 Artem Oganesyan1,2*
Artem Oganesyan1,2* Andrew Gregory3
Andrew Gregory3 Florent Malard4Nerses Ghahramanyan2Mohamad Mohty4
Florent Malard4Nerses Ghahramanyan2Mohamad Mohty4 Dickran Kazandjian5
Dickran Kazandjian5 Arsène Mekinian6,7Yervand Hakobyan1,2 on behalf of MINHEMON
Arsène Mekinian6,7Yervand Hakobyan1,2 on behalf of MINHEMON- 1Department of Hematology and Transfusion Medicine, National Institute of Health, Yerevan, Armenia
- 2Department Of Adult Hematology, Hematology Center after Prof. R. Yeolyan, Yerevan, Armenia
- 3Wayne State University School of Medicine, Detroit, MI, United States
- 4Department of Clinical Hematology and Cellular Therapy, INSERM, Saint-Antoine Research Centre, Assistance Publique-Hôpitaux de Paris, Hôpital Saint Antoine, Paris, France
- 5Myeloma Program, Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, United States
- 6Department of Internal Medicine (DMU i3), Sorbonne University, Assistance Publique-Hôpitaux de Paris, Hôpital Saint Antoine, Paris, France
- 7French-Armenian Clinical Research Center, National Institute of Health, Yerevan, Armenia
Monoclonal gammopathy of clinical significance (MGCS) represents a new clinical entity referring to a myriad of pathological conditions associated with the monoclonal gammopathy of undetermined significance (MGUS). The establishment of MGCS expands our current understanding of the pathophysiology of a range of diseases, in which the M protein is often found. Aside from the kidney, the three main organ systems most affected by monoclonal gammopathy include the peripheral nervous system, skin, and eye. The optimal management of these MGUS-related conditions is not known yet due to the paucity of clinical data, the rarity of some syndromes, and limited awareness among healthcare professionals. Currently, two main treatment approaches exist. The first one resembles the now-established therapeutic strategy for monoclonal gammopathy of renal significance (MGRS), in which chemotherapy with anti-myeloma agents is used to target clonal lesion that is thought to be the culprit of the complex clinical presentation. The second approach includes various systemic immunomodulatory or immunosuppressive options, including intravenous immunoglobulins, corticosteroids, or biological agents. Although some conditions of the MGCS spectrum can be effectively managed with therapies aiming at the etiology or pathogenesis of the disease, evidence regarding other pathologies is severely limited to individual patient data from case reports or series. Future research should pursue filling the gap in knowledge and finding the optimal treatment for this novel clinical category.
Introduction
Monoclonal gammopathy of undetermined significance (MGUS) is a precancerous clonal plasma or lymphoplasmacytic proliferative disorder, which is defined by an asymptomatic appearance of monoclonal immunoglobulin (called M protein) in the serum at a concentration of <3 g/dL as well as less than 10% of bone marrow infiltration with plasma cells (1). MGUS is one of the most common premalignant conditions affecting 1-3% of adults which may lead to multiple myeloma (MM) (2, 3). In the contrast, MM is characterized by malignant plasma cell proliferation that produces M protein (usually >3g/dL) with ≥10% of bone marrow infiltration and is often manifested by end-organ damage commonly known as CRAB criteria (hypercalcemia, renal insufficiency, anemia, bone lytic lesions) (1). On average, MGUS has an annual progression rate of 1-2% (4) and is more prevalent in males and Blacks with increasing incidence in older adults (5). MGUS is mostly sporadic, although genetic predisposition may also play a role (6). There are 3 main clinical subtypes based on the type of M protein present: non-IgM, IgM, and light-chain MGUS. With regards to the risk of transformation into MM, MGUS is categorized as low, intermediate or high risk based on M protein level, type of M protein and free light chain ratio (7). Patients who do not meet the criteria for MM and have no symptoms are usually not treated, but rather monitored every 2-3 years for low-risk MGUS and annually for intermediate and high-risk MGUS for possible disease progression and potential complications, such as fractures, thromboembolic disease, or secondary malignancies (8).
Even in the absence of MM, various types of organ damage in the context of MGUS have been observed, involving neurological, skin, blood, and eye diseases. Importantly, the spectrum of these pathologies may range from single-organ disorders to systemic diseases. This new clinical entity is called monoclonal gammopathy of clinical significance (MGCS). Diagnosis of this disease is complicated by non-specific and alternating symptoms, poor understanding of pathogenesis, as well as complex clinical presentations. Although several disease pathways have been proposed, including monoclonal immunoglobulin deposition in tissues, autoantibody activity of M-protein, cytokine activation, and complement alternate pathway activation; the mechanisms are widely unknown (9). Moreover, the optimal management of these patients is unclear and yet to be determined.
One of the well-discussed examples is monoclonal gammopathy of renal significance (MGRS), in which kidney damage (e.g., tubulopathy, glomerulopathy, glomerulonephritis) caused by M-protein deposits, involving light chains, heavy chains, or intact immunoglobulins, occurs in the absence of MM or lymphoproliferative disorder. Rigorous research has led to an improved understanding of the disease, its diagnosis and management, in which the treatment is rather directed at protein-producing clones and type of pathological injury and not at histopathological features (10). Currently, anti-clonal therapy against either B-cells or plasma cells with novel anti-myeloma regimens (i.e., proteasome inhibitors, monoclonal antibodies, alkylating agents, immunosuppressants) is known to be more effective compared to immunomodulatory treatment commonly used for autoimmune-related renal diseases (11).
The biggest dilemma facing clinicians is the therapeutic target of MGCS. Two main approaches exist (Figure 1), one of which suggests an anti-paraproteinemic strategy that involves reduction or elimination one of which suggests an anti-paraproteinemic strategy that involves reduction or elimination of the aberrant clone-producing M protein. These various chemotherapeutic regimens aim to address the hematological aspect of MGCS suggesting that monoclonal gammopathy is the main etiological driver. The second approach focuses on immune modulation with common therapies, such as systemic corticosteroids, intravenous immunoglobulins (IVIG), or biologic agents, among others. This strategy supposes that immune dysfunction is the primary culprit of the complex disease. The choice between these approaches can be related to the possible pathogenesis of the disease (Table 1).
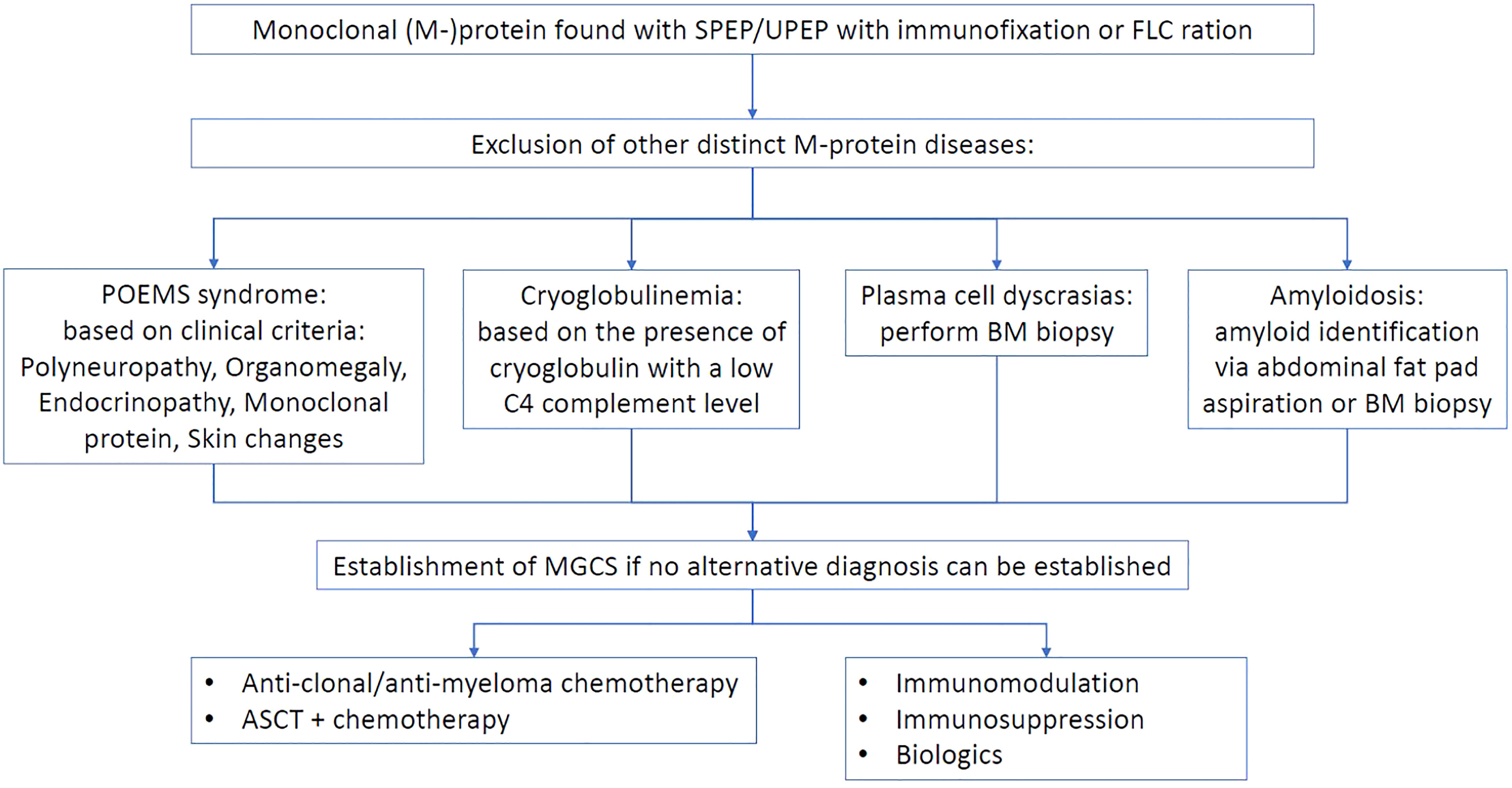
Figure 1 Proposed management approach to monoclonal gammopathy of clinical significance.

Table 1 Potential mechanisms of MGCS-related diseases.
The present narrative literature review is based on an extensive literature search (described in the Supplement) and aims at describing the management of the various clinical disorders (aside from kidney pathologies) constituting the MGCS spectrum. Some clinical entities (e.g., POEMS or cryoglobulinemia) are not discussed in this article as they were addressed in several detailed reviews (13, 14).
MGUS-associated peripheral nervous system involvement
Peripheral neuropathies have been associated with MGUS (most commonly of IgM origin) and were shown to be the most frequent indication for the diagnostic workup in this patient group, compromising almost a fifth of individuals with MGUS (15). The prevalence of M protein in peripheral neuropathies was estimated to range from 3 to 10% (16). Several mechanisms mediated by M protein activity have been proposed, including demyelination, binding to myelin-associated glycoprotein, as well as antiganglioside antibodies (15).
Chronic inflammatory demyelinating polyneuropathy
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a progressive and relapsing immune-mediated inflammation characterized by peripheral muscle weakness and sensory impairment (17). CIDP has multiple subtypes, one of which is associated with MGUS, in which monoclonal IgM antibodies are directed against myelin-associated glycoprotein (MAG) in around half of the patients leading to demyelination of distal large sensorimotor fibers (17). In a population-based study of 17,398 Minnesota residents (603 with MGUS and 16,793 controls), individuals with MGUS were shown to have a six-fold increased risk of CIDP compared to the general population (18). The typical picture of nerve biopsy includes the widening of the myelin lamellae as well as IgM and C3d deposits on myelin sheaths (18, 19). The primary treatment options for MG-associated CIPD involve plasmapheresis (19, 20), IVIG (21, 22), and steroids alone (23–25) or in combination with cyclophosphamide (19, 20, 25) (Table 2). Patients with slow progression and minimal symptoms may not require any intervention (19, 27). Interestingly, CIDP-MGUS is more responsive to plasma exchange (reaching on average 74%) compared to other types of CIDP, such as sensory, multifocal, or diabetes-associated (26). The cumulative efficacy of IVIG across studies is around 60% but ranges from 33% to 76%, likely attributable to other patient characteristics, such as disease severity or the origin of neuropathy (e.g., diabetes) (21–23, 27). Similarly, the success of plasma exchange (average response rate of 74%) and steroids (average response rate of 60%) in clinical improvements may also vary substantially from case to case (20, 38–40). Plasmapheresis can be effective in combination with chemotherapy or immunosuppression (20). The combination of cyclophosphamide with steroids gives a 55% response rate on average (19, 25). Adjunction of immunosuppressant (azathioprine, mycophenolate-mofetil, cyclosporin A) or immunomodulatory (lenalidomide) drugs may be warranted in treatment-resistant situations, but the data in this area is very limited (20, 24, 38–41). A case report on severe CIDP-MGUS showed a sustainable improvement with rituximab (42).
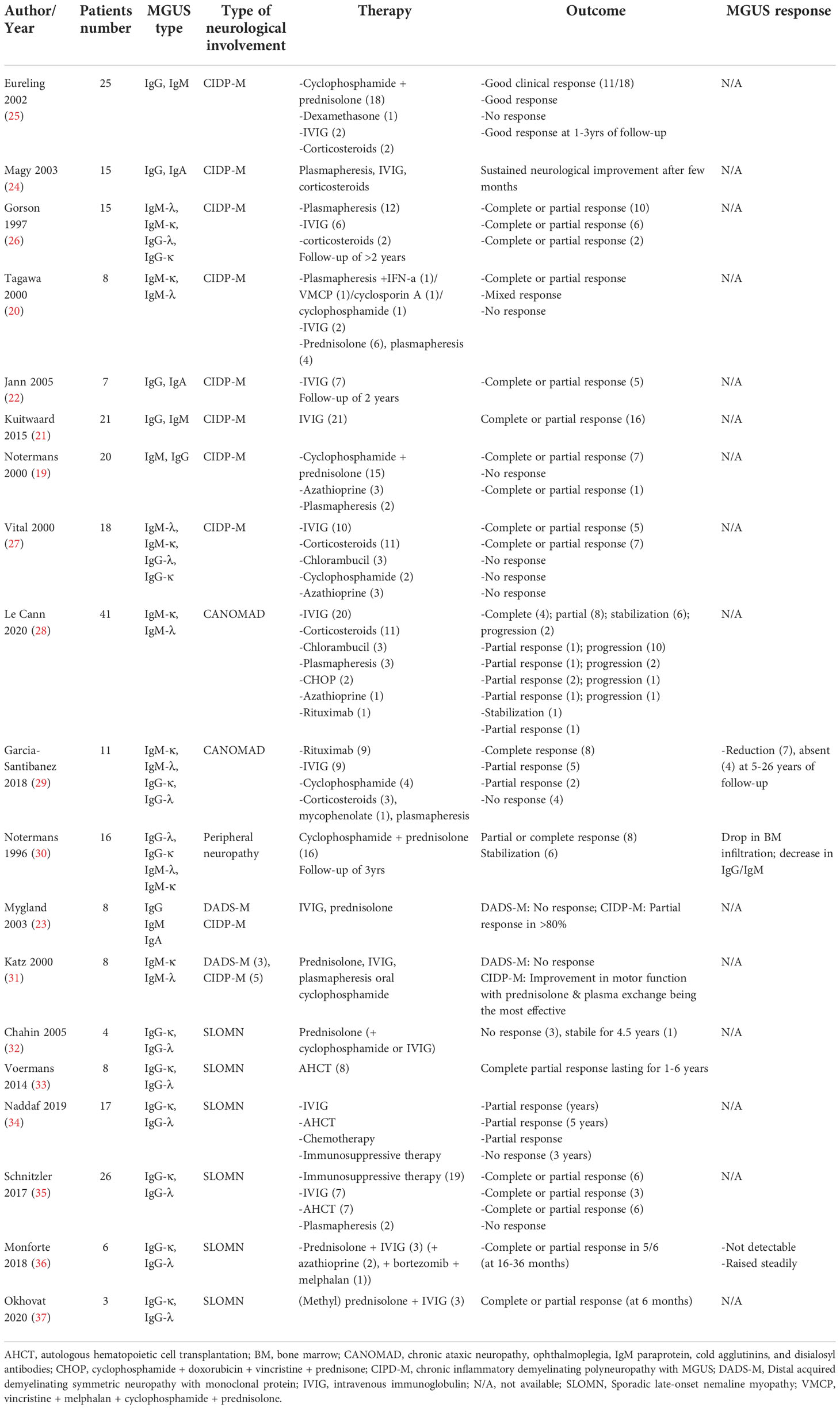
Table 2 Literature review on the management of MGUS-related peripheral nervous system disorders.
