 Valentina Granata1,2
Valentina Granata1,2 Valentina Possetti1,3
Valentina Possetti1,3 Raffaella Parente1
Raffaella Parente1 Barbara Bottazzi1
Barbara Bottazzi1 Antonio Inforzato1,3
Antonio Inforzato1,3 Cristina Sobacchi1,2*
Cristina Sobacchi1,2*- 1IRCCS Humanitas Research Hospital, Rozzano, Italy
- 2Milan Unit, National Research Council - Institute for Genetic and Biomedical Research (CNR-IRGB), Milan, Italy
- 3Department of Biomedical Sciences, Humanitas University, Pieve Emanuele, Italy
Osteomyelitis (OM) is an infectious disease of the bone predominantly caused by the opportunistic bacterium Staphylococcus aureus (S. aureus). Typically established upon hematogenous spread of the pathogen to the musculoskeletal system or contamination of the bone after fracture or surgery, osteomyelitis has a complex pathogenesis with a critical involvement of both osteal and immune components. Colonization of the bone by S. aureus is traditionally proposed to induce functional inhibition and/or apoptosis of osteoblasts, alteration of the RANKL/OPG ratio in the bone microenvironment and activation of osteoclasts; all together, these events locally subvert tissue homeostasis causing pathological bone loss. However, this paradigm has been challenged in recent years, in fact osteoblasts are emerging as active players in the induction and orientation of the immune reaction that mounts in the bone during an infection. The interaction with immune cells has been mostly ascribed to osteoblast-derived soluble mediators that add on and synergize with those contributed by professional immune cells. In this respect, several preclinical and clinical observations indicate that osteomyelitis is accompanied by alterations in the local and (sometimes) systemic levels of both pro-inflammatory (e.g., IL-6, IL-1α, TNF-α, IL-1β) and anti-inflammatory (e.g., TGF-β1) cytokines. Here we revisit the role of osteoblasts in bacterial OM, with a focus on their secretome and its crosstalk with cellular and molecular components of the bone microenvironment and immune system.
Introduction
Osteomyelitis is a severe bone infection arising from hematogenous spread of pathogens, mainly in pediatric patients (1), or direct contamination of the bone after fracture or surgery, more commonly in adults (2). Typically associated with comorbidities, osteomyelitis has a relatively high incidence amongst diabetics, where it develops secondary to vascular and neuropathic complications of hyperglycemia, and may require extreme clinical measures (i.e., limb amputation) (3). Osteomyelitis occurs also in in the absence of known risk factors for invasive infections, which underlines the complexity of its etiopathogenesis. Several pathogens, including bacteria, fungi, and viruses, can cause bone infections, but the most common etiologic agent of the disease is the Gram-positive bacterium Staphylococcus aureus (S. aureus), which is responsible for up to 60% of the cases (4). Different classification schemes have been proposed for osteomyelitis, based on a range of clinical and microbiological characteristics (5). Most commonly, the disease is classified as either acute or chronic, according to its duration, and either hematogenous or contiguous, according to the origin of infection. During a bone infection, the serum levels of several cytokines [e.g., IL-6, IL-8, IL-1β, IL-12(p70)], angiogenic factors (e.g., VEGF), and acute phase proteins (e.g., C reactive protein, CRP) increase, which is of diagnostic value in the clinical handling of the disease; nevertheless, specific infection markers with diagnostic value are still sought for (6, 7).
The first line of defense against invading pathogens, including S. aureus, is represented by cells of the innate immune system (neutrophils, monocytes, macrophages), which detect the pathogens through germline-encoded pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs) and cytoplasmic receptors. PRRs recognize specific microbial components known as pathogen-associated molecular patterns (PAMPs), such as LPS, lipoteichoic acid, lipoproteins, and peptidoglycans, and convey the biochemical signals that are ultimately responsible for activation of the immune cells (8). PRRs are also made by non-immune cells, including cells of the osteoclast and osteoblast lineage in the bone. All TLRs but TLR2 and TLR4 are downregulated during osteoclast differentiation, and relative timing of TLR and RANKL/MCSF stimulation of osteoclast precursors elicit opposite effect on osteoclast formation (9). Very recently it has been reported that TLR2 and TLR9 signaling contributes marginally to the inflammatory bone loss and enhanced osteoclast formation that accompany S. aureus-dependent osteomyelitis (see Osteoclasts and their progenitors) (10). However, it is generally accepted that osteoblasts use TLRs and NODs to recognize and respond to S. aureus, which leads to secretion of the master osteoclastogenic cytokine RANKL as well as other factors (11), as discussed below.
In line with the general paradigm of immunological activation upon PRR engagement, in the context of a bone infection, polymorphonuclear leukocytes (PMNs) and macrophages migrate into the bone microenvironment and therein release several diverse inflammatory mediators, including cytokines, chemokines (e.g., IL-1β, IL-6, and TNF-α, CCL3 and CXCL2), and other factors. The immune reaction that mounts in the bone alters the local homeostasis profoundly, which adds on the direct effects of S. aureus on skeletal cells (8). Proinflammatory mediators promote formation and activation of osteoclasts, thus enhancing bone resorption; in parallel, new, poorly structured bone is deposited at the periosteum (periosteal bone reaction) by osteoblasts to confine the infection. These opposing processes collectively result into progressive disruption of the bone microarchitecture. The crosstalk between immune and skeletal cells in the context of bone infections sustained by S. aureus (a paradigmatic example of osteoimmunology) is schematically depicted in Figure 1.
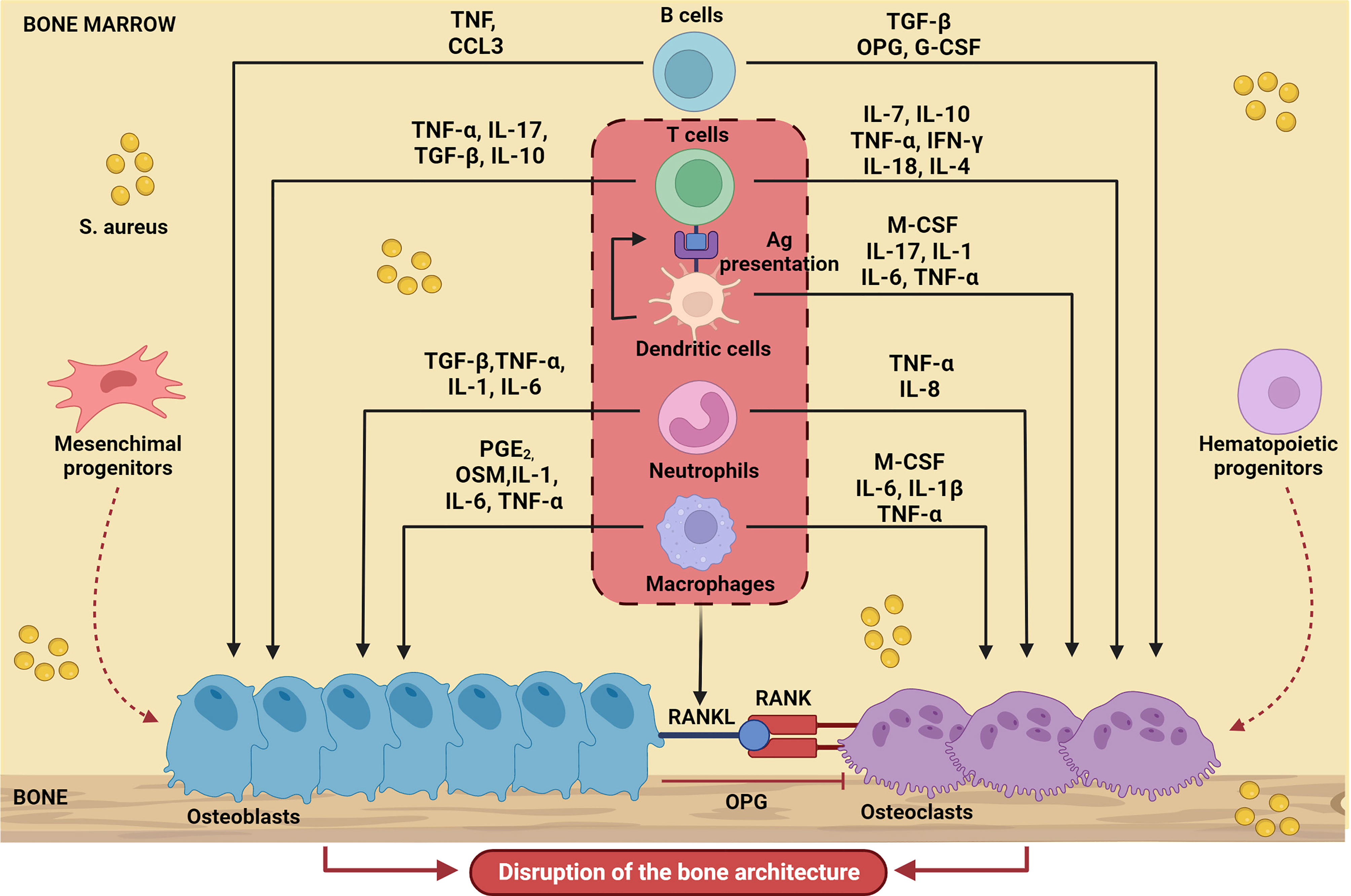
Figure 1 Schematic representation of bone cell regulation by the main soluble factors produced by immune cells during S. aureus-induced osteomyelitis. Upon infection, immune cells migrate into the bone microenvironment and release cytokines (IL-17, IL-10, IL-1, IL-1β, IL-6, IL-7, IL-18, IL-4, IL-8), chemokines (CCL3), growth factors (M-CSF, G-CSF) and other several inflammatory mediators (TNF-α, PGE2, TGFβ, INF-γ, OSM) that influence osteoblast and osteoclast activity. The result is massive disruption of the normal bone architecture. The Figure was created with BioRender.com.
