 Kai R. Plunkett1,2
Kai R. Plunkett1,2 Jesse D. Armitage1,2†
Jesse D. Armitage1,2† Andrisha-Jade Inderjeeth3†
Andrisha-Jade Inderjeeth3† Alison M. McDonnell2†
Alison M. McDonnell2† Jason Waithman1,2†
Jason Waithman1,2† Peter K. H. Lau4,5*†
Peter K. H. Lau4,5*†- 1School of Biomedical Sciences, University of Western Australia, Nedlands, WA, Australia
- 2Telethon Kids Institute, University of Western Australia, Nedlands, WA, Australia
- 3Department of Medical Oncology, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 4Melanoma Discovery Laboratory, Harry Perkins Institute of Medical Research, Nedlands, WA, Australia
- 5Department of Medical Oncology, Sir Charles Gairdner Hospital, Nedlands, WA, Australia
Tissue-resident memory T (TRM) cells have emerged as key players in the immune control of melanoma. These specialized cells are identified by expression of tissue retention markers such as CD69, CD103 and CD49a with downregulation of egress molecules such as Sphingosine-1-Phosphate Receptor-1 (S1PR1) and the lymphoid homing receptor, CD62L. TRM have been shown to be integral in controlling infections such as herpes simplex virus (HSV), lymphocytic choriomeningitis virus (LCMV) and influenza. More recently, robust pre-clinical models have also demonstrated TRM are able to maintain melanoma in a dormant state without progression to macroscopic disease reminiscent of their ability to control viral infections. The discovery of the role these cells play in anti-melanoma immunity has coincided with the advent of immune checkpoint inhibitor (ICI) therapy which has revolutionized the treatment of cancers. ICIs that target programmed death protein-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) have led to substantial improvements in outcomes for patients with metastatic melanoma and have been rapidly employed to reduce recurrences in the resected stage III setting. While ICIs mediate anti-tumor activity via CD8+ T cells, the specific subsets that facilitate this response is unclear. TRM invariably exhibit high expression of immune checkpoints such as PD-1, CTLA-4 and lymphocyte activating gene-3 (LAG-3) which strongly implicates this CD8+ T cell subset as a crucial mediator of ICI activity. In this review, we present pre-clinical and translational studies that highlight the critical role of TRM in both immune control of primary melanoma and as a key CD8+ T cell subset that mediates anti-tumor activity of ICIs for the treatment of melanoma.
Introduction
Immune checkpoint inhibitors that target the PD-1 axis form the backbone of systemic treatment for cutaneous malignancies, namely melanoma (1), cutaneous squamous cell carcinoma (cSCC) (2) and Merkel cell carcinoma (3). Original indications were approved in metastatic melanoma, leading to unprecedented long-term survival outcomes with 5-year overall survival (OS) rates exceeding 50% with combination ipilimumab-nivolumab (anti-CTLA-4 and anti-PD-1) (4). Given these substantial improvements in outcomes for patients with metastatic disease, ICIs were quickly investigated in the resected stage III melanoma setting where adjuvant anti-PD-1 with pembrolizumab was shown to reduce recurrence by approximately 40% compared to placebo (5). Recently, several studies display encouraging activity of ICI in the neoadjuvant preoperative setting, which promises to be a new treatment paradigm for the management of stage III melanoma (6–8).
Given the shift towards adjuvant and neoadjuvant immunotherapy for the treatment of melanoma, a critical understanding of the tumor microenvironment (TME) within the skin is required to inform new therapeutic combinations with anti-PD-1. Notably, a pre-requisite for anti-tumor responses to ICI include the presence of T cells co-located at the tumor margin (9). However, an important sub-class of T cells, tissue-resident memory CD8+ T (TRM) cells are major players in mediating anti-tumor responses to ICI (10, 11). TRM permanently reside in tissues and are unable to circulate due to the lack of lymphoid homing markers CCR7 and CD62L. Additionally, skin TRM are identified by the increased expression of the C-type lectin receptor CD69 and the αE subunit of the αEβ7 integrin CD103 (12–14). Critically, tumor-infiltrating TRM express immune checkpoint molecules including PD-1, CTLA-4, and LAG-3. TRM are also a rich source of type I interferons (IFN), IFN-γ and tumor necrosis factor alpha (TNF-α) which are essential for anti-tumor activity (11, 15).
TRM were originally described as having central roles in immune control of viral infections such as HSV and LCMV (13, 16, 17). Moreover there is a substantial body of evidence that indicates a similar critical role for TRM in both immune control of melanoma and mediating responses to ICIs (12). Seminal pre-clinical models show TRM are vital in controlling cutaneous melanoma by inducing a state of immune-mediated equilibrium which is reminiscent of HSV dormancy where the host immune system suppresses but does not completely eliminate the virus (12, 13). In humans, the presence and abundance of TRM is associated with improved prognosis in patients with melanoma, TRM isolated from human melanoma specimens display high PD-1 and LAG-3 expression corroborating pre-clinical models (10, 18). This finding has critical therapeutic importance given the recent successful development of combination anti-LAG-3 with anti-PD-1 in metastatic melanoma (19). Moreover, tumor-infiltrating TRM correlate with longer disease-free periods and OS in patients of cSCC (20, 21), breast (22), ovarian (23), endometrial (24), pancreatic ductal adenocarcinoma (25) and lung cancer (26, 27) which further exemplifies the importance of this T cell subset in cancer outcomes.
With neoadjuvant trials and the recent success of adjuvant treatment in melanoma, the stage is set for a new raft of immunotherapy studies to further improve patient outcomes. The past 10 years has produced a stunning paradigm shift in the treatment of skin malignancies but the precise immunological mechanisms that underpin ICI-driven anti-tumor responses remain unclear. This review will explore the basic biology of TRM and their role in mediating clinical responses to ICI in cutaneous malignancies.
Hallmarks of tissue-resident memory T cells
Phenotypic features
Over the past two decades our understanding of CD8 memory T cell subsets have evolved considerably. Stem memory T cells (TSCM, defined by CD45RA+CCR7+CD27+CD95+) (28) represent the least differentiated memory subset and display a multipotency potential to differentiate into two major memory populations central memory (TCM) and effector memory (TEM) cells (29, 30). TCM primarily reside in lymphoid organs and are reactivated following antigen stimulation, such as during secondary viral infections. Expression of the chemokine receptor CCR7 and the lymphoid specific L-selectin CD62L allow TCM to migrate from the lymph or blood vessels to enter secondary lymphoid organs (31). In contrast, TEM are memory cells that do not express CCR7 and CD62L, but display chemokine receptors, such as CXCR3 and adhesion molecules, such as intracellular adhesion molecule 1 (ICAM-1), that allow entry into inflamed tissues (32). TEM exert cytotoxic activities on target cells via their ability to release IFN-γ, granzymes and perforins. Moreover, terminally differentiated effector memory (TEMRA) represent a highly differentiated population of memory CD45RA+ T cells with low CD62L and CCR7 low expression alongside high cytolytic capability (30). Both TCM and TEM form the group of circulating memory T (TCIRC) cells as they circulate via the lymphatics and blood stream (33).
More recently, a third major population of CD8+ memory T cells, TRM have been identified. TRM permanently reside in tissue following infection or malignancy and do not re-enter circulation following establishment of a population in the site (12). Originally described in viral models, TRM arise from killer cell lectin-like receptor G1 (KLRG1) negative population of memory precursor cells, which lack the lymphoid homing receptor CD62L and the tissue egress receptor S1PR1 (34). S1PR1 and its ligand, sphinosine-1-phosphate (S1P) is a critical signaling pathway for the egress of effector CD8+ T cells from lymphoid organs to sites of infection (35). Until recently it was unclear which type of memory T cells are progenitors for TRM in the skin. However work by Matos et al. indicates TCM display higher skin tropism than TEM, and are therefore more efficient progenitors for skin TRM (36).
CD8+ TRM can be distinguished from their circulating counterparts largely through expression of the key surface molecules CD69, CD103 and CD49a combined with reduced expression of tissue egress and lymph node homing markers such as S1PR1, CCR7, CD62L (17, 34, 37). CD103 associates with integrin β7 (CD49d) to form the αEβ7 integrin complex which binds the cell adhesion molecule E-cadherin and anchors TRM within barrier tissues (38) (Figure 1). CD103 is most highly expressed by CD8+ TRM in mucosal sites such as the lung and skin where its expression is driven by TGF-β. CD69 is a transmembrane C-type lectin receptor that is constitutively expressed on TRM and contributes to the establishment of tissue residency via inhibition of S1PR1-mediated tissue egress. Loss of S1PR1 on CD8+ T cells leaves T cells unable to respond to S1P secreted by endothelial cells, maintaining their niche within tissues (38, 39) (Figure 2).
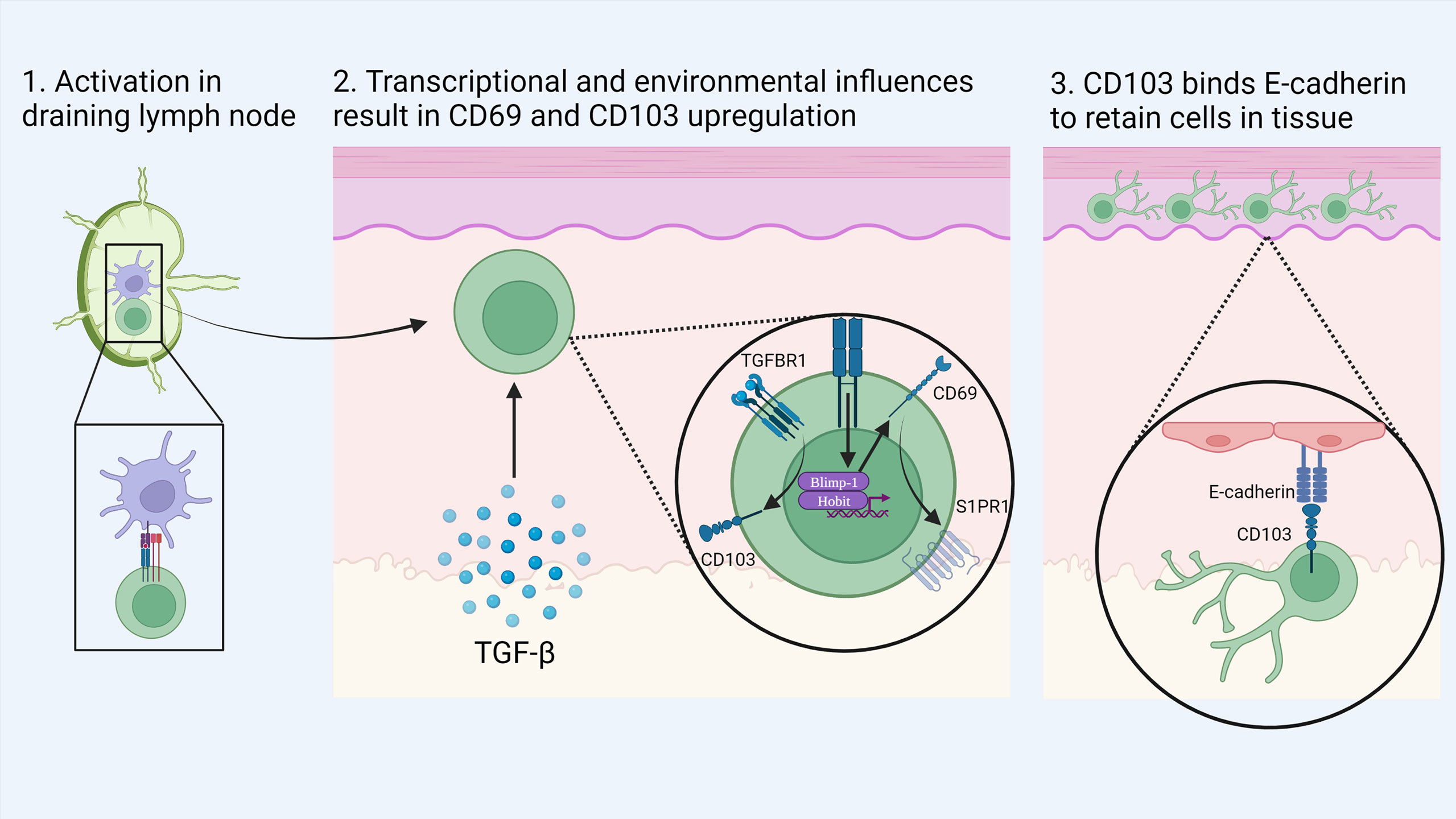
Figure 1 Development of skin TRM: Naïve CD8+ T cells are activated by dendritic cells in the local draining lymph node. Upon migration to the skin, local environmental factors such as TGF-β induce the transition to a TRM phenotype. Activation of the transcription factors Blimp-1 and Hobit drives the expression of CD69 which in turn causes the down regulation of S1PR1. In addition, TGF-β stimulates the upregulation of CD103 which binds to E-cadherin in the epidermis to retain cells in tissue. Created with Biorender.com.
