 Dongdong Zheng1†
Dongdong Zheng1† Jia Liu
Jia Liu Kexiang Liu
Kexiang Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 November 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1039241
This article is part of the Research TopicInteraction Between Metabolism and the NLRP3 InflammasomeView all 9 articles
The endothelium is a single layer of epithelium covering the surface of the vascular system, and it represents a physical barrier between the blood and vessel wall that plays an important role in maintaining intravascular homeostasis. However, endothelial dysfunction or endothelial cell death can cause vascular barrier disruption, vasoconstriction and diastolic dysfunction, vascular smooth muscle cell proliferation and migration, inflammatory responses, and thrombosis, which are closely associated with the progression of several diseases, such as atherosclerosis, hypertension, coronary atherosclerotic heart disease, ischemic stroke, acute lung injury, acute kidney injury, diabetic retinopathy, and Alzheimer’s disease. Oxidative stress caused by the overproduction of reactive oxygen species (ROS) is an important mechanism underlying endothelial cell death. Growing evidence suggests that ROS can trigger endothelial cell death in various ways, including pyroptosis, parthanatos, and ferroptosis. Therefore, this review will systematically illustrate the source of ROS in endothelial cells (ECs); reveal the molecular mechanism by which ROS trigger pyroptosis, parthanatos, and ferroptosis in ECs; and provide new ideas for the research and treatment of endothelial dysfunction-related diseases.
Endothelium is the highly active monolayer of epithelium that covers the surface of blood vessels. Endothelium plays an important role in maintaining vasomotor, coagulation and anticoagulation systems, immune regulation, vascular smooth muscle proliferation and migration (1–3). Reactive oxygen species (ROS) in endothelial cells (EC) are mainly derived from mitochondria, NADPH oxidase (NOXs), eNOS uncoupling and xanthine oxidase (XO) (4, 5). Under physiological conditions, ROS are essential for physiological cellular functions such as host defense, post-translational processing of proteins, cell signaling, regulation of gene expression, and cell differentiation (6). However, ROS overproduction may cause endothelial dysfunction (ED) and endothelial cell death. The impairment of NO synthesis marks the onset of ED, which is mainly mediated by the eNOS uncoupling mechanism (7). In the process of ROS-mediated ED, the expression of various pro-inflammatory cytokines, i.e., interleukin-1β (interleukin-1β), interleukin-18 (interluekin-18, IL-18), and cell adhesion molecules, i.e., intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin may be promoted in endothelial cells. These molecules are closely related to the occurrence of inflammatory responses (8, 9). In addition, ROS can mediate a variety of programmed cell death (PCD) in endothelial cells, such as pyroptosis, parthanatos and ferroptosisis. It is worth noting that endothelial dysfunction or endothelial cell death is closely related to the occurrence and development of various diseases, such as atherosclerosis (10), coronary heart disease (11), hypertension (12), ischemic stroke (13), acute lung injury (14), acute kidney injury (15), diabetic retinopathy (16) and Alzheimer’s disease (17) (Figure 1). This review systematically elucidates the sources of ROS in EC; covers the molecular mechanisms of ROS-induced pyroptosis, parthanatos and ferroptosis in EC cells; and provide new insights for the research and treatment of endothelial cell death-related diseases.
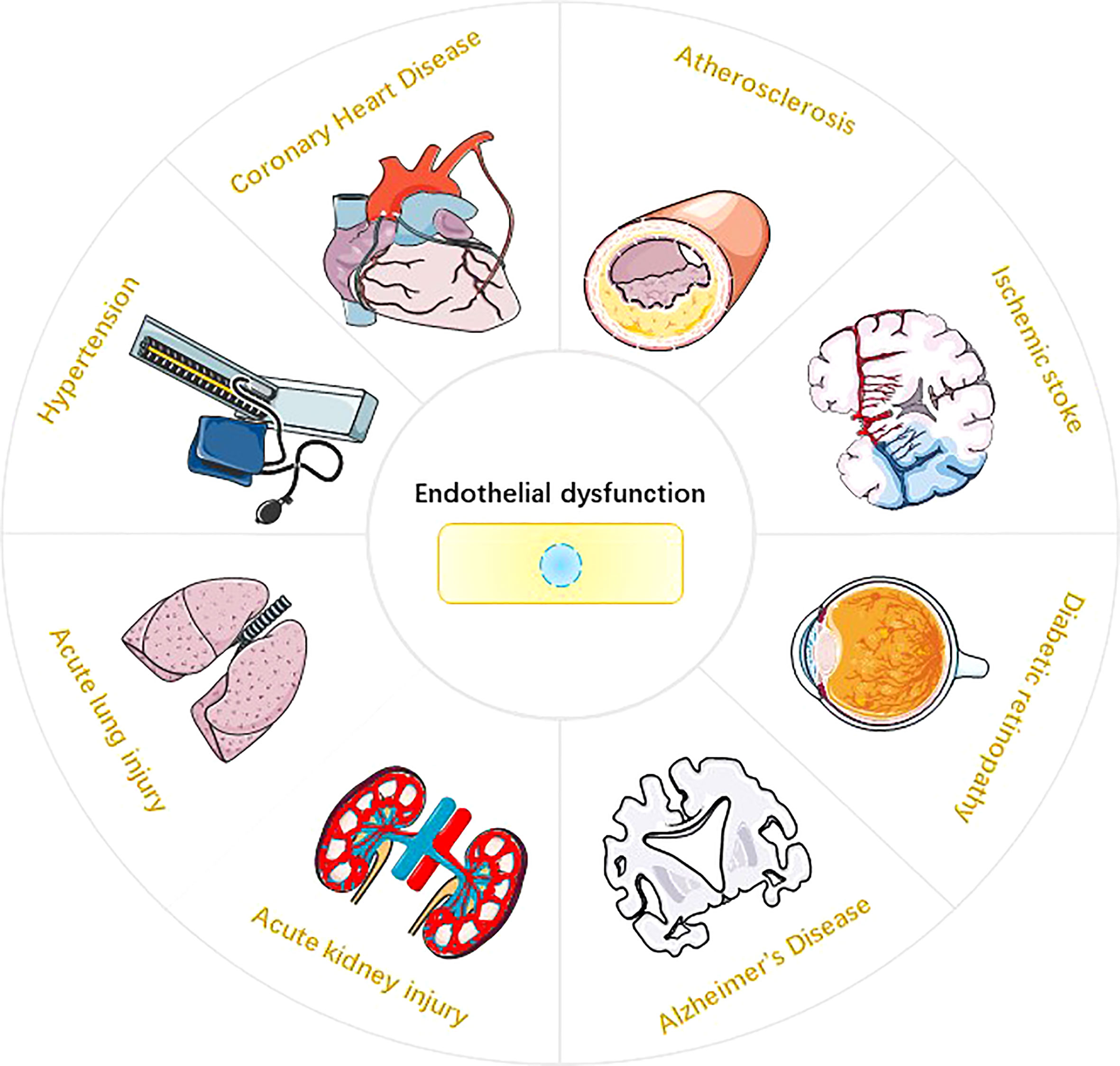
Figure 1 Endothelial dysfunction and Disease. Endothelial dysfunction is involved in the pathophysiological process of various diseases (10-17), such as atherosclerosis, hypertension, coronary atherosclerotic heart disease, ischemic stroke, acute lung injury, acute kidney injury, diabetic retinopathy, and Alzheimer’s disease.
Intracellular ROS are mainly composed of superoxide anions (), hydrogen peroxide (H2O2), and hydroxyl radicals (OH•) (18). O2 forms by capturing an electron, which leads to the generation of other ROS. is unstable in aqueous solutions due to its short half-life; therefore, intracellular is quickly scavenged or converted to other forms of ROS. is cleared or converted mainly via three pathways:1) generates H2O2 through the action of superoxide dismutase (SOD); 2) low concentrations (picomolar range) of interact with nitric oxide (NO) to generate peroxynitrite anion (ONOO•), which occurs even faster than disproportionation to generate H2O2; and 3) high concentrations of generate OH• through the Fenton reaction with H2O2 (18). In addition, OH• reacts with fatty acids to generate lipid free radicals (L•). ROS in ECs are mainly derived from mitochondria, NADPH oxidase (NOX), endothelial NOS (eNOS) uncoupling, and xanthine oxidase (XO) (4, 5) (Figure 2).
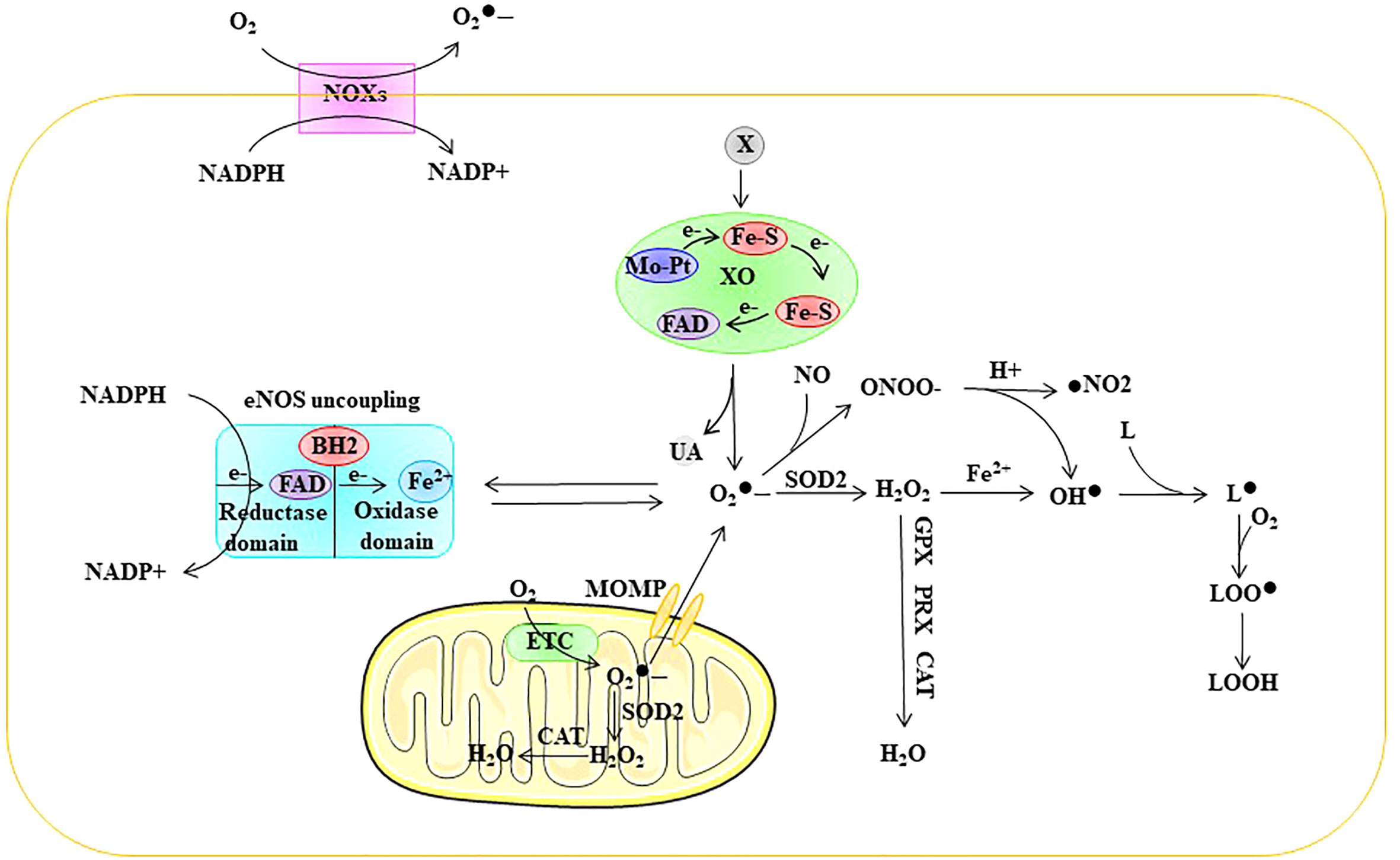
Figure 2 Sources of ROS in ECs. ROS in ECs are mainly derived from mitochondria, NOXs, eNOS uncoupling, and XO. ETC electron transport chain, eNOS endothelial nitric oxide synthase, FAD flavin adenine dinuc-leotide, Fe-S iron-sulfur center, Mo-co molybdenum cofactor, MOMP mi-tochondrial outer membrane permeabilization, NOX NADPH oxidase, SOD superoxide dismutase, GPX glutathione peroxidase, PRX peroxired-oxin, CAT catalase, superoxide, ONOO• peroxynitrite anion, H2O2 hydrogen peroxide, OH• peroxyl radical, L lipid, L• lipid free radical, LOO• lipid peroxy radical, LOOH lipid peroxide, UA uric acid, X xanthine, XO xanthine oxidase.