Distal acquired demyelinating symmetric neuropathy with monoclonal protein
A slightly distinct peripheral nervous system disorder associated with MGUS is distal, acquired, demyelinating, symmetric neuropathy with M protein (DADS-M), which occurs in older males affecting large sensory nerve fibers, resulting in sensory ataxia and diminished sensory response, and motor neurons with decreased motor conduction velocity and prolonged distal latencies (43). Muscles of the face, proximal limbs and trunk are usually intact (44). This syndrome is distinct from the classical DADS by the clinical picture and pathogenesis, which is recognized as a subtype of CIDP. Nonetheless, the treatment, which is largely similar to the one described for CIDP with comparably worse outcomes overall, is based primarily on the severity of neurological symptoms, rather than on the levels of IgM. Moreover, the detection of anti-MAG, which is present in about 50-70% of patients (44, 45), takes a decisive role in the choice of therapy as it is mostly resistant to standard treatment options (28, 41), such as IVIG, plasmapheresis, or systemic glucocorticoids (44, 45). Second-line treatment includes rituximab, lenalidomide, carfilzomib, or cyclophosphamide, the data on which is however limited (40).
Chronic ataxic neuropathy, ophthalmoplegia, IgM paraprotein, cold agglutinins, and disialosyl antibodies
Another complex syndrome observed in patients with MGUS is chronic ataxic neuropathy, ophthalmoplegia, IgM paraprotein, cold agglutinins, and disialosyl antibodies (CANOMAD). The proposed pathophysiology of CANOMAD involves IgM-mediated autoantibodies against gangliosides with disialosyl groups affecting sensory, ocular, and bulbar nerves leading to gait problems, muscle weakness, and paresthesia (28, 29). The disease is progressive in its nature with possible chronic relapses (28). Several effective treatment options exist based on data from case reports and series, with the most supported being IVIG (46–48) and rituximab-based strategies (46, 47, 49, 50). A French multicenter retrospective study of 45 patients with CANOMAD found 53% and 52% clinical response with IVIG and rituximab, respectively, while immunosuppressants were not shown to be particularly beneficial (28). These findings were supported by an earlier series of 11 cases (29), although some case reports claimed the efficacy of steroids (51, 52).
Sporadic late onset nemaline myopathy
Sporadic late-onset nemaline myopathy (SLOMN) is characterized by subacute and progressive muscular weakness, pain, and atrophy involving myofibers of the proximal limbs, neck, and face (36). Clinical presentation typically includes head drop, dysphagia, dysarthria, nemaline bodies in the cytoplasm, heart failure, and severe respiratory insufficiency leading to death within a few years of onset (32, 35, 36). It occurs in middle-aged men and women with more than half of patients having MGUS (34, 35). The exact mechanism and etiology of SLOMN are unclear. For the first-line treatment of SLOMN autologous hematopoietic cell therapy (AHCT) following high-dose melphalan should be considered, which demonstrated high rates of favorable response with hematologic and muscular improvements with over half of patients having positive long-term outcomes (33, 35, 37, 53). Chemotherapy directed against MM (lenalidomide with dexamethasone, rituximab with cyclophosphamide, or bortezomib with cyclophosphamide and dexamethasone) for patients not suitable for AHCT was shown to lead to some positive outcomes, such as disease stabilization and improved muscular performance (33, 53). IVIG is another alternative with comparably modest efficacy (35, 36, 53). Plasmapheresis is only partially effective, whereas steroids and other immunosuppressive therapies showed not promising outcomes for patients with SLOMN (30, 32, 36, 37, 54).
MGUS-associated cutaneous involvement
Skin is one of the most commonly affected organs by a clonal proliferation of lymphocytes or plasma cells (Figure 2). Aside from Waldenström macroglobulinemia and amyloidosis (discussed elsewhere), which are the result of extravascular depositions of M-protein, a myriad of other cutaneous manifestations can be caused by vascular deposits, abnormal cytokine response, or pathological activity of immunoglobulins (62) (Table 3).
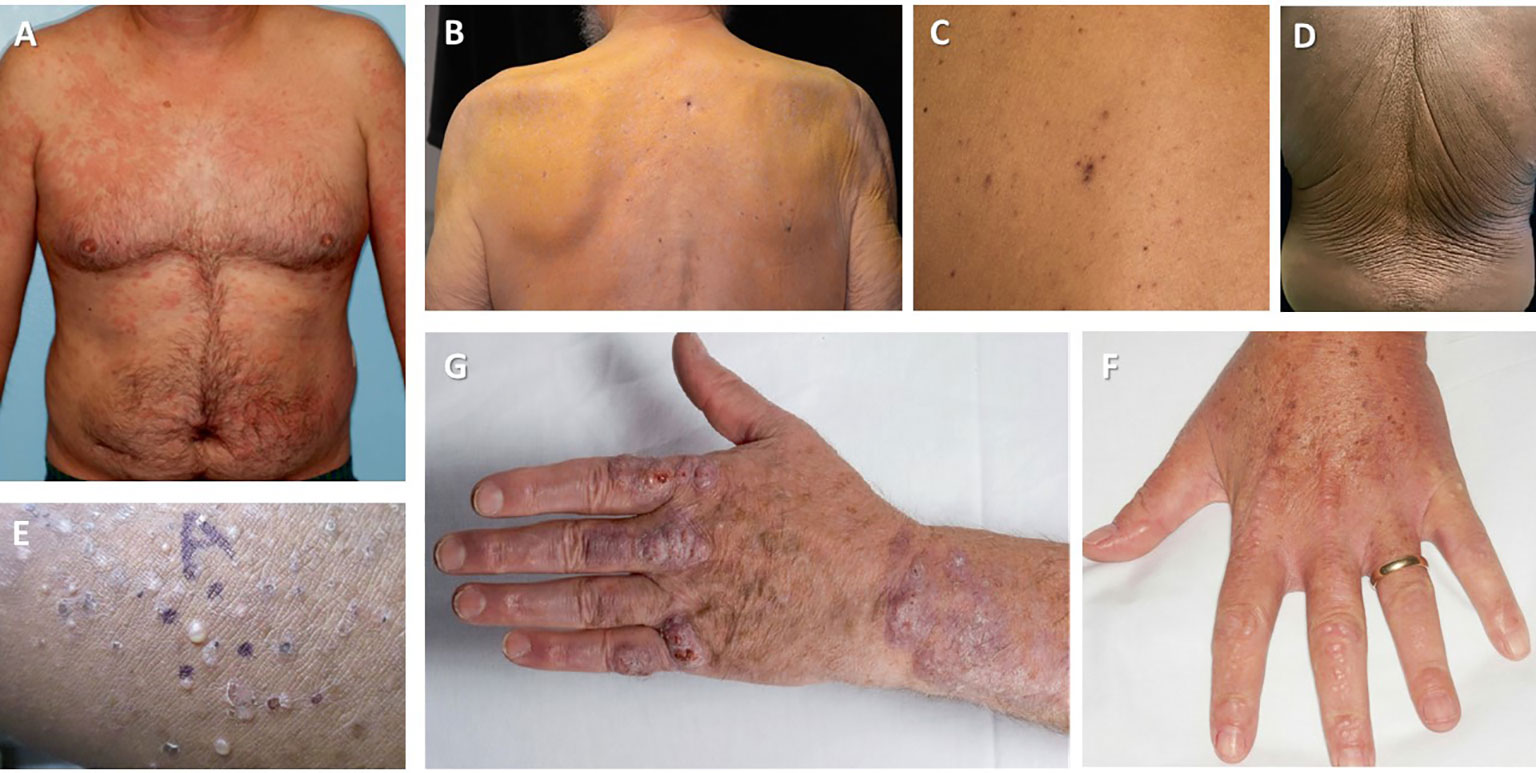
Figure 2 Cutaneous manifestations of MGCS. (A) Schnitzler syndrome (Image courtesy: Wilmas et al. 2018 (55)) (B) Nonhyperlipidemic xanthomatosis (Image courtesy: Cohen et al. 2015 (56)) (C) Telangiectasias in TEMPI syndrome (Image courtesy Khan et al., 2014 (57)) (D) Acquired cutis laxa (Image courtesy: Shalhout et al., 2010 (58)) (E) Subcorneal pustular dermatosis (Image courtesy: Young et al., 2021 (59)) (G) Necrobiotic xanthogranuloma (Image courtesy: Inthasotti et al. 2010 (60)) (F) Scleromyxedema (Image courtesy: Claveau et al. 2022 (61)).
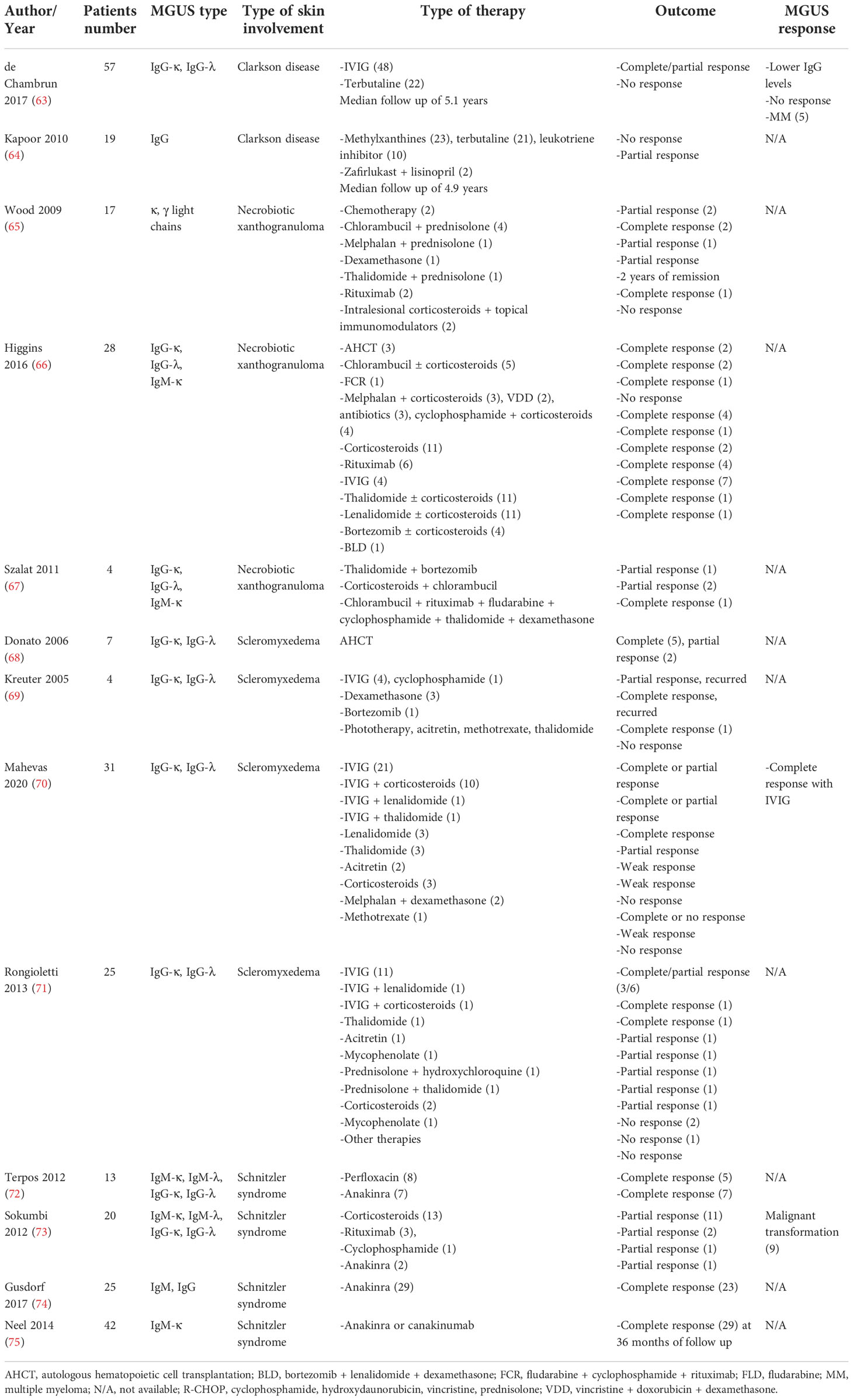
Table 3 Literature review of the management of MGUS-related skin disorders.
Schnitzler syndrome
Schnitzler syndrome is a rare systemic late onset autoinflammatory disease characterized by periodic fever, urticarial rash (neutrophilic urticarial dermatosis), bone pain with osteosclerotic changes, myalgia, lymphadenopathy, and arthralgia, as well as immunoproliferative disorders, such as B-cell lymphoma or MGUS (primarily IgM with kappa component). The disease mainly affects middle-aged adults of all ethnic groups and both sexes (76). Complications include lymphoproliferative disorder and AA amyloidosis, if left treated (76, 77). Schnitzler syndrome is thought to be a result of abnormal activation of the innate immune system with aberrant functioning of cytokines (77). Data from individual cases and clinical trials indicate that the most effective treatment option to date is anakinra (100 mg), the IL-1 receptor antagonist, which targets the key pathogenic mechanism of this disease and leads to complete remission in over 80% of patients (72, 74, 75, 78–82). Other therapies blocking IL-1 include rilonacept (IL-1 inhibitor) and canakinumab (IL-1 beta inhibitor), both of which demonstrated substantial clinical efficacy (83, 84). Second-line therapies, used previously before the introduction of anti-IL-1 treatment, include systemic glucocorticoids, NSAIDs, antihistamines, immunosuppressants, biologics, antimetabolites, showing limited efficacy and unfavorable safety profile overall (72, 73, 80, 81, 85–87). Emerging data also suggests promising use of Bruton tyrosine kinase (BTK) inhibitors (e.g., ibrutinib) as a mode of anti-clonal therapy (88).
