 Payal Aggarwal1†
Payal Aggarwal1† Wen Luo1,2†Katherine C. Pehlivan1†Hai Hoang1
Wen Luo1,2†Katherine C. Pehlivan1†Hai Hoang1 Prajwal Rajappa3Timothy P. Cripe3Kevin A. Cassady3
Prajwal Rajappa3Timothy P. Cripe3Kevin A. Cassady3 Dean A. Lee3
Dean A. Lee3 Mitchell S. Cairo1,2,4,5*
Mitchell S. Cairo1,2,4,5*- 1Department of Pediatrics, New York Medical College, Valhalla, NY, United States
- 2Department of Pathology, Microbiology and Immunology, New York Medical College, Valhalla, NY, United States
- 3Center for Childhood Cancer Research, Abigail Wexner Research Institute at Nationwide Children’s Hospital, Columbus, OH, United States
- 4Department of Medicine, New York Medical College, Valhalla, NY, United States
- 5Department of Cell Biology and Anatomy, New York Medical College, Valhalla, NY, United States
High grade gliomas are identified as malignant central nervous tumors that spread rapidly and have a universally poor prognosis. Historically high grade gliomas in the pediatric population have been treated similarly to adult high grade gliomas. For the first time, the most recent classification of central nervous system tumors by World Health Organization has divided adult from pediatric type diffuse high grade gliomas, underscoring the biologic differences between these tumors in different age groups. The objective of our review is to compare high grade gliomas in the adult versus pediatric patient populations, highlighting similarities and differences in epidemiology, etiology, pathogenesis and therapeutic approaches. High grade gliomas in adults versus children have varying clinical presentations, molecular biology background, and response to chemotherapy, as well as unique molecular targets. However, increasing evidence show that they both respond to recently developed immunotherapies. This review summarizes the distinctions and commonalities between the two in disease pathogenesis and response to therapeutic interventions with a focus on immunotherapy.
1 Introduction
High Grade Gliomas (HGG) represent the most common and aggressive brain tumor noted to have poor outcomes in adults and children (1–3). In the adult population, HGG represents the majority of primary brain tumors and therefore been extensively studied over many years (1–3). By contrast, pediatric high grade gliomas (pHGG) have remained a relatively under-investigated disease, as they represent only a fraction of primary malignant brain tumors in children. Recent genomic and epigenomic profiling studies have revealed key distinctions between adult HGG (aHGG) and pHGG, suggesting they are quite different diseases (1–3).
In fact, the 2021 WHO Classification of Tumors of the Central Nervous System (CNS), also referred to as WHO CNS 5, included major changes to the categorization of high grade gliomas and separated pHGG from aHGG to reflect this concept of their differing biology (4). Another major change in the WHO CNS 5 affecting the diagnostic implications for high grade gliomas is that tumors are now graded within tumor entities, while in past versions, a single diagnostic entity corresponded to specific tumor grade. This slightly complicates the delineation of low grade vs. high grade glioma, as previously it was clear from the tumor diagnostic entity whether it was low grade (generally considered Grade 1 or 2) vs. high grade (generally grade 3 or 4). Furthermore, in this version, both molecular and histologic findings may impact tumor grade within a given entity, with certain molecular alterations automatically designating a high grade tumor regardless of histologic findings (4).
When compared to the WHO CNS 4, published in 2016, WHO CNS 5 shows a continued progression towards an integrated molecular and histological diagnostic approach. Whereas in the WHO CNS 4, glioblastomas (GBMs) included both IDH- mutant and IDH-wild type tumors, adult-type diffuse gliomas are divided into distinct categories on the basis of IDH-mutation status in CNS 5, with the term glioblastoma, applying only to IDH wild-type tumors. IDH-mutant astrocytomas are classified separately and can encompass grade 2, 3, and 4 tumors. Adult tumors with both IDH mutations and 1q/19p co-deletion are classified as oligodendrogliomas, which can be grade 2 or 3, generally have a prolonged OS compared to tumors lacking these molecular changes (5).
In CNS 5, Pediatric-type diffuse gliomas are categorized into four distinct categories, with their histone mutation status playing a central role in their delineation: 1) Diffuse midline glioma, H3 K27-altered, 2) Diffuse hemispheric glioma, H3 G34-mutant, 3) Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype and 4) Infant-type hemispheric glioma (4).
For adults with HGG, standard of care employs the techniques of maximal surgical resection and radiation accompanied by chemotherapy followed by maintenance chemotherapy (6, 7). Most pediatric neuro-oncologists employ a similar treatment strategy for pHGG management for young patients diagnosed with hemispheric tumors who are old enough to receive radiation. Midline gliomas are often not amenable to resection due to their critical locations, and so only biopsy for diagnosis is performed prior to radiotherapy. Despite extensive clinical trials looking at various agents for the treatment of diffuse midline gliomas, thus far no additional therapy has proved to extend overall survival beyond radiation alone. For infant type hemispheric glioma, the concerns of damaging effects of irradiation on developing brains dictate a different treatment approach, to avoid radiation. Infant type hemispheric gliomas, also commonly have mutations which can be targeted with inhibitors, such as ALK, ROS, or NTRK alterations (8). With recent studies revealing deeper insights into gliomagenesis across all age groups and the development of cancer immunotherapy, the distinctions and commonalities of pHGG versus aHGG provide rationale for novel treatment options that are more specific and effective with potential to meaningfully improve the outcomes in patients with these tumor types.
2 Epidemiology
aHGG account for almost 25% of all CNS tumors in adults, with GBM being the most common type. In fact, GBM represents half of all malignant brain tumors diagnosed in adults; every year, there are over 10,000 new cases of GBM that are reported (9, 10). After diagnosis, GBM patients survive on average, 14.6 months and the 5-year survival rate is less than 5% (9, 11). Males are more often affected than females (1.6:1) and whites more often than blacks (2:1) (12, 13). The outcomes in patients over the age of 65 are worse when compared to those younger than 65 at the time of diagnosis (14). The outcomes may be worse because more elderly patients may not be able to tolerate standard of care therapies due to concern of toxicities and comorbid conditions.
When compared with adults, HGG in the pediatric population is less common and makes up only 3-15% primary central nervous system tumors in children (15). An overall survival of 10-73 months has been reported in pHGG. These wide ranges in reported epidemiologic data may be in part due to the changing classifications of HGG in the pediatric population over time, as well as differing inclusion criteria for HGG in pediatric clinical trials. Hemispheric HGG in the pediatric population is seen most frequently between the ages of 15 and nineteen (15), but in younger children, diffuse midline gliomas are recognized more commonly. Although it is unclear why, the disease has a predilection for males in both the pediatric and adult populations (15).
3 Clinical presentations
The challenge with diagnosing HGG includes the nonspecific nature of presenting symptoms. Both adult and pediatric patients can present with headaches and fatigue spanning a few months. In adults, the headache has been described to be similar to that of a tension headache or migraine (16). The headaches may be combined with cognitive deficit and mood changes. However, most patients are unlikely to see a primary care provider due to these symptoms. The most dramatic presentation of hemispheric HGG tends to be seizures. They can be seen at diagnosis in approximately 20% of adults and 30% of children (16, 17). Other symptoms that have been seen in children diagnosed with HGG are failure to thrive, lethargy and emesis, and cranial neuropathies which may be seen in patients with midline gliomas. Genetic predisposition syndromes such as Li-Fraumeni Syndrome, Tuberous Sclerosis, NF-1 and constitutional mismatch repair deficiency are associated with pediatric glioma patients (18–20).
4 Imaging and diagnosis
HGG is most commonly suspected based on imaging findings. Modalities include computed tomography and magnetic resonance imaging (MRI). On initial presentation, patients receive a computed tomography scan due to symptoms that could indicate stroke or other pathology within the brain. Once a bleed or mass in the brain is seen, a contrast enhanced MRI is performed.
In HGGs such as GBM, an infiltrative, heterogenous mass with edema and necrosis is usually seen. Imaging also focuses on areas of necrosis that may have some enhancement. Tumors such as GBM are vascular by nature and imaging is able to magnify this finding.
For hemispheric HGG, maximal safe surgical resection of the gross tumor is recommended. For tumors in midline or other eloquent locations, a biopsy may be obtained for tissue diagnosis. Depending on where the tumor is, there may not be enough tissue to justify the risk of obtaining a biopsy. This is especially true if HGG is located in the corpus callosum, the butterfly region of the brain that interconnects the two cerebral hemispheres. Imaging plays a critical role for surgical planning. Perfusion weighted imaging can characterize the tumor and aid in predicting the tumor grade and multimodal imaging allows the surgeon to determine the ideal target for a biopsy sample (21).
5 Molecular studies
Although aHGG and pHGG are histologically indistinguishable, the molecular biology of these tumors has shown significant differences in their genomic footprints. As underscored by the WHO CNS 5, IDH mutation status plays a key diagnostic and prognostic role in adult-type diffuse gliomas. IDH mutations confer an improved overall survival in adults with high grade gliomas when compared to those with IDH wild-type disease (22). However, with-in IDH mutated astrocytomas, the co-existence of CDK2A/B homozygous deletions are associated with poorer outcomes, and dictate an automatic grade 4 designation. For the IDH-wildtype tumors, several hallmark mutations correlated with poorer outcomes also automatically dictate a grade 4 tumor, regardless of the presence of characteristic histologic features such as microvascular proliferation or necrosis (8). These mutations include amplification of epidermal growth factor receptor (EGFR), TERT promotor mutations, and chromosome 7/10 copy number variants. EGFR encodes a tyrosine kinase receptor that is involved with cell proliferation, differentiation and cancer development (23). EGFR amplification is seen in up to 60% of adult glioblastomas while its occurrence in pediatric high grade tumors is less frequent (24). EGFR alterations represent an important therapeutic target for both small molecule inhibitors as well as immunotherapeutics. The PTEN gene is an important tumor suppressor gene that can inhibit cell invasion, prevent cell adherence to the matrix and formation of blood vessels. PTEN mutations are seen in a large proportion of aHGG patients and are rare to find in pHGGs (24, 25). TP53 is another important tumor suppressor gene, which is mutated in a subset of aHGG. The p53 pathway which also includes CDK2A, MDM2, in addition to TP53 is altered in the majority of adult GBM tumors.
In regards to pHGG, histone alterations play an important biologic and prognostic role. The majority of diffuse midline gliomas harbor a K27M mutation at histone 3.3 or histone 3.1. G34R or G34V mutations of H.3. are seen in hemispheric pHGG. Histone mutant tumors have a poorer OS than those that are wildtype. Given the prevalence of histone mutations in pediatric high grade gliomas, attempts to target these epigenetic pathways are underway (26). Like in aHGG, TP53 mutations are also common in pHGG, often found with co-existing histone mutations. These key molecular findings in aHGGs and pHGGs are summarized in Table 1.
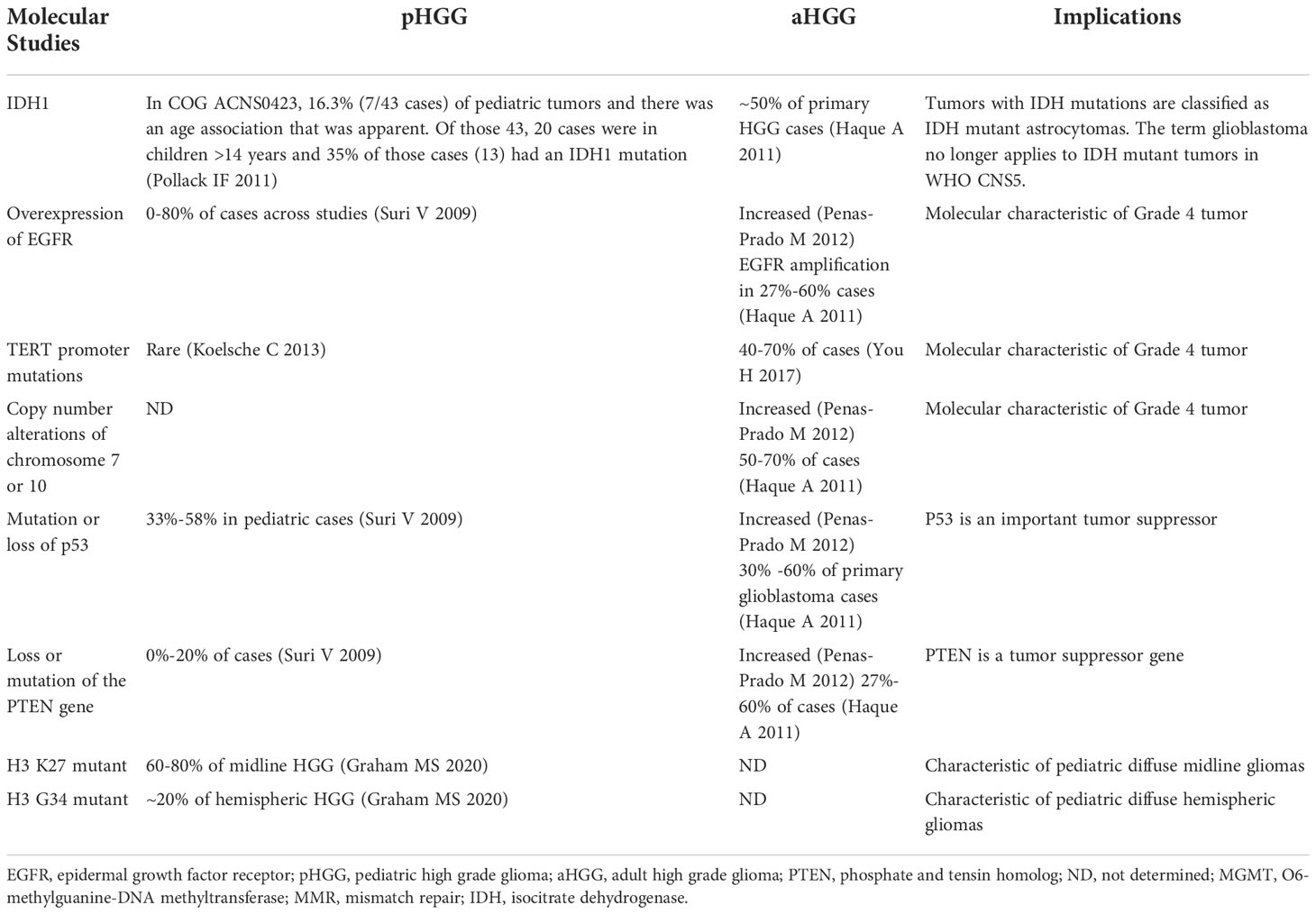
Table 1 Molecular profiling of pediatric and adult high grade glioma.
