
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 November 2022
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1036249
 Edmund Y. M. Chung1*
Edmund Y. M. Chung1* Yuan M. Wang1
Yuan M. Wang1 Karen Keung2
Karen Keung2 Min Hu3
Min Hu3 Hugh McCarthy1,4,5
Hugh McCarthy1,4,5 Germaine Wong1,6Lukas Kairaitis7Bhadran Bose8David C. H. Harris3,6
Germaine Wong1,6Lukas Kairaitis7Bhadran Bose8David C. H. Harris3,6 Stephen I. Alexander1,4
Stephen I. Alexander1,4Primary membranous nephropathy (PMN) is one of the common causes of adult-onset nephrotic syndrome and is characterized by autoantibodies against podocyte antigens causing in situ immune complex deposition. Much of our understanding of the disease mechanisms underpinning this kidney-limited autoimmune disease originally came from studies of Heymann nephritis, a rat model of PMN, where autoantibodies against megalin produced a similar disease phenotype though megalin is not implicated in human disease. In PMN, the major target antigen was identified to be M-type phospholipase A2 receptor 1 (PLA2R) in 2009. Further utilization of mass spectrometry on immunoprecipitated glomerular extracts and laser micro dissected glomeruli has allowed the rapid discovery of other antigens (thrombospondin type-1 domain-containing protein 7A, neural epidermal growth factor-like 1 protein, semaphorin 3B, protocadherin 7, high temperature requirement A serine peptidase 1, netrin G1) targeted by autoantibodies in PMN. Despite these major advances in our understanding of the pathophysiology of PMN, treatments remain non-specific, often ineffective, or toxic. In this review, we summarize our current understanding of the immune mechanisms driving PMN from animal models and clinical studies, and the implications on the development of future targeted therapeutic strategies.
Primary membranous nephropathy (PMN) is one of the commonest causes of adult-onset nephrotic syndrome with an incidence of 12 cases per million per year (1). It is a kidney-limited autoimmune disease characterized by in situ formation of subepithelial glomerular immune deposits containing immunoglobulin G (IgG) autoantibodies and complement (2). While spontaneous disease remission occurs in around 32% of patients, approximately a third ultimately progress to kidney failure needing dialysis or kidney transplantation (1). The podocyte antigens responsible for eliciting an autoimmune response in PMN remained elusive until 2009 when M-type phospholipase A2 receptor 1 (PLA2R) was discovered, followed by thrombospondin type-1 domain-containing protein 7A (THSD7A) in 2014 (3, 4). These discoveries improved the diagnosis of PMN with anti-PLA2R autoantibody testing having a specificity of 99%, which together with the kidney biopsy light microscopy findings of subepithelial “spikes” of the glomerular basement membrane (GBM) on silver methenamine stain, immunofluorescence finding of granular deposits of IgG along the glomerular capillary wall, and electron microscopy finding of exclusive subepithelial localization of electron-dense deposits, differentiate PMN from other forms of nephrotic syndrome (1, 5). However, despite this improved understanding, treatments for PMN remain non-specific. Cytotoxic therapy (chlorambucil or cyclophosphamide) plus glucocorticoids lower the 10-year incidence of kidney failure to approximately 10% (compared to 40% in those receiving supportive treatment) but is associated with significant toxicity such as infertility, infection and malignancy (6–10). Calcineurin inhibitors have proven short-term efficacy though are limited by high relapse rate of 40% and long-term nephrotoxicity (11). The anti-CD20 monoclonal antibody rituximab targeting B cells is the most targeted treatment to date but may not eliminate autoreactive B cells due to changes in the CD20 antigen, does not eliminate high-affinity antibody-producing plasma cells that do not express CD20, and fails to induce complete or partial remission in up to 40% of patients (12, 13). In comparing these existing treatments, a recent trial found cyclophosphamide plus glucocorticoids to be superior to calcineurin inhibitors plus single-dose rituximab (total 1g) though there was a higher anti-PLA2R autoantibody titer at baseline in the calcineurin inhibitor/rituximab group, which together with the low dose of rituximab used, may have contributed to worse outcomes (14). In contrast, rituximab (total 3g) was superior to calcineurin inhibitors (12), but rituximab (total 2g) was comparable to cyclophosphamide plus glucocorticoids (15). Lastly, recurrent membranous nephropathy (MN) after kidney transplantation also occurs in 35-50% of patients and accounts for 50% allograft loss 5 years after diagnosis, highlighting the unmet need for targeted treatments in PMN (16–19).
Antigen-independent treatments targeting autoantibody production is attractive and feasible given the multitude of disease antigens implicated in PMN particularly with improved antibodies targeting plasma cells. Targeted treatments against antigen-specific B cells or antigen-specific T cells helpers directed against antigens linked to PMN, using chimeric antigen receptor (CAR) T cells, chimeric autoantigen receptor expressing (CAAR) T cells, or CAR regulatory T cells (Tregs) to eliminate or suppress antigen-specific effectors are also conceivable. However, these experimental treatments remain untested in human autoimmune disease and rigorous testing in animal models of PMN is required (20). In this review, we compare the immune mechanisms driving PMN and associated animal models, and implications for evaluating novel targeted therapeutics.
Heymann et al. described active Heymann nephritis (AHN) in 1959, where immunization of the proximal tubular antigen Fx1A with complete Freund’s adjuvant in Lewis rats caused development of anti-Fx1A antibodies and in situ glomerular subepithelial immune complex formation after 4 weeks, and proteinuria after 8-10 weeks (21). Passive Heymann nephritis (PHN) is a more limited model induced by administration of sheep anti-rat Fx1A antibodies with cell- and blood-free perfusion to prevent immune complex formation in the circulation (22, 23). While PHN was widely used to study the effector mechanisms caused by glomerular IgG deposition, it does not recapitulate autologous formation of podocyte autoantibodies seen in AHN and which leads to PMN.
The identification of antigens implicated in PMN has led to the development of mouse models where passive administration of anti-PLA2R and anti-THSD7A antibodies into transgenic mice with murine full-length PLA2R on podocytes and wild-type mice respectively were able to induce similar disease to PMN with subepithelial glomerular IgG deposition, C3 deposition and proteinuria (24–27).
Recently, Tomas et al. have described transgenic knock-in mice expressing human PLA2R specifically on podocytes. These mice spontaneously developed anti-human PLA2R autoantibodies after 3 weeks, nephrotic syndrome with glomerular deposition of murine IgG1 (equivalent of human IgG4) and complement activation after 4 weeks, and glomerular subepithelial electron-dense deposits after 6 weeks (28). Overall, this model faithfully recapitulates human PMN.
Passive administration of antibodies against mouse aminopeptidase A (29, 30), dipeptidyl peptidase IV (31), and anti-mouse podocyte antibodies were also able to induce glomerular IgG deposition with proteinuria but without complement activation, unlike human PMN (32, 33). Furthermore, these antigens are not implicated in human PMN.
Intravenous injections of cationic bovine serum albumin (BSA) and immunization of recombinant human non-collagenous domain 1 of α3(IV) collagen (rh-α3NC1) both produced significant albuminuria and subepithelial deposits of IgG and C3 (34, 35). However, glomerular deposition of exogenous antigen (BSA and rh-α3NC1) in these models significantly differs from the pathogenesis of human PMN (35).
Human PMN is characterized by heterogeneous presentation and clinical course with around a third of patients undergoing spontaneous remission, a third experiencing persistent proteinuria, and a third progressing to kidney failure (36). While light microscopy findings on kidney biopsy also exhibit some variability, ranging from a normal appearance early in disease to thickened GBM as disease progresses, granular deposition of IgG and C3 in glomeruli on immunofluorescence and electron-dense subepithelial deposits on electron microscopy are seen at any stage of disease (1, 36).
The pathogenic antigen in Fx1A was identified as a 330-kDa glycoprotein called megalin, which is a low-density lipoprotein receptor involved in protein endocytosis and transcytosis (Table 1) (53–56). Another autoantibody in AHN was subsequently identified targeting the 44-kDa receptor associated protein (RAP), which acts as an endoplasmic reticulum chaperone for megalin (55).
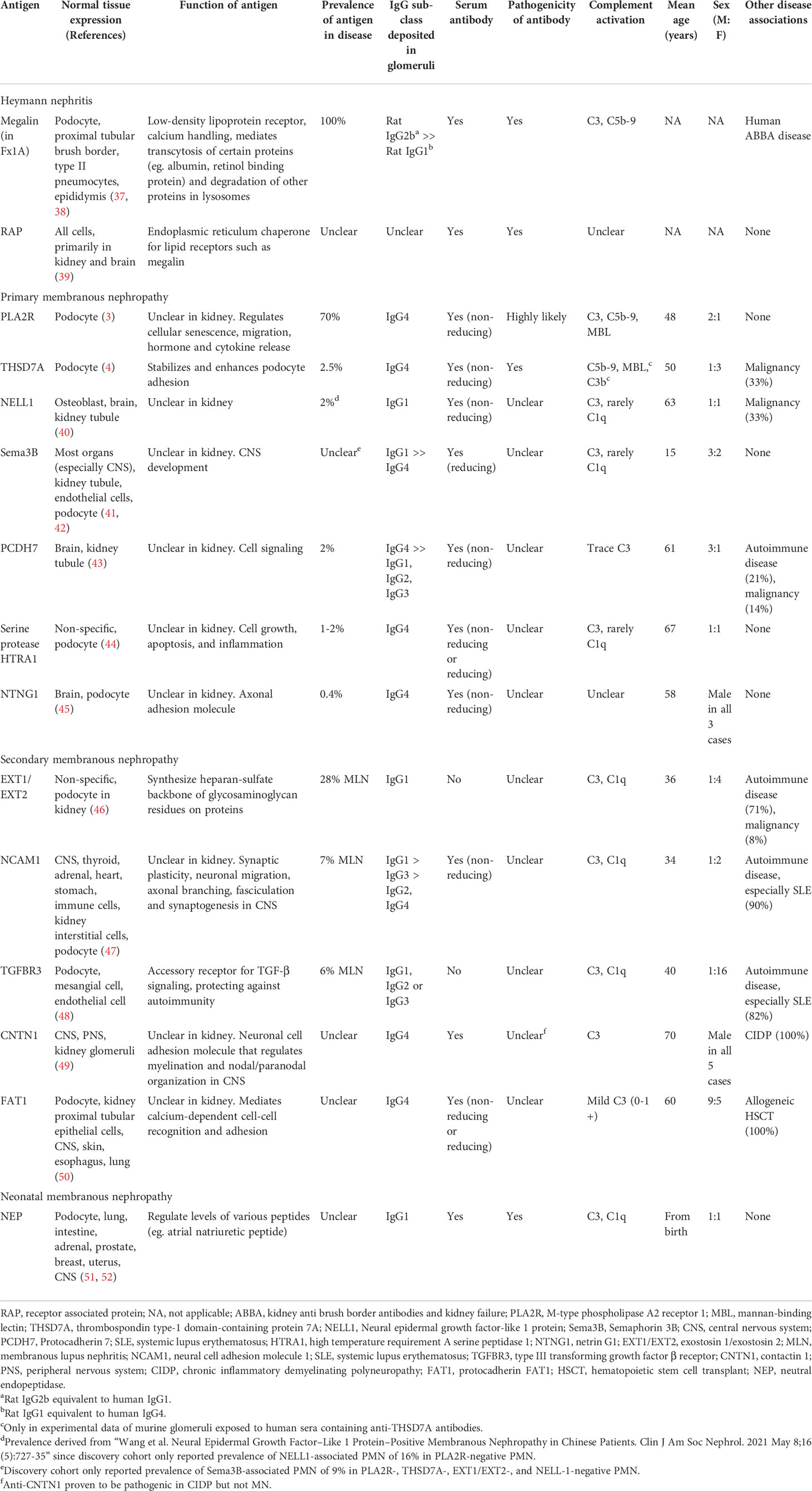
Table 1 Disease antigens implicated in Heymann nephritis and membranous nephropathy.
AHN is characterized by anti-Fx1A autoantibodies without circulating IgG subclass predominance but induce proteinuria via complement-fixing rat IgG2b (equivalent to human IgG1) (Figure 1A.1) (57–59). The main pathogenic epitope on megalin at its N-terminal domain is glycosylation- and conformation-dependent though epitope spreading occurs, similar to PMN, whereby the immune response to a disease antigen may extend to non-cross-reactive peptide sequences on the same antigen (intramolecular spreading) or adjacent antigens (intermolecular spreading) over time (60, 61). Accordingly, anti-RAP autoantibodies likely represent intermolecular epitope spreading due to the association of RAP with megalin (Figure 1A.2).