Besides their function in bone homeostasis, both osteoclasts and osteoblasts exert immune regulatory functions by orchestrating synthesis and release of various inflammatory molecules (12). In the present review, we present and discuss the roles of osteoblasts in the pathogenesis of osteomyelitis, with a focus on osteoblast-derived soluble factors and their contribution to the long-term fate of bone infections (Figure 2).
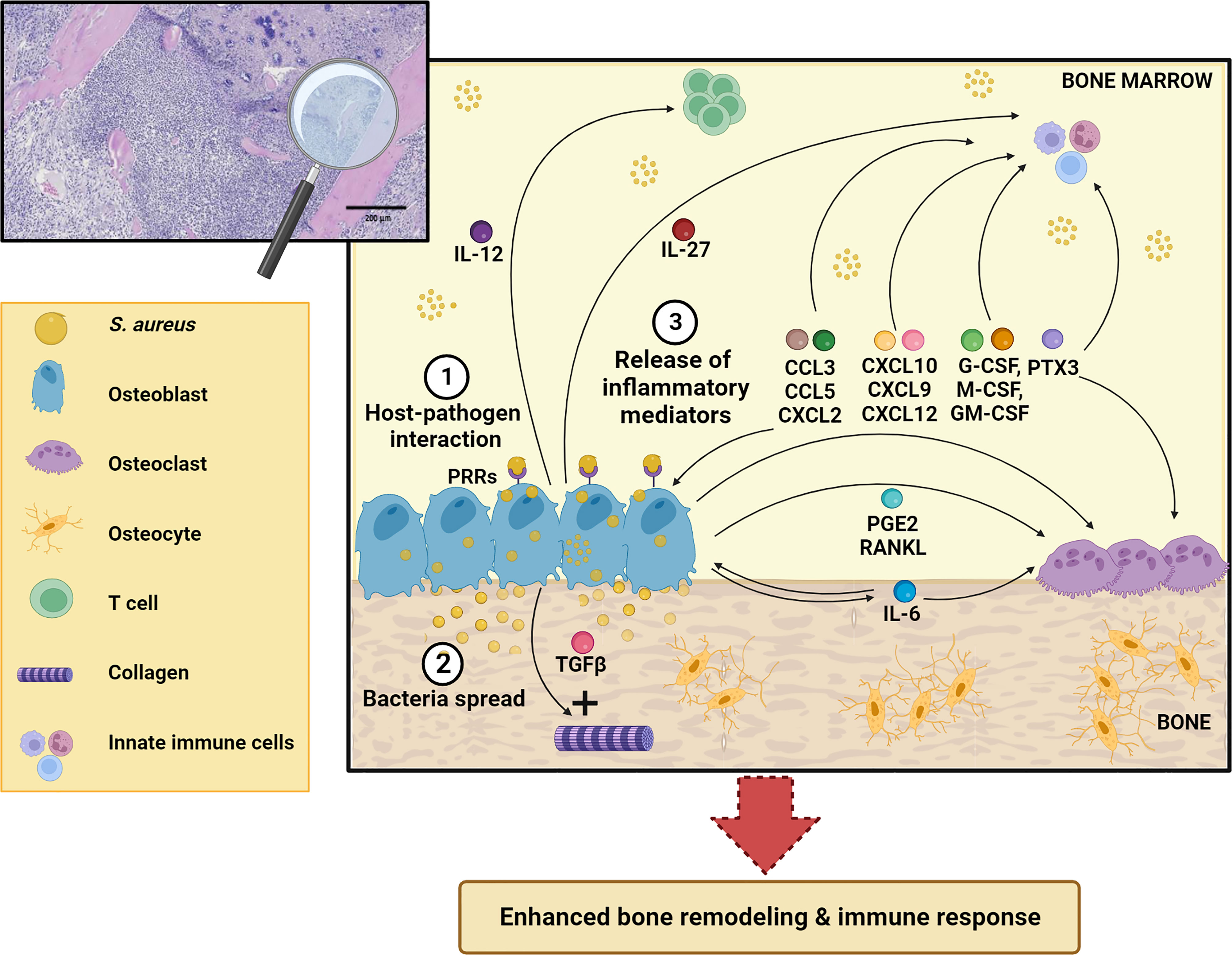
Figure 2 Schematic representation of osteoblast response to S. aureus infection. After osteoblast infection (1), S. aureus can grow and spread further in the bone (2) and induce synthesis and release of different inflammatory mediators (IL-12, IL-27, IL-6, CCL3, CCL5, CXCL2, CXCL10, CXCL9, CXCL12, TGFβ, G-CSF, M-CSF, GM-CSF, PTX3, RANKL, PGE2) (3) that act both on skeletal and immune cells. This leads to enhanced bone remodeling and immune response. See the main text for more details. The Figure was created with BioRender.com.
Mainstays in bone biology and basic microbiology of S. aureus osteomyelitis
Bone is a dynamic tissue that undergoes constant remodeling throughout life (13). Osteoclasts and osteoblasts are the two cell types primarily involved in bone remodeling, responsible for bone resorption and bone formation, respectively. Osteoclasts are of hematopoietic origin, and unique in their ability to degrade the inorganic (hydroxyapatite) and organic (mostly collagen) matrix of the bone (14) through acidification of the resorption lacuna and release of hydrolytic enzymes (e.g., cathepsin K). Conversely, osteoblasts arise from cells of the mesenchymal lineage, and produce the whole set of components of the mineralized extracellular matrix of the bone, which comprises collagen, proteoglycans and several non-collagenous proteins, and an extremely dense hydroxyapatite-based mineral (15). As the matrix grows, some osteoblasts are enclosed in it and become osteocytes. These are terminally differentiated cells that communicate with each other and other cell types by means of cellular processes that penetrate the canaliculi in the bone extracellular matrix.
The crosstalk between osteoclasts and osteoblasts shapes the extracellular matrix. Their mutual interaction relies on a series of membrane-bound molecules and soluble factors (e.g., EFNB2-EPHB4, FASL-FAS and SEMA3A-NRP1). Among the latter, M-CSF and RANKL, produced by osteoblasts and acting on their specific receptors on the osteoclast surface (C-FMS and RANK, respectively), have a pivotal role in bone homeostasis (14). In addition, osteoblasts release OPG, the decoy receptor for RANKL that hinders RANK-RANKL interaction, reducing osteoclast formation and bone resorption. In fact, RANKL/OPG ratio is commonly used as an indicator of the balance between bone formation and resorption (16).
S. aureus can invade, colonize, and thrive in the bone. Microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), such as fibronectin-binding protein A and B (FnBPA and B), collagen adhesin (Cna) and Staphylococcus protein A (SpA) (17), initiate adhesion to the bone extracellular matrix. Once bound to the matrix, S. aureus can activate diverse mechanisms to escape the host immune response, survive in the bone microenvironment, and establish chronic infection. A primary strategy of immune evasion is biofilm formation, a complex process that involves synthesis and release of extracellular polymeric substances (i.e., polysaccharides and extracellular DNA) and is controlled both by microbial (i.e., the Agr quorum-sensing system) (18) and host factors (i.e., mineral components of the bone matrix, such as Mg2+ and Ca2+, oxygen and nutrient availability) (18). Biofilms reduce osteoblast viability and increase RANKL production, thus promoting bone resorption (19). Moreover, in an in vitro dynamic model of biofilm deposition, the supernatant of TNFα-treated osteoblasts was found to affect S. aureus adhesion and biofilm formation (20). Abscess formation is another survival strategy for S. aureus, whereby following upon the engagement of fibrinogen by the bacterial clamping factors A and B (ClfA and B) and its conversion to fibrin (by coagulase, CoA, and von Willebrand factor-binding protein, vWbp), a fibrous pseudocapsule is formed that encases a core of staphylococcal abscess communities (SACs) and is surrounded by a layer of necrotic leukocytes (mostly, neutrophils) (2). Furthermore, S. aureus is able to invade the osteocyte lacunocanalicular network through bacteria deformation (whereby a key role has been proposed for the transpeptidases penicillin-binding proteins 3 and 4), entry and migration into the canaliculi via asymmetric binary fission at the leading edge (21, 22); these mechanisms are particularly relevant to long term bacterial persistence and eradication. Finally, intracellular infection and survival is a bacterial strategy to evade immune surveillance; of note, all skeletal cells are potential targets, as discussed in the following section.