6 Treatment
6.1 Chemotherapy
Currently when patients are diagnosed with HGG, they are offered treatment with a goal to extend their lives as long-term survival from these tumors is exceedingly rare (16). Standard treatment offered at diagnosis for both the adult and pediatric populations diagnosed with HGG include surgical resection of maximal tissue with accompanying chemotherapy and radiation therapy. Temozolomide, an oral alkylating agent, is most commonly used for adjuvant chemotherapy. Stupp et al. showed that a HGG patients who received temozolomide concurrently with radiation had increased progression-free survival and overall survival when compared with those who did not (27). The two year survival in the cohort that received radiotherapy and temozolomide was 26.5%, with a median progression free survival of 6.9 months, compared to the group that only received radiotherapy whose two year survival was 10.4% with a median progression free survival of 5 months. However, apart from one study (28) most trials indicated that temozolomide chemotherapy had no effect on survival in children.
6.2 Molecular targeted therapy
Recently better understanding of the molecular biology of HGG have resulted in the development of a number of molecular targeted therapies with the hope of slowing the progression and ultimately developing a cure for HGG (29–31). These include therapies targeting tyrosine kinase receptors, tumor growth factor pathways, angiogenic pathways and intracellular signaling pathways.
EGFR and PDGFR (Platelet-Derived Growth Factor Receptor) amplification is commonly seen in aHGG (9). The kinase domain of EGFR and PDGFR and the downstream signaling can be selectively inhibited by tyrosine kinase inhibitors. However, Gefitinib and Erlotinib, oral EGFR inhibitors, and Imatinib, PDGFR inhibitor, have shown only limited benefit in clinical outcomes (9). The lack of expected success could be due to the redundancy of tumorigenic pathways in these tumors; blockade of a single pathway had limited effects.
Drugs targeting angiogenesis including bevacizumab, cedarinib, sunitinib and vatalanib have been investigated in patients with HGG as it is a highly vascularized tumor. However, pHGG patients were not as responsive to bevacizumab as the adult patients (32, 33). Combining irinotecan with bevacizumab (NCT00381797) has also not improved the prognosis in pediatric patients (34, 35).
Inhibiting the intracellular signaling pathways such as PI3K and mTOR as targeted therapy has also been evaluated in HGG. However, these have shown minimal efficacy in tumor control at tolerated doses (7). The redundant signaling pathways, drug toxicity, and blood brain barrier (BBB) are hurdles need to be overcome for further development of PI3K or PI3K/mTOR dual inhibitors in clinical trials (36).
6.3 Immunotherapy
The accelerated progress in the field of cancer immunotherapy has brought significant improvements of survival and quality of life for subsets of patients with cancer. In patients with the devastating HGG, harnessing the immune system to become more effective and specific with lower toxicity compared to current standard of care presents the hope of bringing a cure to the patients. However, the low mutational burden, the intra-tumoral heterogeneity, the BBB, and the immunosuppressive tumor microenvironment (TME), are all challenges for immunotherapy development in HGG. Nevertheless, a number of immunotherapeutic modalities are being tested in HGG preclinically and clinically (Table 2) and hold great promise in the future. For pediatric patients, immunotherapies have become viable therapeutic options, some are changing the course of the treatment for pHGG, while others need to be further evaluated.
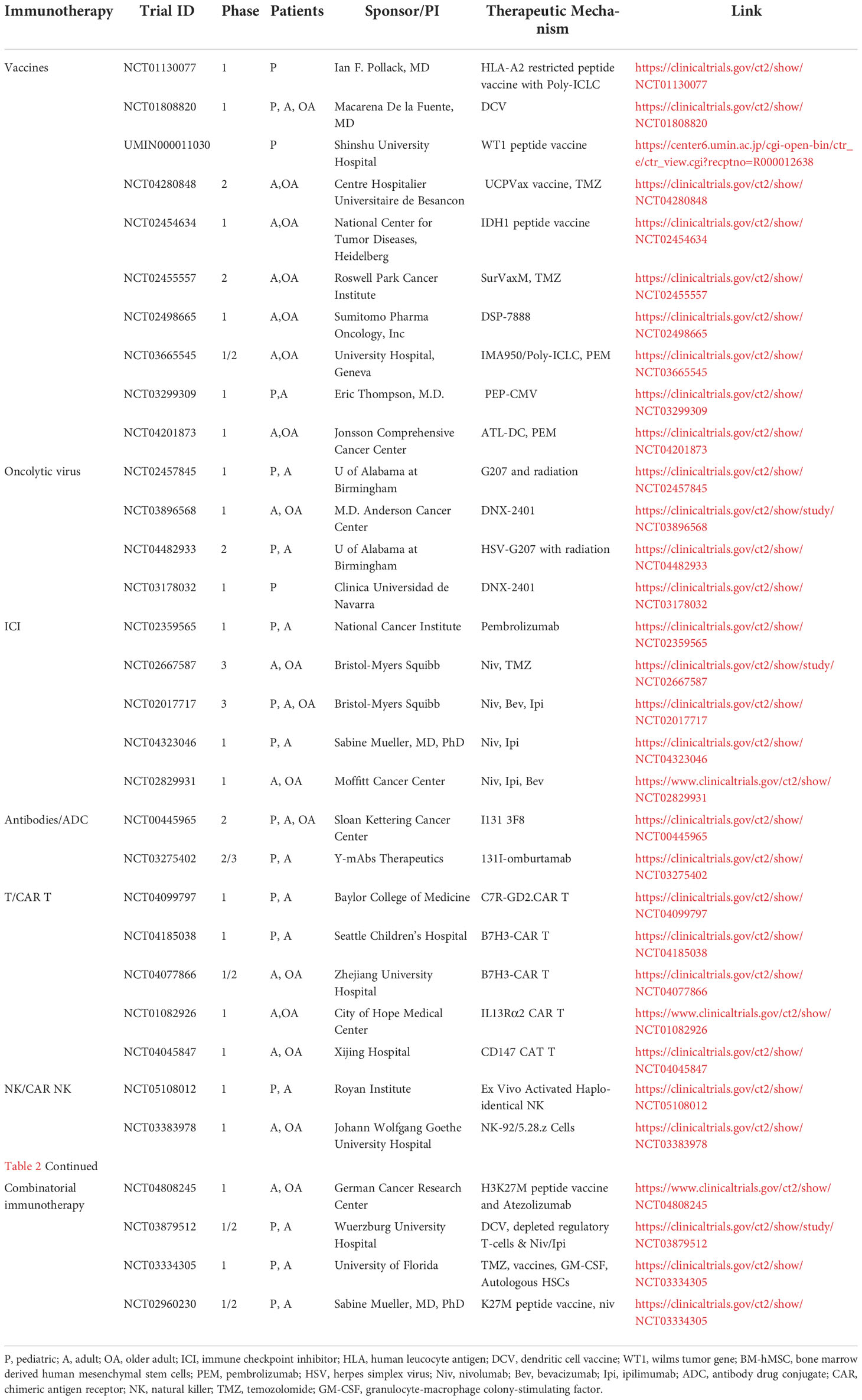
Table 2 List of current immunotherapy clinical trials for adult and pediatric high grade gliomas. .
6.3.1 Vaccines
One type of immunotherapy utilized in HGG is cancer vaccines (Figure 1), which rely on class I presentation of tumor associated antigens. HGG-associated antigens in a vaccination were administered to patients to stimulate an immune response. These antigens are usually small peptides that activate the cytotoxic T lymphocytes (CTLs). Rindopepimut is an injectable peptide which stimulates a response against a specific EGFRvIII antigen, which is expressed in HGG. Clinical trials of adult patients with HGG expressing EGFRvIII have shown positive results with rindopepimut (9, 37–39). However, a recent trial of rindopepimut/temozolomide in patients harboring the EGFRvIII mutation showed disappointing results (40). No significant difference was observed in overall survival in the rindopepimut cohort (20.1 months) compared to the placebo (20.0 months).Other tumor specific antigens for aHGG vaccines include TERT, IDH1, survivin and WT1. Clinical trials evaluating peptide vaccines targeting these antigens in adult GBM patients are ongoing (NCT04280848, NCT02454634, NCT02455557, NCT02498665, NCT03665545) (Table 2). Early results have shown that these vaccines are safe and immunogenic (41–44).
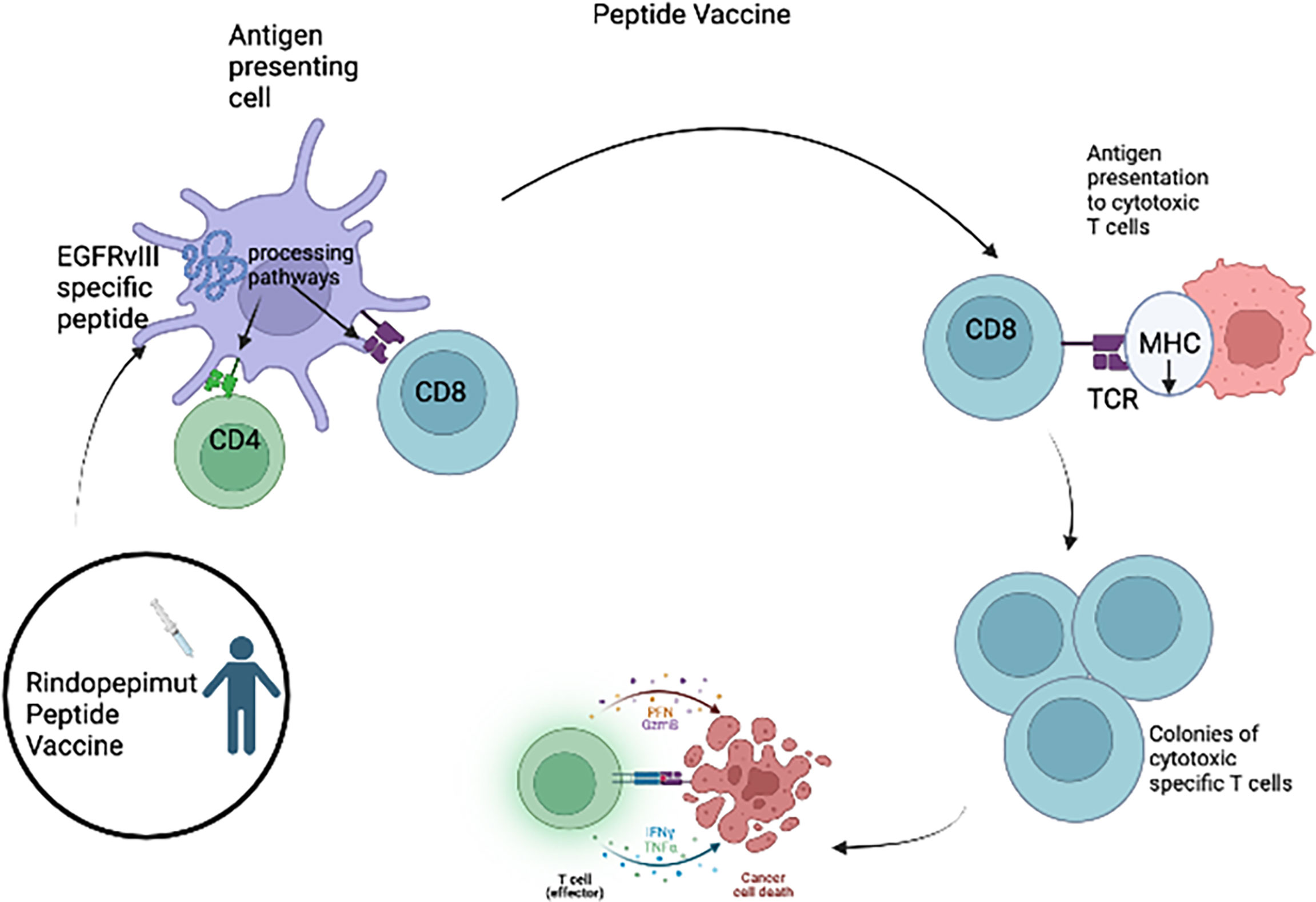
Figure 1 For the Rindopepimut vaccine, EGFRvIII specific peptide is introduced into the bloodstream and the peptide is processed and presented by the APC causing the activation of CD4 helper T cells and CD8+ cytotoxic T cells. The cytotoxic T-cells are then able to cause lysis of the glioma tumor cells.