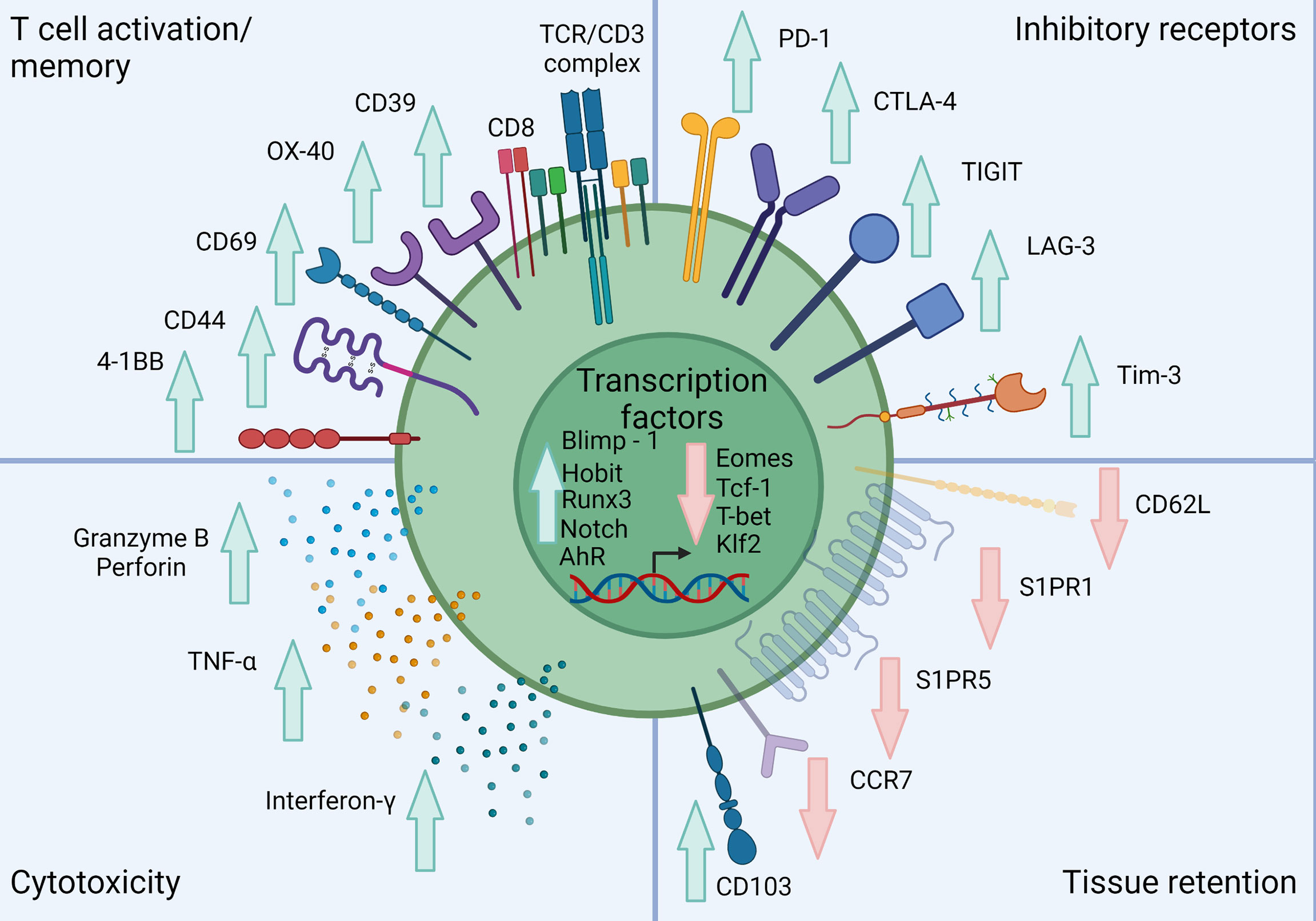
Figure 2 Classical phenotype of skin TRM. Skin TRM up regulate markers of memory such as CD44, CD69 and CD103 while down-regulating the lymphoid homing molecule CCR7 and egress receptors CD62L, S1PR1 and S1PR5. Skin TRM also express a range of inhibitory receptors including PD-1, CTLA-4 and LAG-3. These cells also exhibit a unique transcription factor profile exhibited by increased Blimp-1, Hobit, Runx, AhR and Notch expression with inhibition of Eomes, Tcf-1, T-bet and Klf2. They also show a conventional CD8+ T cell cytolytic profile including Granzyme B, Perforin, TNF-α and IFN-γ expression. Created with Biorender.com.
Several studies indicate that defining TRM with CD69 and CD103 expression is tissue dependent (40). TRM populations have been defined as CD69+ CD103- in different anatomical sites such as intestines (41), secondary lymphoid organs (42), liver (40), kidney and adipose tissue (43). Although TGF-β signals up-regulation of CD103 expression by skin TRM, Tgfbr2-/- mouse models have shown liver TRM can develop independent of TGF-β signaling (40). This indicates the unique immunological niche within each anatomical site plays a key role in the development of tissue-specific TRM.
CD49a is another crucial TRM surface molecule and constitutes the α-subunit of the α1β1 integrin receptor, also known as very late antigen-1 (VLA-1). Similar to CD103, CD49a functions to maintain TRM within tissues (44). In the skin, CD49a binds its cognate ligand collagen IV in the basement membrane which separates the dermis and epidermis to establish tissue residency (44). Both IL-12 and TGF-β can induce expression of CD49a but the former cytokine has been shown to inhibit CD103 expression in vitro (45). Expression of CD49a was found to delineate a functional subset of skin TRM with CD8+CD49a+ producing IFN-γ whereas CD8+CD49- TRM produced IL-17 in psoriatic skin lesions (46). Furthermore CD49a also promotes dendritic extensions by skin TRM which may enhance the cells antigen surveillance capability (45). CD49a expression on TRM is correlated with improved outcomes in patients with melanoma (18).
While CD69, CD103 and CD49a have been used to discriminate TRM from other memory subsets, CD39 has recently been described as a distinguishing marker of tumor specific CD8 TILs (26, 47). Using mass cytometry and T cell receptor (TCR) sequencing of colorectal and lung cancer specimens, CD39 expression was enriched in CD8 tumor infiltrating lymphocytes (TILs) that possessed cancer antigen specificity (47). In contrast CD39- TIL exhibited T cell receptors to non-cancer epitopes including viral antigens such as cytomegalovirus or influenza virus. In a study by Duhen et al, all CD39+ cells also co-expressed both CD69 and CD103, representing a subset of tumor-reactive TRM (26). Gene expression data showed CD39+ TILs exhibited much higher expression of immune checkpoints such as PD-1 and CTLA-4 compared to CD39- TILs (26). Notably the CD39-CD73 axis has also been implicated with immunosuppressive roles (48, 49). CD39 metabolizes extracellular ATP and ADP to AMP, while CD73 converts AMP into adenosine (50). Extracellular adenosine signals through its cognate receptors, A1R, A2AR, A2BR and A3R and causes immunosuppression by proliferation of TREG within the TME, increasing the release of anti-inflammatory cytokines such as IL-10 and TGF-β (51). Pre-clinical models have shown adenosine receptor antagonists can augment anti-tumor activity in combination with anti-PD-1 in pre-clinical models of melanoma (48), breast (48) and colon cancer (52). Although clinical trials of adenosine receptor antagonists are yet to show meaningful activity, CD39 is a marker of melanoma specific TRM that will be explored later in this review (53, 54).
Transcription factors
TRM exhibit a distinct transcriptional program classically defined by the expression of Blimp-1 (PRDM1), Hobit (ZNF683) (55, 56) and RUNX transcription factor family 3 (RUNX3) (Figure 2) (57). This core program enables the maintenance of CD69 expression, while interfering with the expression of egress receptors such as S1PR1 and CCR7 (39). In addition, TRM also express low, but residual levels of T-bet (TBX21), and suppression of Eomesodermin (EOMES) (terminal differentiation), Tcf1 (TCF7) and Krupel-like factor-2 (KLF2) (naïve/central memory phenotype) (58). T-bet and Eomes are T-box transcription factors which have key roles in driving T cell differentiation (59). The suppression of T-bet and Eomes is critical for TRM development as it drives TGF-β signaling (38). In TRM, T-bet is maintained at residual levels to maintain IL-15 signaling to promote long-term survival (38). Eomes expression increases over time as CD8+ T cells undergo differentiation from naïve CD8+ T cells to TEM and maintain homeostasis via IL-15 signaling (60). In TRM however, Eomes expression is completely suppressed, as it is the KLRG1-CD127+ precursors that give rise to TRM (61). This was demonstrated experimentally in a murine model of HSV where Mackay et al. demonstrated that after 4 weeks, skin TRM completely suppressed Eomes expression, while maintaining a residual level of TBX21 (38).
The transcriptional profile promoting TRM differentiation and function is of translational interest. As mentioned previously, TGF-β is critical in the expression of CD103, but also plays a key role in the induction of Notch signaling in TRM (62). Indeed, Notch (58), as well as nuclear receptor 4A1 (NR4A1) (63), are critical for the expression of CD69 and CD103 (64). The up-regulation of RUNX3 also drives CD103 expression as its expression induces a profile of TGF-β response genes through remodeling of chromatin (57). Notch also induces transcription of IFN-γ mRNA, thereby influencing the cytotoxic capacity of TRM (58). Transcriptional analysis of TRM localized within the skin has also identified aryl hydrocarbon receptor (AhR) as a key transcriptional regulator promoting skin residency (65). In an AhR knockout model, no difference in CD8+ T cell infiltration was observed, but their survival decreased dramatically, indicating the role of AhR in long-term persistence (65).
