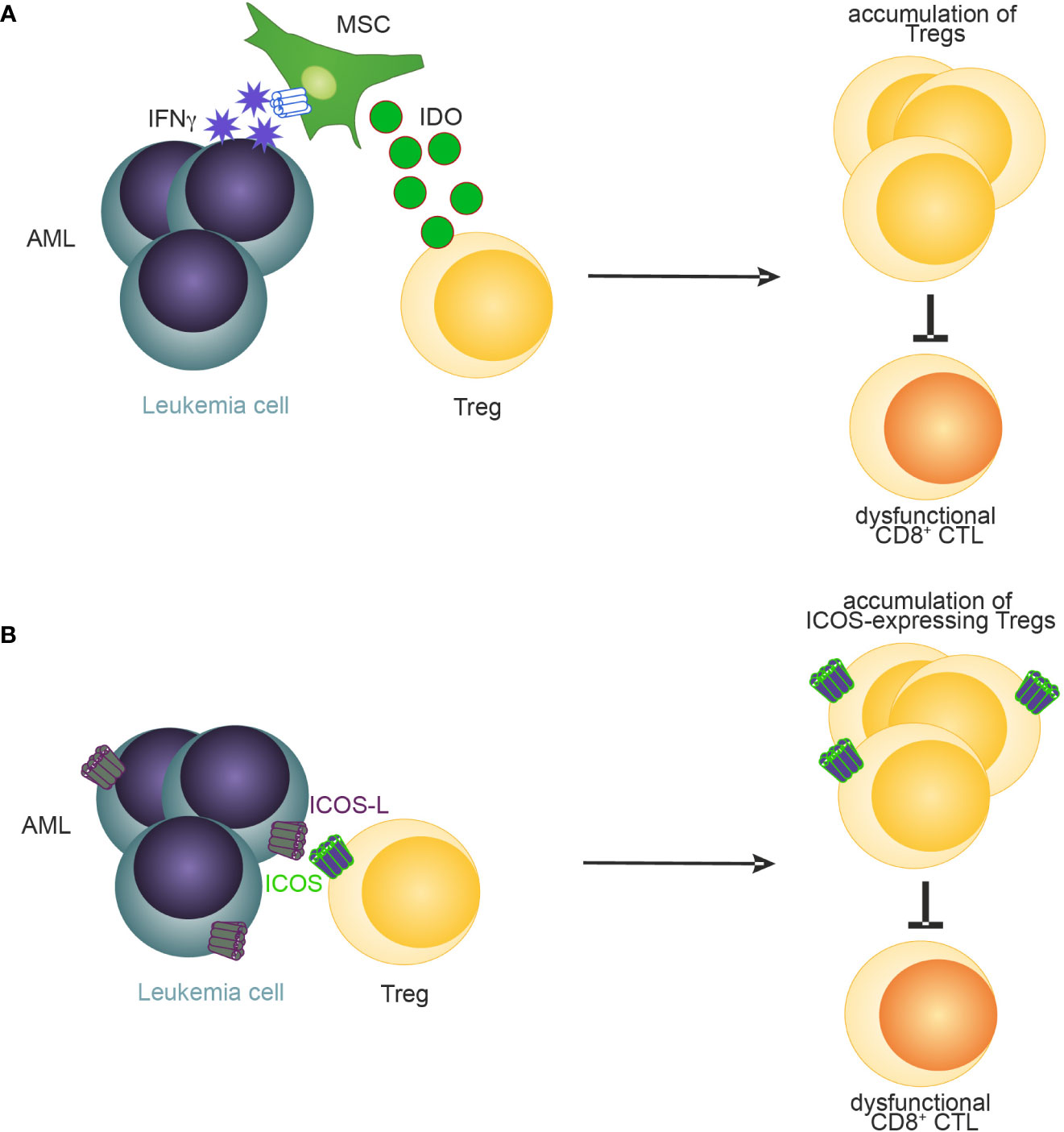
Figure 3 A schematic illustration of reported mechanisms leading to accumulation of Tregs in AML. (A) MSC-derived IDO (94) and (B) ICOS-ICOSL signaling (97) promotes the accumulation of Tregs in AML.
However, the experimental and clinical evidence that directly links Tregs with T-cell anergy/exhaustion in AML is still limited and mostly restricted to pre-clinical mouse models. For example, Tregs have been shown to inhibit the function of adoptively transferred AML-specific CTLs. Depletion of Tregs during adoptive transfer of CTLs improved survival of AML mice (31). Similarly, Treg depletion prior to leukemia induction has been shown to trigger an endogenous anti-leukemic CTL response resulting in prolonged survival of MLL-AF9 AML mice (32). Unpublished data from our group and a recent publication by Hasegewa and co-workers support the notion that Tregs in MLL-AF9 mice limit anti-leukemic immunity only in a situation with minimal leukemia load but not in an advanced stage of the disease (33). Similar to human AML (37), BM Tregs in MLL-AF9 mice have shown to express elevated levels of CXCR4 on the cell surface. Blockade of the CCL3/CCR1-CCR5 and CXCL-12/CXCR4 axis prevented the accumulation of Tregs in BM of MLL-AF9 AML mice (32). These data indicate that Tregs create an immunosuppressive microenvironment in the BM thereby reducing CTL function and immunosurveillance of AML cells. In addition, in vitro experiments revealed that AML cells themselves secret soluble factors that restrain T cell and NK cell function (101). Of note, Hasegawa et al. using the same AML model and RAG-knockout mice demonstrated that CTLs indeed have the capacity to eradicate leukemia at an early stage of the disease (low leukemia load), whereas antigen-specific CTLs are exhausted in animals with advanced leukemia (33).
