 José Pedro Elizalde-Díaz1
José Pedro Elizalde-Díaz1 Clara Leticia Miranda-Narváez2
Clara Leticia Miranda-Narváez2 Juan Carlos Martínez-Lazcano2
Juan Carlos Martínez-Lazcano2 Eduardo Martínez-Martínez1*
Eduardo Martínez-Martínez1*- 1Laboratory of Cell Communication & Extracellular Vesicles, Division of Basic Science, Instituto Nacional de Medicina Genómica, Ciudad de México, Mexico
- 2Laboratorio de Neurofarmacología Molecular y Nanotecnología, Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez, Ciudad de México, Mexico
In the past two years, the world has faced the pandemic caused by the severe acute respiratory syndrome 2 coronavirus (SARS-CoV-2), which by August of 2022 has infected around 619 million people and caused the death of 6.55 million individuals globally. Although SARS-CoV-2 mainly affects the respiratory tract level, there are several reports, indicating that other organs such as the heart, kidney, pancreas, and brain can also be damaged. A characteristic observed in blood serum samples of patients suffering COVID-19 disease in moderate and severe stages, is a significant increase in proinflammatory cytokines such as interferon-α (IFN-α), interleukin-1β (IL-1β), interleukin-2 (IL-2), interleukin-6 (IL-6) and interleukin-18 (IL-18), as well as the presence of autoantibodies against interferon-α (IFN-α), interferon-λ (IFN-λ), C-C motif chemokine ligand 26 (CCL26), CXC motif chemokine ligand 12 (CXCL12), family with sequence similarity 19 (chemokine (C-C motif)-like) member A4 (FAM19A4), and C-C motif chemokine ligand 1 (CCL1). Interestingly, it has been described that the chronic cytokinemia is related to alterations of blood-brain barrier (BBB) permeability and induction of neurotoxicity. Furthermore, the generation of autoantibodies affects processes such as neurogenesis, neuronal repair, chemotaxis and the optimal microglia function. These observations support the notion that COVID-19 patients who survived the disease present neurological sequelae and neuropsychiatric disorders. The goal of this review is to explore the relationship between inflammatory and humoral immune markers and the major neurological damage manifested in post-COVID-19 patients.
Introduction
The pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has increased morbidity and mortality rates worldwide (1, 2). According to various clinical reports and laboratory studies, it is known that the virus can affect different organs such as respiratory tract, lungs, heart, liver, pancreas, kidneys, muscles, and nervous system at different levels (3–5). During the pandemic course, several post COVID-19 effects have been observed that hinder total patient recovery. The World Health Organization (WHO) has denominated these symptoms as long COVID or COVID-19 condition, defining it as a condition that “occurs in individuals with a history of probable or confirmed SARS-CoV-2 infection, usually 3 months from the onset of COVID-19 with symptoms and that last for at least 2 months and cannot be explained by an alternative diagnosis. Symptoms may be new onset following initial recovery from an acute COVID-19 episode or persist from the initial illness. Symptoms may also fluctuate or relapse over time” (6–8).
Several follow-up studies in patients suffering long COVID have documented cardiovascular alterations, fatigue, dyspnea, chest pain, appetite loss and hair loss. Interestingly nervous system seems particularly affected after COVID-19 disease (9, 10). Patients have reported headaches and dizziness, as well as psychiatric disorders and motor discoordination (11–13). In a period of 7 months after viral infection, some patients have presented conditions that are mainly related to neuropsychiatric and neurological deficits, with a prevalence of 19.7% to 36% (4, 14, 15). The characteristic symptoms of these alterations are anosmia, hypogeusia, partial or total hyposmia (16, 17), myalgia, cerebral inflammation, cerebrovascular strokes (18), acute encephalopathy, seizures, Guillain-Barré syndrome (19), neurocognitive disorders, sleep disorders, delirium, memory deficit, concentration deficit, depression, psychosis, hallucinations, paranoia (20), chronic fatigue and partial or total apraxia (21).
Similar to the neurological alterations of SARS-CoV-2 post-infection, there are data from patients who were infected with SARS-CoV-1 and MERS. The clinical follow-up carried out on these patients recorded symptoms of depression, disorder of post-traumatic stress (PTSD), anxiety, sleep disorders, weakness, chronic fatigue and general pain, in a follow-up period covering 6 to 20 months post-infection (22, 23), symptoms set similar to the neurological alterations reported in SARS-CoV-2 post-infection. A meta-analysis of 120,970 patients infected with SARS-CoV-2 revelated that women are more susceptible to present moderate neurological and cardiovascular long-COVID symptoms. It also was reported that age is directly related to a higher incidence of psychiatric, respiratory, digestive and skin conditions. In addition, in a subgroup of 106,284 participants it was observed an incidence of 19.7% of neurological disorders, where the main manifestations included, concentration difficulty (14.6%), headache, disorders of the taste and smell, cognitive impairment, memory deficits, dizziness, and cramps. Furthermore, psychiatric conditions affected 20.3% of the participants, who presented PTSD, depression, sleep disorder and anxiety (14).
The analysis of cerebrospinal fluid (CSF) and peripheral blood samples of 127 patients, who were positive for SARS-CoV-2 and showed neurological damage symptoms after 7 days of infection, revelated that they suffered systemic inflammation and impaired blood-brain barrier (BBB). The neurological manifestations included encephalopathy, altered consciousness, delayed walking reaction, epilepsy-like electroencephalogram (EEG) changes, cerebral ischemia, myelitis, cerebellar ataxia, sensorimotor symptoms of unknown cause, cognitive impairment, peripheral neuropathy, anosmia, headache and nausea (24). Altogether these studies indicate a relationship between SARS-CoV-2 infection and neurological conditions observed in long COVID. The main goal of this review is to elucidate the role of the antiviral dysregulation response by the immune system and its relationship with the sequelae of damage to the central nervous system (CNS) in patients with long COVID.
Relationship between SARS-CoV-2 and nervous system
It has been documented that coronaviruses have the ability to affect the CNS (25). In this context, several investigations have discovered that β-coronaviruses such as MERS-CoV and SARS-CoV-1 can infect the CNS (25–29). Furthermore, traces of SARS-CoV-2 have been detected in the olfactory mucosa, trans olfactory mucosa, neuronal projections and neurons during and after the infection period (30–34). In some COVID-19 cases the first symptoms presented by patients is hyposmia or anosmia. This could be due to the olfactory epithelium damage caused by the coronavirus, which in turn affects the olfactory neural network that is connected with the primary olfactory cortex (35–37). To date there is no precise understanding about the dynamics of the initial antiviral response against SARS-CoV-2 that occur at the level of the olfactory epithelium. However, there are data from nasal samples that showed an increase of proinflammatory cytokines within two days after the first symptoms, compared with samples of same tissue that were taken at longer times (5 or more days after presenting the first symptoms), when the levels of proinflammatory cytokines decreased (17). This could indicate that the immune response produced in the olfactory epithelium associated with nerve cells occurs in a transient manner. However, this response is sufficient to generate some neuronal damage either by a direct action of the virus or by an indirect mechanism that involves the dysregulation of the immune response.
The BBB is the main physiological structural interconnection between the external environment and the brain whose main function is to protect central neurons. It also participates in the selective transit of cells, nutrients and brain cell metabolism toxic byproducts (38). When a systemic inflammation process occurs, the BBB induces a series of brain responses whose main objective is to promote brain survival, which is known as disease behavior (39). This response induces a set of physiological and behavioral changes, coordinated and executed by the brain, which protect the individual from the various phases occurring during an infection. For example, the induction of lethargy allows to fight infection through the induction of fever and anorexia (40, 41).
In patients who succumbed to COVID-19 and who had an exacerbated inflammatory response, presented BBB involvement manifested through multifocal vascular damage caused by autoantibodies. This process that induced serum proteins infiltration into the brain parenchyma, generalized endothelial cell activation, classical complement pathway activation, platelet aggregates and microthrombi adhered to endothelial cells throughout the vascular lumen. In addition, the infiltration of macrophages, T cells and B cells into brain structures has been reported, observing a greater presence of CD8+ T cells in the perivascular region compared to CD4+ cells. There are also reports of astrogliosis in perivascular regions and microglial nodule formation in the hindbrain, which is associated with focal neuronal loss and neuronophagia (42).