Cytomegalovirus (CMV) infects most adults and CMV phosphoprotein 65 (pp65) expresses on greater than 50% of GBM but not on normal brain parenchyma (45). A phase I trial called PRiME (NCT03299309) (Table 2) is ongoing to test pp65 peptide vaccine in both adult and pediatric patients with malignant glioma.
In pediatric glioma patients, peptide vaccines targeting tumor-associated antigens including ephrin type-A receptor 2 (EphA2), interleukin-13 receptor alpha 2 (IL13Rα2), survivin and WT1, have been developed and tested (NCT01130077, UMIN000011030) (Table 2). The vaccination was generally well-tolerated in children and induced modest immune and clinical responses (46–50). An early phase vaccine trial utilizing the immunogenic peptide of the H3K27M mutation (51) in pediatric patients with midline gliomas harboring this mutation is also ongoing (NCT04808245) (Table 2).
Dendritic cell vaccines (DCV) (Figure 2) have been investigated in multiple trials in aHGG patients (52). Dendritic cells loaded with glioma antigens or whole tumor lysate are administered to patients for T cell activation and cytotoxic activity. Early phase clinical trials of DCs pulsed with single antigen including EGFRvIII, WT1, CD133 or IL13Rα2 in adult GBM patients demonstrated these DCVs to be immunogenic with no serious adverse events (38, 53, 54). However, ICT-107, a multi-peptide pulsed DCV which consists of autologous dendritic cells pulsed with six synthetic peptides: melanoma-associated antigen-1 (MAGE-1), antigen isolated from immunoselected melanoma-2 (AIM-2), Her2/neu, tyrosine-related protein-2 (TRP-2), glycoprotein 100 (gp100), and IL-13Rα2 (55), has shown no significant difference in overall survival in the treatment group as compared to controls in a randomized phase II trial in newly diagnosed adult GBM patients (55). Tumor lysate pulsed DCVs in aHGG patients have also shown mixed results. DC-VaxL is under phase III trial and exciting interim results have shown median overall survival of 23.1 months from surgery for the intent-to-treat population (56). However, another tumor lysate pulsed DCV (Audencel) evaluated in a randomized, controlled phase II study of patients with newly diagnosed GBM showed no significant difference in overall survival between the treatment and control groups (57). A phase I trial involving an autologous tumor lysate pulsed DCV (ATL-DC) and pembrolizumab in recurrent adult GBM is currently ongoing (NCT04201873).
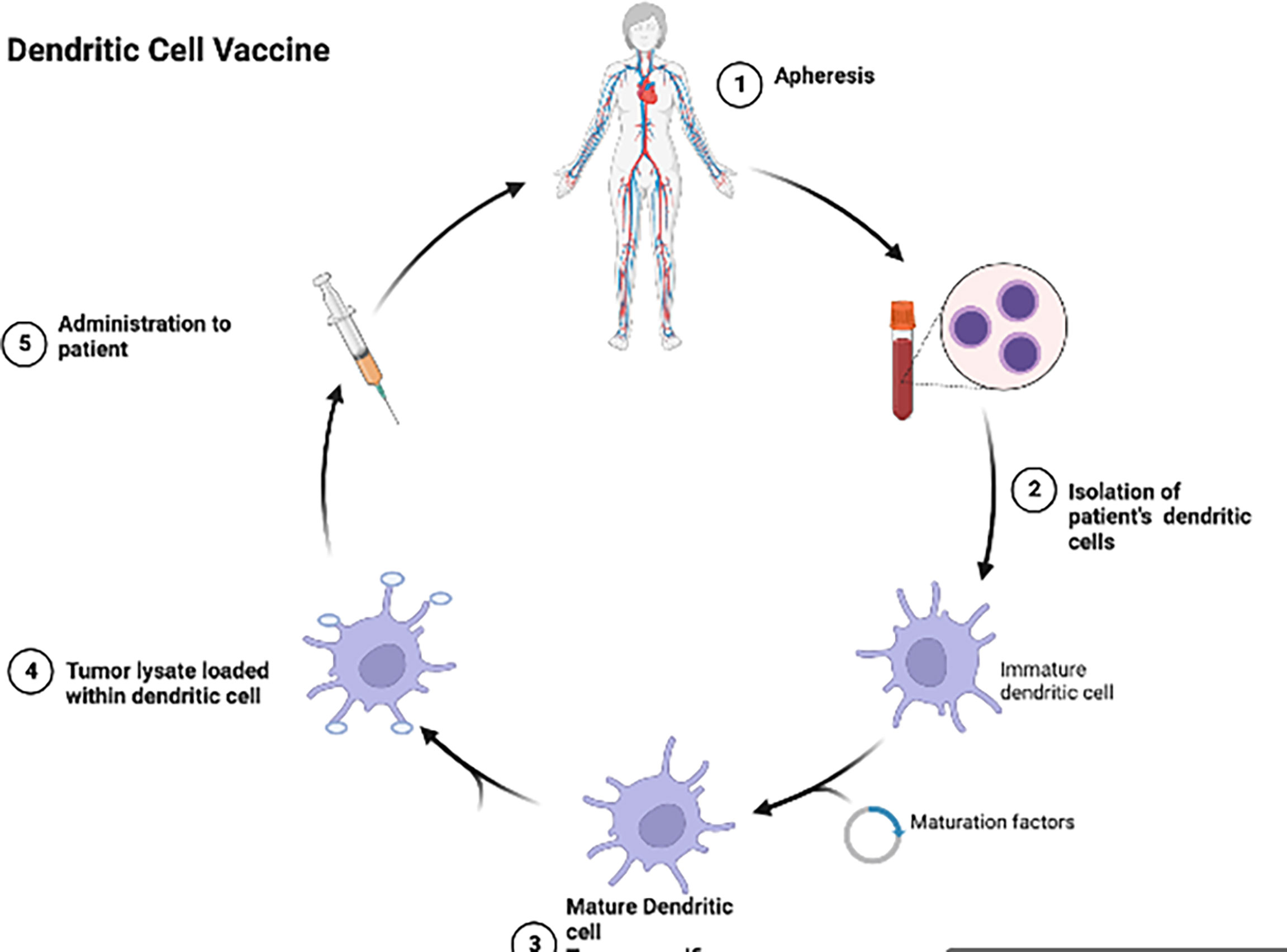
Figure 2 Dendritic cells are isolated from the patient’s blood, allowed to mature and then tumor lysate is loaded into the dendritic cells and given back to the patient as a vaccine.
DCVs have now been moved into the pediatric population. In the HGG-IMMUNO trial, De Vleeschouwer and colleagues evaluated tumor lysate DCV in children and adults with relapsed HGG (58). Improved progression-free survival was seen in the cohort receiving the tumor lysate DCV. It should be noted that younger age was correlated with improved overall survival, although it is unclear whether the difference in survival is caused by different underlying biology and natural course of the tumors in different age groups. A more recent report on DCVs with pulsed tumor lysate in a cohort of pediatric HGG patients with a small sample size also showed promising results (59). Three patients received DC vaccinations and of those two were alive 51 and 40 months from the time of enrollment into the study. The first patient was described to possibly have had a partial response on MRI after dendritic cell vaccination while the second patient was attending school at an age appropriate level and the third patient suffered from mild hemiparesis but was otherwise thriving and successful cognitively. Currently, a number of DCV trials are open and recruiting pHGG and aHGG patients (60) (Table 2).
Although dendritic cell vaccinations are currently in trials, one challenge with this therapeutic modality is HLA downregulation. HLA1 and HLA2 are the most common antigens utilized for cancer vaccines and downregulation or loss of HLA class 1 molecules can prevent tumor cells from being recognized. Zagzag et al. noted that downregulation of major histocompatibility complex (MHC) class I and II antigens allow for the migration and invasion of glioma cells. This downregulation allows for the tumor to go undetected by the immune system and can also explain, in part, the lack of inflammatory infiltrate surrounding glioma cells (61).
6.3.2 Oncolytic virus
Oncolytic viruses have shown promising efficacy in treating cancer (62, 63). They mediate antitumor effects by directly lysing cancer cells while sparing normal cells as well as stimulating immune response and modulating tumor microenvironment towards less immunosuppressive phenotype. Since the first oncolytic viruses -based immunotherapy (T-VEC) (64) has been approved by FDA for treating melanoma patients in 2015, oncolytic viral therapy for other tumor types have also developed to various extent (63). These agents have a particular attraction for brain tumors as they are generally injected intratumorally and thus best for localized disease.
Over the past three decades, various families of viruses, including herpes simplex virus type 1, adenovirus, Newcastle disease virus, reovirus, vaccinia virus and others, have been assessed in preclinical studies for their potential value in the treatment of HGG (65). In a case study in Germany, four aHGG patients were treated with three viruses: Newcastle disease virus, parvovirus, and vaccinia virus. This oncolytic virotherapy was well-tolerated by the patients and induced complete response or stable disease in these patients with overall survival ranging from 4 to 14 years. Importantly, the oncolytic virotherapy improved quality of life of the patients for years after the treatment (66). Multiple early-stage oncolytic viral therapy clinical trials in aHGG patients are currently active in the United States (67). In 2021, Delytact (teserpaturev/G47∆) was approved in Japan as the first oncolytic viral therapy for adult malignant glioma in the world based on the result of a phase II single arm trial showing a survival benefit and good safety profile of G47Δ in patients with residual or recurrent HGG after chemotherapy and radiation therapy (UMIN000015995) (68). In this trial, up to six doses of G47Δ were intratumorally administered in 19 adult patients. The survival rate one year after G47Δ initiation was 84.2%, the median overall survival was 20.2 months after G47Δ initiation and 28.8 months from the initial surgery (68).
Recently, Ring and colleagues reported that compared to aHGG xenografts, patient-derived pHGG xenografts were more sensitive to G207, a genetically engineered herpes simplex type 1 virus (HSV-1) (69). The favorable safety profile and efficacy of G207 in the adult clinical trials resulted in the design of a phase I clinical trial of oncolytic viral therapy with G207 in pediatric patients with HGG. The results of this trial showed acceptable adverse-event profile and evidence of radiographic, neuropathological or clinical responses (70).
Delta-24-RGD is an adenovirus engineered to be competent in replication in tumor cells with a defective RB signaling pathway. It was reported that Delta-24-RGD elicited an antitumor effect and prolonged animal survival in pediatric glioma and diffuse intrinsic pontine glioma (DIPG) mouse models (71). These data, and the proven safety and effectiveness of this virus in adult gliomas, have led to a phase I/II clinical trial of DNX-2401 oncolytic viral therapy in pediatric patients with newly diagnosed DIPG (NCT03178032). In this study, 12 patients between ages 3-12 were enrolled and they received a single infusion of DNX-2401 and subsequent radiotherapy was received by 11 patients. Over a median follow up of 18 months, a reduction in tumor size was assessed via MRI in nine patients; partial response was observed in three patients and stable disease in the other eight; median progression free survival and overall survival were 10.7 and 17.8 months, respectively (72).
6.3.3 Immune checkpoint blockade
Immune checkpoint blockade (ICB), antibody therapy directed against several negative immunologic regulators, cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) and the programmed cell death protein 1 pathway (PD-1/PD-L1), have shown significant success in various malignancies. ICBs such as Ipilimumab (anti-CTLA-4) and pembrolizumab (anti-PD-1), are approved by the FDA for the treatment of a wide spectrum of cancers (73). In aHGG, phase I of the CheckMate 143 trial demonstrated safety and tolerability of nivolumab (anti-PD-1) and ipilumumab (74). However, no single agent therapy demonstrated a survival benefit, including bevacizumab and nivolumab, in patients with recurrent HGG in the phase III trial (75). A number of clinical trials in adults are ongoing to investigate other combinations of ICBs alone and in combination with radiotherapy (Table 2).
Utilization of biomarkers to stratify and identify potentially responsive patients is an important consideration in ICB immunotherapy. PD-L1 expression has been reported to correlate with response to ICBs in adult cancer. Higher level of tumor mutational burden is also associated with an increased expression of neoantigens and enhanced efficacy of ICBs. Interestingly, compared to adult counterparts, pediatric gliomas have been found to have higher levels of microsatellite instability (MSI), a surrogate biomarker for tumor mutational burden (76). Boefett et al. treated two siblings with relapsed HGG with MSI using nivolumab and observed clinically significant responses and a profound radiologic response (77). However, the success of nivolumab was not repeated in another pediatric relapsed HGG case with MSI (78). Several large studies are ongoing to evaluate the efficacy and safety of ICBs in pHGG in a more rigorous manner (Table 2). As with vaccines, ICB rely on expression of tumor associated antigens, which can escape detection with the down regulation of HLA class I expression.