Scleromyxedema
Scleromyxedema is a rare cutaneous mucinosis with some systemic manifestations in association with MG. It usually occurs in middle-aged adults and is characterized by a generalized papular rash with sclerosis, which is the result of fibrosis and mucin deposition (70, 71). Multiorgan involvement (e.g., cardiac, digestive, lung, kidney, musculoskeletal and nervous systems) is the main cause of high morbidity and mortality (71). The etiology and pathogenesis of scleromyxedema are not fully clear but include fibroblast proliferation which might be stimulated by cytokine dysregulation and paraproteins (89). MGUS mostly involves IgG with lambda light chain (90). Based on case series and observational studies, the preferred treatment usually includes IVIG with systemic glucocorticoids, and immunomodulatory drugs (thalidomide or lenalidomide) being a second-line choice. High-dose IVIG (2g per kg given over 5 consecutive days every 4-6 weeks) has been shown to result in clinical remission of the disease for non-severe forms of scleromyxedema (i.e., without cardiac or CNS involvement) (71). The majority of the patients reach at least partial response after 4-6 cycles of this treatment, although improvements might be already visible after just the first two cycles as well (70). Remission may last from a few months to several years; therefore, maintenance therapy (every six to twelve weeks) is often warranted. For severe cases (i.e., refractory to high-dose IVIg or with cardiac and CNS involvement), anti-plasma cell therapies should be advocated.
Alternative therapies include thalidomide or lenalidomide, which are combined with IVIG or used in patients that cannot receive the latter one. Thalidomide. given at a dose of 50-100mg/day and further increased to 200-400mg/day, was shown to lead to improvements in skin lesions and paraprotein concentrations, as well as amelioration of some clinical symptoms within several months to a few years (91–93). Thalidomide adjunction to IVIG may also potentiate the therapeutic effects in complex cases (94). The limitations of the treatment include side effects (peripheral neuropathy) and the length of therapy required until clinical results are seen. Lenalidomide (10-25mg/day for days 1–21 of a 28-day cycle) has a better safety profile but has been tested only in combination with dexamethasone and IVIG (70, 95).
Systemic glucocorticoids (prednisone at 0.5-1 mg/kg per day, prednisolone at 0.3-0.5 mg/kg per day, or oral dexamethasone at 40 mg/day) are another option when the initial therapy has failed. They can be applied either alone or in combination with IVG or thalidomide. The mixed efficacy is based on the data from the case series showing regression of skin manifestations (69), although treatment failure has also been reported (70, 71).
Bortezomib (1.3 mg/m2, on days 1, 4, 8, and 11 every 21 days) in combination with 40 mg dexamethasone has been reported in severe and refractory cases with some success (96, 97) as well as in dermato-neuro syndrome, which is an acute and potentially fatal neurological complication (98). Successful addition of thalidomide to this regimen was also described (99).
Melphalan, a chemotherapeutic agent, has been previously widely used for scleromyxedema but was later abandoned due to severe adverse events, such as sepsis or secondary hematological malignancy (100). Currently, it may be combined with other therapies, such as IVIG, AHCT, or glucocorticoids (68, 100, 101).
Several case reports described high dose melphalan followed by AHCT in selective patients to result in partial remission of systemic manifestations of scleromyxedema (68, 102, 103). Similarly, plasmapheresis has been described to be used in severe and acute cases of scleromyxedema with some efficacy (70).
A myriad of other therapeutic options tried for the treatment of scleromyxedema included retinoids (acitretin and oral isotretinoin) (71, 104), immunosuppressants (mycophenolate mofetil, cyclosporine) (105, 106), and biologics (TNF-alpha inhibitors, interferon-alfa) (107, 108), and chemotherapeutics (methotrexate, cyclophosphamide) (109, 110). The efficacy of these options needs to be further investigated.
Necrobiotic xanthogranuloma
Necrobiotic xanthogranuloma (NXG) is another idiopathic cutaneous pathology associated with paraproteinemia observed in older adults with a mean age of 62 years (111). Classically, it is described as a non–Langerhans cell histiocytosis manifested with papules, plaques, or nodules of various colors most commonly on the periorbital skin, although other regions of the body can also be affected (66). Systemic lesions with ocular, gastrointestinal, cardiac, and respiratory involvement may also occur (65). It has been estimated that 82% of the patients with NXG present with MG, with IgG-kappa being the most common subtype followed by IgG-lambda, IgG-kappa, IgA, and IgM (111).
The optimal treatment of NXG is still unclear providing undefined pathogenesis of the disease. Chemotherapy is reasonable for patients with underlying malignancy (e.g., multiple myeloma or chronic lymphocytic leukemia). Alkylating agents, such as chlorambucil (2-4mg/day) or melphalan (10mg), alone or in combination with other systemic therapies, were shown to result in cutaneous lesion improvements and lesser normalization in paraproteinemia in retrospective observational studies and case reports (65, 67, 112–114). Severe adverse events however limit the applicability of both drugs. Alternatively, oral cyclophosphamide (1 mg/kg per day for six months) can be used alone or in combination with steroids (112, 115, 116). Similarly, bortezomib alone or combined with steroids and/or lenalidomide/thalidomide is another choice for patients for whom chemotherapy is an option as it may lead to improved skin disease (66, 67). Successful application of IVIG inducing a complete or partial clinical response was described in several reports (66, 111, 117). Systemic glucocorticoids (pulsed dexamethasone or prednisone) were associated with symptomatic regression of NXG in a series of cases (65, 118). Some limited data also exist on benefits from the treatment with thalidomide and lenalidomide, including remission of skin lesions and decrease in gammopathy (65, 119, 120). Plasmapheresis or AHCT should be reserved for refractory cases (65, 121). Patients with more localized scenarios may benefit from topical or intralesional preparations, such as immunomodulators or steroids, as well as ultraviolet or radiotherapy (122–125). Surgery is an important component of the management of these patients both from cosmetic and functional point of view (126, 127). Other potential therapy options may include dapsone (128), antimalarials (111), and biologics (125, 129, 130).
Hyperlipidemic and nonhyperlipidemic xanthomatosis
Xanthomatosis is a skin manifestation (cholesterol depositions) of a disturbance in lipid metabolism with or without hyperlipidemia. The condition is often associated with MGUS of IgG lambda or IgG kappa chains (131–134). Data on the treatment of xanthomatosis is lacking, but one report showed regression of xanthomas with probucol combined with topical steroid and oral antihistamines (132). However, others failed to demonstrate any benefits with cholestyramine, gemfibrozil (131), or steroids (67, 124). Chemotherapy for underlying hematological malignancy led to positive outcomes in some patients but did not show results in others (131, 133).
TEMPI syndrome
TEMPI syndrome (Telangiectasias, Erythrocytosis and Erythropoetininemia, Monoclonal Gammopathy, Periphiric fluid accumulation, Intrapulmonary shunting) is a very rare, acquired disease manifested in middle-aged men and women across all ethnicities around the world (135, 136). The disorder has no definitive identified genetic component and is not fully clear in its pathogenesis (although the role of macrophage migration inhibitory factor (MIF) was suggested in the development of disease) (137), and MGUS, seen in all reported patients, is not restricted to any specific type, unlike in other conditions described here (135, 136). As of 2022, there have been a little more than 30 cases reported worldwide (135–147). Therefore, the treatment options are based on individual data only. To date, bortezomib is the most frequently tested treatment revealing mostly positive outcomes in several reports (135, 138–142). Treatment with daratumumab (anti-CD38 monoclonal antibody) was shown to elicit a complete symptomatic remission in two patients (143), but did not help in the management of another individual (144). AHCT was also described and resulted in complete hematological remission in one patient but was unsuccessful in two other cases (138, 145, 146). Similarly, lenalidomide was also attempted with ambiguous clinical results (143, 147).
Acquired cutis laxa
Acquired cutis laxa is a rare form of connective tissue disorder manifested by loose and inelastic skin due to the degradation of cutaneous elastic fibers. Patients of different ages usually present with “premature aging skin”, which has multiple wrinkled lesions (148). It has been often associated with systemic and cutaneous inflammatory conditions, drug exposure, as well as hematological malignancies and MG, which primarily contains IgG lambda or kappa light chains (58, 148–152). Treatment of acquired cutis laxa is targeted at associated hematological or systemic diseases as no specific options (aside from reconstructive surgery or laser tightening) exist for that condition (148, 149). It is also believed that the management of related disorders will lead to dermatological improvement (58, 150–152); however, this hypothesis has not been proven yet.
Neutrophilic dermatosis
Neutrophilic dermatosis is a diverse group of skin disorders characterized by severe infiltrations involving different cutaneous layers and manifests as ulcerations, pustules, ulcers, or nodules (153–155). Although neutrophilic dermatosis is often associated with inflammatory conditions and extracutaneous involvement, the disease itself is not mediated by infections or vasculitis (154). The exact mechanisms of pathologies are not well understood, and histopathology varies from type to type (154, 155). Despite evolving evidence suggesting the role of myeloid dysfunction in neutrophilic dermatosis, some forms of the disease have been associated with MGUS, including pyoderma gangrenosum, erythema elevatum diutinum, subcorneal pustular dermatosis, Sweet’s syndrome, and neutrophilic urticarial dermatosis (153–156). Treatment varies depending on the type of neutrophilic dermatosis. MGUS mostly relates to IgA and rare IgG with kappa or lambda bounds (153–168).
For pyoderma gangrenosum, which is characterized by bullous or pustular painful ulcers, wound management is the key treatment component with application of topical corticosteroids and local calcineurin inhibitors for localized disease (162). Systemic glucocorticoids or cyclosporine are usually considered for more advanced cases (163, 164). In patients with concomitant MG, systemic glucocorticoids and dapsone (with or without minocycline) have been the most commonly used options with mixed results (153, 158). Bortezomib–dexamethasone regimen led to resolution of lesions in one report (161). Other potential options described in the literature include colchicine, splenectomy, thalidomide, cyclophosphamide, clofazimine, methotrexate, IVIG, and azathioprine, the efficacy of which is yet to be determined (153).
Subcorneal pustular dermatosis (also known as Sneddon-Wilkinson disease) is defined by annular flaccid pustules localized to the axial and inguinal regions (165). Standard therapy involves dapsone, systemic glucocorticoids, or phototherapy (166). Acitretin (25 mg/day or higher) was described in several patients with concurrent gammopathy to lead to the resolution of lesions (153, 157, 159, 163). The potential use of biologic agents (infliximab, etanercept, and adalimumab) was also described in another case report (160).
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a systemic inflammatory condition characterized by widespread erythematous papules or plaques with neutrophilic infiltrates as well as arthritis, fever, and neutrophilia. It is classified as idiopathic, drug-induced or malignancy-associated (167). Traditionally, systemic, topical, or intralesional glucocorticoids are the first-line treatment choice (168). Alternative options include colchicine, dapsone (168). The presence of monoclonal gammopathy seems to not alter the treatment approach in these patients (153).
Neutrophilic urticarial dermatosis is a chronic and recurrent condition with erythematous macules and plaques on extremities and trunk usually resolving in 24-48 hours after eruptions without any residual lesions (169). Aside from being a pathologic hallmark of Schnitzler syndrome, it is commonly associated with adult-onset Still’s disease, lupus erythematosus, and cryopyrin-associated periodic syndromes (169). Dapsone, colchicine, topical steroids, and anakinra are linked to clinical improvements (153, 155, 170).
Clarkson disease (systemic capillary leak syndrome)
Clarkson disease or systemic capillary leak syndrome, first described in 1960, is characterized by sporadic and recurrent episodes of hypovolemic shock and anasarca, which are caused by widespread leakage of plasma and proteins into the extravascular compartment of various tissues and subsequent hypoalbuminemia and hemoconcentration (171, 172). Complications include pulmonary edema, compartment syndrome, and ischemic damage of organs (64, 173). MGUS seen in a majority of patients is IgG kappa or lambda light chains (63, 64). Despite the potential pathologic role of elevated levels of vascular permeability factors (174), the exact etiology is unknown, and the severity of the clinical manifestations varies from case to case (63). Judicious fluid resuscitation and hemodynamic support with the aim to restore the perfusion but at the same time avoid potential complications (e.g., pulmonary edema and compartment syndrome) are key for the management of acute episodes of hypovolemic shock (175, 176). A number of reports described successful application of high-dose IVIG (2g/kg) as a prophylactic measure and long-term treatment leading to a decrease in severity and frequency of attacks (63, 175). Terbutaline with aminophylline or theophylline was reported in several patients with variable efficacy for the prevention of subsequent episodes (173, 175–178). In a study of 69 patients with Clarkson disease, treatment with IVIG was associated with lower mortality and fewer recurrence rate compared to terbutaline in a median follow-up of 5 years (63). Limited data also exist regarding treatment with bevacizumab, infliximab, verapamil, thalidomide, leukotriene inhibitors, and glucocorticoids (64, 176, 179–181).
MGUS-associated ocular involvement
Ophthalmologic injury (sometimes referred to as ocular MG) in MGUS involving primarily corneal and retinal layers of the eye is not a common event (Figure 3). MG-associated ocular diseases have been also described in MM, B-cell lymphoma, plasmacytoma, Waldenström macroglobulinemia, and CLL. Therefore, the identification of visual acuity impairment with specific corneal lesions should prompt physicians for the hematological evaluation for MGCS (Table 4). Conversely, ocular dysfunction may be expected in some patients with MG.