Immune checkpoint expression
TRM often display a phenotype consistent with that of exhausted T cells with the expression of a range of immune checkpoints such as PD-1, LAG-3, T cell immunoreceptor with Ig and ITIM domains (TIGIT), OX-40, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) and LAG-3 (10, 27, 66–68) (Figure 2). Exhausted T cells arise from chronic antigen exposure during viral infections and cancer progression (69). When T cells transition to an exhausted phenotype, they lose their cytotoxic and proliferative functions with a decrease in production of IL-2, IFN-γ and TNF alongside an inability to degranulate against target cells (69). Gene expression data shows higher expression of immune checkpoints including PDCD1, CTLA4, LAG3 and TIGIT by TRM compared to TCM or TEM (66). PD-1 was among the first key markers that identified exhausted T cells in patients with chronic infections such as human immunodeficiency virus (70) and viral hepatitis (71). These seminal observations in viral immunology now form the basis of a large understanding of cancer immunity (72).
Cancer cells that express PD-L1 that contact with CD8 bearing PD-1 leads to a signaling cascade that results in reduction of TCR signaling, proliferation and cytotoxicity that prevents anti-tumor activity (Figure 3) (73). Upon binding of PD-L1 to PD-1, phosphorylation of immune receptor tyrosine–based switch motif (ITSM) occurs, which binds to Src homology region 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2) (73). This directly inhibits ZAP70 thereby inhibiting TCR signaling whilst simultaneously downregulating both RAS and PI3K pathways that are required for downstream activation of critical transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nuclear factor of activated T cells (NFAT). Through these complex mechanisms, tumor cells that bear surface PD-L1 effectively halt cytotoxic immune responses by CD8 cells and therefore facilitates “immune escape.” Multiple studies show TRM expression of PD-1 (10, 11, 74, 75) which indicates the cytotoxic potential of this cell type can be unleashed with anti-PD-1 antibodies such as nivolumab or pembrolizumab.
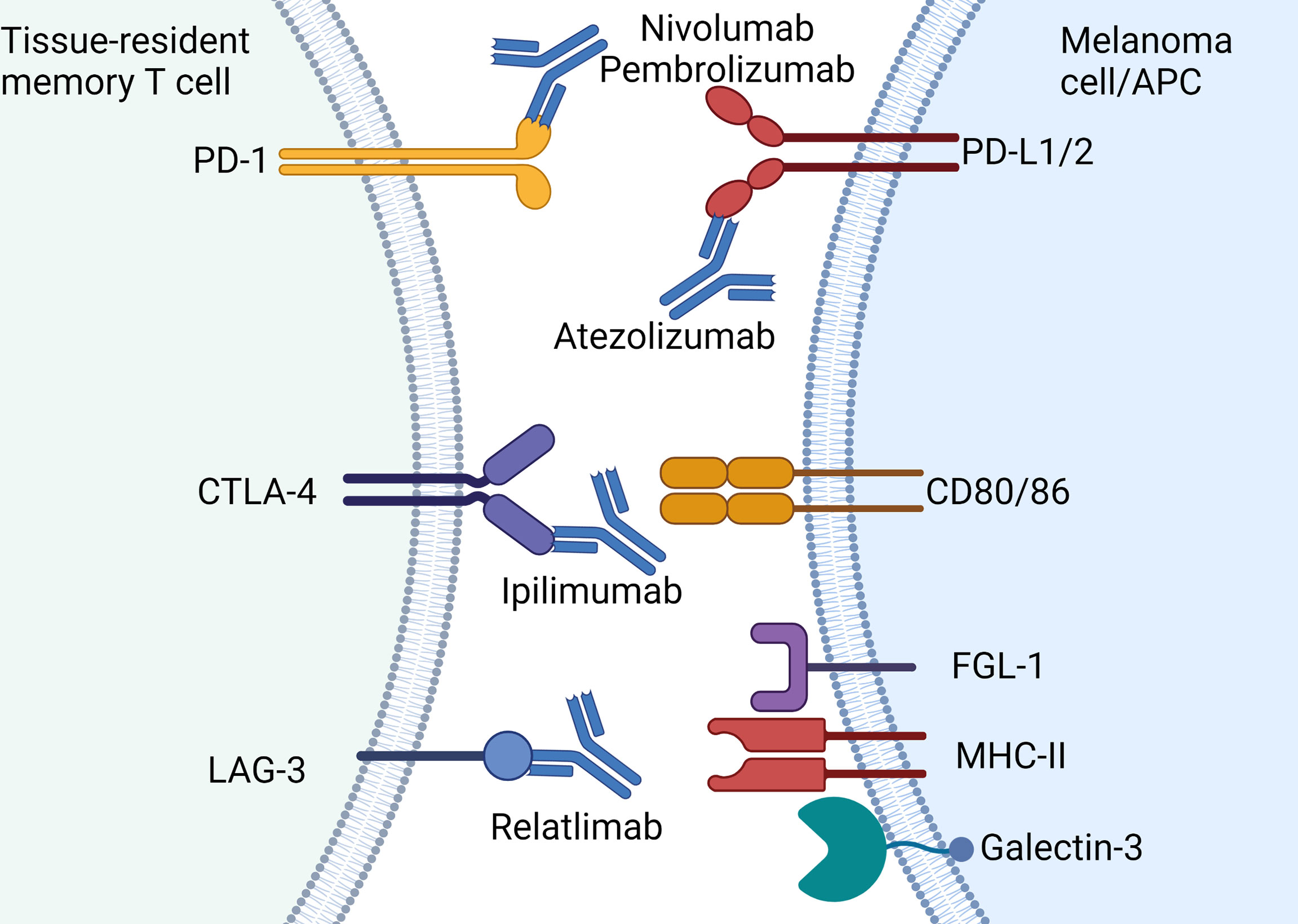
Figure 3 Current approved immune checkpoint inhibitors for the treatment of melanoma: Immune checkpoint molecules expressed by TRM and the approved antibodies including PD-1 (nivolumab, pembrolizumab), PD-L1 (atezolizumab), CTLA-4 (ipilimumab) and LAG-3 (relatlimab). Note atezolizumab is only approved for use in combination with vemurafenib-cobimetinib. Created with Biorender.com.
TRM also express CTLA-4 which was the first immune checkpoint identified that was shown to inhibit anti-cancer responses by CD8 cells (76). Primarily CTLA-4 interactions occur at the time of antigen presentation between dendritic cells and T cells within the draining lymph node. T cell activation is dependent on co-stimulatory molecules namely CD80 and CD86 that bind to CD28. However CTLA-4 competes with CD28 thereby inhibiting costimulatory signaling which subsequently halts T cell activation and proliferation (77, 78). Under chronic TCR stimulation by either infection or malignancy, CTLA-4 expression becomes constitutive, dampening the immune response by induction of T cell anergy (79). TRM from breast cancer samples displayed increased expression of CTLA4 compared to TEM populations (22). However, the role this key checkpoint plays with respect to TRM function remains to be defined.
LAG-3 is highly expressed on skin TRM and also acts as a negative regulator of T cell function (10). This type I transmembrane protein shares approximately 20% amino acid homology with CD4 but its specific mechanism of action and ligands remains unclear (80). LAG-3 migrates to the immunological synapse at the TCR-MHC-II junction and may competitively inhibit TCRs on CD8+ T cells with cross-presenting antigen presenting cells (81) (Figure 3). In addition to MHC class II, LAG-3 also binds with the lectin receptor galectin-3 and fibrinogen-like protein 1 (FGL1) (Figure 3) (82, 83). Upon binding of LAG-3 with galectin-3, IFN-γ production by CD8+ T cells is inhibited (82). While the exact mechanisms of LAG-3 remain controversial, this immune checkpoint has gained prominence with the development of relatlimab (anti-LAG-3 antibody) which has recently been shown to improve anti-tumor activity in combination with nivolumab (19).
Given TRM widely express CTLA-4, PD-1 and LAG-3, which are inhibited by immune checkpoint inhibitors such as ipilimumab, nivolumab, pembrolizumab and relatlimab provides a strong biological rationale this CD8 subset is critical in facilitating anti-tumor responses in melanoma.
TRM are central in melanoma surveillance
Melanoma has been long regarded as an immunological cancer and the presence of tumor infiltrating lymphocytes is recognized as an important prognostic factor for survival (84). Clarke and colleagues described the first TIL classification system with absent, non-brisk and brisk categories (85). The brisk pattern is defined by the presence of lymphocytes throughout the entire tumor whereas non-brisk is typified by scattered, focal groups of lymphocytes which has been shown to be a prognostic factor of improved overall survival compared to absent TILs (86). Further refinement of a classification based upon TIL density and distribution by Azimi and colleagues showed survival in Stage III melanoma patients could be predicted (87). Efforts to refine TIL subsets using CD3, CD8 IHC has also been intensively investigated with greater CD3 and CD8 TIL being a strong predictor of survival (88).
Recent preclinical and translational studies have shown TRM are the key TIL subset that facilitates immune control of melanoma. Using the B16 murine melanoma model, TRM were shown to be essential to maintain melanoma in a state known as ‘immune-mediated equilibrium’ or melanoma dormancy (12). This phenomenon describes the process whereby melanoma cells are maintained as occult lesions in situ rather than continued growth and progression (89). Engraftment of pre-activated T cells were used to over-represent TRM at the site, resulting in more efficient control from melanoma challenge. Using two-photon microscopy, TRM were observed surveilling the skin in regions where melanoma cells were present. Depletion of TCIRC demonstrated that TRM were responsible for the protection from melanoma challenge, in a process driven by TNF-α. This study which substantiated TRM are critical for maintaining immune-mediated equilibrium, may in part account for clinical observations of re-emerging melanoma after years of dormancy. Several human case studies which support the role of TRM in immune control of melanoma have been observed. One case study showed occurrence of a secondary melanoma in breast tissue following a kidney transplant without a primary tumor on the recipient (90) as well as the recurrence of melanoma following 30 to 40 year disease-free periods (91–93), indicating long periods of immune control followed by escape. These data suggest that TRM may play a role in human disease by maintaining occult lesions in the skin, sometimes for decades, before progressing to clinical disease.
However the precise mechanisms of immune escape with progression from “occult to overt” disease are currently unclear. Potentially other cells and factors such as T regulatory cells, myeloid derived suppressor cells and tumor associated macrophages (TAMS) could inhibit TRM function and facilitate immune escape. The presence of TAM identified using CD68 or CD163 have an inverse relationship with survival in primary melanoma (94, 95) and may counter balance TRM function. Moreover, as TRM express PD-1, loss of antigen presentation machinery by melanoma is a well described mechanism of resistance to ICI and therefore may also contribute to immune escape (96, 97). In one study, downregulation of MHC class I, rather than complete loss was associated with de-differentiation of melanoma with increased expression of the receptor tyrosine kinase AXL and downregulation of micropthalmia-associated transcription factor (MITF) (98). Given the breadth of resistance mechanisms of ICI to melanoma including the innate PD-1 Resistance Signature (IPRES) (99, 100), T cell exclusion programs such as beta catenin (101), low tumor mutational burden (102), alternative immune checkpoint expression (103) amongst a host of others indicates the means of immune escape are also likely to be heterogenous.