However, Tregs in AML may not only hamper anti-leukemic T cell immunity but may also directly affect the survival and function of AML blasts LSCs. It was recently demonstrated that Treg-derived IL-10 promotes stemness of murine and human AML LSCs through activation of PI3K/AKT signaling pathway (96). Similarly, the cytokine IL-35 produced by Tregs promotes the proliferation of AML blasts (102).
Even though leukemia-antigen specificity of CD4+ T cells have been documented in several independent studies (7, 56), whether AML antigen–specific Tregs are part of this CD4+ T cell population is still unclear. In general, the evidence for functional tumor antigen–specific Tregs in cancer is very weak due to the lack of adequate MHC class II tetramers, and antigen-specific Tregs have only been documented in a few solid tumors and in B acute lymphoblastic leukemia (51, 103). Nevertheless, recent findings of a pre-clinical study suggests that antigen stimulation may play an important role in the activation and accumulation of Tregs in CML BM (30).
Overall, these studies imply that LSCs reside in an immune-privileged niche in the BM which harbors multiple immune-regulatory mechanisms. It may be assumed that the accumulation of Tregs in the BM represents one immune-regulatory mechanism in leukemia which limits immunosurveillance of LSCs and thereby contributes to disease progression and LSC maintenance. However, why Tregs accumulate in the BM, how this can be controlled, whether they are leukemia antigen-specific and whether selective targeting of Tregs in myeloid neoplasm (e.g. via an agonistic TNFRSF4 antibody) can induce immune control in humans is still unclear.
The elimination of cancer/leukemia cells depends on tumor antigen expression on MHC class I and II. In AML, mutations and immune escape mechanisms may result in the downregulation of MHC expression on leukemia cells. However, reduced, or lost MHC expression has been primarily observed on AML cells after allogeneic HSCT. For example, in 17 out of 34 patients, AML cells displayed a reduced expression of MHC class II at relapse after allogeneic HSCT compared to diagnosis. The downregulation of MHC was triggered by epigenetic mechanisms and could be restored by administration of IFN-y (104). Similarly, another study profiling immune signatures in AML cells after allogeneic HSCT identified the downregulation of MHC class II on a transcriptional level as a major immune escape mechanism (105). Furthermore, impaired processing and loading of leukemia-associated antigen on MHC class II has been described as potential immuno-editing process in AML.
Targeting signaling cascades that promote Treg-mediated immunosuppression in myeloid leukemia or Treg-specific surface antigens may be a viable approach to block Treg function or deplete Tregs and induce anti-leukemic immunity.
The anti-CTLA-4 mAb ipilimumab has been approved for the treatment of metastatic melanoma. However, despite the increased expression of CTLA-4 on Tregs in the tumor microenvironment (TME) of cancer patients and pre-clinical evidence showing the capacity of ipilimumab to deplete Tregs via ADCC-dependent mechanisms, ipilimumab is thought to induce infiltration of intratumoral CD4+ and CD8+ effector T cells without depleting Tregs in the TME in humans (106). In addition, the specificity of CTLA-4 as a Treg restricted target is widely debated because CTLA-4 is widely expressed on activated CD4+ T helper and CD8+ effector T cells and monocyte-derived DCs (72). To ensure a controlled activity of the antibody in the TME and to decouple antitumor efficacy from immunotherapy-related toxicity, a CTLA-4 dual variable domain immunoglobulin (anti-CTLA-4 DVD) was developed where the inner anti-CTLA-4 binding domain is shielded by an outer tumor-targeting domain (106). In the TME, the presence of activated membrane type-serine protease 1 results in cleavage of the outer domain and the liberation of the CTLA-4 binding site. To enhance target specificity, currently bi-specific antibodies targeting CTLA-4 together with other surface receptors with increased expression on Tregs are designed and evaluated. OX-40 is highly expressed on Tregs in AML (60) and CML (30) and is currently studied as a treatment for patients with refractory/relapsed AML as monotherapy or in combination with azacitidine and avelumab (NCT03390296). A human CTLA-4 x OX-40 bispecific antibody (ATOR-1015) which demonstrated efficacy in Treg depletion assays in vitro and syngeneic mouse models in vivo (106) is currently under evaluation in a first-in-human phase I clinical trial (NCT03782467). Similarly, a humanized CTLA-4 x GITR bispecific antibody (ATOR-1144) was shown to deplete Tregs in vitro through ADCC-dependent mechanisms (107).