The SARS-CoV-2 induces a nuclear structure reorganization and the dispersion of the genomic compartments of the cell, which leads to the low expression of the genes ADCY3, CNGA2, GN13, GFY, OMP, LHX2 and ATF5, which are key in the olfactory receptors signaling and this downregulation lead to anosmia (17). It has been proposed that once the virus enters the olfactory receptor neurons, the infection is propagated through the synaptic connections (43). In the case of the olfactory receptor neurons-mitral cells axis, there is an activation of the glial, which in turn promote the release proinflammatory cytokines such as IFN-α, TNF-α, IL-1α, IL-1β, IL-2, IL-6, IL-8, IL-17A, IL-18, CXCL10, CXCL12, CCL1, CCL2, CCL3, CCL4, CCL5, CCL7, CCL11, GM-CSF and B cell-activating factor belonging to the TNF family (BAFF). These cytokines that have been detected at elevated levels in samples of CSF, brain tissue, and serum of peripheral blood from patients with severe COVID-19 (44–49). It should be noted that the upregulated production of these cytokines can cause serious damage to the CNS, since it promotes neuronal stress and apoptosis, as well as the interruption of the BBB (43). In a mild respiratory COVID mouse model, it was observed that these events eventually increase neuroinflammation cascades causing synaptic loss, demyelination, excitotoxicity and transcriptional downregulation of Trem2, Sall3 and Adrb1 genes in microglia, the latter gene being an indicator of white matter degeneration (48). Other cerebral regions can potentially be affected by a similar mechanism. For instance, midbrain dopamine neurons derived from human pluripotent stem cells are selectively permissive to SARS-CoV-2 infection. This triggers an inflammatory response at neuronal level and the expression of the insulin like growth factor binding protein 7 (IGFBP7) and LAMININ B1 genes associated with cellular senescence (32). The expression of these molecules leads to the overactivation of glia and trigger mechanisms of neuronal damage (50). Overall, the neuronal damage associated with the upregulation of proinflammatory cytokines could be the cause of the appearance of neurological symptoms related with long COVID (Table 1).
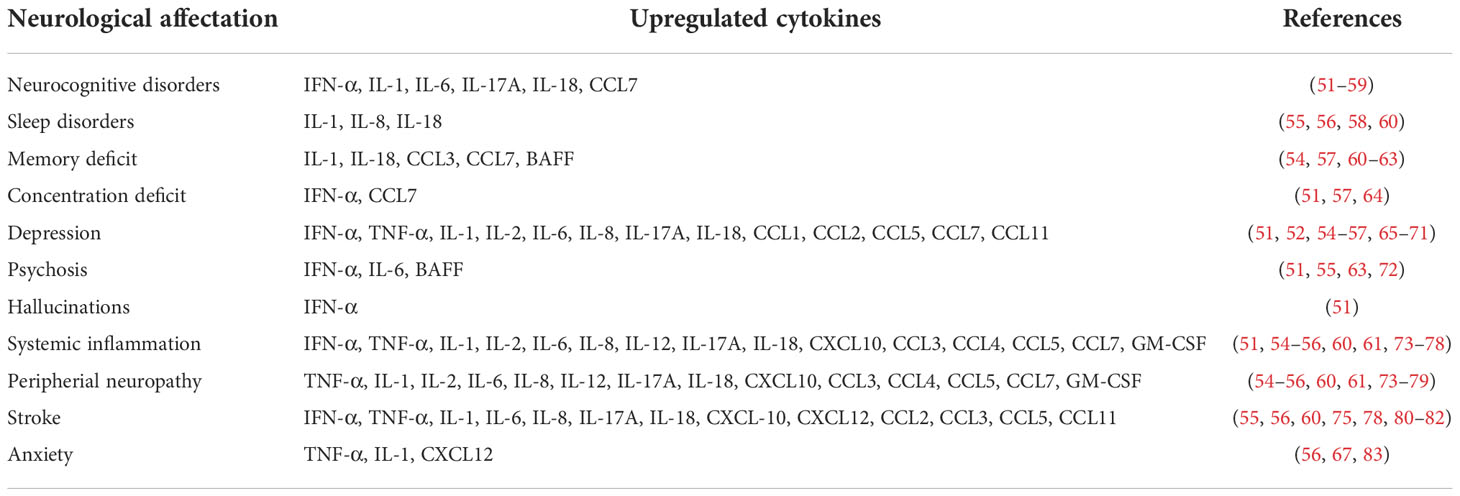
Table 1 Upregulated cytokines associated at neurological damage observed in patients with long COVID.
The effects that SARS-CoV-2 infection induces in brain structures was analyzed on 401 patients who suffered from COVID-19. Using the UK Biobank database, there was a selection of patients with brain imaging studies prior to COVID infection, and all patients were subject to brain imaging 38 months later. All the patients had at least one or more of the following affectations: significant reduction in gray matter thickness and tissue contrast in the orbitofrontal cortex, changes in diffusion measures, which are indicators of tissue damage, increase in CSF volume and overall size brain reduction (37). These changes were consistent and related to previously detected cognitive impairment in the study population. SARS-CoV-2 infection also changes the vasculature of the brain, since one of the damages induced by the virus is ischemic and hemorrhagic cerebrovascular strokes (84). A postmortem study in patients who died from severe COVID-19 revelated the presence of viral inclusion structures, accumulation of inflammatory cells in the vascular endothelium (lymphocytic endotheliitis), and endothelial cell apoptosis (50). All these sequelae of SARS-CoV-2 infection in the CNS has been monitored in the serum and CSF of patients with long COVID who present neurological damage symptoms (encephalopathy, seizures, paraplegia, paresis, Guillain-Barré syndrome, ataxia and dysesthesia). These patients show a slight increase in white blood cells and an increase in the concentrations of total proteins and albumin, which indicates that the virus triggers a systemic dysfunction that can be detected at blood and CSF level (24).
Deciphering the process of neurological damage caused by the exacerbated innate immune response to SARS-CoV-2
Once a virus reaches the nerves and brain tissue, an inflammatory mechanism is activated which aims to limit the infection process, eliminate the virus, or repair cell damage. Depending on the activated immunological pathway and the magnitude with which it is activated, the response can have positive or negative consequences on the physiology and behavior of the individual (85). The complications of exacerbated neuroinflammation can include headache, ischemia, interstitial edema, cerebral vasodilatation, blood vessel injury, vomiting, visual loss, blood stasis, increased cerebral pressure, cognitive problems, and loss of consciousness (86–89). Neuroinflammation characterized by an early and brief inflammatory response is considered neuroprotective, and is initiated by the activation of glial and endothelial cells (90, 91). On the contrary, a prolonged neuroinflammatory activation induces damage to brain structures and tissues, which has been associated with several neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (92, 93).
The role of the microglia during resting conditions is to constantly examine the brain microenvironment to maintain homeostasis through the elimination of cellular waste (94). When there is a damage to neuronal structures, a process known as microglia activation occurs. This process is characterized by the release of cytokines, chemokines, and inflammatory molecules (95). However, when the immune response is dysregulated, the exacerbated release of proinflammatory cytokines occurs, which has been associated with high mortality in patients with COVID-19 (96). This type of patients show microglia hyperactivation through multisystem inflammatory syndrome (97, 98) and systemic inflammatory response syndrome (99).
Dysregulation of the immune response due to the SARS-CoV-2 infection has the ability to downregulate angiotensin converting enzyme 2 (ACE-2) expression, which influences the activation and balance of the inflammatory pathway (100). The decreased expression of ACE-2 increases the concentration of Ang-II favoring the ACE/Ang-II/AT1R pathway. This leads to the activation of the NF-κB transcription factor and the consequent activation of the production and release of proinflammatory cytokines (101). Altered cytokine concentrations have been observed in samples of both patients with acute SARS-CoV-2 and in patients with manifestations associated with long COVID (43, 102, 103). The increase in Ang-II concentration also favors the Ang-II/aminopeptidase-A/Ang-III/aminopeptidase-A/Ang-IV/AT4R pathway (104, 105). The increase in Ang-III concentration induces hormone overproduction such as vasopressin in the hypothalamus and aldosterone in the adrenal gland (105). These alterations result in increased peripheral vascular resistance and blood pressure. Moreover, Ang-III dysregulates Na+/K+ equilibrium which results in vascular damage, stroke and heart attack (106, 107). Both Ang-III and Ang-IV can bind to AT1R, thus induce the activation of this receptor and the activation consequently of the NF-κB transcription factor (105, 108, 109). The increase of Ang-IV dysregulates the vasodilatation process, increases the excretion of sodium, and the release of plasminogen activator inhibitor-1, favoring the development of thrombotic events both in lungs and in the brain (108, 110–113). According to transcriptome databases, ACE2 is expressed in excitatory and inhibitory neurons, astrocytes, oligodendrocytes, and endothelial cells (114). We believe that ACE-2 downregulation induced by SARS-CoV-2 infection, is one of the first pathways responsible for immunological response damage to the CNS.
An additional mechanism associated with pro-inflammatory cytokines induction occurs when the virus infects the cell, and the innate immune system detects viral RNA genome, either as ssRNA or one of dsRNA’s intermediaries through the Toll-Like Receptors including TLR3, TLR7, and TLR8 (115, 116). These receptors are responsible for activation of transcription factors such as IRF3, IRF7, NF-κB, ISRE3, and API. This transcription factors are related to the expression of key proinflammatory cytokines in the antiviral response such as TNF-α, IFN-α, IFN-β and IFN-γ (115, 117). IFN-α and IFN-β activates genes involved in apoptosis processes, in the modulation of immune response, in cellular attraction and adhesion, and genes involved in antiviral and pathogenic detection (118). The balance that exists between IFN-α and IFN-β concentrations is key in the regulation of the inflammatory response. If there is any imbalance in their concentrations, the IFN-γ production is affected and therefore the anti-inflammatory process does not occur. In addition a chronic inflammation is promoted when the humoral response is deficient (119). Interestingly, in samples of respiratory epithelial cells and plasmacytoid dendritic cells from patients with severe COVID-19, there is a decrease of type-I IFNs associated with self-recessive deficiencies in genes that code for the proteins involved in interferon production (e.g. TLR3, UNC93B, TRIF, TBK1, TBK1, IRF3, IRF7, IFNAR1/2, MYD88, GATA2 and IRAK4) (120–126). In CSF samples of patients with acute COVID-19 and sings of neurological damage, it was found a reduced interferon response, expansion of clonal T cells and a depletion of CD4+ T cells (127). Thus, it is possible that the interferon production during and after infection is a key point in the process of regulating systemic and neuronal inflammation.