Mitochondria are the source of cellular power and produce ATP through oxidative phosphorylation (OXPHO), which accounts for approximately 80% of the energy requirements, with glycolysis accounting for the remaining 20%. Mitochondrial ROS production results from oxidative phosphorylation associated with aerobic respiration within the mitochondrial electron transport chain (ETC). Mitochondrial complexes I and III are the major sites for the generation of (19–21). Electron leakage from the ETC results in the reduction of O2 to rather than to H2O. SOD further disproportionates mitochondrial to form H2O2. Approximately 1-2% of O2 entering the ETC is estimated to be converted into ROS (22) (Figure 2). Moreover, mitochondrial ROS overproduction is one of the causes of EC dysfunction. For example, Rao et al. showed that nicotinamide nucleotide transhydrogenase (NNT) knockout resulted in a significant increase in mitochondrial ROS production and glutathione peroxidase activity and a decline in glutathione reductase activity (23).
NAPDH oxidase (NOX) is an important source of ROS in cells. The NOX family includes NOX1, NOX2, NOX3, NOX4, NOX5, and dual oxidases (DUOX1 and DUOX2) (22). NOXs are multi-transmembrane proteins whose C-termini are exposed in the cytoplasm, and they share common domains, including six conserved transmembrane domains, four conserved heme-binding histidines, flavin adenine dinucleotide (FAD)-binding domains, and NADPH-binding domains (24). NOX in turn transfers electrons from NADPH to FAD, the heme group, and then to O2, resulting in and/or H2O2 production (25).
The main subtypes of NOX in ECs include NOX1, NOX2, NOX4, and NOX5 (25, 26). The catalytic product of NOX1, NOX2, and NOX5 is , while the catalytic product of NOX4 is H2O2 (Figure 3). NOX complexes consist of catalytic subunits (NOX) and regulatory subunits, with the exception of NOX5, which consists of only one catalytic subunit (22). NOX2 is the first NOX isoform identified in ECs and represents the most widely and deeply studied isoform; therefore, we first discuss its activation mechanism. Under resting conditions, NOX2 and p22phox are located on the membrane as inactive complexes while p40phox, p67phox, and p47phox subunits are located in the cytoplasm (22). Activation of NOX2 also requires the small GTPase Rac1. Activation of Rac1 initiates NOX2, and Rac1 is recruited to the membrane and then recruits other cytosolic components (27). p47phox is then phosphorylated by protein kinase C (PKC) and transferred to the membrane together with p67phox and p40phox (28). Next, the phosphorylation of p47phox can combine with p22phox to realize the assembly and activation of the NOX2 complex (29). The basal activity of NOX2 in ECs is low, although it is rapidly activated by pathological causative factors, such as hyperlipidemia, hypertension, and hyperglycemia (30). EC injury in the early stages of vascular disease has been reported to be mediated by excess NOX2-derived superoxide (31). Similar to NOX2, NOX1 activation requires the assembly of multiple subunits. During NOX1 activation, the activation function of p67phox is performed by NOXA1 and the organizer function of p47phox is performed by NOXO1 (32, 33). Compared with p47phox, NOXO1 does not contain an auto-inhibitor domain; therefore, the NOX1-NOXO1-NOXA1 complex has high basal activity (29). Reports have indicated that endothelin-1 (ET-1) overexpression in ECs promotes atherosclerosis progression through NOX1 in type 1 diabetes, perivascular oxidative stress, and inflammation (34). Furthermore, NOX1 is involved in eNOS uncoupling in ECs. For example, Youn et al. found that NOX1 activation in streptozotocin-induced diabetic mice is dependent on p47phox and NOXO1 and mediates eNOS uncoupling. NOX1 knockout mice are protected from ED (35). NOX4 is the most highly expressed NOX homolog in ECs. Compared to NOX1 and NOX2, activation of NOX4 requires only p22phox and polymerase delta-interacting protein 2 (Poldip2) (30, 36). Several studies have suggested that NOX4 plays an important role in ED. For example, Jiang et al. found that NOX4 knockdown attenuated pulmonary ROS production in septic mice, attenuated redox-sensitive activation of the CaMKII/ERK1/2/MLCK pathway, and restored the expression of the tight junction proteins ZO-1 and occludin to maintain the integrity of the EC barrier (37). Zhao et al. showed that tert-butyl hydroperoxide (t-BHP) induces EC apoptosis through NOX4 (38). However, there are also reports that NOX4 protects ECs during oxidative stress. This may be related to the generation of H2O2 by NOX4. H2O2 is considered an important signaling intermediate because of its ability to selectively and reversibly oxidize reactive cysteine residues, thereby altering the function of protein targets including phosphatases, kinases, ion channels, and transcription factors (39). In EC, these effects ultimately lead to increased expression and activity of important angioprotective enzymes, including eNOS (40). Furthermore, unlike superoxide, H2O2 does not react appreciably with NO, and thus does not reduce NO bioavailability (39). Unlike NOX1, NOX2, and NOX4, the activation of NOX5 does not depend on other subunits. NOX5 contains an N-terminal calmodulin-like domain with four Ca2+ binding sites (EF hands) (39). Therefore, NOX5 activity can be directly regulated by changes in the intracellular [Ca2+]. Evidence suggests that NOX5 plays an important role in ED. Silva et al. found that lysophosphatidylcholine drives NOX5-dependent ROS production in ECs via calcium influx, leading to ED (40). Elbatreek et al. found that NOX5 overexpression in mice caused eNOS uncoupling, thus leading to ED (41). Therefore, ROS derived from NOXs play an important role in mediating ED.

Figure 3 The structure of NOXs. The main subtypes of NOX in ECs include NOX1, NOX2, NOX4, and NOX5. The catalytic product of NOX1, NOX2, and NOX5 is , while the catalytic product of NOX4 is H2O2.
Nitric oxide (NO) plays an important role in maintaining vascular homeostasis owing to its vasodilatory effects. Nitric oxide synthase (NOS) is synthesized from l-arginine and O2 and represents a key enzyme involved in nitric oxide (NO) synthesis. There are three subtypes of NOS: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) (42). NOS functions as a homodimer during NO biosynthesis. Each monomer has an oxygenase domain at the N-terminus and a reductase domain at the C-terminus. The oxygenase domain consists of binding sites for FAD, FMN, and NADPH and is linked to the reductase domain through a calmodulin recognition site. The reductase domain contains binding sites for heme, tetrahydrobiopterin (BH4), and l-arginine. The formation of NO requires electron flow, which starts at the flavin level in the reductase domain and ends at the heme level in the oxygenase domain (7). Specifically, NADPH releases electrons in the reductase domain and transfers them to heme via FAD and FMN. In the presence of l-arginine and cofactor BH4, electrons can reduce O2 to form NO and l-citrulline (43, 44). The presence of BH4 is critical for NO formation because it is involved in l-arginine binding and electron transfer. During ED, BH4 depletion is considered the main mechanism by which eNOS uncoupling generates ROS (45, 46). In fact, in the absence of BH4, l-arginine cannot bind to its site and the terminal electron acceptor becomes O2, thus forming instead of NO, a process defined as eNOS uncoupling (47, 48). Notably, ROS derived from the NOX system are closely related to the depletion of BH4 (30). Furthermore, reacts with NO to form ONOO•, which can lead to the oxidation of iron-sulfur centers and eNOS core ZnS4 (4, 49). Taken together, these results suggest that eNOS decoupling is closely associated with ED (Figure 2).
Xanthine oxidoreductase (XOR) exists in two different forms, xanthine dehydrogenase (XDH) and XO, and they represent the rate-limiting enzymes in purine metabolism (50). Normally, XOR exists in the cells in the form of XDH. XDH is a homodimer of approximately 300 kDa, with four redox centers in each subunit: a molybdenum cofactor (Mo-co), two iron-sulfur (Fe-S) centers, and a flavin adenine dinucleotide (FAD) domain (51). XDH catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid at the Mo-co site, and electrons shuttle through two Fe-S centers to the FAD binding site, where NAD+ is reduced to NADH (51). Under physiological conditions, XOR is mainly present in ECs in the form of XDH (52). XDH can break down hypoxanthine into uric acid (53) and reduce nitrite to produce NO, which helps regulate vasodilation and blood pressure (53). However, under oxidative stress conditions, ROS can oxidize cystine thiols on XDH, resulting in the conversion of XDH to XO (30, 54). The main difference between XO and XDH is their oxidative substrate affinity, where XO has a reduced affinity for NAD+ and more than 11-fold increased affinity for O2 (54). While promoting the decomposition of hypoxanthine into uric acid, XO generates through one-electron reduction, and H2O2 through two-electron reduction (55). Reports have indicated that XO-induced ED is closely related to its by-products, including ROS and uric acid (56, 57). Intracellular uric acid can exacerbate oxidative stress in ECs, thereby causing ED (52). Therefore, XO is an important source of ROS in ECs and closely related to ED (Figure 2).
Pyroptosis is a type of programmed cell death caused by various stimuli. The molecular features of pyroptosis include inflammasome assembly and activation, membrane pore formation, and pro-inflammatory cytokine maturation and release. Depending on whether pyroptosis requires caspase-1 activation, it can be divided into the classical and non-classical inflammasome pathways.
The inflammasome is composed of the intracellular recognition receptor, adaptor protein apoptosis-associated speck-like protein (ASC), and effector protein caspase-1 (57). The intracellular recognition receptors that constitute the inflammasome include the NOD-like receptor (NLR) protein family of AIM2-like receptors (ALRs) and pyrin, which can directly or indirectly activate ASC to activate caspase-1 (58, 59). Structurally, these intracellular recognition receptors contain a CARD or PYD domain at their N-terminus. ASC contains a PYD structure and a CARD domain, and caspase-1 contains a CARD domain (60). The intracellular recognition receptors NLRP1, NLRP3, NLRP6, AIM2, and pyrin all contain PYD at their N-termini, whereas NLRP1 and NLRC4 contain CARD (57, 61) (Figure 4).