Skeletal cell infection in the context of S. aureus osteomyelitis
Osteoclasts and their progenitors
A growing body of evidence supports the concept of osteoclasts working as the innate immune cells of the bone (23, 24); accordingly, it is expected that they play a role in the framework of osteomyelitis. Bacterial infection has the potential to influence osteoclast formation directly, through receptor ligation on progenitor cells (9), and indirectly, through enhanced cytokine release from neighboring cells (8). In particular, in vitro S. aureus infection of murine bone marrow-derived osteoclast precursors has been shown to induce their differentiation into activated macrophages that actively secrete proinflammatory cytokines, such as CCL5, MIP-1α, MIP-1β, G-CSF, IL-12p40, and MCP-1 (25). These cytokines enhance the bone resorption capacity of uninfected mature osteoclasts and differentiation of uninfected precursors. Moreover, infection of mature osteoclasts directly promotes cell fusion, which contributes to enhance their ability to resorb bone (25). A similar effect has been also demonstrated upon exposure of human monocyte-derived osteoclasts to Staphylococcal superantigen TSST-1, while the Panton-Valentine leukocidin, known as one of the most powerful pore-forming toxins, and hemolysin-α induced osteoclast death, indicating that the clinical presentation and outcome of bone and joint infection could be related, at least partly, to the toxin profile of the S. aureus isolate involved (26). The osteoclast intracellular infection, which has been demonstrated also in vivo (27), has been recently studied exploiting fluorescently labelled bacterial strains visualized by confocal and time-lapse microscopy (28). Intracellular penetration of bacteria occurred in vitro in a short timeframe; bacterial proliferation started two-four hours post-infection, but the bacterial load in the intracellular compartment of infected osteoclasts did not change over time (28). Osteoclast colonization was accompanied by reduced bactericidal potential compared to non-infected osteoclast precursors (29). Moreover, the proliferative capacity of S. aureus within osteoclasts was dependent on the signaling cascade activated by the osteoclastogenic master transcription factor NFATc1, and varied to some extent amongst individual osteoclasts (29).
Osteoblasts and their progeny
S. aureus has been shown to infect osteoblasts and downstream mature cells (osteocytes) both in in vitro and in vivo animal models and clinical biopsies (21, 30–33). The interaction of osteoblasts with S. aureus involves several pathogen-derived factors and components of the osteoblast plasma membrane. For example, a major virulence factor of S. aureus, SpA, interacts directly with osteoblasts via TNFR1, and activates intracellular signaling cascades that result into reduced proliferation, enhanced apoptosis, and impaired mineralizing potential of cultured osteoblasts (34). The binding of S. aureus FnBPA and FnBPB to the extracellular matrix protein fibronectin is recognized as another important process in osteoblast infection. Indeed, fibronectin is believed to act as a “molecular bridge” in linking osteoblasts to the pathogen through α5β1 integrin (35). In this respect, the supramolecular structure of fibronectin in the bone extracellular matrix has been recently shown to play an essential role in bacterial uptake by osteoblasts (36).
Whether bacteria internalization in osteoblasts is achieved passively or through an active process has been debated. It is likely that in fact both mechanisms are in place (37, 38). Intracellular infection has been proposed to occur very rapidly upon exposure to S. aureus. Indeed, bacteria have been detected by immunofluorescence microscopy inside murine calvaria osteoblast-like MC3T3-E1 cells as early as 15 minutes after infection, and the rate of bacteria internalization was found to increase over time (39). Invasion likely requires actin rearrangement at the cell surface; in fact, treatment with the actin depolymerization agent Cytochalasin D significantly reduced S. aureus invasion (40).
Mouton and colleagues recently demonstrated that S. aureus internalization (assessed in vitro through a gentamycin/lysostaphin protection assay that allows eliminating adherent and non-adherent bacteria, while sparing intracellular pathogen) induced an impairment in early osteoblast differentiation by interfering with β1 integrin signaling, leading to decreased expression of RUNX2 and COL1α1 and ALP activity. Accordingly, an internalization defective S. aureus strain lacking fnbpA expression did not elicit this effect (41). Consistent with this, in vivo infection with the S. aureus strain capable of cell internalization altered some bone histomorphometric parameters, supporting the hypothesis that osteoblast functions are impaired upon intracellular bacterial infection (41).
Osteoblast colonization by S. aureus stimulates the focal adhesion kinase (FAK)/epidermal growth factor receptor (EGFR) and c-Src signaling pathways by enhancing their phosphorylation in a time-dependent fashion; on the contrary, inhibition of the EGFR/FAK or c-Src signaling pathways significantly reduces the rate of pathogen internalization (39). Consequently, these pathways could be targeted in parallel to standard antibiotic therapy of chronic S. aureus osteomyelitis.
Osteoblast infection by S. aureus has been confirmed also in the presence of invasive MRSA infection of the human MG-63 osteosarcoma cell line, using imaging flow cytometry (IFC) which is more sensitive and reproducible than conventional cell culture methods (42). Pathogen uptake is known to vary depending on the strain irrespectively of antibiotic resistance (43). Statistical analysis of results obtained by IFC assays demonstrated that intracellular persistence capacity of several different MRSA strains over a 24 hour-timeframe was correlated with the total number of infected cells at 24 hour-post-infection and not with the number of bacteria that managed to enter/replicate in each single cell, defined by spot counting after cell transient permeabilization and pathogen staining with a membrane-impermeable green-fluorochrome vancomycin analogue (42). This would suggest that other factors besides the specific clone define the bacteria ability to internalize and persist inside osteoblasts. Future research is needed on this point that is relevant to chronicization of infection.
The encounter of the pathogen with osteoblasts results into release of cytokines and chemokines (as discussed later on) that recruit and activate immune cells; increased RANKL production that sustains osteoclast activity; impaired bone matrix production and mineralization; and ultimately osteoblast death through upregulation of the cell death signal transducer TRAIL and its cell surface death receptors, and concomitant decrease of the decoy receptor OPG (44, 45). All these events contribute to bone loss. Progression from acute to chronic bone and joint infections is accompanied by phenotypic adaptation of the pathogen to a less virulent form, called “small colony variant”. This is characterized by increased intracellular persistence and antibiotics resistance, and reduced cytokine release induction and immune system stimulation (46, 47). In particular, osteocytes have been recently demonstrated to constitute a reservoir for silent or persistent infection owing to reduced antimicrobial capacity to eliminate intracellular bacteria, and higher pathogen survival in the form of small colony variants (11, 48). Moreover, infected osteocytes can also elicit an inflammatory response that contributes to communication with other skeletal cells, immune cell recruitment and bone disruption (11).
The mechanisms described above are specific for S. aureus and not shared by other opportunistic bacteria involved in orthopedic infections, which points to the evolution of bacterial species-specific ways of interaction with eukaryotic cells that need to be further elucidated.
Osteoblasts as inflammatory cells
The osteoblast secretome in a non-infected setting
The secretory function is highly relevant in the framework of osteoblast activities. It comprises the release of the diverse components of the extracellular matrix, essentially calcium-deficient apatite and trace elements, for the inorganic part; type I collagen and other types of collagens, and non-collagenous proteins (γ-carboxyglutamate-containing proteins, proteoglycans, glycoproteins, and small integrin-binding ligands N-linked glycoproteins), for the organic part (49). Osteoblasts also release a variety of cytokines, chemokines, and growth factors (i.e., the osteoblast secretome) that regulate osteoclast formation and resorptive activity (e.g., M-CSF, RANKL, OPG, WNT5A, WNT16, IL-6, GM-CSF) (14), enhance the osteoblast anabolic function (e.g., IGFs and IGFBPs) (50), support hematopoiesis (e.g., G-CSF, osteopontin, thrombopoietin, angiopoietin 1, CXCL12, SCF and IL-7) (51) and act as immune modulators (e.g., IL-6, GM-CSF, CCL5) (52). In this respect, an intimate crosstalk takes place in the bone between skeletal cells and the immune system, which also involves the release of cytokines and chemokines in diverse pathophysiological contexts (53). Systemic and local factors such as inflammation and infection elicit marked changes in bone cell functions, as further described in the following sections.
The osteoblast secretome in S. aureus osteomyelitis: Cytokines and growth factors
At sites of infection, soluble factors deriving from activated immune and skeletal cells make up an inflammatory milieu that mediates reciprocal regulation of the osteal and immune components and provides host defense against pathogens. In this regard, osteoblasts play an active role, and cannot be regarded as inert niches for bacterial colonization. In fact, not only do they strive to kill intracellular bacteria by increasing the production of reactive oxygen species and oxidative stress, but they also take part in the immune response orchestrated by professional innate immune cells through production of antimicrobial peptides (beta-defensins), as shown in in vitro and in vivo models and ex vivo in human specimens (54, 55). In addition, the osteoblast secretome further boosts the inflammatory response and affect the behavior of skeletal cells (see Figure 2).