6.3.4 Antibody and antibody drug conjugates (ADC)
Recent progress and efficacy of antibody-based immunotherapy (Figure 3) have promoted the searching for clinically relevant tumor antigens as immunotherapy targets in solid tumors. Gangliosides are sphingolipids that contain carbohydrates and are widely expressed in normal tissues, which renders most of them ineffective for cancer therapy. However, the disialoganglioside GD2 subtype is overexpressed in many tumors with minimal expression in normal tissues (79). Over the past few decades, several anti-GD2 monoclonal antibodies (mAbs) have been developed (80–83). Anti-GD2 mAbs target GD2 expressing tumor cells and recruit Fc-receptor-expressing innate immune cells such as natural killer (NK) cells and macrophages to mediate antibody dependent cell mediated cytotoxicity (ADCC) and/or phagocytosis (12). Indeed, anti-GD2 mAbs significantly increased survival rates and transformed the landscape for pediatric patients with primary and relapsed neuroblastoma (84–87). It is also well known that GD2 is expressed on HGG cells. A chimeric anti-GD2 antibody dinutuximab beta induces ADCC and has anti-tumor efficacy against HGG cells. This study provided rationale for GD2 directed immunotherapy against HGG (88). A phase II study of intrathecal I-3F8 anti-GD2 mAb in patients with GD2 expressing CNS neoplasms in all age groups is ongoing (NCT00445965).
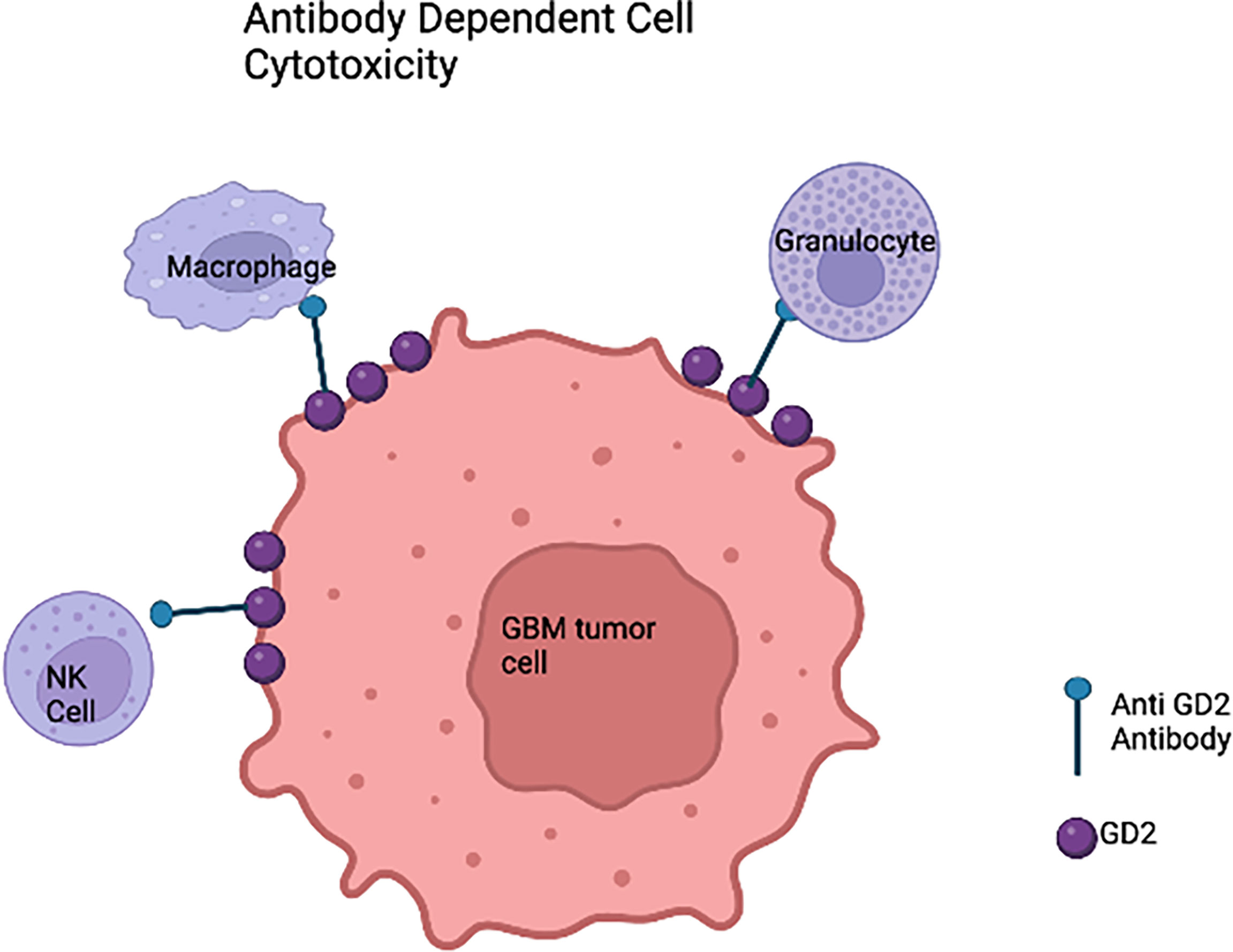
Figure 3 Anti-GD2 immunotherapy of glioblastoma cells based on the innate immune system. In the presence of anti-GD2 monoclonal antibodies,GBM cells would be susceptible to the activity of natural killer (NK) cells that would mediate antibody-dependent cell mediated cytotoxicity (ADCC), granulocyte mediated ADCC, and monocyte macrophage ADCC.
B7-H3, also called CD276 or B7RP-2, belongs to the B7 ligand family. B7-H3 is highly expressed on differentiated tumor cells, metastasis-initiating cells, tumor-associated vasculature and stroma, and has a limited distribution in normal tissues, all which features a favorable target for antibody-based immunotherapy (89). B7-H3 blocking mAbs have demonstrated efficacy in increasing tumor infiltration of CD8+ T cell and NK cell, limiting tumor growth, and/or improving animal survival in mouse models of various types of cancer including melanoma, colorectal cancer and ovarian cancer (90–92). In addition, B7-H3 mAbs have been used as vehicles to selectively deliver radioisotopes to B7-H3+ tumors in clinical trials (NCT03275402, NCT00445965). Convection-enhanced brainstem delivery of 124I-8H9 to DIPG tumors in patients of 2-21 years old has demonstrated to be safe with minimal systemic exposure of radioisotopes and no toxicity (NCT01502917) (93).
Another antibody based immunotherapy with high potential is antibody drug conjugates (ADC), which are composed of a mAb which specifically recognizes a surface protein and a cytotoxic payload (94). Unlike systemically administrated immunotherapy, ADCs directly and specifically deliver the cytotoxic payload to the tumor. This significantly decreases the toxicity of ADC. Furthermore, the additive/synergistic effect of the combination of the antibody and the payload significantly enhances tumor cell killing (95). Phase I and III clinical trials using EGFRvIII ADCs AMG 595 (NCT01475006) and ABT 414 (NCT02573324, NCT02343406), respectively, have shown favorable pharmacokinetics with possible benefit for adult patients with EGFRvIII-mutated HGG (96–98).
6.3.5 Cytotoxic T lymphocytes and chimeric antigen receptor (CAR) T cell therapy
Adoptive cell transfer is also being studied as a novel therapeutic strategy in HGG. Autologous immune cells, mainly CTLs are isolated from peripheral blood mononuclear cells and activated ex vivo with autologous tumor cells (9).
With respect to specificity and independence of MHC molecules, CAR represents a promising approach (Figure 4). A single chain variable fragment of an antibody is connected to the CD3 T cell receptor signaling domain and this helps target the T cell cytolytic activity. The target antigen can be engaged without presentation by the MHC (99). The FDA had originally approved CAR T cells for hematologic malignancies but many studies have shown potential efficacy of CAR T cells for brain tumors such as HGG, medulloblastoma, and ependymomas (11, 100).
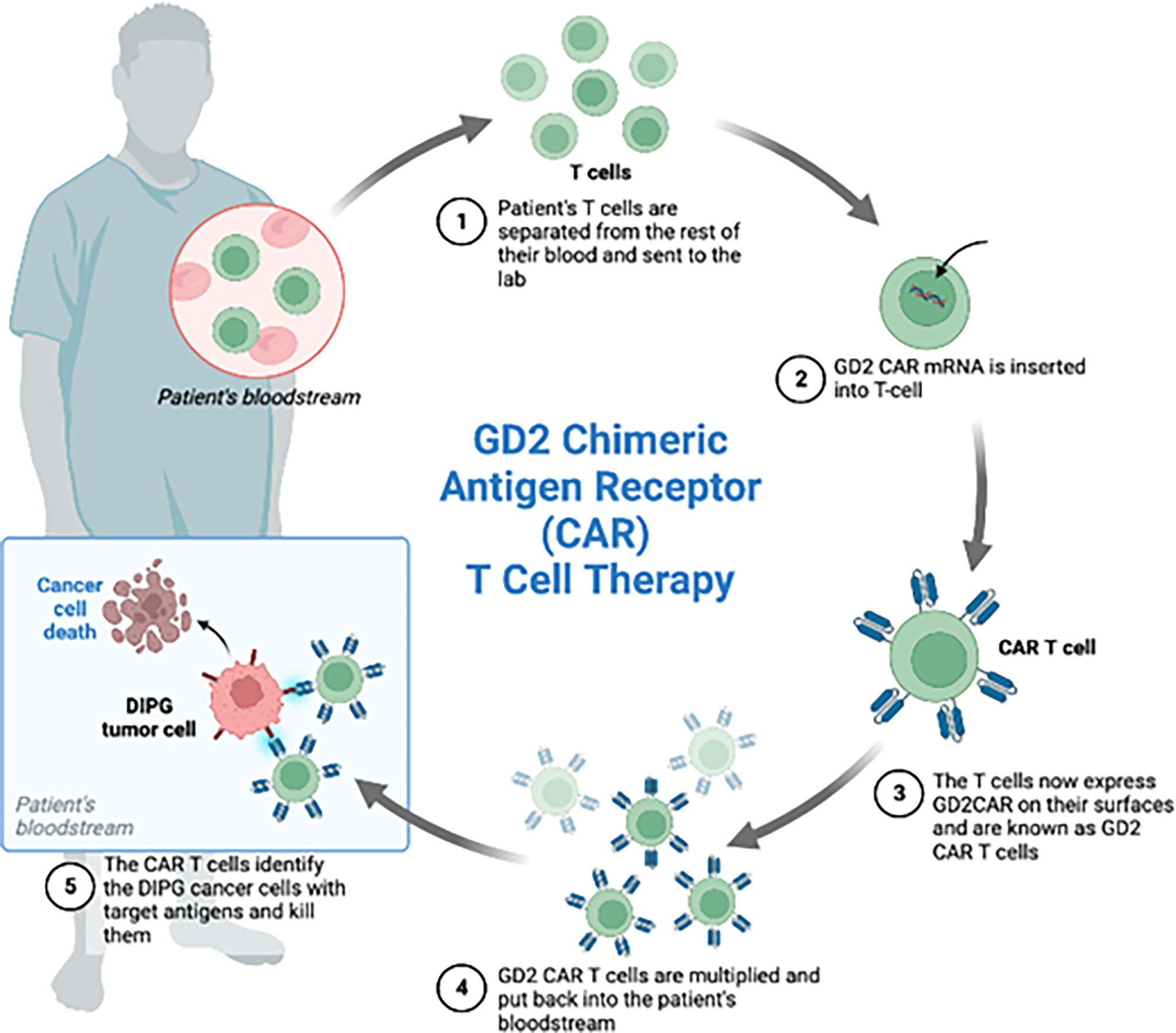
Figure 4 Four patients with the diagnosis of diffuse intrinsic pontine glioma were enrolled in the phase 1 clinical trial of GD2-CAR T cells. Each patient received GD2 CAR T cells. These CAR T cells were generated by isolating the patient’s T-cells from the rest of the blood, inserting GD2 CAR mRNA and administering the GD2 CAR T cells back to the patients. Expansion of CAR T cells, increased inflammation after treatment and increased CAR T cells in the CSF during peak inflammation was observed.
In a pilot first-in-human study in adult patients with recurrent GBM, intracranial infusions of IL13Rα2 CAR T cells have shown excellent efficacy and no severe side effects (NCT01082926) (101). EGFRvIII targeting CAR T cells demonstrated effective control of xenograft tumor growth in a GBM preclinical mouse model (102). However, in a pilot trial in adult patients with GBM, adoptive transfer of EGFRvIII-CAR T cells did not demonstrate clinically meaningful effects (103). The limited efficacy of EGFRvIII CAR T cell therapy is in part due to the heterogeneous expression of EGFRvIII in glioma cells and the immunosuppressive TME in GBM (104, 105). CD147 is a transmembrane glycoprotein that belongs to the immunoglobulin superfamily. CD147 is overexpressed in common tumors including gliomas (106) and is associated with poor prognosis in GBM patients (107, 108). In adult patients with recurrent GBM, an early phase clinical trial of CD147-CAR T cells is currently in progress (NCT04045847).