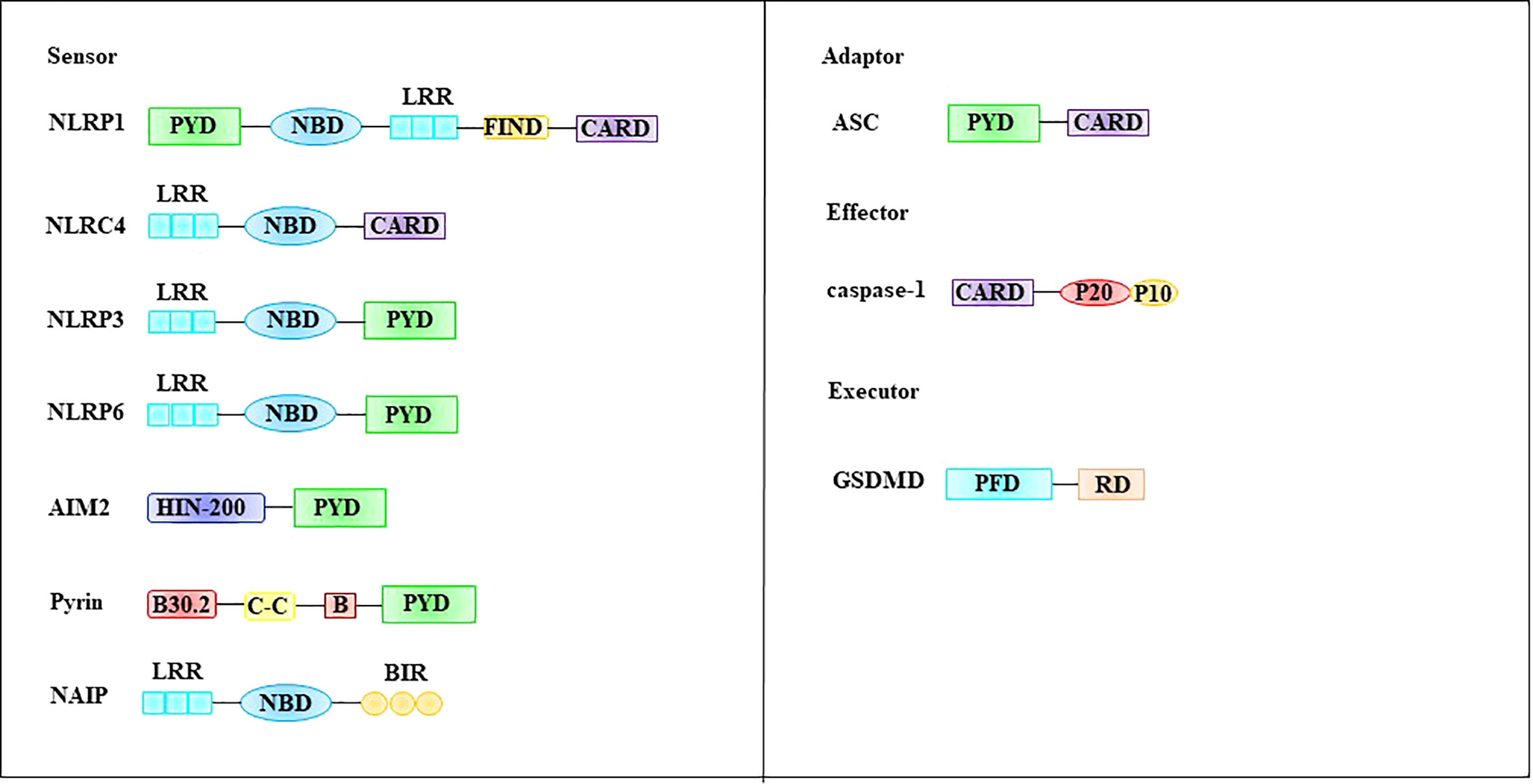
Figure 4 Molecular structures involved in pyroptosis. The inflammasome is composed of the intracellular recognition receptor, adaptor protein ASC, and effector protein caspase-1. GSDMD, a family of pore-forming effector proteins, is thought to be the executor of pyroptosis. ASC apoptosis-associated speck-like protein, CARD, caspase-recruitment domain, FIIND function-to-find domain, GSDMD Gasdermin D, LRR leucine-rich repeat domain, NBD nucleotide-binding domain, PFD pore-forming domain, PYD pyrin domain, RD repressor domain.
Gasdermin D (GSDMD), a family of pore-forming effector proteins, is thought to be the executor of pyroptosis. GSDMD is a member of the gasdermin protein family, which includes GSDMA, GSDMB, GSDMC, GSDMD, GSDME (also known as DFNA5), and PJVK (also known as DFNB59). Most members of this family have been shown to exhibit pore-punching effects (62). Among these, the most extensive and in-depth research has been performed on GSDMD. GSDMD is composed of a pore-forming domain (PFD), linker, and repressor domain (RD) (62, 63) (Figure 4). The PFD (also known as N-GSDMD) is located at the N-terminus and consists of 242 amino acids. This part is an important structure for the GSDMD to perform the punching function. RD (also known as C-GSDMD) is located at the C-terminus and consists of 199 amino acids, which is an important structure for inhibiting GSDMD function (63). The linker between PFD and RD is composed of 43 amino acids, and this part is the switch for GSDMD activation (63). During pyroptosis, the linker of GSDMD can be cleaved by activated caspase-1 or caspase-4/5/11, and C-GSDMD dissociates from GSDMD, releasing its inhibitory effect on N-GSDMD (64, 65). Subsequently, N-GSDMD was integrated into the cell membrane, and approximately 16 PFD monomers were oligomerized to form membrane pores with a diameter of 10-15 nm. The formation of membrane pores causes a loss of cell membrane integrity and breaks the osmotic pressure barrier of the plasma membrane (62, 66). Under normal circumstances, intracellular sodium ions are low and potassium ions are high. However, extracellular fluid is high in sodium ions and low in potassium ions. The formation of this intracellular and extracellular ion concentration difference is dependent on the Na+ pump (Na+-K+-ATPase). Na+-K+-ATPase is widely expressed on the cell membrane surface and acts as a sodium-potassium antiporter. Each Na+-K+-ATPase can transport three sodium ions from the intracellular to extracellular space and two potassium ions into the cell by consuming one molecule of ATP (67). This asymmetric cation transport mechanism plays an important role in maintaining differences in the chemical concentration gradients of sodium and potassium ions inside and outside the cell. Notably, this asymmetric cation transport mechanism mediates cell swelling together with N-GSDMD. Specifically, when N-GSDMD forms pores in the cell membrane, the force generated by the concentration gradient expelling potassium ions out of the cell is roughly offset by the electric field force that pulls potassium ions into the cytoplasm, resulting in the passage of potassium ions through the membrane pores. Therefore, the flux is minimized. In contrast, both the sodium ion concentration gradient and electric field force promote the entry of sodium ions into cells, resulting in a large influx of sodium ions (62). The influx of sodium ions is accompanied by the entry of water molecules, which causes cells to swell or even rupture.
Physiologically, interleukin-1β (IL-1β) and interleukin-18 (IL-18) exist in inactive precursor forms, namely pro-IL-1β and pro-IL-18 (62). However, during pyroptosis, activated caspase-1 cleaves pro-IL-1β and pro-IL-18 to produce mature IL-1β and IL-18 (53). Unlike other cytokines, mature IL-1β and IL-18 are not secreted out of cells via the endoplasmic reticulum-Golgi pathway; rather, this action depends on N-GSDMD (68, 69). Therefore, N-GSDMD is an important channel for the secretion of mature IL-1β and IL-18 into the extracellular space during pyroptosis.
Depending on whether pyroptosis requires caspase-1 activation, it can be divided into the classical and non-classical inflammasome pathways (Figure 5). The classical inflammasome pathway mainly includes the assembly and activation of inflammasomes, formation of porins, and maturation and secretion of IL-1β and IL-18. Specifically, intracellular and extracellular PAMPs or DAMPs (e.g., viral dsDNA, bacterial lipopolysaccharide, extracellular ATP, ox-LDL, and cholesterol crystals) can promote inflammasome assembly and activation. Inflammasomes activate pro-caspase-1 via self-cleavage. Activated caspase-1 cleaves the porin GSDMD to generate mature N-GSDMD (70) and cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18 (70). Compared with the classical inflammasome pathway, activation of the non-canonical inflammasome pathway does not require the assembly and activation of the inflammasome. The bacterial cell wall component lipopolysaccharide can activate caspase-11 (human) or caspase-4/5 (murine) (71, 72). Activated caspase-4/5/11 cleaves GSDMD to generate mature N-GSDMD (64, 65). Subsequently, N-GSDMD is integrated into the cell membrane to form membrane pores that mediate pyroptosis.