Cytokine synthesis and release involve several intracellular signaling pathways including the NF-kB pathway, which regulates the secretion of IL-6 and MCP1 (CCL2; see below) (56), and the JNK pathway, which is activated downstream of S. aureus SpA binding to TNFR1 on the osteoblast plasma membrane and leads to increased TLR2 and RANKL protein levels and TNF-α and IL-6 secretion (56).
The transcriptomic profile of infected osteoblasts has been recently comprehensively investigated by RNAseq analysis of FACS-sorted S. aureus-bearing MG-63 cells, and compared to that of non-infected cells and of a mixed cell population comprising both infected and not-infected MG-63 cells (57). Specifically, this work indicated enhanced immune and inflammatory responses in a model of long-term infection, taking advantage of engineered bacterial strains expressing a fluorescent reporter gene that allowed isolation of infected cells 6 days after infection. Top up-regulated genes included several cytokines such as IL-33, IL-32, IL-6, IL-1β, IL-1α, IL-24, G-CSF, TRAIL and TNFSF14, with higher protein levels in culture supernatants. Accordingly, in the Gene Set Enrichment Analysis, several pathways enriched in FACS-sorted infected MG-63 were related to the immune response, including functional categories such as antigen processing and presentation (of note, CD44 and HLA-DR expression has been reported in cultured human osteoblasts) (58) complement and coagulation cascade, platelet activation, Th17 cell differentiation, IL-17 pathway, NOD-like and TLR signal cascades (known to be involved in bacteria recognition), and cytokine signaling (57).
At the protein level, IL-6 has been found to be upregulated in infected murine osteoblasts (in various experimental settings) and human bone tissues (59, 60). Increased IL-6 induces COX-2 and thereby PGE2 and RANKL, which modulate osteoclast recruitment and differentiation, contributing to progressive inflammatory damage and bone loss (61). Enhanced RANKL production by osteoblasts, without concomitant significant change in OPG expression, occurs also downstream of TLR2 recognition of S. aureus; accordingly, RANKL increase is abrogated in osteoblasts from Tlr2 knockout mice. This mechanism would support pronounced bone resorption and periosteal osteoclast formation in S. aureus-infected bones (62).
IL-12 also is secreted by osteoblasts in S. aureus-induced osteomyelitis (59). It has been proposed to strengthen Th1 immune responses and favor elimination of intracellular bacteria (63), a mechanism that could be potentially used to develop novel strategies for infection prevention (64, 65). Since IL-12 in the bone microenvironment promotes myeloid-derived suppressor cell recruitment, it is plausible that osteoblasts contribute to this mechanism (66).
Expression of the highly conserved anti-inflammatory cytokine TGFβ1 has been reported to change in MG-63 cells infected with four different clinically isolated S. aureus strains (43), with 2 of them causing downregulation in the short and one upregulation in the long timeframe (3 and 24 hours post infection, respectively). Among other functions, TGFβ1 stimulates type I collagen production (67), therefore an increase of this factor might explain the abnormal matrix deposition that occurs in the periosteum during an infection.
IL-1β and TNF-α increased in the bone of animals with S. aureus-induced OM (43, 68, 69), and both cytokines stimulated osteoclast maturation and function. However, some findings regarding to IL-1β are contradictory: in vitro infection of murine primary osteoblasts with S. aureus resulted in increased transcription, but not in increased protein synthesis or secretion. Of note, the same was observed for IL-18 (70), a potent inflammatory molecule, structurally and functionally closely related to IL-1β: in fact, it favors osteoclast differentiation by expanding the inflammatory response and inhibits the osteogenic function (71). Owing to the fine line between intense or exaggerated immune response, it is reasonable that IL-1β and IL-18 production is strictly controlled (70), however their regulation deserves further investigation. In parallel, the specific IL-18 inhibitor IL-18BP was upregulated in vitro in human primary osteoblasts 2 hours after S. aureus infection (https://www.ebi.ac.uk/arrayexpress/E-MTAB-6700), while no data are available regarding IL18-BP protein production by osteoblasts in this specific context, to the best of our knowledge. Therefore, at present, activated immune cells, not osteoblasts, are the most likely source of these ILs in the bone microenvironment during bone infections.
Also, while low levels of TNF-α were detected in in vitro differentiated human osteoblasts and in the MG-63 cell line in basal conditions (72), a marked increase was observed in MG-63 upon infection (43, 73). A recent study described a novel signaling cascade comprising TNF-α/miR-129-5p/endothelial nitric oxide synthase (eNOS) in the pathogenesis of osteomyelitis (74). Briefly, TNF-α and miR-129-5p were upregulated while eNOS was downregulated in S. aureus-infected MC3T3-E1 cells and in osteomyelitis patients’ blood. Accordingly, a TNF-α blocker inhibited miR-129-5p and elevated eNOS expression, likely contributing to rescue the mineralization defect caused by S. aureus infection in MC3T3-E1 cells (74).
IL-27 expression has been recently demonstrated to be induced early (on day 1) in the infected bone in a transtibial model of S. aureus-induced osteomyelitis and upon in vitro infection of MC3T3-E1 cells and primary osteoblasts (75). This cytokine likely contributes to host innate immune response in the early phases of the infection by stimulating local neutrophil recruitment and activation.
Finally, there is preliminary evidence of IFN-β secretion by mature murine osteoblasts in response to S. aureus infection (76). This type I interferon has been reported to be a negative regulator of RANKL-mediated osteoclastogenesis by inhibiting the translation of the critical signaling component c-Fos (77). Based on these data, S. aureus would stimulate osteoblasts to release factors with opposing effects on osteoclastogenesis (78); it can be speculated that the production of IFN-β represents a compensatory response to restore bone homeostasis.
Cytokine production from infected osteoblasts has been shown also in the framework of three-dimensional (3D) models of S. aureus-induced osteomyelitis, which more closely reproduce composition and structure of the natural bone compared to conventional 2D culture systems. For example, in a 3D model of osteomyelitis based on the coculture of MC3T3-E1 cells and S. aureus on magnesium-doped hydroxyapatite/collagen I scaffolds, Tnf-α expression increased over time during infection. The same was observed for the long pentraxin Ptx3, a key pattern recognition molecule with emerging roles in bone pathophysiology, known to be induced by TNF-α. On the contrary, Tgf-β(a reported repressor of Ptx3 transcription) decreased over time. In the conditioned medium of the 3D cocultures, TNF-α was not detected, while PTX3 and OPG levels were stable over time (79). Importantly, the expression of selected osteogenic (Bmp2, Alp, Spp1), and antioxidant (Nrf-2, Ho-1) genes were substantially affected by the applied 3D setting, indicating matrix-dependent effects on osteoblasts during an S. aureus infection (79).
In S. aureus osteomyelitis, mRNA and serum protein levels of G-CSF significantly increase in infected patients, contributing to bone loss through suppression of osteoblast function in favor of osteoclast formation (80), and to enhanced phagocytic activity of immune cells. G-CSF is also released directly by osteoblasts upon S. aureus infection, as well as GM-CSF and M-CSF. In fact, GM-CSF and G-CSF mRNA expression and protein secretion were substantially upregulated in cultured mouse and human osteoblasts following interaction with S. aureus (81). Furthermore, these cytokines were induced in unexposed osteoblasts separated from infected osteoblasts by means of a transwell system, thus pointing to a paracrine-autocrine regulatory mechanism. On the contrary, M-CSF secretion increased only in cultures of infected human osteoblasts (81). These CSFs allow osteoblasts to modulate the cellular composition of the bone microenvironment, favoring differentiation of myeloid progenitors towards osteoclasts and various innate immune cell fates (66).
The osteoblast secretome: Chemokines
In response to S. aureus infection, osteoblasts also produce chemokines, members of the C-X-C-motif chemokine ligand (CXCL) and CC-motif (CCL) families, such as CCL2, CCL5, CCL7, CCL8, CCL10, CCL11, CCL13, CCL20, CCL26, and CXCL1, CXCL2, CXCL3, CXCL5, CXCL6 and CX3CL1 (57). Chemokines are traditionally known to act as immune cell chemoattractants, recruiting and activating components of the innate and adaptive immunity, however skeletal cells also are endowed with autocrine and paracrine chemokine signaling, which modulates bone turnover (82).
CCL3 and CXCL2 are known inflammatory mediators enhancing osteoclast formation and osteolysis (83). Their expression was documented in samples from osteolytic sites of patients with implant-associated infection (83). In vitro experiments showed that human primary osteoblasts released CCL3 and CXCL2 upon S. aureus infection (83). This indicates that, besides monocytes, also osteoblasts contribute to the sustained local production of these factors and the related enhanced RANKL-dependent bone resorption (82), in line with previous evidence in a mouse model (84) of S. aureus-induced osteomyelitis. Overexpression of CXCL2 would also result into inhibition of osteoblast formation through downmodulation of the ERK1/2 signaling upstream of RUNX2 (85). In accordance with a role of the CCL2-CCR2 axis in S. aureus-induced osteomyelitis, Ccr2-deficient mice had a higher bacterial load than wild type mice in a model of implant-associated S. aureus infection in which a bioluminescent bacterial strain was inoculated directly into the knee joint after implantation of an orthopedic-grade titanium pin. The infection was monitored in vivo by means of bioluminescence and ex vivo by colony-forming unit counting in the infected joint tissue (86). While the specific contribution of osteoblast-derived CCR2 could not be established in this model, the higher bacterial burden in Ccr2-deficient mice was ascribed to lower T cell and myeloid cell infiltration and overall reduced host defense against the pathogen in the absence of a functional CCR2/CCL2 axis.