Reinforcing the importance of TRM facilitating immune control of melanoma is the central role these cells play in mediating vitiligo, an auto-immune condition leading to depigmentation of the skin. Patients with vitiligo bear melanocyte-specific CD8+ T cells that are involved with the destruction of melanocytes (104). Two separate groups using different B16 mouse models of vitiligo showed CD8+ TRM identified by CD69+ CD103+ co-expression were critical in mediating vitiligo (105, 106). Moreover, both groups showed TRM produced high levels of IFN-γ implicating their role in melanocyte destruction. Further human studies show CD49a+ CD103+ CD8+ (46) and CD69+ CD103± CD8+ TRM (105) were also central in mediating depigmentation with the latter study showing CXCR3 expression on melanocyte-specific TRM. The relationship of TRM and vitiligo is of clinical importance as skin depigmentation is associated with improved clinical outcomes to anti-PD-1 treatment in metastatic melanoma (107, 108).
In addition, the immunological mechanisms that underpin vitiligo with responses to ICI has been recently demonstrated in an elegant study using patient matched melanoma tumor samples, blood and vitiligo affected skin that were subjected to TCR and single cell sequencing (109). Paired TCR sequencing of TRM from the skin and TEM from blood showed similar T cell clones that highly expressed IFN-γ which were maintained years after treatment with immunotherapy thereby indicating durable immunological memory of these subsets. Furthermore, three distinct populations of TRM were identified using single cell sequencing: TRM-FOS, TRM-TOX and TRM-INFG each displaying different functional profiles. TRM-FOS consisted of a profile relating to TCR signaling but was not shown to correlate with improved patient outcomes. TRM-TOX displayed a more prototypical profile with high expression of immune checkpoints (LAG-3, PD-1 and CTLA-4), cytotoxicity (perforin and granzyme B) and key TRM markers including CD103. The third cluster, TRM-IFNG expressed a strong cytokine profile with increased expression of IFN-γ, and in particular, TNF-α. Unlike TRM-FOS the TRM-TOX and TRM-IFNG populations were associated with improved survival. This data demonstrates that TRM may be reactivated following ICIs and cause the onset of vitiligo, leading to improved patient outcomes. Importantly it highlights the importance of TRM in maintaining long term host immunity which might partly account for the durable efficacy of immune checkpoint inhibitors in metastatic melanoma (4, 110).
The presence of TRM transcriptional signatures are also associated with improved outcomes in various solid cancers such as breast cancer (22), endometrial cancer (24), ovarian cancer (23, 111) and melanoma (18, 112). Analysis of the Cancer Genome Atlas melanoma dataset using a TRM core signature comprising of CD69, ITGAE, CD8A, TNFSRF18, 2B4 genes showed expression of TRM related genes was associated with survival in an ICI naïve population (10). Furthermore, sophisticated computational analysis of the transcriptome of tumor infiltrating lymphocytes (TILs) from metastatic melanoma revealed better prognosis in patients with a TRM signature compared to an early activation signature in those with poorer outcomes (66). Samples exhibiting this TRM signature also showed greater dendritic cell activation and IFN-γ signaling, suggesting increased likelihood of response to anti-PD-1 therapy (66). Collectively, these transcriptional TRM signatures from several groups provide further evidence to the integral role of TRM in immune control of melanoma. Coupled with the high expression of immune checkpoints such as PD-1, CTLA-4 and LAG-3 further implicate TRM as a key potential effector of response to immune checkpoint inhibitors. The next section details the clinical role of immune checkpoint inhibitors in the treatment of metastatic melanoma and translational studies that support TRM as a critical mediator of response to ICI.
Immune-checkpoint inhibitors in treatment of metastatic melanoma
Melanoma was the first tumor type where anti-tumor activity of ICIs, namely anti-CTLA-4 and anti-PD-1 antibodies, was demonstrated. Ipilimumab (anti-CTLA-4) was the first ICI developed and although objective responses were modest at 10-15% (113, 114), approximately 20% of patients demonstrated long term survival to 10 years (115). However, the advent of two anti-PD-1 antibodies, namely pembrolizumab and nivolumab, which were developed in parallel, revolutionized melanoma cancer therapeutics. The Phase III KEYNOTE-006 trial (116) compared two pembrolizumab regimens (10 mg/kg fortnightly; n = 279 or 3 weekly; n = 277) to ipilimumab (3mg/kg every 3 weeks for 4 doses; n = 278) which showed superior progression-free survival (PFS) and objective responses. The two combined pembrolizumab arms exhibited a response rate of 37% versus 13% in the ipilimumab control arm with a median PFS of 5.6 versus 2.8 months respectively (116). Accordingly median OS was 32.7 months versus 15.9 months in favor of pembrolizumab with similarly improved 5-year landmark survival rates at 38.7% compared to 31% for ipilimumab (110) (Table 1).
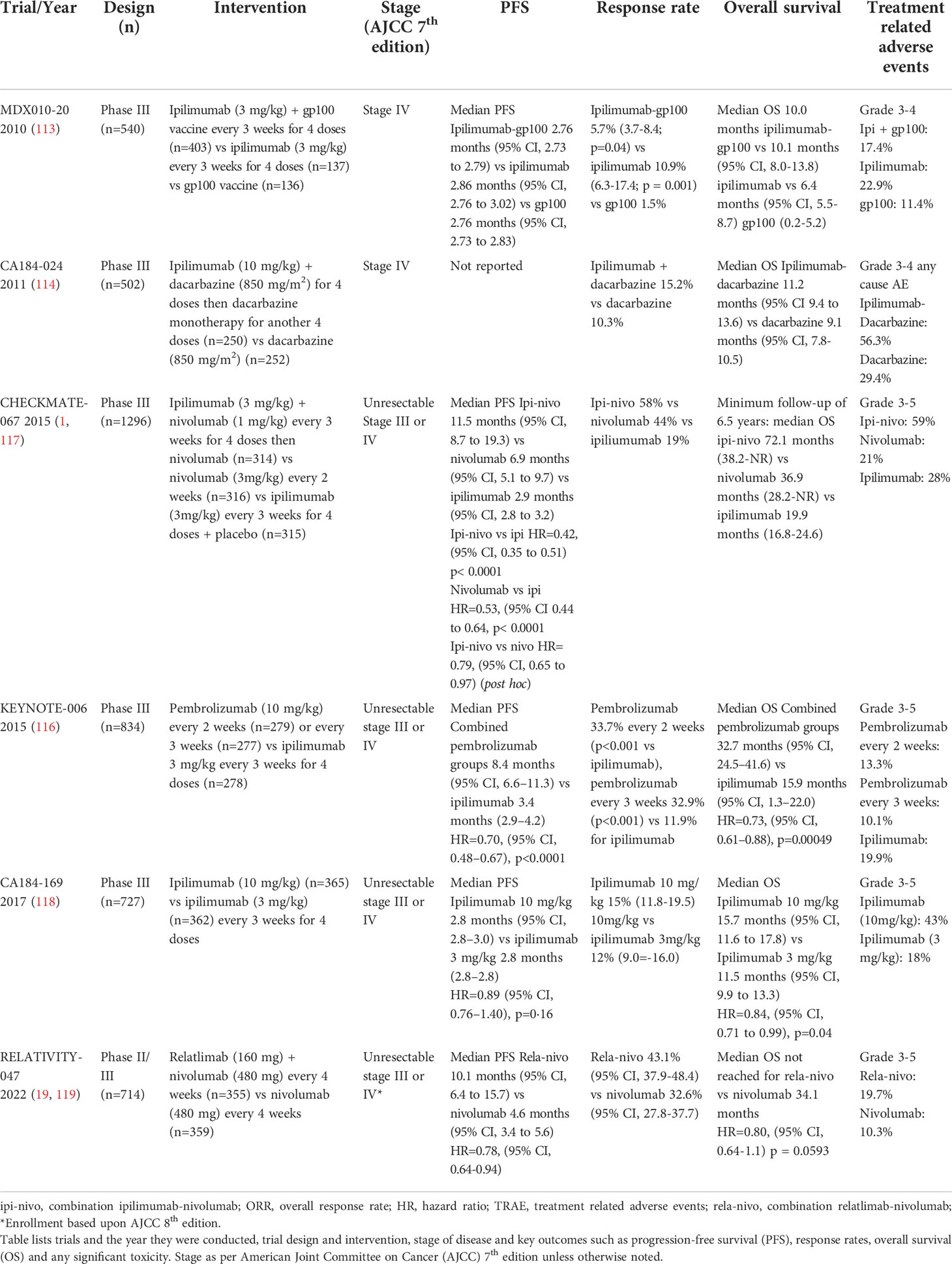
Table 1 Key clinical trials in metastatic/unresectable melanoma.
Further evidence for first line anti-PD-1 monotherapy was demonstrated in the CHECKMATE-067 randomized control trial which investigated nivolumab (n=316) or combination ipilimumab-nivolumab (n=314) versus ipilimumab monotherapy (n=315) (1). Response rates of 57.6%, 43.7% and 19% for ipilimumab-nivolumab, nivolumab monotherapy and ipilimumab monotherapy respectively were observed. Accordingly median PFS was 11.5 months for combination ipilimumab-nivolumab, 6.9 months for nivolumab and 2.9 months for ipilimumab. The hazard ratio (HR) for PFS for combination ipilimumab-nivolumab was 0.42 versus ipilimumab and 0.53 for nivolumab compared to ipilimumab indicating a substantial improvement over anti-CTLA-4 monotherapy.
Whilst the trial was not designed nor powered to formally compare combination ipilimumab-nivolumab over nivolumab monotherapy, the ad hoc HR for PFS of these two arms was 0.79 (95% CI 0.64-0.96) in favor of the combination (4). This translated to a moderate 7% absolute improvement in PFS at the five-year landmark in favor of the combination versus nivolumab monotherapy (36% vs 29%). Whilst ipilimumab-nivolumab may appear to have superior efficacy over anti-PD-1 monotherapy, the combination was less tolerable with treatment related grade 3-4 (severe to life threatening) adverse events of 59% compared to 23% with nivolumab.
Combination anti-LAG3 with nivolumab however has emerged as a more tolerable ICI doublet regimen (19). In the Phase II/III Relativity-047 trial combination relatlimab with nivolumab demonstrated superior median PFS of 10.1 months compared to 4.6 months for nivolumab monotherapy (HR=0.75; 95%CI 0.62-0.92) (Table 1). Landmark PFS at 12 months was 47.7% for relatlimab-nivolumab compared to 36% for nivolumab monotherapy. Objective response rates were 43.1% for relatlimab-nivolumab and 32.6% for nivolumab monotherapy (119). To date superior overall survival for the anti-LAG3 combination is yet to be shown with median overall survival not yet attained for relatlimab-nivolumab and 34.1 months for nivolumab monotherapy (HR=0.80; 95%CI 0.64-1.01, p=0.0593). Importantly the toxicity profile was encouraging with grade 3-4 treatment-related adverse events (TRAE) of 18.9% in the combination arm which compares favorably to ipilimumab-nivolumab. Hence combination anti-LAG3 and anti-PD1 may become the immune checkpoint inhibitor regimen of choice in metastatic melanoma.