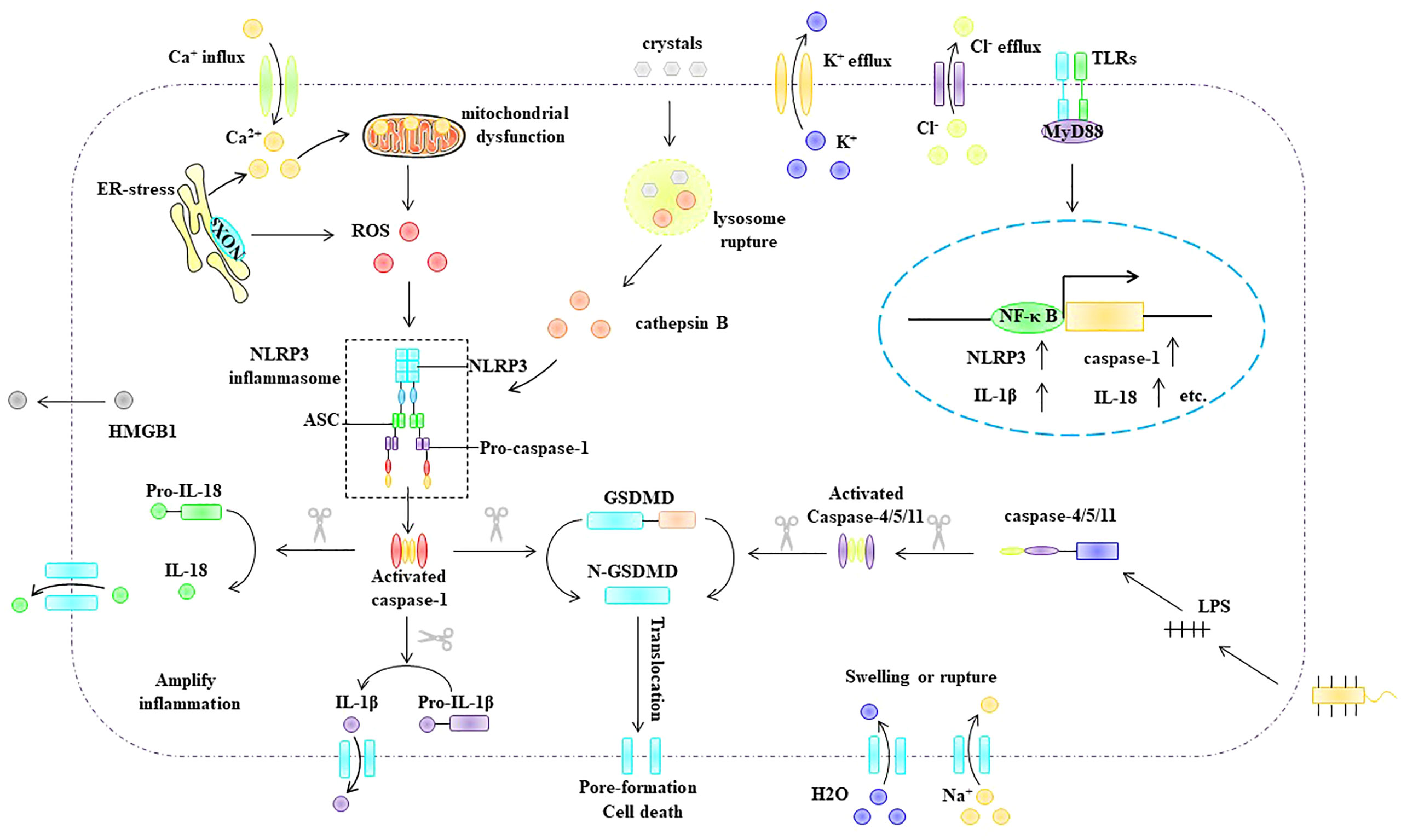
Figure 5 Pyroptosis pathway. Depending on whether pyroptosis requires caspase-1 activation, it can be divided into the classical and non-classical inflammasome pathways. ASC, apoptosis-associated speck-like protein, ER endoplasmic reticulum, GSDMD Gasdermin D, HMGB1 high mobility group box 1, IL-1β interleukin-1β, IL -18 interleukin- 18, MyD88 myeloid differentiation primary response gene 88, NLRP3 NLR-family pyrin domain-containing protein 3NF-κB nuclear factor kappa B, NLRP3 NLR-family pyrin domain-containing protein 3, ox-LDL oxidized low density lipoprotein, ROS reactive oxygen species, TLR Toll-like receptors.
Among the classical inflammasome pathways, NLRP3 inflammasome-mediated pyroptosis is the most extensively studied. The NLRP3 inflammasome is composed of the intracellular sensor protein NLRP3, adaptor protein ASC, and effector protein pro-caspase-1 (70, 73). NLRP3 inflammasome activation is thought to include multiple upstream signals, most of which are not mutually exclusive, including potassium (K+) efflux (74, 75), calcium flux (76), endoplasmic reticulum stress (77), mitochondrial dysfunction (78), ROS (79), and lysosomal disruption (80). Notably, ROS, as upstream signals of NLRP3 inflammasome activation, play an important role in NLRP3 inflammasome activation (81–83). Overall, the mechanism by which ROS activate the NLRP3 inflammasome involves two important processes: the initiation phase and the activation phase. The initiation signal indicates that ROS can upregulate the expression of NLRP3, pro-caspase-1, and pro-IL-1β (79). During the activation stage, ROS can promote the assembly and activation of the NLRP3 inflammasome, and thioredoxin-interacting protein (TXNIP) plays an important role in this process. TXNIP has been identified as a reduced thioredoxin protein (Trx) binding protein. When cells are in a quiescent state, TXNIP interacts with the redox domain of Trx and is considered a negative regulator of Trx. However, when intracellular ROS are increased, Trx is oxidized, thus leading to the dissociation of TXNIP from Trx, which subsequently interacts with NLRP3, leading to the assembly and activation of the NLRP3 inflammasome (84, 85).
Numerous recent studies have shown that NLRP3 inflammasome activation plays an important role in mediating ED (61). Increasing evidence has shown that certain stimuli, such as oxidized low-density lipoprotein, hyperglycemia, and nicotine, can activate the NLRP3 inflammasome in EC, thus leading to endothelial cell death. For example, Wu et al. found that ox-LDL induced the upregulation of NLRP3, caspase-1, and IL-1β in ECs in a dose-dependent manner (86). Hang et al. found that ox-LDL stimulated NLRP3 inflammasome activation, increased IL-1β and IL-18 maturation and secretion, increased intracellular ROS, and increased lactate dehydrogenase (LDH) release in ECs (87). Chen et al. found that ox-LDL could induce increases in ROS and upregulate the expression of ICAM-1, TXNIP, NLRP3, and caspase-1 in ECs (88). Zhuang et al. found that forkhead box P transcription factor 1 (Foxp1) is a negative regulator of NLRP3 inflammasome activation in ECs, and they also revealed found that Foxp1 is significantly downregulated in atherosclerosis-susceptible endothelium and Foxp1 knockout in ApoE-/- mice exacerbates atherosclerosis. Subsequently, NLRP3, caspase-1, and pro-IL-1β were significantly upregulated and IL-1β secretion was increased. The team further demonstrated that Foxp1 is a gatekeeper of vascular inflammation and a transcriptional repressor; moreover, it can inhibit the expression of NLRP3, caspase-1, and pro-IL-1β from the transcription initiation level (60). Numerous studies have shown that NOX4 plays an important role in mediating endothelial dysfunction in type 2 diabetes (89). For example, Liao et al. performed in vitro and in vivo experiments and found that high glucose levels can promote the generation of ROS in ECs by upregulating NOX4 (90). Li et al. found that high levels can upregulate the expression of NOX4, NLRP3, and caspase-1 in ECs and showed that high glucose-induced NLRP3 inflammasome activation was dependent on NOX4 and mediates EC tight junction barrier disruption (91). Dunn et al. found that high glucose levels can upregulate TXNIP expression to induce endothelial dysfunction. In addition, the team found that knockdown of TXNIP could alleviate high glucose-mediated endothelial dysfunction (92). Chen et al. found that silencing NLRP3 could reverse the high glucose-induced upregulation of NLRP3, caspase-1, IL-1β, IL-18, and ICAM-1, and they further found that ROS scavengers could reverse the high glucose-induced upregulation of IL-1β and IL-18 in ECs. Furthermore, the team found that TXNIP knockdown inhibited IL-1β and IL-18 maturation (8). Wu et al. found that nicotine could induce upregulation of NLRP3, caspase-1, ASC, IL-1β, and IL-18 expression in ECs, DNA damage, and LDH release, and they also found that N-acetylcysteine (NAC) could inhibit nicotine-induced inflammasome activation and alleviate DNA damage, indicating that nicotine mediates NLRP3 inflammasome activation in ECs through ROS. The team further found that the knockdown of NLRP3 or ASC could inhibit nicotine-induced activation of the NLRP3 inflammasome in ECs. Similarly, the caspase-1 inhibitor VP-765 also inhibits nicotine-induced activation of the NLRP3 inflammasome in ECs (93). Zhang et al. found that nicotine could activate the NLRP3 inflammasome in EC. The team further found that NLRP3 inflammasome activation can promote the destruction of tight junction proteins between ECs, resulting in increased vascular permeability (94). Cau et al. found that Ang-II induces endothelial dysfunction, vascular remodeling, and hypertension through NLRP3 inflammasome activation (95).
In conclusion, stimulatory factors, such as ox-LDL, hyperglycemia, nicotine, and Ang II, can cause an increase in ROS in ECs. In endothelial cells, ROS acts as a bridge between pathological stimuli such as ox-LDL, hyperglycemia, Ang II, and nicotine and the activation of the NLRP3 inflammasome. The ROS-triggered NLRP3 inflammasome activation process in ECs involves two key steps: the initiation and activation phases. The initiation phase refers to the ROS-induced upregulation of NLRP3, caspase-1, IL-1β, and IL-18 in EC. The activation phase refers to ROS promoting the assembly and activation of the NLRP3 inflammasome through TXNIP. NLRP3 inflammasome activation promotes the maturation of IL-1β and IL-18 and can mediate the formation of porin N-GSDMD, thus causing cell swelling and even rupture, which lead to cell death. In addition, the formation of membrane pores promotes the release of cellular components, including IL-1β, IL-18, and HMGB1, which are involved in inflammatory responses. IL-1β, IL-18, and HMGB1 can bind to the EC surface at IL-1R, IL-18R, and TLR, respectively, and upregulate the expression of ICAM-1 and VCAM-1 via the Myd88/IRAK-1/TRAF-6/NF-κB pathway. In addition, NLRP3 inflammasome activation can disrupt tight junction proteins between EC, resulting in increased vascular permeability (Figure 6).
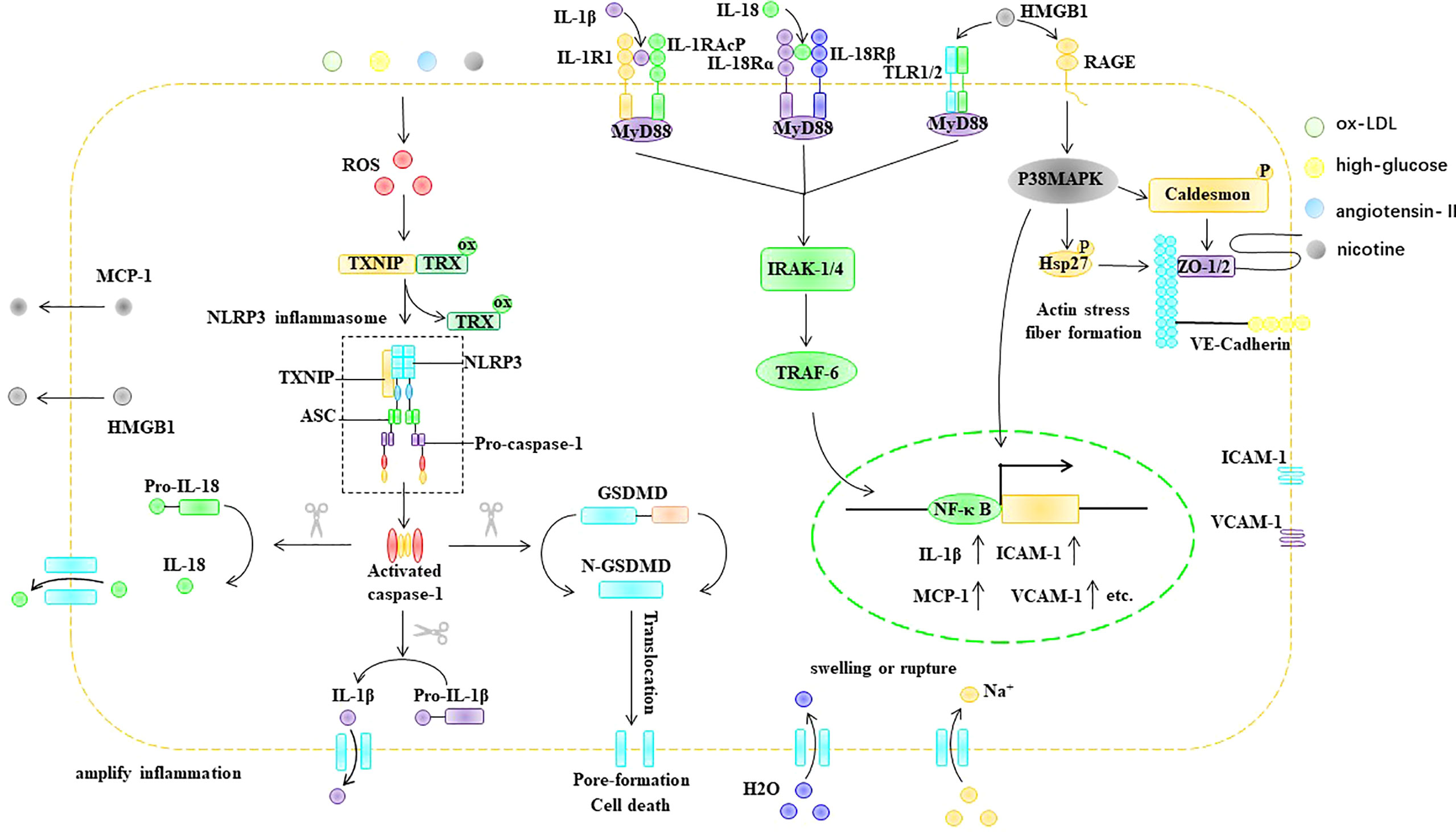
Figure 6 ROS trigger pyroptosis in EC. ASC, apoptosis-associated speck-like protein, ER endoplasmic reticulum, GSDMD Gasdermin D, HMGB1 high mobility group box 1, Hsp heat shock protein, ICAM-1 intercellular adhesion molecule-1, IL-1β interleukin-1β, IL-18 interleukin- 18, IL-1R IL-1 receptor, IL-18R IL-18 receptor, IRAK IL-1R-associated kinase, MCP-1 monocyte chemoattractant protein-1, MyD88 myeloid differentiation primary response gene 88, NF-κB nuclear factor kappa B, NLRP3 NLR-family pyrin domain-containing protein 3, ox-LDL oxidized low density lipoprotein, RAGE the receptor for advance glycation end products, ROS reactive oxygen species, TLR Toll-like receptors, Trx thioredoxin protein, TRAF TNF receptor-associated factor, TXNIP thioredoxin-interacting protein, VCAM-1 intervascular adhesion molecule-1.