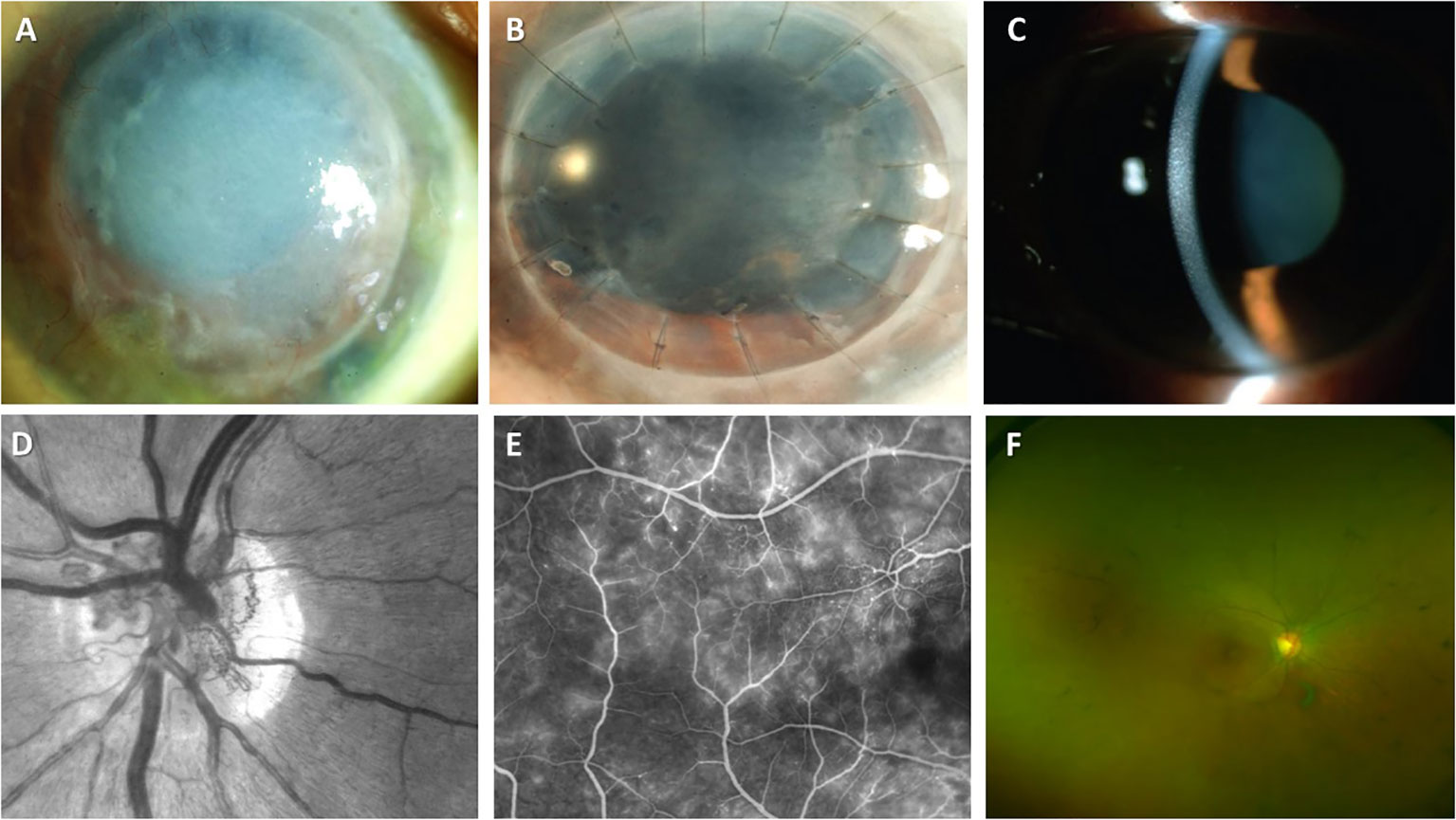
Figure 3 Ocular manifestations of MGCS. (A-C) MGCS-associated kerathopathy with visible deposits on slit-lamp examination (A, B (Image courtesy: Koo, et al., 2011) (182) and C (Image courtesy: Kocabeyoglu et al., 2014) (183)); (D-G) MGC-associated maculopathy. (D) Neovascularization of the disc on the fundal examination (Image courtesy: Gonzales et al., 2021) (184); (E) Fluoroscopic angiography demonstrating telangiectasia of vessels and leakage from retinal capillaries (Image courtesy: Gonzales et al., 2021) (184); (F) Colored fundus examination showing optic disc pallor, attenuation of retinal vessels, and peripheral pigmentation (Image courtesy: Eton et al., 2020) (185).
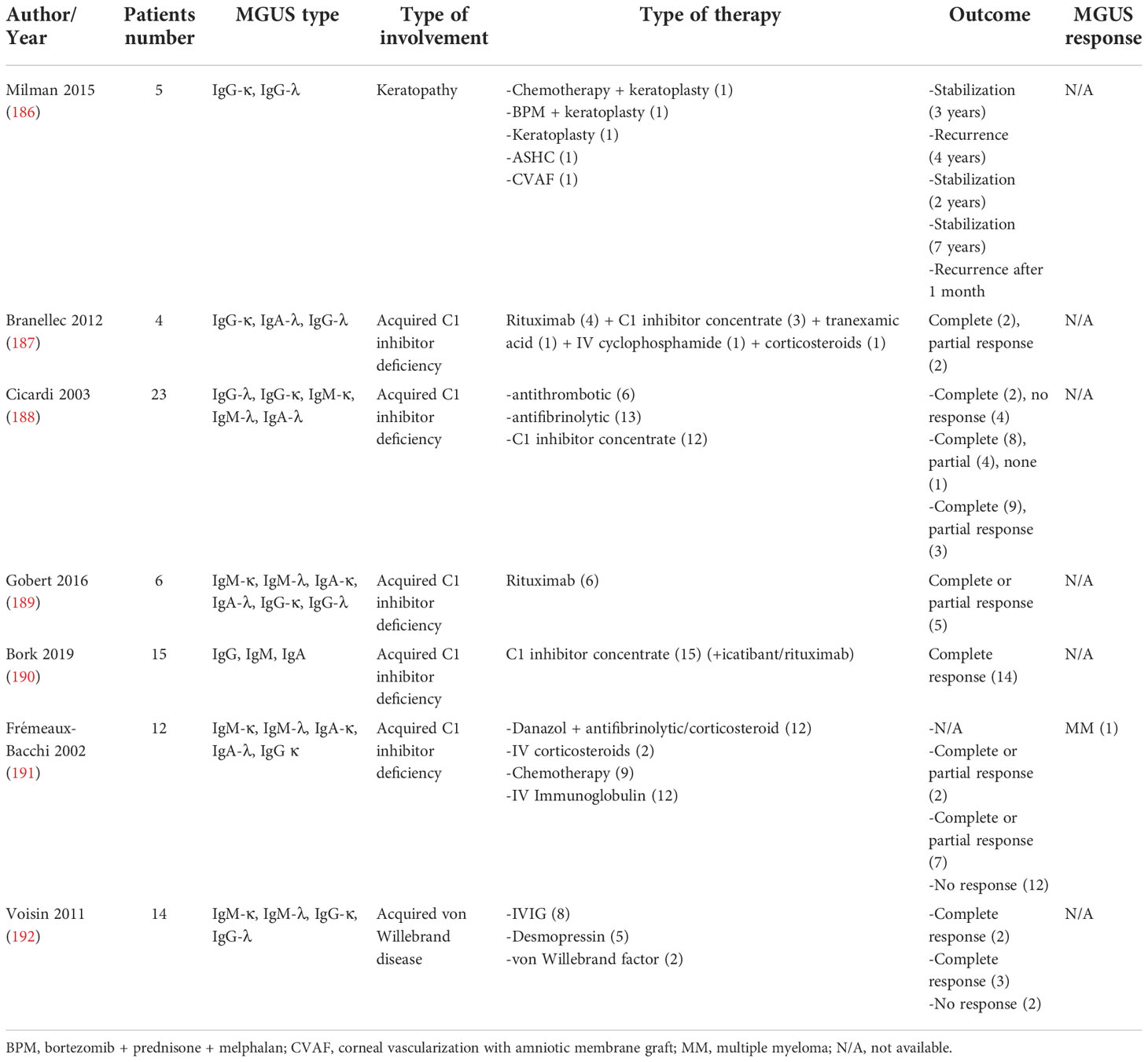
Table 4 Literature review on other MGUS-related disorders.
Paraproteinemic keratopathy
Paraproteinemic keratopathy (also known as keratopathy of monoclonal gammopathy or immunotactoid keratopathy) is caused by bilateral corneal depositions of immunoglobulins mostly of IgG-kappa origin causing distinct opacities and potentially leading to tissue dystrophy with gradual visual acuity loss (183, 193–196). The depositions can be in the form of crystals (called crystalline keratopathy) or non-crystalline (peripheral bands/patches/granules, or lattice) (183, 195, 197). In some cases, immunoglobulin-bound copper depositions (reminding Kayser-Fleischer rings of Wilson disease) can also be seen (198, 199). Therapy consisting of MG-specific treatment and reconstructive surgery depends on the severity of ocular involvement. In some cases, the disease might be asymptomatic requiring no intervention but continuous monitoring of MGUS and visual function (197). In more severe cases of ocular involvement and MGUS progression, chemotherapy and/or AHCT aiming at hematological correction can resolve symptoms and stop the disease progression (186, 200). Keratoplasty or corneal transplantation usually carries a short-term benefit for visual repair as high rates of recurrences have been reported (201–204), while topical agents (steroids and tacrolimus) were not shown to be effective (205).
Maculopathy of monoclonal gammopathy
A much rarer manifestation of ophthalmologic injury in MGUS is maculopathy with the possibility of unilateral or bilateral retinal detachment and subsequent visual loss (206). Clinical presentation may include inflammation of the iris and vitreous body as well as macular detachment (207). Besides immunoglobulin depositions, paraproteins acting as autoantibodies against macula were proposed as potential pathogenesis, however, the exact mechanism of maculopathy in MGUS is unknown (207). Due to a low number of reports, optimal treatment is yet to be discovered. Hematologic targeting (chemotherapy, rituximab, plasmapheresis) may lead to clinical resolution (206, 207), whereas ocular surgery, acetazolamide or eplerenone, and topical agents (glucocorticoids, triamcinolone, dorzolamide, anti-vascular endothelial growth factors) are not always effective with only short-term symptomatic correction of visual function (208, 209).
Other diseases associated with MGUS
Acquired C1 inhibitor deficiency
Acquired C1 inhibitor deficiency, also known as acquired angioedema, is a rare disease manifested with recurrent episodes of angioedema of the skin and mucosa of gastrointestinal and upper respiratory tracts (210). Pathogenesis involves autoantibodies against C1 inhibitor, involvement of bradykinin and cytokines, as well as abnormal activity of the classical complement pathway by neoplastic tissue (211). Clinical presentation is identical to hereditary angioedema with the difference that patients with acquired C1 inhibitor deficiency are older (≥40 years of age) with no family history and usually have associated diseases (in 70-85%), including lymphoproliferative disorders, MGUS, and non-Hodgkin lymphoma (NHL), and autoimmune diseases (188, 212). MGUS (without a specific predominant type) has been reported in 30-40% of patients occurring before, at or after the diagnosis of acquired C1 inhibitor deficiency with a low likelihood of progressing into multiple myeloma (188–191). The disease management focuses on acute treatment for angioedema episodes (i.e., C1 inhibitor replacement, fresh frozen plasma, icatibant, or ecallantide), which may prompt intubation in severe cases (188–191, 213). Prophylactic measures for the prevention of episodes include antifibrinolytics (tranexamic acid), corticosteroids, androgens (danazol), or regular use of C1 concentrate (188, 212, 214). Management of associated diseases can also be of significant clinical benefit (191, 212). Rituximab was reported to be effective in two reports, leading to symptomatic relief in eight out of eleven patients in total (187, 206) (Table 4).
Acquired von Willebrand disease
Acquired von Willebrand disease (aVWD) is the less common type of VWD due to an underlying medical disorder affecting von Willebrand Factor (VWF) (215). It corresponds to only 1-5% of all cases with a similar clinical and laboratory presentation also seen in the inherited type of VWD (i.e., spontaneous major and minor bleeding) (215). This disease has been associated with a number of conditions, including cardiovascular disease (216), Wilms tumor in children (217), hypothyroidism (218), autoimmune disorders (219), drug use (220), and hematological malignancies, such as myeloproliferative neoplasms and lymphoproliferative disorders (221), the latter nearly always having an underlying MGUS (192). The prevailing majority have IgG with only a small proportion carrying IgM paraprotein (192, 222). Clinical management of aVWD is similar to the inherited form and the presence of monoclonal gammopathy does not substantially affect the treatment strategy. Correction of bleeding is the main approach, for which desmopressin (DDAVP), factor-replacing therapy with concentrates of VWF/recombinant activated factor VII, as well as antifibrinolytic therapy have been used with varying degrees of success (222–225). IVIG (1g/kg/day for two days with 3-week interval repeats) can also lead to positive outcomes and is most reasonable in cases of related autoimmune diseases (222, 225–227), and a recent systematic literature review found an 85% response rate in patients with MGUS (228) Successful combination of concentrates with IVIG were also reported (229). Limited data also exist regarding the applications of lenalidomide (230), rituximab (231), daratumumab (232) and plasmapheresis (233) (Table 4). Clinicians should aim at revealing the underlying condition as its management can alleviate the symptom severity and disease progress.
Crystal-storing histiocytosis
Crystal-storing histiocytosis (CSH) is a rare condition, in which abnormal immunoglobulins are accumulated in the form of crystals in lysosomes of histiocytes resulting in single-organ or multiorgan damage, involving the kidney, eye, lungs, bone marrow, gastrointestinal tract, or spleen (234). The disease is thus categorized as an Ig deposition disease along with other pathologies caused by MGUS (primarily IgG with kappa light chain) (234–239). Treatment is based on the underlying condition as well as the severity and progression of the clinical picture. Careful monitoring is optimal for limited pathological lesions (234). In more severe circumstances, different chemotherapeutic regimens (bortezomib-based, daratumumab-based, or R-CHOP) led to considerable clinical improvements in case reports (236–239). AHCT was successful in a complex of ocular and periorbital crystal-storing histiocytosis with Fanconi syndrome (200) (Table 4).
Conclusion
The spectrum of various disorders associated with MGUS, in the absence of MM, Waldenström macroglobulinemia, or other lymphoproliferative disorders, describes a new entity of MGCS, the management of which remains a subject of further research and is yet to be determined. Apart from a few diseases, in which specific etiological or pathogenic therapeutic options are known (e.g., anakinra for Schnitzler syndrome or C1 inhibitor concentrate for acquired C1 inhibitor deficiency), treatment of MGCS involves myeloma-targeting agents or immunosuppressive and immunomodulatory medications, depending on the type of the disorder associated with M protein. Future studies are required to deepen our understanding of the pathogenesis of MGCS, which may guide us through the path of finding the optimal treatment for this complex yet intriguing clinical spectrum concerning multiple medical disciplines.