In vitro S. aureus infection of human primary osteoblasts also causes a strong upregulation and release of CCL5 (also known as RANTES) compared to other cell types, such as endothelial and epithelial cells (87). The levels of this chemokine influence osteoclast and osteoblast formation and function, as highlighted in the CCL5-deficient mouse (88), besides acting as chemoattractant for monocytes/macrophages and T lymphocytes (89).
Additionally, in a mouse model of implant-associated osteomyelitis, CXCL10 and CXCL9 were up-regulated in the infected femurs versus controls at 3- and 14-days post-infection (90), suggesting a possible role of these chemokines (produced also by osteoblasts through TLR4 activation (91, 92)) in the pathological bone turnover occurring during Osteomyelitis. Of note, in clinical samples from S. aureus-infected patients and from a mouse model of MRSA skin infection, CXCL10 and CXCL9 have also been found to enhance the spontaneous release of the virulence factor SpA, however the underlying mechanism and the biological significance of this process are yet to be defined (93). In this regard, there is evidence that extracellular SpA contributes to biofilm formation by S. aureus (39), therefore the ability of certain chemokines to induce its release could paradoxically help the pathogen skip immune recognition via encasing in a protective layer of biofilm (94). A similar process could occur in principle in bone infections, further increasing the complexity of molecular interactions that underpin osteomyelitis.
Moreover, in a mouse model of endodontic infection-induced inflammation that mimics osteomyelitis of the jaw, the chemokines Cxcl5, Cxcl2, and Cxcl13 were among the top upregulated genes in bone lesions (95).
Furthermore, osteoblasts express both CXCL12 (also known as SDF-1) and its receptor CXCR4. This signaling axis has been extensively studied in relation to the bone marrow niche (96) and implicated in skeletal homeostasis (97). Its involvement in bone remodeling during Osteomyelitis is quite likely, though not specifically investigated thus far, to the best of our knowledge.
Finally, the neutrophil chemoattractants CCL5, CXCL1, and CXCL8 and the chemokines related to T cell activation CXCL9, CXCL10, and CXCL11, were strongly upregulated in human-osteocyte-like cells in response to S. aureus invasion, in ex vivo infected human bone and in bone specimens from the infected acetabulum site of patients suffering from periprosthetic joint infection (48). CCL5 and CXCL10 proteins, but not CXCL8 were also confirmed to be secreted by infected osteocyte-like cells.
Limitations of in vitro and in vivo models
Dissection of the mechanisms underlying bone infection is hindered by the involvement of a variety of cells within a complex, not easily accessible microenvironment. In vivo models have contributed significantly to our understanding of osteomyelitis and remain a valuable tool, even though they have inherent limitations (e.g., difficulties related to joint dimension and surgery procedures in small animals; different cortical bone composition and structure), as recently reviewed (98, 99).
Conventional in vitro 2D models and advanced microfluidics systems allow addressing specific hypotheses in a simplified environment (2, 100, 101), however, they also present some drawbacks: for instance, the effort to simplify the model may overlook molecules and/or cellular populations that are of pathogenetic relevance. Moreover, results achieved using cell lines may present inconsistencies with respect data obtained using primary cells. For example, S. aureus internalization has been shown to be 30-fold lower in human primary osteoblasts than in the human osteoblast cell line hFOB. On the other hand, human primary osteoblasts displayed significantly lower cell death and higher cytokine and chemokine production (87). These findings indicate that immortalized cell lines, though widely used, do not (always) faithfully reflect post-invasion and post-infection events occurring in primary cells and raise some doubts on the physiological relevance of cell line-based in vitro infection models. Conversely, the use of primary cells has drawbacks due to limited availability of material and inter-donor variability.
3D models that closely resemble the in vivo conditions have been implemented (79, 102, 103). In this regard, manufacturing of bioactive bone mimetic scaffolds that recapitulate texture and chemistry of the natural bone matrix provide unique experimental tools to study the interface between pathogens (including S. aureus) and both skeletal and immune cells in a tightly controlled setting. Yet, it is problematic to reproduce in 3D models the cellular and molecular complexity of the bone microenvironment.
Overall, consistency and translation of findings from both in vivo and in vitro settings is often problematic, and integration of different models and expertise is warranted.
Conclusions and perspectives
In conclusion, our concise overview shows that osteoblasts are actively involved in the response to infection in S. aureus osteomyelitis. In this framework, they are engaged in complex osteoimmunological interactions. This implies the release of a variety of factors collectively described as the osteoblasts secretome, which, on one hand, recruit and activate immune cells, on the other modulate skeletal cell function. The major source of most inflammatory mediators in the bone marrow are professional innate and adaptive immune cells enrolled from the periphery or differentiated locally in the infected bone (8) (see Figure 1), as cell subsets specifically endowed with this function. Nevertheless, osteoblasts have turned out to be partners in defense (and crime) in S. aureus osteomyelitis through an arsenal of diverse factors (see Figure 2). Based on the established osteoblast-osteoclast bidirectional communication occurring in pathophysiological conditions (14), we would expect bone resorbing cells in turn impact on osteoblast function during infection. This aspect would be worth investigating, also considering the increasing heterogeneity recognized within the osteoclast lineage and its possible translational implications (24, 104).
The osteoblast secretome appears to be specific to the infectious agent, but whether any of its components or combinations of them can be used as specific biomarkers of the bone infection in the clinic remains to be evaluated. The possibility to manipulate the arsenal represented by the OB secretome for therapeutic purposes in addition to standard antibiotics treatments should also be considered. The double-faced nature of several cytokines and chemokines, which foster professional immune cells but sustain bone metabolism overbalance, makes this putative strategy challenging, though worth investigating, as prompted by an unmet medical need.
Author contributions
GV and SC drafted the manuscript. GV and SC generated the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by Fondazione Beppe e Nuccy Angiolini Onlus.
Acknowledgments
GV and PV are supported by Fondazione Beppe e Nuccy Angiolini Onlus. We gratefully acknowledge their generous contribution to our research.