Clinical trials assessing the potential of targeting CTLA-4 in AML are limited. In a phase 1b study, ipilimumab was tested in 522 patients with hematologic malignancies relapsing after allogeneic HSCT. The complete response rate was 23%. In addition, immune-related adverse events and graft-versus-host disease were reported (108). Ipilimumab is currently evaluated in several clinical phase I trials as a treatment for relapsed/refractory AML patients (NCT03912064, NCT03600155, NCT01822509) and as maintenance therapy for AML and MDS patients alone or in combination the anti-PD-1 antibody Nivolumab and/or azacitidine after allogeneic HSCT (NCT02846376) and for its potential to induce graft-versus-malignancy effects after allogeneic HSCT (NCT00060372).
CD25 is highly expressed on activated Tregs in the TME and only marginally on effector T cells (109). Therefore, several therapeutics targeting CD25 have been recently developed and were approved for the prevention of organ transplantation rejection (anti-CD25 mAb Basiliximab) and the treatment of cutaneous T-cell lymphoma (IL-2-diphtheria toxin fusion protein denileukin diftitox). However, clinical studies have demonstrated that denileukin diftitox is ineffective in eliminating Tregs in patients (107). An open-label, multicenter Phase I clinical trial is underway investigating the safety and tolerability of the anti-CD25 depleting antibody RO7296682 in patients with advanced solid tumors (NCT04158583). In addition, the potential of basiliximab conjugated with 1,4,7,10-tetraazacyclododecane tetraacetic acid (DOTA) and radiolabeled with Yttrium-90 given together with fludarabine, melphalan, and total marrow and lymphoid irradiation (TMLI) is evaluated for the treatment of patients with high-risk acute leukemia or myelodysplastic syndrome (NCT05139004). A phase I dose-escalation clinical trial assessed safety and efficacy of Camidanlumab tesirine, an antibody-drug conjugate targeting CD25 in 34 relapsed/refractory AML patients (110). Two patients achieved complete responses with incomplete hematologic recovery. However, the trial was terminated early for reasons other than safety and efficacy.
FOXP3 serves as a lineage-specific transcription factor of Tregs. As FOXP3 is primarily expressed in the nucleus, FOXP3 has never been considered as a viable target to deplete Tregs in cancer. However, the potential of targeting FOXP3 directly has recently gained considerable interest. A T cell receptor mimic antibody recognizing a FOXP3-derived epitope in the context of HLA-A*02:01 was shown to selectively recognize and eliminate FOXP3-expressing Tregs in a PBMC xenograft model (111). Similar results were obtained when the TCR mimic antibody was engineered in a bispecific T-cell engager. Another strategy currently followed to selectively deplete Tregs in cancer are antisense oligonucleotide against FOXP3. The clinical candidate antisense oligonucleotide AZD8701 demonstrated encouraging efficacy in reducing FOXP3 and Treg immunosuppressive function in primary human Tregs in vitro as well as in humanized mouse models in vivo (112). The safety and tolerability of AZD8701 is currently evaluated in a phase 1a/b clinical trial with or without Durvalumab in patients with advanced solid tumors (NCT04504669). However, strategies targeting FOXP3 bear the risk for the development of autoimmune disease as FOXP3 expression is most likely not limited to tumor tissues. Clinical trials targeting FOXP3 in myeloid malignancies are pending.
The BM is a hematopoietic organ that harbors in addition to classical niche cells also a variety of different immune cells such as B and T cells. CD4+ T cells including Tregs significantly shape the cytokine milieu in the BM microenvironment and thereby promote key functions of HSC such as quiescence, self-renewal proliferation and differentiation. These mechanisms evolved to guarantee a constant generation of blood cells from all lineages and to prevent damage and exhaustion of the stem cell pool. LSCs co-opt these evolutionary conserved mechanisms of the HSC niche and at the same time re-model the HSC niche to promote leukemogenesis and LSC function and to reduce the HSC reservoir in the BM. In leukemia, Tregs regulate the expansion of LSCs and leukemia progenitors and contribute to immune evasion of leukemia cells by cell-interactions or through secretion of cytokines. Therefore, targeting Tregs seems to be an attractive therapeutic option and warrants further investigation in humans. So far, however, hardly any clinical information on the potential of targeting Tregs in myeloid leukemia is available. Only a limited number of clinical trials currently addresses this possibility mainly by targeting immune-related surface receptors such as CD25, CTLA-4 and TNFRSF4 which are expressed on Tregs. However, whether these approaches will lead to the elimination of LSCs and thereby cure of leukemia is still unknown. Similarly, our knowledge on the emergence of severe adverse events such as the induction of inflammation or auto-immune disease upon Treg manipulation and their effect on the disease is limited and must be crucially considered prior to initiating and planning clinical trials. Consequently, a better understanding of immune cells including Tregs and their function in the BM microenvironment during disease and homeostasis may contribute to the generation of novel and safe immunotherapeutic strategies aiming at elimination of leukemia at the level of the LSC and the identification of factors and interactions which may translate into the possibility to generate cells of different hematopoietic lineages ex vivo for specific applications, respectively.