Parthanatos is a type of programmed cell death that is dependent on poly (ADP-ribosome) polymerase 1 (PARP-1) (96, 97). PARP-1 is an ADP-ribosyltransferase that transfers ADP ribose from nicotinamide adenine dinucleotide (NAD+) to receptor proteins (98, 99). PARP-1 was originally described as a DNA nick sensor enzyme activated by DNA single- and double-strand breaks (100). DNA damage-induced activation of PARP-1 is considered the classical pathway for the activation of this enzyme. ROS, ionizing radiation, and alkylating agents are common causes of DNA fragmentation (101–103). PARP-1 activation depends on the degree of DNA damage. However, when DNA is extensively damaged, the overactivation of PARP-1 causes the accumulation of poly (ADP-ribose) (PAR), a process that consumes large amounts of NAD+. NAD+ is a direct substrate for the synthesis of PAR and a cofactor in many redox reactions, such as the tricarboxylic acid cycle, glycolysis, and pentose phosphate pathway (104, 105). Furthermore, the translocation of PAR from the nucleus to the mitochondria causes the release of apoptosis-inducing factor (AIF) (106, 107). After AIF leaves the mitochondria, it forms a complex with the macrophage migration inhibitory factor (MIF) in the cytoplasm. Subsequently, nuclear translocation of the AIF/MIF complex causes chromatin condensation and DNA fragmentation, ultimately leading to cell death (108–110) (Figure 7).
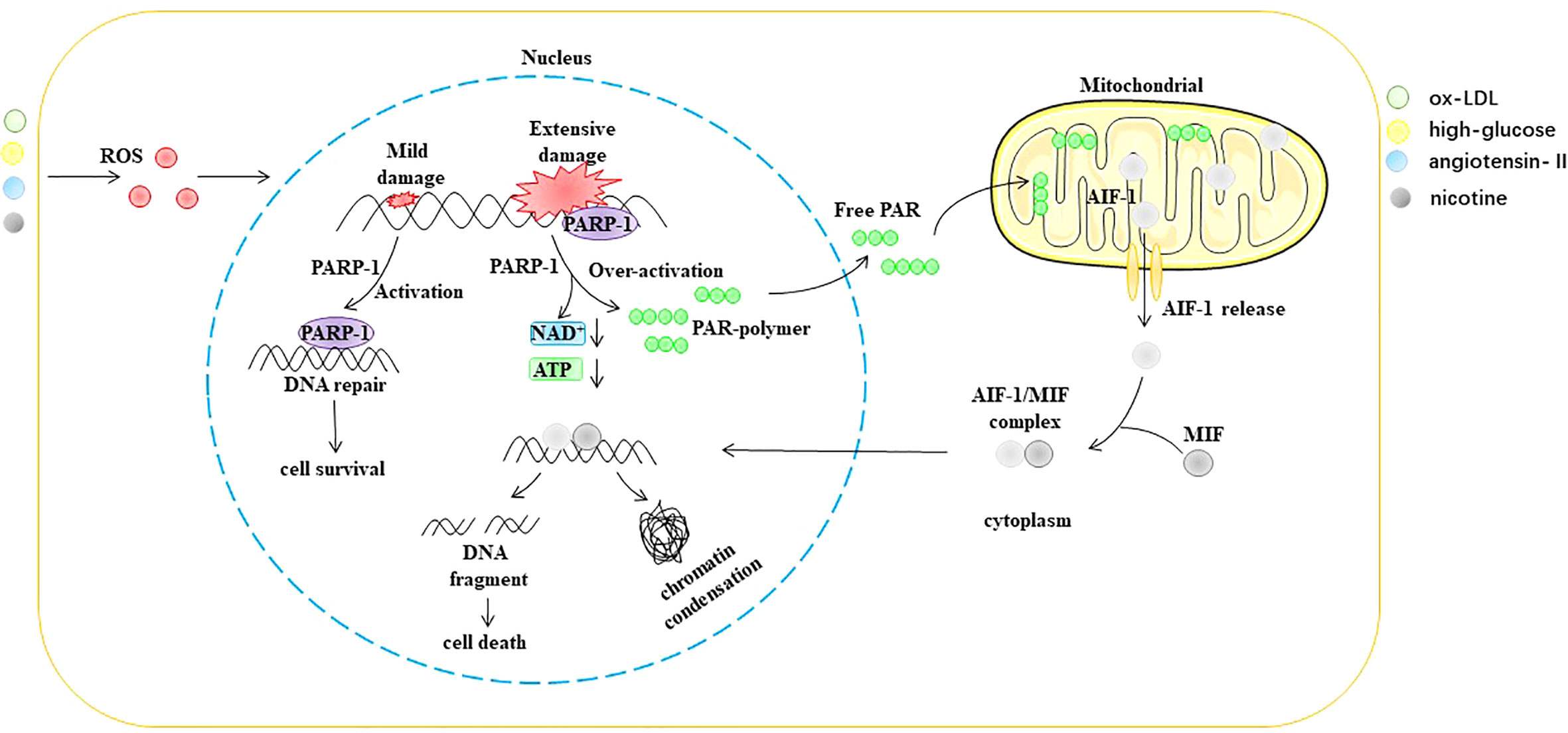
Figure 7 ROS trigger parthanatos in EC. AIF-1 apoptosis-inducing factor 1, ATP Adenosine triphosphate, MIF macrophage migration inhibitory factor, NAD+ nicotinamide adenine dinucleotide, ox-LDL oxidized low density lipoprotein, PAR poly (ADP-ribose), PARP-1 poly (ADP ribosome) polymerase 1, ROS reactive oxygen species.
In recent years, many studies have shown that stimuli such as ROS, Ang II, ox-LDL, and hyperglycemia can trigger the occurrence of parthanatos in ECs. For example, Mathews et al. found that H2O2 and ONOO could activate PARP-1 in ECs, leading to EC death. Knockdown of PARP-1 inhibits H2O2 or ONOO• triggered EC death (111). Liang et al. observed DNA damage and increased PARP-1 expression and activity in a model of Ang II-induced EC oxidative stress (112). Zhang et al. found that ox-LDL can induce coronary EC damage that is independent of caspase but dependent on the nuclear translocation of AIF (113). Wang et al. found that PARP1 was a key factor in the upregulation of arginase II (Arg II) induced by ox-LDL (114). Arg II results in reduced NO synthesis by competing with eNOS for the same substrate, l-arginine (115–117). PARP1 deficiency results in suppressed Arg II expression, enhanced eNOS expression, and improved NO production and endothelial function (114). Choi et al. found that enhanced PARP-1 activity is closely related to coronary artery endothelial dysfunction in mice with type 2 diabetes. The team further found that inhibition of PARP-1 activity restored eNOS phosphorylation and alleviated DNA damage, thereby improving ED (118). Taken together, ROS can cause extensive DNA damage that leads to the hyperactivation of PARP-1 and triggers parthanatos in EC.
Ferroptosis is an iron-dependent process involving programmed cell death. Lipid peroxidation, which is the process by which OH• attacks the carbon-carbon double bonds of lipids, particularly polyunsaturated fatty acids (PUFAs) (119), is an important marker for ferroptosis. Therefore, the production of OH• is a key factor in the lipid peroxidation process. reacts with H2O2 under the catalysis of Fe2+ to form Fe3+, OH•, and OH-, which is called the Fenton reaction. In addition, reacts with Fe3+ to form Fe2+ in a process called the Haber-Weiss cycle. Lipid peroxidation can be divided into three stages: initiation, propagation, and termination. In the initial stage, OH• interacts with lipids to form carbon-centred lipid radicals (L•). L• reacts with oxygen to generate a lipid peroxy radical (LOO•), which further generates a new L• (propagating phase) and lipid hydrogen peroxide (LOOH) from another molecular lipid. L• and LOOH produced during the propagation stage can be terminated by antioxidant molecules of the mevalonate pathway, such as coenzyme Q10 (CoQ 10) and vitamin E (VitE) (120–122). In addition, studies have shown that iron chelators, such as deferoxamine (DFO) and ciclopirox olamine (CPX), can inhibit the occurrence of ferroptosis by inhibiting lipid peroxidation (122–125).
Lipid peroxidation is regulated by the glutathione antioxidant system, which is composed of glutathione (GSH), glutathione peroxidase (GPX), and glutaredoxin (GRX), and it can effectively prevent ROS overgeneration (126, 127). Glutamate, cystine, and glycine are the raw materials used for the synthesis of GSH. Cystine enters the cell through the amino acid antiporter Xc system, which is composed of the light chain subunit SLC7A11 and the heavy chain subunit SLC3A2. The Xc system exchanges cystine with glutamate in such a way that cystine enters the cell and is further reduced to cysteine (128). Glutamate cysteine ligase (GCL) catalyzes the formation of γ-glutamate-cysteine from glutamate and cysteine, which is the rate-limiting step in GSH synthesis. Subsequently, γ-glutamic acid cysteine and glycine are catalyzed by GSH synthase (GSS) to generate GSH (129). GSH effectively maintains Gpx in a reduced state. Gpx can effectively scavenge intracellular hydrogen peroxide and peroxide to maintain intracellular redox homeostasis (130). Eight different Gpxs (Gpx1-8) have been found in humans, and Gpx1-4 and Gpx6 are selenoproteins (131). Compared with other members of the Gpx family, Gpx4 is a lipid peroxidation-repair enzyme, and it can convert lipid peroxides (LOOH) to their corresponding alcohols (LOH) (122, 132). Therefore, Gpx4 is considered a central inhibitor of ferroptosis. Numerous studies have shown that inhibiting the Xc-GSH-Gpx4 antioxidant system can induce ferroptosis in cells. For example, erastin, sulfasalazine (SAS), and sorafenib initiate ferroptosis in cells by inhibiting the Xc system (133, 134). Butionine sulfoxamine (BSO) induces ferroptosis by inhibiting GCL (134), while RSL3 can initiate ferroptosis in cells by inhibiting the activity of Gpx4 (135) (Figure 8).