Author contributions
The manuscript was drafted and edited by AO, who also performed the systematic literature search. The paper was designed and structured by AM. Data extraction was performed by AG. FM, MM, YH, NG, AG, and DK reviewed and edited the article. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1045002/full#supplementary-material
References
1. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol (2014) 15(12):e538–48. doi: 10.1016/S1470-2045(14)70442-5
2. Aguzzi F, Bergami MR, Gasparro C, Bellotti V, Merlini G. Occurrence of monoclonal components in general practice: clinical implications. Eur J haematol (1992) 48(4):192–5. doi: 10.1111/j.1600-0609.1992.tb01584.x
3. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. New Engl J Med (2006) 354(13):1362–9. doi: 10.1056/NEJMoa054494
4. Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. New Engl J Med (2018) 378(3):241–9. doi: 10.1056/NEJMoa1709974
5. Therneau TM, Kyle RA, Melton LJ III, Larson DR, Benson JT, Colby CL, et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clinic Proc (2012) 87(11):1071–9. doi: 10.1016/j.mayocp.2012.06.014
6. Landgren O, Kristinsson SY, Goldin LR, Caporaso NE, Blimark C, Mellqvist UH, et al. Risk of plasma cell and lymphoproliferative disorders among 14621 first-degree relatives of 4458 patients with monoclonal gammopathy of undetermined significance in Sweden. Blood (2009) 114(4):791–5. doi: 10.1182/blood-2008-12-191676
7. Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. International myeloma working group. monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia (2010) 24:1121–27. doi: 10.1038/leu.2010.60
8. van de Donk NW, Palumbo A, Johnsen HE, Engelhardt M, Gay F, Gregersen H, et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European myeloma network. haematologica (2014) 99(6):984. doi: 10.3324/haematol.2013.100552
9. Dispenzieri A. Monoclonal gammopathies of clinical significance. Hematology Am Soc Hematol Educ Program 2020 (2020) 2020(1):380–8. doi: 10.1182/hematology.2020000122
10. Leung N, Bridoux F, Nasr SH. Monoclonal gammopathy of renal significance. New Engl J Med (2021) 384(20):1931–41. doi: 10.1056/NEJMra1810907
11. Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, et al. A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int (2018) 94(1):199–205. doi: 10.1016/j.kint.2018.02.020
12. Fermand JP, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, Leung N, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood (2018) 132(14):1478–85. doi: 10.1182/blood-2018-04-839480
13. Roccatello D, Saadoun D, Ramos-Casals M, Tzioufas AG, Fervenza FC, Cacoub P, et al. Cryoglobulinaemia. Nat Rev Dis Primers (2018) 4(1):1–6. doi: 10.1038/s41572-018-0009-4
14. Elder SA, Kocoglu MH. POEMS syndrome: A review of our patient population. Blood (2019) 134:5516. doi: 10.1182/blood-2019-123414
15. Ravindran A, Lackore KA, Glasgow AE, Drake MT, Hobbs MA, Kourelis T, et al. Monoclonal gammopathy of undetermined significance: Indications for prediagnostic testing, subsequent diagnoses, and follow-up practice at Mayo clinic. Mayo Clinic Proc (2020) 95(5):944–54. doi: 10.1016/j.mayocp.2019.12.033
16. Kelly JJ, Kyle RA, O'Brien PC, Dyck PJ. Prevalence of monoclonal protein in peripheral neuropathy. Neurology (1981) 31(11):1480–. doi: 10.1212/WNL.31.11.1480
17. Farhad K, Traub R, Ruzhansky KM, Brannagan TH III. Causes of neuropathy in patients referred as “idiopathic neuropathy”. Muscle Nerve (2016) 53(6):856–61. doi: 10.1002/mus.24969
18. Bida JP, Kyle RA, Therneau TM, Melton LJ III, Plevak MF, Larson DR, et al. Disease associations with monoclonal gammopathy of undetermined significance: a population-based study of 17,398 patients. Mayo Clinic Proc (2009) 84(8):685–93. doi: 10.4065/84.8.685
19. Notermans NC, Franssen H, Eurelings M, van der Graaf Y, Wokke JH. Diagnostic criteria for demyelinating polyneuropathy associated with monoclonal gammopathy. Muscle Nerve (2000) 23(1):73–9. doi: 10.1002/(SICI)1097-4598(200001)23:1<73::AID-MUS9>3.0.CO;2-5
20. Tagawa Y, Yuki N, Hirata K. Anti-SGPG antibody in CIDP: Nosological position of IgM anti-MAG/SGPG antibody-associated neuropathy. Muscle Nerve (2000) 23(6):895–9. doi: 10.1002/(SICI)1097-4598(200006)23:6<895::AID-MUS8>3.0.CO;2-G
21. Kuitwaard K, Hahn AF, Vermeulen M, Venance SL, van Doorn PA. Intravenous immunoglobulin response in treatment-naïve chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry (2015) 86(12):1331–6. doi: 10.1136/jnnp-2014-309042
22. Jann S, Beretta S, Bramerio MA. Different types of chronic inflammatory demyelinating polyneuropathy have a different clinical course and response to treatment. Muscle Nerve (2005) 32(3):351–6. doi: 10.1002/mus.20391
23. Mygland Å, Monstad P. Chronic acquired demyelinating symmetric polyneuropathy classified by pattern of weakness. Arch Neurol (2003) 60(2):260–4. doi: 10.1001/archneur.60.2.260
24. Magy L, Chassande B, Maisonobe T, Bouche P, Vallat JM, Léger JM. Polyneuropathy associated with IgG/IgA monoclonal gammopathy: a clinical and electrophysiological study of 15 cases. Eur J Neurol (2003) 10(6):677–85. doi: 10.1046/j.1468-1331.2003.00687.x
25. Eurelings M, Notermans N, Wokke J, Bosboom W, Van den Berg L. Sural nerve T cells in demyelinating polyneuropathy associated with monoclonal gammopathy. Acta neuropathol (2002) 103(2):107–14. doi: 10.1007/s004010100437
26. Gorson KC, Allam G, Ropper AH. Chronic inflammatory demyelinating polyneuropathy: clinical features and response to treatment in 67 consecutive patients with and without a monoclonal gammopathy. Neurology (1997) 48(2):321–8. doi: 10.1212/WNL.48.2.321
27. Vital A, Lagueny A, Julien J, Ferrer X, Barat M, Hermosilla E, et al. Chronic inflammatory demyelinating polyneuropathy associated with dysglobulinemia: a peripheral nerve biopsy study in 18 cases. Acta neuropathol (2000) 100(1):63–8. doi: 10.1007/s004010051193
28. Le Cann M, Bouhour F, Viala K, Simon L, Tard C, Rossi C, et al. CANOMAD: a neurological monoclonal gammopathy of clinical significance that benefits from b-cell–targeted therapies. Blood (2020) 136(21):2428–36. doi: 10.1182/blood.2020007092
29. Garcia-Santibanez R, Zaidman CM, Sommerville RB, Lopate G, Weihl CC, Pestronk A, et al. CANOMAD and other chronic ataxic neuropathies with disialosyl antibodies (CANDA). J Neurol (2018) 265(6):1402–9. doi: 10.1007/s00415-018-8853-4
30. Notermans NC, Lokhorst HM, Franssen H, van der Graaf Y, Teunissen LL, Jennekens FG, et al. Intermittent cyclophosphamide and prednisone treatment of polyneuropathy associated with monoclonal gammopathy of undetermined significance. Neurology (1996) 47(5):1227–33. doi: 10.1212/WNL.47.5.1227
31. Katz JS, Saperstein DS, Gronseth G, Amato AA, Barohn RJ. Distal acquired demyelinating symmetric neuropathy. Neurology (2000) 54(3):615–. doi: 10.1212/WNL.54.3.615
32. Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology (2005) 65(8):1158–64. doi: 10.1212/01.wnl.0000180362.90078.dc
33. Voermans NC, Benveniste O, Minnema MC, Lokhorst H, Lammens M, Meersseman W, et al. Sporadic late-onset nemaline myopathy with MGUS: long-term follow-up after melphalan and SCT. Neurology (2014) 83(23):2133–9. doi: 10.1212/WNL.0000000000001047
34. Naddaf E, Milone M, Kansagra A, Buadi F, Kourelis T. Sporadic late-onset nemaline myopathy: clinical spectrum, survival, and treatment outcomes. Neurology (2019) 93(3):e298–305. doi: 10.1212/WNL.0000000000007777
35. Schnitzler LJ, Schreckenbach T, Nadaj-Pakleza A, Stenzel W, Rushing EJ, Van Damme P, et al. Sporadic late-onset nemaline myopathy: clinico-pathological characteristics and review of 76 cases. Orphanet J rare Dis (2017) 12(1):1–2. doi: 10.1186/s13023-017-0640-2
36. Monforte M, Primiano G, Silvestri G, Mirabella M, Luigetti M, Cuccagna C, et al. Sporadic late-onset nemaline myopathy: clinical, pathology and imaging findings in a single center cohort. J Neurol (2018) 265(3):542–51. doi: 10.1007/s00415-018-8741-y
37. Okhovat AA, Nilipour Y, Boostani R, Vahabizad F, Najmi S, Nafissi S, et al. Sporadic late-onset nemaline myopathy with monoclonal gammopathy of undetermined significance: report of four patients. Neuromuscular Disord (2021) 31(1):29–34. doi: 10.1016/j.nmd.2020.11.004
38. Frohman EM, Tusa R, Mark AS, Cornblath DR. Vestibular dysfunction in chronic inflammatory demyelinating polyneuropathy. Ann Neurol (1996) 39(4):529–35. doi: 10.1002/ana.410390415
39. Baek SH, Hong YH, Choi SJ, Ahn SH, Park KH, Shin JY, et al. Electrodiagnostic data-driven clustering identifies a prognostically different subgroup of patients with chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry (2019) 90(6):674–80. doi: 10.1136/jnnp-2018-319758
40. Steiner N, Schwärzler A, Göbel G, Löscher W, Wanschitz J, Gunsilius E. Are neurological complications of monoclonal gammopathy of undetermined significance underestimated? Oncotarget (2017) 8(3):5081. doi: 10.3390/jcm11071848
41. Mathis S, Franques J, Richard L, Vallat JM. Monoclonal gammopathy of undeterminated significance and endoneurial IgG deposition: a case report. Medicine (2016) 95(36):e4807. doi: 10.1097/MD.0000000000004807
42. Posa A, Emmer A, Kornhuber M. Severe CIDP-MGUS responsive to rituximab. Heliyon (2020) 6(6):e04230. doi: 10.1016/j.heliyon.2020.e04230
43. Doneddu PE, Cocito D, Manganelli F, Fazio R, Briani C, Filosto M, et al. Atypical CIDP: diagnostic criteria, progression and treatment response. data from the Italian CIDP database. J Neurol Neurosurg Psychiatry (2019) 90(2):125–32. doi: 10.1136/jnnp-2018-318714
44. Filosto M, Cotelli M, Todeschini A, Broglio L, Vielmi V, Rinaldi F, et al. Clinical spectrum and evolution of monoclonal gammopathy-associated neuropathy: an observational study. neurol (2012) 18(6):378–84. doi: 10.1097/NRL.0b013e31826a99e9
45. Dalakas MC. Pathogenesis and treatment of anti-MAG neuropathy. Curr Treat options Neurol (2010) 12(2):71–83. doi: 10.1007/s11940-010-0065-x
46. Arbogast SD, Khanna S, Koontz DW, Tomsak RL, Katirji B, Leigh RJ. Chronic ataxic neuropathy mimicking dorsal midbrain syndrome. J Neurol Neurosurg Psychiatry (2007) 78(11):1276–7. doi: 10.1136/jnnp.2007.120444
47. Sanvito L, Rajabally YA. Optic neuropathy associated with CANOMAD: description of 2 cases. Muscle Nerve (2011) 44(3):451–5. doi: 10.1002/mus.22157
48. Krenn M, Keir G, Wieshmann UC. CANOMAD responding to weekly treatment with intravenous immunoglobulin (IVIg). Case Rep (2014) 2014:bcr2013202545. doi: 10.1136/bcr-2013-202545
49. Delmont E, Jeandel PY, Hubert AM, Marcq L, Boucraut J, Desnuelle C. Successful treatment with rituximab of one patient with CANOMAD neuropathy. J Neurol (2010) 257(4):655–7. doi: 10.1007/s00415-009-5412-z
50. Löscher WN, Woertz A, Wallnöfer M, Wanschitz JV, Luef G. Successful treatment of CANOMAD with IVIg and rituximab. J Neurol (2013) 260(4):1168–70. doi: 10.1007/s00415-013-6867-5
51. Delval A, Stojkovic T, Vermersch P. Relapsing sensorimotor neuropathy with ophthalmoplegia, antidisialosyl antibodies, and extramembranous glomerulonephritis. Muscle Nerve (2006) 33(2):274–7. doi: 10.1002/mus.20452
52. Oh SJ, Almeida DF, Villa SC. Monosialosyl antibody in a case mimicking CANOMAD syndrome. J Clin Neuromuscular Dis (2019) 21(1):53–4. doi: 10.1097/CND.0000000000000247
53. Voermans NC, Minnema M, Lammens M, Schelhaas HJ, Kooi AV, Lokhorst HM, et al. Sporadic late-onset nemaline myopathy effectively treated by melphalan and stem cell transplant. Neurology (2008) 71(7):532–4. doi: 10.1212/01.wnl.0000310814.54623.6f
54. Montagnese F, Portaro S, Musumeci O, Migliorato A, Moggio M, Fagiolari G, et al. Sporadic late-onset nemaline myopathy in a woman with multiple myeloma successfully treated with lenalidomide/dexamethasone. Muscle Nerve (2015) 51(6):934–5. doi: 10.1002/mus.24545
55. Wilmas K, Aria A, Torres-Cabala CA, Lu H, Duvic M. Schnitzler syndrome in a patient with a family history of monoclonal gammopathy. Dermatol Online J (2018) 24(1):1–5. doi: 10.5070/D3241037926
56. Cohen YK, Elpern DJ. Diffuse normolipemic plane xanthoma associated with monoclonal gammopathy. Dermatol Pract conceptual (2015) 5(4):65. doi: 10.5826/dpc.0504a16
57. Khan J, Sykes DB. Case report: a 37-year-old male with telangiectasias, polycythemia vera, perinephric fluid collections, and intrapulmonary shunting. BMC Hematol (2014) 14(1):1–4. doi: 10.1186/2052-1839-14-11
58. Shalhout SZ, Nahas MR, Drews RE, Miller DM. Generalized acquired cutis laxa associated with monoclonal gammopathy of dermatological significance. Case Rep Dermatol Med (2020), 2020. doi: 10.1155/2020/7480607
59. Young PA, Bae GH, Konia TH. Subcorneal pustular dermatosis associated with IgG monoclonal gammopathy of undetermined significance. Dermatol Online J (2021) 27(4):1–5. doi: 10.5070/D3274053153
60. Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: A case report. Dermatol Res Pract (2011) 2011:1–5. doi: 10.1155/2011/927852
61. Claveau JS, Wetter DA, Kumar S. Cutaneous manifestations of monoclonal gammopathy. Blood Cancer J (2022) 12(4):1–4. doi: 10.1038/s41408-022-00661-1