Conflict of interest
BB receives royalties for reagents related to innate immunity and is inventor of patents related to PTX3 and other innate immunity molecules.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. McNeil JC. Acute hematogenous osteomyelitis in children: Clinical presentation and management. Infection Drug Resistance (2020) 13:4459–73. doi: 10.2147/IDR.S257517
2. Hofstee MI, Muthukrishnan G, Atkins GJ, Riool M, Thompson K, Morgenstern M, et al. Current concepts of osteomyelitis: From pathologic mechanisms to advanced research methods. Am J Pathol (2020) 190(6):1151–63. doi: 10.1016/j.ajpath.2020.02.007
3. Rubitschung K, Sherwood A, Crisologo AP, Bhavan K, Haley RW, Wukich DK, et al. Pathophysiology and molecular imaging of diabetic foot infections. Int J Mol Sci (2021) 22(21):11552. doi: 10.3390/ijms222111552
4. Masters EA, Ricciardi BF, Bentley KLM, Moriarty TF, Schwarz EM, Muthukrishnan G. Skeletal infections: Microbial pathogenesis, immunity and clinical management. Nat Rev Microbiol (2022) 20(7):385–400. doi: 10.1038/s41579-022-00686-0
5. Hotchen AJ, McNally MA, Sendi P. The classification of long bone osteomyelitis: A systemic review of the literature. J Bone Joint Infection (2017) 2(4):167–74. doi: 10.7150/jbji.21050
6. Brinkmann J, Zeissler EC, Scharrenberg JS, Schenk J, Majjouti M, Oberste M, et al. The diagnostic value of cytokines for the discrimination of vertebral osteomyelitis and degenerative diseases of the spine. Cytokine (2022) 150:155782. doi: 10.1016/j.cyto.2021.155782
7. Evans CA, Jellis J, Hughes SP, Remick DG, Friedland JS. Tumor necrosis factor-alpha, interleukin-6, and interleukin-8 secretion and the acute-phase response in patients with bacterial and tuberculous osteomyelitis. J Infect Dis (1998) 177(6):1582–7. doi: 10.1086/515313
8. Brandt SL, Putnam NE, Cassat JE, Serezani CH. Innate immunity to staphylococcus aureus: Evolving paradigms in soft tissue and invasive infections. J Immunol (2018) 200(12):3871–80. doi: 10.4049/jimmunol.1701574
9. Souza PPC, Lerner UH. Finding a toll on the route: The fate of osteoclast progenitors after toll-like receptor activation. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01663
10. Petronglo JR, Putnam NE, Ford CA, Cruz-Victorio V, Curry JM, Butrico CE, et al. Context-dependent roles for toll-like receptors 2 and 9 in the pathogenesis of staphylococcus aureus osteomyelitis. Infection Immun, e0041722. doi: 10.1128/iai.00417-22
11. Garcia-Moreno M, Jordan PM, Gunther K, Dau T, Fritzsch C, Vermes M, et al. Osteocytes serve as a reservoir for intracellular persisting staphylococcus aureus due to the lack of defense mechanisms. Front Microbiol (2022) 13:937466. doi: 10.3389/fmicb.2022.937466
12. Dapunt U, Giese T, Stegmaier S, Moghaddam A, Hänsch GM. The osteoblast as an inflammatory cell: Production of cytokines in response to bacteria and components of bacterial biofilms. BMC Musculoskeletal Disord (2016) 17(1):243. doi: 10.1186/s12891-016-1091-y
13. Siddiqui JA, Partridge NC. Physiological bone remodeling: Systemic regulation and growth factor involvement. Physiol (Bethesda) (2016) 31(3):233–45. doi: 10.1152/physiol.00061.2014
14. Kim JM, Lin C, Stavre Z, Greenblatt MB, Shim JH. Osteoblast-osteoclast communication and bone homeostasis. Cells (2020) 9(9):2073. doi: 10.3390/cells9092073
15. Al-Bari AA, Al Mamun A. Current advances in regulation of bone homeostasis. FASEB bioAdvances (2020) 2(11):668–79. doi: 10.1096/fba.2020-00058
16. Martin TJ, Sims NA. RANKL/OPG; critical role in bone physiology. Rev Endocrine Metab Disord (2015) 16(2):131–9. doi: 10.1007/s11154-014-9308-6
17. Stracquadanio S, Musso N, Costantino A, Lazzaro LM, Stefani S, Bongiorno D. Staphylococcus aureus internalization in osteoblast cells: Mechanisms, interactions and biochemical processes. What did we learn from experimental models? Pathogens (2021) 10(2):239. doi: 10.3390/pathogens10020239
18. Le KY, Otto M. Quorum-sensing regulation in staphylococci–an overview. Front Microbiol (2015) 6. doi: 10.3389/fmicb.2015.01174
19. Sanchez CJ Jr., Ward CL, Romano DR, Hurtgen BJ, Hardy SK, Woodbury RL, et al. Staphylococcus aureus biofilms decrease osteoblast viability, inhibits osteogenic differentiation, and increases bone resorption in vitro. BMC Musculoskelet Disord (2013) 14:187. doi: 10.1186/1471-2474-14-187
20. Reffuveille F, Josse J, Velard F, Lamret F, Varin-Simon J, Dubus M, et al. Bone environment influences irreversible adhesion of a methicillin-susceptible staphylococcus aureus strain. Front Microbiol (2018) 9:2865. doi: 10.3389/fmicb.2018.02865
21. de Mesy Bentley KL, Trombetta R, Nishitani K, Bello-Irizarry SN, Ninomiya M, Zhang L, et al. Evidence of staphylococcus aureus deformation, proliferation, and migration in canaliculi of live cortical bone in murine models of osteomyelitis. J Bone Mineral Res (2017) 32(5):985–90. doi: 10.1002/jbmr.3055
22. de Mesy Bentley KL, MacDonald A, Schwarz EM, Oh I. Chronic osteomyelitis with staphylococcus aureus deformation in submicron canaliculi of osteocytes: A case report. JBJS Case connector (2018) 8(1):e8. doi: 10.2106/JBJS.CC.17.00154
23. Wu Y, Humphrey MB, Nakamura MC. Osteoclasts–the innate immune cells of the bone. Autoimmunity (2008) 41(3):183–94. doi: 10.1080/08916930701693180
24. Madel M-B, Ibáñez L, Wakkach A, de Vries TJ, Teti A, Apparailly F, et al. Immune function and diversity of osteoclasts in normal and pathological conditions. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.01408
25. Trouillet-Assant S, Gallet M, Nauroy P, Rasigade J-P, Flammier S, Parroche P, et al. Dual impact of live staphylococcus aureus on the osteoclast lineage, leading to increased bone resorption. J Infect Dis (2014) 211(4):571–81. doi:10.1093/infdis/jiu386
26. Flammier S, Rasigade J-P, Badiou C, Henry T, Vandenesch F, Laurent F, et al. Human monocyte-derived osteoclasts are targeted by staphylococcal pore-forming toxins and superantigens. PLoS One (2016) 11(3):e0150693. doi: 10.1371/journal.pone.0150693
27. Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol (2009) 17(2):59–65. doi: 10.1016/j.tim.2008.11.005
28. Yu K, Song L, Kang HP, Kwon H-K, Back J, Lee FY. Recalcitrant methicillin-resistant staphylococcus aureus infection of bone cells: Intracellular penetration and control strategies. Bone Joint Res (2020) 9(2):49–59. doi: 10.1302/2046-3758.92.BJR-2019-0131.R1
29. Krauss JL, Roper PM, Ballard A, Shih CC, Fitzpatrick JAJ, Cassat JE, et al. Staphylococcus aureus infects osteoclasts and replicates intracellularly. mBio (2019) 10(5):1128. doi: 10.1128/mBio.02447-19
30. Reilly SS, Hudson MC, Kellam JF, Ramp WK. In vivo internalization of staphylococcus aureus by embryonic chick osteoblasts. Bone (2000) 26(1):63–70. doi: 10.1016/S8756-3282(99)00239-2
31. Bosse MJ, Gruber HE, Ramp WK. Internalization of bacteria by osteoblasts in a patient with recurrent, long-term osteomyelitis. A Case Rep J Bone Joint Surg Am Volume (2005) 87(6):1343–7. doi: 10.2106/jbjs.d.02649
32. Hamza T, Li B. Differential responses of osteoblasts and macrophages upon staphylococcus aureus infection. BMC Microbiol (2014) 14(1):207. doi: 10.1186/s12866-014-0207-5
33. Josse J, Guillaume C, Bour C, Lemaire F, Mongaret C, Draux F, et al. Impact of the maturation of human primary bone-forming cells on their behavior in acute or persistent staphylococcus aureus infection models. Front Cell Infection Microbiol (2016) 6:64. doi: 10.3389/fcimb.2016.00064
34. Claro T, Widaa A, O'Seaghdha M, Miajlovic H, Foster TJ, O'Brien FJ, et al. Staphylococcus aureus protein a binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS One (2011) 6(4):e18748. doi: 10.1371/journal.pone.0018748
35. Sinha B, François PP, Nüsse O, Foti M, Hartford OM, Vaudaux P, et al. Fibronectin-binding protein acts as staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol (1999) 1(2):101–17. doi: 10.1046/j.1462-5822.1999.00011.x
36. Niemann S, Nguyen MT, Eble JA, Chasan AI, Mrakovcic M, Bottcher RT, et al. More is not always better-the double-headed role of fibronectin in staphylococcus aureus host cell invasion. mBio (2021) 12(5):e0106221. doi: 10.1128/mBio.01062-21