The inflammatory response in the CNS system is mediated by resident microglia and astrocytes (128), which detects the presence of an exogenous or pathogenic agent such as SARS-CoV-2 (129). Besides its direct participation in the elimination of an infection, the microglia establish the balance between the innate immune response and the adaptive immune response (130, 131). During acute COVID-19, the exacerbated release of proinflammatory cytokines promotes the production of reactive oxygen species (ROS), which causes stress and cell damage at the systemic level, affecting brain tissue (129). In some COVID-19 patients these cellular events manifest in symptoms such as ischemia, inflammation of brain tissue, obstruction of blood flow, headaches, loss of consciousness, cerebral edema, and neuronal death (131–133).
Previous studies have reported that during influenza virus infection there is an increase in the levels of proinflammatory cytokines, such as IL-1β, IL-6, CXCL8, CXCL9, CXCL10, CCL2, and TNF-α, in the CSF of patients who present neurological alterations such as acute encephalitis and encephalopathy (134, 135). It is also known that patients infected with human orthopneumovirus and presenting neurological symptoms such as encephalitis and encephalopathies, have elevated levels of the proinflammatory cytokines IL-6, IL-8, CCL2, and CCL4 in CSF samples (136, 137). West Nile virus is also known to cause a neuroinvasive disease manifesting meningitis, meningoencephalitis, encephalitis, or acute flaccid paralysis, commonly associated with diarrhea/vomiting, weakness, impaired vision, confusion, or drowsiness, and shows elevated levels of proinflammatory cytokines IL4, IL6, and IL10 in serum samples (138). Finally, Zika virus can infect the CNS and induce microcephaly in fetuses and rare but serious neurological diseases in adults, which are associated with excessive production of IFN-α, IFN-β, IL-6, and TNF-α (139).
Interestingly, these neuroinflammatory pathological processes observed in long COVID patients, resemble those that occur in early phase of Parkinson’s disease (PD and AD (92). For example, high levels of TNF-α and low levels of TNF-β have been detected in CSF samples from patients with mild cognitive impairment who progressed to AD, and the cytokines IL-1β, IL-6, and TNF-α, tend to increase slowly, while the cytokines IL-18, MCP-1, and IP-10 peak at a certain stage of the disease (140, 141). Activation of microglial cells has been detected in the substantia nigra of patients with PD, due to the fact that aggregated α-synuclein is released from the damaged dopaminergic neurons (142). The accumulation of α-synuclein leads microglia to a reactive proinflammatory phenotype in which TNF-α, nitric oxide, and IL-1β are produced, generating a neuroinflammatory state as recently shown in an in vitro model of PD (143).
Role of the dysregulated antibodies response against SARS-CoV-2 infection in neurological disorders
Part of neurological sequelae previously mentioned suffered by SARS-CoV-2 patients, were also reported in individuals who survived SARS-CoV-1 infection in 2004 who presented cerebrovascular disorders such as ischemic stroke (144). These affectations could be caused by abnormalities in coagulation and hyperinflammation promoted by the presence of antiphospholipid autoantibodies (eg. antiphosphatidylserine or antiprothrombin) produced by plasma cells (88, 145, 146). Autoantibodies are a type of antibodies that recognize epitopes present in organs or tissues of the same individual and are related to the development of autoimmune diseases including allergies and oncopathologies (147, 148). Much of the generation of these autoantibodies is caused by genetic mutations, infections or environmental factors (149). The autoantibody generation can result from an altered production of cytokines, stimulation of toll-like receptors, or pattern recognition receptors (150). Furthermore, they can also originate from an inadequate and dysregulated release of autoantigens by cells and tissues, and/or molecular mimicry (150, 151). In the case of COVID-19 infection, various studies indicate that the spike protein of SARS-CoV-2 is the causal agent of inducing the autoantibodies generation, which might be a common characteristic in coronavirus infections (147, 148, 150, 152). It has been reported that the antibodies produced by plasma cells against spike protein or receptor-binding domain of the SARS-CoV-2 can cross-bind with own antigens (153). In a follow-up study of 610 patients after 6 to 12 months post-infection with SARS-CoV-2, there were low concentrations of IgM and IgG3 that correlated with a predisposition to develop long COVID. Moreover, 71% of these patients presented severe COVID-19 and bronchial asthma at the same time (152). Regarding these immunoglobulins, it is known that both are induced by the controlled production of interferons and antagonized by IL-14 (154, 155). In addition, IgMs have a relevant role in the humoral response since it is the first immunoglobulin that participates in pathogen elimination (156). IgMs functions as a powerful complement activator, participate in the activation and regulation of the inflammatory response, opsonization, and destruction of pathogens present in the circulatory system (155, 157). In addition, IgMs are associated with the protective mechanisms of the vasculature and mucous membranes (157). IgG3s, activate the complement system and have a great affinity with Fc receptors (158). The deficiency of IgG3s is related with the development of autoimmune diseases (159). This could indicate that the innate immune response dysregulation directly affects the humoral response activation process, which leads to a deficient, non-specific and delayed production of antibodies against SARS-CoV-2.
In a recent multicenter study it was proposed that a deficient and prolonged immune response in hospitalized severe COVID-19 patients promotes the adaptive immune response that attacks non-structural viral proteins and causes the development of IgG autoantibodies (160). Similarly, a proteomic profiling analysis revealed that the generation of certain autoantibodies (e.g. MUC1 or TNFRSF6B) is associated with the severity of the disease (147). Consistent with this notion, several investigations have also found that patients who had COVID-19 exhibit marked increases in autoantibody reactivity compared with uninfected individuals (160, 161). These individuals show a high prevalence of autoantibodies against immunomodulatory proteins (including cytokines, chemokines, complement components, and cell surface proteins) (162). The main consequence of these autoantibodies is the disruption of the immune function and the impairment of the virologic control by inhibiting immunoreceptor signaling and altering the composition of peripheral immune cells (163, 164).
There are cases where the presence of autoantibodies can be detected prior to any viral infection, suggesting a genetic predisposition to the generation of these autoantibodies (165). This could explain why some COVID-19 patients are more susceptible to produce autoantibodies that promote long COVID (166, 167). Recent studies have shown that some of these autoantibodies have an affinity for blood vessel and nervous system proteins, which could explain the neurological effects of long COVID by two mechanisms (168). First, autoantibodies could potentiate the cellular stress induced by proinflammatory cytokines. Second, autoantibodies could cause specific and long-term damage in patients suffering from post-COVID neurological sequelae (43, 168). In fact, COVID-19 patients with neurological sequelae produce autoantibodies that inhibit the function of key proteins involved in neuroprotection processes, neurite outgrowth, axogenesis, neuronal plasticity, neurotransmission, neuronal survival, and axonal regeneration (Supplementary Table 1) (167). The generation of these autoantibodies may aggravate the neuronal damage.
The dysregulation of the immune response and the deficient elimination of cells infected by SARS-CoV-2 promote the release of autoantigens towards the extracellular space and the consequent generation of autoantibodies (169, 170). The analysis of the “autoantigenicoma” in patients who suffered from COVID-19 through the detection of autoantigens bound to determatan sulfate (autoantigen-DS complex) seems to be helpful to predict the appearance of autoimmune diseases and neurological damage (171, 172). Using this strategy, 751 autoantigen candidates were found, of which 657 are directly altered by infection with SARS-CoV-2. Remarkably, 400 of those autoantigens are related to autoimmune diseases and cancer (162). Regarding the nervous system, 150 autoantigens of proteins are related to axon guidance, neuron projection, myelin sheath, axon growth cone, neuronal cell body, cerebellar Purkinje cell layer, peripheral nervous system axon regeneration, radial glial scaffolds and proteins related to the olfactory bulb. There were also 193 autoantigens of proteins related to neurological diseases such as neuronal infection with Japanese encephalitis virus, neuroblastoma, glioblastoma, neurodegeneration in Down syndrome, AD, schizophrenia, cerebral ischemia induced neurodegenerative diseases, PD, and neurodegeneration (Supplementary Table 2) (172). The mechanism by which coronaviruses could resemble conditions of early events of neurodegeneration should be explored considering the participation of the immune system and the uncontrolled generation of autoantibodies that deteriorate neuronal circuits.
Summary and proposal
The effects of long COVID on the CNS are increasingly evident. For this reason, in the present work we analyzed the role of the immune response against the coronavirus and its impact on neuronal structures. The SARS-CoV-2 infects olfactory epithelial cells through ACE-2 (173). Through genetic rearrangements, the virus downregulates the expression of proteins such as olfactory receptors and ACE-2 (17, 100). The latter is implicated in the production of proinflammatory cytokines (43). When the immune system detects the entry of the virus, it activates the primary response, which is characterized by the release of proinflammatory cytokines and the activation of immune cells. These processes are regulated by type-I INFs and together with IFN-γ (115, 117) induce the generation of antibodies (130, 131). However, due to the downregulation of ACE-2 and mutations in type I INFs, the inflammatory response is dysregulated, provoking the exacerbated release of proinflammatory cytokines (117). This response damages cellular structures and promotes the release of autoantigens (168, 169). At the same time, the dysregulation of the innate immune response affects the activation process of the humoral response (119, 169). This may lead to a nonspecific and delayed production of antibodies against SARS-CoV-2 and the generation of autoantibodies that recognize key proteins involved in neuronal regeneration and repair processes, thereby increasing neurodegeneration (167). We think this generates a cyclical process of recognition and destruction of neuronal structures (Figure 1). Depending on the region that is affected, this promotes the appearance of neurological symptoms observed in patients with long COVID.