62. Lipsker D. Monoclonal gammopathy of cutaneous significance: review of a relevant concept. J Eur Acad Dermatol Venereol (2017) 31(1):45–52. doi: 10.1111/jdv.13847
63. de Chambrun MP, Gousseff M, Mauhin W, Lega JC, Lambert M, Rivière S, et al. Intravenous immunoglobulins improve survival in monoclonal gammopathy-associated systemic capillary-leak syndrome. Am J Med (2017) 130(10):1219–e19. doi: 10.1016/j.amjmed.2017.05.023
64. Kapoor P, Greipp PT, Schaefer EW, Mandrekar SJ, Kamal AH, Gonzalez-Paz NC, et al. Idiopathic systemic capillary leak syndrome (Clarkson's disease): the Mayo clinic experience. Mayo Clinic Proc (2010) 85(10):905–12. doi: 10.4065/mcp.2010.0159
65. Wood AJ, Wagner MV, Abbott JJ, Gibson LE. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol (2009) 145(3):279–84. doi: 10.1001/archdermatol.2008.583
66. Higgins LS, Go RS, Dingli D, Kumar SK, Rajkumar SV, Dispenzieri A, et al. Clinical features and treatment outcomes of patients with necrobiotic xanthogranuloma associated with monoclonal gammopathies. Clin Lymph Myelom Leukemia (2016) 16(8):447–52. doi: 10.1016/j.clml.2016.04.009
67. Szalat R, Arnulf B, Karlin L, Rybojad M, Asli B, Malphettes M, et al. Pathogenesis and treatment of xanthomatosis associated with monoclonal gammopathy. Blood (2011) 118(14):3777–84. doi: 10.1182/blood-2011-05-356907
68. Donato ML, Feasel AM, Weber DM, Prieto VG, Giralt SA, Champlin RE, et al. Scleromyxedema: role of high-dose melphalan with autologous stem cell transplantation. Blood (2006) 107(2):463–6. doi: 10.1182/blood-2004-12-4870
69. Kreuter A, Altmeyer P. High-dose dexamethasone in scleromyxedema: report of 2 additional cases. J Am Acad Dermatol (2005) 53(4):739–40. doi: 10.1016/j.jaad.2005.04.012
70. Mahévas T, Arnulf B, Bouaziz JD, Livideanu CB, Osio A, Servy A, et al. Plasma cell–directed therapies in monoclonal gammopathy–associated scleromyxedema. Blood (2020) 135(14):1101–10. doi: 10.1182/blood.2019002300
71. Rongioletti F, Merlo G, Cinotti E, Fausti V, Cozzani E, Cribier B, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol (2013) 69(1):66–72. doi: 10.1016/j.jaad.2013.01.007
72. Terpos E, Asli B, Christoulas D, Brouet JC, Kastritis E, Rybojad M, et al. Increased angiogenesis and enhanced bone formation in patients with IgM monoclonal gammopathy and urticarial skin rash: new insight into the biology of schnitzler syndrome. haematologica (2012) 97(11):1699. doi: 10.3324/haematol.2012.067306
73. Sokumbi O, Drage LA, Peters MS. Clinical and histopathologic review of schnitzler syndrome: the Mayo clinic experience (1972-2011). J Am Acad Dermatol (2012) 67(6):1289–95. doi: 10.1016/j.jaad.2012.04.027
74. Gusdorf L, Asli B, Barbarot S, Néel A, Masseau A, Puéchal X, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy (2017) 72(2):177–82. doi: 10.1111/all.13035
75. Néel A, Henry B, Barbarot S, Masseau A, Perrin F, Bernier C, et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in schnitzler's syndrome: a French multicenter study. Autoimmun Rev (2014) 13(10):1035–41. doi: 10.1016/j.autrev.2014.08.031
76. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Trans Allergy (2014) 4(1):1–5. doi: 10.1186/2045-7022-4-41
77. de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler syndrome study group. schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis rheum (2007) 37(3):137–48. doi: 10.1016/j.semarthrit.2007.04.001
78. Bashir M, Bettendorf B, Hariman R. A rare but fascinating disorder: case collection of patients with schnitzler syndrome. Case Rep Rheumatol (2018) 2018:1–4. doi: 10.1155/2018/7041576
79. Tinazzi E, Puccetti A, Patuzzo G, Sorleto M, Barbieri A, Lunardi C. Schnitzler syndrome, an autoimmune–autoinflammatory syndrome: Report of two new cases and review of the literature. Autoimmun Rev (2011) 10(7):404–9. doi: 10.1016/j.autrev.2011.01.003
80. Larocca CA, McEvoy JW, Ellis CL, Junkins-Hopkins J, Kolb T, Baer AN, et al. Schnitzler's syndrome associated with pancreatitis: a disease of IL-1 dysregulation. Clin Rheumatol (2012) 31(1):169–74. doi: 10.1007/s10067-011-1804-4
81. Cascavilla N, Bisceglia M, D'arena G. Successful treatment of schnitzler's syndrome with anakinra after failure of rituximab trial. Int J immunopathol Pharmacol (2010) 23(2):633–6. doi: 10.1177/039463201002300226
82. de Koning HD, Bodar EJ, Simon A, van der Hilst JC, Netea MG, van der Meer JW. Beneficial response to anakinra and thalidomide in schnitzler’s syndrome. Ann rheum Dis (2006) 65(4):542–4. doi: 10.1136/ard.2005.045245
83. Krause K, Tsianakas A, Wagner N, Fischer J, Weller K, Metz M, et al. Efficacy and safety of canakinumab in schnitzler syndrome: a multicenter randomized placebo-controlled study. J Allergy Clin Immunol (2017) 139(4):1311–20. doi: 10.1016/j.jaci.2016.07.041
84. Krause K, Weller K, Stefaniak R, Wittkowski H, Altrichter S, Siebenhaar F, et al. Efficacy and safety of the interleukin-1 antagonist rilonacept in schnitzler syndrome: an open-label study. Allergy (2012) 67(7):943–50. doi: 10.1111/j.1398-9995.2012.02843.x
85. Eiling E, Möller M, Kreiselmaier I, Brasch J, Schwarz T. Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol (2007) 57(2):361–4. doi: 10.1016/j.jaad.2007.03.036
86. Kastritis E, Katoulis A, Terpos E, Panayiotides I, Gavriatopoulopu M, Dimopopoulos MA. Schnitzler's syndrome: increased levels of bone formation and angiogenesis factors are reduced after successful pefloxacin treatment. Clin Lymph Myelom (2008) 8(6):359–62. doi: 10.3816/CLM.2008.n.053
87. Murota H, Shoda Y, Ishibashi T, Sugahara H, Matsumura I, Katayama I. Improvement of recurrent urticaria in a patient with schnitzler syndrome associated with b-cell lymphoma with combination rituximab and radiotherapy. J Am Acad Dermatol (2009) 61(6):1070–5. doi: 10.1016/j.jaad.2008.12.040
88. Huang Y, Wang Y, Yu F, Mao X, Wang B, Li J, et al. Case report: therapeutic use of ibrutinib in a patient with schnitzler syndrome. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.894464
89. Kalli F, Cioni M, Parodi A, Altosole T, Ferrera F, Barra G, et al. Increased frequency of interleukin-4 and reduced frequency of interferon-γ and IL-17-producing CD4+ and CD8+ cells in scleromyxedema. J Eur Acad Dermatol Venereol (2020) 34(5):1092–7. doi: 10.1111/jdv.16136
90. Georgakis CD, Falasca G, Georgakis A, Heymann WR. Scleromyxedema.Clinics Dermatol (2006) 24(6):493–7. doi: 10.1016/j.clindermatol.2006.07.011
91. Amini-Adle M, Thieulent N, Dalle S, Balme B, Thomas L. Scleromyxedema: successful treatment with thalidomide in two patients. Dermatology (2007) 214(1):58–60. doi: 10.1159/000096914
92. Guarenti I, Sebastiani V, Pinto G, de Souza PR, de Almeida H. Successful treatment of scleromyxedema with oral thalidomide. Int J Dermatol (2013) 52(5):631–2. doi: 10.1111/j.1365-4632.2011.05030.x
93. Sansbury JC, Cocuroccia B, Jorizzo JL, Gubinelli E, Gisondi P, Girolomoni G. Treatment of recalcitrant scleromyxedema with thalidomide in 3 patients. J Am Acad Dermatol (2004) 51(1):126–31. doi: 10.1016/j.jaad.2004.01.058
94. Efthimiou P, Blanco M. Intravenous gammaglobulin and thalidomide may be an effective therapeutic combination in refractory scleromyxedema: case report and discussion of the literature. Semin Arthritis rheum (2008) 38(3):188–94. doi: 10.1016/j.semarthrit.2007.10.015
95. Brunet-Possenti F, Hermine O, Marinho E, Crickx B, Descamps V. Combination of intravenous immunoglobulins and lenalidomide in the treatment of scleromyxedema. J Am Acad Dermatol (2013) 69(2):319–20. doi: 10.1016/j.jaad.2013.01.028
96. Migkou M, Gkotzamanidou M, Terpos E, Dimopoulos MA, Kastritis E. Response to bortezomib of a patient with scleromyxedema refractory to other therapies. Leukemia Res (2011) 11(35):e209–11. doi: 10.1016/j.leukres.2011.07.021
97. Cañueto J, Labrador J, Román C, Santos-Briz Á, Contreras T, Gutiérrez NC, et al. The combination of bortezomib and dexamethasone is an efficient therapy for relapsed/refractory scleromyxedema: a rare disease with new clinical insights. Eur J haematol (2012) 88(5):450–4. doi: 10.1111/j.1600-0609.2012.01772.x
98. Fett NM, Toporcer MB, Dalmau J, Shinohara MM, Vogl DT. Scleromyxedema and dermato–neuro syndrome in a patient with multiple myeloma effectively treated with dexamethasone and bortezomib. Am J Hematol (2011) 86(10):893–6. doi: 10.1002/ajh.22131
99. Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J haematol (2012) 157(4):411–. doi: 10.1111/j.1365-2141.2012.09088.x
100. Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol (1995) 33(1):37–43. doi: 10.1016/0190-9622(95)90007-1
101. Fleming KE, Virmani D, Sutton E, Langley R, Corbin J, Pasternak S, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J cutaneous Pathol (2012) 39(5):508–17. doi: 10.1111/j.1600-0560.2012.01882.x
102. Lacy MQ, Hogan WJ, Gertz MA, Dispenzieri A, Rajkumar SV, Hayman S, et al. Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation. Arch Dermatol (2005) 141(10):1277–82. doi: 10.1001/archderm.141.10.1277
103. Bos R, de Waal EG, Kuiper H, Hazenberg BP, Vellenga E. Thalidomide and dexamethasone followed by autologous stem cell transplantation for scleromyxoedema. Rheumatology (2011) 50(10):1925–6. doi: 10.1093/rheumatology/ker209
104. Hisler BM, Savoy LB, Hashimoto K. Improvement of scleromyxedema associated with isotretinoin therapy. J Am Acad Dermatol (1991) 24(5):854–7. doi: 10.1016/0190-9622(91)70132-L
105. Kim S, Park TH, Lee SM, Kim YH, Cho MK, Whang KU, et al. Scleromyxedema with multiple systemic involvement: Successful treatment with intravenous immunoglobulin. Dermatol Ther (2020) 33(3):e13378. doi: 10.1111/dth.13378
106. Saigoh S, Tashiro A, Fujita S, Matsui M, Shibata S, Takeshita H, et al. Successful treatment of intractable scleromyxedema with cyclosporin a. Dermatology (2003) 207(4):410–1. doi: 10.1159/000074127
107. Arginelli F, Paganelli A, Rongioletti F, Pellacani G, Conti A. Ineffectiveness of infliximab CT-P13 for the treatment of scleromyxedema: A case report. Dermatologic therapy (2018) 31:e12583. doi: 10.1111/dth.12583
108. Tschen JA, Chang JR. Scleromyxedema: treatment with interferon alfa. J Am Acad Dermatol (1999) 40(2):303–7. doi: 10.1016/S0190-9622(99)70471-8
109. Mehta V, Balachandran C, Rao R. Arndt gottron scleromyxedema: successful response to treatment with steroid minipulse and methotrexate. Indian J Dermatol (2009) 54(2):193. doi: 10.4103/0019-5154.53183
110. Kuldeep CM, Mittal AK, Gupta LK, Paliwal VK, Sharma P, Garg A. Successful treatment of scleromyxedema with dexamethasone cyclophosphamide pulse therapy. Indian J Dermatol Venereol Leprol (2005) 71(1):44–5. doi: 10.4103/0378-6323.13787
111. Nelson CA, Zhong CS, Hashemi DA, Ashchyan HJ, Brown-Joel Z, Noe MH, et al. A multicenter cross-sectional study and systematic review of necrobiotic xanthogranuloma with proposed diagnostic criteria. JAMA Dermatol (2020) 156(3):270–9. doi: 10.1001/jamadermatol.2019.4221
112. Ryan E, Warren LJ, Szabo F. Necrobiotic xanthogranuloma: response to chlorambucil. Australas J Dermatol (2012) 53(2):e23–5. doi: 10.1111/j.1440-0960.2010.00710.x
113. Spraul CW, Wagner P, Lang GK. Bilateral necrobiotic xanthogranuloma of the eyelids with associated paraproteinemia: Case report and review of literature. Klinische Monatsblatter fur Augenheilkunde (2002) 219(1-2):55–8. doi: 10.1055/s-2000-23502
114. Finan MC, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. a review of 22 cases. Medicine (1986) 65(6):376–88. doi: 10.1097/00005792-198611000-00003
115. Khan IJ, Azam NA, Sullivan SC, Habboush HW, Christian A. Necrobiotic xanthogranuloma successfully treated with a combination of dexamethasone and oral cyclophosphamide. Can J Ophthalmol (2009) 44(3):335–6. doi: 10.3129/i09-021
116. Meyer S, Szeimies RM, Landthaler M, Hohenleutner S. Cyclophosphamide–dexamethasone pulsed therapy for treatment of recalcitrant necrobiotic xanthogranuloma with paraproteinemia and ocular involvement. Br J Dermatol (2005) 153(2):443–5. doi: 10.1111/j.1365-2133.2005.06737.x
117. Olson RM, Harrison AR, Maltry A, Mokhtarzadeh A. Periorbital necrobiotic xanthogranuloma successfully treated with intravenous immunoglobulin. Case Rep Ophthalmol (2018) 9(1):76–81. doi: 10.1159/000485913
118. Chave TA, Chowdhury MM, Holt PJ. Recalcitrant necrobiotic xanthogranuloma responding to pulsed high-dose oral dexamethasone plus maintenance therapy with oral prednisolone. Br J Dermatol (2001) 144(1):158–61. doi: 10.1046/j.1365-2133.2001.03967.x
119. Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone. Ann Hematol (2016) 95(4):671. doi: 10.1007/s00277-016-2604-3
120. Efebera Y, Blanchard E, Allam C, Han A, Lee S, Munshi N. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymph Myelom Leukemia (2011) 11(3):298–302. doi: 10.1016/j.clml.2011.03.020
121. Goede JS, Misselwitz B, Taverna C, Schanz U, Dispenzieri A, Hummel Y, et al. Necrobiotic xanthogranuloma successfully treated with autologous stem cell transplantation. Ann Hematol (2007) 86(4):303–6. doi: 10.1007/s00277-006-0231-0
122. Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc (2005) 103:69.