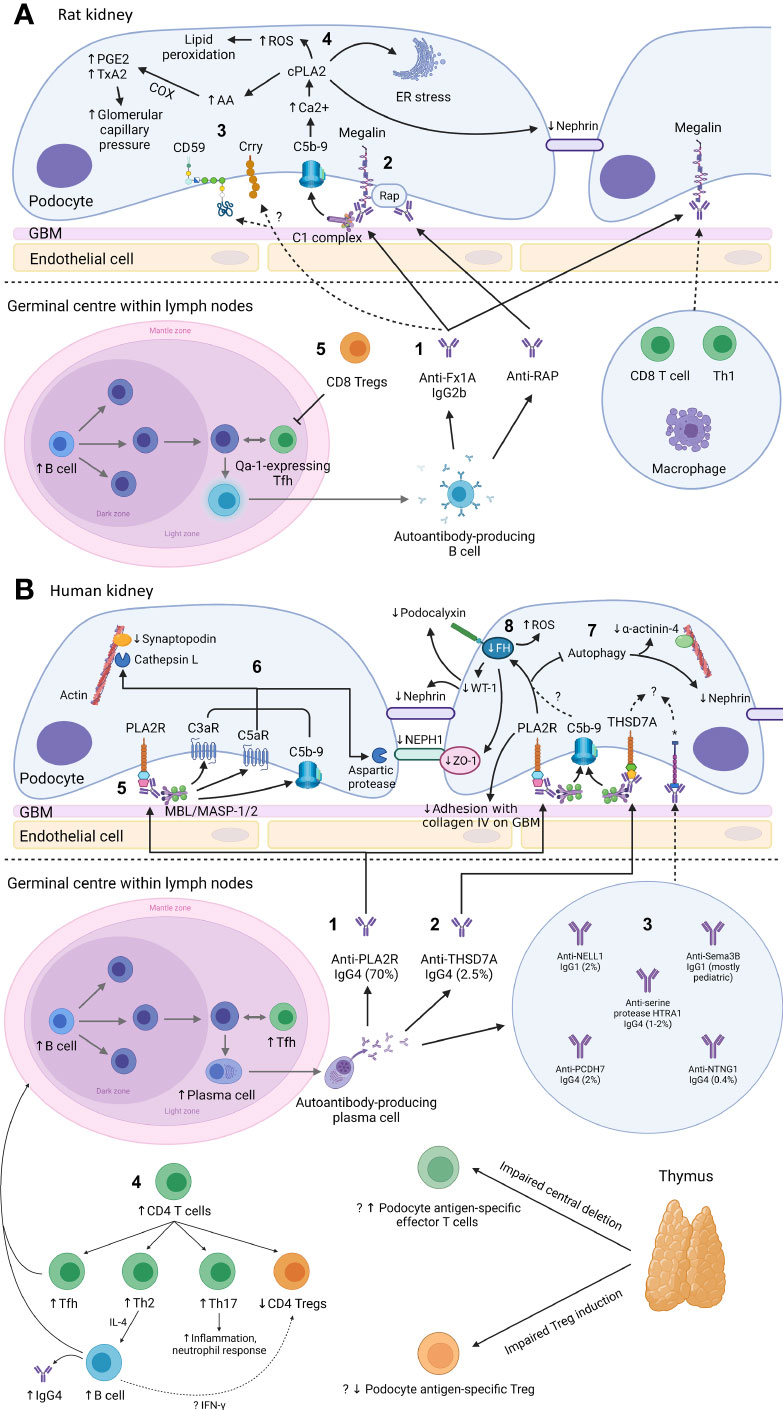
Figure 1 Mechanism of podocyte injury in Heymann nephritis (A) and primary membranous nephropathy (B) Created with BioRender.com. GBM, glomerular basement membrane; ROS, reactive oxygen species; cPLA2, cytosolic phospholipase A2; PGE2, prostaglandin E2; TxA2, thromboxane A2; COX, cyclo-oxygenase; AA, arachidonic acid; ER, endoplasmic reticulum; RAP, receptor associated protein; Treg, regulatory T cell; Th1, CD4+ T helper-1 cell; Tfh, CD4+ T follicular helper cell; FH, fumarate hydratase; WT-1, Wilms tumor-1; PLA2R, M-type phospholipase A2 receptor 1; C3aR, C3a receptor; C5aR, C5a receptor; MBL, mannan-binding lectin; MASP, mannan-binding lectin serine protease; ZO-1, zonula occludens-1; THSD7A, thrombospondin type-1 domain-containing protein 7A; NELL1, neural epidermal growth factor-like 1 protein; Sema3B, semaphorin 3B; PCDH7, protocadherin 7; HTRA1, high temperature requirement A serine peptidase 1; NTNG1, netrin G1; Th2, CD4+ T helper-2 cell; Th17, CD4+ T helper-17 cell; IFN-γ, interferon-γ; *cognate antigen for anti-NELL1, anti-Sema3B, anti-PCDH7, anti-serine protease HTRA1, and anti-NTNG1 autoantibodies respectively. (A): 1. In Heymann nephritis, anti-Fx1A antibodies bind primarily to megalin on podocytes and activate the classical complement pathway (via the IgG2b subclass). 2. Anti-RAP antibodies are also detected and may represent epitope spreading due to the association between megalin and RAP, which assists the transport of megalin from ER to cell surface. 3. Complement activation may be potentially exacerbated by binding of anti-Fx1A antibodies to complement regulatory proteins Crry and CD59. 4. Sublethal C5b-9 injury causes intracellular calcium influx, activating cPLA2, which hydrolyzes membrane phospholipids of the podocyte, ER and nuclear envelope. This causes ER stress, generation of ROS, disruption of the slit diaphragm protein nephrin, and release of AA with subsequent COX-mediated generation of prostanoids (PGE2, TxA2) that increase glomerular filtration pressure, exacerbating proteinuria. Glomerular IgG and complement deposition attract glomerular infiltrates of CD8+ T cells, Th1 cells, and macrophages, potentially via the IgG Fc receptor and anaphylatoxins C3a/C5a. 5. Autoantibody production in Heymann nephritis is at least in part dependent on Qa-1-expressing Tfh cells, which are inhibited by CD8+ Tregs. (B): 1. In primary membranous nephropathy (PMN), autoantibodies are predominantly directed against PLA2R, and 2. less commonly against THSD7A, 3. NELL1, PCDH7, serine protease HTRA1 and NTNG1, except in pediatric PMN where Sema3B is the most common podocyte antigen targeted by autoantibodies. 4. Increased B cells and Tfh cells are observed, which interact in the germinal center of lymph nodes to induce differentiation of B cells into high-affinity antibody-producing plasma cells. Autoantibodies in PMN are primarily of the IgG4 subclass, potentially due to Th2-mediated IL-4 production. Loss of tolerance to podocyte antigens such as PLA2R may be due to loss of thymus-derived podocyte antigen-specific Tregs. 5. Glycosylated anti-PLA2R IgG4 bind the MBL/MASP-1/2 complex to activate the lectin complement pathway. 6. This causes activation of C3aR and C5aR as well as deposition of C5b-9, which activates cathepsin L- and an aspartic protease-mediated proteolysis of the actin cytoskeleton protein synaptopodin and slit diaphragm protein NEPH1 respectively. Anti-PLA2R antibody-containing serum is also associated with reduced FH, impaired autophagy and reduced adhesion to the GBM, though it is unclear whether these effects are mediated via complement activation. 7. Impairment in autophagy results in internalization of nephrin and the actin cytoskeleton protein α-actinin-4. 8. Reduced FH leads to intracellular fumarate accumulation, which is associated with increased generation of ROS, reduced slit diaphragm protein ZO-1 and reduced transcription of WT-1, which normally activates the expression of podocyte proteins nephrin and podocalyxin. The mechanisms of podocyte injury of other autoantibodies associated with PMN remain incompletely understood.
For the passive administration of anti-PLA2R antibodies, transgenic mice expressing murine PLA2R were required due to a lack of PLA2R expression normally on murine podocytes (24). Murine PLA2R has 72% sequence homology with human PLA2R and adoptive transfer of human anti-PLA2R autoantibodies could not bind murine PLA2R. Instead disease induction required transfer of rabbit anti-PLA2R antibodies (24). Therefore, the pathogenicity of human anti-PLA2R antibodies remains to be definitively proven in vivo.
In contrast, murine podocytes normally express THSD7A. Interestingly, human sera containing anti-THSD7A antibodies but not purified human anti-THSD7A antibodies activated the lectin complement pathway on murine podocytes and caused oxidative stress, disruption of nephrin, and rearrangement of the podocyte actin cytoskeleton (25–27). In comparison, rabbit anti-THSD7A antibodies (with or without sera) induced significant proteinuria in the absence of complement activation (25, 27). These discrepant results could be related to lower amounts of injected purified human antibody, alteration of autoantibodies when purified using acid-elution or the presence of other proteins within human sera required for complement activation. Furthermore, it is unclear whether generation of rabbit anti-PLA2R and anti-THSD7A antibodies are predominantly of IgG4 subclass (like their human counterparts) and therefore whether disease pathophysiology caused by passive administration of these antibodies is homologous to human PMN (24, 27).
Full length human PLA2R was expressed on podocytes of these mice and anti-human PLA2R antibodies recognized the cysteine-rich domain as well as C-type lectin domains (CTLD) 1, 7 and 8, which are the same epitopes recognized by anti-PLA2R antibodies in patients with PMN (28, 62–64). Therefore, this represents the ideal model for confirming the pathogenicity of human anti-PLA2R autoantibodies.
While there are many similarities between Heymann nephritis and PMN, they are not homologous primarily due to differences in the disease antigen. Megalin (called low-density lipoprotein receptor-related protein 2 (LRP2) in humans) is 77% homologous between rodents and humans, though human tissue expression is predominantly on proximal tubular brush border and glomerular staining is weak (56, 65). Accordingly, anti-LRP2 antibodies in humans cause ABBA (anti brush border antibody) disease, characterized by tubular immune deposits with segmental glomerular subepithelial deposits, subnephrotic proteinuria, and kidney failure (66).
Unlike Heymann nephritis, IgG4 is the predominant IgG subclass in most patients with PMN though IgG1 is predominantly found in neural epidermal growth factor-like 1 protein (NELL1)- and semaphorin 3B (Sema3B)-associated PMN without significant activation of the classical complement pathway based on rare C1q deposition (40, 41).
PLA2R is the primary disease antigen in 70% of PMN and while the pathogenicity of anti-PLA2R autoantibodies is highly likely due to the strong correlation of antibody titres with disease activity, in vitro evidence of podocyte injury caused by anti-PLA2R sera and lectin pathway activation by anti-PLA2R IgG4, definitive in vivo evidence of pathogenicity is yet to be confirmed (Figure 1B.1) (3, 67–70). The major epitope targeted by anti-PLA2R antibodies is located at the 31-mers peptide sequence of the cysteine-rich domain of PLA2R (specifically the N-terminal linear stretch VIQSES and C-terminal stretch SVLTLENC regions) with additional antibody epitopes identified at CTLD1, CTLD7 and CTLD8 (62, 64, 71). Epitope spreading from the cysteine-rich domain to the CTLD1 and/or 7 domains occurs in 25-66% of patients over the course of 3 months to 2 years (72, 73). However, data are inconsistent regarding whether epitope spreading is associated with treatment resistance (64, 72, 73).
Genome-wide association studies have greatly enhanced our understanding of how self-antigens such as PLA2R trigger an autoimmune response. The risk of PMN was substantially increased in individuals with both single nucleotide polymorphisms (SNP) in class II human leukocyte antigen (HLA-DQA1*0501 in Europeans, -DRB1*1501 in East Asians, and -DRB1*0301 in both ethnicities) on antigen-presenting cells (APC) and SNPs in the PLA2R locus (rs4664308 in Europeans and rs17831251 in both Europeans and East Asians) (74, 75). These non-coding region SNPs were either associated with other SNPs within the PLA2R coding region or an enhancer element resulting in an altered amino acid sequence or increased tissue expression respectively (75). This may enhance antigen presentation by APCs expressing risk HLA variants to their cognate CD4+ T cell, thereby initiating the process of autoimmunity. In silico analysis predicted HLA-DRB1*1501 and HLA-DRB1*0301 preferentially bound to PLA2R peptides in the CTLD1, CTLD7 and between CTLD4 and CTLD5 regions compared to the 31-mers peptide, though experimental validation is required (76).
In recurrent PLA2R-associated MN after kidney transplantation, detectable anti-PLA2R autoantibody titres pre-transplantation and steroid-free immunosuppression are known risk factors (16, 77–79). Positive glomerular staining for PLA2R in these cases also suggest the same antigen is implicated in recurrent MN (78). However, the HLA risk alleles (HLA-DQA1*0501, -DRB1*1501, and -DRB1*0301) associated with PMN were not associated with recurrent MN but rather two non-coding HLA-D SNPs (rs9271550 and rs9271705) and three PLA2R SNPs (rs3828323, rs17830558, and rs3749117)) when present on the donor but not the recipient (80). Another study found an association between recurrent MN and recipient HLA-A3 (81). Discrepancies between studies may be due to the relatively small sample sizes and different diagnostic criteria for recurrent MN. Overall, the pathogenesis of recurrent MN remains poorly understood and further studies are required.
THSD7A was the second podocyte antigen discovered in PMN, implicated in 10% of PLA2R-negative PMN (Figure 1B.2) (4). THSD7A and PLA2R are structurally similar as are their associated autoantibodies, which are predominantly of IgG4 subclass, activate complement with glomerular C5b-9 staining, bind initially to the immunodominant epitope on the N-terminal domain of their target antigen exclusively in non-reducing conditions, and are associated with epitope-spreading (4, 25, 63, 82). Contrary to the prevailing paradigm of a single podocyte antigen being targeted in PMN, dual anti-PLA2R and anti-THSD7A antibodies and staining on biopsy has been reported in 1% of cases (83).
Laser microdissection and mass spectrometry on kidney biopsies of PLA2R-negative PMN has led to the discovery of many more disease antigens such as NELL1, Sema3B, protocadherin 7 (PCDH7), high temperature requirement A serine peptidase 1 (HTRA1), and netrin G1 (NTNG1) (Figure 1B.3). These antigens account for less than 10% of PMN and their associated autoantibodies vary in their predominant IgG subclass, ability to activate complement, and the association of their pathogenic epitope with disulfide bonds based on whether autoantibodies were detected under reducing or non-reducing conditions (Table 1) (40, 41, 43–50). Anti-Sema3B autoantibodies correlated with disease activity in a case report of severe pediatric PMN, suggestive of pathogenicity (84). However, the pathogenicity of other autoantibodies remains to be determined and the mechanism by which these self-antigens trigger an autoimmune response remain unclear. Furthermore, it remains unclear how some of these antigens such as NELL1 and PCDH7 display weak or no staining in normal glomeruli but are implicated in PMN, whereby the subepithelial localization of deposits on electron microscopy in NELL1- and PCDH7-associated MN strongly suggest shedding from podocytes rather than mesangial or endothelial cells. NELL1 and PCDH7 are known to be secretory proteins and highly glycosylated proteins respectively (85), which may impact their expression under healthy conditions. However, further research on the expression and localization of these novel antigens in normal podocytes are required. Overall, it is apparent that PMN is not a single disease entity but rather a group of diseases characterized by autoantibodies against different antigens causing kidney-limited autoimmune disease.