TRM as effectors of immune checkpoint inhibitors
Both KEYNOTE-006 and CHECKMATE-067 established anti-PD-1 as the mainstay treatment for metastatic melanoma which also led to studies investigating the tumor microenvironment and circulating immune subsets associated with resistance or response to therapy. Previous studies showed anti-PD-1 anti-tumor responses were reliant on the localization of CD8+ T cells at the tumor margin (8). Efforts were then aimed at characterization of the CD8+ T cell subset associated with response to ICI. One study showed intratumoral TEM, identified by CD8+CD4-CD45RO+, were expanded in patients treated with pembrolizumab (n=53) (120). In keeping with a “TRM like” phenotype, CCR7 was not expressed by this subset. Other cell populations such as TREG, natural killer and monocytes did not appear to be expanded during PD-1 treatment. Although, this study did not utilize key TRM markers such as CD103 or CD69 in their analyses it did establish memory T cells as being crucial in mediating responses to anti-PD-1.
Further studies by Boddupalli and colleagues demonstrated TRM as a key CD8+ T cell population involved with ICI response (11). Using single cell mass cytometry, comprehensive analyses of TIL from metastatic lesions showed approximately 60% of CD8+ T cells exhibited a TRM phenotype with CD45RO+CD69+CCR7– expression. Further analyses showed approximately 50% of these cells expressed CD103, further demonstrating the TRM phenotype. The variable expression of this key TRM marker is likely due to the heterogeneity of anatomical sites which included skin, soft tissue, lymph node, colon and lung that were analyzed. These TRM also exhibited high expression of immune checkpoints, namely PD-1 and TIM-3. Gene expression data of sorted CD8+CD69+ T cells also displayed enrichment of NR4A2, CTLA-4, SKI-like proto-oncogene (SKIL), regulator of G protein signaling 1 (RGS1) with low expression of CCR7 and S1PR1, thereby reinforcing the identification of the TRM as a prevalent T cell subset in metastatic tissue in this study.
Separately Edwards et al, provided additional evidence for TRM as being a mediator of ICI responses (10). Utilizing multiplex IHC of pre-treatment and early on treatment biopsies of melanoma patients treated with ICI showed that CD8+CD103+ cells were increased in patients that exhibited an objective response to treatment (10). Supporting the findings from Boddupalli et al. (11), analysis of selected melanoma deposits by flow cytometry demonstrated approximately 30% of CD8+ TILs co-expressed CD69 and CD103, consistent with a TRM phenotype in ICI naïve patients. Moreover expression of immune checkpoints such as PD-1 and LAG-3 were expressed highly in CD8+CD69+CD103+ T cells compared to CD8+CD69-CD103- cells (10).
In the largest cohort to date aimed at identifying immune cell populations involved with response to anti-PD-1 (n=63) or combination anti-CTLA-4 and anti-PD-1 in melanoma (n=57) Gide et al. identified a T effector memory signature was associated with response to ICI (103). Transcriptome data showed IFN-γ related genes and T cell infiltrating genes such as CD8A, CD8B, ITGAE (CD103), PDCD1 (PD-1) were associated with response and improved PFS with anti-PD-1 therapy. Utilizing mass cytometry from patient samples collected at baseline and early on treatment, an EOMES+CD69+CD45RO+ effector memory T cell phenotype was identified that correlated with response to ICI (103). Again similar to the findings of Boddupalli (11), approximately 50% of these cells expressed CD103 with very low CCR7 expression indicating a TRM phenotype is involved. The high EOMES expression is not a typical feature of TRM. However, as the tumor biopsies were acquired at an early timepoint approximately 2-weeks after initiation of ICI treatment these cells potentially represents an immature TRM differentiation program. In summary, these translational datasets using a combination of gene expression, immunofluorescence and mass cytometry demonstrates TRM as a key CD8 subset in the tumor microenvironment that mediates anti-tumor response to ICI.
Adjuvant treatment for stage II and III melanoma
Given the impressive long-term survival in advanced disease, adjuvant anti-PD-1 was quickly investigated in patients following surgical excision of the primary melanoma and regional lymph node basin in stage III melanoma. The first trial using adjuvant anti-PD-1 in melanoma was CHECKMATE-238 (121) which compared adjuvant nivolumab 3 mg/kg (n=453) for 12 months to ipilimumab 10 mg/kg (n=453) in resected stage IIIB, IIIC or IV disease. Ipilimumab (10 mg/kg 3 weekly for 4 doses then every 12 weeks) was the comparator arm as it had previously been shown to improve recurrence rates and OS in the EORTC 18071 trial (122). However, given the high grade 3-4 toxicity rate of approximately 55%, adjuvant ipilimumab was not used widely. As expected, nivolumab showed superior recurrence-free survival (RFS) (HR=0.65; 97.56% CI 0.51-0.83) compared to ipilimumab (121). Updated follow-up of Stage IIIB and IIIC melanoma confirmed the superiority of nivolumab compared to ipilimumab (HR=0.71; 95% CI 0.58-0.88) with a 4-year landmark RFS of 51.7% versus 41.2% in favor of PD-1 monotherapy. Importantly nivolumab was more tolerable with treatment related grade 3-4 adverse events of 14.4% compared to 45.9% for ipilimumab. As such anti-PD-1 heralded a new benchmark in adjuvant therapy for melanoma (123) (Table 2).

Table 2 Clinical trials investigating adjuvant ICIs in resected melanoma.
Shortly thereafter the KEYNOTE-054 trial investigated a 12-month course of pembrolizumab (200 mg, 3 weekly) (n=513) compared with placebo control (n=505) in resected stage III melanoma (5). Pembrolizumab reduced recurrences by approximately 40% (HR=0.57; 98.4% CI 0.43-0.74; p<0.001) compared to placebo (5). Distant metastasis free survival (DMFS), a secondary endpoint of this study reported after 5 years was 60.6% in the pembrolizumab arm compared to 44.5% for placebo which strongly justifies anti-PD-1 in this setting (HR=0.62; 95% CI 0.52-0.74) (126). Treatment related grade 3-4 adverse events for pembrolizumab were in line with nivolumab in CHECKMATE-238 at 14.7% with 13.8% of patients discontinuing anti-PD-1 due to toxicity (Table 2).
In addition to anti-PD-1, combination BRAF-MEK inhibitors namely dabrafenib-trametinib are also effective in reducing recurrences in melanoma. Approximately 40% of melanoma harbor a BRAFV600 mutation which is amenable to sequential blockade of the mitogen activated protein kinase pathway with BRAF-MEK inhibitors (130, 131). While the anti-tumor activity of BRAF-MEKi are attributable to inhibition of the BRAFV600 oncogene, this targeted therapy also aids in recruitment of CD8 into the TME (132, 133) via several immunomodulatory properties including enhancement of melanoma differentiation antigens (132, 134, 135) and MHC Class I expression (134–136) alongside reduction of immunosuppressive cytokines such as IL-6 with IL-10 (132, 137). Whether BRAF-MEKi also enhances TRM establishment is currently unclear. In the metastatic setting, combination BRAF-MEK inhibitors are associated with high response rates of 60-70% with median PFS of approximately 11-12 months (138, 139). The COMBI-AD Phase III trial had a similar study design to KEYNOTE-054 which investigated 12 months of dabrafenib-trametinib (n=438) compared to placebo (n=432) in resected stage III melanoma (124). Dabrafenib-trametinib improved RFS with a hazard ratio of 0.57 (95% CI; 0.39–0.58) with an improved 5-year RFS rate of 52% compared to 36% in the placebo arm (125). DMFS at 5 years was also superior for dabrafenib-trametinib at 65% compared to 54% in placebo (HR for distant metastasis or death=0.55; 95% CI; 0.44 to 0.70). Interestingly 25% of patients discontinued dabrafenib-trametinib due to toxicity which is somewhat higher compared to adjuvant anti-PD-1 monotherapy (10-15%). Dabrafenib-trametinib induces drug induced pyrexia which necessitates dose interruption and in some instances discontinuation.
Although anti-PD-1 reduces recurrences in Stage III melanoma, approximately 45% of patients still recur despite adjuvant treatment (126). Given the improved response rate of combination ipilimumab-nivolumab in the metastatic setting, the CHECKMATE-915 (127) trial investigated this immunotherapy doublet compared to nivolumab monotherapy to reduce recurrences in Stage III melanoma (Table 2). Ipilimumab was administered at a lower dose of 1 mg/kg (six weekly) and nivolumab 240 mg (two weekly) for 1 year in the combination arm as tolerability is dose dependent (118). Unfortunately, combination therapy resulted in higher treatment related grade 3-4 adverse events at 33% compared to nivolumab at 13% with no difference in the primary endpoints of RFS (HR=0.92; 97.3% CI, 0.77-1.09); P = 0.26) in the adjuvant setting. As such BRAF-MEK inhibitors or anti-PD-1 monotherapy remain the standard of care for adjuvant treatment in Stage III melanoma.
Building upon the improvements in stage III melanoma, anti-PD-1 has also been investigated to reduce recurrences in Stage II disease. Stage IIB (melanoma thickness < 2.0 – 4.0 mm with ulceration or melanoma thickness > 4.0 mm without ulceration) and IIC melanoma (melanoma thickness > 4.0 mm with ulceration) exhibit 5-year overall survival outcomes of 87% and 82% respectively (140). By comparison Stage IIIB melanoma exhibits 5-year survival of 83% which provides a strong clinical rationale for adjuvant trials particularly in IIC disease. The Phase III KEYNOTE-716 trial compared 1 year of pembrolizumab (200 mg IV 3 weekly) treatment with placebo in resected stage IIB and IIC melanoma (128). Notably 64% (625 out of 976) of enrolled patients had Stage IIB melanoma. In the second interim analysis with a median follow-up period of 20.9 months, recurrences in pembrolizumab treated patients was lower at 15% compared to 24% in the placebo control arm (HR=0.61; 95% CI 0.45–0.82). Treatment related grade 3-4 adverse events in the pembrolizumab arm were in line with prior adjuvant studies at 16%. A further update of the trial with a median follow-up of 37.2 months showed 2 year RFS at 81.2% in the pembrolizumab arm compared to 72.8% for placebo (129). Notably distant metastasis free survival was in favor of pembrolizumab at 88.1% compared to 82.2%. Given this moderate improvement in distant metastasis free survival balanced with the side effect of treatment, further follow-up of the trial is required to ascertain overall potential benefit. In addition, the results of CHECKMATE-76K (NCT04099251) are awaited which investigates nivolumab and may clarify the role of adjuvant anti-PD1 in Stage II disease.