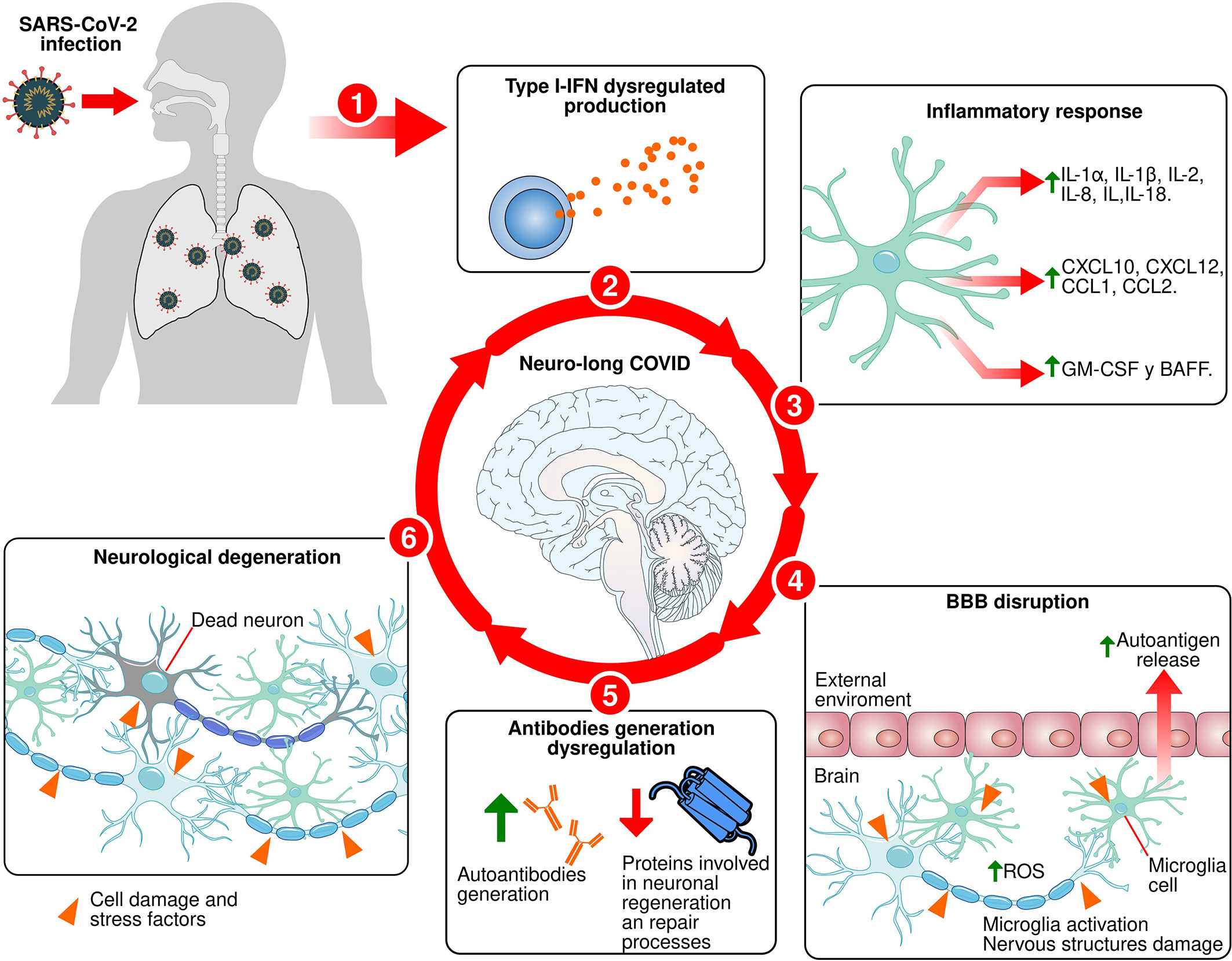
Figure 1 Proposed mechanism for neuro-long COVID. 1: SARS-CoV-2 infects olfactory epithelial and lungs. 2: Type-I IFNs production dysregulated during primary immune response process against SARS-CoV-2 infection. 3: Exacerbated release of proinflammatory cytokines. 4: The exacerbated and dysregulated inflammatory response causes the proinflammatory molecules release that damage the BBB, facilitate the infiltration of immune cells into brain tissue, activate microglia, and damaging brain tissue cells, causing the autoantigens release. 5: Innate immune response dysregulation affects the humoral response activation process and induce a nonspecific and delayed production of antibodies against SARS-CoV-2 and the generation of autoantibodies against key proteins involved in neuronal regeneration and repair processes. 6: Induction of neuronal death in specific areas.
Author contributions
JM-L and CM-N wrote the manuscript. JE-D and EM-M conceived and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Consejo Nacional de Ciencia y Tecnología (CONACYT #64382) and internal funds of the Instituto Nacional de Medicina Genómica.
Acknowledgments
JE-D is a postdoctoral researcher at the Instituto Nacional de Medicina Genómica and received a fellowship from CONACYT in the program “Estancias Posdoctorales por México en Apoyo por SARS CoV2 (COVID-19, 2166969)”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1039427/full#supplementary-material
References
1. WHO. WHO coronavirus (COVID-19) dashboard. Geneva, Switzerland: World Health Organization (2022). Available at: https://covid19.who.int/.
2. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Diseases (2020) 20(5):533–4. doi: 10.1016/S1473-3099(20)30120-1
3. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med (2020) 26(7):1017–32. doi: 10.1038/s41591-020-0968-3
4. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in wuhan, China. JAMA Neurol (2020) 77(6):683–90. doi: 10.1001/jamaneurol.2020.1127
5. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain (2020) 143(10):3104–20. doi: 10.1093/brain/awaa240
6. Dietrich A, Stete K, Hilger H, Götz V, Biever P, Hosp J, et al. Complaints and clinical findings six months after COVID-19: Outpatient follow-up at the university medical center freiburg. Deutsche Medizinische Wochenschrift (2021) 146(17):e65–73. doi: 10.1055/a-1546-4291
7. WHO. A clinical case definition of post COVID-19 condition by a Delphi consensus. Geneva, Switzerland: World Health Organization (2021). Available at: https://www.who.int/publications/i/item/WHO-2019-nCoV-Post_COVID-19_condition-Clinical_case_definition-2021.1.
8. WHO. Coronavirus disease (COVID-19): Post COVID-19 condition. Geneva, Switzerland: World Healt Organization (2021). Available at: https://www.who.int/news-room/questions-and-answers/item/coronavirus-disease-(covid-19)-post-covid-19-condition#:~:text=People%20wth%20post%20COVID%2D19,as%20work%20or%20household%20chores.
9. Carfì A, Bernabei R, Landi F. For the gemelli against c-P-ACSG. persistent symptoms in patients after acute COVID-19. JAMA (2020) 324(6):603–5. doi: 10.1001/jama.2020.1260313
10. Tenforde MW, Kim SS, Lindsell CJ, Billig Rose E, Shapiro NI, Files DC, et al. Symptom duration and risk factors for delayed return to usual health among outpatients with COVID-19 in a multistate health care systems network - united states, march-June 2020. MMWR Morb Mortal Wkly Rep (2020) 69(30):993–8. doi: 10.15585/mmwr.mm6930e1
11. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet (2021) 397(10270):220–32. doi: 10.1016/S0140-6736(20)32656-8
12. Arnold DT, Hamilton FW, Milne A, Morley AJ, Viner J, Attwood M, et al. Patient outcomes after hospitalisation with COVID-19 and implications for follow-up: Results from a prospective UK cohort. Thorax (2021) 76(4):399–401. doi: 10.1136/thoraxjnl-2020-216086
13. Asadi-Pooya AA, Akbari A, Emami A, Lotfi M, Rostamihosseinkhani M, Nemati H, et al. Risk factors associated with long COVID syndrome: A retrospective study. Iranian J Med Sci (2021) 46(6):428.