123. Al-Niaimi FA, Dawn G, Cox NH. Necrobiotic xanthogranuloma without paraproteinaemia: marked improvement with psoralen ultraviolet a treatment. Clin Exp Dermatol: Clin Dermatol (2010) 35(3):275–7. doi: 10.1111/j.1365-2230.2009.03447.x
124. Rodriguez O, Meyers C, Weiss BM, Elenitsas R, Rosenbach M. Necrobiotic xanthogranuloma treated with topical nitrogen mustard (mechlorethamine). JAMA Dermatol (2016) 152(5):589–90. doi: 10.1001/jamadermatol.2015.5151
125. Georgiou S, Tsambaos D. Hypercalcaemia and hypercalciuria after topical treatment of psoriasis with excessive amounts of calcipotriol. Acta dermato-venereol (1999) 79(1):86. doi: 10.1080/000155599750011822
126. Oestreicher J, Dookeran R, Nijhawan N, Kolin A. Necrobiotic xanthogranuloma with predominant periorbital involvement. Ophthal Plast Reconstructive Surg (2010) 26(6):473–5. doi: 10.1097/IOP.0b013e3181d92955
127. Schaudig U, Al-Samir K. Upper and lower eyelid reconstruction for severe disfiguring necrobiotic xanthogranuloma. Orbit (2004) 23(1):65–76. doi: 10.1076/orbi.23.1.65.28989
128. Wei YH, Cheng JJ, Wu YH, Liu CY, Hung CJ, Hsu JD, et al. Necrobiotic xanthogranuloma: response to dapsone. Dermatol Ther (2015) 28(1):7–9. doi: 10.1111/dth.12179
129. Sagiv O, Thakar SD, Morrell G, Tetzlaff MT, Esmaeli B. Rituximab monotherapy is effective in treating orbital necrobiotic xanthogranuloma. Ophthal Plast reconstructive Surg (2018) 34(1):e24–7. doi: 10.1097/IOP.0000000000000988
130. Venencie PY, Le Bras P, Toan ND, Tchernia G, Delfraissy JF. Recombinant interferon alfa-2b treatment of necrobiotic xanthogranuloma with parapoteinemia. J Am Acad Dermatol (1995) 32(4):666–7. doi: 10.1016/0190-9622(95)90370-4
131. Stockman A, Delanghe J, Geerts ML, Naeyaert JM. Diffuse plane normolipaemic xanthomatosis in a patient with chronic lymphatic leukaemia and monoclonal gammopathy. Dermatology (2002) 204(4):351–4. doi: 10.1159/000063384
132. Kourou K, Suga Y, Muramatsu S, Yaguchi H, Ogawa H. A case of diffuse plane normolipemic xanthomatosis associated with pancytopenia and monoclonal gammopathy. J Dermatol (2006) 33(1):64–7. doi: 10.1111/j.1346-8138.2006.00013.x
133. Davies MJ, Whitehead K, Quagliotto G, Wood D, Patheja RS, Sullivan TJ. Adult orbital and adnexal xanthogranulomatous disease. Asia-Pacific J Ophthalmol (2017) 6(5):435–43. doi: 10.22608/APO.2017246
134. Marcoval J, Moreno A, Bordas X, Gallardo F, Peyrí J. Diffuse plane xanthoma: clinicopathologic study of 8 cases. J Am Acad Dermatol (1998) 39(3):439–42. doi: 10.1016/S0190-9622(98)70321-4
135. Sykes DB, Schroyens W, O'Connell C. The TEMPI syndrome–a novel multisystem disease. New Engl J Med (2011) 356:2582–90. doi: 10.1056/NEJMc1106670
136. Sykes DB, O’Connell C, Schroyens W. The TEMPI syndrome. Blood (2020) 135(15):1199–203. doi: 10.1182/blood.2019004216
137. Sun C, Xu J, Zhang B, Huang H, Chen L, Yan H, et al. Whole-genome sequencing suggests a role of MIF in the pathophysiology of TEMPI syndrome. Blood Adv (2021) 5(12):2563–8. doi: 10.1182/bloodadvances.2020003783
138. Rosado FG, Oliveira JL, Sohani AR, Schroyens W, Sykes DB, Kenderian SS, et al. Bone marrow findings of the newly described TEMPI syndrome: when erythrocytosis and plasma cell dyscrasia coexist. Modern Pathol (2015) 28(3):367–72. doi: 10.1038/modpathol.2014.117
139. Kwok M, Korde N, Landgren O. Bortezomib to treat the TEMPI syndrome. New Engl J Med (2012) 366(19):1843–5. doi: 10.1056/NEJMc1202649
140. Jasim S, Mahmud G, Bastani B, Fesler M. Subcutaneous bortezomib for treatment of TEMPI syndrome. Clin Lymph Myelom Leukemia (2014) 14(6):e221–3. doi: 10.1016/j.clml.2014.07.004
141. Pascart T, Herbaux C, Lemaire A, Soncin F, Hachulla E, Hatron PY, et al. Coexistence of rheumatoid arthritis and TEMPI syndrome: New insight in microangiogenic-related diseases. Joint Bone Spine (2016) 83(5):587–8. doi: 10.1016/j.jbspin.2015.06.011
142. Schroyens W, O'Connell C, Sykes DB. Complete and partial responses of the TEMPI syndrome to bortezomib. New Engl J Med (2012) 367(8):778–80. doi: 10.1056/NEJMc1205806
143. Sykes DB, Schroyens W. Complete responses in the TEMPI syndrome after treatment with daratumumab. New Engl J Med (2018) 378(23):2240–2. doi: 10.1056/NEJMc1804415
144. Lor M, Cheng M, Liang B, Cheng CE. TEMPI syndrome with progressive telangiectasias associated with pulmonary deterioration. JAMA Dermatol (2020) 156(12):1379–80. doi: 10.1001/jamadermatol.2020.2668
145. Diral E, Parma M, Renso R, Pezzatti S, Terruzzi E, Perfetti P, et al. A fatal case of TEMPI syndrome, refractory to proteasome inhibitors and autologous stem cell transplantation. Leukemia Res (2020) 97:106441. doi: 10.1016/j.leukres.2020.106441
146. Kenderian SS, Rosado FG, Sykes DB, Hoyer JD, Lacy MQ. Long-term complete clinical and hematological responses of the TEMPI syndrome after autologous stem cell transplantation. Leukemia (2015) 29(12):2414–6. doi: 10.1038/leu.2015.298
147. Liang SH, Yeh SP. Relapsed multiple myeloma as TEMPI syndrome with good response to salvage lenalidomide and dexamethasone. Ann Hematol (2019) 98(10):2447–50. doi: 10.1007/s00277-019-03761-4
148. Berk DR, Bentley DD, Bayliss SJ, Lind A, Urban Z. Cutis laxa: a review. J Am Acad Dermatol (2012) 66(5):842–e1. doi: 10.1016/j.jaad.2011.01.004
149. De Larrea CF, Rovira M, Mascaró JM Jr., Torras A, Sole M, Lloreta J, et al. Generalized cutis laxa and fibrillar glomerulopathy resulting from IgG deposition in IgG-lambda monoclonal gammopathy: pulmonary hemorrhage during stem cell mobilization and complete hematological response with bortezomib and dexamethasone therapy. Eur J haematol (2009) 82(2):154–8. doi: 10.1111/j.1600-0609.2008.01181.x
150. New HD, Callen JP. Generalized acquired cutis laxa associated with multiple myeloma with biphenotypic IgG-λ and IgA-κ gammopathy following treatment of a nodal plasmacytoma. Arch Dermatol (2011) 147(3):323–8. doi: 10.1001/archdermatol.2011.26
151. Silveira L, Torres I, Salvino MA, Follador I, Bittencourt AL. Acquired cutis laxa with an interstitial granulomatous reaction associated with IgG lambda monoclonal gammopathy. Am J Dermatopathol (2013) 35(4):e67–71. doi: 10.1097/DAD.0b013e31827bceaf
152. Lavorato FG, Alves MD, Maceira JM, Unterstell N, Serpa LA, Azulay-Abulafia L. Primary systemic amyloidosis, acquired cutis laxa and cutaneous mucinosis in a patient with multiple myeloma. Anais Brasileiros Dermatol (2013) 88:32–5. doi: 10.1590/abd1806-4841.20132531
153. Szalat R, Monsel G, Le Goff W, Battistella M, Bengouffa D, Schlageter MH, et al. The spectrum of neutrophilic dermatoses associated with monoclonal gammopathy: association with IgA isotype and inflammatory profile. J Am Acad Dermatol (2015) 73(5):809–20. doi: 10.1016/j.jaad.2015.07.031
154. Asahina A, Sakurai N, Suzuki Y, Narushima K. Schnitzler’s syndrome with prominent neutrophil infiltration misdiagnosed as sweet’s syndrome: a typical example of urticarial neutrophilic dermatosis. Clin Exp Dermatol: Exp Dermatol (2010) 35(4):e123–6. doi: 10.1111/j.1365-2230.2009.03746.x
155. Al-Musalhi B, Gerstein W. Monoclonal gammopathy of undetermined significance and neutrophilic dermatosis. Oman Med J (2016) 31(5):394–5. doi: 10.5001/omj.2016.79
156. Kentley J, Marshall C, Bewley A. Erythema elevatum diutinum–associated with loss of the uvula. JAAD Case Rep (2017) 3(3):212–4. doi: 10.1016/j.jdcr.2017.02.007
157. Canpolat F, Akpinar H, Cemil BÇ, Eskioğlu F, Öztürk E. A case of subcorneal pustular dermatosis in association with monoclonal IgA gammopathy successfully treated with acitretin. J Dermatol Treat (2010) 21(2):114–6. doi: 10.3109/09546630902882071
158. Bayle P, Laplanche G, Gorguet B, Oksman F, Boulinguez S, Bazex J. Neutrophilic dermatosis: a case of overlapping syndrome with monoclonal antineutrophil cytoplasmic autoantibody activity. Dermatology (1994) 189(1):69–71. doi: 10.1159/000246788
159. Marliere V, Beylot-Barry M, Beylot C, Doutre MS. Successful treatment of subcorneal pustular dermatosis (Sneddon-Wilkinson disease) by acitretin: report of a case. Dermatology (1999) 199(2):153–5. doi: 10.1159/000018224
160. Versini M, Mantoux F, Angeli K, Passeron T, Lacour JP. Maladie de sneddon-Wilkinson échappant à l’infliximab et à l’étanercept: efficacité de l’adalimumab. Annales Dermatol Vénéréol (2013) 140(12):797–800. doi: 10.1016/j.annder.2013.07.012
161. Velasco-Tamariz V, Carreño-Tarragona G, Tous-Romero F, Gil-de la Cruz E, Martín-Clavero E, Rivera-Díaz R. Dramatic resolution of disseminated pyoderma gangrenosum associated with monoclonal gammopathy after therapy with bortezomib and dexamethasone. Int Wound J (2017) 14(6):1382–4. doi: 10.1111/iwj.12746
162. Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol (2018) 14(3):225–33. doi: 10.1080/1744666X.2018.1438269
163. Maverakis E, Marzano AV, Le ST, Callen JP, Brüggen MC, Guenova E, et al. Pyoderma gangrenosum. Nat Rev Dis Primers (2020) 6(1):1–9. doi: 10.1038/s41572-020-0213-x
164. Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol (2005) 53(2):273–83. doi: 10.1016/j.jaad.2004.10.006
165. Cheng S, Edmonds E, Ben-Gashir M, Yu RC. Subcorneal pustular dermatosis: 50 years on. Clin Exp Dermatol (2008) 33(3):229–33. doi: 10.1111/j.1365-2230.2008.02706.x
166. Watts PJ, Khachemoune A. Subcorneal pustular dermatosis: a review of 30 years of progress. Am J Clin Dermatol (2016) 17(6):653–71. doi: 10.1007/s40257-016-0202-8
167. Nofal A, Abdelmaksoud A, Amer H, Nofal E, Yosef A, Gharib K, et al. Sweet's syndrome: diagnostic criteria revisited. JDDG: J der Deutschen Dermatol Gesellschaft (2017) 15(11):1081–8. doi: 10.1111/ddg.13350
168. Villarreal-Villarreal CD, Ocampo-Candiani J, Villarreal-Martínez A. Sweet syndrome: a review and update. Actas dermo-sifiliograficas (2016) 107(5):369–78. doi: 10.1016/j.ad.2015.12.001
169. Gusdorf L, Lipsker D. Neutrophilic urticarial dermatosis: A review. Annales Dermatol Vénéréol (2018) 145(12):735–40. doi: 10.1016/j.annder.2018.06.010
170. Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. report of 9 new cases and review of the literature. Medicine (2009) 88(1):23–31. doi: 10.1097/MD.0b013e3181943f5e
171. Clarkson B, Thompson D, Horwith M, Luckey EH. Cyclical edema and shock due to increased capillary permeability. Am J Med (1960) 29(2):193–216. doi: 10.1016/0002-9343(60)90018-8
172. Gousseff M, Arnaud L, Lambert M, Hot A, Hamidou M, Duhaut P, et al. The systemic capillary leak syndrome: a case series of 28 patients from a European registry. Ann Internal Med (2011) 154(7):464–71. doi: 10.7326/0003-4819-154-7-201104050-00004
173. Matsumura M, Kakuchi Y, Hamano R, Kitajima S, Ueda A, Kawano M, et al. Systemic capillary leak syndrome associated with compartment syndrome. Internal Med (2007) 46(18):1585–7. doi: 10.2169/internalmedicine.46.0254
174. Xie Z, Ghosh CC, Patel R, Iwaki S, Gaskins D, Nelson C, et al. Vascular endothelial hyperpermeability induces the clinical symptoms of clarkson disease (the systemic capillary leak syndrome). Blood (2012) 119(18):4321–32. doi: 10.1182/blood-2011-08-375816
175. Nakagawa N, Ota H, Tanabe Y, Kabara M, Matsuki M, Chinda J, et al. A case of idiopathic systemic capillary leak syndrome with high serum levels of G-CSF on exacerbation. Internal Med (2011) 50(6):597–600. doi: 10.2169/internalmedicine.50.4857
176. Alkhunaizi AM, Kabbani AH, ElTigani MA. Chronic idiopathic systemic capillary leak syndrome: a case report. Allergy Asthma Clin Immunol (2019) 15(1):1–4. doi: 10.1186/s13223-019-0347-0
177. Correia CP, Guiomar V, Coelho F, Almeida J. Non-occlusive mesenteric ischaemia and acute kidney injury: A case report of severe idiopathic systemic capillary leak syndrome. Eur J Case Rep Internal Med (2019) 6(7):001156. doi: 10.12890/2019_001156
178. Droder RM, Kyle RA, Greipp PR. Control of systemic capillary leak syndrome with aminophylline and terbutaline. Am J Med (1992) 92(5):523–6. doi: 10.1016/0002-9343(92)90749-2
179. Wan XC, Lai A, Kompala T, Ten R. Mimicker of hereditary angioedema: Idiopathic systemic capillary leak syndrome successfully treated with intravenous immunoglobulin. Ann Allergy Asthma Immunol (2017) 118(5):631–2. doi: 10.1016/j.anai.2017.02.013
180. Dhir V, Arya V, Malav IC, Suryanarayanan BS, Gupta R, Dey AB. Idiopathic systemic capillary leak syndrome (SCLS): case report and systematic review of cases reported in the last 16 years. Internal Med (2007) 46(12):899–904. doi: 10.2169/internalmedicine.46.6129
181. Yabe H, Yabe M, Koike T, Shimizu T, Morimoto T, Kato S. Rapid improvement of life-threatening capillary leak syndrome after stem cell transplantation by bevacizumab. Blood (2010) 115(13):2723–4. doi: 10.1182/blood-2009-11-247056
182. Koo H, Oh DH, Chun YS, Kim JC. A case of crystalline keratopathy in monoclonal gammopathy of undetermined significance (MGUS). Korean J Ophthalmol (2011) 25(3):202–5. doi: 10.3341/kjo.2011.25.3.202
183. Kocabeyoglu S, Mocan MC, Haznedaroglu IC, Uner A, Uzunosmanoglu E, Irkec M. In vivo confocal microscopic characteristics of crystalline keratopathy in patients with monoclonal gammopathy: report of two cases. Indian J Ophthalmol (2014) 62(9):938. doi: 10.4103/0301-4738.143933
184. González DP, Iglicki M, Svetitsky S, Bar-On Y, Habot-Wilner Z, Zur D. Occlusive retinal vasculopathy with macular branch retinal artery occlusion as a leading sign of atypical hemolytic uremic syndrome–a case report. BMC Ophthalmol (2021) 21(1):1–7. doi: 10.1186/s12886-021-01820-x
185. Eton EA, Abrams G, Khan NW, Fahim AT. Autoimmune retinopathy associated with monoclonal gammopathy of undetermined significance: a case report. BMC Ophthalmol (2020) 20(1):1–7. doi: 10.1186/s12886-020-01423-y
186. Milman T, Kao AA, Chu D, Gorski M, Steiner A, Simon CZ, et al. Paraproteinemic keratopathy: the expanding diversity of clinical and pathologic manifestations. Ophthalmology (2015) 122(9):1748–56. doi: 10.1016/j.ophtha.2015.05.029
187. Branellec A, Bouillet L, Javaud N, Mekinian A, Boccon-Gibod I, Blanchard-Delaunay C, et al. Acquired C1-inhibitor deficiency: 7 patients treated with rituximab. J Clin Immunol (2012) 32(5):936–41. doi: 10.1007/s10875-012-9691-2
188. Cicardi M, Zingale LC, Pappalardo E, Folcioni A, Agostoni A. Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (2003) 82(4):274–81. doi: 10.1097/01.md.0000085055.63483.09
189. Gobert D, Paule R, Ponard D, Levy P, Frémeaux-Bacchi V, Bouillet L, et al. A nationwide study of acquired C1-inhibitor deficiency in France: characteristics and treatment responses in 92 patients. Medicine (2016) 95(33):e4363. doi: 10.1097/MD.0000000000004363
190. Bork K, Staubach-Renz P, Hardt J. Angioedema due to acquired C1-inhibitor deficiency: spectrum and treatment with C1-inhibitor concentrate. Orphanet J Rare Dis (2019) 14(1):1–1. doi: 10.1186/s13023-019-1043-3
191. Frémeaux-Bacchi V, Guinnepain MT, Cacoub P, Dragon-Durey MA, Mouthon L, Blouin J, et al. Prevalence of monoclonal gammopathy in patients presenting with acquired angioedema type 2. Am J Med (2002) 113(3):194–9. doi: 10.1016/S0002-9343(02)01124-5
192. Voisin S, Hamidou M, Lefrançois A, Sigaud M, Mahé B, Trossaërt M. Acquired von willebrand syndrome associated with monoclonal gammopathy: a single-center study of 36 patients. Medicine (2011) 90(6):404–11. doi: 10.1097/MD.0b013e3182397166
193. Gama I, Almeida L. Lattice-like paraproteinemic keratopathy (PPK) of monoclonal gammopathy of undetermined significance (MGUS). Case Rep (2017) 2017:bcr2016218031. doi: 10.1136/bcr-2016-218031
194. Karakus S, Gottsch JD, Caturegli P, Eghrari AO. Monoclonal gammopathy of “ocular” significance. Am J Ophthalmol Case Rep (2019) 15:100471. doi: 10.1016/j.ajoc.2019.100471
195. Lisch W, Wasielica-Poslednik J, Kivelä T, Schlötzer-Schrehardt U, Rohrbach JM, Sekundo W, et al. The hematologic definition of monoclonal gammopathy of undetermined significance in relation to paraproteinemic keratopathy (An American ophthalmological society thesis). Trans Am Ophthalmol Soc (2016) 114:T7-1–T7-21.