A recent publication demonstrated the potential efficacies of CAR-T cells targeting pediatric diffuse midline gliomas (DMG). GD2 has been investigated in clinical and preclinical trials for neuroblastoma and GD2-CAR T cell therapies have been well tolerated in neuroblastoma (99). High GD2 expression level is correlated with the H3K27M mutation. In the patient derived H3K27M+ DMG orthotopic xenograft models, almost complete clearance of the tumor and improved animal survival were observed after the peripheral administration of GD2 specific CAR T cells (109). Mount et al. studied GD2 directed killing while using patient derived H3K27M-mutant glioma cell cultures. The group generated GD2-targeting CAR T cells and found significant GD2 dependent killing and cytokine release upon exposing to the CAR T cells targeting GD2 but not those targeting CD19 (110). Majzner and Ramakrishna et al. conducted the first Phase I dose escalation trial of GD2 CAR T cells in four children diagnosed with H3K27M+DMG. They found an increased concentration of CAR T cells in the tumor and clinical as well as radiological improvement in patients. Tumor inflammation associated neurotoxicity was the biggest concern in the trial as the inflammation of the brainstem transiently worsened with CAR T cell infusions in each of the patients (11).
B7-H3-CAR T cells with various B7-H3 antibody single chain variable fragments have been created and demonstrated potent in vitro cytotoxicity against HGG and effectively controlled tumor growth and prolonged animal survival in vivo (111, 112). Currently, B7-H3 CAR T cells are under evaluation in clinical trials in adult (NCT04077866) and pediatric patients with HGG (NCT04185038). A phase I trial of C7R-GD2 CAR T cells, which is engineered to express an interleukin-7 receptor (C7R) for constitutive activation of CAR T cells, is ongoing in both adult and pediatric patients with GD2-expressing HGG (NCT04099797).
6.3.6 NK and CAR NK cell therapy
NK cells are as important in initiating cytotoxic activity against malignant tumor cells as CTLs. Unlike T cells, NK cells require no prior sensitization for tumor immunity. Their function and activity is regulated by various NK receptors. One class includes killer inhibitory receptors (KIRs) while the other receptors allow for the activation of NK cells. KIRs are recognized and bound by MHC class I molecules (MHC-I) on tumor cells which render the NK cells ineffective as they become inactivated. NK cells have also been shown to interact with DCs and play an integral role in the immune response along with DCs and CTLs. Administration of in-vitro-activated NK cells as adoptive cellular immunotherapy has shown efficacy in treating malignant tumors (25).
In glioma, very few NK cells infiltrate into the tumor. According to Kmiecik et al. (113), NK cells made up ~2% of all infiltrating immune cells and most of these NKs were CD56dimCD16neg subtype. This phenotype showed activation in other tumors, suggesting although the abundance of NK cells in the glioma microenvironment is low, they have cytotoxic potential. Analysis of the HGG cell surface molecules showed the presence of high levels of MHC-I that interact with the NK inhibitory receptors. Blocking the interaction between the MHC molecules on HGG cells and KIRs on NK cells may aid in bringing more NK cells into the tumor microenvironment (TME) (114). Induction of NKG2D ligand expression by HDACi is another approach to overcoming immunosuppression to NK cells (115) as previously reported by our group and others (116).
Ishikawa et al. achieved decreased tumor volume without neurogenic toxicity in adult malignant brain tumor patients using autologous NK cell therapy. In 56% of the cases they studied, tumor size was reduced. In 19% of the cases, there was a significant reduction of the tumor size compared to what it was during the pretreatment phase (25).
Recent development of CAR NK cell therapy as a novel immunotherapeutic modality has been successful and addressed issues inherent to CAR T cell therapy such as side effects including cytokine release syndrome (CRS) and encephalopathies (ICANS). In Germany, a clinical trial is ongoing with HER2-CAR-NK-92 cells in adult patients suffering from recurrent HER2+ HGG (CAR2BRAIN, NCT03383978) (115). The recently completed dose-escalation study has shown no dose-limiting toxicities at neither of the three applied dose levels. Currently, patients are being recruited for the expansion cohort of the trial.
6.4 Combinatorial therapy
The poor prognosis of HGG is in part attributed to the highly heterogenous, metastatic and angiogenic nature of the tumor, leading to the tumor resistance to therapy. Combinatorial therapy approaches are being developed and tested in preclinical and clinical settings against HGG to overcome therapy resistance.
Temozolomide is the standard chemotherapy for HGG and has been tested extensively in combination with various regimens including cilengitide, bevacizumab, paclitaxel, irinotecan and tarceva preclinically (117–124) and in clinical trials in adult patients (18 years and older) (NCT00686725, NCT00689221, NCT00943826, NCT00262730, NCT00544817, NCT01402063, NCT00805961, NCT00979017, NCT00525525).
In a study on phosphor-tyrosine-mediated signaling pathways activated by EGFRvIII, the c-Met receptor tyrosine kinase was found to be EGFRvIII-cross-activated. This provided a rationale of a combinatorial therapy with an inhibitor of c-Met and an inhibitor of EGFR. The combination had an additive/synergistic anti-tumor effect demonstrated by enhanced cytotoxicity against EGFRvIII+ HGG cells compared to either compound alone, suggesting potential clinical value of the combination of c-Met and EGFR inhibitors in both adult and pediatric EGFRvIII+ HGG patients (125). In aHGG, a study conducted by Montano et al. identified 44% (32 of 73) of aHGG cases as EGFRvIII positive (126). In pHGG, Bax and colleagues found that only 17% (6 of 35) of pHGG cases they studied harbored EGFRvIII mutations (127). However, Li et al. found 44% (4 of 9) of pediatric DIPG cases to be EGFRvIII positive (128). More studies with larger sample size in both aHGG and pHGG are needed to determine if there is a significant difference in incidence of EGFRvIII in these age groups to contribute to a significant difference in response to the combination of c-Met kinase and EGFR inhibitors.
The combination of DC vaccines and ICB has shown effectiveness in the treatment of melanoma patients (129). Human cytomegalovirus (CMV) proteins and nucleic acids were found in the majority of aHGG. In a cohort of 25 pHGGs, approximately 67% of the samples express CMV antigen pp65 or IE1-72 as detected by immunohistochemical staining and in situ hybridization (130). With the hypothesis that nivolumab-induced ICB could increase effectiveness of DC vaccines in HGG, an early phase, single-center, randomized clinical trial to assess the safety of nivolumab combined with CMV pp65 mRNA pulsed DC vaccine in adult patients with recurrent resectable HGG was completed (NCT02529072). Safety of the combination in recurrent HGG patients is at similar level to that of nivolumab alone. However, due to lack of efficacy in improving overall survival in recurrent HGG patients shown in the CheckMate 143 trial, this study was terminated early. Continued evaluation of combination of new IBCs with DC or peptide vaccination in aHGG and pHGG is underway (Table 2) (NCT04808245, NCT03879512, NCT03334305, NCT02960230).
Myeloid-derived suppressor cells (MDSCs) are a very heterogeneous population of bone marrow derived immature myeloid cells. Under normal conditions, these immature myeloid cells have the potential to differentiate into multiple types of cells including macrophages, granulocytes, and DCs. In pathological states of HGG, the subverted differentiation of immature myeloid cells leads to the generation, expansion, activation and recruitment of MDSCs in the tumor bed as well as the peripheral blood (131, 132). MDSCs are present at very low frequencies in healthy human body. In adult patients with HGG, MDSCs constitute greater than 40% of the tumor-infiltrating immune cells and confer immunosuppression by expressing IL-4Rα, inducible nitric oxide synthase (iNOS), arginase, PD-L1, and CD80, and suppressing antigen-specific T cells (133, 134). MDSC depletion or checkpoint blockade was found to enhance the efficacy of HSV-1 thymidine kinase (HSV-1TK) and Fms-like tyrosine kinase ligand (Flt3L) gene therapy in aHGG (134). Only few studies investigated the role of MDSCs specifically in pHGGs. It was reported that high levels of circulating MDSCs correlate with poor prognosis in DIPG patients, indicating a role of MDSCs in immunosuppression and tumor immune evasion mechanism in pHGG (135) and a potential value of combinatorial immunotherapy targeting MDSCs in pHGG.
HGG resistance to NK cell therapy is in large part due to the small number of activated NK cells, poor NK in vivo persistence, and lack of specific tumor targeting (136). To overcome HGG NK resistance, our group combined approaches to increasing NK cell number and activity (ex vivo NK expansion), improving persistence and trafficking (IL-15 superagonist N-803), and enhancing tumor targeting (anti-GD2 mAb dinutuximab). N-803 combined with dinutuximab and ex vivo expanded NK cells significantly prolonged animal survival of GD2+ HGG xenografted NSG mice, providing the rationale for clinical investigation of this combinatorial NK cell therapy in patients with GD2+ HGG (137) as reported by our group.
HGG resistance to NK therapy is also due to an unfavorable and hypoxic TME that suppresses NK cell effector function (138). CD73 is a hypoxic ectoenzyme that drives the accumulation of adenosine, leading to significant impairment of NK cell activity (139). Wang et al. engineered anti-GD2-NKG2D bispecific CAR NK cells to simultaneously harbor a functional domain to inhibit the activity of CD73 independently of CAR to enhance NK cell activity. Compared to mock NK cells, these multifunctional engineered NK cells more potently targeted patient-derived HGG cells and tumors in vitro and in vivo (140). Since GD2 and CD73 are widely expressed on various lines of HGG cells, both from pediatric and adult patients (140), the above mentioned two combinatorial CAR NK cell therapy have potential generalizable clinical value in both pHGG and aHGG.
Oncolytic viruses can be armed with immunostimulatory molecules to enhance their immune-activating characteristics and are highly synergistic when combined with other immunotherapies such as NK cell therapy. In an immunocompetent model of high grade glioma, a virus expressing IL15/IL15Rα combined with off-the-shelf anti-EGFR CAR NK cells have shown synergistic effects on reducing tumor growth and extending animal survival in comparison to single agents by increasing NK and CD8+ T cells intracranial infiltration and activation and improving CAR NK cell persistence (141).
6.5 Barriers to treatment
Although immunotherapy is an attractive therapeutic strategy for HGG, one of the challenges that need to be overcome is the blood-brain barrier (BBB) (142, 143). BBB is the body’s defense of protecting the brain and the CNS system from toxins, inflammation and infectious sources present in the blood. While it prevents pathogens from getting into the CNS system, it also prevents the passage of therapeutic agents into the brain. Although once considered to be an immuno-privileged site, the CNS is no longer considered to be so with regards to the passage of immune cells (142–144). It is now understood that immune cell trafficking across the BBB is closely regulated (145). The immune system keeps surveillance via lymphatics to the deep cervical lymph nodes but immune cells rarely cross the BBB (145). However, when there are excessive inflammatory signals, a plethora of cells including neutrophils, B and T cells and monocytes can enter the CNS to strive to maintain the brain’s homeostasis and avoid damages from the inflammation (145). The macrophages and microglia make up the CNS resident immune system and respond to even minor changes in the CNS homeostasis becoming reactive and causing inflammation (146). There are also immunosuppressive cytokines such as TGFβ or IL-10 that get secreted and encourage further tumor growth making treatment complicated (11).
MRI in adult GBM not only shows the accumulation of normally brain-impermeable material in all GBM, providing evidence of BBB disruption, but also demonstrates that most GBMs have gross tumor burden protected by an intact BBB, suggesting the BBB is still a key barrier to effective treatment (147, 148).
In the case of pediatric tumors, different subgroups affect the BBB differently (149–151). It has been noted that DMGs display little to no contrast enhancement on MRI compared to other pHGGs, suggesting a more intact BBB (149, 150). This is probably due to other pHGGs induce abnormal vessel morphology and BBB disruption while DMGs maintain a vasculature similar to that of normal brainstem (151).
While the BBB poses a challenge, so does the immunosuppressive TME (152, 153). Berghoff and colleagues found that the status of IDH mutation was associated with the immunosuppressive TME of diffuse glioma; IDH mutant diffuse gliomas including GBM had a lower rate of T cell infiltration than the IDH-wildtype cases (154). In addition, the glioma TME is characterized by hypoxic areas and low nutrients which causes autophagy and response to stress that hamper T cell function (155, 156). Indoleamine 2,3 dioxygenase (IDO) and arginase (Arg1) highly expressed by tumor cells and myeloid cells within the TME can cause suppression of T cell functions (157, 158). Tumor cells and macrophages in hypoxic conditions also release immunosuppressive factors such as hypoxia‐inducible factor‐1 alpha (HIF‐1α) and prostaglandin E2 (PGE2) and inhibit T cell proliferation and function (159, 160). Targeting these hypoxia and metabolic pathways may alter the immunosuppressive TME and enhance T or CAR T cell therapy (161).
Tumor-associated macrophages/microglia (TAMs) and MDSCs are important components and main proportion of infiltrating immune cells in both adult and pediatric HGG TME (162, 163). Together with the immune-suppressive cytokines and chemokines, these cells prevent the innate and adaptive immune system from recognizing and eradicating the tumor cells (162, 163). High levels of MDSCs are correlated with poor prognosis in both adult and pediatric patients with HGG (135, 164). Interestingly, a negative correlation was reported between the abundance of TAMs and the overall survival in adult but not pediatric HGG patients (165). Furthermore, TAMs in aHGG and pHGG TME seem to present different phenotypes. Lin et al. recently found that although DIPG tumor cells produce CSF1, which is associated with the M2 phenotype of macrophages, macrophages in DIPG TME do not appear to have the characteristics of M2 macrophages as in aHGG (164, 166). Infiltration of cytotoxic effector immune cells including CD8+ T cells and NK cells has been demonstrated in the TME of aHGG; however, the activity of these infiltrating effector cells is suppressed by mechanisms mediated by TGF-β, LDH5, and galectin-1 among others (163, 167). In contrast, scarce T cell infiltration was demonstrated in pHGG TME (153). The tumors harbor histone mutants K27M and G34R were considerably “cold”, based on the observation of lack of CD8+ T cells in immunohistochemical staining (168). Jha et al. investigated the frequency of T cells in both adult and pediatric DMG TME. It was found that while CD3+ T cell frequency did not exhibit much difference, CD8+ T cells showed a significantly greater frequency in adult compared to pediatric DMG TME (169). The same group also evaluated PD-L1 expression in 126 DMG samples from both adult and pediatric patients and found a significant correlation between PD-L1 expression and number of tumor-infiltrating lymphocytes in both age groups. Patients with low PD-L1 expressing tumors had a longer overall survival compared to those with high PD-L1 expressing tumors in both groups (169). All these findings suggest that pHGG and aHGG TMEs are similar, but each has their own unique features.