14. Di Gennaro F, Belati A, Tulone O, Diella L, Bavaro DF, Bonica R, et al. Long covid: A systematic review and meta-analysis of 120,970 patients. Intern Emerg Med (2022). doi: 10.1007/s11739-022-03164-w
15. Spudich S, Nath A. Nervous system consequences of COVID-19. Science (2022) 375(6578):267–9. doi: 10.1126/science.abm2052
16. Wang TZ, Sell J, Weiss D, Lall R. COVID-19 presenting as anosmia and dysgeusia in new York city emergency departments. medRxiv (2020) 2020.07.06.20147751. doi: 10.1101/2020.07.06.20147751
17. Zazhytska M, Kodra A, Hoagland DA, Fullard JF, Shayya H, Omer A, et al. Disruption of nuclear architecture as a cause of COVID-19 induced anosmia. bioRxiv (2021) 2021.02.09.430314. doi: 10.1101/2021.02.09.430314
18. Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry (2020) 7(10):875–82. doi: 10.1016/S2215-0366(20)30287-X
19. Pezzini A, Padovani A. Lifting the mask on neurological manifestations of COVID-19. Nat Rev Neurol (2020) 16(11):636–44. doi: 10.1038/s41582-020-0398-3
20. Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry (2021) 8(5):416–27. doi: 10.1016/S2215-0366(21)00084-5
21. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med (2021) 27(4):601–15. doi: 10.1038/s41591-021-01283-z
22. Ahmed H, Patel K, Greenwood DC, Halpin S, Lewthwaite P, Salawu A, et al. Long-term clinical outcomes in survivors of severe acute respiratory syndrome and middle East respiratory syndrome coronavirus outbreaks after hospitalisation or ICU admission: A systematic review and meta-analysis. J Rehabil Med (2020) 52(5):1–11. doi: 10.2340/16501977-2694
23. Moldofsky H, Patcai J. Chronic widespread musculoskeletal pain, fatigue, depression and disordered sleep in chronic post-SARS syndrome; a case-controlled study. BMC neurol (2011) 11(1):1–7. doi: 10.1186/1471-2377-11-37
24. Jarius S, Pache F, Körtvelyessy P, Jelčić I, Stettner M, Franciotta D, et al. Cerebrospinal fluid findings in COVID-19: a multicenter study of 150 lumbar punctures in 127 patients. J neuroinflammation (2022) 19(1):1–33. doi: 10.1186/s12974-021-02339-0
25. Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dubé M, et al. Human coronaviruses and other respiratory viruses: Underestimated opportunistic pathogens of the central nervous system? viruses (2019) 12(1):14. doi: 10.3390/v12010014
26. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J virol (2008) 82(15):7264–75. doi: 10.1128/JVI.00737-08
27. Li K, Wohlford-Lenane C, Perlman S, Zhao J, Jewell AK, Reznikov LR, et al. Middle East respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect diseases (2016) 213(5):712–22. doi: 10.1093/infdis/jiv499
28. De Santis G. SARS-CoV-2: a new virus but a familiar inflammation brain pattern. Brain Behav Immun (2020) 87:95. doi: 10.1016/j.bbi.2020.04.066
29. Desforges M, Le Coupanec A, Stodola JK, Meessen-Pinard M, Talbot PJ. Human coronaviruses: Viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res (2014) 194:145–58. doi: 10.1016/j.virusres.2014.09.011
30. Philippens IHCHM, Böszörményi KP, Wubben JA, Fagrouch ZC, van Driel N, Mayenburg AQ, et al. SARS-CoV-2 causes brain inflammation and induces lewy body formation in macaques. bioRxiv (2021) 2021:02.23.432474. doi: 10.1101/2021.02.23.432474
31. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci (2021) 24(2):168–75. doi: 10.1038/s41593-020-00758-5
32. Chen S, Han Y, Yang L, Kim T, Nair M, Harschnitz O, et al. SARS-CoV-2 infection causes dopaminergic neuron senescence. Res Sq (2021) p. rs–51346. doi: 10.21203/rs.3.rs-513461/v1
33. Chertow D, Stein S, Ramelli S, Grazioli A, Chung J-Y, Singh M, et al. SARS-CoV-2 infection and persistence throughout the human body and brain. Res Sq (2021). doi: 10.21203/rs.3.rs-1139035/v1
34. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. New Engl J Med (2020) 383(6):590–2. doi: 10.1056/NEJMc2011400
35. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv (2020) 6(31):eabc5801. doi: 10.1126/sciadv.abc5801
36. Cooper KW, Brann DH, Farruggia MC, Bhutani S, Pellegrino R, Tsukahara T, et al. COVID-19 and the chemical senses: Supporting players take center stage. Neuron (2020) 107(2):219–33. doi: 10.1016/j.neuron.2020.06.032
37. Douaud G, Lee S, Alfaro-Almagro F, Arthofer C, Wang C, McCarthy P, et al. SARS-CoV-2 is associated with changes in brain structure in UK biobank. Nature (2022) 604(7907):697–707. doi: 10.1038/s41586-022-04569-5
38. Benz F, Liebner S. Structure and function of the blood–brain barrier (BBB). Handb Exp Pharmacol (2022) 273:3–31. doi: 10.1007/164_2020_404
39. Harden LM, Kent S, Pittman QJ, Roth J. Fever and sickness behavior: Friend or foe? Brain behavior Immun (2015) 50:322–33. doi: 10.1016/j.bbi.2015.07.012
40. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun (2017) 60:1–12. doi: 10.1016/j.bbi.2016.03.010
41. Galea I. The blood–brain barrier in systemic infection and inflammation. Cell Mol Immunol (2021) 18(11):2489–501. doi: 10.1038/s41423-021-00757-x
42. Lee MH, Perl DP, Steiner J, Pasternack N, Li W, Maric D, et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain (2022) 145(7):2555–68. doi: 10.1093/brain/awac151
43. Haidar MA, Shakkour Z, Reslan MA, Al-Haj N, Chamoun P, Habashy K, et al. SARS-CoV-2 involvement in central nervous system tissue damage. Neural regeneration Res (2022) 17(6):1228. doi: 10.4103/1673-5374.327323
44. Farhadian S, Glick LR, Vogels CBF, Thomas J, Chiarella J, Casanovas-Massana A, et al. Acute encephalopathy with elevated CSF inflammatory markers as the initial presentation of COVID-19. BMC Neurol (2020) 20(1):248. doi: 10.1186/s12883-020-01812-2
45. Bodro M, Compta Y, Llansó L, Esteller D, Doncel-Moriano A, Mesa A, et al. Increased CSF levels of IL-1β, IL-6, and ACE in SARS-CoV-2–associated encephalitis. Neurol - Neuroimmunol Neuroinflammation (2020) 7(5):e821. doi: 10.1212/NXI.0000000000000821
46. Perrin P, Collongues N, Baloglu S, Bedo D, Bassand X, Lavaux T, et al. Cytokine release syndrome-associated encephalopathy in patients with COVID-19. Eur J Neurol (2021) 28(1):248–58. doi: 10.1111/ene.14491
47. Pilotto A, Masciocchi S, Volonghi I, De Giuli V, Caprioli F, Mariotto S, et al. SARS-CoV-2 encephalitis is a cytokine release syndrome: Evidences from cerebrospinal fluid analyses. Clin Infect Dis (2021) 73(9):e3019–26. doi: 10.1093/cid/ciaa1933
48. Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee M-H, Wood J, et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell (2022) 185(14):2452–68.e16. doi: 10.1016/j.cell.2022.06.008
49. Acosta-Ampudia Y, Monsalve DM, Rojas M, Rodríguez Y, Zapata E, Ramírez-Santana C, et al. Persistent autoimmune activation and proinflammatory state in post-coronavirus disease 2019 syndrome. J Infect diseases (2022) 225(12):2155–62. doi: 10.1093/infdis/jiac017
50. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet (2020) 395(10234):1417–8. doi: 10.1093/infdis/jiac017
51. Aw E, Zhang Y, Carroll M. Microglial responses to peripheral type 1 interferon. J Neuroinflammation (2020) 17(1):340. doi: 10.1186/s12974-020-02003-z
52. Chen J, Liu X, Zhong Y. Interleukin-17A: The key cytokine in neurodegenerative diseases. Front Aging Neurosci (2020) 12:566922. doi: 10.3389/fnagi.2020.566922
53. Di Filippo M, Mancini A, Bellingacci L, Gaetani L, Mazzocchetti P, Zelante T, et al. Interleukin-17 affects synaptic plasticity and cognition in an experimental model of multiple sclerosis. Cell Rep (2021) 37(10):110094. doi: 10.1016/j.celrep.2021.110094
54. Drexhage RC, van der Heul-Nieuwenhuijsen L, Padmos RC, van Beveren N, Cohen D, Versnel MA, et al. Inflammatory gene expression in monocytes of patients with schizophrenia: Overlap and difference with bipolar disorder. a study in naturalistically treated patients. Int J Neuropsychopharmacol (2010) 13(10):1369–81. doi: 10.1017/S1461145710000799
55. Espíndola OM, Gomes YCP, Brandão CO, Torres RC, Siqueira M, Soares CN, et al. Inflammatory cytokine patterns associated with neurological diseases in coronavirus disease 2019. Ann Neurol (2021) 89(5):1041–5. doi: 10.1002/ana.26041
56. Konsman JP. Cytokines in the brain and neuroinflammation: We didn’t starve the fire! Pharmaceuticals (2022) 15(2):140. doi: 10.3390/ph15020140
57. Stuart MJ, Singhal G, Baune BT. Systematic review of the neurobiological relevance of chemokines to psychiatric disorders. Front Cell Neurosci (2015) 9:357. doi: 10.3389/fncel.2015.00357
58. Szabo A, O’Connell KS, Ueland T, Sheikh MA, Agartz I, Andreou D, et al. Increased circulating IL-18 levels in severe mental disorders indicate systemic inflammasome activation. Brain Behav Immun (2022) 99:299–306. doi: 10.1016/j.bbi.2021.10.017
59. Williams JA, Burgess S, Suckling J, Lalousis PA, Batool F, Griffiths SL, et al. Inflammation and brain structure in schizophrenia and other neuropsychiatric disorders: A mendelian randomization study. JAMA Psychiatry (2022) 79(5):498–507. doi: 10.1001/jamapsychiatry.2022.0407
60. Alboni S, Cervia D, Sugama S, Conti B. Interleukin 18 in the CNS. J neuroinflammation (2010) 7(1):1–12. doi: 10.1186/1742-2094-7-9
61. Azizi G, Khannazer N, Mirshafiey A. The potential role of chemokines in alzheimer’s disease pathogenesis. Am J Alzheimer's Dis Other Dementias® (2014) 29(5):415–25. doi: 10.1177/1533317513518651
62. Marciniak E, Faivre E, Dutar P, Alves Pires C, Demeyer D, Caillierez R, et al. The chemokine MIP-1α/CCL3 impairs mouse hippocampal synaptic transmission, plasticity and memory. Sci Rep (2015) 5(1):15862. doi: 10.1038/srep15862
63. Sumita Y, Murakawa Y, Sugiura T, Wada Y, Nagai A, Yamaguchi S. Elevated BAFF levels in the cerebrospinal fluid of patients with neuro-behçet’s disease: BAFF is correlated with progressive dementia and psychosis. Scandinavian J Immunol (2012) 75(6):633–40. doi: 10.1111/j.1365-3083.2012.02694.x
64. Nesterova IV, Kovaleva SV, Malinovskaya VV, Chudilova GA, Rusinova TV. Congenital and acquired interferonopathies: Differentiated approaches to interferon therapy. Innate Immun Health Dis (2020). doi: 10.5772/intechopen.91723.