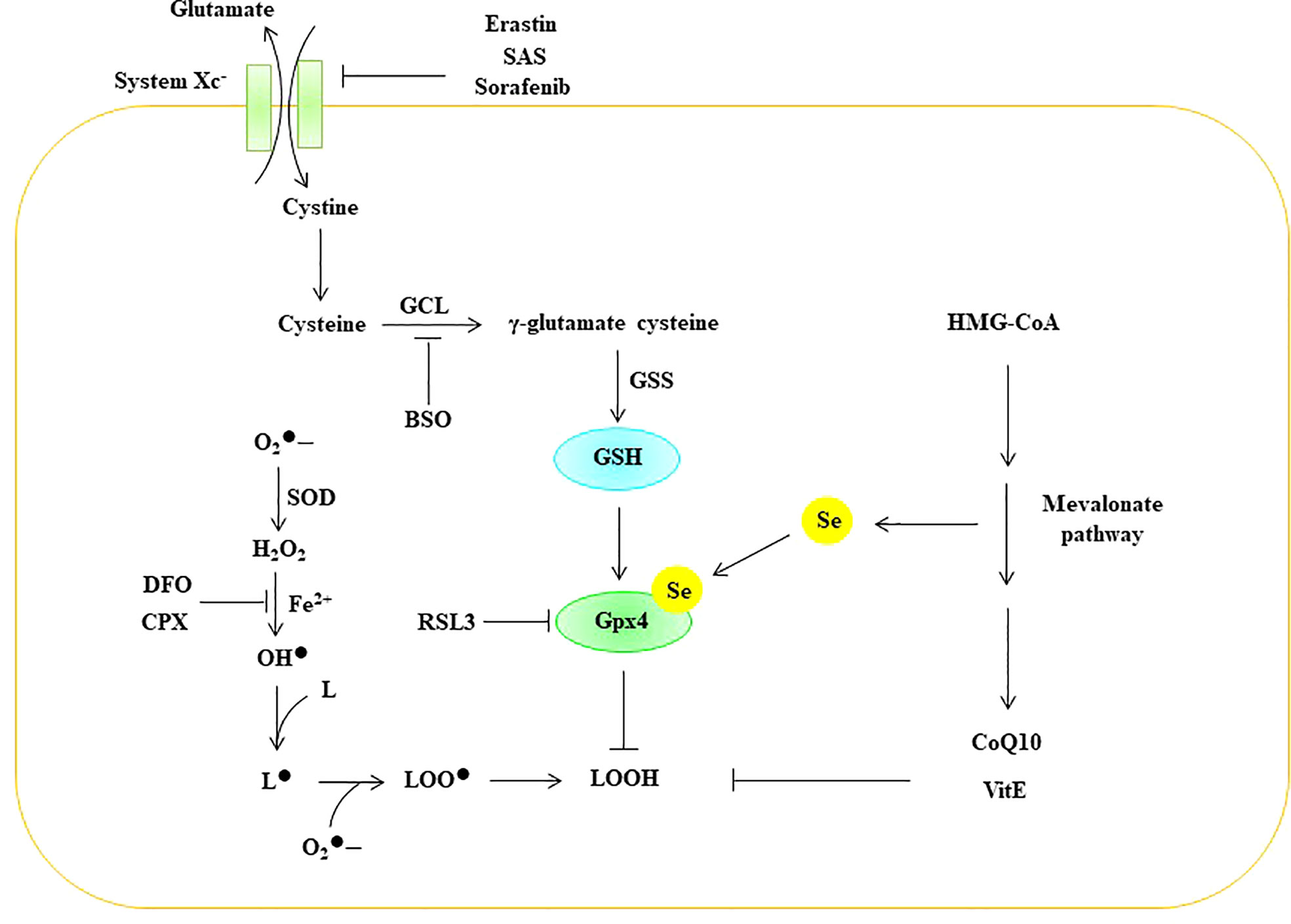
Figure 8 ROS trigger ferroptosis in EC. BSO Butionine sulfoxamine, CoQ 10 coenzyme Q10, CPX ciclopirox olamine, DFO deferoxamine GCL glutamate cysteine ligase, Gpx glutathione peroxidase, GSH glutathione, GSS GSH synthase, HMG-CoA 3-hydroxy-3-methyl-glutaryl-coenzyme A, Se selenium, SOD superoxide dismutase, SAS Sulfasalazine, VitE vitamin E, superoxide, H2O2 hydrogen peroxide, OH• peroxyl radical, L lipid, L• lipid free radical, LOO• lipid peroxy radical, LOOH lipid peroxide.
In recent years, studies have shown that ferroptosis is closely related to endothelial cell death. For example, Qin et al. found that zinc oxide nanoparticles (ZnONPs) could induce iron and lipid peroxidation in ECs in a dose- and time-dependent manner (136). The team further applied the lipid reactive oxygen species scavenger ferrostatin-1 and the iron chelator DFO to alleviate ZnONP-induced ferroptosis in ECs (22). Luo et al. showed that ferroptosis is related to ED and that the p53-xCT-GSH axis can regulate the process of EC ferroptosis (137). Sheng et al. showed that lysophosphatidylcholine (LPC) can induce increased intracellular iron and lipid peroxide levels and mitochondrial atrophy in EC. This process can be reversed by astragaloside IV (AS-IV) (138). Therefore, ferroptosis is an important mechanism by which ROS trigger programmed cell death in EC.
In conclusion, hyperlipemia, hyperglycemia, nicotine and hypertension are common pathogenic factors causing impaired NO synthesis in EC, up-regulated expressions of pro-inflammatory cytokines and intercellular adhesion factors in EC, and EC death. ROS may be the common mechanism for these pathological activities (8, 9, 86, 91, 93, 95). Under pathological conditions, ROS in EC mainly originates from mitochondria, NOXs, eNOS uncoupling, and XO (4, 5). In addition, these pathways may independently or jointly cause excessive accumulation of ROS in EC. ROS can cause impaired NO synthesis through the eNOS uncoupling mechanism, thereby causing vasomotor dysfunction (7). ROS can up-regulate the expression of pro-inflammatory cytokines and intercellular adhesion factors in endothelial cells, such as IL-1β, IL-18, ICAM-1, VCAM-1, and E-selectin, which are participates the process of monocyte-endothelial cell adhesion, increased vascular permeability, and monocyte differentiation to macrophages (8, 9). Therefore, ROS is an important signal that mediates the involvement of EC in inflammatory responses. Furthermore, ROS can trigger endothelial cell death through different molecular mechanisms, including pyroptosis, parthanatos, and ferroptosis, which have been demonstrated in some animal models of disease. For example, Zhuang et al. found that Foxp1 expression was significantly downregulated in atherosclerosis-susceptible endothelium. The team further demonstrated that knockout of Foxp1 in ApoE-/- mice promoted the up-regulation of NLRP3, caspase-1 and Pro-IL-1β, increased IL-1β secretion, and enhanced monocyte adhesion, migration and Infiltrates into the vessel wall of the aortic root, thereby exacerbating the formation of atherosclerotic plaques (60). Wu et al. found that nicotine can mediate the pyroptosis of aortic endothelial cells and exacerbate the formation of atherosclerotic plaques by constructing an ApoE-/- mouse atherosclerosis model (93). Kong et al. found that targeting the P2X7/NLRP3 signaling pathway prevents retinal endothelial cell pyroptosis in diabetic retinopathy (139). Kasson et al. showed that enhanced NF-κB activity impairs vascular function in male type 2 diabetic mice through a PARP-1, Sp-1 and COX-2-dependent mechanism (140). Abdul et al. found that ferroptosis in brain microvascular endothelial cells of diabetic mice is closely related to vascular degeneration and neurovascular remodeling after stroke, and this process can be reversed by DFO (141). Therefore, how to effectively scavenge ROS may be an important target for the treatment of endothelial cell death-related diseases. For example, N-acetylcysteine (NAC) is a potent ROS scavenger. Studies have shown that NAC inhibits pyroptosis, parthanatos, and ferroptosis by scavenging ROS (93, 142, 143). However, the following question about ROS-triggered EC death is unresolved and remains to be further explored: Do pyroptosis, parthanatos, and ferroptosis processes occur independently or simultaneously in the process of ROS-triggered EC death?