MN podocyte antigens exostosin 1/eoxstosin 2 (EXT1/EXT2), neural cell adhesion molecule 1 (NCAM1), and transforming growth factor-β receptor 3 (TGFBR3) are associated with other autoimmune diseases, especially systematic lupus erythematosus (SLE) (46–48). In particular, EXT1/EXT2-associated membranous lupus nephritis confers a better prognosis than EXT1/EXT2-negative membranous lupus nephritis (86). This may be due to EXT1/EXT2-related glycosylation of heparan sulfate, which protects the GBM against immune injury. Contactin 1 (CNTN1) is associated with chronic inflammatory demyelinating polyneuropathy-related MN and protocadherin FAT1 (FAT1) is associated with allogeneic hematopoietic stem cell transplant-related MN (49, 50).
In contrast, while MN secondary to infections such as hepatitis B have been well described and hypothesized to be due to molecular mimicry, the associated podocyte antigen remains unclear (87). Evidence of molecular mimicry due to malignancy is more convincing for THSD7A- and NELL1-associated MN. THSD7A was found in tumor and follicular dendritic cells of tumor-infiltrated lymph nodes in separate cases of gallbladder adeno-neuroendocrine carcinoma and endometrial carcinoma (88, 89) Up to 33% of NELL1-associated MN were secondary to malignancy with NELL1 tumor expression demonstrated in 2 cases (invasive ductal carcinoma of the breast and follicular lymphoma) (90). Lastly, neutral endopeptidase (NEP/CD10) is a podocyte antigen implicated in neonatal MN, which is caused by placental transfer of anti-NEP antibodies to the fetus from NEP-deficient mothers (truncating mutations in the MME gene) who undergo alloimmunization during the pregnancy or from previous miscarriages (51, 91).
IgG4 against aldose reductase (AR) and superoxide dismutase 2 (SOD2) have also been described in multiple PMN cohorts (92, 93). However, these antigens are not expressed on the surface of normal podocytes, antibody titres do not correlate with disease activity, and both AR and SOD2 are involved in oxidative stress, a known downstream effect of podocyte injury (92, 93). Accordingly, in vitro and in vivo evidence demonstrated podocyte membrane expression of SOD2 increasing with intracellular oxidation and after passive transfer of anti-THSD7A IgG respectively (27, 92). These data suggest antibodies targeted AR and SOD2 are a secondary phenomenon to autoantibody-mediated podocyte injury exposing neoantigens rather than autoantibodies driving PMN pathogenesis. More recently, autoantibodies against Formin-like 1 (FMNL1) were detected in patients with PMN as well as other forms of glomerular disease (IgA nephropathy and focal segmental glomerulosclerosis), which recognize FMNL1 expression on macrophages (94). Whether FMNL1 positive macrophages contribute to the pathogenesis of PMN (ie. M1 macrophages) or highlight a reparative role of macrophages (ie. M2 macrophages) in PMN requires further study.
There is a clear T cell role in Heymann nephritis with CD4+ T cell depletion abolishing IgG and C3 deposition as well as proteinuria, demonstrating the essential role of CD4+ T cell-B cell interaction in autoantibody formation (95). In contrast, CD8+ T cell depletion reduced proteinuria with intact glomerular IgG and C3 deposition in AHN (59, 96), and reduced proteinuria in the autologous phase (week 2-4) in PHN but not during the heterologous phase (week 1) where infused anti-Fx1A antibodies cause complement-mediated podocyte injury (97). Therefore, the small CD8+ T cell infiltrate seen in Heymann nephritis likely occurs downstream of podocyte antibody deposition and proteinuria.
Development of anti-Fx1A autoantibodies in AHN suggest loss of immune tolerance, likely due to escape from thymic deletion by megalin-specific effector T cells and/or impaired induction of megalin-specific Foxp3+ CD4+ Tregs, though overall levels of Foxp3+ CD4+ Tregs are unchanged in AHN (98). In contrast, evidence of regulation that dampen autoimmunity in AHN were found to be mediated by thymus-derived CD8+ T cells, which reduced autoantibody production and proteinuria upon adoptive transfer (99, 100). In particular, CD8+ Tregs reduced anti-Fx1A antibody production in AHN by targeting CD4+ T follicular helper (Tfh) cells expressing the non-classical MHC class I molecule Qa-1 (Figure 1A.5). In PMN, CD8+ Tregs have yet to be described while CD4+ Tregs were reduced in patients with active disease prior to receiving immunosuppression (101).
Tomas et al. demonstrated that human PLA2R was not expressed in the thymus of their transgenic mice, explaining the spontaneous formation of anti-human PLA2R antibodies is likely due to a lack of central tolerance (28). In contrast, another study described transgenic knock-in mice with ubiquitous expression of human PLA2R that did not develop a PMN phenotype, likely due to establishment of immune tolerance to human PLA2R, though expression of human PLA2R in the thymus of these mice was not reported (102). Lastly, in the study by Tomas et al, a PMN phenotype did not develop in recombination activating gene (RAG) 2 knock-out mice expressing human PLA2R specifically on podocytes, indicating the requirement of mature T and B cells for the formation of anti-human PLA2R autoantibodies (28).
Study of T cell subsets in PMN have revealed an increased CD4+:CD8+ T cell ratio (103, 104). Further analysis of CD4+ T cells in PMN showed increased Th2 cells without change in Th1 cells, increased Tfh cells, and lower CD4+ Tregs, which were associated with increased IgG4 production, increased plasma cells and a loss of immune tolerance respectively (Figure 1B.4) (101, 103, 105–108). Our understanding of PMN disease antigens and class II HLA linkage suggest that HLA-driven effector T cells and/or the loss of Tregs likely play a role in loss of immune tolerance, similar to Goodpasture disease, another autoimmune kidney disease caused by autoantibodies directed against a glomerular antigen (109). Indeed, reduced CD4+ Treg number and/or function have been described in various autoimmune diseases, demonstrating its critical role in maintaining immune tolerance (110–113). Accordingly, enhanced ability of Tregs to suppress the anti-PLA2R response in PMN has also been hypothesized to be the mechanism underlying the spontaneous remission observed in a third of patients, however data supporting this theory are lacking (114).
More recently, increased Th17 cells have been demonstrated to correlate with disease activity, relapse and thrombotic complications in PMN (115, 116), and IL-17 signaling was differentially expressed in single-cell RNA sequencing of kidney biopsy specimens in patients with PLA2R-associated PMN (117). Whether Th17 activation is triggered by glomerular antibody deposition, proteinuria or tissue injury requires further study. Single-cell RNA sequencing of kidney biopsy specimens in PMN also identified tumor necrosis factor (TNF) signaling and NOD-like receptor signaling in glomerular endothelial cells, pericytes and tubular cells compared to healthy controls, suggesting a role of innate immunity (117). In comparison, bulk RNA sequencing and microarray expression profiles of the glomerular compartment of kidney biopsies revealed a PMN-specific signature significantly enriched in NF-kB1 targets compared to other glomerulopathies, which is consistent with a GWAS finding of a variant at the NF-kB1 locus conferring an increased risk of PMN (75, 118). Regarding the significance of TNF signaling in PMN, circulating TNF receptors correlated with proteinuria and tubular TNF receptor expression but not anti-PLA2R autoantibody titres, which may reflect a downstream effect of heavy proteinuria on stimulating tubular cell chemokine production and infiltration of TNF-α-producing macrophages and lymphocytes (119, 120). Indeed, TNF inhibition reduced kidney inflammatory infiltrates but not proteinuria or immune complex deposition in BSA-induced murine MN, highlighting the need to validate the findings of transcriptomic profiling with in vivo studies to improve our understanding of disease pathophysiology (121).
Analysis of B cell populations in PMN have demonstrated an increase in naive B cells and reduced memory B cells, which could be secondary to Tfh-mediated differentiation of B cells into autoantibody-producing plasma cells or B cell infiltration into the kidney (104, 122). B cell depletion with rituximab was associated with increased CD4+ Tregs, potentially due to B cell expression of interferon-γ, which suppressed CD4+ Tregs in animal studies (Figure 1B.4) (104, 123, 124). Regulatory B cells (Bregs) have been implicated in the suppression of B-cell-mediated autoimmunity through IL-10 production. Breg levels in PMN have varied between studies due to differences in surface markers assessed (101, 125, 126), though overall higher Bregs correlated with better treatment responses (101, 125).
Despite the rapid discovery of antigens targeted by autoantibodies in PMN, identification of antigen-specific T and B cells has yet to be described. T cell receptor (TCR) gene sequencing in patients with PMN revealed lower diversity of the VDJ cassette combination compared to healthy controls, which may represent expansion of antigen-specific T cells (127). The same group analyzed the B cell receptor (BCR) sequences in patients with PMN and found increased somatic hypermutation and length distribution of the third complementarity-determining region of the heavy chain (CDR-H3) of all immunoglobulin isotypes compared to healthy controls, indicative of overall increased B cell activation in PMN (128). Future studies linking TCR and BCR sequences with pathogenic epitopes of PLA2R (or other antigens implicated in MN) may assist in identifying autoreactive T and B cells.
While megalin alone could recapitulate AHN, immunization with Fx1A caused more severe proteinuria suggesting additional antigens targeted by anti-Fx1A autoantibodies contributed to podocyte injury (129). Such antigens include solid-phase complement regulatory proteins such as Crry (murine equivalent of complement receptor 1 (CR1)) and CD59 (Figure 1A.3, Figure 2). Indeed, Crry-deficient Fx1A could not induce AHN without administration of anti-Crry antibodies (130). Lower glomerular CR1 and CD59 are reported in PMN though without evidence of associated autoantibodies and may simply represent podocyte loss (131, 132). Overall, these data suggest complement activation plays an important role in mediating podocyte injury after antibody deposition in Heymann nephritis.
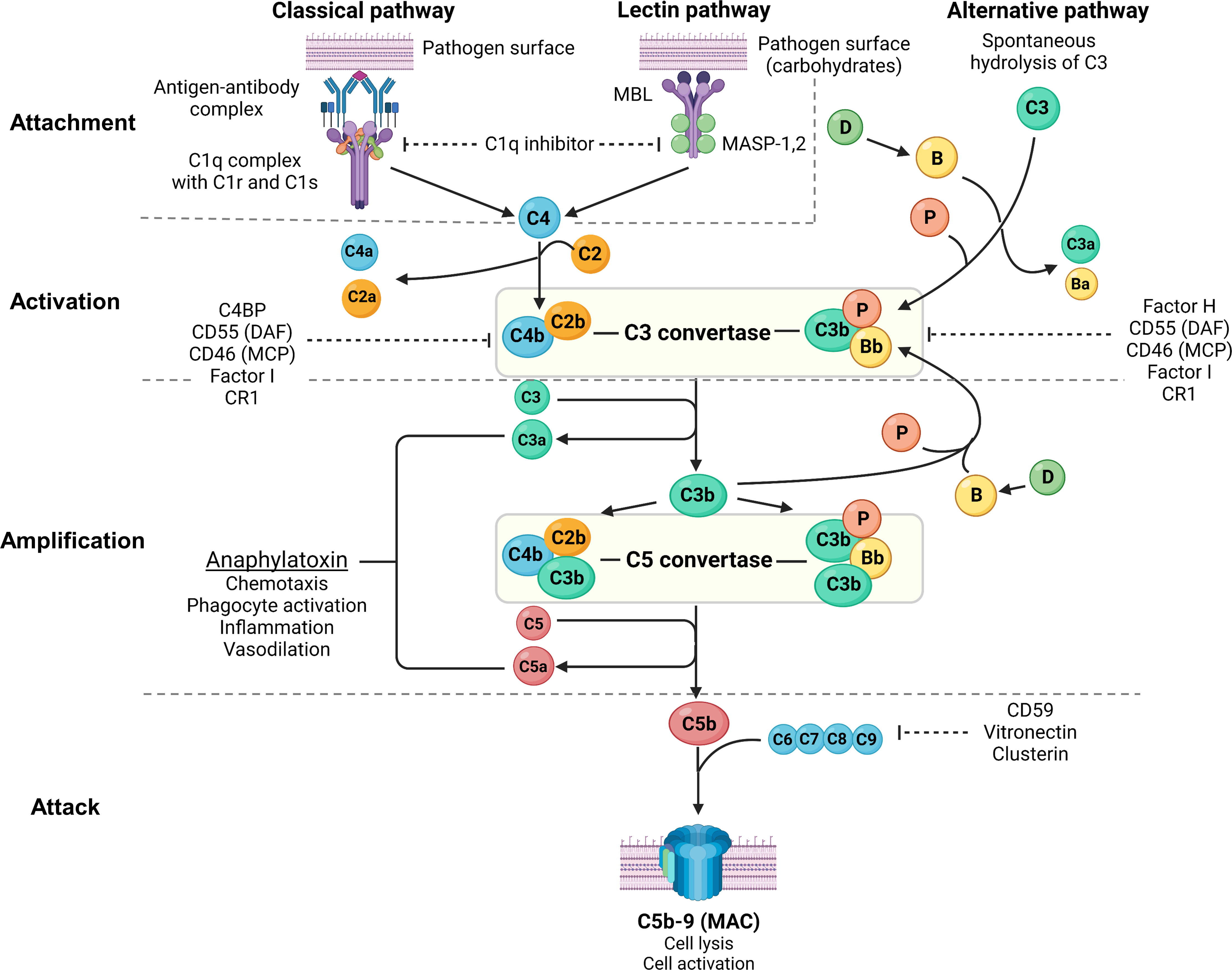
Figure 2 Overview of the complement system. Created with BioRender.com. MBL, mannan-binding lectin; MASP, mannan-binding lectin serine protease; C4BP, C4b-binding protein; DAF, decay-accelerating factor; MCP, membrane cofactor protein; CR1, complement receptor 1; MAC, membrane attack complex; C3aR, C3a receptor; C5aR, C5a receptor; PMN, primary membranous nephropathy; PLA2R, M-type phospholipase A2 receptor 1; THSD7A, thrombospondin type-1 domain-containing protein 7A. The complement system consists of 3 pathways: classical pathway, lectin pathway and alternative pathway, which can be conceptually understood using 4A’s: attachment, activation, amplification, and attack (steps of complement activation in bold text below). In the classical pathway, complement C1q attaches to the Fc receptor of antigen-bound antibodies (IgG1, IgG3 and IgM in humans, and IgG2a and IgG2b in rodents). The lectin pathway is similar to the classical pathway where lectins (such as ficolins, MBLs or collectins) attach to carbohydrates on pathogens, activating MASP-1 and MASP-2 (analogous to C1r and C1s of the classical pathway), which cleave C4 and C2 to form C3 convertase (C4b2b). In the alternative pathway, small amounts of C3 are spontaneously activated (“C3 tickover”) due to a labile thioester bond, forming a C3b fragment that binds to factor B (B), which in turn is cleaved by factor D (D) to form the alternative pathway C3 convertase (C3bBb) that is stabilised by properdin (P), forming C3bBbP. Regardless of the pathway of activation, C3 convertase cleaves C3 into C3a and C3b, which amplifies alternative pathway activation whereby further factor B binds to C3b forming more C3 convertase. Apart from amplifying C3 convertase formation, C3b also acts as an opsonin and binds to C3 convertase to form C5 convertase (C4b2b3b and C3bBbC3bP), which cleaves C5 into C5a and C5b. C3a and C5a act as potent anaphylatoxins via their respective receptors (C3aR and C5aR). Complement attack is initiated by C5b sequentially binding C6, C7, C8, and C9 to form the MAC (C5b-9), which causes transmembrane pores that cause osmotic cell lysis or sublethal cell injury, activating internal cellular processes. The complement system is tightly regulated by proteins present in plasma (fluid-phase) and on cell surfaces including the podocyte (solid-phase). Fluid-phase regulators include C1q inhibitor (binds C1r/C1s and MASP-1/MASP-2 to prevent classical and lectin pathway activation), C4BP (binds C4b to prevent formation of the classical/lectin pathway C3 convertase), factor H (binds C3b to prevent formation of the alternative pathway C3 convertase), factor I (cofactor for factor H and MCP, which both inhibit C3 convertase formation), vitronectin (binds the C5b-7 complex to prevent MAC formation) and clusterin (binds C7, C8, C9 to prevent MAC formation). Solid-phase regulators include DAF (dissociates C2b from C4b and Bb from C3b to inactivate C3 convertase), MCP (binds C3b and C4b to prevent C3 convertase formation), CR1 (binds C3b and C4b to prevent C3 convertase formation) and CD59 (binds C8 and C9 to prevent MAC formation).