65. Carvalho LA, Bergink V, Sumaski L, Wijkhuijs J, Hoogendijk WJ, Birkenhager TK, et al. Inflammatory activation is associated with a reduced glucocorticoid receptor alpha/beta expression ratio in monocytes of inpatients with melancholic major depressive disorder. Trans Psychiatry (2014) 4(1):e344–e. doi: 10.1038/tp.2013.118
66. Ghoryani M, Faridhosseini F, Talaei A, Faridhosseini R, Tavakkol-Afshari J, Moghaddam MD, et al. Gene expression pattern of CCL2, CCL3, and CXCL8 in patients with bipolar disorder. J Res Med Sciences: Off J Isfahan Univ Med Sci (2019) 24:45. doi: 10.4103/jrms.JRMS_763_18
67. Hoseth EZ, Ueland T, Dieset I, Birnbaum R, Shin JH, Kleinman JE, et al. A study of TNF pathway activation in schizophrenia and bipolar disorder in plasma and brain tissue. Schizophr Bulletin (2017) 43(4):881–90. doi: 10.1093/schbul/sbw183
68. Ivanovska M, Abdi Z, Murdjeva M, Macedo D, Maes A, Maes M. CCL-11 or eotaxin-1: an immune marker for ageing and accelerated ageing in neuro-psychiatric disorders. Pharmaceuticals (2020) 13(9):230. doi: 10.3390/ph13090230
69. Kuzior H, Fiebich BL, Yousif NM, Saliba SW, Ziegler C, Nickel K, et al. Increased IL-8 concentrations in the cerebrospinal fluid of patients with unipolar depression. Compr Psychiatry (2020) 102:152196. doi: 10.1016/j.comppsych.2020.152196
70. Lanz TA, Reinhart V, Sheehan MJ, Rizzo SJS, Bove SE, James LC, et al. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: A comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Trans Psychiatry (2019) 9(1):151. doi: 10.1038/s41398-019-0492-8
71. Wang Q, Roy B, Turecki G, Shelton RC, Dwivedi Y. Role of complex epigenetic switching in tumor necrosis factor-α upregulation in the prefrontal cortex of suicide subjects. Am J Psychiatry (2018) 175(3):262–74. doi: 10.1176/appi.ajp.2017.16070759
72. Engh JA, Ueland T, Agartz I, Andreou D, Aukrust P, Boye B, et al. Plasma levels of the cytokines b cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) in schizophrenia, bipolar, and major depressive disorder: A cross sectional, multisite study. Schizophr Bulletin (2022) 48(1):37–46. doi: 10.1093/schbul/sbab106
73. Bedrossian N, Haidar M, Fares J, Kobeissy FH, Fares Y. Inflammation and elevation of interleukin-12p40 in patients with schizophrenia. Front Mol Neurosci (2016) 9:16. doi: 10.3389/fnmol.2016.00016
74. Bido S, Muggeo S, Massimino L, Marzi MJ, Giannelli SG, Melacini E, et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat Commun (2021) 12(1):6237. doi: 10.1038/s41467-021-26519-x
75. Dhaiban S, Al-Ani M, Elemam NM, Maghazachi AA. Targeting chemokines and chemokine receptors in multiple sclerosis and experimental autoimmune encephalomyelitis. J Inflammation Res (2020) 13:619. doi: 10.2147/JIR.S270872
76. Doron H, Amer M, Ershaid N, Blazquez R, Shani O, Lahav TG, et al. Inflammatory activation of astrocytes facilitates melanoma brain tropism via the CXCL10-CXCR3 signaling axis. Cell Rep (2019) 28(7):1785–98.e6. doi: 10.1016/j.celrep.2019.07.033
77. Estevao C, Bowers CE, Luo D, Sarker M, Hoeh AE, Frudd K, et al. CCL4 induces inflammatory signalling and barrier disruption in the neurovascular endothelium. Brain Behavior Immun - Health (2021) 18:100370. doi: 10.1016/j.bbih.2021.100370
78. Hajiannasab R. Investigating the extent of CCL4 and CCL5 chemokine as well as IL17 and IL23 cytokine gene expression in the patients afflicted with multiple sclerosis. Int J Clin Med (2016) 7(9):598–607. doi: 10.4236/ijcm.2016.79066
79. Perrella O, Guerriero M, Izzo E, Soscia M, Carrieri PB. Interleukin-6 and granulocyte macrophage-CSF in the cerebrospinal fluid from HIV infected subjects with involvement of the central nervous system. Arquivos neuro-psiquiatria (1992) 50:180–2. doi: 10.1590/S0004-282X1992000200008
80. Bakr NM, Hashim NA, Awad A, Sarhan A-A. Association between interleukin-18 promoter polymorphisms and risk of ischemic stroke: A case-control study. Egyptian J Med Hum Genet (2018) 19(1):13–8. doi: 10.1016/j.ejmhg.2017.08.014
81. Guyon A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci (2014) 8:65. doi: 10.3389/fncel.2014.00065
82. Muhammad M. Tumor necrosis factor alpha: A major cytokine of brain neuroinflammation. Cytokines (2019) in Behzadi P (eds.) (London: Cytokines, IntechOpen) 861231. doi: 10.5772/intechopen.85476
83. Yang L, Wang M, Guo YY, Sun T, Li YJ, Yang Q, et al. Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain Behav Immun (2016) 56:352–62. doi: 10.1016/j.bbi.2016.03.001
84. Dhamoon MS, Thaler A, Gururangan K, Kohli A, Sisniega D, Wheelwright D, et al. Acute cerebrovascular events with COVID-19 infection. Stroke (2021) 52(1):48–56. doi: 10.1161/STROKEAHA.120.031668
85. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: The devil is in the details. J Neurochem (2016) 139(S2):136–53. doi: 10.1111/jnc.13607
86. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun (2020) 87:18–22. doi: 10.1016/j.bbi.2020.03.031
87. Spence JD, de Freitas GR, Pettigrew LC, Ay H, Liebeskind DS, Kase CS, et al. Mechanisms of stroke in COVID-19. Cerebrovasc Dis (2020) 49(4):451–8. doi: 10.1159/000509581
88. Qi X, Keith KA, Huang JH. COVID-19 and stroke: A review. Brain Hemorrhages (2021) 2(2):76–83. doi: 10.1016/j.hest.2020.11.001
89. Sastry S, Cuomo F, Muthusamy J. COVID-19 and thrombosis: The role of hemodynamics. Thromb Res (2022) 212:51–7. doi: 10.1016/j.thromres.2022.02.016
90. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol (2004) 173(6):3916–24. doi: 10.4049/jimmunol.173.6.3916
91. von Zahn J, Moller T, Kettenmann H, Nolte C. Microglial phagocytosis is modulated by pro- and anti-inflammatory cytokines. Neuroreport (1997) 8(18):3851–6. doi: 10.1097/00001756-199712220-00003
92. Badanjak K, Fixemer S, Smajić S, Skupin A, Grünewald A. The contribution of microglia to neuroinflammation in parkinson’s disease. Int J Mol Sci (2021) 22(9):4676. doi: 10.3390/ijms22094676
93. Muzio L, Viotti A, Martino G. Microglia in neuroinflammation and neurodegeneration: From understanding to therapy. Front Neurosci (2021) 15. doi: 10.3389/fnins.2021.742065
94. Kulkarni A, Chen J, Maday S. Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr Opin Neurobiol (2018) 51:29–36. doi: 10.1016/j.conb.2018.02.008
95. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol (2021) 17(3):157–72. doi: 10.1038/s41582-020-00435-y
96. Que Y, Hu C, Wan K, Hu P, Wang R, Luo J, et al. Cytokine release syndrome in COVID-19: A major mechanism of morbidity and mortality. Int Rev Immunol (2022) 41(2):217–30. doi: 10.1080/08830185.2021.1884248
97. Jiang L, Tang K, Levin M, Irfan O, Morris SK, Wilson K, et al. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect Diseases (2020) 20(11):e276–e88. doi: 10.1016/S1473-3099(20)30651-4
98. Blatz AM, Randolph AG. Severe COVID-19 and multisystem inflammatory syndrome in children in children and adolescents. Crit Care Clinics (2022) 38(3):571–86. doi: 10.1016/j.ccc.2022.01.005
99. Masi P, Hékimian G, Lejeune M, Chommeloux J, Desnos C, Pineton De Chambrun M, et al. Systemic inflammatory response syndrome is a major contributor to COVID-19-Associated coagulopathy: Insights from a prospective, single-center cohort study. Circulation (2020) 142(6):611–4. doi: 10.1161/CIRCULATIONAHA.120.048925
100. Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, et al. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res (2021) 128(9):1323–6. doi: 10.1161/CIRCRESAHA.121.318902