CR conceptualized and wrote the review article.
This work was supported by grants from the Swiss National Science Foundation (310030_179394) and the Stiftung für klinisch-experimentelle Tumorforschung. I thank Adrian F. Ochsenbein for helpful discussions and input during the preparation of the manuscript and Stefan Forster for generating the IHC images. I apologize to the authors whose work was not mentioned due to space limitations.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol (2019) 20(5):303–20. doi: 10.1038/s41580-019-0103-9
2. Campbell C, Rudensky A. Roles of regulatory T cells in tissue pathophysiology and metabolism. Cell Metab (2020) 31(1):18–25. doi: 10.1016/j.cmet.2019.09.010
3. Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol (2017) 19(3):214–23. doi: 10.1038/ncb3475
4. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508(7495):269–73. doi: 10.1038/nature13034
5. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell (2010) 7(3):380–90. doi: 10.1016/j.stem.2010.07.011
6. Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell (2011) 9:298–310. doi: 10.1016/j.stem.2011.09.010
7. Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ (2015) 22(2):187–98. doi: 10.1038/cdd.2014.89
8. Man Y, Yao X, Yang T, Wang Y. Hematopoietic stem cell niche during homeostasis, malignancy, and bone marrow transplantation. Front Cell Dev Biol (2021) 9:621214. doi: 10.3389/fcell.2021.621214
9. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell (2019) 177(7):1915–1932.e16. doi: 10.1016/j.cell.2019.04.040
10. Guarnerio J, Mendez LM, Asada N, Menon AV, Fung J, Berry K, et al. A non-cell-autonomous role for pml in the maintenance of leukemia from the niche. Nat Commun (2018) 9(1):66. doi: 10.1038/s41467-017-02427-x
11. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell (2013) 13(3):285–99. doi: 10.1016/j.stem.2013.06.009
12. Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood (2012) 119(2):540–50. doi: 10.1182/blood-2011-04-348151
13. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell (2018) 22(1):64–77.e6. doi: 10.1016/j.stem.2017.11.006
14. Mead AJ, Neo WH, Barkas N, Matsuoka S, Giustacchini A, Facchini R, et al. Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD–induced myeloproliferation. J Exp Med (2017) 214(7):2005–21. doi: 10.1084/jem.20161418
15. Passaro D, Garcia-Albornoz M, Diana G, Chakravarty P, Ariza-McNaughton L, Batsivari A, et al. Integrated OMICs unveil the bone-marrow microenvironment in human leukemia. Cell Rep (2021) 35(6):109119. doi: 10.1016/j.celrep.2021.109119
16. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell (2006) 124(2):407–21. doi: 10.1016/j.cell.2005.10.041
17. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell (2014) 15(3):365–75. doi: 10.1016/j.stem.2014.06.020
18. Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature (2014) 512(1):78–81. doi: 10.1038/nature13383
19. Zehentmeier S, Pereira JP. Cell circuits and niches controlling b cell development. Immunol Rev (2019) 289(1):142–57. doi: 10.1111/imr.12749
20. Sharara LI, Andersson A, Guy-Grand D, Fischer A, DiSanto JP. Deregulated TCR alpha beta T cell population provokes extramedullary hematopoiesis in mice deficient in the common gamma chain. Eur J Immunol (1997) 27(4):990–8. doi: 10.1002/eji.1830270428/asset/1830270428_ftp.pdf?v=1&t=hygdrznc&s=ba10ebe9016e181ebec8afa71273e494b1105362
21. Broxmeyer HE, Bruns HA, Zhang S, Cooper S, Hangoc G, McKenzie ANJ, et al. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin m. Immunity (2002) 16(6):815–25. doi: 10.1016/S1074-7613(02)00319-9
22. Hernández-Malmierca P, Vonficht D, Schnell A, Uckelmann HJ, Bollhagen A, Mahmoud MAA, et al. Antigen presentation safeguards the integrity of the hematopoietic stem cell pool. Cell Stem Cell (2022) 29(5):760–775.e10. doi: 10.1016/j.stem.2022.04.007
23. Riether C, Gschwend T, Huguenin AL, Schürch CM, Ochsenbein AF. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia (2015) 29:1781–5. doi: 10.1038/leu.2015.26
24. Radpour R, Riether C, Simillion C, Höpner S, Bruggmann R, Ochsenbein AF. CD8 + T cells expand stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. Leukemia (2019) 33:2379–92. doi: 10.1038/s41375-019-0441-9
25. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474(7350):216–9. doi: 10.1038/nature10160
26. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150highBone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell (2018) 22(3):445–453.e5. doi: 10.1016/j.stem.2018.01.017
27. Pierini A, Nishikii H, Baker J, Kimura T, Kwon H-S, Pan Y, et al. Foxp3+ regulatory T cells maintain the bone marrow microenvironment for b cell lymphopoiesis. Nat Commun (2017) 8:15068. doi: 10.1038/ncomms15068
28. Urbieta M, Barao I, Jones M, Jurecic R, Panoskaltsis-Mortari A, Blazar BR, et al. Hematopoietic progenitor cell regulation by CD4+CD25+ T cells. Blood (2010) 115(23):4934–43. doi: 10.1182/blood-2009-04-218826
29. Camacho V, Matkins VR, Patel SB, Lever JM, Yang Z, Ying L, et al. Bone marrow tregs mediate stromal cell function and support hematopoiesis via IL-10. JCI Insight (2020) 5(22):e135681. doi: 10.1172/jci.insight.135681
30. Hinterbrandner M, Rubino V, Stoll C, Forster S, Schnüriger N, Radpour R, et al. Tnfrsf4-expressing regulatory T cells promote immune escape of chronic myeloid leukemia stem cells. JCI Insight (2021) 6(23):e151797. doi: 10.1172/jci.insight.151797
31. Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood (2009) 114(18):3793–802. doi: 10.1182/blood-2009-03-208181
32. Wang R, Feng W, Wang H, Wang L, Yang X, Yang F, et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett (2020) 469:151–61. doi: 10.1016/j.canlet.2019.10.032
33. Hasegawa K, Tanaka S, Fujiki F, Morimoto S, Nakajima H, Tatsumi N, et al. An immunocompetent mouse model for MLL/AF9 leukemia reveals the potential of spontaneous cytotoxic T-cell response to an antigen expressed in leukemia cells. PloS One (2015) 10(12):e0144594. doi: 10.1371/journal.pone.0144594
34. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. XRhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell (2013) 153(5):1025–35. doi: 10.1016/j.cell.2013.04.040
35. Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med (2011) 208(2):261–71. doi: 10.1084/jem.20101688
36. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood (2010) 116(23):4815–28. doi: 10.1182/blood-2009-11-253534
37. Wan Y, Zhang C, Xu Y, Wang M, Rao Q, Xing H, et al. Hyperfunction of CD4 CD25 regulatory T cells in de novo acute myeloid leukemia. BMC Cancer (2020) 20(1):472. doi: 10.1186/s12885-020-06961-8
38. Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell (2011) 20(5):661–73. doi: 10.1016/j.ccr.2011.10.012
39. Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen E, Golan K, et al. Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol (2012) 13(11):1072–82. doi: 10.1038/ni.2408
40. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL-12/CXCR4 signals. Cancer Res (2004) 64(22):8451–5. doi: 10.1158/0008-5472.CAN-04-1987
41. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone marrow and the control of immunity. Cell Mol Immunol (2012) 9:11–9. doi: 10.1038/cmi.2011.47
42. Price PW, Cerny J. Characterization of CD4+ T cells in mouse bone marrow. i. increased activated/memory phenotype and altered TCR vbeta repertoire. Eur J Immunol (1999) 29(3):1051–6. doi: 10.1002/(SICI)1521-4141(199903)29:03<1051::AID-IMMU1051>3.0.CO;2-Y
43. Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol (2010) 184(8):4317–26. doi: 10.4049/jimmunol.0903781
44. Camacho V, McClearn V, Patel S, Welner RS. Regulation of normal and leukemic stem cells through cytokine signaling and the microenvironment. Int J Hematol (2017) 105:566–77. doi: 10.1007/s12185-017-2184-6
45. Kim S, Park K, Choi J, Jang E, Paik DJ, Seong RH, et al. Foxp3+regulatory T cells ensure b lymphopoiesis by inhibiting the granulopoietic activity of effector T cells in mouse bone marrow. Eur J Immunol (2015) 45(1):167–79. doi: 10.1002/eji.201444532
46. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong H-A, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell (2017) 169(6):1119–1129.e11. doi: 10.1016/j.cell.2017.05.002
47. Mathur AN, Zirak B, Boothby IC, Tan M, Cohen JN, Mauro TM, et al. Treg-cell control of a CXCL5-IL-17 inflammatory axis promotes hair-Follicle-Stem-Cell differentiation during skin-barrier repair. Immunity (2019) 50(3):655–667.e4. doi: 10.1016/j.immuni.2019.02.013