The crucial role of complement-mediated glomerular injury in Heymann nephritis was demonstrated by the prevention of proteinuria in PHN with complement depletion using cobra venom factor (133). In AHN, proteinuria was dependent on glomerular deposition of rat complement-fixing IgG2b suggesting classical pathway activation (57, 58), and increased podocyte expression of complement factor H (CFH) in PHN also suggests alternative pathway activation (134). More recently, classical and alternative pathway but not lectin pathway activation was confirmed in PHN with associated increases in plasma C3a and glomerular expression of C3a receptor (C3aR). Accordingly, C3aR antagonism ameliorated disease by inhibiting the downregulation of synaptopodin (actin-associated protein in podocytes) and Bcl2 (apoptosis inhibition gene), and the upregulation of β-catenin on podocytes, which was recapitulated in human podocyte cell lines exposed to PMN sera (135). Interestingly, C3aR antagonism did not alter inflammatory cell infiltration (including CD4+ T cells, CD8+ T cells, and macrophages) in the kidney of PHN rats despite C3a, an anaphylatoxin, being a potent chemoattractant (135).
Glomerular deposition of terminal complement component C5b-9 has been demonstrated in both Heymann nephritis and PMN (70). PVG/c rats with or without C6 deficiency induced with PHN or AHN demonstrated no differences in proteinuria, though C6 depletion using goat anti-rat C6 IgG in Sprague-Dawley rats with PHN significantly reduced proteinuria (97, 136, 137). Nevertheless, C5b-9 has been shown to cause sublethal podocyte injury, leading to activation of cytosolic phospholipase A2, oxidative stress, lipid peroxidation, DNA damage, and disruption of the podocyte slit diaphragm (Figure 1A.4) (138).
Despite glomerular IgG and complement deposition being well-described in PMN, this finding long appeared to be inconsistent with the predominance of non-complement fixing IgG4. While there is a predominance of complement-fixing IgG1 deposition in early stages of PMN (139, 140), the lack of glomerular C1q but almost universal C4d deposition suggested activation of the lectin pathway rather than the classical pathway (Table 2) (141). Indeed, glomerular deposition of mannan-binding lectin (MBL) co-localized with anti-PLA2R IgG4 and correlated with disease activity (142, 143). Recently, Haddad et al. provided important insights into the mechanism of lectin pathway activation in PMN by showing glycosylation patterns on anti-PLA2R IgG4 allow binding of MBL (Figure 1B.5). Such glycosylation patterns were not demonstrated in IgG4 of PLA2R-negative PMN, secondary MN or other glomerular diseases, demonstrating distinct autoimmune responses against different antigens implicated in PMN. Furthermore, anti-PLA2R IgG4 but not IgG4-depleted sera from patients with PLA2R-associated PMN was essential for the proteolysis of two podocyte proteins synaptopodin and NEPH1 via C3aR and C5aR activation on podocytes (Figure 1B.6), which could be prevented by inhibiting MBL-associated serine protease (MASP) or using C6-depleted sera (70). Increased serum C3a and glomerular expression of C3aR have also been demonstrated in PMN patients, which correlated with disease activity and was associated with increased podocyte expression of PLA2R and reduced expression of synaptopodin (135).
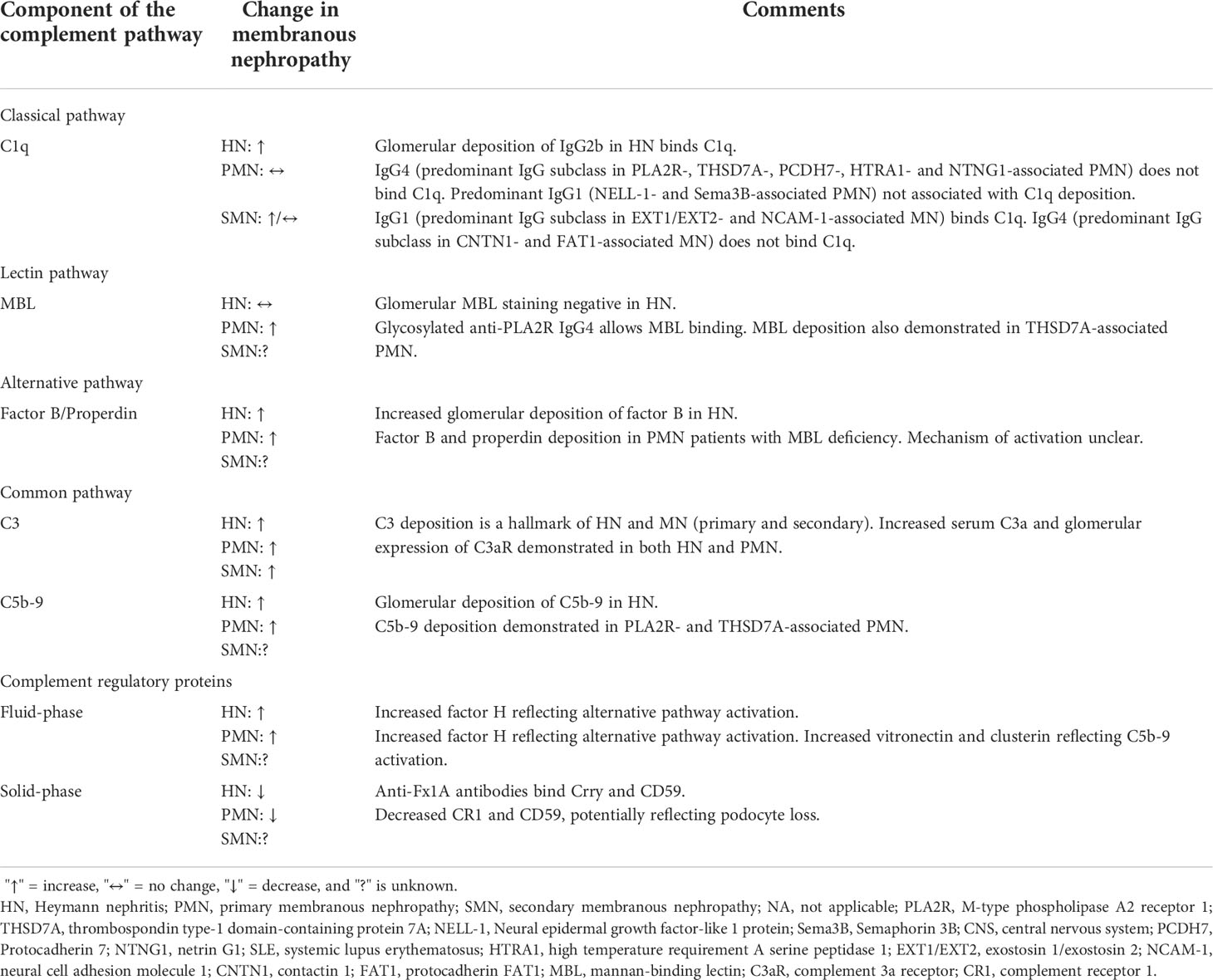
Table 2 Complement system involvement in Heymann nephritis and membranous nephropathy.
Despite growing evidence of lectin pathway activation in PMN, PLA2R-associated PMN has also been described in patients with MBL deficiency in whom disease activity was mediated by alternative pathway activation (144). However, the mechanism of alternative pathway activation in PMN without initial activation of the lectin pathway remains unclear though one could postulate the role of traditional activators of the alternative pathway such as infection, which increase C3a levels and subsequent overexpression of PLA2R on podocytes that predisposes to autoimmunity. Anti-CFH autoantibodies are an unlikely mechanism of alternative pathway activation, detected in only 3% of patients with PMN (145). CFH also binds to human glomeruli via interaction with heparan sulfate, which is downregulated in PMN, AHN and PHN though whether this results in alternative pathway activation remains unproven (146–149).
Impaired podocyte autophagy, fumarate accumulation in podocytes causing oxidative stress, and disruption of podocyte adhesion to collagen IV in the GBM have also been implicated in glomerular injury in PMN. Impairment of podocyte autophagy in PMN causes internalization of nephrin and α-actinin-4 (podocyte actin cytoskeleton protein), which in PLA2R-associated PMN may be mediated by activation of the mTOR/ULK1ser757 signaling pathway (Figure 1B.7) (150–153). Fumarate hydratase, which co-localizes with podocalyxin on podocytes, is reduced in PLA2R-associated PMN in an oxidative stress- and C5b-9-dependent manner (Figure 1B.8) (154). Inhibition of fumarate hydratase using small interfering RNA results in fumarate accumulation within podocytes causing oxidative stress and reduction in zonula occludens-1 (podocyte slit diaphragm protein) and Wilm’s tumor 1 (podocyte transcription factor) (154, 155). Lastly, in vitro evidence suggests human sera containing anti-PLA2R antibodies impair podocyte adhesion to collagen IV on the GBM though the exact mechanism remains unclear (156).
PMN appears to be different disease entities rather than a uniform disease but with a unified immunological phenotype of autoantibody formation against an antigen mostly expressed on podocytes. Therefore, targeting antibody generation through B-cells, plasma cells and Tfh cells is an attractive strategy (Figure 3). While rituximab, a chimeric anti-CD20 monoclonal antibody, is effective at depleting B cells, treatment response in PMN is variable (60-85% complete or partial remission at 24 months) (12, 15). This may be related to autoantibody production by CD20-negative plasma cells, changes in the CD20 antigen that may be restricted to autoreactive B cells, or the development of neutralizing antibodies against rituximab. Ofatumumab and obinutuzumab are humanized anti-CD20 monoclonal antibodies, which have successfully treated patients with PMN who developed serum sickness to rituximab or anti-rituximab antibodies (157, 158), and demonstrated superior B-cell depletion compared to rituximab respectively (159). The efficacy of binutuzumab and belimumab [humanized monoclonal antibody against B-cell activating factor (BAFF)] in lupus nephritis, another disease mediated in part by autoreactive B cells, and CD19 CAR T cells in treatment-refractory SLE are also promising alternative B cell-depleting therapies, which require further evaluation in PMN (160–163).