101. Furuhashi M, Moniwa N, Takizawa H, Ura N, Shimamoto K. Potential differential effects of renin-angiotensin system inhibitors on SARS-CoV-2 infection and lung injury in COVID-19. Hypertension Res (2020) 43(8):837–40. doi: 10.1038/s41440-020-0478-1
102. del Valle-Mendoza J, Tarazona-Castro Y, Merino-Luna A, Carrillo-Ng H, Kym S, Aguilar-Luis MA, et al. Comparison of cytokines levels among COVID-19 patients living at sea level and high altitude. BMC Infect Diseases (2022) 22(1):96. doi: 10.1016/j.ijid.2021.12.138
103. Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes S-S, et al. From online data collection to identification of disease mechanisms: The IL-1ß, IL-6 and TNF-α cytokine triad is associated with post-acute sequelae of COVID-19 in a digital research cohort. Cell Rep Med (2021) 3(6):100663. doi: 10.1016/j.xcrm.2022.100663
104. Cosarderelioglu C, Nidadavolu LS, George CJ, Oh ES, Bennett DA, Walston JD, et al. Brain renin-angiotensin system at the intersect of physical and cognitive frailty. Front Neurosci (2020) 14:586314–. doi: 10.3389/fnins.2020.586314
105. Wright JW, Mizutani S, Harding JW. Focus on brain angiotensin III and aminopeptidase a in the control of hypertension. Int J Hypertension (2012) 2012:124758. doi: 10.1155/2012/124758
106. Palmer BF, Clegg DJ. Extrarenal effects of aldosterone on potassium homeostasis. Kidney360 (2022) 3(3):561. doi: 10.34067/KID.0006762021
107. Pavo N, Prausmüller S, Spinka G, Goliasch G, Bartko PE, Wurm R, et al. Myocardial angiotensin metabolism in end-stage heart failure. J Am Coll Cardiol (2021) 77(14):1731–43. doi: 10.1016/j.jacc.2021.01.052
108. Cure E, Ilcol TB, Cumhur Cure M. III, and IV may be important in the progression of COVID-19. J Renin Angiotensin Aldosterone Syst (2020) 21(4):1470320320972019–. doi: 10.1177/1470320320972019
109. Singh KD, Jara ZP, Harford T, Saha PP, Pardhi TR, Desnoyer R, et al. Novel allosteric ligands of the angiotensin receptor AT1R as autoantibody blockers. Proc Natl Acad Sci (2021) 118(33):e2019126118. doi: 10.1073/pnas.2019126118
110. Mehrabadi ME, Hemmati R, Tashakor A, Homaei A, Yousefzadeh M, Hemati K, et al. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. BioMed Pharmacother (2021) 137:111363–. doi: 10.1016/j.biopha.2021.111363
111. Kramár EA, Krishnan R, Harding JW, Wright JW. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul Peptides (1998) 74(2):185–92. doi: 10.1016/S0167-0115(98)00039-1
112. Yang R, Walther T, Gembardt F, Smolders I, Vanderheyden P, Albiston AL, et al. Renal vasoconstrictor and pressor responses to angiotensin IV in mice are AT1a-receptor mediated. J hypertension (2010) 28(3):487–94. doi: 10.1097/HJH.0b013e3283343250
113. Qiu H, Wu Y, Wang Q, Liu C, Xue L, Wang H, et al. Effect of berberine on PPAR(α)-NO signalling pathway in vascular smooth muscle cell proliferation induced by angiotensin IV. Pharm Biol (2017) 55(1):227–32. doi: 10.1080/13880209.2016.1257642
114. Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol (2021) 11. doi: 10.3389/fneur.2020.573095
115. Manik M, Singh RK. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J Med Virol (2022) 94(3):869–77. doi: 10.1002/jmv.27405
116. Bagheri-Hosseinabadi Z, Rezazadeh Zarandi E, Mirabzadeh M, Amiri A, Abbasifard M. mRNA expression of toll-like receptors 3, 7, 8, and 9 in the nasopharyngeal epithelial cells of coronavirus disease 2019 patients. BMC Infect Diseases (2022) 22(1):448. doi: 10.1186/s12879-022-07437-9
117. Hwang M, Bergmann Cornelia C, Williams Bryan RG. Neuronal ablation of Alpha/Beta interferon (IFN-α/β) signaling exacerbates central nervous system viral dissemination and impairs IFN-γ responsiveness in Microglia/Macrophages. J Virol (2020) 94(20):e00422–20. doi: 10.1128/JVI.00422-20
118. Li Y, Wilson HL, Kiss-Toth E. Regulating STING in health and disease. J Inflammation (2017) 14(1):11. doi: 10.1186/s12950-017-0159-2
119. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? cytokine and anti-cytokine interventions. Autoimmun Rev (2020) 19(7):102567. doi: 10.1016/j.autrev.2020.102567
120. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020), 370(6515):eabd4570. doi: 10.1126/science.abd4570
121. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med (2019) 216(9):2057–70. doi: 10.1084/jem.20182295
122. Schmidt A, Peters S, Knaus A, Sabir H, Hamsen F, Maj C, et al. TBK1 and TNFRSF13B mutations and an autoinflammatory disease in a child with lethal COVID-19. NPJ Genomic Med (2021) 6(1):55. doi: 10.1038/s41525-021-00220-w
123. Asano T, Boisson B, Onodi F, Matuozzo D, Moncada-Velez M, Maglorius Renkilaraj Majistor Raj L, et al. X-Linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci Immunol (2021) 6(62):eabl4348. doi: 10.1126/sciimmunol.abl4348
124. Fallerini C, Daga S, Mantovani S, Benetti E, Picchiotti N, Francisci D, et al. Association of toll-like receptor 7 variants with life-threatening COVID-19 disease in males: Findings from a nested case-control study. eLife (2021) 10:e67569. doi: 10.7554/eLife.67569
125. Solanich X, Vargas-Parra G, van der Made CI, Simons A, Schuurs-Hoeijmakers J, Antolí A, et al. Genetic screening for TLR7 variants in young and previously healthy men with severe COVID-19. Front Immunol (2021) 12:719115–. doi: 10.3389/fimmu.2021.719115
126. Zhang Q, Bastard P, Karbuz A, Gervais A, Tayoun AA, Aiuti A, et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature (2022) 603(7902):587–98. doi: 10.1038/s41586-022-04447-0
127. Heming M, Li X, Räuber S, Mausberg AK, Börsch A-L, Hartlehnert M, et al. Neurological manifestations of COVID-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity (2021) 54(1):164–75.e6. doi: 10.1016/j.immuni.2020.12.011
128. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122(4):1164–71. doi: 10.1172/JCI58644
129. Crunfli F, Carregari VC, Veras FP, Silva LS, Nogueira MH, Antunes ASLM, et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc Natl Acad Sci (2022) 119(35):e2200960119. doi: 10.1073/pnas.2200960119
130. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol (2017) 35(1):441–68. doi: 10.1146/annurev-immunol-051116-052358
131. Rutkai I, Mayer MG, Hellmers LM, Ning B, Huang Z, Monjure CJ, et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat Commun (2022) 13(1):1745. doi: 10.1038/s41467-022-29440-z
132. Soltani Zangbar H, Gorji A, Ghadiri T. A review on the neurological manifestations of COVID-19 infection: A mechanistic view. Mol Neurobiol (2021) 58(2):536–49. doi: 10.1007/s12035-020-02149-0
133. Rhodes R, Love G, Lameira F, Shahmirzadi M, Fox S, Vander Heide R. Reactive and acute inflammatory microvasculopathy in 36 COVID-19 autopsy brains. Res Sq (2022) 1–24. doi: 10.21203/rs.3.rs-1619440/v1
134. Hasegawa S, Matsushige T, Inoue H, Shirabe K, Fukano R, Ichiyama T. Serum and cerebrospinal fluid cytokine profile of patients with 2009 pandemic H1N1 influenza virus-associated encephalopathy. Cytokine (2011) 54(2):167–72. doi: 10.1016/j.cyto.2011.01.006
135. Paiva TM, Theotonio G, Paulino RS, Benega MA, Silva DBB, Borborema SET, et al. (H3N2) strain isolated from cerebrospinal fluid from a patient presenting myelopathy post infectious. J Clin Virol (2013) 58(1):283–5. doi: 10.1016/j.jcv.2013.05.021
136. Sweetman LL, Y-t Ng, IJ B, Bodensteiner JB. Neurologic complications associated with respiratory syncytial virus. Pediatr neurol (2005) 32(5):307–10. doi: 10.1016/j.pediatrneurol.2005.01.010
137. Kawashima H, Kashiwagi Y, Ioi H, Morichi S, Oana S, Yamanaka G, et al. Production of chemokines in respiratory syncytial virus infection with central nervous system manifestations. J Infection Chemother (2012) 18(6):827–31. doi: 10.1007/s10156-012-0418-3