37. Hudson MC, Ramp WK, Nicholson NC, Williams AS, Nousiainen MT. Internalization of staphylococcus aureus by cultured osteoblasts. Microbial Pathogenesis (1995) 19(6):409–19. doi: 10.1006/mpat.1995.0075
38. Jevon M, Guo C, Ma B, Mordan N, Nair SP, Harris M, et al. Mechanisms of internalization of staphylococcus aureus by cultured human osteoblasts. Infection Immunity (1999) 67(5):2677–81. doi: 10.1128/IAI.67.5.2677-2681.1999
39. Ji Z, Su J, Hou Y, Yao Z, Yu B, Zhang X. EGFR/FAK and c-src signalling pathways mediate the internalisation of staphylococcus aureus by osteoblasts. Cell Microbiol (2020) 22(10):e13240. doi: 10.1111/cmi.13240
40. Mohamed W, Sommer U, Sethi S, Domann E, Thormann U, Schütz I, et al. Intracellular proliferation of s. aureus in osteoblasts and effects of rifampicin and gentamicin on s. aureus intracellular proliferation and survival. Eur Cells Materials (2014) 28:258–68. doi: 10.22203/eCM.v028a18
41. Mouton W, Josse J, Jacqueline C, Abad L, Trouillet-Assant S, Caillon J, et al. Staphylococcus aureus internalization impairs osteoblastic activity and early differentiation process. Sci Rep (2021) 11(1):17685. doi: 10.1038/s41598-021-97246-y
42. Bongiorno D, Musso N, Lazzaro LM, Mongelli G, Stefani S, Campanile F. Detection of methicillin-resistant staphylococcus aureus persistence in osteoblasts using imaging flow cytometry. MicrobiologyOpen (2020) 9(5):e1017. doi: 10.1002/mbo3.1017
43. Musso N, Caruso G, Bongiorno D, Grasso M, Bivona DA, Campanile F, et al. Different modulatory effects of four methicillin-resistant staphylococcus aureus clones on MG-63 osteoblast-like cells. Biomolecules (2021) 11(1):72. doi: 10.3390/biom11010072
44. Reott MA Jr., Ritchie-Miller SL, Anguita J, Hudson MC. TRAIL expression is induced in both osteoblasts containing intracellular staphylococcus aureus and uninfected osteoblasts in infected cultures. FEMS Microbiol Letters (2008) 278(2):185–92. doi: 10.1111/j.1574-6968.2007.00988.x
45. Young AB, Cooley ID, Chauhan VS, Marriott I. Causative agents of osteomyelitis induce death domain-containing TNF-related apoptosis-inducing ligand receptor expression on osteoblasts. Bone (2011) 48(4):857–63. doi: 10.1016/j.bone.2010.11.015
46. Trouillet-Assant S, Lelièvre L, Martins-Simões P, Gonzaga L, Tasse J, Valour F, et al. Adaptive processes of staphylococcus aureus isolates during the progression from acute to chronic bone and joint infections in patients. Cell Microbiol (2016) 18(10):1405–14. doi: 10.1111/cmi.12582
47. Tuchscherr L, Kreis CA, Hoerr V, Flint L, Hachmeister M, Geraci J, et al. Staphylococcus aureus develops increased resistance to antibiotics by forming dynamic small colony variants during chronic osteomyelitis. J Antimicrobial Chemother (2016) 71(2):438–48. doi: 10.1093/jac/dkv371
48. Yang D, Wijenayaka AR, Solomon LB, Pederson SM, Findlay DM, Kidd SP, et al. Novel insights into staphylococcus aureus deep bone infections: the involvement of osteocytes. mBio (2018) 9(2):e00415–18. doi: 10.1128/mBio.00415-18
49. Lin X, Patil S, Gao Y-G, Qian A. The bone extracellular matrix in bone formation and regeneration. Front Pharmacol (2020) 11. doi: 10.3389/fphar.2020.00757
50. Govoni KE. Insulin-like growth factor-I molecular pathways in osteoblasts: potential targets for pharmacological manipulation. Curr Mol Pharmacol (2012) 5(2):143–52. doi: 10.2174/1874467211205020143
51. Galán-Díez M, Kousteni S. The osteoblastic niche in hematopoiesis and hematological myeloid malignancies. Curr Mol Biol Rep (2017) 3(2):53–62. doi: 10.1007/s40610-017-0055-9
52. Terashima A, Takayanagi H. The role of bone cells in immune regulation during the course of infection. Semin Immunopathol (2019) 41(5):619–26. doi: 10.1007/s00281-019-00755-2
53. Guder C, Gravius S, Burger C, Wirtz DC, Schildberg FA. Osteoimmunology: A current update of the interplay between bone and the immune system. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00058
54. Varoga D, Tohidnezhad M, Paulsen F, Wruck CJ, Brandenburg L, Mentlein R, et al. The role of human beta-defensin-2 in bone. J Anatomy (2008) 213(6):749–57. doi: 10.1111/j.1469-7580.2008.00992.x
55. Varoga D, Wruck CJ, Tohidnezhad M, Brandenburg L, Paulsen F, Mentlein R, et al. Osteoblasts participate in the innate immunity of the bone by producing human beta defensin-3. Histochem Cell Biol (2009) 131(2):207–18. doi: 10.1007/s00418-008-0522-8
56. Ning R, Zhang X, Guo X, Li Q. Staphylococcus aureus regulates secretion of interleukin-6 and monocyte chemoattractant protein-1 through activation of nuclear factor kappaB signaling pathway in human osteoblasts. Braz J Infect Diseases (2011) 15(3):189–94. doi: 10.1016/s1413-8670(11)70173-8
57. Nicolas A, Deplanche M, Commere P-H, Diot A, Genthon C, Marques da Silva W, et al. Transcriptome architecture of osteoblastic cells infected with staphylococcus aureus reveals strong inflammatory responses and signatures of metabolic and epigenetic dysregulation. Front Cell infection Microbiol (2022) 12. doi: 10.3389/fcimb.2022.854242
58. Reyes-Botella C, Montes MJ, Vallecillo-Capilla MF, Olivares EG, Ruiz C. Expression of molecules involved in antigen presentation and T cell activation (HLA-DR, CD80, CD86, CD44 and CD54) by cultured human osteoblasts. J Periodontol (2000) 71(4):614–7. doi: 10.1902/jop.2000.71.4.614
59. Bost KL, Ramp WK, Nicholson NC, Bento JL, Marriott I, Hudson MC. Staphylococcus aureus infection of mouse or human osteoblasts induces high levels of interleukin-6 and interleukin-12 production. J Infect Diseases (1999) 180(6):1912–20. doi: 10.1086/315138
60. Marriott I, Gray DL, Tranguch SL, Fowler VG, Stryjewski M, Scott Levin L, et al. Osteoblasts express the inflammatory cytokine interleukin-6 in a murine model of staphylococcus aureus osteomyelitis and infected human bone tissue. Am J Pathol (2004) 164(4):1399–406. doi: 10.1016/S0002-9440(10)63226-9
61. Somayaji SN, Ritchie S, Sahraei M, Marriott I, Hudson MC. Staphylococcus aureus induces expression of receptor activator of NF-kappaB ligand and prostaglandin E2 in infected murine osteoblasts. Infection Immunity (2008) 76(11):5120–6. doi: 10.1128/IAI.00228-08
62. Kassem A, Lindholm C, Lerner UH. Toll-like receptor 2 stimulation of osteoblasts mediates staphylococcus aureus induced bone resorption and osteoclastogenesis through enhanced RANKL. PloS One (2016) 11(6):e0156708. doi: 10.1371/journal.pone.0156708
63. Hamza T, Barnett JB, Li B. Interleukin 12 a key immunoregulatory cytokine in infection applications. Int J Mol Sci (2010) 11(3):789–806. doi: 10.3390/ijms11030789
64. Li B, Jiang B, Boyce BM, Lindsey BA. Multilayer polypeptide nanoscale coatings incorporating IL-12 for the prevention of biomedical device-associated infections. Biomaterials (2009) 30(13):2552–8. doi: 10.1016/j.biomaterials.2009.01.042
65. Li B, Jiang B, Dietz MJ, Smith ES, Clovis NB, Rao KMK. Evaluation of local MCP-1 and IL-12 nanocoatings for infection prevention in open fractures. J Orthopaedic Res Off Publ Orthopaedic Res Society (2010) 28(1):48–54. doi: 10.1002/jor.20939
66. Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, Kielian T. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during staphylococcus aureus orthopedic implant infection. J Immunol (2015) 194(8):3861–72. doi: 10.4049/jimmunol.1402689
67. Kasagi S, Chen W. TGF-beta1 on osteoimmunology and the bone component cells. Cell Biosci (2013) 3(1):4. doi: 10.1186/2045-3701-3-4
68. Littlewood-Evans AJ, Hattenberger MR, Lüscher C, Pataki A, Zak O, O'Reilly T. Local expression of tumor necrosis factor alpha in an experimental model of acute osteomyelitis in rats. Infection Immunity (1997) 65(8):3438–43. doi: 10.1128/iai.65.8.3438-3443.1997
69. Garcia-Alvarez F, Navarro-Zorraquino M, Castro A, Grasa JM, Pastor C, Monzon M, et al. Effect of age on cytokine response in an experimental model of osteomyelitis. Biogerontology (2009) 10(5):649–58. doi: 10.1007/s10522-008-9211-1
70. Marriott I, Hughes FM Jr., Bost KL. Bacterial infection of osteoblasts induces interleukin-1beta and interleukin-18 transcription but not protein synthesis. J Interferon Cytokine Res (2002) 22(10):1049–55. doi: 10.1089/107999002760624288
71. Jiang N, An J, Yang K, Liu J, Guan C, Ma C, et al. NLRP3 inflammasome: A new target for prevention and control of osteoporosis? Front Endocrinol (Lausanne) (2021) 12:752546–. doi: 10.3389/fendo.2021.752546