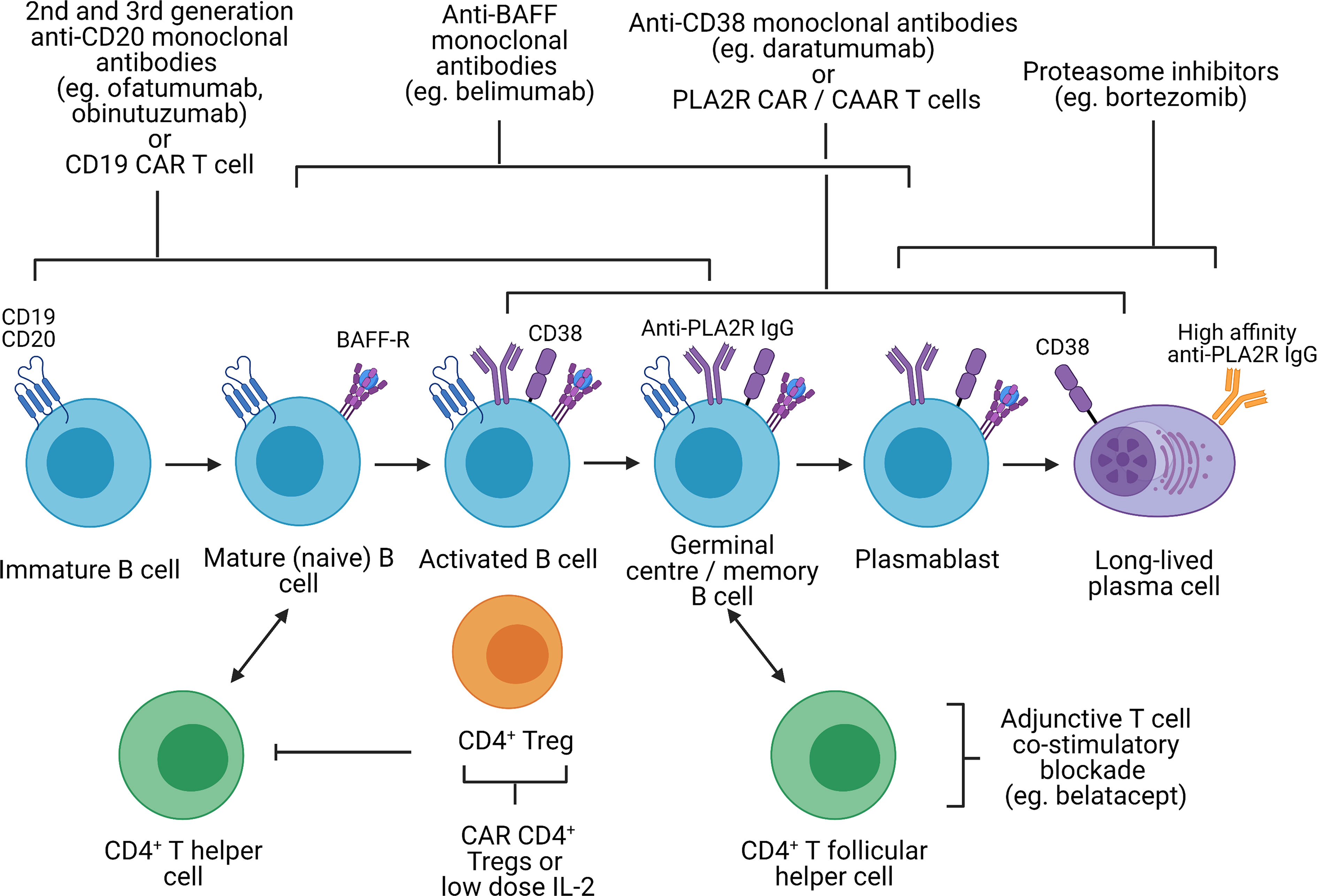
Figure 3 Potential immunological targets for novel therapeutics in primary membranous nephropathy. Created with BioRender.com. BAFF-R, B-cell activating factor receptor; CAR, chimeric antigen receptor; CAAR, chimeric autoantibody receptor; PLA2R, M-type phospholipase A2 receptor 1; Treg, regulatory T cell, In primary membranous nephropathy, autoreactive B cells produce autoantibodies targeting podocyte antigens (represented by PLA2R in this figure but also include thrombospondin type-1 domain-containing protein 7A, neural epidermal growth factor-like 1 protein, semaphorin 3B, protocadherin 7, high temperature requirement A serine peptidase 1, and netrin G1). These autoreactive B cells originate from the bone marrow, mature from immature B cells to mature B cells, become activated upon interaction with CD4+ T helper cells (via cytokines and CD40 ligation) and undergo isotype switching. Activated B cells subsequently migrate to the germinal center of secondary lymphoid organs where they interact with CD4+ T follicular helper cells, resulting in B cell proliferation and somatic hypermutation, ultimately differentiating into plasmablast and long-lived plasma cells which secrete high-affinity autoantibodies. Immature, mature, activated and germinal center B cells express CD19 and CD20, which can be targeted by 2nd and 3rd generation anti-CD20 monoclonal antibodies (eg. ofatumumab and obinutuzumab), which may be superior to rituximab, or CD19 CAR T cells. However, in cases of primary membranous nephropathy resistant to 1st generation anti-CD20 monoclonal antibodies (eg. rituximab) that respond to 2nd or 3rd generation anti-CD20 monoclonal antibodies, this may be due to changes in the CD20 antigen that may be restricted to autoreactive B cells. CD19 and CD20 are also not expressed on plasmablasts or long-lived plasma cells which continue to secrete autoantibodies despite B cell depletion. Monoclonal antibodies targeting BAFF (eg. belimumab), preventing binding to the BAFF receptor, target both B cells and plasmablasts but not long-lived plasma cells. Proteasome inhibitors have been shown to be effective in depleting plasmablasts and plasma cells. Anti-CD38 monoclonal antibodies (eg. daratumumab) may also be effective against plasma cells, plasmablasts and B cells. However, proteasome inhibitors and anti-CD38 monoclonal antibodies are associated significant toxicity and short-duration therapy to deplete plasma cells followed by adjunctive therapy (eg. B cell depletion or T cell co-stimulatory blockade to prevent B cell activation) may be better tolerated. With the discovery of disease antigens underpinning primary membranous nephropathy, CAR T cells or CAAR T cells targeting autoantibodies (eg. anti-PLA2R) on autoreactive B cells, plasmablasts and plasma cells are conceivable. Lastly, harnessing the capacity of CD4+ regulatory T cells to induce immune tolerance at sites of inflammation independent of antigen specificity (infectious tolerance) is attractive. Such strategies may involve CAR regulatory T cells directed to the kidney (eg. targeting PLA2R on the podocyte) or low-dose IL-2, which preferentially expands regulatory T cells not effector T cells.
Treatment resistance to B-cell depletion may also be attributable to persistent memory plasma cells, which produce autoantibodies independent of T and B cell interactions. The proteasome inhibitor bortezomib effectively depletes plasma cells and has been effective in both treatment-resistant and treatment-naïve PMN (164, 165). Plasma cells express CD38 and CD138, which could be targeted by anti-CD38 monoclonal antibodies such as daratumumab. However, safety concerns of neurotoxicity with bortezomib and infection with both bortezomib and daratumumab may limit their use. An alternative approach could be a short course of plasma cell-depleting therapy followed by B cell depletion or T cell co-stimulatory blockade to inhibit the Tfh-mediated B cell differentiation into plasma cells. In an uncontrolled cohort study and a case series of kidney transplant recipients with antibody-mediated rejection, short-duration bortezomib (on average 4 doses of 1.3 mg/m2) followed by adjunctive therapy (including rituximab, plasmapheresis, or intravenous immunoglobulin) or T cell co-stimulatory blockade with belatacept respectively were effective (166, 167). However, controlled clinical trials are needed to confirm the efficacy of these treatment regimens.
The discovery of the disease antigens driving PMN also raises the possibility of antigen-specific therapeutics such as CAR or CAAR T cell therapies against PLA2R-specific B cells. In pemphigus vulgaris, an antibody-mediated autoimmune blistering skin disease, CAAR T cells expressing the disease autoantigen desmoglein 3 was able to eliminate desmoglein 3-specific B cells in a mouse model even in the presence of circulating autoantibodies and without off-target toxicity (168). A similar approach is conceivable in PMN where B cell-mediated autoimmunity is driven by a single autoantigen in most cases.
Inducing immune tolerance without abrogating anti-infection and anti-tumor immunity is the ultimate therapeutic strategy. CAR CD4+ Tregs targeting organ-specific antigens suppressed autoimmunity in animal models of type 1 diabetes, multiple sclerosis and autoimmune colitis (169–171). In particular, CAR Tregs in the autoimmune colitis model were able to suppress disease activity despite being specific for an antigen distinct from the disease antigen (171). This demonstrates the phenomenon of infectious tolerance whereby CD4+ Tregs can confer suppressive activity to conventional T cells via membrane-bound transforming growth factor-β (TGF-β) (172, 173). PLA2R may represent an ideal tissue marker to direct CAR Tregs to the glomerulus in PMN. An alternative strategy may be selective expansion of Tregs using low-dose IL-2, which was effective in animal models of rheumatoid arthritis and autoimmune colitis (174). However, a mechanism of IL-2 delivery to the kidney remains unclear.
Two distinct regulatory T cell subsets also exist to maintain immune tolerance at the germinal center: CD8+ Tregs and T follicular regulatory (Tfr) cells. In a mouse model of multiple sclerosis, yeast libraries have been used to identify peptides that engage the CD8+ Treg TCR and vaccination of these peptides suppressed disease activity (175). Neuritin, a protein produced by Tfr cells to prevent differentiation of B cells into plasma cells has also been shown to suppress autoantibody production in Tfr-deficient mice (176). However, the importance of CD8+ Tregs and Tfr cells in PMN remains to be established and development of these promising though experimental therapies will require rigorous pre-clinical evaluation.
Heymann nephritis has significantly contributed to our understanding of the disease mechanisms causing PMN but distinct differences in the underlying disease antigen, predominant autoantibody IgG subclass, and mechanism of complement activation and glomerular injury remain. Despite this, AHN remains a relevant and feasible model that recapitulates the unifying disease phenotype of PMN and therefore is suitable for testing novel therapeutics targeting autoantibody production. Activation of the alternative complement pathway is also a hallmark of both AHN/PHN and PMN with C3aR antagonism ameliorating disease in PHN (135), and an ongoing trial will evaluate the factor B inhibitor iptacopan (LNP023) compared to rituximab in PMN (NCT04154787).
Models of PMN induced by passive administration of anti-PLA2R and anti-THSD7A autoantibodies may better resemble the mechanism of glomerular injury in human PMN. However, PLA2R and THSD7A in these models were not humanized, reflecting the inability of transferred human anti-PLA2R autoantibodies or purified human anti-THSD7A to recapitulate disease. Furthermore, rabbit anti-THSD7A antibodies lack the ability to activate complement, a hallmark of THSD7A-associated PMN. These animal models also require the passive administration of antibodies and therefore do not reproduce the immunological mechanism of autoantibody production in PMN.
Most recently, transgenic knock-in mice expressing human PLA2R specifically on podocytes faithfully recapitulates human PMN phenotype, representing an important and exciting development that opens opportunities to further understand the disease mechanisms driving PMN and test novel antigen-specific therapeutics such as CAR or CAAR T cells. It remains unclear whether a similar approach can be replicated to model MN associated with non-PLA2R antigens, though developing transgenic mice with podocyte-specific expression of the numerous non-PLA2R MN disease antigens present a significant challenge as well as feasibility issues. An alternative approach would be to immunize susceptible murine strains with a specific PMN-associated antigen and adjuvant. However, while expression of Sema3B and serine protease HTRA1 have been demonstrated in murine podocytes, PCDH7 is not detectable in rat kidneys and it is unclear whether murine podocytes express NELL1 or NTNG1 (42, 177–179).
Our understanding of PMN is rapidly evolving with the promise of targeted therapeutics now emerging. Integrating technologies such as mapping T and B cell receptor sequences to their cognate antigen, HLA-peptide tetramer analysis, and single-cell RNA sequencing of lymphocytes will likely offer greater insights into the immune mechanisms causing PMN leading to the development of more effective and targeted treatments in this challenging disease.
Conceptualized review topic: EC, SA. Participated in the writing of the paper: EC, YW, KK, MH, HM, GW, LK, BB, DH, SA. Supervision: YW, KK, SA. Each author contributed important intellectual content during manuscript drafting or revision and agrees to be personally accountable for the individual’s own contributions and to ensure that questions pertaining to the accuracy or integrity of any portion of the work, even one in which the author was not directly involved, are appropriately investigated and resolved, including with documentation in the literature if appropriate. All authors contributed to the article and approved the submitted version.