48. Liu Z, Hu X, Liang Y, Yu J, Li H, Shokhirev MN, et al. Glucocorticoid signaling and regulatory T cells cooperate to maintain the hair-follicle stem-cell niche. Nat Immunol (2022) 23(7):1086–97. doi: 10.1038/s41590-022-01244-9
49. Shao Q, Gu J, Zhou J, Wang Q, Li X, Deng Z, et al. Tissue tregs and maintenance of tissue homeostasis. Front Cell Dev Biol (2021) 9:717903. doi: 10.3389/fcell.2021.717903
50. Lussana F, Di Ianni M, Rambaldi A. Tregs: Hype or hope for allogeneic hematopoietic stem cell transplantation? Bone Marrow Transpl (2017) 52:1225–32. doi: 10.1038/bmt.2017.30
51. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol (2019) 4(31):eaao4310. doi: 10.1126/sciimmunol.aao4310
52. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest (2003) 112(11):1688–96. doi: 10.1172/JCI17702
53. Joffre O, Gorsse N, Romagnoli P, Hudrisier D, Van Meerwijk JPM. Induction of antigen-specific tolerance to bone marrow allografts with CD4+CD25+ T lymphocytes. Blood (2004) 103(11):4216–21. doi: 10.1182/blood-2004-01-0005
54. Greinix HT, Ladiges WC, Graham TC, Maslan S, Raff RF, Sandmaier BM, et al. Late failure of autologous marrow grafts in lethally irradiated dogs given anti-class II monoclonal antibody. Blood (1991) 78(8):2131–8. doi: 10.1182/blood.V78.8.2131.bloodjournal7882131
55. Huss R, Beckham C, Storb R, Deeg H. Major histocompatibility complex class II expression is required for posttransplant immunological but not hemopoietic reconstitution in mice. Transplantation (1994) 58(12):1366–71.
56. Hsieh Y-C, Kirschner K, Copland M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia (2021) 35(5):1229–42. doi: 10.1038/s41375-021-01238-w
57. Greiner J, Goetz M, Schuler PJ, Bulach C, Hofmann S, Schrezenmeier H, et al. Enhanced stimulation of antigen-specific immune responses against nucleophosmin 1 mutated acute myeloid leukaemia by an anti-programmed death 1 antibody. Br J Haematol (2022) 198(5):866–74. doi: 10.1111/bjh.18326
58. Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight (2018) 3(21):323–43. doi: 10.1172/jci.insight.120974
59. Hughes A, Clarson J, Tang C, Vidovic L, White DL, Hughes TP, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood (2017) 129(9):1166–76. doi: 10.1182/blood-2016-10-745992
60. Brück O, Blom S, Dufva O, Turkki R, Chheda H, Ribeiro A, et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia (2018) 32(7):1643–56. doi: 10.1038/s41375-018-0175-0
61. Harrington P, Dillon R, Radia D, McLornan D, Woodley C, Asirvatham S, et al. Chronic myeloid leukaemia patients at diagnosis and resistant to tyrosine kinase inhibitor therapy display exhausted T-cell phenotype. Br J Haematol. (2022). doi: 10.1111/bjh.18302
62. Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, et al. signatures of CD8 + T cell dysfunction in AML patients and their reversibility with response to chemotherapy. Ref Inf JCI Insight (2018) 3(21):e120974. doi: 10.1172/jci.insight.120974:e120974
63. Lamble AJ, Kosaka Y, Laderas T, Maffit A, Kaempf A, Brady LK, et al. Reversible suppression of T cell function in the bone marrow microenvironment of acute myeloid leukemia. Proc Natl Acad Sci U.S.A. (2020) 117(25):14331–41. doi: 10.1073/pnas.1916206117
64. Radpour R, Stucki M, Riether C, Ochsenbein AF. Epigenetic silencing of immune-checkpoint receptors in bone marrow- infiltrating T cells in acute myeloid leukemia. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.663406
65. Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat Commun (2019) 10(1):1065. doi: 10.1038/s41467-019-08871-1
66. Bilich T, Nelde A, Kowalewski DJ, Peper J, Schemionek M, Kanz L, et al. Mapping the HLA ligandome landscape of chronic myeloid leukemia identifies novel CD8+ and CD4+ T cell-epitopes for immunotherapeutic approaches. Blood (2016) 128(22):4232. doi: 10.1182/blood.V128.22.4232.4232
67. Berlin C, Kowalewski DJ, Schuster H, Mirza N, Walz S, Handel M, et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia (2015) 29:647–59. doi: 10.1038/leu.2014.233
68. Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: A multicentre observational trial. Lancet (2005) 365(9460):657–62. doi: 10.1016/S0140-6736(05)17945-8
69. Bocchia M, Defina M, Aprile L, Ippoliti M, Crupi R, Rondoni M, et al. Complete molecular response in CML after p210 BCR-ABL1-derived peptide vaccination. Nat Rev Clin Oncol (2010) 7:600–3. doi: 10.1038/nrclinonc.2010.141
70. Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, et al. Pan-cancer adaptive immune resistance as defined by the tumor inflammation signature (TIS): results from the cancer genome atlas (TCGA). J Immunother Cancer (2018) 6(1):63. doi: 10.1186/s40425-018-0367-1
71. Davidson-Moncada J, Viboch E, Church S, Warren S, Rutella S. Dissecting the immune landscape of acute myeloid leukemia. Biomed (2018) 6(4):110. doi: 10.3390/biomedicines6040110
72. Daver N. Immune checkpoint inhibitors in acute myeloid leukemia. Best Pract Res Clin Haematol (2021) 34(1):101247. doi: 10.1016/j.beha.2021.101247
73. Khaldoyanidi S, Nagorsen D, Stein A, Ossenkoppele G, Subklewe M. Immune biology of acute myeloid leukemia: Implications for immunotherapy. J Clin Oncol (2021) 39(5):419–32. doi: 10.1200/JCO.20.00475
74. Daver N, Alotaibi AS, Bücklein V, Subklewe M. T-Cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leuk (2021) 35(7):1843–63. doi: 10.1038/s41375-021-01253-x
75. Krönig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol (2014) 92(3):195–203. doi: 10.1111/ejh.12228
76. Isidori A, Cerchione C, Daver N, DiNardo C, Garcia-Manero G, Konopleva M, et al. Immunotherapy in acute myeloid leukemia: Where we stand. Front Oncol (2021) 11:1558. doi: 10.3389/fonc.2021.656218
77. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4+CD25+CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer (2011) 129(6):1373–81. doi: 10.1002/ijc.25791
78. Nadal E, Garin M, Kaeda J, Apperley J, Lechler R, Dazzi F. Increased frequencies of CD4(+)CD25(high) t(regs) correlate with disease relapse after allogeneic stem cell transplantation for chronic myeloid leukemia. Leukemia (2007) 21(3):472–9. doi: 10.1038/sj.leu.2404522
79. Rojas JM, Wang L, Owen S, Knight K, Watmough SJ, Clark RE. Naturally occurring CD4+ CD25+ FOXP3+ T-regulatory cells are increased in chronic myeloid leukemia patients not in complete cytogenetic remission and can be immunosuppressive. Exp Hematol (2010) 38(12):1209–18. doi: 10.1016/j.exphem.2010.09.004
80. Tanaka A, Nishikawa H, Noguchi S, Sugiyama D, Morikawa H, Takeuchi Y, et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J Exp Med (2020) 217(2):e20191009. doi: 10.1084/jem.20191009
81. Najima Y, Yoshida C, Iriyama N, Fujisawa S, Wakita H, Chiba S, et al. Regulatory T cell inhibition by dasatinib is associated with natural killer cell differentiation and a favorable molecular response–the final results of the d-first study. Leuk Res (2018) 66:66–72. doi: 10.1016/j.leukres.2018.01.010
82. Irani YD, Hughes A, Clarson J, Kok CH, Shanmuganathan N, White DL, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br J Haematol (2020) 191(3):433–41. doi: 10.1111/bjh.16718
83. Okada M, Imagawa J, Tanaka H, Nakamae H, Hino M, Murai K, et al. Final 3-year results of the dasatinib discontinuation trial in patients with chronic myeloid leukemia who received dasatinib as a second-line treatment. Clin Lymphoma Myeloma Leuk (2018) 18(5):353–360.e1. doi: 10.1016/j.clml.2018.03.004
84. Fujioka Y, Sugiyama D, Matsumura I, Minami Y, Miura M, Atsuta Y, et al. Regulatory T cell as a biomarker of treatment-free remission in patients with chronic myeloid leukemia. Cancers (Basel) (2021) 13(23):e5904. doi: 10.3390/cancers13235904
85. Hekim C, Ilander M, Yan J, Michaud E, Smykla R, Vähä-Koskela M, et al. Dasatinib changes immune cell profiles concomitant with reduced tumor growth in several murine solid tumor models. Cancer Immunol Res (2017) 5(2):157–69. doi: 10.1158/2326-6066.CIR-16-0061-T
86. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of ido. Nat Med (2011) 17(9):1094–100. doi: 10.1038/nm.2438
87. Williams P, Basu S, Garcia-Manero G, Christopher, Hourigan S, Oetjen KA, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer (2019) 125:1470–81. doi: 10.1002/cncr.31896
88. van Galen P, Hovestadt V, Wadsworth MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell (2019) 176(6):1265–1281.e24. doi: 10.1016/j.cell.2019.01.031
89. Kumar Bansal A, Kumar Sharawat S, Gupta R, Vishnubhatla S, Dhawan D, Bakhshi S. Regulatory T cells in pediatric AML are associated with disease load and their serial assessment suggests role in leukemogenesis. Am J Blood Res (2020) 10:90–6.
90. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer (2011) 129(6):1373–81. doi: 10.1002/ijc.25791
91. Sander FE, Nilsson M, Rydström A, Aurelius J, Riise RE, Movitz C, et al. Role of regulatory T cells in acute myeloid leukemia patients undergoing relapse-preventive immunotherapy. Cancer Immunol Immunother (2017) 66(11):1473–84. doi: 10.1007/s00262-017-2040-9
92. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res (2009) 15(10):3325–32. doi: 10.1158/1078-0432.CCR-08-3010
93. Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of CD4+CD25highregulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol (2005) 75(6):468–76. doi: 10.1111/j.1600-0609.2005.00537.x
94. Corradi G, Bassani B, Simonetti G, Sangaletti S, Vadakekolathu J, Fontana MC, et al. Release of IFNγ by acute myeloid leukemia cells remodels bone marrow immune microenvironment by inducing regulatory T cells. Clin Cancer Res (2022) 28(14):3141–55. doi: 10.1158/1078-0432.CCR-21-3594
95. Dong Y, Han Y, Huang Y, Jiang S, Huang Z, Chen R, et al. PD-L1 is expressed and promotes the expansion of regulatory T cells in acute myeloid leukemia. Front Immunol (2020) 11:1710. doi: 10.3389/fimmu.2020.01710
96. Xu Y, Mou J, Wang Y, Zhou W, Rao Q, Xing H, et al. Regulatory T cells promote the stemness of leukemia stem cells through IL10 cytokine-related signaling pathway. Leukemia (2022) 36(2):403–15. doi: 10.1038/s41375-021-01375-2
97. Han Y, Dong Y, Yang Q, Xu W, Jiang S, Yu Z, et al. Acute myeloid leukemia cells express ICOS ligand to promote the expansion of regulatory T cells. Front Immunol (2018) 9:2227. doi: 10.3389/fimmu.2018.02227/full
98. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol (2017) 8:22. doi: 10.3389/fimmu.2017.01918
99. Ragaini S, Wagner S, Marconi G, Parisi S, Sartor C, Nanni J, et al. An IDO1 -related immune gene signature predicts overall survival in acute myeloid leukemia. Blood Adv (2022) 6(1):87–99. doi: 10.1182/bloodadvances.2021004878
100. Wu L, Amarachintha S, Xu J, Oley F, Du W. Mesenchymal COX2-PG secretome engages NR4A-WNT signalling axis in haematopoietic progenitors to suppress anti-leukaemia immunity. Br J Haematol (2018) 183(3):445–56. doi: 10.1111/bjh.15548
101. Orleans-Lindsay JK, Barber LD, Prentice HG, Lowdell MW. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function–implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol (2001) 126(3):403–11. doi: 10.1046/j.1365-2249.2001.01692.x
102. Tao Q, Pan Y, Wang Y, Wang H, Xiong S, Li Q, et al. Regulatory T cells-derived IL-35 promotes the growth of adult acute myeloid leukemia blasts. UICC Int J Cancer IJC (2015) 137:2384–93. doi: 10.1002/ijc.29563
103. Farrar Pauken A, Williams RT, Jenkins MK, Luke Manlove MS, Berquam-Vrieze KE. Cells by cross-reactive induced regulatory T adaptive immunity to leukemia is inhibited. J Immunol Ref (2022) 195:4028–37. doi: 10.4049/jimmunol.1501291
104. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med (2018) 379(24):2330–41. doi: 10.1056/NEJMoa1808777
105. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med (2019) 25(4):603–11. doi: 10.1038/s41591-019-0400-z
106. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
107. Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother (2005) 28(6):582–92. doi: 10.1097/01.cji.0000175468.19742.10
108. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med (2016) 375(2):143–53. doi: 10.1056/NEJMoa1601202
109. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity (2017) 46(4):577–86. doi: 10.1016/j.immuni.2017.03.013
110. Goldberg AD, Atallah E, Rizzieri D, Walter RB, Chung K-Y, Spira A, et al. Camidanlumab tesirine, an antibody-drug conjugate, in relapsed/refractory CD25-positive acute myeloid leukemia or acute lymphoblastic leukemia: A phase I study. Leuk Res (2020) 95:106385. doi: 10.1016/j.leukres.2020.106385
111. Dao T, Mun SS, Scott AC, Jarvis CA, Korontsvit T, Yang Z, et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology (2019) 8(7):e1570778. doi: 10.1080/2162402X.2019.1570778
Keywords: regulatory T cell (Treg), leukemia stem cell (LSC), immune escape, hematopoietic stem cell, hematopoietic stem cell niche
Citation: Riether C (2022) Regulation of hematopoietic and leukemia stem cells by regulatory T cells. Front. Immunol. 13:1049301. doi: 10.3389/fimmu.2022.1049301
Received: 20 September 2022; Accepted: 20 October 2022;
Published: 02 November 2022.
Edited by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyReviewed by:
Stéphane J. C. Mancini, INSERM UMR1236 Microenvironnement, Différenciation cellulaire, Immunologie et Cancer, FranceCopyright © 2022 Riether. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carsten Riether, Y2Fyc3Rlbi5yaWV0aGVyQGluc2VsLmNo
†ORCID: Carsten Riether, orcid.org/0000-0001-7512-513X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.