196. Wasielica-Poslednik J, Gericke A, Pfeiffer N, Lisch W. Paraproteinämische keratopathie als klinisches zeichen einer monoklonalen gammopathie. Klinische Monatsblätter für Augenheilkunde (2019) 236(03):289–94. doi: 10.1007/s00347-017-0615-7
197. Lisch W, Saikia P, Pitz S, Pleyer U, Lisch C, Jaeger M, et al. Chameleon-like appearance of immunotactoid keratopathy. Cornea (2012) 31(1):55–8. doi: 10.1097/ICO.0b013e31821ddd0c
198. Shah S, Espana EM, Margo CE. Ocular manifestations of monoclonal copper-binding immunoglobulin. Survey Ophthalmol (2014) 59(1):115–23. doi: 10.1016/j.survophthal.2013.03.002
199. Tzelikis PF, Laibson PR, Ribeiro MP, Rapuano CJ, Hammersmith KM, Cohen EJ. Ocular copper deposition associated with monoclonal gammopathy of undetermined significance: case report. Arquivos Brasileiros Oftalmol (2005) 68:539–41. doi: 10.1590/S0004-27492005000400021
200. Duquesne A, Werbrouck A, Fabiani B, Denoyer A, Cervera P, Verpont MC, et al. Complete remission of monoclonal gammopathy with ocular and periorbital crystal storing histiocytosis and fanconi syndrome. Hum Pathol (2013) 44(5):927–33. doi: 10.1016/j.humpath.2012.10.012
201. Chiang HH, Wieland RS, Rogers TS, Gibson PC, Atweh G, McCormick G. Paraproteinemic keratopathy in monoclonal gammopathy of undetermined significance treated with primary keratoprosthesis: Case report, histopathologic findings, and world literature review. Medicine (2017) 96(50). doi: 10.1097/MD.0000000000008649
202. Hall RB, Yeung SN, Yenson PR, Moloney G. A series of 4 cases of distinct keratopathy secondary to dysproteinemia: Immunotactoid keratopathy. Can J Ophthalmol (2014) 49(4):388–91. doi: 10.1016/j.jcjo.2014.04.011
203. Stiefel HC, Sandhu RK, Miller AK, Wilson DJ, Chamberlain WD. Characterization of corneal deposition keratopathy in the setting of blood cell dyscrasia and a minimally invasive technique to clear the cornea in a single case. Am J Ophthalmol Case Rep (2019) 13:83–8. doi: 10.1016/j.ajoc.2018.12.010
204. Wasielica-Poslednik J, Gericke A, Desuki A, Schlötzer-Schrehardt U, Pfeiffer N, Lisch W. Recurrence of paraproteinemic keratopathy after penetrating keratoplasty and its assessment with confocal microscopy. Am J Ophthalmol Case Rep (2018) 11:87–91. doi: 10.1016/j.ajoc.2018.06.014
205. Tainsh LT, Coady PA, Sinard JH, Neparidze N, Meskin SW, Adelman RA, et al. Asymmetric deep stromal keratopathy in a patient with multiple myeloma. Cornea (2017) 36(3):372–4. doi: 10.1097/ICO.0000000000001139
206. Mansour AM, Arevalo JF, Badal J, Moorthy RS, Shah GK, Zegarra H, et al. Paraproteinemic maculopathy. Ophthalmology (2014) 121(10):1925–32. doi: 10.1016/j.ophtha.2014.04.007
207. Saffra N, Rakhamimov A, Solomon WB, Scheers-Masters J. Monoclonal gammopathy of undetermined significance maculopathy. Can J Ophthalmol (2013) 48(6):e168–70. doi: 10.1016/j.jcjo.2013.07.018
208. Leys A, Vandenberghe P. Serous macular detachments in a patient with IgM paraproteinemia: an optical coherence tomography study. Arch Ophthalmol (2001) 119(6):911–3.
209. Smith SJ, Johnson MW, Ober MD, Comer GM, Smith BD. Maculopathy in patients with monoclonal gammopathy of undetermined significance. Ophthalmol Retina (2020) 4(3):300–9. doi: 10.1016/j.oret.2019.09.018
210. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the h ereditary a ngioedema I nternational W orking G roup. Allergy (2014) 69(5):602–16. doi: 10.1111/all.12380
211. Cugno M, Zanichelli A, Foieni F, Caccia S, Cicardi M. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends Mol Med (2009) 15(2):69–78. doi: 10.1016/j.molmed.2008.12.001
212. Cicardi M, Zanichelli A. The acquired deficiency of C1-inhibitor: lymphoproliferation and angioedema. Curr Mol Med (2010) 10(4):354–60. doi: 10.2174/156652410791317066
213. Tohani A, Chua I, Grigoriadou S, Buckland MS, Longhurst HJ. Acquired C1 inhibitor deficiency: should we monitor for associated antibody deficiency? Ann Allergy Asthma Immunol (2014) 112(3):265–7. doi: 10.1016/j.anai.2013.12.024
214. Oostergo T, Prins G, Schrama YC, Leeuwenburgh I. Small bowel angioedema due to acquired C1 inhibitor deficiency: a case report and overview. Eur J Gastroenterol Hepatol (2013) 25(4):507–13. doi: 10.1097/MEG.0b013e32835c94ff
215. Kumar S, Pruthi RK, Nichols WL. Acquired von willebrand's syndrome: a single institution experience. Am J Hematol (2003) 72(4):243–7. doi: 10.1002/ajh.10298
216. Horiuchi H, Doman T, Kokame K, Saiki Y, Matsumoto M. Acquired von willebrand syndrome associated with cardiovascular diseases. J Atheroscl Thromb (2019) 26:303–14. doi: 10.5551/jat.RV17031
217. Jonge Poerink-Stockschläder AB, Dekker I, Risseeuw-Appel IM, Haählen K. Acquired Von willebrand disease in children with a wilms' tumor. Med Pediatr Oncol (1996) 26(4):238–43. doi: 10.1002/(SICI)1096-911X(199604)26:4<238::AID-MPO3>3.0.CO;2-K
218. Stuijver DJ, Piantanida E, Van Zaane B, Galli L, Romualdi E, Tanda ML, et al. Acquired von W illebrand syndrome in patients with overt hypothyroidism: A prospective cohort study. Haemophilia (2014) 20(3):326–32. doi: 10.1111/hae.12275
219. Kasatkar P, Ghosh K, Shetty S. Acquired von willebrand syndrome: a rare disorder of heterogeneous etiology. J Postgraduate Med (2013) 59(2):98. doi: 10.4103/0022-3859.113816
220. Kreuz W, Linde R, Funk M, Meyer-Schrod R, Föll E, Nowak-Göttl U, et al. Valproate therapy induces von willebrand disease type I. Epilepsia (1992) 33(1):178–84. doi: 10.1111/j.1528-1157.1992.tb02303.x
221. Federici AB. Acquired von willebrand syndrome: an underdiagnosed and misdiagnosed bleeding complication in patients with lymphoproliferative and myeloproliferative disorders. Semin Hematol (2006) 43:S48–58. doi: 10.1053/j.seminhematol.2005.11.003
222. Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood (1998) 92(8):2707–11. doi: 10.1182/blood.V92.8.2707
223. Castaman G, Rodeghiero F, Di Bona E, Ruggeri M. Clinical effectiveness of desmopressin in a case of acquired von willebrand's syndrome associated with benign monoclonal gammopathy. Blut (1989) 58(4):211–3. doi: 10.1007/BF00320776
224. Goudemand J, Samor B, Caron C, Jude B, Gosset D, Mazurier C. Acquired type II von willebrand's disease: demonstration of a complexed inhibitor of the von willebrand factor-platelet interaction and response to treatment. Br J haematol (1988) 68(2):227–33. doi: 10.1111/j.1365-2141.1988.tb06194.x
225. Howard C, Lin T, Cunningham M, Lipe B. IgGG kappa monoclonal gammopathy of undetermined significance (MGUS) presenting as acquired type III Von willebrand syndrome. Blood coagulation fibrinolysis (2014) 25(6):631. doi: 10.1097/MBC.0000000000000112
226. Franchini M, Mannucci PM. Acquired von willebrand syndrome: focused for hematologists. Haematologica (2020) 105(8):2032. doi: 10.3324/haematol.2020.255117
227. Agarwal N, Klix MM, Burns CP. Successful management with intravenous immunoglobulins of acquired von willebrand disease associated with monoclonal gammopathy of undetermined significance. Ann Internal Med (2004) 141(1):83–4. doi: 10.7326/0003-4819-141-1-200407060-00037
228. Abou-Ismail MY, Rodgers GM, Bray PF, Lim MY. Acquired von willebrand syndrome in monoclonal gammopathy–a scoping review on hemostatic management. Res Pract Thromb haemostasis (2021) 5(2):356–65. doi: 10.1002/rth2.12481
229. Dane KE, Lindsley JP, Kickler T, Streiff MB, Moliterno A, Yui J, et al. Continuous-infusion von willebrand factor concentrate is effective for the management of acquired von willebrand disease. Blood Adv (2021) 5(14):2813–6. doi: 10.1182/bloodadvances.2021004843
230. Lavin M, Brophy TM, Rawley O, O'Sullivan JM, Hayden PJ, Browne PV, et al. Lenalidomide as a novel treatment for refractory acquired von willebrand syndrome associated with monoclonal gammopathy. J Thromb Haemostasis (2016) 14(6):1200–5. doi: 10.1111/jth.13317
231. Grimaldi D, Bartolucci P, Gouault-Heilmann M, Martin-Toutain I, Khellaf M, Godeau B. Rituximab failure in a patient with monoclonal gammopathy of undetermined significance (MGUS)-associated acquired von willebrand syndrome. Thromb haemostasis (2008) 99(04):782–3. doi: 10.1160/TH07-07-0456
232. Mathew Thomas V, Gilreath JA, Newton S, Smock KJ, Lim MY. Daratumumab as a novel treatment for refractory acquired von willebrand syndrome associated with monoclonal gammopathy. Haemophilia (2021) 27(4):e563–6. doi: 10.1111/hae.14323
233. Nageswara Rao AA, Rodriguez V, Long ME, Winters JL, Nichols WL, Pruthi RK. Transient neonatal acquired von willebrand syndrome due to transplacental transfer of maternal monoclonal antibodies. Pediatr Blood Cancer (2009) 53(4):655–7. doi: 10.1002/pbc.22084
234. Dogan S, Barnes L, Cruz-Vetrano WP. Crystal-storing histiocytosis: report of a case, review of the literature (80 cases) and a proposed classification. Head Neck Pathol (2012) 6(1):111–20. doi: 10.1007/s12105-011-0326-3
235. Aline-Fardin A, Bender S, Fabiani B, Buob D, Brahimi S, Verpont MC, et al. Pseudo-peritoneal carcinomatosis presentation of a crystal-storing histiocytosis with an unmutated monoclonal κ light chain. Medicine (2015) 94(32):e1247. doi: 10.1097/MD.0000000000001247
236. Contejean A, Larousserie F, Bouscary D, Dohan A, Deau-Fischer B, Szwebel TA, et al. A colonic mass revealing a disseminated crystal storing histiocytosis secondary to indolent multiple myeloma: a case report with literature review. BMC Gastroenterol (2020) 20(1):1–5. doi: 10.1186/s12876-020-01364-2
237. Uthamalingam P, Mehta S. Crystal-storing histiocytosis: Report of a rare case presenting with pathological fracture of femur. is there more to the entity? Int J Surg Pathol (2017) 25(5):458–61. doi: 10.1177/1066896917696746
238. Tao Q, Zhang W, Chen Z, Gao L, Yan J, Wang M, et al. Generalized crystal-storing histiocytosis with diffuse large b-cell lymphoma and monoclonal gammopathy in a Chinese elderly woman: a case report. BMC Cancer (2019) 19(1):1–8. doi: 10.1186/s12885-019-5734-x
Keywords: monoclonal gammopathy, monoclonal gammopathy of clinical significance, MGUS, immunotherapy, monoclonal gammopathy of undetermined significance, MGCS
Citation: Oganesyan A, Gregory A, Malard F, Ghahramanyan N, Mohty M, Kazandjian D, Mekinian A and Hakobyan Y (2022) Monoclonal gammopathies of clinical significance (MGCS): In pursuit of optimal treatment. Front. Immunol. 13:1045002. doi: 10.3389/fimmu.2022.1045002
Received: 15 September 2022; Accepted: 07 November 2022;
Published: 23 November 2022.
Edited by:
Maria Giovanna Danieli, Università Politecnica delle Marche, ItalyReviewed by:
Chiara Cardamone, University of Salerno, ItalySeerapani Gopaluni, University of Cambridge, United Kingdom
Chen Wang, USF Health, United States
Copyright © 2022 Oganesyan, Gregory, Malard, Ghahramanyan, Mohty, Kazandjian, Mekinian and Hakobyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Artem Oganesyan, YS50Lm9nYW5lc3lhbkBnbWFpbC5jb20=