EC is supported by the NHMRC Postgraduate Research Scholarship (APP 2014037). GW is supported by the NHMRC Fellowships (APP 1195414 and 1147657). SA is supported by the NHMRC Grants (APP 1183810 and 1161554).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol (2017) 12(6):983. doi: 10.2215/CJN.11761116
2. Ronco P, Debiec H. Molecular pathogenesis of membranous nephropathy. Annu Rev Pathol (2020) 15(1):287–313. doi: 10.1146/annurev-pathol-020117-043811
3. Beck LH Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med (2009) 361(1):11–21. doi: 10.1056/NEJMoa0810457
4. Tomas NM, Beck LH, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med (2014) 371(24):2277–87. doi: 10.1056/NEJMoa1409354
5. Du Y, Li J, He F, Lv Y, Liu W, Wu P, et al. The diagnosis accuracy of PLA2R-AB in the diagnosis of idiopathic membranous nephropathy: a meta-analysis. PloS One (2014) 9(8):e104936. doi: 10.1371/journal.pone.0104936
6. Ponticelli C, Zucchelli P, Imbasciati E, Cagnoli L, Pozzi C, Passerini P, et al. Controlled trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med (1984) 310(15):946–50. doi: 10.1056/NEJM198404123101503
7. Ponticelli C, Zucchelli P, Passerini P, Cagnoli L, Cesana B, Pozzi C, et al. A randomized trial of methylprednisolone and chlorambucil in idiopathic membranous nephropathy. N Engl J Med (1989) 320(1):8–13. doi: 10.1056/NEJM198901053200102
8. Ponticelli C, Zucchelli P, Passerini P, Cesana B. Methylprednisolone plus chlorambucil as compared with methylprednisolone alone for the treatment of idiopathic membranous nephropathy. the Italian idiopathic membranous nephropathy treatment study group. N Engl J Med (1992) 327(9):599–603. doi: 10.1056/NEJM199208273270904
9. Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, Cesana B, et al. A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol (1998) 9(3):444–50. doi: 10.1681/ASN.V93444
10. Jha V, Ganguli A, Saha TK, Kohli HS, Sud K, Gupta KL, et al. A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol (2007) 18(6):1899–904. doi: 10.1681/ASN.2007020166
11. Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, et al. Cyclosporine in patients with steroid-resistant membranous nephropathy: a randomized trial. Kidney Int (2001) 59(4):1484–90. doi: 10.1046/j.1523-1755.2001.0590041484.x
12. Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, et al. Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med (2019) 381(1):36–46. doi: 10.1056/NEJMoa1814427
13. Ruggenenti P, Fervenza FC, Remuzzi G. Treatment of membranous nephropathy: time for a paradigm shift. Nat Rev Nephrol (2017) 13(9):563–79. doi: 10.1038/nrneph.2017.92
14. Fernández-Juárez G, Rojas-Rivera J, Logt AV, Justino J, Sevillano A, Caravaca-Fontán F, et al. The STARMEN trial indicates that alternating treatment with corticosteroids and cyclophosphamide is superior to sequential treatment with tacrolimus and rituximab in primary membranous nephropathy. Kidney Int (2021) 99(4):986–98. doi: 10.1016/j.kint.2020.10.014
15. Scolari F, Delbarba E, Santoro D, Gesualdo L, Pani A, Dallera N, et al. Rituximab or cyclophosphamide in the treatment of membranous nephropathy: The RI-CYCLO randomized trial. J Am Soc Nephrol (2021) 32(4):972–82. doi: 10.1681/ASN.2020071091
16. Grupper A, Cornell LD, Fervenza FC, Beck LH Jr., Lorenz E, Cosio FG. Recurrent membranous nephropathy after kidney transplantation: Treatment and long-term implications. Transplantation (2016) 100(12):2710–16. doi: 10.1097/TP.0000000000001056
17. Stahl R, Hoxha E, Fechner K. PLA2R autoantibodies and recurrent membranous nephropathy after transplantation. N Engl J Med (2010) 363(5):496–8. doi: 10.1056/NEJMc1003066
18. Dabade TS, Grande JP, Norby SM, Fervenza FC, Cosio FG. Recurrent idiopathic membranous nephropathy after kidney transplantation: a surveillance biopsy study. Am J Transpl (2008) 8(6):1318–22. doi: 10.1111/j.1600-6143.2008.02237.x
19. Moroni G, Gallelli B, Quaglini S, Leoni A, Banfi G, Passerini P, et al. Long-term outcome of renal transplantation in patients with idiopathic membranous glomerulonephritis (MN). Nephrol Dial Transpl (2010) 25(10):3408–15. doi: 10.1093/ndt/gfq223
20. Sadeqi Nezhad M, Seifalian A, Bagheri N, Yaghoubi S, Karimi MH, Adbollahpour-Alitappeh M. Chimeric antigen receptor based therapy as a potential approach in autoimmune diseases: How close are we to the treatment? Front Immunol (2020) 11:603237. doi: 10.3389/fimmu.2020.603237
21. Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by freund’s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med (1959) 100(4):660–4. doi: 10.3181/00379727-100-24736
22. Couser W, Steinmuller D, Stilmant M, Salant D, Lowenstein L. Experimental glomerulonephritis in the isolated perfused rat kidney. J Clin Invest (1978) 62(6):1275–87. doi: 10.1172/JCI109248
23. Adler S, Baker PJ, Pritzl P, Couser WG. Detection of terminal complement components in experimental immune glomerular injury. Kidney Int (1984) 26(6):830–7. doi: 10.1038/ki.1984.225
24. Meyer-Schwesinger C, Tomas NM, Dehde S, Seifert L, Hermans-Borgmeyer I, Wiech T, et al. A novel mouse model of phospholipase A2 receptor 1-associated membranous nephropathy mimics podocyte injury in patients. Kidney Int (2020) 97(5):913–9. doi: 10.1016/j.kint.2019.10.022
25. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies against thrombospondin type 1 domain–containing 7A induce membranous nephropathy. J Clin Invest (2016) 126(7):2519–32. doi: 10.1172/JCI85265
26. Wang Z, Wen L, Dou Y, Zhao Z. Human anti-thrombospondin type 1 domain-containing 7A antibodies induce membranous nephropathy through activation of lectin complement pathway. Biosci Rep (2018) 38(3). doi: 10.1042/BSR20180131
27. Tomas NM, Meyer-Schwesinger C, von Spiegel H, Kotb AM, Zahner G, Hoxha E, et al. A heterologous model of thrombospondin type 1 domain-containing 7A-associated membranous nephropathy. J Am Soc Nephrol (2017) 28(11):3262. doi: 10.1681/ASN.2017010030
28. Tomas NM, Dehde S, Meyer-Schwesinger C, Huang M, Hermans-Borgmeyer I, Maybaum J, et al. Podocyte expression of human phospholipase A2 receptor 1 causes immune-mediated membranous nephropathy in mice. Kidney Int (2022). doi: 10.1016/j.kint.2022.09.008
29. Assmann K, Van Son J, Dijkman H, Koene R. A nephritogenic rat monoclonal antibody to mouse aminopeptidase a. induction of massive albuminuria after a single intravenous injection. J Exp Med (1992) 175(3):623–35. doi: 10.1084/jem.175.3.623
30. Dijkman HB, Gerlofs-Nijland ME, van der Laak JA, Wetzels JF, Groenen PJ, Assmann KJ. Podocyte changes after induction of acute albuminuria in mice by anti-aminopeptidase a mAb. Nephron Exp Nephrol (2003) 94(3):e85–93. doi: 10.1159/000072026
31. Natori Y, Hayakawa I, Shibata S. Identification of gp108, a pathogenic antigen of passive heymann nephritis, as dipeptidyl peptidase IV. Clin Exp Immunol (1987) 70(2):434.
32. Meyer-Schwesinger C, Dehde S, Klug P, Becker JU, Mathey S, Arefi K, et al. Nephrotic syndrome and subepithelial deposits in a mouse model of immune-mediated anti-podocyte glomerulonephritis. J Immunol (2011) 187(6):3218–29. doi: 10.4049/jimmunol.1003451
33. Meyer T, Schwesinger C, Wahlefeld J, Dehde S, Kerjaschki D, Becker J, et al. A new mouse model of immune-mediated podocyte injury. Kidney Int (2007) 72(7):841–52. doi: 10.1038/sj.ki.5002450
34. Border WA, Harry JW, Kamil ES, Cohen AH. Induction of membranous nephropathy in rabbits by administration of an exogenous cationic antigen: Demonstration of a pathogenic role for electrical charge. J Clin Invest (1982) 69(2):451–61. doi: 10.1172/JCI110469
35. Zhang J-J, Malekpour M, Luo W, Ge L, Olaru F, Wang X-P, et al. Murine membranous nephropathy: Immunization with α3 (IV) collagen fragment induces subepithelial immune complexes and FcγR-independent nephrotic syndrome. J Immunol (2012) 188(7):3268–77. doi: 10.4049/jimmunol.1103368
36. Lerner GB, Virmani S, Henderson JM, Francis JM, Beck LH Jr. A conceptual framework linking immunology, pathology, and clinical features in primary membranous nephropathy. Kidney Int (2021) 100(2):289–300. doi: 10.1016/j.kint.2021.03.028
37. Chatelet F, Brianti E, Ronco P, Roland J, Verroust P. Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. i. renal distribution. Am J Pathol (1986) 122(3):500.
38. Chatelet F, Brianti E, Ronco P, Roland J, Verroust P. Ultrastructural localization by monoclonal antibodies of brush border antigens expressed by glomeruli. II. extrarenal distribution. Am J Pathol (1986) 122(3):512.
39. Zheng G, Bachinsky DR, Stamenkovic I, Strickland DK, Brown D, Andres G, et al. Organ distribution in rats of two members of the low-density lipoprotein receptor gene family, gp330 and LRP/alpha 2MR, and the receptor-associated protein (RAP). J Histochem Cytochem (1994) 42(4):531–42. doi: 10.1177/42.4.7510321
40. Sethi S, Debiec H, Madden B, Charlesworth MC, Morelle J, Gross L, et al. Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int (2020) 97(1):163–74. doi: 10.1016/j.kint.2019.09.014
41. Sethi S, Debiec H, Madden B, Vivarelli M, Charlesworth MC, Ravindran A, et al. Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int (2020) 98(5):1253–64. doi: 10.1016/j.kint.2020.05.030
42. Guan F, Villegas G, Teichman J, Mundel P, Tufro A. Autocrine class 3 semaphorin system regulates slit diaphragm proteins and podocyte survival. Kidney Int (2006) 69(9):1564–9. doi: 10.1038/sj.ki.5000313
43. Sethi S, Madden B, Casal Moura M, Nasr SH, Klomjit N, Gross L, et al. Hematopoietic Stem Cell Transplant-Membranous Nephropathy Is Associated with Protocadherin FAT1. J Am Soc Nephrol (2022) 33(5):1033–44. doi: 10.1681/ASN.2020081165
44. Al-Rabadi LF, Caza T, Trivin-Avillach C, Rodan AR, Andeen N, Hayashi N, et al. Serine protease HTRA1 as a novel target antigen in primary membranous nephropathy. J Am Soc Nephrol (2021) 32(7):1666–81. doi: 10.1681/ASN.2020101395
45. Reinhard L, Machalitza M, Wiech T, Groene HJ, Lassé M, Rinschen M, et al. Netrin G1 is a novel target antigen in primary membranous nephropathy. J Am Soc Nephrol (2022) 33(10):1823–31. doi: 10.1681/ASN.2022050608
46. Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al. Exostosin 1/Exostosin 2–associated membranous nephropathy. J Am Soc Nephrol (2019) 30(6):1123. doi: 10.1681/ASN.2018080852
47. Caza TN, Hassen SI, Kuperman M, Sharma SG, Dvanajscak Z, Arthur J, et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int (2020) 100(1):171–81. doi: 10.1016/j.kint.2020.09.016
48. Caza TN, Hassen SI, Kenan DJ, Storey A, Arthur JM, Herzog C, et al. Transforming growth factor beta receptor 3 (TGFBR3)-associated membranous nephropathy. Kidney360 (2021) 2(8):1275–86. doi: 10.34067/KID.0001492021
49. Le Quintrec M, Teisseyre M, Bec N, Delmont E, Szwarc I, Perrochia H, et al. Contactin-1 is a novel target antigen in membranous nephropathy associated with chronic inflammatory demyelinating polyneuropathy. Kidney Int (2021) 100(6):1240–9. doi: 10.1016/j.kint.2021.08.014
50. Sethi S, Madden B, Casal Moura M, Nasr SH, Klomjit N, Gross L, et al. Hematopoietic stem cell transplant-membranous nephropathy is associated with protocadherin FAT1. J Am Soc Nephrol (2022) 33(5):1033–44. doi: 10.1681/ASN.2021111488
51. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med (2002) 346(26):2053–60. doi: 10.1056/NEJMoa012895
52. Erhuma M, Köbel M, Mustafa T, Wulfänger J, Dralle H, Hoang-Vu C, et al. Expression of neutral endopeptidase (NEP/CD10) on pancreatic tumor cell lines, pancreatitis and pancreatic tumor tissues. Int J Cancer (2007) 120(11):2393–400. doi: 10.1002/ijc.22252
53. Edgington TS, Glassock RJ, Dixon FJ. Autologous immune complex nephritix induced with renal tubular antigen: I. identification and isolation of the pathogenic antigen. J Exp Med (1968) 127(3):555–72. doi: 10.1084/jem.127.3.555
54. Kerjaschki D, Farquhar MG. The pathogenic antigen of heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci U S A (1982) 79(18):5557–61. doi: 10.1073/pnas.79.18.5557
55. Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol (1995) 6(1):35. doi: 10.1681/ASN.V6135
56. Kukida M, Sawada H, Daugherty A, Lu HS. Megalin: a bridge connecting kidney, the renin-angiotensin system, and atherosclerosis. Pharmacol Res (2020) 151:104537. doi: 10.1016/j.phrs.2019.104537
57. Brüggemann M, Teale C, Clark M, Bindon C, Waldmann H. A matched set of rat/mouse chimeric antibodies. identification and biological properties of rat h chain constant regions mu, gamma 1, gamma 2a, gamma 2b, gamma 2c, epsilon, and alpha. J Immunol (1989) 142(9):3145–50.