TRM in primary and stage III melanoma
As mentioned previously, TILs are a well-known prognostic factor for melanoma, but only until recently have TRM come to the forefront as a specific CD8 subset mediating immune control. Two recent studies utilizing multiplex immunohistochemistry investigated subpopulations of TILs and how they are linked to patient outcomes (53, 54). The first study quantified various populations of intra-tumoral CD8+ T cells in samples from patients with Stage III melanoma prior to adjuvant treatment with anti-PD-1 or combination anti-PD-1 with anti-CTLA-4 on clinical trial (54). Patients who benefited from adjuvant anti-PD-1 harbored greater proportions of CD39+ CD103+ PD-1+ CD8+ T cells with improved RFS. Conversely patients exhibiting a greater proportion of “bystander” cells delineated by CD39-CD103-PD-1-CD8+ had comparatively worse outcomes. Moreover the CD39+ CD103+PD-1+CD8+ were located closer to the tumor margin in comparison to their bystander counterparts which further implicates their role in clinical benefit of adjuvant ICI.
Following up their work in Stage III melanoma, a similar multiplex immunohistochemistry platform was utilized for studies in primary melanomas measuring at least 1 mm thick (53). A TRM population delineated by CD39+CD103+PD-1-CD8+ was found in high proportions as both stromal and intratumoral CD8 T cells. Moreover, this population of cells were shown to have improved recurrence-free survival (RFS) ≥5 years and were also localized to the tumor margin. It is interesting to speculate whether this population of TRM may represent a potential biomarker for clinical benefit for adjuvant anti-PD-1 in stage II melanoma. However, the technical challenges of multiplex immunohistochemistry reserve this as an investigational tool and not currently for clinical use.
Altogether these two published studies further implicate CD39+ as a key TRM biomarker of tumor-reactive cells and as a potential biomarker of clinical benefit for ICI. Notably the key TRM markers CD69 and CD49a were not included in these studies which would be useful to further define their phenotype. Further confirmatory studies are required to establish CD39 as a melanoma specific TRM marker and to elicit the role of other cell types in the tumor microenvironment that shapes the immune response.
Future directions: Adjuvant and neoadjuvant treatment for melanoma
Although adjuvant anti-PD-1 or BRAF-MEK inhibitors reduce the risk of recurrence in Stage II and III melanoma, much work is required to further improve outcomes. As mentioned previously approximately 45% of patients relapse despite adjuvant anti-PD-1 in Stage III melanoma (126). In KEYNOTE-054, the impact of recurrence free survival from pembrolizumab was mostly attributed to a reduction in distant metastases from 53.3% in the placebo group compared to 39.1% for the anti-PD-1 group (126). Adjuvant pembrolizumab reduced the rate of lung (13.2% pembrolizumab arm versus 21.0% placebo), lymph node (12.6% vs 18.0%) and liver (7.8% versus 11.3%) as sites of distant relapse compared to placebo (126). Interestingly pembrolizumab did not greatly reduce local or local regional relapses as the first site of recurrence at 14.4% in the pembrolizumab group and 19.0% in the control arm. This observation highlights the need to target the skin tumor microenvironment to reduce the likelihood of local recurrences. As outlined above, LAG-3 is expressed on skin TRM and therefore blockade of this immune checkpoint might be beneficial in reducing such local recurrences. The RELATIVITY-098 trial (NCT05002569) investigates adjuvant relatlimab-nivolumab in resected stage III melanoma. The improvement in PFS and a favorable toxicity profile shown in the previously mentioned RELATIVITY-047 trial warrants investigation of relatlimab in the both the adjuvant and neoadjuvant (NCT02519322) setting.
The focus of the treatment paradigm in melanoma has shifted earlier to neoadjuvant therapy for stage III disease (6). Neoadjuvant systemic therapy allows histopathological assessment of response to treatment which may allow personalization of post-surgical adjuvant management. A pooled analysis of six neoadjuvant melanoma trials including anti-PD-1 monotherapy (n=37), combination ipilimumab-nivolumab (n=104) as well as BRAF-MEK inhibitors (n=51) in the neoadjuvant setting suggested pathological complete response (pCR) is a reasonable surrogate endpoint for relapse free survival. In this analysis after a median follow-up of 20.9 months, patients who achieved pCR with neoadjuvant treatment demonstrated superior RFS than in patients who did not have a pCR (89% versus 50% at 2 years; p<0.001). Interestingly patients who had a near pCR or partial pathological response (pPR) from immunotherapy had similar 2-year RFS that exceeded 90% as with pCR. Conversely patients treated with targeted therapy who did not achieve pCR (including pPR) had poor outcomes (2-year RFS for pCR of 79% vs non-pCR of 13%), similar to the cohort with pathological no response (pNR) after treatment. The 2-year RFS was 96% for patients treated with immunotherapy who achieved pCR, near pCR or even partial pathological response. Given the encouraging RFS observed with patients who exhibited a pCR or near pCR, neoadjuvant treatment for Stage III melanoma is an area of intensive investigation.
The pivotal OpACIN Phase II dose finding study (n=99) demonstrated combination ipilimumab-nivolumab with two cycles of ipilimumab 1 mg/kg plus nivolumab 3 mg/kg (Arm B) was better tolerated than two cycles of ipilimumab 3 mg/kg plus nivolumab 1 mg/kg once every 3 weeks (Arm A) or two cycles of ipilimumab 3 mg/kg once every 3 weeks directly followed by two cycles of nivolumab 3 mg/kg once every 2 weeks intravenously (Arm C) (7). Treatment related grade 3-4 adverse events was 20% for Arm B compared to 38% and 50% for arms A and C respectively. Importantly pathological complete response with the ipilimumab 1 mg/kg dose (Arm B) was very encouraging at 57%.
Based upon the OpACIN results, the PRADO phase II study (n=99 investigated 2 doses of combination ipilimumab 1 mg/kg and nivolumab 3 mg/kg in stage IIIB-IIID melanoma followed by response directed adjuvant therapy (8). Response assessments were performed after six weeks of neoadjuvant ICI and resection of the index lymph node (ILN) that was defined as the largest lymph node metastasis at baseline. Patients who achieved a major pathological response (≤10% viable tumor) in the ILN, did not proceed with therapeutic lymph node dissection (TLND) nor adjuvant therapy. Patients with a partial pathological response (pPR defined by >10% to ≤50% viable tumor) or pathological non-response (pNR ≥50% viable tumor) proceeded to TLND alone and TLND followed by adjuvant systemic therapy respectively. The primary objective of pathological response rate (pRR) was achieved in 72% (95% CI; 62-80%) of patients with a major pathological response (MPR) in 61% (95% CI; 50-70%). At two years both the RFS and distant metastasis free survival was 64% in patients with a pPR, compared with 93% and 98% in major pathological response respectively. Highly encouraging RFS and DMFS rates at 2 years were maintained at 85% and 89% respectively for all patients who underwent surgery. This approach may potentially lead to reduced surgical and treatment related morbidity without compromising on longer term clinical outcomes. However results of longer term follow up are awaited.
In addition to ipilimumab-nivolumab, combinations of targeted therapy and anti-PD-1 in the neoadjuvant setting have also been investigated. The NeoTrio phase II study investigated stage III BRAF V600 mutant melanoma treated with six weeks of neoadjuvant pembrolizumab monotherapy or in combination with dabrafenib-trametinib (D-T) in two different regimens (141). Patients were randomized 1:1:1 to pembrolizumab (n=20), sequential D-T followed by pembrolizumab (n=20) or concurrent D-T with pembrolizumab (n=20). Neoadjuvant therapy was followed by total lymph node dissection and adjuvant systemic therapy. Preliminary results showed pathological complete responses pCR were highest in concurrent therapy (50%) compared to pembrolizumab monotherapy (30%) and sequential therapy (15%). Rates of MPR for concurrent, pembrolizumab monotherapy and sequential therapy were 55%, 40% and 30% respectively. Landmark 12 month RFS and event free survival was similar across all three groups. However longer term follow up is required to ascertain the influence of pathological response on rates of disease recurrence.
In conclusion, the use of pre-operative immune checkpoint inhibitors for stage III melanoma are under intensive investigation and may improve upon the benchmarks set in the adjuvant resected setting. The side effect of ICI treatment is an important consideration, but the neoadjuvant platform shows very promising relapse free survival. Well characterized TRM pre-clinical models of cutaneous melanoma may also inform neoadjuvant approaches as it provides opportunities to assess both the tumor and lymph node responses to ICI simultaneously as well as test new immunotherapy combinations. Moreover, the recent advent of spatial transcriptomics may provide further insights into the mechanisms of ICI on TRM function alongside assessment of the TME in these pre-clinical neoadjuvant models. Future neoadjuvant clinical studies should include biomarker assessment for clinical benefit but also consideration of the immunological roles TRM play given the recent translational studies highlighting the importance of this subset in both early and metastatic melanoma.
Concluding remarks
The development of immune checkpoint inhibitors has fundamentally changed the systemic management of metastatic cutaneous malignancies and are now employed in earlier lines as adjuvant treatment in Stage II and III melanoma. TRM are implicated in facilitating anti-melanoma immune responses, providing enhanced tumor control and promoting the phenomenon of “immune-mediated equilibrium”. Importantly TRM express an array of immune checkpoints such as PD-1, CTLA-4 and LAG-3, which are all current targets of ICIs in melanoma. Therefore, we propose that TRM are prominent mechanistic targets for ICIs, and the clinical success of these therapies depend on the activity of these cells at the tumor site. Future studies are required to fully unveil the role TRM play in the anti-tumor response, and more importantly how this can be exploited in future therapeutic strategies particularly in the neoadjuvant setting.