A challenge of immunotherapy for HGG is tumor antigen heterogeneity both between patients and within individual tumors. Barish et al. investigated the expression of three clinically relevant tumor antigens, IL13Rα2, HER2, and EGFR, using IHC combined with digital imaging and pixel-level quantitative measurements of DAB (3,3′-Diaminobenzidine) visualized immunoreactivity, on tumor samples from 43 aHGG patients who had not been treated with immunotherapy (170). It was observed that expression of these antigens was highly variable and non-homogeneous across all 43 patient samples. Within individual tumor samples, expression of these antigens was not uniform, but rather mapped into local neighborhoods with high or low antigen expression (170). In pHGG patient-derived orthotopic xenograft samples, the expression of HER2, IL-13Rα2, EphA2, B7-H3, and GD2 was evaluated and demonstrated to be heterogenous by flow cytometry (171). In a small-scale study, GD2 expression was investigated by IHC and showed a varying intensity and staining pattern (cytoplasmic/nuclear, focal/diffuse) in tissue samples from 9 pHGG patients (172). The inter-patient tumor antigen heterogeneity hinders the development of a universal single immunotherapy for HGG while the intra-patient heterogeneity results in the survival of tumor antigen-deficient clones in patients treated with single immunotherapy. Immunotherapies targeting multiple antigens simultaneously are explored to overcome tumor antigen heterogeneity in HGG (173, 174).
With the clinical application of cancer immunotherapy comes the increasing awareness of one inherent limitation of immunotherapy, which is the potentially fatal toxicities including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and on-target off-tumor toxicity, among others. CRS is due to high level immune activation by natural and bispecific antibody or CAR T cell which results in massive release of cytokines including IL-6 and IFN-γ, leading to life-threatening side effects (175, 176). ICANS is a neurologic complication caused by CAR T cells. Patients with severe ICANS demonstrate evidence of endothelial activation and BBB disruption (177, 178). On-target off-tumor toxicity results from antigen-specific attack by the immunotherapy on host tissues when the targeted tumor antigen is also expressed on normal tissues. This toxicity can occur after immune checkpoint blockade treatment or adoptive T cell therapy with genetically engineered T cells (179, 180). These toxicities need to be managed by carefully designing the immunotherapy agents and the clinical trials in both adult and pediatric patients (181, 182).
7 Conclusion
pHGG is characterized by a specific set of genetic and epigenetic aberrations different from those found in aHGG and therefore is a distinctly different biological disease compared to aHGG (4)(Table 1). Molecular targeted therapies that are based on genetic and epigenetic alterations in aHGG are not as effective against pHGG (32–35). A better understanding of the signaling pathways and molecular modulators of pHGG is essential to allow newer specific targeted molecular and other treatment strategies to emerge for pHGG. However, pHGG is a rare disease which poses a challenge to obtain clinical samples for extensive investigation for disease mechanism. However, efforts are underway through research consortiums such as the Children’s Oncology Group and the Childhood Brain Tumor Tissue Network to obtain samples and extensively profile them in order to accelerate research and discovery in this domain. Immunotherapy is a rapidly advancing and promising therapeutic option for HGG. Although pHGG and aHGG are distinctive in terms of molecular biology background, they do harbor some of the same tumor targets such as GD2 (183) and EGFRvIII (128) and have somewhat similar TME with abundant immunosuppressive MDSCs and TAMs and lack of active immune effector cells (135, 162–164, 169), suggesting that certain immunotherapies for aHGG could be extended to pHGG. However, distinctions between aHGG and pHGG as well as the fundamental differences between adult and children need to be recognized when applying adult immunotherapy regimens or developing specific new immunotherapy in pHGG (2, 184, 185). Significant challenges remain, these include delivery of immunotherapy reagents to penetrate BBB, overcoming tumor heterogeneity, managing immunotherapy associated toxicities and circumventing immunosuppressive TME. Carefully designed combinatorial therapy is likely to be the key to addressing these challenges. With rapid scientific advances being made and a number of ongoing and soon-to-follow clinical trials, more effective therapies are promised to change the future course and improve the outcome of the adults and children with these devastating tumors.
Author contributions
PA, WL, KP, HH, PR, TC, KC, DL, and MC wrote, reviewed and revised the manuscript. All authors approved the final manuscript for submission.
Funding
This work was supported in part by grant U54 (CA232561-01/A1) (MC, TC, DL, KC), Pediatric Cancer Research Foundation (MC) and Children’s Cancer Fund (MC).
Conflict of interest
MC serves as a consultant for Jazz, Omeros, Astra Zeneca, Nektar Therapeutics and Novartis and a member of the Speakers Bureau for Jazz, Servier, Amgen, Sanofi and Sobi. DL reports personal fees and other from Kiadis Pharma, CytoSen Therapeutics, Courier Therapeutics, and Caribou Biosciences outside the submitted work; In addition, DL has a patent broadly related to NK cell therapy of cancer with royalties paid to Kiadis Pharma. TC recently served as a one-time consultant to Blueprint, Incyte, Oncopeptides, serves as a DSMB chair for SpringWorks and is a cofounder of Vironexis Biotherapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell (2016) 164(3):550–63. doi: 10.1016/j.cell.2015.12.028
2. Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer (2014) 14(2):92–107. doi: 10.1038/nrc3655
3. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell (2012) 22(4):425–37. doi: 10.1016/j.ccr.2012.08.024
4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
5. Yang Z, Ling F, Ruan S, Hu J, Tang M, Sun X, et al. Clinical and prognostic implications of 1p/19q, IDH, BRAF, MGMT promoter, and TERT promoter alterations, and expression of ki-67 and p53 in human gliomas. Cancer Manag Res (2021) 13:8755–65. doi: 10.2147/CMAR.S336213
6. Fernandes C, Costa A, Osorio L, Lago RC, Linhares P, Carvalho B, et al. Current standards of care in glioblastoma therapy. In: De Vleeschouwer S, editor. Glioblastoma. (Brisbane (AU): Codon Publications) (2017).
7. Nabors LB, Portnow J, Ahluwalia M, Baehring J, Brem H, Brem S, et al. Central nervous system cancers, version 3.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2020) 18(11):1537–70. doi: 10.6004/jnccn.2020.0052
8. Wen PY, Packer RJ. The 2021 WHO classification of tumors of the central nervous system: clinical implications. Neuro Oncol (2021) 23(8):1215–7. doi: 10.1093/neuonc/noab120
9. Wilson TA, Karajannis MA, Harter DH. Glioblastoma multiforme: State of the art and future therapeutics. Surg Neurol Int (2014) 5:64. doi: 10.4103/2152-7806.132138
10. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
11. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature (2022) 603(7903):934–41. doi: 10.1038/s41586-022-04489-4
12. Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol (2016) 134:381–97. doi: 10.1016/B978-0-12-802997-8.00023-2
13. Patel NP, Lyon KA, Huang JH. The effect of race on the prognosis of the glioblastoma patient: a brief review. Neurol Res (2019) 41(11):967–71. doi: 10.1080/01616412.2019.1638018
14. Braun K, Ahluwalia MS. Treatment of glioblastoma in older adults. Curr Oncol Rep (2017) 19(12):81. doi: 10.1007/s11912-017-0644-z
15. Davis ME. Glioblastoma: Overview of disease and treatment. Clin J Oncol Nurs (2016) 20(5 Suppl):S2–8. doi: 10.1188/16.CJON.S1.2-8
16. McKinnon C, Nandhabalan M, Murray SA, Plaha P. Glioblastoma: clinical presentation, diagnosis, and management. BMJ (2021) 374:n1560. doi: 10.1136/bmj.n1560
17. Soomro SH, Ting LR, Qing YY, Ren M. Molecular biology of glioblastoma: Classification and mutational locations. J Pak Med Assoc (2017) 67(9):1410–4.
18. Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol (2017) 35(21):2402–9. doi: 10.1200/JCO.2017.73.0119
19. Guerreiro Stucklin AS, Tabori U, Grotzer MA. The changing landscape of pediatric low-grade gliomas: Clinical challenges and emerging therapies. Neuropediatrics (2016) 47(2):70–83. doi: 10.1055/s-0035-1570491
20. Michaeli O, Tabori U. Pediatric high grade gliomas in the context of cancer predisposition syndromes. J Korean Neurosurg Soc (2018) 61(3):319–32. doi: 10.3340/jkns.2018.0031
21. Clarke JL, Chang SM. Neuroimaging: diagnosis and response assessment in glioblastoma. Cancer J (2012) 18(1):26–31. doi: 10.1097/PPO.0b013e318244d7c8
22. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi: 10.1056/NEJMoa0808710
23. Sabbah DA, Hajjo R, Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr Top Med Chem (2020) 20(10):815–34. doi: 10.2174/1568026620666200303123102
24. Suri V, Das P, Pathak P, Jain A, Sharma MC, Borkar SA, et al. Pediatric glioblastomas: a histopathological and molecular genetic study. Neuro Oncol (2009) 11(3):274–80. doi: 10.1215/15228517-2008-092
25. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res (2004) 24(3b):1861–71.
26. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell (2017) 32(4):520–37 e5. doi: 10.1016/j.ccell.2017.08.017
27. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
28. Cohen KJ, Heideman RL, Zhou T, Holmes EJ, Lavey RS, Bouffet E, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the children's oncology group. Neuro Oncol (2011) 13(4):410–6. doi: 10.1093/neuonc/noq205
29. Da Ros M, De Gregorio V, Iorio AL, Giunti L, Guidi M, de Martino M, et al. Glioblastoma chemoresistance: The double play by microenvironment and blood-brain barrier. Int J Mol Sci (2018) 19(10):2879. doi: 10.3390/ijms19102879
30. Donson AM, Addo-Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer (2007) 48(4):403–7. doi: 10.1002/pbc.20803
31. Penas-Prado M, Armstrong TS, Gilbert MR. Glioblastoma. Handb Clin Neurol (2012) 105:485–506. doi: 10.1016/B978-0-444-53502-3.00004-5
32. Narayana A, Kunnakkat S, Chacko-Mathew J, Gardner S, Karajannis M, Raza S, et al. Bevacizumab in recurrent high-grade pediatric gliomas. Neuro Oncol (2010) 12(9):985–90. doi: 10.1093/neuonc/noq033
33. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res (2007) 13(4):1253–9. doi: 10.1158/1078-0432.CCR-06-2309
34. Gururangan S, Chi SN, Young Poussaint T, Onar-Thomas A, Gilbertson RJ, Vajapeyam S, et al. Lack of efficacy of bevacizumab plus irinotecan in children with recurrent malignant glioma and diffuse brainstem glioma: a pediatric brain tumor consortium study. J Clin Oncol (2010) 28(18):3069–75. doi: 10.1200/JCO.2009.26.8789
35. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol (2009) 27(28):4733–40. doi: 10.1200/JCO.2008.19.8721
36. Zhao HF, Wang J, Shao W, Wu CP, Chen ZP, To ST, et al. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development. Mol Cancer (2017) 16(1):100. doi: 10.1186/s12943-017-0670-3
37. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol (2008) 20(5):267–75. doi: 10.1016/j.smim.2008.04.001
38. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE 2nd, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther (2009) 8(10):2773–9. doi: 10.1158/1535-7163.MCT-09-0124
39. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
40. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
41. Ahluwalia M, Reardon D, Abad A, Curry W, Wong E, Peereboom D, et al. ATIM-41. PHASE II TRIAL OF a SURVIVIN VACCINE (SurVaxM) for newly diagnosed glioblastoma. Neuro Oncol (2018) 20(Suppl 6):vi10–vi11. doi: 10.1093/neuonc/noy148.036
42. Izumoto S, Tsuboi A, Oka Y, Suzuki T, Hashiba T, Kagawa N, et al. Phase II clinical trial of wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg (2008) 108(5):963–71. doi: 10.3171/JNS/2008/108/5/0963
43. Oji Y, Hashimoto N, Tsuboi A, Murakami Y, Iwai M, Kagawa N, et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int J Cancer (2016) 139(6):1391–401. doi: 10.1002/ijc.30182
44. Platten M, Bunse L, Riehl D, Bunse T, Ochs K, Wick W. Vaccine strategies in gliomas. Curr Treat Options Neurol (2018) 20(5):11. doi: 10.1007/s11940-018-0498-1
45. Cuoco JA, Benko MJ, Busch CM, Rogers CM, Prickett JT, Marvin EA. Vaccine-based immunotherapeutics for the treatment of glioblastoma: Advances, challenges, and future perspectives. World Neurosurg (2018) 120:302–15. doi: 10.1016/j.wneu.2018.08.202
46. Hirabayashi K, Yanagisawa R, Saito S, Higuchi Y, Koya T, Sano K, et al. Feasibility and immune response of WT1 peptide vaccination in combination with OK-432 for paediatric solid tumors. Anticancer Res (2018) 38(4):2227–34. doi: 10.21873/anticanres.12465
47. Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Normolle DP, et al. Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: results of a pilot study. J Neurooncol (2016) 130(3):517–27. doi: 10.1007/s11060-016-2245-3
48. Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Potter DM, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol (2014) 32(19):2050–8. doi: 10.1200/JCO.2013.54.0526
49. Sawada A, Inoue M, Kondo O, Yamada-Nakata K, Ishihara T, Kuwae Y, et al. Feasibility of cancer immunotherapy with WT1 peptide vaccination for solid and hematological malignancies in children. Pediatr Blood Cancer (2016) 63(2):234–41. doi: 10.1002/pbc.25792
50. Tsuboi A, Hashimoto N, Fujiki F, Morimoto S, Kagawa N, Nakajima H, et al. A phase I clinical study of a cocktail vaccine of wilms' tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol Immunother (2019) 68(2):331–40. doi: 10.1007/s00262-018-2274-1
51. Ochs K, Ott M, Bunse T, Sahm F, Bunse L, Deumelandt K, et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology (2017) 6(7):e1328340. doi: 10.1080/2162402X.2017.1328340
52. Schaller TH, Sampson JH. Advances and challenges: dendritic cell vaccination strategies for glioblastoma. Expert Rev Vaccines (2017) 16(1):27–36. doi: 10.1080/14760584.2016.1218762
53. Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, Sato Y, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy (2012) 14(6):733–42. doi: 10.3109/14653249.2012.666633
54. Rudnick JD, Fink KL, Landolfi JC, Markert J, Piccioni DE, Glantz MJ, et al. Immunological targeting of CD133 in recurrent glioblastoma: A multi-center phase I translational and clinical study of autologous CD133 dendritic cell immunotherapy. J Clin Oncol (2017) 35:2059–9. doi: 10.1200/JCO.2017.35.15_suppl.2059
55. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res (2019) 25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261
56. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med (2018) 16(1):142. doi: 10.1186/s12967-018-1507-6
57. Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: A phase II randomized trial. Cancers (Basel) (2018) 10(10):372. doi: 10.3390/cancers10100372
58. De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res (2008) 14(10):3098–104. doi: 10.1158/1078-0432.CCR-07-4875
59. Lasky JL 3rd, Panosyan EH, Plant A, Davidson T, Yong WH, Prins RM, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res (2013) 33(5):2047–56.