58. Noble B, Van Liew JB, Andres GA, Brentjens JR. Factors influencing susceptibility of LEW rats to heymann nephritis. Clin Immunol Immunopathol (1984) 30(2):241–54. doi: 10.1016/0090-1229(84)90059-X
59. Penny MJ, Boyd RA, Hall BM. Permanent CD8+ T cell depletion prevents proteinuria in active heymann nephritis. J Exp Med (1998) 188(10):1775–84. doi: 10.1084/jem.188.10.1775
60. Shah P, Tramontano A, Makker SP. Intramolecular epitope spreading in heymann nephritis. J Am Soc Nephrol (2007) 18(12):3060–6. doi: 10.1681/ASN.2007030342
61. Tramontano A, Makker SP. Conformation and glycosylation of a megalin fragment correlate with nephritogenicity in heymann nephritis. J Immunol (2004) 172(4):2367. doi: 10.4049/jimmunol.172.4.2367
62. Fresquet M, Jowitt TA, Gummadova J, Collins R, O’Cualain R, McKenzie EA, et al. Identification of a major epitope recognized by PLA2R autoantibodies in primary membranous nephropathy. J Am Soc Nephrol (2015) 26(2):302. doi: 10.1681/ASN.2014050502
63. Fresquet M, Rhoden SJ, Jowitt TA, McKenzie EA, Roberts I, Lennon R, et al. Autoantigens PLA2R and THSD7A in membranous nephropathy share a common epitope motif in the n-terminal domain. J Autoimmun (2020) 106:102308. doi: 10.1016/j.jaut.2019.102308
64. Reinhard L, Zahner G, Menzel S, Koch-Nolte F, Stahl RAK, Hoxha E. Clinical relevance of domain-specific phospholipase A(2) receptor 1 antibody levels in patients with membranous nephropathy. J Am Soc Nephrol (2020) 31(1):197–207. doi: 10.1681/ASN.2019030273
65. Perez-Gomez MV, Sanchez-Niño MD, Ortiz A. Megalin/lipoprotein receptor-related protein 2 autoimmunity and kidney disease. Clin Kidney J (2020) 13(3):281–6. doi: 10.1093/ckj/sfz171
66. Larsen CP, Trivin-Avillach C, Coles P, Collins AB, Merchant M, Ma H, et al. LDL receptor-related protein 2 (Megalin) as a target antigen in human kidney anti-brush border antibody disease. J Am Soc Nephrol (2018) 29(2):644–53. doi: 10.1681/ASN.2017060664
67. Hofstra JM, Beck LH, Beck DM, Wetzels JF, Salant DJ. Anti-phospholipase A2 receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol (2011) 6(6):1286–91. doi: 10.2215/CJN.07210810
68. Rao SJ, Shen Q, Wang HM, Tang S, Wang XY. The association of anti-PLA2R with clinical manifestations and outcomes in idiopathic membranous nephropathy: a meta-analysis. Int Urol Nephrol (2020) 52(11):2123–33. doi: 10.1007/s11255-020-02588-7
69. Ruggenenti P, Debiec H, Ruggiero B, Chianca A, Pellé T, Gaspari F, et al. Anti-phospholipase A2 receptor antibody titer predicts post-rituximab outcome of membranous nephropathy. J Am Soc Nephrol (2015) 26(10):2545. doi: 10.1681/ASN.2014070640
70. Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1–associated membranous nephropathy. J Clin Invest (2021) 131(5):e140453. doi: 10.1172/JCI140453
71. Fresquet M, Lockhart-Cairns MP, Rhoden SJ, Jowitt TA, Briggs DC, Baldock C, et al. Structure of PLA2R reveals presentation of the dominant membranous nephropathy epitope and an immunogenic patch. Proc Natl Acad Sci U S A (2022) 119(29):e2202209119. doi: 10.1073/pnas.2202209119
72. Seitz-Polski B, Debiec H, Rousseau A, Dahan K, Zaghrini C, Payré C, et al. Phospholipase A2 receptor 1 epitope spreading at baseline predicts reduced likelihood of remission of membranous nephropathy. J Am Soc Nephrol (2018) 29(2):401. doi: 10.1681/ASN.2017070734
73. Seitz-Polski B, Dolla G, Payré C, Girard CA, Polidori J, Zorzi K, et al. Epitope spreading of autoantibody response to PLA2R associates with poor prognosis in membranous nephropathy. J Am Soc Nephrol (2016) 27(5):1517. doi: 10.1681/ASN.2014111061
74. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med (2011) 364(7):616–26. doi: 10.1056/NEJMoa1009742
75. Xie J, Liu L, Mladkova N, Li Y, Ren H, Wang W, et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun (2020) 11(1):1600. doi: 10.1038/s41467-020-15383-w
76. Cui Z, Xie LJ, Chen FJ, Pei ZY, Zhang LJ, Qu Z, et al. MHC class II risk alleles and amino acid residues in idiopathic membranous nephropathy. J Am Soc Nephrol (2017) 28(5):1651–64. doi: 10.1681/ASN.2016020114
77. Debiec H, Martin L, Jouanneau C, Dautin G, Mesnard L, Rondeau E, et al. Autoantibodies specific for the phospholipase A2 receptor in recurrent and de novo membranous nephropathy. Am J Transpl (2011) 11(10):2144–52. doi: 10.1111/j.1600-6143.2011.03643.x
78. Kattah A, Ayalon R, Beck LH Jr., Sethi S, Sandor DG, Cosio FG, et al. Anti-phospholipase A2 receptor antibodies in recurrent membranous nephropathy. Am J Transpl (2015) 15(5):1349–59. doi: 10.1111/ajt.13133
79. Quintana LF, Blasco M, Seras M, Pérez NS, López-Hoyos M, Villarroel P, et al. Antiphospholipase A2 receptor antibody levels predict the risk of posttransplantation recurrence of membranous nephropathy. Transplantation (2015) 99(8):1709–14. doi: 10.1097/TP.0000000000000630
80. Berchtold L, Letouzé E, Alexander MP, Canaud G, Logt AV, Hamilton P, et al. HLA-d and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int (2021) 99(3):671–85. doi: 10.1016/j.kint.2020.08.007
81. Batal I, Vasilescu ER, Dadhania DM, Adel AA, Husain SA, Avasare R, et al. Association of HLA typing and alloimmunity with posttransplantation membranous nephropathy: A multicenter case series. Am J Kidney Dis (2020) 76(3):374–83. doi: 10.1053/j.ajkd.2020.01.009
82. Seifert L, Hoxha E, Eichhoff AM, Zahner G, Dehde S, Reinhard L, et al. The most n-terminal region of THSD7A is the predominant target for autoimmunity in THSD7A-associated membranous nephropathy. J Am Soc Nephrol (2018) 29(5):1536. doi: 10.1681/ASN.2017070805
83. Larsen CP, Cossey LN, Beck LH. THSD7A staining of membranous glomerulopathy in clinical practice reveals cases with dual autoantibody positivity. Mod Pathol (2016) 29(4):421–6. doi: 10.1038/modpathol.2016.32
84. Fila M, Debiec H, Perrochia H, Djouadi N, Verpont MC, Buob D, et al. Recurrence of anti-semaphorin 3B-mediated membranous nephropathy after kidney transplantation. J Am Soc Nephrol (2022) 33(3):503–9. doi: 10.1681/ASN.2021101323
85. Sethi S. New ‘Antigens’ in membranous nephropathy. J Am Soc Nephrol (2021) 32(2):268–78. doi: 10.1681/ASN.2020071082
86. Ravindran A, Casal Moura M, Fervenza FC, Nasr SH, Alexander MP, Fidler ME, et al. In patients with membranous lupus nephritis, exostosin-positivity and exostosin-negativity represent two different phenotypes. J Am Soc Nephrol (2021) 32(3):695–706. doi: 10.1681/ASN.2020081181
87. Xie Q, Li Y, Xue J, Xiong Z, Wang L, Sun Z, et al. Renal phospholipase A2 receptor in hepatitis b virus-associated membranous nephropathy. Am J Nephrol (2015) 41(4-5):345–53. doi: 10.1159/000431331
88. Hoxha E, Wiech T, Stahl PR, Zahner G, Tomas NM, Meyer-Schwesinger C, et al. A mechanism for cancer-associated membranous nephropathy. N Engl J Med (2016) 374(20):1995–6. doi: 10.1056/NEJMc1511702
89. Hoxha E, Beck LH Jr., Wiech T, Tomas NM, Probst C, Mindorf S, et al. An indirect immunofluorescence method facilitates detection of thrombospondin type 1 domain-containing 7A-specific antibodies in membranous nephropathy. J Am Soc Nephrol (2017) 28(2):520–31. doi: 10.1681/ASN.2016010050
90. Caza TN, Hassen SI, Dvanajscak Z, Kuperman M, Edmondson R, Herzog C, et al. NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int (2021) 99(4):967–76. doi: 10.1016/j.kint.2020.07.039
91. Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet (2004) 364(9441):1252–9. doi: 10.1016/S0140-6736(04)17142-0
92. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol (2010) 21(3):507–19. doi: 10.1681/ASN.2008121259
93. Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, et al. Coexistence of different circulating anti-podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol (2012) 7(9):1394–400. doi: 10.2215/CJN.02170312
94. Bruschi M, Cavalli A, Moll S, Candiano G, Scapozza L, Patel JJ, et al. Discovery of anti-formin-like 1 protein (FMNL1) antibodies in membranous nephropathy and other glomerular diseases. Sci Rep (2022) 12(1):13659. doi: 10.1038/s41598-022-17696-w
95. Quiza CG, Leenaerts PL, Hall BM. The role of T cells in the mediation of glomerular injury in heymann’s nephritis in the rat. Int Immunol (1992) 4(4):423–32. doi: 10.1093/intimm/4.4.423
96. Penny MJ, Boyd RA, Hall BM. Role of T cells in the mediation of heymann nephritis. ii. identification of Th1 and cytotoxic cells in glomeruli. Kidney Int (1997) 51(4):1059–68. doi: 10.1038/ki.1997.148
97. Spicer ST, Tran GT, Killingsworth MC, Carter N, Power DA, Paizis K, et al. Induction of passive heymann nephritis in complement component 6-deficient PVG rats. J Immunol (2007) 179(1):172–8. doi: 10.4049/jimmunol.179.1.172
98. Wang YM, Zhang GY, Hu M, Polhill T, Sawyer A, Zhou JJ, et al. CD8+ regulatory T cells induced by T cell vaccination protect against autoimmune nephritis. J Am Soc Nephrol (2012) 23(6):1058. doi: 10.1681/ASN.2011090914
99. de Heer E, Daha MR, Burgers J, van Es LA. Reestablishment of self tolerance by suppressor T-cells after active heymann’s nephritis. Cell Immunol (1986) 98(1):28–33. doi: 10.1016/0008-8749(86)90264-9
100. de Heer E, Daha MR, van Es LA. The autoimmune response in active heymann’s nephritis in Lewis rats is regulated by T-lymphocyte subsets. Cell Immunol (1985) 92(2):254–64. doi: 10.1016/0008-8749(85)90007-3
101. Cantarelli C, Jarque M, Angeletti A, Manrique J, Hartzell S, O'Donnell T, et al. A comprehensive phenotypic and functional immune analysis unravels circulating anti-phospholipase A2 receptor antibody secreting cells in membranous nephropathy patients. Kidney Int Rep (2020) 5(10):1764–76. doi: 10.1016/j.ekir.2020.07.028
102. Jaber S, Goehrig D, Bertolino P, Massemin A, Bihl F, Chabry J, et al. Generation of a conditional transgenic mouse model expressing human phospholipase A2 receptor 1. Sci Rep (2020) 10(1):8190. doi: 10.1038/s41598-020-64863-y
103. Kuroki AKI, Iyoda M, Shibata T, Sugisaki T. Th2 cytokines increase and stimulate b cells to produce IgG4 in idiopathic membranous nephropathy. Kidney Int (2005) 68(1):302–10. doi: 10.1111/j.1523-1755.2005.00415.x
104. Rosenzwajg M, Languille E, Debiec H, Hygino J, Dahan K, Simon T, et al. B- and T-cell subpopulations in patients with severe idiopathic membranous nephropathy may predict an early response to rituximab. Kidney Int (2017) 92(1):227–37. doi: 10.1016/j.kint.2017.01.012
105. Masutani K, Taniguchi M, Nakashima H, Yotsueda H, Kudoh Y, Tsuruya K, et al. Up-regulated interleukin-4 production by peripheral T-helper cells in idiopathic membranous nephropathy. Nephrol Dial Transpl (2004) 19(3):580–6. doi: 10.1093/ndt/gfg572
106. Shi X, Qu Z, Zhang L, Zhang N, Liu Y, Li M, et al. Increased ratio of ICOS+/PD-1+ follicular helper T cells positively correlates with the development of human idiopathic membranous nephropathy. Clin Exp Pharmacol Physiol (2016) 43(4):410–6. doi: 10.1111/1440-1681.12555
107. Zhang Z, Shi Y, Yang K, Crew R, Wang H, Jiang Y. Higher frequencies of circulating ICOS+, IL-21+ T follicular helper cells and plasma cells in patients with new-onset membranous nephropathy. Autoimmunity (2017) 50(8):458–67. doi: 10.1080/08916934.2017.1385775
108. Hirayama K, Ebihara I, Yamamoto S, Kai H, Muro K, Yamagata K, et al. Predominance of type-2 immune response in idiopathic membranous nephropathy. cytoplasmic cytokine analysis. Nephron (2002) 91(2):255–61. doi: 10.1159/000058401
109. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature (2017) 545(7653):243–7. doi: 10.1038/nature22329
110. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199(7):971–9. doi: 10.1084/jem.20031579
111. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med (2004) 200(3):277–85. doi: 10.1084/jem.20040165
112. Crispin JC, Martínez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun (2003) 21(3):273–6. doi: 10.1016/S0896-8411(03)00121-5
113. Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest (2002) 109(1):131–40. doi: 10.1172/JCI0213605
114. Cattran DC, Brenchley PE. Membranous nephropathy: integrating basic science into improved clinical management. Kidney Int (2017) 91(3):566–74. doi: 10.1016/j.kint.2016.09.048
115. Cremoni M, Brglez V, Perez S, Decoupigny F, Zorzi K, Andreani M, et al. Th17-immune response in patients with membranous nephropathy is associated with thrombosis and relapses. Front Immunol (2020) 11:574997. doi: 10.3389/fimmu.2020.574997
116. Dan T, JiGuang G ZJ. Variation of peripheral Th17/Treg imbalance in patients with idiopathic membranous nephropathy after cyclosporin a treatment: A prognostic marker of idiopathic membranous nephropathy. BioMed J Sci Tech Res (2018) 7(5). doi: 10.26717/BJSTR.2018.07.001579
117. Xu J, Shen C, Lin W, Meng T, Ooi JD, Eggenhuizen PJ, et al. Single-cell profiling reveals transcriptional signatures and cell-cell crosstalk in anti-PLA2R positive idiopathic membranous nephropathy patients. Front Immunol (2021) 12:683330. doi: 10.3389/fimmu.2021.683330
118. Sealfon R, Mariani L, Avila-Casado C, Nair V, Menon R, Funk J, et al. Molecular characterization of membranous nephropathy. J Am Soc Nephrol (2022) 33(6):1208–21. doi: 10.1681/ASN.2021060784
119. Lee SM, Yang S, Cha RH, Kim M, An JN, Paik JH, et al. Circulating TNF receptors are significant prognostic biomarkers for idiopathic membranous nephropathy. PloS One (2014) 9(8):e104354. doi: 10.1371/journal.pone.0104354
120. Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med (1998) 339(20):1448–56. doi: 10.1056/NEJM199811123392007
121. Huang YS, Fu SH, Lu KC, Chen JS, Hsieh HY, Sytwu HK, et al. Inhibition of tumor necrosis factor signaling attenuates renal immune cell infiltration in experimental membranous nephropathy. Oncotarget (2017) 8(67):111631–41. doi: 10.18632/oncotarget.22881
122. Wang B, Zuo K, Wu Y, Huang Q, Qin WS, Zeng CH, et al. Correlation between b lymphocyte abnormality and disease activity in patients with idiopathic membranous nephropathy. J Int Med Res (2011) 39(1):86–95. doi: 10.1177/147323001103900111
123. Olalekan SA, Cao Y, Hamel KM, Finnegan A. B cells expressing IFN-γ suppress treg-cell differentiation and promote autoimmune experimental arthritis. Eur J Immunol (2015) 45(4):988–98. doi: 10.1002/eji.201445036
124. Roccatello D, Sciascia S, Di Simone D, Solfietti L, Naretto C, Fenoglio R, et al. New insights into immune mechanisms underlying response to rituximab in patients with membranous nephropathy: A prospective study and a review of the literature. Autoimmun Rev (2016) 15(6):529–38. doi: 10.1016/j.autrev.2016.02.014
125. Ramachandran R, Kaundal U, Girimaji N, Rakha A, Rathi M, Gupta KL, et al. Regulatory b cells are reduced and correlate with disease activity in primary membranous nephropathy. Kidney Int Rep (2020) 5(6):872–8. doi: 10.1016/j.ekir.2020.03.023
126. Dong Z, Liu Z, Dai H, Liu W, Feng Z, Zhao Q, et al. The potential role of regulatory b cells in idiopathic membranous nephropathy. J Immunol Res (2020) 2020:7638365. doi: 10.1155/2020/7638365
127. Zhang Y, Jin Y, Guan Z, Li H, Su Z, Xie C, et al. The landscape and prognosis potential of the T-cell repertoire in membranous nephropathy. Front Immunol (2020) 11:387. doi: 10.3389/fimmu.2020.00387
128. Su Z, Jin Y, Zhang Y, Guan Z, Li H, Chen X, et al. The diagnostic and prognostic potential of the b-cell repertoire in membranous nephropathy. Front Immunol (2021) 12:635326. doi: 10.3389/fimmu.2021.635326
129. Kamata K, Baird L, Erikson M, Collins A, McCluskey R. Characterization of antigens and antibody specificities involved in heymann nephritis. J Immunol (1985) 135(4):2400–8.