72. Bu R, Borysenko CW, Li Y, Cao L, Sabokbar A, Blair HC. Expression and function of TNF-family proteins and receptors in human osteoblasts. Bone (2003) 33(5):760–70. doi: 10.1016/j.bone.2003.07.006
73. Chen Q, Hou T, Wu X, Luo F, Xie Z, Xu J. Knockdown of TNFR1 suppresses expression of TLR2 in the cellular response to staphylococcus aureus infection. Inflammation (2016) 39(2):798–806. doi: 10.1007/s10753-016-0308-4
74. Ma X, Xia W, Zong Y, Jiang C, Shan H, Lin Y, et al. Tumor necrosis factor-α promotes staphylococcus aureus-induced osteomyelitis through downregulating endothelial nitric oxide synthase. J Microbiol Immunol Infection = Wei mian yu gan ran za zhi (2021) 54(6):1018–27. doi: 10.1016/j.jmii.2020.08.002
75. Morita Y, Saito M, Rangel-Moreno J, Franchini AM, Owen JR, Martinez JC, et al. Systemic IL-27 administration prevents abscess formation and osteolysis via local neutrophil recruitment and activation. Bone Res (2022) 10(1):56. doi: 10.1038/s41413-022-00228-7
76. Johnson MB, Suptela SR, Sipprell SE, Marriott I. Substance p exacerbates the inflammatory and pro-osteoclastogenic responses of murine osteoclasts and osteoblasts to staphylococcus aureus. Inflammation (2022) 10:1007. doi: 10.1007/s10753-022-01731-z
77. Hayashida C, Ito J, Nakayachi M, Okayasu M, Ohyama Y, Hakeda Y, et al. Osteocytes produce interferon-β as a negative regulator of osteoclastogenesis. J Biol Chem (2014) 289(16):11545–55. doi: 10.1074/jbc.M113.523811
78. Takayanagi H, Kim S, Taniguchi T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res Ther (2002) 4(3):S227. doi: 10.1186/ar581
79. Parente R, Possetti V, Schiavone ML, Campodoni E, Menale C, Loppini M, et al. 3D cocultures of osteoblasts and staphylococcus aureus on biomimetic bone scaffolds as a tool to investigate the host-pathogen interface in osteomyelitis. Pathog (Basel Switzerland) (2021) 10(7):837. doi: 10.3390/pathogens10070837
80. Hou Y, Qin H, Jiang N, Liu G, Wu H, Bai L, et al. G-CSF partially mediates bone loss induced by staphylococcus aureus infection in mice. Clin Science (2019) 133(12):1297–308. doi: 10.1042/CS20181001
81. Bost KL, Bento JL, Ellington JK, Marriott I, Hudson MC. Induction of colony-stimulating factor expression following staphylococcus or salmonella interaction with mouse or human osteoblasts. Infection Immunity (2000) 68(9):5075–83. doi: 10.1128/IAI.68.9.5075-5083.2000
82. Brylka LJ, Schinke T. Chemokines in physiological and pathological bone remodeling. Front Immunol (2019) 10:2182. doi: 10.3389/fimmu.2019.02182
83. Dapunt U, Maurer S, Giese T, Gaida MM, Hansch GM. The macrophage inflammatory proteins MIP1alpha (CCL3) and MIP2alpha (CXCL2) in implant-associated osteomyelitis: linking inflammation to bone degradation. Mediators Inflammation (2014) 2014:728619. doi: 10.1155/2014/728619
84. Marriott I, Gray DL, Rati DM, Fowler VG Jr., Stryjewski ME, Levin LS, et al. Osteoblasts produce monocyte chemoattractant protein-1 in a murine model of staphylococcus aureus osteomyelitis and infected human bone tissue. Bone (2005) 37(4):504–12. doi: 10.1016/j.bone.2005.05.011
85. Yang N, Liu Y. The role of the immune microenvironment in bone regeneration. Int J Med Sci (2021) 18(16):3697–707. doi: 10.7150/ijms.61080
86. Wang Y, Dikeman D, Zhang J, Ackerman N, Kim S, Alphonse MP, et al. CCR2 contributes to host defense against staphylococcus aureus orthopedic implant-associated infections in mice. J Orthopaedic Res (2022) 40(2):409–19. doi: 10.1002/jor.25027
87. Strobel M, Pfortner H, Tuchscherr L, Volker U, Schmidt F, Kramko N, et al. Post-invasion events after infection with staphylococcus aureus are strongly dependent on both the host cell type and the infecting s. aureus strain. Clin Microbiol Infection (2016) 22(9):799–809. doi: 10.1016/j.cmi.2016.06.020
88. Wintges K, Beil FT, Albers J, Jeschke A, Schweizer M, Claass B, et al. Impaired bone formation and increased osteoclastogenesis in mice lacking chemokine (C-c motif) ligand 5 (Ccl5). J Bone Mineral Res (2013) 28(10):2070–80. doi: 10.1002/jbmr.1937
89. Josse J, Velard F, Gangloff SC. Staphylococcus aureus vs. osteoblast: Relationship and consequences in osteomyelitis. Front Cell Infection Microbiol (2015) 5:85. doi: 10.3389/fcimb.2015.00085
90. Lin Y, Su J, Wang Y, Xu D, Zhang X, Yu B. mRNA transcriptome analysis of bone in a mouse model of implant-associated staphylococcus aureus osteomyelitis. Infection Immun (2021) 89(5):e00814–20. doi: 10.1128/IAI.00814-20
91. Gasper NA, Petty CC, Schrum LW, Marriott I, Bost KL. Bacterium-induced CXCL10 secretion by osteoblasts can be mediated in part through toll-like receptor 4. Infection Immun (2002) 70(8):4075–82. doi: 10.1128/IAI.70.8.4075-4082.2002
92. Huang B, Wang W, Li Q, Wang Z, Yan B, Zhang Z, et al. Osteoblasts secrete Cxcl9 to regulate angiogenesis in bone. Nat Commun (2016) 7:13885. doi: 10.1038/ncomms13885
93. Yung SC, Parenti D, Murphy PM. Host chemokines bind to staphylococcus aureus and stimulate protein a release. J Biol Chem (2011) 286(7):5069–77. doi: 10.1074/jbc.M110.195180
94. Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, et al. Protein a-mediated multicellular behavior in staphylococcus aureus. J Bacteriol (2009) 191(3):832–43. doi: 10.1128/JB.01222-08
95. Sasaki H, Furusho H, Rider DB, Dobeck JM, Kuo WP, Fujimura A, et al. Endodontic infection-induced inflammation resembling osteomyelitis of the jaws in toll-like receptor 2/Interleukin 10 double-knockout mice. J Endodontics (2019) 45(2):181–8. doi: 10.1016/j.joen.2018.10.007
96. Aoki K, Kurashige M, Ichii M, Higaki K, Sugiyama T, Kaito T, et al. Identification of CXCL12-abundant reticular cells in human adult bone marrow. Br J Haematol (2021) 193(3):659–68. doi: 10.1111/bjh.17396
97. Shahnazari M, Chu V, Wronski TJ, Nissenson RA, Halloran BP. CXCL12/CXCR4 signaling in the osteoblast regulates the mesenchymal stem cell and osteoclast lineage populations. FASEB J (2013) 27(9):3505–13. doi: 10.1096/fj.12-225763
98. Meroni G, Tsikopoulos A, Tsikopoulos K, Allemanno F, Martino PA. Soares filipe JF. A journey into animal models of human osteomyelitis: A review. Microorganisms (2022) 10(6):1135. doi: 10.3390/microorganisms10061135
99. Roux KM, Cobb LH, Seitz MA, Priddy LB. Innovations in osteomyelitis research: A review of animal models. Anim Models Exp Med (2021) 4(1):59–70. doi: 10.1002/ame2.12149
100. Lee J-H, Wang H, Kaplan JB, Lee WY. Effects of staphylococcus epidermidis on osteoblast cell adhesion and viability on a Ti alloy surface in a microfluidic co-culture environment. Acta Biomaterialia (2010) 6(11):4422–9. doi: 10.1016/j.actbio.2010.05.021
101. Masters EA, Salminen AT, Begolo S, Luke EN, Barrett SC, Overby CT, et al. An in vitro platform for elucidating the molecular genetics of s. aureus invasion of the osteocyte lacuno-canalicular network during chronic osteomyelitis. Nanomedicine: Nanotechn Biol Med (2019) 21:102039. doi: 10.1016/j.nano.2019.102039
102. Lee J-H, Gu Y, Wang H, Lee WY. Microfluidic 3D bone tissue model for high-throughput evaluation of wound-healing and infection-preventing biomaterials. Biomaterials (2012) 33(4):999–1006. doi: 10.1016/j.biomaterials.2011.10.036
103. Lee J-H, Wang H, Kaplan JB, Lee WY. Microfluidic approach to create three-dimensional tissue models for biofilm-related infection of orthopaedic implants. Tissue Eng Part C: Methods (2010) 17(1):39–48. doi: 10.1089/ten.tec.2010.0285
Keywords: osteomyelitis, infection, bone, osteoblast, staphylococcus aureus, cytokines, chemokines
Citation: Granata V, Possetti V, Parente R, Bottazzi B, Inforzato A and Sobacchi C (2022) The osteoblast secretome in Staphylococcus aureus osteomyelitis. Front. Immunol. 13:1048505. doi: 10.3389/fimmu.2022.1048505
Received: 19 September 2022; Accepted: 03 November 2022;
Published: 22 November 2022.
Edited by:
Marita Troye Blomberg, Stockholm University, SwedenReviewed by:
Fintan Thomas Moriarty, AO Research Institute, SwitzerlandDiana Boraschi, Chinese Academy of Science (CAS), China
Copyright © 2022 Granata, Possetti, Parente, Bottazzi, Inforzato and Sobacchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Sobacchi, Y3Jpc3RpbmEuc29iYWNjaGlAaHVtYW5pdGFzcmVzZWFyY2guaXQ=