60. Foster JB, Madsen PJ, Hegde M, Ahmed N, Cole KA, Maris JM, et al. Immunotherapy for pediatric brain tumors: past and present. Neuro Oncol (2019) 21(10):1226–38. doi: 10.1093/neuonc/noz077
61. Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest (2005) 85(3):328–41. doi: 10.1038/labinvest.3700233
62. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discovery (2015) 14(9):642–62. doi: 10.1038/nrd4663
63. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer (2018) 6(1):140. doi: 10.1186/s40425-018-0458-z
64. Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer (2016) 4:53. doi: 10.1186/s40425-016-0158-5
65. Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J (2012) 18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9
66. Gesundheit B, Ben-David E, Posen Y, Ellis R, Wollmann G, Schneider EM, et al. Effective treatment of glioblastoma multiforme with oncolytic virotherapy: A case-series. Front Oncol (2020) 10:702. doi: 10.3389/fonc.2020.00702
67. Lu VM, Shah AH, Vallejo FA, Eichberg DG, Luther EM, Shah SS, et al. Clinical trials using oncolytic viral therapy to treat adult glioblastoma: a progress report. Neurosurg Focus (2021) 50(2):E3. doi: 10.3171/2020.11.FOCUS20860
68. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47 for residual or recurrent glioblastoma: a phase 2 trial. Nat Med (2022) 28:1630–39. doi: 10.1038/s41591-022-01897-x
69. Ring E MB, Nan L, Etminan T, Markert J, Gillespie GY, Friedman G. Comparison of the sensitivities of pediatric high-grade brain tumor versus adult glioblastoma xenografts to engineered oncolytic herpes simplex virotherapy. Neuro Oncol (2016) 18:iii141. doi: 10.1093/neuonc/now080.09
70. Friedman GK, Johnston JM, Bag AK, Bernstock JD, Li R, Aban I, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med (2021) 384(17):1613–22. doi: 10.1056/NEJMoa2024947
71. Martinez-Velez N, Garcia-Moure M, Marigil M, Gonzalez-Huarriz M, Puigdelloses M, Gallego Perez-Larraya J, et al. The oncolytic virus delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun (2019) 10(1):2235. doi: 10.1038/s41467-019-10043-0
72. Gallego Perez-Larraya J, Garcia-Moure M, Labiano S, Patino-Garcia A, Dobbs J, Gonzalez-Huarriz M, et al. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N Engl J Med (2022) 386(26):2471–81. doi: 10.1056/NEJMoa2202028
73. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
74. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol (2018) 20(5):674–86. doi: 10.1093/neuonc/nox208
75. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-Oncology (2017) 19(suppl_3):iii21. doi: 10.1093/neuonc/nox036.071
76. Viana-Pereira M, Lee A, Popov S, Bax DA, Al-Sarraj S, Bridges LR, et al. Microsatellite instability in pediatric high grade glioma is associated with genomic profile and differential target gene inactivation. PloS One (2011) 6(5):e20588. doi: 10.1371/journal.pone.0020588
77. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol (2016) 34(19):2206–11. doi: 10.1200/JCO.2016.66.6552
78. Zhu X, McDowell MM, Newman WC, Mason GE, Greene S, Tamber MS. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: case report. J Neurosurg Pediatr (2017) 19(2):249–53. doi: 10.3171/2016.8.PEDS16326
79. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
80. Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, et al. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol (1987) 5(9):1430–40. doi: 10.1200/JCO.1987.5.9.1430
81. Saleh MN, Khazaeli MB, Wheeler RH, Dropcho E, Liu T, Urist M, et al. Phase I trial of the murine monoclonal anti-GD2 antibody 14G2a in metastatic melanoma. Cancer Res (1992) 52(16):4342–7.
82. Soman G, Kallarakal AT, Michiel D, Yang X, Saptharish N, Jiang H, et al. Analytical characterization of ch14.18: a mouse-human chimeric disialoganglioside-specific therapeutic antibody. MAbs (2012) 4(1):84–100. doi: 10.4161/mabs.4.1.18566
83. Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, et al. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol (1998) 16(6):2169–80. doi: 10.1200/JCO.1998.16.6.2169
84. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with Recurrent/Refractory neuroblastoma. Clin Cancer Res (2017) 23(21):6441–9. doi: 10.1158/1078-0432.CCR-17-0379
85. Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, et al. A phase II trial of Hu14.18K322A in combination with induction chemotherapy in children with newly diagnosed high-risk neuroblastoma. Clin Cancer Res (2019) 25(21):6320–8. doi: 10.1158/1078-0432.CCR-19-1452
86. Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol (2017) 18(7):946–57. doi: 10.1016/S1470-2045(17)30355-8
87. Mody R, Yu AL, Naranjo A, Zhang FF, London WB, Shulkin BL, et al. Irinotecan, temozolomide, and dinutuximab with GM-CSF in children with refractory or relapsed neuroblastoma: A report from the children's oncology group. J Clin Oncol (2020) 38(19):2160–9. doi: 10.1200/JCO.20.00203
88. Marx S, Wilken F, Wagner I, Marx M, Troschke-Meurer S, Zumpe M, et al. GD2 targeting by dinutuximab beta is a promising immunotherapeutic approach against malignant glioma. J Neurooncol (2020) 147(3):577–85. doi: 10.1007/s11060-020-03470-3
89. Zhou WT, Jin WL. B7-H3/CD276: An emerging cancer immunotherapy. Front Immunol (2021) 12:701006. doi: 10.3389/fimmu.2021.701006
90. Cai D, Li J, Liu D, Hong S, Qiao Q, Sun Q, et al. Tumor-expressed B7-H3 mediates the inhibition of antitumor T-cell functions in ovarian cancer insensitive to PD-1 blockade therapy. Cell Mol Immunol (2020) 17(3):227–36. doi: 10.1038/s41423-019-0305-2
91. Lee YH, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H, et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res (2017) 27(8):1034–45. doi: 10.1038/cr.2017.90
92. Lu H, Shi T, Wang M, Li X, Gu Y, Zhang X, et al. B7-H3 inhibits the IFN-gamma-dependent cytotoxicity of Vgamma9Vdelta2 T cells against colon cancer cells. Oncoimmunology (2020) 9(1):1748991. doi: 10.1080/2162402X.2020.1748991
93. Souweidane MM, Kramer K, Pandit-Taskar N, Zhou Z, Haque S, Zanzonico P, et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: a single-centre, dose-escalation, phase 1 trial. Lancet Oncol (2018) 19(8):1040–50. doi: 10.1016/S1470-2045(18)30322-X
94. Parakh S, Parslow AC, Gan HK, Scott AM. Antibody-mediated delivery of therapeutics for cancer therapy. Expert Opin Drug Deliv (2016) 13(3):401–19. doi: 10.1517/17425247.2016.1124854
95. Anderson MG, Falls HD, Mitten MJ, Oleksijew A, Vaidya KS, Boghaert ER, et al. Targeting multiple EGFR-expressing tumors with a highly potent tumor-selective antibody-drug conjugate. Mol Cancer Ther (2020) 19(10):2117–25. doi: 10.1158/1535-7163.MCT-20-0149
96. Lassman AB, Pugh SL, Wang TJC, Aldape K, Gan HK, Preusser M. Depatuxizumab mafodotin (ABT-414) in EGFR-amplified newly diagnosed GBM: A randomized, double-blind, phase III, international clinical trial. Neuro-Oncology (2022), noac173. doi: 10.1093/neuonc/noac173
97. Parakh S, Nicolazzo J, Scott AM, Gan HK. Antibody drug conjugates in glioblastoma - is there a future for them? Front Oncol (2021) 11:718590. doi: 10.3389/fonc.2021.718590
98. Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, et al. Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody-drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemother Pharmacol (2019) 84(2):327–36. doi: 10.1007/s00280-019-03879-2
99. Patterson JD, Henson JC, Breese RO, Bielamowicz KJ, Rodriguez A. CAR T cell therapy for pediatric brain tumors. Front Oncol (2020) 10:1582. doi: 10.3389/fonc.2020.01582
100. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med (2020) 26(5):720–31. doi: 10.1038/s41591-020-0827-2
101. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol (2014) 16(10):1304–12. doi: 10.1093/neuonc/nou045
102. Chen M, Sun R, Shi B, Wang Y, Di S, Luo H, et al. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. BioMed Pharmacother (2019) 113:108734. doi: 10.1016/j.biopha.2019.108734
103. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
104. Lin YJ, Mashouf LA, Lim M. CAR T cell therapy in primary brain tumors: Current investigations and the future. Front Immunol (2022) 13:817296. doi: 10.3389/fimmu.2022.817296
105. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
106. Xiong L, Edwards CK 3rd, Zhou L. The biological function and clinical utilization of CD147 in human diseases: a review of the current scientific literature. Int J Mol Sci (2014) 15(10):17411–41. doi: 10.3390/ijms151017411
107. Li H, Xi Z, Dai X, Wu W, Li Y, Liu Y, et al. CD147 and glioma: a meta-analysis. J Neurooncol (2017) 134(1):145–56. doi: 10.1007/s11060-017-2499-4
108. Yang M, Yuan Y, Zhang H, Yan M, Wang S, Feng F, et al. Prognostic significance of CD147 in patients with glioblastoma. J Neurooncol (2013) 115(1):19–26. doi: 10.1007/s11060-013-1207-2
109. Aziz-Bose R, Monje M. Diffuse intrinsic pontine glioma: molecular landscape and emerging therapeutic targets. Curr Opin Oncol (2019) 31(6):522–30. doi: 10.1097/CCO.0000000000000577
110. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med (2018) 24(5):572–9. doi: 10.1038/s41591-018-0006-x
111. Nehama D, Di Ianni N, Musio S, Du H, Patane M, Pollo B, et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine (2019) 47:33–43. doi: 10.1016/j.ebiom.2019.08.030
112. Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics (2019) 14:279–87. doi: 10.1016/j.omto.2019.07.002
113. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol (2013) 264(1-2):71–83. doi: 10.1016/j.jneuroim.2013.08.013
114. Golan I, Rodriguez de la Fuente L, Costoya JA. NK cell-based glioblastoma immunotherapy. Cancers (Basel) (2018) 10(12):522. doi: 10.3390/cancers10120522
115. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front Immunol (2019) 10:2683. doi: 10.3389/fimmu.2019.02683
116. Chu Y, Yahr A, Huang B, Ayello J, Barth M, Cairo MS. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology (2017) 6(9):e1341031. doi: 10.1080/2162402X.2017.1341031
117. Iorio AL, da Ros M, Genitori L, Lucchesi M, Colelli F, Signorino G, et al. Tumor response of temozolomide in combination with morphine in a xenograft model of human glioblastoma. Oncotarget (2017) 8(52):89595–606. doi: 10.18632/oncotarget.19875
118. Kim SS, Rait A, Kim E, Pirollo KF, Chang EH. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine (2015) 11(2):301–11. doi: 10.1016/j.nano.2014.09.005
119. Lam FC, Morton SW, Wyckoff J, Vu Han TL, Hwang MK, Maffa A, et al. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat Commun (2018) 9(1):1991. doi: 10.1038/s41467-018-04315-4
120. Lan F, Pan Q, Yu H, Yue X. Sulforaphane enhances temozolomide-induced apoptosis because of down-regulation of miR-21 via wnt/beta-catenin signaling in glioblastoma. J Neurochem (2015) 134(5):811–8. doi: 10.1111/jnc.13174
121. Liu YJ, Ma YC, Zhang WJ, Yang ZZ, Liang DS, Wu ZF, et al. Combination therapy with micellarized cyclopamine and temozolomide attenuate glioblastoma growth through Gli1 down-regulation. Oncotarget (2017) 8(26):42495–509. doi: 10.18632/oncotarget.17205
122. Nitta Y, Shimizu S, Shishido-Hara Y, Suzuki K, Shiokawa Y, Nagane M. Nimotuzumab enhances temozolomide-induced growth suppression of glioma cells expressing mutant EGFR in vivo. Cancer Med (2016) 5(3):486–99. doi: 10.1002/cam4.614
123. Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol (2011) 13(4):384–92. doi: 10.1093/neuonc/noq193