130. Schiller B, He C, Salant DJ, Lim A, Alexander JJ, Quigg RJ. Inhibition of complement regulation is key to the pathogenesis of active heymann nephritis. J Exp Med (1998) 188(7):1353–8. doi: 10.1084/jem.188.7.1353
131. Moll S, Miot S, Sadallah S, Gudat F, Mihatsch MJ, Schifferli JA. No complement receptor 1 stumps on podocytes in human glomerulopathies. Kidney Int (2001) 59(1):160–8. doi: 10.1046/j.1523-1755.2001.00476.x
132. Ravindran A, Madden B, Charlesworth MC, Sharma R, Sethi A, Debiec H, et al. Proteomic analysis of complement proteins in membranous nephropathy. Kidney Int Rep (2020) 5(5):618–26. doi: 10.1016/j.ekir.2020.01.018
133. Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest (1980) 66(6):1339–50. doi: 10.1172/JCI109987
134. Ren G, Doshi M, Hack BK, Alexander JJ, Quigg RJ. Rat glomerular epithelial cells produce and bear factor h on their surface that is up-regulated under complement attack. Kidney Int (2003) 64(3):914–22. doi: 10.1046/j.1523-1755.2003.00188.x
135. Gao S, Cui Z, Zhao MH. Complement C3a and C3a receptor activation mediates podocyte injuries in the mechanism of primary membranous nephropathy. J Am Soc Nephrol (2022) 33(9):1742–56. doi: 10.1681/ASN.2021101384
136. Leenaerts PL, Hall BM, Van Damme BJ, Daha MR, Vanrenterghem YF. Active heymann nephritis in complement component C6 deficient rats. Kidney Int (1995) 47(6):1604–14. doi: 10.1038/ki.1995.224
137. Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol (1989) 135(1):185–94.
138. Cybulsky AV, Quigg RJ, Salant DJ. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol (2005) 289(4):F660–671. doi: 10.1152/ajprenal.00437.2004
139. Huang CC, Lehman A, Albawardi A, Satoskar A, Brodsky S, Nadasdy G, et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol (2013) 26(6):799–805. doi: 10.1038/modpathol.2012.237
140. Hoxha E, Kneißler U, Stege G, Zahner G, Thiele I, Panzer U, et al. Enhanced expression of the m-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int (2012) 82(7):797–804. doi: 10.1038/ki.2012.209
141. Ma H, Sandor DG, Beck LH Jr. The role of complement in membranous nephropathy. Semin Nephrol (2013) 33(6):531–42. doi: 10.1016/j.semnephrol.2013.08.004
142. Zhang Y, Liu Y, Liang L, Liu L, Tang X, Tang L, et al. Effect of glomerular mannose-binding lectin deposition on the prognosis of idiopathic membranous nephropathy. Kidney Blood Press Res (2020) 45(5):713–26. doi: 10.1159/000508665
143. Hayashi N, Okada K, Matsui Y, Fujimoto K, Adachi H, Yamaya H, et al. Glomerular mannose-binding lectin deposition in intrinsic antigen-related membranous nephropathy. Nephrol Dial Transpl (2017) 33(5):832–40. doi: 10.1093/ndt/gfx235
144. Bally S, Debiec H, Ponard D, Dijoud F, Rendu J, Fauré J, et al. Phospholipase A2 receptor–related membranous nephropathy and mannan-binding lectin deficiency. J Am Soc Nephrol (2016) 27(12):3539. doi: 10.1681/ASN.2015101155
145. Sethi A, Miao J, Willrich MAV, Frinack JL, Cattran DC, Fervenza FC, et al. Limited significance of antifactor h antibodies in patients with membranous nephropathy. Clin J Am Soc Nephrol (2021) 16(6):939–41. doi: 10.2215/CJN.16631020
146. Luo W, Olaru F, Miner JH, Beck LH, van der Vlag J, Thurman JM, et al. Alternative pathway is essential for glomerular complement activation and proteinuria in a mouse model of membranous nephropathy. Front Immunol (2018) 9(1433). doi: 10.3389/fimmu.2018.01433
147. Raats C, Luca M, Bakker M, Van der Wal A, Heeringa P, Van Goor H, et al. Reduction in glomerular heparan sulfate correlates with complement deposition and albuminuria in active heymann nephritis. J Am Soc Nephrol (1999) 10(8):1689–99. doi: 10.1681/ASN.V1081689
148. Van Den Born J, van den Heuvel LP, Bakker MA, Veerkamp JH, Assmann KJ, Weening JJ, et al. Distribution of GBM heparan sulfate proteoglycan core protein and side chains in human glomerular diseases. Kidney Int (1993) 43(2):454–63. doi: 10.1038/ki.1993.67
149. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor h is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol (2013) 190(5):2049–57. doi: 10.4049/jimmunol.1201751
150. Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem (2008) 283(35):23731–8. doi: 10.1074/jbc.M801918200
151. Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 120(4):1084–96. doi: 10.1172/JCI39492
152. Meyer-Schwesinger C, Meyer T, Münster S, Klug P, Saleem M, Helmchen U, et al. A new role for the neuronal ubiquitin c-terminal hydrolase-L1 (UCH-L1) in podocyte process formation and podocyte injury in human glomerulopathies. J Pathol (2009) 217(3):452–64. doi: 10.1002/path.2446
153. Yang L, Wu Y, Lin S, Dai B, Chen H, Tao X, et al. sPLA2-IB and PLA2R mediate insufficient autophagy and contribute to podocyte injury in idiopathic membranous nephropathy by activation of the p38MAPK/mTOR/ULK1(ser757) signaling pathway. FASEB J (2021) 35(2):e21170. doi: 10.1096/fj.202001143R
154. Jo HA, Hyeon JS, Yang SH, Jung Y, Ha H, Jeong CW, et al. Fumarate modulates phospholipase A2 receptor autoimmunity-induced podocyte injury in membranous nephropathy. Kidney Int (2021) 99(2):443–55. doi: 10.1016/j.kint.2020.06.031
155. You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol (2016) 27(2):466–81. doi: 10.1681/ASN.2015030302
156. Škoberne A, Behnert A, Teng B, Fritzler MJ, Schiffer L, Pajek J, et al. Serum with phospholipase A2 receptor autoantibodies interferes with podocyte adhesion to collagen. Eur J Clin Invest (2014) 44(8):753–65. doi: 10.1111/eci.12292
157. Podestà MA, Ruggiero B, Remuzzi G, Ruggenenti P. Ofatumumab for multirelapsing membranous nephropathy complicated by rituximab-induced serum-sickness. BMJ Case Rep (2020) 13(1):e232896. doi: 10.1136/bcr-2019-232896
158. Boyer-Suavet S, Andreani M, Lateb M, Savenkoff B, Brglez V, Benzaken S, et al. Neutralizing anti-rituximab antibodies and relapse in membranous nephropathy treated with rituximab. Front Immunol (2019) 10:3069. doi: 10.3389/fimmu.2019.03069
159. Tobinai K, Klein C, Oya N, Fingerle-Rowson G. A review of obinutuzumab (GA101), a novel type II anti-CD20 monoclonal antibody, for the treatment of patients with b-cell malignancies. Adv Ther (2017) 34(2):324–56. doi: 10.1007/s12325-016-0451-1
160. Amoura Z, Remy P, Quintana Porras L, Chiche L, Chauveau D, Roccatello D, et al. Alternative renal response definitions in a randomized, controlled trial of obinutuzumab for proliferative lupus nephritis [abstract]. Arthritis Rheumatol (2020) 72(suppl 10):104. doi: 10.1136/annrheumdis-2020-eular.2983
161. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med (2020) 383(12):1117–28. doi: 10.1056/NEJMoa2001180
162. Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N Engl J Med (2021) 385(6):567–9. doi: 10.1056/NEJMc2107725
163. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med (2022) 28:2124–32. doi: 10.3410/f.742323959.793595617
164. Geara AS, Bhoj V, Hogan JJ. Bortezomib treatment for refractory PLA2R-positive membranous nephropathy. Glomerular Dis (2021) 1(1):40–3. doi: 10.1159/000515087
165. Hartono C, Chung M, Kuo SF, Seshan SV, Muthukumar T. Bortezomib therapy for nephrotic syndrome due to idiopathic membranous nephropathy. J Nephrol (2014) 27(1):103–6. doi: 10.1007/s40620-013-0028-x
166. Kizilbash S, Claes D, Ashoor I, Chen A, Jandeska S, Matar RB, et al. Bortezomib in the treatment of antibody-mediated rejection in pediatric kidney transplant recipients: A multicenter Midwest pediatric nephrology consortium study. Pediatr Transpl (2017) 21(3):e12873. doi: 10.1111/petr.12873
167. Jain D, Rajab A, Young JS, Yin D, Nadasdy T, Chong AS, et al. Reversing donor-specific antibody responses and antibody-mediated rejection with bortezomib and belatacept in mice and kidney transplant recipients. Am J Transpl (2020) 20(10):2675–85. doi: 10.1111/ajt.15881
168. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353(6295):179–84. doi: 10.1126/science.aaf6756
169. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
170. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi: 10.1186/1742-2094-9-112
171. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology (2008) 134(7):2014–24. doi: 10.1053/j.gastro.2008.02.060
172. Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med (2002) 196(2):255–60. doi: 10.1084/jem.20020394
173. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med (2008) 205(9):1975–81. doi: 10.1084/jem.20080308
174. Ottolenghi A, Bolel P, Sarkar R, Greenshpan Y, Iraqi M, Ghosh S, et al. Life-extended glycosylated IL-2 promotes treg induction and suppression of autoimmunity. Sci Rep (2021) 11(1):7676. doi: 10.1038/s41598-021-87102-4
175. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature (2019) 572(7770):481–7. doi: 10.1038/s41586-019-1467-x
176. Gonzalez-Figueroa P, Roco JA, Papa I, Núñez Villacís L, Stanley M, Linterman MA, et al. Follicular regulatory T cells produce neuritin to regulate b cells. Cell (2021) 184(7):1775–1789.e1719. doi: 10.1016/j.cell.2021.02.027
177. Rinschen MM, Gödel M, Grahammer F, Zschiedrich S, Helmstädter M, Kretz O, et al. A multi-layered quantitative In vivo expression atlas of the podocyte unravels kidney disease candidate genes. Cell Rep (2018) 23(8):2495–508. doi: 10.1016/j.celrep.2018.04.059
178. Sano K, Tanihara H, Heimark RL, Obata S, Davidson M, St John T, et al. Protocadherins: a large family of cadherin-related molecules in central nervous system. EMBO J (1993) 12(6):2249–56. doi: 10.1002/j.1460-2075.1993.tb05878.x
Keywords: membranous nephropathy, Heymann nephritis, autoantibody, immunology, podocyte, treatment
Citation: Chung EYM, Wang YM, Keung K, Hu M, McCarthy H, Wong G, Kairaitis L, Bose B, Harris DCH and Alexander SI (2022) Membranous nephropathy: Clearer pathology and mechanisms identify potential strategies for treatment. Front. Immunol. 13:1036249. doi: 10.3389/fimmu.2022.1036249
Received: 04 September 2022; Accepted: 19 October 2022;
Published: 02 November 2022.
Edited by:
Miroslav Sekulic, Columbia University, United StatesReviewed by:
Linda Reinhard, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2022 Chung, Wang, Keung, Hu, McCarthy, Wong, Kairaitis, Bose, Harris and Alexander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edmund Y. M. Chung, ZWRtdW5kLmNodW5nQGhlYWx0aC5uc3cuZ292LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.