KL, DZ and JL jointly completed the conception and writing of the review. HP, ZZ and RW completed the picture drawing of the review. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China Grant 81970399.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ASC apoptosis-associated speck-like protein; ALR AIM2-like receptors; Ang II angiotension; AIF apoptosis inducing factor; Arg II arginase II; ATP Adenosine triphosphate; BSO Butionine sulfoxamine; CARD caspase-recruitment domain; CoQ 10 coenzyme Q10; CPX ciclopirox olamine; DFO deferoxamine; DUOX dual oxidases; EC endothelial cell; ED endothelial dysfuction; ER endoplasmic reticulum; ET-1 endothelin-1; ETC electron transport chain; FAD flavin adenine dinucleotide; FIIND function-to-find domain; Forkhead box P transcription factor 1 Foxp1; GSDMD Gasdermin D; Gpx glutathione peroxidase; Grx glutaredoxin; GSH glutathione; GSS GSH synthase; HMG-CoA 3-hydroxy-3-methyl-glutaryl-coenzyme A; HMGB1 high mobility group box 1; Hsp heat shock protein; ICAM-1 intercellular adhesion molecule-1; IL-1β interleukin-1β; IL-18 interleukin-18; IL-1R IL-1 receptor; IL-18R IL-18 receptor; IRAK IL-1R-associated kinase; LDH lactate dehydrogenase; LPS lysophosphatidylcholine; MIF macrophage migration inhibitory factor; Mo-co molybdenum cofactor; MyD88 myeloid differentiation primary response gene 88; NAC N-acetylcysteine; NAD+ nicotinamide adenine dinucleotide; NBD nucleotide-binding domain; NLRs NOD-like receptor proteins; NLRP3 NLR-family pyrin domain-containing protein 3; NNT nicotinamide nucleotide transhydrogenase; NO nitric oxide; NOX NAPDH oxidase; NOS nitric oxide synthase; ox-LDL oxidized low density lipoprotein; OXPHO oxidative phosphorylation; PUFAs polyunsaturated fatty acids; PAR poly (ADP-ribose); PARP-1 poly (ADP-ribosome) polymerase 1; PFD pore-forming domain; PKC protein kinase C; PYD pyrin domain; Poldip2 polymerase delta-interacting protein 2; RD repressor domain; ROS reactive species oxygen; SOD superoxide dismutase; SAS Sulfasalazine; Se selenium; TRAF TNF receptor-associated factor; Trx reduced thioredoxin protein; TXNIP thioredoxin-interacting protein; t-BHP tert-butyl hydroperoxide; XO xanthine oxidase; XDH xanthine dehydrogenase; XOR xanthine oxidoreductase; VCAM-1 intervascular adhesion molecule-1; VitE vitamin E; ZnONPs zinc oxide nanoparticles

1. Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol (2017) 37(9):e108–14. doi: 10.1161/ATVBAHA.117.309813
2. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res (2017) 120(4):713–35. doi: 10.1161/CIRCRESAHA.116.309326
3. Abu Nabah YN, Losada M, Estellés R, Mateo T, Company C, Piqueras L, et al. CXCR2 blockade impairs angiotensin II-induced CC chemokine synthesis and mononuclear leukocyte infiltration. Arterioscler Thromb Vasc Biol (2007) 27(11):2370–6. doi: 10.1161/ATVBAHA.107.147009
4. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
5. El Assar M, Angulo J, Rodríguez-Mañas L. Oxidative stress and vascular inflammation in aging. Free Radic Biol Med (2013) 65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003
6. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87(1):245–313. doi: 10.1152/physrev.00044.2005
7. Rochette L, Lorin J, Zeller M, Guilland JC, Lorgis L, Cottin Y, et al. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther (2013) 140(3):239–57. doi: 10.1016/j.pharmthera.2013.07.004
8. Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflammation Res (2017) 66(2):157–66. doi: 10.1007/s00011-016-1002-6
9. Limb GA, Hickman-Casey J, Hollifield RD, Chignell AH. Vascular adhesion molecules in vitreous from eyes with proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci (1999) 40(10):2453–7. Available at: https://iovs.arvojournals.org/article.aspx?articleid=2162545
10. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res (2016) 118(4):620–36. doi: 10.1161/CIRCRESAHA.115.306301
11. Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, et al. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet (2014) 383(9932):1933–43. doi: 10.1016/S0140-6736(14)60107-0
12. Ferro CJ, Webb DJ. Endothelial dysfunction and hypertension. Drugs (1997) 53 Suppl 1:30–41. doi: 10.2165/00003495-199700531-00006
13. Hu X, De Silva TM, Chen J, Faraci FM. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ Res (2017) 120(3):449–71. doi: 10.1161/CIRCRESAHA.116.308427
14. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci (2020) 21(22):8793. doi: 10.3390/ijms21228793
15. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. (2011) 121(11):4210–21. doi: 10.1172/JCI45161
16. Gui F, You Z, Fu S, Wu H, Zhang Y. Endothelial dysfunction in diabetic retinopathy. Front Endocrinol (Lausanne). (2020) 11:591. doi: 10.3389/fendo.2020.00591
17. Yamazaki Y, Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of alzheimer's disease. Int J Mol Sci (2017) 18(9):1965. doi: 10.3390/ijms18091965
19. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
20. Bretón-Romero R, Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol (2014) 2:529–34. doi: 10.1016/j.redox.2014.02.005
21. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
22. Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol (2009) 554:165–81. doi: 10.1007/978-1-59745-521-3_11
23. Rao KNS, Shen X, Pardue S, Krzywanski DM. Nicotinamide nucleotide transhydrogenase (NNT) regulates mitochondrial ROS and endothelial dysfunction in response to angiotensin II. Redox Biol (2020) 36:101650. doi: 10.1016/j.redox.2020.101650
24. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol (2020) 17(3):170–94. doi: 10.1038/s41569-019-0260-8
25. Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene (2001) 269(1-2):131–40. doi: 10.1016/s0378-1119(01)00449-8
26. Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal (2009) 11(4):791–810. doi: 10.1089/ars.2008.2220
27. Mizuno T, Kaibuchi K, Ando S, Musha T, Hiraoka K, Takaishi K, et al. Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J Biol Chem (1992) 267(15):10215–8. doi: 10.1016/S0021-9258(19)50005-9
28. Kitada M, Koya D, Sugimoto T, Isono M, Araki S, Kashiwagi A, et al. Translocation of glomerular p47phox and p67phox by protein kinase c-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes (2003) 52(10):2603–14. doi: 10.2337/diabetes.52.10.2603
29. Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell (2003) 113(3):343–55. doi: 10.1016/s0092-8674(03)00314-3
30. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab (2014) 25(9):452–63. doi: 10.1016/j.tem.2014.06.012
31. Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discovery (2011) 10(6):453–71. doi: 10.1038/nrd3403
32. Schröder K, Weissmann N, Brandes RP. Organizers and activators: Cytosolic nox proteins impacting on vascular function. Free Radic Biol Med (2017) 109:22–32. doi: 10.1016/j.freeradbiomed.2017.03.017
33. Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med (2014) 76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046
34. Ouerd S, Idris-Khodja N, Trindade M, Ferreira NS, Berillo O, Coelho SC, et al. Endothelium-restricted endothelin-1 overexpression in type 1 diabetes worsens atherosclerosis and immune cell infiltration via NOX1. Cardiovasc Res (2021) 117(4):1144–53. doi: 10.1093/cvr/cvaa168
35. Youn JY, Gao L, Cai H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia (2012) 55(7):2069–79. doi: 10.1007/s00125-012-2557-6
36. Dolmatova EV, Forrester SJ, Wang K, Ou Z, Williams HC, Joseph G, et al. Endothelial Poldip2 regulates sepsis-induced lung injury via rho pathway activation. Cardiovasc Res (2021) 16:cvab295. doi: 10.1093/cvr/cvab295
37. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol (2020) 36:101638. doi: 10.1016/j.redox.2020.101638
38. Zhao W, Feng H, Sun W, Liu K, Lu JJ, Chen X. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: Roles of NOX4 and mitochondrion. Redox Biol (2017) 11:524–34. doi: 10.1016/j.redox.2016.12.036
39. Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res (2000) 86(3):347–54. doi: 10.1161/01.res.86.3.347
40. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, et al. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation (2011) 124(6):731–40. doi: 10.1161/CIRCULATIONAHA.111.030775
41. da Silva JF, Alves JV, Silva-Neto JA, Costa RM, Neves KB, Alves-Lopes R, et al. Lysophosphatidylcholine induces oxidative stress in human endothelial cells via NOX5 activation - implications in atherosclerosis. Clin Sci (Lond). (2021) 135(15):1845–58. doi: 10.1042/CS20210468
42. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J (2012) 33(7):829–37. doi: 10.1093/eurheartj/ehr304
43. Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med (2013) 188(6):639–46. doi: 10.1164/rccm.201304-0686PP
44. Chapple SJ, Cheng X, Mann GE. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol (2013) 1(1):319–31. doi: 10.1016/j.redox.2013.04.001
45. Wu Y, Ding Y, Ramprasath T, Zou MH. Oxidative stress, GTPCH1, and endothelial nitric oxide synthase uncoupling in hypertension. Antioxid Redox Signal (2021) 34(9):750–64. doi: 10.1089/ars.2020.8112
46. Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, et al. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens (2002) 15(4 Pt 1):326–32. doi: 10.1016/s0895-7061(01)02317-2
47. Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. (1997) 99(1):41–6. doi: 10.1172/JCI119131
48. Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. (1998) 95(16):9220–5. doi: 10.1073/pnas.95.16.9220
49. Takaya T, Hirata K, Yamashita T, Shinohara M, Sasaki N, Inoue N, et al. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arterioscler Thromb Vasc Biol (2007) 27(7):1632–7. doi: 10.1161/ATVBAHA.107.142182
50. Schmidt HM, Kelley EE, Straub AC. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol (2019) 21:101072. doi: 10.1016/j.redox.2018.101072
51. Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med (2002) 33(6):774–97. doi: 10.1016/s0891-5849(02)00956-5
52. Polito L, Bortolotti M, Battelli MG, Bolognesi A. Xanthine oxidoreductase: A leading actor in cardiovascular disease drama. Redox Biol (2021) 48:102195. doi: 10.1016/j.redox.2021.102195
53. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev (2011) 22(4):189–95. doi: 10.1016/j.cytogfr.2011.10.001
54. Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc Natl Acad Sci U S A. (2000) 97(20):10723–8. doi: 10.1073/pnas.97.20.10723
55. Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A. (2001) 98(26):15215–20. doi: 10.1073/pnas.221292098
56. Daiber A, Andreadou I, Oelze M, Davidson SM, Hausenloy DJ. Discovery of new therapeutic redox targets for cardioprotection against ischemia/reperfusion injury and heart failure. Free Radic Biol Med (2021) 163:325–43. doi: 10.1016/j.freeradbiomed.2020.12.026
57. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
58. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol (2019) 40(11):1035–52. doi: 10.1016/j.it.2019.09.005
59. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity (2019) 50(6):1352–64. doi: 10.1016/j.immuni.2019.05.020
60. Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, et al. Endothelial Foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ Res (2019) 125(6):590–605. doi: 10.1161/CIRCRESAHA.118.314402
61. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis (2020) 11(9):776. doi: 10.1038/s41419-020-02985-x
62. Kovacs SB, Miao EA. Gasdermins: Effectors of pyroptosis. Trends Cell Biol (2017) 27(9):673–84. doi: 10.1016/j.tcb.2017.05.005
63. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
64. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
65. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
66. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
67. Faller LD. Mechanistic studies of sodium pump. Arch Biochem Biophys (2008) 476(1):12–21. doi: 10.1016/j.abb.2008.05.017
68. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
69. Martín-Sánchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ (2016) 23(7):1219–31. doi: 10.1038/cdd.2015.176
70. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