Author contributions
KP, A-JI and PL contributed to the conception of the manuscript. KP, A-JI and PL wrote the first draft of the manuscript. JA, AM, JW and PL provided critical appraisal of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
PL has received honoraria from Bristol Myers Squibb and Pfizer. PL received institutional funding for the purposes of clinical trials from Ambrx, AstraZeneca, Beigene, Bristol Myers Squibb, Pimera, MSD and Roche Genentech. These companies did not provide funding for the publication of this article.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
2. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med (2018) 379(4):341–51. doi: 10.1056/NEJMoa1805131
3. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med (2016) 374(26):2542–52. doi: 10.1056/NEJMoa1603702
4. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
5. Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med (2018) 378(19):1789–801. doi: 10.1056/NEJMoa1802357
6. Menzies AM, Amaria RN, Rozeman EA, Huang AC, Tetzlaff MT, van de Wiel BA, et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the international neoadjuvant melanoma consortium (INMC). Nat Med (2021) 27(2):301–9. doi: 10.1038/s41591-020-01188-3
7. Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol (2019) 20(7):948–60. doi: 10.1016/S1470-2045(19)30151-2
8. Reijers ILM, Menzies AM, van Akkooi ACJ, Versluis JM, van den Heuvel NMJ, Saw RPM, et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: the PRADO trial. Nat Med (2022) 28(6):1178–88. doi: 10.1038/s41591-022-01851-x
9. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
10. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) tumor-resident CD8(+) T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res: an Off J Am Assoc Cancer Res (2018) 24:3036–45. doi: 10.1158/1078-0432.CCR-17-2257
11. Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight (2016) 1(21):e88955. doi: 10.1172/jci.insight.88955
12. Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature (2019) 565(7739):366–71. doi: 10.1038/s41586-018-0812-9
13. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10(5):524–30. doi: 10.1038/ni.1718
14. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Trans Med (2015) 7(279):279ra39. doi: 10.1126/scitranslmed.3010302
15. Banchereau R, Chitre AS, Scherl A, Wu TD, Patil NS, de Almeida P, et al. Intratumoral CD103+ CD8+ T cells predict response to PD-L1 blockade. J immunotherapy cancer. (2021) 9(4):e002231. doi: 10.1136/jitc-2020-002231
16. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev (2018) 283(1):54–76. doi: 10.1111/imr.12650
17. Gebhardt T, Mackay LK. Skin-resident memory T cells keep herpes simplex virus at bay. Immunol Cell Biol (2013) 91(7):441–2. doi: 10.1038/icb.2013.26
18. Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, Donda A, et al. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol (2016) 7:573. doi: 10.3389/fimmu.2016.00573
19. Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med (2022) 386(1):24–34. doi: 10.1056/NEJMoa2109970
20. Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell (2020) 182(2):497–514.e22.doi: 10.1016/j.cell.2020.05.039
21. Lai C, Coltart G, Shapanis A, Healy C, Alabdulkareem A, Selvendran S, et al. CD8+CD103+ tissue-resident memory T cells convey reduced protective immunity in cutaneous squamous cell carcinoma. J Immunother Canc (2021) 9(1):e001807. doi: 10.1136/jitc-2020-001807
22. Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med (2018) 24(7):986–93. doi: 10.1038/s41591-018-0078-7
23. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res (2014) 20(2):434–44. doi: 10.1158/1078-0432.CCR-13-1877
24. Workel HH, Komdeur FL, Wouters MC, Plat A, Klip HG, Eggink FA, et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur J Canc (2016) 60:1–11. doi: 10.1016/j.ejca.2016.02.026
25. Başoğlu T, Akar KE, Bağcı P, Akgül Babacan N, Öztürk MA, Öztürk FE, et al. Prognostic value of tissue-resident memory T cells and tumor microenvironmental features in resected pancreatic adenocarcinoma. Balkan Med J (2022) 39(1):12–20. doi: 10.5152/balkanmedj.2021.21122
26. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-Expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun (2018) 9(1):2724. doi: 10.1038/s41467-018-05072-0
27. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol (2015) 194(7):3475–86. doi: 10.4049/jimmunol.1402711
28. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi: 10.1038/nm.2446
29. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi: 10.1038/44385
30. Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity (2018) 48(2):214–26. doi: 10.1016/j.immuni.2018.02.010
31. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol (2013) 43(11):2797–809. doi: 10.1002/eji.201343751
32. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22(1):745–63. doi: 10.1146/annurev.immunol.22.012703.104702
33. Park SL, Gebhardt T, Mackay LK. Tissue-resident memory T cells in cancer immunosurveillance. Trends Immunol (2019) 40(8):735–47. doi: 10.1016/j.it.2019.06.002
34. Benechet AP, Menon M, Xu D, Samji T, Maher L, Murooka TT, et al. T Cell-intrinsic S1PR1 regulates endogenous effector T-cell egress dynamics from lymph nodes during infection. Proc Natl Acad Sci USA (2016) 113(8):2182–7. doi: 10.1073/pnas.1516485113
35. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301. doi: 10.1038/ni.2744
36. Matos TR, Gehad A, Teague JE, Dyring-Andersen B, Benezeder T, Dowlatshahi M, et al. Central memory T cells are the most effective precursors of resident memory T cells in human skin. Sci Immunol (2022) 7(70):eabn1889. doi: 10.1126/sciimmunol.abn1889
37. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA (2012) 109(18):7037–42. doi: 10.1073/pnas.1202288109
38. Mackay Laura K, Wynne-Jones E, Freestone D, Pellicci Daniel G, Mielke Lisa A, Nman Dane M, et al. T-Box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43(6):1101–11. doi: 10.1016/j.immuni.2015.11.008
39. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-Phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194(5):2059–63. doi: 10.4049/jimmunol.1402256
40. Christo SN, Evrard M, Park SL, Gandolfo LC, Burn TN, Fonseca R, et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat Immunol (2021) 22(9):1140–51. doi: 10.1038/s41590-021-01004-1
41. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol (2015) 16(4):406–14. doi: 10.1038/ni.3108
42. Schenkel JM, Fraser KA, Masopust D. Cutting edge: Resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol (2014) 192(7):2961–4. doi: 10.4049/jimmunol.1400003
43. Crowl JT, Heeg M, Ferry A, Milner JJ, Omilusik KD, Toma C, et al. Tissue-resident memory CD8+ T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat Immunol (2022) 23(7):1121–31. doi: 10.1038/s41590-022-01229-8
44. Reilly EC, Emo KL, Buckley PM, Reilly NS, Smith I, Chaves FA, et al. TRM integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci (2020) 117(22):12306–14. doi: 10.1073/pnas.1915681117
45. Bromley SK, Akbaba H, Mani V, Mora-Buch R, Chasse AY, Sama A, et al. CD49a regulates cutaneous resident memory CD8(+) T cell persistence and response. Cell Rep (2020) 32(9):108085. doi: 10.1016/j.celrep.2020.108085
46. Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity (2017) 46(2):287–300. doi: 10.1016/j.immuni.2017.01.009
47. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature (2018) 557(7706):575–9. doi: 10.1038/s41586-018-0130-2
48. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA (2013) 110(36):14711–6. doi: 10.1073/pnas.1308209110
49. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
50. Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci (2018) 19(12):3837. doi: 10.3390/ijms19123837
51. Zhang J, Yan W, Duan W, Wüthrich K, Cheng J. Tumor immunotherapy using A(2A) adenosine receptor antagonists. Pharm (Basel). (2020) 13(9):237. doi: 10.3390/ph13090237
52. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res (2015) 3(5):506–17. doi: 10.1158/2326-6066.CIR-14-0211
53. Attrill GH, Lee H, Tasker AT, Adegoke NA, Ferguson AL, da Silva IP, et al. Detailed spatial immunophenotyping of primary melanomas reveals immune cell subpopulations associated with patient outcome. Front Immunol (2022) 13:979993. doi: 10.3389/fimmu.2022.979993
54. Attrill GH, Owen CN, Ahmed T, Vergara IA, Colebatch AJ, Conway JW, et al. Higher proportions of CD39+ tumor-resident cytotoxic T cells predict recurrence-free survival in patients with stage III melanoma treated with adjuvant immunotherapy. J Immunother Cancer (2022) 10(6):e004771. doi: 10.1136/jitc-2022-004771
55. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352(6284):459–63. doi: 10.1126/science.aad2035
56. Parga-Vidal L, Taggenbrock RLRE, Beumer-Chuwonpad A, Aglmous H, Kragten NAM, Behr FM, et al. Hobit and blimp-1 regulate TRM abundance after LCMV infection by suppressing tissue exit pathways of TRMprecursors. Eur J Immunol 52(7):1095–1111. doi: 10.1002/eji.202149665
57. Fonseca R, Burn TN, Gandolfo LC, Devi S, Park SL, Obers A, et al. Runx3 drives a CD8+ T cell tissue residency program that is absent in CD4+ T cells. Nat Immunol (2022) 23(8):1236–45. doi: 10.1038/s41590-022-01273-4
58. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol (2016) 17(12):1467–78. doi: 10.1038/ni.3589
59. McLane LM, Banerjee PP, Cosma GL, Makedonas G, Wherry EJ, Orange JS, et al. Differential localization of T-bet and eomes in CD8 T cell memory populations. J Immunol (2013) 190(7):3207–15. doi: 10.4049/jimmunol.1201556
60. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
61. Luoma AM, Suo S, Wang Y, Gunasti L, Porter CBM, Nabilsi N, et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell (2022) 185(16):2918–35.e29. doi: 10.1016/j.cell.2022.06.018
62. Hasan F, Chiu Y, Shaw RM, Wang J, Yee C. Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight (2021) 6(10):e138970. doi: 10.1172/jci.insight.138970
63. Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, et al. ABC Transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest (2016) 126(10):3905–16. doi: 10.1172/JCI85329
64. Milner JJ, Goldrath AW. Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr Opin Immunol (2018) 51:162–9. doi: 10.1016/j.coi.2018.03.017
65. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA (2014) 111(14):5307–12. doi: 10.1073/pnas.1322292111
66. Jaiswal A, Verma A, Dannenfelser R, Melssen M, Tirosh I, Izar B, et al. An activation to memory differentiation trajectory of tumor-infiltrating lymphocytes informs metastatic melanoma outcomes. Cancer Cell (2022) 40(5):524–44 e5. doi: 10.1016/j.ccell.2022.04.005
67. Salek-Ardakani S, Moutaftsi M, Sette A, Croft M. Targeting OX40 promotes lung-resident memory CD8 T cell populations that protect against respiratory poxvirus infection. J Virol (2011) 85(17):9051–9. doi: 10.1128/JVI.00619-11
68. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty acid oxidation controls CD8(+) tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res (2020) 8(4):479–92. doi: 10.1158/2326-6066.CIR-19-0702
69. Jiang Y, Li Y, Zhu B. T-Cell exhaustion in the tumor microenvironment. Cell Death Disease (2015) 6(6):e1792–e. doi: 10.1038/cddis.2015.162
70. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi: 10.1038/nature05115
71. Watanabe T, Bertoletti A, Tanoto TA. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J Viral Hepatitis (2010) 17(7):453–8. doi: 10.1111/j.1365-2893.2010.01313.x
72. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
73. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
74. Galvez-Cancino F, Lopez E, Menares E, Diaz X, Flores C, Caceres P, et al. Vaccination-induced skin-resident memory CD8(+) T cells mediate strong protection against cutaneous melanoma. Oncoimmunology (2018) 7(7):e1442163. doi: 10.1080/2162402X.2018.1442163
75. Edwards J, Tasker A, Pires da Silva I, Quek C, Batten M, Ferguson A, et al. Prevalence and cellular distribution of novel immune checkpoint targets across longitudinal specimens in treatment-naive melanoma patients: Implications for clinical trials. Clin Cancer Res: an Off J Am Assoc Cancer Res (2019) 25(11):3247–58. doi: 10.1158/1078-0432.CCR-18-4011
76. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
77. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity (1994) 1(9):793–801. doi: 10.1016/S1074-7613(94)80021-9
78. van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med (1997) 185(3):393–403. doi: 10.1084/jem.185.3.393
79. Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol (2017) 49:14–9. doi: 10.1016/j.coi.2017.07.014
80. Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Canc (2020) 8(2):e001014. doi: 10.1136/jitc-2020-001014
81. Guy C, Mitrea DM, Chou P-C, Temirov J, Vignali KM, Liu X, et al. LAG3 associates with TCR–CD3 complexes and suppresses signaling by driving co-receptor–lck dissociation. Nat Immunol (2022) 23(5):757–67. doi: 10.1038/s41590-022-01176-4
82. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3(4):412–23. doi: 10.1158/2326-6066.CIR-14-0150
83. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176(1-2):334–47.e12. doi: 10.1016/j.cell.2018.11.010
84. Mihm MC Jr., Mule JJ. Reflections on the histopathology of tumor-infiltrating lymphocytes in melanoma and the host immune response. Cancer Immunol Res (2015) 3(8):827–35. doi: 10.1158/2326-6066.CIR-15-0143
85. Clark WH Jr., Elder DE, Guerry Dt, Braitman LE, Trock BJ, Schultz D, et al. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst (1989) 81(24):1893–904. doi: 10.1093/jnci/81.24.1893
86. Clemente CG, Mihm MC Jr., Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer (1996) 77(7):1303–10. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5
87. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol (2012) 30(21):2678–83. doi: 10.1200/JCO.2011.37.8539
88. Fu Q, Chen N, Ge C, Li R, Li Z, Zeng B, et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: a systematic review and meta-analysis. Oncoimmunology (2019) 8(7):1593806. doi: 10.1080/2162402X.2019.1593806
89. Park SL, Mackay LK, Waithman J, Gebhardt T. Tissue-resident memory T cells orchestrate tumour-immune equilibrium. Cell Stress. (2019) 3(5):162–4. doi: 10.15698/cst2019.05.187
90. MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med (2003) 348(6):567–8. doi: 10.1056/NEJM200302063480620
91. Miller JJ, Lofgren KA, Hughes SR, Cash SE, Kenny PA. Genomic analysis of melanoma evolution following a 30-year disease-free interval. J Cutaneous Pathol (2017) 44(9):805–8. doi: 10.1111/cup.12989
92. Saleh D, Peach AH. Ultra-late recurrence of malignant melanoma after 40 years of quiescent disease. J Surg Oncol (2011) 103(3):290–1. doi: 10.1002/jso.21821
93. Terhorst D, Radke C, Trefzer U. Ultra-late recurrence of malignant melanoma after a disease-free interval of 41 years. Clin Exp Dermatol (2010) 35(3):e20–1. doi: 10.1111/j.1365-2230.2009.03330.x
94. Jensen TO, Schmidt H, Moller HJ, Hoyer M, Maniecki MB, Sjoegren P, et al. Macrophage markers in serum and tumor have prognostic impact in American joint committee on cancer stage I/II melanoma. J Clin Oncol: Off J Am Soc Clin Oncol (2009) 27(20):3330–7. doi: 10.1200/JCO.2008.19.9919
95. Piras F, Colombari R, Minerba L, Murtas D, Floris C, Maxia C, et al. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-d-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer (2005) 104(6):1246–54. doi: 10.1002/cncr.21283
96. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
97. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
98. Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat Commun (2020) 11(1):1897. doi: 10.1038/s41467-020-15726-7
99. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165(1):35–44. doi: 10.1016/j.cell.2016.02.065
100. Lau PKH, Feran B, Smith L, Lasocki A, Molania R, Smith K, et al. Melanoma brain metastases that progress on BRAF-MEK inhibitors demonstrate resistance to ipilimumab-nivolumab that is associated with the innate PD-1 resistance signature (IPRES). J Immunother Cancer (2021) 9(10):e002995. doi: 10.1136/jitc-2021-002995
101. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
102. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
103. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/Anti-CTLA-4 combined therapy. Cancer Cell (2019) 35(2):238–55 e6. doi: 10.1016/j.ccell.2019.01.003
104. van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol (2009) 129(9):2220–32. doi: 10.1038/jid.2009.32
105. Boniface K, Jacquemin C, Darrigade AS, Dessarthe B, Martins C, Boukhedouni N, et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol (2018) 138(2):355–64. doi: 10.1016/j.jid.2017.08.038
106. Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol (2019) 139(4):769–78. doi: 10.1016/j.jid.2018.10.032
107. Nakamura Y, Tanaka R, Asami Y, Teramoto Y, Imamura T, Sato S, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: A multi-institutional retrospective study. J Dermatol (2017) 44(2):117–22. doi: 10.1111/1346-8138.13520
108. Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol (2016) 152(1):45–51. doi: 10.1001/jamadermatol.2015.2707
109. Han J, Zhao Y, Shirai K, Molodtsov A, Kolling FW, Fisher JL, et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat Canc (2021) 2(3):300–11. doi: 10.1038/s43018-021-00180-1
110. Robert C, Ribas A, Schachter J, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol (2019) 20(9):1239–51. doi: 10.1016/S1470-2045(19)30388-2
111. Komdeur FL, Wouters MC, Workel HH, Tijans AM, Terwindt AL, Brunekreeft KL, et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget (2016) 7(46):75130–44. doi: 10.18632/oncotarget.12077
112. Menares E, Gálvez-Cancino F, Cáceres-Morgado P, Ghorani E, López E, Díaz X, et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun (2019) 10(1):4401. doi: 10.1038/s41467-019-12319-x
113. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
114. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
115. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol: Off J Am Soc Clin Oncol (2015) 33(17):1889–94. doi: 10.1200/JCO.2014.56.2736
116. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
117. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J Clin Oncol: Off J Am Soc Clin Oncol (2022) 40(2):127–37. doi: 10.1200/JCO.21.02229
118. Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol (2017) 18(5):611–22. doi: 10.1016/S1470-2045(17)30231-0
119. Tawbi HA, Hodi FS, Lipson EJ, Schadendorf D, Ascierto PA, Matamala L, et al. Nivolumab (NIVO) + relatlimab (RELA) versus NIVO in previously untreated metastatic or unresectable melanoma: OS and ORR by key subgroups from RELATIVITY-047. J Clin Oncol (2022) 40(16_suppl):9505–. doi: 10.1200/JCO.2022.40.16_suppl.9505
120. Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res (2016) 4(3):194–203. doi: 10.1158/2326-6066.CIR-15-0210
121. Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med (2017) 377(19):1824–35. doi: 10.1056/NEJMoa1709030
122. Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): A randomised, double-blind, phase 3 trial. Lancet Oncol (2015) 16(5):522–30. doi: 10.1016/S1470-2045(15)70122-1
123. Ascierto PA, Del Vecchio M, Mandala M, Gogas H, Arance AM, Dalle S, et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-c and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol (2020) 21(11):1465–77. doi: 10.1016/S1470-2045(20)30494-0
124. Long GV, Hauschild A, Santinami M, Atkinson V, Mandala M, Chiarion-Sileni V, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med (2017) 377(19):1813–23. doi: 10.1056/NEJMoa1708539
125. Dummer R, Hauschild A, Santinami M, Atkinson V, Mandalà M, Kirkwood JM, et al. Five-year analysis of adjuvant dabrafenib plus trametinib in stage III melanoma. N Engl J Med (2020) 383(12):1139–48. doi: 10.1056/NEJMoa2005493
126. Eggermont AMM, Kicinski M, Blank CU, Mandala M, Long GV, Atkinson V, et al. Five-year analysis of adjuvant pembrolizumab or placebo in stage III melanoma. NEJM Evidence (2022). doi: 10.1056/EVIDoa2200214
127. Weber JS, Schadendorf D, Del Vecchio M, Larkin J, Atkinson V, Schenker M, et al. Adjuvant therapy of nivolumab combined with ipilimumab versus nivolumab alone in patients with resected stage IIIB-d or stage IV melanoma (CheckMate 915). J Clin Oncol: Off J Am Soc Clin Oncol (2022):JCO2200533. doi: 10.1200/JCO.22.00533
128. Luke JJ, Rutkowski P, Queirolo P, Del Vecchio M, Mackiewicz J, Chiarion-Sileni V, et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): a randomised, double-blind, phase 3 trial. Lancet (2022) 399(10336):1718–29. doi: 10.1016/S0140-6736(22)00562-1
129. Long GV, Luke JJ, Khattak M, de la Cruz Merino L, Del Vecchio M, Rutkowski P, et al. Distant metastasis-free survival with pembrolizumab versus placebo as adjuvant therapy in stage IIB or IIC melanoma: The phase 3 KEYNOTE-716 study. J Clin Oncol (2022) 40(17_suppl):LBA9500–LBA. doi: 10.1200/JCO.2022.40.17_suppl.LBA9500
130. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature (2017) 545(7653):175–80. doi: 10.1038/nature22071
131. Lyle M, Haydu LE, Menzies AM, Thompson JF, Saw RP, Spillane AJ, et al. The molecular profile of metastatic melanoma in Australia. Pathology (2016) 48(2):188–93. doi: 10.1016/j.pathol.2015.12.008
132. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res: an Off J Am Assoc Cancer Res (2013) 19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630
133. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res: An Off J Am Assoc Cancer Res (2012) 18(5):1386–94. doi: 10.1158/1078-0432.CCR-11-2479
134. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70(13):5213–9. doi: 10.1158/0008-5472.CAN-10-0118
135. Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res (2015) 3(6):602–9. doi: 10.1158/2326-6066.CIR-15-0030
136. Lau PKH, Cullinane C, Jackson S, Walker R, Smith LK, Slater A, et al. Enhancing adoptive cell transfer with combination BRAF-MEK and CDK4/6 inhibitors in melanoma. Cancers (2021) 13(24). doi: 10.3390/cancers13246342
137. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med (2006) 203(7):1651–6. doi: 10.1084/jem.20051848
138. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med (2015) 372(1):30–9. doi: 10.1056/NEJMoa1412690
139. Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2018) 19(5):603–15. doi: 10.1016/S1470-2045(18)30142-6
140. Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, et al. Melanoma staging: Evidence-based changes in the American joint committee on cancer eighth edition cancer staging manual. CA Cancer J Clin (2017) 67(6):472–92. doi: 10.3322/caac.21409
141. Long GV, Carlino MS, Au-Yeung G, Spillane AJ, Shannon KF, Gyorki DE, et al. NeoTrio: Randomized trial of neoadjuvant (NAT) pembrolizumab (Pembro) alone, in sequence (SEQ) with, or concurrent (CON) with dabrafenib plus trametinib (D+T) in resectable BRAF-mutant stage III melanoma to determine optimal combination of therapy. J Clin Oncol (2022) 40(16_suppl):9503–. doi: 10.1200/JCO.2022.40.16_suppl.9503
Keywords: tissue-resident memory CD8(+) T cells, cancer immunotherapy, melanoma, immune checkpoints, neoadjuvant immunotherapy, adjuvant immunotherapy, tumor microenvironment
Citation: Plunkett KR, Armitage JD, Inderjeeth A-J, McDonnell AM, Waithman J and Lau PKH (2022) Tissue-resident memory T cells in the era of (Neo) adjuvant melanoma management. Front. Immunol. 13:1048758. doi: 10.3389/fimmu.2022.1048758
Received: 20 September 2022; Accepted: 13 October 2022;
Published: 16 November 2022.
Edited by:
Ester Lozano, Department of Cell Biology, University of Barcelona, SpainReviewed by:
Matthew J. Atherton, University of Pennsylvania, United StatesXiaodong Zheng, Anhui Medical University, China
Copyright © 2022 Plunkett, Armitage, Inderjeeth, McDonnell, Waithman and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter K. H. Lau, cGV0ZXIubGF1MkBoZWFsdGgud2EuZ292LmF1
†ORCID: Jesse D. Armitage, orcid.org/0000-0002-6558-5914
Andrisha-Jade Inderjeeth, orcid.org/0000-0001-8888-6640
Alison M McDonnell, orcid.org/0000-0002-1471-2151
Jason Waithman, orcid.org/0000-0003-4940-0155
Peter K. H. Lau, orcid.org/0000-0003-1071-7956