124. Wang H, Cai S, Bailey BJ, Reza Saadatzadeh M, Ding J, Tonsing-Carter E, et al. Combination therapy in a xenograft model of glioblastoma: enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J Neurosurg (2017) 126(2):446–59. doi: 10.3171/2016.1.JNS152513
125. Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A (2007) 104(31):12867–72. doi: 10.1073/pnas.0705158104
126. Montano N, Cenci T, Martini M, D'Alessandris QG, Pelacchi F, Ricci-Vitiani L, et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia (2011) 13(12):1113–21. doi: 10.1593/neo.111338
127. Bax DA, Gaspar N, Little SE, Marshall L, Perryman L, Regairaz M, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res (2009) 15(18):5753–61. doi: 10.1158/1078-0432.CCR-08-3210
128. Li G, Mitra SS, Monje M, Henrich KN, Bangs CD, Nitta RT, et al. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol (2012) 108(3):395–402. doi: 10.1007/s11060-012-0842-3
129. van Willigen WW, Bloemendal M, Boers-Sonderen MJ, de Groot JWB, Koornstra RHT, van der Veldt AAM, et al. Response and survival of metastatic melanoma patients treated with immune checkpoint inhibition for recurrent disease on adjuvant dendritic cell vaccination. Oncoimmunology (2020) 9(1):1738814. doi: 10.1080/2162402X.2020.1738814
130. Wakefield A, Pignata A, Ghazi A, Ashoori A, Hegde M, Landi D, et al. Is CMV a target in pediatric glioblastoma? expression of CMV proteins, pp65 and IE1-72 and CMV nucleic acids in a cohort of pediatric glioblastoma patients. J Neurooncol (2015) 125(2):307–15. doi: 10.1007/s11060-015-1905-z
131. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
132. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
133. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight (2016) 1(2):e85841. doi: 10.1172/jci.insight.85841
134. Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, et al. Immunosuppressive myeloid cells' blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol Ther (2017) 25(1):232–48. doi: 10.1016/j.ymthe.2016.10.003
135. Mueller S, Taitt JM, Villanueva-Meyer JE, Bonner ER, Nejo T, Lulla RR, et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. J Clin Invest (2020) 130(12):6325–37. doi: 10.1172/JCI140378
136. Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol (2019) 9:51. doi: 10.3389/fonc.2019.00051
137. Chu Y, Nayyar G, Jiang S, Rosenblum JM, Soon-Shiong P, Safrit JT, et al. Combinatorial immunotherapy of n-803 (IL-15 superagonist) and dinutuximab with ex vivo expanded natural killer cells significantly enhances in vitro cytotoxicity against GD2(+) pediatric solid tumors and in vivo survival of xenografted immunodeficient NSG mice. J Immunother Cancer (2021) 9(7):e002267. doi: 10.1136/jitc-2020-002267
138. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife (2020) 9:e52176. doi: 10.7554/eLife.52176
139. Wang J, Matosevic S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J Mol Med (Berl) (2018) 96(9):903–13. doi: 10.1007/s00109-018-1679-9
140. Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci U.S.A. (2021) 118(45):e2107507118. doi: 10.1073/pnas.2107507118
141. Ma R, Lu T, Li Z, Teng KY, Mansour AG, Yu M, et al. An oncolytic virus expressing IL15/IL15Ralpha combined with off-the-Shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res (2021) 81(13):3635–48. doi: 10.1158/0008-5472.CAN-21-0035
142. Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer (2020) 20(1):26–41. doi: 10.1038/s41568-019-0205-x
143. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
144. Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci (2012) 15(8):1096–101. doi: 10.1038/nn.3161
145. Marchetti L, Engelhardt B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc Biol (2020) 2(1):H1–H18. doi: 10.1530/VB-19-0033
146. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol (2021) 39:251–77. doi: 10.1146/annurev-immunol-093019-110159
147. Nowosielski M, DiFranco MD, Putzer D, Seiz M, Recheis W, Jacobs AH, et al. An intra-individual comparison of MRI, [18F]-FET and [18F]-FLT PET in patients with high-grade gliomas. PloS One (2014) 9(4):e95830. doi: 10.1371/journal.pone.0095830
148. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? a critical assessment of existing clinical data. Neuro Oncol (2018) 20(2):184–91. doi: 10.1093/neuonc/nox175
149. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol (2012) 2:205. doi: 10.3389/fonc.2012.00205
150. Warren KE. Beyond the Blood:Brain barrier: The importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front Oncol (2018) 8:239. doi: 10.3389/fonc.2018.00239
151. Wei X, Meel MH, Breur M, Bugiani M, Hulleman E, Phoenix TN. Defining tumor-associated vascular heterogeneity in pediatric high-grade and diffuse midline gliomas. Acta Neuropathol Commun (2021) 9(1):142. doi: 10.1186/s40478-021-01243-1
152. Gargini R, Segura-Collar B, Sanchez-Gomez P. Cellular plasticity and tumor microenvironment in gliomas: The struggle to hit a moving target. Cancers (Basel) (2020) 12(6):1622. doi: 10.3390/cancers12061622
153. Ross JL, Velazquez Vega J, Plant A, MacDonald TJ, Becher OJ, Hambardzumyan D. Tumour immune landscape of paediatric high-grade gliomas. Brain (2021) 144(9):2594–609. doi: 10.1093/brain/awab155
154. Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol (2017) 19(11):1460–8. doi: 10.1093/neuonc/nox054
155. Howie D, Waldmann H, Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol (2014) 5:409. doi: 10.3389/fimmu.2014.00409
156. Jawhari S, Ratinaud MH, Verdier M. Glioblastoma, hypoxia and autophagy: a survival-prone 'menage-a-trois'. Cell Death Dis (2016) 7(10):e2434. doi: 10.1038/cddis.2016.318
157. Hosseinalizadeh H, Mahmoodpour M, Samadani AA, Roudkenar MH. The immunosuppressive role of indoleamine 2, 3-dioxygenase in glioblastoma: mechanism of action and immunotherapeutic strategies. Med Oncol (2022) 39(9):130. doi: 10.1007/s12032-022-01724-w
158. Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, Kleinschmidt-DeMasters BK, et al. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res (2011) 17(22):6992–7002. doi: 10.1158/1078-0432.CCR-11-1107
159. Fanelli GN, Grassini D, Ortenzi V, Pasqualetti F, Montemurro N, Perrini P, et al. Decipher the glioblastoma microenvironment: The first milestone for new groundbreaking therapeutic strategies. Genes (Basel) (2021) 12(3):445. doi: 10.3390/genes12030445
160. Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, et al. Hypoxia potentiates glioma-mediated immunosuppression. PloS One (2011) 6(1):e16195. doi: 10.1371/journal.pone.0016195
161. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev (2019) 290(1):60–84. doi: 10.1111/imr.12773
162. Antonucci L, Canciani G, Mastronuzzi A, Carai A, Del Baldo G, Del Bufalo F. CAR-T therapy for pediatric high-grade gliomas: Peculiarities, current investigations and future strategies. Front Immunol (2022) 13:867154. doi: 10.3389/fimmu.2022.867154
163. Kamran N, Alghamri MS, Nunez FJ, Shah D, Asad AS, Candolfi M, et al. Current state and future prospects of immunotherapy for glioma. Immunotherapy (2018) 10(4):317–39. doi: 10.2217/imt-2017-0122
164. Lakshmanachetty S, Cruz-Cruz J, Hoffmeyer E, Cole AP, Mitra SS. New insights into the multifaceted role of myeloid-derived suppressor cells (MDSCs) in high-grade gliomas: From metabolic reprograming, immunosuppression, and therapeutic resistance to current strategies for targeting MDSCs. Cells (2021) 10(4):893. doi: 10.3390/cells10040893
165. Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PloS One (2012) 7(8):e43339. doi: 10.1371/journal.pone.0043339
166. Lin GL, Nagaraja S, Filbin MG, Suva ML, Vogel H, Monje M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun (2018) 6(1):51. doi: 10.1186/s40478-018-0553-x
167. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest (2017) 97(5):498–518. doi: 10.1038/labinvest.2017.19
168. Mackay A, Burford A, Molinari V, Jones DTW, Izquierdo E, Brouwer-Visser J, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell (2018) 33(5):829–42 e5. doi: 10.1016/j.ccell.2018.04.004
169. Jha P, Manjunath N, Singh J, Mani K, Garg A, Kaur K, et al. Analysis of PD-L1 expression and T cell infiltration in different molecular subgroups of diffuse midline gliomas. Neuropathology (2019) 39(6):413–24. doi: 10.1111/neup.12594
170. Barish ME, Weng L, Awabdeh D, Zhai Y, Starr R, D'Apuzzo M, et al. Spatial organization of heterogeneous immunotherapy target antigen expression in high-grade glioma. Neoplasia (2022) 30:100801. doi: 10.1016/j.neo.2022.100801
171. Haydar D, Houke H, Chiang J, Yi Z, Ode Z, Caldwell K, et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro Oncol (2021) 23(6):999–1011. doi: 10.1093/neuonc/noaa278
172. de Billy E, Pellegrino M, Orlando D, Pericoli G, Ferretti R, Businaro P, et al. Dual IGF1R/IR inhibitors in combination with GD2-CAR T-cells display a potent anti-tumor activity in diffuse midline glioma H3K27M-mutant. Neuro Oncol (2022) 24(7):1150–63. doi: 10.1093/neuonc/noab300
173. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2018) 20(4):506–18. doi: 10.1093/neuonc/nox182
174. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi: 10.1172/JCI83416
175. Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med (2018) 66(2):50–2. doi: 10.1016/j.retram.2018.03.003
176. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi: 10.1182/blood-2014-05-552729
177. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
178. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with b-cell acute lymphoblastic leukemia. Cancer Discovery (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
179. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5(197):197ra03. doi: 10.1126/scitranslmed.3006034
180. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
181. Crotty E, Downey K, Ferrerosa L, Flores C, Hegde B, Raskin S, et al. Considerations when treating high-grade pediatric glioma patients with immunotherapy. Expert Rev Neurother (2021) 21(2):205–19. doi: 10.1080/14737175.2020.1855144
182. Petersen CT, Krenciute G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front Oncol (2019) 9:69. doi: 10.3389/fonc.2019.00069
183. Golinelli G, Grisendi G, Prapa M, Bestagno M, Spano C, Rossignoli F, et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther (2020) 27(7-8):558–70. doi: 10.1038/s41417-018-0062-x
184. Kattner P, Strobel H, Khoshnevis N, Grunert M, Bartholomae S, Pruss M, et al. Compare and contrast: pediatric cancer versus adult malignancies. Cancer Metastasis Rev (2019) 38(4):673–82. doi: 10.1007/s10555-019-09836-y
Keywords: high grade glioma, glioblastoma, pediatric, adult, immunotherapy
Citation: Aggarwal P, Luo W, Pehlivan KC, Hoang H, Rajappa P, Cripe TP, Cassady KA, Lee DA and Cairo MS (2022) Pediatric versus adult high grade glioma: Immunotherapeutic and genomic considerations. Front. Immunol. 13:1038096. doi: 10.3389/fimmu.2022.1038096
Received: 06 September 2022; Accepted: 07 November 2022;
Published: 22 November 2022.
Edited by:
Barbara Rolfe, The University of Queensland, AustraliaReviewed by:
Akane Yamamichi, University of California, San Francisco, United StatesJoshua John Breunig, Cedars Sinai Medical Center, United States
Giedre Krenciute, St. Jude Children’s Research Hospital, United States
Copyright © 2022 Aggarwal, Luo, Pehlivan, Hoang, Rajappa, Cripe, Cassady, Lee and Cairo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitchell S. Cairo, TWl0Y2hlbGxfQ2Fpcm9AbnltYy5lZHU=
†These authors share first authorship