71. Zamyatina A, Heine H. Lipopolysaccharide recognition in the crossroads of TLR4 and caspase-4/11 mediated inflammatory pathways. Front Immunol (2020) 11:585146. doi: 10.3389/fimmu.2020.585146
72. Matikainen S, Nyman TA, Cypryk W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol (2020) 204(12):3063–9. doi: 10.4049/jimmunol.2000373
73. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev (2015) 265(1):35–52. doi: 10.1111/imr.12286
74. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science (1996) 272(5262):735–8. doi: 10.1126/science.272.5262.735
75. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
76. Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol (2014) 35(6):253–61. doi: 10.1016/j.it.2014.02.007
77. Chen X, Guo X, Ge Q, Zhao Y, Mu H, Zhang J. ER stress activates the NLRP3 inflammasome: A novel mechanism of atherosclerosis. Oxid Med Cell Longev (2019) 2019:3462530. doi: 10.1155/2019/3462530
78. Rovira-Llopis S, Apostolova N, Bañuls C, Muntané J, Rocha M, Victor VM. Mitochondria, the NLRP3 inflammasome, and sirtuins in type 2 diabetes: New therapeutic targets. Antioxid Redox Signal (2018) 29(8):749–91. doi: 10.1089/ars.2017.7313
79. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal (2015) 22(13):1111–29. doi: 10.1089/ars.2014.5994
80. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9(8):847–56. doi: 10.1038/ni.1631
81. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17(8):588–606. doi: 10.1038/nrd.2018.97
82. Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol (2019) 26:101254. doi: 10.1016/j.redox.2019.101254
83. Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10(3):210–5. doi: 10.1038/nri2725
84. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
85. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
86. Wu Q, He X, Wu LM, Zhang RY, Li LM, Wu CM, et al. MLKL aggravates ox-LDL-Induced cell pyroptosis via activation of NLRP3 inflammasome in human umbilical vein endothelial cells. Inflammation (2020) 43(6):2222–31. doi: 10.1007/s10753-020-01289-8
87. Hang L, Peng Y, Xiang R, Li X, Li Z. Ox-LDL causes endothelial cell injury through ASK1/NLRP3-mediated inflammasome activation via endoplasmic reticulum stress. Drug Des Devel Ther (2020) 14:731–44. doi: 10.2147/DDDT.S231916
88. Chen M, Li W, Zhang Y, Yang J. MicroRNA-20a protects human aortic endothelial cells from ox-LDL-induced inflammation through targeting TLR4 and TXNIP signaling. BioMed Pharmacother. (2018) 103:191–7. doi: 10.1016/j.biopha.2018.03.129
89. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci (2019) 20(15):3775. doi: 10.3390/ijms20153775
90. Liao Y, Gou L, Chen L, Zhong X, Zhang D, Zhu H, et al. NADPH oxidase 4 and endothelial nitric oxide synthase contribute to endothelial dysfunction mediated by histone methylations in metabolic memory. Free Radic Biol Med (2018) 115:383–94. doi: 10.1016/j.freeradbiomed.2017.12.017
91. Li XX, Ling SK, Hu MY, Ma Y, Li Y, Huang PL. Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic Biol Med (2019) 145:175–86. doi: 10.1016/j.freeradbiomed.2019.09.015
92. Dunn LL, Simpson PJ, Prosser HC, Lecce L, Yuen GS, Buckle A, et al. A critical role for thioredoxin-interacting protein in diabetes-related impairment of angiogenesis. Diabetes (2014) 63(2):675–87. doi: 10.2337/db13-0417
93. Wu X, Zhang H, Qi W, Zhang Y, Li J, Li Z, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis (2018) 9(2):171. doi: 10.1038/s41419-017-0257-3
94. Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of cathepsin b-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. Eur J Pharmacol (2019) 865:172795. doi: 10.1016/j.ejphar.2019.172795
95. Cau SB, Bruder-Nascimento A, Silva MB, Ramalho FNZ, Mestriner F, Alves-Lopes R, et al. Angiotensin-II activates vascular inflammasome and induces vascular damage. Vascul Pharmacol (2021) 139:106881. doi: 10.1016/j.vph.2021.106881
96. Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
97. Fatokun AA, Dawson VL. Dawson TM. parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol (2014) 171(8):2000–16. doi: 10.1111/bph.12416
98. Hegedűs C, Virág L. Inputs and outputs of poly(ADP-ribosyl)ation: Relevance to oxidative stress. Redox Biol (2014) 2:978–82. doi: 10.1016/j.redox.2014.08.003
99. Zhou Y, Liu L, Tao S, Yao Y, Wang Y, Wei Q, et al. Deng y. parthanatos and its associated components: Promising therapeutic targets for cancer. Pharmacol Res (2021) 163:105299. doi: 10.1016/j.phrs.2020.105299
100. Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell (2004) 119(6):803–14. doi: 10.1016/j.cell.2004.11.002
101. Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol (2019) 10:306. doi: 10.3389/fphar.2019.00306
102. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev (2019) 99(4):1765–817. doi: 10.1152/physrev.00022.2018
103. Bárány T, Simon A, Szabó G, Benkő R, Mezei Z, Molnár L, et al. Oxidative stress-related parthanatos of circulating mononuclear leukocytes in heart failure. Oxid Med Cell Longev (2017) 2017:1249614. doi: 10.1155/2017/1249614
104. Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci (2008) 1147:233–41. doi: 10.1196/annals.1427.014
105. Heeres JT, Hergenrother PJ. Poly(ADP-ribose) makes a date with death. Curr Opin Chem Biol (2007) 11(6):644–53. doi: 10.1016/j.cbpa.2007.08.038
106. Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal (2011) 4(167):ra20. doi: 10.1126/scisignal.2000902
107. Cohausz O, Blenn C, Malanga M, Althaus FR. The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol Life Sci (2008) 65(4):644–55. doi: 10.1007/s00018-008-7516-5
108. Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science (2016) 354(6308):aad6872. doi: 10.1126/science.aad6872
109. Liu L, Li J, Ke Y, Zeng X, Gao J, Ba X, et al. The key players of parthanatos: opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell Mol Life Sci (2022) 79(1):60. doi: 10.1007/s00018-021-04109-w
110. Jiang HY, Yang Y, Zhang YY, Xie Z, Zhao XY, Sun Y, et al. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol (2018) 14:361–70. doi: 10.1016/j.redox.2017.10.002
111. Mathews MT, Berk BC. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler Thromb Vasc Biol (2008) 28(4):711–7. doi: 10.1161/ATVBAHA.107.156406
112. Liang ES, Bai WW, Wang H, Zhang JN, Zhang F, Ma Y, et al. PARP-1 (Poly[ADP-ribose] polymerase 1) inhibition protects from ang II (Angiotensin II)-induced abdominal aortic aneurysm in mice. Hypertension (2018) 72(5):1189–99. doi: 10.1161/HYPERTENSIONAHA.118.11184
113. Zhang W, Li D, Mehta JL. Role of AIF in human coronary artery endothelial cell apoptosis. Am J Physiol Heart Circ Physiol (2004) 286(1):H354–8. doi: 10.1152/ajpheart.00579.2003
114. Wang Q, Zhao T, Zhang W, Yu W, Liu B, Wang Z, et al. Poly (ADP-ribose) polymerase 1 mediated arginase II activation is responsible for oxidized LDL-induced endothelial dysfunction. Front Pharmacol (2018) 9:882. doi: 10.3389/fphar.2018.00882
115. Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res (2006) 99(9):951–60. doi: 10.1161/01.RES.0000247034.24662.b4
116. Pandey D, Bhunia A, Oh YJ, Chang F, Bergman Y, Kim JH, et al. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ Res (2014) 115(4):450–9. doi: 10.1161/CIRCRESAHA.115.304262
117. Wei SJ, Cheng L, Liang ES, Wang Q, Zhou SN, Xu H, et al. Poly(ADP-ribose) polymerase 1 deficiency increases nitric oxide production and attenuates aortic atherogenesis through downregulation of arginase II. Clin Exp Pharmacol Physiol (2017) 44(1):114–22. doi: 10.1111/1440-1681.12685
118. Choi SK, Galán M, Kassan M, Partyka M, Trebak M, Matrougui K. Poly(ADP-ribose) polymerase 1 inhibition improves coronary arteriole function in type 2 diabetes mellitus. Hypertension (2012) 59(5):1060–8. doi: 10.1161/HYPERTENSIONAHA.111.190140
119. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
120. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy (2021) 17(9):2054–81. doi: 10.1080/15548627.2020.1810918
121. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
122. Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol (2016) 26(3):165–76. doi: 10.1016/j.tcb.2015.10.014
123. Zhang Y, Fan BY, Pang YL, Shen WY, Wang X, Zhao CX, et al. Neuroprotective effect of deferoxamine on erastininduced ferroptosis in primary cortical neurons. Neural Regener Res (2020) 15(8):1539–45. doi: 10.4103/1673-5374.274344
124. Sharma A, Flora SJS. Positive and negative regulation of ferroptosis and its role in maintaining metabolic and redox homeostasis. Oxid Med Cell Longev (2021) 2021:9074206. doi: 10.1155/2021/9074206
125. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther (2021) 6(1):49. doi: 10.1038/s41392-020-00428-9
126. Lv H, Zhen C, Liu J, Yang P, Hu L, Shang P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid Med Cell Longev (2019) 2019:3150145. doi: 10.1155/2019/3150145
127. Bajic VP, Van Neste C, Obradovic M, Zafirovic S, Radak D, Bajic VB, et al. Glutathione "Redox homeostasis" and its relation to cardiovascular disease. Oxid Med Cell Longev (2019) 2019:5028181. doi: 10.1155/2019/5028181
128. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med (2009) 30(1-2):42–59. doi: 10.1016/j.mam.2008.05.005
129. Wang L, Ahn YJ, Asmis R. Sexual dimorphism in glutathione metabolism and glutathione-dependent responses. Redox Biol (2020) 31:101410. doi: 10.1016/j.redox.2019.101410
130. Ferguson GD, Bridge WJ. The glutathione system and the related thiol network in caenorhabditis elegans. Redox Biol (2019) 24:101171. doi: 10.1016/j.redox.2019.101171
131. Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal (2011) 15(7):1957–97. doi: 10.1089/ars.2010.3586
132. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol (2020) 30(6):478–90. doi: 10.1016/j.tcb.2020.02.009
133. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
134. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
135. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
136. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy (2021) 17(12):4266–85. doi: 10.1080/15548627.2021.1911016
137. Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao YY, et al. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J Diabetes. (2021) 12(2):124–37. doi: 10.4239/wjd.v12.i2.124
138. Sheng S, Xu J, Liang Q, Hong L, Zhang L. Astragaloside IV inhibits bleomycin-induced ferroptosis in human umbilical vein endothelial cells by mediating LPC. Oxid Med Cell Longev (2021) 2021:6241242. doi: 10.1155/2021/6241242
139. Kong H, Zhao H, Chen T, Song Y, Cui Y. Targeted P2X7/NLRP3 signaling pathway against inflammation, apoptosis, and pyroptosis of retinal endothelial cells in diabetic retinopathy. Cell Death Dis (2022) 13(4):336. doi: 10.1038/s41419-022-04786-w
140. Kassan M, Choi SK, Galán M, Bishop A, Umezawa K, Trebak M, et al. Enhanced NF-κB activity impairs vascular function through PARP-1-, SP-1-, and COX-2-dependent mechanisms in type 2 diabetes. Diabetes (2013) 62(6):2078–87. doi: 10.2337/db12-1374
141. Abdul Y, Li W, Ward R, Abdelsaid M, Hafez S, Dong G, et al. Deferoxamine treatment prevents post-stroke vasoregression and neurovascular unit remodeling leading to improved functional outcomes in type 2 Male diabetic rats: Role of endothelial ferroptosis. Transl Stroke Res (2021) 12(4):615–30. doi: 10.1007/s12975-020-00844-7
142. Li D, Kou Y, Gao Y, Liu S, Yang P, Hasegawa T, et al. Oxaliplatin induces the PARP1-mediated parthanatos in oral squamous cell carcinoma by increasing production of ROS. Aging (Albany NY). (2021) 13(3):4242–57. doi: 10.18632/aging.202386
Keywords: reactive oxygen species, pyroptosis, parthanatos, ferroptosis, endothelial cells
Citation: Zheng D, Liu J, Piao H, Zhu Z, Wei R and Liu K (2022) ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 13:1039241. doi: 10.3389/fimmu.2022.1039241
Received: 07 September 2022; Accepted: 17 October 2022;
Published: 01 November 2022.
Edited by:
Benoit Pourcet, Université de Lille, FranceReviewed by:
Aleksandr E. Vendrov, University of Michigan, United StatesCopyright © 2022 Zheng, Liu, Piao, Zhu, Wei and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kexiang Liu, a3hsaXU2NEBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.