138. Constant O, Barthelemy J, Nagy A, Salinas S, Simonin Y. West Nile Virus neuroinfection in humans: Peripheral biomarkers of neuroinflammation and neuronal damage. Viruses (2022) 14(4):756. doi: 10.3390/v14040756
139. Kung P-L, Chou T-W, Lindman M, Chang NP, Estevez I, Buckley BD, et al. Zika virus-induced TNF-α signaling dysregulates expression of neurologic genes associated with psychiatric disorders. J Neuroinflammation (2022) 19(1):100. doi: 10.1186/s12974-022-02460-8
140. Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of alzheimer’s disease. J Neurology Neurosurg Psychiatry (2003) 74(9):1200–5. doi: 10.1136/jnnp.74.9.1200
141. Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and alzheimer’s disease: A comparative overview. Mol Neurobiol (2014) 50(2):534–44. doi: 10.1007/s12035-014-8657-1
142. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early parkinson's disease. Ann Neurol (2005) 57(2):168–75. doi: 10.1002/ana.20338
143. Rojanathammanee L, Murphy EJ, Combs CK. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J Neuroinflamm (2011) 8(1):44. doi: 10.1186/1742-2094-8-44
144. Umapathi T, Kor AC, Venketasubramanian N, Lim CCT, Pang BC, Yeo TT, et al. Large Artery ischaemic stroke in severe acute respiratory syndrome (SARS). J Neurol (2004) 251(10):1227–31. doi: 10.1007/s00415-004-0519-8
145. Emmenegger M, Kumar SS, Emmenegger V, Malinauskas T, Buettner T, Rose L, et al. Anti-prothrombin autoantibodies enriched after infection with SARS-CoV-2 and influenced by strength of antibody response against SARS-CoV-2 proteins. PloS Pathogens (2021) 17(12):e1010118. doi: 10.1371/journal.ppat.1010118
146. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-Vessel stroke as a presenting feature of covid-19 in the young. New Engl J Med (2020) 382(20):e60. doi: 10.1056/NEJMc2009787
147. Juanes-Velasco P, Landeira Viñuela A L, García-Vaquero M, Lecrevisse Q, Herrero R, Ferruelo A, et al. SARS-CoV-2 infection triggers auto-immune response in ARDS. Front Immunol (2022) 73. doi: 10.3389/fimmu.2022.732197
148. Juanes-Velasco P, Landeira-Viñuela A, Acebes-Fernandez V, Hernández Á-P, Garcia-Vaquero ML, Arias-Hidalgo C, et al. Deciphering human leukocyte antigen susceptibility maps from immunopeptidomics characterization in oncology and infections. Front Cell Infection Microbiol (2021) 11:642583. doi: 10.3389/fcimb.2021.642583
149. Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol (2008) 4(9):491–8. doi: 10.1038/ncprheum0895
150. Díez P, Pérez-Andrés M, Bøgsted M, Azkargorta M, García-Valiente R, Dégano RM, et al. Dynamic intracellular metabolic cell signaling profiles during Ag-dependent b-cell differentiation. Front Immunol (2021) 12:637832. doi: 10.3389/fimmu.2021.637832
151. Castanares-Zapatero D, Chalon P, Kohn L, Dauvrin M, Detollenaere J, Maertens de Noordhout C, et al. Pathophysiology and mechanism of long COVID: A comprehensive review. Ann Med (2022) 54(1):1473–87. doi: 10.1080/07853890.2022.2076901
152. Cervia C, Zurbuchen Y, Taeschler P, Ballouz T, Menges D, Hasler S, et al. Immunoglobulin signature predicts risk of post-acute COVID-19 syndrome. Nat Commun (2022) 13(1):446. doi: 10.1038/s41467-021-27797-1
153. Briquez PS, Rouhani SJ, Yu J, Pyzer AR, Trujillo J, Dugan HL, et al. Severe COVID-19 induces autoantibodies against angiotensin II that correlate with blood pressure dysregulation and disease severity. Sci Adv (2022) 8(40):eabn3777. doi: 10.1126/sciadv.abn3777
154. Morris SC, Gause WC, Finkelman FD. IL-4 suppression of in vivo T cell activation and antibody production. J Immunol (2000) 164(4):1734–40. doi: 10.4049/jimmunol.164.4.1734
155. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: Enhancement of antibody responses through direct stimulation of b and T cells by type I IFN. J Immunol (2006) 176(4):2074–8. doi: 10.4049/jimmunol.176.4.2074
156. Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14(4):461–70. doi: 10.1016/S1074-7613(01)00126-1
158. Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CAM, Kammeijer GSM, et al. Hinge-region O-glycosylation of human immunoglobulin G3 (IgG3)*[S]. Mol Cell proteomics (2015) 14(5):1373–84. doi: 10.1074/mcp.M114.047381
159. Bhat NM, Kshirsagar MA, Bieber MM, Teng NNH. IgG subclasses and isotypes of VH4-34 encoded antibodies. Immunol investigations (2015) 44(4):400–10. doi: 10.3109/08820139.2015.1015682
160. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun (2021) 12(1):1–15. doi: 10.1038/s41467-021-25509-3
161. Feng A, Yang E, Moore A, Dhingra S, Chang S, Yin X, et al. Autoantibodies targeting cytokines and connective tissue disease autoantigens are common in acute non-SARS-CoV-2 infections. Res Sq (2022). doi: 10.21203/rs.3.rs-1233038/v1
162. Wang JY, Roehrl MW, Roehrl VB, Roehrl MH. A master autoantigen-ome links alternative splicing, female predilection, and COVID-19 to autoimmune diseases. J Trans Autoimmunity (2022) 5:100147. doi: 10.1016/j.jtauto.2022.100147
163. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in wuhan, China: A descriptive study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
164. Asfuroglu Kalkan E, Ates I. A case of subacute thyroiditis associated with covid-19 infection. J endocrinological Invest (2020) 43(8):1173–4. doi: 10.1007/s40618-020-01316-3
165. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
166. Dotan A, David P, Arnheim D, Shoenfeld Y. The autonomic aspects of the post-COVID19 syndrome. Autoimmun Rev (2022) 21(5):103071. doi: 10.1016/j.autrev.2022.103071
167. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse functional autoantibodies in patients with COVID-19. Nature (2021) 595(7866):283–8. doi: 10.1038/s41586-021-03631-y
168. Khamsi R. Rogue antibodies could be driving severe COVID-19. Nature (2021) 590(7844):29–31. doi: 10.1038/d41586-021-00149-1
169. Saheb Sharif-Askari N, Saheb Sharif-Askari F, Ahmed SBM, Hannawi S, Hamoudi R, Hamid Q, et al. Enhanced expression of autoantigens during SARS-CoV-2 viral infection. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.686462
170. Burbelo PD, Iadarola MJ, Keller JM, Warner BM. Autoantibodies targeting intracellular and extracellular proteins in autoimmunity. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.548469
171. Lee J, Rho J-h, Roehrl MH, Wang JY. Dermatan sulfate is a potential master regulator of IgH via interactions with pre-BCR, GTF2I, and BiP ER complex in pre-b lymphoblasts. bioRxiv (2021) 2021:01.18.427153. doi: 10.1101/2021.01.18.427153
172. Wang JY, Zhang W, Roehrl VB, Roehrl MW, Roehrl MH. An autoantigen atlas from human lung HFL1 cells offers clues to neurological and diverse autoimmune manifestations of COVID-19. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.831849
Keywords: long COVID syndrome, SARS-CoV-2, inflammatory response, neurodegeneration, autoantobodies, autoantigens
Citation: Elizalde-Díaz JP, Miranda-Narváez CL, Martínez-Lazcano JC and Martínez-Martínez E (2022) The relationship between chronic immune response and neurodegenerative damage in long COVID-19. Front. Immunol. 13:1039427. doi: 10.3389/fimmu.2022.1039427
Received: 08 September 2022; Accepted: 28 November 2022;
Published: 16 December 2022.
Edited by:
Omid Mirmosayyeb, University at Buffalo, United StatesReviewed by:
Chung-Hsing Chou, Tri-Service General Hospital, TaiwanZhaoqi Yan, Gladstone Institutes, United States
Copyright © 2022 Elizalde-Díaz, Miranda-Narváez, Martínez-Lazcano and Martínez-Martínez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Martínez-Martínez, ZW1hcnRpbmV6QGlubWVnZW4uZ29iLm14